

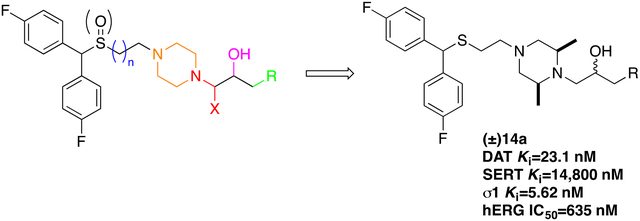
Abstract
Atypical dopamine transporter (DAT) inhibitors have shown therapeutic potential in preclinical models of psychostimulant abuse. In rats, 1-(4-(2-((bis(4-fluorophenyl)methyl)sulfinyl)ethyl)-piperazin-1-yl)-propan-2-ol (3b) was effective in reducing the reinforcing effects of both cocaine and methamphetamine, but did not exhibit psychostimulant behaviors itself. While further development of 3b is ongoing, diastereomeric separation, as well as improvements in potency and pharmacokinetics were desirable for discovering pipeline drug candidates. Thus, a series of bis(4-fluorophenyl)methyl)sulfinyl)alkyl alicyclic amines, where the piperazine-2-propanol scaffold was modified, were designed, synthesized and evaluated for binding affinities at DAT, as well as the serotonin transporter and sigma1 receptors. Within the series, 14a showed improved DAT affinity (Ki=23 nM) over 3b (Ki=230 nM), moderate metabolic stability in human liver microsomes, and a hERG/DAT affinity ratio = 28. While 14a increased locomotor activity relative to vehicle, it was significantly lower than activity produced by cocaine. These results support further investigation of 14a as a potential treatment for psychostimulant use disorders.
Keywords: Modafinil, atypical dopamine uptake inhibitors, dopamine transporter, serotonin transporter, σ1 receptors, hERG channel, psychostimulants, cocaine
Grapical Abstract
INTRODUCTION
Deaths related to drug overdose within the United States are rising rapidly, with less than 17,000 deaths in 1999, and reaching 70,237 deaths in 2017.1 Despite warnings by the Drug Enforcement Agency (DEA) of a national rebound in cocaine initiates and cocaine poisoning deaths, it has now become the third highest-killing illicit substance, behind synthetic opioids, such as fentanyl, and heroin.1, 2 Regrettably, pharmacotherapeutic treatments for those addicted to psychostimulants, like cocaine and methamphetamine, have yet to be approved by the FDA, and thus, psychostimulant use disorders are currently treated exclusively with behavioral therapies.3 An overwhelming need for the development of a medication-assisted treatment (MAT) for psychostimulant use disorders has been exemplified by the superior outcomes in MAT programs for opioid users versus solely behaviorally-based treatments.4–7
Cocaine binds to the dopamine transporter (DAT), blocking the reuptake of dopamine (DA), enhancing dopaminergic neurotransmission, and resulting in feelings of euphoria and reward that can lead to abuse.8–13 Drug development efforts to design a small molecule that competitively blocks cocaine at DAT, while possessing low abuse potential, have led to once promising compounds like JHW00714–17 and GBR12909 (vanoxerine).18–20 Owing to its favorable results in animal models of cocaine use disorders, GBR12909 was advanced into Phase I clinical trials before failing, due to rate-dependent QTc elongation in healthy subjects.18, 21–23
Modafinil (1, Figure 1) and its (R)-enantiomer are FDA approved as prescription medications to promote wakefulness in those suffering from narcolepsy, shift work sleep disorder, and obstructive sleep apnea.24–26 Modafinil is a DA uptake blocker, but does not exhibit cocaine-like abuse potential in animal models.25, 27 Similar to treatment of attention deficit hyperactivity disorder (ADHD) using DAT ligands (amphetamine and methylphenidate), 1 is used off-label as a cognitive enhancer but exhibits limited addiction liability in humans.25 Results of studies investigating 1 as a treatment for psychostimulant use disorders in humans have shown mixed results. When cocaine-dependent subjects were treated with 1 coupled to cognitive behavioral therapy vs. placebo, a reduction in cocaine use was observed, as measured by cocaine-negative urine samples; however, in a larger study, there were no significant differences between 1 and placebo.28 More recently, a large clinical trial in cocaine-dependent subjects showed no significant difference between 1 and placebo; however, a subpopulation of non-alcohol abusing cocaine-dependent subjects exhibited an increase in cocaine-free days and an overall reduction in cocaine cravings when treated with 1.29 For these reasons, our group has focused on chemically modifying modafinil in the search for a safe and more effective pharmacotherapy for the treatment of psychostimulant use disorders.30–32 Of note, others have also expanded on DAT SAR with thiazole analogues of modafinil and are investigating them in rodent models of memory enhancement.33, 34
Figure 1.
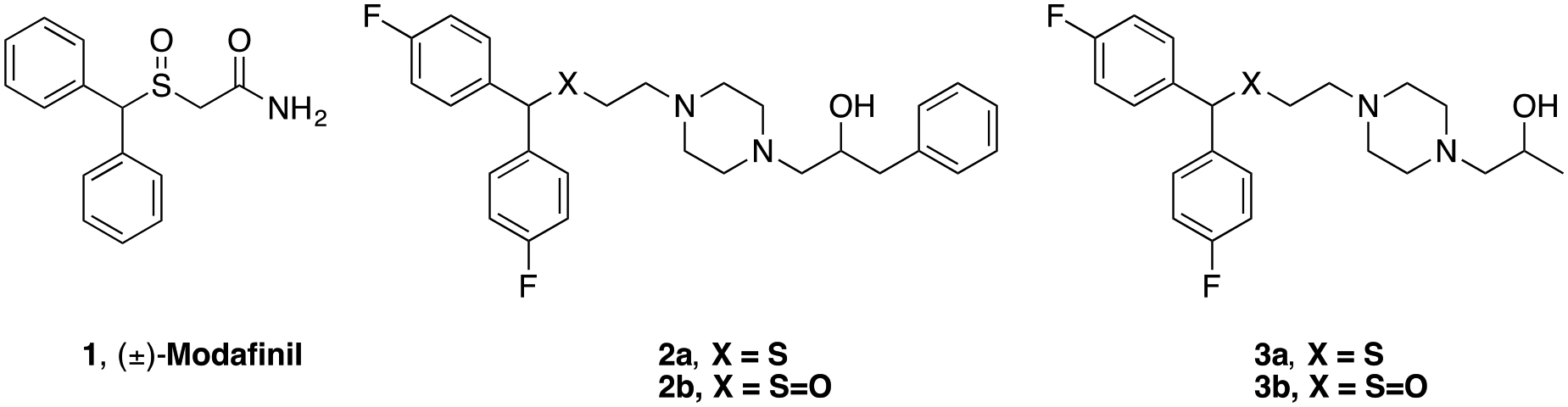
Chemical structures of DAT inhibitors (±)-modafinil and recently discovered analogues
Two compounds resulting from our efforts, 2b and 3b (Figure 1), were found to bind with higher affinity both at rat and human DAT (rDAT and hDAT, respectively) compared to 1.32 Binding assays with a Y156F mutant of hDAT confirmed that both compounds bind DAT differently than cocaine, showing a binding profile more similar to JHW007.27, 35–37 Curiously, in rodent models, 2b displayed a cocaine-like behavioral profile, whereas 3b did not.38 In contrast, pretreating rodents with 3b decreased self-administration of cocaine and reduced cocaine-induced reinstatement to drug seeking behaviors.37, 38 Based on these findings,27, 31, 39, 40 2b and 3b were characterized for their binding modes in the central ligand binding site of hDAT using molecular dynamics (MD) simulations38 Interestingly, DAT preferred an outward open conformation in the presence of 2b, similar to cocaine, whereas DAT preferred a more occluded conformation when 3b was bound, providing an explanation, at the molecular level, for the observed behavioral profiles.32, 41–43
While inhibition of DAT has hitherto been our strategy in developing potential psychostimulant use disorders, dual inhibition of σ1 receptors and DAT has been shown to significantly reduce cocaine’s reinforcing effects in animal models of cocaine abuse.44, 45 One hypothesis posits that this is the result of sigma1 receptor (σ1R) modulation of DAT conformation.46 DAT inhibitors with high affinity for σ1R have also been reported to attenuate the reinforcing actions of methamphetamine measured in self-administration tests.37, 47 Moreover, a recent study reported that methamphetamine might activate σ1R, leading to oxidation of a cysteine residue on VMAT2, which would be responsible for a reduction in the vesicular transport of DA, thus potentiating the dopaminergic actions of methamphetamine.48 However, recent studies also found that activation (not blockade) of σ1R would attenuate methamphetamine induced ambulatory activity, motivated behaviors, and enhancement of brain reward stimulation.49 As there are evident relationships between DAT and σ1R, understanding how our compounds bind σ1R will prove beneficial.
Generally, within the bis(4-fluorophenyl)methyl)sulfinyl)ethylpiperazines series,32 the sulfoxide analogues have lower affinity for DAT than their sulfide counterparts. For example, the sulfide analogue of 3b, 3a, has higher DAT affinity than 3b (DAT Ki = 37.8 ± 8.72 nM vs. 230 ± 40.5 nM), however, 3a was determined to be metabolically unstable, with a half-life of 14 min in rat liver microsomes, whereas 3b had a half-life of 126 min.37 Hence, our focus has primarily been on the development of sulfoxides. Furthermore, the sulfoxide analogues tend to possess lower potency at hERG,37 a highly beneficial characteristic for drugs vying for clinical trials.50–53
hERG, the human ether-à-go-go-related potassium channel, is involved in the repolarization of the cardiac action potential.21, 50–54 Mutation or drug inhibition of this channel leads to delays in repolarization, which then leads to prolonged QT syndrome and the lethal cardiac arrhythmia torsade de pointes.51–53 The hERG channel is highly promiscuous and binds a wide range of molecules, including those that possess aromatic groups, primarily because of its tyrosine 652 and phenylalanine 656 residues, and those with positively charged groups (e.g., protonatable amines), due to the strong electrostatic environment within the pore.51–53 Numerous drugs have been withdrawn from the market due to cardiotoxicity related to hERG inhibition, and although not all hERG blockers lead to lethal cardiotoxicity, there are strong correlations.55 Thus it is a metric that the FDA mandates reporting prior to progressing to clinical trials.55
While 3b is currently being developed as a medication candidate for psychostimulant use disorder, we sought to improve its DAT affinity, metabolic stability and hERG profiles through chemical modifications of the N-terminus. The objective of this series was to find analogues of 2a, 2b, 3a and 3b that displayed higher affinity for DAT and potentially σ1R, were metabolically stable, and showed an acceptable hERG/DAT affinity ratio.
While the piperazine scaffold is a widely used and effective building block in drug design, it is also highly vulnerable to various drug metabolizing enzymes and often binds to hERG, due to its electron rich nature.56 To potentially bypass these problems, while attempting to preserve high DAT affinity, we set out to explore the effects imparted by (1) the amidation of the terminal piperazine nitrogen (Figure 2, red), (2) the oxidation and reduction of the terminal alcohol moiety (Figure 2, pink), (3) varying the substitution on the terminal phenyl group of 2 with electron donating and withdrawing groups (Figure 2, green), (4) varying the alicyclic amine linker (Figure 2, orange), and (5) resolving the secondary alcohol chiral center (Figure 2, pink). The cis-2,6-dimethylpiperazine and 2,8-diazaspiro[4.5]decane scaffolds inspired by compounds listed in Figure 3 were introduced as alternative alicyclic amine moieties. The cis-2,6-dimethylpiperazine group of rimcazole (5), was chosen to attenuate the pKa of the terminal tertiary amine on the piperazine, which should decrease hERG affinity. As rimcazole is a good DAT inhibitor (Ki = 97.7 ± 12 nM), the additional methyl groups were not anticipated to decrease binding of the new compounds at DAT. The 2,8-diazaspiro[4.5]decane group, shown recently in a series of highly selective D3 receptor antagonists (6),57, 58 not only decreases lipophilicity, thus potentially decreasing hERG affinity,50 but also increases metabolic stability as the piperazine scaffold is replaced.56
Figure 2.
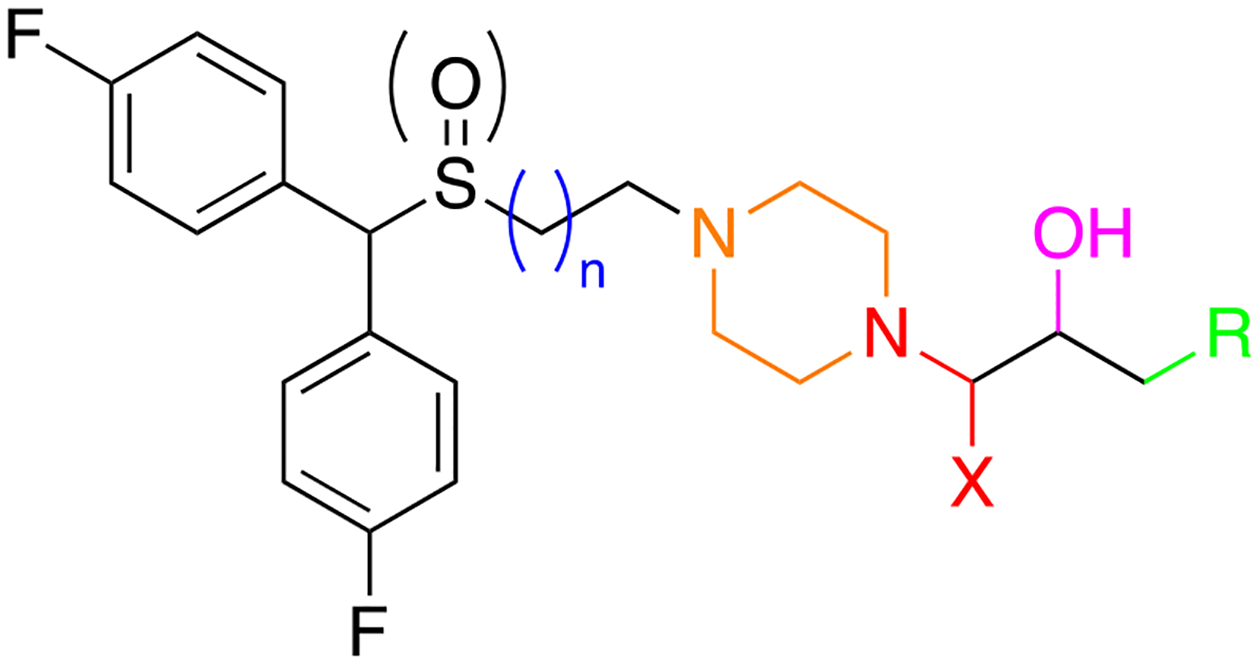
Sites of modifications made to 2 and 3.
Figure 3.
