

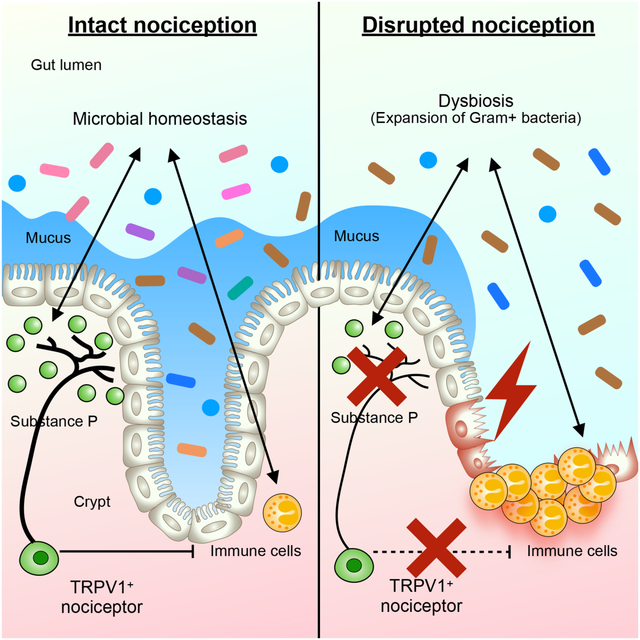
SUMMARY
Nociceptive pain is a hallmark of many chronic inflammatory conditions including inflammatory bowel diseases (IBD), however whether pain-sensing neurons influence intestinal inflammation remains poorly defined. Employing chemogenetic silencing, adenoviral-mediated colon-specific silencing and pharmacological ablation of TRPV1+ nociceptors, we observed more severe inflammation and defective tissue-protective reparative processes in a murine model of intestinal damage and inflammation. Disrupted nociception led to significant alterations in the intestinal microbiota and a transmissible dysbiosis, while mono-colonization of germ-free mice with Gram+ Clostridium spp. promoted intestinal tissue protection through a nociceptor-dependent pathway. Mechanistically, disruption of nociception resulted in decreased levels of substance P, and therapeutic delivery of substance P promoted tissue-protective effects exerted by TRPV1+ nociceptors in a microbiota-dependent manner. Finally, dysregulated nociceptor gene expression was observed in intestinal biopsies from IBD patients. Collectively, these findings indicate an evolutionarily conserved functional link between nociception, the intestinal microbiota, and the restoration of intestinal homeostasis.
Graphical Abstract
INTRODUCTION
Pain is an evolutionarily conserved sensory and emotional response that can serve as an early warning signal to protect the body from tissue damage or injury (Baral et al., 2019; Basbaum et al., 2009; Chu et al., 2020; Huh and Veiga-Fernandes, 2020; Scholz and Woolf, 2002; Veiga-Fernandes and Mucida, 2016; Yoo and Mazmanian, 2017). However, chronic pain produces severe distress and impairs the quality of life of an estimated 50 million patients in the US who are diagnosed with chronic inflammatory diseases including rheumatoid arthritis, multiple sclerosis, and inflammatory bowel diseases (IBD), with the estimated cost of treating chronic pain exceeding $600 billion annually (Dahlhamer et al., 2018; Ng et al., 2017).
IBD represents a group of chronic inflammatory diseases characterized by ongoing inflammation of all or part of the gastrointestinal tract (Abraham and Cho, 2009; Baumgart and Sandborn, 2012; Chang, 2020; Danese and Fiocchi, 2011; Friedrich et al., 2019; Neurath, 2014; Torres et al., 2017; Uhlig and Powrie, 2018; Ungaro et al., 2017). Abdominal pain is a common symptom of IBD, which is thought to arise from different mechanisms including gut distention, intestinal inflammation and microbial dysbiosis (Baral et al., 2019; Bielefeldt et al., 2009). Pain-sensing neurons, that innervate peripheral tissues, including the skin, lung, and gastrointestinal tract, transduce environmental signals including thermal stimulation, noxious stimuli and mechanical damage to the central nervous system and are indispensable for effective pathogen defense and tissue homeostasis (Baral et al., 2019; Basbaum et al., 2009; Chu et al., 2020; Foster et al., 2017).
Nociceptors are a specialized subset of sensory neurons expressing transient receptor vanilloid 1 (TRPV1), a non-selective cation channel that can be activated by a wide variety of stimuli including heat, capsaicin and inflammatory mediators (Basbaum et al., 2009; Scholz and Woolf, 2002; Veiga-Fernandes and Mucida, 2016; Woolf and Ma, 2007; Yoo and Mazmanian, 2017). Upon activation, the nerve terminals of nociceptors release calcitonin gene-related peptide (CGRP, encoded by Calca), substance P (encoded by Tac1) and other neuropeptides, which either amplify or inhibit the downstream inflammatory cascade (Chu et al., 2020; Mashaghi et al., 2016; Yano and Artis, 2022). Despite emerging advances in understanding neuro-immune interactions, it remains unclear whether nociceptors play a role in regulating the development, severity, and/or resolution of chronic inflammatory diseases such as IBD.
Here we visualized TRPV1+ nociceptor innervation in mouse intestines during homeostasis and inflammation. Targeted chemogenetic silencing, adenoviral-mediated colon-specific silencing or pharmacological ablation of TRPV1+ nociceptors resulted in enhanced susceptibility in a murine model of intestinal damage and inflammation, indicating that gut-innervating TRPV1+ nociceptors are tissue-protective in these contexts. Transient silencing or permanent ablation of TRPV1+ nociceptors resulted in alterations in the composition of the intestinal microbiota, and transplantation of the microbiota from mice with dysregulated nociception resulted in exacerbated intestinal damage and inflammation in wild-type recipient mice. Disruption of TRPV1+ nociceptor-mediated tissue-protective functions were associated with alterations in Gram+ bacteria and selective colonization of nociceptor-sufficient, but not nociceptor-deficient, germ-free mice with a consortium of Gram+ Clostridium species promoted tissue protection. Critically, levels of nociceptor-derived substance P were reduced upon chemogenetic silencing or pharmacological ablation of TRPV1+ nociceptors and therapeutic delivery of substance P limited severe inflammation in mice with disrupted nociception. Lastly, we detected dysregulated patterns of TRPV1+ nociceptor innervation and altered expression of nociceptor-associated genes including TRPV1 and TAC1 in intestinal biopsies from IBD patients compared to healthy controls, indicating dysregulation of nociception is an evolutionarily conserved feature of chronic intestinal inflammation. Taken together, these results highlight the role of gut-innervating nociceptors in regulating the composition of the microbiota required to limit inflammation and promote intestinal tissue protection.
RESULTS
Chemogenetic silencing of TRPV1+ nociceptors results in impaired tissue protection in a murine model of intestinal damage and inflammation
The gastrointestinal tract is densely innervated by both intrinsic and extrinsic neurons including components of the sensory nervous system (Veiga-Fernandes and Mucida, 2016; Zeisel et al., 2018). To assess whether TRPV1+ nociceptors innervate the mouse colon, we first crossed Trpv1-Cre mice with a cell lineage reporter mouse line tdTomatofl/stop/fl to examine the distribution of TRPV1+ nociceptors in naïve and inflamed mouse colons. TRPV1-tdTomato, in conjunction with a pan-neuronal marker βIII-tubulin, marked TRPV1+ nociceptor innervation in the crypts and muscularis in the colon at steady state as well as following exposure to dextran sodium sulphate (DSS), a murine model of intestinal damage and inflammation (Monticelli et al., 2015; Troy et al., 2009; Wirtz et al., 2017) (Figure 1A and 1B, yellow arrows).
Figure 1. Chemogenetic silencing of TRPV1+ nociceptors lead to enhanced susceptibility to DSS-induced colitis.
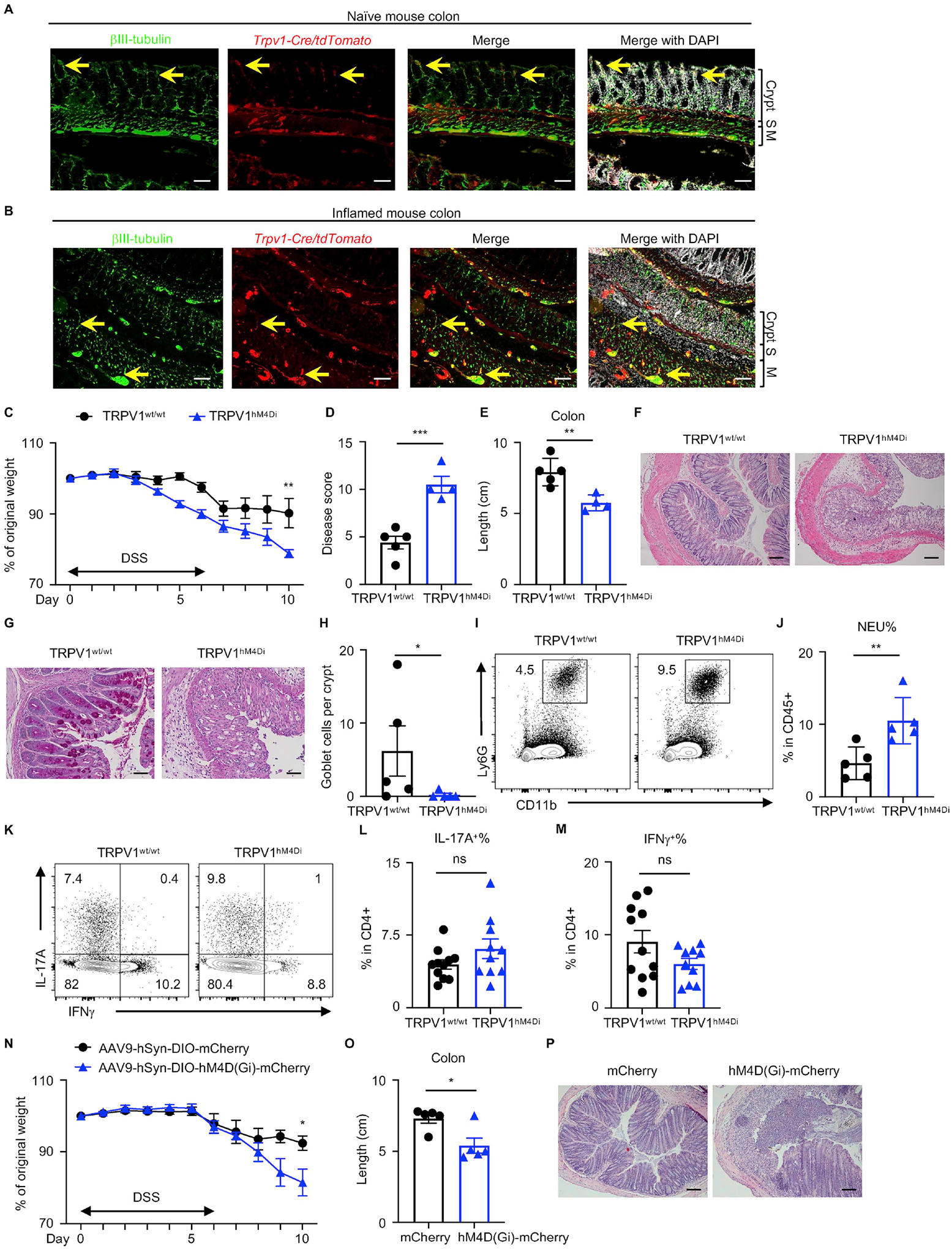
(A, B) Naïve and inflamed mouse colons from Trpv1-Cre/tdTomato reporter mice, stained for βIII-tubulin (green) and DAPI (white). S, submucosa; M, muscularis. Yellow arrows show representative TRPV1+ nociceptor innervation. (C to H) Disease and recovery of DSS-treated TRPV1hM4Di and their littermate control mice were monitored by daily weight loss (C), clinical disease score (D), colon length (E) and H&E staining of the distal colon on day 10 (F). (G, H) PAS staining of the distal colon and analysis of PAS+ goblet cells per crypt. (I, J) Frequency of colonic neutrophils from mice in (C) on day 10, pre-gated on live, CD45+ events. (K to M) Flow cytometric analysis of T cell cytokine production from mice in (C) on day 10, pre-gated on live, CD45+ CD4+ events. (N to P) Disease and recovery of DSS-treated Trpv1-Cre mice infected with AAV9 viruses were monitored by daily weight loss (N), colon length (O) and H&E staining of the distal colon on day 10 (P). Scale bars = 50 μm in (A), (B), (F) and (P), and 100 μm in (G). Data are representative of three independent experiments with n=4 to 5 mice per group in (C to J, N to Q). Data are pooled from three independent experiments with n=4–5 mice per group in each experiment in (L) and (M). Data are Mean ± SEM. ns, not significant, * P < 0.05, ** P < 0.01, *** P < 0.001.
To interrogate the potential functional impact of gut-innervating TRPV1+ nociceptors on intestinal damage and inflammation, we employed a chemogenetic approach to acutely silence TRPV1+ nociceptors in vivo by generating TRPV1hM4Di mice in which TRPV1+ nociceptors express the inhibitory DREADD (Designer Receptors Exclusively Activated by Designer Drugs) human M4 muscarinic receptor (hM4Di) (Ferguson et al., 2011; Urban and Roth, 2015) (Figure S1A). Expression of hM4Di was detected by immunofluorescence microscopy in the dorsal root ganglia (DRG, where the cell bodies of TRPV1+ nociceptors innervating the intestine are located (Riol-Blanco et al., 2014; Woolf and Ma, 2007)) of TRPV1hM4Di but not littermate control TRPV1wt/wt mice (Figure S1B, yellow arrows). Delivery of the inactive clozapine analog clozapine-N-oxide (CNO), a synthetic ligand for the engineered receptor hM4Di, resulted in transient silencing of TRPV1+ nociceptors as indicated by significantly reduced phosphorylated mitogen-activated protein kinase 3/1 immunoreactive neurons (Figure S1C and S1D), which can be induced in DRG neurons by intense stimulation or under chronic pathological conditions (Guo et al., 2020). The DREADD-mediated acute silencing of TRPV1+ nociceptors did not affect baseline body mass (Figure S1E), food and water intake (Figure S1F and S1G) or total gastrointestinal transit time following DSS administration (Figure S1H). However, DSS-exposed TRPV1hM4Di mice exhibited significantly more body weight loss (Figure 1C), a significant increase in clinical disease score including parameters of general morbidity, rectal bleeding, and diarrhea (Figure 1D), and significantly reduced colon length (Figure 1E) compared to littermate control TRPV1wt/wt mice. Further, when littermate control mice began recovering from DSS-induced intestinal damage, TRPV1hM4Di mice exhibited increased inflammatory cell infiltration, intestinal edema, the loss of epithelial crypt structure (Figure 1F) and significantly reduced numbers of goblet cells in the distal colon (Figure 1G and 1H), indicative of defective tissue repair machinery following acute silencing of TRPV1+ nociceptors.
We next analyzed intestinal immune cell responses upon TRPV1+ nociceptor silencing during DSS-induced colitis. TRPV1+ nociceptor-silenced mice exhibited a significant increase in neutrophil infiltration in the colon compared to control littermates, suggesting more severe intestinal inflammation (Figure 1I and 1J). However, the percentages of colonic IL-17A- or IFNγ-producing CD4+ T cells (Figure 1K–M) and regulatory T cells (Treg cells) (Figure S1I–K) were comparable between TRPV1hM4Di and littermate control TRPV1wt/wt mice. Taken together, these data suggest that TRPV1+ nociceptors play a critical role in regulating tissue damage, inflammation and repair in the intestine, without substantially impacting the adaptive immune response in this setting.
To specifically manipulate TRPV1+ nociceptors in the intestine, we injected a Cre-dependent AAV9 virus carrying the inhibitory DREADD into the distal colons of Trpv1-Cre mice and treated these mice with CNO to acutely silence colon-innervating TRPV1+ nociceptors following DSS administration (Figure S1L) (Gore et al., 2019; Skorput et al., 2022). Expression of mCherry was detected by immunofluorescence microscopy in the lumbar DRG (L5/L6/S1) (Figure S1M), with significantly more mCherry+ neurons labeled in the lumbar DRG compared to the nodose vagal ganglion (Figure S1N and S1O), which is consistent with previous findings that vagal neurons predominantly innervate the proximal colon whereas DRG neurons mainly innervate the jejunum and distal colon in mice (Guo et al., 2019b; Kyloh et al., 2011; Meerschaert et al., 2020; Tan et al., 2008). Compared to Trpv1-Cre mice with distal colonic injection of control virus (AAV9-hSyn-DIO-mCherry), Trpv1-Cre mice injected with AAV9 virus carrying the inhibitory DREADD (AAV9-hSyn-DIO-hM4D(Gi)-mCherry) exhibited significantly more weight loss (Figure 1N), significantly increased colon shortening (Figure 1O), and significantly more crypt loss in the distal colon (Figure 1P). Together, these data suggest that gut-innervating TRPV1+ nociceptors are critical in mediating tissue protection during intestinal damage and inflammation.
In addition to loss-of-function chemogenetic approaches, we sought to investigate whether activation of TRPV1+ nociceptors impacted DSS-induced intestinal damage and inflammation. We employed a gain-of-function chemogenetic approach to activate TRPV1+ nociceptors in vivo by generating TRPV1hM3Dq mice in which TRPV1+ nociceptors expressed the excitatory human M3 muscarinic receptor (hM3Dq) (Ferguson et al., 2011). Chemogenetic activation of TRPV1+ nociceptors following DSS administration resulted in significantly less body weight loss (Figure S1P). There were significantly decreased disease activity (Figure S1Q), less colon shortening (Figure S1R), improved histopathology of the distal colon (Figure S1S) and reduced neutrophil infiltration in the colon (Figure S1T) upon chemogenetic activation of TRPV1+ nociceptors following DSS administration, further supporting a tissue-protective role exerted by TRPV1+ nociceptors in this mouse model of intestinal inflammation.
Pharmacological ablation of TRPV1+ nociceptors exacerbates DSS-induced colitis
We employed a complementary pharmacological strategy to permanently ablate TRPV1+ nociceptors in adult mice in vivo. Subcutaneous injections of resiniferatoxin (RTX), a potent TRPV1 channel agonist, result in systemic ablation of TRPV1+ nociceptors (Elekes et al., 2007; Mishra and Hoon, 2010) (Figure S2A), allowing us to investigate the influence of nociceptors on intestinal damage, inflammation and repair in diverse mouse models and experimental settings. RTX-mediated TRPV1+ nociceptor ablation in wild-type B6 mice was confirmed by diminished expression of nociceptor marker gene including Trpv1, Tac1 and Scn10a in the DRG (Figure 2A). Importantly, RTX treatment resulted in significantly fewer TRPV1+ neurons in the DRG (Figure 2B and 2C), suggesting successful TRPV1+ nociceptor ablation.
Figure 2. TRPV1+ nociceptor ablation exacerbates DSS-induced colitis.
