

Abstract
Intracellular pathogens pose a significant threat to animals. In defense, innate immune sensors attempt to detect these pathogens using pattern recognition receptors that either directly detect microbial molecules or indirectly detect their pathogenic activity. These sensors trigger different forms of regulated cell death, including pyroptosis, apoptosis, and necroptosis, which eliminate the infected host cell niche while simultaneously promoting beneficial immune responses. These defenses force intracellular pathogens to evolve strategies to minimize or completely evade the sensors. In this review, we discuss recent advances in our understanding of the cytosolic pattern recognition receptors that drive cell death, including NLRP1, NLRP3, NLRP6, NLRP9, NLRC4, AIM2, IFI16, and ZBP1.
Keywords: innate immunity, pattern recognition receoptors, pyroptosis, necroptosis, inflammasome, caspase
INTRODUCTION
Many pathogens attempt to hide from extracellular defenses by replicating inside host cells. Such intracellular pathogens replicate either in the vacuolar compartment or in the cytosolic/nuclear compartment. To counteract intracellular pathogens, host cells have a strategy to trigger regulated cell death (RCD) pathways, including apoptosis, pyroptosis, and necroptosis. Killing the infected cell removes the replicative niche of the pathogen, allowing the host immune system to clear the pathogen before it can enter and successfully commandeer the next cell. However, the host–pathogen interface is constantly evolving; if the host evolves an RCD program that eliminates intracellular infection, pathogens are now under pressure to evolve evasion strategies. This never-ending “Red Queen’s race” (1) results in increasingly complex host–pathogen interactions, making it challenging to know which side is “winning” at any particular point.
A complex array of cytosolic sensors monitors cells for infection and can trigger RCD (Table 1). Pattern recognition receptors (PRRs) bind directly to pathogenic molecules or sense the direct consequences of pathogen virulence properties. Here, we discuss PRRs that sense intracellular pathogens and induce RCD. Numerous inflammasome pathways trigger pyroptosis, and a single cytosolic PRR is known to trigger necroptosis. Other pathways that are not cytosolic or not PRRs, including tumor necrosis factor/Toll-like receptor (TNF/TLR)-driven and BH3 family–driven RCD, are not covered here.
Table 1.
Regulated cell death sensors and their downstream signaling partners.
| Gene | Priming steps | Activator | Direct outcome | Directly signals to | |||
|---|---|---|---|---|---|---|---|
| Human | Mouse | Transcriptional regulation | Posttranslational regulation | ||||
| Activation | Inhibition | ||||||
| Inflammasomes | |||||||
| NLRP1 | Nlrp1a, Nlrp1b | ATF4 | Ubiquitination, FIIND cleavage | DPP9 | Functional degradation, MDP, dsRNA, VbP | Oligomerization | ASC, caspase-1 |
| CARD8 | Absent | ANRIL | ND | DPP9 | VbP | Oligomerization | ASC, caspase-1 |
| NLRP3 | Nlrp3 | NF-κB | Dephosphorylation, phosphorylation, deubiquitination, others | Phosphorylation, dephosphorylation, ubiquitination, nitrosylation, itaconation, others | NEK7 licensing, nigericin, pores, dsRNA via DHX33, many more | Oligomerization | ASC |
| NLRC4 | Nlrc4 | IRF8, PU.1, and Brd4 | Phosphorylation | Dephosphorylation | NAIP | Oligomerization | ASC, caspase-1 |
| NLRP6 | Nlrp6 | TNF | Ubiquitination | Deubiquitination | Taurine, LTA, dsRNA via DHX15 | Liquid-liquid phase separation | ASC |
| NLRP9 | Nlrp9a, Nlrp9b, Nlrp9c | Reproductive organs, lower GI tract | ND | ND | dsRNA via DHX9 | Oligomerization | ASC |
| NAIP | Naip1, Naip2, Naip5, Naip6 | IRF8, PU.1, and Brd4; Naip2 by IFN | ND | ND | T3SS rod, T3SS needle, flagellin | Confirmational Change | NLRC4 |
| AIM2 | Aim2 | Constitutive, upregulated by IFNs | ND | p202, POP3 | dsDNA | Oligomerization | ASC |
| IFI16 | Ifi16 | IFNs | ND | Ubiquitination | dsDNA | Oligomerization | ASC |
| MEFV (Pyrin) | Mefv (Pyrin) | IFNs, NF-κB | ND | Phosphorylation | Inactivation of Rho GTPase | Oligomerization | ASC |
| Adaptor | |||||||
| ASC | Asc | Constitutive | Phosphorylation, ubiquitination | Dephosphorylation, deubquitination, POP1, POP2 | NLRs, ALRs, Pyrin | Polymerization | Caspase-1 (caspase-8) |
| Caspases | |||||||
| CASP1 | Casp1 | Constitutive | Phosphorylation | Ubiquitination, nitrosylation, CARD16, CARD17, CARD18 | ASC, NLRP1, NLRC4 | Polymerization, protease activation | GSDMD, IL-1β, IL-18 (BID) |
| CASP4, CASP5 | Casp11 | Casp11 IFN inducible, CASP4 constitutive | Indirectly enhanced by GBPs/IRGs | ND | LPS | Polymerization, protease activation | GSDMD |
| Gasdermins | |||||||
| GSDMA | Gsdma1, Gsdma2, Gsdma3 | Keratinocytes, upper GI cells | ND | Phosphorylation | ND | Oligomerization, pore formation | NA |
| GSDMB | Absent | Lung, GI cells | ND | ND | Granzyme A | pore formation | NA |
| GSDMC | Gsdmc1, Gsdmc2, Gsdmc3, Gsdmc4 | GI cells, TGF inducible | ND | ND | Caspase-8 | ND | NA |
| GSDMD | Gsdmd | Constitutive, upregulated by IFNs | ND | Caspase-3 | Caspase-1, caspase-11 (caspase-8) | Oligomerization, pore formation | NA |
| GSDME | Gsdme | Specific cell types or inducible | ND | Phosphorylation | Caspase-3, granzyme B | Oligomerization, pore formation | NA |
| Cytokines | |||||||
| IL1B | Il1b | Constitutive in some cells, induced by NF-κB in others | Deubiquitination | Ubiquitination | Caspase-1 | NA | IL-1R |
| IL18 | Il18 | Constitutive and inducible | ND | ND | Caspase-1 | NA | IL-18R |
| Necroptosis inducer | |||||||
| ZBP1 | Zbp1 | IFNs | Ubiquitination | Deubiquitination | Z-RNA, Z-DNA | ND | RIPK3 |
Abbreviations: ALR, AIM2-like receptor; ANRIL, antisense non coding RNA in the INK4 locus; ASC, apoptosis-associated speck-like protein; ATF4, activating transcription factor 4; CARD, caspase activation and recruitment domain; dsDNA, double-stranded DNA; GBP, guanylate-binding protein; GI, gastrointestinal; GSDM, gasdermin; IFN, interferon; IRF8, IFN regulatory factor 8; IRG, immunity-related guanosine triphosphatase; LPS, lipopolysaccharide; LTA, lipoteichoic acid; NA, not applicable; ND, not determined; NF-κB, nuclear factor κB; NLR, NOD-like receptor; POP, PYD-only protein; TGF, transforming growth factor; TNF, tumor necrosis factor; T3SS, type III secretion system; VbP, Val-boroPro.
Parentheses In the two rightmost columns are used for slower output pathways that function as backups when primary pathways are absent.
PYROPTOSIS
Inflammasomes are multiprotein complexes containing sensor proteins, which detect virulence traits of pathogens either directly, by sensing pathogen-associated molecular patterns (PAMPs), or indirectly, by sensing virulence factor activities (2). Inflammasomes all cause proinflammatory cytokine release and cell death. Inflammasomes have different domain architecture, but all contain either a pyrin domain (PYD) or a caspase activation and recruitment domain (CARD) (3).
CARD-containing inflammasomes directly activate caspase-1 via CARD-CARD interactions and are amplified by the apoptosis-associated speck-like protein (ASC) adapter, a PYD-CARD protein. In contrast, PYD-containing inflammasomes require ASC to signal, recruiting it via PYD-PYD interactions that trigger ASC polymerization (forming ASC specks), which serve as a hub for caspase-1 activation (4, 5). This activated caspase-1 cleaves the linker of the pore-forming protein gasdermin D (GSDMD), which liberates the N-terminal pore-forming domain, which form pores in the plasma membrane (6–9). These pores trigger cell surface Ninjurin1 oligomerization and subsequent plasma membrane rupture (i.e., pyroptosis) (9a). At the same time, caspase-1 cleaves the proinflammatory cytokines IL-1β and IL-18 to their active forms, which escape the cytosol through the GSDMD pore or by other means (10, 11). Indeed, the charge properties of the GSDMD pore preferentially attract the cleaved forms of the cytokines for release (12). Inflammasome activation also triggers the production of eicosanoids and releases intracellular damage-associated molecular pattern (DAMPs), which drive further inflammation (13). The highly inflammatory nature of pyroptosis can clear pathogens, but it can also lead to immunopathology (14, 15).
NLRP1
Many Nod-like receptors (NLRs) form inflammasomes. The mechanism of activation of the first inflammasome, NLRP1 (16), was recently elucidated.
Activation
NLRP1 diverges from the direct PAMP-binding paradigm set by the TLR family. Instead, NLRP1 monitors for virulence factor activity. This detection is enabled by a domain architecture not present in other NLRs. NLRP1 proteins have the typical NACHT and leucine-rich repeat (LRR) domains, but these are followed by a function to find domain (FIIND) and a CARD that are key to its function (17, 18) (Figure 1). Additionally, the human NLRP1 paralog contains an N-terminal PYD; however, unlike other NLRPs, this PYD is not essential.
Figure 1.
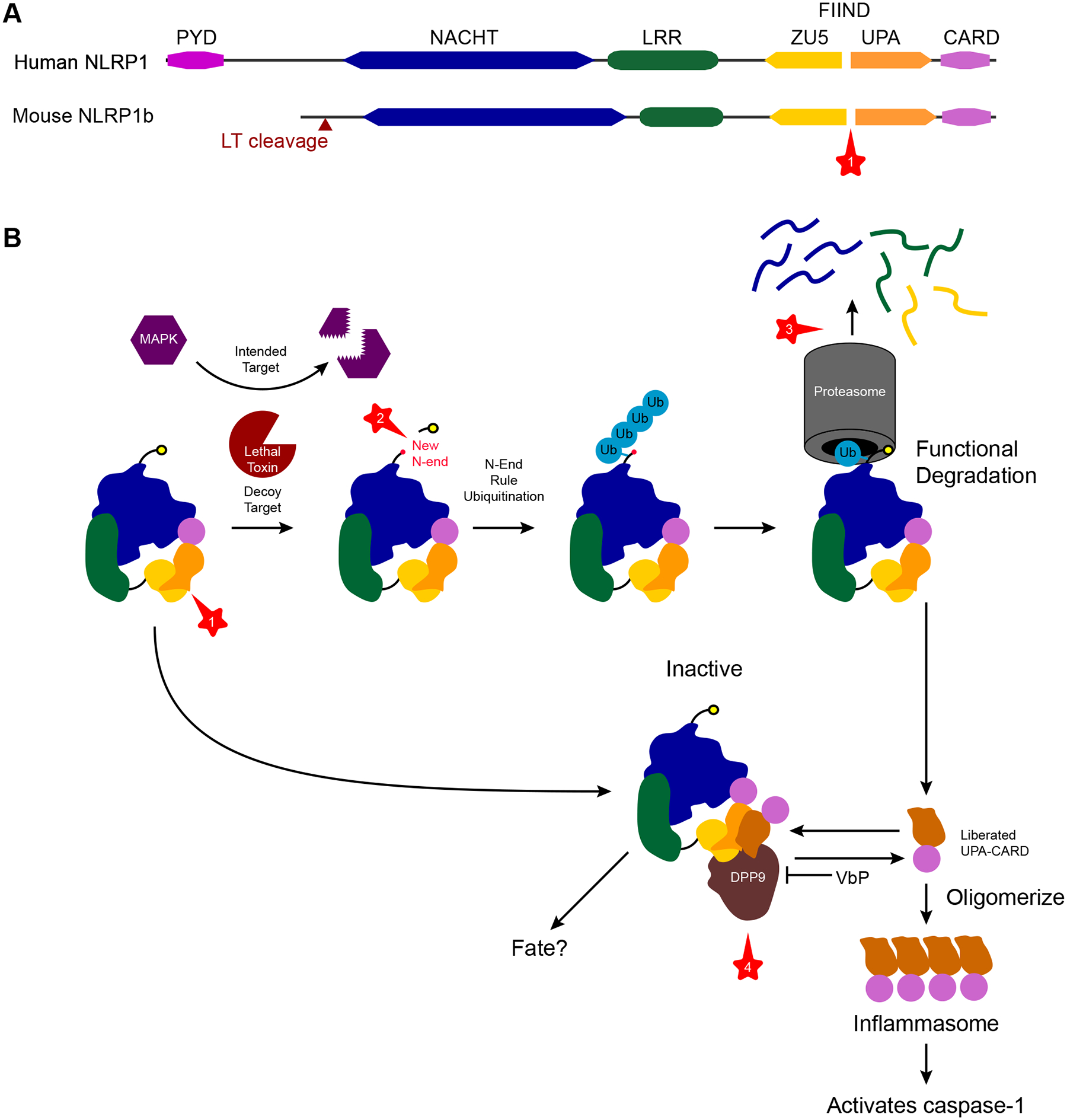
NLRP1 regulation by a series of proteolytic events. NLRP1 is regulated and activated by a series of protease steps (①–④). (a) NLRP1 is composed of NACHT and LRR domains, which are typical of NLR proteins, but it also has a unique FIIND and CARD at its C terminus. Additionally, human NLRP1 has a PYD that is not essential for its function. The FIIND exerts autoproteolytic activity and cleaves itself in the middle (①), generating a ZU5 and an UPA fragment. (b) Bacillus anthracis uses the pathogen protease LT to cleave MAP kinases. However, murine NLRP1b has a decoy sequence that lures LT to cleave NLRP1b in its N terminus (②). This process results in a novel nonmethionine N terminus that is detected by the N-end rule ubiquitinases, which target the protein to the proteasome, leading to functional degradation of NLRP1b (③). The proteasome degrades the NACHT, LRR, and ZU5 domains, but, because of the lack of a peptide bond between ZU5 and UPA, the UPA-CARD C-terminal fragment escapes the proteasome. This UPA-CARD is now free to oligomerize into an inflammasome that activates caspase-1. However, if the UPA-CARD is not generated in sufficient quantities, its activity is quenched. The dipeptidyl protease DPP9 normally cleaves proteins with specificity for cleaving after any amino acid followed by a proline at the N terminus of a protein. DPP9 organizes a ternary complex among itself, an intact NLRP1, and an UPA-CARD. DPP9 binds the intact NLRP1 by its ZU5 and UPA domains, and the UPA-CARD by its UPA domain, preventing the UPA-CARD from forming an inflammasome. Although the protease activity of DPP9 is essential for this inhibition (④), whether DPP9 cleaves the UPA-CARD is unknown. Abbreviations: CARD, caspase activation and recruitment domain; DPP, dipeptyl protease; FFIND, function to find domain; LRR, leucine-rich repeat; LT, lethal toxin; NLR, Nod-like receptor; PYD, pyrin domain; VbP, Val-boroPro.