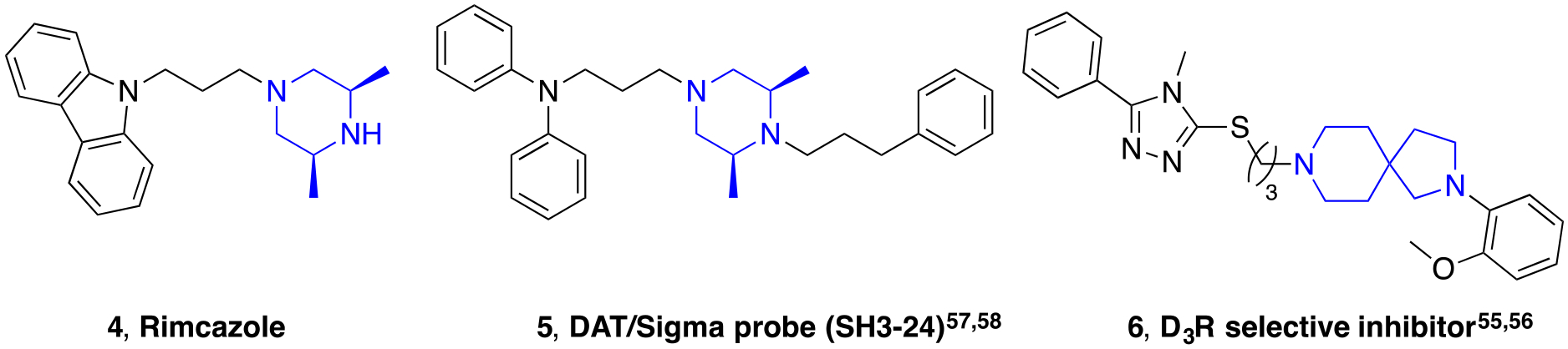
Chemical structures of compounds possessing other alicyclic amine linkers.57–60
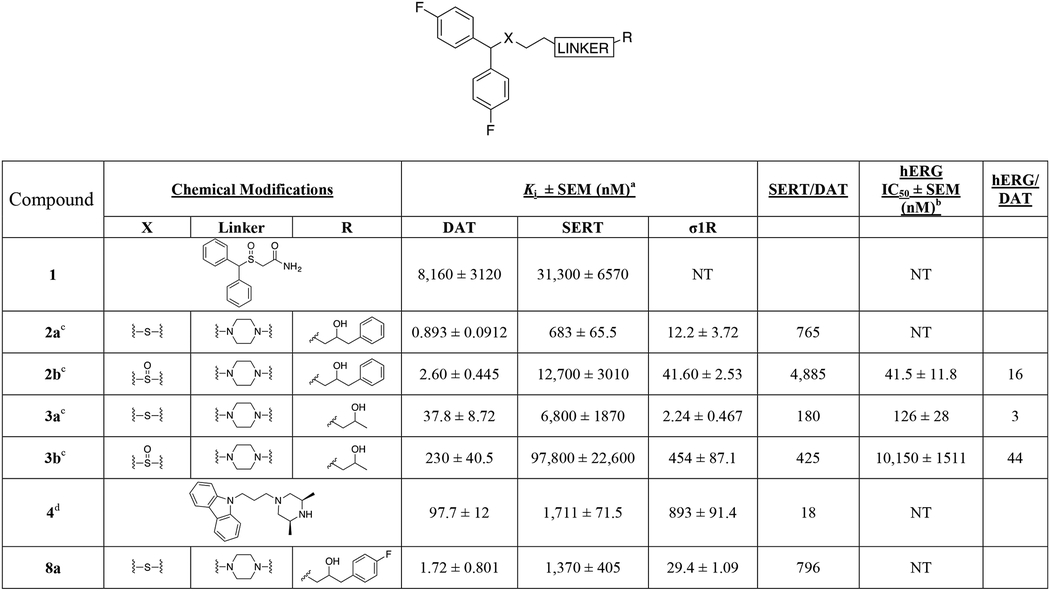
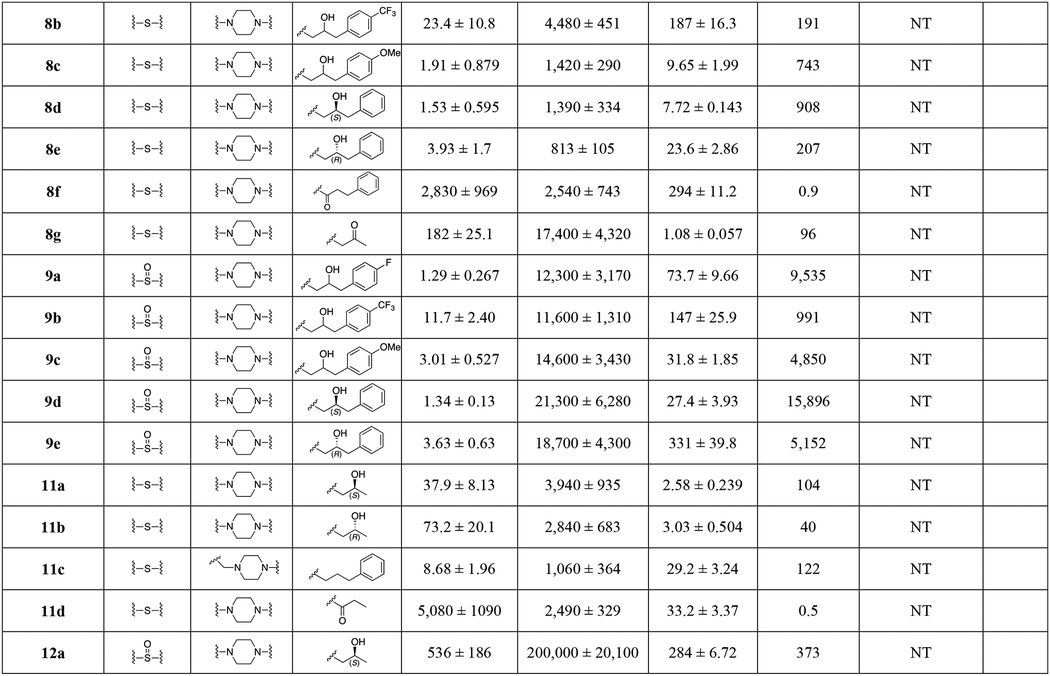
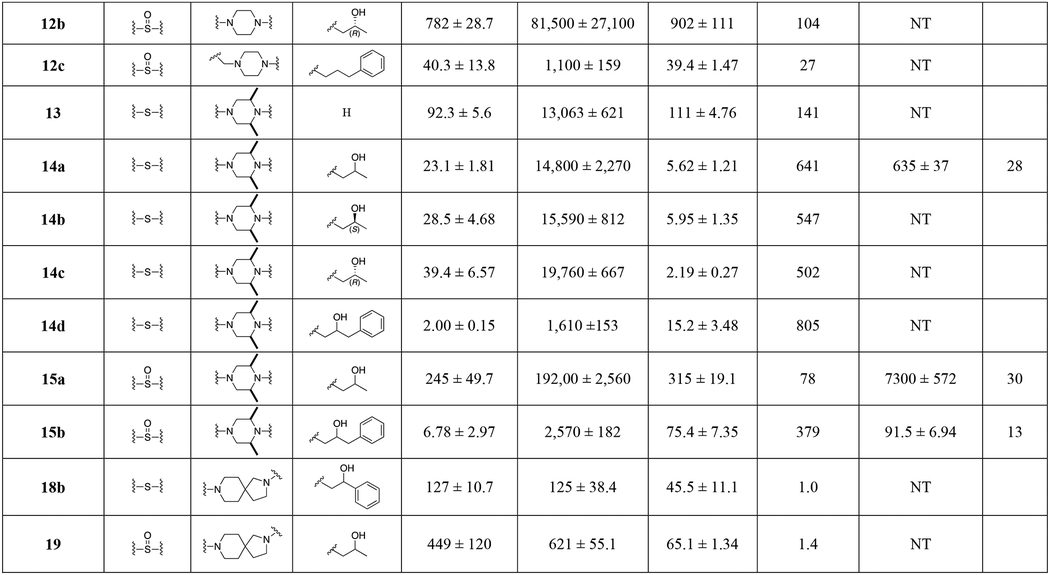
All final compounds were evaluated for DAT and SERT binding in rat brain (Table 1; NET binding for select compounds is reported in Table S3). σ1R binding in guinea pig brain was also evaluated for continued investigation of a previous hypothesis, that compounds with dual DAT/σ1R inhibitor profiles may be more therapeutically beneficial over DAT selective inhibitors.44, 59–63 In addition, a subset of compounds were tested for their hERG binding profile. Furthermore, a subset of analogues was also tested on cell lines overexpressing DAT and the Y156F mutant. The affinity ratio between the binding affinity to WT DAT and its Y156F mutant has previously been used to predict if a compound demonstrates a classical or more atypical DAT binding profile, as reported for (R)-modafinil, 2b and 3b.27 Based on these findings, the stereoisomers of 2b, 3b, and several of the analogues described herein were evaluated for metabolic stability in mouse liver microsomes, as well as hERG affinity. Finally, compound 14a and its diastereomers (14b, c), were chosen as new lead compounds and compared to cocaine for their effects on locomotor activity in mice.
Table 1.
Binding Data for Sulfenyl-, and Sulfinylalkylamine Analogues.a
|
Each Ki value represents data from at least three independent experiments, each performed in triplicate. Ki values were analyzed by PRISM. Binding assay procedures are described in detail in Experimental Methods.
Concentration (log) response curves were fitted to a logistic equation (three parameters assuming complete block of the current at very high test compound concentrations) to generate estimates of the 50% inhibitory concentration (IC50). The concentration-response relationship of each compound was constructed from the percentage reductions of current amplitude by sequential concentrations. NT; not tested.
data from ref. 32.
data from ref. 62.
CHEMISTRY
Syntheses of novel sulfenylalkylamine (8a-8g, 11a-11d) and sulfinylalkylamine analogues (9a-9e, 12a-12c) were achieved as depicted in Scheme 1 and Scheme 2. Epoxide ring opening using the appropriate oxiranes with 1-(2-((bis(4-fluorophenyl)methyl)thio)ethyl)piperazine 7 gave alcohols 8a-8c in quantitative yield.32 Alkylation of 7 with the appropriate halides provided compounds 8d, 8e and 8g in 66%−84% yield. Amidation of the appropriate carboxylic acids using 1,1’-carbonyldiimidazole (CDI) gave amide 8f in 86% yield. To understand the effect of stereochemistry on DAT, the stereoisomers of 2b were resolved using commercially available (2S)- or (2R)-1-chloro-3-phenylpropan-2-ol to give 9d and 9e.
Scheme 1.
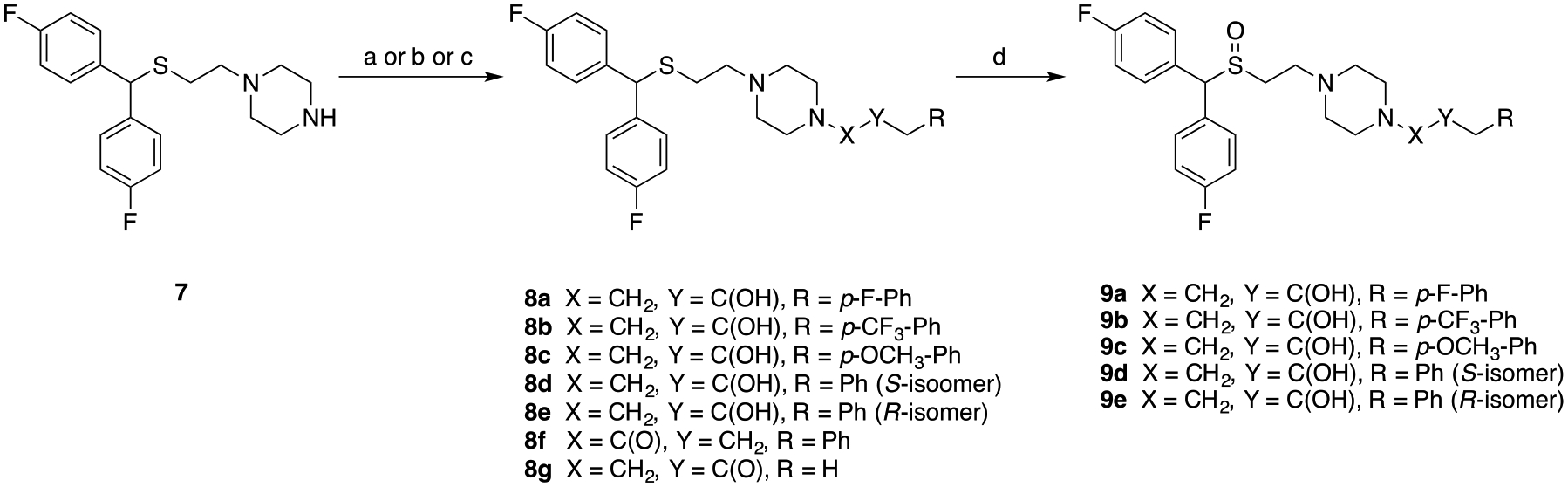
Synthesis of compounds 8a-g and 9a-e.a
aReagents and conditions: (a) appropriate oxirane, isopropanol, reflux, overnight; (b) appropriate halide, K2CO3, acetonitrile, reflux; (c) CDI, 3-phenylpropanoic acid, THF, RT, overnight; (d) H2O2, CH3COOH/CH3OH, 40 °C, overnight.
Scheme 2.
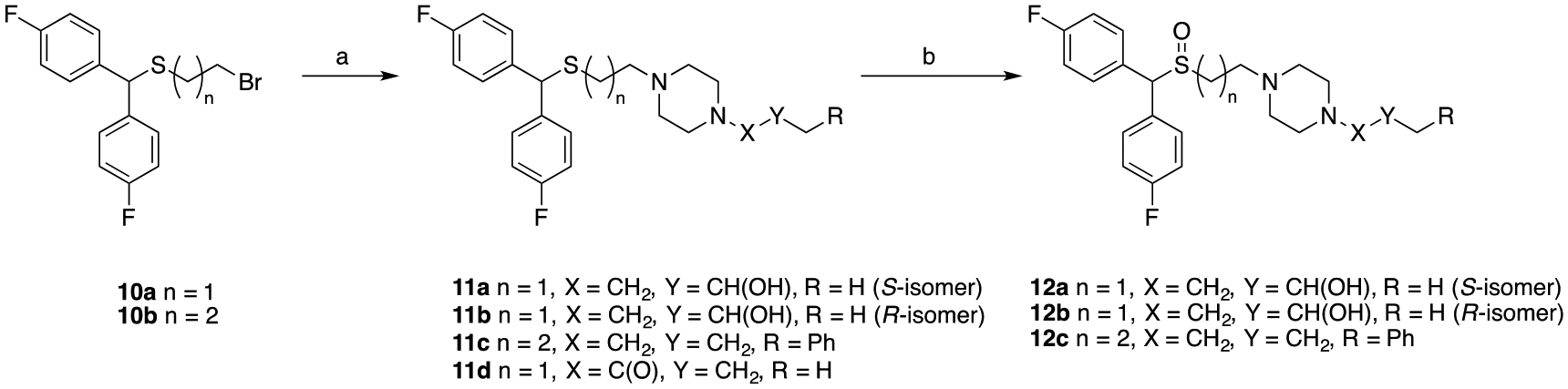
Synthesis of compounds 11a-d and 12a-c.a
aReagents and conditions: (a) appropriate piperazine, K2CO3, acetonitrile, reflux, overnight; (b) H2O2, CH3COOH/CH3OH, 40 °C, overnight.
Synthesis of compound 10b was achieved using methods similar to 10a.32 Dehydration of bis(4-fluorophenyl)methanol with 3-mercaptopropan-1-ol in the presence of trifluoroacetic acid, followed by treatment with K2CO3 in H2O/acetone provided the sulfide alcohol in quantitative yield. Bromination via Appel conditions gave 10b in 98% yield. Alkylation of bromides 10a32 and 10b, with the appropriate piperazines provided compounds 11a-11d. Lastly, oxidation of all sulfenylamine compounds (8a-8e, 11a-11c) using hydrogen peroxide in an acetic acid/methanol solution gave the sulfinylamine compounds (9a-9e, 12a-12c) in 35–60% yield. Similarly, the stereoisomers of 3b (12a and 12b) were synthesized using either commercially available (S)- or (R)-propylene oxide.
The cis-dimethylpiperazine series was achieved through similar methods as the piperazine analogues (Scheme 3). Alkylation of cis-2,6-dimethylpiperazine with 10a afforded intermediate 13, and the sulfenyl products 14a-14d were obtained through ring epoxide opening with the appropriate oxiranes. Subsequent oxidation of the sulfenyl compounds into the sulfinyl products gave 15a and 15b. As 14a showed promising pharmacological properties for our purposes, its stereoisomers 14b and 14c were also synthesized using commercially available (S)- or (R)-propylene oxide for comparison.
Scheme 3.
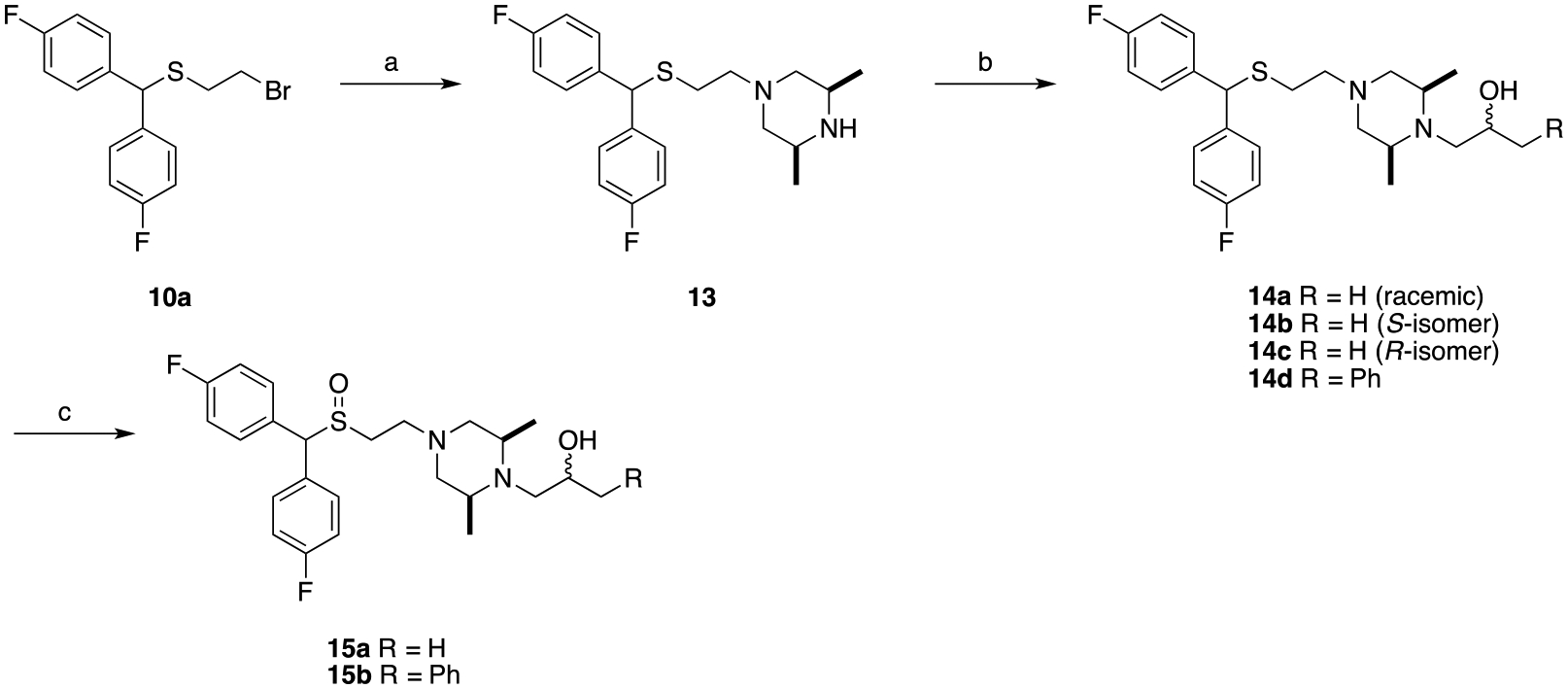
Synthesis of compounds 14a-d and 15a-b.a
aReagents and conditions: (a) cis-2,6-dimethylpiperazine, K2CO3, acetonitrile, reflux, overnight; (b) appropriate oxirane, isopropyl alcohol, reflux, overnight; (c) H2O2, CH3COOH/CH3OH, RT, 48 h.