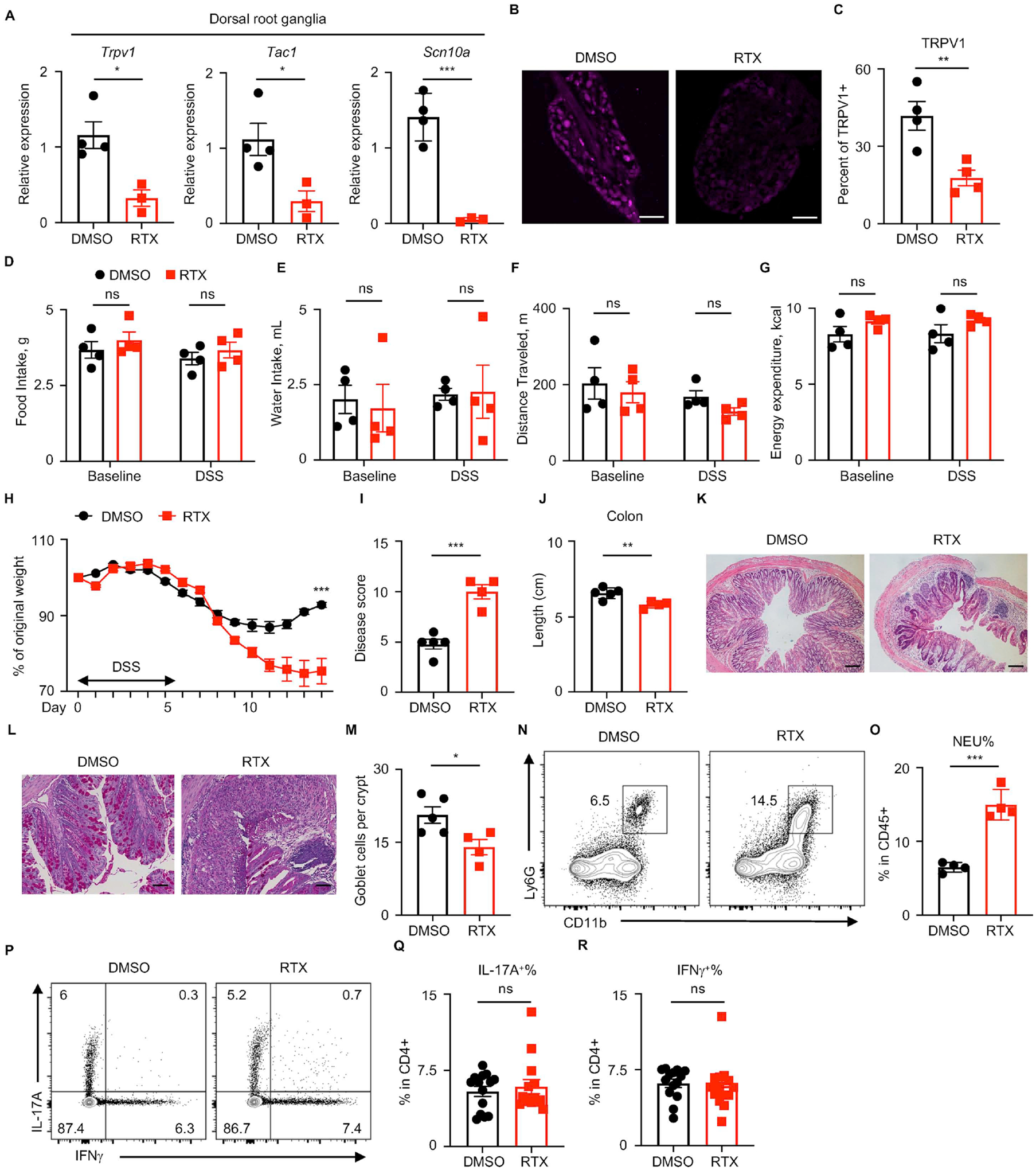
(A) DRG nociceptor marker gene expression was assessed in DRGs isolated from DMSO- or RTX-treated B6 mice at steady state. (B, C) Immunofluorescence microscopy and quantification of TRPV1+ DRGs isolated from DMSO- or RTX-treated B6 mice at day 10 post DSS administration. (D to G) The Promethion Metabolic Cage System was used to measure daily food (D) and water intake (E), locomotion (F) and energy expenditure of each mouse (G) treated with DMSO or RTX. (H to M) Disease and recovery of DSS-treated DMSO- and RTX-treated B6 mice were monitored by daily weight loss (H), clinical disease score (I), colon length (J) and H&E staining of the distal colon (K) at day 14. (L, M) PAS staining of the distal colon and analysis of PAS+ goblet cells per crypt. (N, O) Frequency of colonic neutrophils from mice in (H) on day 10, pre-gated on live, CD45+ events. (P to R) Flow cytometric analysis of T cell cytokine production from mice in (H) on day 10, pre-gated on live, CD45+ CD4+ events. Scale bars = 200 μm in (B) and 50 μm in (K) and 100 μm in (L). Data are representative of three independent experiments with n=3 to 5 mice per group in (A to O). Data are pooled from three independent experiments with n=4–5 mice per group in each experiment in (P to R). Data are Mean ± SEM. ns, not significant, * P < 0.05, ** P < 0.01, *** P < 0.001.
To examine the consequences of nociceptor ablation under conditions of intestinal damage and inflammation, DMSO- and RTX-treated wild-type mice were exposed to DSS, and parameters of clinical disease were monitored (Figure S2A). Notably, RTX-mediated ablation of TRPV1+ nociceptors did not affect baseline body mass (Figure S2B), baseline total gastrointestinal transit time (Figure S2C), food and water intake (Figure 2D and 2E), locomotion (Figure 2F), energy expenditure (Figure 2G). In comparison to DMSO-treated mice, RTX-treated mice exhibited overall heightened disease activity as reflected by significantly more weight loss (Figure 2H), a significant increase in clinical disease score (Figure 2I) and significantly reduced colon length (Figure 2J). Furthermore, histological examination of the distal colon tissue revealed that RTX-treated mice, similar to TRPV1hM4Di mice, exhibited more severe histopathological changes within the intestinal mucosa including prominent tissue ulceration, edema, increased inflammatory cell infiltration (Figure 2K) and significantly reduced numbers of goblet cells (Figure 2L and 2M) relative to DMSO-treated mice.
Next, we sought to investigate whether immune responses were influenced by RTX-mediated ablation of nociceptors. At steady state, there were no significant changes in most immune cell populations in the colon lamina propria upon RTX-mediated nociceptor ablation (Figure S2D–L). However, consistent with chemogenetic silencing of TRPV1+ nociceptors, following exposure to DSS, nociceptor-ablated mice exhibited a significant increase in colonic neutrophil frequency relative to control mice, a hallmark of more severe intestinal inflammation (Figure 2N and 2O). The percentages of colonic IL-17A- and IFNγ-producing CD4+ T cells were comparable (Figure 2P–R) and there were no significant differences in the percentages of colonic Treg cells (Figure S2M–O) between DMSO- and RTX-treated mice.
Since TRPV1+ nociceptors innervating the mouse intestine are either from DRG or nodose ganglion (Chavan et al., 2017), we next determined whether DRG nociceptors contributed to tissue protection following DSS-induced colitis using intrathecal injection of DMSO or RTX. Following DSS administration, mice ablated of TRPV1+ DRG nociceptors exhibited overall increased disease severity as reflected by significantly more weight loss (Figure S2P), a significant increase in clinical disease score (Figure S2Q), and more severe histopathology in the distal colon (Figure S2R). Collectively, by using both transient chemogenetic silencing and permanent pharmacological ablation of TRPV1+ nociceptors, we identify that gut-innervating TRPV1+ nociceptors are necessary to limit inflammation and promote tissue repair in the context of intestinal damage.
Disruption of TRPV1+ nociceptors results in a transmissible microbial dysbiosis
The intestinal microbiota interact with the immune system and the nervous system and participate in the regulation of multiple inflammatory disorders including asthma and IBD as well as neurological disorders such as neurodegenerative diseases and chronic pain (Chu et al., 2019; Fung et al., 2017; Hooper et al., 2012; Lu and Stappenbeck, 2022; Ni et al., 2017; Veiga-Fernandes and Mucida, 2016; Vuong et al., 2017). To investigate whether there is crosstalk between the intestinal microbiota and TRPV1+ nociceptors that influences the maintenance of intestinal homeostasis, we first analyzed bacterial communities by 16S ribosomal RNA (rRNA) gene sequencing from fecal DNA samples isolated from DMSO- and RTX-treated mice at steady state. Principal coordinates analysis (PCoA) demonstrated that samples clustered largely by treatment (Figure 3A), indicating significant changes in the composition of the intestinal microbiota in RTX-treated mice compared to DMSO-treated mice. Specifically, the relative abundance of Bacteroidetes spp. was significantly decreased while that of Firmicutes spp. was increased in RTX-treated mice (Figure 3B and 3C), indicating a potential role of TRPV1+ nociceptors play in regulating the composition of the microbiota.
Figure 3. TRPV1+ nociceptor ablation leads to a transmissible dysbiosis.
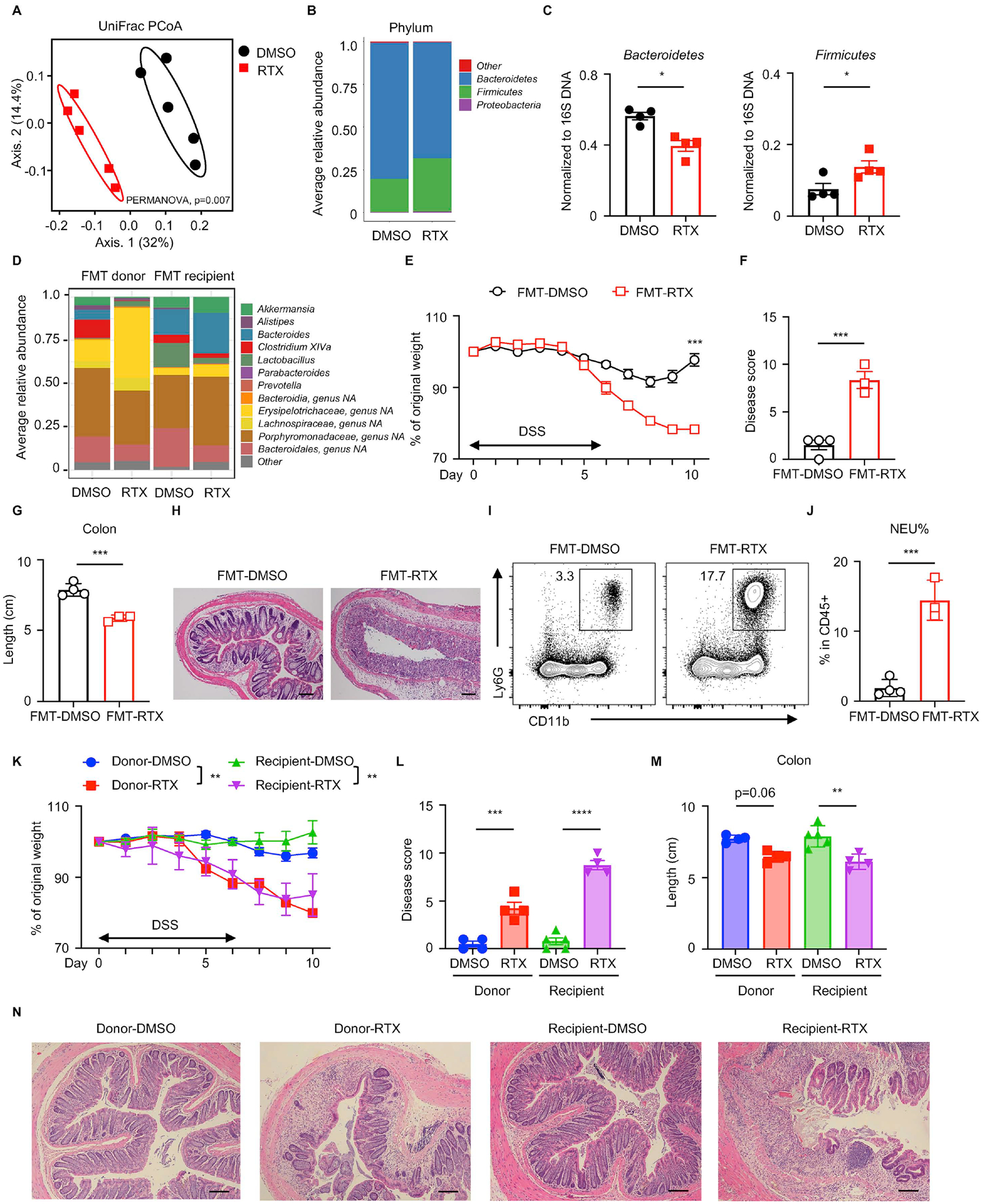
(A) Fecal microbial composition was analyzed by 16S rRNA gene sequencing with principal coordinates analysis in DMSO- or RTX-treated mice. (B) Averaged relative abundance at the phylum level. (C) Phylum-specific qPCR of fecal samples from DMSO- or RTX-treated mice. (D) FMT donor and recipient fecal microbial composition was analyzed by 16S rRNA gene sequencing, presented as averaged relative abundance at the genus level. (E to H) Disease and recovery of DSS-treated FMT mice were monitored by daily weight loss (E), clinical disease score (F), colon length (G) and H&E staining of distal colon (H) at day 10. (I, J) Frequency of colonic neutrophils from mice in (E) on day 10, pre-gated on live, CD45+ events. (K to N) Disease and recovery of DSS-treated cohoused mice were monitored by daily weight loss (K), clinical disease score (L), colon length (M), and H&E staining of distal colon (N) at day 10. Scale bars = 50 μm in (H) and (N). Data are representative of two independent experiments with n=3–5 per group. Data are Mean ± SEM. * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
Given that microbial dysbiosis is a key factor associated with human IBD and murine models of intestinal damage and inflammation (Abraham and Cho, 2009; Brown et al., 2019; Kaser et al., 2010; Saleh and Elson, 2011; Uhlig and Powrie, 2018), we sought to test whether the dysbiotic intestinal microbiota elicited by ablation of TRPV1+ nociceptors could influence intestinal inflammation or tissue protection in vivo. To examine whether RTX-induced dysbiosis could transfer disease phenotypes, we employed fecal microbiota transfer (FMT) from DMSO- or RTX-treated wild-type specific pathogen-free (SPF) mice into wild-type germ-free (GF) recipients (labeled ‘FMT-DMSO’ or ‘FMT-RTX’) by oral gavage of cecal content-derived material from donor mice (Figure S3A). In the FMT donor groups, upon either systemic RTX-mediated ablation or intrathecal RTX-mediated ablation of TRPV1+ nociceptors, bacterial 16S rRNA gene sequencing revealed notable changes in the composition of the intestinal microbiota at the family and genus level, including a reduction in the relative abundance of the Bacteroides genus, the Clostridium XIVa genus, the Parabacteroides genus, the Porphyromonadaceae family and the Bacteroidales order, as well as an increase in the relative abundance of the Lactobacillus genus and the Erysipelotrichaceae family (Figure 3D, FMT donor and Figure S3B). Six weeks following FMT, bacterial 16S rRNA gene sequencing of fecal contents from recipient mice revealed preserved alterations in the abundance of certain taxa between DMSO- or RTX-treated donor mice and their corresponding recipient mice, including reduced levels of Clostridium XIVa, Parabacteroides and Bacteroidales, as well as increased level of Erysipelotrichaceae family (Figure 3D, FMT recipient). These observations indicate that the microbial dysbiosis from RTX-treated mice could be transferred and maintained in GF wild-type recipient mice.
Following FMT, recipient mice were then exposed to DSS and parameters of clinical disease were monitored (Figure S3A). While FMT-DMSO mice recovered from DSS-induced intestinal damage, FMT-RTX mice displayed persistent weight loss (Figure 3E), significantly elevated clinical disease activity (Figure 3F) and significantly shortened colon length (Figure 3G), along with more severe histopathologic features including immune cell infiltration and loss of crypt structure (Figure 3H). Increased neutrophil influx was also detected in the FMT-RTX mice compared to FMT-DMSO mice (Figure 3I and 3J), suggesting a role for the intestinal microbiota in driving the inflammatory response in the context of a disruption in nociceptive sensing. We performed additional natural microbiota reconstitution experiments by cohousing germ-free mice (recipient mice) with either DMSO- or RTX-treated SPF mice (donor mice) for 4 weeks and exposed both donor and recipient mice to DSS (Figure S3C). As expected, in the donor group, RTX treatment (Donor-RTX) resulted in exacerbated disease severity compared to DMSO treatment (Donor-DMSO) (Figure 3K). Consistent with the FMT experiments by oral gavage, germ-free B6 mice cohoused with RTX-treated mice (recipient-RTX) exhibited significantly increased weight loss (Figure 3K), higher clinical disease score (Figure 3L), increased colon shortening (Figure 3M), and more severe histopathology (Figure 3N) compared to germ-free mice cohoused with DMSO-treated mice (recipient-DMSO). Taken together, FMT and cohousing experiments suggest that RTX-mediated ablation of TRPV1+ nociceptors results in a transmissible dysbiosis that drives defective tissue protection upon intestinal damage.
Similar to RTX-mediated TRPV1+ nociceptor ablation, DREADD-mediated acute silencing of TRPV1+ nociceptors resulted in significant alterations in the composition of the intestinal microbiota following DSS administration (Figure S3D and S3E). To investigate the role of the intestinal microbiota in TRPV1+ nociceptor-mediated tissue protection in the chemogenetic mouse model, we exposed either TRPV1wt/wt mice and TRPV1hM4Di mice (separately housed by genotype), or cohoused TRPV1wt/wt and TRPV1hM4Di mice to DSS in drinking water and monitored disease progression (Figure S3F). Separately housed TRPV1hM4Di mice were more susceptible to DSS-induced colitis compared to separately housed TRPV1wt/wt mice, while TRPV1hM4Di mice cohoused with TRPV1wt/wt control mice exhibited similar susceptibility compared to cohoused TRPV1wt/wt control mice, as reflected by similar weight loss (Figure S3G), slightly higher clinical disease score (Figure S3H), comparable colon length (Figure S3I), comparable neutrophil infiltration (Figure S3J) and similar histopathology in the distal colon (Figure S3K). Although separately housed and cohoused TRPV1wt/wt mice exhibited similar weight loss and colon shortening, clinical disease activity and severe histopathology was notable in the cohoused TRPV1wt/wt mice compared to separately housed TRPV1wt/wt mice, indicating that TRPV1+ nociceptor silencing-mediated dysbiosis is transmissible to wild-type controls cohoused in the same cage. Collectively, these data demonstrate that RTX-mediated ablation and chemogenetic silencing of TRPV1+ nociceptors lead to a transmissible dysbiosis which is sufficient to confer enhanced susceptibility to DSS-induced intestinal damage and inflammation.
Vancomycin-sensitive Gram+ bacteria are required for the exacerbated intestinal inflammation upon nociceptor ablation
To test whether the intestinal microbiota is required to drive the enhanced susceptibility to DSS-induced colitis in SPF mice upon RTX-mediated ablation of TRPV1+ nociceptors, we employed established approaches (Alenghat et al., 2013; Bessman et al., 2020; Hill et al., 2010; Rakoff-Nahoum et al., 2004; Tang et al., 2015) to deliver either broad spectrum antibiotics (ABX) or single ABX to deplete specific bacterial communities (Figure 4A). Broad spectrum ABX treatment eliminated RTX-mediated exacerbation of DSS-induced intestinal damage and inflammation, as both ABX-treated DMSO groups and ABX-treated RTX groups exhibited similar weight loss (Figure 4B), comparable clinical disease score (Figure 4C) and colon length (Figure 4D), and similar histological architecture in the distal colon (Figure 4E). These findings indicate that following ablation of TRPV1+ nociceptors, signals derived from the intestinal microbiota promote severe intestinal inflammation.
Figure 4. Manipulation of the intestinal microbiota improves DSS-induced colitis susceptibility upon TRPV1+ nociceptor ablation.
