

SUMMARY
Nuclear pores are essential for nuclear-cytoplasmic transport. Whether and how cells change nuclear pores to alter nuclear transport and cellular function is unknown. Here, we show that rat heart muscle cells (cardiomyocytes) undergo a 63% decrease in nuclear pore numbers during maturation, which changes their responses to extracellular signals. The maturation-associated decline in nuclear pore numbers is associated with lower nuclear import of signaling proteins, such as Mitogen-activated Protein Kinase (MAPK). Experimental reduction of nuclear pore numbers decreased nuclear import of signaling proteins, resulting in decreased expression of immediate-early genes. In a mouse model of high blood pressure, reduction of nuclear pore numbers improved adverse heart remodeling and reduced progression to lethal heart failure. The decrease in nuclear pore numbers in cardiomyocyte maturation and resulting functional changes demonstrate how terminally differentiated cells permanently alter their handling of information flux across the nuclear envelope and, with that, their behavior.
Keywords: Nuclear pore, cardiomyocyte, nuclear transport, cardiac remodeling, cardiac hypertrophy, MAP kinase
eTOC Blurb:
Nuclear pores are essential for nuclear-cytoplasmic transport of proteins. Han et al. show that heart muscle cells undergo a substantial decrease in nuclear pore numbers during maturation. Lower nuclear pore numbers reduce nuclear import of proteins, which reduces stress response in cardiomyocytes and improves heart function in mice.
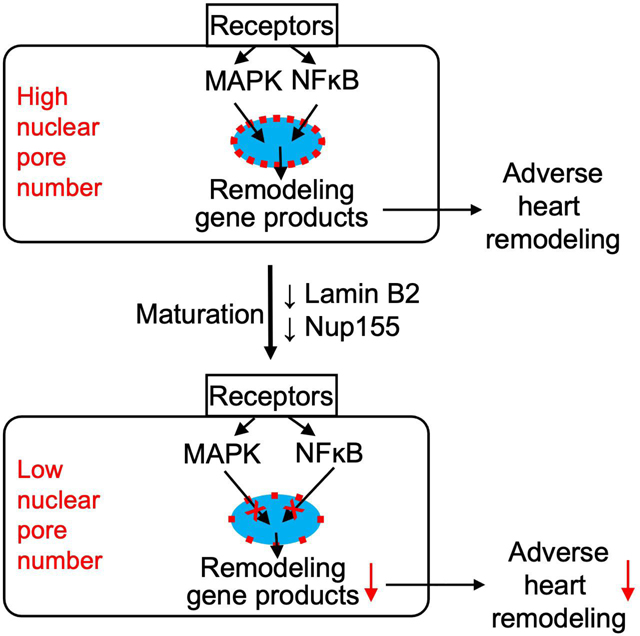
INTRODUCTION
The nuclear envelope is a double phospholipid bilayer separating the nucleus from the cytoplasm. Nuclear pores (NP) transport macromolecules into and out of the nucleus at a rate of several hundred molecules per second, and this includes signal transduction proteins to regulate gene transcription (Knockenhauer and Schwartz, 2016). Whether and how cells alter NPs to modulate the access of signal transduction proteins into nuclei and, with that, their transcriptional and functional responses, is unknown(Miller et al., 1991).
One property that can be assessed is the number of NPs per nucleus. NP numbers vary widely between cell types and species: whereas yeast have approximately 100 NP per nucleus, newt heart cells have approximately 12,000 (Maul and Deaven, 1977). NP numbers are constant in adult rat liver and brain cells, suggesting that cells establish the NP numbers for their lifetime when they terminally differentiate (D’Angelo et al., 2009). We present the observation that heart muscle cells (cardiomyocytes) decrease the number of their NPs during maturation toward the post-mitotic state, and we provide evidence for the significance of this decrease for nuclear transport and for the response of the heart to stress.
After mammalian cardiomyocytes enter the terminally differentiated state and become post-mitotic, they grow by enlargement (hypertrophy). In mice and rats, this transition occurs within weeks after birth and is indicated by the formation of binucleated cardiomyocytes (Kirillova et al., 2021). Although hypertrophy is a mechanism of developmental heart growth, in terminally differentiated cardiomyocytes, cardiac stress induces maladaptive hypertrophy, which leads to pathological remodeling of the heart, ultimately resulting in heart failure. Cardiac remodeling due to high blood pressure is a significant public health problem(Heineke and Molkentin, 2006). Cardiac maladaptive remodeling is initiated by extracellular factors that activate G-protein coupled receptors (GPCR) and receptor tyrosine kinases (RTK), which activate intracellular signaling pathways, chiefly, the mitogen-activated protein kinase (MAPK) pathways, leading to gene transcription changes(Heineke and Molkentin, 2006). Although cardiac maladaptive signaling at the level of GPCR, RTK, MAPK, and gene transcription is well understood, the fundamental mechanisms that could alter the propagation of these signals into the nucleus to determine the maladaptive response in cardiomyocytes are unknown. We have developed a hypothesis based on our observation that the number of NPs decreases during cardiomyocyte maturation. We present testing of the hypothesis that the decrease of NP numbers decreases nuclear import of signaling proteins, which changes adaptive gene transcription, and, with that, improves the cardiac response to stress.
RESULTS
Cardiomyocytes decrease their nuclear pore (NP) numbers during maturation
NPs are cylindrical macromolecular complexes of approximately 120 – 180 nm outer diameter. Multiples of ∼34 different proteins, called nucleoporins (Nups), are assembled into NPs consisting of ∼1,000 Nups. Nups are arranged in 8-fold symmetry around a ∼5 nm central opening(Lin and Hoelz, 2019). Due to their very large size, individual NPs can be visualized with microscopy(Knockenhauer and Schwartz, 2016). Using immunofluorescence confocal microscopy, we visualized NPs in less mature (fetal and neonatal) and in terminally differentiated adult rat ventricular cardiomyocyte nuclei (Fig. 1a, Suppl. Video S1). Quantification showed that the number of NPs per nucleus decreased from 1,856 ± 66 NPs in fetal (E14.5) to 1,040 ± 53 in neonatal (P2), and further to 678 ± 32 in adult (P60) rat cardiomyocytes (Fig. 1b). This overall 2.7-fold decrease corresponds to a 63% decline of the NP number.
Figure 1. Cardiomyocytes decrease their number of nuclear pores (NP) during maturation.

Cardiomyocytes from fetal (E14.5), neonatal (P2), and adult (P60) rats were analyzed. (a) Nuclear pores detected with antibody mAb 414. Confocal microscopy photomicrographs of nuclei of the indicated developmental stages. The lower row are corresponding 3D reconstructions shown in Supplemental Video S1. Scale bars 2 μm. (b) Decrease of NP numbers with advancing cardiomyocyte differentiation. Three experiments with n=44 fetal, n=45 neonatal, n=47 adult cardiomyocytes. (c) NP counts in binucleated cardiomyocytes are lower than in mononucleated cardiomyocytes. (d) Linear regression shows slope of 1.16, indicating same NP numbers in both nuclei of binucleated cardiomyocytes. R2, regression coefficient. (e) Freeze-fracture electron microscopy photograph of fetal (E14.5) and neonatal (P2) rat cardiomyocyte nuclei. Scale bar 200 nm. (f) Quantification of electron microscopy photographs. N=15 nuclei (3 fetal rat hearts), N= 8 nuclei (3 neonatal rat hearts). (g) Heat map of nucleoporin (Nup) mRNA expression in single mouse cardiomyocytes. Nups were grouped according to their subcomplexes(Raices and D’Angelo, 2012). Each symbol represents one cardiomyocyte or nucleus (b-d, f). Statistical significance tested with Student’s t-test (f) and ANOVA followed by multiple comparison (b, c). Mean ± SEM are indicated with horizontal lines.
Given that, in mice and rats, 80–90% of cardiomyocytes become binucleated after birth, and they are considered terminally differentiated (Kirillova et al., 2021), we compared NP numbers between mono- and binucleated cardiomyocytes. In neonatal rats, binucleated cardiomyocytes had significantly lower NP numbers than mononucleated cardiomyocytes (Fig. 1c). The two nuclei in binucleated cardiomyocytes showed no differences in NP numbers (Fig. 1d).
We cross-validated the quantification of NP obtained by freeze-fracture replica electron microscopy (EM, Fig. 1e, f), the method used for a classical NP quantification study (Maul and Deaven, 1977). As this method visualizes only a small portion of a nucleus, NP area densities rather than total number of NPs per nucleus have to be compared. Fetal (E14.5) rat cardiomyocyte nuclei showed 7.3 ± 0.5 NP/μm2 and neonatal (P2) 4.6 ± 0.2 NP/μm2. These results agree with our quantifications by confocal microcopy (7.1 ± 0.1 NP/μm2 in fetal and 5.9 ± 0.2 NP/μm2 in neonatal cardiomyocyte nuclei, see Fig. 1b). The freeze-fracture approach did not yield sufficient material for investigating adults. Taken together, these results support the conclusion that NP numbers and area densities decrease during cardiomyocyte maturation (Fig. 1b, e, f).
We compared nucleoporin (Nup) expression in different maturation stages of cardiomyocytes. To examine Nup mRNA changes, we analyzed our published deep transcriptional profiles of single cardiomyocytes(Han et al., 2020; Liu et al., 2019). This dataset was generated from single mouse cardiomyocytes [embryonic day 14.5 (E14.5), postnatal day 5 (P5), postnatal day 19 (P19)], which were analyzed using the Eberwine method to accurately represent low-abundance transcripts (Kim et al., 2011a). The majority of Nup mRNAs decreased during cardiomyocyte maturation, with the exception of TPR and Nup45 (Fig. 1g). Largely in agreement with the single-cell transcriptome data, Western blotting results of selected Nups (Suppl. Fig. S1) showed that Nup107 and Nup98 decreased significantly from fetal to neonatal cardiomyocytes. Nup62 protein levels were preserved in neonatal compared to fetal cardiomyocytes, whereas Nup153 was higher in the neonatal cardiomyocytes. Adult cardiomyocytes expressed very low level of all the selected Nups (Nup107, Nup98, Nup62 and Nup153). In conclusion, cardiomyocytes decrease Nup expression at the mRNA and protein level during maturation.
Lower NP numbers are associated with lower nuclear import of signaling proteins
To determine the functional significance of the decrease in NP numbers, we examined nuclear import of signaling proteins. We opted to study proteins with a molecular mass > 40 kDa, i.e., ones that require active nuclear transport (Knockenhauer and Schwartz, 2016). We assayed nuclear import of extracellular signal-regulated kinase (ERK) using a previously validated photoactivatable Dronpa-ERK1 reporter (Ando et al., 2004). Fetal rat cardiomyocytes (E14.5) showed a significantly higher Dronpa-ERK1 import rate than neonatal (P2) cardiomyocytes (Fig. 2a-e, Suppl. Video S2). In conclusion, the maturation-associated decline in NP numbers is associated with a decline in nuclear import rate of the Dronpa-ERK1 reporter.
Figure 2. Higher NP signals are associated with higher nuclear translocation of MAPK and NFĸB signaling, and upregulated expression of an immediate early stress gene.

(a, b) Live cell imaging of nuclear import of Dronpa-ERK in cultured rat cardiomyocytes. (c) Experiments begin with photobleaching the entire cardiomyocyte (green outline), followed by photoactivation of the indicated region of cytoplasm (purple rectangles, t=0 s), and acquisition in the nucleus at 15 fps for 180 s (red circle). (d) Dronpa fluorescent intensity after activation shows that a neonatal cardiomyocyte (P2) has lower import than a fetal (E14.5) cardiomyocyte. See also Supplemental Video S2. (e) Population analysis showed significantly decreased nuclear import rate at P2 (n = 310) compared to E14.5 cardiomyocytes (n=240). (f) Photomicrographs for analysis of p-ERK and NP intensities in neonatal rat ventricular cardiomyocytes. NPHigh, ERKHigh and NPLow, ERKLow nuclei indicated with white and yellow arrowheads, respectively. (g) Zoom of NPHigh, ERKHigh and NPLow, ERKLow nuclei indicated in (f). (h-j) Quantitative analysis shows that nuclear import of p-ERK is higher in NPHigh nuclei under basal conditions (n=1,142, h) and stimulation with endothelin 1 (ET-1, 100 nM, n=686, i) or fibroblast growth factor 1 (FGF1, 50 ng/mL, n=879, j). (k-n) Nuclear import of p38 mitogen-activated kinase (MAPK) is higher in NPHigh nuclei under basal conditions (n= 2,142, l) and stimulation with endothelin 1 (ET-1, 100 nM, n=2,381, m) or fibroblast growth factor 1 (FGF1, 50 ng/mL, n=2,080, n). (o-q) Nuclear import of nuclear factor kappa B (NFĸB) is higher in NPHigh nuclei under basal conditions (n= 2,410, p) and stimulation with ET-1 (100 nM, n=2,824, q). (r-t) Photomicrographs and quantitative analysis show that nuclear import of cFOS is higher in NPHigh nuclei under basal conditions (n= 2,261, s) and stimulation with ET-1 (100 nM, n=2,079, t). Scale bars 10 μm (f, k, o, r), 2 μm (g). Whiskers indicate 10 and 90th percentiles. Statistical significance tested with Wilcoxon rank sum test (e) and t-test (h-j, l-n, p, q, s, t). Mean ± SEM are indicated with horizontal lines.
