

Abstract
Regeneration and tissue homeostasis require accurate production of missing cell lineages. Cell production is driven by changes to gene expression, which is shaped by multiple layers of regulation. Here, we find that the ubiquitous mRNA base‐modification, m6A, is required for proper cell fate choice and cellular maturation in planarian stem cells (neoblasts). We mapped m6A‐enriched regions in 7,600 planarian genes and found that perturbation of the m6A pathway resulted in progressive deterioration of tissues and death. Using single‐cell RNA sequencing of >20,000 cells following perturbation of the m6A pathway, we identified an increase in expression of noncanonical histone variants, and that inhibition of the pathway resulted in accumulation of undifferentiated cells throughout the animal in an abnormal transcriptional state. Analysis of >1,000 planarian gene expression datasets revealed that the inhibition of the chromatin modifying complex NuRD had almost indistinguishable consequences, unraveling an unappreciated link between m6A and chromatin modifications. Our findings reveal that m6A is critical for planarian stem cell homeostasis and gene regulation in tissue maintenance and regeneration.
Keywords: differentiation, m6A, planarian, regeneration, stem cells
Subject Categories: Chromatin, Transcription & Genomics; Development
Somatic stem cell specification and regeneration of the flatworm are safeguarded by the m6A mRNA modification pathway.
Introduction
Tissue maintenance and regeneration depend on the production of the necessary cell types. At a tissue scale, cell signaling balances cell production and contributes to selection of cell fates. Yet, at the scale of an individual cell, it is the regulation of gene expression that determines cell identity. Mechanisms that regulate gene expression of stem cells and other progenitors (e.g., dedifferentiated cells) are therefore central in the study of regeneration and tissue maintenance.
Recently, biochemical modifications to RNA molecules have emerged as regulators of gene expression. N6‐methylation of adenosine (m6A) is a widespread modification to mRNAs, which is conserved from yeast to mammals. m6A modulates major aspects of RNA metabolism, including RNA stability, localization, and splicing (Yang et al, 2018). The mRNAs are decorated with methyl groups installed by a methyltransferase complex (MTC), composed of the methyltransferase METTL3 and accessory proteins, including METTL14, KIAA1429, WTAP, Hakai, RBM15/RBM15B, and ZC3H13 (Yang et al, 2018). The methylated sites are recognized by several reader proteins including YTH‐domain containing proteins affecting gene expression (Patil et al, 2018). Studies in multiple organisms have shown that m6A and many components of the m6A pathway are essential for diverse processes: meiosis in the yeast Saccharomyces cerevisiae (Schwartz et al, 2013), oogenesis and sex determination in Drosophila melanogaster (Hongay & Orr‐Weaver, 2011; Kan et al, 2017), early development in mammals (Geula et al, 2015), B‐cell development (Zheng et al, 2020), and hematopoiesis (Zhang et al, 2017). m6A is therefore a key regulator of dynamic processes involving cellular proliferation and differentiation, and thus it could play an important role in tissue regeneration.
Planarians are flatworms that can regrow and support any adult tissue using a heterogeneous population of stem cells known as neoblasts (Rink, 2013). Following injury, neoblasts rapidly divide and grow a blastema that forms missing tissues. During homeostasis, neoblasts support existing tissues by continuously replacing them. The plasticity of individual neoblasts requires stringent regulation of gene expression (Wagner et al, 2012; Rink, 2013; Adler & Sánchez Alvarado, 2015). Consequently, neoblast gene expression determines lineage selection (van Wolfswinkel et al, 2014; Molinaro & Pearson, 2018), cell cycle progression (Raz et al, 2021), and cellular migration (Sahu et al, 2021). Transcriptional regulation has been studied at multiple resolutions in planarians: extracellular signaling and other systemic signals that regulate neoblast biology have been studied in depth over the past decade. Epigenetic regulators that modify the chromatin landscape and alter fate determination were discovered (Duncan et al, 2015). Transcription factor expression that is associated with differentiation to distinct planarian lineages has been described (van Wolfswinkel et al, 2014). However, molecular and functional studies of mRNA methylation have not been reported in planarians, and their impact on tissue homeostasis and regeneration is unknown.
Here, we analyzed the functions of planarian genes encoding components of the m6A‐modification pathway (m6A genes) and studied the consequence of their inhibition. We mapped m6A‐enriched regions in 7,600 genes expressed in a diversity of cell types and conditions. We identified multiple roles for the m6A pathway in neoblast biology, including regulation of cell fate choice, cell cycle control, and production of polyadenylated histone variants. Inhibition of m6A genes resulted in coordinated overexpression of adjacent genes in repetitive genomic regions, which were transcriptionally silenced in control animals. Systematic reanalysis of >1,000 planarian gene expression datasets revealed striking molecular similarities following silencing of the nucleosome remodeling and deacetylase complex (NuRD) component CHD4, suggesting a functional association between m6A and chromatin regulation. Single‐cell RNA sequencing (scRNAseq) and confocal imaging revealed an accumulation of a previously undescribed class of cells following inhibition of m6A genes and of NuRD. Our study reveals that m6A is ubiquitous in planarians, establishes assays for studying m6A on a genomic scale, and reveals critical functions of m6A genes that are fundamental to regeneration and homeostasis.
Results
Components of m6A pathway are broadly expressed in planarians
We identified planarian genes that encode putative components of the MTC by searching for genes in the planarian genome encoding proteins similar to known MTC components in human, yeast, and fruit fly (Materials and Methods). We detected sequences corresponding to all of the canonical MTC components (Figs 1A and EV1A; Appendix Tables S1 and S2). We identified six putative genes encoding m6A readers in the planarian genome by searching the YTH‐domain signature in the predicted planarian proteome (Figs 1A and EV1B). Orthologs of these putative YTH‐encoding genes were found in six additional planarian species (Materials and Methods). By comparison, Drosophila melanogaster has two YTH‐containing genes and vertebrates have five genes encoding a YTH‐domain (Lence et al, 2017), suggesting that the YTH gene family is expanded in planarians.
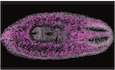
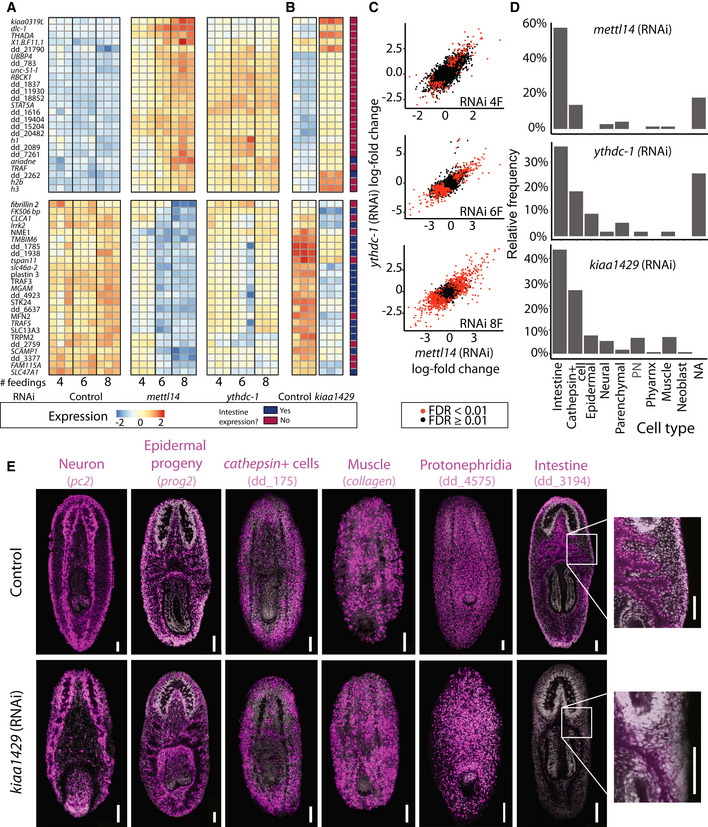
Figure 1. Expression of m6A genes is required for planarian regeneration and viability.
-
AComponents of the m6A MTC (left) and readers (right) that were identified in the planarian genome (Fig EV1A and B; Materials and Methods).
-
BEffect of RNAi on planarian homeostasis and regeneration. Gene expression was inhibited by feeding the animals with dsRNA against the target gene (Materials and Methods). Phenotypes were observed in kiaa1429 (RNAi) animals following five dsRNA feedings in homeostasis ((i); 10/10 animals), in regeneration ((ii); 10/10 animals; shown 10 days post amputation), and following nine feedings of mettl14 (iii) or ythdc‐1 dsRNA feedings (iv). Matching controls did not show phenotypes (n = 10 per control group). Experiments were performed at least three times with over 10 animals per experiment in each experimental group. Scale = 1 mm.
-
C, DThe relative abundance of m6A was estimated by using an anti‐m6A‐antibody (Materials and Methods), showing similar m6A levels compared to recent quantification of m6A in mammals (Liu et al, 2020b); (C) Quantification of m6A on RNA isolated from kiaa1429 (RNAi) and control animals following five RNAi feedings (left), or from mettl14 (RNAi) and control animals following nine RNAi feedings (right), revealed a strong reduction in m6A levels following inhibition of gene expression (Student's t‐test P < 0.05). (D) Comparison of m6A levels on polyadenylated and non‐polyadenylated RNA showed that polyadenylated RNA was highly enriched in m6A (Student's t‐test *P < 0.05).
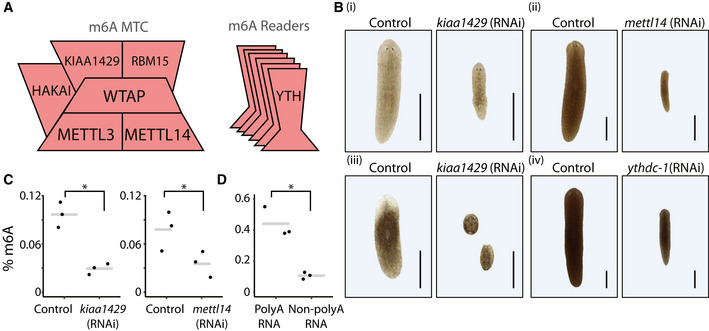
Figure EV1. m6A genes are conserved and functional in planarians.
-
AConservation of the genes encoding the MTC is shown across diversity of organisms. Annotation for A. thaliana was obtained from a previous analysis (Růžička et al, 2017).
-
BDomain analysis of putative YTH‐domain (red block) containing genes.
-
C, DThe gene expression of planarian MTC and reader‐encoding genes, across different cell types and lineages, was extracted from the planarian scRNAseq resource (Plass et al, 2018) (blue to red, low and high expression levels, respectively). The expression of MTC‐encoding genes is widespread in different cell types, including in the stem cell compartment (black arrow, indicated in the mettl3 panel). The expression of reader‐encoding genes is not widespread, except for ythdc‐1, which is expressed across multiple cell types and conditions.
-
EInhibition of m6A genes resulted in lysis of the animals and eventually death. Shown are representative control and kiaa1429 (RNAi) animals. Scale = 1 mm.
-
FInhibition of m6A genes resulted in defects in food uptake. Shown are animals following feeding with calf liver mixed with a red food color. Control animals show normal food uptake (top) compared with kiaa1429 (RNAi) animals, which stopped eating. Size measurements were performed on separate animals, which were subjected to the same RNAi treatment. Student's t‐test; ****P < 0.0001. Scale = 1 mm.
-
GInhibition of mettl14 and ythdc‐1 by RNAi has resulted in size reduction. Shown are representative images (left) and measurement of animal sizes following nine RNAi feedings. Significance was calculated using one‐way ANOVA followed by Dunnett's multiple comparison test; ***P < 0.001; *P < 0.05. Scale = 1 mm.
-
HqPCR analysis showed that the gene expression of mettl3 (top) and wtap (bottom) is not downregulated significantly following RNAi, which could likely explain the lack of penetrant phenotypes in these conditions. Error bars indicate the 95% confidence interval. Samples include two technical replicates and at least two biological replicates.
-
ISchematic of the design of two non‐overlapping gene fragments that were used for synthesis of dsRNA targeting the kiaa1429 gene. The sequence used for the experiments presented in the manuscript was labeled “original kiaa1429 clone.”
-
JdsRNA that was produced using a second cloned fragment of kiaa1429, labeled “non‐overlapping kiaa1429 (RNAi),” produced phenotypes that were similar to the phenotypes observed in a non‐overlapping clone. This further indicated that the kiaa1429 (RNAi) phenotype resulted from inhibition of kiaa1429 gene expression and not because of an off‐target effect of the RNAi. Student's t‐test ***P < 0.001. Scale = 1 mm.
We analyzed the expression of the genes that encode m6A‐pathway components (m6A genes) across cell types in the planarian cell lineage atlas (Plass et al, 2018). We found that m6A genes are expressed across multiple cell types and particularly in neoblasts (Fig EV1C and D). Genes that encode YTH‐domain were expressed in largely nonoverlapping cell types, suggesting that these putative readers might have distinct functions, which are activated in a cell‐type‐specific manner (Fig EV1D). Only one YTH‐domain gene, ythdc‐1, was expressed broadly and across cells in multiple stages of differentiation. Analysis of MTC gene expression showed that MTC‐encoding genes were expressed during the differentiation of multiple lineages and in multiple cell types (Fig EV1C). This suggests that m6A has a role in cellular differentiation in planarians, and that it might modulate different aspects of RNA metabolism using distinct m6A readers.
m6A pathway activity is required for planarian regeneration and homeostasis
We studied the function of the m6A pathway in planarians by systemically inhibiting the m6A genes using RNAi (Materials and Methods). Inhibition of components of the MTC encoded by kiaa1429 and mettl14 resulted in a smaller body size compared with control animals, failure to uptake food, and eventually in lysis (Figs 1B and EV1E–G). Inhibition of other MTC‐encoding genes did not result in a penetrant phenotype, yet inefficient inhibition of gene expression could explain this result (Batista et al, 2014; e.g., of mettl3; Fig EV1H). Inhibition of the predicted m6A reader, ythdc‐1, resulted in a similar phenotype (Figs 1B and EV1G). Interestingly, ythdc‐1 is the only m6A reader gene that is broadly expressed across cell types and stages of lineage differentiation (Fig EV1D). Next, we tested the ability of animals to regenerate following inhibition of the m6A pathway by feeding the animals with dsRNA and amputating them. Inhibition of both MTC‐encoding genes and ythdc‐1 eliminated the ability of the animals to regenerate. For example, amputated kiaa1429 (RNAi) animals did not produce a blastema and consequently died (Fig 1B). We tested if this rapid effect was an outcome of depletion of m6A or a nonspecific off‐target effect of the RNAi. We tested the specificity of the kiaa1429 (RNAi) phenotype by repeating the experiment using a nonoverlapping dsRNA construct and found comparable phenotypes (Fig EV1I and J; Materials and Methods). Next, we quantified the m6A levels of RNA from kiaa1429 (RNAi), mettl14 (RNAi), and control animals using an anti‐m6A‐antibody (Materials and Methods). We detected 3.3‐ and 2.2‐fold reduction (Student's t‐test P < 0.05) in the level of m6A (Fig 1C) following inhibition of kiaa1429 and mettl14, respectively. Next, we tested if m6A was found on planarian mRNA or on functional non‐polyadenylated transcripts. Polyadenylated RNA was over fourfold enriched with m6A (Fig 1D; P < 0.05). These results demonstrate that (i) planarian m6A genes are required for regeneration and tissue homeostasis, (ii) that the MTC genes are required for installing m6A, and (iii) that m6A is enriched on mRNA.