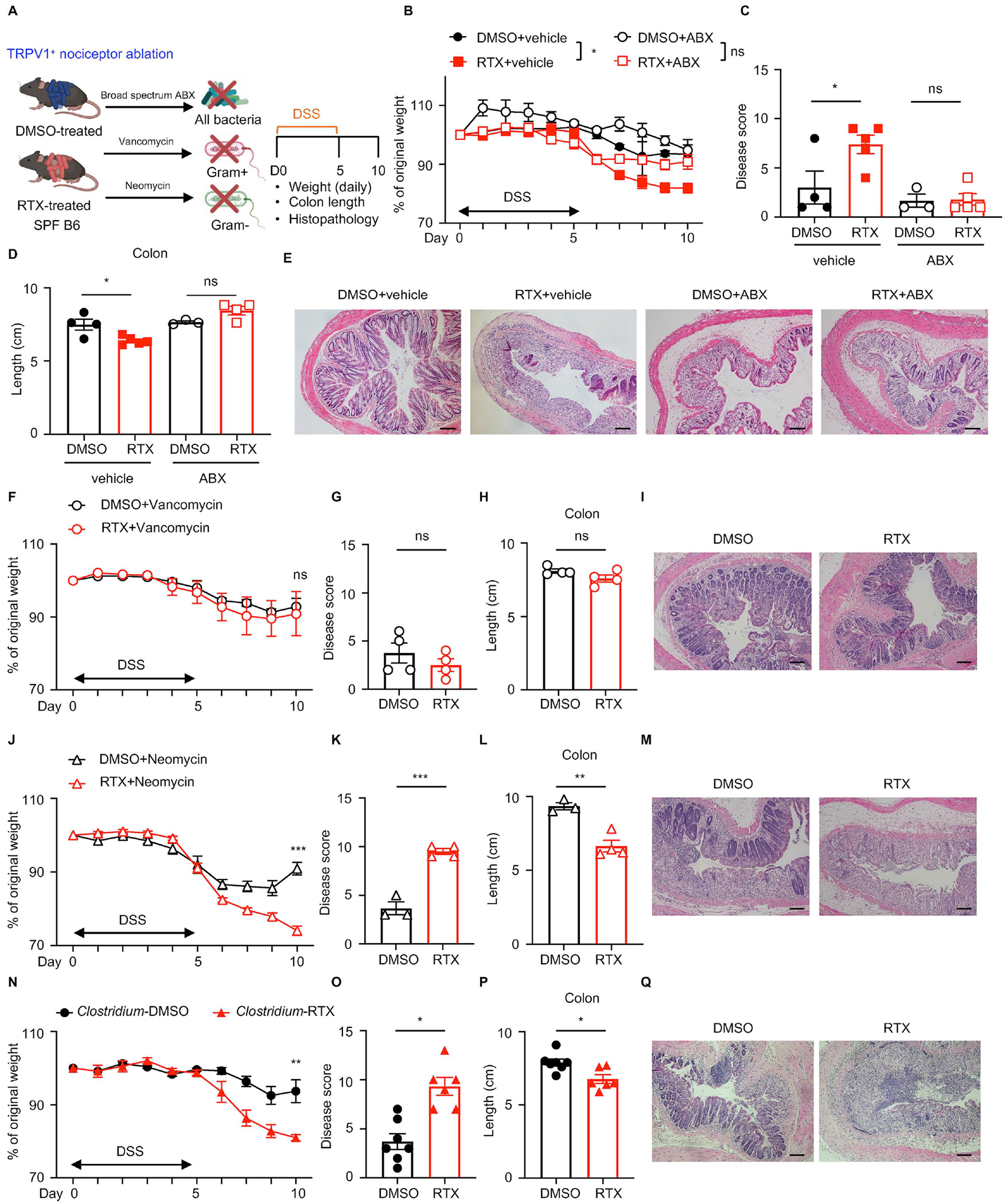
(A) DMSO- or RTX-treated mice administered with vehicle or broad spectrum antibiotic cocktail (ABX), vancomycin, or neomycin were exposed to DSS for 5 days and disease and recovery were monitored daily. (B to E) Daily weight loss (B), clinical disease score (C), colon length (D), and H&E staining of the distal colon (E) in vehicle or ABX-treated mice at day 10. (F to I) Daily weight loss (F), clinical disease score (G), colon length (H) and H&E staining of the distal colon (I) in vancomycin-treated mice at day 10. (J to M) Daily weight loss (J), clinical disease score (K), colon length (L) and H&E staining of the distal colon (M) in neomycin-treated mice at day 10. (N to Q) Daily weight loss (N), clinical disease score (O), colon length (P) and H&E staining of the distal colon (Q) in Clostridium spp.-colonized ex-GF mice at day 10. Scale bars = 50 μm in (E), (I), (M) and (Q). Data are representative of two independent experiments with n=3–7 per group. Data are Mean ± SEM. ns, not significant, * P < 0.05, ** P < 0.01, *** P < 0.001.
We next employed individual ABX to determine the specific microbial communities that influence TRPV1+ nociceptor-mediated tissue protection during DSS-induced colitis. Following vancomycin treatment (which preferentially depletes Gram+ bacteria), RTX-mediated susceptibility to DSS was comparable to DMSO treatment group, as indicated by similar weight loss (Figure 4F), similar clinical disease score (Figure 4G), comparable colon length (Figure 4H), and similar histological architecture in the distal colon (Figure 4I). In contrast, following neomycin treatment (which preferentially depletes Gram− bacteria), RTX-treated mice exhibited enhanced susceptibility to DSS, as reflected by significantly more weight loss (Figure 4J), significantly increased clinical disease score (Figure 4K), significantly reduced colon length (Figure 4L), and more severe histopathological changes within the intestinal mucosa, including prominent crypt loss and edema compared to control mice (Figure 4M). Together, these data suggest that vancomycin-sensitive bacteria are required for the enhanced susceptibility to DSS-induced colitis upon nociceptor ablation. It is possible that vancomycin and neomycin treatment result in alterations in relative abundance and functionality of the remaining vancomycin- or neomycin-resistant bacterial communities, which might also account for the disease phenotype upon RTX-mediated nociceptor ablation.
Colonization with a Clostridium consortium is sufficient to drive TRPV1+ nociceptor-mediated tissue protection
Since we observed profound alterations in the composition of the intestinal microbiota upon RTX-mediated nociceptor ablation in the SPF setting (Figure 3D), we sought to test whether specific bacterial species are sufficient to drive exacerbated intestinal inflammation following RTX-mediated ablation of TRPV1+ nociceptors using different vendor-sourced mice and established gnotobiotic mouse models. First, we employed RTX-mediated TRPV1+ nociceptor ablation in Taconic-sourced B6 mice (which have been reported to carry high levels of SFB) versus JAX-sourced B6 mice (that are SFB-negative) (Ivanov et al., 2009) to test whether TRPV1+ nociceptors mediate tissue protection in mice that carry vendor-specific microbiota. Compared to DMSO-treated Taconic B6, RTX-treated Taconic B6 were more susceptible to DSS-induced colitis, characterized by significantly more weight loss (Figure S4A), significantly elevated clinical disease score (Figure S4B), significantly increased colon shortening (Figure S4C), and more severe histopathology (Figure S4D). Thus, consistent with results from SFB-negative JAX B6 mice (Figure 2), ablation of nociceptors lead to enhanced susceptibility to DSS-induced colitis in SFB-positive Taconic B6 mice.
Second, based on results from the bacterial 16S rRNA gene sequencing showing the abundance of specific bacteria taxa including the Bacteroides genus, the Clostridium XIVa genus, and the Erysipelotrichaceae family (Figure 3D) were altered upon nociceptor disruption and the antibiotic treatment experiments (Figure 4E–L), we utilized gnotobiotic mouse models with GF mice colonized with defined consortia of microbial communities, specifically (i) the Clostridium spp. consortia (Gram+) (Atarashi et al., 2013; Atarashi et al., 2011), (ii) a combination of Bacteroides thetaiotaomicron (Gram−) and Enterococcus faecium (Gram+) (Bt+Ef), or (iii) altered Schaedler flora (ASF), a well-established minimal microbial consortium in mice (Schaedler and Dubos, 1962) (Figure S4E). Bt+Ef colonized ex-GF B6 mice (Figure S4F–I) and ASF mice (Figure S4J–M) exhibited similar colitis susceptibility between DMSO- and RTX-treated groups, indicating that these bacterial communities were not sufficient to drive the enhanced susceptibility to DSS-induced intestinal pathology observed in RTX-treated SPF mice. In contrast, RTX-mediated ablation of TRPV1+ nociceptors in Clostridium spp. consortia-colonized ex-GF mice resulted in a similar colitis phenotype to that observed in SPF B6 mice (Figure 4N–Q), indicating that Clostridium spp. colonization is sufficient to drive enhanced susceptibility to DSS-induced colitis in the absence of TRPV1+ nociceptors.
To further investigate whether certain bacteria contribute to TRPV1+ nociceptor-mediated tissue protection, we focused on several bacterial species that exhibited altered abundance following RTX treatment in SPF B6 mice. We investigated this by using nociceptor-sufficient GF B6 mice mono-colonized with Gram− bacteria including Bacteroides fragilis and Prevotella copri, or Gram+ bacteria including Clostridium innocuum and Clostridium sporogenes, (Figure S4N). Compared to GF B6 mice, Gram+ bacterial colonization resulted in less weight loss and increased survival rate (Figure S4O and S4P), indicating a tissue-protective role played by Gram+ Clostridium spp. in ex-GF mice with intact nociception. Taken together, results from ABX and gnotobiotic experiments demonstrate that vancomycin-sensitive Gram+ bacteria are required to drive enhanced susceptibility to DSS-induced colitis in the absence of TRPV1+ nociceptors. Of note, mono-colonization with a consortium of Clostridium spp. in nociceptor-deficient ex-GF mice was sufficient to drive exacerbated intestinal inflammation. In contrast, mono-colonization of nociceptor-sufficient ex-GF mice with Clostridium spp. promoted intestinal tissue protection through a nociceptor-dependent pathway. Although RTX-mediated nociceptor ablation resulted in overall increased abundance of Firmicutes spp., most of which are Gram+, there were reduced levels of Clostridium spp. while increased levels of Erysipelotrichaceae within this phylum (Figure 3B–D), indicating that TRPV1+ nociceptors play a critical role in maintaining the balance of protective and pathogenic microbes to promote tissue protection during intestinal damage (Atarashi et al., 2013; Jin et al., 2022).
The nociceptive neuropeptide substance P mediates tissue protection during DSS-induced intestinal damage
Activation of the TRPV1 ion channel on nociceptors results in the release of vesicles containing nociceptive neuropeptides including CGRP and substance P from the activated nerve terminals (Baral et al., 2019; Suvas, 2017). Analysis of a single-cell RNA-Seq dataset of the murine nervous system, which enables the examination of gene expression across different tissues, revealed that two of the most highly expressed neuropeptides from TRPV1+ nociceptors are substance P (encoded by Tac1) and CGRP (encoded by Calca), whereas intrinsic enteric neurons highly express neuropeptides including VIP and NMU (Figure S5A) (Zeisel et al., 2018). Tac1 has also been shown to be expressed in enteric neuron subsets and enteroendocrine cells in the mouse colon (Billing et al., 2019; Drokhlyansky et al., 2020; Morarach et al., 2021). Therefore, we assessed the mRNA expression of Trpv1 and Tac1 across multiple tissues in SPF B6 mice at steady state and detected the highest expression in the DRG (Figure S5B and S5C). In addition, Tac1 expression was minimally detected in either the lamina propria or the muscularis layer, whereas Nmu expression was significantly enriched in the muscularis layer compared to the lamina propria in the colons of SPF B6 mice at steady state (Figure S5D). Taken together, these results indicate that while Nmu is highly expressed in intrinsic enteric neurons, Tac1 is primarily expressed in the DRG.
Next, we sought to investigate if there are any alterations of substance P or CGRP levels upon nociceptor disruption. Chemogenetic silencing (Figure 5A–D) and RTX-mediated ablation (Figure 5E and 5F) of TRPV1+ nociceptors resulted in significantly reduced colonic substance P and CGRP levels following DSS administration, indicating that TRPV1+ nociceptors are a major source of substance P and CGRP in the inflamed mouse colon. Substance P has been reported to play a protective role in preserving epithelial barrier function (Suvas, 2017), thus we first investigated whether administration of substance P is sufficient for TRPV1+ nociceptor-mediated tissue protection via gain-of-function experiments. Substance P administration attenuated disease severity in RTX-treated B6 mice as reflected by significantly less body weight loss (Figure 5G), reduced clinical disease score (Figure 5H), less colon shortening (Figure 5I), and improved histopathology in the distal colon (Figure 5J). In addition, administration of substance P upon DREADD-mediated silencing of TRPV1+ nociceptors following exposure to DSS resulted in less severe weight loss (Figure 5K), attenuated disease activity (Figure 5L), reduced colon shortening (Figure 5M) and improved histopathology in the distal colon (Figure 5N). Consistently, substance P administration ameliorated disease severity observed in Trpv1-Cre mice injected with AAV9 virus carrying the inhibitory DREADD (AAV9-hSyn-DIO-hM4D(Gi)-mCherry) following DSS administration (Figure S5E–H). Collectively, these data indicate that one mechanism through which TRPV1+ nociceptors promote tissue protection is via the nociceptive neuropeptide substance P.
Figure 5. Substance P is required for tissue protection mediated by TRPV1+ nociceptors.
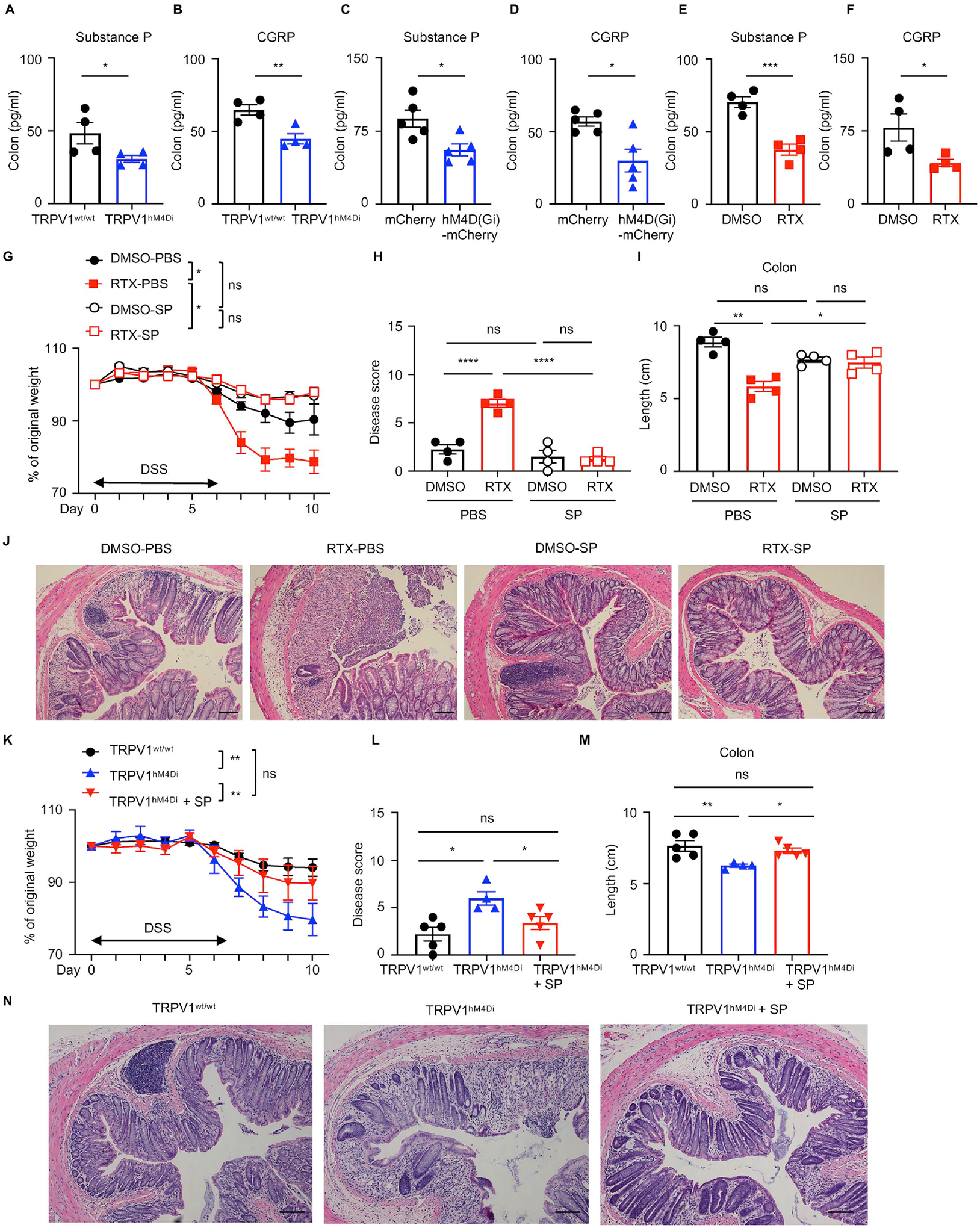
(A, B) Colon substance P and CGRP levels in DSS-treated TRPV1hM4Di or their littermate control mice. (C, D) Colon substance P and CGRP levels in DSS-treated TRPV1wt/wt mice infected with AAV9 viruses. (E, F) Colon substance P and CGRP levels in DSS-treated DMSO- or RTX-treated mice. (G to J) Disease and recovery of DSS-treated DMSO- or RTX-treated mice administered with substance P were assessed from day 0 to day 10 by daily weight loss (G), clinical disease score (H), colon length (I) and H&E staining of the distal colon (J) at day 10. (K to N) Disease and recovery of DSS-treated TRPV1hM4Di or their littermate control administered with substance P were assessed by daily weight loss (K), clinical disease score (L), colon length (M) and H&E staining of the distal colon (N) at day 10. Scale bars = 50 μm in (J) and (N). Data are representative of two independent experiments with n=4–5 per group. Data are shown as mean ± SEM. ns, not significant, * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
We also utilized a genetic loss-of-function approach to further investigate the role of substance P in mediating tissue protection during DSS-induced intestinal damage and inflammation by using the Tac1−/− mouse line. At steady state, Tac1−/− mice exhibited similar percentages of most immune cells in the colon (Figure S6A–I). Substance P deficiency did not affect baseline body mass (Figure S6J), food and water intake (Figure S6K and S6L), or total gastrointestinal transit time (Figure S6M). However, mice lacking substance P demonstrated enhanced susceptibility to DSS-induced colitis compared to wild-type control mice (Tac1+/+), as reflected by significantly more body weight loss (Figure 6A), a significant increase in clinical disease score (Figure 6B), and significantly reduced colon length (Figure 6C). Furthermore, when wild-type control mice began recovering from DSS-induced intestinal damage, Tac1−/− mice exhibited severe intestinal damage and loss of tissue-reparative processes characterized by absence of epithelial crypt structure with inflammatory cell infiltration and intestinal edema in the distal colon (Figure 6D). The neutrophil infiltration in the colon was comparable between Tac1−/− mice and wild-type control mice (Tac1+/+) following exposure to DSS (Figure S6N and S6O). Similarly, there were no significant differences in the percentages of colonic Treg cells (Figure S6P–R). There was comparable proportion of colonic IL-17A-producing CD4+ T cells but increased proportion of IFNγ-producing CD4+ T cells in Tac1−/− mice compared to wild-type control mice (Figure S6S–U). Critically, following DSS administration, DMSO- and RTX-treated Tac1−/− mice exhibited comparable weight loss (Figure 6E), clinical disease score (Figure 6F), colon length (Figure 6G) and histopathology of the distal colon (Figure 6H), indicating that substance P is required to integrate the tissue-protective functions of TRPV1+ nociceptors upon intestinal damage. Taken together, these data suggest that TRPV1+ nociceptor-derived substance P controls susceptibility to DSS-induced intestinal damage and inflammation.
