

ABSTRACT
Immune cell-type composition changes with age, potentially weakening the response to infectious diseases. Profiling epigenetics marks of immune cells can help us understand the relationship with disease severity. We therefore leveraged a targeted DNA methylation method to study the differences in a cohort of pneumonia patients (both COVID-19 positive and negative) and unaffected individuals from peripheral blood.
This approach allowed us to predict the pneumonia diagnosis with high accuracy (AUC = 0.92), and the PCR positivity to the SARS-CoV-2 viral genome with moderate, albeit lower, accuracy (AUC = 0.77). We were also able to predict the severity of pneumonia (PORT score) with an R2 = 0.69. By estimating immune cellular frequency from DNA methylation data, patients under the age of 65 positive to the SARS-CoV-2 genome (as revealed by PCR) showed an increase in T cells, and specifically in CD8+ cells, compared to the negative control group. Conversely, we observed a decreased frequency of neutrophils in the positive compared to the negative group. No significant difference was found in patients over the age of 65. The results suggest that this DNA methylation-based approach can be used as a cost-effective and clinically useful biomarker platform for predicting pneumonias and their severity.
KEYWORDS: DNA methylation, pneumonia, SARS-CoV-2, targeted bisulfite sequencing, cell-type deconvolution, biomarkers
Introduction
Covid-19 infection was declared a global pandemic by WHO on 11 March 2020 [1]. It is transmitted from person to person through droplets and progresses asymptomatically in 70% of the infected individuals. By contrast, in the symptomatic group it may manifest itself with mild or severe symptoms [2]. In cases with mild symptoms, upper respiratory tract symptoms such as fever, dry cough, and fatigue may develop, and abnormal chest CT findings may also be present. In cases with severe symptoms, dyspnoea, diarrhoea, severe pneumonia, acute respiratory distress syndrome (ARDS) or multiple-organ failure develop, and mortality rates vary between 4.3% and 15% according to different reports [3,4].
The most widely reported risk factor for developing severe COVID-19 symptoms is chronological age [5]. Immune cell composition changes with age, and it can potentially compromise the immune response, including the adaptive immune response, to infectious disease [6]. Cell type composition is reflected in epigenetic studies that have elucidated molecular changes underlying cancer and infectious diseases [7,8]. Evaluating the DNA methylation of immune cells during and after infection can help explain how the epigenome reflects disease severity. Previous work has suggested the vulnerability of the elderly to severe Covid-19 may be related to the effect of the epigenome on viral entry [5]. Epigenetic profiling might help elucidate molecular changes induced by viruses as well as host–virus interactions, including genetic factors that contribute to the protective or pathogenic host responses [9].
To date, only a few studies have assessed the DNA methylation levels in COVID-19 infected subjects [10–13]. All the studies used array-based genome-wide approaches (Infinium MethylationEPIC Array) to identify the DNA methylation sites associated with COVID-19 disease or susceptibility to it. In these studies, all of the significantly different DNA methylation sites identified between COVID-19 positive and the control group are directly or indirectly associated with the interferon signalling pathway and the viral response.
In contrast to the abovementioned studies that utilized DNA methylation arrays (testing the methylation levels of approximately 850ʹ000 CpG sites) for de novo discovery purposes, we decided to leverage a cost-effective targeted DNA methylation approach [14] to provide insights into the epigenetic effects that distinguish patients with respiratory diseases in comparison to unaffected individuals based on a custom panel. We first constructed a panel of sites that were likely to be impacted by viral infection based on previous literature (mostly based on transcriptomic studies). Moreover, we included sites able to discriminate between immune cell types and epigenetic-age predictors, based on DNA methylation data present in the literature. We then used this panel to perform targeted bisulfite sequencing allowing us to measure the methylation with high accuracy at approximately 5000 regions. Among our most interesting findings, we show that the changes in methylation at these regions are associated with the variability of immune cell composition across our cohort, as well as the severity of pneumonia. In this study, we demonstrate that our approach informs about the changes related to respiratory diseases and it can be used to predict disease state, despite not interrogating comprehensively the CpG methylation levels in the genome. Thus, DNA methylation profiling provides insights into immune responses and enables the creation of clinically useful biomarkers.
Results
Demographic and clinical features
Demographic and clinical features of our study cohort are shown in Table 1.
Table 1.
Clinical and demographical data of the groups.
| Control group (N = 30) |
Covid-19 Infection (N = 69) |
Atypical Pneumonia (N = 21) |
Bronnchopneumonia (N = 8) |
p-Value | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gender N (%) |
Male | 23 (57.5%) | 37 (53.6%) | 12 (57.1%) | 6 (75%) | *0.752 | ||||
| Female | 17 (42.5%) | 32 (46.4%) | 9 (42.9%) | 2 825%) | ||||||
| Mean ± SD | Median (IQR) |
Mean ± SD | Median (IQR) |
Mean ± SD | Median (IQR) |
Mean ± SD | Median (IQR) |
|||
| Age | 60.1 ± 18.6 | 62.5 (52.2–75) |
47.1 ± 19.6 | 44 (31–63) |
59.2 ± 19.2 | 63 (38.5–75) |
55.1 ± 20.4 | 58 (33–70.7) |
0.005 | |
| Symptom Onset (Day) | 5.4 ± 5.6 | 3 (2–6.7) |
6.1 ± 7.9 | 3 (1.75–7.7) |
3 ± 2.28 | 2.5 (1–4.7) |
0.478 | |||
| Fever (0C) | 36.4 ± 0.5 | 36.3 (36.2–36.5) |
36.8 ± 0.6 | 36.7 (36.5–37.1) |
36.9 ± 0.7 | 36.7 (36.4–37.3) |
37.8 ± 0.6 | 372 (36.8–38.1) |
0.0001 | |
| sPO2 | 96 ± 1.7 | 96 (95–98) |
95.1 ± 4.4 | 96 (94–98) |
93.3 ± 6.4 | 95 (93–96.5) |
95.7 ± 2.7 | 96.5 (94.2–98) |
0.401 | |
| Systolic Blood Pressure (mm/Hg) | 124.8 ± 11.9 | 127.5 (120–130) |
123.1 ± 20.8 | 120 (110–131) |
131 ± 32.5 | 133 (110–147) |
125.5 ± 8.4 | 123 (118.5–133.25) |
0.451 | |
| Diastolic Blood Pressure (mm/Hg) | 78.3 ± 9.6 | 80 (70–89) |
75.2 ± 10.2 | 80 (70–80) |
78 ± 20.5 | 80 (67–86) |
77.1 ± 6.2 | 79.5 (70–82.25) |
0.458 | |
*p-value is derived from the Fisher exact test. Other p-values are derived from the Kruskal-Wallis test.