As for the diazaspiro compounds (Scheme 4), the sulfenyl products 18a and 18b were again obtained through similar methods as previous products. Alkylation of 10a with tert-butyl 2,8-diazaspiro[4.5]decane-2-carboxylate, followed by Boc deprotection afforded 17. The terminal alcohol moieties were installed using the appropriate oxiranes to afford 18a and 18b, and final oxidation of 18a gave the sulfinyl product 19.
Scheme 4.
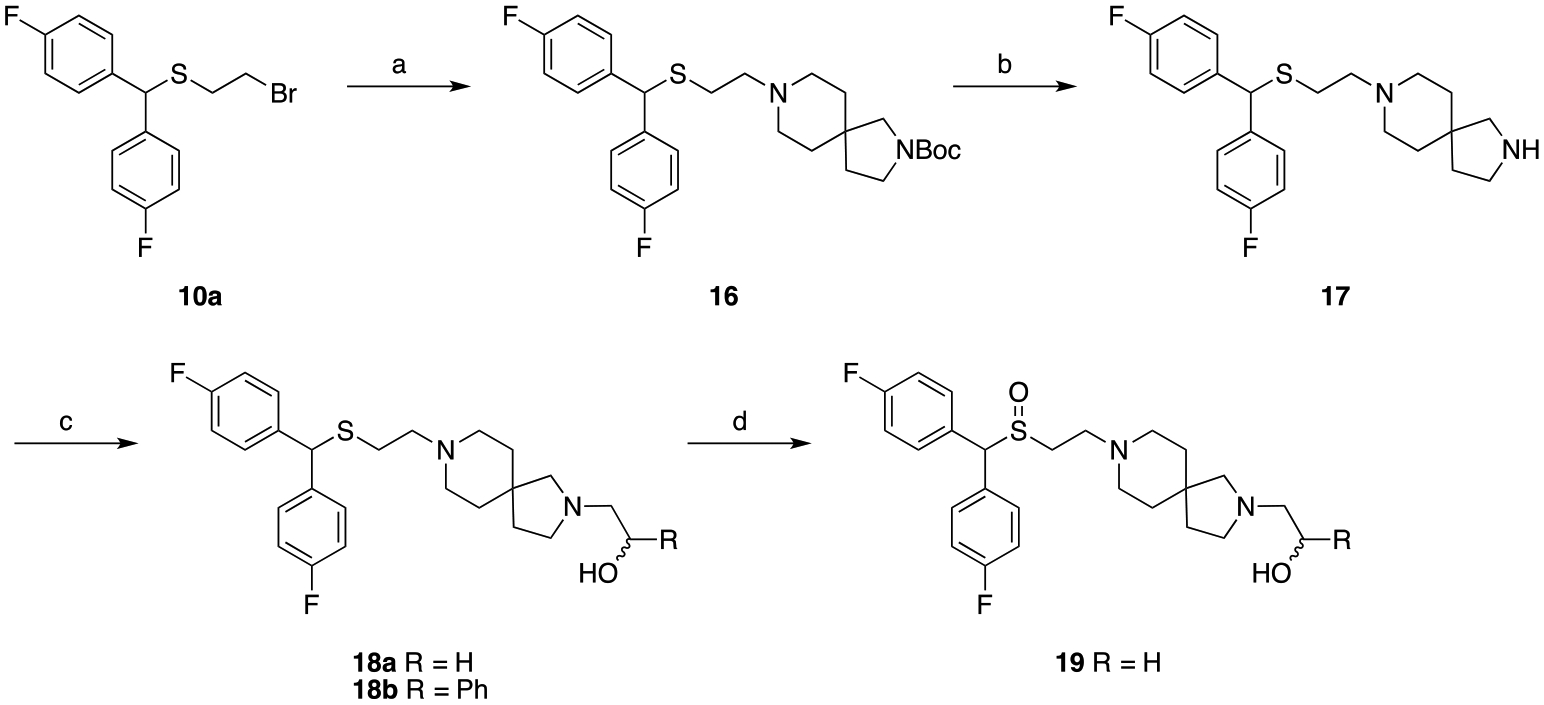
Synthesis of diazaspiro analoguesa
aReagents and conditions: (a) tert-butyl 2,8-diazaspiro[4.5]decane-2-carboxylate, K2CO3, acetonitrile, reflux, overnight; (b) TFA, DCM, RT, 5 h; (c) appropriate oxirane, isopropyl alcohol, reflux, 18–48 h; (d) H2O2, CH3COOH/CH3OH, RT, 48 h.
BIOLOGICAL RESULTS AND DISCUSSION
SAR at DAT, SERT and σ1R.
All final compounds (8, 9, 11, 12, 14, 15, 18 and 19) were evaluated for binding at DAT and SERT in rat brain membranes and σ1R in guinea pig brain membranes, and compared to known DAT inhibitors 1−4 (Figure 1, Figure 3). The binding affinities (Ki values) are presented in Table 1.
In general, the sulfinylamine compounds were less potent DAT inhibitors than their sulfenylamine precursors, with the exception of 8b and 9b, although the difference between the two was negligible. Unsurprisingly, the compounds with a phenyl-2-propanol terminus had the lowest DAT Ki values and higher affinity when compared to their non-aromatic counterparts, similar to 2 versus 3. 8b and 9b had the lowest DAT affinities within that group, likely because of the compounding bulk and electron withdrawing effects of the p-CF3 group, whereas 8a, 8c, 8d and 8e were comparable to 2a, and 9a, 9c, 9d and 9e were comparable to 2b. Modifying the terminal amine of the piperazine to an amide, with or without a phenyl terminus (8f and 11d respectively), resulted in DAT Ki values in the low micromolar range, confirming the MD simulation findings that the terminal tertiary amine is crucial for DAT binding.38 Oxidizing the 2-propanol of 3 to 2-propanone (8g) increased the DAT Ki, decreasing DAT affinity by ~5 fold compared to 3a, which also agreed with the MD simulation of 3b in DAT, where the hydroxyl group in the 2-propanol end participates in binding.38 Converting the piperazine linker to the cis-2,6-dimethylpiperazine scaffold did not alter the DAT Ki’s significantly, as anticipated.
As our goal in modifying the alicyclic amine linker was aimed at lowering the rate of metabolism and decreasing hERG affinity, the lack of change in DAT binding was ideal. The diazaspiro compounds (18b and 19), however, showed relatively lower DAT binding affinities compared to their parent analogues (3a and 3b respectively), and were not selective for DAT over SERT. Interestingly, both compounds showed a binding preference for σ1R over either DAT or SERT. The extension of the alkyl chain between the sulfur and piperazine and removal of the alcohol (11c and 12c) increased DAT Ki values when compared to 2a and 2b.
The resolved diastereomers of 3b (12a and 12b) showed no appreciable enantioselectivity at DAT or SERT, and the same was observed for the diastereomers of 2b and 14a, 9d and 9e, and 14b and 14c, respectively. Although the (S)-diastereomer in all cases (8d, 9d, 11a, 12a and 14b) had lower Ki’s than the (R)- diastereomers (8e, 9e, 11b, 12b and 14c), the differences were minimal.
In most cases, these novel analogues were highly DAT selective when compared to SERT, with the exception of (as mentioned) amides 8f and 11d, and diazaspiro compounds 18b and 19, where DAT and SERT Ki’s were equivalent.
With respect to σ1R binding, compounds without a phenyl terminus showed high affinity binding, which is the reverse trend when compared to DAT binding, however, in general the sulfides had higher affinities for σ1R than the sulfoxides, as seen at DAT. Compounds 8f, 11d, and 8g were σ1R selective compared to either monoamine transporter, with 8g being the best σ1R inhibitor amongst all the compounds shown (Ki = 1.08 nM). Interestingly, while stereochemistry had very little effect on DAT binding, a significant difference between the σ1R Ki of sulfoxides 9e and 9d, and to a lesser extent, 12b and 12a was observed. This difference was not observed between the stereoisomers of their sulfide precursors.
Compounds 14a, 15a and 15b were tested for their potencies in blocking hERG. As predicted, the cis-2,6-dimethylpiperazine analogue of 3b, 15a, had the lowest affinity for hERG amongst this series of compounds, while 15b had the highest, reflecting the literature and hERG results of their predecessors.21, 32, 50–55 Importantly, the hERG/DAT affinity ratio of the sulfenyl compound 14a was much higher than that of its parent compound 3a (28 and 3 respectively) and comparable to 15a (30). Fortuitously, compounds with an affinity ratio >30-fold are unlikely to produce a QT prolongation effect.64 With a significantly higher DAT affinity than 3b and an acceptable hERG/DAT affinity ratio, 14a has development potential as a MAT for psychostimulant use disorders.
Molecular pharmacology and mutagenesis studies
We next evaluated the binding ratios of compounds 14a, 15a, 15b and 18b in DAT wildtype (WT) and the Y156F mutant to preliminarily assess the nature of DAT binding in vitro, as has been done previously with earlier generation analogues of benztropine and modafinil.27, 32, 40 In DAT WT, the OH-group of Tyr156 is suggested to generate an H-bond to Asp79, which initiates occlusion of the binding site to the extracellular environment after substrate binding. Molecular docking simulations65 and X-ray crystallography66 have suggested that cocaine prefers to bind DAT in an outward facing conformation that does not interfere with the H-bond formation. Hence there is little binding affinity difference between the WT and Y156F mutant DAT for cocaine and cocaine-like compounds.32 In contrast, binding of the atypical DAT inhibitors induce a more occluded conformation, which depends on the Tyr156-Asp79 H-bond formation.27, 38, 39, 65, 66 Hence, mutation of Y156 typically decreases the binding affinity of atypical DAT inhibitors resulting in a >2-fold decrease in Ki between DAT WT and the DAT Y156F mutation (see Table 2, affinity ratio).27, 36
Table 2.
Ki values for inhibition of [3H]WIN35,428 binding by indicated compounds to DAT WT and the Y156F mutant.a
| Compound | WT Ki (nM) | n | Y156F Ki (nM) | n | Affinity Ratio |
|---|---|---|---|---|---|
| Cocaine b | 450 [340;590] | 3 | 350 [280; 450] | 5 | 0.8 |
| WIN35,428 b | 13 [12;14] | 10 | 13 [9.6–17] | 6 | 1 |
| 14a | 58.3 [48.6; 69.9] | 4 | 269 [251; 289] | 4 | 4.6 |
| 15a | 406 [398; 414] | 3 | 3030 [2390; 3830] | 3 | 7.5 |
| 15b | 3.83 [3.02; 4.87] | 4 | 19.1 [12.2; 29.9] | 3 | 5.8 |
| 18b | 104 [88; 123] | 3 | 239 [199; 288] | 3 | 2.3 |
[3H]WIN35,428 binding is performed on intact COS7 cells transiently expressing DAT WT or the Y156F mutant. Data are shown as mean [SE interval] and are calculated from pIC50 and the SE interval from pIC50 ± SE.
data from ref. 27.
Inhibition of [3H]WIN35,428 binding on COS7 cells transiently expressing WT DAT or Y156F was determined. All tested analogues showed a significant decrease in binding affinity for Y156F relative to WT DAT in contrast to cocaine or its higher affinity analogue, WIN35,428 (Table 2). We have previously reported that 2b, based on the Y156F binding assay assessment, may be an atypical DAT inhibitor, however, pre-clinical behavioral evaluation demonstrated it had a more cocaine-like behavioral profile.38 Thus, although the Y156F/WT DAT ratio suggests that these compounds bind differently than cocaine, it does not always predict their behavioral profiles. Of note, Ki values (nM) for inhibition of [3H]DA uptake were obtained in this same cell line for 14a = 100 [79.9; 125]; 15a = 552 [490; 621]; 15b = 11.1 [5.70; 21.8] and 18b = 268 [228;315] using methods previously described.32 These data closely correspond to the Ki values found in the WT DAT binding assay (Table 2.)
Metabolic Stability in Mouse Microsomes.
A subset of analogues (8a, 8d, 8e, 9a-e, 12c, 14a, 15a, 15b, 18b) were tested for phase I metabolism following procedures previously described67 to predict the susceptibility to Phase I metabolism following in vivo administration. As seen in Table S2, similar to 2b, compounds with a phenyl-propanol terminus (8a, 8d, 8e, 9a-e, 12c and 15b) were highly susceptible to phase I metabolism, with almost no compound present after 60 min. In contrast, the stereoisomers of 3b, 12a and 12b, both exhibit exceptional and similar metabolic stability, more so than 3b (t1/2 = 126 min).
Within the modified alicyclic amine compound series (Table S2 and Figure S1), compounds 15a and 18b exhibited moderate stability with 30–44% of both compounds remaining in mouse microsomes fortified with NADPH. In contrast, compound 14a and 15b underwent substantial phase I metabolism with <10% remaining in microsomes fortified with NADPH at 1 h, suggesting CYP dependent metabolism of the compounds, and relatively short in vitro t1/2. Since 18b contains a diazaspiro linker instead of the classic piperazine, the metabolism result was not surprising as the typical piperazine degrading phase I metabolic enzymes would be ineffective against that moiety.56 Furthermore, the sulfinyl compound 15a was more stable than its sulfenyl counterpart 14a, a preserved trend when compared to 2b and 3b versus 2a and 3a. However, the cis-2,6-dimethylpiperazine scaffold did not improve metabolic stability in mice as observed with 14a and 15a.
At a glance, the poor metabolic stability of 14a seems problematic (Figure 4), however, further metabolite identification studies revealed 13 as one of the major metabolites for 14a (Figure S1). Interestingly, 13 is an active metabolite with moderately high affinity for DAT and σ1R (Ki = 92.3 ± 5.6 nM and 111 ± 4.76 respectively), hence, this rapid metabolism may not be disadvantageous. Furthermore, the (R)-enantiomer (14c), showed higher stability than the (S)-enantiomer (14b) at 30% and 10%, respectively (Table S2). Additionally, metabolism in mice is typically quite rapid and not necessarily a reflection of higher species. Indeed, human liver microsome studies showed modest stability >30% of 14a and its enantiomers (14b, 14c) remaining after 60 min suggesting that 14a, 14b and 14c remain useful tools for in vivo studies (Figure S2).
Figure 4. Metabolic stability in mouse microsomes.