m6A is abundant on planarian mRNAs
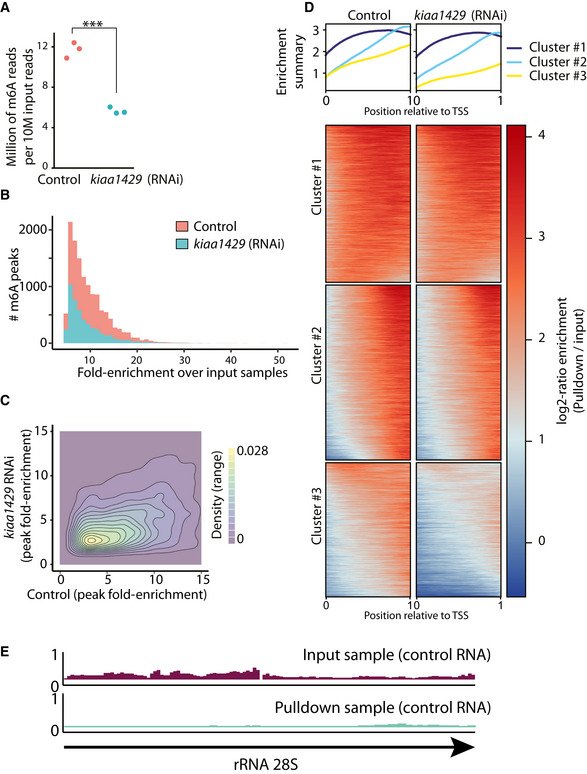
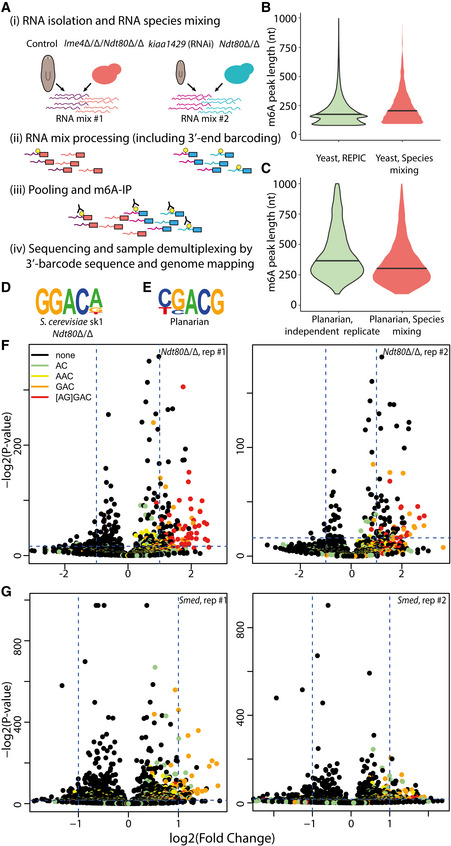
We next mapped m6A‐enriched regions in planarian RNAs by using m6A‐seq2 (Dierks et al, 2021). m6A‐seq2 allows multiplexed antibody‐based m6A profiling of multiple samples in a single tube, thereby also permitting quantitative comparison of methylation levels across diverse samples (Fig 2A; Materials and Methods; Appendix Table S3). RNA was extracted from control animals, having normal m6A levels, and from kiaa1429 (RNAi) animals, having depleted m6A (Figs 1C and 2A). m6A‐seq2 confirmed that mRNA originating from kiaa1429 (RNAi) animals was enriched substantially and significantly less than the control counterparts (Fig EV2A), and that far fewer “peaks,” and of decreased intensity, were detected in the kiaa1429 (RNAi) animals in comparison to the controls (Fig EV2A–D; Materials and Methods). We mapped the sequencing data to the planarian genome and detected regions enriched following IP in comparison to input (“m6A peaks”) via MeTPeak (Cui et al, 2016; Fig 2A and B; Materials and Methods). The read mapping was similar across all libraries (84–90%, SD = 1.5%; Appendix Table S4), without a significant difference between the mapping of m6A‐enriched libraries and their input controls and between kiaa1429 (RNAi) and their controls (Materials and Methods). Reads were aligned to >20 k genes, producing a comprehensive map of the planarian m6A transcriptome (Dataset EV1). We detected 7,600 genes having m6A peaks enriched by >5‐fold over input in the control m6A libraries (MeTPeak score <1E‐4). Many peaks were detectable following inhibition of kiaa1429, but their enrichment score was significantly reduced (Figs 2B and EV2B–D; Dataset EV1; P = 2.5E‐6). We divided the genes to bins based on their expression in the input libraries and examined m6A‐enriched regions in each bin. The most highly expressed genes (bin 10) had fewer m6A peaks compared with genes in bins 6–9 (Fig 2C), an observation consistent with findings in mammalian systems (Dominissini et al, 2012; Schwartz et al, 2014). Interestingly, m6A was not detected on rRNA (Fig EV2E), in contrast to mammalian systems where two sites are installed via a mechanism independent of METTL3, which could contribute to the lower m6A detection in non‐polyA RNA (Fig 1D). m6A peaks were strongly biased toward the 3′ end of transcripts, similar to the case in mammalian systems, a trend that was not observed in the input samples (Fig 2B). Interestingly, the peak length distribution in planarians was longer than observed in other organisms (Liu et al, 2020c; Fig 2D). We validated this result by mixing planarian and yeast (S. cerevisiae sk1) RNA and processing the mixed sample according to the m6A‐seq2 protocol (Fig EV3, Dataset EV1; Materials and Methods). The mixed RNA sample processing included RNA fragmentation, 3′‐end barcoding, m6A enrichment, library preparation, and sequencing, therefore reducing many of the technical aspects that could contribute to the increased fragment length observed in planarian m6A peaks. We mapped the sequenced mixed libraries to the planarian and yeast genome and performed m6A peak detection (Fig EV3A–E; Materials and Methods). Yeast m6A peak lengths (median = 198 nt, s.d. = 222) were similar to those previously published in the REPIC database (Liu et al, 2020c; Fig EV3B) and were significantly shorter compared with planarian m6A peaks detected in this experiment (median 298 nt, s.d. = 222; Student's t‐test = 2.2E‐16). The distribution of planarian m6A peaks was similar to our initial mapping experiment (Fig EV3C) and longer than observed in other organisms.
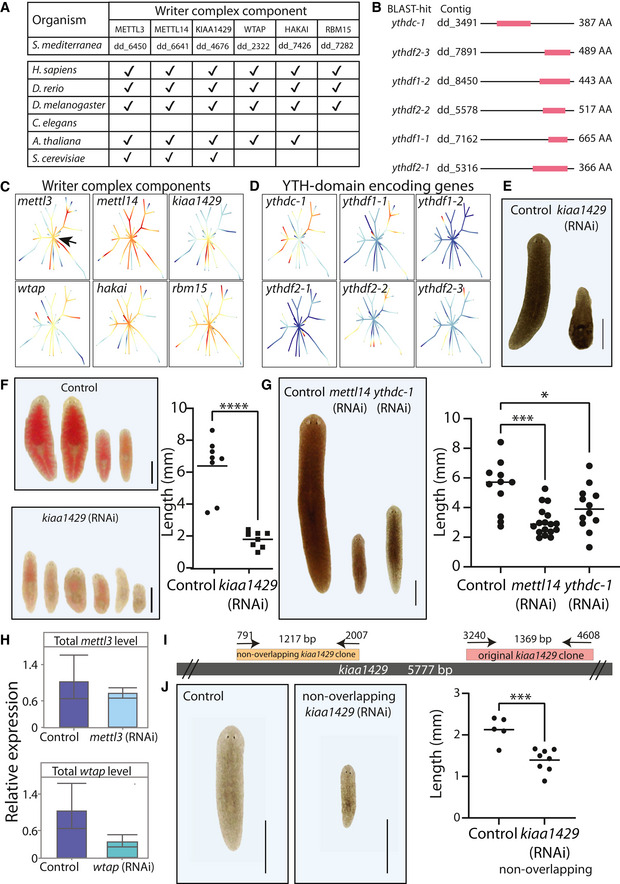
Figure 2. Transcriptome‐wide mapping of m6A in planarians.
- Outline of mapping of m6A on planarian RNA. RNA was extracted from control or kiaa1429 (RNAi) animals in triplicates, with at least 10 animals in each replicate (i). Then, RNA was molecularly tagged with a barcoded RNA oligo (ii). Samples of barcoded RNA were mixed, and methylated RNA was enriched using an anti‐m6A‐antibody; input samples were sequenced without anti‐m6A‐antibody enrichment (iii). RNA was used for RNAseq library preparation, and resultant libraries were sequenced. Sequencing data was mapped to the planarian genome, and m6A‐enriched regions (peaks) were detected using MeTPeak (Cui et al, 2016); read coverage across Smed‐ast8 is shown as normalized RPKM that was scaled to 0–1 across samples (iv).
- m6A peaks are highly enriched at the 3′ end of the transcript. X‐axis shows the relative position to the transcription start site (TSS); transcript lengths were normalized to 1,000 nt. Y‐axis is the average peak expression (normalized by RPKM) across 740 high‐confidence peaks (≥10 fold‐enriched). Plot produced using the deepTools2 package (Ramírez et al, 2016) (Materials and Methods).
- The number of m6A peaks, which were determined using MetPeak (Cui et al, 2016), is shown (top) as a function of gene expression in each bin (bottom, box plot).
- The distribution of the m6A peak length as detected by MeTPeak; peak length was limited to up to 1,000 nt for visualization. Black line represents the median peak length. Processed data for organisms other than planarian were obtained from REPIC (Liu et al, 2020c).
Figure EV2. m6A is abundant on planarian mRNA and depleted following kiaa1429 RNAi.
- The number of RNAseq reads for each m6A‐seq2 pulldown library per 10 M of the corresponding input reads is shown. The number of reads in each pulldown library in the m6A‐seq2 protocol correlates with the abundance of m6A (Dierks et al, 2021) (red and blue dot, control and kiaa1429 (RNAi) sample, respectively. ***Student's t‐test P < 0.001).
- The number of m6A‐rich regions (peaks) was larger in control samples and depleted following inhibition of kiaa1429. Moreover, the fold‐enrichment of the peaks in the control samples over the gene expression observed in the input samples is greater, in comparison to the kiaa1429 (RNAi) samples.
- A 2d‐density plot showing the correlation between m6A‐enriched regions in control and kiaa1429 (RNAi) animals. The density plot shows that m6a peaks were more highly enriched in the control samples compared to kiaa1429 (RNAi) samples, demonstrating the depletion of m6A following inhibition of kiaa1429.
- Profile of m6A‐enriched regions across genes in control and kiaa1429 (RNAi) showed enrichment toward the 3′‐end. The length of transcripts with detectable m6A‐enriched regions was normalized to 1,000 nt. Then, the expression across the transcript was computed by generating bins of gene expression (Materials and Methods) and calculating the log‐fold change between the anti‐m6A‐antibody pulldown library and the input sample. K‐means was used to separate three profiles of m6A‐enriched regions.
- Shown is the RPKM normalized expression of the planarian rRNA 28S (block arrow) in the m6A pulldown library and in control. There was no detectable m6A peak on planarian rRNA.
Figure EV3. Analysis of m6A peak length by species mixing experiment.
-
ARNA was extracted (i) from control or (ii) kiaa1429 (RNAi) animals in duplicates, with at least 10 animals in each replicate, (iii) from meiosis‐blocked S. cerevisiae strain with high levels of m6A (Ndt80Δ/Δ) (Dierks et al, 2021), and (iv) from S. cerevisiae strain devoid of m6A, due to deletion of the methyltransferase (Ime4Δ/Δ/Ndt80Δ/Δ). RNA was combined into two pools (Materials and Methods), and was processed according to the m6A‐seq2 protocol (Dierks et al, 2021). Following sequencing, reads were associated with the original samples based on their 3′‐end barcode (red and blue, representing mix #1 and mix #2, respectively), and then based on their mapping to either the planarian transcriptome or the yeast genome (Materials and Methods).
-
BViolin plot comparing the length of m6A peaks in yeast from either the REPIC database (Liu et al, 2020c), or from the data collected in this experiment; m6A regions larger than 1,000 bp were excluded from the plot, horizontal line indicates the median length.
-
CComparison of planarian m6A peaks in our initial m6A‐seq2 experiment, and in the m6a‐seq2 profiling in the species RNA mixing experiment. Shown are peaks shorter than 1,000 bp. To avoid comparison of lowly enriched peaks, shown are peaks with fold‐enrichments that are larger than the median fold‐enrichment.
-
D, EShown is the most frequent sequence motif detected in our mixed m6A‐seq2 experiment (Materials and Methods), as detected by HOMER (Heinz et al, 2010) for yeast (D) and planarian (E).
-
F, GShown are the fold change and associated P‐values for each k‐mer. DRACH‐like k‐mers are colored as in the figure legend. Data is shown for yeast (F; panels represent two biological replicates) and planarian (G; panels represent two biological replicates).
The increased length of planarian m6A peaks rendered the detection of the m6A installment motif DRACH (D = A/G/U; R = A/G; H = A/C/U) (Linder et al, 2015) challenging. We therefore used high‐confidence m6A‐enriched regions shorter than 250 bp (Fig EV3D–G; Materials and Methods; top 500 enriched peaks). Using this approach, CGACG, which is similar to the DRACH motif, was found to be the most enriched sequence. These results were recapitulated on the basis of a separate analysis pipeline (Schwartz et al, 2014; Dierks et al, 2021), which we applied to both yeast and planarian data. This analysis also revealed an enrichment for “GAC” harboring motifs in planarians and the GGACA motif in yeast (Fig EV3F and G). Thus, m6A maps in planarians exhibit many of the classical hallmarks of m6A: they are enriched toward the ends of genes, depleted from highly expressed genes, are enriched in a “GAC” motif, and are highly depleted upon inhibition of kiaa1429. The increased length of planarian m6A‐enriched regions may suggest that planarian m6A sites are found in spatial proximity in RNA molecules, and higher‐resolution mapping of m6A sites could be used to further test this hypothesis.
m6A genes regulate neoblast gene expression
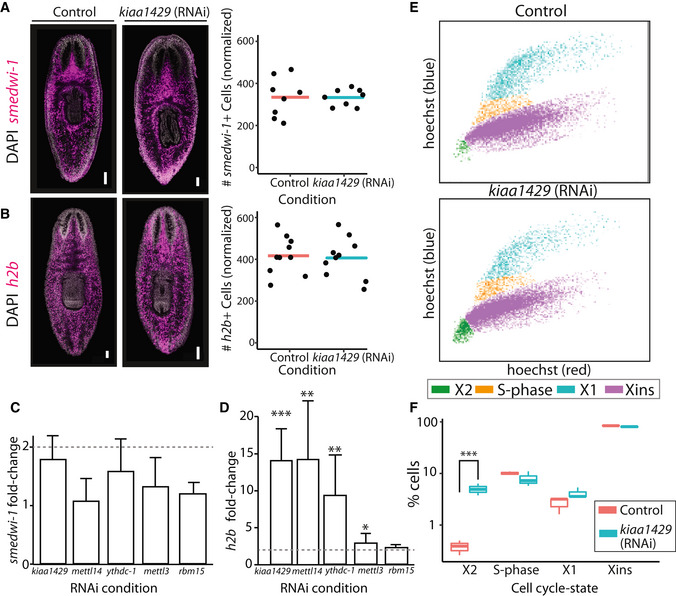
The animal size reduction and lysis following inhibition of expression of m6A genes suggested that the animals were unable to produce cells normally. Failure to maintain and generate tissue is often a consequence of neoblast depletion or failure of neoblasts to differentiate (Reddien et al, 2005; Lin & Pearson, 2014; Zhu et al, 2015). We tested whether neoblasts were depleted following m6A‐pathway inhibition by counting neoblasts by fluorescent in situ hybridization (FISH; Materials and Methods) using two canonical neoblast markers, smedwi‐1 (Reddien et al, 2005) and h2b (Guo et al, 2006). The number of smedwi‐1 + or h2b + cells per unit area was similar following inhibition of kiaa1429 gene expression (Fig 3A and B). Therefore, the phenotypes were not a consequence of neoblast depletion. Our FISH approach allowed counting neoblasts using neoblast markers, but not to measure gene expression levels. We used quantitative reverse transcription PCR (qPCR) for measuring smedwi‐1 and h2b expression level following RNAi on polyadenylated RNA (Fig 3C and D; Materials and Methods). We saw no significant change in smedwi‐1 gene expression in all of the tested RNAi conditions. By contrast, we found a significant upregulation of up‐to 14‐fold in h2b expression following inhibition of m6A genes. h2b was overexpressed even in conditions that did not produce a lysis phenotype (Fig 3D). The overexpression of h2b suggested that changes to neoblast gene expression preceded the development of the RNAi phenotype.