The individuals that participated in our study can be further stratified in five groups depending on the positivity to the PCR test for SARS-CoV-2 genome (positive or negative), or the CT scan (COVID-19 compatible, atypical, bacterial, normal) (Supplementary Figure 1). In this study, we tested the ability of our assay to discriminate positive and negative individuals in three main comparisons:
- Respiratory Diagnosis (Figure 1a): either positive to the PCR test or abnormal CT scan (viral, bacterial, atypical), or both (groups I, II, III, IV) vs. PCR test negative and normal CT scan (group V);
Figure 1.
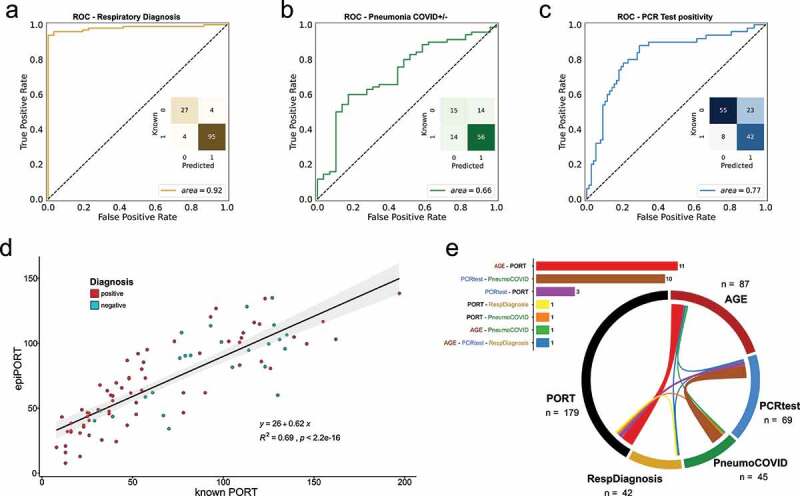
a,b,c) ROC curve of the LOOCV penalized logistic regression models (refer to Supplementary Figure 1 for group definition). a) Respiratory Diagnosis (positive: groups I, II, III, and IV vs. negative: group V). b) COVID-19 Diagnosis within the pneumonia group (positive: groups I, II, and III vs. negative: group IV). c) PCR test positivity (positive: groups II, and III vs. negative: groups I, IV, and IV). d) epiPORT model (LOOCV penalized linear regression) based on COVID-19 diagnosis vs. non COVID-19 pneumonia (positive: groups I, II, and III vs. negative: group IV). Individuals with a positive diagnosis are indicated in teal, while negative in red. e) Predictive CpG sites shared among the models (see Supplementary Table 5). The barplot (top) shows the number of shared predictive CpG sites. In the chord diagram (bottom) the shared predictive sites are displayed as ribbons (the thickness is proportional to the number of shared sites) connecting the models (the numbers represent the predictive CpG sites for each model). The overlap was calculated using the VIB/UGent Venn webtool (http://bioinformatics.psb.ugent.be/webtools/Venn/).
- Pneumonia COVID ± (Figure 1b, d): non-COVID pneumonia (group IV) vs. COVID-19 clinical diagnosis (either positive to the PCR test or COVID-19-compatible CT scan) (groups I, II, III). In this comparison, individuals from the control group (group V) were not included;
- PCR test positivity (Figures 1c, 2e and 3): PCR test positive (groups II, III) vs. PCR test negative individuals (groups I, IV, V). In this comparison, the grouping of individuals is independent of the CT scan results.
Figure 2.
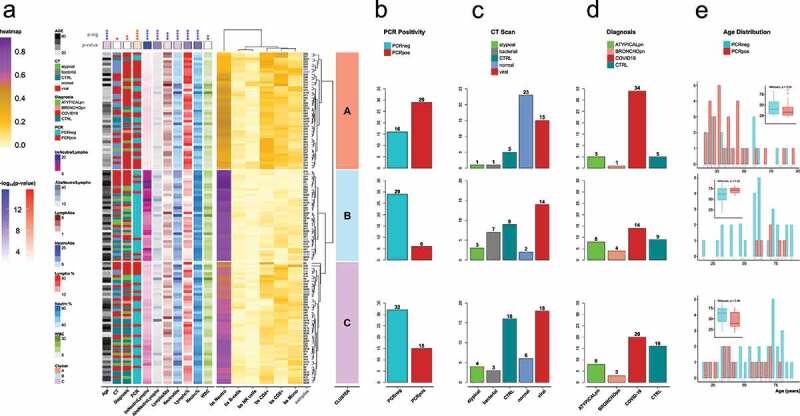
a) Heatmap of the individual samples based on the values of 5meC-calculated cell types: neutrophils (bs Neutro); B cells (bs B-cells); NK cells (bs NK cells); CD4 T cells (bs CD4+); CD8 T cells (bs CD8+); monocytes (bs Mono). Samples are divided in three groups based on the hierarchical clustering: cluster A (salmon); cluster B (light blue); cluster C (orchid). Technical replicates are labelled in blue. The annotations on the left refers to: age (Age); CT scan classification (CT): atypical, bacterial, non-pneumonia control (CTRL), pneumonia normal (normal), viral; diagnosis classification (Diagnosis): atypical pneumonia (ATYPICALpn), bronchopneumonia (BRONCHOpn), COVID-19 pneumonia (COVID-19), non-pneumonia control (CTRL); PCR test positivity (PCR): negative (PCRneg), positive (PCRpos); ratio between 5meC-calculated Neutrophils and Lymphocytes (bsNeutroLympho); ratio between Neutrophil and Lymphocyte counts (AbsNeutroLympho); Lymphocyte counts (LymphoAbs); Neutrophil counts (NeutroAbs); Lymphocyte percentage (Lympho %); Neutrophil percentage (Neutro %); white blood cell counts (WBC). Statistical analysis of the distribution of the annotation features among the three clusters: blue for continuous values (Kruskal-Wallis); red for categorical values (Fisher’s exact test): Diagnosis (controls, COVID-19 pneumonia, non-COVID-19 pneumonia); CT (control, viral, non-viral); PCR (positive, negative); statistical significance (p-sig; ns = p-val >0.05; * = p-val <0.05; ** = p-val <0.01; *** = p-val <0.001; **** = p-val <0.0001). The p-value coloured boxes below the p-sig, and above the annotations represents the -log10(p-value). b,c,d) Bar Plot of counts for each category of PCR test positivity (b), CT scan results (c), and Diagnosis (d) in each cluster. (e) Age distribution grouped by PCR test positivity for each cluster. Positive: groups II, and III vs. negative: groups I, IV, and IV (refer to Supplementary Figure 1).