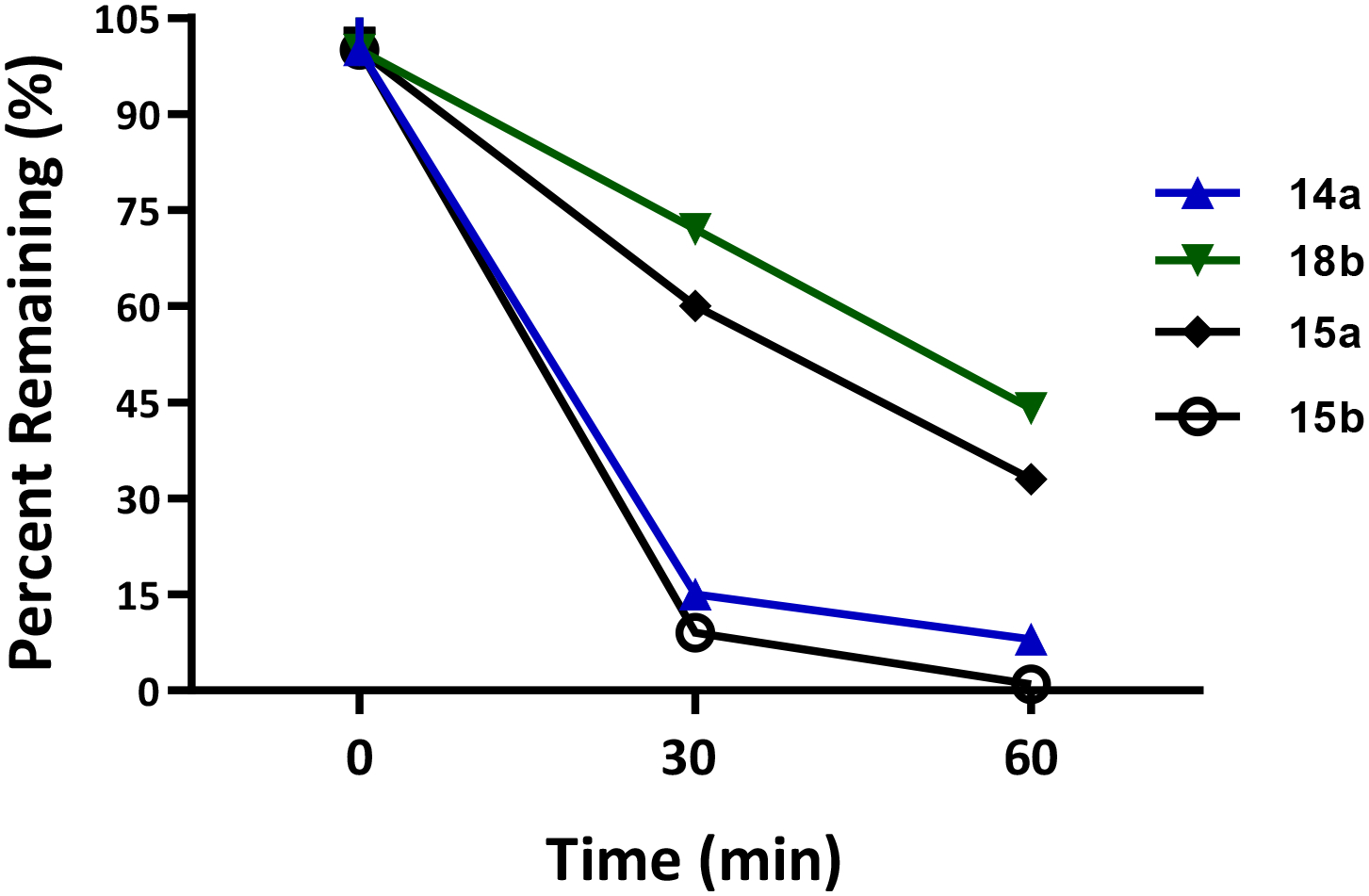
Compounds 14a and 15b showed susceptibility to phase I metabolism in mouse liver microsomes. Compounds 15a and 18b showed modest stability, with 18b being the most stable with 44% remaining after 60 min incubation.
Locomotor activity of 14a-c compared to cocaine in mice
While 15b had the highest DAT affinity in this subset, with a Y156F/WT DAT affinity ratio of 5.8, our recent discovery that its parent compound, 2b, despite a similarly high Y156F/WT DAT ratio, was indeed cocaine-like behaviorally,38 dampened our enthusiasm for further evaluating this compound in vivo. Thus, based on the overall profiles in vitro, 14a-c were chosen as the best candidates for in vivo characterization, starting with locomotor activity studies in mice. Figure 5 shows the effects of 14a-c on locomotor activity compared to cocaine and 3b (data for 3b were reanalyzed from Keighron et al. 2019,68 see methods for details). Systemic administration (i.p.) of cocaine (3, 10, 17 and 30 mg/kg) produced an increase in ambulatory activity, measured as maximum distance traveled in 30 minutes, that was significantly higher from that produced by 14a (1, 3, 10 mg/kg, and 30 mg/kg) and 3b (3, 10, 30 and 56 mg/kg) (Two-way ANOVA, main effect Drug: F2,75= 53.18, p<0.01; main effect Dose: F4,75=12.87, p<0.01; Drug by Dose interaction: F8,75=6.41, p<0.01). Mice injected with 14a showed an increase in locomotion that was significantly higher than the effect exhibited by 3b (p<0.05), but not significantly different than vehicle (p>0.05). The enantiomers of 14a, 14b and 14c, showed similar effects when compared to 14a (two-way ANOVA, main effect Drug, F2,45=0.08, NS; main effect Dose, F2,45=3.1, NS; Drug by Dose interaction, F4,45=0.16, NS). It is worth noting that compound 14a and its enantiomers showed levels of behavioral activity similar to those reported by Desai et al.69 and Schmeichel et al.70 for an atypical DAT inhibitor with potential therapeutic efficacy for ADHD. In the latter report, administration of the benztropine analogue AHN 2–005 (N-allyl-3α-[bis(4’-fluorophenyl)methoxy]tropane) did not significantly increase quadrant entering (an index of ambulatory activity) or rearing, but only modestly increased quiet wake.70
Figure 5.
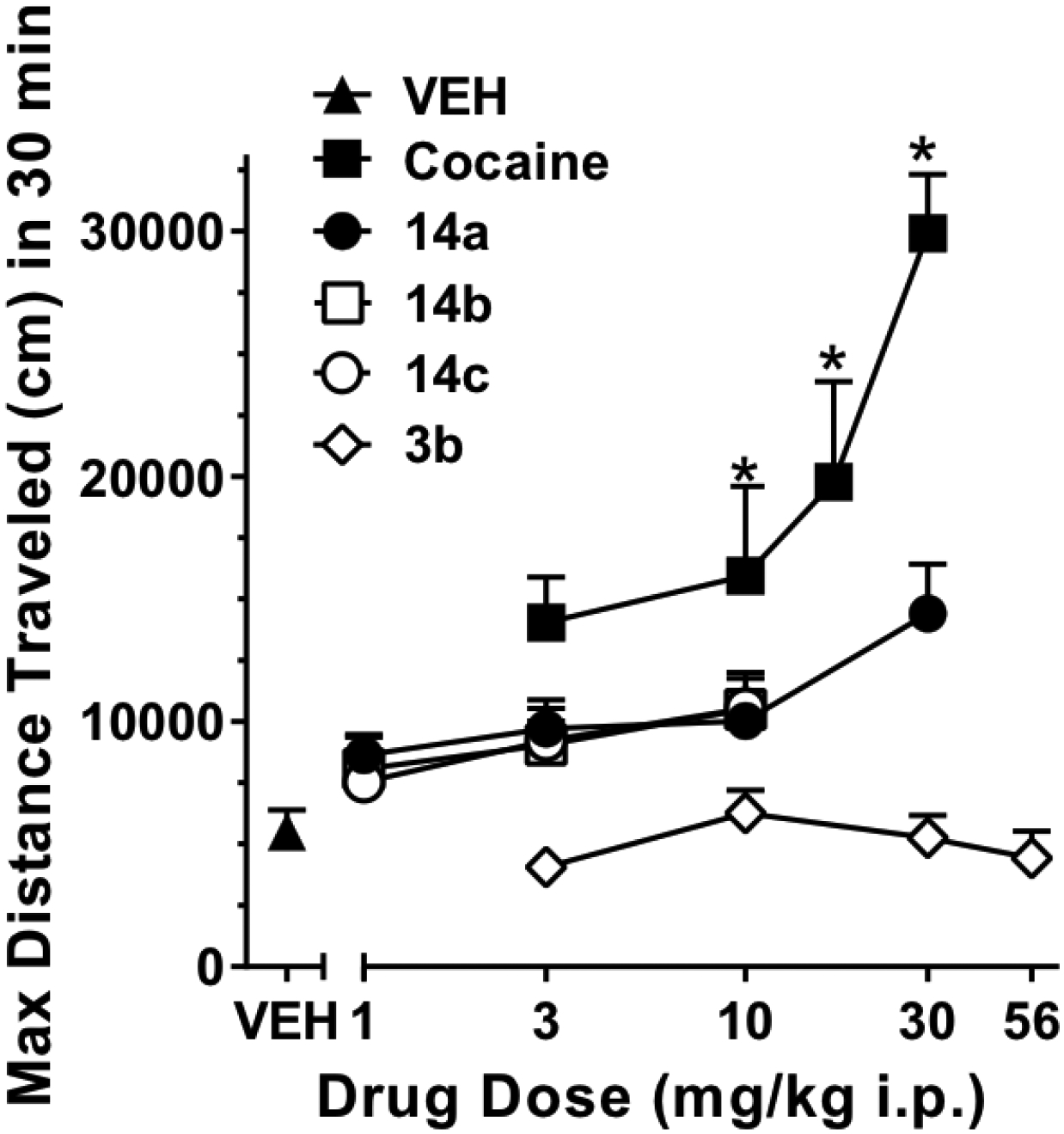
Effects of 14a-c compared to 3b and cocaine on mouse locomotion. Data are expressed as the maximum distance traveled in 30 min during a 2 h observation period, as a function of drug dose. * = p<0.05 VS vehicle (VEH) treated mice. N=6 for all groups.
CONCLUSION
In summary, herein we have synthesized a series of novel analogues of 2 and 3 that have a range of DAT affinities and selectivities over SERT. Specifically, we have extended SAR at the DAT by manipulating the ethylpiperazine linker, propanol terminus, and substitution on the phenyl ring of 2. All compounds were tested for binding to DAT and SERT, as well as σ1R. In addition, a subset of compounds (3b, 14a, 15a, 15b) were tested for NET and other off-target binding interactions (Table S3–S9). There were no significant interactions observed at a concentration of 100 nM for any of these compounds at any off target tested and most of these analogues were also inactive at 10 μM. When tested against hERG, 15a had the lowest affinity, whereas 15b had the highest (Table 1). Interestingly, the hERG/DAT affinity ratio of 14a was similar to that of 15a (28 and 30 respectively). We tested a subset of analogues (14a, 14d, 15a, and 18b) in binding assays for affinities at both the WT and Y156F DAT mutant to determine if they demonstrated atypical binding profiles, as previously reported for (±)-modafinil, 2b and 3b.27 These data suggest that 14a did indeed bind the DAT in a conformation that was more similar to atypical DAT inhibitors (affinity ratio = 4.6) supporting further development.
Phase I metabolism studies showed that 14a was rapidly metabolized in mouse liver microsomes (t1/2 ≈ 20 min), although its major metabolite (13) was also a DAT inhibitor. However, 14a and its enantiomers were more stable in human liver microsomes (t1/2 > 40 min). Given that the major metabolite is also active at DAT and based on these collective in vitro data, compound 14a was chosen as the lead candidate for further in vivo investigation. We discovered, that similar to previously reported atypical DAT inhibitors,15, 26, 32, 69–74 compound 14a produced only moderate locomotor stimulation in mice and was substantially less efficacious than cocaine. These results are consistent with an atypical DAT inhibitor profile and suggest that 14a may be a potential lead for development as a therapeutic for psychostimulant abuse. Further investigations of this compound in rodent models of cocaine abuse are underway to extend testing of the atypical DAT inhibitor hypothesis, as well as to further investigate the role of σ1 receptors in the behavioral profile of these agents.32, 37 As 14a also binds with high affinity to σ1 receptors (Ki = 5.62 nM) and has ~4-fold higher binding affinity than at DAT, its dual DAT/σ1 receptor targeting may prove to be an important mechanistic underpinning of its potential therapeutic profile.
EXPERIMENTAL METHODS
Synthesis.
All chemicals and solvents were purchased from chemical suppliers unless otherwise stated, and used without further purification. 1H and 13C NMR spectra were acquired using a Varian Mercury Plus 400 spectrometer at 400 MHz and 100 MHz, respectively. Chemical shifts are reported in parts-per-million (ppm) and referenced according to deuterated solvent for 1H NMR spectra (CDCl3, 7.26 or acetone-d6, 2.05) and 13C NMR spectra (CDCl3, 77.2 or acetone-d6, 29.8 and 206.0). Gas chromatography-mass spectrometry (GC/MS) data were acquired (where obtainable) using an Agilent Technologies (Santa Clara, CA) 7890B GC equipped with an HP-5MS column (cross-linked 5% PH ME siloxane, 30 m × 0.25 mm i.d. × 0.25 μm film thickness) and a 5977B mass-selective ion detector in electron-impact mode. Ultrapure grade helium was used as the carrier gas at a flow rate of 1.2 mL/min. The injection port and transfer line temperatures were 250 and 280 °C, respectively, and the oven temperature gradient used was as follows: the initial temperature (70°C) was held for 1 min and then increased to 300°C at 20°C/min over 11.5 min, and finally maintained at 300°C for 4 min. All column chromatography was performed using a Teledyne Isco CombiFlash RF flash chromatography system. Combustion analyses were performed by Atlantic Microlab, Inc. (Norcross, GA) and agree with ± 0.4% of calculated values. All melting points were determined on an OptiMelt automated melting point system and are uncorrected. On the basis of NMR and combustion data, all final compounds are >95% pure.
1-(4-(2-((Bis(4-fluorophenyl)methyl)thio)ethyl)piperazin-1-yl)-3-(4-fluorophenyl)propan-2-ol (8a).
Compound 8a was prepared using the method that was previously described32 using 7 (455mg, 1.3 mmol) and 2-(4-fluorobenzyl)oxirane (198 mg, 1.3 mmol) to give the product (650 mg, 100% yield) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.33–7.36 (m, 4H), 7.12–7.19 (m, 2H), 6.94–7.02 (m, 6H), 5.18 (s, 1H), 2.75–3.88 (m, 1H), 2.45–2.73 (m, 16H); 13C NMR (100 MHz, CDCl3) δ 163.1, 162.8, 160.6, 160.4, 137.0, 136.9, 133.9, 130.7, 130.6, 129.8, 129.7, 115.7, 115.4, 115.2, 115.0, 67.1, 63.3, 57.8, 53.1, 52.9, 40.4, 29.4. The free base was converted to the oxalate salt and recrystallized from methanol to give a white solid. Mp 210–211°C. Anal. (C28H31F3N2OS · 2C2H2O4 · 0.25H2O) C, H, N.
1-(4-(2-((Bis(4-fluorophenyl)methyl)thio)ethyl)piperazin-1-yl)-3-(4-(trifluoromethyl)phenyl)propan-2-ol (8b).
Compound 8b was prepared as described for 8a using 7 (455mg, 1.3 mmol) and 2-(4-(trifluoromethyl)benzyl)oxirane (264 mg, 1.3 mmol) to give the product 8b (700 mg, 98% yield) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.53–7.55 (m, 2H), 7.33–7.36 (m, 6H), 6.96–7.10 (m, 4H), 5.18 (s, 1H), 2.82–3.92 (m, 1H), 2.27–2.79 (m, 16H); 13C NMR (100 MHz, CDCl3) δ 163.2, 163.1, 142.5, 137.0, 136.7, 129.8, 129.7, 129.6, 125.3, 125.2, 125.2, 122.9, 115.7, 115.6, 115.5, 115.4, 66.7, 63.3, 57.7, 53.1, 52.9, 52.8, 41.0, 29.4, 29.3. The free base was converted to the oxalate salt and recrystallized from methanol to give a white solid. Mp 209–210°C. Anal. (C29H31F5N2OS · 2C2H2O4) C, H, N.
1-(4-(2-((Bis(4-fluorophenyl)methyl)thio)ethyl)piperazin-1-yl)-3-(4-methoxyphenyl)propan-2-ol (8c).
Compound 8c was prepared as described for 8a using 7 (455 mg, 1.3 mmol) and 2-(4-methoxybenzyl)oxirane (195 mg, 1.3 mmol) to give the product (650 mg, 98% yield) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.34–7.36 (m, 4H), 7.12–7.14 (m, 2H), 7.97–7.02 (m, 4H), 6.82–6.84 (m, 2H), 5.18 (s, 1H), 3.82–3.86 (m, 1H), 3.78 (s, 3H), 2.25–2.75 (m, 16H); 13C NMR (100 MHz, CDCl3) δ 163.1, 160.6, 158.1, 137.0, 136.9, 130.2, 129.8, 129.7, 115.7, 115.6, 115.4, 113.8, 67.3, 63.3, 57.8, 55.2, 53.1, 52.9, 40.4, 29.4. The free base was converted to the oxalate salt and recrystallized from methanol to give a white solid. Mp 207–208 °C. Anal. (C29H34F2N2O2S · 2C2H2O4 · 0.25H2O) C, H, N.
(S)-1-(4-(2-((Bis(4-fluorophenyl)methyl)thio)ethyl)piperazin-1-yl)-3-phenylpropan-2-ol (8d)
A mixture of 7 (348 mg, 1.00 mmol), commercially available (S)-1-chloro-3-phenylpropan-2-ol (204 mg, 1.20 mmol), K2CO3 (552 mg, 4.0 mmol) and KI (catalytic) in acetonitrile (30 mL) was refluxed overnight. The solvent was removed, H2O (50 mL) was added to the residue, and the aqueous mixture was extracted with ethyl acetate (3 × 50 ml). The organic layer was dried over MgSO4, the solvent was removed in vacuo, and the crude product was purified by flash column chromatography (ethyl acetate/TEA = 95:5) to give 8d (320 mg, 66% yield) as a yellow oil. +24.79 (MeOH, c = 0.73); 1H NMR (400 MHz, CDCl3) δ 7.18–7.36 (m, 9H), 6.96–7.01 (m, 4H), 5.18 (s, 1H), 3.85–3.92(m, 1H), 2.76–2.82 (m, 1H), 2.56–2.68 (m, 3H), 2.26–2.54 (m, 12H); 13C NMR (100 MHz, CDCl3) δ 163.1, 160.6, 138.2, 137.0, 129.8, 129.7, 129.3, 128.3, 126.3, 115.6, 115.3, 67.2, 63.4, 57.8, 53.1, 52.9, 41.3, 29.4. The free base was converted to the oxalate salt and recrystallized from methanol to give a white solid. Mp 208–209°C. Anal. (C28H32F2N2OS · 2C2H2O4 · 0.25H2O) C, H, N.
