

Summary
Potent and biostable inhibitors of the main protease (Mpro) of SARS-CoV-2 were designed and synthesized based on an active hit compound 5h (2). Our strategy was based not only on the introduction of fluorine atoms into the inhibitor molecule for an increase of binding affinity for the pocket of Mpro and cell membrane permeability but also on the replacement of the digestible amide bond by a surrogate structure to increase the biostability of the compounds. Compound 3 is highly potent and blocks SARS-CoV-2 infection in vitro without a viral breakthrough. The derivatives, which contain a thioamide surrogate in the P2-P1 amide bond of these compounds (2 and 3), showed remarkably preferable pharmacokinetics in mice compared with the corresponding parent compounds. These data show that compounds 3 and its biostable derivative 4 are potential drugs for treating COVID-19 and that replacement of the digestible amide bond by its thioamide surrogate structure is an effective method.
Subject areas: Health sciences, Pharmaceutical science, Medical biochemistry, Virology
Graphical abstract
Highlights
-
•
Potent main protease inhibitors, which block SARS-CoV-2 infection, were developed
-
•
Our strategy adopted fluorine scan and amide surrogate replacement in molecules
-
•
The antiviral activity of the top compound is higher than that of Nirmatrelvir
-
•
Some compounds have remarkably preferable pharmacokinetics in mice
Health sciences; Pharmaceutical science; Medical biochemistry; Virology.
Introduction
The pandemic of the novel COVID-19, which is produced by the positive-strand RNA virus SARS-CoV-2, has been continuing for more than two years.1,2,3,4 Vaccination has been pervasive worldwide and has been very effective in suppression of infection as well as aggravation, but the spread of SARS-CoV-2 has not relented because of frequent breakthrough infections in vaccinated people and the numbers of people who have avoided vaccination,5,6 and drugs are needed to treat infected patients. Initially, Remdesivir, a repositioning inhibitor of RNA-dependent RNA polymerase (RdRp), which had been administered to patients of Ebola hemorrhagic fever, was used as the first FDA-authorized anti-COVID-19 drug.7,8 Other inhibitors of this type, including inhibitors such as Molnupiravir,9,10,11 which are specific for SARS-CoV-2 RdRp have been developed. Subsequently, Nirmatrelvir/PF-07321332 (1), aka Paxlovid, a novel inhibitor of the main protease (Mpro) of SARS-CoV-2, was authorized by FDA in 2021 (Figure 1).12
Figure 1.

The structures of SARS-CoV-2 Mpro inhibitors, Nirmatrelvir/PF-07321332 (1) and 5h (2)
The genome of SARS-CoV-2 encodes two large overlapping polyproteins, pp1a and pp1ab, which are processed principally by two viral proteases, Mpro and a papain-like protease (PLpro), to generate the virally functional proteins.13,14,15,16,17 Mpro is classified as a member of the cysteine protease family. The genome of SARS-CoV-2 has approximately 80% nucleotide identity with the genome of SARS-CoV-1. The Mpros of both SARS-CoV-2 and SARS-CoV-1 have 96% amino acid sequence identity18 and near identity in their tertiary structures and they form almost the same active center of Mpro.19 As Mpro is essential for viral replication and there is no human enzyme closely homologous with Mpro, it is an important and attractive drug discovery target for the treatment of COVID-19. Another Mpro inhibitor, S-217622, which is apparently not correlated with covalent bond formation with Mpro, has been developed.20 In addition, several other Mpro inhibitors have been developed to date.21,22,23,24,25,26,27 As the development of additional several drugs is required for a repertory of drug choice, we have tried to develop other Mpro inhibitors. Previously, we characterized a hit compound, 5h (2), as a SARS-CoV-2 Mpro inhibitor19 among a panel of known compounds, which had originally shown inhibitory activity against SARS-CoV (Figure 1).28,29,30 As the inhibitory activity or biological stability of compound 5h (2) is insufficient, we have attempted in this study to develop more effective inhibitors with increased activity and biological stability based on compound 5h (2) and are reporting the results.
Results
Concept of compound design
Compound 5h (2) is a tripeptide mimic, with an electrophilic ketone group as a warhead. It functions as a tightly binding reversible-covalent inhibitor of SARS-CoV-2 and has a Ki value of 17.6 nM.19 In common with Nirmatrelvir/PF-07321332 (1), compound 2 has P3, P2, P1, and P1′ sites (Figure 1). The warhead electrophilic carbonyl group at the P1′ site of compound 2 can trap the nucleophilic thiol group of Cys145 at the active center of Mpro with a reversible covalent bond, which produces a hemithioketal structure as an intermediate that can inactivate Mpro.19,30 In order to increase the potency of Mpro inhibitors such as compound 5h (2), fluorine atoms were introduced to inhibitor molecules in an attempt to realize a fluorine-associated interaction31 with the pocket of Mpro, and an increase of the cell membrane permeability.32,33 To date, this fluorine scan strategy has been applied to the design of several compounds.34,35,36,37,38,39 Practically, the effectiveness of introduction of fluorine into inhibitor molecules, including enhancement of hydrophobic interaction because of the formation of bonds between a halogen atom and the target enzyme and an increase of intracellular uptake has been revealed by the development of highly potent HIV-1 protease inhibitors.40,41,42 Hydrolysis of the P1-P2 amide bond of compound 5h (2) in the liver, on the other hand might lead to its low availability.30 Thus, the P2-P1 amide bond was replaced by a thioamide, a surrogate structure. Using a sulfur atom as an alternative to an oxygen atom, a thioamide structure is a useful biomimetic for an amide bond in natural peptides because thioamide bonds are resistant to normal peptidases, which can cleave peptide bonds.43,44 A thioamide-containing small drug, ethionamide has been utilized as an antibiotic to treat tuberculosis for over 60 years.45
Synthesis of compounds 3 and 4
The purity of the final synthesized compounds was measured by analytical HPLC or NMR and was >95%. Experimental procedures of the synthesis of all of the compounds including characterization data are provided in the supplemental information – SI (Data S1). The synthesis of the representative compounds (3 and 4) is shown in Figures 2 and 3. A derivative in which the P1-P2 amide bond was replaced by thioamide, was synthesized as compound 4 (Figure 3).46,47 Coupling of the tert-butyl ester hydrochloride of Leu (5) with 7-fluoro-4-methoxyindole-2-carboxylic acid (6) by 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) hydrochloride, 1-hydroxybenzotriazole (HOBt) monohydrate and N,N-diisopropylethylamine (DIPEA) provided the amide (7), and this was followed by deprotection of the tert-butyl group with hydrochloride to give the acid (8). Treatment of the Nα-Boc-derivative of methyl (S)-2-amino-3-((S)-2-oxopyrrolidin-3-yl)propanoate (9) by 4-fluorobenzothiazole pretreated with n-butyllitium produced a ketone (10), and the subsequent deprotection of the Nα-Boc group with tetrafluoroboric acid diethyl ether complex gave an amine (11). Condensation of the acid (8) and the amine (11) with 1-[(1-(cyano-2-ethoxy-2-oxoethylideneaminooxy)dimethylaminomorpholino)]uronium hexafluorophosphate (COMU) in the presence of DIPEA yielded the target compound (3). Coupling of Boc-Leu-OH (12) with 4-nitro-1,2-phenylenediamine by a mixed anhydride reaction using isobutylchloroformate and N-methylmorpholine gave the amide (13). Treatment of 13 with phosphorus pentasulfide in the presence of sodium carbonate produced the thioamide (14),46,47 and this was followed by treatment with sodium nitrite in the presence of aqueous acetic acid to yield the triazole (15). Condensation of the triazole (15) with the amine (11) in the presence of DIPEA led to a thioamide (16), and this was followed by deprotection of the Nα-Boc group with tetrafluoroboric acid diethyl ether complex to give the amine (17). Condensation of the acid (6) with the amine (17) by COMU in the presence of DIPEA yielded the target compound (4).
Figure 2.

Synthetic scheme for compound 3
Figure 3.

Synthetic scheme for compound 4
Structure-activity relationship studies on derivatives modified at the P1′ site
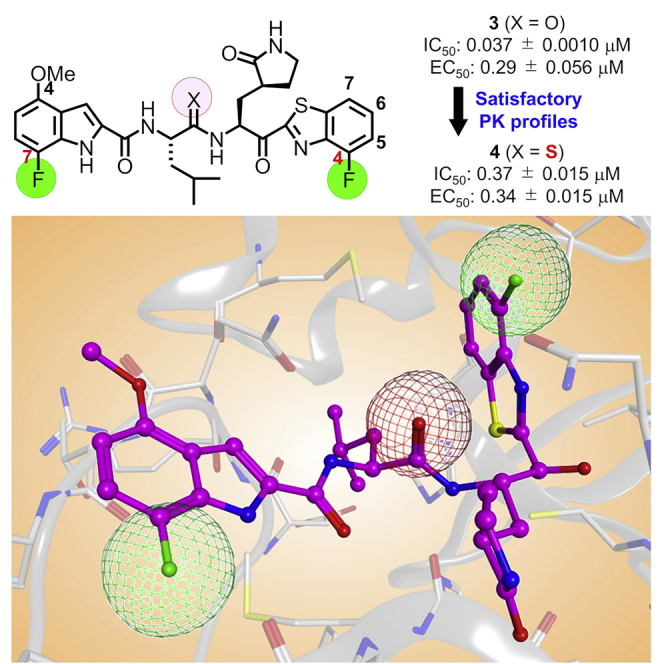
The structure of compound 5h (2) is shown in Figure 1. Fluorobenzothiazolyl ketones function as a warhead with increased electrophilicity. Initially, the benzothiazole moiety at the P1′ site was modified by the introduction of fluorine atoms (Figure 4). The EC50 and CC50 values of the products were determined with RNA-qPCR and WST-8 assays, respectively, using VeroE6 cells (see the experimental model and subject details section).19 The introduction of a fluorine atom at position 4 or 5 of the benzothiazole moiety maintained the antiviral activity (compounds 18/19) but the introduction of a fluorine atom at position 6 (compound 20) slightly decreased this activity. The introduction of a cyclopropane moiety at position 4 of the benzothiazole moiety gave compound 21 but did not affect the potency substantially. Derivatives bearing alkyl esters with one or two trifluoromethyl group(s) (22 or 23, respectively) failed to show significant antiviral activity suggesting that trifluoromethyl-containing alkyl esters when compared with those with the original benzothiazolyl ketone moiety are not suitable as units at the P1′ site. A 4- or 5-fluorobenzothiazole moiety or a normal benzothiazole moiety is suitable as a unit at the P1′ site. According to the X-ray crystal structure of Mpro and 5h (Figure 5B),19 there is enough space close to position 4 or 5 of the benzothiazole moiety at the P1′ site to introduce groups such as a fluorine atom.
Figure 4.

The structures of derivatives modified at the P1′ site
EC50 and CC50 values were determined with RNA-qPCR and WST-8 assays, respectively, using VeroE6 cells.19 The numbers represent the average EC50 value ± SD (μM) and CC50 value ± SD (μM) from at least two independent experiments. Fold changes to 5h (2) are shown. n.a., not applicable.
Figure 5.

