

Abstract
Positron emission tomography (PET) is a powerful imaging technology that could visualize and measure metabolic processes in vivo and/or obtain unique information about drug candidates at early stages. Identification of new and improved molecular probes plays a critical role in PET, but its progress is limited in many situations due to the lack of efficient and simple labeling methods to modify biologically active small molecules and/or drugs. Although various approaches have been reported, current methods to radiofluorinate unactivated arenes are still relatively limited, especially in a simple and site-selective way. Here we disclose a method for constructing C–18F bonds through direct halide/18F conversion in electron-rich halo(hetero)arenes. [18F]F− is efficiently introduced into a broad spectrum of readily available aryl halide precursors in a site-selective manner under mild photoredox conditions. Notably, we demonstrate that our direct 19F/18F exchange method enables rapid PET probe diversification through the preparation and evaluation of a [18F]-labeled O-methyl tyrosine library, leading to the discovery of new cancer imaging agents. This strategy also results in the high-yielding synthesis of the widely used PET agent L-[18F]FDOPA from a readily available L-FDOPA analog. Taken together, our photoredox-mediated halide/18F interconversion strategy offers a new chemical tool for preparing new and clinically significant 18F-labeled PET agents.
Introduction
Positron emission tomography (PET) is a routinely-used non-invasive imaging technique for the real-time diagnosis and monitoring of human diseases, most notably being oncological and neurological disorders1,2. Advances in radiotracer development have accelerated the clinical adoption of PET and fostered a growing interest in robust methodologies for synthesizing PET agents via the late-stage installation of short-lived radionuclides3. Because many small molecule pharmaceuticals and therapeutics contain aromatic or heteroaromatic systems within their framework4–7, it is highly desirable to introduce radionuclides on these moieties in a synthetically facile and efficient manner. Late stage radiolabeling is especially preferred considering the decay of short lived PET isotopes8–11. Fluorine-18 (18F) is arguably the most widely used short-lived PET isotope (t1/2 ~110 min) due to its excellent imaging properties, wide availability and ideal half-life. A common strategy used for constructing aryl C(sp2)–18F bonds is nucleophilic aromatic substitution (SNAr), which substitutes a (pseudo)halide with 18F fluoride (18F−) (Fig.1A). This strategy is routinely used for the synthesis of PET agents with high molar activity12–15 but is limited to electron-deficient (hetero)aromatic systems16. Consequently, systems for the nucleophilic radiofluorination of electron-neutral and -rich aromatics have been investigated over the past decade3,17. Progress towards this goal have focused on either transition-metal mediated methods18,19,20–24 or the development of specialized nucleofuges25–29, with several of the aforementioned strategies adapted for automation18,20–25,28,30,31, which streamlines their translation to clinical use. As a major substrate class for arene functionalization, aryl (pseudo)halides are commonly used intermediates on route to synthesizing organometallic or prefunctionalized arene precursors for radiofluorination (such as aryl-palladium18/nickel complexes19, aryl boronic acids22/esters20, aryl stannanes23 and aryl iodonium salts/ylides21,24,25) (Fig.1B). Clearly, methods that could directly radiofluorinate electron-rich aryl halides are highly desired due to its simplicity. However, there are scarce radiofluorination examples for unactivated aryl (pseudo)halides except a recent method disclosed by the Scott and Sanford groups which allowed the ligand-directed conversion of aryl halides to radiofluorinated arenes using an NHC-copper complex31. While the installation of 18F in electron-neutral and -rich arenes is impressive, the requisite substrate directing group could limit the generality of this approach (Fig.1C). Additionally, this method is limited to aryl iodides and bromides, with chloro-arenes being minimally reactive and no 19F/18F exchange product observed. Given the dearth of methods for the direct radiofluorination of electron-rich aryl chlorides and fluorides, developing a simple strategy to directly radiofluorinate these motifs is highly desired, especially considering their stability and abundance in therapeutics7,32,33. Importantly, the success of this approach would also enable the direct conversion of readily available fluorinated therapeutics into 18F-labeled radiopharmaceuticals through simple late-stage 19F/18F exchange34–36.
Fig.1 |. Nucleophilic arene 18F-fluorination.
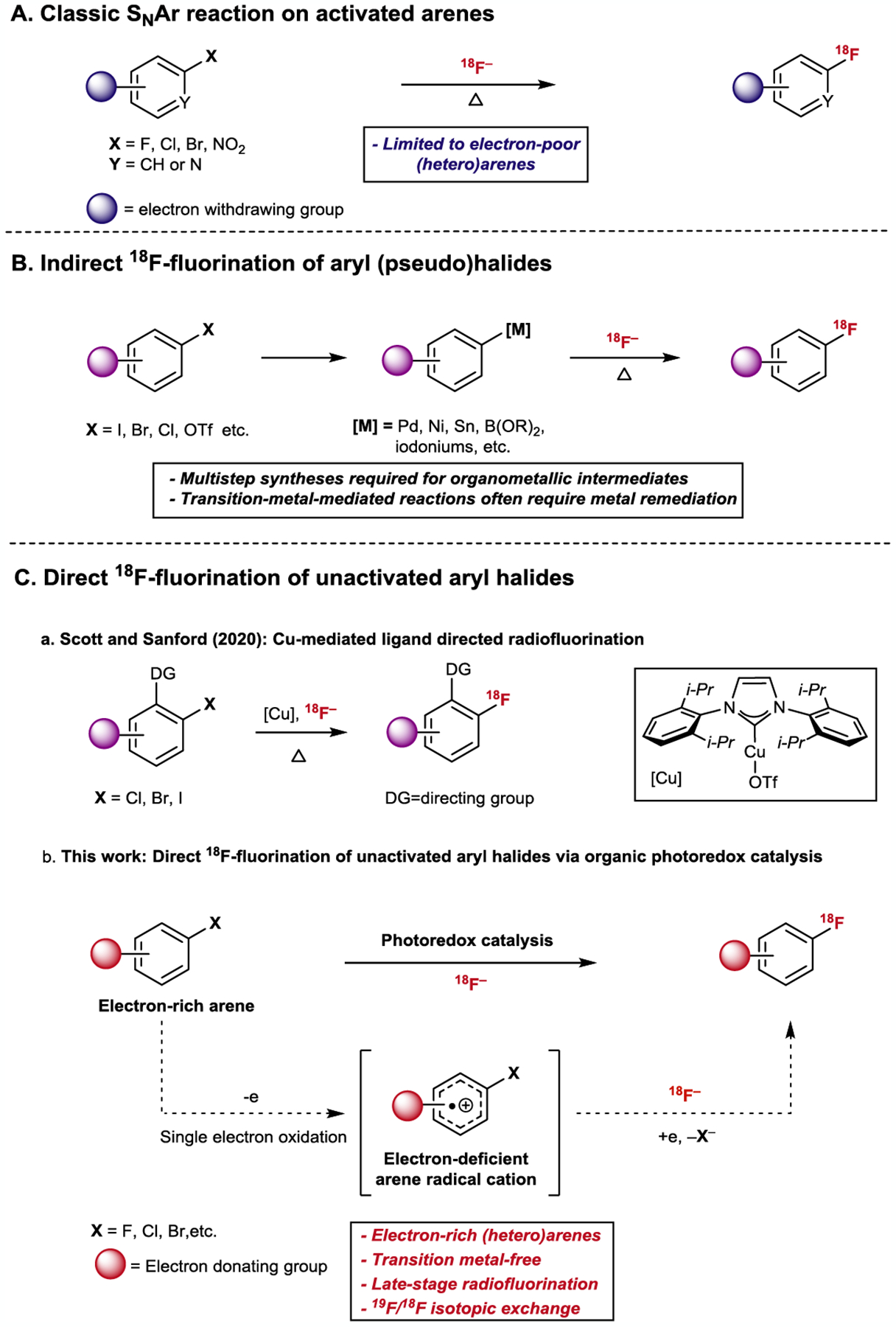
(A) SNAr reaction on activated (electron-deficent) arenes (B) Indirect 18F-fluorination of aryl(pseudo) halides (C) Direct 18F-fluorination of aryl halides
We recently disclosed two acridinium photoredox-mediated methods for arene radiofluorination in which the photoactivated acridinium oxidizes arenes to arene cation radicals, thus enabling site-selective radiofluorination of C(sp2)–H37 and C(sp2)–O29 bonds. Inspired by our previous success, we explored the feasibility of directly converting an aryl halide into their 18F-labeled congeners for electron-rich arenes. Upon single electron oxidation, we envisioned the resulting electron-deficient cation radical would capture the 18F− at the halide-bearing carbon. Subsequent reduction and expulsion of the halide nucleofuge would furnish the desired radiofluorinated arene (Fig. 1C). This radiofluorination strategy would obviate the need for lengthy, multi-step precursor synthesis, greatly simplifying product isolation, and open up new ways to prepare clinically significant PET agents.