Figure 3. m6A pathway expression is not required for neoblast viability.
-
A, BThe number of neoblasts did not change significantly following inhibition of the MTC‐gene kiaa1429. Shown is FISH of smedwi‐1 (A) and h2b (B) in control and kiaa1429 (RNAi) animals (left). smedwi‐1 + or h2b + cells were counted following FISH and showed no significant difference in expression (right; bar represents average normalized cell count; Materials and Methods). Scale = 100 μm.
-
C, DGene expression analysis of smedwi‐1 (C) and h2b (D). The expression of smedwi‐1 and h2b was determined in the different RNAi conditions compared to the expression of these genes in control animals (Materials and Methods). Dashed line marks a twofold change in expression. Measurements performed on biological triplicates. P‐value was calculated by two‐sided Student's t‐test followed by Bonferroni correction for multiple hypotheses. Lack of asterisk indicated a non‐significant change in gene expression. Error bars indicate the 95% confidence interval (***P < 0.001, **P < 0.01, *P < 0.05).
-
ERepresentative FACS plots of cells isolated from control (top) and kiaa1429 (RNAi) animals (bottom) show an increase in cell abundance in the X2 gate (green; Materials and Methods).
-
FQuantification of the cells in each FACS gate in control and kiaa1429 (RNAi) animals showed a sixfold increase in the abundance of cells in the X2 gate (Student's t‐test ***P < 0.005). FACS experiments were performed in biological triplicates. Boxes represent the IQR, whiskers represent the 1.5 × IQR, and central band represents the median.
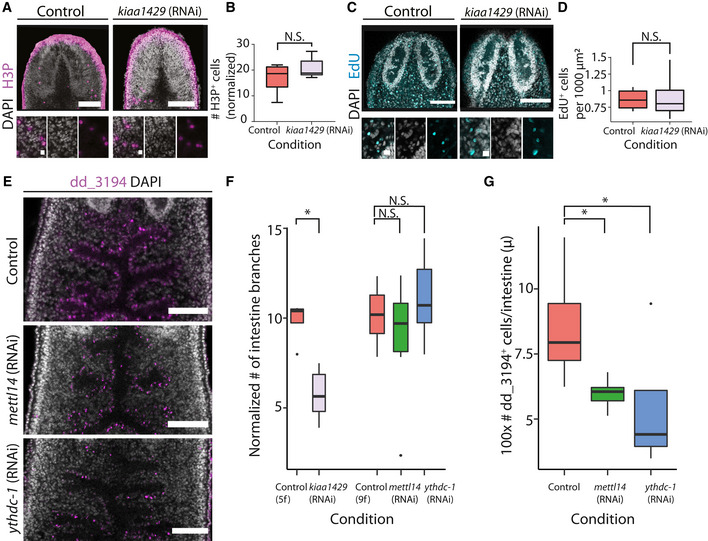
Expression of h2b is a hallmark of neoblast cell‐cycle progression (Guo et al, 2006; Wenemoser & Reddien, 2010; Solana et al, 2012). We therefore tested whether the expression of m6A genes was required for normal cell cycle regulation. The relative abundance of cell cycle states was analyzed by fluorescence‐activated cell sorting (FACS) of Hoechst‐labeled planarian cells from control and kiaa1429 (RNAi) animals (Fig 3E and F; Materials and Methods). G1 neoblasts and recently divided post‐mitotic progenitors are found specifically in a FACS gate called X2 (Hayashi et al, 2006). Our FACS analysis (Fig 3E and F) revealed a sixfold increase in the number of cells in the X2 gate following kiaa1429 RNAi and more minor changes in the number of cells in the G2/M gate (X1) or in fully differentiated cells (Xins). The increase in the X2 cell population could be a consequence of an increase (i) in G1 neoblasts; (ii) in post‐mitotic progenitors; or (iii) in both. We considered these options. The FACS analysis revealed that there was a minor change in the abundance of S‐phase and G2 cells (Fig 3E and F). In addition, FISH revealed that there was no absolute change in neoblast number following kiaa1429 (RNAi), as determined by smedwi‐1 + or h2b + cell labeling (Fig 3A). Taken together, these data suggested that the population size of G1/S/G2 neoblasts did not change dramatically. We further tested this hypothesis using two approaches. First, we used an anti‐H3P antibody to label mitotic cells and counted H3P+ cells found in a defined region anterior to the pharynx (Fig EV4A and B; Materials and Methods). We found no significant increase (P = 0.59) in the number of H3P+ cells following inhibition of kiaa1429, which indicated that the neoblast population mitotic rate is unperturbed. In addition, we estimated the number of cells going through S‐phase in kiaa1429 (RNAi) and control animals by metabolic labeling using the thymidine analog, F‐ara‐EdU. Animals were soaked in F‐ara‐EdU for 16 h and were then immediately fixed for further processing. Following F‐ara‐EdU detection, the F‐ara‐EdU+ cells were counted in a ventral sub‐epidermal layer that is distinguishable from the intestine (Fig EV4C and D; Materials and Methods). We found no significant difference in the number F‐ara‐EdU+ cells between the kiaa1429 (RNAi) and control animals, which further indicated that in the conditions tested here, there was no detectable defect in neoblast cell cycle progression. Therefore, the increase in X2 cell abundance was most likely a consequence of an increase in the number of immature smedwi‐1 − post‐mitotic progenitors, and not of G1 neoblasts, which are smedwi‐1 +.
Figure EV4. Reduction in intestinal cells following inhibition of m6A genes.
-
A, BShown is H3P labeling using an anti‐H3P‐antibody in kiaa1429 (RNAi) and in control animals (scale = 100 and 10 μm, top and bottom panels, respectively; top panels show a maximal intensity projection of the H3P and DAPI signal; Materials and Methods). (B) The number of H3P+ cells was counted in a region anterior to the pharynx and posterior to the brain, in a z‐stack and was normalized to the area of the counting (Materials and Methods). There was an insignificant difference in the number of H3P+ cells between the conditions (Student's t‐test; n = 6 biological replicates). Boxes represent the IQR, whiskers represent the 1.5 × IQR, and central band represents the median.
-
C, DShown are confocal images of kiaa1429 (RNAi) and control animals, which were soaked in F‐ara‐EdU for 16 h (scale = 100 and 10 μm, top and bottom panels; Materials and Methods). (D) The number of F‐ara‐EdU+ cells was counted in a region anterior of the pharynx and posterior to the brain and was normalized to the rectangle size (Materials and Methods). There was an insignificant difference in the number of F‐ara‐EdU+ cells between the tested conditions (Student's t‐test; n = 5 biological replicates). Boxes represent the IQR, whiskers represent the 1.5 × IQR, and central band represents the median.
-
E–GThe effect of inhibition of m6A pathway‐encoding genes on the intestine integrity and cells was examined by FISH. (E, F) Inhibition of mettl14 (RNAi) or ythdc‐1 (RNAi) by 9 dsRNA feedings did not result in significant differences in the morphology of the intestine, as estimated by counting the number of intestine branches in confocal images (Materials and Methods). A significant difference in the number of intestine branches was observed following kiaa1429 (RNAi) following five RNAi feedings compared to its control. Scale = 100 μm. (E, G) A significant reduction in the number of intestine cells labeled by dd_3194 was found following either mettl14 (RNAi) or ythdc‐1 (RNAi) (two‐sided t‐test controlled by Bonferroni's correction). The dd_3194+ cells were counted in confocal images and the counting was normalized to the length of the intestine branches (Materials and Methods). *P < 0.05.
m6A genes regulate cell‐type‐specific gene expression programs
The increase in post‐mitotic progenitors following the inhibition of m6A pathway components may have affected multiple cellular lineages and gene expression programs. We profiled the emergence of gene expression changes following inhibition of mettl14 and ythdc‐1 after four, six, and eight RNAi feedings (Fig 4A; Dataset EV2; Materials and Methods). The phenotype of kiaa1429 (RNAi) animals developed more rapidly, and therefore, we measured gene expression changes only following five RNAi feedings (Fig 4B). The gene expression changes following mettl14 (RNAi) and ythdc‐1 (RNAi) were remarkably similar, with correlation >0.7 between all time points (Fig 4A–C). Comparison of mettl14 (RNAi) or ythdc‐1 (RNAi) with kiaa1429 (RNAi) resulted in high correlation as well, 0.56 and 0.46, with mettl14 (RNAi) or ythdc‐1 (RNAi), respectively (Appendix Fig S1A and B), despite the time point differences associated with the rapid development of the kiaa1429 (RNAi) phenotype. The sequencing validated the specificity of our RNAi and corroborated our qPCR results (Fig 4A and B; Appendix Fig S1C–F). This indicated that the phenotypes were a consequence of m6A depletion and not m6A‐independent functions of the MTC.
Figure 4. Gene expression changes following inhibition of m6A genes.
-
A, BHeatmap of genes that were differentially expressed following inhibition of m6A genes. Shown is the expression of the top 25 overexpressed and 25 underexpressed genes (top and bottom, respectively; Dataset EV2) following six mettl14 dsRNA feedings, at different conditions and time points. Rows and columns represent genes and samples, respectively. Blue and red, low to high gene expression (row‐normalized z‐score). Genes that were previously determined to be expressed in the intestine (Fincher et al, 2018) were highlighted in dark blue, or otherwise in dark red.
-
CCorrelation of gene expression changes between mettl14 (RNAi) and ythdc‐1 (RNAi) compared to their controls. Each colored dot represents a gene (expression >1 transcripts per million, TPM; red and black, significant and nonsignificant change in gene expression compared to controls, respectively).
-
DThe proportion of genes that were downregulated following RNAi and were found to be expressed in a single cell type. Shown is an analysis based on the top 300 genes that were downregulated following RNAi (six dsRNA feedings for mettl14 or ythdc‐1).
-
EFISH for detection of major planarian cell types following inhibition of kiaa1429 by RNAi using previously established cell‐type specific gene expression markers (Fincher et al, 2018). The distribution of most major planarian cell types was comparable between the control (top) and kiaa1429 (RNAi) animals (bottom). A striking exception was the reduction in intestine cells (right; see Fig EV4E–G for quantification), which severely affected the intestine branching morphology in kiaa1429 (RNAi) animals. Scale = 100 μm.
The presence of m6A has been previously associated with a high transcript turnover rate (Lee et al, 2020; Zaccara & Jaffrey, 2020). Therefore, we predicted that inhibition of the m6A pathway would result in an overexpression of transcripts at early stages of the phenotype emergence. Indeed, following four RNAi feedings, most differentially expressed genes were upregulated: 77 and 83% for mettl14 (RNAi) and ythdc‐1 (RNAi), respectively (Appendix Fig S1G; Dataset EV2). By contrast, at late time points, most differentially expressed genes were downregulated (Appendix Fig S1G), which likely reflected the indirect effects of m6A gene inhibition, such as depletion of a cell type.
We annotated differentially expressed genes using the planarian cell‐type transcriptome atlas (Fincher et al, 2018). The majority of the upregulated genes following RNAi were not cell‐type‐specific (Dataset EV2), nor did they share a common function (Rozanski et al, 2019) or have association with the injury response (Wurtzel et al, 2015). By contrast, a major fraction of the downregulated genes were cell‐type specific and were normally expressed in the planarian intestine and intestine‐associated neoblasts (Fincher et al, 2018; Plass et al, 2018; Fig 4A, B and D). For example, the transcription factor nkx‐2.2, which is required for specification of intestine cells (Forsthoefel et al, 2012), was downregulated following RNAi, and similarly, activin‐1 expression was reduced. The reduction in intestine‐specific gene expression was observed already following four RNAi feedings, but it was exacerbated at later time points (Fig 4A and B; Dataset EV2).
To test whether the reduction in intestinal gene expression represented downregulation of a gene expression program or a reduction in intestine cell number, we analyzed by FISH kiaa1429 (RNAi), mettl14 (RNAi), and ythdc‐1 (RNAi) and control animals using a riboprobe labeling the planarian intestine (Figs 4E and EV4E–G). We found a severe defect in intestinal branching morphology following kiaa1429 (RNAi) (Figs 4E and EV4F, Materials and Methods), which suggested that the RNAi resulted in depletion of intestine cells. Indeed, following inhibition of kiaa1429, animals did not uptake food (Fig EV1F). Inhibition of mettl14 (RNAi) and ythdc‐1 (RNAi) resulted in a significant decrease of 43% and 57% in the number intestinal cells, respectively, without loss of intestine morphology (Fig EV4E–G) at the time point tested (Bonferroni corrected Student's t‐test P < 0.05 for both conditions). This result was consistent with the observation that inhibition of kiaa1429 resulted in a more rapidly developing phenotype. To test whether there was a major depletion in other cell types, we performed FISH using five other cell type markers for muscle, neuron, epidermal lineage, cathepsin + cells, and protonephridia on kiaa1429 (RNAi) animals (Fig 4E). The general defect in kiaa1429 (RNAi) body morphology (Fig 1B) limited the ability to quantify the abundance of individual cell types. Yet, cell‐type‐specific FISH labeling demonstrated that all major cell types were distributed across the animal, with the exception of intestine cells (Fig 4E). Importantly, cell types and lineages other than the intestine might have been affected by the RNAi, yet other factors, such as the cell‐type‐specific turnover rate, might have caused the intestine to develop a significant defect more rapidly.
m6A indirectly represses polyadenylated histone gene expression and silences repetitive DNA
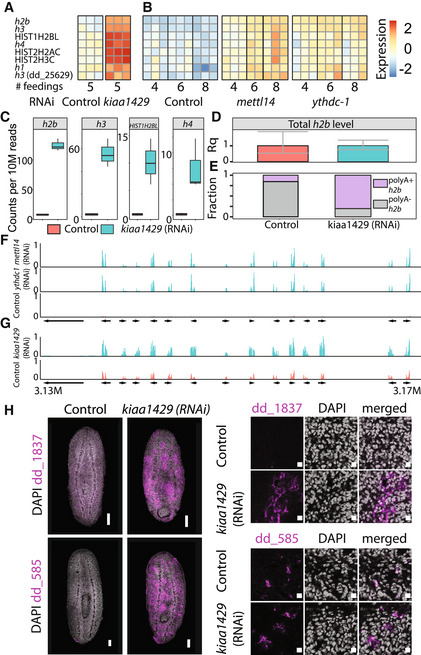
The reduction in intestine cells together with the increase in the immature progenitor population that we observed by FACS in kiaa1429 (RNAi) animals suggested that there was a defect in cell production or maturation. We examined the identity of the overexpressed genes in our RNAseq data, which could potentially explain the overabundance of immature progenitors. Previous analysis of cycling planarian cells shows that histone gene expression changes dramatically during neoblast specialization (Labbé et al, 2012). We found that many histone‐encoding genes were overexpressed, indicating a broad change to neoblast cell state. For example, seven and eight such genes were upregulated by over twofold in kiaa1429 (RNAi) and mettl14 (RNAi) animals, respectively (Fig 5A and B; Dataset EV2). Particularly, h2b and h3 were strongly overexpressed in all conditions (Fig 5A and B; Appendix Fig S1E).