The X-ray crystal structure of SARS-CoV-2 Mpro and 5h (2) 7JKV
(A) The two protomers of SARS-CoV-2 Mpro are shown in black and gray surface presentations, and 5h (2) is shown in cyan sticks and mesh (PDB: 7JKV).19 Hydrogen, nitrogen, oxygen, and sulfur atoms are shown in gray, blue, red, and yellow, respectively.
(B and C) The binding pocket of Mpro, which interacts with 5h (2), is shown. The benzothiazole moiety at the P1′ site is shown in the center (B). The 4-methoxyindole moiety at the P3 site, which is surrounded by hydrophobic residues, is shown in the center (C).
Structure-activity relationship studies on derivatives modified at the P3 site
Next, the indole moiety at the P3 site was modified (Figure 6). The introduction of a fluorine atom at position 7 of the indole moiety (compound 24) maintained or slightly decreased the antiviral activity of 5h. The introduction of a fluorine atom at position 7 and the replacement of the 4-methoxy group by a fluorine atom gave compound 25, which mostly retained the antiviral activity of 5h, indicating that the pattern of 4,7-difluoro-substituents is acceptable and that an electron-withdrawing group as well as an electron-donating group is suitable as the 4-substituent group. Other difluoro-substitutions failed to cause any positive effects: 4,5-difluoro-substitution (compound 26) slightly decreased the antiviral activity of 5h, and 4,6-difluoro-substitution (compound 27) largely decreased the antiviral activity of 5h. The replacement of the 4-methoxy group by a trifluoromethoxy group (compound 28) mostly maintained the potency. According to the X-ray crystal structure of Mpro and 5h, there is enough space close to positions 4–7 of the indole moiety at the P3 site to introduce groups such as a fluorine atom, a methoxy group, or a trifluoromethoxy group. Positions 4–7 of the indole moiety are located in a solvent contact area (Figure 5C). The replacement of the indole moiety by a benzyloxy group (compound 29) caused a remarkable decrease of the potency. As a result, it was concluded that 7-fluoro-4-methoxy-substituents, 4,7-difluoro-substituents, and 4-trifluoromethoxy-substituents of the indole moiety are suitable for high antiviral activity. Therefore, to perform structure-activity relationship studies on the benzothiazole moiety, 7-fluoro-4-methoxy-substituents, 4,7-difluoro-substituents, or 4-trifluoromethoxy-substituents of the indole moiety were fixed at the P3 site.
Figure 6.

The structures of derivatives modified at the P3 site
Structure-activity relationship studies on compound 24 derivatives with a 7-fluoro-4-methoxyindole moiety at the P3 site
First, 7-fluoro-4-methoxy-substituents of the indole moiety at the P3 site were used, and the benzothiazole moiety at the P1′ site was derivatized (Figure 7). As a 4- or 5-fluorobenzothiazole moiety is suitable as a unit at the P1′ site of derivatives of 5h, which have 4-methoxyindole at the P3 site, compound 3, which has a 4-fluorobenzothiazole moiety at the P1′ site and a 7-fluoro-4-methoxyindole moiety at the P3 site, and compound 30, which has a 5-fluorobenzothiazole moiety at the P1′ site and a 7-fluoro-4-methoxyindole moiety at the P3 site, were designed and synthesized. Compound 3 showed remarkably higher antiviral activity than 5h, and compound 30 mostly maintained the potency of 5h, suggesting that a 4-fluorobenzothiazole moiety is more suitable than a 5-fluorobenzothiazole moiety as a unit at the P1′ site. According to the X-ray crystal structure of Mpro and compound 3, the complex structure of compound 3 is similar to that of 5h (Figure 8). In the complex with Mpro, the warhead carbonyl group at the P1′ site of compound 3, similar to that of 5h, forms a covalent bond with the side chain thiol group of the Cys145 residue producing its hemithioketal structure, and the entire structure of compound 3 has hydrogen bond interactions as well as hydrophobic interactions with amino acid residues in the pocket of the active site. There is enough space close to positions 4 and 5 of the benzothiazole moiety at the P1′ site to introduce a fluorine atom (Figure 8A) as well as sufficient space close to positions 4–7 of the indole moiety at the P3 site to introduce a fluorine atom or a methoxy group. Positions 4–7 of the indole moiety are located in a solvent contact area (Figure 8B). Next, the replacement of the fluorine atom at position 4 of the benzothiazole moiety by a chlorine atom, a bromine atom, a methoxy group, or a cyclopropane group was investigated. Compound 24 derivatives with a 4-chlorobenzothiazole moiety, a 4-methoxybenzothiazole moiety or a 4-cyclopropylbenzothiazole moiety, (31, 33 and 34, respectively) obviously decreased the potency of compound 3 but largely maintained the potency of 5h (2). A derivative of compound 24 with a 4-bromobenzothiazole moiety (32), maintained the same or slightly less potency than compound 3. In addition, the replacement of the fluorine atom at position 5 of the benzothiazole moiety of compound 30 by a trifluoromethoxy group and a methyl group was investigated. Compound 24 derivatives with a 5-trifluoromethoxybenzothiazole moiety (35) or a 5-methylbenzothiazole moiety (36) reduced the potency of compound 3, and are more potent than compound 30, with a 5-fluorobenzothiazole moiety. The replacement of the fluorine atom at position 5 of the benzothiazole moiety by a hydroxymethyl group was investigated. A derivative of compound 24 with a 5-hydroxymethylbenzothiazole moiety (37), significantly reduced the potency of compound 3, and mostly maintained the potency of compound 30. Judged by the X-ray crystal structure of Mpro with 5h (2) or compound 3 (Figures 5 and 7), introduction of hydrophilic groups such as hydroxymethyl at position 5 of the benzothiazole moiety is not suitable because a hydrogen bond acceptor could affect the hydrogen bond between the sidechain hydroxy group of Thr25 and the mainchain carbonyl group of Cys44 of Mpro and might cause the disadvantageous formation of the inhibitor-Mpro complex in the binding pocket. The normal benzothiazole moiety of 5h (2) and the 4F-benzothiazole moiety of compound 3 have no significant effect on the hydrogen bond between Thr25 and Cys44 of Mpro (SI, Figure S1). The 5-OCF3 group on the benzothiazole ring of compound 35 plausibly forms a hydrogen bond with the sidechain hydroxy group of Thr25 and a halogen bond with the mainchain carbonyl group of Cys44 simultaneously. These interactions might contribute to a favorable formation of the inhibitor-Mpro complex in the binding pocket. The introduction of a fluorine atom at position 7 of the benzothiazole moiety was examined. Compound 24 derivatives having a 7-fluorobenzothiazole moiety (38) or a 4,7-difluorobenzothiazole moiety (39) showed significantly lower antiviral activity than compound 3, which has a 4-fluorobenzothiazole moiety. This suggests that the introduction of the fluorine atom at position 7 of the benzothiazole moiety is not compatible with high potency although the introduction of the fluorine atom at position 4 is critical. The replacement of the benzothiazolyl ketone moiety by an alkylester group or an amide group was also examined. Compound 24 derivatives with alkyl esters and one or two trifluoromethyl group(s) (40 or 41, respectively), showed moderate or insignificant antiviral activity, indicating that trifluoromethyl-containing alkyl esters are not suitable as units at the P1′ site compared with the original benzothiazolyl ketone moiety, as has been seen in compounds 22 and 23. A derivative of compound 24 with an added N-methoxy-N-methyl amide (a Weinreb amide) group (42) failed to show significant antiviral activity, suggesting that this amide group is not suitable as a unit at the P1′ site.
Figure 7.

The structures of compound 24 derivatives with a 7-fluoro-4-methoxyindole moiety at the P3 site
n.d., not determined.
Figure 8.

The superimposed structure of SARS-CoV-2 Mpro with 5h (2) (PDB: 7JKV) or compound 3 (PDB: 8DOY)
The binding pocket of Mpro is shown in ribbon/surface presentation in gray, and 5h (2) and compound 3 are shown in overlay images of cyan/magenta sticks, respectively. Hydrogen, nitrogen, oxygen, fluorine, and sulfur atoms are shown in gray, blue, red, green and yellow, respectively.
(A) The binding pocket of Mpro, which interacts with 5h (2)/compound 3.
(B) The formation of a hydrogen bond between the Nα-amino group of L-leucine at the P2 site of 5h (2)/compound 3 and the sidechain carbonyl group of Gln189 is in the center.
(C) No interaction between the carbonyl group of L-leucine at the P2 site of 5h (2)/compound 3 (red mesh) and Mpro is shown in the center.
(D) The formation of a hydrogen bond between the Nα-amino group of (S)-2-amino-3-((S)-2-oxopyrrolidin-3-yl)propanoic acid at the P1 site of 5h (2) or compound 3 and the mainchain carbonyl group of His164 is shown in the center.
Structure-activity relationship studies on compound 25 derivatives having a 4,7-difluoroindole moiety at the P3 site
Next, 4,7-difluoro-substituents of the indole moiety at the P3 site were examined, and the benzothiazole moiety at the P1′ site was derivatized (Figure 9). As a 4- or 5-fluorobenzothiazole moiety is suitable as a unit at the P1′ site of 5h derivatives, compound 43, with a 4-fluorobenzothiazole moiety at the P1′ site and a 4,7-difluoroindole moiety at the P3 site, and compound 44, which has a 5-fluorobenzothiazole moiety at the P1′ site and a 4,7-difluoroindole moiety at the P3 site, were designed and synthesized. Compound 43 showed remarkably higher antiviral activity than 5h and compound 44 showed slightly higher or equivalent potency compared with 5h, indicating that as observed in compounds 3 and 30, a 4-fluorobenzothiazole moiety is more suitable than a 5-fluorobenzothiazole moiety as a unit at the P1′ site. The introduction of a fluorine atom at position 7 of the benzothiazole moiety was tested. A compound 25 derivative with a 7-fluorobenzothiazole moiety (45) showed significantly lower antiviral activity than compound 43 with a 4-fluorobenzothiazole moiety, and almost the same potency as compound 44 with a 5-fluorobenzothiazole moiety. This suggests that the introduction of a fluorine atom at position 7 of the benzothiazole moiety does not encourage high potency although the introduction of the fluorine atom at position 4 is critical. Next, the replacement of the fluorine atom at position 4 of the benzothiazole moiety by a chlorine or bromine atom or a methoxy group was investigated. Derivatives of compound 25 with 4-chlorobenzothiazole and 4-bromobenzothiazole moieties (46 and 47, respectively) mostly retained the potency of compound 43, suggesting that a 4-chlorobenzothiazole or 4-bromobenzothiazole moiety is suitable as a unit at the P1′ site as a 4-fluorobenzothiazole moiety. A derivative of compound 25 with a 4-methoxybenzothiazole moiety (48) obviously reduced the potency of compound 43, although it mostly maintained the potency of 5h, indicating that a 4-methoxybenzothiazole moiety is not suitable as a unit at the P1′ site as had been observed in compound 33. The replacement of the fluorine atom at position 5 of the benzothiazole moiety by a trifluoromethoxy group or a methyl group was investigated. A derivative of compound 25 with a 5-trifluoromethoxybenzothiazole moiety (49) slightly decreased the potency of compound 43, and is much more active than compound 44 with the 5-fluorobenzothiazole moiety, suggesting that a 5-trifluoromethoxybenzothiazole moiety is suitable as a unit at the P1′ site as observed in compound 35. A derivative of compound 25 with a 5-methylbenzothiazole moiety (50) reduced the potency of compound 43, indicating that a 5-methylbenzothiazole moiety is not suitable as a unit at the P1′ site as it is not consistent with the result observed in compound 36. Furthermore, the replacement of the benzothiazole moiety by a benzyl group was investigated. A derivative of compound 25 with a benzyl group (51) showed significantly lower antiviral activity than compound 25 with a benzothiazole moiety. This suggests that a benzyl group at the P1′ site is less suitable for high potency than a benzothiazole moiety.
