

The complex genomic landscape of RAS-mutant tumors is reflective of selection processes in a cancer lineage–specific and context-dependent manner, highlighting differential therapeutic vulnerabilities that can be clinically translated.
Abstract
The RAS family of small GTPases represents the most commonly activated oncogenes in human cancers. To better understand the prevalence of somatic RAS mutations and the compendium of genes that are coaltered in RAS-mutant tumors, we analyzed targeted next-generation sequencing data of 607,863 mutations from 66,372 tumors in 51 cancer types in the AACR Project GENIE Registry. Bayesian hierarchical models were implemented to estimate the cancer-specific prevalence of RAS and non-RAS somatic mutations, to evaluate co-occurrence and mutual exclusivity, and to model the effects of tumor mutation burden and mutational signatures on comutation patterns. These analyses revealed differential RAS prevalence and comutations with non-RAS genes in a cancer lineage-dependent and context-dependent manner, with differences across age, sex, and ethnic groups. Allele-specific RAS co-mutational patterns included an enrichment in NTRK3 and chromatin-regulating gene mutations in KRAS G12C-mutant non–small cell lung cancer. Integrated multiomic analyses of 10,217 tumors from The Cancer Genome Atlas (TCGA) revealed distinct genotype-driven gene expression programs pointing to differential recruitment of cancer hallmarks as well as phenotypic differences and immune surveillance states in the tumor microenvironment of RAS-mutant tumors. The distinct genomic tracks discovered in RAS-mutant tumors reflected differential clinical outcomes in TCGA cohort and in an independent cohort of patients with KRAS G12C-mutant non–small cell lung cancer that received immunotherapy-containing regimens. The RAS genetic architecture points to cancer lineage–specific therapeutic vulnerabilities that can be leveraged for rationally combining RAS-mutant allele-directed therapies with targeted therapies and immunotherapy.
Significance:
The complex genomic landscape of RAS-mutant tumors is reflective of selection processes in a cancer lineage–specific and context-dependent manner, highlighting differential therapeutic vulnerabilities that can be clinically translated.
Introduction
The RAS family of small GTPases represent the most commonly activated oncogenes, with mutations in KRAS, NRAS, and HRAS found in approximately one-third of all human cancers (1, 2). RAS proteins function as binary switches that cycle from an inactive GDP-bound state to an active GTP-bound state, in response to upstream signaling, typically from receptor tyrosine kinases. The majority of RAS mutations affect codons 12, 13, or 61 near the nucleotide binding pocket (3), resulting in loss of GTPase activity and constitutive activation of RAS (4). These hotspot mutations promote oncogenic transformation across human cancers (5–8) with the position and type of substitution show a tumor type–dependent distribution (9). KRAS codon 12 mutations are most commonly seen in pancreatic adenocarcinoma (PAC), lung adenocarcinoma (LUAD), and colorectal carcinoma (10–12) and may be linked with differential clinical outcomes (13, 14). NRAS Q61 mutations are more prevalent in melanoma (15), while NRAS G12 mutations are more frequently seen in hematologic malignancies (16). HRAS mutations are overall less frequent and predominantly occur in head and neck squamous cell carcinoma and bladder cancer (17, 18).
Nevertheless, tissue- and context-specific prevalence and comutation patterns of RAS genes remain incompletely understood. To this end, elucidating the patterns of RAS mutations may reveal biologically relevant differences that point to differential therapeutic vulnerabilities. This is particularly timely given the development of novel inactive state-selective inhibitors that trap KRAS G12C in its GDP-bound state (19, 20), as well as pan-RAS inhibitors of the active GTP-bound form of RAS (21). The clinical development of different classes of RAS-targeting agents highlights the need for comprehensive studies evaluating the prevalence of RAS mutations across tumor types and assessing for potential dependencies between RAS and non-RAS mutations. Here, we sought to understand the distribution and heterogeneity of mutant RAS alleles in a comprehensive pan-cancer manner and developed novel analytic frameworks for assessment of prevalence and comutation patterns of RAS genes leveraging next-generation sequencing (NGS) data and >600,000 mutations from >66,000 individuals across 51 cancer types in the AACR Project GENIE Registry (version 6.1; ref. 22). Our analyses highlight the context-dependent genomic diversity of RAS and may inform the development of effective tissue-specific targeted therapeutic strategies.
Materials and Methods
Cohort description
We extracted targeted NGS data from the AACR Project GENIE Registry (version 6.1), encompassing 66,372 patients across 51 cancer types and 19 study centers (Supplementary Table S1). Of these patients, 2,959 had multiple tumor samples sequenced and 343 had multiple tumor types. For each patient with multiple samples sequenced, a single tumor sample was selected for inclusion in subsequent prevalence and co-occurrence analyses. While RAS prevalence analyses and comutation of RAS with copy-number aberrations and fusions in non-RAS genes were performed utilizing all available patients (n = 66,372), only cases with matched tumor and normal DNA sequencing were included in the comutation analyses of non-RAS sequence variants to avoid bias related to single nucleotide polymorphisms (SNP) and other germline alterations. To this end, 27,257 patients across 49 cancer types and three study centers were utilized for comutation analyses with sequence variants (Supplementary Table S2). For a subset of patients with non–small cell lung cancer (NSCLC), clinical metadata was retrieved and clinical outcomes with immunotherapy-only and immunotherapy-containing regimens were assessed (n = 209; Supplementary Table S3). For The Cancer Genome Atlas (TCGA) cohort, whole-exome sequencing derived multicenter mutation calls from TCGA pan-cancer atlas (23) were retrieved from the NCI Genomic Data Commons for 10,217 tumor samples [https://gdc.cancer.gov/about-data/publications/mc3–2017; adrenocortical carcinoma (ACC) = 92, bladder urothelial carcinoma (BLCA) = 411, breast invasive carcinoma (BRCA) = 1,026, cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC) = 291, cholangiocarcinoma (CHOL) = 36, colon adenocarcinoma (COAD) = 406, lymphoid neoplasm diffuse large B-cell lymphoma (DLBC) = 37, esophageal carcinoma (ESCA) = 185, glioblastoma multiforme (GBM) = 400, head and neck squamous cell carcinoma (HNSCC) = 509, kidney chromophobe (KICH) = 66, kidney renal clear cell carcinoma (KIRC) = 370, kidney renal papillary cell carcinoma (KIRP) = 282, acute myeloid leukemia (LAML) = 140, brain lower grade glioma (LGG) = 525, liver hepatocellular carcinoma (LIHC) = 365, LUAD = 517, lung squamous cell carcinoma (LUSC) = 485, mesothelioma (MESO) = 82, ovarian serous cystadenocarcinoma (OV) = 411, pancreatic adenocarcinoma (PAAD) = 178, pheochromocytoma and paraganglioma (PCPG) = 184, prostate adenocarcinoma (PRAD) = 498, rectum adenocarcinoma (READ) = 150, sarcoma (SARC) = 239, skin cutaneous melanoma (SKCM) = 468, stomach adenocarcinoma (STAD) = 439, testicular germ cell tumors (TGCT) = 134, thyroid carcinoma (THCA) = 500, thymoma (THYM) = 123, uterine corpus endometrial carcinoma (UCEC) = 531, uterine carcinosarcoma (UCS) = 57, uveal melanoma (UVM) = 80] and filtered to keep nonsynonymous alterations. Clinical annotations of tumors were accessed using TCGA clinical data resource (24).
NGS, tumor mutation burden, and mutational spectra
Various NGS panels covering a median of 78 kb of coding sequence per sample [interquartile range (IQR): 22 kb–1,181 kb] were utilized, yielding a total of 607,863 sequence alterations, 980,850 copy-number aberrations and 6,514 fusions. A median of 4 (IQR: 2–8) sequence genomic alterations were detected per tumor sample. We considered all KRAS-, NRAS-, and HRAS-mutant alleles at codons 12, 13, or 61 and estimated their prevalence by gene, codon, and allele. Non-RAS sequence mutations were further characterized as hotspots [recurring ≥ 10 times in COSMIC version 92.1.0 (25)], driver missense mutations [utilizing a CHASMplus score of ≥ 0.75 (26)], putative loss-of-function sequence alterations (frameshift, nonsense, splice site mutations) and considered as single features as well as in the context of gene families and pathways. Non-RAS structural alterations were also considered and were limited to fusions, deep deletions and high-level amplifications to include structural aberrations most likely to be oncogenic. With each build of GENIE, a bed file is released by Synapse (http://www.synapse.org) that provides the set of HUGO gene symbols targeted by each assay. In a separate file, information is provided that maps the sequencing ID for a sample to the assay ID. By merging these two files, we determined for each gene pair the set of platforms and corresponding samples available for analysis. For a given gene pair, the denominator was derived by the number of samples sequenced on the platforms that included both genes. We performed a very limited assessment of mutation cellular fractions for a pair of presumed clonal alterations (KRAS/STK11) and a pair of putatively clonal/subclonal alterations (KRAS/PIK3CA) assuming the same purity and allelic ploidy. Overall, the data modalities available through the AACR project GENIE registry were inadequate to accurately computationally derive mutation cellular fractions as allele-specific copy numbers could not be computed. Estimation of the number of nonsynonymous somatic mutations per Mb (tumor mutation burden; TMB) was calibrated using ATGC, a machine learning model that incorporates positional and sequence related contexts to identify somatic variants (27). For mutation signature analyses, we restricted the dataset to 19,057 patients with a TMB of at least 1 mutation/Mb. Mutational signatures per tumor sample were determined on the basis of the fraction of coding single-nucleotide variants in each of 96 trinucleotide contexts using the deconstructSigs R package (28).
Log-linear models for mutation frequencies
To jointly model RAS mutation prevalence in multiple cancer types, we implemented a Bayesian hierarchical logistic model with diffuse priors using JAGS version 4.3.0 (29). The hierarchical model shrinks the empirical cancer-specific prevalence to the average prevalence from all available cancer types in the AACR Project GENIE Registry. For rare cancer types, the amount of shrinkage can be large while for well-represented cancers the posterior mean from the Bayesian model will be nearly equivalent to the empirical fraction of patients with a RAS variant (30). The sampling distribution for the number of patients yi with a RAS mutation in cancer type i is given by
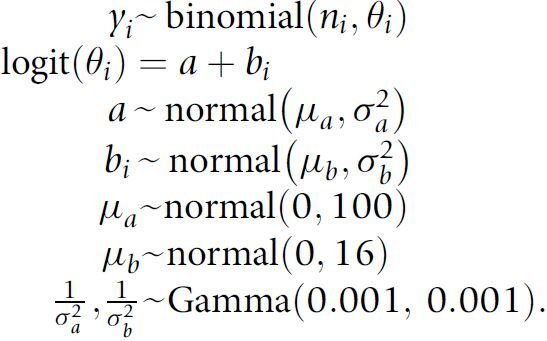 |
This model was fit independently for each of the 20 different RAS hotspot mutations and for each of the different amino acid substitutions (e.g., G12A, G12C, etc.) using all 66,372 tumor samples. Posterior summaries were based on 30,000 Markov chain Monte Carlo (MCMC) iterations from three independent chains following a burn-in of 500 simulations. In addition to the marginal cancer-specific prevalence of RAS mutations, we also estimated prevalence stratified by sex, race, and age. To allow for a nonlinear relationship between prevalence and age, we stratified patients into four age strata: <40, 40–50, 51–65, and >65. For a given cancer type, we evaluated the above model with i denoting the stratum for sex, race, or age. To report the difference between the marginal (overall) prevalence and the prevalence within sex, race, or age, we fit two independent beta-binomial models using beta (0.5, 0.5) priors and summarized the difference in the two posterior distributions by the 0.025, 0.5, and 0.975 quantiles.