Results
To evaluate whether acridinium photocatalysts could promote halide/18F exchange in electron-rich arenes, we adapted our previously-reported radiodeoxyfluorination conditions to 1-chloro-4-methoxybenzene (1-Cl) with the intent of promoting halide extrusion. Irradiating a reaction solution containing the substrate, [18F]n-tetrabutylammonium fluoride ([18F]TBAF) and acridinium S1 in a multicomponent DCE/t-BuOH/MeCN solvent system with a 450 nm laser under consistent air bubbling for 30 min leads to the formation of 1-18F, albeit in 1.7% radiochemical conversion (RCC) as calculated by HPLC isolation. In our previous (radio)deoxyfluorination study29, we found that methoxy groups were ineffective due to the competitive deprotonation of acidic O-methyl C–H bonds upon formation of the arene cation radical — this deleterious pathway competes with extrusion of halide under aerobic conditions38. To circumvent this potential side-reaction, the radiofluorination of 1-Cl was conducted under a nitrogen atmosphere, which successfully increased the isolation yield of 1-18F to 12.8 ± 0.3% (n=3) with 71.25 GBq/μmol molar activity (Am).
With these preliminary results, we then screened other nucleofuges commonly used in SNAr reactions (Fig 2). The radiofluorination of haloarene 1-bromo-4-methoxybenzene (1-Br) yields a comparable RCC with 1-Cl whereas 1-iodo-4-methoxybenzene (1-I) was surprisingly less reactive, with a ten-fold lower RCC observed. Interestingly, 1-fluoro-4-methoxybenzene (1-F) was found to provide the highest yield of radiofluorinated product through direct 19F/18F exchange (79.7%). This observation represents a significant breakthrough in the field because it allows simple and efficient conversion of electron-rich fluorinated bioactive compounds or pharmaceuticals to 18F-labelled PET radiotracers directly. Prior reports of fluoroarene 19F/18F exchange have been restricted to electron-deficient compounds and generally require relatively high temperatures36,39. A potential limitation of this strategy is the decreased molar activity (Am) by virtue of using the HPLC-inseparable 19F-fluoroarene. However, Am values can be significantly improved by limiting the amount of aryl fluoride, which has been demonstrated previously36a,40. Next, we found that aryl triflate 1-OTf was successfully radiofluorinated, albeit with lower yields. Interestingly, 1-methoxy-4-nitrobenzene (1-NO2) was unreactive despite the well-documented substitution of nitro nucleofuges in traditional SNAr.
Fig.2 |. Reaction scope of direct 18F-fluorination of aryl halides via halide/18F interconversion.
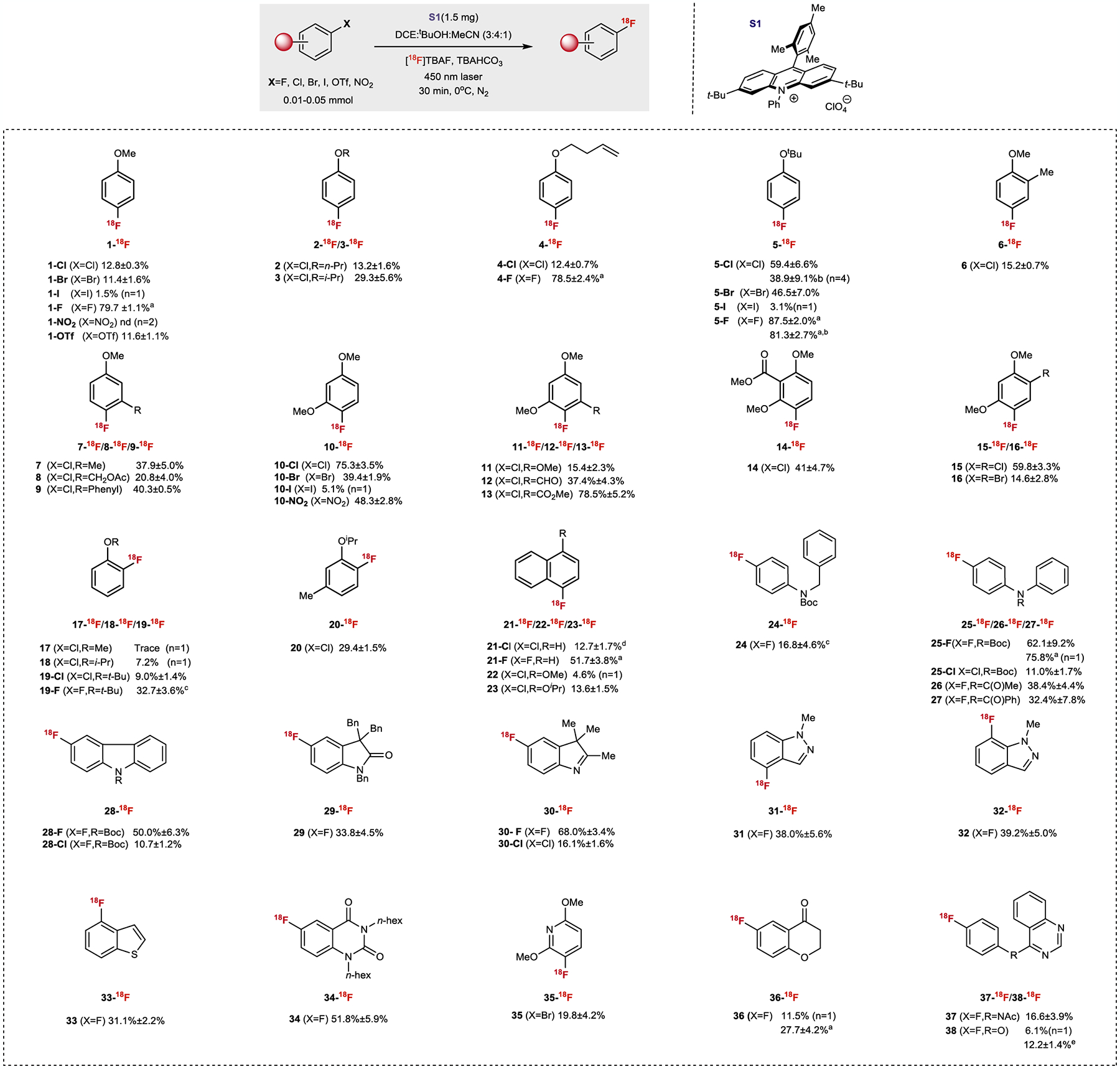
All radiochemical conversions (RCCs) were calculated by HPLC isolation and averaged over 3 experiments unless otherwise noted. 0.37–1.11 GBq [18F]TBAF were generally used for the labeling. 0.05 mmol substrate were used for all the labeling reactions except Ar-F which used 0.01 mmol unless otherwise noted. a0.05 mmol substrate. bBlue LED instead of laser. c0.02 mmol substrate. d0.1 mmol substrate. eReaction was performed under air.
To better understand the effect of substitution pattern on the halogen/18F interconversion, we evaluated a range of aromatic and heteroaromatic substrates (Fig 2). Increasing alkylation at the α-carbon relative to oxygen in O-alkylated 4-chlorophenol derivatives resulted in a 2- to 4-fold RCC increase, suggesting that labile C–H bonds adjacent to the O-atom be inhibitory to the reaction (2, 3, 4-Cl, 5-Cl). This effect is less pronounced for 19F to 18F conversion, where minimal RCC differences were observed. For example, both 4-F and 5-F were obtained in excellent RCC based on HPLC isolation. We also performed the halide/18F interconversion reactions with blue LED instead of laser on substrates 5-Cl and 5-F. We were pleased to find that the Cl/18F conversion proceeded on 5-Cl albeit with lower RCC. Meanwhile, the 19F/18F isotopic exchange on 5-F afforded comparable RCC with LED irradiation. A meta-methyl substituent (6) resulted in moderate RCC improvement, while substituents ortho to the chlorine nucleofuge resulted in more efficient halide/18F conversion (7–9). This observation is tentatively attributed to the enhanced stability of a putative captodative cation radical intermediate invoked in our previous findings29,41. 2,4-Dimethoxy-substituted aryl halides (10-Cl, 10-Br, 10-I) were labelled with 18F− leading to 10-18F in good, moderate and low RCCs respectively. Interestingly, 10-NO2 which is more electron rich than mono-methoxy 1-NO2, was successfully radiofluorinated with 48.3% RCC. This observation suggests that the lack of reactivity with 1-NO2 is likely due to a mismatch in redox potentials between 1-NO2 and the acridinium catalyst. The radiofluorination of 2,4,6- and 2,3,4-substituted chlorobenzenes (11–14) was accomplished with moderate to excellent RCCs. Desymmetrization of dihalogenated aromatics (15 and 16) was also demonstrated, although the reduced solubility of dibrominated 16 resulted in a lower RCC than the more soluble dichlorinated analog (15). More pronounced changes in radiofluorination efficiency were observed O-alkylated haloarenes bearing ortho-nucleofuges (17–20). Chloro- and fluoro-naphthalenes and their alkoxy-substituted derivatives were also successfully radiofluorinated (21-23). Aryl fluorides containing protected amines (24, 25-F, 26–27) undergo 19F/18F conversion with moderate to good RCC under the standard labelling conditions, with aryl chloride 25-Cl demonstrating lower radiofluorination efficiency. Chloro- and fluoro-substituted heterocycles were also successfully radiofluorinated via Cl/18F or 19F/18F conversion, as demonstrated for carbazoles 28-F and 28-Cl, N-benzyl indolinone 29, 3,3-dimethy-3H-indoles 30-F and 30-Cl, N-methyl indazoles 31 and 32, benzo[b]thiophene 33, quinazoline-2,4(1H,3H)-dione 34, pyridine 35, and chromanone 36. Additionally, 4-(arylamino)quinazoline fragment 37 and its analog 38, common pharmacophores in kinase inhibitors42, were found to be competent substrates for direct 19F/18F exchange. Several substrate limitations exist for this methodology (Supplementary Figure 138). Generally, the presence of an activated benzylic group or α-heteroatom methylene unit leads to decreased yield or results in no reaction, likely because the deprotonation by fluoride is kinetically favored. Additionally, substates bearing more oxidizable functional groups other than the halogenated aromatic ring under light irradiation, for example, unprotected amines or alcohols would lead to competing electron transfer pathways and thus inhibit the halogen-18F exchange process. Lastly, substrates where the halogen and methoxy are meta to each other, tend to give low or no yield under current conditions.