Figure 9.

The structures of compound 25 derivatives having a 4,7-difluoroindole moiety at the P3 site
Structure-activity relationship studies on compounds with other indole derivatives at the P3 site
Other indole derivatives at the P3 site were investigated (Figure 10). Compound 52, which has a 4-fluorobenzothiazole moiety at the P1′ site and a 4-fluoro-7-methoxyindole moiety at the P3 site, showed less antiviral activity than 5h, and clearly lower potency than compound 3, which has a 7-fluoro-4-methoxyindole moiety at the P3 site. Therefore, the opposite disposition of a fluorine atom and a methoxy group as substituents on the indole moiety is unsuitable. Compound 53, which has a 5-fluorobenzothiazole moiety at the P1′ site and a 4,5-difluoroindole moiety at the P3 site, and compound 54, with a 5-fluorobenzothiazole moiety at the P1′ site and a 4,6-difluoroindole moiety at the P3 site, showed almost the same potency as compound 44, which has a 5-fluorobenzothiazole moiety at the P1′ site and a 4,7-difluoroindole moiety at the P3 site. Compounds 53, 54 and 44 showed slightly higher or equal potency as 5h, indicating that the disposition of two fluorine atoms as substituents on the indole moiety did not cause any significant effect on antiviral activity. Compound 55, which has a 4-fluorobenzothiazole moiety at the P1′ site and a 4-trifluoromethoxyindole moiety at the P3 site, showed almost the same antiviral activity as compound 18, which has a 4-fluorobenzothiazole moiety at the P1′ site and a 4-methoxyindole moiety at the P3 site. Compound 56, which has a 5-fluorobenzothiazole moiety at the P1′ site and a 4-trifluoromethoxyindole moiety at the P3 site, showed almost the same antiviral activity as compound 19, which has a 5-fluorobenzothiazole moiety at the P1′ site and a 4-methoxyindole moiety at the P3 site. Therefore, a 4-methoxyindole moiety and 4-trifluoromethoxyindole moiety at the P3 site have almost the same effect on the potency.
Figure 10.

The structures of compounds with other indole derivatives at the P3 site
Structure-activity relationship studies on derivatives, in which the l-Leucine residue at the P2 site was modified
Next, the l-leucine residue at the P2 site was modified (Figure 11) because the amide bond between the P2 and P1 sites is susceptible to hydrolysis in the liver according to the metabolic analysis of 5h performed by Konno et al.30 Nα-Methylation of amino acid residues is simple and useful for modification of amide bonds. Nα-Methylation of (S)-2-amino-3-((S)-2-oxopyrrolidin-3-yl)propanoic acid corresponding to the P1 site is described below. Firstly, Nα-methyl-L-leucine corresponding to the P2 site was used to prepare compounds 57, 58 and 59. Compound 57 is a derivative of 5h (2) with Nα-methyl-l-leucine at the P2 site, and both compounds 57 and 5h (2) have a benzothiazole moiety at the P1′ site and a 4-methoxyindole moiety at the P3 site. Compound 57 shows remarkably lower antiviral activity than 5h (2), suggesting that Nα-methylation of the l-leucine residue at the P2 site does not maintain or increase the potency. According to the X-ray crystal structure of Mpro and 5h (2) or compound 3 (Figure 8B), the α-amino hydrogen atom of the l-leucine residue at the P2 site interacts with the carbonyl oxygen atom of the side chain of Gln189 of Mpro by hydrogen bonding. Compound 58 is a derivative of compound 30 with Nα-methyl-l-leucine at the P2 site, and compounds 58 and 30 both have a 5-fluorobenzothiazole moiety at the P1′ site and a 7-fluoro-4-methoxyindole moiety at the P3 site. Compound 58 showed significantly lower antiviral activity than compound 30. This is consistent with the observation that Nα-methylation of the l-leucine residue at the P2 site does not maintain or increase the potency. Compound 59 is a derivative of compound 44 with Nα-methyl-l-leucine at the P2 site, and both compounds 59 and 44 have a 5-fluorobenzothiazole moiety at the P1′ site and a 4,7-difluoroindole moiety at the P3 site. Compound 59 mostly maintains the antiviral activity of compound 44, but shows significant cytotoxicity and CC50 = 96 μM. The reason why Nα-methylation of the L-leucine residue at the P2 site is suitable for the maintenance of potency only in the case of compound 59 is unclear, but compound 59 is not appropriate as a lead because of its significant cytotoxicity. A thioamide structure being analogous to a carboxyamide bond, a dipeptide surrogate corresponding to the P2-P1 site, l-Leu-ψ[C(S)-NH]-(S)-2-amino-3-((S)-2-oxopyrrolidin-3-yl)propanoic acid, was synthesized and used to prepare compounds 4, 60, 61 and 62. Compound 60 is a derivative of 5h (2) with l-Leu-ψ[C(S)-NH]-(S)-2-amino-3-((S)-2-oxopyrrolidin-3-yl)propanoic acid at the P2-P1 site, and both compounds 60 and 5h (2) have a benzothiazole moiety at the P1′ site and a 4-methoxyindole moiety at the P3 site. Compound 60 mostly maintained the antiviral activity of 5h. As a result, the thioamide structure of the l-leucine residue at the P2 site was deemed to be suitable for the maintenance of the potency of 5h. This suggests that, according to the X-ray crystal structure of the complex of 5h (2) and Mpro (Figure 8C), as there is no interaction between Mpro and the carbonyl group of the l-leucine residue at the P2 site, this carbonyl group can be changed to a surrogate such as a thiocarbonyl group. Compound 60 is appropriate as a lead compound of biostable inhibitors, and it was subjected to in vivo stability testing. Compounds 4, 61 and 62 are derivatives of compounds 3, 44 and 56, respectively, with l-Leu-ψ[C(S)-NH]-(S)-2-amino-3-((S)-2-oxopyrrolidin-3-yl)propanoic acid at the P2-P1 site and the corresponding benzothiazole moieties at the P1′ site and indole moieties at the P3 site. The thiocarbonyl type derivatives 4, 61 and 62 mostly maintained or enhanced the antiviral activity of the corresponding parent carbonyl compounds 3, 44 and 56, respectively. The replacement of the carbonyl structure of the l-leucine residue at the P2 site by a thiocarbonyl structure is also applicable in the case of compounds 3, 44 and 56, although compounds 61 and 62 showed significant cytotoxicity, with CC50 = 38 ± 1.1 μM and 50 ± 1.1 μM, respectively. Similar to compound 60, compound 4 is appropriate as a lead compound for biostable inhibitors, and was submitted to in vivo stability tests. The replacement of the l-leucine residue at the P2 site by other amino acid residues was explored. Compounds 63 and 64 are a derivative of compound 30 with 1-aminocyclopentane-1-carboxylic acid and a derivative of compound 3 with 1-amino-3,3-difluorocyclopentane-1-carboxylic acid, respectively, at the P2 site, a 5- or 4-fluorobenzothiazole moiety, respectively, at the P1′ site, and a 7-fluoro-4-methoxyindole moiety at the P3 site. Compounds 63 and 64 have a remarkably reduced antiviral activity compared with compounds 30 and 3. As a result, the substitution of a 1-aminocyclopentane-1-carboxylic acid derivative at the P2 site was deemed to be unsuitable for the maintenance of the potency of compounds 30 and 3. Compounds 65 and 66 are derivatives of compounds 30 and 55, respectively, with l-trifluoromethylalanine at the P2 site, the corresponding benzothiazole moieties at the P1′ site and indole moieties at the P3 site. Compounds 65 and 66 mostly retained the antiviral activity of the parent compounds 30 and 55, respectively. The substitution of l-trifluoromethylalanine at the P2 site is applicable. Compounds 67 and 68 are derivatives of compounds 3 and 43, respectively, with l-trifluoroethylalanine at the P2 site, the corresponding benzothiazole moieties at the P1′ site and indole moieties at the P3 site. Compounds 67 and 68 had a reduced antiviral activity of the parent compounds 3 and 43, respectively. The substitution of l-trifluoroethylalanine at the P2 site fails to maintain the potency. Compounds 69 and 70 are derivatives of compounds 3 and 43, respectively, with l-difluoromethylalanine at the P2 site, the corresponding benzothiazole moieties at the P1′ site and indole moieties at the P3 site. Compounds 69 and 70 have lower antiviral activity than the parent compounds 3 and 43, respectively. In contrast to the substitution of l-trifluoroethylalanine, substitution of l-difluoromethylalanine at the P2 site fails to maintain the potency of the compound. Concerning the replacement of the l-leucine residue at the P2 site by other amino acid residues, the replacement by l-trifluoromethylalanine might be applicable, but the replacement by a 1-aminocyclopentane-1-carboxylic acid derivative, l-trifluoroethylalanine, or l-difluoromethylalanine is unsuitable. This suggests that considering the space between the S2 site of Mpro and the side chain group of the l-leucine residue at the P2 site as shown in the X-ray crystal structure of the complex of compound 3 and Mpro, the side chain group of l-trifluoromethylalanine might be suitable in size for the S2 site of Mpro, but the side chain groups of a 1-aminocyclopentane-1-carboxylic acid derivative, l-trifluoroethylalanine, and l-difluoromethylalanine are too large to fit the S2 site of Mpro (Figure S2).
Figure 11.

The structures of derivatives, in which the l-leucine residue at the P2 site was modified
Structure-activity relationship studies on derivatives, in which the (S)-2-amino-3-((S)-2-oxopyrrolidin-3-yl)propanoic acid residue at the P1 site was modified
Finally, the (S)-2-amino-3-((S)-2-oxopyrrolidin-3-yl)propanoic acid residue at the P1 site was modified (Figure 12). Nα-Methylation of (S)-2-amino-3-((S)-2-oxopyrrolidin-3-yl)propanoic acid corresponding to the P1 site was investigated because, as mentioned above, the amide bond between the P2 and P1 sites is susceptible to hydrolysis. Compound 71 is a derivative of 5h with Nα-methyl-(S)-2-amino-3-((S)-2-oxopyrrolidin-3-yl)propanoic acid at the P1 site, and both compounds 71 and 5h have a benzothiazole moiety at the P1′ site and a 4-methoxyindole moiety at the P3 site. Compound 71 had a significantly reduced antiviral activity compared with 5h, suggesting that Nα-methylation of the (S)-2-amino-3-((S)-2-oxopyrrolidin-3-yl)propanoic acid residue at the P1 site is not suitable for the maintenance or increase of potency. According to the X-ray crystal structure of Mpro and 5h (Figure 8D), the α-amino hydrogen atom of the (S)-2-amino-3-((S)-2-oxopyrrolidin-3-yl)propanoic acid residue at the P1 site enjoys hydrogen bonding with the carbonyl oxygen atom of His164 of Mpro. Substitution of other amino acid residues at the P1 site was examined. Compound 72 is a derivative of 5h with l-pyridin-4-ylalanine at the P1 site, and both compounds 72 and 5h have a benzothiazole moiety at the P1′ site and a 4-methoxyindole moiety at the P3 site. Compound 72 reduced the antiviral activity of 5h by about 50%. Compound 73 is a derivative of compound 44 with l-pyridin-4-ylalanine at the P1 site, and both compounds 73 and 44 have a 5-fluorobenzothiazole moiety at the P1′ site and a 4,7-difluoroindole moiety at the P3 site. Compound 73 failed to show antiviral activity below 100 μM. Compound 74 is a derivative of compound 44 with l-pyridin-3-ylalanine at the P1 site, and has a 5-fluorobenzothiazole moiety at the P1′ site and a 4,7-difluoroindole moiety at the P3 site. Compound 74 showed significantly reduced antiviral activity compared with compound 44. This indicates that the substitution of l-pyridin-3- or 4-ylalanine at the P1 site is not applicable. The (S)-2-amino-3-((S)-2-oxopyrrolidin-3-yl)propanoic acid residue at the P1 site has been widely used in the design of human rhinovirus 3C protease inhibitors,48 and might be indispensable.
