

Abstract
Cellular senescence is a stress response elicited by different molecular insults. Senescence results in cell cycle exit and is characterised by multiple phenotypic changes such as the production of a bioactive secretome. Senescent cells accumulate during ageing and are present in cancerous and fibrotic lesions. Drugs that selectively kill senescent cells (senolytics) have shown great promise for the treatment of age‐related diseases. Senescence plays paradoxical roles in cancer. Induction of senescence limits cancer progression and contributes to therapy success, but lingering senescent cells fuel progression, recurrence, and metastasis. In this review, we describe the intricate relation between senescence and cancer. Moreover, we enumerate how current anticancer therapies induce senescence in tumour cells and how senolytic agents could be deployed to complement anticancer therapies. “One‐two punch” therapies aim to first induce senescence in the tumour followed by senolytic treatment to target newly exposed vulnerabilities in senescent tumour cells. “One‐two punch” represents an emerging and promising new strategy in cancer treatment. Future challenges of “one‐two punch” approaches include how to best monitor senescence in cancer patients to effectively survey their efficacy.
Keywords: cellular senescence, chemotherapy, oncogene‐induced senescence, radiotherapy, senolytics, therapy‐induced senescence
Conventional cancer therapies induce senescence. While senescence contributes to therapy outcome, accumulation of senescent cells can promote cancer progression, metastasis and therapy resistance. One‐two punch approaches aim to combine cancer therapies with senolytics to remove senescent tumour cells. Here, we review senescence in the context of cancer and discuss future challenges of senotherapies for cancer treatment.
Abbreviations
- 17‐AAG
17‐allylamino‐17‐demethoxygeldanamycin
- 17‐DMAG
17‐dimethylaminoethylamino‐17‐demethoxygeldanamycin
- ADCC
antibody‐dependent cell‐mediated cytotoxicity
- AML
acute myeloid leukaemia
- AMPK
AMP‐activated protein kinase
- AURK
aurora kinase
- BET
bromodomain and extraterminal domain
- BETd
bromodomain and extraterminal domain degrader
- CAR T cell
chimeric antigen receptor T cell
- CDK
cyclin‐dependent kinase
- CG
cardiac glycoside
- cGAMP
cGMP‐AMP
- cGAS
cyclic GMP‐AMP synthase
- CML
chronic myeloid leukaemia
- D +Q
dasatinib and quercetin
- DDR
DNA damage response
- DNA‐SCARS
DNA segments with chromatin alterations reinforcing senescence
- DNMT
DNA methyltransferase
- DRI
D‐Retro inverso
- EMT
epithelial‐mesenchymal transition
- GMD
galactose‐modified duocarmycin
- HCC
hepatocellular carcinoma
- HDAC
histone deacetylase
- HNSCC
head and neck squamous cell carcinoma
- HR
hormone receptor
- HSP
heat shock protein
- HUVEC
human umbilical vein endothelial cell
- ICI
immune checkpoints inhibitors
- ICSA
International Cell Senescence Association
- IKK
IkB kinase
- KAT
histone lysine acetyltransferase
- LSEC
liver sinusoidal endothelial cell
- MEF
mouse embryonic fibroblast
- MMP
matrix metalloproteinase
- NK
natural killer
- NSCLC
non‐small cell lung cancer
- OIS
oncogene‐induced senescence
- OSCC
oral squamous cell carcinoma
- PARP
poly(ADP‐ribose) polymerase
- PARPi
PARP inhibitor
- PASP
p21‐activated secretory phenotype
- PDAC
pancreatic ductal adenocarcinoma
- PI3K
phosphatidylinositol tri‐phosphate
- PICS
PTEN loss‐induced cellular senescence
- PIN
prostate intraepithelial neoplasia
- PLK
polo‐like kinase
- PROTAC
proteolysis targeting chimera
- ROS
reactive oxygen species
- RTK
receptor tyrosine kinase
- SAHA
suberoyanilide hydroxamic acid
- SASP
senescence‐associated secretory phenotype
- SA‐β‐gal
senescence‐associated β‐galactosidase
- STING
stimulator of interferon genes
- suPAR
soluble form of uPAR
- TIS
therapy‐induced senescence
- TKI
tyrosine kinase inhibitor
- TME
tumour microenvironment
- uPAR
urokinase plasminogen activator receptor
Introduction
During their lifetime, cells are subjected to a variety of damages. Depending on the nature and strength of those damages, cells can repair them. When this is not possible, they activate death signalling pathways to avoid the impact that damage cells could have on tissue homeostasis. An alternative to trigger cell death is cellular senescence. Cellular senescence is a highly stable cell cycle arrest initiated in response to a variety of stress signals to prevent the replication of old, damaged or preneoplastic cells [1]. The implementation of the senescence growth arrest depends on the activation of the p21CIP1/WAF1/p53 and p16 INK4A/RB tumour suppressor pathways [2].
Besides undergoing a highly‐stable cell cycle arrest, senescent cells reprogram their metabolism, suffer structural changes, epigenetic modifications and macromolecular (DNA, proteins, lipids) damage and induce a bioactive secretome, termed the senescence‐associated secretory phenotype (SASP) [1, 3, 4, 5]. How the SASP is regulated has been reviewed elsewhere [6, 7]. Briefly, the SASP is linked to the DNA damage response (DDR) [8] and its induction requires sensing macromolecular damage by elements of the innate immune pathways (such as cGAS/STING or the inflammasome) [9, 10, 11]. Eventually, these signals are integrated and activate key factors such as mTOR [12, 13], GATA4 [14] or p38 MAPK [15]. Ultimately, the sensors and mediators of the SASP induction converge to the activation of key transcriptional factors such as NF‐κB and C/EBPβ that control the expression of key inflammatory factors such as IL‐1β, IL‐6 and IL‐8 [16, 17, 18]. The secretion of these factors creates a feedback loop that reinforce the SASP phenotype [19].