We examined the association between the natural variations of NP numbers in neonatal rat ventricular cardiomyocytes (NRVM) and corresponding nuclear ERK levels with immunofluorescence microscopy. Cardiomyocyte nuclei with lower NP signals had lower nuclear phosphorylated ERK (p-ERK) levels (Fig. 2f-h). To examine nuclear import of increased p-ERK signals, we stimulated cardiomyocytes with endothelin 1 (ET-1), a GPCR agonist that activates ERK. This showed lower p-ERK in cardiomyocyte nuclei with lower NP numbers and higher p-ERK in nuclei with higher NP numbers (Fig. 2i). Next, we stimulated with fibroblast growth factor 1 (FGF1), an agonist of the fibroblast growth factor receptor, a receptor tyrosine kinase, that activates ERK. This showed lower p-ERK signals in nuclei with low NP and higher p-ERK intensity in NP-high nuclei (Fig. 2j). We then examined p38 mitogen-activated kinase (p38 MAPK), which is activated by ET-1 and FGF1 (Fig. 2k-n). In addition to MAPK nuclear import, we investigated Nuclear factor-κB (NF-κB), a rapidly acting transcription factor that does not require protein synthesis for activation. Since ET-1 activates NF-κB nuclear translocation(Purcell et al., 2001), we compared nuclear NFĸB intensity in NP-low and NP-high cells. NF-ĸB signals, both at basal and ET-1-stimulated conditions, were lower in nuclei with lower NP intensity and higher in those with higher NP intensity (Fig. 2o-q). To study the consequence of different import of signaling molecules, we assessed transcriptional regulation by comparing levels of an early stress gene (c-FOS), which is directly regulated by MAPK signaling, between NP-high and NP-low cardiomyocytes. After treating NRVMs with ET-1, the c-FOS protein levels in the cytoplasmic fraction were significantly upregulated (Suppl. Fig. S2). At the same time, immunofluorescence microscopy showed that high levels of c-FOS appear in NP-high cells whereas NP-low cells exhibit lower c-FOS signal, indicating that the c-FOS level in the nucleus is determined by the NP number (Fig. 2r-t). In summary, using the natural variation of NP numbers and examination under three different extracellular conditions (control, ET-1, FGF1), and using four different read-outs (ERK, p38, NFĸB, cFOS), we demonstrate a direct relationship between NP numbers and nuclear import of signaling proteins. In conclusion, these results close the connection between NP number differences, via changes in nuclear transport, to gene transcription changes.
Modulating Lamin B2 expression levels changes NP numbers
After establishing that the maturation-related decrease in NP numbers is associated with decreased import of signaling proteins, we experimentally modified NP numbers and examined the consequences for nuclear import. For this, we drew on the fact that NPs are formed from pre-pores located in the endoplasmic reticulum (ER), whose phospholipid bilayers are continuous with the nuclear envelope. At the end of M-phase, cells form a new nuclear envelope from ER sheets engulfing the daughter chromosomes(Anderson and Hetzer, 2007). Pre-pores travel with these ER sheets to the nuclear envelope(Chou et al., 2021). Consequently, to experimentally reduce NP numbers, we inhibited M-phase progression with the goal of reducing pre-pore traffic from the ER to the nuclear envelope. We have previously demonstrated that decreased expression of the nuclear lamina filament, Lamin B2 (Lmnb2), reduces M-phase progression in cardiomyocytes(Han et al., 2020). Thus, decreased Lmnb2 expression levels should prevent Mphase NP insertion. To test this possibility, we suppressed Lmnb2 expression by previously validated siRNA knockdown(Han et al., 2020), which decreased NP numbers from 1,096 ± 41 to 899 ± 25 in NRVMs (Fig. 3a).
Figure 3. Altering Lmnb2 gene expression levels induces corresponding changes of nuclear pore (NP) numbers.

NP detected with mAb414 and visualized with confocal microscopy (a, h) and STORM (b, f, i). (a) Lmnb2 siRNA knockdown decreases NP numbers in the nuclear envelope in NRVMs. Scale bar: 2 μm. Three independent experiments with n=35 for scrambled siRNA; n=45 for Lmnb2 siRNA. (b) STORM in cardiomyocytes from αMHC-cre+/− (Ctr) and αMHC-cre+/−, Lmnb2fl/fl (Lmnb2 KO) P2 mice. Nuclei outlined with red dots. Boxes indicate zoomed areas. (c-e) Lmnb2 KO decreases NP numbers (c), NP area density in nuclei (d) and ratio of nucleo-cytoplasm NP density (e). (f) Colocalization analysis of NP and endoplasmic reticulum (ER, calnexin) by STORM in neonatal (P2) ventricular cardiomyocytes from Tnnt2-Cre+/− (Ctr) and Tnnt2-Cre+/−, Lmnb2f/f (Lmnb2 KO) mice. Boxes indicate zoomed areas shown in lower panels. (g) Digital colocalization analysis of two independent experiments. (h) Confocal microscopy of NPs in NRVMs transduced with control and Lmnb2 adenoviruses. n=45 for control and Lmnb2 groups. (i-l) Visualization with STORM (i) and digital quantification (j-l) show that Lmnb2 gene transfer increases total number of NPs per nucleus (j) and NP area density in the NE (k) without altering NP size (l). Boxes indicate areas shown at higher magnifications (bottom). Scale bars 2 μm (a, b, f lower panels, h, i) 10 μm (f upper panels). Each symbol represents one cardiomyocyte. Statistical significance tested with t-test. Mean ± SEM are indicated with horizontal lines.
We next quantified NP numbers after Lmnb2flox gene inactivation using previously validated cardiomyocyte-specific Lmnb2 knockout mice [αMHC-Cre; Lmnb2flox/flox (Coffinier et al., 2011; Han et al., 2020)]. To visualize individual NPs, we utilized Stochastic Optical Reconstruction Microscopy (STORM), a super-resolution fluorescent imaging technique(Rust et al., 2006). Using STORM, we found that Lmnb2 KO cardiomyocytes exhibited lower NP numbers per nucleus (1166 ± 63) compared to control (1499 ± 90), which corresponded to a decrease in area density from 6.4 ± 0.3 μm−2 in control to 5.1 ± 0.2 μm−2 of NP in Lmnb2 KO (Fig. 3b-d). To investigate the mechanisms leading to this decrease, we quantified NPs outside of the nucleus, where NP-generation begins, by STORM(Anderson and Hetzer, 2007; Eisenhardt et al., 2014; Knockenhauer and Schwartz, 2016; Vollmer et al., 2012). The area density ratio of NP in the nucleus to outside of the nucleus was lower in Lmnb2 KO, indicating that, although Lmnb2 KO cardiomyocytes have fewer pores in their nuclei, they have more pores outside of their nuclei (Fig. 3e). The higher signal of NP outside of Lmnb2 KO nuclei suggested that Lmnb2 KO reduced NP traffic. We investigated this by examining the co-localization of NP outside of the nucleus with markers of the endoplasmic reticulum (ER), where pre-pores reside and their trafficking begins(Anderson and Hetzer, 2007; Eisenhardt et al., 2014; Knockenhauer and Schwartz, 2016; Vollmer et al., 2012). We utilized previously validated crosses of Tnnt2-Cre mice with Lmnb2flox, which inactivate the Lmnb2 gene earlier in heart development than αMHC-Cre mice(Coffinier et al., 2011) (Fig. 3f). We applied digital colocalization analysis of NP and the ER marker, calnexin, on STORM photomicrographs, which revealed decreased colocalization of NPs and ER in Lmnb2 KO (Fig. 3g). These results show that the ER contains fewer pre-pores that could be trafficked to the nuclear envelope and supports the conclusion that decreased nuclear pore trafficking could decrease the NP numbers in Lmnb2 KO. In addition, this suggests a previously unrecognized function of Lmnb2 in NP traffic from the ER to the nuclear envelope.
Cardiomyocytes decrease Lmnb2 expression during maturation(Han et al., 2020). We have previously shown that increasing the expression of Lmnb2 rescues this decline and overcomes the developmentally inhibited progression from prophase to prometaphase in neonatal cardiomyocytes(Han et al., 2020). This suggested that the Lmnb2 overexpression-induced increased prophase to prometaphase progression could increase NP numbers. Accordingly, we transduced NRVM with a previously validated adenovirus expressing Lmnb2 (Ad-Lmnb2, Fig. 3h), which increased the NP numbers from 1,040 ± 53 to 1,281 ± 45 per nucleus (Fig. 3h). Consistently, STORM (Fig. 3i) showed that increasing Lmnb2 expression increased the NP numbers from 1,097 ± 90 in control to 1,378 ± 71 after transduction with AdV-Lmnb2 (Fig. 3j). Corresponding NP area density in nuclei was increased by Lmnb2 overexpression (Fig. 3k). However, STORM showed that NP size was not changed (Fig. 3l), indicating that increasing Lmnb2 levels did not alter the basic inner structure of NP. This supports the conclusion that increasing Lmnb2 expression levels increases NP numbers in the nuclear envelope.
Experimentally decreased NP numbers reduce nuclear import of ERK
To determine the functional consequence of the lower NP numbers in Lmnb2 KO nuclei, we examined nuclear p-ERK by immunofluorescence microscopy in neonatal cardiomyocytes (Fig. 4a). Cardiomyocytes from Tnnt2-Cre; Lmnb2flox mice (P2) exhibited lower nuclear p-ERK intensity (Fig. 4b, c), suggesting decreased nuclear import. We then assayed the nuclear import with the live reporter, Dronpa-ERK (Fig. 4d-f). Lmnb2 KO cardiomyocytes exhibited significantly lower peak Dronpa-ERK than control (Fig. 4g-i), consistent with the results in Figure 4b, c. In conclusion, decreasing the NP numbers with Lmnb2 KO decreases nuclear import of ERK.
Figure 4. Altering NP numbers by modifying Lmnb2 expression induces corresponding changes of ERK nuclear import.

(a) Immunofluorescence microscopy shows high NP signal and high nuclear p-ERK in control (Tnnt2-Cre+/−, top) and low NP signal and low nuclear p-ERK in Lmnb2 KO (Tnnt2-Cre+/−; Lmnb2fl/fl, bottom). (b) Quantification shows lower nuclear p-ERK in Lmnb2 KO. Each symbol represents one nucleus (3 independent experiments). (c) Histogram of results shown in b. Lmnb2 KO in black bars fitted with red line. Control in white bars fitted with black line. (d, e) Live-cell nuclear import assay of Dronpa-ERK1. Green outline indicates 3 control cardiomyocytes (Tnnt2-Cre+/−) before photobleaching. Nuclei were identified with siR700 staining (e). Corresponding regions of Dronpa-ERK photoactivation in cytoplasm (rectangles) and nuclear read-out (circles) are outlined with same colors. (f) Post-hoc immunofluorescence microscopy corresponding to panel d confirms cardiomyocyte identity with troponin I antibody. (g, h) Dynamic nuclear import of Dronpa-ERK analyzed by two-parameter Michaelis-Menten model. (i) Comparison between control (Tnnt2-Cre+/−) and Lmnb2 KO (Tnnt2-Cre+/−, Lmnb2f/f) shows that Dronpa-ERK nuclear import is significantly lower in Lmnb2 KO cardiomyocytes. (j, l) Western blots of p-ERK and total ERK in nuclear and cytoplasmic fractions of control (Scrmb = scrambled) and Lmnb2 knockdown (Lmnb2 siRNA) NRVMs at baseline (no treatment: NT) and stimulation with ET-1 (100 nM, 30 min). Histone H3 served as loading control for nuclear (j) and GAPDH for cytoplasmic fractions (l). Results from one of three independent experiments shown. (k, m) Quantifications of Western blots of nuclear (k) and cytoplasmic (m) fractions. (n) Immunofluorescence microscopy shows lower nuclear p-ERK in control and higher nuclear p-ERK in NRVMs transduced with Lmnb2 adenovirus. (o) Quantitative analysis of photomicrographs shows higher nuclear p-ERK in Lmnb2 overexpressing NRVMs. Each symbol represents one cardiomyocyte nucleus (3 independent experiments). Scale bars: 2 μm (a), 50 μm (f) and 10 μm (n), Statistical tests: t-test (b, o), mixed effect analysis (i), ANOVA (k, m). Mean ± SEM are indicated with horizontal lines.