NLRP1 sensing involves the activity of multiple different proteolytic steps: autoprocessing, pathogen proteases, proteasomal functional degradation, and dipeptyl protease (DPP) regulation. The first protease is the FIIND, which cleaves itself (autoprocessing), generating two fragments: the ZU5, which remains covalently tied to the NACHT-LRR, and the UPA, which remains covalently tied to the CARD (19). Autoprocessing occurs constitutively but not with immediate efficiency; whether this step is regulated remains unclear. Importantly, the UPA-CARD fragment remains noncovalently associated with NACHT-LRR-ZU5 (17–19). Once cleaved, NLRP1 is primed and ready to sense virulence factors in the cytosol.
The nonobligatory pathogen protease step can be confusing, as some NLRP1 activators skip this step. However, because the protease lethal toxin (LT) was key to the discovery of the NLRP1 activation mechanism, we describe it first (20). LT delivers a protease toxin (called lethal factor) that cleaves and inactivates host MAP kinases (21). However, NLRP1b has evolved what appears to be a lure for this activity; thus, LT also cleaves NLRP1b. This process generates a novel N terminus of NLRP1b, which starts the activation cascade.
NLRP1 activation uniquely requires proteasomal degradation (22, 23). In the case of LT, the novel N terminus triggers ubiquitination by the cellular N-end rule pathway, which is one of the normal regulatory systems for protein homeostasis (24–28). Normally, proteins begin with a methionine; the N-end rule recognizes other N-terminal residues as a marker of abnormality, and ubiquitinates the protein to instigate proteasomal degradation (25). As N-terminal NLRP1 is fed into the proteasome, the prior FIIND autoprocessing permits the C-terminal fragment to escape. This liberated UPA-CARD becomes an inflammasome that activates caspase-1 (26–28). Thus, functional degradation occurs when most of the NLRP1 protein is degraded by the proteasome; meanwhile, the escaping UPA-CARD is the functional inflammasome (26) (Figure 1). Another example of this functional degradation is found in several picornavirus 3C proteases that cleave NLRP1 (29, 30).
NLRP1 can also detect nonprotease virulence factors. The Shigella type III secretion system (T3SS) translocated effector IpaH7.8 is a ubiquitin ligase that directly ubiquitinates NLRP1b, again resulting in functional degradation (26). Therefore, NLRP1 can sense virulence factors that have diverse enzymatic activities (proteases or ubiquitinases), which converge upon functional degradation. The common theme is that NLRP1 appears to act as a decoy that evolved to be targeted by virulence factors, a concept first established in plant NLR-like sensors (31).
NLRP1 was also recently shown to respond to double-stranded RNA (dsRNA), again following the functional degradation pathway (32). The structural basis underlying dsRNA recognition and how it causes functional degradation remain to be described.
NLRP1 is also regulated by the endogenous protease DPP. The DPP inhibitor Val-boroPro triggers NLRP1 activation (33, 34). DPPs cleave N-terminal dipeptides and generate a novel N terminus of their substrates (35), and inhibition of DPP8/9 causes NLRP1 activation (34, 36). The critical role of DPP in preventing aberrant NLRP1 activation is illustrated by recent structural studies that revealed a ternary complex among DPP9, a FIIND of an intact NLRP1, and a UPA of a second functionally degraded NLRP1 (37, 38). It is not known why DPP9 evolved to dock with two NLRP1 proteins in order to inhibit inflammasome formation, or why the DPP9 enzymatic function is essential (36).
Pathogens detected by NLRP1
Bacillus anthracis is a gram-positive bacterium that causes anthrax in livestock and, occasionally, humans. Bacterial spores sense the macrophage phagosome, triggering germination and intracellular growth. However, after this first round of intracellular replication, B. anthracis replicates primarily in the extracellular space. B. anthracis secretes toxins, including LT, to support vacuolar survival (39). Only some murine Nlrp1b alleles are able to detect LT (129S, BALB/c); other Nlrp1b alleles cannot (C57BL/6J) (20). Successful LT detection by Nlrp1b makes mice resistant to in vivo infection (40). Whether NLRP1b is important for closing an intracellular niche by inducing pyroptosis, or for attacking an extracellular niche, remains to be determined. In this regard, IL-1β could be useful against both niches by attracting neutrophils to attack either extracellular bacteria or bacteria trapped within the remnants of pyroptotic macrophage pore-induced intracellular traps (PITs) (41).
Viruses are obligate intracellular cytosolic pathogens, and thus pyroptosis could close the intracellular niche. The picornavirus of single-stranded RNA (ssRNA) viruses includes enteroviruses (coxsackieviruses, polioviruses, echoviruses) and rhinoviruses (42). Picornaviruses use a viral 3C protease to cleave their viral polyprotein into mature proteins (43, 44). The 3C protease from multiple picornaviruses also cleaves human NLRP1 and mouse NLRP1B, triggering activation (29, 30). This 3C cleavage site in NLRP1 is rapidly evolving, with variation among primates and even within the human population (30) suggesting an evolutionary conflict between 3C and NLRP1. Whether NLRP1 helps clear these viruses in vivo has not been established.
Shigella species are intracellular cytosolic pathogens; however, whether the detection of IpaH7.8 by NLRP1 drives clearance of the bacteria in vivo has not been studied (26). This process may be facilitated by recent descriptions of mouse models of Shigella flexneri infection (45–47).
Toxoplasma gondii is an intracellular parasite that establishes a vacuolar niche for both replication and long-term persistence. In mice, T. gondii can trigger NLRP1 activation, and Casp1/11−/−, Asc−/−, and Nlrp1−/− mice have increased susceptibility (48). How NLRP1 detects T. gondii could be elucidated by further study inspired by the functional degradation model.
CARD8
CARD8 is a primate-specific inflammasome that is not an NLR but instead consists of a short N terminus followed by a FIIND-CARD. Like NLRP1, CARD8 is regulated by DPP8/9 and activated by Val-boroPro (36, 38, 49). Whether pathogens are detected by CARD8 and whether it has a role in clearing intracellular infection remain to be explored.
NLRP3
NLRP3 is the most-studied inflammasome and is activated by diverse stimuli, which lead to hallmarks of cellular catastrophe (such as damaged organelles or cytosolic ion fluxes) in both infectious and noninfectious contexts (50). The full mechanism of NLRP3 activation remains a conundrum, despite extensive study.
Activation
NLRP3 has the typical NLR domains (PYD-NACHT-LRR) and requires the ASC adaptor to activate caspase-1. Inactive NLRP3 was recently shown to exist in a double ring cage structure held together by the LRR domains, which holds the PYD in the sequestered interior of the cage (50a). NLRP3 activation is strictly regulated in at least three steps: (a) priming, (b) activation, and (c) licensing (50, 51) (Figure 2). The first priming step can be mediated by diverse pathways in response to PAMPs, DAMPs, or cytokines. Priming occurs in two ways: (a) through an increase in the amount of NLRP3 protein, typically by nuclear factor κB (NF-κB) signaling, and/or (b) through posttranslational modifications such as ubiquitylation or phosphorylation (50). After priming, other agonists can cause NLRP3 activation and oligomerization. The underlying mechanisms remain unclear but may include loss of intracellular ions (potassium or calcium), release of oxidized mitochondrial DNAs, lysosomal disruption, and organelle dysfunction (mitochondria or Golgi apparatus). During the oligomerization process, a third licensing step is required, wherein NIMA-related kinase 7 (NEK7), which is normally involved in mitosis, plays an essential role in assembling the inflammasome oligomer (52–54) by connecting two adjacent NLRP3 monomers to stabilize the inflammasome disk (55).
Figure 2.
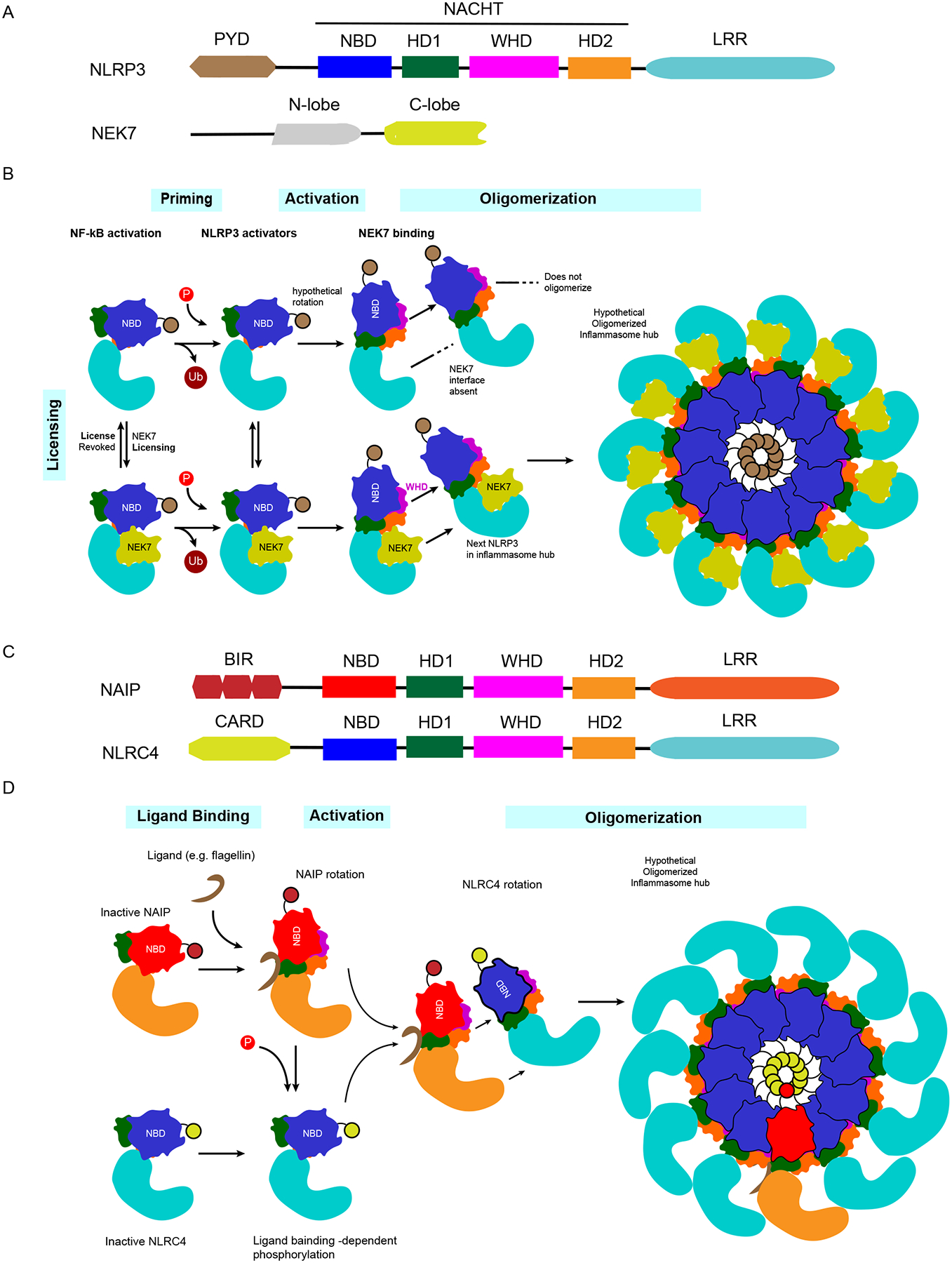
Regulation of NLRP3 and NLRC4 activation. NLRP3 activation requires three distinct stimulatory priming/licensing events, whereas NAIP/NLRC4 activation can proceed from exposure to a single activating agonist. (a) NLRP3 is composed of PYD, NACHT, and LRR domains. The NACHT domain includes four subdomains: NBD, HD1, WHD, and HD2. The licensing partner of NLRP3 is NEK7, which is composed of an N-lobe and a C-lobe. (b) NLRP3 activation requires three steps: priming, activation, and licensing. Under NF-κB signaling, the priming step occurs by multiple posttranslational modifications, whose structural functions and interdependence remain to be elucidated (e.g., deubiquitylation and phosphorylation). After priming, activation occurs by exposure to NLRP3 agonists, followed by (hypothetical) rotation of the NBD-HD1 module. Licensing requires NEK7, whose C-lobe connects two adjacent NLRP3 monomers to enable oligomerization into an inflammasome complex. (c) NAIPs and NLRC4 have closely related structures. Both include similar NACHT-LRR domains; however, NAIPs have BIR domains, while NLRC4 has a CARD in each N terminus. (d) Once bacterial ligands bind their cognate NAIP, the NAIP undergoes rotational activation, followed by NLRC4 phosphorylation by unknown mechanisms. The activated NAIP interacts with an NLRC4 monomer and induces NLRC4 rotational activation, which enables NLRC4 to oligomerize. Only 1 activated NAIP is required to recruit and activate 10 NLRC4 monomers and oligomerize in a domino-like reaction, resulting in the formation of an inflammasome disk complex. Abbreviations: CARD, caspase activation and recruitment domain; LRR, leucine-rich repeat; NAIP, NLR family apoptosis inhibitory protein; NEK7, NIMA-related kinase 7; NF-κB, nuclear factor κB; NLR, Nod-like receptor; NLRC4, NLR family CARD domain–containing 4; PYD, pyrin domain.