Senescence was initially described in 1961 by Hayflick et al. [20] who revealed that normal cells stop to replicate after a finite number of passages. Nowadays, we refer to this phenomenon as replicative senescence. It is attributed to the end replication problem that shortens telomere, provokes persistent DNA damage and induces the downstream activation of senescence [21, 22]. Importantly, other insults such as oncogene activation, genotoxic and oxidative stress or mitochondrial defects also lead to senescence independently of telomeric shortening (referred collectively as premature senescence) [23]. For example, expression of oncogenic RAS in primary cells is associated with a permanent cell cycle arrest phenotypically indistinguishable from cellular senescence (oncogene‐induced senescence, OIS) [24].
Importantly, senescence plays multiple roles in health and disease. Transient senescence activation is part of normal tissue development through embryogenesis (developmental senescence) [25, 26] to adulthood (where it maintains tissue homeostasis) [27]. Induction of senescence limits the replication of damaged cells and elicits their elimination by the immune system in a SASP‐dependent fashion. In this context, acute senescence is beneficial, and for example contributes to limit fibrosis [28] and cancer initiation [29]. However, when senescent cells linger in a tissue they often play detrimental roles, contributing to ageing and many diseases, including paradoxically cancer progression [30].
Identifying senescent cells is key to better understand their roles in vivo. This is often achieved by assaying for senescence‐associated β‐galactosidase activity (SA‐β‐gal), upregulated in senescent cells due to their increase in lysosomal biogenesis [31]. Other markers such as expression of the cell cycle regulators p16INK4A, reduced levels of Lamin B1, absence of proliferation or induction of SASP components are also used to identify senescence. Due to the heterogeneity of senescence, a combination of those markers is needed to assess senescence, as summarised in the consensus position from the International Cell Senescence Association (ICSA) [1].
Senescent cells in tumours
Different types of senescent cells are present in the tumour microenvironment (TME) during cancer initiation, progression and in response to therapy. Many preneoplastic lesions, including lung adenomas [32], melanocytic nevi [33], lymphomas [34] or prostate intraepithelial neoplasia (PIN) [35] are enriched in senescent cells. This is because activation of oncogenes (e.g., RAS in lung or BRAF in nevi) or loss of tumour suppressor (e.g., PTEN in the prostate) induces senescence, what restrains tumour progression. Another contributor to senescence induction in the context of tumorigenesis are anticancer treatments, as radiotherapy, conventional chemotherapy and some targeted therapies: that cause so‐called therapy‐induced senescence (TIS) in the tumour cells [36]. Cancer therapies can also induce senescence in cells other than the tumour cells. Indeed, induction of senescence in normal tissues has been suggested to cause some of the side effects associated with chemotherapy [37]. Finally, other cells in the TME might also undergo senescence [38]. Stromal senescent cells are an emerging factor contributing to tumorigenesis and promoting cancer drug resistance [39, 40]. Senescent cells in the TME can also arise in a paracrine fashion, as factors secreted by tumour (senescent) cells can induce senescence in the stroma or render infiltrating immune cells senescent. For example, implanting different tumour cells grafts (breast, pancreas, endometria, lung) in a p16INK4a luciferase reporter mice results in luciferase activity arising in the tumour‐associated stroma, demonstrating the ability of tumours to induce senescence in their surroundings [41].
Paradoxical roles of senescence in cancer
Senescence exerts multiple and sometimes opposing effects in tumorigenesis. The oncogenic activation events involved in cancer initiation trigger OIS in preneoplastic lesions and limit their progression. Consequently, mutations that disable senescence are needed for tumours to progress to more malignant stages. Most cancer therapies work, at least in part, by triggering senescence (TIS). But TIS in non‐cancerous cells has been linked to some of the side effects associated with chemotherapy [37]. And lingering senescent cells present in the tumour and TME contribute to sustain cancer development and progression [42].
Antitumorigenic roles of senescence
Events that drive cancer initiation, such as activation of oncogenes (e.g., RAS or BRAF) or loss of tumour suppressors (e.g., PTEN), trigger OIS (in the case of PTEN loss also known as PTEN loss‐induced cellular senescence or PICS). OIS restricts tumorigenesis by imposing a stable cell cycle arrest in preneoplastic cells [32, 33, 34] that facilitates the subsequent elimination of these cells by the immune system, thus contributing to tumour clearance [43]. Surveillance by the immune system is an important part of the antitumour senescence response. Senescence immunosurveillance is initiated upon secretion of immunomodulatory cytokines by senescent cells as part of the SASP. For example, premalignant senescent hepatocytes are cleared in vivo through a CD4+ T cells response requiring monocytes and macrophages to actively eliminate the senescent cells [29]. This clearance is dependent of expression of IL1, CCL2 and other SASP factors [9]. The initiation of immune surveillance precedes full senescence: induction of p21 is sufficient to promote a secretome (termed PASP or p21‐activated secretory phenotype) that can attract macrophages [44]. The SASP, produced by hepatocytes [44] or senescent stellate cells [45], induces M1 polarisation in macrophages, favouring clearance of senescent cells. On the contrary, p53‐deficient stellate cells promote an M2 polarisation, creating a pro‐tumorigenic microenvironment which stimulates the proliferation of cancer cells [45]. Mouse liver carcinomas undergoing senescence recruit NK cells in a way that promotes the recruitment of NK cells, and the elimination of senescent tumour cells through a NKG2D‐dependent response [43, 46]. In summary, senescence limits cancer progression by triggering arrest of cancerous cells and activating different components of the immune system.