Figure 6. Substance P mediates tissue protection from DSS-induced colitis in a microbiota-dependent manner.
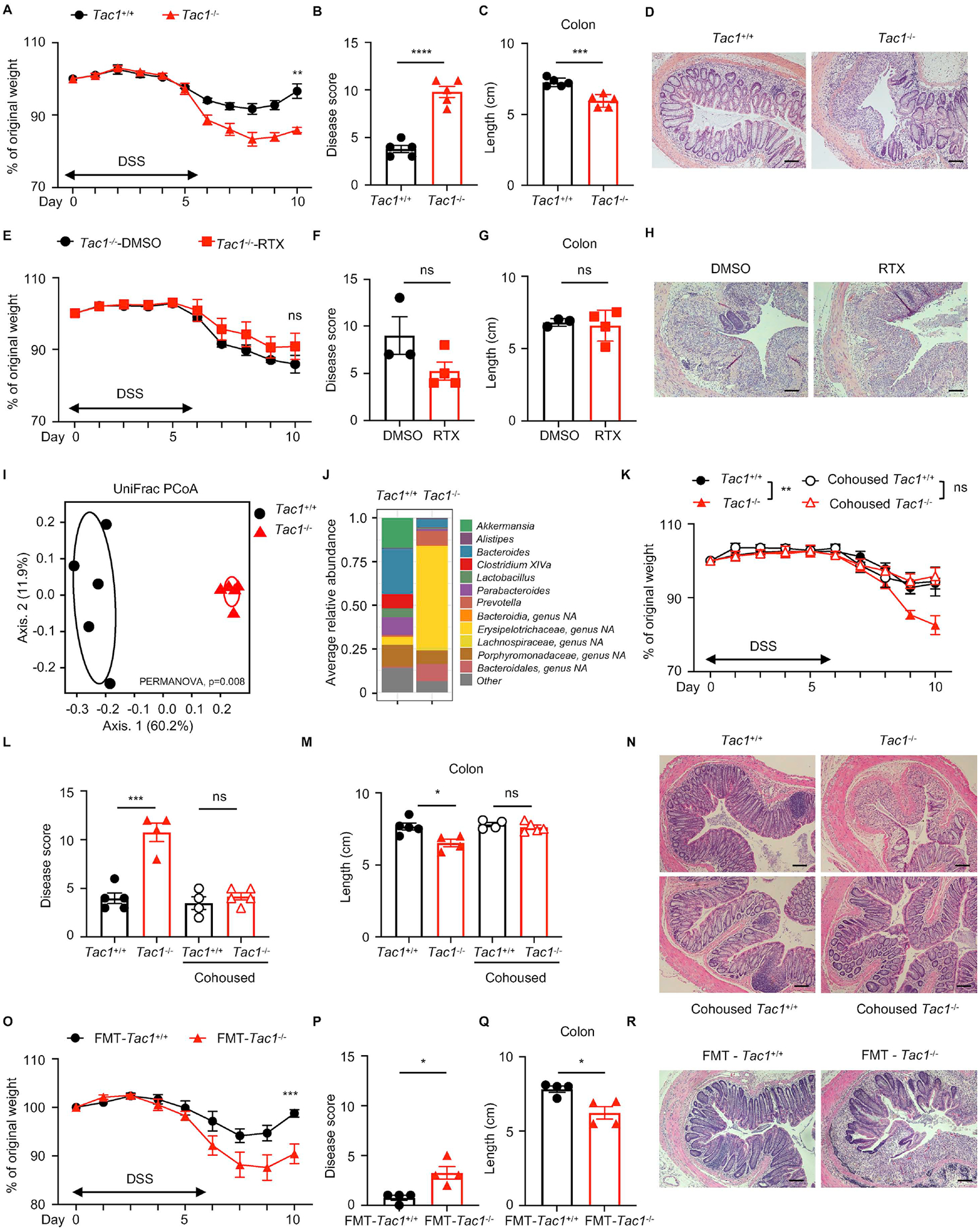
(A to D) Disease and recovery of DSS-treated Tac1+/+ and Tac1−/− mice were monitored by daily weight loss (A), clinical disease score (B), colon length (C), and H&E staining of the distal colon (D) at day 10. (E to H) Disease and recovery of DSS-treated DMSO- or RTX-treated Tac1−/− mice were monitored by daily weight loss (E), clinical disease score (F), colon length (G) and H&E staining of the distal colon (H) at day 10. (I, J) Principal coordinates analysis (I) and averaged relative abundance at the genus level (J) of feces from Tac1+/+ and Tac1−/− mice at steady state. (K to N) Disease and recovery of DSS-treated single housed or cohoused Tac1+/+ and Tac1−/− mice were monitored by daily weight loss (K), clinical disease score (L), colon length (M), and H&E staining of the distal colon (N). (O to R) Disease and recovery of DSS-treated FMT mice were monitored by daily weight loss (O), clinical disease score (P), colon length (Q) and H&E staining of distal colon (R) at day 10. Scale bars = 50 μm in (D), (H), (N) and (R). Data are representative of three independent experiments with n=3–5 per group. Data are Mean ± SEM. ns, not significant, * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
Substance P regulates the intestinal microbiota to mediate tissue protection
To investigate whether substance P regulates the composition of the intestinal microbiota, we analyzed fecal bacterial communities by 16S rRNA gene sequencing in samples isolated from Tac1+/+ and Tac1−/− mice at steady state. The PCoA plot demonstrated that samples clustered largely by genotype, indicating that substance P plays a role in regulating the composition of the intestinal microbiota (Figure 6I). Changes in specific taxa were shared between RTX-mediated TRPV1+ nociceptor ablation and genetic deletion of substance P, including reduced levels of Bacteroides, Porphyromonadaceae, Parabacteroides and Clostridium_XIVa, and increased level of Erysipelotrichaceae (Figure 3D and 6J).
To investigate the causal relationship between altered intestinal microbiota and impaired tissue protection upon genetic deletion of substance P, we cohoused Tac1+/+ mice and Tac1−/− mice and monitored disease progression following DSS administration. Enhanced susceptibility to DSS-induced colitis observed in Tac1−/− mice was abolished following cohousing with Tac1+/+ mice, reflected by comparable body weight loss (Figure 6K), similar disease activity (Figure 6L), comparable colon length shortening (Figure 6M) and comparable histopathology (Figure 6N) between cohoused Tac1+/+ mice and Tac1−/− mice. Further, transplantation of the intestinal microbiota from Tac1−/− mice into ex-GF B6 mice resulted in significantly more severe DSS-induced colitis compared to ex-GF mice receiving intestinal microbiota from Tac1+/+ mice, with significantly more weight loss (Figure 6O), significantly higher clinical disease score (Figure 6P), significantly shortened colon length (Figure 6Q) and more severe histopathology in the distal colon (Figure 6R). Together, these results suggest that substance P exerts tissue-protective effects by modulating the composition of the intestinal microbiota.
Dysregulated nociceptor activity in the context of intestinal inflammation and dysbiosis
To investigate whether TRPV1+ nociceptors are regulated by intestinal inflammation and dysbiosis, we measured levels of substance P and CGRP in naïve and inflamed colons of DSS-exposed mice and observed significantly increased levels of these neuropeptides following DSS administration (Figure 7A and 7B), suggesting that nociceptive neurons are actively functioning in the context of intestinal inflammation. To investigate whether there is crosstalk between the microbiota and nociceptors, we measured levels of substance P and CGRP in the colons of FMT recipient mice following DSS administration. Compared to the FMT-DMSO group, the FMT-RTX group exhibited significantly reduced levels of substance P but similar levels of CGRP in the colon (Figure 7C and 7D). There were also significantly decreased levels of substance P, but not CGRP, in the colons of germ-free B6 mice compared to SPF B6 mice following DSS administration (Figure 7E and 7F). Together, these data suggest that specific bacterial species modulated by TRPV1+ nociceptors provide positive feedback by activating nociceptors, further promoting substance P production and subsequent tissue protection.
Figure 7. Nociceptive neuropeptide expression is altered in intestinal inflammation and dysbiosis in mouse and human.
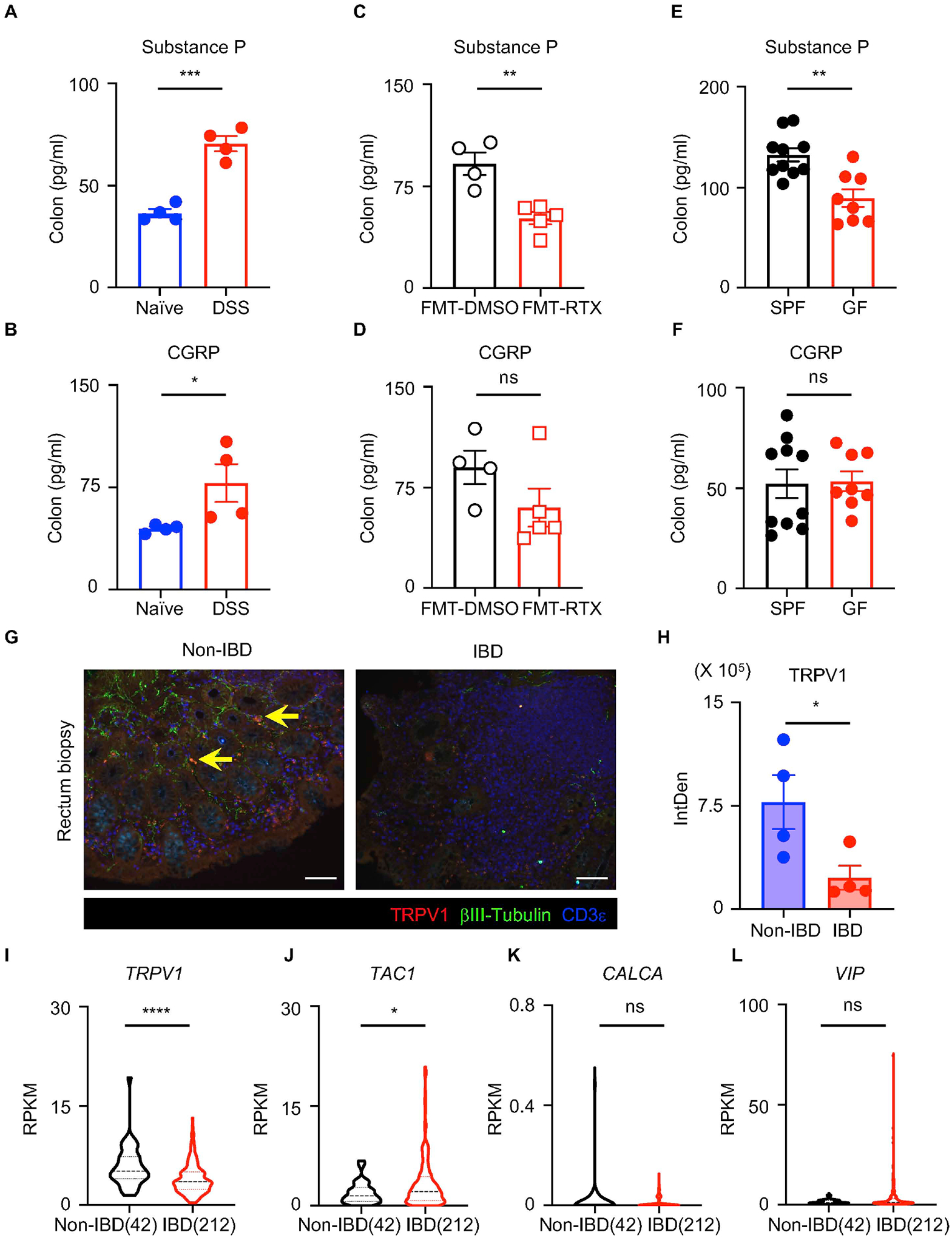
(A, B) Colon substance P and CGRP levels in naïve or DSS-treated mice. (C, D) Colon substance P and CGRP levels in DSS-treated FMT-DMSO or FMT-RTX mice. (E, F) Colon substance P and CGRP levels in DSS-treated SPF or GF mice. (G, H) Representative sections and quantification of TRPV1 fluorescence intensity of pediatric non-IBD and IBD colon biopsy samples, stained for TRPV1 (red), βIII-tubulin (green) and CD3ε (blue). Yellow arrows show representative TRPV1+ nociceptor innervation. Scale bars = 100 μm in (G). (I to L) Analysis of RNA-Seq dataset of pediatric IBD biopsy samples with violin plots of TRPV1, TAC1, CALCA and VIP expression. Data are pooled from three independent experiments with n=3–4 mice per group in (E, F). Data are Mean ± SEM. ns, not significant, * P < 0.05, *** P < 0.001, *** P < 0.005, **** P < 0.0001.
Dysregulated nociceptor activity in the inflamed human intestine
Finally, examination of TRPV1+ nociceptors in human intestinal biopsies revealed notable differences between healthy (non-IBD) and IBD patients. Immunofluorescence microscopy revealed TRPV1+ neuronal innervation in the mucosal layer of healthy intestines, which was confirmed by co-staining with a pan-neuronal marker βIII-tubulin (Figure 7G, yellow arrows). TRPV1+ nociceptor innervation was significantly diminished in inflamed colon tissues isolated from IBD patients compared to healthy colon tissues (inflammation indicated by CD3ε+ T cell density) (Figure 7G and 7H), indicating that the regulatory networks between gut-innervating TRPV1+ nociceptors and environmental signals such as the intestinal microbiota are dysregulated in human IBD. To explore the potential functional relevance of nociceptors in the context of human IBD, we analyzed expression of genes encoding nociceptor-associated neuropeptides in intestinal tissues isolated from healthy individuals versus IBD patients (Haberman et al., 2014). The expression of nociceptor-related genes such as TRPV1 and TAC1 (Yissachar et al., 2017) was significantly altered in the intestinal biopsy samples isolated from IBD patients relative to non-IBD healthy controls (Figure 7I and 7J). In contrast, the expression levels of CALCA and VIP were comparable (Figure 7K and 7L), indicating there is dysregulated nociceptor activity, including that mediated by substance P, in the context of ongoing chronic inflammation in the human intestine.
DISCUSSION
Mammals have coevolved with their microbiota over thousands of years, providing a hospitable environmental niche and sources of nutrients for the microorganisms while the microbiota provides many advantages for the host, including nutritional and metabolic benefits and protection from infection and tissue injury (Ansaldo et al., 2021; Belkaid and Hand, 2014; Brestoff and Artis, 2013; Hill and Artis, 2010; Hooper et al., 2012). The importance of this host-microbiota relationship is illustrated by the well-characterized consequences of a breakdown of this dialogue and its association with multiple metabolic, neurological, and chronic inflammatory disorders (Ansaldo et al., 2021; Blander et al., 2017; Clemente et al., 2012; Dabke et al., 2019; Kamada et al., 2013; Lu and Stappenbeck, 2022; Morais et al., 2021; Rooks and Garrett, 2016; Tilg et al., 2020; Vuong et al., 2017). In the present study, we demonstrate that TRPV1+ nociceptors mediate tissue protection following DSS-induced intestinal damage and inflammation in part through regulating the composition of the intestinal microbiota. In the absence of TRPV1+ nociceptors, vancomycin-sensitive Gram+ bacterial communities lead to enhanced susceptibility to DSS-induced colitis in mice (Figure S7A and S7B). Further, mono-colonization of a consortium of Gram+ Clostridium spp. in GF mice with disrupted nociception was sufficient to drive exacerbated intestinal inflammation, while mono-colonization of nociceptor-sufficient GF mice with Clostridium spp. was tissue-protective (Figure S7C and S7D). Mechanistically, TRPV1+ nociceptors-derived substance P limited intestinal inflammation in a microbiota-dependent manner. These findings highlight a previously unrecognized role for TRPV1+ nociceptors in regulating the composition of the intestinal microbiota through substance P, which is required for tissue protection and the restoration of intestinal homeostasis in the context of ongoing inflammation.
Previous studies identified that TRPV1+ nociceptors induce vasodilation, increasing blood flow and edema independently of the immune response through release of neuropeptides (Brain and Williams, 1989; Edvinsson et al., 1987; McCormack et al., 1989; Saria, 1984). TRPV1+ nociceptors also contribute to cutaneous skin inflammation by modulating the IL-23-IL-17 axis (Cohen et al., 2019; Kashem et al., 2015; Riol-Blanco et al., 2014) and drive pulmonary allergic reactions through regulating type 2 innate lymphoid cells (Talbot et al., 2015). In other contexts, TRPV1+ nociceptors can perform immunoregulatory functions including suppression of the recruitment and activity of neutrophils during bacterial infection that lead to increased bacterial survival in the skin and lungs (Baral et al., 2018; Pinho-Ribeiro et al., 2018). In the present report, we extend these studies to identify previously unrecognized functions for TRPV1+ nociceptors in regulating intestinal inflammation and tissue homeostasis. Therefore, TRPV1+ nociceptors can perform both pro-inflammatory and anti-inflammatory functions in a context-dependent and tissue-specific manner.
Mechanistically, we identify a role of nociceptor-derived substance P in regulating the composition of the intestinal microbiota and mediating tissue protection following acute intestinal injury. Inflammatory pain can be elicited by inflammation and intestinal dysbiosis resulting in hyperalgesia (Guo et al., 2019a). In this context, specific pathogenic bacteria have been shown to directly activate nociceptors and elicit pain and inflammation (Chiu et al., 2013; Pinho-Ribeiro et al., 2018). Several RORγt+ Treg cell- or Th17 cell-inducing microbes have been reported to directly modulate TRPV1+ nociceptor activity and regulate Tac1 expression in nociceptors (Yissachar et al., 2017). Based on these findings, further studies will also be required to determine whether specific types of beneficial bacteria or inflammatory mediators such as IL-6 and TNFα can modulate nociceptor activation and substance P release during intestinal damage and inflammation.
Substance P is widely distributed throughout the nervous system has been implicated in neuro-immune interactions during tissue injury and cutaneous type 2 inflammation (Perner et al., 2020; Zhang et al., 2019). Substance P released from nociceptors also promotes the development of allergic type 2 inflammation by inducing mast cell degranulation in the skin and lung (Cyphert et al., 2009; Serhan et al., 2019) and has been shown to regulate macrophage polarization in in vitro cultures and in a mouse model of spinal cord injury (Lim et al., 2017; Sun et al., 2008). However, the role of substance P in intestinal inflammation has remained elusive. The findings of the present study provide insights into the role of nociceptor-derived substance P in regulating the intestinal microbiota and in promoting tissue protection following acute intestinal damage. Further studies will be necessary to dissect the molecular mechanisms that underlying this neuronal-microbiota crosstalk and the extent to which substance P acts in parallel and/or synergy with other neurotransmitters and inflammatory mediators.
Nociceptor-derived CGRP has been shown to limit recruitment of neutrophils and γδT cells during acute bacterial infection in the skin and lung as well as SFB levels and M cell function in the ileum following experimental Salmonella infection (Baral et al., 2018; Lai et al., 2020; Pinho-Ribeiro et al., 2018). In addition, we and others have previously shown that CGRP negatively regulates pulmonary type 2 immune responses during helminth infection through effects on ILC2 effector function (Nagashima et al., 2019; Wallrapp et al., 2019; Xu et al., 2019). In addition to changes in substance P levels, we detected significantly reduced levels of CGRP and goblet cell responses in the colons of mice with disrupted nociception following DSS administration, indicating a potential role of TRPV1+ nociceptor-derived CGRP in regulating goblet cells, mucus production and barrier function, which could further contribute to tissue reparative processes (Paone and Cani, 2020). Therefore, it remains to be determined whether nociceptor-derived CGRP and substance P act cooperatively at different stages of intestinal damage and inflammation to limit inflammation and promote tissue protection and repair.