Figure 3.
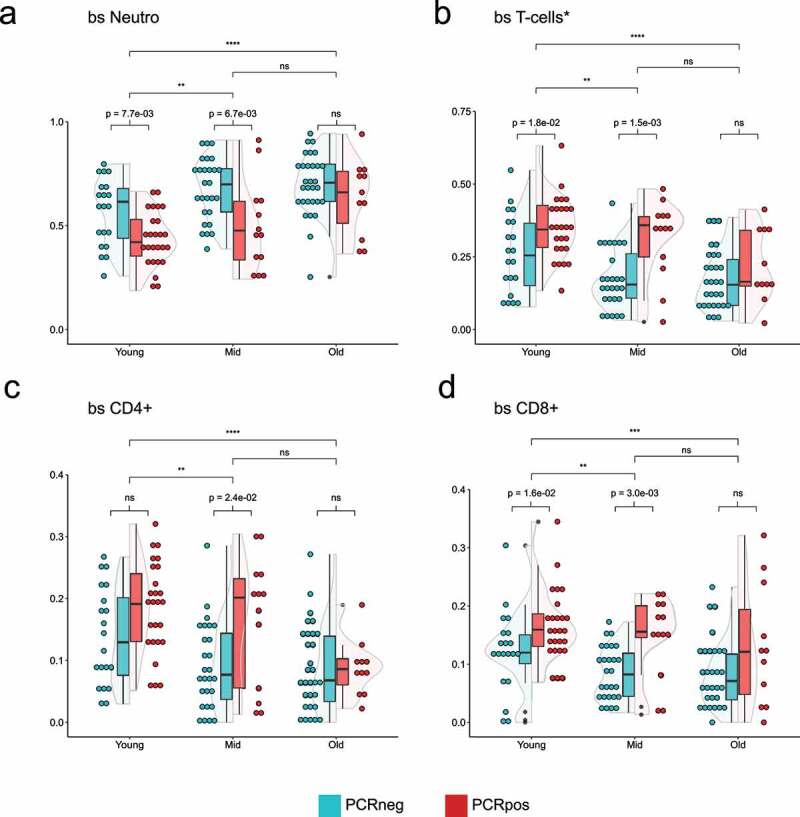
Violin plots showing the distribution of 5meC-calculated neutrophils (a), T-cells (b, sum of CD4+, and CD8+), CD4 + T cells (c), CD8 + T cells (d) in three different age groups split by the PCR positivity test. Teal: PCR test negative; red: PCR test positive. Wilcoxon rank sum test was used to compare the means. Top bars are comparing age groups (ns = p-val >0.05; * = p-val <0.05; ** = p-val <0.01; *** = p-val <0.001; **** = p-val <0.0001), while the bottom bars are comparing PCR positive and negative within each group (ns = p-val >0.05; if the p-val <0.05, the value is indicated).
Targeted bisulfite Sequencing (TBS-seq) panel design
The TBS-seq panel targets a total of 4426 regions selected from sites that are used as DNA methylation-based age predictors; sites that are present in the promoters of viral-response genes or in the exons of COVID-19-associated infection (ACE2 and TMPRSS2); immune cell type specific methylation sites. The criteria for the selection of the regions are listed in the Material and Methods section and their coordinates in Supplementary Table 2. We carried out targeted bisulfite sequencing (TBS-seq) of these regions using a previously described protocol [14,15].
TBS-seq can predict pneumonia-related traits
Initially, we asked whether DNA methylation covering the targeted regions could be used to distinguish healthy controls and patients with respiratory diseases by training Leave One Out Cross Validated (LOOCV) penalized logistic regression models. In a cohort of 130 samples, 122 samples were correctly predicted as healthy or diagnosed with COVID-19 or non-COVID-19 pneumonia (27 and 95, respectively), while only 8 samples (6%) were mis-predicted, with an AUC of 0.92 (Figure 1a).
We then tried to differentiate between patients diagnosed with COVID-19 vs. non-COVID-19 pneumonia (n = 68, and 29, respectively). An individual is diagnosed with COVID-19 based on the results of the CT scan, positivity using the PCR molecular test, and levels of ferritin and D-Dimer. The classification showed a reduced AUC (0.66) compared to the prediction of healthy vs. pneumonia-diagnosed individuals (Figure 1b). The 28 mis-classified individuals were equally distributed between false positives and false negatives (14 and 14, respectively). The individuals in the false-negative group (predicted negative but diagnosed COVID-19 positive) are all negative for the PCR molecular test, whereas the false-positive group (predicted positive, but diagnosed with non-COVID-19 pneumonia) shows an equal distribution of PCR positive and PCR negative patients (Supplementary Table 3). Given these results, we also attempted to predict the PCR positivity status of the cohort. We were able to predict the PCR positivity with a moderate degree of accuracy (AUC = 0.77, Figure 1c). We therefore focused on this PCR positivity-based classification for the rest of the study.
In addition to classification models, we fit a LOOCV-penalized regression model to predict PORT score, a continuous index used for mortality prediction in community-acquired pneumonia (CAP) patients. The PORT score is calculated using age, gender, chronic diseases, mental status, in addition to several vitals and blood test results. A DNA methylation-based model (epiPORT) can predict the PORT score with high accuracy (R2 = 0.69; Figure 1d). The PORT score values are only calculated for patients with COVID-19 or non-COVID-19 pneumonia (respiratory diseases) and we don’t find any significant differences between predicted and actual PORT scores between COVID-19 and non-COVID-19 individuals for both PCR test positivity (groups II, III vs. I, IV, V) and respiratory diagnosis (groups I, II, III vs. IV, V) classifications (Supplementary Figure 2). We constructed an epigenetic clock using penalized regression that predicts the age of each individual based on their DNA methylation profiles. However, we don’t observe a significant difference in the epigenetic age acceleration (i.e., difference between predicted and expected epigenetic age) between PCR positive and PCR negative individuals (Supplementary Figure 3).