Disabling senescence is needed for tumour progression and resistance to therapy
Although the senescence cell‐cycle arrest is considered as irreversible, it is believed that cells that have undergone senescence could re‐entry cell cycle [47]. This might explain how tumours progress or become resistant to anticancer therapies. There is evidence of senescent cells escaping from replicative senescence [47] and OIS [48]. Gorgoulis' group recently defined the concept of the escape from oncogene‐induced senescence (EOIS) helped by cellular models where deregulation of oncogenic signalling results in EOIS [48]. Upon CDC6 expression in epithelial cells, an early chromosomal reorganisation (inversion of chromosome 3) induces the circadian transcription factor Basic Helix–Loop–Helix Family Member E40 BHLHE40 activation (circadian clock machinery regulation). This is associated with senescent cells' re‐entry in cell cycle, likewise demonstrating a link between genomic instability and senescence escape [48].
Cancer cells can also escape TIS. MCF‐7 breast cancer cell clones treated with therapeutic doses of doxorubicin, after a burst of senescence induction, can recover and regain their proliferative ability that is associated with CDC2 overexpression [49]. H1229 non‐small cell lung cancer cells exposed to a variety of chemotherapeutic agents can also escape from TIS by overexpressing CDC2/CDK1, what is a very rare event [50]. Induction of senescence has been also shown to induce reprogramming. Interestingly lymphoma cells that have undergone senescence show an upregulation of reprogramming and display enhanced aggressiveness [51]. A recent study on primary acute myeloid leukaemia (AML) patients' cells revealed that even if chemotherapy induces a senescence‐like phenotype, this phenotype is transient and contributes to cancer relapse by promoting aggressiveness and stem cell potential of tumour cells [52]. Cancer cells that escape from TIS are often polyploid [53] and express markers of stemness and aggressiveness [54]. Senescent tumour cells can represent a form of “cell dormancy”, and thus also contributes to disease recurrence [55].
Pro‐tumorigenic roles of senescence
While senescence can act as an intrinsic or evoked antitumour response, in the long term, the persistence of senescent cells has detrimental effects on tumorigenesis, promoting tumour progression, dissemination and recurrence [37, 56].
Tumour cell senescence can lead to the emergence of more aggressive variants. Particularly, chemotherapy‐induced senescence can reprogram cancer cells which acquired stemness properties, thus enhancing their aggressiveness, resistance to therapy and relapse [51]. TIS can also promote cancer relapse by preventing the induction of apoptosis by chemotherapeutics. In the context of wild‐type p53, doxorubicin failed to induce apoptosis of mammary tumours in mouse model, while p53 mutant cells did not undergo cell cycle arrest and were killed due to mitotic abnormalities [56]. This indicates that p53 status is an important factor influencing cancer therapy, in part by affecting senescence induction.
Senescent cells communicate with adjacent cells via ligands/receptors interactions to modulate their state and their microenvironment [6, 7]. Senescent cells can promote the proliferation of adjacent tumour cells in a paracrine manner via the SASP [57]. For example, in a model of paediatric craniopharyngioma, oncogene activation in pituitary stem cells leads to senescence and the SASP drives tumorigenesis in a paracrine manner [58]. The SASP can also reinforce senescence or cause paracrine senescence [9, 19]. During therapy‐induced senescence (TIS), especially upon radiotherapy, there is a bystander signalling mediated by inflammatory mediators and gap junctions that can be related to paracrine senescence [59]. Whether the reinforced senescent phenotype will allow a better immune clearance of the cells or in the contrary, confer a higher pro‐tumorigenic environment could depend on the context. The SASP of senescent cells lingering in the tumour microenvironment contributes to create a chronic inflammatory environment [60]. The SASP of senescent fibroblasts can promote tumour growth and dissemination, in part through the induction of epithelial‐mesenchymal transition (EMT) in adjacent cells [61]. The SASP also influences immune responses beyond the positive effects of senescence immunosurveillance. While SASP production stimulates the clearance of senescent cells by the immune system [9, 29], when senescence is bypassed or if the senescent cells are not fully eliminated, senescent cancer cells can reshape the immune microenvironment promoting immune escape. For example, in the context of liver cancer, the SASP can recruit CCR2+ immature myeloid cells (iMCs) that inhibit NK cell action, promoting cancer progression [62]. Similarly, senescence observed in PTEN‐null prostate cancer results in immature myeloid cell infiltration that antagonises senescence, promoting tumour progression [63].
A senescent stroma plays detrimental role in cancer. Co‐injection of senescent fibroblasts with skin, breast or prostate cancer cells favours the aggressiveness of the generated tumours [39, 64, 65]. The tumour‐promoting effect of senescent fibroblasts is likely to be mediated by the secretion of SASP factors that can foster a growth‐stimulating and/or pro‐angiogenic microenvironment [66]. Senescent endothelial cells are also present in the tumour microenvironment and secrete factors that influence the tumour behaviour [67]. For example, SASP factors like CXCL11 secreted by senescent endothelial cells contribute to the cancer aggressiveness of breast cancer [68].