To probe the chemo- and regioselectivity of the halide/18F conversion, we studied substrates bearing more than one potential halogen nucleofuge (Fig. 3a). Radiofluorination of compound 39 provides both Br/18F and Cl/18F conversion products (15-18F, 16-18F) in 1:1.4 ratio in slight favor of chlorine substitution. When the bromide was shifted to the meta position relative to the methoxy groups (40), selective Cl/18F conversion (40-18F) was observed. These results suggest that 1) chlorine is a better nucleofuge than bromine, and 2) the site of SNAr is largely dependent on arene electronics. Next, we explored the selectivity between Cl/18F conversion (SNAr) and bimolecular nucleophilic substitution (SN2), which is a commonly employed strategy to introduce 18F into alkyl groups in PET radiotracer design3. Compound 41 is a substrate containing both aryl and alkyl chlorides (Fig. 3b). Selective Cl/18F conversion (SNAr) (41-18F) is observed under photoredox conditions while heating the reaction to 100 °C results in exclusive [18F]F− displacement of the alkyl chloride (41-18F-a). Next, we probed the chemoselectivity between electron-rich and electron-deficient arenes within one molecule (Fig. 3c). Using compound 42 as our model substrate, we obtained exclusive 19F/18F conversion (42-18F) with no traditional SNAr product (42-18F-a)43. Taken together, these results suggest that our halide exchange strategy is selective for electron-rich substrates using photoredox conditions without 18F− substitution of alkyl halides and electron-deficient aryl halides.
Fig.3 |. Chemo-and regioselectivity study of aryl halide/18F interconversion.
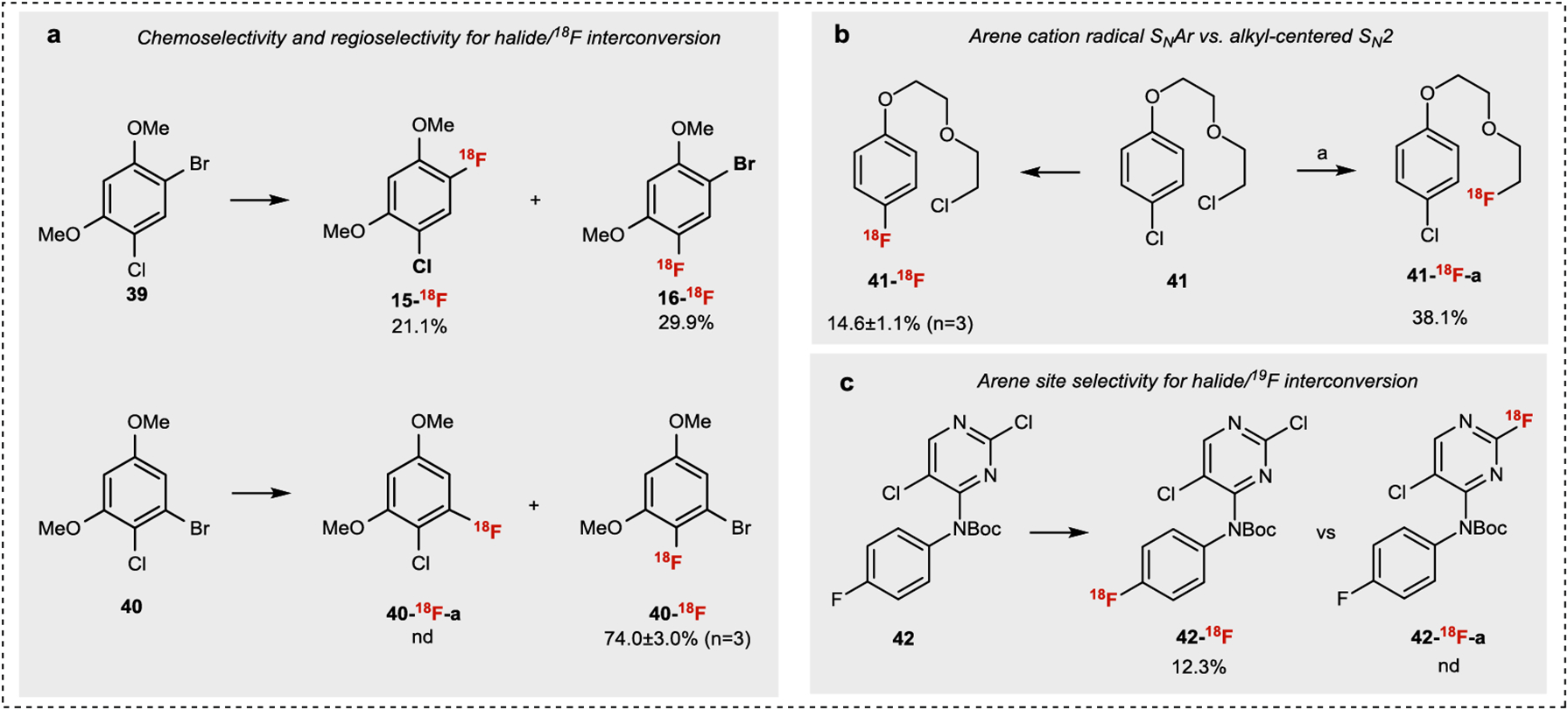
All halide/18F interconversion reactions were conducted under standard conditions listed in Fig.2. a. Comparison of reactivity between ArBr and ArCl. b. Comparison of reactivity between SNAr and SN2 under light condition. c. Comparison of reactivity between electron rich and electron deficient aromatic rings. a[18F]TBAF, MeCN, 100°C, 10 min (See SI for details).
We next applied our halide/18F interconversion strategy towards the radiolabeling of known pharmaceuticals and bioactive molecules (Fig. 4a). Clofibrate (43) and Boc-protected atomoxetine (44) were directly converted to 18F-fluorinated analogs (43-18F, 44-18F) through aryl-Cl/18F exchange while 18F-labelled flurbiprofen methyl ester (45) and diflunisal (46) were obtained (45-18F, 46-18F) through direct 19F/18F conversion. Mono-Boc protected fluorodopamine (47-F) was efficiently labelled through 19F/18F conversion (47-18F) in excellent RCC. Fluorinated dopamine 47-18F could also be synthesized through Cl/18F exchange with a lower RCC but higher molar activity. 18F-labelled 2-phenoxyaniline derivatives have been investigated as translocator protein (TSPO)-specific PET agents for neuroinflammation imaging44. Using our aryl-Cl/18F conversion method, 18F successfully replaced the chlorides in 2-phenoxyaniline derivatives, leading to potential new imaging agents (48-18F, 49-18F, and 50-18F) targeting TSPO. Additionally, [18F]fluorouracil (53-18F), an important PET agent in oncology, was readily obtained through aryl-Cl/18F conversion followed by a simple deprotection (Fig. 4b). The key pyrimidine intermediate 52-18F obtained from easily available chloroarene 52 showed much higher labeling efficiency than previously reported aryl nickel (II) complex30 and aryl iodonium ylide25a. Moreover, the precursor can be obtained in fewer synthetic steps than the diarylether analog which we previously used for radiodeoxyfluorination29. Taken together, our Cl/18F exchange strategy offers a synthetically accessible alternative to [18F]fluorouracil with high Am45.