In summary, this study identifies a previously unrecognized pathway through which gut-innervating TRPV1+ nociceptors regulate the composition of the intestinal microbiota to mediate tissue-protective processes in the context of ongoing intestinal damage and inflammation. The identification of a microbiota-dependent and immuno-modulatory role for the gut-innervating nociceptors could have implications for the development of neuromodulation-based therapies and pain management strategies for IBD and other chronic inflammatory disorders.
Limitations of the Study
Although our study provides a comprehensive understanding of the role of TRPV1+ nociceptors in regulating intestinal inflammation, several limitations should be mentioned. First, our study demonstrated that Clostridium spp. is sufficient to drive TRPV1+ nociceptor-mediated tissue protection in mice with intact nociception. However, further metabolomic and transcriptomic studies are required to investigate the mechanisms underlying the regulation of Clostridium spp. by TRPV1+ nociceptors in this setting. Second, while the chemogenetic manipulation was specific to colon-innervating TRPV1+ nociceptors, future studies will be required to distinguish between DRG and vagus nerve populations. Thirdly, for the microbiological studies we primarily adopted the approach using chemical ablation of TRPV1+ nociceptors. While we also observed significant alterations in the composition of the intestinal microbiota upon acute silencing of TRPV1+ nociceptors in the chemogenetic system following DSS administration, future studies with novel gnotobiotic and chemogenetic mouse models will be needed to address the nociceptor-microbiota crosstalk following acute neuronal manipulation. Appropriate controls for the fidelity and stability of microbial communities in the context of FMT will also be important in these studies. Finally, loss-of-function experiments related to substance P were not limited to TRPV1+ nociceptors in this study and future neuronal cell lineage-specific genetic manipulations will be required to investigate this functionally at a cell-lineage-specific level. Thus, the development of new mouse genetic tools and subsequent future studies will be necessary to investigate the interactions between the gut-innervating nociceptors, TRPV1+ nociceptor-derived substance P and CGRP and specific members of the intestinal microbiota.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, David Artis (dartis@med.cornell.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All data including raw sequencing reads are uploaded to the SRA and are publicly available as of the date of publication. The accession numbers are listed in the Key resources table. The paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request. Microscopy data reported in this paper will be shared by the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Acquisition of human intestine biopsy samples
De-identified intestinal biopsies from the terminal ileum and rectum of pediatric individuals with Ulcerative Colitis or controls who did not have inflammatory bowel disease were obtained following Institutional-Review-Board-approved protocol from the JRI IBD Live Cell Bank Consortium at Weill Cornell Medicine. Informed consent was obtained for all subjects. Subject baseline characteristics for these samples are listed in Tables S3.
Animal models
C57BL/6 (B6) (Jax no.000664), Trpv1-Cre (Jax no.017769), tdTomatofl/stop/fl (Jax no.007914), hM4Difl-stop-fl (Jax no.026219), hM3Dqfl-stop-fl (Jax no.026220) and Tac1−/− (Jax no.004103) mice were purchased from The Jackson Laboratories and bred at specific pathogen-free facilities at Weill Cornell Medicine. Taconic C57BL/6 were purchased from Taconic Biosciences. Germ-free C57BL/6J were maintained at the Weill Cornell Medicine Gnotobiotic Mouse Facility. Germ-free C57BL/6J and microbial colonized ex-GF mice were maintained in sterile isocages during all experimental procedures. All mice used were between 6 and 16 weeks old and were age- and sex-matched for each experiment, maintained on a 12 hour light–dark cycle, and provided food and water ad libitum. Male and female mice were used for experiments; no differences were observed within the parameters analyzed. Littermates of the same sex were randomly assigned to experimental groups. All protocols were approved by the Weill Cornell Medicine Institutional Animal Care and Use Committee (IACUC), and all experiments were performed according to the guidelines of the IACUC.
METHOD DETAILS
Murine acute colitis induction and clinical scoring
If not mentioned specifically, 2.5% colitis-grade dextran sulfate sodium salt (DSS) with average molecular weight of 36,000–50,000 Da (MP Biomedicals) was added to drinking water for 6 days, followed by drinking waters for 4 to 7 days. Mice were monitored daily for morbidity (piloerection, lethargy), weight loss and rectal bleeding. Severity of disease was scored as follows: weight loss (no change, 0; less than 5%, 1; 6–10%, 2; 11–20%, 3; more than 20%, 4); feces (normal, 0; pasty, semi-formed, 1; sticky, 2; sticky with some blood, 3; completely liquid, bloody or unable to defecate after 10min, 4), rectal bleeding (no blood, 0; visible blood in rectum, 1; visible blood on fur, 2); general appearance (normal, 0; piloerect, 1; lethargic and piloerect, 2; lethargic and hunched, 3; motionless and sickly, 4).
Antibiotic treatment
Ampicillin (0.5 g/L), vancomycin (0.25 g/L), metronidazole (0.5 g/L), neomycin (0.5 g/L) and gentamicin (0.5 g/L) were supplemented with 1 packet of artificial sweetener (Sweet’ N Low) per 250 ml of drinking water as previously described (Bessman et al., 2020; Chu et al., 2019; Hill et al., 2012). Vehicle (Sweet’ N Low only) or antibiotics were administered beginning 2 weeks prior to 2% DSS administration, and antibiotic treatment continued throughout the DSS experiment.
Microbial colonization
Germ-free B6 mice were colonized with indicated bacteria consortia by oral gavage for 2 weeks before RTX-mediated ablation of TRPV1+ nociceptors and maintained within sterile vinyl isolators at the Weill Cornell Gnotobiotic Mouse Facility. After 4 weeks of nociceptor ablation, mice were used for DSS-induced colitis experiments in the isolators with 2% DSS in drinking water.
Fecal microbiota transplantation
For FMT studies, donor colon and cecum contents were collected, pooled by treatment group, weighed, re-suspended at 150 mg/ml in PBS supplemented with 10% glycerol, and frozen. Thawed fecal suspension (200 μL per mouse) was then administered to recipient germ-free B6 mice by a single oral gavage. Germ-free B6 mice were maintained within sterile vinyl isolators at the Weill Cornell Gnotobiotic Mouse Facility and monitored for germ-free status by weekly aerobic and anaerobic culturing. After FMT, recipient mice were housed in sterile cages. After 4 to 6 weeks of transplantation, mice were used for DSS-induced colitis experiments in the isolators.
Chemogenetic manipulation of TRPV1+ nociceptors
To perform chemogenetic manipulation of TRPV1+ nociceptors, we bred Trpv1-Cre homozygous mice to hM4Difl-stop-fl mice (DREADD for inhibition) or hM3qifl-stop-fl mice (DREADD for activation), generating TRPV1hM4Di, TRPV1hM3Dq or littermate control TRPV1wt/wt mice. To perform silencing of nociceptors during DSS-induced acute colitis, TRPV1hM4Di and littermate control TRPV1wt/wt mice were treated with CNO (1 mg/kg) from day 0 to day 10 every other day. To perform excitation of nociceptors during DSS-induced acute colitis, TRPV1hM3Dq and littermate control TRPV1wt/wt mice were treated with CNO (0.1 mg/kg) from day 0 to day 6 every other day.
Ablation of TRPV1+ nociceptors
Resiniferatoxin (RTX), a capsaicin analogue, was injected subcutaneously into indicated mice in three escalating (30 μg/kg, 70 μg/kg, and 100 μg/kg) doses on consecutive days. Control mice were treated with vehicle solution (DMSO in PBS). Mice were allowed to rest for 4 weeks before DSS-induced acute colitis. For targeted ablation of the dorsal root ganglion (DRG) TRPV1+ nociceptors, indicated mice were injected intrathecally with RTX (50 ng/mouse) or vehicle solution (DMSO in PBS) while under isoflurane on two consecutive days in the L5-L6 region using a 30G needle and Gastight syringe (Hamilton) (Mishra and Hoon, 2010). Ablation efficiency was confirmed by measurement of nociceptor markers genes in the DRG and quantification of TRPV1+ neurons in the DRG by immunofluorescence microscopy.
Colon injection of AAV9 viruses carrying DREADD
Mice were anesthetized with 3% isoflurane followed by 1% isoflurane to maintain anesthesia. To prevent dehydration of eyes, artificial tear was used. After shaving and sterilization of the abdomen, mice were placed on a sterile surgical pad and covered with a sterile surgical drape. The colon was exposed by making a midline incision through the abdominal wall. 2 μL of AAV9 virus (500 nL * 4 sites) was injected at a flow rate of 100 nL/minute to enhance convection with a pulled glass pipette, and the needle was left in place for at least 5 minutes to prevent reflux.
After injection, the abdominal wall was sutured, and skin was closed using surgical sutures. Antibiotic ointment was applied to the closed surgical site. To perform silencing of nociceptors during DSS-induced acute colitis, mice injected with AAV9 viruses were treated with CNO (1 mg/kg) from day 0 to day 10 every day. Tissues were collected for analysis at least 3 weeks after AAV injections. AAV-hSyn-DIO-mCherry and AAV-hSyn-DIO-hM4D(Gi)-mCherry viruses were a gift from Bryan Roth (Addgene plasmid # 50459; Addgene plasmid #44362) (Krashes et al., 2011).
In vivo administration of neuropeptides
Substance P was purchased from Tocris and administered intraperitoneally every other day at dose of 2 μg per mouse starting from day 0 to day 10 during DSS-induced colitis.
Metabolic Analyses
Metabolic analyses on DMSO- and RTX-treated mice were performed by the Metabolic Phenotyping Center at Weill Cornell Medicine. Animals were individually housed in a temperature controlled Promethion Metabolic Screening System (Sable Systems International, NV). Food and water intake and body mass were recorded continuously by gravimetric measurements within the cages. Physical activity was determined according to beam breaks within a grid of infrared sensors built into each cage. Energy expenditure was calculated using the Weir equation (Energy expenditure = 3.941 kcal/L × VO2 + 1.106 kcal/L × VCO2) (Weir, 1949). Energy expenditure is displayed as the total kcal per specified periods of time, with values adjusted by ANCOVA (Speakman, 2013) for body mass or corrected body mass using VassarStats. Mice were acclimated to this environment on a 12 hour light/dark cycle for 48 hour before the 24 hour recording period began. When not analyzed in metabolic cages, food and water intake were analyzed in cages holding 4–5 mice each and averaged per mouse. Food and water intake was analyzed by daily weighing of food and water.
Gastrointestinal motility measurements
For measurement of total gastrointestinal transit time, mice were given an oral gavage of 6% carmine red (Sigma-Aldrich) dissolved in 0.5% methylcellulose (prepared in sterile 0.9% NaCl). Total intestinal transit time was measured as the time from oral gavage it took for mice to pass a fecal pellet that contained carmine.
Murine intestinal tissue isolation
For intestinal lamina propria lymphocyte preparations, colon was removed, cleaned from remaining fat and washed in ice-cold PBS. Intestinal tissue was opened longitudinally and washed in ice-cold PBS. Dissociation of epithelial cells was performed by incubation on a shaker at 37 °C in HBSS (Sigma-Aldrich) containing 1% FBS and 5 mM EDTA (Thermo Fisher Scientific) two times for 15 min. After each step, samples were vortexed, and the epithelial fraction was filtered through a 100 μm filter. Afterwards, remaining tissue was chopped into small pieces and enzymatic digestion was performed using dispase (0.4 U/ml; Thermo Fisher Scientific), collagenase type III (1.5 mg/ml; Worthington) and DNase I (20 μg/ml; Sigma-Aldrich) in RPMI (Corning) containing 5% FBS for 60 min on a shaker at 37°C. Leukocytes were filtered through a 70 μm cell strainer and further enriched by a 40% Percoll (GE Healthcare) gradient centrifugation.
Flow cytometry
Mouse single-cell suspensions were incubated on ice with anti-CD16/CD32 (93) antibody (Biolegend) and the following conjugated antibodies in PBS. Dead cells were routinely excluded with Fixable Aqua Dead Cell Stain (Thermo Fisher Scientific). For surface staining, B220 (RA3–6B2), CD45 (30-F11), CD3ε (145–2C11), CD4 (RM4–5), CD5 (53–7.3), CD8α (53–6.7), CD11b (M1–70), CD11c (N418), CD127 (A7R34), CD90.2 (30-H12), Fc0εR1α (MAR-1), Gr1 (RB6–8C5) and TCRγ/δ (GL3), Ly6G (1A8), MHCII (I-A/I-E) (M5/114.15.2), CD64 (X54–5/7.1), SiglecF (E50–2440) and F4/80 (BM8) were used. Cytokine production was induced by stimulation of cells for 4 h at 37 °C with the presence of 50 ng/ml phorbol 12-myristate 13-acetate (Sigma), 500 ng/ml ionomycin and GolgiStop (BD Bioscience) in complete RPMI-1640 medium (containing 10% FBS, 50 mM 2-mercaptoethanol, 1 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin). IL-17A (TC11–18H10.1) and IFNγ (XMG1.2) were stained using the BD Cytofix/Cytoperm and Perm/Wash solutions. GATA3 (TWAJ), FoxP3 (FJK-16S), T-bet (4B10), Helios (22F6) and RORγt (B2D) were stained using the Foxp3 transcription factor staining buffer set (Thermo Fisher Scientific). All antibodies used in flow cytometry were purchased from Thermo Fisher Scientific, Biolegend or BD Biosciences. Flow cytometry experiments were acquired using a custom configuration Fortessa flow cytometer and analyzed with FlowJo software (TreeStar).
Immunofluorescence
Tissues were harvested in standard fashion and fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) for 4 hours at 4 °C before washing 3 times in PBS. Tissues were then dehydrated overnight at 4 °C in 30% sucrose dissolved in PBS. Dehydrated tissues were cryoprotected in Tissue-Tek OCT compound (Sakura) and stored at −80 °C until sectioning at a thickness of 10 μm using a cryotome (Leica Instruments) and immobilization on SuperFrost Plus slides (VWR). Immobilized tissues were then stored at −20 °C until immunostaining. For mouse sections, slides were allowed to come to room temperature and excess OCT medium was washed off in PBS and tissue sections were then blocked in PBS with 5% normal goat serum, 5% normal donkey serum (both Jackson Immunoresearch) and 0.1% Triton X-100 (Sigma-Aldrich) for 30 minutes. For Figure 1A, 1B, S1B, S1N and S1M, tissue sections were then stained with the following primary antibodies diluted in blocking buffer for 1h at room temperature: anti-βIII-tubulin-eFluor660 (2G10, eBioscience). For Figure S1C and 2B, tissue sections were stained with the following primary antibodies diluted in blocking buffer for overnight at 4 °C: rabbit anti-phospho-p44/42 mitogen-activated protein kinase (Thr202/Tyr204) monoclonal antibody (CST) or guinea pig anti-Capsaicin receptor antibody (Millipore). Tissue sections were then washed 3 times in PBS, incubated with secondary antibody anti-rabbit IgG Alexa555 or anti-guinea pig IgG Alexa647 (Invitrogen) for 1h at room temperature. Tissue sections were then washed 3 times in PBS, incubated with DAPI (Invitrogen) for 5 minutes prior to a final wash in PBS, and mounted with Prolong Gold antifade reagent (Invitrogen). Representative images were captured under an inverted Nikon Eclipse Ti microscope (Nikon).
For human sections, slides were allowed to come to room temperature and excess OCT medium was washed off in PBS and tissue sections were fixed in ice-cold acetone for 20 minutes and washed in PBS. After air dry, tissue sections were then blocked in PBS with 10% normal horse serum (Jackson Immunoresearch) and 1% BSA (Sigma-Aldrich) for 30 minutes and treated with Streptavidin/Biotin blocking kit (Vector laboratories). Tissue sections were then stained with the following primary antibodies diluted in blocking buffer for 1h at room temperature anti-TRPV1 (BS-1931R, Bioss) and biotinylated anti-CD3ε (OKT3, Biolegend). Sections were washed 3 times with PBS and then incubated with secondary antibodies including anti-βIII-tubulin-AF488 (2G10, eBioscience), anti-rabbit IgG-TRITC (Jackson Immunoresearch), Streptavidin IgG-AF647 (Invitrogen) in blocking buffer for 1h at room temperature. Tissue sections were then washed 3 times in PBS, incubated with DAPI (Invitrogen) for 5 minutes prior to a final wash in PBS, and mounted with Prolong Gold antifade reagent (Invitrogen). Representative images were captured under an inverted Nikon Eclipse Ti microscope (Nikon).
Histology
To detect mucin in intestinal tissue sections, proximal intestinal tissues were fixed with 4% paraformaldehyde (bioWORLD), embedded in paraffin, and 5 μm sections stained with PAS combined with Alcian Blue by IDEX BioResearch. Images were acquired using Nikon Eclipse Ti microscope (Nikon). The number of goblet cells in villus was calculated from the number of goblet cells in 20 crypts per mouse.
Enzyme-Linked Immunosorbent Assay (ELISA)
5 cm of distal colons were harvested from mice and homogenized by sonication in PBS on ice. After sonication, supernatants were purified by centrifugation and used for ELISA analysis. Mouse Substance P and CGRP were detected using competitive mouse substance P and CGRP ELISA kits (both from Cayman Chemical). All procedures were carried out according to manufacturer’s instructions. Samples were assayed on Softmax Pro Software in duplicates and concentrations were determined from a standard curve.