The subcellular location at which NLRP3 activates has been a topic of debate in the literature and may be the key to its activation mechanism. Early publications argued for mitochondria or mitochondria-associated endoplasmic reticulum membranes (50), whereas more recent publications advocate for dispersed trans-Golgi networks and the microtubule-organizing center (50a, 56–58). The discovery of how these locations fit into an overall mechanism of NLRP3 activation will undoubtedly spark additional studies.
Pathogens detected by NLRP3
Pathogens that cause cellular crisis could activate NLRP3. Indeed, a broad spectrum of pathogens, including bacteria, viruses, and fungi, can activate NLRP3 (50, 59). NLRP3 activates in response to virulence factors that cause cytosolic ion fluxes, including ionophores (nigericin), bacterial pore-forming toxins, and viral pore-forming proteins (viroporins) (50). Many other cell death pathways can also cause NLRP3 to activate. NLRP3 also senses PAMPs such as intracellular dsRNA or DNA/RNA hybrids (50). This activation is indirect, as the DExD/H-box RNA helicase DHX33 binds dsRNA from various pathogens, after which DHX33 interacts with and activates NLRP3 (60). The in vivo utility of RNA detection by NLRP3 is illustrated by West Nile virus infection, where Nlrp3-deficent mice show increased mortality (61). Conversely, RNA detection is not useful against vesicular stomatitis virus in mouse models of infection, despite the observation that NLRP3 can detect this virus in vitro (62).
One of the most-studied viral infections in the NLRP3 literature is influenza A virus (IAV), which can activate NLRP3 by multiple mechanisms, including dsRNA sensing (63) and the M2 viroporin (64). However, IAV can also inhibit NLRP3 activation by using nonstructural protein 1 (NS1) (65, 66). Conflicting reports suggest that NLRP3-deficient mice may or may not have increased mortality after IAV infection or may have altered adaptive responses (67–69). These IAV infection phenotypes also change in different genetic backgrounds (70). Therefore, more studies are needed to understand the complex interactions between IAV and NLRP3.
Evasion of NLRP3
Some pathogen-derived proteins can inhibit NLRP3 activation in each step. In the priming step, enterovirus 71 (EV71) proteases 2A and 3C directly cleave and inactivate NLRP3 (71). Human parainfluenza virus 3C interacts with NLRP3 and promotes its ubiquitination and degradation (72). In the activation step, measles virus and paramyxovirus V protein as well as IAV NS1 directly interact with and inhibit NLRP3 (73, 74). PB1-F2, another IAV protein, can attenuate the mitochondrial membrane potential, thereby inhibiting NLRP3 (75), although other isoforms of PB1-F2 induce excessive NLRP3 activation (76, 77).
NAIP/NLRC4
Together, NLR family apoptosis inhibitory proteins (NAIPs) and NLR family CARD domain–containing 4 (NLRC4) detect the activity of bacterial T3SS and T4SS, both of which inject effector molecules into the host cells and can promote a variety of effects, including bacterial invasion and intracellular replication. (Note that NAIPs are poorly named and are not apoptosis inhibiors.)
Activation
Humans encode a single NAIP, while mice encode four functional NAIPs. The NAIP directly binds bacterial ligands. In mice, NAIP1 and NAIP2 detect T3SS needle and inner rod proteins, respectively, while NAIP5 and NAIP6 detect flagellin (78–84). In humans, these functions are condensed into a single NAIP (81, 84–86). After ligand recognition, the activated NAIP undergoes a confirmational change that triggers NLRC4 oligomerization into an inflammasome hub consisting of 1 NAIP plus 10 NLRC4s (87–91) (Figure 2). NAIPs and NLRC4 are constitutively expressed in myeloid cells and intestinal epithelial cells under interferon-regulator factor 8–related transcriptional factors (92, 93), with the exception of mouse NAIP1, which is expressed only in peritoneal macrophages (83). NLRC4 may be posttranslationally regulated by S533 phosphorylation by protein kinase Cδ and LRRK2 (94–96); however, another study found that S533 phosphorylation is dispensable (97). Thus, the importance of this regulatory mechanism requires further investigation.
Pathogens detected by NAIP/NLRC4
NAIP/NLRC4-deficient mice often show increased bacterial burdens during infection with T3SS/T4SS pathogens (98). For example, our group discovered that an environmental pathogen, Chromobacterium violaceum, killed Nlrc4−/− mice even after 100 CFU infection, whereas wild-type mice survived even after 1,000,000 CFU infection, which is the strongest phenotype within known NLRC4-activating pathogens (59, 99). We also identified a milder phenotype during systemic Burkholderia thailandensis infection, where Nlrc4−/− mice maintain bacterial burdens for 3 days after wild-type mice clear the infection (100). Similarly, Nlrc4−/− or Casp1–/– mice have higher burdens of Salmonella enterica sv. Typhimurium during oral infection and succumb earlier than wild-type mice (98, 101–103). S. flexneri is another important gastrointestinal pathogen of humans, but mice are naturally resistant to infection. A recent paper showed that Naip1-6Δ/Δ mice as well as Nlrc4−/− mice become susceptible to oral Shigella infection (45). In pulmonary Burkholderia pseudomallei and Legionella pneumophila infection, both Nlrc4−/− mice and Nlrp3−/− mice have increased susceptibility to infection compared with wild-type mice (98, 104, 105).
Evasion of NAIP/NLPR4.
In Salmonella, all the T3SS proteins known to activate NAIP/NLRC4 (PrgI, PrgJ, and FliC) are secreted by the SPI1 T3SS (106). During intracellular replication, S. Typhimurium represses SPI1 and switches to a SPI2 T3SS (107, 108) whose rod protein evades NLRC4 (80). This observation suggests that S. Typhimurium evades NLRC4 during its intracellular replication phase (109). Similarly, Listeria monocytogenes represses flagellin during in vivo infection, enabling NLRC4 evasion (110, 111), which may also be relevant for nonmotile pathogens such as Shigella. After SPI1 is repressed, NLRP3 can detect S. Typhimurium, but NLRP3 fails to clear the infection on its own, as C57BL/6 mice still succumb. A low-virulence M525P-derivative strain enables prolonged infections, revealing that NLRP3 can promote late-protective Th1 immune responses (112). Pseudomonas aeruginosa effectors ExoU and ExoS along with phospholipase A2 activity inhibit NLRC4 (113), although the mechanism is still unknown. Overall, it is surprising that more bacteria have not developed systems to directly inhibit the NAIP/NLRC4 inflammasome; perhaps such evasion strategies have yet to be discovered.
NLRP6
The sequence and structure of NLRP6 are similar to those of NLRP3; however, its mechanism of activation and its function in combating intracellular pathogens are not well established. NLRP6 has both inflammasome and noninflammasome functions.
Activation.
Since its early identification (as PYPAF5), NLRP6 has been known to signal through ASC to activate caspase-1 (114, 115). In vivo, Nlrp6−/− mice have less serum IL-18 basally and during dextran sulfate sodium–induced colitis (116). Multiple agonists or enhancers for NLRP6 have been proposed, including the metabolite taurine, as well as cell wall lipids from gram-positive (lipoteichoic acid) or gram-negative [lipopolysaccharide (LPS)] bacteria, and dsRNA (115, 117, 118, 124). Upon activation, NLRP6 undergoes liquid-liquid phase separation as part of its activation process (118a). Whereas most studies suggest that NLRP6 signals through ASC to caspase-1, the Núñez group (117) observed that NLRP6 signals through ASC to both caspase-11 and caspase-1, but this did not result in pyroptosis. Further studies of the NLRP6 signaling pathway could elucidate how different agonists can result in different downstream pathways and whether LPS detection occurs redundantly by NLRP6 and caspase-11 (see caspase-11 section below).
NLRP6 is regulated by posttranslational modification, but, in contrast to what occurs with NLRP3, ubiquitination promotes NLRP6 signaling (119). How this ubiquitination is regulated awaits further study.
Pathogens detected by NLRP6
L. monocytogenes infection as well as cytosolic lipoteichoic acid activates NLRP6 to release IL-1β and IL-18 in vitro. However, other publications report that inflammasome activation occurs through combinations of NLRC4, NLRP3, and AIM2 in response to L. monocytogenes (120). In mice, NLRP6 and caspase-11 were reported not to protect but rather to exacerbate L. monocytogenes infection (117). In contrast, earlier publications found that Casp1–/–Casp11–/– mice were more susceptible (121) and that Casp11–/– mice had normal susceptibility (122). The detrimental effects of NLRP6 were dependent on IL-18 secretion (117); however, one publication reported that this same IL-18-driven detrimental effect occurred through NLRP3 (123). Further confounding the picture is the finding that IL-18 exerts antimicrobial effects against L. monocytogenes infection (99). Resolving these conflicts will require further investigation.
NLRP6 can also combat ssRNA viruses, including murine norovirus 1 and encephalomyocarditis virus, during in vivo oral infection, although the mechanism is independent of caspase-1 (124). Mechanistically, NLRP6 binds viral RNA via DHX15 to activate mitochondrial antiviral-signaling protein (MAVS) and initiate a type I/III interferon response. Interestingly, this DHX15-NLRP6-MAVS axis is analogous to the DHX33-NLRP3-MAVS axis (63) but differs in its ability to activate caspase-1 through this pathway. How NLRP6 signals through ASC to caspase-1/11 (or, alternatively, through MAVS) requires further study.
NLRP6 has been reported to have inflammasome-independent functions, such as mucin-granule exocytosis and coordinating autophagy in intestinal goblet cells (125). NLRP6 also influences colonic commensal bacteria (116, 126), and, in turn, microbiota-associated metabolites shape NLRP6 activation (115). However, this effect of NLRP6 upon the microbiota remains contentious (127, 128). Much remains to be learned about NLRP6.
NLRP9
Early studies indicated that NLRP9 is expressed mainly in reproductive organs and is associated with reproductive health (129, 130). More recent studies indicate that NLRP9 is an inflammasome (131). In comparison to humans, mice have expanded NLRP9 into three paralogs (Nlrp9a–Nlrp9c), and fully Nlrp9-deficient mice have defects in blastocyst development due to an unknown mechanism (132). Nlrp9b was recently found to be expressed specifically in ileal intestinal epithelial cells (IECs), and both Nlrp9b−/− mice and IEC-specific knockouts are susceptible to rotavirus (131). In contrast, Nlrp9a and Nlrp9c are not expressed in IECs and are not important for rotavirus defense. This rotavirus clearance required GSDMD, suggesting that IEC pyroptosis and/or extrusion clears rotavirus infection. Mechanistically, the RNA helicase DHX9 binds rotavirus dsRNA, which mediates NLRP9b recognition (131). Human NLRP9 can also bind rotavirus RNA. Whether human NLRP9 is a functional homolog that combines the murine developmental and viral defense functions remains to be determined.
AIM2
Cytosolic DNA may arise from pathogens or from host mitochondria DNA. Absent in melanoma 2 (AIM2) directly binds either self or nonself dsDNA and forms an inflammasome. AIM2 is the prototype of the AIM2-like receptors (ALRs).
Activation.
AIM2 is expressed basally by many cell types and can be further induced by type I interferon (120). AIM2 consists of an N-terminal PYD and a C-terminal HIN domain not found in NLR family inflammasomes. This HIN domain binds directly to dsDNA that is at least 80 bp long (133). Cryo–electron microscopy structural studies show that in the resting state the HIN inhibits the PYD, but once the HIN domain detects dsDNA, the PYD nucleates the polymerization of ASC (133).
AIM2 has several inhibitory mechanisms. p202, a mouse ALR composed of two HIN domains without a PYD, inhibits the activity of AIM2 by binding like a decoy ALR (134). Similarly, IFI16-β, a human transcript isoform of IFN-γ-inducible protein 16 (IFI16), inhibits AIM2 inflammasome (135). Furthermore, the human PYD-only protein 3 was found to specifically bind the AIM2 PYD and thereby inhibit its interaction with ASC (136).
Pathogens detected by AIM2
Although AIM2 can detect dsDNA from bacteria, virus, and fungi, the pathway required to liberate dsDNA is different. Bacteria must escape from the vacuole and undergo bacteriolysis, which can be caused by cytosolic GTPases such as guanylate-binding proteins (GBPs) (137, 138) and immunity-related GTPase family member b10 (IRGB10) (139). These are interferon-stimulated genes. Francisella novicida and the vaccine strain of Francisella tularensis were the first bacteria reported to activate AIM2 (140, 141). In F. novicida infection, GBP2, GBP5, and IRGB10 all localize to F. novicida and cause bacteriolysis, exposing the bacterial DNA (137, 138). Irgb10−/− mice are more susceptible to F. novicida infection; this phenotype is similar to Gbpchr3 knockout, Aim2–/–, and Casp1–/– mice (139). Other intracellular bacteria, such as L. monocytogenes, Legionella pneumophila, and Mycobacterium species, also activate AIM2 in vitro (120).
Several DNA viruses activate AIM2, including mouse cytomegalovirus (MCMV) and vaccinia virus (138, 141–143). MCMV-infected mice exhibited AIM2- and ASC-dependent IL-18 production and viral clearance (141). Vaccinia virus–infected macrophages and human papillomavirus–infected keratinocytes release IL-1β or IL-18 in an AIM2-dependent manner (141, 144). EV71-infected neuronal cells also activate AIM2, which restricts viral replication in vitro (145). AIM2 also plays a role in fungal infections, such as Aspergillus fumigatus or Candida albicans; however, the mechanism of releasing DNA remains undetermined (146).
Evasion of AIM2
Among wide varieties of DNA viruses, only a subset has been found to activate AIM2, perhaps due to evasion strategies. Indeed, herpes simplex virus 1 evades by inhibiting DNA–AIM2 interaction using VP22 (147). Another herpesvirus, human cytomegalovirus, uses pUL83 to interact with and disrupt the AIM2 inflammasome (148). Viral DNA is typically shielded by nucleoproteins that uncoat only in the nucleus, thus creating a natural evasion strategy. Whether AIM2 detects only defective viral particles, or whether viral DNA is deliberately liberated by host cell machinery in the cytosolic space, is a topic for further investigation.
IFI16
IFI16 is an ALR composed of a PYD and two HIN domains. The activation mechanism of IFI16 often appears to be parallel to that of AIM2; indeed, IFI16 can directly interact with dsDNA (133). Human IFI16 can detect Kaposi sarcoma–associated herpesvirus and release IL-1β (149). IFI16 detects HIV-1 infection in CD4 T cells and induces pyroptosis (150). However, IFI16 does not substitute for AIM2 in Aim2–/– cells or mice; therefore, the two must have distinct properties. How a host differentially uses IFI16, AIM2, and other, similar ALRs to defend against pathogens requires further elucidation.