Fostering immunosuppression is another way by which senescent cells can promote tumour progression. For example, in a model of aged skin, senescent stromal cells recruit suppressive myeloid cells and secrete SASP factors like IL‐6 to reinforce immunosuppression. The same observation was made in cancer patients where senescent stromal cells are adjacent to immune cells, thus creating a tumour permissive microenvironment [39]. Other strategy used by senescent cells to escape immune surveillance is the matrix metalloproteinase (MMP)‐mediated shedding of NKG2D ligands [69]. Elimination of senescent cells by NK cells involves the recognition of NKG2D ligands at their cell surface. While the expression of NKG2D ligands is induced during senescence, senescent cells also secrete MMPs, that cleave NKG2D ligands, avoiding NK surveillance.
Cancer therapy and senescence‐inducing drugs
Many cancer therapeutics induce senescence. Examples include drugs that trigger DNA damage, i.e., genotoxic chemotherapy or radiotherapy (Table 1). Various targeted therapies, such as CDK4/6 inhibitors, can also induce senescence without being genotoxic (Table 2). Some of these senescence‐inducing drugs can cause senescence not only in the tumour itself but also in the TME. Thus, induction of senescence by cancer therapies plays key role in cancer treatment but can also cause unwanted side effects associated with lingering senescent cells.
Table 1.
Senescence inducers: conventional chemotherapy and radiotherapy.
| Class | Drug | Cancer | Model |
|---|---|---|---|
| Alkylating agents | Cisplatin | Fibrosarcoma | HT1080 [70] |
| Melanoma | A375, B16F10 [71] | ||
| Nasopharyngeal carcinoma | CNE1 [72] | ||
| Cyclophosphamide | B‐cell lymphoma | Eμ‐Myc;ectopic Bcl2 mouse model [73] | |
| Temozolomide | Glioma | U87MG [74, 75]; GaMG [74]; U87, LN229 [76]; LN229 [77] | |
| Melanoma | MM200, IgR3, SK‐MEL‐28, Mel‐FH [78] | ||
| Topoisomerase inhibitors | Doxorubicin | B‐cell lymphoma | Eμ‐Nras G12V;ectopic Bcl2 mouse model [34] |
| Breast cancer | MCF‐7 [70, 79, 80]; Reporter Mouse model p16‐3MR‐MMTV‐PyMT grafts [37]; patients [79] | ||
| Cervical carcinoma | HeLa [70] | ||
| Colon cancer | HCT‐116, SW480 [70]; LS174T, HCA‐7 [79] | ||
| Fibrosarcoma | HT1080 [70] | ||
| Glioma | U251 [70] | ||
| HCC | HepG2 [70] | ||
| Larynx carcinoma | Hep‐2 [70] | ||
| Lung cancer | NCI‐H460, A549 [81] | ||
| Osteosarcoma | Saos2 [70] | ||
| Ovarian cancer | A2780 [70, 79] | ||
| Prostate cancer | LNCaP [70, 82]; DU145 [82] | ||
| Daunorubicin | T‐cell lymphoma | Jurkat T cells [83] | |
| Etoposide | Fibrosarcoma | HT1080 [70] | |
| HCC | HepG2, U2OS [84] | ||
| Irinotecan | Colorectal cancer | HCT‐116 [85, 86, 87]; LS174T [86, 87] | |
| Microtubule inhibitors | Docetaxel | Prostate cancer | LNCaP, DU145 [82]; Pten pc−/− mouse model [88] |
| Paclitaxel | Breast cancer | MCF‐7 [89, 90]; MDA‐MB‐231 [90] | |
| Colorectal cancer | HCT‐116 [90] | ||
| Melanoma | A549 [90] | ||
| Neuroblastoma | SH‐SY5Y [90] | ||
| Antimetabolites | Gemcitabine | Pancreatic carcinoma | Panc1 [91, 92]; Miapaca‐2 [92] |
| Methotrexate | Breast cancer | MCF‐7 [93] | |
| Cytotoxic antibiotic | Bleomycin | Lung cancer | A549 [94, 95] |
| Radiotherapy | Breast cancer | MCF‐7 [96] | |
| Glioma | U87 [97] |
Table 2.
Senescence inducers: targeted therapies.