Modeling co-occurrence
To characterize co-occurrence of RAS hotspot mutations with other non-RAS mutations, we aggregated non-RAS mutations to the gene level. For sequence mutations, we limited our analyses to patients with sequenced matched normal DNA to avoid spurious associations with germline variants (study centers MSK, UHN, and UCSF). We limited co-occurrence analyses to cancers with at least 50 RAS variants and 50 variants of the non-RAS gene. We used a Poisson log-linear model to estimate the cancer-specific rates of co-occurrence between RAS variants and variants in other genes. Representing the data as a K-dimensional array of 2 × 2 tables (K denotes the number of cancers with at least 20 patients), yijk denotes the number of patients with cancer type k with RAS variant i and non-RAS gene variant j. For each gene, the log-linear model includes a main effect for the RAS variant, a main effect for the gene variant, and an interaction term to quantify whether RAS and non-RAS variants were more likely to co-occur or appear mutually exclusive. This approach, while points to positive selection, does not imply an evolutionary order of mutations, rather, comutation indicates whether two variants occur in tumors more often or less often than expected under an independence model. We used a Bayesian hierarchical implementation to share information between cancer types and to estimate the overall association between two genes. As in the prevalence model, codependency estimates for rare cancer types were pulled toward the overall association while posterior means and medians of the regression coefficient for the interaction term for well-represented cancer types approximate empirical estimates. The full model for the frequencies of co-occurrences of RAS mutations with non-RAS variants using the total number of subjects of cancer type k, yk, as an offset is given by
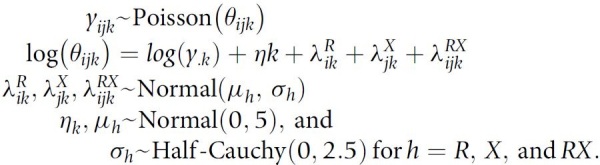 |
The coefficient
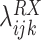 is the log OR for an association between RAS and gene X. Nonzero values of
is the log OR for an association between RAS and gene X. Nonzero values of
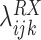 indicate a departure from independence, positive values indicate co-occurrence more often than expected from the marginal prevalence of these variants, and negative values suggest co-occurrence less often than expected with mutual exclusivity as an extreme. Upper-level parameters μRX and σRX describe the overall mean association and the heterogeneity of the association across the K strata. Co-occurrence models were implemented using Stan and rstan (31, 32). Hypothesizing that for some genes the co-occurrence with RAS hotspots variants may only be evident among inactivating mutations, copy-number alterations, or rearrangements, we additionally evaluated co-occurrence of RAS hotspot variants with these other mutation types. In total, we evaluated 48,564 inactivating mutations across 500 genes, 26,172 homozygous deletions, 66,920 high copy amplifications, and 6,514 gene–gene fusions. As structural changes in coding regions resulting from high copy amplifications, homozygous deletions, and gene–gene fusions are more likely to be somatic, all 66,372 patients were utilized for these analyses.
indicate a departure from independence, positive values indicate co-occurrence more often than expected from the marginal prevalence of these variants, and negative values suggest co-occurrence less often than expected with mutual exclusivity as an extreme. Upper-level parameters μRX and σRX describe the overall mean association and the heterogeneity of the association across the K strata. Co-occurrence models were implemented using Stan and rstan (31, 32). Hypothesizing that for some genes the co-occurrence with RAS hotspots variants may only be evident among inactivating mutations, copy-number alterations, or rearrangements, we additionally evaluated co-occurrence of RAS hotspot variants with these other mutation types. In total, we evaluated 48,564 inactivating mutations across 500 genes, 26,172 homozygous deletions, 66,920 high copy amplifications, and 6,514 gene–gene fusions. As structural changes in coding regions resulting from high copy amplifications, homozygous deletions, and gene–gene fusions are more likely to be somatic, all 66,372 patients were utilized for these analyses.
Statistical significance and multiple testing
While our analysis is Bayesian and we use posterior credible intervals from the hierarchical models to evaluate evidence of comutation dependencies, we also provide frequentist P values as one-number summaries that can be compared with conservative thresholds for statistical significance based on the number of non-RAS genes evaluated and whether the non-RAS gene is a known cancer driver. As the posterior distribution of the regression coefficients λ were approximately normal, two-sided P values were obtained by 2 × (1 − Φ(|
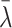 |/sλ) where Φ denotes the standard normal cumulative distribution function,
|/sλ) where Φ denotes the standard normal cumulative distribution function,
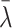 is the posterior mean, and sλ the posterior SD. We report P values on the negative log10 scale. To evaluate statistical significance of the nominal P values, we also computed the maximum P value that would be statistically significant following Bonferroni correction for multiple hypothesis testing. Among N0 non-RAS genes not known to be cancer drivers and N1 known cancer drivers, we computed cutoffs for statistical significance as
is the posterior mean, and sλ the posterior SD. We report P values on the negative log10 scale. To evaluate statistical significance of the nominal P values, we also computed the maximum P value that would be statistically significant following Bonferroni correction for multiple hypothesis testing. Among N0 non-RAS genes not known to be cancer drivers and N1 known cancer drivers, we computed cutoffs for statistical significance as
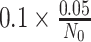 and
and
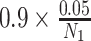 , respectively, thereby controlling the overall familywise error rate at 0.05 and allowing a 9-fold greater prior weight for variants in genes that are more likely to influence positive or negative selection at the cellular level. This approach is likely conservative as shrinkage from the hierarchical model would curtail statistical significance of any unusual patterns, particularly for comparisons involving cancers that were rare in the AACR Project GENIE Registry.
, respectively, thereby controlling the overall familywise error rate at 0.05 and allowing a 9-fold greater prior weight for variants in genes that are more likely to influence positive or negative selection at the cellular level. This approach is likely conservative as shrinkage from the hierarchical model would curtail statistical significance of any unusual patterns, particularly for comparisons involving cancers that were rare in the AACR Project GENIE Registry.
Simulation of co-occurrence frequencies
As mutual exclusivity of KRAS and EGFR mutations is well established, we used the empirical co-occurrence frequencies of these two genes in NSCLC, colorectal carcinoma, and melanoma as the basis of our simulations. For each of these cancers, we calculated the coefficients for a saturated log-linear model. For a new cancer (simulated) with n patients, we sampled coefficients from a normal distribution centered at the average coefficients from the observed cancers. For example, the interaction term was simulated as λRX∗ ∼ normal (λRX, σλ), such that deviations from the true population-level parameter λRX for a given simulation is controlled by σλ. To simulate the co-occurrence frequencies for this cancer, we specified the number of available patients (n = 50, 100, or 150), σλ (σλ = 0.1, 0.2, or 0.5), and sampled co-occurrence frequencies from a multinomial distribution with probabilities calculated from η∗, λR∗, λX∗, and λRX∗ (adding another level of sampling variability to the simulated co-occurrence frequencies). For each combination of σλ and n, we independently simulated 100 datasets, generating a total of 900 co-occurrence datasets. The Bayesian hierarchical model was fit independently to each of the 900 simulated datasets. For each evaluated model, we performed a burnin of 5,000 MCMC simulations and 10,000 simulations following burnin. A thinning parameter of 10 was used to reduce autocorrelation of the MCMC chains. A quantile-based 95% credible interval for λRX and the posterior median were obtained for each simulated dataset. For comparison, we computed ORs and 95% confidence intervals for Fisher exact and χ2 tests of co-occurrence in the simulated cancer. For each combination of σλ and n and each of the three methods evaluated (Bayesian hierarchical model, Fisher exact, and χ2), we calculated the proportion of 100 simulations where the credible interval (or confidence interval) for the interaction coefficient spanned the true value. A 95% confidence interval for the proportion of spanning intervals was obtained from a beta-binomial with a uniform beta prior. As the above simulations assume that the relationship between KRAS and EGFR in the simulated cancer is likely to be similar to the relationship in other cancers, we also simulated datasets where the log-linear model coefficients were completely independent from the three GENIE cancers listed above. In particular, the interaction coefficient was simulated from a normal distribution centered at zero with SD 0.5. Given the true parameter values that were independent of GENIE, we then simulated λ*coefficients and co-occurrence frequencies from a multinomial distribution as previously described. The Bayesian hierarchical model estimates co-occurrence for the simulated cancer using data from all four cancers, while χ2 and Fisher exact tests were computed for the simulated cancer alone. For each of the three models, we calculated the proportion of 100 simulated datasets that the 95% credible interval (Bayesian model) or confidence interval (χ2 and Fisher exact) spans the true value of the interaction coefficient λRX.
Comparison of Bayesian hierarchical model to existing approaches for mutation co-occurrence detection
We compared our approach for identifying co-occurrence and mutual exclusivity with SELECT v1.6 (33) and DISCOVER (34). We tested 3,513 NSCLC samples with matched tumor-normal NGS data from the GENIE cohort for co-occurrence and mutual exclusivity, limiting our analyses to comutations of KRAS codons 12, 13, and 61 with other non-RAS genes that were mutated in 50 or more samples. DISCOVER was run by setting alternative parameter to less for mutual exclusivity and greater for co-occurrence. These analyses are summarized in Supplementary Table S4.
Stratification of co-occurrence models by potential confounders
TMB, mutation signatures, age, race, and sex were considered as potential confounders of the co-occurrence estimates. To assess whether co-occurrence was associated with any of these characteristics, we evaluated a similar model as in a co-occurence model but with k denoting strata of the confounding variable of interest. For the assessment of TMB as a potential confounder, we stratified patients into 5 quintiles of TMB with k indexing the quintile. We excluded patients in the first TMB quintile (fewer than < 1 mutation/Mb) from these analyses. The interaction coefficient in this model, λ(RX)ijk, quantifies the co-occurrence of RAS and gene X among patients with similar TMB. The parameter μRX describes the overall association between RAS and gene X and the heterogeneity of this association between TMB strata. Similarly, we implemented the hierarchical model for stratification of co-occurrence by race, sex, age, and tumor type. To highlight interactions with potential confounding, we selected comutations for which one or more of the strata-specific 95% credible intervals did not overlap the 95% credible interval in the multicancer model. In addition, we limited the stratified analyses to comutations and cancer types having at least 100 RAS and non-RAS mutations.
RNA-sequencing analyses
After identifying candidate mutations in tumors from TCGA set, the following procedure was applied iteratively across all cancer types. First, the TCGAbiolinks R package was used to retrieve harmonized raw RNA-sequencing counts data from the NCI Genomic Data Commons within the target cancer type (35). Both the mutations and counts data were then filtered to remove tumors without both data modalities. A total of 9,258 tumor samples with both whole-exome mutation calls and RNA sequence data were subsequently analyzed (COAD, n = 404; LUAD, n = 511; PAAD, n = 170; READ, n = 146; SKCM, n = 466; UCEC, n = 526; ACC, n = 79; BLCA, n = 406; BRCA, n = 1014; CESC, n = 288; CHOL, n = 36; DLBC, n = 37; ESCA, n = 161; GBM, n = 159; HNSCC, n = 495; KICH, n = 65; KIRC, n = 365; KIRP, n = 279; LAML, n = 67; LGG, n = 521; LIHC, n = 360; LUSC, n = 482; MESO, n = 81; OV, n = 255; PCPG, n = 183; PRAD, n = 495; READ, n = 146; SARC, n = 237; SKCM, n = 466; STAD, n = 373; TGCT, n = 151; THCA, n = 495; THYM, n = 118; UCS, n = 56; and UVM, n = 80). For each pair or triplet of RAS with non-RAS mutations, tumors were categorized as double/triple mutants (all RAS and non-RAS mutations present), RAS mutants (only RAS mutations present), target mutants (only non-KRAS mutations present), or wild type (no mutations in RAS or non-RAS genes considered present). Cases with less than 10 mutations per comparison group were excluded, this filter restricted the downstream analyses to 1,308 COAD, LAD, PAAD, READ, SKCM, and UCEC tumors. DESeq2 was used to test for gene expression differences between double/triple mutants versus RAS-mutant tumors are well as non–RAS-mutant tumors versus wild-type tumors within single cancer types (36). The resulting differential expression was then analyzed by gene set enrichment analysis (GSEA) using fgsea with a curated list of genes from the Molecular Signatures Database (37) using −log(FDR) * sign(FC) as the ranking metric where FDR is the FDR and FC is the fold change (38). To visualize results of GSEA across multiple comparisons for LUAD, uniform manifold approximation and projection (UMAP) dimensionality reduction and k means clustering were performed. Normalized enrichment scores were drawn from all TCGA-LUAD cohort comparisons involving a KRAS G12C mutation for input into UMAP using the uwot R package with default settings. Clustering was performed with the k means R function with 10 centers and 50 random starts.
Survival analyses
The Kaplan–Meier method was used to estimate the survival function and the survival curves were compared using the nonparametric log-rank test. All P values were based on two-sided testing and differences were considered significant at P < 0.05. Statistical analyses were done using R version 3.6 and higher (http://www.R-project.org/).
Availability of code
We used the R package workflow to facilitate reproducibility of tables and figures (39) and Github to host the open source code (https://github.com/cancer-genomics/genie_ras_hallmarks). All results and visualizations can be accessed at http://www.ras-hallmarks.jhmi.edu/.
Results
Approach
To better understand the prevalence of somatic RAS mutations in an unbiased pan-cancer manner, we utilized targeted NGS data from the AACR Project GENIE Registry version 6.1, encompassing 607,863 mutations from 66,372 individuals across 51 cancer types and 19 study centers (Supplementary Table S1; Supplementary Fig. S1; ref. 22). We estimated the cancer type–specific prevalence of KRAS-, NRAS-, and HRAS-mutant alleles at codons 12, 13, and 61 using a Bayesian hierarchical model in the overall population and stratified by age, race, and sex (Materials and Methods). These analyses enable posterior distributions for the prevalence of RAS mutations and allow information to be shared across cancer types and across age, sex, and ethnic groups through the hierarchical structure. We then investigated the compendium of genes with RAS-dependent comutations and performed a comprehensive pan-cancer analysis of RAS comutation patterns with mutations, copy-number alterations, and rearrangements in non-RAS genes in a context-dependent manner (Materials and Methods). We restricted comutation analyses with sequence variants to 27,257 individuals for whom matched tumor-normal DNA-targeted NGS data were available to avoid bias related to analysis of germline SNPs. These analyses encompassed 190,490 mutations across 49 cancer types and three study centers (Supplementary Table S2; Supplementary Fig. S1) and revealed unique patterns of RAS co-occurrence and mutual exclusivity with non-RAS somatic mutations. We modeled RAS/non-RAS co-occurrence using a Bayesian hierarchical model and compared its performance by simulation of co-occurrence frequencies as well as by comparison with existing methods (Materials and Methods; refs. 33, 34), showing that the Bayesian hierarchical model was superior to other approaches (Supplementary Figs. S2 and S3; Supplementary Table S4). As tumor genomic features, including TMB, and mutational signatures could confound the comutation analyses, we evaluated comutation patterns stratified by these genomic characteristics (Materials and Methods). To investigate differences in cancer hallmark activation in tumors with RAS comutations, we integrated genomic and transcriptomic sequence data from 10,217 tumors from TCGA. The association of RAS allele comutation profiles with clinical outcomes was interrogated in the pan-cancer cohort from TCGA as well as in a cohort of patients with NSCLC from the AACR project GENIE registry who received immunotherapy-containing regimens.