We used Lmnb2 siRNA knockdown in NRVM to obtain sufficient quantities of lysate for a different approach of measuring nuclear ERK import using Western blotting. We activated ERK by adding ET-1. Quantification of the Western blot results showed that stimulation with ET-1 increased nuclear p-ERK by 1.6 to 7.1-fold in control (Fig. 4j, k). Lmnb2 siRNA reduced nuclear p-ERK, but not cytoplasmic p-ERK (Fig. 4j-m), nor total ERK level in the nucleus or cytoplasm (Suppl. Fig. S3), which supports the conclusion that decreasing the NP numbers with Lmnb2 siRNA decreases nuclear import of ET-1-activated ERK. We also investigated the effects of experimentally increasing NP numbers by Lmnb2 overexpression on p-ERK nuclear translocation. We found that the p-ERK signal was significantly higher in cardiomyocytes transduced with AdV-Lmnb2 compared to control (Fig. 4n, o), demonstrating with an independent line of evidence that NP numbers determine nuclear import of signaling molecules.
We next sought to reduce NPs numbers by suppressing the expression of one of the Nups, Nup155, which was previously shown to reduce NP numbers(Mitchell et al., 2010). In NRVMs, Nup155 siRNA significantly decreased NP area density (Suppl. Fig. S4a, b). We next semi-quantitively assessed Nup and p-ERK intensity in the same nuclei. Consistent with the NP area density decrease, Nup155 knockdown reduced nuclear Nup intensity and nuclear p-ERK intensity (Suppl. Fig. S4c-e). To investigate the gene regulatory consequences, we examined the transcription of multiple stress genes stimulated by ET-1, and found that cJun, cFos, Nppa and Nppb upregulation was lower after decreasing NP numbers with Nup155 siRNA (Suppl. Fig. S4f). Because ET-1 is a hypertrophic stimulus for cardiomyocytes, we compared the level of the sarcomere proteins, β-MHC and α-MHC, which are typically up- and down-regulated, respectively, in cardiac stress. Our results showed that β-MHC, both at the gene (Myh7) and protein levels, was upregulated by ET-1, and this was substantially suppressed by Nup155 siRNA (Suppl. Fig. 4g, i, k). α-MHC protein levels, reduced by ET-1, were increased by Nup155 knockdown (Fig. 4g, i, k). Although, under the experimental conditions, ET-1 did not stimulate enlargement of NRVM area size in culture, we show that ET-1 increased β-MHC both at mRNA (MYH7) and protein levels. This was inhibited by knocking down Nup155, which demonstrates that the reduction of NP numbers alters cardiomyocyte remodeling gene regulation. Taken together, these results demonstrate that NP numbers govern information flux into the nucleus, through which they transcriptionally regulate downstream gene targets.
Reduction in NP numbers improves survival and reduces adverse myocardial remodeling after pressure overload in vivo
Adult mammalian hearts adapt to pressure overload – e.g., from elevated blood pressure – by tissue remodeling, which involves gene transcription changes that lead to dilation of the ventricles with increased mass of the heart (hypertrophy), tissue fibrosis, and culminates in heart failure. Mechanistically, pressure overload activates paracrine signaling in the myocardium, leading to the stimulation of GPCR and RTK in cardiomyocytes(Heineke and Molkentin, 2006). Because the previous results showed that lower NP numbers decrease signal propagation, we hypothesized that, by experimentally reducing NP numbers, we could alter the maladaptive response to cardiac pressure overload. To induce pressure overload, we performed transverse aortic constriction (TAC) surgery in mice, which mimics maladaptive cardiac remodeling due to high blood pressure in humans. We used the mouse model of Lmnb2 gene inactivation-induced decrease of NP numbers to test the hypothesis by comparing Control (Tnnt2-cre+/−) and Lmnb2 KO (Tnnt2-cre+/−, Lmnb2fl/fl) adult mice.
Lmnb2 KO mice had a significantly lower mortality rate, resulting a higher cumulative survival of 69% one month after TAC versus 48% in control mice (Mantel-Cox test, Fig. 5a). This suggested that the Lmnb2 KO-induced NP decrease diminishes the maladaptive response following cardiac pressure overload, and we used MRI to determine structural and functional changes in these hearts. Both control and Lmnb2 KO mice showed normal ejection fraction at baseline prior to TAC (Pre-TAC Control 62.2 ± 1.2%; KO 64.3 ± 1.1%, Fig. 5b-d, Suppl. Video S3). Since pathological cardiac remodeling in the TAC model begins during the first week following surgery, we focused on the early maladaptive cardiac response in this period. One week post-TAC, control mice showed a decreased ejection fraction (Fig. 5b, d, 33.3 ± 5.0%), whereas Lmnb2 KO mice exhibited a nearly normal ejection fraction (Fig. 5c, d, 56.1 ± 3.4%). These results demonstrate a better-preserved heart function in Lmnb2 KO mice after pressure overload.
Figure 5. Mice with Lmnb2 KO-induced lower NP numbers in cardiomyocyte nuclei show improved survival and heart function and reduced cardiac remodeling after pressure overload in vivo.

Tnnt2-cre, Lmnb2f/f (Lmnb2 KO) and Tnnt2-cre (Control) mice were subjected to transverse aortic constriction (TAC) surgery. (a) Kaplan-Meier curves show that Lmnb2 KO increases survival rate after TAC. Control n=27, Lmnb2 KO n=26. (b-f) Cardiac MRI to determine ejection fraction and myocardial volumes 3 days before (Pre) and 7 days after (Post) TAC. MRI slices from the same heart (b, c) at the same distance from the apex taken at end-systole (ES) and end-diastole (ED). Quantitative MRI results show preserved ejection fraction (EF, d), less ventricular wall thinning (e), and less ventricular dilation (f) in Lmnb2 KO mice at 7 dpi. See Supplemental Video S3. (g) Heart weight to body weight ratios are lower in Lmnb2 KO mice, indicating less hypertrophy at 7 dpi. (h) AFOG staining shows less myocardial fibrosis in Lmnb2 KO mice at 7 days post TAC. Scale bars 1 mm (top row) and 100 μm (bottom row). Images from two different hearts were shown in each group. Boxes indicate zoomed areas. (i) Quantification of AFOG-stained sections shows less fibrosis in Lmnb2 KO mice. Each symbol represents the mean of 3 cross-sections from one heart. (j) Mason-Trichrome staining shows less fibrosis (blue staining) in heart tissue in Lmnb2 KO mice at 7dpi post TAC. Scale bars: 100 μm. Images from two different hearts shown in each group. (k) Western blot of nuclear and cytoplasmic protein extracts from heart tissue of control and Lmnb2 KO mice. The two lanes shown for each genotype were from two different animals. (l) Quantification of Western blots shows lower nuclear p-ERK signaling in Lmnb2 KO compared with control. Each symbol represents one heart. Statistical significance tested with Log-rank (Mantel-Cox) test (a); two-way ANOVA analysis followed by Sidak’s multiple comparison test (d, e, f, g); and t-test (i, l). Mean ± SEM are indicated with horizontal lines.
To determine whether the preserved heart function in Lmnb2 KO mice resulted from reduced cardiac remodeling, we analyzed the myocardium with MRI to look for signs of cardiac hypertrophy and with histology to assess myocardial fibrosis. At 7 days after TAC, Lmnb2 KO mice showed a significantly smaller increase in myocardial volume than control mice (Fig. 5e, 89.6 ± 3.0 mm3 and 112.5 ± 6.9 mm3, respectively, P <0.0002), indicating less cardiac hypertrophy. Moreover, Lmnb2 KO mice developed less ventricular dilation after TAC (Fig. 5f, control 146.2 ± 6.5 mm3; KO 117.7 ± 3.2 mm3, P <0.0001), indicating less remodeling. At 7 days post-TAC, Lmnb2 KO mice displayed a significantly lower heart-weight to body-weight ratio than control (Fig. 5g), indicating less cardiac hypertrophy. Myocardial fibrosis, the deposition of collagen in the interstitial space, is a characteristic tissue change of maladaptive remodeling, and Lmnb2 KO mice had 21% less myocardial fibrosis than control (Fig. 5h-j).
We next examined ERK activation and nuclear import in nuclear and cytoplasmic fractions of heart lysate after TAC. Western blot showed that nuclear p-ERK was higher in control than in Lmnb2 KO (Fig. 5k). Normalizing nuclear p-ERK to histone H3 and cytoplasmic p-ERK to GAPDH showed that the nuclear-to-cytoplasmic ratio of p-ERK in Lmnb2 KO was lower than in control (Fig. 5l), in agreement with the results shown in Figure 4. This supports the conclusion that Lmnb2 KO hearts have less nuclear import of p-ERK, which is consistent with less severe adverse remodeling. Taken together, lower NP numbers, induced by Lmnb2 gene inactivation, reduced cardiac maladaptation after pressure overload.
Mice with lower NP numbers show less regulation of cardiac remodeling gene pathways
The maladaptation of mammalian hearts to pressure overload occurs through changes of gene transcription, which are directed by a network of extracellular signals and intracellular signaling pathways (Heineke and Molkentin, 2006). We hypothesized that the lower NP numbers present in Lmnb2 KO mice could alter maldadaptive gene transcription pathways. We tested this with RNAseq analysis after TAC. There was no significant difference of gene expression between control and Lmnb2 KO without pressure overload (Fig. 6a). This is consistent with our finding that Lmnb2 KO hearts showed no structural or functional differences before pressure overload (Fig. 5b-g) and suggests that, without extracellular triggers, lower NP numbers in Lmnb2 KO mice alone do not alter gene transcription. Given that the Lmnb2 KO mice showed less adverse remodeling after pressure overload (Fig. 5f), we hypothesized that Lmnb2 KO and control mice differ in their regulation of just a subset of genes, namely those involved in cardiac hypertrophy and remodeling. Volcano plots showed that pressure overload induced significant expression changes of many genes (P<0.05, > 2-fold, Fig. 6a). Pressure overload downregulated 569 (Fig. 6b) and upregulated 445 genes (Fig. 6c) that were shared between control and Lmnb2 KO mice. However, control hearts decreased the expression of 1,302 (Fig. 7b) and increased the expression of 1,039 unique genes (Fig. 6c) that Lmnb2 KO did not regulate. In contrast, Lmnb2 KO downregulated 233 (Fig. 6b) and upregulated the expression of 215 unique genes (Fig. 6c). Taken together, Lmnb2 KO mice retained regulation of 1,014 genes (common in control and Lmnb2 KO, 26.7%), lost regulation of 2,341 genes (61.6%), and gained regulation of 448 genes (11.8%) after pressure overload (Fig. 6d). Thus, mice with lower NP numbers (Lmnb2 KO) regulated 5.2-fold fewer genes than control after pressure overload (Fig. 6d).
Figure 6. Mice with Lmnb2 KO-induced lower NP numbers in cardiomyocyte nuclei show less remodeling-related gene expression changes after pressure overload in vivo.