RNA isolation and quantitative real-time PCR
Mouse tissues including dorsal root ganglia (DRG), duodenum, jejunum, ileum, colon, mesenteric lymph nodes and lung were harvested and homogenized in Trizol (Thermo Fisher Scientific). RNA was extracted with chloroform and RNA concentration was determined using a Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific). For the colon muscularis layer dissection, after euthanasia the colon was cleaned, cut open and washed in HBSS and the muscularis region was carefully dissected from the underlying mucosa (lamina propria). Each region was then homogenized, and RNA was extracted using the RNeasy Plus Mini Kit (QIAGEN). Reverse transcription of total RNA was performed using the High Capacity cDNA Reverse Transcription kit according to the protocol provided by the manufacturer (Thermo Fisher Scientific). Quantitative real-time PCR was performed using SYBR green chemistry (Invitrogen) and primers listed in Key Resources Table and Table S1 on a QuantStudio 6 Flex Real-Time PCR (Thermo Fisher Scientific). Gene expression was normalized as n-fold difference to the housekeeping gene Actb.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| beta-3 Tubulin Monoclonal Antibody (2G10-TB3), Alexa Fluor 488, eBioscience | Thermo Fisher Scientific | Cat# 53-4510-82, RRID: AB_2574421 |
| beta-3 Tubulin Monoclonal Antibody (2G10-TB3), eFluor 660, eBioscience | Thermo Fisher Scientific | Cat# 50-4510-82, RRID: AB_2574213 |
| Anti-Capsaicin Receptor Antibody, CT | Millipore | Cat# AB5566 |
| TRPV1/VR1 Polyclonal Antibody | Bioss | Cat# BS-1931R |
| Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (D13.14.4E) XP Rabbit mAb | Cell Signaling Technology | Cat# 4370 |
| Biotin anti-human CD3 Antibody | Biolegend | Cat# 317320, RRID: AB_10916519 |
| Streptavidin, Alexa Fluor 647 Conjugate | Thermo Fisher Scientific | Cat# S32357 |
| Rhodamine (TRITC) AffiniPure Donkey Anti-Rabbit IgG (H+L) | Jackson Immunoresearch | Cat# 711-025-152, RRID: AB_2340588 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 | Thermo Fisher Scientific | Cat# A-31572, RRID: AB_162543 |
| Goat anti-Guinea Pig IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-21450, RRID: AB_2735091 |
| TruStain FcX™ (anti-mouse CD16/32) (93) | Biolegend | Cat# 101320, RRID: AB_1574975 |
| Anti-mouse CD45 Brilliant Violet 785 (30-F11) | Biolegend | Cat# 103149, RRID: AB_2564590 |
| Anti-mouse CD4 BUV395 (RM4-5) | Thermo Fisher Scientific | Cat# 363-0042-82, RRID: AB_2920941 |
| Anti-mouse CD8α PE-ef610 (53-6.7) | Thermo Fisher Scientific | Cat# 61-0081-82, RRID: AB_2574524 |
| Anti-mouse CD3ε PerCP-Cy5.5 (145-2C11) | Biolegend | Cat# 45-0031-82, RRID: AB_1107000 |
| Anti-mouse CD5 PerCP-Cy5.5 (53-7.3) | Biolegend | Cat# 100624, RRID: AB_2563433 |
| Anti-mouse CD11b PerCP-Cy5.5 (M1-70) | Biolegend | Cat# 101228, RRID: AB_893232 |
| Anti-mouse CD11c PerCP-Cy5.5 (N418) | Biolegend | Cat# 117328, RRID: AB_2129641 |
| Anti-mouse CD64 BV605 (X54-5/7.1) | Biolegend | Cat# 139323, RRID: AB_2629778 |
| Anti-mouse CD127 PECy7 (A7R34) | Biolegend | Cat# 351320, RRID: AB_10897098 |
| Anti-mouse CD90.2 Alexa Fluor 700 (30-H12) | Biolegend | Cat# 105320, RRID: AB_493725 |
| Anti-mouse CD11b BV650 (M1-70) | Biolegend | Cat# 117328, RRID: AB_2129641 |
| Anti-mouse MHCII (I-A/I-E) Alexa Fluor 780 (M5/114.15.2) | Thermo Fisher Scientific | Cat# 47-5321-82, RRID AB_1548783 |
| Anti-mouse CD11c FITC (N418) | Biolegend | Cat# 117306, RRID: AB_313775 |
| Anti-mouse F4/80 APC (BM8) | Biolegend | Cat# 123116, RRID: AB_893481 |
| Anti-mouse SiglecF BV421 (E50-2440) | BD Biosciences | Cat# 565934, RRID: AB_2722581 |
| Anti-mouse FcεR1α PerCP-Cy5.5 (MAR-1) | Biolegend | Cat# 134320, RRID: AB_10641135 |
| Anti-mouse Gr1 PerCP-Cy5.5 (RB6-8C5) | Biolegend | Cat# 108428, RRID: AB_893558 |
| Anti-mouse Ly6G BUV395 (1A8) | BD Biosciences | Cat# 563978, RRID: AB_2716852 |
| Anti-mouse B220 PerCP-Cy5.5 (RA3-6B2) | Biolegend | Cat# 103236, RRID: AB_893354 |
| Anti-mouse TCRγ/δ PerCP-Cy5.5 (GL3) | Biolegend | Cat# 118118, RRID: AB_10612756 |
| Anti-mouse CD3ε Alexa Fluor 780 (145-2C11) | Thermo Fisher Scientific | Cat# 47-0031-82, RRID: AB_11149861 |
| Anti-mouse IFNγ PECy7 (XMG1.2) | Biolegend | Cat# 505826, RRID: AB_2295770 |
| Anti-mouse IL-17A PE (TC11-18H10.1) | Biolegend | Cat# 506904, RRID: AB_315464 |
| Anti-mouse GATA3 eFluor660 (TWAJ) | Thermo Fisher Scientific | Cat# 50-9966-42, RRID: AB_10596663 |
| Anti-mouse RORγt PE (B2D) | Thermo Fisher Scientific | Cat# 12-6981-82, RRID: AB_10807092 |
| Anti-mouse T-bet BV605 (4B10) | Biolegend | Cat# 644817, RRID: AB_11219388 |
| Anti-mouse FoxP3 AF488 (FJK-16s) | Thermo Fisher Scientific | Cat# 53-5773-82, RRID: AB_763537 |
| Anti-mouse Helios Pacific Blue (22F6) | Biolegend | Cat# 137220, RRID: AB_10690535 |
| Bacterial and virus strains | ||
| pAAV9-hSyn-DIO-hM4D(Gi)-mCherry | Bryan Roth Lab | Addgene plasmid #44362; RRID: Addgene_44362 |
| pAAV9-hSyn-DIO-mCherry | Bryan Roth Lab | Addgene plasmid #50459; RRID: Addgene_50459 |
| Altered Schaedler Flora (ASF) | N/A | (Gomes-Neto et al., 2017) |
| Clostridium species | N/A | (Atarashi et al., 2017) |
| Bacteroides thetaiotaomicron VPI-5482 | ATCC | Cat# 29148 |
| Enterococcus faecium | Chun-Jun Guo Lab | N/A |
| Bacteroides fragilis 3_1_12 | BEI | Cat# HM-20 |
| Prevotella copri DSM 18205 | DSMZ | Cat# DSM18205 |
| Clostridium sporogenes ATCC 15579 | ATCC | Cat# 15579 |
| Clostridium innocuum DSM 22910 | DSMZ | Cat# DSM22910 |
| Biological samples | ||
| Human intestinal biopsy samples | JRI IBD Live Cell Bank Consortium at Weill Cornell Medicine | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Clozapine-N-oxide | AK Scientific Inc. | Cat# AMTA056 |
| DAPI | Thermo Fisher Scientific | Cat# D1306 |
| Dextran Sulfate Sodium Salt, colitis grade | MP Biochemical | Cat# 160110 |
| Substance P | Tocris | Cat# 1156 |
| Resiniferatoxin | Alomone labs | Cat# R-400 |
| Dimethyl sulfoxide (DMSO) | Sigma | Cat# D8418 |
| Dithiothreitol (DTT) | Sigma | Cat# 43815 |
| DNase I | Sigma | Cat# D5025 |
| Dispase | Thermo Fisher Scientific | Cat# 17105041 |
| Collagenase Type III | Worthington Chemicals | Cat# LS004183 |
| 4% paraformaldehyde | bioWORLD | Cat# 30450002 |
| Triton X-100 | Sigma | Cat# T8787 |
| Tissue-Tek O.C.T. Compound, Sakura Finetek | VWR | Cat# 25608-930 |
| Prolong Gold antifade reagent | Thermo Fisher Scientific | Cat# P36961 |
| 1X Dulbecco’s Phosphate-Buffered Saline (PBS) | Corning | Cat# 21-031-CV |
| RPMI 1640 with L-glutamine | Corning | Cat# 10-040-CV |
| 2-mercaptoethanol | Sigma | Cat# M6250 |
| L-glutamine | Thermo Fisher Scientific | Cat# 25030081 |
| Penicillin Streptomycin solution | Thermo Fisher Scientific | Cat# 15140122 |
| Hanks’ Balanced Salt solution (HBSS) | Sigma | Cat# H6648 |
| Fetal Bovine Serum (FBS) | Peak Serum | Cat# PS-FB1 |
| Normal Horse Serum | Jackson Immunoresearch | Cat# 008-000-121 |
| Normal Goat Serum | Jackson Immunoresearch | Cat# 005-000-121 |
| Normal Donkey Serum | Jackson Immunoresearch | Cat# 017-000-121 |
| Bovine Serum Albumin (BSA) | Sigma | Cat# A6003-25G |
| HEPES | Thermo Fisher Scientific | Cat# 15630-080 |
| Ionomycin | Sigma | Cat# I0634 |
| Phorbol 12-myristate 13-acetate (PMA) | Sigma | Cat# P8139 |
| Protein Transport Inhibitor (Containing Monensin) GolgiStop | BD Biosciences | Cat# 554724 |
| Percoll | GE Healthcare | Cat# GE17-0891-01 |
| 0.5M EDTA, pH 8.0 | Thermo Fisher Scientific | Cat# 15575020 |
| Ampicillin Trihydrate | Santa Cruz Biotechnology | Cat# sc-254945A |
| Vancomycin Hydrochloride | Chem-Impex International | Cat# 00315 |
| Metronidazole | Sigma | Cat# M1547 |
| Neomycin Trisulfate Salt Hydrate | Sigma | Cat# N6386 |
| Gentamicin Sulfate | Gemini BioProducts | Cat# 400-100P |
| Carmine | Sigma | Cat# C1022 |
| Trizol | Thermo Fisher Scientific | Cat# 15596026 |
| Critical commercial assays | ||
| Live/Dead Fixable Aqua Dead Cell Stain Kit | Thermo Fisher Scientific | Cat# L34957 |
| Foxp3 transcription factor staining buffer set | Thermo Fisher Scientific | Cat# 00-5523-00 |
| BD Fixation/Permeabilization Solution Kit (Cytofix/Cytoperm) | BD Biosciences | Cat# 554714 |
| CGRP ELISA Kit | Cayman Chemical | Cat# 589001 |
| Substance P ELISA Kit | Cayman Chemical | Cat# 583751 |
| SYBR Green Master Mix | Thermo Fisher Scientific | Cat# 4368577 |
| DNeasy PowerSoil Kit | QIAGEN | Cat# 12888 |
| Streptavidin/Biotin Blocking Kit | Vector Laboratories | Cat# SP-2002 |
| RNeasy Plus Mini Kit | QIAGEN | Cat# 74134 |
| High Capacity cDNA Reverse Transcription kit | Thermo Fisher Scientific | Cat# 4368814 |
| Deposited data | ||
| 16S microbiota sequencing | This paper | SRA: PRJNA744090 |
| Experimental models: Organisms/strains | ||
| Mouse: Inhouse B6 | The Jackson Laboratory | N/A |
| Mouse: C57BL/6 | The Jackson Laboratory | RRID: IMSR_JAX:000664 |
| Mouse: Trpv1-Cre | The Jackson Laboratory | RRID: IMSR_JAX:017769 |
| Mouse: tdTomatofl/stop/fl | The Jackson Laboratory | RRID: IMSR_JAX:007914 |
| Mouse: hM4Difl-stop-fl | The Jackson Laboratory | RRID: IMSR_JAX:026219 |
| Mouse: hM3Dqfl-stop-fl | The Jackson Laboratory | RRID: IMSR_JAX:026220 |
| Mouse: Tac1−/− | The Jackson Laboratory | RRID: IMSR_JAX:004103 |
| Mouse: C57BL/6 | Taconic | Stock No. Black 6 (B6NTac) |
| Mouse: Germ-free C57BL/6 | This paper | N/A |
| Oligonucleotides | ||
| Primers for tissue quantitative real-time PCR, see Table S1 | This paper | N/A |
| Mm_Nmu_1_SG QuantiTect Primer Assay (QT00133091) | QIAGEN | Cat# 249900 |
| Primers for fecal bacterial colonization quantitative real-time PCR, see Table S2 | This paper | N/A |
| Software and algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Bowtie2 | Johns Hopkins University | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| FlowJo V9.9.3 software | Tree Star | https://www.flowjo.com/solutions/flowjo/downloads |
| FACS Diva software | BD Biosciences | https://www.flowjo.com/solutions/flowjo/downloads |
| GraphPad Prism 9.0 software | GraphPad | www.graphpad.com |
Fecal 16S amplicon sequencing
Library preparation, sequencing, and operational taxonomic unit (OTU) table generation were performed by the Microbiome Core Lab at Weill Cornell Medicine. Each fecal pellet was deposited into a Qiagen PowerBead glass 0.1 mm tube (13118–50). Using a Promega Maxwell RSC PureFood GMO and Authentication Kit (AS1600), 1 ml of CTAB buffer and 20 μl of RNase A Solution were added to the PowerBead tube containing the sample. The sample/buffer were mixed for 10 seconds on a Vortex Genie2 and then incubated at 95 °C for 5 min on an Eppendorf ThermoMixer F2.0, shaking at 1500 rpm. The tube was removed and clipped to a horizontal microtube attachment on a Vortex Genie2 (SI-H524) and vortexed at high-speed for 20 min. The sample was then removed from the Vortex and centrifuged in an Eppendorf Centrifuge 5430R at 4 °C, 12700 rpm for 10 min. Upon completion, the sample was centrifuged again for an additional 10 min to eliminate foam. The sample tube cap was removed, and the sample checked for foam and particulates. If foam or particulates were found in the sample, they were carefully removed using a P1000 pipette. The opened tube was then added to a Promega MaxPrep Liquid Handler tube rack. The Liquid Handler instrument was loaded with proteinase K tubes, lysis buffer, elution buffer, 1000 ml tips, 50 ml tips, 96-sample deep-well plate, and Promega Maxwell RSC 48 plunger tips. The Promega MaxPrep Liquid Handler instrument was programmed to use 300 μl of sample and transfer all sample lysate into a Promega Maxwell RSC 48 extraction cartridge for DNA extraction. Upon completion, the extraction cartridge was loaded into Promega Maxwell RSC 48 for DNA extraction and elution. DNA was eluted in 100 μl and transferred to a standard 96-well plate. DNA was quantified using a Quant-iT dsDNA High Sensitivity Assay Kit using a Promega GloMax plate reader on a microplate (655087).
Libraries were prepared according to the protocol of the Earth Microbiome Project (https://earthmicrobiome.org/protocols-and-standards/16s/). Amplicon libraries were washed using Beckman Coulter AMPure XP magnetic beads. Library quality and size verification was performed using a PerkinElmer LabChip GXII instrument with DNA 1K Reagent Kit (CLS760673). Library concentrations were quantified using Quant-iT dsDNA High Sensitivity Assay Kit using a Promega GloMax plate reader on a microplate (655087). Library molarity was calculated based on library peak size and concentration. Libraries were normalized to 2nM using the PerkinElmer Zephyr G3 NGS Workstation (133750) and pooled together using the same volume across all normalized libraries into a 1.5 ml Eppendorf DNA tube (022431021). Pooled libraries were sequenced on the Illumina MiSeq instrument at loading concentration of 8pM with 10% PhiX, using MiSeq Reagent Kit v2, 500-cycles (MS-102-2003), producing paired 250 bp reads.
Raw reads were processed and clustered into operational taxonomic units (OTUs) using USEARCH version 11.0.667 (Edgar, 2010). Specifically, reads were demultiplexed and read pairs merged, with a maximum of five mismatching bases in the overlap region, as well as a minimum sequence agreement of 80%. PhiX contaminant sequences were removed, and merged sequences were filtered according to FASTQ quality scores using a maximum expected nominal error number of 1.0 per read. Filtered sequences were clustered into OTUs at a 97% identity threshold using the USEARCH cluster_otus command with default settings. Unfiltered merged reads were mapped to the OTU representative sequences, generating an OTU table. Taxonomic classification of OTU representative sequences was performed with the USEARCH SINTAX command with a confidence score of 0.8, using version 16 of the RDP 16S training set (Cole et al., 2014). A phylogenetic tree was generated from the OTU representative sequences using the USEARCH -cluster_aggd command with default settings. Diversity estimation and principal coordinates analysis (PCoA) ordination were performed using the phyloseq R package version 1.30.0 (McMurdie and Holmes, 2013) after subsampling the OTU table to even depth. To test for clustering of samples by genotype or treatment, the PERMANOVA test was applied using the adonis function of the vegan R package version 2.5.6.
Fecal bacterial colonization analysis
For analysis of bacterial colonization by qPCR, fecal DNA was purified using the PowerSoil kit (Qiagen). Bacterial colonization was quantified using species-specific primers normalized to universal 16S primers which amplify all eubacteria. All primers are listed in Table S2.
QUANTIFICATION AND STATISTICAL ANALYSIS
Analysis of published IBD-seq data
RNAseq gene expression data from a study of the ileal transcriptome in pediatric Crohn disease patients were downloaded from NCBI GEO using the accession number GSE57945 (Haberman et al., 2014). Specifically, expression levels measured in reads per kilobase per million reads mapped (RPKM) as well as sample metadata were downloaded using the R package GEOquery (Davis and Meltzer, 2007). Statistical tests (Kruskal-Wallis with Tukey post-hoc test for differential expression and Spearman correlation test) were performed in R.
Statistical analysis
P value of data sets was determined by unpaired two-tailed Student’s t-test with 95% confidence interval. Normal distribution was assumed. If equal variances between two groups could not be assumed, Welch’s correction was performed. All statistical tests were performed with GraphPad Prism. Error bars depict the SEM.
Data reporting
No statistical methods were used to predetermine sample size. The experiments were not randomized, and the investigators were not blinded to allocation during experiments and outcome assessment. No animals were excluded from the analysis unless clearly indicated.
Supplementary Material
(A) TRPV1hM4Di and their littermate control mice were given DSS in drinking water for 6 days and treated with CNO every other day from day 0 through day 10. (B) Microscopic immunofluorescent visualization of TRPV1hM4Di DRG cells bodies for the pan-neuronal marker βIII-tubulin (green), hM4Di-mCitrine (red) and DAPI (blue). Yellow arrows show representative TRPV1+ DRG cell bodies. (C, D) Immunofluorescence microscopy and quantification of phospho-ERK+ DRGs isolated from TRPV1hM4Di or their littermate control mice treated with CNO at day 10 post DSS administration. (E) Baseline body mass. (F, G) Averaged food intake and water intake per mouse per day during DSS administration. (H) Total GI transit was measured at day 6 post DSS administration. (I to K) Representative plots of colon lamina propria cells isolated from TRPV1hM4Di or their littermate control mice and analyzed for regulatory T cells by flow cytometry on day 10, pre-gated on live, CD45+ CD4+ events. (L) Trpv1-Cre mice were infected with AAV9 viruses carrying the empty vector (mCherry) or inhibitory DREADD (hM4D(Gi)-mCherry) via intra-colonic injection, given DSS in drinking water for 6 days and treated with CNO every day from day 0 through day 10. (M) Microscopic immunofluorescent visualization of DRG cells bodies stained with the pan-neuronal marker βIII-tubulin (green) and mCherry (magenta) from mice in (L). (N, O) Immunofluorescence microscopy and quantification of mCherry+ neurons in lumbar DRG (L5/L6/S1) and nodose vagal ganglion from mice in (L). (P to T) TRPV1hM3Dq and their littermate control mice were given DSS in drinking water for 6 days and treated with CNO every other day from day 0 to day 6. Disease and recovery were evaluated on day 10 with body weight (P), clinical disease score (Q), colon length (R), H&E staining of the distal colon (S) and colonic neutrophil frequency (T). Scale bars = 200 μm in (B), (C), (M) and (N). Scale bars = 50 μm in (S). Data are representative of three independent experiments with n=3–5 mice per group. Data are Mean ± SEM. ns, not significant, * P < 0.05, ** P < 0.01.