(R)-1-(4-(2-((Bis(4-fluorophenyl)methyl)thio)ethyl)piperazin-1-yl)-3-phenylpropan-2-ol (8e).
Compound 8e was prepared as described for 8d using 7 (348 mg, 1.00 mmol), commercially available (R)-1-chloro-3-phenylpropan-2-ol (204 mg, 1.20 mmol) to give the product (350 mg, 73% yield) as a yellow oil. −24.80 (MeOH, c = 0.75). 1H NMR (400 MHz, CDCl3) δ 7.19–7.36 (m, 9H), 6.95–7.01 (m, 4H), 5.18 (s, 1H), 3.87–3.91(m, 1H), 2.77–2.82 (m, 1H), 2.27–2.69 (m, 15H); 13C NMR (100 MHz, CDCl3) δ 163.1, 160.6, 138.2, 137.0, 129.8, 129.7, 129.3, 128.3, 126.3, 115.7,115.6, 115.4,115.3, 67.2, 63.4, 57.8, 53.1, 52.9, 41.3, 29.4. The free base was converted to the oxalate salt and recrystallized from methanol to give a white solid. Mp 208–209°C. Anal. (C28H32F2N2OS · 2C2H2O4 · 0.25H2O) C, H, N.
1-(4-(2-((Bis(4-fluorophenyl)methyl)thio)ethyl)piperazin-1-yl)-2-phenylethan-1-one (8f).
A mixture of CDI (243 mg, 1.50 mmol), and 3-phenylpropanoic acid (225 mg, 1.50 mmol) in THF (12 mL) was stirred at room temperature under argon. After 2 h, 7 (523 mg, 1.50 mmol) in THF (7 mL) was added and the reaction mixture was stirred overnight at room temperature. Solvent was removed and the reaction residue was purified by flash column chromatography (ethyl acetate/TEA = 95:5) to give 8f (620 mg, 86% yield) as a yellow oil. 1H NMR (CDCl3) δ 7.18–7.36 (m, 9H), 6.97–7.01 (m, 4H), 5.16 (s, 1H) 3.58–3.59 (m, 2H), 3.34–3.35 (m, 2H), 2.92–2.95 (m, 2H), 2.49–2.60 (m, 6H), 2.22–2.33 (m, 4H); 13C NMR (CDCl3) δ 170.5, 163.1, 160.7, 141.2, 136.9, 136.8, 129.8, 129.7, 128.5, 128.4, 126.2, 115.7, 115.6, 115.5, 115.4, 57.5, 53.0, 52.9, 52.5, 45.3, 41.3, 34.9, 31.5, 29.3. The free base was converted to the oxalate salt and recrystallized from hot isopropanol to give a white solid. Mp 190–191°C. Anal. (C28H30F2N2OS · C2H2O4) C, H, N.
1-(4-(2-((Bis(4-fluorophenyl)methyl)thio)ethyl)piperazin-1-yl)propan-2-one (8g).
Compound 8g was prepared as 8d using 1-bromopropan-2-one (137.0 mg, 0.73 mmol) to give the product (340 mg, 84% yield) as yellow oil. 1H NMR (CDCl3) δ 7.32–7.36 (m, 4H), 6.96–7.00 (m, 4H), 5.18 (s, 1H), 3.12–3.17 (m, 2H), 2.46–2.56 (m, 12H), 2.12–2.13 (m, 3H); 13C NMR (CDCl3) δ 206.3, 163.1, 160.6, 137.0, 129.8, 129.7, 115.7, 115.6, 115.5, 115.4, 68.1, 58.3, 57.8, 53.3, 52.9, 52.8, 52.7, 32.4, 32.3, 32.0, 29.3, 29.2, 27.7. The free base was converted to the oxalate salt and recrystallized from hot isopropanol to give a white solid. Mp 222–223°C. Anal. (C22H26F2N2OS · 2C2H2O4) C, H, N.
1-(4-(2-((Bis(4-fluorophenyl)methyl)sulfinyl)ethyl)piperazin-1-yl)-3-(4-fluorophenyl)propan-2-ol (9a).
Compound 9a was prepared as previously described32 using 8a (360 mg, 0.72 mmol) to give the product (140 mg, 38% yield) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.35–7.42 (m, 4H), 6.93–7.12 (m, 8H), 4.91 (s, 1H), 3.82–3.88 (m, 1H), 2.25–2.82 (m, 16H); 13C NMR (100 MHz, CDCl3) δ 164.0, 163.7, 162.0, 161.5, 161.3, 160.4, 133.9, 131.7, 131.0, 130.9, 130.7, 130.5, 130.3, 116.4, 116.2, 115.8, 115.6, 115.2, 115.0, 69.8, 67.1, 63.3, 53.1, 50.8, 48.2, 40.4. The free base was converted to the oxalate salt and recrystallized from methanol to give a white solid. Mp 199–200°C (dec.). Anal. (C28H31F3N2O2S · 2C2H2O4) C, H, N.
1-(4-(2-((Bis(4-fluorophenyl)methyl)sulfinyl)ethyl)piperazin-1-yl)-3-(4-(trifluoromethyl)phenyl)propan-2-ol (9b).
Compound 9b was prepared as previously described32 using 8b (360 mg, 0.72 mmol) to give the product (150 mg, 41% yield) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.52–7.55 (m, 2H), 7.33–7.42 (m, 6H), 7.04–7.11 (m, 4H), 4.91 (s, 1H), 3.87–3.93 (m, 1H), 2.31–2.78 (m, 16H); 13C NMR (100 MHz, CDCl3) δ 164.0, 163.8, 161.6, 161.3, 142.5, 131.7, 131.0, 130.9, 130.5, 130.3, 130.0, 129.1, 129.1, 128.8, 128.5, 116.4, 116.2, 116.1, 115.9, 115.6, 69.8, 66.8, 63.3, 53.1, 50.8, 48.2, 41.1, 41.0, 40.9. The free base was converted to the oxalate salt and recrystallized from methanol to give a white solid. Mp 198–199°C (dec.). Anal. (C29H31F5N2O2S · 2C2H2O4 · 0.25H2O) C, H, N.
1-(4-(2-((Bis(4-fluorophenyl)methyl)sulfinyl)ethyl)piperazin-1-yl)-3-(4-methoxyphenyl)propan-2-ol (9c).
Compound 9c was prepared as previously described32 using 8c (340 mg, 0.66 mmol) to give the product (160 mg, 59% yield) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.36–7.42 (m, 4H), 7.04–7.13 (m, 6H), 6.82–6.84 (m, 2H), 4.92 (s, 1H), 3.79–3.86 (m, 1H), 3.78 (s, 3H), 2.29–2.77 (m, 16H); 13C NMR (100 MHz, CDCl3) δ 164.0, 163.7, 161.6, 161.3, 158.2, 131.7, 131.0, 130.9, 130.5, 130.3, 130.2, 116.4, 116.2, 115.8, 115.6, 113.8, 69.7, 67.4, 63.3, 55.2, 53.1, 50.8, 48.2, 40.3. The free base was converted to the oxalate salt and recrystallized from methanol to give a white solid. Mp 197–198°C (dec.). Anal. (C29H34F2N2O3S · 2C2H2O4 · 0.25H2O) C, H, N.
(S)-1-(4-(2-((Bis(4-fluorophenyl)methyl)sulfinyl)ethyl)piperazin-1-yl)-3-phenylpropan-2-ol (9d).
Compound 9d was prepared as previously described32 using 8d (430 mg, 0.89 mmol) to give the product (160 mg, 36% yield) as a yellow oil. +21.29 (MeOH, c = 0.70). 1H NMR (400 MHz, CDCl3) δ 7.19–7.42 (m, 9H), 7.04–7.11 (m, 4H), 4.93 (s, 1H), 3.88–3.92 (m, 1H), 2.28–2.83 (m, 16H); 13C NMR (100 MHz, CDCl3) δ 164.0, 163.8, 161.5, 161.3, 138.2, 131.8, 131.0, 130.9, 130.6, 130.5, 130.3, 129.3, 128.4, 126.3, 116.4, 116.2, 115.9, 115.6, 69.8, 67.3, 63.4, 53.1, 50.8, 48.2, 41.3. The free base was converted to the oxalate salt and recrystallized from methanol to give a white solid. Mp 196–197°C. Anal. (C28H32F2N2O2S · 2C2H2O4) C, H, N.
(R)-1-(4-(2-((Bis(4-fluorophenyl)methyl)sulfinyl)ethyl)piperazin-1-yl)-3-phenylpropan-2-ol (9e).
Compound 9e was prepared as previously described32 using 8e (740 mg, 1.53 mmol) to give the product (360 mg, 47% yield) as a yellow oil. −22.75 (MeOH, c = 0.80). 1H NMR (400 MHz, CDCl3) δ 7.20–7.42 (m, 9H), 7.05–7.11 (m, 4H), 4.92 (s, 1H), 3.88–3.92 (m, 1H), 2.32–2.84 (m, 16H); 13C NMR (100 MHz, CDCl3) δ 164.0, 163.8, 131.0, 130.9, 130.5, 130.3, 129.3, 128.4, 126.3, 116.4, 116.2, 115.9, 115.6, 69.8, 67.3, 63.4, 53.1, 50.8, 48.2, 41.3. The free base was converted to the oxalate salt and recrystallized from methanol to give a white solid. Mp 196–197°C. Anal. (C28H32F2N2O2S · 2C2H2O4 · 0.25H2O) C, H, N.
(Bis(4-fluorophenyl)methyl)(3-bromopropyl)sulfane (10b).
Compound 10b was prepared as 10a which was previously published32 using 3-mercaptopropan-1-ol to give the product as yellow oil. The product yield over two steps is 98%. 1H NMR (400 MHz, CDCl3) δ 7.36–7.38 (m, 4H), 6.99–7.03 (m, 4H), 5.13 (s, 1H), 3.44–3.47 (m, 2H), 2.51–2.55 (m, 2H), 2.03–2.07 (m, 2H); GC/MS (EI) m/z 358 (M+)
(S)-1-(4-(2-((Bis(4-fluorophenyl)methyl)thio)ethyl)piperazin-1-yl)propan-2-ol (11a)
Compound 11a was prepared as described for 8d using 10a (1.05 g, 3.00 mmol), commercially available 1-(S)-(piperazin-1-yl)propan-2-ol (433 mg, 3 mmol) to give the product (1.07 g, 88% yield) as a yellow oil. +24.56 (MeOH, c = 0.90). 1H NMR (400 MHz, CDCl3) δ 7.32–7.37 (m, 4H), 6.96–7.02 (m, 4H), 5.19 (s, 1H), 3.77–3.82 (m, 1H), 2.18–2.68 (m, 14H), 1.10–1.13 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 163.1, 160.7, 137.0, 129.8, 129.7, 115.7, 115.5, 115.4, 65.5, 62.2, 57.8, 53.2, 53.1, 53.0, 52.8, 29.4, 29.3, 20.0. The free base was converted to the oxalate salt and recrystallized from methanol to give a white solid. Mp 213–214°C. Anal. (C22H28F2N2OS · 2C2H2O4 · 0.75H2O) C, H, N.
(R)-1-(4-(2-((Bis(4-fluorophenyl)methyl)thio)ethyl)piperazin-1-yl)propan-2-ol (11b).
Compound 11b was prepared as described for 8d using 10a (1.05 g, 3.00 mmol), commercially available 1-(R)-(piperazin-1-yl)propan-2-ol (433 mg, 3 mmol) to give the product (680 mg, 56% yield) as a yellow oil. −24.77 (MeOH, c=0.90). 1H NMR (400 MHz, CDCl3) δ 7.31–7.37 (m, 4H), 6.94–7.02 (m, 4H), 5.19 (s, 1H), 3.78–3.82 (m, 1H), 2.18–2.70 (m, 14H), 1.11–1.16 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 163.1, 160.7, 137.0, 129.8, 129.7, 115.7, 115.5, 115.4, 65.5, 62.2, 57.8, 53.2, 53.1, 52.8, 52.9, 29.4, 20.0. The free base was converted to the oxalate salt and recrystallized from methanol to give a white solid. Mp 200–201°C. Anal. (C22H28F2N2OS · 2C2H2O4) C, H, N.
1-(3-((Bis(4-fluorophenyl)methyl)thio)propyl)-4-(3-phenylpropyl)piperazine (11c).
Compound 11c was prepared as described for 8d using 10b (985 mg, 2.76 mmol), commercially available 1-(3-phenylpropyl)piperazine (564 mg, 2.76 mmol) to give the product (1.1 g, 83% yield) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.33–7.37 (m, 4H), 7.25–7.29 (m, 2H), 7.17–7.19 (m, 3H), 6.98–7.01 (m, 4H), 5.13 (s, 1H), 2.61–2.65 (m, 2H), 2.36–2.40 (m, 14H), 1.71–1.86 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 163.1, 160.6, 142.0, 137.0, 129.8, 129.7, 128.4, 128.3, 125.8, 115.5, 115.3, 58.0, 57.2, 53.0, 52.5, 33.7, 30.1, 28.5, 26.2. The free base was converted to the oxalate salt and recrystallized from methanol to give a white solid. Mp 202–203 °C (dec.). Anal. (C29H34F2N2S · 2C2H2O4) C, H, N.
1-(4-(2-((Bis(4-fluorophenyl)methyl)thio)ethyl)piperazin-1-yl)propan-1-one (11d).
Compound 11d was prepared as 11a, using 10b (515 mg, 1.5 mmol), commercially available 1-(piperazin-1-yl)propan-1-one (213.0 mg, 1.50 mmol) to give the product (600 mg, 100% yield) as yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.32–7.36 (4H, m), 6.96–7.10 (m, 4H), 5.16 (s, 1H), 3.56–3.59 (m, 2H), 3.40–3.58 (m, 2H), 2.47–2.55 (m, 4H), 2.27–2.36 (m, 6H), 1.09–1.13 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 172.2, 163.1, 160.7, 136.9, 129.8, 129.7, 115.7, 115.6, 115.4, 57.6, 53.2, 52.9, 52.6, 45.2, 41.4, 29.4, 26.4, 9.4. The free base was converted to the oxalate salt and recrystallized from methanol to give a white solid. Mp 168–170°C. Anal. (C22H26F2N2OS · C2H2O4 · 0.25H2O) C, H, N.
(S)-1-(4-(2-((Bis(4-fluorophenyl)methyl)sulfinyl)ethyl)piperazin-1-yl)propan-2-ol (12a)
Compound 12a was prepared as previously described32 using 11a (420mg, 1.03 mmol) to give the product (220 mg, 51% yield) as a yellow oil. +22.00 (MeOH, c = 1.00). 1H NMR (400 MHz, CDCl3) δ 7.37–7.42 (m, 4H), 7.04–7.11 (m, 4H), 4.93 (s, 1H), 3.78–3.83 (m, 1H), 2.19–2.83 (m, 14H), 1.11–1.12 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 164.0, 163.8, 161.6, 161.3, 131.8, 131.7, 131.0, 130.9, 130.5, 130.4, 130.3, 116.4, 116.2, 115.9, 115.6, 69.8, 65.5, 62.3, 53.1, 50.8, 48.2, 20.0. The free base was converted to the oxalate salt and recrystallized from methanol to give a white solid. Mp 194–195°C. Anal. (C22H28F2N2O2S · 2C2H2O4) C, H, N.
(R)-1-(4-(2-((Bis(4-fluorophenyl)methyl)sulfinyl)ethyl)piperazin-1-yl)propan-2-ol (12b).
Compound 12b was prepared as previously described32 using 11b (470mg, 1.16 mmol) to give the product (170 mg, 35% yield) as a yellow oil. −23.06 (MeOH, c = 0.85). 1H NMR (400 MHz, CDCl3) δ 7.38–7.43 (m, 4H), 7.05–7.12 (m, 4H), 4.93 (s, 1H), 3.78–3.83 (m, 1H), 2.19–2.84 (m, 14H), 1.11–1.16 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 164.0, 163.8, 161.6, 161.3, 131.8, 131.7, 131.0, 130.9, 130.5, 130.4, 130.3, 116.4, 116.2, 115.9, 115.6, 69.8, 65.5, 62.2, 53.1, 50.8, 48.2, 20.0. The free base was converted to the oxalate salt and recrystallized from methanol to give a white solid. Mp 188–190°C (dec.). Anal. (C22H28F2N2O2S · 2C2H2O4) C, H, N.
1-(3-((Bis(4-fluorophenyl)methyl)sulfinyl)propyl)-4-(3-phenylpropyl)piperazine (12c)
Compound 12c was prepared as previously described32 using 11c (600mg, 1.25 mmol) to give the product (370 mg, 60% yield) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.37–7.41 (m, 4H), 7.25–7.29 (m, 2H), 7.16–7.19 (m, 3H), 7.05–7.11 (m, 4H), 4.77 (s, 1H), 2.32–2.65 (m, 16H), 1.76–1.96 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 164.0, 163.7, 161.5, 161.3, 142.1, 131.8, 131.0, 130.9, 130.5, 130.3, 130.2, 128.4, 128.3, 125.7, 116.4, 116.2, 115.9, 115.6, 70.1, 58.0, 56.6, 53.1, 53.0, 48.8, 33.7, 28.6, 19.9. The free base was converted to the oxalate salt and recrystallized from methanol to give a white solid. Mp 213–214°C. Anal. (C29H34F2N2OS · 2C2H2O4) C, H, N.