We define the ‘predictive CpG’ sites for each model as the CpG sites that have non-zero coefficients in 90% of the models (see Materials and Methods). We then compared the ‘predictive CpG’ sites used to build the various models described above to measure the overlap among the predictions (Supplementary Table 4). Figure 1e shows the overlap among the sites used to build each model. The number of shared sites is modest in general, with only one CpG site in common among three predictions (age, COVID vs. other respiratory diseases, and respiratory diagnosis) (Supplementary Table 5). The higher number of overlapping sites is seen between the age and PORT score prediction (n = 11). This is expected, since age plays a substantial role in the PORT score calculation. Not surprisingly, the second highest number of sites shared between predictions (n = 10) is between COVID-19 PCR test positivity (positivity criteria as in Figure 1c) and COVID-19 vs. non COVID-19 pneumonia diagnosis (positivity criteria as in Figure 1b). A better overlap among the models is seen if the CpG-associated genes are tested for shared members (Supplementary Table 6).
In addition to our multivariate models, we also identified differentially methylated regions (DMRs, Supplementary Table 7) within the loci covered by our assay (Supplementary Table 8). In the majority of the comparisons evaluated, the hypomethylated regions are enriched in immune response terms such as lymphocyte, leukocytes and T cell activation (Supplementary Table 9). For the hypermethylated regions, only one comparison (positive vs. negative respiratory diagnosis) shows enriched terms related to metabolic processes. The overlap of the DMRs obtained by pairwise comparisons is more extensive (n = 327 of 3232 metilene-defined regions) than the one observed by the CpG sites used for the predictions (n = 28 of 393 total predictive CpG sites, or of 6268 total variable CpG sites) (Supplementary Table 10, and Supplementary Table 5, respectively).
Cell type differences are associated with COVID positivity
DNA methylation patterns are affected by both epigenetic changes within specific cell types, as well as changes in the relative abundance of cell types. In addition, the observed enrichment of leukocyte-related genes in the DMR analysis prompted us to investigate the changes in cell type abundance across our cohort. We did not observe differences in white-blood cell counts (WBC) between PCR positive and negative subjects, but there is a significant difference for both neutrophils and lymphocytes counts (Supplementary Figure 4). We were able to estimate additional cell type percentages of each type of lymphocyte (B cells, CD4 + T cells, CD8 + T cells, and NK cells) as well as monocytes in each sample by using cell-specific DNA methylation loci as previously described (see Methods, Supplementary Table 12, Supplementary Table 13, and Supplementary Table 14). The methylation estimated percentages are well correlated with the clinical cell count data for lymphocytes and neutrophils percentages (r = 0.65, r = 0.65, respectively, Supplementary Figure 5). The fact that the predicted cell type abundances are highly correlated with the measured values in this study further demonstrates the robustness of our previously validated deconvolution approach. We then asked whether the cell type percentages could be used to predict the PCR test positivity status. As seen in Supplementary Figure 6, the prediction using methylation-estimated cell types (AUC = 0.65) is better than the one using clinical cell counts data (AUC = 0.60), although not as accurate as the one based solely on DNA methylation levels.
To study changes in cell-type percentage due to COVID infection we clustered the samples using the methylation-estimated cell-type distributions for neutrophils, B cells, NK cells, CD4+, CD8+, and monocytes, and grouped the samples in 3 distinct clusters (Figure 2a). Cluster A, which is enriched in patients diagnosed with COVID-19 (Figure 2d, top panel), shows a high level of lymphocytes (mainly CD4, CD8, and NK cells) and the lowest level of neutrophils. Two-thirds of the samples in cluster A are COVID positive based on the PCR test (Figure 2b, top panel). By contrast, cluster B, shows the highest level of neutrophils and the lowest level of lymphocytes (Figure 2a). The vast majority of the samples in this cluster are negative to the PCR test (Figure 2b, middle panel), a third of which are diagnosed as COVID-19 positive because of a positive CT scan (Figures 2c and 2d, middle panel). Samples in cluster C have intermediate neutrophil and lymphocyte levels with the majority of the samples being PCR negative (Figure 2b, bottom panel). This cluster also has the majority of non-pneumonia samples (e.g., CTRL; Figures 2c, 2d, bottom panel). Although there are no significant age differences within each cluster between PCR positive and negative individuals (Figure 2e), there is a significant difference between clusters (Kruskal-Wallis, p = 1.35 x 10−5), with group B having the older population, followed by C, then A with the youngest.
Differences in cell type distributions between PCR positive and negative individuals are age dependent
To study the interdependence of cell-type percentages, COVID positivity and age, samples were divided into three age groups with a similar number of individuals: young (0–40 years; n = 46), mid (41–63 years; n = 40), and old (older than 64; n = 41). We first examined the distribution of 5meC-derived neutrophils in the three age groups and found that, although the young group shows significant differences from the mid and old groups, the difference between the mid and old age groups was not significant (Figure 3a). Although the neutrophil clinical counts do not show any statistically significant difference, the clinical neutrophil percentage shows significant differences among age groups (Supplementary Figure 7A, 7B). Similar results were obtained for the distribution of T-cells and their subtypes CD4+ and CD8+ (Figures 3b, 3c, and 3d). We observed significant differences in both the neutrophils and the T-cell percentages within the young and the mid-age groups between PCR positive and PCR negative individuals (Figures 3a, and 3b). In particular, the CD8 + T-cell population shows significant differences in both the young and mid-age groups, while CD4+ distribution is significant only in the mid-age group (Figures 3c, and 3d). By contrast, the older group doesn’t show any significant difference. Clinical leukocyte counts show differences among age groups, but not within age groups (based on PCR positivity) (Supplementary Figure 7C). Clinical lymphocyte percentage shows a significant difference in the mid-age group between PCR positive and negative patients, in addition to age-related differences (Supplementary Figure 7D). When performing the same analysis restricted to COVID-19 and non-COVID-19 pneumonia patients, significant differences are only seen in the mid-age group for the cell types examined (Supplementary Figure 8). Differences in the PORT score or the epiPORT score are statistically significant between age groups, but not between PCR positive or negative individuals (Supplementary Figure 9).
Discussion
In this study, we explore the relationship between DNA methylation and clinical features of Covid-19 infection. Our study population included several groups: those that were PCR positive and negative for Sars-Cov-2, and within each of these two groups, individuals that were positive or negative for pneumonia. This allowed us to test whether Covid-19 pneumonia led to a different epigenetic profile than pneumonia caused by other infections. The average age of our patient group was 53.9, but the ages ranged across five decades, allowing us to test the effect of age on the epigenetic response.
All samples in our cohort were collected in the first wave of the Covid-19 pandemic, in the spring of 2020. At the onset of the pandemic, CT results were used for the diagnosis of Covid-19. Bilateral, lobular, peripherally localized, widespread patched ground glass opacities are reported as the characteristic thoracic CT finding of COVID-19 pneumonia. In addition, high levels of ferritin and D-Dimer were also used in the diagnosis. D-dimer levels increase in severe cases. Patients with a low total lymphocyte count at the onset of the disease usually have a poor prognosis. In severe patients, the number of peripheral blood lymphocytes gradually decreases. For COVID-19 cases, blood lymphocyte count <800/μl, serum CRP> 40 mg/l, ferritin> 500 ng/ml, D-Dimer> 1000 ng/ml can be summarized as poor prognostic factors. Later during the course of the pandemic, Covid-19 was diagnosed exclusively with RT-PCR tests and CT results.