Myocardial mRNA from adult Tnnt2-cre (Control) and Tnnt2-cre, Lmnb2fl/fl (Lmnb2 KO) mice without (sham) and with pressure-overload (TAC) was subjected to RNAseq (2 hearts per condition). (a) Volcano plots show that without pressure overload (Sham), very few genes are differentially expressed between control and Lmnb2 KO (>2-fold, P<0.05). Vertical interrupted lines indicate ± 2-fold change and horizontal interrupted lines indicate P = 0.05. Each gene indicated by one symbol color-coded according to legend. (b, c) Venn diagrams show that pressure overload downregulates (b) and upregulates fewer genes (c) in Lmnb2 KO than in control. Areas of Venn diagrams and overlaps are proportionate to gene numbers. (d) Pie chart shows that, despite overlapping regulation of 26.7% of all regulated genes, fewer genes are uniquely regulated genes in Lmnb2 KO than in control. (e) Pathway enrichment analysis shows that control hearts upregulate protein biosynthesis pathways (indicated on the right), while Lmnb2 KO upregulates the pathways of Positive Regulation of Developmental Process and Developmental Maturation (italicized), consistent with the predicted more differentiated phenotype induced by decreased NP numbers in Lmnb2 KO. (f, g) Chord diagrams indicating the genes corresponding to selected regulated pathways in control (f) and Lmnb2 KO hearts (g), supporting the conclusion that Lmnb2 KO hearts show less expression of adverse remodeling and maintain expression of antihypertrophy genes.
Figure 7. The number of NPs per nucleus may be a regulated barrier for intracellular signal transduction.

(a) Summary of the quantitative changes downstream of the induced changes of NP numbers. NP number decrease reduces nuclear import of gene regulatory pathways (measured with pERK, p38, NFκB, cFos), which synergistically induce larger changes of gene transcription (measured with cJun, cFos, Nppa, Nppb, Myh7, Myh6, corresponding to Suppl. Fig. S4) and protein expression (α-MHC, β-MHC). Decreases of >100% indicate decreases below control values. (b) The presented results identify the NP number (indicated in red) as additional level at which signal transduction could be broadly regulated. This is demonstrated in cardiomyocytes at the example of 2 receptors belonging to 2 different classes (1G protein-coupled receptor and 1 receptor tyrosine kinase) and 4 cytoplasmic signal transducers (ERK, p38, NFκB, c-Fos).
We sought to connect the regulation of fewer genes in Lmnb2 KO to the marked differences in cardiac remodeling between control and Lmnb2 KO. To this end, we used gene ontology pathway enrichment analysis, which showed upregulation of pathways associated with protein synthesis (Structural constituent of ribosome, Ribosomal large subunit biogenesis, Regulation of transcription from RNA-polymerase II, Large ribosomal subunit, and Cytosolic large ribosomal subunit) in control, but not in Lmnb2 KO (Fig. 6e). The upregulation of many ribosomal genes is consistent with the cardiac hypertrophy in control mice (Fig. 6f). Control mice upregulated Elongin B and Cullin-2, which collaborate in transcriptional regulation (Fig. 6f). A large group of upregulated genes mapped to the proteasome. The lack of expected retrieval of GO pathways related to MAPK signaling in control could be explained by the broad effects of altered nuclear transport on many transcriptional pathways, which may prevent enrichment.
Lmnb2 KO showed upregulation of genes related to Developmental Maturation (Fig. 6g), consistent with the notion that Lmnb2 KO may advance differentiation. The developmental maturation genes were expressed at lower normalized enrichment scores (NES) in Lmnb2 KO than the NES of upregulated genes in control, consistent with broadly restricted gene regulation in Lmnb2 KO. Lmnb2 KO maintained expression of genes with a protective function against hypertrophy (for example, Mapt, Hsf1, Hspb6, Rreb1, RBM15C, Cecam 1, Fig. 6g). Lmnb2 KO maintained expression of genes supporting fatty acid metabolism (for example, Ppard, Gpam, Fig. 6g), which is relevant because the progression to heart failure decreases physiologic fatty acid metabolism. Several pro-angiogenic genes expressed in endothelial cells (for example, Pdgfb, Sema6a, Epha4, Eng, Plxnd1, Sox18, Arhgef15, Plxnb1, Cdh5, Anks1, Scarf1, Fig. 6g), were upregulated, suggesting that their beneficial upregulation in Lmnb2 KO hearts did not occur in cardiomyocytes, but rather in endothelial cells, which supports the overall conclusion that lower nuclear transport restricts gene regulation in Lmnb2 KO cardiomyocytes. In conclusion, the RNAseq analysis showed that Lmnb2 KO hearts exhibit less pathological gene regulation and maintain beneficial gene expression signatures.
DISCUSSION
The main conclusion is that mammalian cardiomyocytes reduce their NP numbers during maturation, which decreases nuclear import of signal transduction molecules, reduces regulated gene expression, and improves adverse myocardial remodeling in vivo. The conclusion that lower NP numbers in mature cardiomyocytes alter nuclear import capacity builds on several independent lines of investigation demonstrating that altering NP numbers changes the transmission of cytoplasmic signaling pathways into the nucleus and, with that, gene transcription and cardiac remodeling (Fig. 7a). Only three prior reports have evaluated NP in cardiomyocytes(Chahine et al., 2015; Perez-Terzic et al., 1999; 2001). Although these reports have related nuclear transport changes to cardiomyocyte remodeling, all three of them have built on the abandoned model of molecular movements of a central plug as an internal gating mechanism of NP(Perez-Terzic et al., 1996; Strubing and Clapham, 1999).
An active question is what functional advantages terminal differentiation could bring to cardiomyocytes, besides the disadvantage of a lower proliferative capacity. Our previous report showed that Lmnb2 KO reduces cell cycle potential and cardiomyocyte regeneration, however, the underlying changes remained unclear (Han et al., 2020). Our latest results suggest that, by decreasing NP numbers, Lmnb2 KO reduces their susceptibility to adverse remodeling. Thus, decreased NP numbers could reduce access of both mitogenic and hypertrophic signals to the nucleus.
This report rigorously characterizes the effect of altered NP numbers on nuclear import of four signaling proteins and its functional significance at the cellular level, and for the cardiac stress response (Fig. 7a). Traditional modular thinking about signal propagation from the extracellular space to regulation of gene expression in the nucleus considers sequential stimulation of cell membrane-spanning receptors, activation of intracytoplasmic signal transducers, and intranuclear regulation of gene transcription (Fig. 7b). Our results suggest that, at least for cardiomyocytes, an additional module, namely the NP number, should be considered as a regulatory mechanism for inducing changes of gene transcription in response to extracellular signals (Fig. 7b). Because NP are the only known pathways into nuclei, this effects all signals that go into the nucleus. The simulataneous effect of altered NP numbers on many signaling pathways could synergize at the level of gene transcription (Fig. 7a), and our results support this possibility because moderate reductions in NP numbers cause profound changes in cardiac stress response (Fig. 7a).
Although NPs have been quantified in only few cell types, the conclusion was that different cell types have a set number of NPs, i.e., the NP number per nucleus is constant within specific cell types(Maul and Deaven, 1977). Our finding of decreasing NP numbers during cardiomyocyte maturation advances this concept. The decreased NP numbers in terminally differentiated cardiomyocytes and corresponding functional changes could represent a paradigm in which post-mitotic cells permanently alter their handling of information flux across the nuclear envelope and, with that, their behavior. Because cardiomyocytes are prototypical post-mitotic cells, the proposed model in cardiomyocytes could potentially be extended to other post-mitotic cells. Our results suggest that post-mitotic cells may have lower NP numbers than other cell types, especially rapidly proliferating ones. The previously reported progressive deterioration of NP structure and function with increasing age in neurons (D’Angelo et al., 2009) is significant in this context, because the NP number could become limiting when post-mitotic cells age.
Whether and how cells regulate nuclear transport has remained a mystery. In the search for answers, scientists have envisioned that NPs function like ion channels, and have therefore searched for gating mechanisms. We show here how cardiomyocytes regulate nuclear transport by altering NP numbers. Changes in NP numbers are akin to changes in the number of ion channels in a cell. Similar to the regulation of ion transport by different abundances of channels, the results we presented indicate a paradigm in which differentiated cells decrease nuclear transport of signaling molecules by decreasing the abundance of NPs.
Limitations of this study
Although we have previously shown that Lmnb2 gene inactivation increases the barrier against cardiomyocyte proliferation and heart regeneration by increasing the formation of polyploid nuclei(Han et al., 2020), lack of an approach to quantify ploidy and NP numbers on the same nuclei prevented testing whether polyploid nuclei have different NP numbers than diploid nuclei.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Bernhard Kuhn, Bernhard.kuhn2@CHP.edu.
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Data and Code availability
The datasets supporting the current study have not been deposited in a public repository because of author preferences and are available from the corresponding author upon request.
The RNA-Seq data generated during this study have been deposited at GEO and are publicly available at GEO Series accession number GSE186949 as of the date of publication.
This paper does not report original code.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Mice
Animals were housed under a 12-h light, 12-h dark cycle in the Laboratory Animal Research Center at the Children’s Hospital Rangos Research Center. Animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC). Animals were housed in a pathogen-free barrier facility with free access to autoclaved water and irradiated, pelleted food. Caging and bedding were autoclaved prior to use and changed once per week. A maximum of 4 adult mice were housed per cage. Animals were monitored daily for health concerns. The study design, including number of animals, type, and time points of analyses, were predefined by the investigators. The mice used were backcrossed and maintained on a C57BL/6N background. Both male and female mice from 10–12 weeks were used for Transverse Aortic Constriction (TAC) surgery. Mice used for other analysis before and after weaning were also sex balanced and analyzed at indicated time points. Littermates of the same sex were randomly assigned to experimental groups. Assignment of TAC surgery was performed by L.H. TAC surgery was independently performed by N.A. in a blinded manner without knowledge of genotypes. Cardiac MRI was performed and analyzed in a blinded manner by S.K.W and Y.W. Outliers were not excluded from results. The number of biological and technical replicates is provided in the figure legends.
METHOD DETAILS
Inactivation of Lmnb2flox gene in vivo and genotyping
The Lmnb2flox gene was inactivated using αMHC-Cre(Agah et al., 1997) and Tnnt2-Cre(Jiao et al., 2003). For Cre-induced Lmnb2 gene inactivation, we bred Cre+/−; Lmnb2flox/wt mice with Lmnb2flox/wt. Both male and female mice were used for experiments. Littermate controls were used for experiments and in data analysis.
Embryonic (E14.5) and neonatal rat and mouse cardiomyocyte isolation
Embryonic rat ventricular cardiomyocytes were isolated from 14.5 days old (E14.5), neonatal p2 rats and p2 mice. Neomyts cardiomyocyte isolation kit (Cellutron Life Technologies NC9073658) was used according to manufacturer protocols. The final cell pellet was resuspended in 500 μL of PBS, supplemented with 10% FBS, and filtered through a 70 μm Nylon mesh to remove remaining tissue chunks. Cells were cultured at 37°C 5% CO2 in DMEM supplemented with 10% FBS until time of experimentation.
Knockdown of Lmnb2 and NUP 155 genes in vitro
Neonatal cardiomyocytes were cultured in DMEM containing 10% FBS at 37° 5% CO2. After 24hours cells were treated with a final concentration of 0.75nM of scramble siRNA, siRNA NUP 155 or siRNA Lmnb2 for 6 days. Cells were cultured in DMEM without FBS for the first 48hrs. 200 ul of DMEM containing 10% FBS was added after 48hrs to prevent cell death. Cells were cultured until time of experimentation. Select wells were incubated with ET-1 (100nM) for 30mins or 24hrs towards the end of siRNA incubation.
Overexpression of Lmnb2 gene in vitro
Neonatal cardiomyocytes were cultured in DMEM containing 10% FBS at 37° 5% CO2. After 24hours, cells were treated with 1ul of 8 × 1013 Adv-Lmnb2-GAP in DMEM 10% FBS and incubated for 48–72hrs. Cells were fixed in 4% Paraformaldehyde for 20 mins at room temperature followed by immunofluorescent staining. Cells were imaged with a Nikon A1 confocal microscope with 40x oil objective lens. For each coverslip, Z-stack images were taken with 0.5 μm step size to cover the entire nuclear depth and analyzed with Nikon Elements. For quantification, images were converted to max intensity projection to obtain binary threshold under the DAPI channel, which was then converted to ROIs. Nuclei were omitted from counts if they were binucleated, Troponin I negative, or if the entire nucleus and surrounding cytoplasm were not visible. Mean intensity for pERK for each nucleus was recorded.