Figure 12.

The structures of derivatives, in which the (S)-2-amino-3-((S)-2-oxopyrrolidin-3-yl)propanoic acid residue at the P1 site was modified
In terms of cytotoxicity, the CC50 values of all of the synthesized compounds, with the exception of compounds 29, 40, 51, 59, 61 and 62 are over 100 μM, suggesting absence of significant cytotoxicity.
Enzymatic inhibitory activity of synthesized compounds
As the antiviral activity of the synthesized compounds was evaluated against SARS-CoV-2 in VeroE6 cells and several compounds showed significant antiviral activity, enzymatic inhibitory activity of selected hit compounds against Mpro was investigated for target validation (Table 1) (experimental model and subject details section).19 In most compounds, antiviral activity (EC50) and enzymatic inhibitory activity (IC50) are positively correlated, and the Mpro inhibitory activity is approximately one order greater than the antiviral activity according to IC50 and EC50 values observed with the parent compound 5h. Compound 24, a 5h derivative with a 7-fluoroindole moiety at the P3 site, showed potent Mpro inhibitory activity in view of its antiviral activity whereas compound 28, a 5h derivative with a 4-trifluoromethoxyindole moiety at the P3 site, showed low Mpro inhibitory activity compared with its antiviral activity, suggesting that compound 28 might easily penetrate cells and is biologically stable (see investigation of pharmacokinetics (PK)). Compound 3, which has a 4-fluorobenzothiazole moiety at the P1′ site and a 7-fluoroindole moiety at the P3 site, compound 43, which has a 4-fluorobenzothiazole moiety at the P1′ site and a 4,7-difluoroindole moiety at the P3 site, and compound 44, which has a 5-fluorobenzothiazole moiety at the P1′ site and a 4,7-difluoroindole moiety at the P3 site, all showed Mpro inhibitory activity comparable with their antiviral activity. Compound 54, which has a 5-fluorobenzothiazole moiety at the P1′ site and a 4,6-difluoroindole moiety at the P3 site, showed Mpro inhibitory activity similar to its antiviral activity. The Mpro inhibitory activity of the thiocarbonyl type derivatives 4, 60, and 61 was almost equal to their antiviral activity, according to the IC50 and EC50 values. This suggests that compared with the corresponding parent carbonyl compounds 3, 5h, and 44, such thiocarbonyl type derivatives might show significant activity in cells relative to their enzyme inhibitory activity, possibly because of enhanced cellular penetration or biological stability. The clinically used drug Nirmatrelvir/PF-07321332 (1) exhibits potent Mpro inhibitory activity comparable with its antiviral activity. In order to confirm that the anti-SARS-CoV-2 activity of the compounds could not be because of non-specific cytotoxicity, immunostaining experiments were performed.19 These experiments showed that compounds 3 and 4 have no significant cytotoxicity at concentrations up to 100 μM (SI, Figure S3).
Table 1.
Enzymatic inhibitory activity of synthesized compounds
| Compound | IC50 |
EC50 |
CC50 |
|---|---|---|---|
| (μM) | (μM) | (μM) | |
| 5h (2) | 0.36 ± 0.0050 (1.0)a | 2.2 ± 0.53 (1.0) | >100 |
| 24 | 0.054 ± 0.0015 (0.15) | 3.4 ± 0.15 (1.2) | >100 |
| 28 | 2.5 ± 0.023 (7.1) | 2.9 ± 0.15 (1.0) | >100 |
| 3 | 0.037 ± 0.0010 (0.10) | 0.29 ± 0.056 (0.026) | >100 |
| 43 | 0.050 ± 0.0070 (0.14) | 0.40 ± 0.090 (0.2) | >100 |
| 44 | 0.12 ± 0.010 (0.34) | 2.7 ± 0.15 (0.7) | >100 |
| 54 | 0.54 ± 0.020 (1.5) | 2.8 ± 0.20 (1.1) | >100 |
| 60 | 0.99 ± 0.060 (2.8) | 2.7 ± 0.15 (1.1) | >100 |
| 4 | 0.37 ± 0.015 (1.0) | 0.34 ± 0.015 (0.16) | >100 |
| 61 | 0.67 ± 0.050 (1.9) | 0.49 ± 0.060 (0.2) | 38 ± 1.1 |
| PF-07321332 (1) | 0.028 ± 0.0050 (0.080) | 1.6 ± 0.51 (0.55) | >100 |
| Remdesivir | not tested | 5.0 ± 0.050 (1.5) | >100 |
The inhibitory activity of each compound against Mpro (IC50 value) was determined using 3CL Protease, Untagged (SARS-CoV-2) Assay Kit (BPS Bioscience: Catalog#: 78042-1).19
EC50 values were determined with RNA-qPCR and CC50 values with WST-8 assays, using VeroE6 cells.
Numbers represent the average IC50 value ± SD (μM), EC50 value ± SD (μM), and CC50 value ± SD (μM) from two independent experiments. EC50 and CC50 values for 24, 28, 3, 43, 44, 54, 60, 4, and 61 are from Figures 6, 7, 9, 10, and 11.
The numbers in parentheses show fold changes to 5h.
Detailed antiviral activity of compounds 3 and 4 against SARS-CoV-2
The antiviral activity of the hit compounds (3, 4) against SARS-CoV-2 was investigated in detail (Table 2). The EC75 and EC99 values of these compounds were determined and compared with the values from 5h (2) and PF-07321332 (1). Compound 3 showed the most potent antiviral activity among the four compounds with an EC99 of <1 μM. In the EC50, EC75, and EC99 values, compound 3 is superior to PF-07321332 (1) by approximately one order of magnitude. A thiocarbonyl type compound (4) also showed more potent antiviral activity than PF-07321332 (1) in each of these three indexes. Thus, compounds 3 and 4 are useful lead compounds as anti-SARS-CoV-2 drugs.
Table 2.
Detailed antiviral activity of compounds 3 and 4 against SARS-CoV-2
| Compound | EC50 |
EC75 |
EC99 |
CC50 |
|---|---|---|---|---|
| (μM) | (μM) | (μM) | (μM) | |
| 5h (2) | 1.9 ± 0.50 | 4.3 ± 0.58 | 8.4 ± 0.23 | >100 |
| 3 | 0.23 ± 0.030 | 0.46 ± 0.020 | 0.97 ± 0.0020 | >100 |
| 4 | 0.28 ± 0.045 | 0.59 ± 0.030 | 4.1 ± 3.1 | >100 |
| PF-07321332 (1) | 1.9 ± 0.18 | 4.4 ± 0.21 | 9.7 ± 0.015 | >100 |
EC50, EC75, and EC99 values were determined with RNA-qPCR and CC50 values were determined with WST-8 assays, using VeroE6 cells.
Numbers represent the average EC50 value ± SD (μM) and CC50 value ± SD (μM) from two independent experiments.
Antiviral activity of compound 3 against some SARS-CoV-2 mutants
Initially, we investigated the effects of the synthesized compounds against a WK-521 strain of SARS-CoV-2 (ancestral Wuhan strain),19 which is classified into various Wuhan strains. Although Mpro, especially its active center, has common structures in different mutants, various SARS-CoV-2 mutants were tested in the present study (Table 3). The parent compound, 5h (2), showed significant antiviral activity against QHN001 (α), TY8-612 (β), TY7-501 (γ), K1734 (δ), K5356 (κ), 00012 (ο, BA.1), and 02037 (ο, BA.2) strains at micromolar levels when tested against a WK-521 strain, and a lead compound (3) exhibited approximately one order of magnitude more potent antiviral activity than 5h (2) against these mutant strains. This universal effectivity is an impactful advantage of compound 3.
Table 3.
Antiviral activity of compound 3 against some SARS-CoV-2 mutants
| Strains | 5h (2) |
3 |
|---|---|---|
| EC50 (μM) | EC50 (μM) | |
| WK-521 (ancestral Wuhan strain) | 2.6 ± 0.070 | 0.28 ± 0.02 |
| QHN001 (α) | 2.3 ± 0.090 | 0.34 ± 0.09 |
| TY8-612 (β) | 2.2 ± 0.050 | 0.22 ± 0.035 |
| TY7-501 (γ) | 2.4 ± 0.050 | 0.28 ± 0.060 |
| K1734 (δ), Exp. 1 | 2.7 ± 0.075 | 0.35 ± 0.15 |
| K1734 (δ), Exp. 2 | not tested | 0.59 ± 0.24 |
| K5356 (κ) | 2.4 ± 0.13 | 0.29 ± 0.065 |
| 00012 (ο, ΒΑ.1) | Not tested | 0.47 ± 0.050 |
| 02037 (ο, ΒΑ.2) | Not tested | 0.33 ± 0.10 |
EC50 values were determined with RNA-qPCR and CC50 values with WST-8 assays, using VeroE6 cells.
Numbers represent the average EC50 value ± SD (μM) and CC50 value ± SD (μM) from two independent experiments.
Exp.: experiment.