Figure 5. Overexpression of polyadenylated histone‐encoding transcripts.
-
A, BHeatmap showing eight histone‐encoding genes that were upregulated following inhibition of m6A‐pathway‐encoding genes. Rows and columns represent genes and samples, respectively. Blue and red, low to high gene expression (row‐normalized z‐score).
-
CBoxplots showing the normalized number of reads that map to a histone gene body and are paired with a read pair containing a polyA (n = 3 biological replicates). Inhibition of kiaa1429 resulted in a dramatic increase in the mapping to polyA in histone‐encoding genes. Boxes represent the IQR, whiskers represent the 1.5 x IQR, and central band represents the median.
-
DQuantification of the entire h2b transcript pool including polyA+ and polyA− transcripts using qPCR (Materials and Methods). No significant change in the total h2b expression was observed following inhibition of kiaa1429. Error bars represent the 95% confidence interval. Experiment performed in biological triplicates.
-
EComparison of the polyA+ and polyA− h2b transcripts showed an overabundance of polyA+ h2b in kiaa1429 (RNAi) animals compared with controls.
-
F, GShown is a summary of gene mapping to a representative overexpressed gene neighborhood (contig: dd_Smes_g4_15). Arrows represent gene annotation, which was extracted from PlanMine (Rozanski et al, 2019). Panels show expression in TPM scaled to 0–1.
-
HThe overexpression of two representative genes from the upregulated genomic neighborhoods was validated by FISH. Cells expressing these genes were found throughout the animal in sub‐epidermal and parenchymal layers (Left, Scale = 100 μm); higher magnification shows perinuclear expression (right, scale = 10 μm).
Canonical histone‐encoding genes are transcribed without a polyA tail (Dávila López & Samuelsson, 2008). Our RNAseq library preparation protocol selects polyadenylated RNA by using poly‐dT beads (Materials and Methods). Therefore, non‐polyadenylated RNA should have been vastly excluded from the RNAseq libraries. We hypothesized that the high prevalence of histone‐encoding gene transcripts might have represented an aberrant expression of polyadenylated histone isoforms, which were undetectable when the m6A pathway functioned normally.
We tested this hypothesis by searching for polyadenylated histone transcripts in the RNAseq data. Our kiaa1429 (RNAi) and control libraries were sequenced in a paired‐end configuration (Appendix Fig S2A). We reasoned that if kiaa1429 inhibition resulted in activation of polyadenylated histone transcription, we could detect RNAseq read pairs, where the first read in the pair mapped to a histone gene body, and the second read of the pair had a sequence that includes a polyT, which is the reverse‐complement of the polyA tail (Appendix Fig S2A; Materials and Methods). By contrast, in control samples, we expected to find fewer read pairs of this kind (Fig 5C; Appendix Fig S2A–C). Indeed, polyadenylated histone transcripts were detectable almost exclusively in kiaa1429 (RNAi) RNAseq compared with the control (Fig 5C). For example, we found a >17‐fold increase in polyA read mapping to h2b following kiaa1429 (RNAi).
The overexpression of polyadenylated histone RNAs following kiaa1429 (RNAi) could represent an absolute increase in histone gene expression, or alternatively, a relative increase in polyadenylated histone transcripts compared with non‐polyadenylated transcripts. We compared the total expression of h2b in kiaa1429 (RNAi) and control animals using cDNA produced with random hexamers, which were used to reverse transcribe both polyadenylated and non‐polyadenylated RNAs. We found that h2b expression levels were essentially identical (Fig 5D; Materials and Methods). This result indicated that the observed increase in h2b expression in the RNAseq data resulted from presence of polyadenylated h2b transcripts and not from an absolute increase in h2b gene expression.
We estimated the proportion of polyadenylated h2b transcripts in RNA extracted from control or kiaa1429 (RNAi) animals. We used cDNA produced with either random hexamers or a poly‐dT primer and validated that cDNA production using both methods was similarly efficient (Appendix Fig S2D and E). In control animals, 15.4% of the h2b transcripts were polyadenylated. By comparison, following RNAi of kiaa1429 80.2% of the h2b transcripts were polyadenylated (Fig 5E; Appendix Fig S2F and G). The shift toward production of polyA+ histone transcripts might reflect changes to cell cycle state, which is often associated with histone transcription (Duronio & Marzluff, 2017; Marzluff & Koreski, 2017). Importantly, we did not detect peaks of m6A in h2b and h3 transcripts (Dataset EV1), which suggested that m6A was not a direct regulator of polyadenylated histone gene expression.
The majority of overexpressed genes following inhibition of the m6A pathway were not associated with a distinct function (Fig 4A and B; Dataset EV2). However, some of the most overexpressed transcripts were annotated as pseudogenes (Dataset EV2). We mapped the RNAseq data to the planarian genome (Rozanski et al, 2019) and inspected regions containing these overexpressed genes. Remarkably, overexpressed genes were highly enriched within genomic clusters (Fig 5F and G). These gene clusters were not expressed in control samples, but were dramatically induced following RNAi (Fig 5F and G; Dataset EV2). Importantly, the intergenic regions in these genomic neighborhoods were almost completely silenced, and therefore, this overexpression was not a consequence of lack of transcriptional regulation over the entire genome segment (Fig 5F and G). Several genes in these clusters had detectable m6A‐enriched regions in our m6A‐seq2 datasets (Dataset EV1) and encoded ubiquitin‐like domain (Dataset EV2). We searched for similar sequences in other planarian transcriptomes (Rozanski et al, 2019) using BLAST and found similar sequences in the Schmidtea polychroa transcriptome, but not in other planarian species. We cloned two of these overexpressed genes and performed FISH on kiaa1429 (RNAi) and control animals (Fig 5H). FISH showed that these genes were indeed expressed only following kiaa1429 RNAi, validating that expression of m6A genes was required for their silencing. We hypothesized that nuclear packaging might be altered. Analysis of nuclear labeling by DAPI of kiaa1429 (RNAi) and control animals indicated that chromatin density was indeed different (Appendix Fig S3H), with extraneous unlabeled nuclear regions, suggesting a change to nuclear packaging. In summary, inhibition of m6A genes resulted in broad changes to gene expression, including a coordinated overexpression of gene clusters that are normally silenced, together with an overabundance of polyadenylated histone transcripts, indicating an emergence of irregular cell states following inhibition of m6A genes.
m6A pathway activity regulates cellular maturation
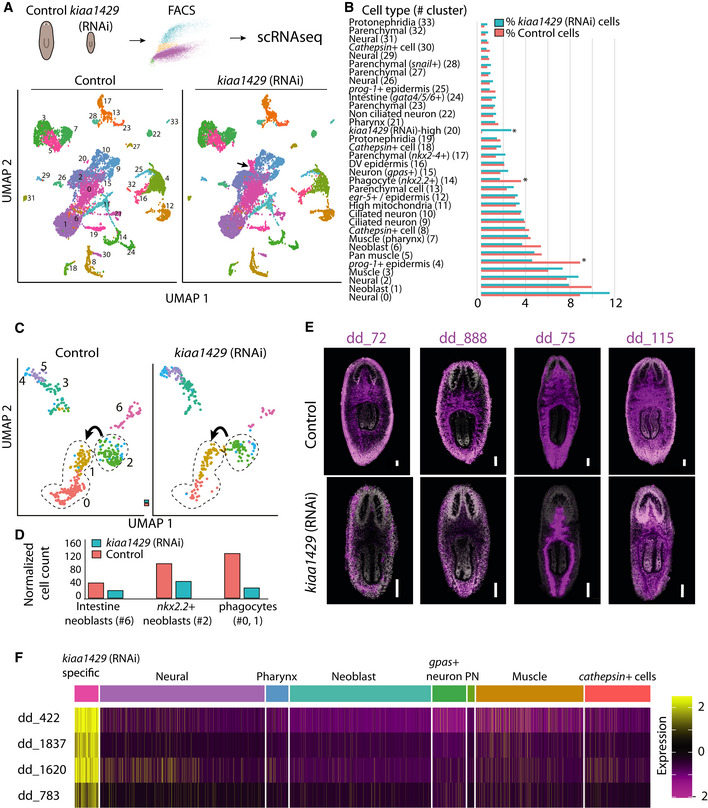
Our results indicated that m6A genes had different roles in different cell populations. We dissected the cell‐type‐specific roles of m6A genes by using scRNAseq of >20,000 cells isolated from control and kiaa1429 (RNAi) animals (Figs 6A and EV5A–D; Materials and Methods). Then, we used the Seurat framework (Stuart et al, 2019) and the planarian cell type atlas (Fincher et al, 2018; Plass et al, 2018) for assigning cell type identities for all of the sequenced cells (Dataset EV3). We assigned the cells to 34 major groups, annotated the major planarian cell types, and analyzed gene expression changes following kiaa1429 (RNAi) for each cell type (Fig 6A and B; Dataset EV3). We focused on several aspects of the m6A depletion phenotype: (i) changes to neoblast gene expression and state; (ii) molecular characterization of the cells expressing the repetitive gene clusters; and (iii) the effect of the RNAi on intestine cells.
Figure 6. Cell type‐specific regulation of gene expression by m6A.
-
AOutline of scRNAseq experiment. Cells were isolated from dissociated animals, purified by FACS and subjected to scRNAseq (top; Materials and Methods). Analysis of cell populations detected in the scRNAseq and visualized by UMAP (dot represents cell, and color represents cluster association). Comparison of scRNAseq of kiaa1429 (RNAi) and control revealed an expansion of a molecularly defined cell cluster (#20, black arrow).
-
BRelative representation of clusters between kiaa1429 (RNAi) and control animals. Most clusters were similarly represented between samples. However, depletion of phagocytic intestine cells, epidermal progenitors, and expansion of a kiaa1429 (RNAi) cell cluster was detected (*>90% difference in detectable the relative cell population size between conditions).
-
C, DAnalysis of cells from the intestine lineage showed depletion in intestine neoblasts (gamma, #6), nkx‐2.2 + progenitors (#2), and differentiated intestine cells (#0, #1). Arrow indicates transition to differentiated cell state. Normalized cell count of intestine lineage cells are summarized (bottom).
- E
-
FscRNAseq heatmap produced using Seurat (Stuart et al, 2019) of four representative genes from the kiaa1429 (RNAi)‐overexpressed genomic clusters. The major planarian cell types did not express these four genes. By contrast, a kiaa1429 (RNAi)‐specific cell cluster is characterized by high expression of these genes. Rows and columns represent genes and cells, respectively. Purple to yellow, low to high gene expression. Top row shows assignment of cells to cell types based on gene expression (Dataset EV3).
Figure EV5. Analysis of kiaa1429 (RNAi) phenotypes by scRNAseq.
-
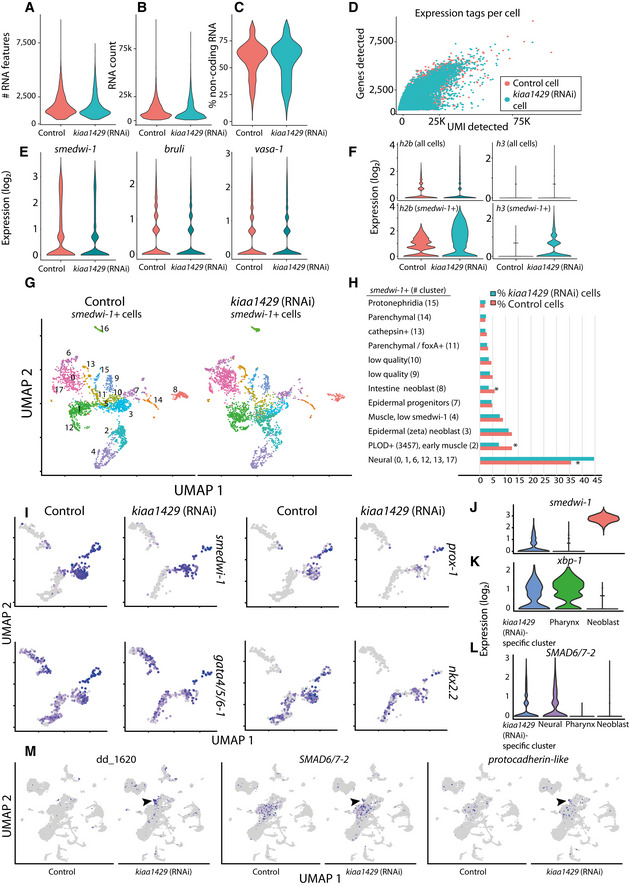
A–DQuality measurements of scRNAseq libraries prepared from control and kiaa1429 (RNAi) cells. The quality of the libraries produced from both conditions was assessed using the Seurat package (Stuart et al, 2019). The number of expressed genes (A); unique molecular tags (B); non‐coding gene expression (C); and the correlation between the number of expressed genes and the unique molecular tags were highly similar between the libraries (D). This indicated that the quality of libraries was comparable.
-
EExpression levels of canonical neoblast markers were highly similar in control and kiaa1429 (RNAi).
-
FThe polyadenylated transcript expression of h2b and h3 was much higher in kiaa1429 (RNAi) animals compared to controls. The expression was detectable in neoblasts (bottom), and to a much lesser degree in the entire cell population (top), which includes neoblasts as well.
-
G, HUMAP representation of neoblasts (color dots, left panel), and their identity (right panel). Neoblast identity was assigned based on expression of previously published gene expression markers (Fincher et al, 2018). Most neoblast clusters were not affected by the RNAi, yet several lineages (right, asterisk) were differentially represented following kiaa1429 (RNAi).
-
IExpression of neoblast (smedwi‐1) and specialized intestine neoblast gene expression markers was overlaid on UMAP plots of the intestine lineage (cells represented by dots; gray and purple, low to high ranked expression).
-
J–LComparison of gene expression between several cell clusters. Importantly, pharynx cells are post‐mitotic, and therefore show minimal smedwi‐1 expression (J). Post‐mitotic cells express xbp‐1, which was previously shown to be expressed in differentiating and differentiated cells (Raz et al, 2021) (K). SMAD6/7–2 is expressed in the kiaa1429 (RNAi) specific‐cluster and in neural cells (L).
-
MHighly expressed genes in the kiaa1429 (RNAi)‐specific cluster compared to control are shown in UMAP plots. Contig dd_1620 is highly expressed in the kiaa1429 (RNAi)‐specific cluster. The genes SMAD6/7–2 and protocadherin‐like (dd_15376, gene model SMESG000067388) are expressed in the kiaa1429 (RNAi)‐specific cluster (black arrowhead); both genes are also expressed in cells of the neural clusters (e.g., cluster 2).