Pan-cancer prevalence of mutant RAS alleles
Consistent with previous studies (1), the posterior median prevalence and 95% credible interval (2.550.097.5 percentiles) for RAS codon 12, 13, and 61 mutations varied among cancer types, ranging from 72.374.075.7% in PAC, 42.343.544.7% in colorectal carcinoma, 28.829.730.6% in NSCLC, 23.725.326.9% in melanoma, 19.120.922.7% in cancer of unknown primary (CUP), 4.65.97.3% in myelodysplastic/myeloproliferative syndrome, and 1.21.51.8% in central nervous system tumors (Supplementary Fig. S4; Supplementary Table S5). RAS mutations were less common in prostate, breast, kidney cancer, and mesothelioma with mutation rates affecting ≈1% of individuals. KRAS mutations occurred at higher frequency in a wide range of cancers, including gastrointestinal tumors, lung cancer, and gynecologic malignancies (Fig. 1), while NRAS was more frequently mutated in melanoma, thyroid cancer, and hematologic malignancies and HRAS was overall less frequently mutated (Supplementary Fig. S5). A prevalence analysis at the mutant allele level confirmed that the type of amino acid substitution at the codon level showed tissue specificity, such that NSCLC predominantly harbored KRAS G12C in contrast to colorectal carcinoma and PAC where G12C represented 10% and 1% of codon 12 mutations, respectively. In PAC, hepatobiliary, ampullary, colorectal carcinoma, small bowel, and appendiceal cancer, KRAS G12D was the predominant KRAS mutation; however, G12D comprised only 17% of codon 12 mutations in NSCLC. KRAS G12R mutations were rare in colorectal carcinoma and NSCLC (1%–2%), but were the third most frequent alteration in PAC (Fig. 1; Supplementary Table S6). KRAS G13D was the third most frequent RAS alteration in colorectal carcinoma, small bowel, ampullary, and appendiceal cancer as well as in leukemia, whereas KRAS Q61H followed codon 12 mutations in PAC and hepatobiliary cancer. In gynecologic malignancies, KRAS G12V and G12D were the most prevalent alterations, followed by NRAS Q61R and KRAS G12A in ovarian cancer and KRAS G13D and G12A in uterine cancer. Interestingly, the RAS genetic heterogeneity of CUP resembled both colorectal carcinoma/APC and NSCLC tumors, with recurring mutations in KRAS G12D, G12V, G12C, and G13D. A similar genomic heterogeneity at the codon level was noted for NRAS-driven tumors, such as melanoma, thyroid cancer, and leukemia (Supplementary Fig. S6). NRAS Q61R was the dominant alteration for melanoma and thyroid cancer and the leading NRAS alteration in ovarian cancer, while NRAS G12D was more frequent in leukemia. NRAS Q61K was the second most frequent NRAS alteration in melanoma and thyroid cancer, while NRAS G13D and Q61R were more frequent in leukemia (Supplementary Fig. S6). The most prevalent HRAS mutations were Q61R in thyroid cancer, head and neck cancer, and bladder cancer, with a lower prevalence of HRAS Q61K mutations detected in thyroid cancer (Supplementary Fig. S7).
Figure 1.

Prevalence of KRAS codon 12, 13, and 61 and G12C mutations is tumor type and context dependent. A, Prevalence of KRAS mutations at codons 12, 13, and 61; the empirical estimate of the KRAS prevalence is shown as solid gray points, while the posterior median prevalence is shown as clear blue circles. KRAS codon 12 sequence alterations were dominant in PAC (65.367.269.1%), ampullary cancer (37.046.355.9%), appendiceal cancer (37.642.948.1%), small bowel cancer (25.431.437.8%), colorectal carcinoma (28.930.031.1%), NSCLC (24.225.025.9%), CUP (14.115.717.4%), uterine cancer (11.012.313.8%), ovarian cancer (7.58.69.8%), hepatobiliary cancer (6.98.29.8%), cervical cancer (4.97.210.1%), neuroendocrine tumors (4.36.59.3%), germ cell tumors (4.35.77.4%), and bladder cancer (3.34.25.2%), while KRAS codon 13 and 61 mutations were found at lower frequencies in these cancers. KRAS codon 13 sequence alterations were detected in 7.27.98.5% of colorectal carcinoma, 3.15.69.2% of small bowel cancer, 1.33.88.4% of ampullary cancer, 2.13.86.1% of appendiceal cancer, 1.32.85.3% of B-lymphoblastic leukemia/lymphoma, 1.72.32.9% of uterine cancer and 1.82.12.3% of NSCLC, whereas KRAS codon 61 mutations were detected in 4.75.56.4% of PAC, 1.13.40.8% of ampullary cancer, 0.92.34.8% of small bowel cancer, 1.51.82.2% of colorectal carcinoma, 1.51.82.0% of NSCLC, and 0.91.52.3% of thyroid cancer. KRAS G12C mutations were detected in 111213% of NSCLC, 1.53.56.3% of small bowel cancer, 0.83.27.1% of ampullary cancer, 2.73.13.6% of colorectal carcinoma, 2.33.03.8% of CUP, 1.12.54.3% of appendiceal cancer, 0.71.11.6% of uterine cancer, and less than 1% of PAC, hepatobiliary cancer, small cell lung cancer, neuroendocrine tumors, and bladder cancer. B, Posterior median prevalence estimates for mutant KRAS alleles at codons 12, 13, and 61 varied based on the tumor type interrogated. As a representative example, NSCLC predominantly harbored KRAS G12C mutations in contrast to gastrointestinal tumors and pancreatic cancer that predominantly harbored KRAS G12D and G12V mutations. Similarly, posterior median prevalence estimates for mutant NRAS alleles at codons 12, 13, and 61 showed an enrichment in NRAS Q61R and Q61K mutations in melanoma and thyroid cancer. Error bars, 95% credible intervals. CRC, colorectal cancer; BLL, B-lymphoblastic leukemia; MDS/MPN, myelodysplastic syndrome/myeloproliferative neoplasm; SCLC, small cell lung cancer; CNS, central nervous system tumor.
Prevalence of mutant RAS alleles is context dependent
We subsequently tailored the prevalence analyses to investigate differences in the distribution of mutant RAS alleles by age, sex, and race (Fig. 2; Supplementary Table S7). Notably, when compared to the overall population prevalence estimates, RAS mutations were less prevalent in younger patients with melanoma (7.311.815.7% lower), CUP (7.312.516.5% lower), NSCLC (7.611.014.2% lower), and PAC (7.714.621.7% lower). Conversely, RAS mutations were more prevalent in younger patients with ovarian cancer (8.915.823.5% higher) and B-lymphoblastic leukemia/lymphoma (0.28.216.7% higher) compared with the overall population (Fig. 2). The prevalence of KRAS codon 12 mutations was lower in younger patients with PAC (7.714.822.0% lower), CUP (9.012.615.2% lower), and NSCLC (7.010.113.0% lower). At the mutant allele level, these differences were driven by the lower prevalence in younger patients of NRAS Q61 L in melanoma, KRAS G12C and G12V in CUP, KRAS G12C and G12V in NSCLC, and KRAS G12D, G12R, and G12V in PAC (Fig. 2; Supplementary Fig. S7). In contrast, the prevalence of NRAS G12D was higher in younger patients with B-lymphoblastic leukemia/lymphoma, whereas the KRAS G12C-, G12D-mutant alleles were more prevalent in younger patients with ovarian cancer. KRAS G12V mutations were less frequent in younger patients with colorectal carcinoma, whereas KRAS G13D mutations were more frequent in younger patients with leukemia (Supplementary Fig. S7).
Figure 2.

Prevalence of mutant RAS alleles at codons 12, 13, or 61 stratified by age, race, and sex reveals differential host-dependent frequencies. A–F, Prevalence of KRAS-mutant alleles varies by age group, race, and sex for patients with cancer of unknown primary (A), NSCLC (B), pancreatic adenocarcinoma (C), colorectal cancer (D), melanoma (E), and ovarian cancer (F). G–I, Cancer-specific estimates of RAS prevalence obtained from a single hierarchical model (y-axis) were compared with the overall prevalence from 51 separate cancer-specific models, where the effects of age (G), race (H), and sex (I) were modeled hierarchically (x-axis). For several cancers including NSCLC, the prevalence of RAS mutations in demographic subgroups differed significantly from the overall prevalence estimates. Specifically, when compared with the overall population prevalence estimates, RAS mutations were less prevalent in patients younger than 40 years with melanoma (7.311.815.7% lower) and CUP (7.312.516.5% lower), and patients younger than 50 years with NSCLC (7.611.014.2% lower) and PAC (7.714.621.7% lower). RAS mutations were more prevalent in patients younger than 40 years with ovarian cancer (8.915.823.5% higher) and B-lymphoblastic leukemia/lymphoma (0.28.216.7% higher) compared with the overall population. A negative difference in prevalence indicates higher prevalence in the subset compared with the overall population prevalence. BLL, B-lymphoblastic leukemia; CRC, colorectal cancer.
Similarly, we observed a sex-based cancer type–dependent dimorphism of mutant RAS prevalence: colorectal carcinoma and NSCLC tumors in women more frequently harbored RAS mutations compared with men (1.43.65.7% and 1.12.64.1% higher, respectively), whereas melanoma tumors in women had fewer RAS mutations (0.43.36.2% lower). In colorectal carcinoma, KRAS codon 12 mutations were more frequent in women (0.42.44.4% higher) and similarly KRAS codon 12 and in particular KRAS G12C mutations were more frequent in women with NSCLC (0.92.33.7% and 0.21.22.3% higher, respectively; Supplementary Table S7; Fig. 2; Supplementary Fig. S9). We then evaluated RAS-mutant allele selection in different racial backgrounds, defined by self-reported race (Supplementary Table S8; Supplementary Fig. S10). Among patients with colorectal carcinoma, an enrichment for RAS mutations was noted in colorectal carcinoma tumors of Black individuals relative to other racial groups (6.611.115.5% higher). Black patients with colorectal carcinoma harbored a higher number of KRAS codon 12 mutations (2.26.510.8% higher), specifically G12V, and KRAS codon 13 mutations, specifically G13D (1.74.47.5% higher). In NSCLC, RAS mutations occurred less frequently in Asian patients (15.918.621.1% lower), who harbored a lower number of KRAS codon 12 mutations (13.816.318.6% lower), in particular KRAS G12C mutations (6.98.610.0% lower). Smaller differences from the overall prevalence were noted for KRAS G12A (1.21.92.3% lower), G12D (0.41.82.9% lower), G12V (2.53.84.7% lower), and G13D (6.98.610.0% lower) in NSCLC of Asian individuals. RAS mutations, specifically KRAS G12A, were less common among Black individuals with uterine cancer (4.79.613.4% lower). Black individuals with HNSCC showed an enrichment for HRAS G13R mutations, whereas NRAS Q61K and Q61R were more frequent in thyroid cancer in Black patients. In the melanoma group, a lower prevalence for NRAS Q61 L mutations was noted for Black and Asian individuals (Supplementary Fig. S10). Taken together, these novel findings support the context-dependent distribution of mutant RAS alleles that extends past tissue of origin and should be considered in target population selection and the rational design of clinical trials.
RAS sequence and structural comutations depend on cancer lineage
To assess the compendium of RAS comutations, we modeled dependencies between RAS and non-RAS genes hierarchically across cancer types with a Bayesian model (Materials and Methods). The hierarchical model allowed for information on co-occurrence to be shared across cancer types, permitted a formal assessment of the heterogeneity of co-occurrence between different cancers, and shrunk estimates for rarer cancer types toward the overall mean. These analyses revealed co-occurrence and mutual exclusivity of RAS and non-RAS mutations in a cancer type–dependent manner. RAS hotspot mutations co-occurred with mutations in ATM, KEAP1, MAX, NKX2-1, RBM10, STK11, and U2AF1 in NSCLC (−log10P > 12), CDKN2A in PAC (−log10P > 9), PIK3CA in colorectal carcinoma (−log10P > 6), ARID1A and PTEN in uterine cancer (−log10P > 8), and TERT in melanoma (−log10P > 12, Fig. 3A; Supplementary Table S9). RAS mutations were mutually exclusive with BRAF and RNF43 in colorectal carcinoma (−log10P > 8), BRAF in melanoma (−log10P > 13) and thyroid cancer (−log10P > 9.9), EGFR in NSCLC (−log10P > 51) and DAXX and MEN1 in PAC (−log10P > 13). A cancer type–specific co-occurrence pattern was noted for RAS and TP53 genomic alterations, with a statistically significant enrichment in TP53 mutations in RAS-mutant PAC (−log10P = 51.6) but a significant underrepresentation of TP53 mutations in RAS-mutant ovarian cancer (−log10P = 12.3), NSCLC (−log10P = 28.3), uterine cancer (−log10P = 9.3), and colorectal carcinoma (−log10P = 13.5, Fig. 3B; Supplementary Table S9). RAS/RAS comutations were infrequent (less than 3%) in pancreatic cancer, thyroid cancer, hepatobiliary cancer, NSCLC, colorectal carcinoma, melanoma, and CUP. Notably, we found co-occurring RAS-RAS mutations in 4.4% of ovarian cancers, 4.9% of uterine cancers, and 11.7% of bladder cancers. While infrequent in most cancers, HRAS/NRAS comutations were found in bladder cancer (5.9%), while KRAS/NRAS and KRAS/KRAS comutations were found in low frequencies in ovarian cancer (1.8%). Similarly, KRAS/KRAS and NRAS/NRAS comutations were found in 1.5% of uterine cancers (Supplementary Table S10).