Investigation of pharmacokinetics (PK)
The pharmacokinetics of synthesized compounds PF-07321332 (1), 5h (2), 24, 28, 60, 44, 3, 43, and 4 in mice were investigated. Compounds PF-07321332 (1), 5h (2), 24, 28, 60, 44, 3, 43, and 4 were administered intravenously (i.v.) at 2.0 mg/kg into mice. Blood samples were collected at various time points after administration (15, 30, 60, 120, and 240 min) and the plasma was obtained by centrifugation. All of the compounds were detected by LC-MS/MS. (Figures 13A and 13B). The half-life values were calculated using a non-compartment model (for details, see the experimental model and subject details section) (Figure 13E). As a result, the half-life values of PF-07321332 (1) and 5h (2) were <23 min, relatively shorter than those of the other compounds. The half-life values of compounds 24 and 44 (∼30 min) were also short, suggesting that the introduction of fluorine atoms in the indole moiety at the P3 site might not significantly extend the half-life. The half-life values of compounds 28 and 60 (∼50–60 min) are approximately 2–3 times longer than that of the parent compound, indicating that the introduction of a 4-trifluoromethoxy group in the indole moiety at the P3 site and the replacement of the P2-P1 amide bond by a thioamide structure might prolong the half-life. High blood concentrations of compound 60 were maintained after 60 min, and even 240 min had passed. The positive effect of the thioamide structure of compound 60 is reasonable because hydrolysis of the P2-P1 amide bond of 5h (2) with human and rat cryopreserved hepatocytes might underlie its low biological stability.30 The positive effect of the 4-trifluoromethoxy group in the indole moiety at the P3 site of compound 28 is also reasonable because metabolism of the 4-methoxy group of 5h (2) with cryopreserved hepatocytes, producing a hydroxyl group was detected.30 The half-lives of potent compounds 3, 4 and 43 (32–37 min) were also short, suggesting the introduction of fluorine atoms in the benzothiazole moiety at the P1′ site as well as in the indole moiety at the P3 site might not significantly prolong the half-life, and that the replacement of the P2-P1 amide bond of compound 3 by a thioamide structure did not have any significant effect on prolongation of the half-life although higher blood concentrations of compound 4 were maintained compared with those of compound 3 after 60 min, and even 240 min. Compounds 3, 4, and 28, which showed relatively satisfactory PKs upon i.v. administration, were administered orally (p.o.) at 2.00 mg/kg and intraperitoneally (i.p.) at 20 mg/kg into mice. Blood samples were collected at various time points after administration (15, 30, 60, 120, 240, 480 and 1,440 min) (Figures 13C, 13D and 13F). Although the half-life of compound 4 after p.o. administration (2.85 h) was shorter than that of compound 3 (4.33 h), compound 4 maintained remarkably higher blood concentrations for the first day than compound 3. The initial concentration of compound 3 did not increase compared with that of compound 4, and a relatively higher value of the half-life of compound 3 was calculated. The half-life value of compound 4 after i.p. administration (3.84 h) was longer than that of compound 3 (2.72 h), and compound 4 maintained remarkably higher blood concentrations than compound 3, although after 1 day, the concentration of compound 3 was slightly higher than that of compound 4. In p.o. and i.p. administrations, the replacement of the P2-P1 amide bond of compound 3 by a thioamide structure might effectively extend the half-life. The PK profiles of compound 28 in p.o. and i.p. administrations were not satisfactory when compared with those of compound 4. As a result, the replacement of the P2-P1 amide bond of these inhibitors by a thioamide structure was thought to possibly lead to favorable PK profiles. Although compounds 28 and 60 are suitable compounds in i.v. administration, taken together with antiviral activity compound 4 is a more suitable compound, particularly when administered p.o. or i.p.
Figure 13.

PK profiles of synthesized compounds in the mouse blood
(A–D) Plots of concentrations of synthesized compounds in the mouse blood after administration. (A) i.v. administration of compounds PF-07321332 (1), 5h (2), 24, 60, 44, and 28 at 2.0 mg/kg into mice; (B) i.v. administration of compounds 3, 4, and 43 at 2.0 mg/kg into mice; (C) p.o. administration of compounds 3, 4, and 28 at 20 mg/kg into mice; (D) i.p. administration of compounds 3, 4, and 28 at 20 mg/kg into mice. number: mouse number, ex. 1-1 and 1-2: two different mice with administration of compound 1.
(E and F) Calculated values of the half-lives (min) of the test compounds in i.v. administration (E, 240 min) and in p.o. and i.p. administrations (F, 1,440 min) from each data points in (A)–(D).
Discussion
As the development of new drugs in addition to existing drugs is necessary to support a repertory of chemotherapeutic choice, we attempted to develop novel Mpro inhibitors based on a previously characterized compound, 5h (2). In this study we developed more effective inhibitors with increased activity and biological stability compared with compound 5h (2). Our process of development of lead compounds included the introduction of fluorine atoms into the inhibitor molecules to increase the binding affinity for the pocket of Mpro and the cell membrane permeability, and the replacement of the unstable amide bond by surrogates such as a thioamide structure. As a result, several potent inhibitors were developed through detailed structure–activity relationships focused on the P3, P2, P1, and P1′ sites of compound 5h (2). Compound 3 effectively blocks SARS-CoV-2 infection in vitro without significant cytotoxicity. Similar to compound 5h (2), compound 3 forms a covalent bond with the side chain thiol of the Cys145 residue of Mpro and according to the X-ray crystal structure analysis, enjoys hydrogen bond interactions and hydrophobic interactions with amino acid residues in the active site pocket of Mpro. Compound 3 also has highly potent enzymatic inhibitory activity against Mpro at the IC50 value of 0.037 ± 0.0010 μM. This is true of the other selected hit compounds, which have significant anti-SARS-CoV-2 activity in cells and possess remarkable enzymatic inhibitory activity, showing that there is a positive correlation between anti-SARS-CoV-2 activity and the enzymatic inhibitory activity against Mpro. Compound 3 showed one-order higher antiviral activity than 5h (2) and PF-07321332 (1), even in the EC99 values. Compound 3 exhibits high antiviral activity against several mutant strains such as QHN001 (α), TY8-612 (β), TY7-501 (γ), K1734 (δ), K5356 (κ), and 00012 (ο, BA.1), and 02037 (ο, BA.2) strains as against a WK-521 strain (ancestral Wuhan strain). This universal effectivity is the strong point of compound 3. In future, the justification of testing compound 3 against SARS-CoV-2 variants would be expanded for plausible conquest of drug resistance to these Mpro inhibitors,49,50 and the possible mutation in Mpro to confer viral evasion of known Mpro inhibitors, Nirmatrelvir/PF-07321332 (1) and 5h (2), was investigated in detail.51 Furthermore, compound 4, which has a thioamide structure in place of the amide bond between the P2 and P1 sites of compound 3, has remarkable anti-SARS-CoV-2 activity, comparable with that of compound 3 in terms of EC50 and EC75 values, although the enzymatic inhibitory activity of compound 4 against Mpro is significantly lower than that of compound 3. Thus, thioamide derivatives might express significant activity in cells in proportion and relative to enzyme inhibitory activity, possibly because of enhanced cell membrane permeability or biological stability compared with the corresponding parent amide-type compounds. Significantly, compound 4 showed remarkably preferable pharmacokinetics in mice, especially in p.o. and i.p. administrations, compared with the corresponding parent compound 3, suggesting that replacement of the hydrolyzable amide bond by its thioamide surrogate structure can increase the biostability of the compound. Our data show that compounds 3 and 4 are useful drug candidates for Mpro inhibitors, and further development based on these compounds is continuing.
Limitations of the study
We have developed highly potent Mpro inhibitors that block SARS-CoV-2 infection in vitro without viral breakthrough. Some compounds showed remarkably preferable pharmacokinetics in mice compared with the parent compounds. Future experiments should evaluate the antiviral activity of these compounds in animal models of SARS-CoV-2 infection, and develop more effective inhibitors for drugs treating COVID-19 based on the present data.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| primary antibodies: convalescent IgG fraction | a convalescent COVID-19 individual, reference S1 | N/A |
| secondary antibody: goat polyclonal anti-human-IgG-Alexa Fluor 488 Fab fragment antibody | Jackson ImmunoResearch Laboratories, Inc | Cat#109-547-003; RRID: AB_2337854 |
| Texas Red™-X dye conjugated Phalloidin | Thermo Fisher Scientific | Cat#T7471 |
| Bacterial and virus strains | ||
| JPN/TY/WK-521 (SARS-CoV-2WK-521) | National Institute of Infectious Diseases (Tokyo, Japan) | GISAID Accession ID; EPI_ISL_408667 |
| B.1.1.7 (alpha) strain [hCoV-19/Japan/QHN001/2020 (SARS-CoV-2QHN001)] | clinically isolated, National Institute of Infectious Diseases | GISAID Accession ID; EPI_ISL_804007) |
| B.1.351 (beta) strain [hCoV-19/Japan/TY8-612-P0/2021 (SARS-CoV-2TY8-612)] | clinically isolated, National Institute of Infectious Diseases | GISAID Accession ID; EPI_ISL_1123289 |
| P.1 (gamma) strain [hCoV-19/Japan/TY7-501-P0/2021 (SARS-CoV-2TY7-501)] | clinically isolated, National Institute of Infectious Diseases | GISAID Accession ID; EPI_ISL_833366 |
| B.1.617.2 (delta) strain [hCoV-19/Japan/TKYK01734/2021 (SARS-CoV-2K1734)] | clinically isolated, Tokyo Metropolitan Institute of public Health | GISAID Accession ID; EPI_ISL_2080609 |
| B.1.617.1 (kappa) strain [TKYTK5356_2021 (SARS-CoV-25356)] | clinically isolated, Tokyo Metropolitan Institute of public Health | GISAID Accession ID; EPI_ISL_2378733 |
| BA.1.1.529 (omicron BA.1) strain [hCoV-19/Japan/TKYX00012/2021 (SARS-CoV-2000129)] | clinically isolated, Tokyo Metropolitan Institute of public Health | GISAID Accession ID; EPI_ISL_8559478 |
| BA.1.1.529 (omicron BA.2) strain [hCoV-19/Japan/TKYS02037/2022 (SARS-CoV-202037)] | clinically isolated, Tokyo Metropolitan Institute of public Health | GISAID Accession ID; EPI_ISL_9397331 |
| Biological samples | ||
| BL21-CodonPlus(DE3)-RIL strain | Agilent | Cat#230245 |
| Chemicals, peptides, and recombinant proteins | ||
| SARS-CoV-2 Mpro/3CLpro | Hattori et al.19 | Accession #: MN908947 |
| Nirmatrelvir | MedChemExpress | HY-138687 |
| Remdesivir | Selleck | S8932 |
| Critical commercial assays | ||
| 3CL Protease, Untagged (SARS-CoV-2) Assay Kit | BPS Bioscience | Cat#78042-1 |
| QIAamp Viral RNA Mini QIAcube Kit | Qiagen | Cat#52926 |
| One Step PrimeScript III RT-qPCR mix | TaKaRa Bio | Cat# RR600B |
| Cell Counting Kit-8 | Dojindo | Cat# 341-08001 |
| Deposited data | ||
| SARS-CoV-2 Mpro-compound 3 co-crystal structure | This paper | PDB: 8DOY |
| Experimental models: Cell lines | ||
| VeroE6 cell | ATCC https://www.atcc.org/products/crl-1586 | Cat# ATCC CRL-1586 |
| Experimental models: Organisms/strains | ||
| Jcl:ICR (ICR) female mice | CLEA Japan | Cat# Jcl:ICR https://www.clea-japan.com/products/outbred/item_a0340 |
| Oligonucleotides | ||
| 5′-AAATTTTGGGGACCAGGAAC-3′ (forward) | SARS-CoV-2 nucleocapsid | Shirato et al.52 |
| 5′- TGGCAGCTGTGTAGGTCAAC-3′ (reverse) | SARS-CoV-2 nucleocapsid | Shirato et al.52 |
| 5′-FAM-ATGTCGCGCATTGGCATGGA-black hole quencher 1 (BHQ1)-3′ | SARS-CoV-2 nucleocapsid | Shirato et al.52 |
| Recombinant DNA | ||
| pGEX-4T1 vector (cloned into SARS-CoV2 Mpro-encoding sequence) | https://www.genscript.com/ | The sequence of SARS-CoV 3CL Protease was optimized and synthesized at GensSript and cloned into PGEX-4T1 vectors for E. Coli expression |
| Software and algorithms | ||
| QuantAnalysis Ver2.2.1 | Bruker Daltonics | Cat#1836735 |
| R software (version 4.0.5) | R Foundation for Statistical Computing | https://www.r-project.org/ |
| MolRep | Vagin and Teplyakov56 | https://www.ccp4.ac.uk/html/molrep.html |
| REFMAC5 | Murshudov et al.57 | https://www2.mrc-lmb.cam.ac.uk/groups/murshudov/ |
| Dundee PRODRG2 server | Schüttelkopf and van Aalten58 | http://davapc1.bioch.dundee.ac.uk/cgi-bin/prodrg |
| ChemDraw Professional 18.0 | PerkinElmer | https://www.perkinelmer.com/category/chemdraw |
| GraphPad Prism 9 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Other | ||
| preparative HPLC: JASCO PU-2086 plus/UV-2075 plus | JASCO Corporation | https://www.jasco.co.jp/jpn/product/index.html#LC |
| preparative HPLC: JASCO PU-2087 plus/UV-2075 plus | JASCO Corporation | https://www.jasco.co.jp/jpn/product/index.html#LC |
| preparative HPLC: JASCO PU-4086-Binary/UV-4075 | JASCO Corporation | https://www.jasco.co.jp/jpn/product/index.html#LC |
| analytical HPLC: JASCO PU-2089 plus/UV-2075 plus | JASCO Corporation | https://www.jasco.co.jp/jpn/product/index.html#LC |
| automatic silica gel flash column chromatography system (Isolera One) | Biotage | https://www.biotage.com/isolera-1-flash-chromatography-instrument |
| Pure C-815 | Buchi | https://www.buchi.com/en/products/instruments/pure-chromatography-systems |
| NMR: Bruker AVANCE III 400 spectrometer | Bruker Daltonics | https://www.bruker.com/en/products-and-solutions/mr/nmr.html |
| NMR: Bruker AVANCE 500 | Bruker Daltonics | https://www.bruker.com/en/products-and-solutions/mr/nmr.html |
| NMR: JNM-ECA500 | JEOL | https://www.jeol.co.jp/en/products/category_nmr.html |
| SPring-8 BL41XU | SPring-8 | http://www.spring8.or.jp/wkg/BL41XU/instrument/lang-en/INS-0000000328?set_language=en&cl=en |
| MS: micrOTOF focus | Bruker Daltonics | https://www.bruker.com/en/products-and-solutions/mass-spectrometry.html |
| LC-MS/MS: impact II | Bruker Daltonics | https://www.bruker.com/en/products-and-solutions/mass-spectrometry.html |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by Hirokazu Tamamura (tamamura.mr@tmd.ac.jp).