Fig.4 |. Application of the halide/18F interconversion to late-stage radiofluorination.
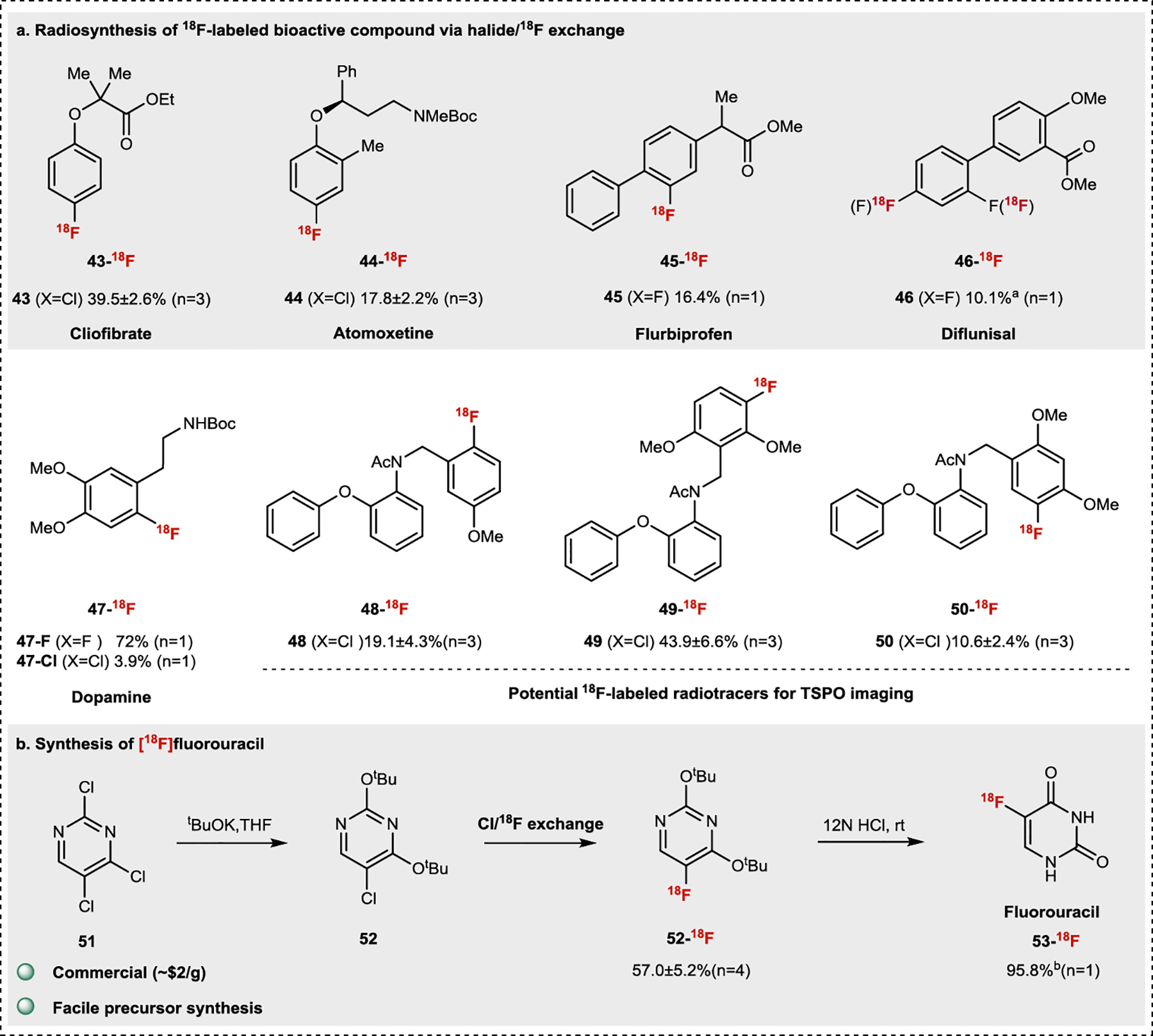
All halide/18F exchange reactions were conducted under standard conditions listed in Fig.2. a. Labeling of bioactive compounds. b. Synthesis of [18F]fluorouracil. a0.05 mmol substrate. bDeprotection yield.
We further evaluated the application of direct 19F/18F conversion within biologically-relevant electron rich arenes due to its exceptional efficiency and simplicity. Although the resulting PET agents may have reduced Am, it still represents a broadly useful technology for studying the pharmacokinetics/pharmacodynamics of fluorine-containing drugs34–36 or imaging transporter-mediated processes, such as synthesizing fluorinated amino acid agents for large neutral amino acid transporter (LAT1) imaging46. Amino acid metabolism represents another important class of pathways in cancer progression in addition to glucose metabolism. We were particularly interested in synthesizing 18F-labeled tyrosine analogs46. In a proof of principle study, our direct 19F/18F exchange method allowed us to develop new 18F-labeled tyrosine probes47 via facile installation of 18F within the aromatic core of a small library of O-methyl fluorotyrosine derivatives (54–63) (Fig. 5a). Good to excellent RCCs were observed when the fluorine nucleofuge is located at the ortho or para position relative to the methoxy group (54, 56-60, 62, 63). Lower but significant labelling was observed when the fluorine is positioned meta to the methoxy group (55, 61). After a simple deprotection (See SI for details), ten 18F-labelled O-methyl tyrosines were obtained and evaluated as potential PET agents in MCF7 breast cancer models. While radiotracers 54–58, 60–62 are constitutional isomers, the relative position of 18F and methoxy substitution significantly impacts their tumor uptake and clearance profiles (Fig. 5b). Most of the PET tracers demonstrated initial prominent tumor uptake at 1h post-injection followed by washout at 3h (54-18F-COOH, 55-18F-COOH, 56-18F-COOH, 58-18F-COOH, 59-18F-COOH, 62-18F-COOH, 63-18F-COOH). In contrast, PET agents 57-18F-COOH, 60-18F-COOH, 61-18F-COOH showed high and persistent retention in the MCF-7 tumor within the same timeframe. While amino acid analogs with initial high uptake and clearance (leading to high contrast) are great candidates for imaging applications, other analogs with prolonged tumor retention provides amino acid backbones for potential therapy applications in which radioiodinated (131I) or boronated (10B) analogs can be used as cancer treatments via radioactive iodine therapy48 or boron neutron capture therapy49, respectively. Although most of the 18F-labelled tyrosine analogs demonstrated apparent pancreatic uptake, introducing one extra fluorine to the arene ring (difluorinated 63-18F-COOH) greatly reduced the uptake in pancreas while still maintaining prominent tumor uptake. The effect of amino acid stereodefinition was also studied using 61-18F-COOH, which demonstrated high and persistent tumor retention. Although the contrast between the two enantiomers remained comparable at 1h post-injection (p.i.), using L-61-18F-COOH doubled the tumor uptake with increased tumor retention at 3h p.i. compared with D-61-18F-COOH. Taken together, these results suggest that our 19F/18F conversion strategy can be used to systematically investigate new PET agents with different pharmacokinetic properties for cancer imaging. Further evaluation of other O-methyl tyrosine enantiomers and other amino acid scaffolds are currently underway.
Fig.5 |. Screening of 18F-labeled O-methyl tyrosines in a MCF7 (breast cancer) tumor model system.