Langendorff isolation of adult mouse ventricular cardiomyocytes
Ventricular cardiomyocytes were isolated from 2–3 months old mice. Hearts were retrogradely perfused through the aorta. The atria were removed, and the ventricles were placed in 5 mL of digestion buffer (40 ml Perfusion buffer, 2 μg/mL Collagenase II, 10 units/50 mL Neutral Protease) prewarmed to 37°C and incubated for approximately 40 mins at room temperature. Ventricular tissue was gently dispersed into cell suspension along with 5 mL of a stop buffer (perfusion buffer (113 nM NaCl, 4.7 mM KCL, 0.6 mM potassium phosphate monobasic, 0.7 mM sodium phosphate monobasic, 1.2 mM MgSO4, 12 mM sodium bicarbonate, 10 mM potassium bicarbonate, 10 mM HEPES, 30 mM taurine, 10 mM BDM, 5.5 mM glucose supplemented with 10% FBS and 12.5 μM CaCl2). The cell suspension was filtered using a 100-μm filter and centrifuged at 50 x g for 3 minutes. Cells were cultured in DMEM supplemented with 10% FBS at 37 °C 5% CO2 until time of experimentation.
Immunofluorescence microscopy of isolated cardiomyocytes
Isolated cardiomyocytes were fixed at room temperature in 4% paraformaldehyde for 20 min, rinsed 3x in PBS, permeabilized at room temperature with 0.5% Triton-X100/PBS (Fisher BP151–500) for 10 mins, followed by blocking with 3% BSA in PBS (Fisher BP9706–100) at room temperature for 2 hrs. Cells were subsequently treated with primary antibodies (1:200–1:400) diluted in blocking solution and incubated at 4° C overnight. Cells were rinsed 3x in PBS for 5 mins at room temperature followed by secondary antibodies diluted in PBS at 1:400 for 2 hrs. at room temperature. Nuclei were stained with Hoechst 33342 (Invitrogen H3570) diluted in PBS and incubated for 5 minutes at room temperature. The cells were mounted with glycerol containing 1% n-propyl gallate (Sigma P3130–100G). Small strips of Parafilm were placed on each slide to prevent distortion of the nuclei prior to mounting with glass coverslip.
Quantification of nuclear pore numbers with laser-scanning confocal microscopy
We applied a well characterized monoclonal antibody (mAB 414) against Nup62, Nup153, Nup214 and Nup358, which all face the central opening of NPs(Boehmer et al., 2003; D’Angelo et al., 2009; Davis and Blobel, 1987; Pulupa et al., 2020; Yu et al., 2012b). Using immunofluorescence confocal microscopy detection of mAB 414 bound to Nups, nuclei of isolated cardiomyocytes were imaged with a Nikon A1R confocal microscope with a 100x objective lens. Z-stack images were taken using a pixel resolution of 2,048 × 2,048 and step size of 0.3 μm to cover the entire nuclear depth and analyzed with Nikon Elements. Nuclear pore quantification of each 3D image was converted to max projection intensity to obtain binary threshold under the DAPI channel. The binary was converted to ROI and transferred to the original 3D image. To determine the average number of nuclear pores, the top and bottom section of each z-stack showing pores on the surface and along the nuclear envelope were counted using spot detection (Nikon Elements) with a setting of 190 nm for the nuclear pore diameter(Knockenhauer and Schwartz, 2016). Total number of pores for each section were added to obtain the total number of pores per nucleus. Area density of pores was determined by dividing total number by area of ROI. Geometric changes from fetal, neonatal, and adult nuclei were not taken into account. 3-D reconstruction were derived from confocal z-stack images using volume view application in Nikon Elements.
Immunofluorescence antibody staining for STORM imaging
To perform immunostaining, cells were fixed with 4% PFA for 15 minutes and permeabilized with 0.2% Triton X-100 for 10 minutes. After being washed once with PBS, the cells were blocked with 3% BSA for 2 hours and were incubated with primary antibodies diluted in 3% BSA at 4 °C overnight. The cells were washed 3 times with PBS for 5 minutes per wash, and Alexa647-conjugated secondary antibodies diluted in 3% BSA were added to the cells and incubated for 2 hours at room temperature, protected from light. Cells were washed again 3 times with PBS and post-fixed with 4% PFA for 10 minutes. After being washed once with PBS, the samples were ready for STORM imaging. Immediately before imaging, the buffer was switched to the STORM imaging buffer containing 10% (w/v) glucose (Sigma-Aldrich), 0.56 mg/mL glucose oxidase (Sigma-Aldrich), 0.17 mg/mL catalase (Sigma-Aldrich), and 0.14 M β-mercaptoethanol (Sigma-Aldrich).
STORM image acquisition
STORM images were acquired using our custom-built system on the frame of an Olympus IX71 inverted microscope frame with a 100x, NA=1.4 oil immersion objective (UPLSAPO 100XO, Olympus). The imaging system has been described in detail previously(Xu et al., 2018; Xu et al., 2020). For dSTORM imaging, 30,000 frames were acquired at an exposure time of 20 ms per frame. During the image acquisition, a low activation power (∼1 μW) for the 405 nm laser was added at the 3,001st frame and the power of 405 nm laser was gradually increased at a rate of 0.2% per 1,000 frames. Online drift correction was independently performed every 200 frames (∼ 4 s) with fluorescent beads (Thermo Fisher Scientific, F8803) as fiduciary markers (excited with 488 nm laser) throughout the image acquisition process, based on our established method(Ma et al., 2017).
STORM image reconstruction and quantitative analysis
The reconstruction of the STORM image was performed using our custom-written program in MATLAB 2019 (MathWorks) and described in detail in our previous publication (Ma et al., 2017). The reconstructed super-resolution image was rendered by accumulating all the valid molecules with a pixel size of 10 nm followed by a Gaussian smoothing filter (σ = 10 nm). The localized point coordinates that form STORM images of nuclear pores were segmented based on Gaussian clustering method to calculate the size of the nuclear pore and density as described previously(Ma et al., 2017). Gaussian mixed model was used to fit each of the identified nuclear pores and its size was defined as full width half maximum (FWHM). The area density of nuclear pores was defined as the number of nuclear pores divided by the area of the nucleus derived from the STORM image of each nucleus.
Electron Microscopy imaging and quantification of nuclear pores
Fetal and neonatal hearts were resected, atria removed and fixed in 2.5% glutaraldehyde (25% glutaraldehyde stock EM grade, Polysciences,) in 0.01 M PBS, pH 7.3 at 4 °C overnight and then placed in 0.01 M PBS. Hearts were set in brass sample carriers and rapidly frozen in liquid methane. Samples were placed in a JEOL JFDV Freeze fracture then knife fractured, angled at 45°, and coated with approximately 70 angstroms of platinum followed by 100 angstroms of carbon for support. Platinum replica was floated off the sample in a 50% Clorox bleach in H2O solution and the tissue was allowed to dissolve for up to 4 days. Using a wire loop, samples were pulled out of the bleach solution and floated on several washes of DDH2O. Platinum replicas were picked up on formvar coated 200 mesh copper TEM grids and examined on JEOL 1400 Flash transmission electron microscope (JEOL Peabody, MA) with a low mount BIOSPRINT AMT 12k digital camera (Advanced Microscopy Techniques, Danvers, MA). Number of nuclear pores were counted and divided by the size of the nuclear area to obtain nuclear pore density.
Quantification of nuclear ERK1/2, p38, and NUP 414, cJUN, cFOS, and NFkB
Nuclei from isolated cardiomyocytes were double stained with either NUP 414 and Phospho-ERK1/2, NUP414 and p38, NUP 414 and cFOS, NUP414 and cJUN, or NUP 414 and NFkB antibodies. Cells were imaged with a Nikon A1 confocal microscope with 40x oil objective lens. For each sample, 3 fields of 3×3 stitch Z-stack images were taken with 0.5 μm step size to cover the entire nuclear depth and analyzed with Nikon Elements. For quantification, images were converted to max intensity projection to obtain binary threshold under the DAPI channel, which was then converted to ROIs. Nuclei were omitted from counts if they were binucleated, Troponin I negative, or if the entire nucleus and surrounding cytoplasm were not visible. Sum and mean intensity for p38, ERK1/2, cJUN, cFOS, NFkB and NUP 414 for each nucleus was recorded.
Measurement of real-time nuclear import of Dronpa-ERK1
Nuclear import of Dronpa-tagged ERK1 was analyzed as described previously(Ando et al., 2004). The isolated cardiomyocytes were transduced with adenovirus encoding Dronpa fused to ERK1 at a multiplicity of infection (MOI) of 50. Following 24 hr of viral infection, cells were treated with 1 μL of 35 nM siR700 dye for 30 mins to highlight nuclei. Cells were then imaged with a Nikon confocal microscope equipped with a 40x objective lens. The imaging procedure was set up with Nikon Elements in combination with JOBS software. The stimulation regions (photobleaching and photoactivation) were defined based on the initial image acquisition. Then, the fluorescence of Dronpa was photobleached at 488 nm (40% laser power) for 5 loops with one frame per second, followed by photoactivation at selected cytoplasmic regions by excitation at 405 nm (30% laser power) for 2 loops with 4 frames per second. An increase in the fluorescence of Dronpa at the predetermined nuclear ROI was monitored under excitation at 488 nm (0.36% of laser power) for 7 minutes. The nuclear import rate was calculated from the initial increase rate of the nuclear fluorescence. Cells were confirmed as cardiomyocytes by staining with Troponin I post-hoc and imaged with Nikon A1R confocal microscope.
Quantitative analysis of live Dronpa-ERK nuclear import
To model the dynamics of Dronpa-ERK nuclear import, we first normalized the nuclear florescent intensities as percentages of the maximum cytoplasmic florescent intensity after photo-activation for each cardiomyocyte, then, we used a two parameter Michaelis-Menten function to smooth the high frequency data. Two parameters of interest in the Michaelis-Menten model are Vmax, the normalized maximum nuclear florescent intensity, and K, time to reach half of Vmax, for each cardiomyocyte. After this data processing step, these two quantities were compared between the control and Lmnb2 KO groups by a mixed effect model, adjusting for the within pup correlation and batch effect. The data processing and analysis was performed with R version 4.0.3.
Acid Fuchsin Orange G (AFOG) Staining
Hearts were resected, atria were removed, and fixed with 4% PFA and placed on a rocker for 2 hr at room temperature, followed by 3x PBS washes for 5 min and immersion in 30% sucrose at 4°C for 48 hr. Ventricles were embedded in optimal cutting temperature (OCT) compound and sectioned into 10 μm thick cross-sections using a Leica CM1950 cryostat. Three slides with 6 sections per slide from 5 hearts from each group (Control and Lmnb2 KO) were analyzed. Selected slides were fixed with 3.7% formaldehyde for 15 minutes followed by two 5 min washes in 1x PBS. Slides were covered with Bouin fixative for 1 hour at 56 °C followed by a 30-minute wash with running tap water. Slides were subsequently dipped in 1% phosphomolybdic acid (5 min), distilled water (5 min), Acid Fuchsin Orange G (AFOG) solution (10 min) and quickly rinsed with water before dehydration with 95% ethanol (4 min), 100% ethanol (4 min), and Citrosol (4 min). Sections were mounted with Cytoseal and sealed with nail polish. Photomicrographs were taken on a Nikon Eclipse Ti microscope (objective lens 10x) with scale bar inclusion. Areas of healthy myocardium (stained red) and fibrosis (stained blue) were quantified using ImageJ/Fiji (version 2.1.0). Color thresholds were utilized to calculate fibrosis and total section areas (μm2). Total volume and fibrotic volume for each section were calculated, respectively, by multiplying total/fibrotic area x 10 μm section thickness x (8 total slides / number of slides analyzed) x (6 sections per slide / number of sections analyzed per slide). Total heart volume and total fibrosis volume were acquired by adding volume segments from sections belonging to the same heart. The readout is the percentage of fibrotic volume of the total myocardium.