| Class | Drug | Cancer | Model |
|---|---|---|---|
| CDK4/6 inhibitors | Palbociclib | Breast cancer | V720 [98]; MCF‐7, T47D [99] |
| Gastric cancer | AGS, MKN‐45 [100] | ||
| Glioblastoma | DBTRG‐05MG, LN229, U87MG [101] | ||
| HNSCC | CAL27, HN31, PCI15B [102] | ||
| HCC | Huh7, SK‐Hep1 [103] | ||
| Liposarcoma | U2OS [104]; LS8107 [105] | ||
| Lung cancer | A549, H2030, H460 [106]; LSL‐KrasG12V;RERT +/ERT mouse model [107] | ||
| Melanoma | SK‐MEL‐103, SK‐MEL‐103 xenografts [108]; MEL‐10 [109]; SKMEL2 [98] | ||
| OSCC | SAS, OECM1 [110] | ||
| Osteosarcoma | U2OS [98] | ||
| Abemaciclib | Breast cancer | MD‐MB‐453 [111, 112, 113]; BT474 [113]; MCF‐7 [114]; MMTV‐rtTA/tetO‐HER2 mouse model [113] | |
| Ribociclib | Ewing sarcoma | SKNEP‐1 [115] | |
| Neuroblastoma | BE2C, IMR5, EBC1 [116] | ||
| Ovarian cancer | Hey1 [117] | ||
| Epigenetic modulators | 5‐azacytidine | HCC | HepG2 [118, 119]; Huh‐7 [119] |
| Prostate cancer | LNCaP, C4‐2B [82] | ||
| 5‐aza‐2′‐deoxycytidine/decitabine | CML | K‐562, MEG‐01, KBM‐5 [120] | |
| Lung mesothelioma | H28 [121] | ||
| Osteosarcoma | U2OS [122] | ||
| HDACi | SAHA/vorinostat | AML | MOLM‐7, HL‐60, JURK‐MK1 [123] |
| Colorectal cancer | HCT‐116 [124] | ||
| T‐cell lymphoma | MyLa, MJ [125] | ||
| Sirtinol | Breast cancer | MCF‐7 [126, 127] | |
| CML | K562 [128] | ||
| Lung cancer | H1299 [126] | ||
| AURK inhibitors | Alisertib | Colorectal carcinoma | HCT‐116, HCT‐116 xenografts [129] |
| Glioblastoma | GB169 [130] | ||
| Lung cancer | A549 [131] | ||
| Pancreatic cancer | AsPC‐1, BxPC‐3, MIA PaCa‐2, PANC‐1 [132] | ||
| Barasertib | Lung cancer | A549 [131] | |
| Melanoma | A375 [133] | ||
| Danusertib | Glioblastoma | GBM2, G166 [134] | |
| Tozasertib | Lung cancer | A549 [131] | |
| PLK inhibitors | BI‐2536 | Colorectal carcinoma | HCT‐116 [135, 136]; SW480, HCT‐116 xenografts [135] |
| Lung cancer | A549 [135] | ||
| BI‐6727/volasertib | Colorectal carcinoma | HCT‐116 [136] | |
| Lung cancer | A549 [137] | ||
| CFI‐400945 | HCC | MHCC97L [138] | |
| Ovarian cancer | OCC1, ES2 [139] | ||
| Prostate cancer | 22Rv1, DU145, PC‐3, LNCaP, C4‐2 [140] | ||
| PARP inhibitors | Olaparib | AML | KASUMI and NB4‐LR2 [141] |
| Breast cancer | MDA‐MB‐231, OV4453, OV1946 and MDA‐MB‐231 xenografts [142] | ||
| Ovarian cancer | OV1369(R2), OV90, OV4453, OV1946 [142]; SKVO3, A2780, OVCAR‐3 [143] | ||
| Prostate cancer | LNCaP, C4‐2B [144] | ||
| Rucaparib | Prostate cancer | LNCaP, C4‐2B, PC‐3 [145] | |
| Niraparib/Talazoparib | Ovarian cancer | OV1369(R2) [142] | |
| ABT‐888/veliparib | Breast cancer | MCF‐7, MCF‐7 xenografts [146] | |
| Cdc7 inhibitor | XL413 | HCC | Hep3B, Huh7, MHCC97H, PLC/PRF/5 p53 mutant cells [147] |
| Hormone therapy | Tamoxifen | Breast cancer | MCF‐7 [148, 149] |
| ADT | Prostate cancer | LNCaP [150, 151]; LAPC4; LuCaP [151]; Patient biopsies [151] | |
| Bicalutamide | Prostate cancer | LNCaP, LAPC4 [152] | |
| Immunotherapy | Rituximab | B cell lymphoma | EHEB, RC‐48, SD1 [153] |
| Trastuzumab & pertuzumab | Breast cancer | SK‐BR‐3 [154] | |
| VEGF/VEGFR inhibitors | Bevacizumab | Colorectal cancer | MIP101, RKO, SW620, SW480 [155] |
| AZD‐2171 | Colorectal cancer | HCT‐116 [156] | |
| MEK inhibitor | Trametinib | Melanoma | A375, D04 [157]; DMBC11, DMBC12, DMBC21, DMBC28, DMBC17 [158] |
| Lung cancer | A549, H2030, H460 [106] | ||
| B‐Raf inhibitor | Vemurafenib | Melanoma | DMBC11, DMBC12, DMBC21, DMBC28, DMBC17 [158]; M14, M19‐Mel, Malme 3 M, SK‐MEL‐28, UACC‐62, Mel2a, FM88 [159] |
| SKP2 inhibitor | Lung cancer | A549, H1299 [160] | |
| Prostate cancer | PC‐3 [161, 162] |
Chemotherapies
Cytotoxic chemotherapy includes a wide range of drugs that inhibit mitosis or induce DNA damage in cancer cells. Pioneering studies showed that besides killing them, agents such as cisplatin [72] and doxorubicin [70] can also induce senescence in cancer cells. Lower drug dose and chronic administration often result in senescence rather than apoptosis and cell death [163].
Alkylating agents, mainly represented by cisplatin and derivates, alkylate and bind covalently the DNA to form DNA crosslinks. These structures are susceptible to breakage during DNA replication and may lead to the activation of DDR. In the same way, topoisomerase inhibitors like doxorubicin or etoposide affect the ability of the DNA to replicate, by preventing the repair of DNA breaks required to reduce the tension of the unwound DNA strand during the replication. Thus, DNA damage triggers activation of p53/p21 [164] or p16 [73] and causes the subsequent proliferation arrest and senescence of cancer cells. Treatment with different chemotherapeutic agents also results in SASP induction.