The number of smedwi‐1+ cells was similar in control and kiaa1429 (RNAi) animals. Expression of canonical neoblast genes, such as smedwi‐1+, Smed‐vasa, and Smed‐bruli, was comparable (Fig EV5E; Dataset EV3) indicating that the neoblast population indeed remained intact. By contrast, polyadenylated histone transcripts were broadly overexpressed in highly expressing smedwi‐1 cells following kiaa1429 inhibition (Fig EV5F), suggesting a broad impact of the RNAi on the neoblast population transcriptional state. Neoblasts express lineage‐specific genes during differentiation (Reddien, 2013, 2018). We reclustered the neoblasts and compared lineage‐specific gene expression in control and kiaa1429 (RNAi) cells (Fig EV5G and H). We found two neoblast populations that were reduced by ~40%: intestine‐producing neoblast (nkx‐2.2 +) and PLOD + neoblasts (Fig EV5G and H), a class of neoblast that is transcriptionally associated with planarian muscle (Fincher et al, 2018). By contrast, there was an increase of 39–63% in the number of neural‐associated neoblasts, including in the tspan‐1+ neoblasts (Zeng et al, 2018; Raz et al, 2021; Fig EV5G and H). These results suggested that m6A is required for lineage resolution, which if correct would impact the representation of differentiated cell populations. Examination of differentiated cells showed that several lineages were depleted in kiaa1429 (RNAi) animals (Fig 6B; Dataset EV3). The strongest reduction in lineage representation (≥40%) was detected for intestinal phagocytes, consistent with the reduced representation of intestinal gene expression in bulk RNAseq (Fig 6C and D, and 4A and B, Chi‐squared P < 1E‐15). Using FISH, we saw a reduction in multiple intestinal cell types (Fig 6E; Appendix Fig S3A) and disruption to intestinal morphology (Fincher et al, 2018; Forsthoefel et al, 2020). We quantified the reduction of the intestine markers dd_72, dd_115, and dd_888 (Fig 6E; Appendix Fig S3A) by FISH and found a highly significant reduction in the number of dd_72+ and dd_115+ cells (Appendix Fig S3A), with only qualitative difference in the expression of dd_888. The intestine lineage was depleted at all stages of the lineage in the scRNAseq data (Figs 6C and D, and EV5I; Chi‐squared P < 0.001; corrected with Bonferroni's correction), starting at the intestine neoblasts (gamma), continuing with nkx‐2.2 +/smedwi‐1 + progenitors, and finally with the intestine phagocytes (Figs 6C and D, and EV5I). The dramatic decrease in differentiated cells (~75%) following inhibition of kiaa1429, in comparison to a smaller decrease in intestine neoblasts (~50%), might reflect an accumulated defect in cellular production. Indeed, the expression of intestine‐specific genes declined progressively in our RNAseq time courses (Fig 4A and B; Dataset EV2). We tested whether there were indeed less recently produced progenitors integrated to the intestine. Recently integrated intestine cells were detected by co‐labeling kiaa1429 (RNAi) and control animals with anti‐SMEDWI‐1 antibody and with a combination of FISH probes that detects intestine‐specific transcription factors (i.e., nkx‐2.2, gata4/5/6–1, and hnf4; intestine mix). SMEDWI‐1+/intestine mix+ cells were counted in the intestine anterior to the pharynx. We found a significant decrease of >50% (two‐way Student's t‐test P = 0.02) in the number of recently integrated progenitors normalized to the size of the animal (Appendix Fig S4B and C). Therefore, despite no detectable change in the number of mitoses, as indicated by H3P and F‐ara‐EdU labeling (Fig EV4A–D), there was a decrease in the number of newly integrated cells in the intestine.
The increase in neural‐associated neoblasts did not result in an overabundance of differentiated neural cells (Fig 6A and B; Dataset EV3). However, we identified a molecularly defined population, which was overrepresented by >27‐fold following kiaa1429 inhibition (Fig 6B). This kiaa1429 (RNAi)‐specific cell cluster had characteristics of post‐mitotic progenitors: (i) low‐level, but detectable, expression of smedwi‐1 (Fig EV5J); (ii) expression of genes associated with differentiation (e.g., xbp‐1, Fig EV5K) (Raz et al, 2021); and (iii) expression of lineage‐specific genes that are expressed early in differentiation (Fig EV5L; Dataset EV3). The scRNAseq analysis indicated that cells in the cluster highly expressed genes that are associated with neurons and glia, such as Smed‐smad6/7–2 and a protocadherin‐like gene (dd_15376, gene model SMESG000067388) (González‐Sastre et al, 2012; Fincher et al, 2018; Plass et al, 2018; Fig EV5L‐M). These kiaa1429 (RNAi)‐specific cells were distinguished from essentially all other cells by expression of the repetitive gene clusters (Fig 6F, Dataset EV3). This result is consistent with the FISH analysis, which demonstrated a body‐wide increase in cells expressing genes localized to the repetitive genomic clusters following inhibition of kiaa1429 (Fig 5H). Taken together, these results support the hypothesis that inhibition of the m6A pathway leads to a decrease in differentiation of several cellular lineages and the expansion of a molecularly defined progenitor population.
NuRD complex inhibition recapitulates the m6A inhibition phenotype
Multiple lines of evidence indicated that m6A gene expression was required for normal lineage choice and differentiation. m6A is a broad regulator of cellular function, and the inhibition of the m6A pathway could produce this phenotype by affecting other processes directly or indirectly. To better understand the mechanistic basis for the m6A phenotypes, we searched for conditions that induce similar changes to gene expression. We mapped 1,080 published planarian RNAseq libraries to the planarian transcriptome (Rozanski et al, 2019) and then compared gene expression changes in the published data with the RNAseq data that we generated following inhibition of the m6A genes (Appendix Fig S4; Materials and Methods; Table EV1).
Remarkably, this analysis revealed that inhibition of Smed‐CHD4, which encodes a canonical component of the Nucleosome Remodeling and Deacetylase complex (NuRD), recapitulated all of the major aspects of the m6A phenotype (Dataset EV4): (i) overexpression of the repetitive gene clusters that were identified following RNAi of m6A genes (Fig 7A); (ii) CHD4 RNAi resulted in overexpression of polyadenylated histone‐encoding transcripts (Fig 7B and D; Appendix Fig S5A), as we observed in kiaa1429 (RNAi) animals; (iii) The expression of smedwi‐1 was unperturbed, indicating that the neoblast population size was unchanged (Appendix Fig S5A); and (iv) Strikingly, CHD4 (RNAi) animals had reduced intestine gene expression, including transcription factors that are essential for intestinal cell differentiation, morphogenesis, and function (Forsthoefel et al, 2012, 2020), as we observed following inhibition of the m6A genes (Fig 7E). We inhibited a different NuRD complex‐encoding gene, RbAp48, which also resulted in an overexpression of polyadenylated h2b transcripts (Appendix Fig S5B). This further suggested that the molecular similarities detected following inhibition of m6A genes and CHD4 could be generalizable to other NuRD components, which is consistent with the roles of NuRD in transcriptional regulation (Basta & Rauchman, 2015). Interestingly, we analyzed previously published gene expression data from human and mouse of NuRD inhibition by shRNA or by conditional deletion and identified an overexpression of histone genes, such as h2b (Wilczewski et al, 2018; Marques et al, 2020; Appendix Fig S5C and D; Materials and Methods). This indicated that regulation of histone gene expression by CHD4 is found in diverse animals.
Figure 7. Inhibition of CHD4 recapitulates effects of the m6A‐pathway inhibition.
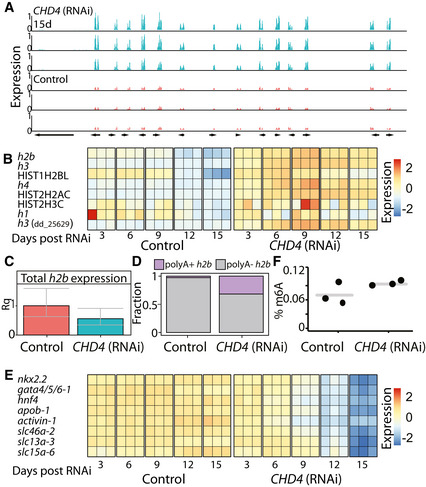
- Overexpressed gene neighborhood (contig: dd_Smes_g4_15:3.13–3.17 Mbp). Shown is the normalized and scaled gene expression in three CHD4 (RNAi) samples, 15 days post feedings (top, blue) and in three corresponding control samples (bottom, red).
- Heatmap showing the eight histone‐encoding genes that were upregulated following inhibition of CHD4, at different time points, in published dataset (Tu et al, 2015). Rows and columns represent genes and samples, respectively. Blue and red, low to high gene expression (row normalized z‐score).
- Quantification of the entire h2b transcript pool including polyA+ and polyA− transcripts using qPCR (Materials and Methods). No significant change in the absolute expression of h2b inhibition of CHD4 (Bonferroni corrected Student's t‐test). Error bars represent the 95% confidence interval (At least two biological replicates, with two technical replicates for each biological replicate).
- Comparison of the polyA+ and polyA− h2b expression level shows an increase in polyA+ h2b in CHD4 (RNAi) animals compared to control animals.
- Heatmap showing the gene expression levels of intestine‐specific TFs and genes following CHD4 RNAi at different time points. Rows and columns represent genes and samples, respectively. Blue and red, low and high gene expression (row normalized z‐score).
- Quantification of m6A on RNA isolated from CHD4 (RNAi) or control animals showed no significant change in abundance of m6A methylation (Student's t‐test).
We considered potential associations of m6A and NuRD, which might explain the similarities in the phenotypes: (i) NuRD and m6A regulate similar processes independently; (ii) NuRD regulates m6A gene expression or m6A pathway activity; or (iii) m6A regulates NuRD. We first examined whether NuRD activity was required for the expression of m6A genes and found that the expression of m6A genes was unperturbed following inhibition of CHD4 (Dataset EV4). Then, we tested whether CHD4 was required for m6A formation and found that inhibition of CHD4 did not deplete m6A (Fig 7F), indicating that NuRD is not necessary for m6A formation. Therefore, NuRD activity is unlikely to be required for the activity of the MTC. We next tested whether m6A genes regulated NuRD gene expression. Inhibition of m6A genes did not result in downregulation of CHD4 or any other NuRD‐encoding components (Dataset EV2; Materials and Methods). This, of course, does not eliminate the possibility that the production or activity of one or more of NuRD components is regulated posttranscriptionally by m6A. Interestingly, CHD4 (RNAi) animals developed phenotypes more rapidly than following inhibition of m6A genes. For example, lysis and changes to gene expression were observed following three RNAi feedings, in contrast to the 8–10 RNAi feedings that were required for lysis following mettl14 (RNAi). It is possible that inhibition of the m6A pathway progressively deteriorates NuRD activity, directly or indirectly. However, it is also possible that the m6A pathway and NuRD function independently and that their inhibition results in similar phenotypes.
Discussion
m6A has been implicated in myriad regulatory roles in diverse biological systems (Dominissini et al, 2012; Batista et al, 2014; Kan et al, 2017; Zhang et al, 2017). Given the essentiality of the MTC, research in mammalian systems has been limited to studying the role of this complex either in cell lines (Dominissini et al, 2012; Batista et al, 2014; Geula et al, 2015; Liu et al, 2020b), or upon selective depletion of this complex in specific tissues or cell types, which has provided a partial and fragmented view on the roles played by this modification. Obtaining a global view on the functions and underlying mechanisms of action of m6A can hugely benefit from systemic, organism‐wide perturbations of the MTC, as have been conducted in a highly limited number of model organisms to date, most of which in species evolutionary distant from human such as yeast and plants (Schwartz et al, 2013, 2014; Kan et al, 2017; Bhat et al, 2018; Baumgarten et al, 2019). Our study adds to these efforts and introduces a systematic analysis of the MTC function in an adult Lophotrochozoan, planarian, which is a widely used model for studying regeneration.
Among the major questions for which we lacked a general understanding to date is the nature of the relationship between methylation writers and readers. All organisms surveyed to date have only a single “writer” machinery, but organisms vary widely in terms of the number of readers that they have, ranging from one detected reader in yeast (Schwartz et al, 2013) to 13 in plants (Reichel et al, 2019). Oftentimes, deletion of the writers is associated with a severe phenotype, whereas readers are associated with much milder defects. In some cases, this has been attributed to redundancy between readers (Kontur et al, 2020; Zaccara & Jaffrey, 2020; Lasman et al, 2020b), but in most cases it cannot be ruled out that the full function of the writers is not mediated by readers, but instead by other machineries (Reichel et al, 2019), potentially also methylation‐independent ones. In this regard, our findings mark a relatively rare exception—alongside with a previous report in Drosophila (Kan et al, 2017), and some recently reported findings in mouse embryonic stem cells (mESC)—in which the full phenotype associated with inhibition of a writer is phenocopied via inhibition of one reader. This strongly suggests a methylation‐dependent function and demonstrates that this function is likely to be mediated entirely via one reader. In parallel, these findings raise questions regarding the role played by the additional readers encoded in the planarian genome, whose disruption fails to give rise to a discernible phenotype. The absence of a phenotype may either be technical, or due to functional redundancy, and will be assessed in future studies.
We profiled the distribution of m6A in the planarian transcriptome and identified 7,600 enriched regions in a diversity of cell types. Our profiling approach included mRNA enrichment and was therefore focused on the detection of m6A enrichment in mRNAs. The m6A‐enriched regions were found mostly near the 3′‐end of the transcripts, as previously reported in other systems (Dominissini et al, 2012; Schwartz et al, 2014). This suggested that the role of m6A in planarians is similar to other systems. However, we identified a unique feature of planarian m6A‐enriched regions: they are longer, on average, than reported m6A‐enriched regions in other organisms (Liu et al, 2020c). This might be explained by lack of antibody specificity, yet we did not observe similar outcomes in our yeast samples. The detection of longer m6A‐enriched regions might be a consequence of individual RNA molecules having multiple methylated adenosine sites in proximity, which could be therefore detected as a broad m6A‐rich region. Single‐nucleotide resolution‐based approaches for detection of m6A, such as miCLIP (Grozhik et al, 2017), could further elucidate these findings.
Importantly, we found essentially identical phenotypes following inhibition of the MTC‐encoding genes mettl14, kiaa1429, and of the putative m6A‐reader, ythdc‐1. The phenotypes were reminiscent of a stem cell deficiency in a regenerative adult organism (Wagner et al, 2012): decline in animal size, failure to regenerate, and eventually death. In a developmental system, similar defects would be detrimental. Indeed, elimination of m6A in developmental contexts has revealed that m6A is critical for early development, cell maturation, and differentiation (Batista et al, 2014; Geula et al, 2015; Lasman et al, 2020a).
Integration of our analysis of the m6A phenotypes showed that m6A is required for production of the planarian intestine with more minor impact on other lineages. Defects in differentiation to certain lineages, but not necessarily to all, have been reported in embryonic stem cells (ESC) (Batista et al, 2014). However, in addition to the defect in differentiation, the lack of m6A in planarians resulted in an emergence of a molecularly defined cell type throughout the planarian body. These cells were undetectable in control animals, and they lacked characteristics of neoblasts: they expressed low levels of neoblast markers and had different morphology. Moreover, these abnormal cells expressed several genes that are broadly expressed in neural progenitors, such as a protocadherin‐like gene (dd_15376, gene model SMESG000067388) and Smed‐SMAD6/7–2. The scRNAseq clustering analysis positioned these cells adjacent to other neural cells, yet further in vivo evidence is required for determining their identity. Therefore, m6A, in planarians, is required for resolving the identity of the differentiating progenitors.