Caspase-11
Murine caspase-11 cleaves GSDMD and causes pyroptosis without activating IL-1β and IL-18 directly (151). The CARDs of caspase-11 and its human paralogs, caspase-4 and −5, directly bind the lipid A motif of LPS when it is present in the cytosol (152–154). This process results in caspase-11 oligomerization and catalytic activation (155, 156). The structure of binding of lipid A by the CARD of caspase-11 has yet to be determined.
Activation
Many intracellular gram-negative bacteria activate caspase-11 (120). Some of them are bona fide cytosol-invasive pathogens (e.g., B. thailandensis and B. pseudomallei), whereas others are vacuolar pathogens whose LPS is normally not detected. When vacuolar pathogens such as S. Typhimurium are mutated to aberrantly enter the cytosol (sifA mutants), increased caspase-11 detection results (157). Alternatively, cytosolic LPS detection could occur if vacuolar bacteria accidentally invade the cytosol due to high multiplicities of infection or long-term endpoints in vitro or if high doses of LPS or outer membrane vesicles are used. Although aberrant activation can occur and can sometimes be detrimental (LPS-induced sepsis), the evolved function of caspase-11 is to clear bacteria that efficiently invade the cytosol.
In some cases, the outer membrane of bacteria may not naturally expose the lipid moieties of LPS, creating a type of evasion strategy. GBP proteins and IRGB10 liberate LPS in certain cytosolic bacteria (139, 158). During bacterial infection, interferon induces expression of caspase-11, GBPs, and immunity-related guanosine triphosphatases (IRGs) (139, 158–160). Recent studies have shown that GBPs liberate or expose LPS for detection. There are 7 GBPs in humans and 11 in mice (161). GBPs work in sequence when exposing LPS: Human GBP1 rapidly polymerizes on the surface of bacteria and initiates recruitment of GBP2–4 to assemble a GBP coat (162, 163). Subsequently, GBP1 functions as a surfactant that disrupts the O-antigen barrier, thereby exposing buried lipid A for detection (164). In mice, IRGB10 resides near the GBPs and promotes LPS liberation (139), although the underlying mechanism remains to be described. Thus, GBPs and IRGs have been implicated in exposing bacterial PAMPs to both caspase-11 and AIM2.
Pathogens detected by Caspase-11
Many intracellular gram-negative bacteria activate caspase-4, −5, and −11 in vitro. Among them, the environmental bacterium B. thailandensis escapes from the vacuole and invades the cytosol using a T3SS that is closely related to that of Shigella species (165). In a systemic infection model, wild-type mice were completely resistant to challenge by even 20,000,000 CFUs of B. thailandensis, which cleared in only 1 day. In contrast, Casp11–/– and Gsdmd–/– mice were highly susceptible to B. thailandensis, succumbing to even 100 bacteria (100, 157, 166, 167). We recently demonstrated why NLRC4 detection of the T3SS was not sufficient to clear these bacteria in a single Casp11–/– mouse (167, 168). Interestingly, using casp11-conditional knockout mice, we revealed that neutrophil caspase-11 is important to clear B. thailandensis. Mechanistically, caspase-11 triggered pyroptosis, whereas NLRC4 and caspase-1 failed to cause pyroptosis in neutrophils, suggesting that caspase-11-dependent neutrophil pyroptosis is essential for defense against B. thailandensis in vivo (167, 168). How NLRC4/caspase-1 in neutrophils fails to trigger pyroptosis remains to be determined, but this process has been observed for NLRP3 inflammasomes as well (169). Although the six GBP genes on chromosome 3 are not required to clear B. thailandensis (100), it is unknown whether the GBPs on chromosome 5 are sufficient.
Numerous bacteria that are not cytosol invasive have been shown to trigger caspase-11 in vitro, most recently P. aeruginosa (170). P. aeruginosa is primarily an extracellular pathogen; however, it has been proposed to escape the vacuole into the cytosol (171). Whether this caspase-11 detection is beneficial or detrimental to the host in vivo remains to be determined. Similarly, the vacuolar pathogen S. Typhimurium was not cleared by caspase-11 (172), but an aberrantly cytosol-invasive ΔsifA mutant was cleared by a caspase-11/GSDMD pathway (157, 173).
In one study, Casp11–/– mice with L. pneumophila infection had increased bacterial burdens, which correlated not with pyroptosis but instead with modulated trafficking of the pathogen-containing vacuole in vitro (174). However, other groups found that caspase-11 causes pyroptosis in vitro in response to L. pneumophila (175, 176) or in response to sdhA mutant L. pneumophila that aberrantly escapes the vacuole (157). The in vivo phenotype was not replicated in another study of Casp11–/– mice, although those mice had a mixed genetic background, where instead NLRC4 was important (177). These conflicting studies may inspire further investigation.
Unlike the T3SS-expresing B. thailandensis, whether Burkholderia cenocepacia is a cytosol-invasive bacterium is unclear. Nevertheless, during B. cenocepacia infection, caspase-11 and GSDMD responses were found not to cause pyroptosis but instead to induce mitochondrial reactive oxygen species production, pathogen-containing vacuole trafficking, and autophagy in macrophages (178, 179). How caspase-11 regulates these nonpyroptotic functions requires further study. In parallel, a B. cenocepacia T6SS effector that attacks RhoA can be detected by the pyrin inflammasome (180). Whether there is redundancy remains to be determined.
Evasion of caspase-11
There appears to be strong evolutionary pressure for cytosol-invasive bacteria to evade caspase-11. The cytosol-invasive Francisella species include some of the most virulent bacteria. Francisella modifies its lipid A to contain only four acyl chains, thereby evading caspase-11 detection (152). Shigella is a bona fide cytosol-invasive bacterium that uses the OspC3 T3SS effector to inhibit human caspase-4 (162, 181, 181a, 181b). B. pseudomallei and Burkholderia mallei remain highly virulent despite sharing the T3SS of B. thailandensis; therefore, they probably use an as-yet-undiscovered mechanism to evade caspase-11.
Pyrin
Pyrin appears to detect mostly extracellular pathogens. Pyrin senses inhibition of Rho GTPases and, thus, detects virulence factors that block phagocytosis. Because this review focuses on intracellular pathogens, we refer readers to earlier reviews (e.g., 98) for more detail.
CELL TYPE–SPECIFIC EFFECTS OF CASPASE-1/11
Macrophages express most inflammasome-associated proteins and have been intensively studied. After GSDMD activation, the macrophage membrane ruptures, releasing all soluble cytosolic content, which is the definition of pyroptosis. Larger particles, including organelles and bacteria, remain trapped within the macrophage corpse, which we term a PIT (41). A PIT is conceptually parallel to a neutrophil extracellular trap (NET) (182) in that both detain bacteria. Meanwhile, IL-1β, IL-18, and eicosanoids promote neutrophil recruitment and subsequent efferocytosis of both the PIT and its entrapped bacteria (41, 183).
Neutrophils undergo a unique form of cell death called NETosis, which releases chromatin to trap extracellular pathogens (182). Recent studies revealed that when caspase-11 or neutrophil serine proteases cleave GSDMD in neutrophils, NETs form in vitro (173, 184). The in vivo relevance of GSDMD-dependent NETs awaits further study, as does the mechanism by which caspase-11 causes NETosis while caspase-1 does not. Whether neutrophils undergo pyroptosis after GSDMD activation remains puzzling, as caspase-11 can cause pyroptosis but, in contrast, NLRC4- or NLRP3-activated caspase-1 fails to do so (167–169).
IECs cover the large surface of the intestine and as such are exposed to copious numbers of microbes. IECs also express many inflammasomes and can undergo pyroptosis; however, prior to pyroptosis they rapidly extrude into the intestinal lumen, effectively removing the infected cell from the body. The extrusion pathway might occur via multiple pathways downstream of the inflammasome. GSDMD seems sufficient to cause extrusion, but backup pathways signaling from ASC to activate caspase-8 are effective in its absence (185). During oral S. Typhimurium infection, NAIP/NLRC4 in IECs contribute to IEC extrusion and to a reduction in bacterial burden (185, 186). Interestingly, while systemic S. Typhimurium infection is not affected by caspase-11 (172), oral S. Typhimurium infection exhibits a higher bacterial burden in Casp11–/– mice (187). Whether IEC pyroptosis is important after IEC extrusion in combating intracellular pathogens is unknown.
Neurons are an important cell type that must be protected from intracellular infection yet can be difficult or impossible to replace. In contrast, macrophages, neutrophils, and IECs are easily and rapidly replaced. Neurons are often highly resistant to RCD; nevertheless, they express both apoptotic and inflammatory caspases, as well as inflammasome components (including NLRP3, AIM2, and GSDMD) (188, 189). In the setting of irreversible compromise during intracellular infection, neurons can undergo pyroptosis. Zika virus has tropism to infect neural progenitor cells in the central nervous system (190, 191), and infection of these cells by Zika virus in the neonatal brain results in caspase-1/GSDMD-dependent pyroptosis, which is linked to developmental defects (192). Even in the mature central nervous system, West Nile virus–infected cortical neurons release IL-1β, which inhibits neuronal viral replication (61).
ZBP1, A NUCLEIC ACID SENSOR THAT INDUCES NECROPTOSIS
Necroptosis is a lytic form of RCD triggered either by transmembrane death receptors (e.g., TNF receptor) or by PRRs that are either transmembrane (TLRs) or cytosolic [Z-DNA-binding protein 1 (ZBP1)]. When triggered, these pathways activate receptor-interacting kinase 3 (RIPK3), which phosphorylates pseudokinase mixed lineage kinase domain–like protein (MLKL). MLKL then forms pores on the plasma membrane, leading to the lytic RCD termed necroptosis (193). Below, we discuss ZBP1, a cytosolic PRR that causes necroptosis. The function of other necroptosis pathways is reviewed elsewhere (194).
Activation
ZBP1 contains two Z α domains and two RIP homotypic interaction motif (RHIM) domains (195). It has been identified biochemically as a Z-DNA-binding protein. DNA adopts the Z-form when rapid replication or unwinding occurs faster than endogenous helicase activities can unwind and relax the DNA. Z-DNA is left-handed with a zigzag backbone, whereas DNA typically adopts the B-form, which is right-handed with a symmetrical backbone (Figure 3). Although initially ZBP1 was proposed to be a stimulator of type I interferon expression, this function was later assigned to cyclic GMP–AMP synthase (195). Now, ZBP1 is accepted as a necroptosis-inducing PRR that senses Z-DNA and Z-RNA.
Figure 3.
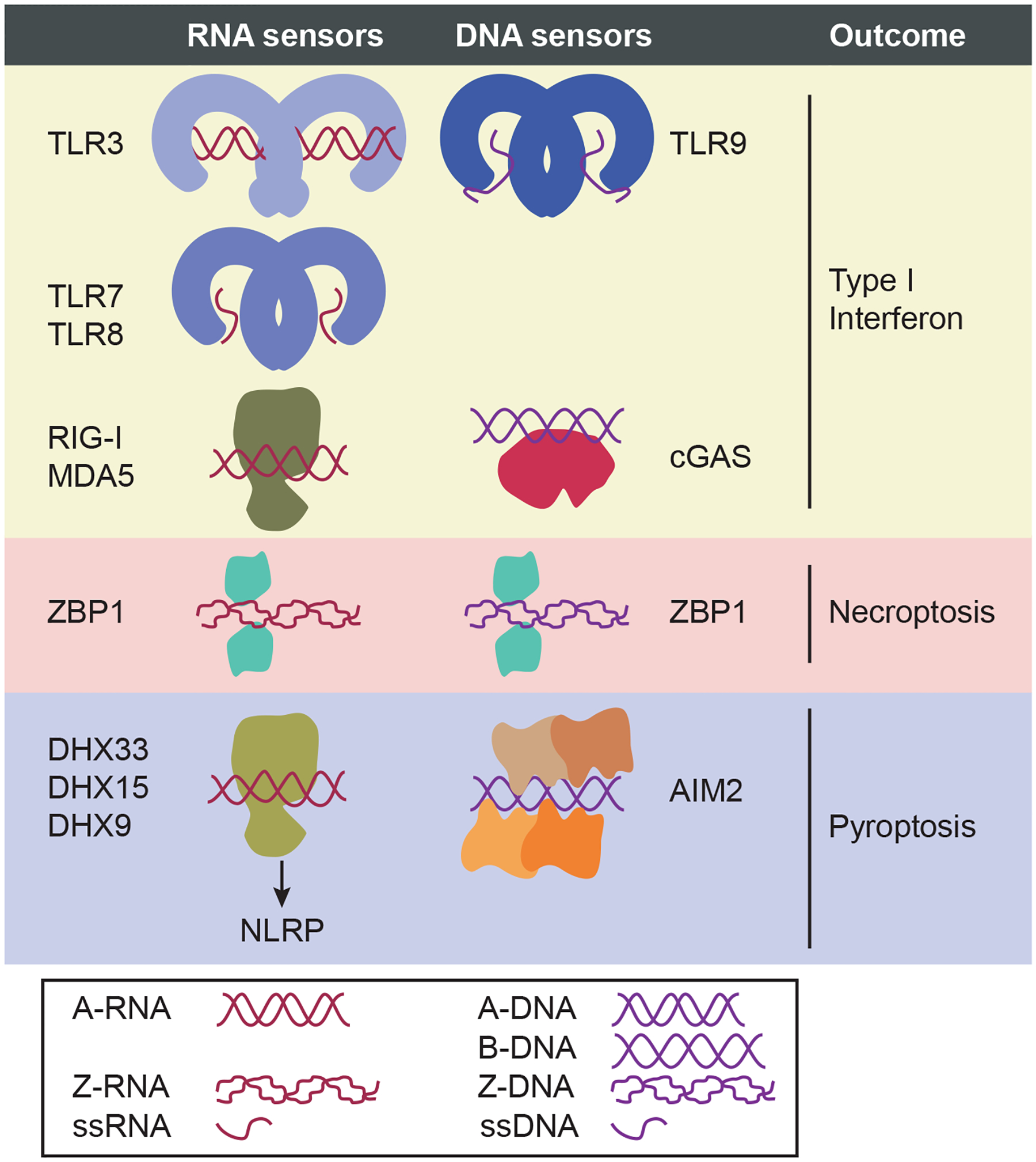
Nucleic acid sensors. Type I IFN–inducing RNA sensors include three TLRs (TLR3, TLR7, and TLR8) and two RLRs (RIG-I and MDA5) (195c). These three RNA-sensing TLRs are localized predominantly to endosomes, where TLR3 recognizes dsRNA (which typically adopts the A-form, A-RNA), while TLR7 and TLR8 recognize ssRNA. The two RLRs are cytosolic RNA sensors that drive IFN responses by sensing A-RNA; RIG-I binds to short A-RNA with 5′ di- or triphosphates, while MDA-5 binds to long A-RNA. Type I IFN–inducing DNA sensors include TLR9 and cGAS. Like the RNA-sensing TLRs, TLR9 is also localized to endosomes; however, its ligand is ssDNA. cGAS recognizes cytosolic B-DNA. ZBP1, a necroptosis-inducing Z-form nucleic acid sensor, recognizes both Z-DNA and Z-RNA as markers of viral infection. Inflammasome sensors that activate caspase-1 also respond to cytosolic DNA and RNA ligands. RNA detection is mediated by three DHX family members (DHX33, DHX15, and DHX9), which are cytosolic A-RNA sensors that activate NLRP inflammasomes (NLRP3, NLRP6, and NLRP9, respectively) to induce pyroptosis by binding with A-RNA. NLRP1 (not shown) also responds to cytosolic RNA, but DHX partnership is not known to be required. The structural relationships between these NLRs and their partners or RNA are not yet established. Finally, the AIM2 inflammasome directly binds to dsDNA. The structures of A-form, B-form, and Z-form nucleic acids are shown. Whereas A-form and B-form are right-handed helices, Z-form adopts a left-handed helix. Genomic DNA exists in B-form, but in contrast, dsRNA does not typically adopt B-form. Z-forms occur during replication or unwinding of nucleic acids that occurs too rapidly for helicases to relax the structure (195a, 195b). Abbreviations: AIM2, absent in melanoma 2; cGAS, cyclic GMP–AMP synthase; DHX, DExD/H-box helicase; dsRNA, double-stranded RNA; NLR, Nod-like receptor; ssRNA, single-stranded RNA; TLR, Toll-like receptor; ZBP1, Z-DNA-binding protein 1.