Previous studies found that in COVID-19 patients the total blood lymphocyte count, and in particular that of T cells, is lower than in healthy controls [16]. In severe cases, both CD4+ and CD8 + T cell blood counts are further decreased compared with moderate cases [17–23][–]. Notably, a lower lymphocyte count was found to be a clinical predictor of mortality due to Covid-19 infection [24,25]. By contrast, in our cohort we don’t observe lymphopenia in COVID-19 (PCR) positive patients.
Using this approach, we were able to delineate one of our most interesting findings: that blood cell type relative abundances vary based on age and positivity to the PCR test. Specifically, we divided our cohort into three clusters based on the abundance of their cell types: the first cluster has the highest number of PCR test positive individuals and it shows a high percentage of lymphocytes (mainly CD4, CD8, and NK cells) and the lowest level of neutrophils. The other two clusters show higher levels of neutrophils and lower levels of lymphocytes and are enriched in patients negative to the PCR test and controls.
These results suggest that the PCR-based assessment is more strongly associated with cell-type differences than the clinical diagnosis based on multiple parameters.
Several studies have shown that both CD4+ and CD8 + T cells from severe COVID-19 patients present a dysregulated status of activation and function [26–30]. Thus, our findings that T cell percentages are strongly associated with disease status and age, support the critical role of these cells in effective immune response to the virus.
Similar observations were made in a recent multi-omic study [11] in which cellular deconvolution analysis identified granulocytes, B cells, NK cells, and monocytes as important cell types involved in the COVID-19 DNA methylation signature. Finally, in our dataset we see a significant difference between the group positive and negative to the PCR test for B cell (Kruskal-Wallis, p = 1.79 x 10−2), but not for the distribution of NK cells (Kruskal-Wallis, p = 0.261).
Despite being covered by our assay, neither ACE2 nor TMPRSS2 genes, identified in the literature as key players for COVID-19 infection, showed differentially methylated regions in their proximity if comparing cases vs. controls. The same conclusion was reached in a study by Misra et al., despite a different tissue being interrogated [31].
Our approach is able to predict the respiratory diagnosis and PCR positivity status with a high degree of accuracy (AUC = 0.92 – Figures 1a, and 0.77 – Figure 1c, respectively). The sites used for most of the iterations to build the various models (‘predictive CpG sites’) do not show enrichment for any particular sets of genes, but some of them are involved in immune and antiviral functions, such as the leukocyte immunoglobulin-like receptors LILRB2, and LILRB1; IFNLR1 which is a class II cytokine receptor that binds cytokines (IL28A, IL28B, IL29) which expression is induced by viral infection; the cytidine deaminase APOBEC3D and interferon-mediated response genes SLC15A4 and PARP9; and the T-cell receptor regulators CUX1 and UBASH3A.
Genes associated with differentially methylated regions are significantly enriched in terms of the immune response, and specifically to T-cell activity, which supports our findings that T cells play a significant role in the age and disease dependent immune response to COVID.
Other genes have been identified in previously published methylation-based screenings of COVID-19 positive patients [10,11,13][,, such as:
- Interferon-related genes are not covered extensively by our assay, but some are selected in the logistic regression models (e.g., IFNLR1, SLC15A4, PARP9). Select genes that are not covered by our assay but are reported in the literature include IRF7 (interferon regulatory factor 7), OAS1 (interferon-induced 2’-5’-Oligoadenylate Synthetase 1 that activates the viral RNA nuclease RNaseL), MX1 (MX Dynamin Like GTPase 1, and involved in the cellular antiviral response), DTX3L (a E3 ubiquitin ligase that works in association with PARP-9), and IFIT3 (an interferon-induced protein that inhibits cellular and viral processes)
- AIM2, which expression is induced by interferon gamma, is a protein that recognizes cytosolic dsDNA that was identified from an EWAS study [12], and that we find it to be associated with the epiPORT score prediction.
- the major histocompatibility complex HLA-C that is not covered by our assay, but it is bound by the leukocyte immunoglobulin-like receptors LILRB1/2, that we see as both differentially methylated and associated with predictive CpG sites in our models.
- the ADP-ribosyltransferase PARP-9 that, together with DTX3L, plays an important role in interferon-mediated antiviral defence. In our assay, PARP-9 is associated with a CpG site selected for PCR positivity and COVID-19 diagnosis prediction.
We believe that the addition of probes capturing these genes-associated CpG sites/regions could improve our assay in its accuracy and overall performance with only a minimal increase of the assay associated costs.
Even though we can use our methylation data to build an accurate epigenetic clock, we do not see evidence of age acceleration in patients positive for the PCR test. This may be due to the fact that the sample collection occurred relatively early in the COVID-19 disease progression and the effects are not yet visible, or that an increased epigenetic age might correlate with the severity of the disease (not tested in our dataset) [32]. Future longitudinal studies may also reveal that the epigenetic ageing consequences of COVID infection appear later in the disease progression.
In conclusion, our approach based on a customizable and cost-effective platform to assess the DNA methylation levels of a few thousand loci is able to distinguish between pneumonia and control individuals with high accuracy and, with slightly less accuracy, COVID-19 vs. non-COVID-19 pneumonia patients. The reduced ability to distinguish individuals within the pneumonia class can be due to the heterogeneous nature of non-COVID-19 pneumonia, to the limited number of patients analysed and to the relatively low number of CpG sites assayed. Our approach is also able to calculate the epiPORT score to assess the severity of community-acquired pneumonia. This suggests that disease severity does impact epigenomes, and can therefore be used for the development of biomarkers. Our data also shows that neutrophils and T cells vary significantly between COVID-19 positive and negative individuals, particularly in young and middle-aged subjects. T-cell and leukocyte terms are enriched in differentially methylated regions and interferon-related genes are associated with the CpG sites used by the logistic regression models built in this study. Thus, the epigenome allows us to capture the age-related decline in the adaptive immune response, which likely underlies the increase in disease severity with age [33].
Limitations of the study
This study has a few limitations. First, it is a non-longitudinal and single-centre study. Moreover, the samples are labelled positive or negative (based on either the diagnosis or the PCR test), but not based on the severity. Nonetheless, we can predict with high accuracy the PORT score that can be considered a risk factor for community-acquired pneumonia (CAP). The control patients are not healthy individuals, but they were admitted to the emergency room with a different pathology than pulmonary-related diseases.