Recently, an analysis of mESCs has found that deletion of Mettl3 promotes an open state of the chromatin by regulating a class of chromatin associated RNAs (Liu et al, 2020a). In addition, Mettl3 regulates self‐renewal of neural stem cells by destabilizing transcripts that encode histone modifiers (Wang et al, 2018). Analysis of planarian nuclei following inhibition of kiaa1429 suggested that chromatin packaging was altered, which similarly to the findings in the mammalian system, may have profound effect on chromatin accessibility, and could be tested in future experiments. Moreover, we found that some of the most highly overexpressed genes, following inhibition of m6A genes, were localized to distinct genomic regions characterized by repetitive DNA. These regions were not expressed in control animals. Incorporation of the planarian cell type gene expression atlas (Fincher et al, 2018) with our m6A mapping data showed that 248 neoblast‐associated transcripts are highly methylated, including many transcriptional regulators (Dataset EV1). Most importantly, inhibition of CHD4, a component of the NuRD complex, has revealed striking molecular similarities, including reduction in intestine cells and upregulation of the repetitive genomic regions. Interestingly, p66, a DNA‐binding component of the NuRD complex, is known to suppress the production of photoreceptor neurons in planarians, suggesting of a link between NuRD and production of the neural lineage (Vásquez‐Doorman & Petersen, 2016). Potentially, m6A could affect the activity of chromatin regulators (Fig ), or alternatively, regulate similar processes indirectly (Fig 8B).
Figure 8. Potential models for interaction of m6A and NuRD.
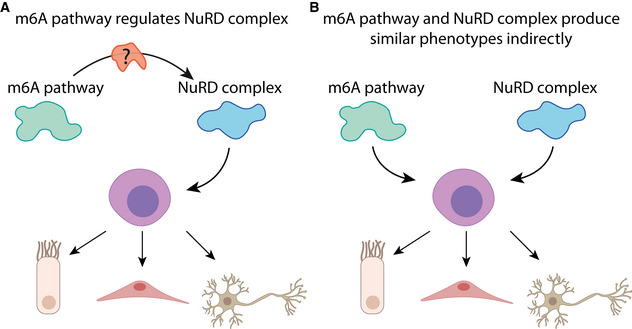
- m6A might regulate neoblast homeostasis by directly regulating NuRD or through a mediator that modulates NuRD function.
- In the alternative model m6A and NuRD regulate similar processes, in parallel, without an association between the pathways.
We did not detect NuRD complex genes that were highly methylated by m6A. Furthermore, inhibition of the m6A pathway did not affect NuRD complex gene expression. Similarly, inhibition of CHD4 did not affect the levels of m6A on planarian RNAs and did not result in a change to gene expression of canonical MTC components. Therefore, the striking similarities between the roles of m6A and NuRD, in planarians, could not be ascribed to simple transcriptional regulation. We therefore suggest two different models for the regulatory association of m6A and NuRD (Fig 8). In the first, m6A is required for proper activity of NuRD, potentially through a mediating molecule that is dependent on m6A methylation (Fig 8A). Alternatively, m6A and NuRD regulate similar processes, such as chromatin accessibility or emergence of a new cell state (Benham‐Pyle et al, 2021), independently (Fig 8B). Further study of neoblast‐specific RNAs that are modified by m6A might reveal the identity of a mediating molecule or distinguish between the two models.
In summary, we have mapped the distribution of m6A in the planarian transcriptome, characterized the roles of RNA‐level modifications in planarian tissue maintenance and regeneration, and demonstrated that m6A is indispensable for regulation of neoblast homeostasis. Our study lays the foundation to a mechanistic analysis of planarian gene expression regulation and consequently a system‐level understanding of planarian stem cell homeostasis.
Materials and Methods
Identification of putative m6A‐pathway and NuRD complex encoding genes
Sequences of genes encoding the MTC complex were extracted from the human, yeast, and fly genomes. Reciprocal BLAST search was performed with tblastx (Camacho et al, 2009) with e‐value <1 × 10−5 using the planarian transcriptome assembly (Liu et al, 2013). Putative reader‐encoding genes were identified by searching YTH domains in silico using hmmscan V3 with parameters [‐‐noali ‐‐cut_nc ‐‐acc ‐‐notextw] (Eddy, 2011). The resultant sequences were then searched in other planarian species in PlanMine (Rozanski et al, 2019). Contig IDs predicted to encode the planarian NuRD complex (GO:0016581) were extracted from PlanMine (Rozanski et al, 2019).
Synthesis of double‐stranded RNA for RNAi experiments and RNAi feedings
Double‐stranded RNA (dsRNA) was synthesized as previously described (Rouhana et al, 2013). Briefly, in vitro transcription (IVT) templates were prepared by PCR amplification of cloned target sequences using primers with 5′ flanking T7 promoter sequences. dsRNA was synthesized using the TranscriptAid T7 High Yield Transcription Kit (CAT K0441; Thermo Scientific). Reactions were incubated overnight at 37°C and then supplemented with RNase‐free DNase for 30 min. RNA was purified by ethanol precipitation and finally resuspended in 25 μl of ddH2O. RNA was analyzed on 1% agarose gel and quantified by Qubit 4 (Thermo scientific) for validating a concentration higher than 5 μg/μl. Animals were starved for at least 7 days prior to RNAi experiments. Schmidtea mediterranea, asexual, were used for all experiments. Animals were fed with dsRNA mixed with beef liver twice a week as previously described (Wurtzel et al, 2017).
cDNA synthesis from total RNA or polyadenylated RNA for qPCR
RNA samples were converted to cDNA and amplified using RevertAid H Minus First Strand cDNA Synthesis Kit (Thermo scientific; K1631). Random hexamers or oligo(dT) primers were used for obtaining cDNA from total RNA or from polyadenylated RNA, respectively. Resultant cDNA was used for qPCR analysis.
Fluorescence‐activated cell sorting (FACS)
Planarians were cut into small fragments using a blade. Tissue fragments were collected into calcium‐free, magnesium‐free medium plus 0.1% BSA (CMFB) and dissociated with collagenase l (1 mg/ml) by pipetting for 5–10 min. To reduce clumping at downstream steps, dissociated cells were strained through a 40 μm filter. Cells were centrifuged at 315 g for 5 min at 4°C and resuspended in CMFB containing Hoechst 33342 (40 μl/ml) for 45 min at RT in the dark. Before loading into FACSAria II flow cytometer (Becton Dickinson), cells were labeled with propidium iodide (5 μg/ml) for cell viability detection. FACS gating was performed as previously described for planarian cell populations (Hayashi et al, 2006).
Preparation of RNA sequencing libraries
RNAseq libraries for mettl14 (RNAi), ythdc‐1 (RNAi), and their control samples were prepared using Kapa stranded mRNAseq (KK8420) according to the manufacturer's protocol.
Planarian fixation for whole‐mount assays
Fixation was performed as previously described (King & Newmark, 2013). Animals were killed with 5% N‐acetyl‐cysteine in PBS for 5 min, then fixed with 4% formaldehyde in PBSTx 0.1 for 20 min. Animals were then washed in PBSTx, 50:50 PBSTx:methanol and stored in methanol at −20°C until further analysis.
Whole‐mount fluorescence in situ hybridization
Fluorescence in situ hybridization (FISH) was performed as previously described (King & Newmark, 2013) with minor modifications. Briefly, fixed animals were bleached and treated with proteinase K (2 μg/ml) in 1× PBSTx. Following overnight hybridizations, samples were washed twice in pre‐hyb solution, 1:1 pre‐hyb‐2× SSC, 2× SSC, 0.2× SSC, PBSTx. Subsequently, blocking was performed in 0.5% Roche Western Blocking Reagent and 5% inactivated horse serum in 1× PBSTx. Animals were incubated in an antibody overnight at 4°C (anti‐DIG‐POD, 1:1,500). Post‐antibody washes and tyramide development were performed as previously described (King & Newmark, 2013). Specimens were counterstained with DAPI overnight at 4°C (Sigma, 1 μg/ml in PBSTx).
Metabolic labeling by F‐ara‐EdU
Animals were soaked for 16 h in 2.5 mg/ml F‐ara‐EdU (Sigma‐Aldrich T511293) diluted in planarian water. Then, animals were fixed in NAC and bleached with formamide and H2O2 for 2 h on a light table. Animals were treated with 0.2 μg/ml Proteinase K for 10 min, 4% FA for 10 min, and were then washed three times in 3% PBSB (PBS supplemented with 3% BSA). Click reaction was performed using baseclick kit (cat. Back‐edu488) and was followed by nuclear staining (DAPI, 1:5,000) overnight in 4°C. Samples were washed three times in PBSTx (0.1%) for 10 min and then mounted for imaging.
H3P labeling
H3P labeling was performed based on published protocols (Wenemoser & Reddien, 2010; LoCascio et al, 2017) on animals following fixation with NAC. Briefly, following fixation blocking was performed with 10% heat inactivated horse serum (HIHS). Anti‐phospho‐Histone H3 Antibody (sigma 04817) was added 1:100 overnight in 4°C. Samples were washed seven times with PBSTx (0.1% triton) for 20 min and were labeled using goat anti‐rabbit‐HRP secondary antibody (Abcam ab6721, 1:300) overnight at 4°C in block. Samples were developed using rhodamine tyramide diluted 1:1,000 in PBSTi.
Image processing for figure assembly
Fluorescent and confocal images were acquired using confocal microscope (Zeiss LSM800). Live images were taken using stereomicroscope (Leica S9i). Images were cropped and rotated, and masks of planarian outline were defined in Adobe Photoshop using the lasso and the object selection tools. Background color, outside the defined mask, was standardized in Adobe Photoshop or in Adobe Illustrator as light blue and black for live and fluorescent images, respectively.
Cell counting following FISH in whole‐mount planarian samples
Images of samples labeled FISH, immunofluorescence, or F‐ara‐EdU were collected using a confocal microscope (Zeiss LSM800). Cell counting was performed on controls and experimental condition. Positive cells were counted using the “Cell Counter” component in ImageJ (Rueden et al, 2017), and the normalized positive cell count was calculated by measuring the area used for cell counting in each animal. Unless mentioned otherwise, the cells were counted in a rectangle starting at the posterior part of the brain and ending at the anterior of the pharynx in ventrally mounted animals.
Cell counting in microscopy images
Counting of H3P+ cells was performed in a rectangle starting anterior to the pharynx and ending at the posterior end of the brain, excluding the epidermis, which had nonspecific labeling background, and includes no neoblasts. Cell counting was performed in z‐stack images in slices of 4 μm. Cell count was normalized to the area of quantification.
Counting of intestinal phagocytes was performed on mettl14 (RNAi) and ythdc‐1 (RNAi) animals. Intestinal phagocytes were labeled using a specific marker by FISH (dd_3194). Animals were imaged by confocal microscopy, and the z‐position showing the largest number of intestinal phagocytes anterior to the pharynx, as determined by two researchers, was selected for counting. Cells were counted using the Cell Counter module in ImageJ (Schindelin et al, 2012; Rueden et al, 2017). Then, the length of the intestinal branches was measured by tracing the intestine based on DAPI labeling next to the intestine lumen by using the segmented line tool in ImageJ. The normalized number of cells per micron of intestine branch was calculated based on these measurements.
F‐ara‐EdU+ cells were counted in a rectangle starting anterior to the pharynx and ending at the posterior end of the brain, in a subepidermal layer that does not include intestine branches, which had nonspecific labeling background. The number of F‐ara‐EdU+ cells was normalized to the area of the region.
Statistical analysis
Statistical tests were performed using GraphPad Prism (v9.2) and the R (v4.0) environment. Differentially expressed genes were determined using edgeR (3.30.3) with functions glmQLFit and glmQLFTest (McCarthy et al, 2012) and with DESeq2 (Love et al, 2014). Threshold for considering an effect significant in hypothesis testing was 0.05, unless stated otherwise. Multiple hypothesis testing was controlled using the Benjamini–Hochberg Procedure or by applying the Bonferroni correction.
Gene cloning and transformation
Genes were amplified from planarian cDNA using gene‐specific primers and cloned into pGEM‐t vector using the manufacturer's protocol (Promega; CAT #A1360). Vectors were transformed into E. coli TOP10 (Thermo Fisher Scientific) by the heat‐shock method. Briefly, 100 μl of bacteria was mixed with 5 μl of each of the cloned vectors, incubated on ice for 30 min, and then placed at 42°C for 45 s. Then, the transformed bacteria were supplemented with 350 μl of SOC medium, and following 1 h of recovery at 37°C, the bacteria were plated on agarose plates containing 1:2,000 Ampicillin, 1:200 Isopropylthio‐b‐D‐galactoside (IPTG), and 1:625 5‐bromo‐4‐chloro‐3‐indolyl‐β‐D‐galactopyranoside (X‐gal). Colonies were grown overnight at 37°C, and colonies were screened by colony PCR using M13F and M13R primers with the following PCR program: (i) 5 min at 95°C; (ii) 34 cycles of 45 s at 95°C, 60 s at 55°C, and 2:30 min at 72°C; (iii) 10 min at 72°C; (iv) hold at 10°C. Reactions were analyzed by gel electrophoresis, and correctly sized gene products were grown overnight in Luria Broth (LB) medium, supplemented with 1:2,000 Ampicillin at 37°C. Plasmids were purified from overnight cultures with the NucleoSpin Plasmid Miniprep Kit (Macherey‐Nagel; CAT #740588). Cloned gene sequences were sequenced by Sanger sequencing.
Measurement of m6A level in purified RNA
m6A RNA methylation level was measured by an m6A RNA methylation assay kit (Abcam, ab185912) following the manufacturer's protocol. Total of 200 ng of RNA isolated from control, mettl14 (RNAi), and kiaa1429 (RNAi) animals was used to determine the fraction of m6A in the RNA. Separately, m6A was measured on polyadenylated RNA and non‐polyadenylated RNA from wild‐type planarians. PolyA+ RNA was separated from polyA‐ RNA using the NEXTFLEX Poly(A) Beads 2.0 kit (PerkinElmer). Following anti‐m6A‐antibody capture, the absorbance was measured at 450 nm using a microplate reader, and the percentage of m6A in the RNA was calculated using the kit supplied controls.
RNA extraction
Samples were collected into 700 μl TRI Reagent (Sigma; CAT #9424) and then homogenized using 0.5 mm zirconium beads in a bead beating homogenizer (Allsheng; Bioprep‐24) for two cycles of 45 s at 3,500 RPM followed by incubation of 5 min at room temperature. Then, 140 μl of chloroform was added to each tube. Tubes were shaken for 15 s and then incubated at room temperature for 3 min. Then, samples were centrifuged at 4°C, 12,000 g for 25 min. The upper phase was separated into a new tube. Then, 500 μl of isopropanol was added, and tubes were inverted five times and then incubated for 10 min at room temperature. Samples were centrifuged at 4°C (12,000 g) for 45 min. The supernatant was removed and the pellet was washed twice with 75% EtOH, centrifuged at 4°C 7,500 g for 5 min, and air‐dried for 10 min. RNA was resuspended in 30 μl of H2O. RNA concentration was measured by Qubit (Invitrogen; Q33226) according to the manufacturer's protocol.
mRNA m6A immunoprecipitation library preparation
m6A immunoprecipitation was performed as previously described (Dierks et al, 2021). RNA samples were extracted from kiaa1429 (RNAi) animals or control, unc22 (RNAi), animals in triplicates, following five dsRNA feedings. Each replicate included at least nine animals. RNA was quantified using Qubit (Invitrogen, Q33226), and 20 μg of kiaa1429 (RNAi) and 30 μg of unc22 (RNAi) RNA were used per sample per replicate. mRNA was enriched by two rounds of polyA selection (Dynabeads, CAT #61011) according to the manufacturer's protocol. mRNA‐enriched samples were quantified by spectrophotometry (SpectraMax QuickDrop, Molecular Devices) and fragmented by RNA Fragmentation Reagents (Ambion, AM8740) for 2 min. Fragmented RNA was purified by MyOne Silane beads (Thermo, 37002D). Then, the purified RNA was treated with Turbo DNase (Thermo Fisher Scientific, AM2238), and the RNA was treated with FastAP (Fermentas, EF0651) and T4 PNK (CAT M0201L), according to the m6A pulldown protocol. Each RNA sample was barcoded by ligation to a unique 3′ adapter, which was flanked by a universal sequence that was used subsequently for reverse transcription. Then, the barcoded RNA samples were pooled by mixing, and 10% of the pooled sample volume was separated and used as control (input) for the m6A pulldown experiment. Immunoprecipitation of m6A was performed by using an anti‐m6A‐antibody (Synaptic Systems) according to protocol. RNAseq libraries were prepared from the immunoprecipitated RNA and input RNA, separately, following the published protocol (Dierks et al, 2021). Library integrity was analyzed by fragment analysis (Agilent TapeStation 4000). Libraries were sequenced on an Illumina (NextSeq 550) machine at the Tel Aviv University NGS unit.