Necroptosis induced by ZBP1 is RIPK3 dependent and does not require the signaling components of other necroptosis pathways (196), although the situation may be more complex (197, 198). Although identified as a Z-DNA-binding protein, ZBP1 also binds Z-RNA, which is structurally similar to Z-DNA. Z-RNA may occur when viruses rapidly synthesize RNA. ZBP1 thus detects RNA viruses such as IAV (199, 200). Surprisingly, Z-RNA is also the ligand during DNA viral infection, including with MCMV (201, 202), vaccinia virus (203–205), and herpes simplex virus (206). This finding suggests that ZBP1 may be a sensor not for genomic nucleic acids but rather for excessive transcription states that generate hyperwound messenger RNA due to excessively fast viral transcription rates.
Pathogens detected by ZBP1
One group showed that, in response to IAV, ZBP1 induces apoptosis and necroptosis (199, 200, 207), whereas another group proposed that ZBP1 additionally activates pyroptosis (208–210). Published studies suggest that ZBP1 both protects against and is detrimental during IAV infection in vivo. Further research will be required to understand the relationship between ZBP1 and IAV. Note that the ZBP1 knockout mice generated by the Akira group (211) and widely used by others have a mixed genetic background that affects natural killer (NK) cells and thereby MCMV infection, potentially making wild-type C57BL/6 mice a problematic control (212, 213).
ZBP1 Evasion
Because necroptosis clears intracellular niches for viral replication, many viruses encode proteins to antagonize necroptosis. For example, MCMV encodes viral inhibitor of RIP activation, a RHIM-containing protein, to target the ZBP1-RIPK3 complex (214). Vaccinia virus encodes the E3 protein to compete with ZBP1 for Z-RNA, thus inhibiting necroptosis (204, 205). A recent study discovered that vaccinia virus also encodes a protein called viral inducer of RIPK3 degradation to trigger degradation of RIPK3 and inhibit necroptosis (215). Similarly, herpes simplex virus 1 evades necroptosis in humans by using ICP6 to antagonize RIPK1 and RIPK3 (217). Intriguingly, this virulence function backfires when this human specific virus infects mice; here ICP6 inadvertently triggers RIPK1 and RIPK3 to cause necroptosis in murine cells (216). This suggests that necroptotic signaling may prevent viruses from crossing species barriers, allowing, in this case, mice to be protected from this human specific virus.
INTERPLAY BETWEEN PYROPTOSIS AND APOPTOSIS
If a pathogen inactivates GSDMD, then inflammasomes fail to execute pyroptosis. Indeed, the EV71 3C protease cleaves GSDMD within its pore-forming domain, abolishing pyroptosis and promoting viral replication (218). Perhaps the host evolved NLRP1 to detect 3C (29, 30) but subsequently some viruses evolved 3C to cleave the downstream GSDMD, thereby negating pyroptosis. This situation is mimicked in Gsdmd–/– cells, and it could be induced physiologically if specific cell types do not express GSDMD. To ensure cell death, GSDMD-deficient cells undergo apoptosis via two different backup pathways. First, activated caspase-1 can cleave and thereby activate BID, which then drives caspase-9-dependent apoptosis (219, 220). Second, ASC can activate caspase-8-dependent apoptosis (185, 221, 222). More research is required to fully understand whether these pathways compensate for the loss of pyroptosis during intracellular infection.
In contrast to pyroptosis, apoptosis is usually said to cause silent, noninflammatory cell death. Once apoptotic signaling has been achieved, apoptotic caspases provide feedback to inhibit other forms of cell death. Caspase-3 cleaves GSDMD within its pore-forming domain, inactivating it and preventing accidental pyroptosis (223, 224). Similarly, caspase-8 cleaves and inactivates RIPK1 and RIPK3 to prevent accidental necroptosis (194, 225). In contrast, certain cell types or stimulation conditions can rewire cells in ways that cause apoptotic caspases to trigger pyroptosis. This process occurs when cells express another gasdermin, GSDME, with an activation linker that is cleaved by caspase-3, causing this normally apoptotic caspase to trigger pyroptosis (226, 227). Some cells express GSDME constitutively, while other cells can be induced to express it. Thus, this apoptosis-to-pyroptosis switch may always be toggled in some cells and inducibly toggled in others (226, 227). Another way to switch from apoptosis to pyroptosis is that caspase-8 can also cleave and activate GSDMD, albeit more slowly than caspase-1 (224, 228, 229). This could serve as a fail-safe timer, such that if apoptosis fails to be completed within a certain time, caspase-8 will attempt to initiate pyroptosis instead (224, 228, 229).
NK cells and cytotoxic T lymphocytes (NK/CTLs) deliver granzyme B to trigger apoptosis, which clears intracellular pathogens including MCMV, lymphocytic choriomeningitis virus, and L. monocytogenes (99). Another gasdermin, GSDMB, is cleaved by granzyme A, delivered from NK/CTLs, and causes pyroptosis (230). Thus, NK/CTL attack can toggle from apoptosis to pyroptosis. However, at least one pathogen antagonizes this pathway; GSDMB is countered by the Shigella IpaH7.8 T3SS effector, which ubiquitinates and degrades GSDMB (231). This study also proposed, in contrast to another report, that GSMDB cannot cause host cell pyroptosis but instead targets the bacterial membrane (231). Interestingly, this same IpaH7.8 effector was also recently shown to degrade GSDMD (231a).
Integrating these observations, we find that hosts have evolved several pathways to switch between apoptosis and pyroptosis. On one hand, GSDME directs both cell-intrinsic apoptosis and NK/CTL attack into pyroptosis (226, 227, 232). On the other hand, GSDMB expression leaves cell-intrinsic apoptosis intact but redirects NK/CTL attack into pyroptosis (230). Importantly, GSDME/B pyroptosis can occur independently of GSDMD, allowing hosts to overcome suppression of inflammasome-dependent pyroptosis by pathogens. The mode of cell death is determined mostly by the GSDME/B expression profile of the targeted cell. However, NK/CTLs might alter their granzyme A/B profile to take control of the cell death decision. Establishing the physiologic relevance of these interwoven pathways in vivo against intracellular pathogens requires additional study.
CLOSING REMARKS
Among the innate sensing proteins, NLRs, ALRs, and ZBP1 have evolved as RCD-inducing sensors for intracellular pathogens. Hosts use these death-inducing sensors to detect many pathogens, but meanwhile host-adapted intracellular pathogens fight back against these sensors. Multiple backup pathways interact in different cell types and under different stimulations to create a complex network of RCD signals. Can all of these diverse and interwoven networks of pathways be methods that evolved to ensure RCD and to alternate between apoptotic and pyroptotic outcomes? We consider this philosophical question in the context of the evolutionary Red Queen hypothesis (1) as it applies to the interface between hosts and pathogens. Every evolutionary step taken by a host provokes an evolutionary response by pathogens, and a subsequent evolutionary response by the host, and so on. How complicated can the processes of RCD become over time? RCD occurs both in the roundworm Caenorhabditis elegans and in humans, which are separated by more than 700 million years of evolution. That is a considerable amount of time to develop complexity, as hosts continuously evolve in competition with intracellular pathogens. Over this time, it seems that RCD pathways have become interwoven in order to ensure that the infected cell dies by pyroptosis, apoptosis, or necroptosis, all of which could eliminate infected cell niches in a redundant manner. Viewed from this perspective, it is somewhat surprising to find single-gene phenotypes where the loss of one RCD pathway has dominant phenotype. It may be that the many more strong in vivo phenotypes will be uncovered as researchers study mice that have multiple RCD pathways simultaneously deleted to prevent the immune response from accomplishing any form of RCD. Such studies could reveal the elegant evolutionary dance between hosts and pathogens that plays out against a background of intricate signaling pathways leading to diverse modes of cell death.
ACKNOWLEDGMENTS
The writing of this review was supported by National Institutes of Health grants AI133236, AI139304, AR072694, and AI136920.
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Van Valen L 1973. A new evolutionary law. Evol. Theory 1:1–30 [Google Scholar]
- 2.Davis BK, Wen H, Ting JP-Y. 2011. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol 29:707–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ting JP-Y, Lovering RC, Alnemri ES, Bertin J, Boss JM, et al. 2008. The NLR gene family: a standard nomenclature. Immunity 28(3):285–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, et al. 2014. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 156(6):1193–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cai X, Chen J, Xu H, Liu S, Jiang Q-X, et al. 2014. Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell 156(6):1207–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi J, Zhao Y, Wang K, Shi X, Wang Y, et al. 2015. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526(7575):660–65 [DOI] [PubMed] [Google Scholar]
- 7.Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, et al. 2015. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526(7575):666–71 [DOI] [PubMed] [Google Scholar]
- 8.Ding J, Wang K, Liu W, She Y, Sun Q, et al. 2016. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535(7610):111–16 [DOI] [PubMed] [Google Scholar]
- 9.Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, et al. 2016. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535(7610):153–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9a.Kayagaki N, Kornfeld OS, Lee BL, Stowe IB, O’Rourke K, et al. 2021. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature. 591(7848):131–36 [DOI] [PubMed] [Google Scholar]
- 10.Monteleone M, Stanley AC, Chen KW, Brown DL, Bezbradica JS, et al. 2018. Interleukin-1β maturation triggers its relocation to the plasma membrane for gasdermin-D-dependent and -independent secretion. Cell Rep. 24(6):1425–33 [DOI] [PubMed] [Google Scholar]
- 11.Schneider KS, Groß CJ, Dreier RF, Saller BS, Mishra R, et al. 2017. The inflammasome drives GSDMD-independent secondary pyroptosis and IL-1 release in the absence of caspase-1 protease activity. Cell Rep. 21(13):3846–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xia S, Zhang Z, Magupalli VG, Pablo JL, Dong Y, et al. 2021. Gasdermin D pore structure reveals preferential release of mature interleukin-1. Nature 593(7860):607–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.von Moltke J, Trinidad NJ, Moayeri M, Kintzer AF, Wang SB, et al. 2012. Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature 490(7418):107–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, et al. 2010. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol 11(12):1136–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jorgensen I, Rayamajhi M, Miao EA. 2017. Programmed cell death as a defence against infection. Nat. Rev. Immunol 17(3):151–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martinon F, Burns K, Tschopp J. 2002. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 10(2):417–26 [DOI] [PubMed] [Google Scholar]
- 17.Finger JN, Lich JD, Dare LC, Cook MN, Brown KK, et al. 2012. Autolytic proteolysis within the function to find domain (FIIND) is required for NLRP1 inflammasome activity. J. Biol. Chem 287(30):25030–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frew BC, Joag VR, Mogridge J. 2012. Proteolytic processing of Nlrp1b is required for inflammasome activity. PLOS Pathog. 8(4):e1002659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.D’Osualdo A, Weichenberger CX, Wagner RN, Godzik A, Wooley J, Reed JC. 2011. CARD8 and NLRP1 undergo autoproteolytic processing through a ZU5-like domain. PLOS ONE 6(11):e27396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boyden ED, Dietrich WF. 2006. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat. Genet 38(2):240–44 [DOI] [PubMed] [Google Scholar]
- 21.Duesbery NS, Webb CP, Leppla SH, Gordon VM, Klimpel KR, et al. 1998. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 280(5364):734–37 [DOI] [PubMed] [Google Scholar]
- 22.Tang G, Leppla SH. 1999. Proteasome activity is required for anthrax lethal toxin to kill macrophages. Infect. Immun 67(6):3055–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fink SL, Bergsbaken T, Cookson BT. 2008. Anthrax lethal toxin and Salmonella elicit the common cell death pathway of caspase-1-dependent pyroptosis via distinct mechanisms. PNAS 105(11):4312–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wickliffe KE, Leppla SH, Moayeri M. 2008. Killing of macrophages by anthrax lethal toxin: involvement of the N-end rule pathway. Cell. Microbiol 10(6):1352–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Varshavsky A 2011. The N-end rule pathway and regulation by proteolysis. Protein Sci. 20(8):1298–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sandstrom A, Mitchell PS, Goers L, Mu EW, Lesser CF, Vance RE. 2019. Functional degradation: a mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes. Science 364(6435):eaau1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chui AJ, Okondo MC, Rao SD, Gai K, Griswold AR, et al. 2019. N-terminal degradation activates the NLRP1B inflammasome. Science 364(6435):82–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu H, Shi J, Gao H, Liu Y, Yang Z, et al. 2019. The N-end rule ubiquitin ligase UBR2 mediates NLRP1B inflammasome activation by anthrax lethal toxin. EMBO J. 38(13):e101996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robinson KS, Teo DET, Tan KS, Toh GA, Ong HH, et al. 2020. Enteroviral 3C protease activates the human NLRP1 inflammasome in airway epithelia. Science 370(6521):eaay2002 [DOI] [PubMed] [Google Scholar]
- 30.Tsu BV, Beierschmitt C, Ryan AP, Agarwal R, Mitchell PS, Daugherty MD. 2021. Diverse viral proteases activate the NLRP1 inflammasome. eLife 10:e60609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones JDG, Vance RE, Dangl JL. 2016. Intracellular innate immune surveillance devices in plants and animals. Science 354(6316):aaf6395 [DOI] [PubMed] [Google Scholar]