(A) SPF B6 mice were treated with DMSO or RTX and rested for 4 weeks for downstream experimental analysis. (B) Baseline body mass. (C) Total GI transit time at steady state. (D to F) Representative plots of colon lamina propria cells isolated from DMSO- and RTX-treated mice and analyzed for regulatory T cells by flow cytometry on day 10 post DSS administration, pre-gated on live, CD45+ CD4+ events. (G to O) Colon lamina propria cells were isolated from DMSO- and RTX-treated mice at steady state and analyzed by flow cytometry for intestinal innate lymphoid cells (G, H), pre-gated on live, CD45+ Lineage− CD127+ CD90+ events; intestinal macrophages (I, J) and neutrophils (K, L), pre-gated on live, CD45+ events; regulatory T cells (M to O), pre-gated on live, CD45+ CD4+ events. (P to R) SPF B6 mice were treated intrathecally with DMSO or RTX and then exposed to DSS for 6 days and disease and recovery were monitored by daily weight loss (P), clinical disease score (Q) and H&E staining of the distal colon (R). Scale bars = 50 μm in (R). Data are representative of three independent experiments with n=3–5 per group. Data are Mean ± SEM. ns, not significant, * P < 0.05, ** P < 0.01.
(A) Fresh cecum content was then harvested from DMSO- and RTX-treated SPF B6 mice and administered to wild-type GF B6 mice by a single oral gavage. After 6 weeks of colonization, fecal microbiota transplant recipient mice were given DSS in drinking water for 6 days and disease parameters were monitored. (B) Averaged relative abundance at the genus level in SPF B6 mice treated intrathecally with DMSO or RTX. (C) GF B6 mice were cohoused with either DMSO- or RTX-treated SPF B6 mice for 4 weeks and DSS administration was then initiated for 6 days. Disease and recovery were monitored. (D) Fecal microbial composition was analyzed by 16S rRNA gene sequencing and principal coordinates analysis in TRPV1hM4Di or their littermate control mice treated with CNO post DSS administration. (E) Averaged relative abundance at the genus level in mice in (D). (F to K) Separately housed and cohoused TRPV1hM4Di and their littermate control mice were given DSS in drinking water for 6 days and treated with CNO every other day from day 0 through day 10 (F). Disease and recovery were monitored by daily weight loss (G), clinical disease score (H), colon length (I), colonic neutrophil frequency (J), and H&E staining of the distal colon (K) at day 10. Scale bars = 50 μm in (K). Data are representative of two independent experiments with n=4–6 mice per group. Data are Mean ± SEM. ns, not significant, * P < 0.05, ** P < 0.01.
(A to D) DMSO- or RTX-treated Taconic B6 mice were exposed to DSS for 6 days with disease and recovery monitored by daily weight loss (A), clinical disease score (B), colon length (C) and H&E staining of the distal colon (D) at day 10. (E) GF B6 mice were colonized with a combination of Clostridium spp., B. theta and E. faecium, or the ASF consortia for 2 weeks, and treated with DMSO or RTX, then rested for 4 weeks before exposure to DSS for 5 days. Disease and recovery were monitored. (F to I) Weight loss (F), clinical disease score (G), colon length (H) and H&E staining of the distal colon (I) in B. theta and E. faecium-colonized ex-GF mice. (J to M) Weight loss (J), clinical disease score (K), colon length (L) and H&E staining of the distal colon (M) in the ASF mice. (N) GF mice and ex-GF mice mono-colonized with either Bacteroides fragillis, Prevotella copri, Clostridium sporogenes, or Clostridium innocuum for 2 weeks were exposed to DSS for 5 days and disease and recovery were monitored daily. (O, P) Daily weight loss (O) and survival (P) in mice in (N). Scale bars = 50 μm in (D), (I) and (M). Data are representative of two independent experiments with n=3–5 per group. Data are Mean ± SEM. ns, not significant, * P < 0.05, ** P < 0.01.
(A) Expression of nociceptive genes and neuropeptides in populations of DRG neurons and intrinsic enteric neurons from a single-cell RNA-Seq dataset of the mouse nervous system, displayed as a heatmap (Zeisel et al., 2018). (B, C) Trpv1 and Tac1 gene expression was assessed in different tissues from SPF B6 mice at steady state. (D) Tac1 and Nmu expression was assessed in the lamina propria and muscularis layers of colons from SPF B6 mice. (E to H) Trpv1-Cre mice were infected with AAV9 viruses carrying the empty vector (mCherry) or inhibitory DREADD (hM4D(Gi)-mCherry) via intra-colonic injection and given DSS in drinking water for 6 days and treated with CNO and substance P (SP) every day from day 0 through day 10. Disease and recovery were monitored by weight loss (E), clinical disease score (F), colon length (G) and H&E staining of the distal colon (H) at day 10. Data are representative of two independent experiments with n=3–5 per group. Scale bars = 50 μm in (H). Data are shown as mean ± SEM. * P < 0.05, ** P < 0.01.
(A to I) Colon lamina propria cells from Tac1+/+ and Tac1−/− mice were isolated and analyzed by flow cytometry at steady state for macrophages (A, B) and neutrophils (C, D), pre-gated on live, CD45+ events; innate lymphoid cells (E, F), pre-gated on live, CD45+ Lineage− CD127+ CD90+ events; regulatory T cells (G to I), pre-gated on live, CD45+ CD4+ events. (J to L) Tac1+/+ and Tac1−/− mice were housed in single cages with free access to food and water, given DSS in drinking water for 6 days. (J) Baseline body mass. (K, L) Averaged food and water intake per mouse per day. (M) Total GI transit was measured at day 6 post DSS administration. (N to U) Tac1+/+ and Tac1−/− mice were exposed to DSS for 6 days. Colon lamina propria neutrophils (N, O) were analyzed by flow cytometry on day 10, pre-gated on live, CD45+ events. Colon lamina propria regulatory T cells (P to R) and T cell cytokine production (S to U) were analyzed by flow cytometry on day 10, pre-gated on live, CD45+ CD4+ events. Data are representative of three independent experiments with n=3–5 per group in (A to R). Data are pooled from three independent experiments with n=4–5 mice per group in each experiment in (T, U). Data are Mean ± SEM. ns, not significant, ** P < 0.01.
A. At steady state, TRPV1+ nociceptors maintain intestinal microbial homeostasis via substance P. B. Chemogenetic silencing or pharmacological ablation of TRPV1+ nociceptors lead to dysbiosis that results in exacerbated inflammation and defective tissue repair upon intestinal damage. C. Clostridium spp. colonization is sufficient to drive exacerbated intestinal inflammation in mice with disrupted nociception. D. Clostridium spp. colonization mediates tissue protection in mice with intact nociception upon intestinal damage.
ACKNOWLEDGEMENTS
We thank the Artis lab members for discussion and critical reading of the manuscript.
Funding:
This work was supported by the grants from the National Institutes of Health (AI172027, AI151599, AI095466, AI095608, AR070116, DK126871, DK132244), LEO foundation, CURE for IBD, Jill Roberts Institute, the Sanders Family, the Rosanne H. Silbermann Family Foundation (all to D.A.), the Crohn’s and Colitis Foundation Research Fellowship Award (901000 to W.Z.; 527125 to A.M.T.), the Sackler Brain and Spine Institute Research Grant (to C.C), the National Institutes of Health (F32AI124517) (to N.J.B.), the National Institutes of Health (DP2 HD101401-01), AGA Research Foundation and The W. M. Keck Foundation (all to C.J.G.), the National Institutes of Health (DK103901, AR077183, and AA027065) (all to H.H.). We also thank patients who donated samples for this study and members of the JRI IBD Live Cell Bank Consortium for patient recruitment and collection of human samples (see below). Diagrams in the figures were created with BioRender.com.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONSORTIA
The members of the JRI Live Cell Bank consortium are: David Artis, Randy Longman, Gregory Sonnenberg, Ellen Scherl, Robbyn Sockolow, Dana Lukin, Robert Battat, Thomas Ciecierega, Aliza Solomon, Elaine Barfield, Kimberley Chien, Johanna Ferriera, Jasmin Williams, Shaira Khan, Peik Sean Chong, Samah Mozumder, Lance Chou, Wenqing Zhou, Anees Ahmed, Connie Zhong, Ann Joseph, Sanchita Kashyap, Joseph Gladstone and Samantha Jensen.
DECLARATION OF INTERESTES
D.A. has contributed to scientific advisory boards at Pfizer, Takeda and the KRF. The other authors declare no competing interests.
REFERENCES
- Abraham C, and Cho JH (2009). Inflammatory bowel disease. N Engl J Med 361, 2066–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alenghat T, Osborne LC, Saenz SA, Kobuley D, Ziegler CG, Mullican SE, Choi I, Grunberg S, Sinha R, Wynosky-Dolfi M, et al. (2013). Histone deacetylase 3 coordinates commensal-bacteria-dependent intestinal homeostasis. Nature 504, 153–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansaldo E, Farley TK, and Belkaid Y (2021). Control of Immunity by the Microbiota. Annu Rev Immunol 39, 449–479. [DOI] [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K, et al. (2013). Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 500, 232–236. [DOI] [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, Cheng G, Yamasaki S, Saito T, Ohba Y, et al. (2011). Induction of colonic regulatory T cells by indigenous Clostridium species. Science 331, 337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baral P, Udit S, and Chiu IM (2019). Pain and immunity: implications for host defence. Nat Rev Immunol 19, 433–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baral P, Umans BD, Li L, Wallrapp A, Bist M, Kirschbaum T, Wei Y, Zhou Y, Kuchroo VK, Burkett PR, et al. (2018). Nociceptor sensory neurons suppress neutrophil and gammadelta T cell responses in bacterial lung infections and lethal pneumonia. Nat Med 24, 417–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basbaum AI, Bautista DM, Scherrer G, and Julius D (2009). Cellular and molecular mechanisms of pain. Cell 139, 267–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgart DC, and Sandborn WJ (2012). Crohn’s disease. Lancet 380, 1590–1605. [DOI] [PubMed] [Google Scholar]
- Belkaid Y, and Hand TW (2014). Role of the microbiota in immunity and inflammation. Cell 157, 121–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessman NJ, Mathieu JRR, Renassia C, Zhou L, Fung TC, Fernandez KC, Austin C, Moeller JB, Zumerle S, Louis S, et al. (2020). Dendritic cell-derived hepcidin sequesters iron from the microbiota to promote mucosal healing. Science 368, 186–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielefeldt K, Davis B, and Binion DG (2009). Pain and inflammatory bowel disease. Inflamm Bowel Dis 15, 778–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billing LJ, Larraufie P, Lewis J, Leiter A, Li J, Lam B, Yeo GS, Goldspink DA, Kay RG, Gribble FM, et al. (2019). Single cell transcriptomic profiling of large intestinal enteroendocrine cells in mice - Identification of selective stimuli for insulin-like peptide-5 and glucagon-like peptide-1 co-expressing cells. Mol Metab 29, 158–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blander JM, Longman RS, Iliev ID, Sonnenberg GF, and Artis D (2017). Regulation of inflammation by microbiota interactions with the host. Nat Immunol 18, 851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brain SD, and Williams TJ (1989). Interactions between the tachykinins and calcitonin gene-related peptide lead to the modulation of oedema formation and blood flow in rat skin. Br J Pharmacol 97, 77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brestoff JR, and Artis D (2013). Commensal bacteria at the interface of host metabolism and the immune system. Nat Immunol 14, 676–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EM, Kenny DJ, and Xavier RJ (2019). Gut Microbiota Regulation of T Cells During Inflammation and Autoimmunity. Annu Rev Immunol 37, 599–624. [DOI] [PubMed] [Google Scholar]
- Chang JT (2020). Pathophysiology of Inflammatory Bowel Diseases. N Engl J Med 383, 2652–2664. [DOI] [PubMed] [Google Scholar]
- Chavan SS, Pavlov VA, and Tracey KJ (2017). Mechanisms and Therapeutic Relevance of Neuro-immune Communication. Immunity 46, 927–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu IM, Heesters BA, Ghasemlou N, Von Hehn CA, Zhao F, Tran J, Wainger B, Strominger A, Muralidharan S, Horswill AR, et al. (2013). Bacteria activate sensory neurons that modulate pain and inflammation. Nature 501, 52–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C, Artis D, and Chiu IM (2020). Neuro-immune Interactions in the Tissues. Immunity 52, 464–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C, Murdock MH, Jing D, Won TH, Chung H, Kressel AM, Tsaava T, Addorisio ME, Putzel GG, Zhou L, et al. (2019). The microbiota regulate neuronal function and fear extinction learning. Nature 574, 543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemente JC, Ursell LK, Parfrey LW, and Knight R (2012). The impact of the gut microbiota on human health: an integrative view. Cell 148, 1258–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JA, Edwards TN, Liu AW, Hirai T, Jones MR, Wu J, Li Y, Zhang S, Ho J, Davis BM, et al. (2019). Cutaneous TRPV1(+) Neurons Trigger Protective Innate Type 17 Anticipatory Immunity. Cell 178, 919–932 e914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, Brown CT, Porras-Alfaro A, Kuske CR, and Tiedje JM (2014). Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res 42, D633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyphert JM, Kovarova M, Allen IC, Hartney JM, Murphy DL, Wess J, and Koller BH (2009). Cooperation between mast cells and neurons is essential for antigen-mediated bronchoconstriction. J Immunol 182, 7430–7439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabke K, Hendrick G, and Devkota S (2019). The gut microbiome and metabolic syndrome. J Clin Invest 129, 4050–4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlhamer J, Lucas J, Zelaya C, Nahin R, Mackey S, DeBar L, Kerns R, Von Korff M, Porter L, and Helmick C (2018). Prevalence of Chronic Pain and High-Impact Chronic Pain Among Adults - United States, 2016. MMWR Morb Mortal Wkly Rep 67, 1001–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danese S, and Fiocchi C (2011). Ulcerative colitis. N Engl J Med 365, 1713–1725. [DOI] [PubMed] [Google Scholar]
- Davis S, and Meltzer PS (2007). GEOquery: a bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 23, 1846–1847. [DOI] [PubMed] [Google Scholar]
- Drokhlyansky E, Smillie CS, Van Wittenberghe N, Ericsson M, Griffin GK, Eraslan G, Dionne D, Cuoco MS, Goder-Reiser MN, Sharova T, et al. (2020). The Human and Mouse Enteric Nervous System at Single-Cell Resolution. Cell 182, 1606–1622 e1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. [DOI] [PubMed] [Google Scholar]
- Edvinsson L, Ekman R, Jansen I, McCulloch J, and Uddman R (1987). Calcitonin gene-related peptide and cerebral blood vessels: distribution and vasomotor effects. J Cereb Blood Flow Metab 7, 720–728. [DOI] [PubMed] [Google Scholar]
- Elekes K, Helyes Z, Nemeth J, Sandor K, Pozsgai G, Kereskai L, Borzsei R, Pinter E, Szabo A, and Szolcsanyi J (2007). Role of capsaicin-sensitive afferents and sensory neuropeptides in endotoxin-induced airway inflammation and consequent bronchial hyperreactivity in the mouse. Regul Pept 141, 44–54. [DOI] [PubMed] [Google Scholar]
- Ferguson SM, Eskenazi D, Ishikawa M, Wanat MJ, Phillips PE, Dong Y, Roth BL, and Neumaier JF (2011). Transient neuronal inhibition reveals opposing roles of indirect and direct pathways in sensitization. Nat Neurosci 14, 22–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster SL, Seehus CR, Woolf CJ, and Talbot S (2017). Sense and Immunity: Context-Dependent Neuro-Immune Interplay. Front Immunol 8, 1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich M, Pohin M, and Powrie F (2019). Cytokine Networks in the Pathophysiology of Inflammatory Bowel Disease. Immunity 50, 992–1006. [DOI] [PubMed] [Google Scholar]
- Fung TC, Olson CA, and Hsiao EY (2017). Interactions between the microbiota, immune and nervous systems in health and disease. Nat Neurosci 20, 145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore R, Riedl MS, Kitto KF, Fairbanks CA, and Vulchanova L (2019). AAV-Mediated Gene Delivery to the Enteric Nervous System by Intracolonic Injection. Methods Mol Biol 1950, 407–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo CJ, Mack MR, Oetjen LK, Trier AM, Council ML, Pavel AB, Guttman-Yassky E, Kim BS, and Liu Q (2020). Kallikrein 7 Promotes Atopic Dermatitis-Associated Itch Independently of Skin Inflammation. J Invest Dermatol 140, 1244–1252 e1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo R, Chen LH, Xing C, and Liu T (2019a). Pain regulation by gut microbiota: molecular mechanisms and therapeutic potential. Br J Anaesth 123, 637–654. [DOI] [PubMed] [Google Scholar]
- Guo T, Bian Z, Trocki K, Chen L, Zheng G, and Feng B (2019b). Optical recording reveals topological distribution of functionally classified colorectal afferent neurons in intact lumbosacral DRG. Physiol Rep 7, e14097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberman Y, Tickle TL, Dexheimer PJ, Kim MO, Tang D, Karns R, Baldassano RN, Noe JD, Rosh J, Markowitz J, et al. (2014). Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J Clin Invest 124, 3617–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill DA, and Artis D (2010). Intestinal bacteria and the regulation of immune cell homeostasis. Annu Rev Immunol 28, 623–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill DA, Hoffmann C, Abt MC, Du Y, Kobuley D, Kirn TJ, Bushman FD, and Artis D (2010). Metagenomic analyses reveal antibiotic-induced temporal and spatial changes in intestinal microbiota with associated alterations in immune cell homeostasis. Mucosal Immunol 3, 148–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill DA, Siracusa MC, Abt MC, Kim BS, Kobuley D, Kubo M, Kambayashi T, Larosa DF, Renner ED, Orange JS, et al. (2012). Commensal bacteria-derived signals regulate basophil hematopoiesis and allergic inflammation. Nat Med 18, 538–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper LV, Littman DR, and Macpherson AJ (2012). Interactions between the microbiota and the immune system. Science 336, 1268–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh JR, and Veiga-Fernandes H (2020). Neuroimmune circuits in inter-organ communication. Nat Rev Immunol 20, 217–228. [DOI] [PubMed] [Google Scholar]
- Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, et al. (2009). Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin WB, Li TT, Huo D, Qu S, Li XV, Arifuzzaman M, Lima SF, Shi HQ, Wang A, Putzel GG, et al. (2022). Genetic manipulation of gut microbes enables single-gene interrogation in a complex microbiome. Cell 185, 547–562 e522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada N, Seo SU, Chen GY, and Nunez G (2013). Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol 13, 321–335. [DOI] [PubMed] [Google Scholar]
- Kaser A, Zeissig S, and Blumberg RS (2010). Inflammatory bowel disease. Annu Rev Immunol 28, 573–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashem SW, Riedl MS, Yao C, Honda CN, Vulchanova L, and Kaplan DH (2015). Nociceptive Sensory Fibers Drive Interleukin-23 Production from CD301b+ Dermal Dendritic Cells and Drive Protective Cutaneous Immunity. Immunity 43, 515–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krashes MJ, Koda S, Ye C, Rogan SC, Adams AC, Cusher DS, Maratos-Flier E, Roth BL, and Lowell BB (2011). Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J Clin Invest 121, 1424–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyloh M, Nicholas S, Zagorodnyuk VP, Brookes SJ, and Spencer NJ (2011). Identification of the visceral pain pathway activated by noxious colorectal distension in mice. Front Neurosci 5, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai NY, Musser MA, Pinho-Ribeiro FA, Baral P, Jacobson A, Ma P, Potts DE, Chen Z, Paik D, Soualhi S, et al. (2020). Gut-Innervating Nociceptor Neurons Regulate Peyer’s Patch Microfold Cells and SFB Levels to Mediate Salmonella Host Defense. Cell 180, 33–49 e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JE, Chung E, and Son Y (2017). A neuropeptide, Substance-P, directly induces tissue-repairing M2 like macrophages by activating the PI3K/Akt/mTOR pathway even in the presence of IFNgamma. Sci Rep 7, 9417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q, and Stappenbeck TS (2022). Local barriers configure systemic communications between the host and microbiota. Science 376, 950–955. [DOI] [PubMed] [Google Scholar]