Figure 3.

Co-occurrence of RAS hotspot mutations with sequence and structural non-RAS mutations is cancer lineage specific. A, Volcano plots of the posterior median log OR (x-axis) versus negative log10P value (y-axis) for the association between RAS mutations and non-RAS variants. Mutations in non-RAS genes with log OR greater than 0 co-occur with RAS mutations as opposed to non-RAS mutations with an interaction coefficient less than 0, which are comutated with RAS at a rate lower than expected or are mutually exclusive. The further to the right on the x axis the closest to true co-occurrence and the further to the left of the axis the closest to true mutual exclusivity, with the statistical significance of the difference from 0 (which indicates independence) plotted on the y-axis. B, For statistically significant associations, 95% posterior credible intervals of the log OR are indicated by error bars. For genes associated with RAS mutations in multiple cancers, multiple vertically offset error bars are displayed. C and D, Volcano plots and posterior credible intervals for the association between RAS mutations and inactivating non-RAS mutations. E, Volcano plots of the posterior median log OR (x-axis) versus negative log10P (y-axis) for the association between RAS mutations with deep deletions, high copy amplifications, and gene fusions. F, 95% posterior credible intervals of the log OR for RAS/non-RAS fusions. Despite the small number of fusions included in the analyses that precluded firm statistical conclusions, fusions involving ALK (EML4-ALK) and RET (RET-KIF5B) genes showed a pattern of mutual exclusivity with RAS mutations. CRC, colorectal cancer.
We subsequently evaluated co-occurrence of RAS codon 12, 13, and 61 mutations with inactivating mutations in non-RAS genes (Materials and Methods). Inactivating mutations in STK11, ATM, RBM10, and KEAP1 co-occurred with RAS mutations in NSCLC (P < 0.0001), whereas sequence alterations in RB1 and KMT2D were mutually exclusive (−log10P > 4.9; Fig. 3C and D; Supplementary Table S11). In PAC, RAS mutations co-occurred with inactivating mutations in CDKN2A and SMAD4 (P < 0.002), but were mutually exclusive with inactivating alterations in MEN1 (P < 0.0001; Fig. 3C; Supplementary Table S11). RAS comutation patterns were driven by inactivating mutations in APC and AMER1 in colorectal carcinoma (P < 0.008), and ARID1A in uterine cancer (P = 0.0003), while mutual exclusivity was noted for inactivating mutations in RNF43 in colorectal carcinoma (P = 0.0003; Fig. 3C and D; Supplementary Table S11). While the majority of comutation patterns were driven by KRAS, NRAS codon 61 mutations co-occurred with TERT sequence alterations (−log10P = 12.3) and were mutually exclusive with BRAF (−log10P = 10.5) and NF1 (−log10P = 5.8) sequence alterations in melanoma. NRAS codon 61 mutations, specifically Q61R, were mutually exclusive with BRAF—predominantly class I—sequence alterations in thyroid cancer (−log10P = 5.73).
We expanded the comutation analysis framework to include copy-number aberrations and similar to the sequence comutation analyses, we identified cancer lineage–specific RAS dependencies (Materials and Methods; Fig. 3E and F). RAS mutations, predominantly codon 12 and G12C, were mutually exclusive with CDKN2A and CDKN2B homozygous deletions in NSCLC (−log10P = 6.3) but NRAS codon 61 and CDKN2A homozygous deletions co-occurred in melanoma (−log10P = 2.9; Supplementary Table S12). KRAS hotspot mutations co-occurred with SMAD4 homozygous deletions in PAC (−log10P = 2.4), but were found to be mutually exclusive of RB1 homozygous deletions in uterine cancer (P = 0.01). In NSCLC, KRAS hotspot mutations co-occurred less frequently with EGFR, ERBB2, FGFR1, and MDM2 high copy-number amplifications (−log10P > 4.4). In colorectal carcinoma, KRAS codon 12 mutations were mutually exclusive with ASXL1 (−log10P = 7), BCL2L1 (−log10P = 7.7), DNMT3B (−log10P = 4.7), EGFR (−log10P = 4.1), FGFR1 (−log10P = 4.8), and PTPRT (−log10P = 4.6) high copy amplifications (Supplementary Table S12). Interestingly, when all amplifications were considered, co-occurring KRAS codon 12, particularly G12C, mutations and KRAS gene amplifications were noted in NSCLC (−log10P = 8.4) and CUP (−log10P = 7.3) and similarly, co-occurring NRAS codon 61 mutations and NRAS gene amplifications were detected in melanoma (−log10P = 5.3). Comutation analyses of RAS genes with fusions in non-RAS genes were limited by the relatively small number of tumors harboring rearrangements.
Most RAS comutation patterns were driven by KRAS (Supplementary Figs. S11 and S12; Supplementary Tables S9–S11). Interestingly, NF1 mutations were less frequent in KRAS codon 12–mutant NSCLC compared with KRAS codon 13–mutant NSCLC, suggesting codon-specific RAS oncogenicity (40), which may result in positive selection of NF1 mutations in non-codon 12 KRAS-mutant NSCLC (9, 41). We then sought to determine whether KRAS comutations were present on the same cancer clone and utilized mutant allele fractions to infer clonality estimates for KRAS and non-KRAS variants, assuming similar ploidy at these loci and in the context of a given tumor purity (Materials and Methods). We interrogated KRAS-STK11 as a representative clonal pair and KRAS-PIK3CA as a putative clonal-subclonal pair; mutant allele frequencies of KRAS and STK11 variants were highly correlated (Pearson R = 0.73), suggesting that these variants may indeed be contained in the same cancer cell clonal populations and supporting the notion that these mutations are acquired early during carcinogenesis. In contrast, KRAS/PIK3CA alterations distributed a looser correlation (Pearson R = 0.5) that could be explained by branching PIK3CA mutations at later stages of tumor evolution of KRAS-mutant cancers (Supplementary Fig. S13). Nevertheless, these differences may be explained by non-euploid allelic copy number of the loci considered and clonality inferences can be inaccurate in the absence of allele-specific copy numbers.
Pan-cancer comutation analyses at the mutant RAS allele level reveal distinct genomic landscapes
To capture the genomic landscapes of RAS-mutant tumors, we evaluated all KRAS-, NRAS-, and HRAS-mutant alleles at codons 12, 13, and 61 and considered co-occurrence with non-RAS mutations (any mutation, hotspot mutation, or inactivating mutation) and copy number (deep deletion or high-level amplification) grouped by gene, gene family, or pathway. In NSCLC, KRAS G12C mutations were found to co-occur with ATM, RBM10, KEAP1, NTRK3, EPHA5, AMER1, and STK11 sequence alterations (−log10P > 4.3; Fig. 4) and inactivating mutations in MGA (−log10P = 3.51), while KRAS G12C mutations were mutually exclusive with EGFR mutations (−log10P = 19.4; Supplementary Tables S13 and S14). RBM10-inactivating mutations were also comutated with KRAS G12A and G12D (−log10P > 3.03). KRAS G12D was more frequently comutated with NKX2-1 and MAX (−log10P > 3.36) and KRAS G12V was more frequently co-mutated with NKX2–1 (−log10P > 3.99). Interestingly, a weaker comutation pattern emerged for KRAS G12V and STK11 (−log10P = 2.89), while KRAS G12D was not significantly comutated with STK11 in NSCLC. All mutant KRAS alleles were less frequently detected in TP53 and EGFR-mutant NSCLC tumors (−log10P > 3.61; Supplementary Table S13). Histology driven comutation analyses showed that these findings were driven by nonsquamous histology, with the exception of KRAS/TP53 comutations; KRAS G12D mutations were less frequently comutated with TP53 in non-squamous NSCLC (−log10P = 2.25; Supplementary Table S15).
Figure 4.

Differential comutation patterns in KRAS- and NRAS-mutant tumors point to RAS/non-RAS gene dependencies based on host features and cancer lineage. Differential RAS-mutant allele comutation patterns per tumor type and by age, race, and sex for KRAS/ATM in NSCLC (A), KRAS/RBM10 in NSCLC (B), KRAS/NTRK3 in NSCLC (C), KRAS/STK11 in NSCLC (D), KRAS/KEAP1 in NSCLC (E), KRAS/EGFR in NSCLC (F), KRAS/PIK3CA in colorectal carcinoma (G), KRAS/FBXW7 in colorectal carcinoma (H), KRAS/RNF43 in colorectal carcinoma (I), KRAS/SF3B1 in colorectal carcinoma (J), KRAS/BRAF in colorectal carcinoma (K), KRAS/TP53 in colorectal carcinoma (L), KRAS/TP53 in PAC (M), KRAS/SMAD4 in PAC (N), KRAS/CDKN2A in PAC (O), KRAS/DAX in PAC (P), KRAS/MEN1 in PAC (Q), KRAS/CASP8 in colorectal carcinoma (R), NRAS/BRAF in melanoma (S), NRAS/TERT in melanoma (T), KRAS/NF1 in melanoma (U), KRAS/PTEN in uterine cancer (V), KRAS/TP53 in uterine cancer (W), and NRAS/BRAF in thyroid cancer (X). Log OR is plotted for each stratified RAS/non-RAS comutation. Red, co-occurrence; blue, mutual exclusivity or occurrence less frequently than expected under independence. CRC, colorectal cancer.
Mutant allele-specific comutations emerged in colorectal carcinoma, where KRAS G13D was preferentially comutated with PIK3CA, FBXW7, CASP8, SF3B1, and BRCA1 (−log10P > 3.93) and co-occurred with inactivating APC mutations (−log10P = 2.5; Fig. 4). KRAS G12D and G12V were mutually exclusive with BRAF—predominantly class I—mutations (−log10P > 3.67) and were less frequent in TP53-mutant tumors (−log10P > 4.53), while KRAS G12V was also less frequently mutated in RNF43 and KMT2D-mutant colorectal carcinoma (−log10P > 3.74). Interestingly, when the primary versus metastatic origin of the tumor sample was considered, PTEN and SF3B1 mutations more frequently co-occurred in metastatic colorectal carcinoma lesions (−log10P > 2.18, Fig. 4; Supplementary Table S16), potentially pointing towards the subclonal origin of these alterations, that may be unique to the metastatic clone.
KRAS G12D was strongly comutated with TP53 in PAC (−log10P = 15.18) predominantly in metastatic tumors, while KRAS G12V and G12R also co-occurred with TP53 mutations though to a lesser degree (−log10P = 8.43 and P = 3.18 respectively; Fig. 4). CDKN2A mutations were predominantly found in KRAS G12D-mutant tumors (−log10P = 3.97), whereas SMAD4 was comutated with KRAS G12R in PAC (−log10P = 4.32). DAXX and MEN1 were mutually exclusive with KRAS G12D and G12V (−log10P > 2.62). PTEN mutations co-occurred with KRAS G12D in uterine cancer (−log10P = 3.33), while TP53 mutations less frequently co-occurred with KRAS G12D and G12V mutations (−log10P > 3.01). In contrast to the genetic diversity of KRAS-mutant NSCLC, colorectal carcinoma, and PAC, in melanoma NRAS Q61K and Q61R were uniformly comutated with TERT and mutually exclusive with NF1 and BRAF—predominantly class I—mutations (−log10P > 2.71; Fig. 4). Comutation analyses between RAS and hotspot mutations in non-RAS genes were consistent with the comutations described above (Supplementary Table S17).
Given the differences in prevalence of RAS-mutant alleles based on age, sex, and race, we postulated that comutation patterns may be differentially distributed in patient subpopulations and performed stratified analyses to further explore this hypothesis (Fig. 4; Supplementary Fig. S14). Women with KRAS G12D colorectal carcinoma tumors were less likely to harbor RNF43 mutations than men, while men with KRAS G13D-mutant colorectal carcinoma tumors were more likely to harbor CASP8 mutations than women. KRAS G12V-mutant colorectal carcinoma in Asian individuals less frequently co-occurred with TP53 mutations. In melanoma, NRAS Q61K mutations more frequently co-occurred with TERT mutations and NRAS Q61R were more frequently comutated with DICER1 in women. In NSCLC, KRAS G12C was more frequently comutated with FAT1 and PDGFRA in women, while KRAS G12C-mutant NSCLC infrequently carried TP53 mutations in men (Supplementary Table S16). In uterine cancer, KRAS G12D more frequently co-occurred with PTEN alterations in Black individuals (Fig. 4). Taken together, these findings support a lineage- and context-specific distribution of RAS comutations in human cancers.