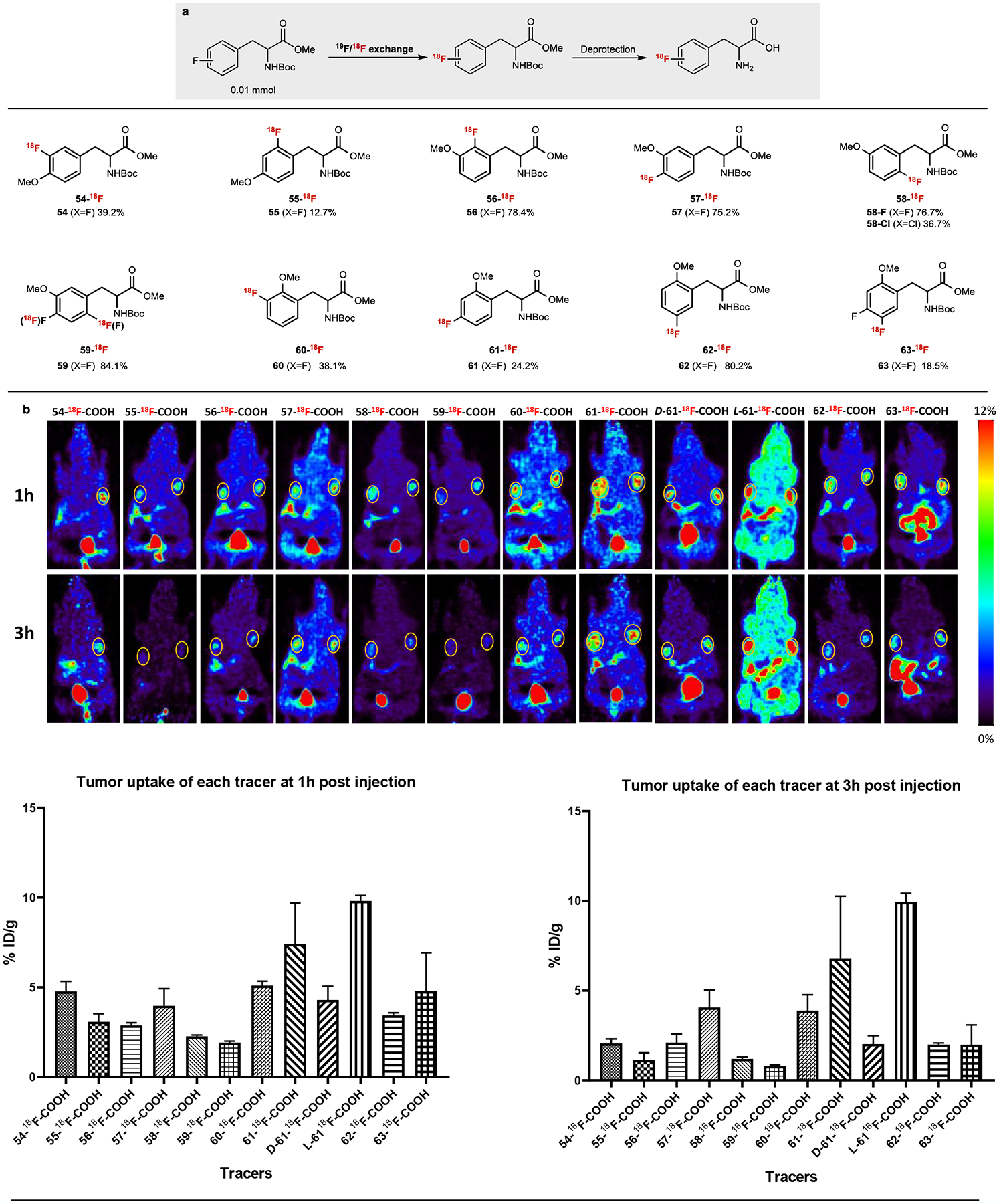
All halide/18F exchange reactions were conducted under standard conditions listed in Fig 2. a. Radiosynthesis of 18F-labeled tyrosines, b. PET imaging of 8F-labeled tyrosines in the MCF7 tumor model system (circled in the pictures)
In addition to developing new PET agents, we are interested in applying our halide/18F interconversion method to streamline the preparation of existing PET agents (Fig. 6). For example, 6-[18F]Fluoro-L-DOPA ([18F]FDOPA) is employed as an important radiopharmaceutical for Parkinson’s disease (PD), brain cancer, and other diseases since the 1980s50. Due to its medical significance, multiple syntheses have been developed to produce [18F]FDOPA for the clinical use20a,51. Given the readily available precursor and simply reaction procedure of our halide/18F exchange approach, we evaluated our method for [18F]FDOPA synthesis using O-methylated DOPA precursors L-64-Cl and L-64-F. We were pleased to find that radiofluorinated proceeds in 4.2% and 73.4% RCC for L-64-Cl and L-64-F, respectively. The resulting product L-64-18F was easily deprotected to L-[18F]FDOPA with 97.1% RCC and >99% enantiomeric excess (ee). No racemization was observed under our photoredox labeling conditions. We also found the 19F/18F conversion in L-64-F remains highly efficient after replacing the laser with more readily available LEDs, performing the reaction without ice cooling, reducing precursor concentration, or shortening the reaction to 5 min at room temperature. Although these conditions would lead to decreased yield, they may be more suitable for automated synthesis. The methoxymethyl (MOM)-protected analog (L-65) and three constitutional isomers of DOPA (66–68) were efficiently labeled via Cl/18F and/or direct 19F/18F conversion as well. Encouraged by the initial successes of our method, we explored the feasibility of synthesizing L-[18F]FDOPA on clinically-relevant scales. Starting from 0.93–1.11 GBq [18F]TBAF, [18F]FDOPA was isolated in 42% and 37.5% n.d.c. RCY (non-decay corrected radiochemical yield) when 0.01 and 0.005 mmol of L-64-F were used respectively (Fig. 6b). Further increasing the scale to ~37 GBq [18F]F− leaded to > 11 GBq of [18F]FDOPA with >30% n.d.c. RCY (> 99% ee, 1.51GBq/μmol) in 100 min (Fig. 6c). Compared with the well-established methods reported by Luxen and Lemaire51b, our radiosynthesis has decreased Am but features the use of a stable and readily available precursor that significantly shortens the post-radiofluorination reaction sequence. Although high Am is not mandatory for the investigation of the neuronal dopaminergic metabolism51a,52, we anticipate that it could be further improved by reducing precursor loading and starting from higher amounts of activity. Overall, our 19F/18F exchange strategy for electron-rich arenes enables a new preparation of [18F]FDOPA with high enantiomeric purity from the readily available isotopic precursor. Future work will be focused on improving and automating this radiolabeling process to meet cGMP compliance standards of clinical applications.
Fig.6 |. Synthesis of [18F]FDOPA.
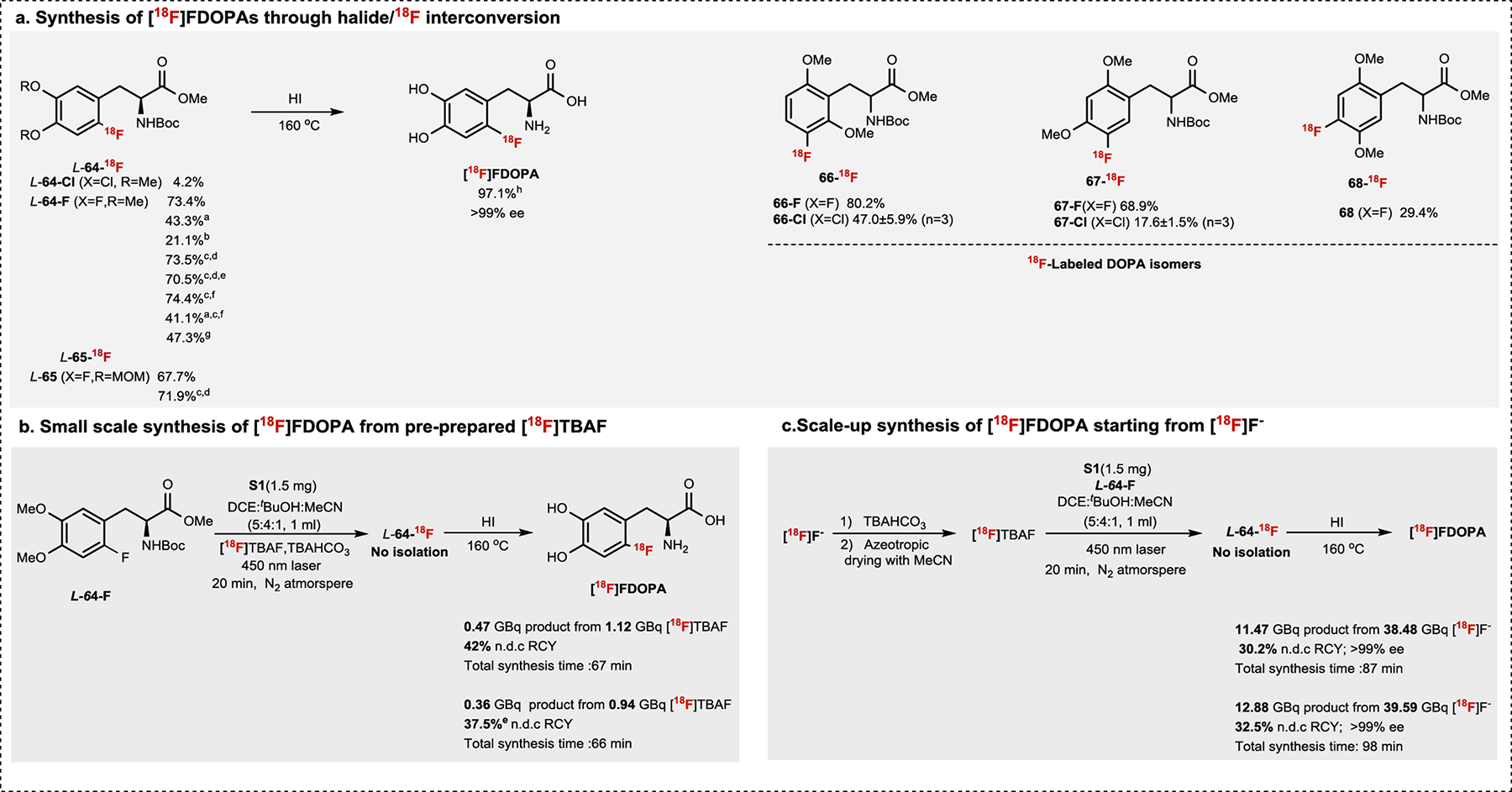
All halide/18F interconversion reactions were conducted under standard condition listed on Fig.2 unless otherwise noted. Radiochemical conversions (RCCs) were calculated by HPLC isolation. 0.05 mmol substrates were used for Cl/18F exchange labeling reactions; 0.01 mmol substrate used for 19F/18F exchange reaction unless otherwise noted. aBlue LED was used instead of laser. bNo DCE were added in the reaction. cNo ice cooling and 500 μl DCE were used. dReaction ran 20 min. e0.005 mmol substrate. fReaction ran 5 min. gair bubbling instead of N2. hDeprotection yield.