Materials availability
This study did not generate new materials or reagents.
Experimental model and subject details
Cells, viruses and test compounds
VeroE6 cells were obtained from the ATCC (Manassas, VA, USA) and were maintained in Dulbecco’s modified Eagle’s medium (D-MEM) supplemented with 10% fetal bovine serum (FBS), 100 μg/mL of penicillin, and 100 μg/mL of streptomycin. SARS-CoV-2 strain JPN/TY/WK-521 (SARS-CoV-2WK-521) was obtained from the National Institute of Infectious Diseases (Tokyo, Japan). Five clinically isolated SARS-CoV-2 mutant strains were used in the current study: a B.1.1.7 (alpha) strain [hCoV-19/Japan/QHN001/2020 (SARS-CoV-2QHN001, GISAID Accession ID; EPI_ISL_804007)], a B.1.351 (beta) strain [hCoV-19/Japan/TY8-612-P0/2021 (SARS-CoV-2TY8-612)] and a P.1 (gamma) strain [hCoV-19/Japan/TY7-501-P0/2021 (SARS-CoV-2TY7-501)] were obtained from National Institute of Infectious Diseases, Tokyo, Japan. A B.1.617.2 (delta) strain [hCoV-19/Japan/TKYK01734/2021 (SARS-CoV-2K1734, GISAID Accession ID; EPI_ISL_2080609)], a B.1.617.1 (kappa) strain [TKYTK5356_2021 (SARS-CoV-25356, DDBJ Accession ID; LC633761)], a BA.1.1.529 (omicron BA.1) strain [hCoV-19/Japan/TKYX00012/2021 (SARS-CoV-200012, GISAID Accession ID; EPI_ISL_8559478)] and a BA.1.1.529 (omicron BA.2) strain [hCoV-19/Japan/TKYS02037/2022 (SARS-CoV-202037, GISAID Accession ID; EPI_ISL_9397331)] were provided from Tokyo Metropolitan Institute of public Health, Tokyo, Japan. Each variant was shown to contain each VOC-specific amino acid substitutions before the assays conducted in the present study (vide infra). An antiviral agent Nirmatrelvir (1) (PF-07321332)12 was purchased from MedChemExpress (Monmouth Junction, NJ). 5h (2) was synthesized by Arun K. Ghosh (Purdue University, USA).21 Remdesivir7,8 was purchased from Selleck (Sylvanfield Drive, Houston). Each compound was dissolved in DMSO at 20 mM as stock solutions.
Method details
Compound synthesis
The synthetic methods for representative compounds 3 and 4 are described in Figures 2 and 3. The purity of all of the final compounds, measured by analytical HPLC or NMR is >95%. Experimental procedures including characterization data are provided in Data S1.
Anti-SARS-CoV-2 assay and cytotoxicity assay
For antiviral assay, cells were seeded in a 96-well plate (2 × 104 cells/well) and incubated. After 1 day, virus was inoculated into cells at each multiplicity of infection (MOI): SARS-CoV-2WK-521, 0.33; SARS-CoV-2QHN001(alpha), 25; SARS-CoV-2TY8-612(beta), 25; SARS-CoV-2TY7-501 (gamma), 20; SARS-CoV-2K1734 (delta), 20; SARS-CoV-2K5356 (kappa), 20; SARS-CoV-200012 (omicron BA.1), 32; SARS-CoV-202037 (omicron BA.2), 33. After additional 3 days, cell culture supernatants were harvested and viral RNA was extracted using a QIAamp viral RNA minikit (Qiagen, Hilden, Germany), and quantitative RT-PCR (RT-qPCR) was then performed using One Step PrimeScript III RT-qPCR mix (TaKaRa Bio, Shiga, Japan) following the instructions of the manufacturers. The primers and probe used for detecting SARS-CoV-2 nucleocapsid52 were 5′-AAATTTTGGGGACCAGGAAC-3′ (forward), 5′- TGGCAGCTGTGTAGGTCAAC-3′ (reverse), and 5′-FAM-ATGTCGCGCATTGGCATGGA-black hole quencher 1 (BHQ1)-3’ (probe). To determine the cytotoxicity of each compound, cells were seeded in a 96-well plate (2 × 104 cells/well). One day later, various concentrations of each compound were added, and cells were then incubated for additional 3 days. The 50% cytotoxic concentrations (CC50) values were determined using the WST-8 assay and Cell Counting Kit-8 (Dojindo, Kumamoto, Japan).
SARS-CoV-2 Mpro inhibition assays
The inhibitory activity (IC50) of each compound against Mpro was determined using 3CL Protease, Untagged (SARS-CoV-2) Assay Kit (BPS Bioscience: Catalog#: 78042-1). SARS-CoV-2 Mpro/3CLpro (Accession #: MN908947) with the authentic N- and C-terminal residues that are released after cleavage from the polyprotein was used for all assays. The details for expression and purification of this fully active Mpro construct was described.53 In general, inhibition of Mpro by a test compound was assessed using a continuous fluorescence assay and the FRET-based substrate UIVT3 (HiLyte Fluor488 TM–ESATLQSGLRKAK-QXL520 TM-NH2) (Anaspec, Fremont, CA). The assay buffer consisted of 50 mM HEPES pH 7.50, 0.1 mg/mL BSA, 0.01% Triton X-100, 2 mM DTT, 1% DMSO and a final enzyme concentration of 200 nM. The assays were performed in Costar 3694 EIA/RIA 96-well half-area, flat bottom, black polystyrene plates (Corning, Corning, NY) at 25 °C. The increase in fluorescence intensity of Mpro catalyzed reactions was measured at a wavelength of 528 nm (20 nm bandwidth) using an excitation wavelength of 485 nm (bandwidth 20 nm) using either a CLARIOstar Plate Reader (BMG Labtech, Cary, NC) or a Synergy H1 hybrid multi-mode plate reader (BioTek, Winooski, VT). The Relative Fluorescence Units (RFU) was measured and plotted to calculate IC50 values.19
Crystallization of Mpro and compound 3
The Mpro solution was concentrated to 3 mg/mL and incubated with 300 μM of 3 for 1 h before crystallization. Crystals were grown using a hanging drop vapour diffusion method at 20 °C. The reservoir solution contained 0.1 M MES pH 6.0, 15% polyethyene glycol (PEG) 6000 and 3% DMSO. Crystals were soaked briefly in a cryoprotection solution containing 0.1 M MES pH 6.0, 35% PEG 400 and 5% DMSO. X-ray data were collected at SPring-8 BL41XU (Hyogo, Japan) using the automatic data collection system ZOO54 and processed by DIALS using xia2 incorporated in ccp4i2.55 The source wavelength for the data collection was 1.0 Å. Data collection statistics are shown in SI, Table S1. The phase problem was solved by molecular replacement using MolRep56 using the 2.16 Å structure of Mpro (PDB: 6LU7 or 7JKV) as a model. All water molecules and ligand atoms were omitted from the starting model. Subsequent cycles of refinement to 1.25 Å resolution were performed in REFMAC5.57 Structure file of compound 3 was generated using the Dundee PRODRG2 server58 and manually fitted to the electron density. All structural figures were produced with MOE (ver. 2020.0901). The data were deposited into the PDB under the PDB: 8DOY.
Pharmokinetics investigations
Animal experiments59
Jcl:ICR (ICR) female mice were purchased from CLEA Japan (Tokyo, Japan). A test compound was administrated by intravenous (2 mg/kg), intraperitoneal (20 mg/kg) or oral (20 mg/kg) injection to ICR mice with body weights ranging from 20 to 30 g (n = 2 or 3). At various time points after administration (i.v.: 15, 30, 60, 120 and 240 min; i.p., p.o.: 0.25, 0.5, 1, 2, 4, 8 and 24 h), blood samples were collected from the retro-orbital venous plexus under sevoflurane anesthesia and centrifuged at 3,000 rpm for 15 min to obtain plasma.
All mice were housed in an air-conditioned animal room at 23 ± 2 °C with a relative humidity of 40–60% under specific pathogen-free conditions, with a 12 h light/dark cycle (08:00−20:00/20:00−08:00). All mice were fed a standard rodent CE-2 diet (CLEA Japan) and had ad libitum access to water. All animal experiments were approved by the President of the National Center for Global Health and Medicine (NCGM) following consideration by the Institutional Animal Care and Use Committee of the NCGM (approval ID no. 21057) and were carried out in accordance with institutional procedures, national guidelines, and the relevant national laws on the protection of animals.
Sample preparation for LC-MS/MS
5 μL of plasma samples was added to 20 μL of MeCN and kept at 4 °C for 15 min for protein precipitation. After centrifugation, the obtained supernatant was added to TFA to give a final concentration of 0.05% for LC-MS/MS analysis.