We next considered convergence of comutations in cancer hallmarks and signaling pathways, including RAS/Raf/MAPK, PI3K/AKT, chromatin regulation, cell-cycle progression, NRF2 pathway/oxidative stress response, Wnt and DNA damage repair gene sets (Supplementary Tables S18 and S19; Fig. 5A). These analyses revealed mutual exclusivity of non-RAS mutations in the RAS/Raf/MAPK pathway with KRAS G12A (−log10P = 5.59), G12C (−log10P = 4.5), G12D (−log10P = 8.52) and less with G12V (−log10P = 2.83) in NSCLC, with NRAS Q61K (−log10P = 3.59) and Q61R (−log10P = 6.6) in melanoma and NRAS Q61R in thyroid cancer (−log10P = 5.57); however, these patterns were not found in colorectal carcinoma or PAC. Interestingly, mutations in DNA damage repair genes (−log10P = 8.21) and genes involved in chromatin regulation (−log10P = 4.12) were enriched in KRAS G12C-mutant NSCLC, alongside the NRF2/oxidative stress response pathway (−log10P = 3.25). A different comutation pattern was noted for colorectal carcinoma, where KRAS G12D and G13D we found to be comutated with genes in the PI3K/AKT pathway (−log10P > 2.43). An enrichment in mutations in the NRF2/oxidative stress response pathway was also identified for KRAS G13D-mutant colorectal carcinoma (−log10P = 2.33). In contrast, mutations in the PI3K/AKT pathway were less frequent in KRAS G12D and G12V PAC (−log10P > 2.97). Unique to PAC, KRAS G12D mutations co-occurred with mutations in genes affecting cell-cycle progression (−log10P = 3.17). These findings were confirmed when only hotspot alterations in non-RAS genes were considered (Supplementary Table S20).
Figure 5.

Convergence of RAS/non-RAS comutations in cancer hallmarks and signaling pathways, accounting for TMB and mutational spectra. A, KRAS- and NRAS-mutant alleles were mutually exclusive with mutations in the RAS/RAF/MAPK pathway, while they co-occurred with mutations in the NRF2 pathway in NSCLC and colorectal carcinoma, PI3K/AKT pathway in colorectal carcinoma, cell-cycle progression in PAC, and chromatin regulation and DNA damage response in NSCLC. B, Triple mutations in KRAS G12C-mutant NSCLC, uterine, PAC, colorectal carcinoma, and CUP tumors. C, Concordance between RAS/non-RAS gene associations between the multicancer model and a TMB-stratified model. Vertical error bars indicate the IQR of the overall association across TMB strata. Highlighted are RAS/non-RAS associations that were statistically significant in the multicancer model but have an overall association in the TMB-stratified model near 0, suggesting that the associations identified in the multicancer model were confounded by TMB. D, Examples of comutations confounded by TMB include RAS codon 12, 13, and 61 mutations and APC mutations in colorectal carcinoma, KRAS codon 12, 13, and 61 mutations in colorectal carcinoma, KRAS codon 12, 13, and 61 mutations and KEAP1 and STK11 mutations in NSCLC. While KRAS/APC and TP53 co-occurrence was influenced by higher TMB in colorectal carcinoma, an inverse association was noted in NSCLC, where KRAS/STK11 and KEAP1 comutations were positively selected in TMB-low tumors. CRC, colorectal cancer.
We next investigated co-occurrence of triple mutations involving KRAS G12C and non-RAS genes in NSCLC, colorectal carcinoma, CUP, PAC, and uterine cancers. Triple comutations followed cancer cell lineage–specific distributions, such that KRAS/KEAP1/STK11 emerged in NSCLC, KRAS/CDKN2A/TP53, KRAS/SMAD4/TP53, KRAS/CDKN2A/SMAD4 in PAC and KRAS/ARID1A/PIK3CA and KRAS/PIK3CA/PTEN in uterine cancer (Fig. 5B). Notably, a number of triple mutations were detected in CUP, including KRAS/KEAP1/STK11, KRAS/KEAP1/SMARC4, KRAS/KEAP1/TP53, KRAS/STK11/SMARC4, KRAS/KEAP1/PTPRD, KRAS/PTPRD/TP53, and KRAS/STK11/TP53. The predominant triple mutation in colorectal carcinoma was KRAS/APC/TP53, followed by KRAS/APC/SMAD4, KRAS/APC/PIK3CA, KRAS/FBXW7/APC, KRAS/APC/SOX9, and KRAS/PIK3CA/TP53 (Fig. 5B). Given the differential impact of ATM inactivation on KRAS-dependent carcinogenesis in the context of TP53 proficiency (42), we also investigated whether ATM, KRAS (all codons), and TP53 mutations co-occurred. The proportion of triple mutant KRAS-positive tumors with inactivating mutations in ATM and TP53 ranged from 0.7310% in NSCLC to 71733% in colorectal carcinoma and 0.6726% in PAC, suggesting that ATM loss and TP53 deficiency in KRAS-driven tumors are positively selected in a cancer type dependent manner.
TMB and mutation spectra may confound RAS comutation patterns
Apparent comutation patterns can be confounded by passenger mutations that accumulate during tumor evolution without conferring a fitness advantage to the cancer cells, especially in the setting of tumors with high TMB. These passenger mutations may cluster in hotspot positions due to underlying mutational processes rather than indicating a positive selection evolutionary process (43). Furthermore, mutational signatures attributed to exposure to specific carcinogens or endogenous mutational processes can be differentially susceptible to mutation (17); co-occurrence of genomic alterations may thus reflect mutational spectra as opposed to positive selection. To derive a TMB-adjusted measure of the dependency between RAS and non-RAS mutations, we stratified samples by quintiles of the predicted genome-wide TMB (Materials and Methods; Supplementary Table S21). Modeling co-occurrence across TMB strata hierarchically, we derived posteriors for the co-occurrence within each TMB quintile as well as the marginal associations. Overall, the multicancer model and TMB-stratified model were highly concordant (Fig. 5C); however, the strength of specific RAS dependencies was attenuated in the context of different TMB backgrounds. The most susceptible co-occurrence patterns included KRAS mutations and STK11, KEAP1, or TP53 mutations in NSCLC and TP53 and inactivating mutations in APC in colorectal carcinoma (Fig. 5D). While higher TMB drove the stronger comutation associations noted between KRAS G12D and G12V and TP53 mutations and KRAS G13D and APC mutations in colorectal carcinoma, lower TMB levels were more strongly associated with KRAS G12C/STK11 and KRAS G12C/KEAP1 mutations, pointing toward a positive selection of these comutations in tumors with lower TMB (Supplementary Table S22). A similar positive selection pattern in TMB-low NSCLC was identified when KRAS G12C/STK11/KEAP1 mutations were considered in combination. Notably, KRAS G12C, G12D, and G12V TMB-high NSCLC tumors were less likely to also harbor TP53 alterations (Supplementary Table S22).
Similarly, to account for unique mutational processes that may introduce bias toward specific mutations (44), we extracted the mutational signature with the highest contribution per tumor, and modeled the co-occurrence across mutational signatures hierarchically for each cancer type (Materials and Methods). We detected associations between mutational processes related to APOBEC mutagenesis and smoking and specific RAS/non-RAS driver gene comutations. In particular, RAS codon 12, 13, and 61 mutations co-occurred less frequently with AMER1, KEAP1, STK11, and TP53 in NSCLC tumors harboring a C>A rich smoking related signature (Supplementary Fig. S15). Consistent with the TMB-stratified analyses in NSCLC, KRAS G12C, and KEAP1 or STK11 comutations were less frequent in tumors harboring a smoking-associated mutational signature. KRAS G12C and STK11 comutations were also less frequent in tumors harboring an APOBEC mutational signature, while KRAS G12C-mutant NSCLC with an APOBEC mutation signature more frequently harbored mutations in NTRK3. KRAS G12C and G12V and TP53 mutations co-occurred less frequently in NSCLC tumors harboring a mutational smoking signature (Supplementary Table S22). In colorectal carcinoma, KRAS codon 12, 13, and 61 and PIK3CA mutations co-occurred more frequently in tumors that harbored an APOBEC mutational signature (Supplementary Fig. S15). Mutual exclusivity between KRAS G12D and RNF43 was more pronounced in colorectal carcinoma tumors harboring a mismatch repair deficiency mutational signature (Supplementary Table S22).
RAS comutations are linked with distinct transcriptomic tracks
Next, we hypothesized that the genomic heterogeneity of RAS-mutant tumors at a mutant allele resolution would be reflected in differential cancer cell signaling and that positive selection of RAS comutations may be driven by immune surveillance states that can be captured in the tumor microenvironment (TME). To explore whether distinct comutation patterns are linked with differential cancer hallmarks, we leveraged transcriptomic sequence data from 9,258 tumors from TCGA and performed gene set enrichment analyses stratified by RAS comutations (Materials and Methods). These analyses revealed discrete transcriptional programs enriched in RAS-mutant tumors in a cancer lineage–dependent manner. In LUAD, co-occurrence of KRAS codon 12 and NTRK3 mutations was associated with upregulation of E2F-dependent proliferation pathways, DNA maintenance and repair and G2–M cell-cycle checkpoint gene sets (Supplementary Fig. S16; Supplementary Table S23). Transcriptomic tracks of KRAS G12C/KEAP1 and KRAS G12C/RBM10-mutant LUADs were enriched for pathways linked with oxidative phosphorylation, reactive oxygen species, and xenobiotic metabolism, with concurrent downregulation of inflammatory responses for KRAS G12C/KEAP1-mutant tumors (Fig. 6; Supplementary Fig. S16; Supplementary Table S23). Oxidative phosphorylation, Krebs cycle and active oxygen species gene sets were similarly upregulated in KRAS G12C/KEAP1/STK11 triple mutant LUAD, with a downregulation of conserved inflammatory responses, IFNγ and IFNα gene sets (Supplementary Table S23). In contrast, KRAS G12C/PTPRD-mutant LUADs were enriched for expression of epithelial-to-mesenchymal transition gene sets, while a more inflamed TME was denoted with an upregulation of IFNγ inflammatory response, TNFa signaling, MHC antigen presentation, and T-cell receptor signaling gene sets (Supplementary Fig. S16). Notably, in investigating KRAS/TP53 comutant LUAD, we identified a dominant upregulation of key immune pathways linked with proinflammatory responses for both KRAS G12C and G12V alongside cell-cycle progression gene sets (Fig. 6; Supplementary Fig. S16; Supplementary Table S23). While similar differential gene expression profiles were observed in NTRK3-only and KEAP1-only LUADs, the KRAS/RBM10, PTPRP, and TP53 comutant tumors had distinct expression patterns compared with non–RAS-mutant tumors, suggesting that in the latter the differentially regulated hallmarks were uniquely related to the comutation rather than the non-RAS mutation alone (Supplementary Table S24; Supplementary Fig. S17).
Figure 6.

Differential expression profiles driven by KRAS G12C comutation status in LUADs. Comutation-driven GSEA leveraging transcriptomic profiles from RNA sequencing revealed marked differences in gene expression programs depending on KRAS G12C comutations. A, Normalized enrichment scores from GSEA in KRAS G12C-mutant LUADs harboring different comutations were used as an input for UMAP dimensionality reduction, which revealed convergence of gene sets in distinct clusters related to immune/inflammatory response, metabolism, sustained/mitogenic signaling, oxidative phosphorylation, apoptosis, DNA maintenance, replication and repair, and cell-cycle progression. B and C,KRAS G12C/KEAP1 comutant LUADs showed a downregulation of gene sets related to inflammatory responses, while showing an enrichment in metabolism, oxidative phosphorylation reactive oxygen species pathway gene sets. The continuous significance score (Signif) indicates the −log10(Padj) * sign(fold-change) from the GSEA. Red, upregulation; blue, downregulation. D and E, A prominent upregulation of inflammatory response related gene expression programs was noted in the TME of KRAS G12C/TP53 comutant LUADs, together with gene sets related to cell-cycle progression and E2F-driven proliferation. Quantile-quantile plots were generated to visually compare the ranks of genes in the pathway to ranks that were sampled from a discrete uniform distribution. Adjusted P values for gene set differential expression are provided for comparison of KRAS G12C/non–RAS-mutant LUAD to KRAS G12C-mutant LUAD. HM, Hallmark; KG, Kyoto Encyclopedia of Genes and Genomes, KEGG.
Similar patterns of differential gene expression programs determined by the KRAS-mutant allele, emerged in COAD, where KRAS G12D and APC comutations were associated with an upregulation of cell-cycle progression, MYC-dependent proliferation and chromosome maintenance and DNA replication and repair gene sets, processes that were not apparent for KRAS G12V/APC or KRAS G13D/APC-mutant COAD (Supplementary Fig. S18; Supplementary Table S25). KRAS G13D/APC-mutant COAD tumors were less inflamed, with a downregulation of inflammatory response and B-cell receptor signaling gene sets. We found an enrichment for E2F- and MYC-dependent proliferation and oxidative phosphorylation pathways in KRAS G12V/PIK3CA and G13D/PIK3CA-mutant COAD tumors (Supplementary Fig. S18; Supplementary Table S25). An epithelial-to-mesenchymal phenotype characterized KRAS G12V/MAPK pathway comutant COAD, notably together with a downregulation of cell-cycle progression, E2F-dependent proliferation, and oxidative phosphorylation expression programs and a modest upregulation of inflammatory response gene expression programs (Supplementary Fig. S18; Supplementary Table S25). These expression patterns were distinct from those of non-RAS gene-only mutant compared to wild type tumors (Supplementary Fig. S19; Supplementary Table S26).