So‐called antimicrotubule agents, such as the taxanes paclitaxel and docetaxel, and vinca‐alkaloids, are a class of cancer drugs that interfere with normal mitosis by causing microtubule dysfunction. Paclitaxel has been described to induce senescence of breast cancer [89], melanoma and neuroblastoma cells [90]. Docetaxel has been shown to induce senescence in different models of prostate cancer, where it is used therapeutically [82, 88]. Antimetabolites, that impair the incorporation of purines and pyrimidines to the DNA during the S phase, also induce senescence. Gemcitabine and methotrexate have been described as senescence inducers, in a p21‐dependent fashion [91, 92]. Methotrexate treatment seems to induce a p53‐dependent senescence without DNA damage [93] that could be linked to reduced DNA synthesis [165]. Finally, bleomycin, a cytotoxic antibiotic used to treat many cancer types, induces a p21‐dependent senescence response [94, 95].
Radiotherapy
Radiotherapy is used to treat many cancer types. Like chemotherapies, irradiation not only causes cell death but can also result in senescence. Either γ‐irradiation [70] or exposure to x‐ray [166] triggers a senescent response. Induction of the senescent phenotype is dependent on the p53 function and induction of DDR in telomeres but independent of telomere shortening [167]. One of the advantages of radiotherapy when compared with cytotoxic chemotherapies is their local administration, which reduces side effects. Despite that, irradiation of surrounding healthy tissue can cause side effects such as radiation‐induced lung fibrosis (RILF), a severe side effect of radiotherapy observed in lung cancer patients [168]. Senescence of cells in the TME (macrophages, pneumocytes and fibroblasts) is known to be part of this complication [169, 170, 171]. Interestingly, FLASH irradiation (consisting in the delivery of large doses of radiation in a fraction of second) minimises the DNA damage of normal lung cells in vitro and of irradiated mice lung in vivo compared to conventional radiotherapy, thus preserving normal lung from radio‐induced senescence and associated lung fibrosis [172].
CDK4/6 inhibitors
Cyclin‐dependent kinases 4 and 6 (CDK4/6) are responsible for the cell cycle transition from G1 to S phase by phosphorylating RB family proteins. Cancer cells often present a hyperactivated CDK4/6 to sustain their proliferation. Therefore, CDK4/6 inhibitors are a promising new cancer therapy [173]. The first CDK4/6 inhibitor, palbociclib, was described in 2004 and later approved by the FDA as a treatment for hormone receptor (HR) + HER2‐ breast cancer [174]. Two other CDK4/6 inhibitors abemaciclib [175] and ribociclib [176] are currently used in the clinic, while others are under evaluation [177]. CDK4/6 activity is needed for cell cycle progression and is often upregulated in tumours, frequently in absence of genetic alterations but due to the upregulation of mitogenic signalling [111]. CDK4/6 inhibitors inhibit E2F transcriptional activity leading to an RB‐dependent cell proliferative arrest. While CDK4/6 inhibitors can induce quiescence, they also can trigger senescence in various cancer cells [178, 179]. Acquired resistance to CDK4/6 inhibitors occurs in highly responsive HR‐positive breast cancer. Loss of RB and overexpression of cyclin E are associated with palbociclib resistance [180, 181, 182]. Pandey et al. also demonstrated the overexpression of cyclin E in palbociclib‐resistant cells. Cyclin E‐CDK2 interaction could be targeted by inhibition of CDK2, and combined inhibition of CDK2‐CDK4/6 synergistically overcomes CDK4/6 resistance and enhances senescence, highlighting the therapeutic relevance of senescence [183].
AURK/PLK inhibitors
Aurora kinases (AURK) are essential serine/threonine kinases that control spindle formation and mitotic progression [184]. Aurora kinases are overexpressed in a broad range of human tumours, including gastrointestinal, breast, ovarian and pancreatic cancer [185]. The expression of AURK is linked to genomic instability and cancer, and due to their role as cell cycle regulators, their inhibition is of great interest for cancer therapy [186]. Inhibitors of Aurora kinases have been proven effective as cancer treatments [187]. Interestingly, the Aurora A inhibitor alisertib, Aurora B inhibitor barasertib and the pan Aurora A/B/C inhibitors danusertib and tozasertib are potent inducers of senescence in cancer cells [129, 131].
Polo‐like kinases (PLK) are another family of serine/threonine kinases essential for cell mitosis [188]. PLK1 is overexpressed in a wide range of tumours [189]. Studies demonstrated the therapeutic value of PLK1 inhibition, particularly in endocrine‐resistant breast cancer, as overexpression of PLK1 is observed in the patient with cancer relapse [190]. The PLK1 inhibitor BI2536 enhances the effect of paclitaxel on MCF‐7 and T‐47D breast cancer cells, by inducing their apoptosis, whereas it prevents tamoxifen‐induced senescence [191]. On the contrary, PLK1 inhibition by MLN0905 promotes senescence in a variety of cancer cells [135]. Interestingly CFI‐400945, an inhibitor of the PLK family member PLK4, induces senescence in cancer cells through cytosolic DNA accumulation and activation of the cGAS/STING pathway [138, 139].