- 32.Bauernfried S, Scherr MJ, Pichlmair A, Duderstadt KE, Hornung V. 2021. Human NLRP1 is a sensor for double-stranded RNA. Science 371(6528):eabd0811 [DOI] [PubMed] [Google Scholar]
- 33.Okondo MC, Johnson DC, Sridharan R, Go EB, Chui AJ, et al. 2017. DPP8 and DPP9 inhibition induces pro-caspase-1-dependent monocyte and macrophage pyroptosis. Nat. Chem. Biol 13(1):46–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okondo MC, Rao SD, Taabazuing CY, Chui AJ, Poplawski SE, et al. 2018. Inhibition of Dpp8/9 activates the Nlrp1b inflammasome. Cell Chem. Biol 25(3):262–67.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Geiss-Friedlander R, Parmentier N, Möller U, Urlaub H, Van den Eynde BJ, Melchior F. 2009. The cytoplasmic peptidase DPP9 is rate-limiting for degradation of proline-containing peptides. J. Biol. Chem 284(40):27211–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhong FL, Robinson K, Teo DET, Tan K-Y, Lim C, et al. 2018. Human DPP9 represses NLRP1 inflammasome and protects against autoinflammatory diseases via both peptidase activity and FIIND domain binding. J. Biol. Chem 293(49):18864–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hollingsworth LR, Sharif H, Griswold AR, Fontana P, Mintseris J, et al. 2021. DPP9 sequesters the C terminus of NLRP1 to repress inflammasome activation. Nature 592(7856):778–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang M, Zhang X, Toh GA, Gong Q, Wang J, et al. 2021. Structural and biochemical mechanisms of NLRP1 inhibition by DPP9. Nature 592(7856):773–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tonello F, Zornetta I. 2012. Bacillus anthracis factors for phagosomal escape. Toxins 4(7):536–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chavarría-Smith J, Vance RE. 2015. The NLRP1 inflammasomes. Immunol. Rev 265(1):22–34 [DOI] [PubMed] [Google Scholar]
- 41.Jorgensen I, Zhang Y, Krantz BA, Miao EA. 2016. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J. Exp. Med 213(10):2113–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zell R 2018. Picornaviridae—the ever-growing virus family. Arch. Virol 163(2):299–317 [DOI] [PubMed] [Google Scholar]
- 43.Solomon T, Lewthwaite P, Perera D, Cardosa MJ, McMinn P, Ooi MH. 2010. Virology, epidemiology, pathogenesis, and control of enterovirus 71. Lancet Infect. Dis 10(11):778–90 [DOI] [PubMed] [Google Scholar]
- 44.Sun D, Chen S, Cheng A, Wang M. 2016. Roles of the picornaviral 3C proteinase in the viral life cycle and host cells. Viruses 8(3):82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mitchell PS, Roncaioli JL, Turcotte EA, Goers L, Chavez RA, et al. 2020. NAIP-NLRC4-deficient mice are susceptible to shigellosis. eLife 9:e59022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Medeiros PHQS, Ledwaba SE, Bolick DT, Giallourou N, Yum LK, et al. 2019. A murine model of diarrhea, growth impairment and metabolic disturbances with Shigella flexneri infection and the role of zinc deficiency. Gut Microbes 10(5):615–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Singer M, Sansonetti PJ. 2004. IL-8 is a key chemokine regulating neutrophil recruitment in a new mouse model of Shigella-induced colitis. J. Immunol 173(6):4197–206 [DOI] [PubMed] [Google Scholar]
- 48.Gorfu G, Cirelli KM, Melo MB, Mayer-Barber K, Crown D, et al. 2014. Dual role for inflammasome sensors NLRP1 and NLRP3 in murine resistance to Toxoplasma gondii. mBio 5(1):01117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johnson DC, Taabazuing CY, Okondo MC, Chui AJ, Rao SD, et al. 2018. DPP8/DPP9 inhibitor-induced pyroptosis for treatment of acute myeloid leukemia. Nat. Med 24(8):1151–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Swanson KV, Deng M, Ting JP-Y. 2019. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol 19(8):477–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50a.Andreeva L et al. NLRP3 cages revealed by full-length mouse NLRP3 structure control pathway activation. Cell 184, 6299–6312.e22 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nozaki K, Miao EA. 2019. A licence to kill during inflammation. Nature 570(7761):316–17 [DOI] [PubMed] [Google Scholar]
- 52.He Y, Zeng MY, Yang D, Motro B, Núñez G. 2016. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530(7590):354–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shi H, Wang Y, Li X, Zhan X, Tang M, et al. 2015. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat. Immunol 17(3):250–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schmid-Burgk JL, Chauhan D, Schmidt T, Ebert TS, Reinhardt J, et al. 2016. A genome-wide CRISPR (clustered regularly interspaced short palindromic repeats) screen identifies NEK7 as an essential component of NLRP3 inflammasome activation. J. Biol. Chem 291(1):103–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sharif H, Wang L, Wang WL, Magupalli VG, Andreeva L, et al. 2019. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature 570(7761):338–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li X, Thome S, Ma X, Amrute-Nayak M, Finigan A, et al. 2017. MARK4 regulates NLRP3 positioning and inflammasome activation through a microtubule-dependent mechanism. Nat. Commun 8:15986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen J, Chen ZJ. 2018. PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature 564(7734):71–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Magupalli VG, Negro R, Tian Y, Hauenstein AV, Di Caprio G, et al. 2020. HDAC6 mediates an aggresome-like mechanism for NLRP3 and pyrin inflammasome activation. Science 369(6510):eaas8995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maltez VI, Miao EA. 2016. Reassessing the evolutionary importance of inflammasomes. J. Immunol 196(3):956–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mitoma H, Hanabuchi S, Kim T, Bao M, Zhang Z, et al. 2013. The DHX33 RNA helicase senses cytosolic RNA and activates the NLRP3 inflammasome. Immunity 39(1):123–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ramos HJ, Lanteri MC, Blahnik G, Negash A, Suthar MS, et al. 2012. IL-1β signaling promotes CNS-intrinsic immune control of West Nile virus infection. PLOS Pathog. 8(11):e1003039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rajan JV, Rodriguez D, Miao EA, Aderem A. 2011. The NLRP3 inflammasome detects encephalomyocarditis virus and vesicular stomatitis virus infection. J. Virol 85(9):4167–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chakrabarti A, Banerjee S, Franchi L, Loo Y-M, Gale M Jr., et al. 2015. RNase L activates the NLRP3 inflammasome during viral infections. Cell Host Microbe 17(4):466–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ichinohe T, Pang IK, Iwasaki A. 2010. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat. Immunol 11(5):404–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chung W-C, Kang H-R, Yoon H, Kang S-J, Ting JP-Y, Song MJ. 2015. Influenza A virus NS1 protein inhibits the NLRP3 inflammasome. PLOS ONE 10(5):e0126456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Moriyama M, Chen I-Y, Kawaguchi A, Koshiba T, Nagata K, et al. 2016. The RNA- and TRIM25-binding domains of influenza virus NS1 protein are essential for suppression of NLRP3 inflammasome–mediated interleukin-1β secretion. J. Virol 90(8):4105–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, et al. 2009. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 30(4):556–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thomas PG, Dash P, Aldridge JR Jr., Ellebedy AH, Reynolds C, et al. 2009. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity 30(4):566–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. 2009. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J. Exp. Med 206(1):79–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pillai PS, Molony RD, Martinod K, Dong H, Pang IK, et al. 2016. Mx1 reveals innate pathways to antiviral resistance and lethal influenza disease. Science 352(6284):463–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang H, Lei X, Xiao X, Yang C, Lu W, et al. 2015. Reciprocal regulation between enterovirus 71 and the NLRP3 inflammasome. Cell Rep. 12(1):42–48 [DOI] [PubMed] [Google Scholar]
- 72.Shil NK, Pokharel SM, Banerjee AK, Hoffman M, Bose S. 2018. Inflammasome antagonism by human parainfluenza virus type 3C protein. J. Virol 92(4):e01776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Noritaka K, Takeshi I, Minako I, Yusuke Y. 2011. Measles virus V protein inhibits NLRP3 inflammasome-mediated interleukin-1β secretion. J. Virol 85(24):13019–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Komatsu T, Tanaka Y, Kitagawa Y, Koide N, Naiki Y, et al. 2018. Sendai virus V protein inhibits the secretion of interleukin-1β by preventing NLRP3 inflammasome assembly. J. Virol 92(19):e00842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yoshizumi T, Ichinohe T, Sasaki O, Otera H, Kawabata S-I, et al. 2014. Influenza A virus protein PB1-F2 translocates into mitochondria via Tom40 channels and impairs innate immunity. Nat. Commun 5:4713. [DOI] [PubMed] [Google Scholar]
- 76.McAuley JL, Tate MD, MacKenzie-Kludas CJ, Pinar A, Zeng W, et al. 2013. Activation of the NLRP3 inflammasome by IAV virulence protein PB1-F2 contributes to severe pathophysiology and disease. PLOS Pathog. 9(5):e1003392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Solbak SMØ, Sharma A, Bruns K, Röder R, Mitzner D, et al. 2013. Influenza A virus protein PB1-F2 from different strains shows distinct structural signatures. Biochim. Biophys. Acta Proteins Proteom 1834(2):568–82 [DOI] [PubMed] [Google Scholar]
- 78.Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, et al. 2006. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1β via Ipaf. Nat. Immunol 7(6):569–75 [DOI] [PubMed] [Google Scholar]
- 79.Franchi L, Amer A, Body-Malapel M, Kanneganti T-D, Özören N, et al. 2006. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1β in Salmonella-infected macrophages. Nat. Immunol 7(6):576–82 [DOI] [PubMed] [Google Scholar]
- 80.Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, et al. 2010. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. PNAS 107(7):3076–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhao Y, Yang J, Shi J, Gong Y-N, Lu Q, et al. 2011. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477(7366):596–600 [DOI] [PubMed] [Google Scholar]
- 82.Kofoed EM, Vance RE. 2011. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 477(7366):592–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rayamajhi M, Zak DE, Chavarria-Smith J, Vance RE, Miao EA. 2013. Mouse NAIP1 detects the type III secretion system needle protein. J. Immunol 191(8):3986–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yang J, Zhao Y, Shi J, Shao F. 2013. Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. PNAS 110(35):14408–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kortmann J, Brubaker SW, Monack DM. 2015. Inflammasome activation in primary human macrophages is dependent on flagellin. J. Immunol 195(3):815–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Reyes Ruiz VM, Ramirez J, Naseer N, Palacio NM, Siddarthan IJ, et al. 2017. Broad detection of bacterial type III secretion system and flagellin proteins by the human NAIP/NLRC4 inflammasome. PNAS 114(50):13242–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hu Z, Yan C, Liu P, Huang Z, Ma R, et al. 2013. Crystal structure of NLRC4 reveals its autoinhibition mechanism. Science 341(6142):172–75 [DOI] [PubMed] [Google Scholar]
- 88.Zhang L, Chen S, Ruan J, Wu J, Tong AB, et al. 2015. Cryo-EM structure of the activated NAIP2-NLRC4 inflammasome reveals nucleated polymerization. Science 350(6259):404–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hu Z, Zhou Q, Zhang C, Fan S, Cheng W, et al. 2015. Structural and biochemical basis for induced self-propagation of NLRC4. Science 350(6259):399–404 [DOI] [PubMed] [Google Scholar]
- 90.Tenthorey JL, Haloupek N, López-Blanco JR, Grob P, Adamson E, et al. 2017. The structural basis of flagellin detection by NAIP5: a strategy to limit pathogen immune evasion. Science 358(6365):888–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yang X, Yang F, Wang W, Lin G, Hu Z, et al. 2017. Structural basis for specific flagellin recognition by the NLR protein NAIP5. Cell Res. 28(1):35–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Karki R, Lee E, Place D, Samir P, Mavuluri J, et al. 2018. IRF8 regulates transcription of Naips for NLRC4 inflammasome activation. Cell 173(4):920–33.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dong X, Hu X, Bao Y, Li G, Yang X-D, et al. 2021. Brd4 regulates NLRC4 inflammasome activation by facilitating IRF8-mediated transcription of Naips. J. Cell Biol 220(3):e202005148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Qu Y, Misaghi S, Izrael-Tomasevic A, Newton K, Gilmour LL, et al. 2012. Phosphorylation of NLRC4 is critical for inflammasome activation. Nature 490(7421):539–42 [DOI] [PubMed] [Google Scholar]
- 95.Matusiak M, Van Opdenbosch N, Vande Walle L, Sirard J-C, Kanneganti T-D, Lamkanfi M. 2015. Flagellin-induced NLRC4 phosphorylation primes the inflammasome for activation by NAIP5. PNAS 112(5):1541–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Liu W, Liu X, Li Y, Zhao J, Liu Z, et al. 2017. LRRK2 promotes the activation of NLRC4 inflammasome during Salmonella typhimurium infection. J. Exp. Med 214(10):3051–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tenthorey JL, Chavez RA, Thompson TW, Deets KA, Vance RE, Rauch I. 2020. NLRC4 inflammasome activation is NLRP3- and phosphorylation-independent during infection and does not protect from melanoma. J. Exp. Med 217(7):e20191736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lacey CA, Miao EA. 2020. Programmed cell death in the evolutionary race against bacterial virulence factors. Cold Spring Harb. Perspect. Biol 12(2):a036459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Maltez VI, Tubbs AL, Cook KD, Aachoui Y, Liana Falcone E, et al. 2015. Inflammasomes coordinate pyroptosis and natural killer cell cytotoxicity to clear infection by a ubiquitous environmental bacterium. Immunity 43(5):987–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Aachoui Y, Kajiwara Y, Leaf IA, Mao D, Ting JP-Y, et al. 2015. Canonical inflammasomes drive IFN-γ to prime caspase-11 in defense against a cytosol-invasive bacterium. Cell Host Microbe 18(3):320–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Carvalho FA, Nalbantoglu I, Aitken JD, Uchiyama R, Su Y, et al. 2012. Cytosolic flagellin receptor NLRC4 protects mice against mucosal and systemic challenges. Mucosal Immunol. 5(3):288–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Franchi L, Kamada N, Nakamura Y, Burberry A, Kuffa P, et al. 2012. NLRC4-driven production of IL-1β discriminates between pathogenic and commensal bacteria and promotes host intestinal defense. Nat. Immunol 13(5):449–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Broz P, Newton K, Lamkanfi M, Mariathasan S, Dixit VM, Monack DM. 2010. Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J. Exp. Med 207(8):1745–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pereira MSF, Morgantetti GF, Massis LM, Horta CV, Hori JI, Zamboni DS. 2011. Activation of NLRC4 by flagellated bacteria triggers caspase-1-dependent and -independent responses to restrict Legionella pneumophila replication in macrophages and in vivo. J. Immunol 187(12):6447–55 [DOI] [PubMed] [Google Scholar]