Conclusion and Outlook
Photoredox-mediated halide/18F conversion offers new inroads for the radiofluorination of electron-rich haloarenes — a limitation of current SNAr methodology. The success of this method is most pronounced for aryl chlorides and fluorides, with the latter transformation enabling direct 19F/18F exchange. Its application is demonstrated by the successful radiofluorination of electron-rich halo(hetero)arenes, commercial pharmaceuticals and physiologically active compounds. Its utility for the synthesis of new PET agents is demonstrated by the rapid preparation of 18F-labeled O-methyl tyrosine analogues and the characterization of their efficacy towards cancer imaging in breast cancer model. Lastly, direct 19F/18F exchange enables a scalable, late-stage radiosynthesis of [18F]F-L-DOPA, with up to 11 GBq of the radiotracer obtained. Further improvements to this methodology, especially future work on automating this radiosynthesis protocol should enable access to new and/or clinically significant PET agents. Overall, the photoredox-mediated 18F-labeling of unactivated aryl halides opens up new strategies for radiotracer synthesis via SNAr disconnections.
Methods
General procedure for the photoredox-mediated halide/18F interconversion.
Reactions were set up according to a modified literature procedure29. The substrate (0.01–0.05 mmol) and photocatalyst (S1, 1.5 mg) were weighed into a 1.5 ml eppendorf tube and transferred (with solvent when the substrate is liquid or oil) into a 5 ml V-vial via pipette. DCE (300 μl), anhydrous MeCN (45–65 μl), tBuOH(400 μl) and 25 μl of n-tetrabutylammonium bicarbonate (TBAHCO3) in MeCN solution (~60 mg/ml) were sequentially added to the V-vial. Then a 10–30 μl aliquot of [18F]TBAF in MeCN (typically 10–30 mCi) was added to the reaction vial via pipette. The reaction V-vial was then fixed either on an iron support and cooled using an ice bath or on a block without cooling. A needle connected to an N2 filled balloon was inserted to the bottom of the V-vial and the reaction medium was continuously sparged throughout the entire reaction time. The reaction was then irradiated top-down with a laser (MDL-D-450, 450 nm, 3.5 W after fibre coupling) (Supplementary Figure 6) or a A160WE Tuna Blue Kessil LED lamp (Supplementary Figure 7) for 30 min. The resulting solution was diluted and evenly mixed with MeCN (0.5–1ml). An aliquot of the reaction mixture (typically 300–1000 μCi) was taken for radio-HPLC analysis. The activity injected into HPLC was measured (this activity was denoted by α) and the time was recorded. The fraction corresponding to radiolabeled product was collected and the activity was measured (this activity was denoted by β) and the time was recorded. The decay-corrected β could be calculated from the recorded isolation time of each substrate. The radiochemical conversion (RCC) was obtained by dividing the decay-corrected β by α. Co-injection of the purified 18F-labelled compound with commercial or synthesized 19F standard via HPLC was used to confirm the identity of the radiolabeled compound.
Radio-HPLC analysis and characterization for 18F-radiolabeled arenes.
All 18F-labeling reactions were performed according to the general procedure unless otherwise noted. Each labeling reaction, starting activity ([18F]TBAF), injected and collected activities, isolation time, decay corrected activity and calculated radiochemical conversion (RCC) are summarized in a table for each substrate. All 18F-labelled products were analyzed and characterized according to the general HPLC conditions listed in supplementary information at section 3.3. Crude radio-HPLC traces of each reaction (labeled with reaction number), HPLC traces of purification and co-injection were listed. The collected 18F-labeled product from crude HPLC analysis may require further HPLC-purification before co-injection with its corresponding 19F standard. The red HPLC traces in the following spectra were obtained with a UV signal at 212 nm unless otherwise noted. The black HPLC traces represent the radio signal.
Synthesis of [18F]FDOPA from preformed [18F]TBAF.
FDOPA precursor L-64-F (0.01 or 0.005 mmol) and Photocatalyst S1 (1.5 mg) were dissolved in the solution of DCE/tBuOH/MeCN in a 5 ml V-vial. After addition of the [18F]TBAF and TBAHCO3 (25 μl), the resulted solution (~1 ml) was top-down irradiated for 20 min under 450 nm laser (450 nm, 3.5 W after fibre coupling) with a N2 balloon sparge at room temperature. The resulting reaction solution was diluted with 1 ml MeCN and passed through an aluminum cartridge (preconditioned with 5 ml DI water) to remove the unconverted 18F-fluoride. Rinse the reaction vial with another 1 ml MeCN which was then passed through the same aluminum cartridge. The elution was collected in another 5 ml V-Vial and caped with a Teflon-lined septum screw cap equipped with a vent needle. The solvent was removed under 100 °C with argon stream. Argon flow was then stopped and the vent needle was removed. 200 μl HI (57 wt.% in H2O) were then added into the V-vial and the mixture was heated under 160 °C for 10 min. A vent needle was then equipped before water (300 μl) and saturated NaHCO3 solution (400 μl) was slowly added to the V-vial. The resulting aqueous solution was passed through a HPLC filter to remove the insoluble catalyst residue. The collected solution was then purified on HPLC to give the product [18F]FDOPA.
Scale-up synthesis of [18F]FDOPA starting from [18F]F−.
The aqueous solution of [18F]F− fluoride produced via the 18O(p,n)18F reaction by proton irradiation (40 μA) was delivered to a hot cell equipped with manipulators and collected in a 5 ml V-vial containing 5 μl TBAHCO3 (20%) water solution. This aqueous solution was azeotropically dried with anhydrous MeCN (1 ml × 5) under a stream of Argon at 100 °C. After removing the water, the V-vial was removed from the heater. The solution of precursor L-64-F (0.01 mmol) and photocatalyst (S1, 1.5 mg) in DCE/tBuOH/MeCN (5/4/1, 1 ml) was then added into the V-vial. The [18F]FDOPA was then obtained after the same reaction and purification procedure as the synthesis starting from preformed [18F]TBAF.
Supplementary Material
Acknowledgments
This work was supported in part by the National Institutes of Health (NIBIB) R01EB029451 (Z.L. and D.A.N.), 5R01CA233904 (Z.L.), UNC LCCC pilot grant (Z.L. and D.A.N.), 1S10OD023611 (Z.L.) and the startup fund from UNC Department of Radiology, Biomedical Research Imaging Center, and UNC Lineberger Comprehensive Cancer Center (Z.L.)., N.E.S.T. and V.A.P are grateful for NSF Graduate Research Fellowships. We thank Dr. Gerald T. Bida for assistance with cyclotron operation, and the University of North Carolina’s Department of Chemistry Mass Spectrometry Core Laboratory, especially Diane Weatherspoon, for their assistance with mass spectrometry analysis.
Footnotes
Competing interests
The authors have filed a provisional US patent on the basis of the research in this manuscript.
Data availability
All the data generated or analysed during this study are included in this article (and its Supplementary Information files).