Material and methods
The present study is a prospective case-control study, and the required approval was obtained from the Ethics Committee of Pamukkale University (60,116,787–020/31,834).
RT-PCR assay
SARS CoV-2 Double Gene RT-qPCR Kit (BS-SY-WCOR-307-1000, Bio-Speedy) was used for diagnosis of the Covid-19 positivity.
Study population
After the required information concerning the study was provided both to the patient group and to the healthy control group, the written consent forms were obtained from all the subjects who agreed to participate in the study.
These subjects were assessed in accordance with the inclusion and exclusion criteria. Patients who were diagnosed with SARS-CoV-2 infection according to WHO (https://www.who.int/publications/i/item/clinical-management-of-SARS-CoV-2) guideline as a result of clinical evaluation in the emergency department and whose diagnosis was confirmed by RT-PCR were included in the study. The grouping of the patients is detailed in the Supplementary Figure 1. 67 patients were diagnosed with COVID-19: 31 patients CT (-) SARS-CoV-2 (+) infection (group III); 19 patients CT(+) SARS-CoV- (+) pneumonia (group II); 18 patients with RT-PCR (-) detected although thorax CT findings were compatible with suspected COVID-19 pneumonia (group I). The RSNAEC (Radiological Society of North America Expert Consensus) guidelines were followed to evaluate the CT scans. 29 patients were negative (-) to the RT-PCR test, but the CT scan results were not compatible with COVID-19 pneumonia (group IV). 30 healthy volunteers were included in the study as the control group (group V). The exclusion criteria for this group consisted of recent history of infection, diagnosis of kidney and liver failure, acute pulmonary embolism, chronic inflammatory disease history (rheumatological disease, autoimmune disease), pregnancy, presence of any cancer diagnosis, chronic obstructive pulmonary disease, asthma disease, and history of cerebrovascular disease.
Data collection
Demographic data, medical history, vital findings (fever, blood pressure, sPO2), laboratory findings (complete blood count; C-reactive protein (CRP), D-dimer, Ferritin and hsTnT parameters) and radiological findings, time to onset of symptoms, Comorbid diseases, hospitalization location of the patients (service or ICU), clinical scores, CT severity scores were recorded in the data set.
CT evaluation
Chest CT performed at the time of admission of the patients to the ED was assessed under the criteria of the RSNAEC (Radiological Society of North America Expert Consensus) by an emergency physician who followed up the patient clinically. The pneumonia cases were classified in line with these criteria and recorded in the clinical classification dataset [34].
Clinical Evaluation
The clinical assessment of the subjects was performed in accordance with COVID-19 diagnosis and treatment guidelines of the Turkish Ministry of Health (https://covid19rehberi.com/wp-content/uploads/2020/08/COVID-19_REHBERI_ERISKIN_HASTA_TEDAVISI.pdf, Accessed August 26th). As this guide was updated, the patient management algorithm was also edited. The Pneumonia Severity Index and CURB-65 scores of the subjects were calculated as suggested in the literature and then recorded in the dataset [35,36].
Blood samples and laboratory parameters
Complete blood count (CBC), C-reactive protein (CRP), creatinine, urea, d-dimer, and ferritin parameters, which are routinely checked during admission to the ED, were recorded in the dataset. In the control group, on the other hand, after 3 cc of blood was drawn into a dry tube, and another 3 cc of blood was placed into the EDTA tube, was analysed through the same methods in the same laboratory. The laboratory parameters of the blood samples requested from the patients in the ED for examination were recorded in the dataset.
Targeted bisulfite Sequencing (TBS-seq)
Probe design
The design of the probe panel used in the Targeted bisulfite Sequencing (TBS-seq) is based on the selection of CpG sites meeting one or more of the following criteria:
• It is described in the literature as part of DNA methylation-based age estimators [37];
• It has blood cell-type specific DNA methylation profiles (referred as CellFi sites);
• Present in the promoters (defined as −1000 bp/+250 bp from the TSS) of viral-response genes and genes involved in SARS and influenza infections (response to virus GO:0009615) [38,39] and covered by the TruSeq Methyl Capture EPIC kit (Illumina, Inc.);
• Present in exons of genes previously associated with SARS-CoV2 infection: ACE2 and TMPRSS2.
Biotinylated probes covering the selected CpG sites have been synthesized by IDT (NGS Discovery Pools). The targeted region coordinates (GRCh38) are listed in Supplementary Table 2.
Library preparation and data generation
Genomic DNA was isolated from the individuals by standard phenol-chloroform extraction method [40]. 500 ng of extracted DNA were used for TBS-seq library preparation as described in Chang et al., with minor modifications [15]. Briefly, fragmented DNA was subject to end repair, dA-tailing and adapter ligation using the NEBNext Ultra II Library prep kit using custom pre-methylated adapters (IDT) [14]. Purified libraries were hybridized to the biotinylated probes according to the manufacturer’s protocol. Captured DNA was treated with bisulfite prior to PCR amplification using KAPA HiFi Uracil+ with the following conditions: 2 min at 98°C; 14 cycles of (98°C for 20 sec; 60°C for 30 sec; 72°C for 30 sec); 72°C for 5 minutes; hold at 4°C. Library QC was performed using the High-Sensitivity D1000 Assay on a 2200 Agilent TapeStation. Libraries were sequenced on a NovaSeq6000 (S1 lane) as paired-end 150 bases.
Data processing
Demultiplexed Fastq files were subject to adapter removal using cutadapt (v2.10) [41] and aligned to the GRCh38 genome using BSBolt Align (v1.3.0) [42]. PCR duplicates were removed using samtools markdup function (samtools version 1.9) [43] before calling methylation using BSBolt CallMethylation function. A DNA methylation matrix containing all the common CpG sites covered by at least 20 reads in all the samples is created using BSBolt AggregateMatrix.
LOOCV models
The aggregate methylation matrix was filtered by removing sites that exhibited low variation between samples (≤0.00254) before model training, resulting in a methylation matrix with 9,935 CpG sites and 130 samples. The methylation values for each row were scaled to have a zero mean and unit variance using the scikit-learn [44] preprocessing.scale module. Using the processed methylation matrix Leave One Out Cross Validated (LOOCV) penalized logistic regression models and penalized linear regression models were trained against sample Covid-19 PCR testing status (n = 128) and PORT assessment score (n = 88) respectively. Briefly, for each sample a separate regression model was trained with the respective sample left out of model training, sample data were then used to predict the sample Covid-19 PCR status or PORT score. Identical model parameters were used between folds (https://github.com/NuttyLogic/EpigeneticAlterationsInCovid19Infections). Within folds 115 and 81 CpG sites had non-zero coefficients in 90% of the port and PCR status models representing 41.2% and 14.3% of the modelled CpG sites with non-zero coefficients among all folds.