LC-MS/MS analysis
To quantify the compounds in prepared samples for LC-MS/MS, analysis was done using a quadrupole-time-of-flight (QTOF) mass spectrometer equipped with a Captive Spray electrospray ionization platform in the positive mode (impact II, Bruker Daltonics, Bremen, Germany) with liquid chromatography (Ultimate 3000 HPLC, Thermo Fisher Scientific, MA). An injected sample was concentrated on an Acclaim PepMap100 C18 trap column (Thermo Fisher Scientific) at flow rate of 20 μL/min. For sample separation, reverse-phase chromatography was conducted using an Acclaim PepMap100 C18 LC column (0.075 mm × 150 mm, 2 μm particle) (Thermo Fisher Scientific) in conditions of isocratic mode of 95% MeCN and 0.1% formic acid for 7 min, a flow rate of 300 nL/min and a temperature of 35 °C. Multiple reaction monitoring mode was used in LC-MS/MS analysis and quantitative analysis was performed by using QuantAnalysis Ver2.2.1 (Bruker Daltonics). A non-compartment model was used for the pharmacokinetic analysis. Each parameter was calculated using the R software (version 4.0.5, R Foundation for Statistical Computing, Vienna, Austria) with the “PK” package.60
Quantification and statistical analysis
Numbers represent the average IC50 value ± SD (μM), EC50 value ± SD (μM), and CC50 value ± SD (μM) from two independent experiments. A non-compartment model was used for the pharmacokinetic analysis, and each parameter was calculated using the R software (version 4.0.5, R Foundation for Statistical Computing, Vienna, Austria) with the “PK” package.60
Acknowledgments
The authors thank Prof. Arun K. Ghosh, Purdue University, for his gift of 5h (2). This work was supported in part by Research Projects on COVID-19, Japan Agency for Medical Research and Development (AMED) 20fk0108510 (S.M., H.M., H.T.), JSPS KAKENHI Grant Numbers 20H03362 (H.T.), and AMED under Grant Numbers JP21am0101098 and JP22ama121043 (Platform Project for Supporting Drug Discovery and Life Science Research, BINDS) (H.T.). This research is based on the Cooperative Research Project of Research Center for Biomedical Engineering. The synchrotron radiation experiments at the SPring-8 beamline BL41XU were approved by the Japan Synchrotron Radiation Research Institute (Proposal No. 2021A2725).
Author contributions
#K.T., T.I., and T.K. contributed equally to the study.
K.T., T.I., T.K, N.H.-K., C.A., M.N., T.O., H.N., N.W., M.H., K.S., Y.M., T.K., S.H., N.T., N.K., T.O., K.N.: investigation; H.H., H.B., D.D., J.S.: analysis; S.M., H.M., H.T.: supervision; K.T., T.I., T.K., H.T.: writing.
All authors have given approval to the final version of the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: November 18, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105365.
Supplemental information
Data and code availability
Data reported in this article will be shared by the lead contact on request. The data of the X-ray crystallographic analysis of the co-crystal structure of SARS-CoV-2 Mpro and compound 3 were deposited into the PDB under the ID: 8DOY. Any additional information required to reanalyze the data reported in this article is available from the lead contact on request. Structural representation of X-ray co-crystal structures of SARS-CoV-2 Mpro and 5h/3, immunostaining experimental data, PK data, X-ray data set and synthetic procedures were provided as Figures S1–S3, Tables S1 and S2 and Data S1 (PDF).
References
- 1.Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mitsuya H., Kokudo N. Sustaining containment of COVID-19: global sharing for pandemic response. Glob. Health Med. 2020;2:53–55. doi: 10.35772/ghm.2020.01040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Livingston E., Bucher K. Coronavirus disease 2019 (COVID-19) in Italy. JAMA. 2020;323:1335. doi: 10.1001/jama.2020.4344. [DOI] [PubMed] [Google Scholar]
- 4.Li Q., Guan X., Wu P., Wang X., Zhou L., Tong Y., Ren R., Leung K.S.M., Lau E.H.Y., Wong J.Y., et al. Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N. Engl. J. Med. 2020;382:1199–1207. doi: 10.1056/NEJMoa2001316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lopez Bernal J., Andrews N., Gower C., Gallagher E., Simmons R., Thelwall S., Stowe J., Tessier E., Groves N., Dabrera G., et al. Effectiveness of Covid-19 vaccines against the B.1.617.2 (delta) variant. N. Engl. J. Med. 2021;385:585–594. doi: 10.1056/NEJMoa2108891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marcec R., Majta M., Likic R. Will vaccination refusal prolong the war on SARS-CoV-2? Postgrad. Med. J. 2021;97:143–149. doi: 10.1136/postgradmedj-2020-138903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pardo J., Shukla A.M., Chamarthi G., Gupte A. The journey of Remdesivir: from Ebola to COVID-19. Drugs Context. 2020;9:2020–2024. doi: 10.7573/dic.2020-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.U.S. Food & Drug Administration FDA approves first treatment for COVID-19. https://www.fda.gov/news-events/pressannouncements/fda-approves-first-treatment-covid-19
- 9.Merck . Merck & Co., Inc.; 2021. Merck and Ridgeback’s Investigational Oral Antiviral Molnupiravir Reduced the Risk of Hospitalization or Death by Approximately 50% Compared to Placebo for Patients with Mild or Moderate COVID-19 in Positive Interim Analysis of Phase 3 Study. [Google Scholar]
- 10.World Health Organization WHO updates its treatment guidelines to include molnupiravir. https://www.who.int/news/item/03-03-2022-molnupiravir
- 11.McIntosh J.A., Benkovics T., Silverman S.M., Huffman M.A., Kong J., Maligres P.E., Itoh T., Yang H., Verma D., Pan W., et al. Engineered ribosyl-1-kinase enables concise synthesis of molnupiravir, an antiviral for COVID-19. ACS Cent. Sci. 2021;7:1980–1985. doi: 10.1021/acscentsci.1c00608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Owen D.R., Allerton C.M.N., Anderson A.S., Aschenbrenner L., Avery M., Berritt S., Boras B., Cardin R.D., Carlo A., Coffman K.J., et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science. 2021;374:1586–1593. doi: 10.1126/science.abl4784. [DOI] [PubMed] [Google Scholar]
- 13.Hilgenfeld R. From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014;281:4085–4096. doi: 10.1111/febs.12936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muramatsu T., Kim Y.-T., Nishii W., Terada T., Shirouzu M., Yokoyama S. Autoprocessing mechanism of severe acute respiratory syndrome coronavirus 3C-like protease (SARS-CoV 3CLpro) from its polyproteins. FEBS J. 2013;280:2002–2013. doi: 10.1111/febs.12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li C., Qi Y., Teng X., Yang Z., Wei P., Zhang C., Tan L., Zhou L., Liu Y., Lai L. Maturation mechanism of severe acute respiratory syndrome (SARS) coronavirus 3C-like proteinase. J. Biol. Chem. 2010;285:28134–28140. doi: 10.1074/jbc.M109.095851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lindner H.A., Fotouhi-Ardakani N., Lytvyn V., Lachance P., Sulea T., Ménard R. The papain-like protease from the severe acute respiratory syndrome coronavirus is a deubiquitinating enzyme. J. Virol. 2005;79:15199–15208. doi: 10.1128/JVI.79.24.15199-15208.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barretto N., Jukneliene D., Ratia K., Chen Z., Mesecar A.D., Baker S.C. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J. Virol. 2005;79:15189–15198. doi: 10.1128/JVI.79.24.15189-15198.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghosh A.K., Xi K., Grum-Tokars V., Xu X., Ratia K., Fu W., Houser K.V., Baker S.C., Johnson M.E., Mesecar A.D. Structure-based design, synthesis, and biological evaluation of peptidomimetic SARS-CoV 3CLpro inhibitors. Bioorg. Med. Chem. Lett. 2007;17:5876–5880. doi: 10.1016/j.bmcl.2007.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hattori S.I., Higashi-Kuwata N., Hayashi H., Allu S.R., Raghavaiah J., Bulut H., Das D., Anson B.J., Lendy E.K., Takamatsu Y., et al. A small compound with an indole moiety inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 2021;12:668. doi: 10.1038/s41467-021-20900-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Unoh Y., Uehara S., Nakahara K., Nobori H., Yamatsu Y., Yamamoto S., Maruyama Y., Taoda Y., Kasamatsu K., Suto T., et al. Discovery of S-217622, a noncovalent oral SARS-CoV-2 3CL protease inhibitor clinical candidate for treating COVID-19. J. Med. Chem. 2022;65:6499–6512. doi: 10.1021/acs.jmedchem.2c00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoffman R.L., Kania R.S., Brothers M.A., Davies J.F., Ferre R.A., Gajiwala K.S., He M., Hogan R.J., Kozminski K., Li L.Y., et al. Discovery of ketone-based covalent inhibitors of coronavirus 3CL proteases for the potential therapeutic treatment of COVID-19. J. Med. Chem. 2020;63:12725–12747. doi: 10.1021/acs.jmedchem.0c01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qiao J., Li Y.-S., Zeng R., Liu F.-L., Luo R.-H., Huang C., Wang Y.-F., Zhang J., Quan B., Shen C., et al. SARS-CoV-2 Mpro inhibitors with antiviral activity in a transgenic mouse model. Science. 2021;371:1374–1378. doi: 10.1126/science.abf1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bai B., Belovodskiy A., Hena M., Kandadai A.S., Joyce M.A., Saffran H.A., Shields J.A., Khan M.B., Arutyunova E., Lu J., et al. Peptidomimetic α-acyloxymethylketone warheads with six-membered lactam P1 glutamine mimic: SARS-CoV-2 3CL protease inhibition, coronavirus antiviral activity, and in vitro biological stability. J. Med. Chem. 2022;65:2905–2925. doi: 10.1021/acs.jmedchem.1c00616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xia Z., Sacco M., Hu Y., Ma C., Meng X., Zhang F., Szeto T., Xiang Y., Chen Y., Wang J. Rational design of hybrid SARS-CoV-2 main protease inhibitors guided by the superimposed cocrystal structures with the peptidomimetic inhibitors GC-376, telaprevir, and boceprevir. ACS Pharmacol. Transl. Sci. 2021;4:1408–1421. doi: 10.1021/acsptsci.1c00099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma C., Xia Z., Sacco M.D., Hu Y., Townsend J.A., Meng X., Choza J., Tan H., Jang J., Gongora M.V., et al. Discovery of di- and trihaloacetamides as covalent SARS-CoV-2 main protease inhibitors with high target specificity. J. Am. Chem. Soc. 2021;143:20697–20709. doi: 10.1021/jacs.1c08060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang H., Pei R., Li X., Deng W., Xing S., Zhang Y., Zhang C., He S., Sun H., Xiao S., et al. The structure-based design of peptidomimetic inhibitors against SARS-CoV-2 3C like protease as Potent anti-viral drug candidate. Eur. J. Med. Chem. 2022;238:114458. doi: 10.1016/j.ejmech.2022.114458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma Y., Yang K.S., Geng Z.Z., Alugubelli Y.R., Shaabani N., Vatansever E.C., Ma X.R., Cho C.-C., Khatua K., Xiao J., et al. A multi-pronged evaluation of aldehyde-based tripeptidyl main protease inhibitors as SARS-CoV-2 antivirals. Eur. J. Med. Chem. 2022;240:114570. doi: 10.1016/j.ejmech.2022.114570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thanigaimalai P., Konno S., Yamamoto T., Koiwai Y., Taguchi A., Takayama K., Yakushiji F., Akaji K., Chen S.E., Naser-Tavakolian A., et al. Development of potent dipeptide-type SARS-CoV 3CL protease inhibitors withNovel P3 scaffolds: design, synthesis, biological evaluation, and docking studies. Eur. J. Med. Chem. 2013;68:372–384. doi: 10.1016/j.ejmech.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Konno S., Thanigaimalai P., Yamamoto T., Nakada K., Kakiuchi R., Takayama K., Yamazaki Y., Yakushiji F., Akaji K., Kiso Y., et al. Design and synthesis of new tripeptide-type SARS-CoV 3CL protease inhibitors containing an electrophilic arylketone moiety. Bioorg. Med. Chem. 2013;21:412–424. doi: 10.1016/j.bmc.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Konno S., Kobayashi K., Senda M., Funai Y., Seki Y., Tamai I., Schäkel L., Sakata K., Pillaiyar T., Taguchi A., et al. 3CL protease inhibitors with an electrophilic arylketone moiety as anti-SARS-CoV-2 agents. J. Med. Chem. 2022;65:2926–2939. doi: 10.1021/acs.jmedchem.1c00665. [DOI] [PubMed] [Google Scholar]