References
- 1.Ametamey SM, Honer M & Schubiger PA Molecular imaging with PET. Chem. Rev 108, 1501–1516 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Pike VW PET radiotracers: crossing the blood-brain barrier and surviving metabolism. Trends Pharmacol. Sci 30, 431–440 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deng XY et al. Chemistry for Positron Emission Tomography: Recent Advances in 11C-, 18F-, 13N-, and 15O-Labeling Reactions. Angew. Chem. Int. Ed 58, 2580–2605 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aldeghi M, Malhotra S, Selwood DL & Chan AW Two- and three-dimensional rings in drugs. Chem. Biol. Drug Des 83, 450–461 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taylor RD, MacCoss M & Lawson AD Rings in drugs. J. Med. Chem 57, 5845–5859 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Meanwell NA Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. Journal of Medicinal Chemistry 61, 5822–5880 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Zhou Y et al. Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II–III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev 116, 422–518 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Jacobson O, Kiesewetter DO & Chen XY Fluorine-18 Radiochemistry, Labeling Strategies and Synthetic Routes. Bioconjugate Chem. 26, 1–18 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Preshlock S, Tredwell M & Gouverneur V 18F-Labeling of Arenes and Heteroarenes for Applications in Positron Emission Tomography. Chem. Rev 116, 719–766 (2016). [DOI] [PubMed] [Google Scholar]
- 10.van der Born D et al. Fluorine-18 labelled building blocks for PET tracer synthesis. Chem. Soc. Rev 46, 4709–4773 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Krishnan HS, Ma LL, Vasdev N & Liang SH 18F-Labeling of Sensitive Biomolecules for Positron Emission Tomography. Chem.-Eur. J 23, 15553–15577 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ding YS et al. Synthesis of High Specific Activity 6-[18F]Fluorodopamine for Positron Emission Tomography Studies of Sympathetic Nervous-Tissue. J. Med. Chem 34, 861–863 (1991). [DOI] [PubMed] [Google Scholar]
- 13.Cai LS, Lu SY & Pike VW Chemistry with [18F]fluoride ion. Eur. J. Org. Chem 2008, 2853–2873 (2008). [Google Scholar]
- 14.Quednow BB et al. Assessment of serotonin release capacity in the human brain using dexfenfluramine challenge and [18F]altanserin positron emission tomography. Neuroimage 59, 3922–3932 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Cole EL, Stewart MN, Littich R, Hoareau R & Scott PJH Radiosyntheses using Fluorine-18: The Art and Science of Late Stage Fluorination. Curr. Top. Med. Chem 14, 875–900 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adams DJ & Clark JH Nucleophilic routes to selectively fluorinated aromatics. Chem. Soc. Rev 28, 225–231 (1999). [Google Scholar]
- 17.Brooks AF, Topczewski JJ, Ichiishi N, Sanford MS & Scott PJ Late-stage [18F]Fluorination: New Solutions to Old Problems. Chem. Sci 5, 4545–4553 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee E et al. A Fluoride-Derived Electrophilic Late-Stage Fluorination Reagent for PET Imaging. Science 334, 639–642 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee E, Hooker JM & Ritter T Nickel-Mediated Oxidative Fluorination for PET with Aqueous [18F] Fluoride. J. Am. Chem. Soc 134, 17456–17458 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Tredwell M et al. A General Copper-Mediated Nucleophilic 18F Fluorination of Arenes. Angew. Chem. Int. Ed 53, 7751–7755 (2014). [DOI] [PubMed] [Google Scholar]; (b) Taylor NJ et al. Derisking the Cu-Mediated 18F-Fluorination of Heterocyclic Positron Emission Tomography Radioligands. J. Am. Chem. Soc 139, 8267–8276 (2017). [DOI] [PubMed] [Google Scholar]; (c) Guibbal F et al. Manual and automated Cu-mediated radiosynthesis of the PARP inhibitor [18F]olaparib. Nat. Protoc 15, 1525–1541 (2020). [DOI] [PubMed] [Google Scholar]
- 21.(a) Chun JH, Lu SY, Lee YS & Pike VW Fast and High-Yield Microreactor Syntheses of ortho-Substituted [F-18]Fluoroarenes from Reactions of [18F]Fluoride Ion with Diaryliodonium Salts. J. Org. Chem 75, 3332–3338 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ichiishi N et al. Copper-Catalyzed [18F]Fluorination of (Mesityl)(aryl)iodonium Salts. Org. Lett 16, 3224–3227 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mossine AV et al. Synthesis of [18F]Arenes via the Copper-Mediated [18F]Fluorination of Boronic Acids. Org. Lett 17, 5780–5783 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Makaravage KJ, Brooks AF, Mossine AV, Sanford MS & Scott PJH Copper-Mediated Radiofluorination of Arylstannanes with [18F]KF. Org. Lett 18, 5440–5443 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McCammant MS et al. Cu-Mediated C–H 18F-Fluorination of Electron-Rich (Hetero)arenes. Org. Lett 19, 3939–3942 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.(a) Rotstein BH, Stephenson NA, Vasdev N & Liang SH Spirocyclic hypervalent iodine(III)-mediated radiofluorination of non-activated and hindered aromatics. Nat. Commun 5, 4365 (2014). [DOI] [PubMed] [Google Scholar]; (b) Liang SH; Wang L; Stephenson NA; Rotstein BH; Vasdev N Facile 18F Labeling of Non-Activated Arenes via a Spirocyclic Iodonium(III) Ylide Method and Its Application in the Synthesis of the mGluR5 PET Radiopharmaceutical [18F] FPEB. Nat. Protoc 14, 1530–1545 (2019). [DOI] [PubMed] [Google Scholar]
- 26.(a) Sander K et al. Sulfonium Salts as Leaving Groups for Aromatic Labelling of Drug-like Small Molecules with Fluorine-18. Sci. Rep 5, 9941 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gendron T et al. Ring-Closing Synthesis of Dibenzothiophene Sulfonium Salts and Their Use as Leaving Groups for Aromatic 18F-Fluorination. J. Am. Chem. Soc 140, 11125–11132 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neumann CN, Hooker JM & Ritter T Concerted nucleophilic aromatic substitution with 19F− and 18F−. Nature 534, 369–373 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Xu P et al. Site-Selective Late-Stage Aromatic [18F]Fluorination via Aryl Sulfonium Salts. Angew. Chem. Int. Ed 59, 1956–1960 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tay NES et al. 19F- and 18F-arene deoxyfluorination via organic photoredox-catalysed polarity-reversed nucleophilic aromatic substitution. Nat. Catal 3, 734–742 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoover AJ et al. A Transmetalation Reaction Enables the Synthesis of [18F]5-Fluorouracil from [18F]Fluoride for Human PET Imaging. Organometallics 35, 1008–1014 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharninghausen LS et al. NHC-Copper Mediated Ligand-Directed Radiofluorination of Aryl Halides. J. Am. Chem. Soc 142, 7362–7367 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilcken R, Zimmermann MO, Lange A, Joerger AC & Boeckler FM Principles and Applications of Halogen Bonding in Medicinal Chemistry and Chemical Biology. J. Med. Chem 56, 1363–1388 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Fang WY et al. Synthetic approaches and pharmaceutical applications of chloro-containing molecules for drug discovery: A critical review. Eur. J. Med. Chem 173, 117–153 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.For silicon/boron-centered 19F/18F exchange, see:; (a) Schirrmacher R et al. 18F-Labeling of Peptides by means of an Organosilicon-Based Fluoride Acceptor. Angewandte Chemie International Edition 45, 6047–6050 (2006). [DOI] [PubMed] [Google Scholar]; (b) Liu Z et al. An Organotrifluoroborate for Broadly Applicable One-Step 18F-Labeling. Angew. Chem. Int. Ed 53, 11876–11880 (2014). [DOI] [PubMed] [Google Scholar]; (c) Liu S et al. Lewis Acid-Assisted Isotopic 18F-19F Exchange in BODIPY Dyes: Facile Generation of Positron Emission Tomography/Fluorescence Dual Modality Agents for Tumor Imaging. Theranostics 3, 181–189 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) An F et al. One-Step, Rapid, 18F–19F Isotopic Exchange Radiolabeling of Difluoro-dioxaborinins: Substituent Effect on Stability and In Vivo Applications. J. Med. Chem 63, 12693–12706 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.For SuFEx-based strategies see:; (a) Zheng Q et al. Sulfur [18F]Fluoride Exchange Click Chemistry Enabled Ultrafast Late-Stage Radiosynthesis. J. Am. Chem. Soc 143, 3753–3763 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jeon MH et al. Late-Stage 18F/19F Isotopic Exchange for the Synthesis of 18F-Labeled Sulfamoyl Fluorides. Org. Lett. ASAP (2021). [DOI] [PubMed] [Google Scholar]