Trichrome Staining
Hearts were resected, atria were removed, and fixed with 4% PFA and placed on a rocker for 2 hr at room temperature, followed by 3x PBS washes for 5 min and immersion in 30% sucrose at 4°C for 48 hr. Ventricles were embedded in optimal cutting temperature (OCT) compound and sectioned into 10 μm thick cross-sections using a Leica CM1950 cryostat. Sections from control and Lmnb2 KO hearts were stained and imaged. Slides were fixed with Bouin fixative for 2 hours at 56 °C followed by a 10-minute wash with running tap water. A working solution of Weigert’s Iron Hematoxylin, made according to manufacturer’s protocol, was added directly to tissue sections for 5 mins and rinsed for 5 mins under running tap water. Slides were treated with Biebrich scarlet-acid fuchsin solution for 5 mins, and subsequently washed by dipping in distilled water 3–5 times. Phosphomolybdic-phosphotungstic acid solution, made by mixing phosphomolybdic acid solution, phosphotungstic acid solution and distilled water (1:1:2), was added to each slide for 15mins followed by addition of aniline blue solution for 5 mins. Slides were rinsed by dipping in distilled water 3–5 times. 1% acetic acid solution was applied to slides for 1 min followed by 3–5 dips in distilled water. Slides were subsequently dipped in 85% ethanol (5s), 100% ethanol (5s), and xylene for (5s). Sections were mounted with Cytoseal and sealed with nail polish. Photomicrographs were taken on a Nikon Eclipse Ti microscope (objective lens 40x) with scale bar inclusion to visualize areas of healthy myocardium (stained red) and fibrosis (stained blue).
Nuclear-cytoplasmic protein extraction
Extraction was conducted using NE-PER™ Nuclear and Cytoplasmic Extraction Reagents (ThermoFisher Scientific No. 78833). Cardiomyocytes were washed with 4 °C PBS, scraped off cell plates, and centrifuged for 5 minutes at 21.3 x g. Cytoplasmic Extraction Reagent I (CER l) and Nuclear Extraction Reagent (NER) were pretreated each with Halt Protease Inhibitor (ThermoFisher No. 78430, 1:100) and Halt Phosphatase Inhibitor Cocktail (ThermoFisher No 1862495, 1:100). Pellets were homogenized in 200 μL CER l mixture by vortexing for 15 seconds and left to incubate at 4 °C for 10 minutes. Cytoplasmic Extraction Reagent II (11 ul CER II) was added to each sample followed by 1 minute incubation at 4 °C. Samples were vortexed for 5 seconds and centrifuged at 4 °C at 21.1 x g. Supernatants (cytoplasmic extracts) were transferred to pre-chilled tubes for storage. Pellets (nuclear extract) were re-homogenized with CER I mixture and centrifuged for 5 minutes at 4 °C at 21.1 x g. Supernatants were discarded. 100 μL NER mixture was added to each pellet. Samples were vortexed for 15 seconds every 10 minutes for a total of 40 minutes. Nuclear and cytoplasmic extracts were homogenized with a hand-held homogenizer prior to being separated into 3 aliquots for storage at −80 ºC.
Tissue protein extraction
Extraction was conducted using NE-PER™ Nuclear and Cytoplasmic Extraction Reagents (ThermoFisher Scientific No. 78833). Heart tissue from sham and TAC-injured mice (5 control and 5 KO) were weighed and placed on dry ice. Cytoplasmic Extraction Reagent I (CER l) and Nuclear Extraction Reagent (NER) were pretreated each with Halt Protease Inhibitor (ThermoFisher No. 78430, 1:100) and Halt Phosphatase Inhibitor Cocktail (ThermoFisher No 1862495, 1:100). 200 μL CER l mixture was added to each tissue sample followed by homogenization using PRO Scientific’s 7mm x 95mm saw-tooth generator probe. 4–5 brief pulses at 75% strength were delivered per sample until lysates were homogenous. Samples were left to incubate at 4 °C for 10 minutes. Cytoplasmic Extraction Reagent II (11 ul CER II) was added to each sample followed by 1 minute incubation at 4 °C. Samples were vortexed for 5 seconds and centrifuged at 4 °C at 21.1 x g. Supernatants (cytoplasmic extracts) were transferred to pre-chilled tubes for storage. Pellets (nuclear extract) were re-homogenized with CER I mixture and centrifuged for 5 minutes at 4 °C at 21.1 x g. Supernatants were discarded. 100 μL NER mixture was added to each pellet. Samples were vortexed for 15 seconds every 10 minutes for a total of 40 minutes. Nuclear and cytoplasmic extracts were homogenized with a 20 μL Hamilton syringe prior to being separated into 3 aliquots for storage at −80 °C.
Whole cell protein extraction, quantification of protein concentrations, Western blot, and quantitative analysis
Embryonic (E14.5), neonatal (P1) and adult (P60) were collected with Pierce RIPA lysis buffer (Thermo Fisher, 89900) with Halt Protease Inhibitor (ThermoFisher, 78430, 1:100) and Halt Phosphatase Inhibitor Cocktail (ThermoFisher, 1862495, 1:100). Protein concentrations were determined using Bicinchoninic Acid assay (BCA, Pierce Thermo Scientific No. 23227). NuPAGE™ LDS Sample Buffer (4X) was premixed with mercaptoethanol (9:1) before being added to nuclear and cytoplasmic extracts by volume (1:3). Samples were heated (95 °C for 5 min) and loaded into 4–15% pre-cast gradient SDS-PAGE gels (Bio-Rad 4561084). Gels were transferred to nitrocellulose or polyvinylidene difluoride (PVDF) membranes using the Trans-Blot Turbo transfer system (Bio-Rad 1704158) for 7 minutes. Membranes were blocked with blocking buffer (1x TBS, 0.1% Tween 20, 5% Non-fat milk) for 1 hr. Blots were probed overnight with primary antibodies (see Key Resources Table) diluted in blocking buffer (1:1,000). Membranes were washed with TBST 3x for 5 minutes followed by incubation with secondary antibodies (1:1,000 in blocking buffer) (See Key Resource Table) for 2 hours at room temperature. Membranes were washed again with TBST 3x for 5 minutes each and developed using ECL Western blotting substrate (Pierce Thermo Scientific 32106). Blots were imaged using the ChemiDoc MP imaging system (Bio-Rad) and analyzed using intensity readouts normalized to Histone-3/GAPDH band intensities.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| P38 MAPK (D13E1) XP Rabbit mAb#8690 | Cell Signaling | Cat# 8690S; Lot# 9 RRID:AB_10999090 |
| Anti-Cardiac Troponin I antibody | Abcam | Cat# ab56357; Lot# GR313459-8 RRID:AB_880622 |
| phospho-p38 antibody, mouse |
Cell Signaling | Cat#; 9216S Lot# 27 RRID:AB_331296 |
| Hoechst 33342, Trihydrochloride, Trihydrate - 10 mg/mL Solution in Water | Invitrogen | Cat# H3570 Lot#1860907; RRID: N/A |
| Anti-GAPDH antibody produced in rabbit | Sigma | Cat# G9545; Lot# 127M4814V RRID:AB_796208 |
| Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (D13.14.4E) XP® Rabbit | Cell Signaling | Cat#: 4370S Lot# 28 RRID:AB_2315112 |
| p44/42 MAPK (Erk1/2) (L34F12) Mouse | Cell Signaling | Cat#; 4696 Lot# 23 RRID:AB_ AB_390780 |
| Anti-Lamin B2 antibody [LN43] | Abcam | Cat# ab8983; Lot# GR141423-12 RRID:AB_306912 |
| NUP Complex Protein Antibody mAb 414 | Abcam | Cat#; ab24609 Lot# GR-3235621-11 RRID:AB_ 448181 |
| Anti-NUP98 mAb | Abcam | Cat#; ab50610 Lot# GR3212026-6[ RRID:AB_ 881769 |
| Anti-NUP107 polyclonal anitbody | Abcam | Cat#; ab73290 Lot# GR6666-28 RRID:AB_1269604 |
| Anti-NUP155 polyclonal anitbody | Novus Biologicals | Cat#; NBP2-19609 Lot# 40429 RRID:AB_2915906 |
| Anti-NUP133 mAb | Santa Cruz | Cat#; sc376763 Lot# E2317 RRID:AB_2889360 |
| c-Jun (60A8) Rabbit mAb | Cell Signaling | Cat#; 9165 Lot# 13 RRID:AB_2130165 |
| c-Jun (60A8) Rabbit mAb (Alexa Fluor 647 conjugate) | Cell Signaling | Cat#; 40502S Lot# 3 RRID:AB_2909794 |
| c-Fos (9F6) Rabbit mAB | Cell Signaling | Cat#; 2250S Lot# 12 RRID:AB_2247211 |
| NF-kB p65 (D14E12) XP Rabbit mAB | Cell Signaling | Cat#; 8242S Lot# 16 RRID: AB_10859369 |
| MYH7-specific Polyclonal Antibody | Proteintech | Cat#; 22280-1-AP Lot# NA RRID: AB_2736821 |
| Myosin Cardiac Heavy Chain Antibody (MYH6) | Genway Biotech | Cat#; GWB-452D0F |
| Lot# NA RRID: AB_10281249 |
||
| Donkey anti-Goat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Thermo Fisher | Cat# A21447 Lot# 2045332 RRID: AB_141844 |
| Donkey anti-Mouse IgG (H+L) Highly Cross- Adsorbed Secondary Antibody, Alexa Fluor 488 |
Thermo Fisher | Cat# A21202 Lot# 2018296 RRID: AB_141607 |
| Donkey anti-Mouse IgG (H+L) Highly Cross- Adsorbed Secondary Antibody, Alexa Fluor 568 |
Thermo Fisher | Cat# A10037 Lot# 2110843 RRID: AB_2534013 |
| Donkey anti-Mouse IgG (H+L) Highly Cross- Adsorbed Secondary Antibody, Alexa Fluor 594 |
Thermo Fisher | Cat# A21203 Lot# 1722995 RRID: AB_141633 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross- Adsorbed Secondary Antibody, Alexa Fluor 488 |
Thermo Fisher | Cat# A21206 Lot# 2072687 RRID: AB_2535792 |
| Donkey Anti-Rabbit IgG H&L (Alexa Fluor® 555) | Abcam | Cat# ab150074 Lot# GR3241278-3 RRID: AB_2636997 |
| Bacterial and Virus Strains | ||
| Ad-Dronpa-ERK 1 | This Paper | NA |
| Ad-Lmnb2 | (Han et al., 2020) | NA |
| Chemicals, Peptides, and Recombinant Proteins | ||
| LMNB2 siRNA | GE Dharmacon | Cat# L-005290-00-0005 |
| Control siRNA | GE Dharmacon | NC1269625 |
| NUP 155 siRNA | GE Dharmacon | Cat# E-095619-00-0020 |
| Critical Commercial Assays | ||
| Cellutron Neomyts cardiomyocyte isolation kit | Thermofisher | Cat# NC9073658 |
| Neonatal Cardiomyocyte isolation kit | Milteni Biotec | Cat# 130-100-825 |
| RNeasy Plus micro kit | Qiagen | Cat# 74034 |
| NE-PER™ Extraction Kit | Thermofisher | Cat# 78833 |
| Deposited Data | ||
| RNAseq | GEO repository | GSE186949 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Lmnb2fl/fl: Lmnb2tm1.1Sgy/J | Laboratory of Dr. Stephen Young, UCLA |
JAX stock #032559 |
| Mouse: Tnnt2-Cre: Tg(Tnnt2-cre)5Blh/j | Jackson lab | JAX stock #024240 |
| Mouse: αMHC-cre: Tg(Myh6-cre)2182Mds/j | Jackson lab | JAX stock #011038 |
| Mouse: αMHC-cre; Lmnb2fl/fl | (Han et al., 2020) | N/A |
| Mouse: Tnnt2-cre; Lmnb2fl/fl | (Han et al., 2020) | N/A |
| Oligonucleotides | ||
| Primer: Gapdh
Forward: GTGGACCTGACCTGCCGTCT Reverse: GGAGGAGTGGGTGTCGCTGT |
This paper | N/A |
| Primer: Actb
Forward: GGAGGAGTGGGTGTCGCTGT Reverse: GTCATACTCCTGCTTGCTGAT |
This paper | N/A |
| Primer: Lmnb2
Forward: GAGGACATTGCCTACAAGTTCAC Reverse: TTCCACACAAGGGTTGATG |
This paper | N/A |
| Primer: ACTB
Forward: CCAACCGCGAGAAGATGA Reverse: CCAGAGGCGTACAGGGATAG |
This paper | N/A |
| Primer: Tnnt2 Forward: TTGGCACCCAATGCAGACTCCTGTTT Reverse: CCCGCACTATGCCAGC AATAAA |
This paper | N/A |
| Primer: MYH6- rat
Forward: GCCCTTTGACATCCGCACAGAGT Reverse: TCTGCTGCATCACCTGGTCCTCC |
This paper | N/A |
| Primer: MYH7- rat
Forward: TGGCACCGTGGACTACAATA Reverse: CTACAGGTGCATCAGCTCCA |
This paper | N/A |
| Primer: NUP 155-rat
Forward: GGCAAGTTCAGAGTAAGAGCG Reverse: CGCAGCAGTAAGAGACAGTTC |
This paper | N/A |
| Primer: cJun-rat
Forward: CGACCCCCACTCAGTTCTTGT Reverse: GCAGCGTATTCTGGCTATGCA |
This paper | N/A |
| Primer: cFos- rat
Forward: CCAAGCGGAGACAGATCAACTT Reverse: TCCAGTTTTTCCTTCTCTTTCAGTAGAT |
This paper | N/A |
| Primer: Nppa- rat
Forward: CGTATACAGTGCGGTGTCCAAC Reverse: CATCTTCTCCTCCAGGTGGTCTAG |
This paper | N/A |
| Primer: Nppb- rat
Forward: AAGTCCTAGCCAGTCTCCAGAACA Reverse: TTGAGAGCTGTCTCTGAGCCATT |
This paper | N/A |
| Software and Algorithms | ||
| NIS-Elements | Nikon Instruments | N/A |
| Nikon Element 3D analysis module | CBI Univ. Pittsburgh | N/A |
| Fiji/Image J | NIH | RRID:SCR_002285 https://imagej.nih.gov/ij/ |
| GraphPad Prism 7 | GraphPad Software | N/A |
| NCBI Primer Blast | NCBI | RRID:SCR_004870 |
| Integrated DNA Technologies (IDT) PrimerQuest software | Integrated DNA technologies (IDT) | https://www.idtdna.com › primerquest |
| R | The R Project for Statistical Computing |
https://www.r-project.org/ |
PCR
Total RNAs were extracted from NRVMs using the RNeasy Mini Kit (Qiagen, 74104) according to the manufacture’s protocol. Reverse transcription was performed using SuperScript™ IV VILO™ Master Mix (Thermo Fisher Scientific, 11756050). Quantitative PCR for the cDNA library (RT-PCR) was set up with iTaq™ Universal SYBR® Green Supermix (Bio-Rad, 172–5124) and measured by CFX96 Touch Real-Time PCR Detection System. Relative quantification to corresponding control was obtained using the threshold cycle (Ct) method with GAPDH as the housekeeping gene and relative expression calculated as 2-ΔΔCt. Primer sequences used in this study are listed in the Key Resources Table.