Processing of m6A‐seq2 library data
m6A‐seq2 libraries were pooled and sequenced in paired‐end mode (Dierks et al, 2021). The resultant sequencing reads were then demultiplexed (i.e., separated to individual libraries) based on an inline library adapter that was ligated to each library according to the protocol (Dierks et al, 2021). Demultiplexing was performed by using cutadapt ‐‐pair‐filter = any ‐‐minimum‐length 24 (Martin, 2011) by supplying internal barcode sequences, followed by trimming of the 10 nt sequence of the barcode from the barcoded sequencing read (Read #2). The resultant fastq files were pre‐processed and mapped to the planarian transcriptome as described in the RNAseq library analysis method section. m6A‐enriched regions (m6a peaks) were determined using MeTPeak with default parameters (Cui et al, 2016). Plots of m6A peak profiles and peak heatmaps were produced using deepTools2 (Ramírez et al, 2016). Peak profiles were generated by merging BAM files of the three replicates of the control or kiaa1429 (RNAi) libraries using the samtools merge command (Li et al, 2009). Then, mapping coverage was calculated for the merged control or kiaa1429 (RNAi) BAM file using the deepTools2 command bamCoverage with parameters [‐‐normalizeUsing RPKM ‐‐binSize 30]. The resultant files were then used for generating peak matrices with the computeMatrix scale‐regions command, which was followed by producing the profile plots with the plotProfile command with parameters [‐‐plotType lines ‐‐perGroup]. Peak heatmaps were produced by taking all of the detectable m6A peaks that were enriched by at least fivefold in the pulldown libraries over the input. Then, the computeMatrix scale‐regions command was used followed by the command plotHeatmap with parameters [‐‐kmeans 3 ‐‐t “#2166 ac,#d1e5f0,#ef8a62,#b2182b”]. Mixed species m6A‐seq2 experiment was performed by using RNA from yeast (Saccharomyces cerevisiae sk1) and from planarian (Fig EV3). RNA was mixed following RNA isolation and polyA‐enrichment, prior to other processing to reduce biases that are associated with handling of RNA. Two pools of RNA were produced, in duplicates by mixing 80% of planarian RNA with 20% of yeast RNA: (i) Control planarian RNA mixed with RNA from S. cerevisiae lacking m6A, due to genetic mutation Ime4Δ/Δ/Ndt80Δ/Δ (Dierks et al, 2021); and (ii) planarian RNA from kiaa1429 (RNAi) animals, which have a reduced m6A level, with RNA from S. cerevisiae having high level of m6A because genetic background (Ndt80Δ/Δ) (Dierks et al, 2021). The pools were then processed according to the m6A‐seq2 protocol. Briefly, the barcoded planarian‐yeast pools were combined, and immunoprecipitation of m6A was performed using an anti‐m6A antibody. Library preparation and Illumina sequencing were performed according to the m6A‐seq2 protocol. Sequencing reads were assigned to respective planarian‐yeast pool based on the sequence of their molecular 3′‐end barcode (Appendix Table S3) and were then assigned to the planarian or yeast genome by mapping the sequences of each pool to a combined genome sequence of planarian (Rozanski et al, 2019), and S. cerevisiae (Dierks et al, 2021), or by mapping reads to a combined transcriptome sequence of planarian and the S. cerevisiae genome sequence. Enrichment in m6A sequence was performed by MeTPeak with default parameters (Cui et al, 2016), and motif finding was performed on regions enriched with m6A that are shorter than 250 bp using HOMER with parameters [‐size given ‐len 5 using the 500 most enriched m6A regions] (Heinz et al, 2010), and using an orthogonal m6A‐associated k‐mer enrichment detection strategy (Schwartz et al, 2014; Dierks et al, 2021).
qPCR analysis of gene expression following RNAi
RNA concentration of each sample was measured using Qubit 4 fluorometer (Invitrogen; Q33226). cDNA synthesis was performed on 1 μg of RNA from each sample using RevertAid H Minus First Strand cDNA Synthesis Kit (Thermo scientific; K1631), following these steps: RNA template was mixed with an oligo‐dT primer and then incubated at 65°C for 5 min. Then, cDNA synthesis proceeded according to the manufacturer's protocol: samples were incubated at 42°C for 60 min and then at 70°C for 5 min. The cDNA sample concentrations were normalized to 1 ng/μl. The expression of the target genes was measured using the QuantStudio 3 Real‐Time PCR system (Applied Biosystems) using the following program [95°C (20 s), 40 cycles (95°C 1 s, 60°C 20 s)], with two technical replicates per sample and at least two biological replicates per sample. The relative gene expression fold‐change was calculated by the ΔΔC t method with gapdh used as an endogenous control. Forward and reverse primers were designed for each target gene. Primer efficiency was tested prior to the experiment by using the standard curve method for five decreasing cDNA concentrations.
Analysis of RNA sequencing gene expression changes
RNAseq sequencing files were preprocessed by trimming the Illumina adapter sequences using trimmomatic V0.38 (Bolger et al, 2014) [ILLUMINACLIP:TruSeq3‐PE.fa:2:30:10:2:keepBothReads LEADING:0 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:30]. Then, the trimmed RNAseq sequences were mapped to the planarian transcriptome (ddV4) (Liu et al, 2013) and genome (Rozanski et al, 2019) using bowtie2 (Langmead & Salzberg, 2012) and HISAT2 (Kim et al, 2019), respectively. Mapping of transcriptome assembly contig IDs to planarian gene models was fetched from PlanMine (Rozanski et al, 2019). Gene expression matrix was then produced using the featureCounts command from the subread‐2.0 package with parameters [‐M ‐s 0] (Liao et al, 2014) for the analyzed RNAseq libraries. Differentially expressed genes were detected using edgeR (3.30.3) with functions glmQLFit and glmQLFTest (McCarthy et al, 2012), and with DESeq2 (1.28.1) (Love et al, 2014).
Detection of polyadenylated transcripts in RNAseq data
Paired‐end RNAseq libraries were preprocessed with trimmomatic V0.38 (Bolger et al, 2014) with parameters [ILLUMINACLIP:TruSeq3‐PE.fa:2:30:10:2:keepBothReads LEADING:0 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:30]. For RNAseq libraries, reads in the Read #2 files were derived from antisense paired‐reads. Sequencing reads from Read #2 files, which contained homopolymers (>10 nt), were discarded, but their read pair (Read #1) was kept. The retained Read #1 file was then mapped to the planarian transcriptome (Rozanski et al, 2019) using bowtie2 V2.3.5.1 with default parameters (Langmead & Salzberg, 2012). Reads that mapped to histone‐encoding genes were isolated from the resultant read mapping BAM file (Li et al, 2009). Then, the unmapped read‐pair (Read #2) of the isolated mapped‐reads (Read #1) was selected from the raw paired‐end RNAseq fastq files. An unmapped Read #2 sequence that included at least 10 consecutive T nucleotides (i.e., the reverse complement of the polyA) was counted as a polyA‐containing read. The number of polyA‐containing reads was summarized per library and normalized by the library size.
Quantification of polyadenylated h2b fraction by qPCR
RNA was isolated from kiaa1429 (RNAi) and from control animals. Purified RNA was converted to cDNA by using random hexamers or poly dT primers. The cDNA samples concentrations were normalized to 1 ng/μl. qPCR was performed using QuantStudio 3 Real‐Time PCR system (Applied Biosystems) with the following settings [95°C (20 s), 40 cycles (95°C 1 s, 60°C 20 s)], with two technical replicates per sample and at least two biological replicates per sample. The relative fold gene expression was calculated by the ΔΔC t method with gapdh used as the endogenous control.
Preparation of scRNAseq libraries following kiaa1429 (RNAi)
Animals were fed five times with unc22 (control) and kiaa1429 dsRNA. Animals were starved for a week following RNAi feedings. Cells were purified for scRNAseq from either control or kiaa1429 (RNAi) animals by FACS using hoechst‐labeling, as previously described (Fincher et al, 2018). Cells were counted and collected by centrifugation (300 g, 5 min). The supernatant was discarded, and the cells were resuspended in 1× PBS with 0.5% BSA to achieve an optimal cell concentration of approximately 1,000 cells per μl. Cells were counted automatically using the Invitrogen Countess II (Thermo Fisher Scientific), and the cell viability was assessed by labeling with trypan blue (0.4%) and counting positive cells.
Libraries were then prepared at the Genomic Research Unit at Tel Aviv University using the 10× Genomics Chromium Controller in conjunction with the single‐cell 3′ v3.1 kit, protocol revision D. Briefly, cell suspensions were diluted in nuclease‐free water according to the manufacturer's protocol to 10,000 cells in each sample. The cDNA synthesis, barcoding, and library preparation were carried out according to the manufacturer's instructions. cDNA libraries were sequenced on Illumina NextSeq 550 at Tel Aviv University.
Systematic gene expression analysis of published planarian RNAseq data
A table of previously published planarian high‐throughput sequencing libraries was extracted from the Sequence Read Archive (Leinonen et al, 2011). The table was manually curated and libraries were divided into biological groups, in each experiment, based on the library description in the Short Read Archive (Table EV1). Then, each library was downloaded using sra‐tools (Leinonen et al, 2011) and was processed on a computational cluster using the computational pipeline used for the RNAseq libraries that were produced in this project. The resultant differential gene expression analysis tables, which were produced for the previously published planarian data, were used for searching for the genes that were differentially expressed in the libraries produced in this project with the following thresholds (FDR <0.0001, log2 > 1 or log2 < −1). We tested a range of parameters for calling differentially expressed genes in the published dataset (FDR range from 0.1 to 1E‐10; minimal gene expression 1–20 transcript per million, TPM) and searched for overlap of gene expression with our dataset. The significance of overlap of differentially expressed genes was robust to these thresholds. To minimize spurious potential correlations, we used conservative thresholds requiring corrected FDR < 1E‐5 and TPM > 10, in order to include a gene in the list of differentially expressed genes for this analysis. The CHD4 (RNAi) libraries at 9, 12, 15 days following RNAi had the most significant overlap of differentially expressed genes with the list of differentially expressed genes in our datasets with P‐value, estimated by hypergeometric hypothesis test, of <1E‐15 and often P‐value <1E‐60, with the highest ranking of overlap (Appendix Fig S4A). For example, 31 out of the top 50 differentially expressed genes in following mettl14 (RNAi) were differentially expressed in the CHD4 (RNAi) 15 days condition, compared with a median overlap of four differentially expressed genes in all of the tested libraries. Furthermore, we selected up to 200 genes with the lowest adjusted P‐value, which meet the thresholds for differential expression, from each condition and computed the Jaccard similarity coefficient (JI). The median JI between our libraries and each of the libraries in this analysis was <0.02, compared with JI >0.14 at 9 and 15 days following CHD4 (RNAi) with the kiaa1429 (RNAi) library. The JI for the CHD4 (RNAi) and our libraries was significantly larger than JIs calculated for the other groups as estimated by two‐sided Wilcoxon rank sum (P = 0.0002).
Analysis of human and mouse gene expression data from CHD4 inhibition or conditional knockout
RNAseq libraries of control and conditional knockout of CHD4 from mouse (Wilczewski et al, 2018) and from control and shRNA‐treated samples against CHD4 from human (Marques et al, 2020) were obtained from the sequence read archive using the fasterq‐dump command (Leinonen et al, 2011). The raw fastq files were trimmed using Trimmomatic‐0.38 (Bolger et al, 2014) [ILLUMINACLIP:TruSeq3‐PE.fa:2:30:10:2:keepBothReads LEADING:0 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:30]. Mouse and human samples were mapped to genome assemblies mm10 and hg38, respectively, using HISAT2 with default parameters (Kim et al, 2019). Gene expression was estimated using featureCounts (Liao et al, 2014) using the included human or mouse annotations. Finally, differential gene expression analysis was performed using DESeq2 (Love et al, 2014) with the DEseq pipeline. Resultant differential gene expression tables were annotated with the bioconductor annotation packages org.Mm.eg.db and org.Hs.eg.db, for mouse and human, respectively (Huber et al, 2015).
Author contributions
Yael Dagan: Conceptualization; data curation; formal analysis; investigation; visualization; methodology; writing – original draft; writing – review and editing. Yarden Yesharim: Conceptualization; data curation; formal analysis; investigation; visualization; methodology; writing – original draft; writing – review and editing. Ashley R Bonneau: Conceptualization; investigation; methodology. Tamar Frankovits: Data curation; investigation. Schraga Schwartz: Software; investigation; methodology; writing – review and editing. Peter W Reddien: Conceptualization; resources; formal analysis; funding acquisition; writing – review and editing. Omri Wurtzel: Conceptualization; resources; data curation; software; formal analysis; supervision; funding acquisition; investigation; visualization; methodology; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix S1
Expanded View Figures PDF
Table EV1
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
PDF+
Acknowledgements
We thank Sarit Edelheit for support with the m6A‐seq2 protocol. We thank Rami Khosravi for assistance with scRNAseq, and Hila Kobo for assistance with Illumina sequencing at the Tel Aviv University core facilities. We thank Jenny Barboy‐Smoliarenko for assistance in illustration. We thank the Wurtzel lab for critical input. O.W. is supported by the Israel Science Foundation (grant 2039/18) and the European Research Council (no. 853640). O.W. is a Zuckerman Faculty Scholar. P.W.R. acknowledges NIH support (R01GM080639). P.W.R. is an investigator of the Howard Hughes Medical Institute and an associate member of the Broad Institute of Harvard, and MIT. S.S. is supported by the European Research Council (ERC) under the European Union‘s Horizon 2020 research and innovation program (grant agreement No. 714023). A.R.B. was supported by grant F32‐GM128411 by the National Institute of General Medical Sciences.
The EMBO Journal (2022) 41: e109895.
See also: BW Benham-Pyle (November 2022)
Data availability
High‐throughput sequencing data produced in this project were deposited to the Sequence Read Archive (Leinonen et al, 2011). The data are available under BioProject accession PRJNA747686.