In KRAS/TP53 comutant COAD and PAC tumors, we identified distinct transcriptional programs depending on the mutant allele, pointing to differential activation of cell-cycle progression, proliferation and DNA repair programs (Supplementary Fig. S20; Supplementary Tables S25 and S27). While an upregulation of antigen presentation, TNFA signaling, neutrophil degranulation and T-cell receptor signaling gene sets characterized KRAS G12V, G12D, and G12R/TP53 comutant PAC, KRAS G13D/TP53 comutant COAD tumors showed a downregulation of inflammatory responses (Supplementary Fig. S20; Supplementary Tables S25 and S27). In assessing TME phenotypes in KRAS-mutant PAC tumors with co-occurring mutations in chromatin regulating genes, we noted an upregulation of key proinflammatory pathways, especially in KRAS G12D-mutant PAC (Supplementary Fig. S21; Supplementary Table S27). In contrast, a downregulation of IFNγ and inflammatory response gene sets was noted in KRAS G12D and G12R/MAPK pathway comutant PAC. KRAS G12D/SMAD4 comutant PAC tumors were characterized by a downregulation of cell-cycle progression, E2F- and MYC-dependent proliferation and DNA replication and repair gene expression programs (Supplementary Fig. S21). These gene expression profiles were distinct from the transcriptomic tracks of non-RAS gene-only mutant compared with wild-type tumors (Supplementary Fig. S22; Supplementary Table S28). Taken together, our findings support differential cancer cell fates in the context of their TME and suggest biologically distinct phenotypes of KRAS-mutant tumors that may reflect clinical outcomes and therapeutic vulnerabilities.
KRAS comutations are linked with differential clinical outcomes
We next evaluated the association between the distinct genomic tracks of RAS-mutant tumors and clinical outcomes. We first assessed 10,217 tumors from TCGA and evaluated differences in overall survival of individuals with tumors harboring RAS comutations. Patients with LUAD harboring KRAS G12C and KEAP1, NTRK3, PIK3CA, or TP53 comutations had a significantly shorter overall survival compared with the group without KRAS comutations (log-rank P = 0.02, P = 0.003, P = 0.015, and P = 0.0003, respectively; Supplementary Fig. S23). KRAS G12V mutations also conferred a worse prognosis in the presence of RBM10 and STK11 mutations (log-rank P = 0.018 and P = 0.03, respectively; Supplementary Fig. S23). In COAD, KRAS G12A/APC and KRAS G12V/AMER1 comutations were associated with shorter overall survival (long-rank P = 0.012 and P = 0.048, respectively; Supplementary Fig. S24) and notably among patients with KRAS G12C-mutant COAD, those also harboring APC and TP53 mutations attained longer overall survival (log-rank P = 0.0087; Supplementary Fig. S24). For patients with pancreatic adenocarcinoma, KRAS G12D and TP53 comutations defined a group of individuals with shorter overall survival (log-rank P = 0.001; Supplementary Fig. S24).
Given the significant differences in gene expression programs pointing to differential activation of cancer hallmarks but also differences in tumor immune surveillance, we next evaluated the impact of KRAS G12C mutations on clinical outcomes with immunotherapy-containing regimens for a cohort of 209 patients with advanced/metastatic KRAS G12C-mutant NSCLC from the AACR project GENIE registry. Leveraging the comutation pairs identified by the pan-cancer model, we performed survival analyses tailored to KRAS G12C lineage tracks and found that KRAS G12C/STK11 comutations conferred shorted overall survival with immunotherapy-containing regimens (log-rank P = 0.003 and P = 0.037, respectively; Fig. 7; Supplementary Fig. S25). In contrast, patients with NSCLC harboring KRAS G12C and TP53 hotspot alterations attained longer overall survival with immunotherapy-containing regimens (log-rank P = 0.05; Fig. 7). A trend towards longer overall survival was noted for patients with NSCLC harboring KRAS G12C and mutations in chromatin regulating genes and specifically ARID1A (log-rank P = 0.14 and P = 0.12, respectively; Supplementary Fig. S25). In contrast, KRAS G12C/AMER1-mutant and KRAS G12C/KEAP1 comutant NSCLC were found to be resistant to immunotherapy-containing regimens (log-rank P = 0.029 and P = 0.13; Fig. 7; Supplementary Fig. S25). Similarly, KRAS G12C and PIK3CA comutations conferred a worse prognosis for patients with NSCLC treated with immunotherapy (log-rank P = 0.028; Fig. 7). Collectively, our findings provide insights in the hallmarks of RAS-mutant cancers and integrate RAS genomic diversity with clinical outcomes and therapeutic vulnerabilities at a RAS-mutant allele resolution.
Figure 7.

Survival analyses for differentially comutated KRAS G12C-mutant NSCLC in a cohort of patients treated with chemotherapy and immunotherapy-containing regimens. A, Patients with NSCLC harboring KRAS G12C and STK11 comutations had a shorter overall survival with immunotherapy (n = 24 vs. n = 82, median survival 5.65 vs. 23.52 months, log-rank P = 0.003, HR: 2.31; 95% confidence interval, CI: 1.31–4.07). B, Conversely, co-occurrence of KRAS G12C and TP53 hotspot alterations was associated with longer overall survival with immunotherapy immunotherapy-containing regimens (n = 56 vs. n = 76, median survival 23.52 vs. 10.09 months, log-rank P = 0.05, HR: 0.63, 95% CI: 0.39–1.01). C,KRAS G12C/AMER1 comutations conferred a worse prognosis for patients with metastatic NSCLC treated with immunotherapy-containing regimens (n = 4 vs. n = 128, median survival of 4.55 vs. 12.85 months, log-rank P = 0.029, HR: 2.93, 95% CI: 1.06–8.07). D, Patients with KRAS G12C/PIK3CA-mutant NSCLC (n = 5) had a significantly shorter overall survival compared with patients with KRAS G12C-mutant tumors who received first-line immunotherapy (n = 25, 4.4 months vs. not reached, log-rank P = 0.028, HR: 3.48, 95% CI: 1.07–11.34). IO, immunotherapy.
Discussion
RAS genes act as prototypic oncogenes in human cancer that drive tumor initiation, proliferation, and progression by oncogenic signaling though the MAPK pathway, suppression of apoptosis as well as by rewiring the metabolic landscape and TME (5, 45). The recent success of the KRAS G12C inhibitors sotorasib and adagrasib in targeting KRAS-mutant NSCLC represents a breakthrough in precision oncology (19, 46, 47); however, the emergence of acquired resistance highlights the importance of understanding the genomic landscape of KRAS-driven tumors that may point to combination treatment strategies (48–51). Leveraging NGS data and >600,000 genomic alterations from >66,000 cancer patients and 51 tumor types in the AACR Project GENIE Registry, we have comprehensively studied the prevalence and genomic diversity of RAS in human cancers with a mutant allele resolution and considered host contexts determined by age, self-reported race and sex as well as tumor backgrounds determined by cell lineage, TMB, and mutational signatures. In investigating transcriptomic differences in 9,258 tumors from TCGA with differential RAS comutations, we discovered distinct RAS-mutant allele-driven gene expression programs pointing to differential recruitment of cancer hallmarks in RAS-mutant tumors. Ultimately, we linked the distinct molecular tracks of RAS-mutant tumors with differential clinical outcomes. Our study is the first one—to the best of our knowledge—to comprehensively assess the genomic landscape of RAS-mutant tumors in a pan-cancer manner and with a mutant allele-specific resolution, considering gene–gene interactions individually as well within gene families and pathways and accounting for host and tumor global genomic features that cooperate toward positive selection of genotypically distinct cancer cell clones, in the context of differential TME phenotypes. Our findings, may thus serve as a blueprint for mapping therapeutic vulnerabilities of RAS-mutant tumors (Fig. 8).
Figure 8.

RAS comutations involve cancer hallmarks in a cancer lineage–dependent manner. RAS comutation patterns point to dependencies of mutant RAS on oncogenic signaling under cancer hallmark pathways. Master regulators of different cancer hallmarks are comutated or are mutually exclusive with RAS-activating mutation depending on the tumor tissue origin. A representative example is that of the cell-cycle regulators CDKN2A and CDKN2B that are comutated with RAS in pancreatic cancer and melanoma, while less frequently comutated in NSCLC. Similarly, cancer hallmarks are differentially recruited in RAS-mutant tumors depending on the tumor tissue lineage. TP53 alterations co-occurred with RAS mutations in pancreatic cancer but were comutated less frequently than expected in RAS-mutant uterine, ovarian cancer, colorectal cancer, and NSCLC. This context-dependent genomic radial of RAS-mutant tumors may point to potential targets for combination therapeutic interventions.
Historically, RAS codon 12, 13, and 61 alterations have been considered equivalent in terms of their oncogenic potential and engagement of effectors and downstream signaling. Nevertheless, the mutated RAS isoform along with the position and type of substitution show a tumor type–dependent distribution (9) that together with the differential outcomes of patients with RAS-mutant tumors point toward a mutant allele selection (reviewed in ref. 1). While RAS proteins can exhibit functional redundancy, codon usage and protein expression depend on the RAS isoform (52); therefore oncogenic RAS signaling depends on the RAS isoform mutated, the codon altered and co-occurring mutations that are selected for in a cancer lineage–dependent manner. The codon-specific selection process during mutagenesis can be modeled in MNU/urethane induced tumors in mice, where not all oncogenic RAS mutations were recovered, despite being tumorigenic in other settings (53). Global activation of KRAS leads to a defined set of cancers, suggesting that activating mutations in KRAS are not universally tumorigenic in the absence of co-occurring perturbations (54). Furthermore, RAS isoforms are concentrated in different cell compartments, which allows for each RAS isoform to come into close contact with different regulators and effectors (55). While hotspot mutations in RAS codons 12, 13, and 61 all affect GTP hydrolysis, resulting in activation of downstream RAF/MEK/ERK signaling, the mechanism through which this is achieved varies by mutation (56). For instance, G13D but not G12V mutations accelerate intrinsic and GEF-mediated exchange; while G12V shows a reduced affinity for RAF yet more potently induces GTPase activity compared with G12D (9). KRAS G12V more efficiently recruits CRAF to the plasma membrane and promotes stem cell proliferation and isoform-specific oncogenicity may impact the subcellular localization and effector binding of RAS isoforms, further enhancing oncogenic signaling (9). Consistent with this notion, we found that while KRAS codon 12 mutations were the leading RAS alterations for PAC, ampullary, appendiceal cancer, colorectal carcinoma, NSCLC, small bowel cancer, CUP, and uterine cancer, while NRAS codon 61 mutations were dominant in melanoma, thyroid cancer, and hematologic malignancies. These findings are in concordance to previously reported RAS isoform frequencies at the codon level (1) and suggest tumor type–dependent selection of RAS oncogenic activation and signaling. To add to the body of knowledge of RAS isoform prevalence, we generated a RAS-mutant allele-specific atlas per cancer lineage, pointing again toward mutant allele-specific positive section in human cancers.
The prevalence of RAS mutations differs by sex and ethnic group (57–59), with an enrichment in KRAS G12C mutations reported in White females with NSCLC and colorectal carcinoma (59). In line with these findings, we detected a higher frequency of KRAS codon 12 in females with colorectal carcinoma and KRAS G12C mutations in females with NSCLC. We found a higher prevalence of KRAS codon 12 mutations in black patients with colorectal carcinoma, while Asian patients overall harbored a lower frequency of RAS mutations. Importantly, we found that codon 12 RAS mutations were less prevalent in younger patients with melanoma, CUP, NSCLC, and PAC but more prevalent in younger patients with ovarian cancer and B-cell leukemia/lymphoma. These findings point toward differential selection processes and genomic backgrounds based on age, sex, and ethnicity for patients with cancer, supporting distinct positive selection trajectories that may be related to genetic ancestries and differences in immune surveillance during tumor evolution.
Consistent with previous studies, the vast majority of KRAS G12C mutations were detected in NSCLC, with lower frequencies in colorectal carcinoma, ampullary, appendiceal cancer, and PAC. Interestingly, 3% of CUP harbor KRAS G12C mutations, which may expand the therapeutic options for patients with CUP. The recent success of KRAS G12C inhibitors across KRAS G12C-mutant cancers, has reinvigorated the enthusiasm in directly targeting KRAS. Furthermore, while KRAS G12C inhibitors have been on the forefront of mutant KRAS inhibition, alternate KRAS codon 12 and 13 alleles are the main oncogenic drivers in non-NSCLC tumors and development of allele-selective inhibitors beyond KRAS G12C is well underway (60). These clinical efforts, highlight the importance of understanding the genomic architecture and TME of non-G12C KRAS-mutant tumors that can in turn enhance effective RAS targeting in cancer types other than NSCLC.
Similar to RAS allele prevalence, comutation patterns between RAS and non-RAS genes occur in a context-dependent manner. This is exemplified in the genomic diversity of RAS-mutant NSCLC that may translate into differential clinical outcomes and reflect different therapeutic sensitivity. Patients with NSCLC with KRAS/KEAP1/NFE2L2 comutations may have shorter overall survival and duration of response with platinum-based chemotherapy (14, 61). STK11 and KRAS comutations have also been linked with inferior outcomes in NSCLC (61, 62) and in PD-L1–positive NSCLC, STK11, and KRAS comutations have been shown to confer resistance to anti–PD-1/PD-L1 therapy with significantly lower objective response rates, shorter progression-free survival and overall survival (62). Importantly, sotorasib and adagrasib may induce a proinflammatory TME in KRAS G12C-mutant NSLC (46, 63), which is in line with by preliminary results from the CodeBreak100 clinical trial, showing responses with single-agent sotorasib in patients with NSCLC harboring KRAS G12C/STK11 comutations (20).