- 105.Ceballos-Olvera I, Sahoo M, Miller MA, Del Barrio L, Re F. 2011. Inflammasome-dependent pyroptosis and IL-18 protect against Burkholderia pseudomallei lung infection while IL-1β is deleterious. PLOS Pathog. 7(12):e1002452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.LaRock DL, Chaudhary A, Miller SI. 2015. Salmonellae interactions with host processes. Nat. Rev. Microbiol 13(4):191–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cummings LA, Wilkerson WD, Bergsbaken T, Cookson BT. 2006. In vivo, fliC expression by Salmonella enterica serovar Typhimurium is heterogeneous, regulated by ClpX, and anatomically restricted. Mol. Microbiol 61(3):795–809 [DOI] [PubMed] [Google Scholar]
- 108.Laughlin RC, Knodler LA, Barhoumi R, Payne HR, Wu J, et al. 2014. Spatial segregation of virulence gene expression during acute enteric infection with Salmonella enterica serovar Typhimurium. mBio 5(1):e00946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Miao EA, Rajan JV. 2011. Salmonella and caspase-1: a complex interplay of detection and evasion. Front. Microbiol 2:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sauer J-D, Pereyre S, Archer KA, Burke TP, Hanson B, et al. 2011. Listeria monocytogenes engineered to activate the Nlrc4 inflammasome are severely attenuated and are poor inducers of protective immunity. PNAS 108(30):12419–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Warren SE, Duong H, Mao DP, Armstrong A, Rajan J, et al. 2011. Generation of a Listeria vaccine strain by enhanced caspase-1 activation. Eur. J. Immunol 41(7):1934–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tourlomousis P, Wright JA, Bittante AS, Hopkins LJ, Webster SJ, et al. 2020. Modifying bacterial flagellin to evade Nod-like receptor CARD 4 recognition enhances protective immunity against Salmonella. Nat. Microbiol 5(12):1588–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, Flavell RA. 2007. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J. Exp. Med 204(13):3235–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Grenier JM, Wang L, Manji GA, Huang WJ, Al-Garawi A, et al. 2002. Functional screening of five PYPAF family members identifies PYPAF5 as a novel regulator of NF-κB and caspase-1. FEBS Lett. 530(1–3):73–78 [DOI] [PubMed] [Google Scholar]
- 115.Levy M, Thaiss CA, Zeevi D, Dohnalová L, Zilberman-Schapira G, et al. 2015. Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell 163(6):1428–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, et al. 2011. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145(5):745–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hara H, Seregin SS, Yang D, Fukase K, Chamaillard M, et al. 2018. The NLRP6 inflammasome recognizes lipoteichoic acid and regulates gram-positive pathogen infection. Cell 175(6):1651–64.e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Leng F, Yin H, Qin S, Zhang K, Guan Y, et al. 2020. NLRP6 self-assembles into a linear molecular platform following LPS binding and ATP stimulation. Sci. Rep 10:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118a.Shen C et al. Phase separation drives RNA virus-induced activation of the NLRP6 inflammasome. Cell 184, 5759–5774.e20 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mukherjee S, Kumar R, Lenou ET, Basrur V, Kontoyiannis DL, et al. 2020. Deubiquitination of NLRP6 inflammasome by Cyld critically regulates intestinal inflammation. Nat. Immunol 21(6):626–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jorgensen I, Miao EA. 2015. Pyroptotic cell death defends against intracellular pathogens. Immunol. Rev 265(1):130–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tsuji NM, Tsutsui H, Seki E, Kuida K, Okamura H, et al. 2004. Roles of caspase-1 in Listeria infection in mice. Int. Immunol 16(2):335–43 [DOI] [PubMed] [Google Scholar]
- 122.Mueller NJ, Wilkinson RA, Fishman JA. 2002. Listeria monocytogenes infection in caspase-11-deficient mice. Infect. Immun 70(5):2657–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Clark SE, Schmidt RL, McDermott DS, Lenz LL. 2018. A Batf3/Nlrp3/IL-18 axis promotes natural killer cell IL-10 production during Listeria monocytogenes infection. Cell Rep. 23(9):2582–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wang P, Zhu S, Yang L, Cui S, Pan W, et al. 2015. Nlrp6 regulates intestinal antiviral innate immunity. Science 350(6262):826–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wlodarska M, Thaiss CA, Nowarski R, Henao-Mejia J, Zhang J-P, et al. 2014. NLRP6 inflammasome orchestrates the colonic host–microbial interface by regulating goblet cell mucus secretion. Cell 156(5):1045–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gálvez EJC, Iljazovic A, Gronow A, Flavell R, Strowig T. 2017. Shaping of intestinal microbiota in Nlrp6- and Rag2-deficient mice depends on community structure. Cell Rep. 21(13):3914–26 [DOI] [PubMed] [Google Scholar]
- 127.Mamantopoulos M, Ronchi F, Van Hauwermeiren F, Vieira-Silva S, Yilmaz B, et al. 2017. Nlrp6- and ASC-dependent inflammasomes do not shape the commensal gut microbiota composition. Immunity 47(2):339–48.e4 [DOI] [PubMed] [Google Scholar]
- 128.Lemire P, Robertson SJ, Maughan H, Tattoli I, Streutker CJ, et al. 2017. The NLR protein NLRP6 does not impact gut microbiota composition. Cell Rep. 21(13):3653–61 [DOI] [PubMed] [Google Scholar]
- 129.Tian X, Pascal G, Monget P. 2009. Evolution and functional divergence of NLRP genes in mammalian reproductive systems. BMC Evol. Biol 9:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kufer TA, Sansonetti PJ. 2011. NLR functions beyond pathogen recognition. Nat. Immunol 12(2):121–28 [DOI] [PubMed] [Google Scholar]
- 131.Zhu S, Ding S, Wang P, Wei Z, Pan W, et al. 2017. Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature 546(7660):667–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kanzaki S, Tamura S, Ito T, Wakabayashi M, Saito K, et al. 2020. Involvement of Nlrp9a/b/c in mouse preimplantation development. Reproduction 160(2):181–91 [DOI] [PubMed] [Google Scholar]
- 133.Jin T, Perry A, Jiang J, Smith P, Curry JA, et al. 2012. Structures of the HIN domain:DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity 36(4):561–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Roberts TL, Idris A, Dunn JA, Kelly GM, Burnton CM, et al. 2009. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 323(5917):1057–60 [DOI] [PubMed] [Google Scholar]
- 135.Wang P-H, Ye Z-W, Deng J-J, Siu K-L, Gao W-W, et al. 2018. Inhibition of AIM2 inflammasome activation by a novel transcript isoform of IFI16. EMBO Rep. 19(10):e45737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Khare S, Ratsimandresy RA, de Almeida L, Cuda CM, Rellick SL, et al. 2014. The PYRIN domain–only protein POP3 inhibits ALR inflammasomes and regulates responses to infection with DNA viruses. Nat. Immunol 15(4):343–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Meunier E, Wallet P, Dreier RF, Costanzo S, Anton L, et al. 2015. Guanylate-binding proteins promote activation of the AIM2 inflammasome during infection with Francisella novicida. Nat. Immunol 16(5):476–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Man SM, Karki R, Malireddi RKS, Neale G, Vogel P, et al. 2015. The transcription factor IRF1 and guanylate-binding proteins target activation of the AIM2 inflammasome by Francisella infection. Nat. Immunol 16(5):467–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Man SM, Karki R, Sasai M, Place DE, Kesavardhana S, et al. 2016. IRGB10 liberates bacterial ligands for sensing by the AIM2 and caspase-11-NLRP3 inflammasomes. Cell 167(2):382–96.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Fernandes-Alnemri T, Yu J-W, Juliana C, Solorzano L, Kang S, et al. 2010. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat. Immunol 11(5):385–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Rathinam VAK, Jiang Z, Waggoner SN, Sharma S, Cole LE, et al. 2010. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol 11(5):395–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, et al. 2009. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458(7237):514–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Fernandes-Alnemri T, Yu J-W, Datta P, Wu J, Alnemri ES. 2009. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458(7237):509–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Reinholz M, Kawakami Y, Salzer S, Kreuter A, Dombrowski Y, et al. 2013. HPV16 activates the AIM2 inflammasome in keratinocytes. Arch. Dermatol. Res 305(8):723–32 [DOI] [PubMed] [Google Scholar]
- 145.Yogarajah T, Ong KC, Perera D, Wong KT. 2017. AIM2 inflammasome–mediated pyroptosis in enterovirus A71–infected neuronal cells restricts viral replication. Sci. Rep 7:5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Karki R, Man SM, Malireddi RKS, Gurung P, Vogel P, et al. 2015. Concerted activation of the AIM2 and NLRP3 inflammasomes orchestrates host protection against Aspergillus infection. Cell Host Microbe 17(3):357–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Maruzuru Y, Ichinohe T, Sato R, Miyake K, Okano T, et al. 2018. Herpes simplex virus 1 VP22 inhibits AIM2-dependent inflammasome activation to enable efficient viral replication. Cell Host Microbe 23(2):254–65.e7 [DOI] [PubMed] [Google Scholar]
- 148.Huang Y, Ma D, Huang H, Lu Y, Liao Y, et al. 2017. Interaction between HCMV pUL83 and human AIM2 disrupts the activation of the AIM2 inflammasome. Virol. J 14(1):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kerur N, Veettil MV, Sharma-Walia N, Bottero V, Sadagopan S, et al. 2011. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi sarcoma–associated herpesvirus infection. Cell Host Microbe 9(5):363–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Monroe KM, Yang Z, Johnson JR, Geng X, Doitsh G, et al. 2014. IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science 343(6169):428–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, et al. 2011. Non-canonical inflammasome activation targets caspase-11. Nature 479(7371):117–21 [DOI] [PubMed] [Google Scholar]
- 152.Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. 2013. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341(6151):1250–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, et al. 2013. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341(6151):1246–49 [DOI] [PubMed] [Google Scholar]
- 154.Shi J, Zhao Y, Wang Y, Gao W, Ding J, et al. 2014. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514(7521):187–92 [DOI] [PubMed] [Google Scholar]
- 155.Lee BL, Stowe IB, Gupta A, Kornfeld OS, Roose-Girma M, et al. 2018. Caspase-11 auto-proteolysis is crucial for noncanonical inflammasome activation. J. Exp. Med 215(9):2279–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ross C, Chan AH, Von Pein J, Boucher D, Schroder K. 2018. Dimerization and auto-processing induce caspase-11 protease activation within the non-canonical inflammasome. Life Sci. Alliance 1(6):e201800237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, et al. 2013. Caspase-11 protects against bacteria that escape the vacuole. Science 339(6122):975–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Meunier E, Dick MS, Dreier RF, Schürmann N, Kenzelmann Broz D, et al. 2014. Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature 509(7500):366–70 [DOI] [PubMed] [Google Scholar]
- 159.Schauvliege R, Vanrobaeys J, Schotte P, Beyaert R. 2002. Caspase-11 gene expression in response to lipopolysaccharide and interferon-γ requires nuclear factor-κB and signal transducer and activator of transcription (STAT) 1. J. Biol. Chem 277(44):41624–30 [DOI] [PubMed] [Google Scholar]
- 160.Rathinam VAK, Vanaja SK, Waggoner L, Sokolovska A, Becker C, et al. 2012. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 150(3):606–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Olszewski MA, Gray J, Vestal DJ. 2006. In silico genomic analysis of the human and murine guanylate-binding protein (GBP) gene clusters. J. Interferon Cytokine Res 26(5):328–52 [DOI] [PubMed] [Google Scholar]
- 162.Wandel MP, Kim B-H, Park E-S, Boyle KB, Nayak K, et al. 2020. Guanylate-binding proteins convert cytosolic bacteria into caspase-4 signaling platforms. Nat. Immunol 21(8):880–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Santos JC, Boucher D, Schneider LK, Demarco B, Dilucca M, et al. 2020. Human GBP1 binds LPS to initiate assembly of a caspase-4 activating platform on cytosolic bacteria. Nat. Commun 11:3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kutsch M, Sistemich L, Lesser CF, Goldberg MB, Herrmann C, Coers J. 2020. Direct binding of polymeric GBP1 to LPS disrupts bacterial cell envelope functions. EMBO J. 39(13):e104926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wiersinga WJ, van der Poll T, White NJ, Day NP, Peacock SJ. 2006. Melioidosis: insights into the pathogenicity of Burkholderia pseudomallei. Nat. Rev. Microbiol 4(4):272–82 [DOI] [PubMed] [Google Scholar]
- 166.Wang J, Deobald K, Re F. 2019. Gasdermin D protects from melioidosis through pyroptosis and direct killing of bacteria. J. Immunol 202(12):3468–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kovacs SB, Oh C, Maltez VI, McGlaughon BD, Verma A, et al. 2020. Neutrophil caspase-11 is essential to defend against a cytosol-invasive bacterium. Cell Rep. 32(4):107967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Chen KW, Groß CJ, Sotomayor FV, Stacey KJ, Tschopp J, et al. 2014. The neutrophil NLRC4 inflammasome selectively promotes IL-1β maturation without pyroptosis during acute Salmonella challenge. Cell Rep. 8(2):570–82 [DOI] [PubMed] [Google Scholar]