1-(2-((Bis(4-fluorophenyl)methyl)thio)ethyl)-3,5-cis-dimethylpiperazine (13).
A mixture of 10a (750 mg, 2.19 mmol), cis-2,6-dimethylpiperazine (1.00 g, 8.75 mmol) and K2CO3 (1.21 g, 8.76 mmol) in acetonitrile (43.8 mL) was refluxed overnight. The solvent was removed, resuspended in CH2Cl2 (30 mL) and water (20 mL). The aqueous layer was extracted with CH2Cl2 (3 × 8 mL). The organic layer was dried over Na2SO4, and the solvent was removed in vacuo. The crude product was purified by flash column chromatography (CH2Cl2/MeOH/NH4OH = 100:0:0–85:13.5:1.5) to give 13 (666 mg, 81%) as a pale yellow oil. 1H NMR (400 MHz, acetone-d6) δ 7.53–7.50 (m, 4H), 7.12–7.08 (m, 4H), 5.49 (s, 1H), 2.80–2.75 (m, 2H), 2.62 (d, J = 10.4 Hz, 2H), 2.50 (s, 4H), 1.46 (m, 2H), 0.93 (d, J = 6.0 Hz, 6H).
1-(4-(2-((Bis(4-fluorophenyl)methyl)thio)ethyl)-2,6-cis-dimethylpiperazin-1-yl)propan-2-ol (14a).
A mixture of 13 (200 mg, 0.53 mmol) and commercially available propylene oxide (372 μL, 5.31 mmol) in iPrOH (5.3 mL) was refluxed overnight. The solvent was removed and the crude oil was purified by flash column chromatography (CH2Cl2/MeOH/NH4OH = 100:0:0–85:13.5:1.5) to give 14a (179 mg, 78%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.39–7.28 (m, 4H), 7.03–6.87 (m, 4H), 5.18 (s, 1H), 3.63 (dq, J = 7.6, 6.1 Hz, 1H), 2.57 (m, 4H), 2.50–2.33 (m, 6H), 1.85–1.65 (m, 2H), 1.07 (d, J = 6.1 Hz, 3H), 0.99 (dd, J = 6.2, 2.6 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 163.0, 160.6, 137.0, 137.0, 129.8, 129.7, 115.5, 115.3, 65.4, 60.8, 58.5, 58.3, 57.6, 56.2, 52.8, 29.2, 20.4, 19.3, 19.1. The free base was converted to the oxalate salt and recrystallized from methanol to give a white solid. Mp 148–149°C. Anal. (C24H32F2N2OS ∙ 2.5C2H2O4 ∙ 0.5H2O) C, H, N.
(S)-1-(−4-(2-((Bis(4-fluorophenyl)methyl)thio)ethyl)-2,6-cis--dimethylpiperazin-1-yl)propan-2-ol (14b).
Compound 14b was prepared as described for 14a, using commercially available (S)-propylene oxide (610 μL, 8.76 mmol) to give 14b (124 mg, 32%) as a colorless oil. 11.105 (MeOH, c = 8.10). 1H NMR (400 MHz, CDCl3) δ 7.37 – 7.34 (m, 4H), 7.02 – 6.97 (m, 4H), 5.20 (s, 1H), 3.69–3.61 (m, 1H), 2.64–2.55 (m, 4H), 2.47 (s, 4H), 2.41 (d, J = 6.8 Hz, 2H), 1.80–1.73 (m, 2H), 1.09 (d, J = 6.0 Hz, 3H), 1.02 (t, J = 2.8 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 163.1, 160.7, 137.0, 137.0, 129.8, 129.7, 115.6, 115.4, 65.4, 60.9, 58.5, 58.4, 57.6, 56.2, 52.9, 29.3, 20.3, 19.4, 19.1. The free base was converted to the oxalate salt and recrystallized from methanol to give a white solid. Mp 133–134°C. Anal. (C24H32F2N2OS ∙ 2C2H2O4 ∙ 2H2O) C, H, N.
(R)-1-(4-(2-((bis(4-fluorophenyl)methyl)thio)ethyl)-2,6-cis-dimethylpiperazin-1-yl)propan-2-ol (14c).
Compound 14c was prepared as described for 14a, using commercially available (R)-propylene oxide (420 μL, 6.02 mmol) to give 14c (278 mg, 53%) as a colorless oil. −11.187 (MeOH, c = 7.15). 1H NMR (400 MHz, CDCl3) δ 7.36–7.33 (m, 4H), 7.00–6.96 (m, 4H), 5.19 (s, 1H), 3.69–3.61 (m, 1H), 2.63–2.54 (m, 4H), 2.46 (s, 4H), 2.41 (d, J = 6.4 Hz, 2H), 1.79–1.73 (m, 2H), 1.08 (d, J = 5.2 Hz, 3H), 1.01 (t, J = 2.8 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 163.1, 160.6, 137.0, 137.0, 129.8, 129.7, 115.6, 115.3, 65.4, 60.8, 58.5, 58.4, 57.6, 56.2, 52.9, 29.3, 20.4, 19.3, 19.1. The free base was converted to the oxalate salt and recrystallized from methanol to give a white solid. Mp 132–134°C. Anal. (C24H32F2N2OS ∙ 2C2H2O4 ∙ 2H2O) C, H, N.
1-(4-(2-((Bis(4-fluorophenyl)methyl)thio)ethyl)-2,6-cis-dimethylpiperazin-1-yl)-3-phenylpropan-2-ol (14d).
Compound 14d was prepared as described for 14a, using commercially available (2,3-epoxypropyl)benzene (1.95 mL, 5.31 mmol) to give 14d (124 mg, 45%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.35 (dd, J = 8.4, 5.4 Hz, 4H), 7.25 (m, 5H), 6.99 (t, J = 8.5 Hz, 4H), 5.20 (s, 1H), 3.78 (dq, J = 10.5, 5.5 Hz, 1H), 3.54 (bs, 1H), 2.80 (dd, J = 13.7, 6.9 Hz, 1H), 2.76–2.53 (m, 7H), 2.51–2.35 (m, 4H), 1.74 (dd, J = 10.4, 7.6 Hz, 2H), 0.99–0.97 (m, 6H). 13C NMR (100 MHz, CDCl3) δ 163.1, 160.6, 138.4, 137.0, 137.0, 129.9, 129.8, 129.4, 129.3, 128.4, 128.4, 126.2, 115.6, 115.4, 70.1, 60.9, 60.8, 58.1, 57.6, 56.1, 52.9, 41.9, 29.2, 19.2, 19.0. The free base was converted to the oxalate salt and recrystallized from methanol to give a white solid. Mp 150–152°C. Anal. (C30H36F2N2OS ∙ 2.5C2H2O4 ∙ 0.5H2O) C, H, N.
1-(4-(2-((Bis(4-fluorophenyl)methyl)sulfinyl)ethyl)-2,6-cis-dimethylpiperazin-1-yl)propan-2-ol (15a)
Compound 15a was prepared as previously described32 using 14a (47.1 mg, 0.10 mmol) to give the product (36.9 mg, 82% yield) as a colorless oil (82% yield, 0.082 mmol). 1H NMR (400 MHz, CDCl3) δ 7.58–7.50 (m, 4H), 7.24–7.15 (m 4H), 5.07 (s, 1H), 3.84–3.77 (m, 1H), 3.58 (bs, 1H), 3.87–2.63 (m, 8H), 2.57 (s, 2H), 2.06–1.95 (m, 2H), 1.23–1.20 (m, 3H), 1.18–1.13 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 163.9, 163.7, 161.4, 161.2, 131.0, 130.9, 130.3, 130.3, 116.3, 116.1, 115.8, 115.5, 69.6, 65.3, 60.9, 60.2, 58.2, 56.1, 50.5, 48.2, 48.1, 20.4. The free base was converted to the oxalate salt and recrystallized from methanol to give an amorphous solid. Anal. (C24H32F2N2O2S ∙ 2C2H2O4 ∙ H2O) C, H, N.
1-(4-(2-((Bis(4-fluorophenyl)methyl)sulfinyl)ethyl)-2,6-cis-dimethylpiperazin-1-yl)-3-phenylpropan-2-ol (15b).
Compound 15b was prepared as previously described32 using 14d (34.3 mg, 0.067 mmol) to give the product (33.2 mg, 94% yield) as a colorless oil (82% yield, 0.082 mmol). The free base was converted to the oxalate salt and recrystallized from methanol to give an amorphous solid. Anal. (C30H36F2N2O2S ∙ 2C2H2O4 ∙ 2H2O) C, H, N. 1H NMR (400 MHz, CD3OD) δ 7.55–7.52 (m, 4H), 7.32–7.21 (m, 5H), 7.17–7.09 (m, 4H), 5.10 (s, 1H), 4.22–4.16 (m, 1H), 3.75–3.65 (m, 1H), 3.53–3.42 (m, 1H), 3.31–3.16 (m, 3H), 3.01–2.84 (m, 5H), 2.79–2.72 (m, 2H), 2.69–2.62 (m, 1H), 2.47–2.32 (m, 2H), 1.29–1.06 (m, 6H). 13C NMR (100 MHz, CDCl3) δ 163.6, 161.6, 161.4, 137.0, 131.4, 131.3, 130.5, 130.4, 129.2, 128.4, 126.5, 115.9, 115.7, 115.3, 115.1, 69.5, 68.7, 67.7, 67.7, 60.0, 60.0, 58.1, 49.5, 49.5, 41.9, 23.9, 23.8, 23.8, 20.5, 14.1, 14.0, 13.9, 13.8.
tert-Butyl-8-(2-((bis(4-fluorophenyl)methyl)thio)ethyl)-2,8-diazaspiro[4.5]decane-2-carboxylate (16).
A mixture of 10a (428 mg, 1.25 mmol), commercially available tert-butyl 2,8-diazaspiro[4.5]decane-2-carboxylate (300 mg, 1.25 mmol) and K2CO3 (2.59 g, 1.88 mmol) in acetonitrile (6.25 mL) was refluxed overnight. The solvent was removed, resuspended in CH2Cl2 (10 mL) and water (8 mL). The aqueous layer was extracted with CH2Cl2 (3 × 5 mL). The organic layer was dried over Na2SO4, and the solvent was removed in vacuo. The crude product was purified by flash column chromatography (CH2Cl2/MeOH/NH4OH = 100:0:0–85:13.5:1.5) to give 16 (577 mg, 89%) as a pale yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.41–7.32 (m, 4H), 7.04–6.92 (m, 4H), 5.22 (s, 1H), 3.35 (dt, J = 20.6, 7.0 Hz, 2H), 3.17 (s, 1H), 3.09. (s, 1H), 2.51 (s, 4H), 2.46–2.32 (m, 2H), 2.29–2.18 (m, 2H), 1.66–1.61 (m, 2H), 1.59–1.49 (m, 4H), 1.46 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 162.9, 160.5, 154.5, 137.1, 137.0, 129.8, 129.7, 115.4, 115.2, 78.9, 78.8, 77.5, 58.0, 52.7, 50.7, 44.1, 43.8, 40.3, 39.4, 34.6, 29.6, 29.4, 28.4.
8-(2-((Bis(4-fluorophenyl)methyl)thio)ethyl)-2,8-diazaspiro[4.5]decane (17).
A mixture of 16 (800 mg, 1.59 mmol) and trifluoroacetic acid (1.5 mL) in CH2Cl2 (7 mL) was stirred for 5h at room temperature. The mixture was diluted with CH2Cl2 (8 mL) and neutralized with 15% NH4OH (aq, 10 mL). The aqueous layer was extracted with CH2Cl2 (3 × 5 mL). The organic layer was dried over Na2SO4, and the solvent was removed in vacuo. The crude product was purified by flash column chromatography (CH2Cl2/MeOH/NH4OH = 100:0:0–85:13.5:1.5) to give 17 (589 mg, 92%) as a pale yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.39–7.35 (m, 4H), 7.01–6.96 (m, 4H), 5.21 (s, 1H), 3.37–3.33 (m, 4H), 2.93–2.89 (m, 2H), 2.65 (s, 2H), 2.32 (s, 4H), 1.56–1.50 (m, 4H), 1.55–1.51 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 162.7, 162.6, 160.2, 136.7, 129.5, 129.4, 129.3, 115.1, 114.9, 57.7, 57.7, 52.4, 50.9, 49.4, 45.5, 40.7, 40.7, 37.5, 35.7, 29.1.
2-(8-(2-((Bis(4-fluorophenyl)methyl)thio)ethyl)-2,8-diazaspiro[4.5]decan-2-yl)-1-phenylethan-1-ol (18b).
Compound 18b was prepared as described for 14a, using commercially available styrene oxide (5.22 mL, 75.6 mmol) to give 18b (149 mg, 38%) as a pale yellow oil. The free base was converted to the oxalate salt and recrystallized from methanol to give an amorphous solid. Anal. (C31H36F2N2OS ∙ 2C2H2O4 ∙ 0.5H2O) C, H, N. 1H NMR (400 MHz, CD3OD) δ 7.46–7.43 (m, 4H), 7.31–7.19 (m, 5H), 7.07–7.03 (m, 4H), 5.37 (s, 1H), 4.19–4.13 (m, 1H), 3.55–3.43 (m, 2H), 3.42–3.15 (m, 9H), 2.80–2.2.77 (m, 2H), 2.75–2.71 (m, 2H), 2.04–2.00 (t, J = 6.8 Hz, 2H), 2.01–1.87 (m, 4H). 13C NMR (100 MHz, CD3OD) δ 165.2, 163.2, 160.8, 137.0, 136.8, 136.7, 129.8, 129.8.9, 129.2, 128.2, 126.3, 115.2, 115.0, 67.3, 60.2, 55.3, 53.7, 51.6, 49.6, 49.5, 41.4, 38.7, 32.2, 25.3.
1-(8-(2-((Bis(4-fluorophenyl)methyl)sulfinyl)ethyl)-2,8-diazaspiro[4.5]decan-2-yl)propan-2-ol (19).
Compound 19 was prepared as described for 14a followed by 15a, using commercially available propylene oxide (2 mL) to give 19 (24.0 mg, 21% yield over two steps) as a colorless oil. The free base was converted to the oxalate salt and recrystallized from methanol to give a white solid. Mp 166–167°C. Anal. (C26H34F2N2O2S ∙ 2C2H2O4) C, H, N. 1H NMR (400 MHz, CD3OD) δ 7.48–7.44 (m, 4H), 7.08–7.34 (m, 4H), 5.38 (s, 1H), 4.10–4.07 (m, 1H), 3.54–3.08 (m, 12H), 2.77–2.72 (m, 2H), 2.07–1.94 (m, 6H), 1.20 (d, J = 6.4 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 165.2, 163.3, 160.8, 136.8, 136.7, 129.9, 129.8, 115.2, 115.0, 62.4, 61.8, 55.3, 53.4, 51.6, 49.6, 49.5, 38.8, 32.2, 25.3, 20.0.
Radioligand Binding Studies
DAT Binding Assay.
Frozen striatum membranes dissected from male Sprague−Dawley rat brains (supplied on ice by Bioreclamation, Hicksville, NY) were homogenized in 20 volumes (w/v) of ice cold modified sucrose phosphate buffer (0.32 M sucrose, 7.74 mM Na2HPO4, and 2.26 mM NaH2PO4, pH adjusted to 7.4) using a Brinkman Polytron (Setting 6 for 20 s) and centrifuged at 48,400 × g for 10 min at 4°C. The resulting pellet was resuspended in buffer, recentrifuged, and suspended in ice cold buffer again to a concentration of 20 mg/mL, original wet weight (OWW). Experiments were conducted in 96-well polypropylene plates containing 50 μL of various concentrations of the inhibitor, diluted using 30% DMSO vehicle, 300 μL of sucrose phosphate buffer, 50 μL of [3H]WIN 35,42875 (final concentration 1.5 nM; Kd = 28.2 nM; PerkinElmer Life Sciences, Waltham, MA), and 100 μL of tissue (2.0 mg/well OWW). All compound dilutions were tested in triplicate and the competition reactions started with the addition of tissue, and the plates were incubated for 120 min at 0–4°C. Nonspecific binding was determined using 10 μM indatraline.
SERT Binding Assay.
Frozen stem membranes dissected from male Sprague−Dawley rat brains (supplied on ice by Bioreclamation, Hicksville, NY) were homogenized in 20 volumes (w/v) of 50 mM Tris buffer (120 mM NaCl and 5 mM KCl, adjusted to pH 7.4) at 25°C using a Brinkman Polytron (at setting 6 for 20 s) and centrifuged at 48,400 × g for 10 min at 4°C. The resulting pellet was resuspended in buffer, recentrifuged, and suspended in buffer again to a concentration of 20 mg/mL, OWW. Experiments were conducted in 96-well polypropylene plates containing 50 μL of various concentrations of the inhibitor, diluted using 30% DMSO vehicle, 300 μL of Tris buffer, 50 μL of [3H]citalopram (final concentration 1.5 nM; Kd = 6.91 nM; PerkinElmer Life Sciences, Waltham, MA), and 100 μL of tissue (2.0 mg/well OWW). All compound dilutions were tested in triplicate and the competition reactions started with the addition of tissue, and the plates were incubated for 60 min at room temperature. Nonspecific binding was determined using 10 μM fluoxetine.