DNA methylation-based cell composition estimation
A reference-based cell estimation approach was utilized to estimate proportions of six blood cell types: neutrophils, monocytes, CD4 + T cells, CD8 + T cells, B cells, and NK cells, as previously described [45–47]. In summary, whole genome bisulfite sequencing (WGBS) methylomes were obtained from the Blueprint Epigenome Project [48]. In total, 37 methylation profiles were analysed from venous blood-derived cell types (Supplementary Table 12). First, to process the reference dataset, we used a sliding window to aggregate the methylation values into regions composed of at least two CpG loci that have similar methylation (≤ 25% methylation difference) and are within 500bp distance from each other. Second, cell-specific regions were selected that were uniquely hypomethylated in one cell type by at least 30% than all other cell types. An exception to these criteria were CD4+ and CD8 + T cells which because of their similarity led to a dearth of unique regions, therefore additional regions specific to T cells as a whole and regions at least 30% methylation difference between CD4+ and CD8 + T cells were also included. As a result, over 100 cell-specific hypomethylated regions were selected representing over 450 CpG loci (Supplementary Table 13). The validation of the deconvolution method was carried out using known cell mixtures in vitro, as described in Nadel et al. [49]. Briefly, combinations of 6 isolated immune cells (neutrophils, monocytes, NK cells, B cells, and CD4+ and CD8 + T cells) were mixed together and whole-genome bisulfite sequencing on the extracted DNA was performed. The results are shown in Supplementary Figure 10. Second, a non-negative least squares regression was performed on the methylation values of the cell-specific regions of the references and samples to estimate the proportion of each cell type within the samples. The resulting cell-type deconvolution data can be found in Supplementary Table 14.
Differentially Methylated Regions (DMRs)
Differentially methylated regions were identified using metilene [50] with the following parameters: -M 300 -m 2 -d 0.05; where -M = the allowed nt distance between two CpGs within a DMR; -m = the minimum # of CpGs in a DMR; -d = the minimum mean methylation difference for calling DMRs. Only regions with a q-value < 0.1 were considered for further analysis. DMRs are further divided into hypermethylated or hypomethylated based on the Δ methylation value (condition positive – condition negative).
Gene association rules
Individual CpG sites and genomic regions (DMRs) were bookmarked with the nearest gene features up and downstream of the assessed site or region using custom python implementation and NCBI RefSeq annotations (hg38).
GO enrichment analysis
The enrichment analysis is performed using g:GOst Functional Profiling (g:Profiler) [51] with the following customizations: multiquery (genes associated with differentially methylated regions and divided in hyper-, and hypo-methylated); Custom domain over annotated genes (genes associated with the background regions defined by metilene); Significance: Benjamini-Hochberg FDR with a threshold of 0.05; Gene Ontology sources: GO biological process, and No electronic GO annotations; Biological pathways: all; Regulatory motifs: all; Protein databases: all.
Supplementary Material
Funding Statement
This work was supported by the National Institutes of Health [T32CA201160].
Data availability
The data is deposited to Gene Expression Omnibus (GEO) and assigned the accession GSE192702.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].CDC . COVID-19 and Your Health. 2021. Available from: https://www.cdc.gov/coronavirus/2019-ncov/prevent-getting-sick/how-covid-spreads.html
- [2].Cascella M, Rajnik M, Aleem A, et al. . Features, Evaluation, and Treatment of Coronavirus (COVID-19). Internet - Treasure Island (FL): StatPearls Publishing; 2021. . [PubMed] [Google Scholar]
- [3].Liu J, Li S, Liu J, et al. Longitudinal characteristics of lymphocyte responses and cytokine profiles in the peripheral blood of SARS-CoV-2 infected patients. EBioMedicine. 2020;55:102763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rothan HA, Byrareddy SN.. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J Autoimmun. 2020;109:102433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Holt NR, Neumann JT, McNeil JJ, et al. Implications of COVID-19 for an ageing population. Med J Aust. 2020;213(8):342–4.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zheng Y, Liu X, Le W, et al. A human circulating immune cell landscape in aging and COVID-19. Protein Cell. 2020;11(10):740–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28(10):1057–1068. [DOI] [PubMed] [Google Scholar]
- [8].Obata Y, Furusawa Y, Hase K. Epigenetic modifications of the immune system in health and disease. Immunol Cell Biol. 2015;93(3):226–232. [DOI] [PubMed] [Google Scholar]
- [9].El Baba R, Herbein G. Management of epigenomic networks entailed in coronavirus infections and COVID-19. Clin Epigenetics. 2020;12(1):118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Balnis J, Madrid A, Hogan KJ, et al. Blood DNA methylation and COVID-19 outcomes. Clin Epigenetics. 2021;13(1):118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bernardes JP, Mishra N, Tran F, et al. Longitudinal multi-omics analyses identify responses of Megakaryocytes, erythroid cells, and plasmablasts as hallmarks of severe COVID-19. Immunity. 2020;53(6):1296–314.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Castro de Moura M, Davalos V, Planas-Serra L, et al. Epigenome-wide association study of COVID-19 severity with respiratory failure. EBioMedicine. 2021;66:103339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Corley M J et al . (2021). Genome‐wide DNA methylation profiling of peripheral blood reveals an epigenetic signature associated with severe COVID‐19. J Leukocyte Bio, 110(1), 21–26. DOI: 10.1002/JLB.5HI0720-466R [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Morselli M, Farrell C, Rubbi L, et al. Targeted bisulfite sequencing for biomarker discovery. Methods. 2020;187:13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chang Y-L, Rossetti M, Gjertson DW, et al. Human DNA methylation signatures differentiate persistent from resolving MRSA bacteremia. Proc Natl Acad Sci U S A. 2021;118(10). DOI: 10.1073/pnas.2000663118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mortaz E, Tabarsi P, Varahram M, et al. The Immune Response and Immunopathology of COVID-19. Front Immunol. 2020;11. DOI: 10.3389/fimmu.2020.02037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Li R, Tian J, Yang F, et al. Clinical characteristics of 225 patients with COVID-19 in a tertiary hospital near Wuhan, China. J Clin Virol. 2020;127:104363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhang G, Hu C, Luo L, et al. Clinical features and short-term outcomes of 221 patients with COVID-19 in Wuhan, China. J Clin Virol. 