Epigenetic modulators
Several drugs that target epigenetic regulators cause senescence. For example, 5‐azacytidine and its deoxy derivate 5‐aza‐2′‐deoxycytidine (decitabine) are analogues of the nucleoside cytidine and used in cancer therapy as inhibitors of DNA methyltransferase [192]. While developed as cytostatic drugs, particularly in haematological cancers, the response in cancer cells was further investigated to demonstrate that senescence induction also contributes to their antiproliferative effects [82, 121]. The mechanism of senescent induction and its ability to induce senescence or apoptosis depends on context. 5‐aza‐2′‐deoxycytidine but not 5‐azacytidine induces profound DNA damage leading to senescence. On the contrary, 5‐azacytidine, known to block RNA synthesis, decreases p53 protein accumulation and leads to the apoptosis of HepG2 cells [118]. Histone deacetylase (HDAC) inhibitors such as vorinostat (suberoyanilide hydroxamic acid, SAHA) are used for the treatment of different cancers such as cutaneous T‐cell lymphomas. HDAC inhibitors cause senescence in different cancer types [124, 125]. The SIRT1 inhibitor sirtinol also causes senescence, although the precise mechanism responsible is unclear [126, 127, 128]. More recently, an inhibitor of the histone lysine acetyltransferases (KATs) KAT6A and KAT6B, has been shown to induce INK4/ARF‐dependent senescence in cancer cell lines, explaining their ability to hinder lymphoma progression [193].
PARP inhibitors
Poly(ADP‐ribose) polymerase (PARP) is involved in the detection of single‐strand breaks (SSBs) and plays important roles in regulating DNA repair and genomic stability. BRCA1 or BRCA2 mutant tumours are sensitive to PARP inhibitors (PARPi) due to their defects on DNA repair [194]. Other tumours with DNA repair defects also display synthetic lethality with PARPi. In other cases, treatment with PARPi causes senescence. For example, treatment of MCF‐7 breast cancer cells with veliparib increases the number of DNA damage foci observed in response to radiation, inducing senescence [146]. This suggested that DNA damage sensitises the cancer cells to the senescence induced by PARPi. Indeed, KASUMI and NB4‐LR2 AML cells, bearing compromised DDR genetic alterations, are sensitive to olaparib‐induced senescence, in contrary to THP‐1 cells [141]. Similar responses have been observed in ovarian [143] and prostate cancers [144, 195]. In ovarian cancer, PARPi induces a reversible senescent‐like phenotype that caused a BCL‐xL‐mediated resistance to apoptosis [142].
Other senescence‐inducing drugs
A number of other drugs and antibodies can induce senescence in cancer cells. In hormone‐dependant cancers, hormone‐receptor antagonists like tamoxifen in breast cancer or bicalutamide in prostate cancer not only inhibit tumour growth but can also induce senescence [148, 149, 150, 151, 152]. In breast cancer, targeting HER2/neu with trastuzumab or pertuzumab also causes senescence [154]. In melanoma, BRAF‐activating mutations are approximatively present in 50% of all tumours [196]. MAPK2K1 and MAPK2K1 encoding MEK1/2 are frequently mutated in melanoma, overall leading to the hyperactivation of MAP kinase pathway [197]. The development of BRAF and MEK inhibitors (respectively vemurafenib and trametinib, among others), shows great antitumorigenic effect in patients while also inducing senescence and contributing to radiosensitise BRAF‐mutated melanoma cells but also to explain the absence of complete response to vemurafenib [157, 158, 159]. VEGFR2 inhibition that results in AKT downregulation also induces p21 and triggers senescence in colorectal cancer cells [156]. Similar results were obtained with the VEGF inhibitor bevacizumab [155]. The SKP2 E3‐ubiquitin ligase is described as an oncogene in many cancers. SKP2 inhibitors exhibit antitumour activities and leads to a p53‐independent, p21/p27‐dependent induction of senescence in lung and prostate cancers [160, 161, 162]. Finally, rituximab anti‐CD20 immunotherapy exerts antilymphoma activity but also displays a pro‐senescence activity towards B‐cell lymphoma, thus sensitising lymphoma cells to conventional chemotherapies [153].
Senolytics and other senotherapeutics
Senescence has both detrimental and beneficial roles in disease (and cancer) progression. That paradoxical behaviour made that for many years it was not obvious what the best way to exploit senescence therapeutically was. That changed with the development of mouse models (such as the INK‐ATTAC mice) allowing for the selective ablation of senescent cells (senolysis). In a series of seminal studies, it was shown that eliminating senescent cells improved lifespan, healthspan and ameliorates many age‐related diseases [198, 199, 200]. That prompted the search for drugs (so‐called senolytics) that selectively kill senescent cells and have the potential to be used against a myriad of age‐related diseases, including cancer (Table 3).
Table 3.
Uses of senolytic drugs in preclinical models of cancer.