σ1 Receptor Binding Assay.
σ1 receptor binding was performed as previously reported.76 Frozen cortex membranes dissected from male guinea pig brains (supplied on ice by Bioreclamation, Hicksville, NY) were homogenized in 10 volumes (w/v) of ice cold modified sucrose Tris buffer (10 mM Tris-HCl with 0.32 M sucrose, adjusted to pH 7.4) with a glass and Teflon homogenizer. The homogenate was centrifuged at 1,240 × g for 10 min at 4 °C. The supernatant was collected into a clean centrifuge tube, and the remaining pellet was resuspended in 10 volumes (w/v) of buffer and centrifuged again at 48,400 × g for 15 min at 4 °C. The resulting pellet was resuspended in ice cold buffer to 50 mg/mL, OWW. Experiments were conducted in 96-well polypropylene plates containing 50 μL of various concentrations of the inhibitor, diluted using 30% DMSO vehicle, 300 μL of modified sucrose Tris buffer, 50 μL of [3H](+)-pentazocine (final concentration 3 nM; Kd = 5.18 nM; PerkinElmer Life and Analytical Sciences, Waltham, MA) and 100 μL of tissue (5.0 mg/well OWW). All compound dilutions were tested in triplicate and the competition reactions started with the addition of tissue, and the plates were incubated for 120 min at room temperature. Nonspecific binding was determined using 10 μM of either PRE084 or (+)-pentazocine.
For all binding assays, incubations were terminated by rapid filtration through Perkin Elmer Uni-Filter-96 GF/B (DAT, SERT and σ1R) or Whatman GF/B filters (NET), presoaked in either 0.3% (SERT and NET) or 0.05% (DAT and σ1R) polyethylenimine, using a Brandel 96-Well Plates Harvester Manifold or Brandel R48 filtering manifold (Brandel Instruments, Gaithersburg, MD). The filters were washed a total of 3 times with 3 mL (3 × 1 mL/well or 3 × 1 mL/tube) of ice cold binding buffer. For DAT, SERT and σ1R binding experiment 65 μL Perkin Elmer MicroScint 20 Scintillation Cocktail was added to each filter well. For NET binding experiment, the filters were transferred in 24-well scintillation plates and 600 μL of CytoScint was added to each well. All the plates/filters were counted using a Perkin Elmer MicroBeta Microplate Counter. IC50 values for each compound were determined from inhibition curves and Ki values were calculated using the Cheng-Prusoff equation.77 When a complete inhibition could not be achieved at the highest tested concentrations, Ki values have been extrapolated by constraining the bottom of the dose-response curves (= 0% residual specific binding) in the non-linear regression analysis. These analyses were performed using GraphPad Prism version 8.00 for Macintosh (GraphPad Software, San Diego, CA). Kd values for the radioligands were determined via separate homologous competitive binding or radioligand binding saturation experiments. Ki values were determined from at least 3 independent experiments performed in triplicate and are reported as mean ± SEM, and the results were rounded to the third significant figure.
Molecular Pharmacology
Site-directed mutagenesis.
The Y156F mutation was introduced with QuickChange (adapted from Stratagene, La Jolla, CA) on DAT WT cDNA cloned into the pcDNA3 expression vector. Clones carrying the mutation were detected by DNA sequencing (Eurofins Genomics, DE) and plasmids were amplified by transformation (XL1 blue cells (Stratagene)) and harvested using the maxi prep kit (Qiagen) according to the manufacturer’s manual.
Cell Culture and Transfection.
COS7 cells were grown in Dulbecco’s modified Eagle’s medium 041 01885 supplemented with 10% fetal calf serum, 2 mM L-glutamine and 0.01 mg/mL gentamicin at 37 °C in 10% CO2. DAT WT and Y156F were transiently transfected into COS7 cells with Lipo2000 (Invitrogen) according to manufacturer’s manual using a cDNA:Lipo2000 ratio of 3:6.
[3H]WIN35,428 binding experiments.
Binding assays were carried out essentially as described.78 Transfected COS7 cells were plated in 24-well dishes (105 cells/well) coated with poly-ornithine (Sigma). 48 h after transfection, cells were washed with 500 μl uptake buffer (UB) (25 mM HEPES, 130 mM NaCl, 5.4 mM KCl, 1.2 mM CaCl2, 1.2 mM MgSO4, 1 mM L-ascorbic acid, 5 mM D-glucose, and 1 μM of the catechol-O-methyltransferase inhibitor Ro 41–0960 (Sigma), pH 7.4), and the non-labeled compound was added to the cells in a total volume of 500 μl UB. The assay was initiated by the addition of 10 nM [3H]WIN35,428 (82 Ci/mmol) (Novandi Chemistry, SE). The reactions were incubated at 5°C until equilibrium was obtained (>90 min). Then cells were washed twice with 500 μL of ice cold UB, lysed in 250 μL of 1% SDS and left for >30 min at RT on gentle shaking. All experiments were carried out with 10 concentrations of unlabeled ligand within a concentration range from 1 nM to 1 mM, performed in triplicates.
All samples were transferred to 24-well counting plates (Perkin Elmer, Waltham, MA), 500 μL (24 well) of Opti-phase Hi Safe 3 scintillation fluid (Perkin Elmer) was added followed by counting of the plates in a Wallac Tri-Lux β-scintillation counter (Perkin Elmer).
Phase I Metabolism in Mouse Liver Microsomes
The phase I metabolic stability assay was conducted in mouse liver microsomes as previously described with minor modifications.67 In brief, the reaction was carried out with 100 mM potassium phosphate buffer, pH 7.4, in the presence of NADPH regenerating system, (compound final concentration was 10 μM; and 0.5 mg/mL microsomes). Positive controls for phase I metabolism (buprenorphine) were also evaluated. Compound disappearance was monitored over time using a liquid chromatography and tandem mass spectrometry (LC/MS/MS) method. All reactions were performed in triplicate.
Chromatographic analysis was performed using an Accela™ ultra-high-performance system consisting of an analytical pump and an autosampler coupled with a TSQ Vantage mass spectrometer (Thermo Fisher Scientific Inc., Waltham, MA). Chromatographic separation was achieved at ambient temperature using Agilent Eclipse Plus column (100 × 2.1mm i.d.) packed with a 1.8 μm C18 stationary phase. The mobile phase consisted of 0.1% formic acid in acetonitrile and 0.1% formic acid in H2O with gradient elution, starting with 10% (organic) linearly increasing to 99% (0–2 min), maintaining at 99% (2–2.5 min) and re-equilibrating to 10% by 2.7 min. The total run time for each analyte was 4.5 min. The mass transitions used for compounds for LC/MS/MS analysis are given in Supporting Information.
Metabolite Identification in Mouse Liver Microsomes
Metabolite identification (MET-ID) was performed on a Dionex ultra high-performance LC system coupled with Q Exactive Focus orbitrap mass spectrometer (Thermo Fisher Scientific Inc., Waltham MA). Separation was achieved using Agilent Eclipse Plus column (100 × 2.1mm i.d; maintained at 35°C) packed with a 1.8 μm C18 stationary phase. The mobile phase consisted of 0.1% formic acid in water and 0.1% formic acid in acetonitrile. Pumps were operated at a flow rate of 0.3 mL/min for 7 min using gradient elution. The mass spectrometer controlled by Xcalibur software 4.0.27.13 (Thermo Scientific) was operated with a HESI ion source in positive ionization mode. Metabolites were identified in the full-scan mode (from m/z 50 to 1600) by comparing t = 0 samples with t = 60 min samples and structures were proposed based on the accurate mass information.
Locomotor Activity Studies in Mice
Approximately two h before the behavioral test, animals were transferred from the animal facility vivarium to the behavioral laboratory. After the acclimatization period, mice were injected i.p. with test compounds or vehicle and then immediately placed into the open field cages. These were clear acrylic testing chambers (40 cm3) (Med Associates, St. Albans, VT) provided with infrared light beam sources spaced 2.5 cm apart along two perpendicular walls directed at light sensitive detectors mounted on the opposing walls of the open field. Ambulatory activity in mice was detected by interruption of the light beams, which occurrence during a 2 h session was automatically recorded and then transformed in distance traveled. The data obtained for each animal and dose of compounds (cocaine, 3, 10, 17 and 30 mg/kg; 14a, 1, 3, 10, 30 mg/kg; 14b, 1, 3, 10 mg/kg; 14c, 1, 3, 10 mg/kg; 3b, 3, 10, 30, 56 mg/kg) was averaged to obtain the group mean (n=6 for all groups) and correspondent SEM. In Figure 5, data have been expressed as the maximum distance traveled in cm/30 min over a 2 h session, as a function of drug and dose. Data were analyzed by two-way ANOVA with “Dose” and “Drug” as factors. Tuckey’s post-hoc test was employed to compare the activity produced by administration of different drugs and also to compare differences in activity between different doses of test compounds and vehicle. Mice were used only once. Data for 3b were reanalyzed from results shown in Keighron et al. 2019,68 which were obtained under identical conditions and with the same open field chambers used to assess the behavioral activity shown in the present study.
Supplementary Material
ACKNOWLDGEMENTS
Support for this research was provided by the National Institute on Drug Abuse - Intramural Research Program (Z1A DA000389). CJL is supported by the Danish Council for Independent Research (0602-02100B and 4183-00581). Care of the animals was in accordance with the guidelines of the National Institutes of Health and the National Institute on Drug Abuse Intramural Research Program Animal Care and Use Program, which is fully accredited by AAALAC International.
ABBREVIATIONS
- ADHD
Attention-deficit/hyperactivity disorder
- ANOVA
Analysis of variance
- Boc
tert-Butyloxycarbonyl
- CDI
Carbonyldiimidazole
- CFT
β-Carbomethoxy-3-β-(4-fluorophenyl)tropane
- CYP
Cytochrome P450
- DA
Dopamine
- DAT
Dopamine transporter
- DCM
Dichloromethane
- DEA
Drug Enforcement Administration
- dec
Decomposed
- DMPK
Drug metabolism and pharmacokinetics
- EI
Electron ionization
- FDA
Food and Drug Administration
- GC
Gas chromatography
- hDAT
Human dopamine transporter
- hERG
Human Ether-a-go-go-related Gene
- HESI
Heated electrospray ionization
- i.p.
Intraperitoneal
- IC50
Half maximal inhibitory concentration
- Kd
Dissociation constant
- Ki
Inhibitor constant
- LC
Liquid chromatography
- MAT
Medication assisted treatment
- MD
Molecular dynamic
- MeOH
Methanol
- MET-ID
Metabolite identification
- MS
Mass spectrometry
- NADPH
Nicotinamide adenine dinucleotide phosphate
- NET
Norepinephrine transporter
- NMR
Nuclear magnetic resonance
- NT
Not tested
- OWW
Original wet weight
- ppm
parts-per-million
- rDAT
Rat dopamine transporter
- RT
Room temperature
- σ1R
Sigma 1 receptor
- SEM
Standard error of the mean
- SERT
Serotonin transporter
- t1/2
Half-life
- TEA
Triethylamine
- TFA
Trifluoroacetic acid
- THF
Tetrahydrofuran
- TSQ
Triple quadrupole
- WT
Wild type
Footnotes
Supporting Information
The Supporting Information is available free of charge.
Microanalysis data on all final compounds, Phase 1 metabolic stability data in mouse liver microsomes, Metabolite ID, Metabolic stability data in human liver microsomes, off target binding data on selected compounds and SMILES data.
REFERENCES
- 1.NIDA. Overdose Death Rates https://www.drugabuse.gov/related-topics/trends-statistics/overdose-death-rates (Accessed Apr 10, 2019).
- 2.DEA. 2017. National Drug Threat Assessment https://www.dea.gov/documents/2017/10/01/2017-national-drug-threat-assessment (Accessed Apr 15, 2019).