Transverse aortic constriction (TAC) surgery
TAC was performed on Tnnt2-Cre Lmnb2flox/flox (Lmnb2 KO) and Tnnt2-Cre (Control) mice at the age of 10 weeks – 12 weeks. The mice were anesthetized in a chamber with 4% isoflurane. A midline incision was made in the anterior neck exposing the suprasternal notch. From there, the sternum was cut open to the 2nd intercoastal rib space to reach the aortic arch by separating the thymus and connective tissues. An 8.0 silk suture was passed under the aortic arch and tied over a blunt 27½ gauge needle. The chest wall was then closed with 6.0 Nylon suture. Bupivacaine (8 μg/g body weight) was administered through subcutaneous injection. The mice were recovered from anesthesia. Bupivacaine was administered subcutaneously for the following 3 days post injury.
In-vivo cardiac MRI
Animal preparation
Anesthesia induction was first achieved by 3–4% isoflurane mixed with oxygen in an induction box for a few minutes. The depth of anesthesia was monitored by the toe reflex, extension of limbs, and spine positioning. Once the necessary depth of anesthesia was established, the mouse was placed on a designated animal bed for imaging and the anesthesia was maintained by 1.0 to 1.5 % isoflurane with 100% oxygen via a nose cone. The body temperature was monitored by an optical rectal temperature probe which is used for feedback control of a warm air blower around the animal to maintain the core temperature 37 °C ± 0.5 °C. The respiration was continuously monitored by placing a small pneumatic pillow under the animal’s diaphragm which was connected to a magnet-comparable pressure transducer feeding to a physiological monitoring computer equipped with respiration-waveform measuring software (SA Instruments, Stony Brook, NY). The respiration waveform was automatically processed to detect inspiration, expiration and respiration rate. ECG was monitored with two subcutaneous MR-comparable Pt needle electrodes connected with MR-compatible ECG monitoring and gating system for flow measurements.
In vivo CMR acquisition
In-vivo cardiac MRI (CMR) was carried out on a Bruker Biospec 7T/30 system (Bruker BioSpin MRI, Billerica, MA) with the 35-mm quadrature coil for both transmission and reception. Free-breathing-no-gating cine MRI with retrospective navigators was acquired with the Bruker Intragate module.
Cine CMR for assessment of cardiac function
White-blood multi-planar cine movies with 20 cardiac phases were acquired for short-axis and long-axis 4-chamber views with the following parameters: Field of view (FOV) = 2.5 cm X 2.5 cm, slice thickness = 1.2 mm, inter-slice gap = 0, acquisition matrix = 256 X 256, in-plane resolution = 98 μm, flip angle (FA) = 10 degrees, echo time (TE) = 3.059 msec, repetition time (TR) = 5.653 msec, number of repetition = 250, total scan time = 3 min 2 sec.
Phase-contrast MRI for flow velocity measurements
Blood flow measurements were performed using ECG- and respiration-triggered FLOWMAP sequence with bipolar gradients for flow velocity encoding with the following parameters: velocity encoding (VENC) ranges 1.47 cm/sec to 250 cm/sec, TE/TR = 5.56/17 msec, matrix = 512 X 512, in-plane resolution = 49 μm, 20 cardiac phases averaging over multiple cardiac cycles with cardiac and respiration gating to cover the R-R interval of ∼280–320 msec, depending on the heart rates, resulting in an equivalent temporal resolution of ∼14 −16 msec.
In vivo CMR analysis
Cardiac function from cine CMR
The left ventricular endocardium and epicardium boundaries of each imaging slice at end-systole (ES) and the end-diastole (ED) were manually traced by a blinded operator on the Paravision 5.1 Xtip software (Bruker BioSpin MRI, Billerica, MA) to calculate the following functional parameters: left ventricular blood volume (LVV), left ventricular wall volume (LV wall), LV mass, stroke volume (SV), ejection fraction (EF), heart rate (HR), cardiac output (CO), longitudinal shortening, and radial shortening. LVV is calculated by summation of blood volume of all the short-axis slices at the end-diastole. , where the internal left ventricle area of slice i at end diastole, and hi is the thickness of each short-axis slice. The ejection fraction (EF) was calculated using the following equation: , where is the internal left ventricle area of slice i at end systole, the internal left ventricle area of slice 𝑖 at the end diastole, and hi is the thickness of each slice. The LV wall volume was calculated by summation of LV wall volume (LVW) of all short-axis slices at the end diastole. , where ( is the area encompassed by the epicardial boundary, is the area encompassed by the endocardial boundary at the end-diastole and hi is the thickness of each slice. The left ventricle wall thickening was calculated through equation where des and ded are the wall thickness measured at the end-systole (des) and the end diastole (ded). LV wall mass was calculated from the end-diastolic LV wall volume times the specific myocardial tissue density 1.055 g/ml(Nahrendorf et al., 2006; Vinnakota and Bassingthwaighte, 2004).
Aortic flow velocity from phase-contrast flow map
Flow velocity vector and magnitude maps were calculated from phase contrast data using the Paravision 5.1 Xtip software (Bruker BioSpin MRI, Billerica, MA). Blood flow velocity and vectors were measured for each cardiac phase at 2 locations on the ascending aorta: before and after the TAC.
Tissue processing for RNAseq
Hearts from sham and TAC-injured adult mice (both control and KO) were resected and atria were removed. The ventricular myocardium was divided horizontally into three parts of approximately equal size. The middle part was used for mRNA extraction using RNeasy Mini Kit (Qiagen, Cat# 74106) followed by library preparation for sequencing.
RNAseq and bioinformatics analysis
The transcriptome profiling of ventricular myocardium from mouse hearts were sequenced in triplicate. The libraries were sequenced by the sequencing core facility at Children’s Hospital of Pittsburgh. An average of 25 ± 3 million reads were produced for each sample, in accordance with the standard requirements for RNA-seq analysis.
Quality controlled FASTQ files were aligned to the Ensembl Mus musculus genome (GRCm38) using STAR aligner (version 2.5.1). Reads that mapped to the coding genes were quantified using featureCounts(Liao et al., 2014), and genes with count values of zero were filtered out before the downstream analysis. Differential gene expression analysis between the different conditions was done using DESeq2(Love et al., 2014) using a model based on the negative binomial distribution. The resulting p-values were then adjusted using the Benjamini and Hochberg approach for controlling the false discovery rate, and differentially expressed genes were determined at the 5% threshold. Gene set enrichment analysis was used to assess the statistical enrichment of gene ontologies, and pathways(Subramanian et al., 2005), and visualization of the significant functional enrichment results was facilitated by clusterProfiler (Yu et al., 2012a).
QUANTIFICATION AND STATITICAL ANALYSIS
Numerical results are presented as means ± SEM unless otherwise indicated. Statistical testing was performed with Student’s t-test, Fisher’s exact test, and analysis of variance (ANOVA, followed by Bonferroni or Sidak post-hoc testing), as appropriate and indicated. Correlation was assessed by calculation of correlation coefficient analysis. Details of statistical analyses of experiments can be found in the figures and figure legends, including exact value of n, what n represents and the statistical tests used. Normal distribution of values was not tested. Statistical significance was achieved with a two-sided P value ≤ 0.05. Statistical analyses were performed with GraphPad Prism.
Supplementary Material
Supplementary Video S1. Rotational 3D videos show decreasing NP numbers from fetal to adult cardiomyocyte nuclei. Related to Figure 1a. Representative videos of 3D reconstructions of fetal, neonatal, and adult cardiomyocyte nuclei to visualize nuclear pores. The videos were reconstructed from z-stack confocal photomicrographs taken with 100x magnification, 0.3 μm for each step. Nuclear pores (mAb414) in red, nuclei (DAPI) in blue, and sarcomeres (troponin I) in grey. This video corresponds to Figure 1a.
Supplementary Video S2. Movies of Dronpa fluorescent intensity in a fetal (E14.5) and neonatal (P2) rat cardiomyocyte. Related to Figure 2c. Representative videos of live cell imaging of Dronpa-ERK fluorescent intensity (green) monitored for 180 s after cytoplasmic photoactivation in a fetal (E14.5) and neonatal (P2) rat cardiomyocyte. Each experiment begins with identification of the cardiomyocyte of interest and its nucleus in bright field microscopy. The entire cardiomyocyte (green outline) is then photobleached, followed by photoactivation of the indicated region of cytoplasm (purple rectangles, t=0 s), and acquisition in the nucleus at 15 fps for 180 s (red or orange circle). This video corresponds to Figure 2c.
Supplementary Video S3. Movies of cardiac MRIs show less adverse remodeling in Lmnb2 KO mice after pressure overload. Related to Figure 6b&c. Representative movies of cine MRI frames show decreased remodeling and better contractile function. Movies were reconstructed from cine MRIs obtained at multiple levels in short-axis and one slide in long-axis 3 days before (Pre) and 7 days after (Post) introduction of pressure overload by TAC. MRI slices from the same hearts at the same distance from the apex are shown.
Highlights:
Mammalian cardiomyocytes decrease number of nuclear pores (NP) during maturation.
Reducing NP numbers lowers nuclear transport, which alters gene regulation.
Lower NP numbers reduce adverse remodeling in mouse model of cardiac stress.