- 31.Zhou P., Zou J., Tian F., Shang Z. Fluorine bonding — how does it work in protein−ligand interactions? J. Chem. Inf. Model. 2009;49:2344–2355. doi: 10.1021/ci9002393. [DOI] [PubMed] [Google Scholar]
- 32.Gillis E.P., Eastman K.J., Hill M.D., Donnelly D.J., Meanwell N.A. Applications of fluorine in medicinal chemistry. J. Med. Chem. 2015;58:8315–8359. doi: 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]
- 33.Shah P., Westwell A.D. The role of fluorine in medicinal chemistry. J. Enzym. Inhib. Med. Chem. 2007;22:527–540. doi: 10.1080/14756360701425014. [DOI] [PubMed] [Google Scholar]
- 34.Smart B.E. Fluorine substituent effects (on bioactivity) J. Fluor. Chem. 2001;109:3–11. doi: 10.1016/S0022-1139(01)00375-X. [DOI] [Google Scholar]
- 35.Schweizer E., Hoffmann-Röder A., Schärer K., Olsen J.A., Fäh C., Seiler P., Obst-Sander U., Wagner B., Kansy M., Diederich F. A fluorine scan at the catalytic center of thrombin: C–F, C–OH, and C–OMe bioisosterism and fluorine effects on pKa and log D values. ChemMedChem. 2006;1:611–621. doi: 10.1002/cmdc.200600015. [DOI] [PubMed] [Google Scholar]
- 36.Morgenthaler M., Aebi J.D., Grüninger F., Mona D., Wagner B., Kansy M., Diederich F. A fluorine scan of non-peptidic inhibitors of neprilysin: fluorophobic and fluorophilic regions in an enzyme active site. J. Fluor. Chem. 2008;129:852–865. doi: 10.1016/j.jfluchem.2008.02.004. [DOI] [Google Scholar]
- 37.Hyohdoh I., Furuichi N., Aoki T., Itezono Y., Shirai H., Ozawa S., Watanabe F., Matsushita M., Sakaitani M., Ho P.-S., et al. Fluorine scanning by nonselective fluorination: enhancing Raf/MEK inhibition while keeping physicochemical properties. ACS Med. Chem. Lett. 2013;4:1059–1063. doi: 10.1021/ml4002419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Giroud M., Harder M., Kuhn B., Haap W., Trapp N., Schweizer W.B., Schirmeister T., Diederich F. Fluorine scan of inhibitors of the cysteine protease human cathepsin L: dipolar and quadrupolar effects in the π-stacking of fluorinated phenyl rings on peptide amide bonds. ChemMedChem. 2016;11:1042–1047. doi: 10.1002/cmdc.201600132. [DOI] [PubMed] [Google Scholar]
- 39.Meanwell N.A. Fluorine and fluorinated motifs in the design and application of bioisosteres for drug design. J. Med. Chem. 2018;61:5822–5880. doi: 10.1021/acs.jmedchem.7b01788. [DOI] [PubMed] [Google Scholar]
- 40.Ghosh A.K., Rao K.V., Nyalapatla P.R., Kovela S., Brindisi M., Osswald H.L., Sekhara Reddy B., Agniswamy J., Wang Y.-F., Aoki M., et al. Design of highly potent, dual-acting and central-nervous-system-penetrating HIV-1 protease inhibitors with excellent potency against multidrug-resistant HIV-1 variants. ChemMedChem. 2018;13:803–815. doi: 10.1002/cmdc.201700824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bulut H., Hattori S.I., Aoki-Ogata H., Hayashi H., Das D., Aoki M., Davis D.A., Rao K.V., Nyalapatla P.R., Ghosh A.K., Mitsuya H. Single atom changes in newly synthesized HIV protease inhibitors reveal structural basis for extreme affinity, high genetic barrier, and adaptation to the HIV protease plasticity. Sci. Rep. 2020;10:10664. doi: 10.1038/s41598-020-65993-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hattori S.I., Hayashi H., Bulut H., Rao K.V., Nyalapatla P.R., Hasegawa K., Aoki M., Ghosh A.K., Mitsuya H. Halogen bond interactions of novel HIV-1 protease inhibitors (PI) (GRL-001-15 and GRL-003-15) with the flap of protease are critical for their potent activity against wild-type HIV-1 and multi-PI-resistant variants. Antimicrob. Agents Chemother. 2019;63:02635-18. doi: 10.1128/AAC.02635-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen X., Mietlicki-Baase E.G., Barrett T.M., McGrath L.E., Koch-Laskowski K., Ferrie J.J., Hayes M.R., Petersson E.J. Thioamide substitution selectively modulates proteolysis and receptor activity of therapeutic peptide hormones. J. Am. Chem. Soc. 2017;139:16688–16695. doi: 10.1021/jacs.7b08417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Phan H.A.T., Giannakoulias S.G., Barrett T.M., Liu C., Petersson E.J. Rational design of thioamide peptides as selective inhibitors of cysteine protease cathepsin L. Chem. Sci. 2021;12:10825–10835. doi: 10.1039/d1sc00785h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schless J.M., Allison R.F., Inglis R.M., White E.F., Topperman S. The use of ethionamide in combined drug regimens in the re-treatment of isoniazid resistant pulmonary tuberculosis. Am. Rev. Respir. Dis. 1965;91:728–737. doi: 10.1164/arrd.1965.91.5.728. [DOI] [PubMed] [Google Scholar]
- 46.Liu C., Barrett T.M., Chen X., Ferrie J.J., Petersson E.J. Fluorescent probes for studying thioamide positional effects on proteolysis reveal insight into resistance to cysteine proteases. Chembiochem. 2019;20:2059–2062. doi: 10.1002/cbic.201900115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shalaby M.A., Grote C.W., Rapoport H. Thiopeptide synthesis. α-Amino thionoacid derivatives of nitrobenzotriazole as thioacylating agents. J. Org. Chem. 1996;61:9045–9048. doi: 10.1021/jo961245q. [DOI] [PubMed] [Google Scholar]
- 48.Dragovich P.S., Prins T.J., Zhou R., Webber S.E., Marakovits J.T., Fuhrman S.A., Patick A.K., Matthews D.A., Lee C.A., Ford C.E., et al. Structure-based design, synthesis, and biological evaluation of irreversible human rhinovirus 3C protease inhibitors. 4. Incorporation of P1 lactam moieties as l-glutamine replacements. J. Med. Chem. 1999;42:1213–1224. doi: 10.1021/jm9805384. [DOI] [PubMed] [Google Scholar]
- 49.Ullrich S., Ekanayake K.B., Otting G., Nitsche C. Main protease mutants of SARS-CoV-2 variants remain susceptible to nirmatrelvir. Bioorg. Med. Chem. Lett. 2022;62:128629. doi: 10.1016/j.bmcl.2022.128629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hu Y., Lewandowski E.M., Tan H., Morgan R.T., Zhang X., Jacobs L.M.C., Butler S.G., Mongora M.V., Choy J., Chen Y., et al. Naturally occurring mutations of SARS-CoV-2 main protease confer drug resistance to nirmatrelvir. bioRxiv. 2022 doi: 10.1101/2022.06.28.497978. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang K.S., Leeuwon S.Z., Xu S., Liu W.R. Evolutionary and structural insights about potential SARS-CoV-2 evasion of nirmatrelvir. J. Med. Chem. 2022;65:8686–8698. doi: 10.1021/acs.jmedchem.2c00404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shirato K., Nao N., Katano H., Takayama I., Saito S., Kato F., Katoh H., Sakata M., Nakatsu Y., Mori Y., et al. Development of genetic diagnositic methods for novel coronavirus 2019 (nCoV-2019) in Japan. Jpn. J. Infect. Dis. 2020;73:304–307. doi: 10.7883/yoken.JJID.2020.061. [DOI] [PubMed] [Google Scholar]
- 53.Anson B.J., Chapman M.E., Lendy E.K., Pshenychnyi S., D’Aquila R.T., Satchell K.J.F., Mesecar A.D. Broad-spectrum inhibition of coronavirus main and papainlike proteases by HCV drugs. Res. Square. 2020 doi: 10.21203/rs.3.rs-26344/v1. [DOI] [Google Scholar]
- 54.Hirata K., Yamashita K., Ueno G., Kawano Y., Hasegawa K., Kumasaka T., Yamamoto M. ZOO: an automatic data-collection system for high-throughput structure analysis in protein microcrystallography. Acta Crystallogr. D Struct. Biol. 2019;75:138–150. doi: 10.1107/S2059798318017795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Winter G., Waterman D.G., Parkhurst J.M., Brewster A.S., Gildea R.J., Gerstel M., Fuentes-Montero L., Vollmar M., Michels-Clark T., Young I.D., et al. DIALS: implementation and evaluation of a new integration package. Acta Crystallogr. D Struct. Biol. 2018;74:85–97. doi: 10.1107/S2059798317017235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vagin A., Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 2010;66:22–25. doi: 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- 57.Murshudov G.N., Skubák P., Lebedev A.A., Pannu N.S., Steiner R.A., Nicholls R.A., Winn M.D., Long F., Vagin A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schüttelkopf A.W., van Aalten D.M.F. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 59.Higashi-Kuwata N., Hayashi S., Kumamoto H., Ogata-Aoki H., Das D., Venzon D., Hattori S.I., Bulut H., Hashimoto M., Otagiri M., et al. Identification of a novel long-acting 4’-modified nucleoside reverse transcriptase inhibitor against HBV. J. Hepatol. 2021;74:1075–1086. doi: 10.1016/j.jhep.2020.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jaki T., Wolfsegger M.J. Estimation of pharmacokinetic parameters with the R package PK. Pharmaceut. Statist. 2011;10:284–288. doi: 10.1002/pst.449. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data reported in this article will be shared by the lead contact on request. The data of the X-ray crystallographic analysis of the co-crystal structure of SARS-CoV-2 Mpro and compound 3 were deposited into the PDB under the ID: 8DOY. Any additional information required to reanalyze the data reported in this article is available from the lead contact on request. Structural representation of X-ray co-crystal structures of SARS-CoV-2 Mpro and 5h/3, immunostaining experimental data, PK data, X-ray data set and synthetic procedures were provided as Figures S1–S3, Tables S1 and S2 and Data S1 (PDF).