- 3.Czoty PW; Stoops WW; Rush CR Evaluation of the “pipeline” for development of medications for cocaine use disorder: A review of translational preclinical, human laboratory, and clinical trial research. Pharmacol. Rev 2016, 68, 533–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mattick RP; Breen C; Kimber J; Davoli M Methadone maintenance therapy versus no opioid replacement therapy for opioid dependence. Cochrane Db. Syst. Rev 2009. [Google Scholar]
- 5.Mattick RP; Breen C; Kimber J; Davoli M Buprenorphine maintenance versus placebo or methadone maintenance for opioid dependence. Cochrane Db. Syst. Rev 2014. [Google Scholar]
- 6.Schwartz RP; Gryczynski J; O’Grady KE; Sharfstein JM; Warren G; Olsen Y; Mitchell SG; Jaffe JH Opioid agonist treatments and heroin overdose deaths in Baltimore, Maryland, 1995–2009. Am. J. Public Health 2013, 103, 917–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jordan CJ; Cao J; Newman AH; Xi ZX Progress in agonist therapy for substance use disorders: Lessons learned from methadone and buprenorphine. Neuropharmacology 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balster RL; Kuhar MJ; Schuster CR Pharmacological Aspects of Drug Dependence : Toward an Integrated Neurobehavioral Approach Springer: Berlin; New York, 1996; p xxv, 658 p. [Google Scholar]
- 9.Schmitt KC; Zhen J; Kharkar P; Mishra M; Chen N; Dutta AK; Reith MEA Interaction of cocaine-, benztropine-, and GBR12909-like compounds with wildtype and mutant human dopamine transporters: molecular features that differentially determine antagonist binding properties. J. Neurochem 2010, 115, 296–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmitt KC; Reith MEA Regulation of the dopamine transporter Aspects relevant to psychostimulant drugs of abuse. Ann. Ny. Acad. Sci 2010, 1187, 316–340. [DOI] [PubMed] [Google Scholar]
- 11.Fleckenstein AE; Volz TJ; Riddle EL; Gibb JW; Hanson GR New insights into the mechanism of action of amphetamines. Annu. Rev. Pharmacol 2007, 47, 681–698. [DOI] [PubMed] [Google Scholar]
- 12.Wood S; Sage JR; Shuman T; Anagnostaras SG Psychostimulants and cognition: A continuum of behavioral and cognitive activation. Pharmacol. Rev 2014, 66, 193–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Madras B; Kuhar MJ The Effects of Drug Abuse on the Human Nervous System First edition. ed.; Elsevier: Amsterdam, 2014; p xiii, 609 pages. [Google Scholar]
- 14.Agoston GE; Wu JH; Izenwasser S; George C; Katz J; Kline RH; Newman AH Novel N-substituted 3 alpha-[bis(4’-fluorophenyl)methoxy]tropane analogues: selective ligands for the dopamine transporter. J. Med. Chem 1997, 40, 4329–4339. [DOI] [PubMed] [Google Scholar]
- 15.Desai RI; Kopajtic TA; Koffarnus M; Newman AH; Katz JL Identification of a dopamine transporter ligand that blocks the stimulant effects of cocaine. J. Neurosci 2005, 25, 1889–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Desai RI; Grandy DK; Lupica CR; Katz JL Pharmacological characterization of a dopamine transporter ligand that functions as a cocaine antagonist. J. Pharmacol. Exp. Ther 2014, 348, 106–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Velazquez-Sanchez C; Ferragud A; Murga J; Carda M; Canales JJ The high affinity dopamine uptake inhibitor, JHW 007, blocks cocaine-induced reward, locomotor stimulation and sensitization. Eur. Neuropsychopharmacol 2010, 20, 501–508. [DOI] [PubMed] [Google Scholar]
- 18.Preti A New developments in the pharmacotherapy of cocaine abuse. Addict. Biol 2007, 12, 133–151. [DOI] [PubMed] [Google Scholar]
- 19.Vanderzee P; Koger HS; Gootjes J; Hespe W Aryl 1,4-dialk(en)ylpiperazines as selective and very potent inhibitors of dopamine uptake. Eur. J. Med. Chem 1980, 15, 363–370. [Google Scholar]
- 20.Rothman RB; Mele A; Reid AA; Akunne HC; Greig N; Thurkauf A; Decosta BR; Rice KC; Pert A GBR12909 antagonizes the ability of cocaine to elevate extracellular levels of dopamine. Pharmacol. Biochem. Be 1991, 40, 387–397. [DOI] [PubMed] [Google Scholar]
- 21.Obejero-Paz CA; Bruening-Wright A; Kramer J; Hawryluk P; Tatalovic M; Dittrich HC; Brown AM Quantitative profiling of the effects of vanoxerine on human cardiac ion channels and its application to cardiac risk. Sci. Rep 2015, 5, 17623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vocci FJ; Acri J; Elkashef A Medication development for addictive disorders: the state of the science. Am. J. Psychiatry 2005, 162, 1432–1440. [DOI] [PubMed] [Google Scholar]
- 23.Rothman RB; Baumann MH; Prisinzano TE; Newman AH Dopamine transport inhibitors based on GBR12909 and benztropine as potential medications to treat cocaine addiction. Biochem. Pharmacol 2008, 75, 2–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwartz JR Modafinil in the treatment of excessive sleepiness. Drug Des. Devel. Ther 2009, 2, 71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mereu M; Bonci A; Newman AH; Tanda G The neurobiology of modafinil as an enhancer of cognitive performance and a potential treatment for substance use disorders. Psychopharmacology 2013, 229, 415–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reith ME; Blough BE; Hong WC; Jones KT; Schmitt KC; Baumann MH; Partilla JS; Rothman RB; Katz JL Behavioral, biological, and chemical perspectives on atypical agents targeting the dopamine transporter. Drug Alcohol Depend 2015, 147, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loland CJ; Mereu M; Okunola OM; Cao JJ; Prisinzano TE; Mazier S; Kopajtic T; Shi L; Katz JL; Tanda G; Newman AH R-Modafinil (Armodafinil): A unique dopamine uptake inhibitor and potential medication for psychostimulant abuse. Biol. Psychiat 2012, 72, 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dackis CA; Kampman KM; Lynch KG; Plebani JG; Pettinati HM; Sparkman T; O’Brien CP A double-blind, placebo-controlled trial of modafinil for cocaine dependence. J. Subst. Abuse. Treat 2012, 43, 303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anderson AL; Reid MS; Li SH; Holmes T; Shemanski L; Slee A; Smith EV; Kahn R; Chiang N; Vocci F; Ciraulo D; Dackis C; Roache JD; Salloum IM; Somoza E; Urschel HC 3rd; Elkashef AM Modafinil for the treatment of cocaine dependence. Drug Alcohol Depend 2009, 104, 133–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cao J; Prisinzano TE; Okunola OM; Kopajtic T; Shook M; Katz JL; Newman AH Structure-activity relationships at the monoamine transporters for a novel series of modafinil (2-[(diphenylmethyl)sulfinyl]acetamide) analogues. ACS Med. Chem. Lett 2010, 2, 48–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okunola-Bakare OM; Cao JJ; Kopajtic T; Katz JL; Loland CJ; Shi L; Newman AH Elucidation of Structural Elements for Selectivity across Monoamine Transporters: Novel 2-[(Diphenylmethyl)sulfinyl]acetamide (Modafinil) Analogues. J. Med. Chem 2014, 57, 1000–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cao J; Slack RD; Bakare OM; Burzynski C; Rais R; Slusher BS; Kopajtic T; Bonifazi A; Ellenberger MP; Yano H; He Y; Bi GH; Xi ZX; Loland CJ; Newman AH Novel and high affinity 2-[(diphenylmethyl)sulfinyl]acetamide (modafinil) analogues as atypical aopamine transporter inhibitors. J. Med. Chem 2016, 59, 10676–10691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kalaba P; Aher NY; Ilic M; Dragacevic V; Wieder M; Miklosi AG; Zehl M; Wackerlig J; Roller A; Beryozkina T; Radoman B; Saroja SR; Lindner W; Gonzalez EP; Bakulev V; Leban JJ; Sitte HH; Urban E; Langer T; Lubec G Heterocyclic analogues of modafinil as novel, atypical dopamine transporter inhibitors. J. Med. Chem 2017, 60, 9330–9348. [DOI] [PubMed] [Google Scholar]
- 34.Kristofova M; Aher YD; Ilic M; Radoman B; Kalaba P; Dragacevic V; Aher NY; Leban J; Korz V; Zanon L; Neuhau W; Wieder M; Langer T; Urban E; Sitte HH; Hoeger H; Lubec G; Aradska J A daily single dose of a novel modafinil analogue CE-123 improves memory acquisition and memory retrieval. Behav. Brain Res 2018, 343, 83–94. [DOI] [PubMed] [Google Scholar]
- 35.Tanda G; Newman AH; Katz JL Discovery of drugs to treat cocaine dependence: behavioral and neurochemical effects of atypical dopamine transport inhibitors. Adv Pharmacol 2009, 57, 253–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Loland CJ; Desai RI; Zou MF; Cao J; Grundt P; Gerstbrein K; Sitte HH; Newman AH; Katz JL; Gether U Relationship between conformational changes in the dopamine transporter and cocaine-like subjective effects of uptake inhibitors. Mol. Pharmacol 2008, 73, 813–823. [DOI] [PubMed] [Google Scholar]
- 37.Tunstall BJ; Ho CP; Cao JJ; Vendruscolo JCM; Schmeichel BE; Slack RD; Tanda G; Gadiano AJ; Rais R; Slusher BS; Koob GF; Newman AH; Vendruscolo LF Atypical dopamine transporter inhibitors attenuate compulsive-like methamphetamine self-administration in rats. Neuropharmacology 2018, 131, 96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Newman AH; Cao J; Keighron JD; Jordan CJ; Bi GH; Liang Y; Abramyan AM; Avelar AJ; Tschumi CW; Beckstead MJ; Shi L; Tanda G; Xi ZX Translating the atypical dopamine uptake inhibitor hypothesis toward therapeutics for treatment of psychostimulant use disorders. Neuropsychopharmacol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bisgaard H; Larsen MAB; Mazier S; Beuming T; Newman AH; Weinstein H; Shi L; Loland CJ; Gether U The binding sites for benztropines and dopamine in the dopamine transporter overlap. Neuropharmacology 2011, 60, 182–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zou MF; Cao JJ; Abramyan AM; Kopajtic T; Zanettini C; Guthrie DA; Rais R; Slusher BS; Shi L; Loland CJ; Newman AH Structure-activity relationship studies on a series of 3 alpha-[bis(4-fluorophenyl)methoxy]tropanes and 3 alpha-[bis(4-fluorophenyl)methylamino]tropanes as novel atypical dopamine transporter (DAT) inhibitors for the treatment of cocaine use disorders. J. Med. Chem 2017, 60, 10172–10187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abramyan AM; Stolzenberg S; Li Z; Loland CJ; Noe F; Shi L The isomeric preference of an atypical dopamine transporter inhibitor contributes to its selection of the transporter conformation. ACS Chem. Neurosci, 2017, 8, 1735–1746. [DOI] [PubMed] [Google Scholar]
- 42.Razavi AM; Khelashvili G; Weinstein H A Markov state-based quantitative kinetic model of sodium release from the dopamine transporter. Sci. Rep, 2017, 7, 40076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheng MH; Kaya C; Bahar I Quantitative assessment of the energetics of dopamine translocation by human dopamine transporter. J. Phys. Chem. B 2018, 122, 5336–5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hiranita T; Soto PL; Kohut SJ; Kopajtic T; Cao JJ; Newman AH; Tanda G; Katz JL Decreases in cocaine self-administration with dual inhibition of the dopamine transporter and Sigma receptors. J. Pharmacol. Exp. Ther 2011, 339, 662–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hiranita T; Hong WC; Kopajtic T; Katz JL Sigma receptor effects of N-substituted benztropine analogs: implications for antagonism of cocaine self-administration. J. Pharmacol. Exp. Ther 2017, 362, 2–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hong WC; Yano H; Hiranita T; Chin FT; McCurdy CR; Su TP; Amara SG; Katz JL The Sigma-1 receptor modulates dopamine transporter conformation and cocaine binding and may thereby potentiate cocaine self-administration in rats. J. Biol. Chem 2017, 292, 11250–11261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hiranita T; Kohut SJ; Soto PL; Tanda G; Kopajtic TA; Katz JL Preclinical efficacy of N-substituted benztropine analogs as antagonists of methamphetamine self-administration in rats. J. Pharmacol. Exp. Ther 2014, 348, 174–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hedges DM; Obray JD; Yorgason JT; Jang EY; Weerasekara VK; Uys JD; Bellinger FP; Steffensen SC Methamphetamine induces dopamine release in the nucleus accumbens through a Sigma receptor-mediated pathway. Neuropsychopharmacol 2018, 43, 1405–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sambo DO; Lin M; Owens A; Lebowitz JJ; Richardson B; Jagnarine DA; Shetty M; Rodriquez M; Alonge T; Ali M; Katz J; Yan L; Febo M; Henry LK; Bruijnzeel AW; Daws L; Khoshbouei H The sigma-1 receptor modulates methamphetamine dysregulation of dopamine neurotransmission. Nat. Commun 2017, 8, 2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jamieson C; Moir EM; Rankovic Z; Wishart G Medicinal chemistry of hERG optimizations: Highlights and hang-ups. J. Med. Chem 2006, 49, 5029–5046. [DOI] [PubMed] [Google Scholar]
- 51.Perrin MJ; Kuchel PW; Campbell TJ; Vandenberg JI Drug binding to the inactivated state is necessary but not sufficient for high-affinity binding to human ether-a-go-go-related gene channels. Mol. Pharmacol 2008, 74, 1443–1452. [DOI] [PubMed] [Google Scholar]
- 52.Gintant G; Sager PT; Stockbridge N Evolution of strategies to improve preclinical cardiac safety testing. Nat. Rev. Drug. Discov 2016, 15, 457–471. [DOI] [PubMed] [Google Scholar]
- 53.Kalyaanamoorthy S; Barakat KH Binding modes of hERG blockers: an unsolved mystery in the drug design arena. Expert. Opin. Drug. Discov 2018, 13, 207–210. [DOI] [PubMed] [Google Scholar]
- 54.Fant AD; Wacker S; Jung J; Guo JQ; Abramyan AM; Duff HJ; Newman AH; Noskov SY; Shi L Toward reducing hERG affinities for DAT inhibitors with a combined machine learning and molecular modeling approach. Biophys. J 2019, 116, 562a–562a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Johannesen L; Vicente J; Mason JW; Erato C; Sanabria C; Waite-Labott K; Hong M; Lin J; Guo P; Mutlib A; Wang J; Crumb WJ; Blinova K; Chan D; Stohlman J; Florian J; Ugander M; Stockbridge N; Strauss DG Late sodium current block for drug-induced long QT syndrome: Results from a prospective clinical trial. Clin. Pharmacol. Ther 2016, 99, 214–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bolleddula J; DeMent K; Driscoll JP; Worboys P; Brassil PJ; Bourdet DL Biotransformation and bioactivation reactions of alicyclic amines in drug molecules. Drug. Metab. Rev 2014, 46, 379–419. [DOI] [PubMed] [Google Scholar]
- 57.Reilly SW; Griffin S; Taylor M; Sahlholm K; Weng CC; Xu K; Jacome DA; Luedtke RR; Mach RH Highly selective dopamine D3 receptor antagonists with arylated diazaspiro alkane cores. J. Med. Chem 2017, 60, 9905–9910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reilly SW; Riad AA; Hsieh CJ; Sahlholm K; Jacome DA; Griffin S; Taylor M; Weng CC; Xu K; Kirschner N; Luedtke RR; Parry C; Malhotra S; Karanicolas J; Mach RH Leveraging a low-affinity diazaspiro orthosteric fragment to reduce dopamine D3 receptor (D3R) ligand promiscuity across highly conserved aminergic G-protein-coupled receptors (GPCRs). J. Med. Chem 2019. [DOI] [PubMed] [Google Scholar]
- 59.Husbands SM; Izenwasser S; Kopajtic T; Bowen WD; Vilner BJ; Katz JL; Newman AH Structure-activity relationships at the monoamine transporters and sigma receptors for a novel series of 9-[3-(cis-3, 5-dimethyl-1-piperazinyl)propyl]carbazole (rimcazole) analogues. J. Med. Chem 1999, 42, 4446–4455. [DOI] [PubMed] [Google Scholar]
- 60.Husbands SM; Izenwasser S; Loeloff RJ; Katz JL; Bowen WD; Vilner BJ; Newman AH Isothiocyanate derivatives of 9-[3-(cis-3,5-dimethyl-1-piperazinyl)propyl]-carbazole (rimcazole): Irreversible ligands for the dopamine transporter. J. Med. Chem 1997, 40, 4340–4346. [DOI] [PubMed] [Google Scholar]
- 61.Cao JJ; Kulkarni SS; Husbands SM; Bowen WD; Williams W; Kopajtic T; Katz JL; George C; Newman AH Dual probes for the dopamine transporter and sigma(1) receptors: novel piperazinyl alkyl-bis(4 ‘-fluorophenyl)amine analogues as potential cocaine-abuse therapeutic agents. J. Med. Chem 2003, 46, 2589–2598. [DOI] [PubMed] [Google Scholar]
- 62.Cao JJ; Kopajtic T; Katz JL; Newman AH Dual DAT/sigma 1 receptor ligands based on 3-(4-(3-(bis(4-fluorophenyl) amino) propyl) piperazin-1-yl)-1-phenylpropan-1-ol. Bioorg. Med. Chem. Lett 2008, 18, 5238–5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Katz JL; Hong WC; Hiranita T; Su TP A role for sigma receptors in stimulant self-administration and addiction. Behav. Pharmacol 2016, 27, 100–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Redfern WS; Carlsson L; Davis AS; Lynch WG; MacKenzie I; Palethorpe S; Siegl PK; Strang I; Sullivan AT; Wallis R; Camm AJ; Hammond TG Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc. Res 2003, 58, 32–45. [DOI] [PubMed] [Google Scholar]
- 65.Beuming T; Kniazeff J; Bergmann ML; Shi L; Gracia L; Raniszewska K; Newman AH; Javitch JA; Weinstein H; Gether U; Loland CJ The binding sites for cocaine and dopamine in the dopamine transporter overlap. Nat. Neurosci 2008, 11, 780–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang KH; Penmatsa A; Gouaux E Neurotransmitter and psychostimulant recognition by the dopamine transporter. Nature 2015, 521, 322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rais R; Thomas AG; Wozniak K; Wu Y; Jaaro-Peled H; Sawa A; Strick CA; Engle SJ; Brandon NJ; Rojas C; Slusher BS; Tsukamoto T Pharmacokinetics of oral D-serine in D-amino acid oxidase knockout mice. Drug Metab. Dispos 2012, 40, 2067–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Keighron JD; Quarterman JC; Cao J; DeMarco EM; Coggiano MA; Gleaves A; Slack RD; Zanettini C; Newman AH; Tanda G Effects of (R)-modafinil and modafinil analogues on dopamine dynamics assessed by voltammetry and microdialysis in the mouse nucleus accumbens shell. ACS Chem. Neurosci 2019, 10, 2012–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Desai RI; Kopajtic TA; French D; Newman AH; Katz JL Relationship between in vivo occupancy at the dopamine transporter and behavioral effects of cocaine, GBR 12909 [1-{2-[bis-(4-fluorophenyl) methoxy]ethyl}−4-(3-phenylpropyl)piperazine], and benztropine analogs. J. Pharmacol. Exp. Ther 2005, 315, 397–404. [DOI] [PubMed] [Google Scholar]
- 70.Schmeichel BE; Zemlan FP; Berridge CW A selective dopamine reuptake inhibitor improves prefrontal cortex-dependent cognitive function: potential relevance to attention deficit hyperactivity disorder. Neuropharmacology 2013, 64, 321–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Newman AH; Allen AC; Izenwasser S; Katz JL Novel 3-alpha-(diphenylmethoxy) tropane analogs - Potent dopamine uptake inhibitors without cocaine-like behavioral profiles. J. Med. Chem 1994, 37, 2258–2261. [DOI] [PubMed] [Google Scholar]
- 72.Katz JL; Izenwasser S; Kline RH; Allen AC; Newman AH Novel 3 alpha-diphenylmethoxytropane analogs: Selective dopamine uptake inhibitors with behavioral effects distinct from those of cocaine. J. Pharmacol. Exp. Ther 1999, 288, 302–315. [PubMed] [Google Scholar]
- 73.Hiranita T; Wilkinson DS; Hong WMC; Zou MF; Kopajtic TA; Soto PL; Lupica CR; Newman AH; Katz JL 2-Isoxazol-3-phenyltropane derivatives of cocaine: molecular and atypical system effects at the dopamine transporter. J. Pharmacol. Exp. Ther 2014, 349, 297–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hong WMC; Kopajtic TA; Xu LF; Lomenzo SA; Jean B; Madura JD; Surratt CK; Trudell ML; Katz JL 2-Substituted 3 beta-aryltropane cocaine analogs produce atypical effects without inducing inward-facing dopamine transporter conformations. J. Pharmacol. Exp. Ther 2016, 356, 624–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Scheffel U; Boja JW; Kuhar MJ Cocaine receptors - In vivo labeling with H-3 (−)cocaine, H-3 Win-35,065–2, and H-3 Win-35,428. Synapse 1989, 4, 390–392. [DOI] [PubMed] [Google Scholar]
- 76.Hiranita T; Soto PL; Tanda G; Kopajtic TA; Katz JL Stimulants as specific inducers of dopamine-independent Sigma agonist self-administration in rats. J. Pharmacol. Exp. Ther 2013, 347, 20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cheng Y; Prusoff WH Relationship between inhibition constant (K1) and concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic-reaction. Biochemical Pharmacology 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- 78.Pedersen AV; Andreassen TF; Loland CJ A conserved salt bridge between transmembrane segments 1 and 10 constitutes an extracellular gate in the dopamine transporter. J. Biol. Chem 2014, 289, 35003–35014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.