Acknowledgements
We thank Stephen Young (University of California, Los Angeles) for providing Lmnb2flox mice. We are grateful for technical advice from Simon Watkins (Center for Biological Imaging, University of Pittsburgh) and technical support from Samuel K. Wyman of the Animal Imaging Core at Rangos Research Center (University of Pittsburgh). We thank Kyla Holbrook and Katelyn Parsons for technical assistance (Children’s Hospital of Pittsburgh). We thank members of the Kuhn laboratory and Michael Tsang (University of Pittsburgh) for support, helpful discussions, and critical reading of the manuscript. This research was supported by the Richard King Mellon Foundation Institute for Pediatric Research (UPMC Children’s Hospital of Pittsburgh), HeartFest, a Transatlantic Network of Excellence grant by the Leducq Foundation (15CVD03), NIH grants R01HL151415, R01 HL151386, and R01HL155597, and a grant from the UPMC Aging Institute (to B.K.). This project was supported by AHA Career Development Awards (to L.H. and Y.L.W.) and by an NIH-Training Grant (to Y.L., T32HL129949). Y.L.W. received support from NIH (EB023507, NS121706-01), AHA (18CDA34140024), and DoD (W81XWH1810070, W81XWH-22-1-0221). This research was supported through S10OD011967 (to D.B.S.). J.-H. J., and B.K. received support from the Clinical and Translational Science Institute at the University of Pittsburgh, which is supported by Clinical and Translational Science Award (CTSA) grant UL1 TR001857.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Agah R, Frenkel PA, French BA, Michael LH, Overbeek PA, and Schneider MD (1997). Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J Clin Invest 100, 169–179. 10.1172/JCI119509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DJ, and Hetzer MW (2007). Nuclear envelope formation by chromatin-mediated reorganization of the endoplasmic reticulum. Nat Cell Biol 9, 1160–1166. 10.1038/ncb1636. [DOI] [PubMed] [Google Scholar]
- Ando R, Mizuno H, and Miyawaki A (2004). Regulated fast nucleocytoplasmic shuttling observed by reversible protein highlighting. Science 306, 1370–1373. 10.1126/science.1102506. [DOI] [PubMed] [Google Scholar]
- Boehmer T, Enninga J, Dales S, Blobel G, and Zhong H (2003). Depletion of a single nucleoporin, Nup107, prevents the assembly of a subset of nucleoporins into the nuclear pore complex. Proc Natl Acad Sci U S A 100, 981–985. 10.1073/pnas.252749899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahine MN, Mioulane M, Sikkel MB, O’Gara P, Dos Remedios CG, Pierce GN, Lyon AR, Foldes G, and Harding SE (2015). Nuclear pore rearrangements and nuclear trafficking in cardiomyocytes from rat and human failing hearts. Cardiovasc Res 105, 31–43. 10.1093/cvr/cvu218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou YY, Upadhyayula S, Houser J, He K, Skillern W, Scanavachi G, Dang S, Sanyal A, Ohashi KG, Di Caprio G, et al. (2021). Inherited nuclear pore substructures template postmitotic pore assembly. Dev Cell 56, 1786–1803 e1789. 10.1016/j.devcel.2021.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffinier C., Jung HJ., Nobumori C., Chang S., Tu Y., Barnes RH 2nd., Yoshinaga Y, de Jong PJ, Vergnes L, Reue K, et al. (2011). Deficiencies in lamin B1 and lamin B2 cause neurodevelopmental defects and distinct nuclear shape abnormalities in neurons. Molecular biology of the cell 22, 4683–4693. 10.1091/mbc.E11-06-0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angelo MA, Raices M, Panowski SH, and Hetzer MW (2009). Age-dependent deterioration of nuclear pore complexes causes a loss of nuclear integrity in postmitotic cells. Cell 136, 284–295. 10.1016/j.cell.2008.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis LI, and Blobel G (1987). Nuclear pore complex contains a family of glycoproteins that includes p62: glycosylation through a previously unidentified cellular pathway. Proc Natl Acad Sci U S A 84, 7552–7556. 10.1073/pnas.84.21.7552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhardt N, Redolfi J, and Antonin W (2014). Interaction of Nup53 with Ndc1 and Nup155 is required for nuclear pore complex assembly. Journal of cell science 127, 908–921. 10.1242/jcs.141739. [DOI] [PubMed] [Google Scholar]
- Han L, Choudhury S, Mich-Basso JD, Ammanamanchi N, Ganapathy B, Suresh S, Khaladkar M, Singh J, Maehr R, Zuppo DA, et al. (2020). Lamin B2 Levels Regulate Polyploidization of Cardiomyocyte Nuclei and Myocardial Regeneration. Dev Cell 53, 42–59 e11. 10.1016/j.devcel.2020.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heineke J, and Molkentin JD (2006). Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol 7, 589–600. [DOI] [PubMed] [Google Scholar]
- Jiao K, Kulessa H, Tompkins K, Zhou Y, Batts L, Baldwin HS, and Hogan BL (2003). An essential role of Bmp4 in the atrioventricular septation of the mouse heart. Genes Dev 17, 2362–2367. 10.1101/gad.1124803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirillova A, Han L, Liu H, and Kuhn B (2021). Polyploid cardiomyocytes: implications for heart regeneration. Development. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knockenhauer KE, and Schwartz TU (2016). The Nuclear Pore Complex as a Flexible and Dynamic Gate. Cell 164, 1162–1171. 10.1016/j.cell.2016.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Smyth GK., and Shi W. (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Lin DH, and Hoelz A (2019). The Structure of the Nuclear Pore Complex (An Update). Annu Rev Biochem 88, 725–783. 10.1146/annurev-biochem-062917-011901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Zhang CH, Ammanamanchi N, Suresh S, Lewarchik C, Rao K, Uys GM, Han L, Abrial M, Yimlamai D, et al. (2019). Control of cytokinesis by beta-adrenergic receptors indicates an approach for regulating cardiomyocyte endowment. Sci Transl Med 11. 10.1126/scitranslmed.aaw6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Xu J, Jin J, Huang Y, and Liu Y (2017). A Simple Marker-Assisted 3D Nanometer Drift Correction Method for Superresolution Microscopy. Biophys J 112, 2196–2208. 10.1016/j.bpj.2017.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maul GG, and Deaven L (1977). Quantitative determination of nuclear pore complexes in cycling cells with differing DNA content. J Cell Biol 73, 748–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M, Park MK, and Hanover JA (1991). Nuclear pore complex: structure, function, and regulation. Physiological reviews 71, 909–949. 10.1152/physrev.1991.71.3.909. [DOI] [PubMed] [Google Scholar]
- Mitchell JM, Mansfeld J, Capitanio J, Kutay U, and Wozniak RW (2010). Pom121 links two essential subcomplexes of the nuclear pore complex core to the membrane. J Cell Biol 191, 505–521. 10.1083/jcb.201007098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahrendorf M., Streif JU., Hiller KH., Hu K., Nordbeck P., Ritter O., Sosnovik D., Bauer L., Neubauer S., Jakob PM., et al. (2006). Multimodal functional cardiac MRI in creatine kinase-deficient mice reveals subtle abnormalities in myocardial perfusion and mechanics. Am J Physiol Heart Circ Physiol 290, H2516–2521. 10.1152/ajpheart.01038.2005. [DOI] [PubMed] [Google Scholar]
- Perez-Terzic C, Gacy AM, Bortolon R, Dzeja PP, Puceat M, Jaconi M, Prendergast FG, and Terzic A (1999). Structural plasticity of the cardiac nuclear pore complex in response to regulators of nuclear import. Circ Res 84, 1292–1301. 10.1161/01.res.84.11.1292. [DOI] [PubMed] [Google Scholar]
- Perez-Terzic C, Gacy AM, Bortolon R, Dzeja PP, Puceat M, Jaconi M, Prendergast FG, and Terzic A (2001). Directed inhibition of nuclear import in cellular hypertrophy. J Biol Chem 276, 20566–20571. 10.1074/jbc.M101950200. [DOI] [PubMed] [Google Scholar]
- Perez-Terzic C, Pyle J, Jaconi M, Stehno-Bittel L, and Clapham DE (1996). Conformational states of the nuclear pore complex induced by depletion of nuclear Ca2+ stores. Science 273, 1875–1877. [DOI] [PubMed] [Google Scholar]
- Pulupa J, Prior H, Johnson DS, and Simon SM (2020). Conformation of the nuclear pore in living cells is modulated by transport state. eLife 9. 10.7554/eLife.60654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell NH, Tang G, Yu C, Mercurio F, DiDonato JA, and Lin A (2001). Activation of NF-kappa B is required for hypertrophic growth of primary rat neonatal ventricular cardiomyocytes. Proc Natl Acad Sci U S A 98, 6668–6673. 10.1073/pnas.111155798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raices M, and D’Angelo MA (2012). Nuclear pore complex composition: a new regulator of tissue-specific and developmental functions. Nat Rev Mol Cell Biol 13, 687–699. 10.1038/nrm3461. [DOI] [PubMed] [Google Scholar]
- Rust MJ, Bates M, and Zhuang X (2006). Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat Methods 3, 793–795. 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strubing C, and Clapham DE (1999). Active nuclear import and export is independent of lumenal Ca2+ stores in intact mammalian cells. J Gen Physiol 113, 239–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha VK., Mukherjee S., Ebert BL., Gillette MA., Paulovich A., Pomeroy SL., Golub TR., Lander ES., and Mesirov JP. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinnakota KC, and Bassingthwaighte JB (2004). Myocardial density and composition: a basis for calculating intracellular metabolite concentrations. Am J Physiol Heart Circ Physiol 286, H1742–1749. 10.1152/ajpheart.00478.2003. [DOI] [PubMed] [Google Scholar]
- Vollmer B, Schooley A, Sachdev R, Eisenhardt N, Schneider AM, Sieverding C, Madlung J, Gerken U, Macek B, and Antonin W (2012). Dimerization and direct membrane interaction of Nup53 contribute to nuclear pore complex assembly. EMBO J 31, 4072–4084. 10.1038/emboj.2012.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Ma H, Jin J, Uttam S, Fu R, Huang Y, and Liu Y (2018). Super-Resolution Imaging of Higher-Order Chromatin Structures at Different Epigenomic States in Single Mammalian Cells. Cell Reports 24, 873–882. 10.1016/j.celrep.2018.06.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Ma H, Ma H, Jiang W, Mela CA, Duan M, Zhao S, Gao C, Hahm E-R, Lardo SM, et al. (2020). Super-resolution imaging reveals the evolution of higher-order chromatin folding in early carcinogenesis. Nat. Commun. 11, 1899. 10.1038/s41467-020-15718-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, Han Y, and He QY (2012a). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SH, Boyce M, Wands AM, Bond MR, Bertozzi CR, and Kohler JJ (2012b). Metabolic labeling enables selective photocrosslinking of O-GlcNAc-modified proteins to their binding partners. Proc Natl Acad Sci U S A 109, 4834–4839. 10.1073/pnas.1114356109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Video S1. Rotational 3D videos show decreasing NP numbers from fetal to adult cardiomyocyte nuclei. Related to Figure 1a. Representative videos of 3D reconstructions of fetal, neonatal, and adult cardiomyocyte nuclei to visualize nuclear pores. The videos were reconstructed from z-stack confocal photomicrographs taken with 100x magnification, 0.3 μm for each step. Nuclear pores (mAb414) in red, nuclei (DAPI) in blue, and sarcomeres (troponin I) in grey. This video corresponds to Figure 1a.
Supplementary Video S2. Movies of Dronpa fluorescent intensity in a fetal (E14.5) and neonatal (P2) rat cardiomyocyte. Related to Figure 2c. Representative videos of live cell imaging of Dronpa-ERK fluorescent intensity (green) monitored for 180 s after cytoplasmic photoactivation in a fetal (E14.5) and neonatal (P2) rat cardiomyocyte. Each experiment begins with identification of the cardiomyocyte of interest and its nucleus in bright field microscopy. The entire cardiomyocyte (green outline) is then photobleached, followed by photoactivation of the indicated region of cytoplasm (purple rectangles, t=0 s), and acquisition in the nucleus at 15 fps for 180 s (red or orange circle). This video corresponds to Figure 2c.
Supplementary Video S3. Movies of cardiac MRIs show less adverse remodeling in Lmnb2 KO mice after pressure overload. Related to Figure 6b&c. Representative movies of cine MRI frames show decreased remodeling and better contractile function. Movies were reconstructed from cine MRIs obtained at multiple levels in short-axis and one slide in long-axis 3 days before (Pre) and 7 days after (Post) introduction of pressure overload by TAC. MRI slices from the same hearts at the same distance from the apex are shown.
Data Availability Statement
The datasets supporting the current study have not been deposited in a public repository because of author preferences and are available from the corresponding author upon request.
The RNA-Seq data generated during this study have been deposited at GEO and are publicly available at GEO Series accession number GSE186949 as of the date of publication.
This paper does not report original code.