- Mashaghi A, Marmalidou A, Tehrani M, Grace PM, Pothoulakis C, and Dana R (2016). Neuropeptide substance P and the immune response. Cell Mol Life Sci 73, 4249–4264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack DG, Mak JC, Coupe MO, and Barnes PJ (1989). Calcitonin gene-related peptide vasodilation of human pulmonary vessels. J Appl Physiol (1985) 67, 1265–1270. [DOI] [PubMed] [Google Scholar]
- McMurdie PJ, and Holmes S (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8, e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meerschaert KA, Adelman PC, Friedman RL, Albers KM, Koerber HR, and Davis BM (2020). Unique Molecular Characteristics of Visceral Afferents Arising from Different Levels of the Neuraxis: Location of Afferent Somata Predicts Function and Stimulus Detection Modalities. J Neurosci 40, 7216–7228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra SK, and Hoon MA (2010). Ablation of TrpV1 neurons reveals their selective role in thermal pain sensation. Mol Cell Neurosci 43, 157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monticelli LA, Osborne LC, Noti M, Tran SV, Zaiss DM, and Artis D (2015). IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc Natl Acad Sci U S A 112, 10762–10767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morais LH, Schreiber H.L.t., and Mazmanian SK (2021). The gut microbiota-brain axis in behaviour and brain disorders. Nat Rev Microbiol 19, 241–255. [DOI] [PubMed] [Google Scholar]
- Morarach K, Mikhailova A, Knoflach V, Memic F, Kumar R, Li W, Ernfors P, and Marklund U (2021). Diversification of molecularly defined myenteric neuron classes revealed by single-cell RNA sequencing. Nat Neurosci 24, 34–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagashima H, Mahlakoiv T, Shih HY, Davis FP, Meylan F, Huang Y, Harrison OJ, Yao C, Mikami Y, Urban JF Jr., et al. (2019). Neuropeptide CGRP Limits Group 2 Innate Lymphoid Cell Responses and Constrains Type 2 Inflammation. Immunity 51, 682–695 e686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neurath MF (2014). Cytokines in inflammatory bowel disease. Nat Rev Immunol 14, 329–342. [DOI] [PubMed] [Google Scholar]
- Ng SC, Shi HY, Hamidi N, Underwood FE, Tang W, Benchimol EI, Panaccione R, Ghosh S, Wu JCY, Chan FKL, et al. (2017). Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet 390, 2769–2778. [DOI] [PubMed] [Google Scholar]
- Ni J, Wu GD, Albenberg L, and Tomov VT (2017). Gut microbiota and IBD: causation or correlation? Nat Rev Gastroenterol Hepatol 14, 573–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paone P, and Cani PD (2020). Mucus barrier, mucins and gut microbiota: the expected slimy partners? Gut 69, 2232–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perner C, Flayer CH, Zhu X, Aderhold PA, Dewan ZNA, Voisin T, Camire RB, Chow OA, Chiu IM, and Sokol CL (2020). Substance P Release by Sensory Neurons Triggers Dendritic Cell Migration and Initiates the Type-2 Immune Response to Allergens. Immunity 53, 1063–1077 e1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinho-Ribeiro FA, Baddal B, Haarsma R, O’Seaghdha M, Yang NJ, Blake KJ, Portley M, Verri WA, Dale JB, Wessels MR, et al. (2018). Blocking Neuronal Signaling to Immune Cells Treats Streptococcal Invasive Infection. Cell 173, 1083–1097 e1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, and Medzhitov R (2004). Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 118, 229–241. [DOI] [PubMed] [Google Scholar]
- Riol-Blanco L, Ordovas-Montanes J, Perro M, Naval E, Thiriot A, Alvarez D, Paust S, Wood JN, and von Andrian UH (2014). Nociceptive sensory neurons drive interleukin-23-mediated psoriasiform skin inflammation. Nature 510, 157–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooks MG, and Garrett WS (2016). Gut microbiota, metabolites and host immunity. Nat Rev Immunol 16, 341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh M, and Elson CO (2011). Experimental inflammatory bowel disease: insights into the host-microbiota dialog. Immunity 34, 293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saria A (1984). Substance P in sensory nerve fibres contributes to the development of oedema in the rat hind paw after thermal injury. Br J Pharmacol 82, 217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaedler RW, and Dubos RJ (1962). The fecal flora of various strains of mice. Its bearing on their susceptibility to endotoxin. J Exp Med 115, 1149–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz J, and Woolf CJ (2002). Can we conquer pain? Nat Neurosci 5 Suppl, 1062–1067. [DOI] [PubMed] [Google Scholar]
- Serhan N, Basso L, Sibilano R, Petitfils C, Meixiong J, Bonnart C, Reber LL, Marichal T, Starkl P, Cenac N, et al. (2019). House dust mites activate nociceptor-mast cell clusters to drive type 2 skin inflammation. Nat Immunol 20, 1435–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skorput AGJ, Gore R, Schorn R, Riedl MS, Marron Fernandez de Velasco E, Hadlich B, Kitto KF, Fairbanks CA, and Vulchanova L (2022). Targeting the somatosensory system with AAV9 and AAV2retro viral vectors. PLoS One 17, e0264938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speakman JR (2013). Measuring energy metabolism in the mouse - theoretical, practical, and analytical considerations. Front Physiol 4, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Ramnath RD, Zhi L, Tamizhselvi R, and Bhatia M (2008). Substance P enhances NF-kappaB transactivation and chemokine response in murine macrophages via ERK1/2 and p38 MAPK signaling pathways. Am J Physiol Cell Physiol 294, C1586–1596. [DOI] [PubMed] [Google Scholar]
- Suvas S (2017). Role of Substance P Neuropeptide in Inflammation, Wound Healing, and Tissue Homeostasis. J Immunol 199, 1543–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot S, Abdulnour RE, Burkett PR, Lee S, Cronin SJ, Pascal MA, Laedermann C, Foster SL, Tran JV, Lai N, et al. (2015). Silencing Nociceptor Neurons Reduces Allergic Airway Inflammation. Neuron 87, 341–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan LL, Bornstein JC, and Anderson CR (2008). Distinct chemical classes of medium-sized transient receptor potential channel vanilloid 1-immunoreactive dorsal root ganglion neurons innervate the adult mouse jejunum and colon. Neuroscience 156, 334–343. [DOI] [PubMed] [Google Scholar]
- Tang C, Kamiya T, Liu Y, Kadoki M, Kakuta S, Oshima K, Hattori M, Takeshita K, Kanai T, Saijo S, et al. (2015). Inhibition of Dectin-1 Signaling Ameliorates Colitis by Inducing Lactobacillus-Mediated Regulatory T Cell Expansion in the Intestine. Cell Host Microbe 18, 183–197. [DOI] [PubMed] [Google Scholar]
- Tilg H, Zmora N, Adolph TE, and Elinav E (2020). The intestinal microbiota fuelling metabolic inflammation. Nat Rev Immunol 20, 40–54. [DOI] [PubMed] [Google Scholar]
- Torres J, Mehandru S, Colombel JF, and Peyrin-Biroulet L (2017). Crohn’s disease. Lancet 389, 1741–1755. [DOI] [PubMed] [Google Scholar]
- Troy AE, Zaph C, Du Y, Taylor BC, Guild KJ, Hunter CA, Saris CJ, and Artis D (2009). IL-27 regulates homeostasis of the intestinal CD4+ effector T cell pool and limits intestinal inflammation in a murine model of colitis. J Immunol 183, 2037–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlig HH, and Powrie F (2018). Translating Immunology into Therapeutic Concepts for Inflammatory Bowel Disease. Annu Rev Immunol 36, 755–781. [DOI] [PubMed] [Google Scholar]
- Ungaro R, Mehandru S, Allen PB, Peyrin-Biroulet L, and Colombel JF (2017). Ulcerative colitis. Lancet 389, 1756–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban DJ, and Roth BL (2015). DREADDs (designer receptors exclusively activated by designer drugs): chemogenetic tools with therapeutic utility. Annu Rev Pharmacol Toxicol 55, 399–417. [DOI] [PubMed] [Google Scholar]
- Veiga-Fernandes H, and Mucida D (2016). Neuro-Immune Interactions at Barrier Surfaces. Cell 165, 801–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuong HE, Yano JM, Fung TC, and Hsiao EY (2017). The Microbiome and Host Behavior. Annu Rev Neurosci 40, 21–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallrapp A, Burkett PR, Riesenfeld SJ, Kim SJ, Christian E, Abdulnour RE, Thakore PI, Schnell A, Lambden C, Herbst RH, et al. (2019). Calcitonin Gene-Related Peptide Negatively Regulates Alarmin-Driven Type 2 Innate Lymphoid Cell Responses. Immunity 51, 709–723 e706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir JB (1949). New methods for calculating metabolic rate with special reference to protein metabolism. J Physiol 109, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirtz S, Popp V, Kindermann M, Gerlach K, Weigmann B, Fichtner-Feigl S, and Neurath MF (2017). Chemically induced mouse models of acute and chronic intestinal inflammation. Nat Protoc 12, 1295–1309. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, and Ma Q (2007). Nociceptors--noxious stimulus detectors. Neuron 55, 353–364. [DOI] [PubMed] [Google Scholar]
- Xu H, Ding J, Porter CBM, Wallrapp A, Tabaka M, Ma S, Fu S, Guo X, Riesenfeld SJ, Su C, et al. (2019). Transcriptional Atlas of Intestinal Immune Cells Reveals that Neuropeptide alpha-CGRP Modulates Group 2 Innate Lymphoid Cell Responses. Immunity 51, 696–708 e699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano H, and Artis D (2022). Neuronal regulation of innate lymphoid cell responses. Curr Opin Immunol 76, 102205. [DOI] [PubMed] [Google Scholar]
- Yissachar N, Zhou Y, Ung L, Lai NY, Mohan JF, Ehrlicher A, Weitz DA, Kasper DL, Chiu IM, Mathis D, et al. (2017). An Intestinal Organ Culture System Uncovers a Role for the Nervous System in Microbe-Immune Crosstalk. Cell 168, 1135–1148 e1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo BB, and Mazmanian SK (2017). The Enteric Network: Interactions between the Immune and Nervous Systems of the Gut. Immunity 46, 910–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel A, Hochgerner H, Lonnerberg P, Johnsson A, Memic F, van der Zwan J, Haring M, Braun E, Borm LE, La Manno G, et al. (2018). Molecular Architecture of the Mouse Nervous System. Cell 174, 999–1014 e1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Liu R, Che D, Pundir P, Wang N, Han S, Cao J, Lv Y, Dong H, Fang F, et al. (2019). A Mast Cell-Specific Receptor Is Critical for Granuloma Induced by Intrathecal Morphine Infusion. J Immunol 203, 1701–1714. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) TRPV1hM4Di and their littermate control mice were given DSS in drinking water for 6 days and treated with CNO every other day from day 0 through day 10. (B) Microscopic immunofluorescent visualization of TRPV1hM4Di DRG cells bodies for the pan-neuronal marker βIII-tubulin (green), hM4Di-mCitrine (red) and DAPI (blue). Yellow arrows show representative TRPV1+ DRG cell bodies. (C, D) Immunofluorescence microscopy and quantification of phospho-ERK+ DRGs isolated from TRPV1hM4Di or their littermate control mice treated with CNO at day 10 post DSS administration. (E) Baseline body mass. (F, G) Averaged food intake and water intake per mouse per day during DSS administration. (H) Total GI transit was measured at day 6 post DSS administration. (I to K) Representative plots of colon lamina propria cells isolated from TRPV1hM4Di or their littermate control mice and analyzed for regulatory T cells by flow cytometry on day 10, pre-gated on live, CD45+ CD4+ events. (L) Trpv1-Cre mice were infected with AAV9 viruses carrying the empty vector (mCherry) or inhibitory DREADD (hM4D(Gi)-mCherry) via intra-colonic injection, given DSS in drinking water for 6 days and treated with CNO every day from day 0 through day 10. (M) Microscopic immunofluorescent visualization of DRG cells bodies stained with the pan-neuronal marker βIII-tubulin (green) and mCherry (magenta) from mice in (L). (N, O) Immunofluorescence microscopy and quantification of mCherry+ neurons in lumbar DRG (L5/L6/S1) and nodose vagal ganglion from mice in (L). (P to T) TRPV1hM3Dq and their littermate control mice were given DSS in drinking water for 6 days and treated with CNO every other day from day 0 to day 6. Disease and recovery were evaluated on day 10 with body weight (P), clinical disease score (Q), colon length (R), H&E staining of the distal colon (S) and colonic neutrophil frequency (T). Scale bars = 200 μm in (B), (C), (M) and (N). Scale bars = 50 μm in (S). Data are representative of three independent experiments with n=3–5 mice per group. Data are Mean ± SEM. ns, not significant, * P < 0.05, ** P < 0.01.
(A) SPF B6 mice were treated with DMSO or RTX and rested for 4 weeks for downstream experimental analysis. (B) Baseline body mass. (C) Total GI transit time at steady state. (D to F) Representative plots of colon lamina propria cells isolated from DMSO- and RTX-treated mice and analyzed for regulatory T cells by flow cytometry on day 10 post DSS administration, pre-gated on live, CD45+ CD4+ events. (G to O) Colon lamina propria cells were isolated from DMSO- and RTX-treated mice at steady state and analyzed by flow cytometry for intestinal innate lymphoid cells (G, H), pre-gated on live, CD45+ Lineage− CD127+ CD90+ events; intestinal macrophages (I, J) and neutrophils (K, L), pre-gated on live, CD45+ events; regulatory T cells (M to O), pre-gated on live, CD45+ CD4+ events. (P to R) SPF B6 mice were treated intrathecally with DMSO or RTX and then exposed to DSS for 6 days and disease and recovery were monitored by daily weight loss (P), clinical disease score (Q) and H&E staining of the distal colon (R). Scale bars = 50 μm in (R). Data are representative of three independent experiments with n=3–5 per group. Data are Mean ± SEM. ns, not significant, * P < 0.05, ** P < 0.01.
(A) Fresh cecum content was then harvested from DMSO- and RTX-treated SPF B6 mice and administered to wild-type GF B6 mice by a single oral gavage. After 6 weeks of colonization, fecal microbiota transplant recipient mice were given DSS in drinking water for 6 days and disease parameters were monitored. (B) Averaged relative abundance at the genus level in SPF B6 mice treated intrathecally with DMSO or RTX. (C) GF B6 mice were cohoused with either DMSO- or RTX-treated SPF B6 mice for 4 weeks and DSS administration was then initiated for 6 days. Disease and recovery were monitored. (D) Fecal microbial composition was analyzed by 16S rRNA gene sequencing and principal coordinates analysis in TRPV1hM4Di or their littermate control mice treated with CNO post DSS administration. (E) Averaged relative abundance at the genus level in mice in (D). (F to K) Separately housed and cohoused TRPV1hM4Di and their littermate control mice were given DSS in drinking water for 6 days and treated with CNO every other day from day 0 through day 10 (F). Disease and recovery were monitored by daily weight loss (G), clinical disease score (H), colon length (I), colonic neutrophil frequency (J), and H&E staining of the distal colon (K) at day 10. Scale bars = 50 μm in (K). Data are representative of two independent experiments with n=4–6 mice per group. Data are Mean ± SEM. ns, not significant, * P < 0.05, ** P < 0.01.
(A to D) DMSO- or RTX-treated Taconic B6 mice were exposed to DSS for 6 days with disease and recovery monitored by daily weight loss (A), clinical disease score (B), colon length (C) and H&E staining of the distal colon (D) at day 10. (E) GF B6 mice were colonized with a combination of Clostridium spp., B. theta and E. faecium, or the ASF consortia for 2 weeks, and treated with DMSO or RTX, then rested for 4 weeks before exposure to DSS for 5 days. Disease and recovery were monitored. (F to I) Weight loss (F), clinical disease score (G), colon length (H) and H&E staining of the distal colon (I) in B. theta and E. faecium-colonized ex-GF mice. (J to M) Weight loss (J), clinical disease score (K), colon length (L) and H&E staining of the distal colon (M) in the ASF mice. (N) GF mice and ex-GF mice mono-colonized with either Bacteroides fragillis, Prevotella copri, Clostridium sporogenes, or Clostridium innocuum for 2 weeks were exposed to DSS for 5 days and disease and recovery were monitored daily. (O, P) Daily weight loss (O) and survival (P) in mice in (N). Scale bars = 50 μm in (D), (I) and (M). Data are representative of two independent experiments with n=3–5 per group. Data are Mean ± SEM. ns, not significant, * P < 0.05, ** P < 0.01.
(A) Expression of nociceptive genes and neuropeptides in populations of DRG neurons and intrinsic enteric neurons from a single-cell RNA-Seq dataset of the mouse nervous system, displayed as a heatmap (Zeisel et al., 2018). (B, C) Trpv1 and Tac1 gene expression was assessed in different tissues from SPF B6 mice at steady state. (D) Tac1 and Nmu expression was assessed in the lamina propria and muscularis layers of colons from SPF B6 mice. (E to H) Trpv1-Cre mice were infected with AAV9 viruses carrying the empty vector (mCherry) or inhibitory DREADD (hM4D(Gi)-mCherry) via intra-colonic injection and given DSS in drinking water for 6 days and treated with CNO and substance P (SP) every day from day 0 through day 10. Disease and recovery were monitored by weight loss (E), clinical disease score (F), colon length (G) and H&E staining of the distal colon (H) at day 10. Data are representative of two independent experiments with n=3–5 per group. Scale bars = 50 μm in (H). Data are shown as mean ± SEM. * P < 0.05, ** P < 0.01.
(A to I) Colon lamina propria cells from Tac1+/+ and Tac1−/− mice were isolated and analyzed by flow cytometry at steady state for macrophages (A, B) and neutrophils (C, D), pre-gated on live, CD45+ events; innate lymphoid cells (E, F), pre-gated on live, CD45+ Lineage− CD127+ CD90+ events; regulatory T cells (G to I), pre-gated on live, CD45+ CD4+ events. (J to L) Tac1+/+ and Tac1−/− mice were housed in single cages with free access to food and water, given DSS in drinking water for 6 days. (J) Baseline body mass. (K, L) Averaged food and water intake per mouse per day. (M) Total GI transit was measured at day 6 post DSS administration. (N to U) Tac1+/+ and Tac1−/− mice were exposed to DSS for 6 days. Colon lamina propria neutrophils (N, O) were analyzed by flow cytometry on day 10, pre-gated on live, CD45+ events. Colon lamina propria regulatory T cells (P to R) and T cell cytokine production (S to U) were analyzed by flow cytometry on day 10, pre-gated on live, CD45+ CD4+ events. Data are representative of three independent experiments with n=3–5 per group in (A to R). Data are pooled from three independent experiments with n=4–5 mice per group in each experiment in (T, U). Data are Mean ± SEM. ns, not significant, ** P < 0.01.
A. At steady state, TRPV1+ nociceptors maintain intestinal microbial homeostasis via substance P. B. Chemogenetic silencing or pharmacological ablation of TRPV1+ nociceptors lead to dysbiosis that results in exacerbated inflammation and defective tissue repair upon intestinal damage. C. Clostridium spp. colonization is sufficient to drive exacerbated intestinal inflammation in mice with disrupted nociception. D. Clostridium spp. colonization mediates tissue protection in mice with intact nociception upon intestinal damage.
Data Availability Statement
All data including raw sequencing reads are uploaded to the SRA and are publicly available as of the date of publication. The accession numbers are listed in the Key resources table. The paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request. Microscopy data reported in this paper will be shared by the lead contact upon request.