In addition to confirming known co-occurrence between KRAS G12C and STK11/KEAP1 mutations in NSCLC as well as their transcriptomic profiles and prognostic/predictive value with immunotherapy-containing regimens, we discovered additional comutation patterns reflecting distinct cellular tracks and immune surveillance states ultimately impacting clinical outcomes. KRAS G12C/NTRK3 comutations were identified in NSCLC, and these tumors showed an upregulation of proliferation and cell-cycle progression pathways. While KRAS/NTRK3 co-occurrence has not been reported as a mechanism of resistance to KRAS G12C inhibitors, TRK receptor signaling may activate the RAS/MAPK pathway (64) and it is therefore possible that these comutations may confer differential clinical efficacy with KRAS inhibition. Our findings may also inform rational combinations of KRAS and NTRK targeted therapies.
The differential response rates seen with the KRAS G12C inhibitor sotorasib suggest cancer type–specific oncogenic KRAS signaling that are potentially reflected in/mediated by comutation profiles with non-RAS genes (19, 20, 65). Early studies investigating the genomic landscape of acquired resistance to KRAS G12C inhibitors point towards heterogeneous polyclonal in nature escape mechanisms (51, 66, 67). In KRAS G12C-mutant colorectal carcinoma cell lines, KRAS G12C inhibition induced rebound activation of the MAPK pathway via upstream activation of EGFR signaling (68). More recently the CodeBreak 100 clinical trial failed reach the benchmark overall response rate for patients with KRAS G12C-mutant colorectal carcinoma, which further exemplifies the context-dependent efficacy of KRAS-directed therapy (65). Notably, we reported mutual exclusivity between KRAS and MAPK pathway mutations in most tumor types, with the exception of colorectal carcinoma and PAC tumors. Given the role of secondary mutations in the MAPK axis in the emergence of acquired resistance to KRAS G12C inhibition (51, 66), it is plausible that co-occurring KRAS G12C and MAPK pathway alterations in colorectal carcinoma may be mediating primary resistance to KRAS G12C inhibitors, reflected in the inferior response rates seen in the CodeBreak100 clinical trial. To this end, our findings may inform rational combinations of KRAS and MAPK pathway inhibitors to prevent or overcome resistance to KRAS targeted therapy in a cancer lineage–dependent manner.
We showed that the context dependent comutation architecture shapes differential cancer hallmark recruitment and immune surveillance states and these may in turn sculpt clinical outcomes with RAS targeted therapies. Our findings can be leveraged to inform rational combinations of targeted therapies in a cancer lineage–dependent manner based on RAS comutations. In colorectal carcinoma, where KRAS is comutated with PIK3CA and genes in the PI3K pathway, combined MEK or ERK with PI3K inhibition may be effective for KRAS codon 12–mutant—G12D and G13D in particular—colorectal carcinoma. While RAS mutations were predominantly mutually exclusive with alterations in receptor tyrosine kinases, NF1 and BRAF class I mutations, combination of RAS targeted therapies with SHP2 or SOS1 inhibition that reduce KRAS-GTP loading and switching to its active state may be particularly effective for the subset of cancers with increased upstream signaling via receptor tyrosine kinases, BRAF class III mutations or in the setting of NF1 loss (60). Given the combined dependency of pancreatic tumors on mutant KRAS signaling and cell-cycle progression via loss of CDKN2A, CDK4/6 inhibitors such as palbociclib may synergize with KRAS inhibitors in improving clinical outcomes in tumors harboring KRAS/CDKN2A comutations. In addition to potential vulnerabilities to targeted therapies, our work highlighted differences in tumor immunoediting of RAS-mutant tumors. We found that KRAS-mutant tumors are not uniformly surrounded by an immunosuppressed TME, with KRAS G12C or G12V/TP53 comutant NSCLC as a representative example of a RAS-mutant subset with an inflamed TME. Importantly, immune surveillance states assessed by TME transcriptomic profiles were reflected in clinical outcomes with immunotherapy-containing regimens, as KRAS G12C/TP53 or KRAS G12C/chromatin regulator comutant NSCLCs were found to attain favorable clinical outcomes. In contrast, the TME of KRAS/MAPK comutant pancreatic cancers was found to be depleted in IFNγ and inflammatory response gene sets, which may explain the limited clinical efficacy of immunotherapy in this tumor type. Collectively, our work exemplified the molecular heterogeneity of RAS-mutant tumors and supports the notion that one-size-fits-all RAS targeted treatment strategies are unlikely to be clinical effective. Our findings can be leveraged for rational combinations of KRAS targeted therapies and enhance the responses to KRAS inhibitors or point to combinations of KRAS targeted therapy with standard of care immunotherapy approaches in a KRAS comutation informed manner.
From a methodologic standpoint, existing approaches for estimating prevalence and co-occurrence either estimate the overall relationship for a specific cancer type without accounting for potential confounding of TMB, mutation signature, sex, race, and age, or fit a series of independent models to each stratum ignoring information between subgroups (69–71). While the first approach can lead to biased estimates as it does not adjust for potential confounders such as TMB and host features, the second approach ignores the possible similarities in gene–gene relationships between subgroups and can be highly sensitive to spurious associations when the number of samples in the subgroups is small. Furthermore, many of the existing methods only assess mutual exclusivity and do not assess dependencies that may reflect positive selection. To address these challenges, we employed Bayesian hierarchical models for prevalence and comutation analyses that allow information to be shared across cancer types and between strata of age, sex, ethnicity, TMB, or mutation signatures through shared parameters at higher levels of these models. For rare cancer types in the AACR Project GENIE Registry, our model shrinks estimates of prevalence or comutations to the overall mean, while for common cancer types prevalence and comutation are nearly independent of other cancers. Implicitly, our model assumes that mutation rates for rarer tumor types could be improved by sharing information between cancers or that co-occurrence for individuals with a single cancer and a specific mutation profile could be informed by the co-occurrence patterns from other mutation profiles from individuals with the same cancer. The availability of posterior distributions for regression coefficients quantifying gene–gene dependencies and the variance of these effects between strata of potential confounders provide a rich inferential framework for modeling and evaluating mutation patterns. Extensions of the log-linear model include the interaction of confounding variables. Our Bayesian model can be applied to additional datasets, used to explore additional non-RAS gene/gene interactions and organically updated with future data, including patients from cancer types not originally included in the model, as the AACR Project GENIE Registry is expanding.
An important consideration in performing comutation analyses is the fact that mutation frequencies may be affected by the total TMB, which in turn may be attributed to key biological processes, such as mismatch repair deficiency. Furthermore, cancer genomes are shaped by exogenous carcinogens and endogenous mutagenic processes that are reflected in their mutational spectra. Comutation patterns may therefore be attributed to the underlying mutational signatures rather than point to a biologically important selection of comutated driver genes. Such “passenger hotspots” do not confer a fitness advantage to the cancer cells, rather occur at high frequency because of being optimal substrates (43). For instance, APOBEC mutational signatures, have been linked with recurrent mutations in DNA stem-loops, with PIK3CA mutations being a representative example of recurrent mutations in the context of substrate optimization (43, 72). Chromatin state also impacts mutation rate as open chromatin is associated with increased DNA repair (73) and ARID1A deficiency has been shown to impair mismatch repair (74), which would in turn cause an increased mutation burden. An enrichment in chromatin regulating genes has been previously described in the context of tumor rejection with immune checkpoint blockade (75), which was consistent with our findings in patients with NSCLC harboring KRAS G12C and hotspot mutations in chromatin regulating genes, that attained longer overall survival with immunotherapy-containing regimens.
While multicenter databases such as the AACR Project GENIE Registry enable large-scale genomic analyses that would not otherwise be possible, our study has several limitations. First, the differences in targeted sequencing platforms, bioinformatic filters, and mutation callers between study centers can artificially inflate the heterogeneity of prevalence and measures of association in co-occurrence models. To address these limitations, we assessed the heterogeneity by reframing the hierarchical model to measure prevalence for a specific cancer hierarchically across study centers. We used this same strategy to assess biological sources of confounding such as age, race, gender, tumor type, mutation burden, and mutation signatures. Furthermore, our analyses were focused on characterization of comutations of hotspot RAS mutations across cancers; future studies will evaluate RAS alterations other than codon 12, 13, and 61 mutations, including KRAS amplifications and rare mutations as well as experimentally validate our findings.
Taken together, our comprehensive assessment of RAS comutations, revealed the context-dependent genomic footprint of RAS-mutant tumors that may enable recognition of dysregulated pathways and potential targets for therapeutic intervention (Fig. 8). Our work unravels the complex genomic profile of RAS-mutant tumors and supports a selection process for specific RAS mutations and comutation patterns in a context-dependent manner. Our findings suggest that oncogenic RAS signaling differs in different tumor types and RAS mutation and comutation patterns lead to different neoplastic phenotypes and potentially point to differential therapeutic vulnerabilities and clinical outcomes. Our study is the first one—to the best of our knowledge—to comprehensively assess the genomic landscape of RAS-mutant tumors in a pan-cancer manner and with a mutant allele-specific resolution, considering gene–gene interactions individually as well within gene families and pathways and considering host and tumor global genomic features that cooperate toward positive selection of genotypically distinct cancer cell clones, in the context of differential TME phenotypes. Our findings can be leveraged for rationally combining RAS directed therapies with targeted therapies and immunotherapy in a context-dependent manner.
Supplementary Material
Acknowledgments
The authors would like to acknowledge the American Association for Cancer Research and its financial and material support in the development of the AACR Project GENIE registry, as well as members of the consortium for their commitment to data sharing. This work was supported by Amgen Inc. and in part by the U.S. NIH grants CA121113 (V. Anagnostou), CA062924 (R.B. Scharpf), and CA006973 (R.B. Scharpf.), the V Foundation (V. Anagnostou.), and the LUNGevity Foundation (V. Anagnostou.).
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors' Disclosures
R.B. Scharpf reports personal fees from Delfi Diagnostics outside the submitted work and Johns Hopkins University owns equity in Delfi Diagnostics. R.B. Scharpf is a founder of and holds equity in Delfi Diagnostics. R.B. Scharpf also serves as the Head of Data Science. This arrangement has been reviewed and approved by the Johns Hopkins University in accordance with its conflict of interest policies. A. Balan reports grants from AstraZeneca grants, Bristol Myers Squibb grants, and Delfi Diagnostics grants during the conduct of the study. J. Fiksel reports other support from Janssen Pharmaceuticals and Vertex Pharmaceuticals outside the submitted work. C. Cherry reports personal fees from C M Cherry Consulting, LLC, outside the submitted work. M.L. LeNoue-Newton reports other support from American Association of Cancer Research during the conduct of the study; other support from GE Healthcare and non-financial support and other support from American Association of Cancer Research outside the submitted work; in addition, M.L. LeNoue-Newton has a patent for 607172-WO-1 pending. H.A. Rizvi reports subsequent to the completion of this work, H.A. Rizvi became an employee at AstraZeneca. J.R. White reports other support from Resphera Biosciences LLC outside the submitted work. L. Raskin reports other support from Amgen during the conduct of the study; other support from Amgen and Abbvie outside the submitted work. A.S. Park reports personal fees from Amgen Inc. during the conduct of the study; personal fees from Amgen Inc. and AbbVie Inc. outside the submitted work. H. Hsu reports personal fees from Amgen Inc. during the conduct of the study; personal fees from Amgen Inc. outside the submitted work. K.C. Arbour reports other support from Mirati, Genetech, Revolution Medicines, and personal fees from Sanofi-Genzyme outside the submitted work. M.M. Awad reports personal fees from AbbVie, Neon, Maverick, Nektar, ArcherDx, Mirati, Gritstone, Novartis, EMD Serono, and NextCure; grants and personal fees from Bristol-Myers Squibb and Genentech; grants from Lilly and AstraZeneca outside the submitted work. G.J. Riely reports grants from Amgen during the conduct of the study; grants from Mirati, Roche, Novartis, Takeda, Pfizer, and Merck outside the submitted work. C.M. Lovly reports other support from AACR GENIE project during the conduct of the study; personal fees from Amgen, Arrivent, AstraZeneca, Blueprints Medicine, Cepheid, D2G Oncology, Daiichi Sankyo, Eli Lilly, EMD Serono, Foundation Medicine, Genentech, Janssen, Medscape, Novartis, Pfizer, Puma, Syros, and Takeda and other support from Roche outside the submitted work. V. Anagnostou reports grants from AstraZeneca, Bristol Myers Squibb, and Delfi Diagnostics during the conduct of the study. No disclosures were reported by the other authors.
Authors' Contributions
R.B. Scharpf: Conceptualization, formal analysis, supervision, validation, investigation, visualization, methodology, writing-original draft, writing-review and editing. A. Balan: Formal analysis. B. Ricciuti: Data curation, writing-review and editing. J. Fiksel: Data curation, methodology, writing-original draft. C. Cherry: Formal analysis, validation, visualization. C. Wang: Data curation, formal analysis. M.L. Lenoue-Newton: Data curation. H.A. Rizvi: Data curation. J.R. White: Formal analysis, writing-original draft. A.S. Baras: Resources, formal analysis. J. Anaya: Formal analysis. B.V. Landon: Data curation, writing-original draft. M. Majcherska-Agrawal: Writing-original draft. P. Ghanem: Writing-original draft. J. Lee: Resources, data curation, project administration. L. Raskin: Funding acquisition, writing-review and editing. A.S. Park: Funding acquisition, writing-review and editing. H. Tu: Funding acquisition, writing-review and editing. H. Hsu: Resources, funding acquisition, writing-review and editing. K.C. Arbour: Data curation, writing-review and editing. M.M. Awad: Conceptualization, writing-review and editing. G.J. Riely: Conceptualization, writing-review and editing. C.M. Lovly: Conceptualization, writing-original draft, writing-review and editing. V. Anagnostou: Conceptualization, formal analysis, supervision, validation, investigation, visualization, methodology, writing-original draft, writing-review and editing.