References
- Adler CE, Sánchez Alvarado A (2015) Types or states? Cellular dynamics and regenerative potential. Trends Cell Biol 25: 687–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basta J, Rauchman M (2015) The nucleosome remodeling and deacetylase complex in development and disease. Transl Res 165: 36–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista PJ, Molinie B, Wang J, Qu K, Zhang J, Li L, Bouley DM, Lujan E, Haddad B, Daneshvar K et al (2014) M(6)a RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 15: 707–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgarten S, Bryant JM, Sinha A, Reyser T, Preiser PR, Dedon PC, Scherf A (2019) Transcriptome‐wide dynamics of extensive m6A mRNA methylation during Plasmodium falciparum blood‐stage development. Nat Microbiol 4: 2246–2259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benham‐Pyle BW, Brewster CE, Kent AM, Mann FG, Chen S, Scott AR, Box AC, Sánchez Alvarado A (2021) Identification of rare, transient post‐mitotic cell states that are induced by injury and required for whole‐body regeneration in Schmidtea mediterranea . Nat Cell Biol 23: 939–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat SS, Bielewicz D, Jarmolowski A, Szweykowska‐Kulinska Z (2018) N6‐methyladenosine (m6A): revisiting the old with focus on new, an Arabidopsis thaliana centered review. Genes (Basel) 9: 596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL (2009) BLAST+: architecture and applications. BMC Bioinformatics 10: 421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui X, Meng J, Zhang S, Chen Y, Huang Y (2016) A novel algorithm for calling mRNA m6A peaks by modeling biological variances in MeRIP‐seq data. Bioinformatics 32: i378–i385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dávila López M, Samuelsson T (2008) Early evolution of histone mRNA 3′ end processing. RNA 14: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dierks D, Garcia‐Campos MA, Uzonyi A, Safra M, Edelheit S, Rossi A, Sideri T, Varier RA, Brandis A, Stelzer Y et al (2021) Multiplexed profiling facilitates robust m6A quantification at site, gene and sample resolution. Nat Methods 18: 1060–1067 [DOI] [PubMed] [Google Scholar]
- Dominissini D, Moshitch‐Moshkovitz S, Schwartz S, Salmon‐Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob‐Hirsch J, Amariglio N, Kupiec M et al (2012) Topology of the human and mouse m6A RNA methylomes revealed by m6A‐seq. Nature 485: 201–206 [DOI] [PubMed] [Google Scholar]
- Duncan EM, Chitsazan AD, Seidel CW, Sánchez Alvarado A (2015) Set1 and MLL1/2 target distinct sets of functionally different genomic loci in vivo . Cell Rep 13: 2741–2755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duronio RJ, Marzluff WF (2017) Coordinating cell cycle-regulated histone gene expression through assembly and function of the Histone Locus Body. RNA Biol 14: 726–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR (2011) Accelerated profile HMM searches. PLoS Comput Biol 7: e1002195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fincher CT, Wurtzel O, de Hoog T, Kravarik KM, Reddien PW (2018) Cell type transcriptome atlas for the planarian Schmidtea mediterranea . Science 360: eaaq1736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsthoefel DJ, James NP, Escobar DJ, Stary JM, Vieira AP, Waters FA, Newmark PA (2012) An RNAi screen reveals intestinal regulators of branching morphogenesis, differentiation, and stem cell proliferation in planarians. Dev Cell 23: 691–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsthoefel DJ, Cejda NI, Khan UW, Newmark PA (2020) Cell‐type diversity and regionalized gene expression in the planarian intestine. eLife 9: e52613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geula S, Moshitch‐Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon‐Divon M, Hershkovitz V, Peer E, Mor N, Manor YS et al (2015) m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science 347: 1002–1006 [DOI] [PubMed] [Google Scholar]
- González‐Sastre A, Molina MD, Saló E (2012) Inhibitory Smads and bone morphogenetic protein (BMP) modulate anterior photoreceptor cell number during planarian eye regeneration. Int J Dev Biol 56: 155–163 [DOI] [PubMed] [Google Scholar]
- Grozhik AV, Linder B, Olarerin‐George AO, Jaffrey SR (2017) Mapping m6A at individual‐nucleotide resolution using crosslinking and immunoprecipitation (miCLIP). Methods Mol Biol 1562: 55–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo T, Peters AHFM, Newmark PA (2006) A Bruno‐like gene is required for stem cell maintenance in planarians. Dev Cell 11: 159–169 [DOI] [PubMed] [Google Scholar]
- Hayashi T, Asami M, Higuchi S, Shibata N, Agata K (2006) Isolation of planarian X‐ray‐sensitive stem cells by fluorescence‐activated cell sorting. Dev Growth Differ 48: 371–380 [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK (2010) Simple combinations of lineage‐determining transcription factors prime cis‐regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hongay CF, Orr‐Weaver TL (2011) Drosophila inducer of MEiosis 4 (IME4) is required for notch signaling during oogenesis. Proc Natl Acad Sci USA 108: 14855–14860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T et al (2015) Orchestrating high‐throughput genomic analysis with Bioconductor. Nat Methods 12: 115–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan L, Grozhik AV, Vedanayagam J, Patil DP, Pang N, Lim K‐S, Huang Y‐C, Joseph B, Lin C‐J, Despic V et al (2017) The m6A pathway facilitates sex determination in Drosophila . Nat Commun 8: 15737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C, Salzberg SL (2019) Graph‐based genome alignment and genotyping with HISAT2 and HISAT‐genotype. Nat Biotechnol 37: 907–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King RS, Newmark PA (2013) In situ hybridization protocol for enhanced detection of gene expression in the planarian Schmidtea mediterranea . BMC Dev Biol 13: 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontur C, Jeong M, Cifuentes D, Giraldez AJ (2020) Ythdf m6A readers function redundantly during zebrafish development. Cell Rep 33: 108598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbé RM, Irimia M, Currie KW, Lin A, Zhu SJ, Brown DDR, Ross EJ, Voisin V, Bader GD, Blencowe BJ et al (2012) A comparative transcriptomic analysis reveals conserved features of stem cell pluripotency in planarians and mammals. Stem Cells 30: 1734–1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasman L, Hanna JH, Novershtern N (2020a) Role of m6a in embryonic stem cell differentiation and in gametogenesis. Epigenomes 4: 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasman L, Krupalnik V, Viukov S, Mor N, Aguilera‐Castrejon A, Schneir D, Bayerl J, Mizrahi O, Peles S, Tawil S et al (2020b) Context‐dependent functional compensation between Ythdf m6A reader proteins. Genes Dev 34: 1373–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Choe J, Park OH, Kim YK (2020) Molecular mechanisms driving mRNA degradation by m6A modification. Trends Genet 36: 177–188 [DOI] [PubMed] [Google Scholar]
- Leinonen R, Sugawara H, Shumway M, International Nucleotide Sequence Database Collaboration (2011) The sequence read archive. Nucleic Acids Res 39: D19–D21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lence T, Soller M, Roignant J‐Y (2017) A fly view on the roles and mechanisms of the m6A mRNA modification and its players. RNA Biol 14: 1232–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930 [DOI] [PubMed] [Google Scholar]
- Lin AYT, Pearson BJ (2014) Planarian yorkie/YAP functions to integrate adult stem cell proliferation, organ homeostasis and maintenance of axial patterning. Development 141: 1197–1208 [DOI] [PubMed] [Google Scholar]
- Linder B, Grozhik AV, Olarerin‐George AO, Meydan C, Mason CE, Jaffrey SR (2015) Single‐nucleotide‐resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods 12: 767–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SY, Selck C, Friedrich B, Lutz R, Vila‐Farré M, Dahl A, Brandl H, Lakshmanaperumal N, Henry I, Rink JC (2013) Reactivating head regrowth in a regeneration‐deficient planarian species. Nature 500: 81–84 [DOI] [PubMed] [Google Scholar]
- Liu J, Dou X, Chen C, Chen C, Liu C, Xu MM, Zhao S, Shen B, Gao Y, Han D et al (2020a) N6‐methyladenosine of chromosome‐associated regulatory RNA regulates chromatin state and transcription. Science 367: 580–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Li K, Cai J, Zhang M, Zhang X, Xiong X, Meng H, Xu X, Huang Z, Peng J et al (2020b) Landscape and regulation of m6A and m6Am Methylome across human and mouse tissues. Mol Cell 77: 426–440.e6 [DOI] [PubMed] [Google Scholar]
- Liu S, Zhu A, He C, Chen M (2020c) REPIC: a database for exploring the N6‐methyladenosine methylome. Genome Biol 21: 100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoCascio SA, Lapan SW, Reddien PW (2017) Eye absence does not regulate planarian stem cells during eye regeneration. Dev Cell 40: 381–391.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques JG, Gryder BE, Pavlovic B, Chung Y, Ngo QA, Frommelt F, Gstaiger M, Song Y, Benischke K, Laubscher D et al (2020) NuRD subunit CHD4 regulates super‐enhancer accessibility in rhabdomyosarcoma and represents a general tumor dependency. Elife 9: e54993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011) Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet J 17: 10 [Google Scholar]
- Marzluff WF, Koreski KP (2017) Birth and death of histone mRNAs. Trends Genet 33: 745–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DJ, Chen Y, Smyth GK (2012) Differential expression analysis of multifactor RNA‐Seq experiments with respect to biological variation. Nucleic Acids Res 40: 4288–4297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinaro AM, Pearson BJ (2018) Myths vs. FACS: what do we know about planarian stem cell lineages? Int J Dev Biol 62: 527–535 [DOI] [PubMed] [Google Scholar]
- Patil DP, Pickering BF, Jaffrey SR (2018) Reading m6A in the transcriptome: m6A‐binding proteins. Trends Cell Biol 28: 113–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plass M, Solana J, Wolf FA, Ayoub S, Misios A, Glažar P, Obermayer B, Theis FJ, Kocks C, Rajewsky N (2018) Cell type atlas and lineage tree of a whole complex animal by single‐cell transcriptomics. Science 360: eaaq1723 [DOI] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, Manke T (2016) deepTools2: a next generation web server for deep‐sequencing data analysis. Nucleic Acids Res 44: W160–W165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz AA, Wurtzel O, Reddien PW (2021) Planarian stem cells specify fate yet retain potency during the cell cycle. Cell Stem Cell 28: 1307–1322.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddien PW (2013) Specialized progenitors and regeneration. Development 140: 951–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddien PW (2018) The cellular and molecular basis for planarian regeneration. Cell 175: 327–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddien PW, Oviedo NJ, Jennings JR, Jenkin JC, Sánchez Alvarado A (2005) SMEDWI‐2 is a PIWI‐like protein that regulates planarian stem cells. Science 310: 1327–1330 [DOI] [PubMed] [Google Scholar]
- Reichel M, Köster T, Staiger D (2019) Marking RNA: m6A writers, readers, and functions in Arabidopsis. J Mol Cell Biol 11: 899–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rink JC (2013) Stem cell systems and regeneration in planaria. Dev Genes Evol 223: 67–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouhana L, Weiss JA, Forsthoefel DJ, Lee H, King RS, Inoue T, Shibata N, Agata K, Newmark PA (2013) RNA interference by feeding in vitro‐synthesized double‐stranded RNA to planarians: methodology and dynamics. Dev Dyn 242: 718–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozanski A, Moon H, Brandl H, Martín‐Durán JM, Grohme MA, Hüttner K, Bartscherer K, Henry I, Rink JC (2019) PlanMine 3.0‐improvements to a mineable resource of flatworm biology and biodiversity. Nucleic Acids Res 47: D812–D820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueden CT, Schindelin J, Hiner MC, DeZonia BE, Walter AE, Arena ET, Eliceiri KW (2017) ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics 18: 529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Růžička K, Zhang M, Campilho A, Bodi Z, Kashif M, Saleh M, Eeckhout D, El‐Showk S, Li H, Zhong S et al (2017) Identification of factors required for m6 a mRNA methylation in Arabidopsis reveals a role for the conserved E3 ubiquitin ligase HAKAI. New Phytol 215: 157–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu S, Sridhar D, Abnave P, Kosaka N, Dattani A, Thompson JM, Hill MA, Aboobaker A (2021) Ongoing repair of migration‐coupled DNA damage allows planarian adult stem cells to reach wound sites. eLife 10: e63779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al (2012) Fiji: an open‐source platform for biological‐image analysis. Nat Methods 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Agarwala SD, Mumbach MR, Jovanovic M, Mertins P, Shishkin A, Tabach Y, Mikkelsen TS, Satija R, Ruvkun G et al (2013) High‐resolution mapping reveals a conserved, widespread, dynamic mRNA methylation program in yeast meiosis. Cell 155: 1409–1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, Bushkin GG, Mertins P, Ter‐Ovanesyan D, Habib N, Cacchiarelli D et al (2014) Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep 8: 284–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scimone ML, Meisel J, Reddien PW (2010) The mi‐2‐like Smed‐CHD4 gene is required for stem cell differentiation in the planarian Schmidtea mediterranea . Development 137: 1231–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solana J, Kao D, Mihaylova Y, Jaber‐Hijazi F, Malla S, Wilson R, Aboobaker A (2012) Defining the molecular profile of planarian pluripotent stem cells using a combinatorial RNAseq, RNA interference and irradiation approach. Genome Biol 13: R19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, Satija R (2019) Comprehensive integration of single‐cell data. Cell 177: 1888–1902.e21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu KC, Cheng L‐C, T K Vu H, Lange JJ, McKinney SA, Seidel CW, Sánchez Alvarado A (2015) Egr‐5 is a post‐mitotic regulator of planarian epidermal differentiation. eLife 4: e10501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner DE, Ho JJ, Reddien PW (2012) Genetic regulators of a pluripotent adult stem cell system in planarians identified by RNAi and clonal analysis. Cell Stem Cell 10: 299–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li Y, Yue M, Wang J, Kumar S, Wechsler‐Reya RJ, Zhang Z, Ogawa Y, Kellis M, Duester G et al (2018) N6‐methyladenosine RNA modification regulates embryonic neural stem cell self‐renewal through histone modifications. Nat Neurosci 21: 195–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenemoser D, Reddien PW (2010) Planarian regeneration involves distinct stem cell responses to wounds and tissue absence. Dev Biol 344: 979–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilczewski CM, Hepperla AJ, Shimbo T, Wasson L, Robbe ZL, Davis IJ, Wade PA, Conlon FL (2018) CHD4 and the NuRD complex directly control cardiac sarcomere formation. Proc Natl Acad Sci USA 115: 6727–6732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wolfswinkel JC, Wagner DE, Reddien PW (2014) Single‐cell analysis reveals functionally distinct classes within the planarian stem cell compartment. Cell Stem Cell 15: 326–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vásquez-Doorman C, Petersen CP (2016) The NuRD complex component p66 suppresses photoreceptor neuron regeneration in planarians. Regeneration 3: 168–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurtzel O, Cote LE, Poirier A, Satija R, Regev A, Reddien PW (2015) A generic and cell‐type‐specific wound response precedes regeneration in planarians. Dev Cell 35: 632–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurtzel O, Oderberg IM, Reddien PW (2017) Planarian epidermal stem cells respond to positional cues to promote cell‐type diversity. Dev Cell 40: 491–504.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Hsu PJ, Chen Y‐S, Yang Y‐G (2018) Dynamic transcriptomic m6A decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res 28: 616–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccara S, Jaffrey SR (2020) A unified model for the function of YTHDF proteins in regulating m6A‐modified mRNA. Cell 181: 1582–1595.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng A, Li H, Guo L, Gao X, McKinney S, Wang Y, Yu Z, Park J, Semerad C, Ross E et al (2018) Prospectively isolated Tetraspanin+ neoblasts are adult pluripotent stem cells underlying planaria regeneration. Cell 173: 1593–1608.e20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Chen Y, Sun B, Wang L, Yang Y, Ma D, Lv J, Heng J, Ding Y, Xue Y et al (2017) m6A modulates haematopoietic stem and progenitor cell specification. Nature 549: 273–276 [DOI] [PubMed] [Google Scholar]
- Zheng Z, Zhang L, Cui X‐L, Yu X, Hsu PJ, Lyu R, Tan H, Mandal M, Zhang M, Sun H‐L et al (2020) Control of early B cell development by the RNA N6‐Methyladenosine methylation. Cell Rep 31: 107819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu SJ, Hallows SE, Currie KW, Xu C, Pearson BJ (2015) A mex3 homolog is required for differentiation during planarian stem cell lineage development. eLife 4: e07025 [DOI] [PMC free article] [PubMed] [Google Scholar]