2020;127:104364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Liu R, Wang Y, Li J, et al. T cell populations contribute to the increased severity of COVID-19. Clin Chim Acta. 2020;508:110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Jiang M, Guo Y, Luo Q, et al. Counts in peripheral blood can be used as discriminatory biomarkers for diagnosis and severity prediction of coronavirus disease 2019. J Infect Dis. 2020;222(2):198–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Qin C, Zhou L, Hu Z, et al. Dysregulation of immune response in patients with coronavirus 2019 (COVID-19) in Wuhan, China. Clin Infect Dis. 2020;71(15):762–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Huang W, Berube J, McNamara M, et al. Lymphocyte subset counts in COVID −19 patients: a Meta-Analysis. Cytometry A. 2020;97(8):772–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Liu Z, Long W, Tu M, et al. Lymphocyte subset (CD4+, CD8+) counts reflect the severity of infection and predict the clinical outcomes in patients with COVID-19. J Infect. 2020;81(2):318–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhou F. China Japan friendship hospital, Department of pulmonary and critical care medicine, Center of respiratory medicine, national clinical research center for respiratory diseases, Institute of respiratory medicine, Chinese academy of medical sciences, Peking union medical college, Beijing, China. Clinical course and risk factors for mortality of adult in patients with COVID-19 in Wuhan, China: a retrospective cohort study. J Med Study Res 2020;1–2. [Google Scholar]
- [25].Luo M, Liu J, Jiang W, et al. IL-6 and CD8+ T cell counts combined are an early predictor of in-hospital mortality of patients with COVID-19. JCI Insight. 2020;5(13): 10.1172/jci.insight.139024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chen Z, John Wherry E. T cell responses in patients with COVID-19. Nat Rev Immunol. 2020;20(9):529–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Adamo S, Chevrier S, Cervia C, et al. Profound dysregulation of T cell homeostasis and function in patients with severe COVID-19. Allergy. 2021;76(9):2866–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kalfaoglu B, Almeida-Santos J, Tye CA, et al. Paralysis in severe COVID-19 infection revealed by single-Cell Analysis. Front Immunol. 2020;11:589380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kalfaoglu B, Almeida-Santos J, Tye CA, et al. T-cell dysregulation in COVID-19. Biochem Biophys Res Commun. 2021;538:204–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bergamaschi L, Mescia F, Turner L, et al. Longitudinal analysis reveals that delayed bystander CD8+ T cell activation and early immune pathology distinguish severe COVID-19 from mild disease. Immunity. 2021;54(6):1257–75.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Misra P, Mukherjee B, Negi R, et al. DNA methylation and gene expression pattern of ACE2 and TMPRSS2 genes in saliva samples of patients with SARS-CoV-2 infection. medRxiv. 2020. doi: 10.1101/2020.10.24.20218727 [DOI] [Google Scholar]
- [32].Kuo C-L, Pilling LC, Atkins JC, et al. COVID-19 severity is predicted by earlier evidence of accelerated aging. medRxiv. Preprint. 2020. Preprint. doi: 10.1101/2020.07.10.20147777. [DOI] [Google Scholar]
- [33].Mueller AL, McNamara MS, Sinclair DA. Why does COVID-19 disproportionately affect older people? Aging (Albany NY). 2020;12(10):9959–9981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Simpson S, Kay FU, Abbara S, et al. Radiological society of North America expert consensus statement on reporting chest CT findings related to COVID-19. Endorsed by the society of thoracic radiology, the American college of radiology, and RSNA - secondary publication. J Thorac Imaging. 2020;35(4):219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lim WS. Defining community acquired pneumonia severity on presentation to hospital: an international derivation and validation study. Thorax. 2003;58(5):377–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ioachimescu OC, Ioachimescu AG, Iannini PB. Severity scoring in community-acquired pneumonia caused by Streptococcus pneumoniae: a 5-year experience. Int J Antimicrob Agents. 2004;24(5):485–490. [DOI] [PubMed] [Google Scholar]
- [37].Horvath S, Raj K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat Rev Genet. 2018;19(6):371–384. [DOI] [PubMed] [Google Scholar]
- [38].Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271–80.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lukassen S, Chua RL, Trefzer T, et al. SARS-CoV-2 receptor ACE2 and TMPRSS2 are primarily expressed in bronchial transient secretory cells. EMBO J. 2020;39:e105114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Guha P, Das A, Dutta S, et al. A rapid and efficient DNA extraction protocol from fresh and frozen human blood samples.J Clin Lab Anal. 2018;32(1):e22181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011;17(1):10–12. [Google Scholar]
- [42].Farrell C, Thompson M, Tosevska A, et al. BiSulfite bolt: a bisulfite sequencing analysis platform. Gigascience. 202110 5 :giab033. doi: 10.1093/gigascience/giab033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Li H, Handsaker B, Wysoker A, et al. 1000 genome project data processing subgroup. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Pedregosa F, Varoquaux G, Gramfort A, et al. Scikit-learn: machine learning in python. J Mach Learn Res. 2011;12:2825–2830. [Google Scholar]
- [45].Chen P-Y, Chu A, Liao -W-W, et al. Prenatal growth patterns and birthweight are associated with differential DNA methylation and gene expression of cardiometabolic risk genes in human placentas: a Discovery-Based approach. Reprod Sci. 2018;25(4):523–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Olivera-Perez HM, Lam L, Dang J, et al. Omega-3 fatty acids increase the unfolded protein response and improve amyloid-β phagocytosis by macrophages of patients with mild cognitive impairment. FASEB J. 2017;31(10):4359–4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Orozco LD, Farrell C, Hale C, et al. Epigenome-wide association in adipose tissue from the METSIM cohort. Hum Mol Genet. 2018;27(14):2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Martens JHA, Stunnenberg HG. BLUEPRINT: mapping human blood cell epigenomes. Haematologica. 2013;98(10):1487–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Nadel BB, Lopez D, Montoya DJ, et al. The Gene Expression Deconvolution Interactive Tool (GEDIT): accurate cell type quantification from gene expression data. Gigascience. 2021;10(2): doi: 10.1093/gigascience/giab002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Jühling F, Kretzmer H, Bernhart SH, et al. metilene: fast and sensitive calling of differentially methylated regions from bisulfite sequencing data. Genome Res. 2016;26(2):256–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Raudvere U, Kolberg L, Kuzmin I, et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019;47(W1):W191–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data is deposited to Gene Expression Omnibus (GEO) and assigned the accession GSE192702.