- 36.For selected examples of 19F/18F exchange on arenes via SNAr for the synthesis of radiopharmaceuticals, see:; (a) Langer O et al. Synthesis of fluorine-18-labeled Ciprofloxacin for PET studies in humans. Nucl. Med. Biol 30, 285–291 (2003). [DOI] [PubMed] [Google Scholar]; (b) Rokka J et al. 19F/18F exchange synthesis for a novel [18F]S1P3-radiopharmaceutical. J. Label. Compd. Radiopharm 56, 385–391 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Chen W et al. Direct arene C–H fluorination with 18F− via organic photoredox catalysis. Science 364, 1170–1174 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zweig Arnold., Hodgson, W. G. & Jura, W. H. The Oxidation of Methoxybenzenes. J. Am. Chem. Soc 86, 4124–4129 (1964). [Google Scholar]
- 39.(a) Blom E, Karimi F & Langstrom B [18F]/19F exchange in fluorine containing compounds for potential use in 18F -labelling strategies. J. Label. Compd. Radiopharm 52, 504–511 (2009). [Google Scholar]; (b) Wagner FM, Ermert J & Coenen HH Three-Step, “One-Pot” Radiosynthesis of 6-Fluoro-3,4-Dihydroxy-L-Phenylalanine by Isotopic Exchange. J. Nucl. Med 50, 1724–1729 (2009). [DOI] [PubMed] [Google Scholar]; (c) Weiss PS, Ermert J, Melean JC, Schafer D & Coenen HH Radiosynthesis of 4-[18F]fluoro-L-tryptophan by isotopic exchange on carbonyl-activated precursors. Bioorg. Med. Chem 23, 5856–5869 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Liu Z et al. An organotrifluoroborate for broadly applicable one-step 18F-labeling. Angew. Chem. Int. Ed 53, 11876–11880 (2014). [DOI] [PubMed] [Google Scholar]
- 41.(a) Tay NES & Nicewicz DA Cation Radical Accelerated Nucleophilic Aromatic Substitution via Organic Photoredox Catalysis. J. Am. Chem. Soc 139, 16100–16104 (2017). [DOI] [PubMed] [Google Scholar]; (b) Holmberg-Douglas N & Nicewicz DA Arene Cyanation via Cation-Radical Accelerated-Nucleophilic Aromatic Substitution. Org. Lett 21, 7114–7118, (2019). [DOI] [PubMed] [Google Scholar]; (c) Venditto NJ & Nicewicz DA Cation Radical-Accelerated Nucleophilic Aromatic Substitution for Amination of Alkoxyarenes. Org. Lett 22, 4817–4822, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shewchuk L et al. Binding mode of the 4-anilinoquinazoline class of protein kinase inhibitor: X-ray crystallographic studies of 4-anilinoquinazolines bound to cyclin-dependent kinase 2 and p38 kinase. J. Med. Chem 43, 133–138, (2000). [DOI] [PubMed] [Google Scholar]
- 43.Olberg DE et al. Synthesis and in vitro evaluation of small-molecule [18F] labeled gonadotropin-releasing hormone (GnRH) receptor antagonists as potential PET imaging agents for GnRH receptor expression. Bioorg. Med. Chem. Lett 24, 1846–1850 (2014). [DOI] [PubMed] [Google Scholar]
- 44.(a) Vivash L & O’Brien TJ Imaging Microglial Activation with TSPO PET: Lighting Up Neurologic Diseases? J. Nucl. Med 57, 165–168 (2016). [DOI] [PubMed] [Google Scholar]; (b) Alam MM, Lee J & Lee SY Recent Progress in the Development of TSPO PET Ligands for Neuroinflammation Imaging in Neurological Diseases. Nucl. Med Mol. Imaging 51, 283–296 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Werry EL et al. Recent Developments in TSPO PET Imaging as A Biomarker of Neuroinflammation in Neurodegenerative Disorders. Int. J. Mol. Sci 20, 3161 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Historical syntheses of [18F]fluorouracil involve direct fluorination of uracil with [18F]F2; see; Oberdorfer F, Hofmann E & Maierborst W Preparation of F-18-Labeled 5-Fluorouracil of Very High-Purity. J. Label. Compd. Radiopharm 27, 137–145 (1989). [Google Scholar]
- 46.(a) Fuchs BC & Bode BP Amino acid transporters ASCT2 and LAT1 in cancer: Partners in crime? Seminars in Cancer Biology 15, 254–266 (2005). [DOI] [PubMed] [Google Scholar]; (b) Wang Q & Holst J L-type amino acid transport and cancer: targeting the mTORC1 pathway to inhibit neoplasia. Am. J. Cancer Res 5, 1281–1294 (2015). [PMC free article] [PubMed] [Google Scholar]; (c) Qi YQ, Liu XH, Li J, Yao HQ & Yuan SH Fluorine-18 labeled amino acids for tumor PET/CT imaging. Oncotarget 8, 60581–60588 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Sun AX, Liu X & Tang GH Carbon-11 and Fluorine-18 Labeled Amino Acid Tracers for Positron Emission Tomography Imaging of Tumors. Front. Chem 5, 124 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.(a) Langen KJ et al. O-(2-[18F]fluoroethyl)-L-tyrosine: uptake mechanisms and clinical applications. Nucl. Med. Biol 33, 287–294 (2006). [DOI] [PubMed] [Google Scholar]; (b) Zoghbi SS et al. PET imaging of the dopamine transporter with 18F-FECNT: A polar radiometabolite confounds brain radioligand measurements. J. Nucl. Med 47, 520–527 (2006). [PubMed] [Google Scholar]; (c) Franck D et al. Investigations into the synthesis, radiofluorination and conjugation of a new [18F]fluorocyclobutyl prosthetic group and its in vitro stability using a tyrosine model system. Bioorg. Med. Chem 21, 643–652 (2013). [DOI] [PubMed] [Google Scholar]; (d) Kuchar M & Mamat C Methods to Increase the Metabolic Stability of 18F-Radiotracers. Molecules 20, 16186–16220 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Stegmayr C, Willuweit A, Lohmann P & Langen KJ O-(2-[18F]-Fluoroethyl)-L-Tyrosine (FET) in Neurooncology: A Review of Experimental Results. Curr. Radiopharm 12, 201–210 (2019). [DOI] [PubMed] [Google Scholar]
- 48.Lee SL Radioactive iodine therapy. Curr. Opin. Endocrinol. Diabetes Obes 19, 420–428 (2012). [DOI] [PubMed] [Google Scholar]
- 49.Barth RF, Mi P & Yang W Boron delivery agents for neutron capture therapy of cancer. Cancer Comm. 38, 35 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garnett ES, Firnau G & Nahmias C Dopamine Visualized in the Basal Ganglia of Living Man. Nature 305, 137–138 (1983). [DOI] [PubMed] [Google Scholar]
- 51.For a minireview, see:; (a) Pretze M, Wängler C & Wängler B 6-[18F]Fluoro-L-DOPA: A Well-Established Neurotracer with Expanding Application Spectrum and Strongly Improved Radiosyntheses. BioMed Research International 2014, e674063 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; For specific methods, see:; (b) Libert LC et al. Production at the Curie Level of No-Carrier-Added 6-18F-Fluoro-L-Dopa. J. Nucl. Med 54, 1154–1161 (2013). [DOI] [PubMed] [Google Scholar]; (c) Lemaire C et al. Automated production at the curie level of no-carrier-added 6-[18F]fluoro-L-dopa and 2-[18F]fluoro-L-tyrosine on a FASTlab synthesizer. J. Label. Compd. Radiopharm 58, 281–290 (2015). [DOI] [PubMed] [Google Scholar]; (d) Preshlock S et al. Enhanced copper-mediated 18F-fluorination of aryl boronic esters provides eight radiotracers for PET applications. Chem. Commun 52, 8361–8364 (2016). [DOI] [PubMed] [Google Scholar]; (e) Luurtsema G et al. Improved GMP-compliant multi-dose production and quality control of 6-[18F]fluoro-L-DOPA. EJNMMI Radiopharm. Chem 1, 7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Zarrad F, Zlatopolskiy BD, Krapf P, Zischler J & Neumaier B A Practical Method for the Preparation of 18F-Labeled Aromatic Amino Acids from Nucleophilic [18F]Fluoride and Stannyl Precursors for Electrophilic Radiohalogenation. Molecules 22, 2231 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Mossine AV et al. One-pot synthesis of high molar activity 6-[18F]fluoro-L-DOPA by Cu-mediated fluorination of a BPin precursor. Org. Biomol. Chem 17, 8701–8705, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Mossine AV et al. Synthesis of high-molar-activity [18F]6-fluoro-L-DOPA suitable for human use via Cu-mediated fluorination of a BPin precursor. Nature Protocols 15, 1742–1759 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Orlovskaya V, Fedorova O, Kuznetsova O & Krasikova R Cu-Mediated Radiofluorination of Aryl Pinacolboronate Esters: Alcohols as Solvents with Application to 6-L-[18F]FDOPA Synthesis. Eur. J. Org. Chem 2020, 7079–7086 (2020). [Google Scholar]; For a review on Cu-mediated radiofluorination of arylboronates and arylstannanes for the synthesis of [18F]FDOPA, see; (j) Krasikova RN Nucleophilic Synthesis of 6-L-[18F]FDOPA. Is Copper-Mediated Radiofluorination the Answer? Molecules 25, 4365 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Luxen A et al. Production of 6-[18F]Fluoro-L-Dopa and its metabolism in vivo - a critical-review. Int. J. Rad. Appl. Instrum. B 19, 149–158 (1992). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data generated or analysed during this study are included in this article (and its Supplementary Information files).