References
- 1. Prior IA, Hood FE, Hartley JL. The frequency of Ras mutations in cancer. Cancer Res 2020;80:2969–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res 2012;72:2457–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, Westover KD. Biochemical and structural analysis of common cancer-associated KRAS mutations. Mol Cancer Res 2015;13:1325–35. [DOI] [PubMed] [Google Scholar]
- 4. Simanshu DK, Nissley DV, McCormick FRAS. Proteins and their regulators in human disease. Cell 2017;170:17–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer 2011;11:761–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Omholt K, Platz A, Kanter L, Ringborg U, Hansson J. NRAS and BRAF mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Clin Cancer Res 2003;9:6483–8. [PubMed] [Google Scholar]
- 7. Weir BA, Woo MS, Getz G, Perner S, Ding L, Beroukhim R, et al. Characterizing the cancer genome in lung adenocarcinoma. Nature 2007;450:893–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Goretzki PE, Lyons J, Stacy-Phipps S, Rosenau W, Demeure M, Clark OH, et al. Mutational activation of RAS and GSP oncogenes in differentiated thyroid cancer and their biological implications. World J Surg 1992;16:576–81. [DOI] [PubMed] [Google Scholar]
- 9. Li S, Balmain A, Counter CM. A model for RAS mutation patterns in cancers: finding the sweet spot. Nat Rev Cancer 2018;18:767–77. [DOI] [PubMed] [Google Scholar]
- 10. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487:330–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511:543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cancer Genome Atlas Research Network; Albert Einstein College of Medicine; Analytical Biological Services; Barretos Cancer Hospital; Baylor College of Medicine; Beckman Research Institute of City of Hope, et al. Integrated genomic and molecular characterization of cervical cancer. Nature 2017;543:378–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov 2015;5:860–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Arbour KC, Jordan E, Kim HR, Dienstag J, Yu HA, Sanchez-Vega F, et al. Effects of co-occurring genomic alterations on outcomes in patients with KRAS-mutant non-small cell lung cancer. Clin Cancer Res 2018;24:334–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Way GP, Sanchez-Vega F, La K, Armenia J, Chatila WK, Luna A, et al. Machine learning detects pan-cancer Ras pathway activation in The Cancer Genome Atlas. Cell Rep 2018;23:172–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hobbs GA, Der CJ, Rossman KL. RAS isoforms and mutations in cancer at a glance. J Cell Sci 2016;129:1287–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013;499:214–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Robertson AG, Kim J, Al-Ahmadie H, Bellmunt J, Guo G, Cherniack AD, et al. Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell 2018;174:1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRAS(G12C) inhibition with sotorasib in advanced solid tumors. N Engl J Med 2020;383:1207–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, et al. Sotorasib for lung cancers with KRAS p.G12C mutation. N Engl J Med 2021;384:2371–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nichols RJ, Cregg J, Schulze CJ, Wang Z, Yang K, Jiang J, et al. A next generation tri-complex KRASG12C(ON) inhibitor directly targets the active, GTP-bound state of mutant RAS and may overcome resistance to KRASG12C(OFF) inhibition [abstract]. In:Proceedings of the American Association for Cancer Research Annual Meeting 2021; 2021 Apr 10–15 and May 17–21. Philadelphia (PA): AACR; Cancer Res 2021;81(13_Suppl):Abstract nr 1261. [Google Scholar]
- 22. AACR Project GENIE Consortium. AACR project GENIE: powering precision medicine through an International Consortium. Cancer Discov 2017;7:818–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ellrott K, Bailey MH, Saksena G, Covington KR, Kandoth C, Stewart C, et al. Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst 2018;6:271–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell 2018;173:400–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, et al. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res 2015;43:D805–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tokheim C, Karchin R. CHASMplus reveals the scope of somatic missense mutations driving human cancers. Cell Syst 2019;9:9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Anaya J, Sidhom JW, Cummings CA, Baras A, Consortium APG. Aggregation Tool for Genomic Concepts (ATGC): a deep learning framework for sparse genomic measures and its application to tumor mutational burden. bioRxiv 2020. [Google Scholar]
- 28. Rosenthal R, McGranahan N, Herrero J, Taylor BS, Swanton C. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol 2016;17:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Plummer M. JAGS: a program for analysis of Bayesian graphical models using Gibbs sampling; 2003. Availablr from: https://www.r-project.org/conferences/DSC-2003/Proceedings/Plummer.pdf.
- 30. Busemeyer J, Wang Z, Townsend J, Eidels A. The Oxford handbook of computational and mathematical psychology. New York, NY: Oxford University Press; 2015. [Google Scholar]
- 31. Team SD. Stan modeling language users guide and reference manual; 2021.
- 32. Team SD. RStan: the R interface to Stan; 2020. R package version 2.21.2. 2020.
- 33. Mina M, Iyer A, Tavernari D, Raynaud F, Ciriello G. Discovering functional evolutionary dependencies in human cancers. Nat Genet 2020;52:1198–207. [DOI] [PubMed] [Google Scholar]
- 34. Canisius S, Martens JW, Wessels LF. A novel independence test for somatic alterations in cancer shows that biology drives mutual exclusivity but chance explains most co-occurrence. Genome Biol 2016;17:261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Colaprico A, Silva TC, Olsen C, Garofano L, Cava C, Garolini D, et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res 2016;44:e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 2015;1:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Korotkevich G, Sukhov V, Sergushichev A. Fast gene set enrichment analysis. bioRxiv 2019. [Google Scholar]
- 39. Blischak JD, Carbonetto P, Stephens M. Creating and sharing reproducible research code the workflowr way. F1000Res 2019;8:1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Winters IP, Chiou SH, Paulk NK, McFarland CD, Lalgudi PV, Ma RK, et al. Multiplexed in vivo homology-directed repair and tumor barcoding enables parallel quantification of Kras variant oncogenicity. Nat Commun 2017;8:2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rabara D, Tran TH, Dharmaiah S, Stephens RM, McCormick F, Simanshu DK, et al. KRAS G13D sensitivity to neurofibromin-mediated GTP hydrolysis. Proc Natl Acad Sci U S A 2019;116:22122–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schmitt A, Knittel G, Welcker D, Yang TP, George J, Nowak M, et al. ATM deficiency is associated with sensitivity to PARP1- and ATR inhibitors in lung adenocarcinoma. Cancer Res 2017;77:3040–56. [DOI] [PubMed] [Google Scholar]
- 43. Buisson R, Langenbucher A, Bowen D, Kwan EE, Benes CH, Zou L, et al. Passenger hotspot mutations in cancer driven by APOBEC3A and mesoscale genomic features. Science 2019;364:eaaw2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature 2013;500:415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mukhopadhyay S, Vander Heiden MG, McCormick F. The metabolic landscape of RAS-driven cancers from biology to therapy. Nat Cancer 2021;2:271–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019;575:217–23. [DOI] [PubMed] [Google Scholar]
- 47. Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, et al. The KRAS(G12C) inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov 2020;10:54–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hata AN, Shaw AT. Resistance looms for KRAS(G12C) inhibitors. Nat Med 2020;26:169–70. [DOI] [PubMed] [Google Scholar]
- 49. Fedele C, Li S, Teng KW, Foster CJR, Peng D, Ran H, et al. SHP2 inhibition diminishes KRASG12C cycling and promotes tumor microenvironment remodeling. J Exp Med 2021;218:e20201414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ryan MB, Fece de la Cruz F, Phat S, Myers DT, Wong E, Shahzade HA, et al. Vertical pathway inhibition overcomes adaptive feedback resistance to KRAS(G12C) inhibition. Clin Cancer Res 2020;26:1633–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tanaka N, Lin JJ, Li C, Ryan MB, Zhang J, Kiedrowski LA, et al. Clinical acquired resistance to KRAS(G12C) inhibition through a novel KRAS switch-II pocket mutation and polyclonal alterations converging on RAS-MAPK reactivation. Cancer Discov 2021;11:1913–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lampson BL, Pershing NL, Prinz JA, Lacsina JR, Marzluff WF, Nicchitta CV, et al. Rare codons regulate KRas oncogenesis. Curr Biol 2013;23:70–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Westcott PM, Halliwill KD, To MD, Rashid M, Rust AG, Keane TM, et al. The mutational landscapes of genetic and chemical models of Kras-driven lung cancer. Nature 2015;517:489–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Guerra C, Mijimolle N, Dhawahir A, Dubus P, Barradas M, Serrano M, et al. Tumor induction by an endogenous K-ras oncogene is highly dependent on cellular context. Cancer Cell 2003;4:111–20. [DOI] [PubMed] [Google Scholar]
- 55. Choy E, Chiu VK, Silletti J, Feoktistov M, Morimoto T, Michaelson D, et al. Endomembrane trafficking of ras: the CAAX motif targets proteins to the ER and Golgi. Cell 1999;98:69–80. [DOI] [PubMed] [Google Scholar]
- 56. Scheffzek K, Ahmadian MR, Kabsch W, Wiesmuller L, Lautwein A, Schmitz F, et al. The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science 1997;277:333–8. [DOI] [PubMed] [Google Scholar]
- 57. Sun Y, Ren Y, Fang Z, Li C, Fang R, Gao B, et al. Lung adenocarcinoma from East Asian never-smokers is a disease largely defined by targetable oncogenic mutant kinases. J Clin Oncol 2010;28:4616–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Reinersman JM, Johnson ML, Riely GJ, Chitale DA, Nicastri AD, Soff GA, et al. Frequency of EGFR and KRAS mutations in lung adenocarcinomas in African Americans. J Thorac Oncol 2011;6:28–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nassar AH, Adib E, Kwiatkowski DJ. Distribution of KRAS (G12C) somatic mutations across race, sex, and cancer type. N Engl J Med 2021;384:185–7. [DOI] [PubMed] [Google Scholar]
- 60. Hofmann MH, Gerlach D, Misale S, Petronczki M, Kraut N. Expanding the reach of precision oncology by drugging all KRAS mutants. Cancer Discov 2022;12:924–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Scheffler M, Ihle MA, Hein R, Merkelbach-Bruse S, Scheel AH, Siemanowski J, et al. K-ras mutation subtypes in NSCLC and associated co-occuring mutations in other oncogenic pathways. J Thorac Oncol 2019;14:606–16. [DOI] [PubMed] [Google Scholar]
- 62. Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov 2018;8:822–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Briere DM, Li S, Calinisan A, Sudhakar N, Aranda R, Hargis L, et al. The KRAS(G12C) inhibitor MRTX849 reconditions the tumor immune microenvironment and sensitizes tumors to checkpoint inhibitor therapy. Mol Cancer Ther 2021;20:975–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cocco E, Scaltriti M, Drilon A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol 2018;15:731–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Fakih MG, Kopetz S, Kuboki Y, Kim TW, Munster PN, Krauss JC, et al. Sotorasib for previously treated colorectal cancers with KRAS(G12C) mutation (CodeBreaK100): a prespecified analysis of a single-arm, phase 2 trial. Lancet Oncol 2022;23:115–24. [DOI] [PubMed] [Google Scholar]
- 66. Zhao Y, Murciano-Goroff YR, Xue JY, Ang A, Lucas J, Mai TT, et al. Diverse alterations associated with resistance to KRAS(G12C) inhibition. Nature 2021;599:679–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Suzuki S, Yonesaka K, Teramura T, Takehara T, Kato R, Sakai H, et al. KRAS inhibitor resistance in MET-amplified KRAS (G12C) non-small cell lung cancer induced by RAS- and non-RAS-mediated cell signaling mechanisms. Clin Cancer Res 2021;27:5697–707. [DOI] [PubMed] [Google Scholar]
- 68. Amodio V, Yaeger R, Arcella P, Cancelliere C, Lamba S, Lorenzato A, et al. EGFR blockade reverts resistance to KRAS(G12C) inhibition in colorectal cancer. Cancer Discov 2020;10:1129–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ciriello G, Cerami E, Sander C, Schultz N. Mutual exclusivity analysis identifies oncogenic network modules. Genome Res 2012;22:398–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mina M, Raynaud F, Tavernari D, Battistello E, Sungalee S, Saghafinia S, et al. Conditional selection of genomic alterations dictates cancer evolution and oncogenic dependencies. Cancer Cell 2017;32:155–68. [DOI] [PubMed] [Google Scholar]
- 71. Vandin F, Upfal E, Raphael BJ. De novo discovery of mutated driver pathways in cancer. Genome Res 2012;22:375–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Temko D, Tomlinson IPM, Severini S, Schuster-Bockler B, Graham TA. The effects of mutational processes and selection on driver mutations across cancer types. Nat Commun 2018;9:1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Adar S, Hu J, Lieb JD, Sancar A. Genome-wide kinetics of DNA excision repair in relation to chromatin state and mutagenesis. Proc Natl Acad Sci U S A 2016;113:E2124–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Shen J, Ju Z, Zhao W, Wang L, Peng Y, Ge Z, et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat Med 2018;24:556–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Anagnostou V, Niknafs N, Marrone K, Bruhm DC, White JR, Naidoo J, et al. Multimodal genomic features predict outcome of immune checkpoint blockade in non-small-cell lung cancer. Nat Cancer 2020;1:99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.