- 169.Karmakar M, Minns M, Greenberg EN, Diaz-Aponte J, Pestonjamasp K, et al. 2020. N-GSDMD trafficking to neutrophil organelles facilitates IL-1β release independently of plasma membrane pores and pyroptosis. Nat. Commun 11:2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Balakrishnan A, Karki R, Berwin B, Yamamoto M, Kanneganti T-D. 2018. Guanylate binding proteins facilitate caspase-11-dependent pyroptosis in response to type 3 secretion system–negative Pseudomonas aeruginosa. Cell Death Discov. 4:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Garai P, Berry L, Moussouni M, Bleves S, Blanc-Potard A-B. 2019. Killing from the inside: intracellular role of T3SS in the fate of Pseudomonas aeruginosa within macrophages revealed by mgtC and oprF mutants. PLOS Pathog. 15(6):e1007812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, et al. 2012. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature 490(7419):288–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Chen KW, Monteleone M, Boucher D, Sollberger G, Ramnath D, et al. 2018. Noncanonical inflammasome signaling elicits gasdermin D–dependent neutrophil extracellular traps. Sci. Immunol 3(26):eaar6676 [DOI] [PubMed] [Google Scholar]
- 174.Akhter A, Caution K, Abu Khweek A, Tazi M, Abdulrahman BA, et al. 2012. Caspase-11 promotes the fusion of phagosomes harboring pathogenic bacteria with lysosomes by modulating actin polymerization. Immunity 37(1):35–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Case CL, Kohler LJ, Lima JB, Strowig T, de Zoete MR, et al. 2013. Caspase-11 stimulates rapid flagellin-independent pyroptosis in response to Legionella pneumophila. PNAS 110(5):1851–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Casson CN, Copenhaver AM, Zwack EE, Nguyen HT, Strowig T, et al. 2013. Caspase-11 activation in response to bacterial secretion systems that access the host cytosol. PLOS Pathog. 9(6):e1003400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Cerqueira DM, Pereira MSF, Silva ALN, Cunha LD, Zamboni DS. 2015. Caspase-1 but not caspase-11 is required for NLRC4-mediated pyroptosis and restriction of infection by flagellated Legionella species in mouse macrophages and in vivo. J. Immunol 195(5):2303–11 [DOI] [PubMed] [Google Scholar]
- 178.Krause K, Caution K, Badr A, Hamilton K, Saleh A, et al. 2018. CASP4/caspase-11 promotes autophagosome formation in response to bacterial infection. Autophagy 14(11):1928–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Estfanous S, Krause K, Anne MNK, Eltobgy M, Caution K, et al. 2021. Gasdermin D restricts Burkholderia cenocepacia infection in vitro and in vivo. Sci. Rep 11:12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Xu H, Yang J, Gao W, Li L, Li P, et al. 2014. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 513(7517):237–41 [DOI] [PubMed] [Google Scholar]
- 181.Kobayashi T, Ogawa M, Sanada T, Mimuro H, Kim M, et al. 2013. The Shigella OspC3 effector inhibits caspase-4, antagonizes inflammatory cell death, and promotes epithelial infection. Cell Host Microbe 13(5):570–83 [DOI] [PubMed] [Google Scholar]
- 181a.Li Z et al. Shigella evades pyroptosis by arginine ADP-riboxanation of caspase-11. Nature 599, 290–295 (2021). [DOI] [PubMed] [Google Scholar]
- 181b.Oh C, Verma A, Hafeez M, Hogland B & Aachoui Y Shigella OspC3 suppresses murine cytosolic LPS sensing. Iscience 24, 102910 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Papayannopoulos V 2017. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol 18(2):134–47 [DOI] [PubMed] [Google Scholar]
- 183.Jorgensen I, Lopez JP, Laufer SA, Miao EA. 2016. IL-1β, IL-18, and eicosanoids promote neutrophil recruitment to pore-induced intracellular traps following pyroptosis. Eur. J. Immunol 46(12):2761–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, et al. 2018. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol 3(26):eaar6689 [DOI] [PubMed] [Google Scholar]
- 185.Rauch I, Deets KA, Ji DX, von Moltke J, Tenthorey JL, et al. 2017. NAIP-NLRC4 inflammasomes coordinate intestinal epithelial cell expulsion with eicosanoid and IL-18 release via activation of caspase-1 and −8. Immunity 46(4):649–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Sellin ME, Müller AA, Felmy B, Dolowschiak T, Diard M, et al. 2014. Epithelium-intrinsic NAIP/NLRC4 inflammasome drives infected enterocyte expulsion to restrict Salmonella replication in the intestinal mucosa. Cell Host Microbe 16(2):237–48 [DOI] [PubMed] [Google Scholar]
- 187.Knodler LA, Crowley SM, Sham HP, Yang H, Wrande M, et al. 2014. Noncanonical inflammasome activation of caspase-4/caspase-11 mediates epithelial defenses against enteric bacterial pathogens. Cell Host Microbe 16(2):249–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Lammert CR, Frost EL, Bellinger CE, Bolte AC, McKee CA, et al. 2020. AIM2 inflammasome surveillance of DNA damage shapes neurodevelopment. Nature 580(7805):647–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Heneka MT, McManus RM, Latz E. 2018. Inflammasome signalling in brain function and neurodegenerative disease. Nat. Rev. Neurosci 19(10):610–21 [DOI] [PubMed] [Google Scholar]
- 190.Li H, Saucedo-Cuevas L, Regla-Nava JA, Chai G, Sheets N, et al. 2016. Zika virus infects neural progenitors in the adult mouse brain and alters proliferation. Cell Stem Cell 19(5):593–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Tang H, Hammack C, Ogden SC, Wen Z, Qian X, et al. 2016. Zika virus infects human cortical neural progenitors and attenuates their growth. Cell Stem Cell 18(5):587–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.He Z, An S, Chen J, Zhang S, Tan C, et al. 2020. Neural progenitor cell pyroptosis contributes to Zika virus–induced brain atrophy and represents a therapeutic target. PNAS 117(38):23869–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Sun L, Wang H, Wang Z, He S, Chen S, et al. 2012. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148(1/2):213–27 [DOI] [PubMed] [Google Scholar]
- 194.Galluzzi L, Kepp O, Chan FK-M, Kroemer G. 2017. Necroptosis: mechanisms and relevance to disease. Annu. Rev. Pathol. Mech. Dis 12:103–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Maelfait J, Liverpool L, Rehwinkel J. 2020. Nucleic acid sensors and programmed cell death. J. Mol. Biol 432(2):552–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195a.Wittig B, Dorbic T, Rich A. 1991. Transcription is associated with Z-DNA formation in metabolically active permeabilized mammalian cell nuclei. PNAS 88(6):2259–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195b.Potaman VN, Sinden RR. 2013. DNA: Alternative Conformations and Biology. Austin, TX: Landes Biosci. [Google Scholar]
- 195c.Liu G & Gack MU Distinct and Orchestrated Functions of RNA Sensors in Innate Immunity. Immunity 53, 26–42 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Upton JW, Kaiser WJ, Mocarski ES. 2010. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe 7(4):302–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Lin J, Kumari S, Kim C, Van T-M, Wachsmuth L, et al. 2016. RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature 540(7631):124–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Newton K, Wickliffe KE, Maltzman A, Dugger DL, Strasser A, et al. 2016. RIPK1 inhibits ZBP1-driven necroptosis during development. Nature 540(7631):129–33 [DOI] [PubMed] [Google Scholar]
- 199.Thapa RJ, Ingram JP, Ragan KB, Nogusa S, Boyd DF, et al. 2016. DAI senses influenza A virus genomic RNA and activates RIPK3-dependent cell death. Cell Host Microbe 20(5):674–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Zhang T, Yin C, Boyd DF, Quarato G, Ingram JP, et al. 2020. Influenza virus Z-RNAs induce ZBP1-mediated necroptosis. Cell 180(6):1115–29.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Sridharan H, Ragan KB, Guo H, Gilley RP, Landsteiner VJ, et al. 2017. Murine cytomegalovirus IE3-dependent transcription is required for DAI/ZBP1-mediated necroptosis. EMBO Rep. 18(8):1429–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Maelfait J, Liverpool L, Bridgeman A, Ragan KB, Upton JW, Rehwinkel J. 2017. Sensing of viral and endogenous RNA by ZBP1/DAI induces necroptosis. EMBO J. 36(17):2529–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Cho YS, Challa S, Moquin D, Genga R, Ray TD, et al. 2009. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137(6):1112–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Koehler H, Cotsmire S, Langland J, Kibler KV, Kalman D, et al. 2017. Inhibition of DAI-dependent necroptosis by the Z-DNA binding domain of the vaccinia virus innate immune evasion protein, E3. PNAS 114(43):11506–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Koehler H, Cotsmire S, Zhang T, Balachandran S, Upton JW, et al. 2021. Vaccinia virus E3 prevents sensing of Z-RNA to block ZBP1-dependent necroptosis. Cell Host Microbe 29(8):1266–76.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Guo H, Gilley RP, Fisher A, Lane R, Landsteiner VJ, et al. 2018. Species-independent contribution of ZBP1/DAI/DLM-1-triggered necroptosis in host defense against HSV1. Cell Death Dis. 9(8):1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Nogusa S, Thapa RJ, Dillon CP, Liedmann S, Oguin TH 3rd, et al. 2016. RIPK3 activates parallel pathways of MLKL-driven necroptosis and FADD-mediated apoptosis to protect against influenza A virus. Cell Host Microbe 20(1):13–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Kuriakose T, Man SM, Malireddi RKS, Karki R, Kesavardhana S, et al. 2016. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol. 1(2):aag2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Kesavardhana S, Kuriakose T, Guy CS, Samir P, Malireddi RKS, et al. 2017. ZBP1/DAI ubiquitination and sensing of influenza vRNPs activate programmed cell death. J. Exp. Med 214(8):2217–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Kesavardhana S, Malireddi RKS, Burton AR, Porter SN, Vogel P, et al. 2020. The Zα2 domain of ZBP1 is a molecular switch regulating influenza-induced PANoptosis and perinatal lethality during development. J. Biol. Chem 295(24):8325–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Ishii KJ, Kawagoe T, Koyama S, Matsui K, Kumar H, et al. 2008. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature 451(7179):725–29 [DOI] [PubMed] [Google Scholar]
- 212.Upton JW, Kaiser WJ, Mocarski ES. 2012. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11(3):290–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Koehler HS, Feng Y, Mandal P, Mocarski ES. 2020. Recognizing limits of Z-nucleic acid binding protein (ZBP1/DAI/DLM1) function. FEBS J. 287(20):4362–69 [DOI] [PubMed] [Google Scholar]
- 214.Upton JW, Kaiser WJ, Mocarski ES. 2012. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11(3):290–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Liu Z, Nailwal H, Rector J, Rahman MM, Sam R, et al. 2021. A class of viral inducer of degradation of the necroptosis adaptor RIPK3 regulates virus-induced inflammation. Immunity 54(2):247–58.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Huang Z, Wu S-Q, Liang Y, Zhou X, Chen W, et al. 2015. RIP1/RIP3 binding to HSV-1 ICP6 initiates necroptosis to restrict virus propagation in mice. Cell Host Microbe 17(2):229–42 [DOI] [PubMed] [Google Scholar]
- 217.Guo H, Omoto S, Harris PA, Finger JN, Bertin J, et al. 2015. Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe 17(2):243–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Lei X, Zhang Z, Xiao X, Qi J, He B, Wang J. 2017. Enterovirus 71 inhibits pyroptosis through cleavage of gasdermin D. J. Virol 91(18):e01069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Tsuchiya K, Nakajima S, Hosojima S, Nguyen DT, Hattori T, et al. 2019. Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat. Commun 10:2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Doerflinger M, Deng Y, Whitney P, Salvamoser R, Engel S, et al. 2020. Flexible usage and interconnectivity of diverse cell death pathways protect against intracellular infection. Immunity 53(3):533–47.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Pierini R, Juruj C, Perret M, Jones CL, Mangeot P, et al. 2012. AIM2/ASC triggers caspase-8-dependent apoptosis in Francisella-infected caspase-1-deficient macrophages. Cell Death Differ. 19(10):1709–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Lee BL, Mirrashidi KM, Stowe IB, Kummerfeld SK, Watanabe C, et al. 2018. ASC- and caspase-8-dependent apoptotic pathway diverges from the NLRC4 inflammasome in macrophages. Sci. Rep 8:3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Taabazuing CY, Okondo MC, Bachovchin DA. 2017. Pyroptosis and apoptosis pathways engage in bidirectional crosstalk in monocytes and macrophages. Cell Chem. Biol 24(4):507–14.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Chen KW, Demarco B, Heilig R, Shkarina K, Boettcher A, et al. 2019. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J. 38(10):e101638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Orning P, Lien E. 2021. Multiple roles of caspase-8 in cell death, inflammation, and innate immunity. J. Leukoc. Biol 109(1):121–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Wang Y, Gao W, Shi X, Ding J, Liu W, et al. 2017. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547(7661):99–103 [DOI] [PubMed] [Google Scholar]
- 227.Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. 2017. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun 8:14128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Orning P, Weng D, Starheim K, Ratner D, Best Z, et al. 2018. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 362(6418):1064–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, et al. 2018. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. PNAS 115(46):E10888–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Zhou Z, He H, Wang K, Shi X, Wang Y, et al. 2020. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 368(6494):eaaz7548 [DOI] [PubMed] [Google Scholar]
- 231.Hansen JM, de Jong MF, Wu Q, Zhang L-S, Heisler DB, et al. 2021. Pathogenic ubiquitination of GSDMB inhibits NK cell bactericidal functions. Cell 184(12):3178–91.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231a.Luchetti G, Roncaioli JL, Chaves RA, Schubert AF, Kofoed EM, et al. 2021. Shigella ubiquitin ligase IpaH7.8 targets gasdermin D for degradation to prevent pyroptosis and enable infection. Cell Host Microbe 29, 1521–1530.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Zhang Z, Zhang Y, Xia S, Kong Q, Li S, et al. 2020. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 579(7799):415–20 [DOI] [PMC free article] [PubMed] [Google Scholar]