| Class | Drug | Cancer | Senescence induction | Model |
|---|---|---|---|---|
| BH3 mimetics | ABT‐263/navitoclax | Breast cancer | Irradiation, paclitaxel | 4226, Cal51 [201] |
| Doxorubicin | MDA‐MB‐231, MDA‐MB‐231 xenograft [202] | |||
| Colorectal cancer | Etoposide | HCT‐116 [203] | ||
| Glioblastoma | Irradiation | GBM39, GBM76, GBM6, GBM123 [204] | ||
| Temozolomide | GBM39, GBM76 [204] | |||
| Lung cancer | Etoposide, irradiation | A549, A549 xenograft [202] | ||
| Breast, lung cancer, osteosarcoma | Doxorubicin | 4226, SKBR7, Cal51, U2OS, A549 [201] | ||
| Lung, breast, colorectal, hepatocellular cancers | Alisertib or etoposide | A549, MDA‐MB‐231, SUM159, RKO, PC9, MCF‐7, Huh7, Hep3B, H358, T47D, HepG2 [203]. | ||
| Ovarian and breast cancers | Olaparib | OV1946, OV4453, MDA‐MB‐231 [142] | ||
| ABT‐199/venetoclax | Breast cancer | Palbociclib | MCF‐7, patient tumour organoid, PDX [205] | |
| Glioblastoma | Temozolomide | LN‐229 [206] | ||
| Sarcoma | Irradiation | STS117, STS93, STS109 [207] | ||
| ABT‐263/navitoclax + S63845 MCL‐1 inhibitor | Breast cancer | Doxorubicin | HCC712, MDA‐MB‐175, MCF‐7, 747D [201] | |
| Irradiation, paclitaxel | MDA‐MB‐175 [201] | |||
| S63845 MCL‐1 inhibitor | Prostate cancer | Docetaxel | PC3 Luc‐ShTimp1 xenografts [208] | |
| Cardiac glycosides | Ouabain | Craniopharyngioma | Oncogenic β‐Catenin | Hesx1Cre/+;Ctnnb1lox(ex3)/+ mouse model [209] |
| Colorectal cancer | Doxorubicin | HCT‐116 [209] | ||
| Preneoplastic hepatocytes | NRASG12V expression | NRASG12V hydrodynamic injection in mice [209] | ||
| Hepatocellular carcinoma, lung cancer | Etoposide, Barasertib, Alisertib, Tozasertib, Palbociclib | SK‐Hep1 and A549 [209] [210] | ||
| Lung cancer | Etoposide, doxorubicin | A549 [210] | ||
| Bleomycin, Gemcitabine, Doxorubicin, Etoposide, Palbociclib | A549 [211] | |||
| Melanoma | Palbociclib | SK‐MEL‐103 [211] | ||
| Digitoxin | Hepatocellular carcinoma, lung cancer | Etoposide, Barasertib, Alisertib, Tozasertib, Palbociclib | SK‐Hep1 and A549 [209] | |
| Digoxin | Breast cancer | Doxorubicin | PDX [211] | |
| Lung cancer | Bleomycin | A549 [211] | ||
| Lung cancer | Gemcitabine | A549 xenografts [211] | ||
| Melanoma | Palbociclib | SK‐MEL‐103 [211] | ||
| Proscillaridin A | Lung cancer | Bleomycin | A549 [211] | |
| Galacto‐conjugates | Gal‐NP(Nav) | Breast cancer | Palbociclib | Xenograft [212] |
| Nav‐Gal | Lung cancer | Cisplatin | A549 + xenograft [213] | |
| Galactose‐modified duocarmycin | Craniopharyngioma | Oncogenic β‐Catenin | Hesx1Cre/+;Ctnnb1lox(ex3)/+ [214] | |
| BET inhibitor | ARV825 | Colorectal carcinoma | Doxorubicin | HCT‐116 [215] |
| Hepatocellular carcinoma | Obesity‐induced HCC mouse model [215] | |||
| HDAC inhibitor | Panobinostat | HNSCC | Taxol, cisplatin | FaDu, UMSCC47 [216] |
| Lung cancer | Taxol, cisplatin | A549, H460 [216] | ||
| FOXO4 peptidomimetics | Melanoma | Doxorubicin | A375, A375 grafts [217] | |
| mTOR inhibitors | AZD8055 and AZD2014 | Hepatocellular carcinoma | XL413 (CDC7 inhibitor) | Huh7, Hep3B; Huh7 and MHCC97H xenografts; MycOE;Trp53KO‐HCC bearing mice [147] |
BH3 mimetics
Many stresses (such as irradiation or chemotherapy) can either induce senescence or trigger cell death. Consequently, for cells to undergo senescence they not only need to exit the cell cycle but also inhibit apoptosis. One mechanism by which senescent cells prevent cell death is by upregulating anti‐apoptotic BCL‐2 family proteins [218]. BH3 mimetics are a class of drugs that inhibit BCL2 family inhibitors. ABT‐737 was one of the first BH3 mimetic drugs developed and has selectivity towards BCL‐2, BCL‐xL and BCL‐W [219]. Other drugs, such as ABT‐263/navitoclax (orally bioavailable) [220] and ABT‐199/venetoclax (a selective BCL‐2 inhibitor) were derived from it [221].
Three pioneering studies showed that ABT‐263 and ABT‐737 can selectively eliminate senescent cells [222, 223, 224]. Upregulation of anti‐apoptotic BCL‐2 family proteins BCL‐2, BCL‐xL and BCL‐W during senescence was suggested as the basis for this sensitivity to BH3 mimetics [218]. Besides eliminating a wide‐range of mouse and human senescent cells, navitoclax (ABT‐263) can rejuvenate the haematopoietic system of irradiated mice [223] and ABT‐737 selectively eliminated senescent cells in lung and skin [218]. Importantly, in most cell types ABT‐263/navitoclax (a BCL‐2 and BCL‐xL inhibitor) but not ABT‐199/venetoclax (a selective inhibitor for BCL‐2) is senolytic, although there are exceptions. Navitoclax causes severe thrombocytopenia, what has limited its clinical use and encouraged the development of more selective BCL‐2 inhibitors, such as ABT‐199/venetoclax [225].
Nevertheless, not all senescent cells are sensitive to ABT‐263. For example, a recent study of senescent cancer cells showed that LoVo cells undergoing senescence in response to alisertib or etoposide treatment are insensitive to ABT‐263 treatment [203]. Similarly, senescent prostate cancer cells treated with antiandrogens such as enzalutamide are resistant to ABT‐263 treatment [226]. Multiple factors are likely responsible of the different sensitivity to BH3 mimetics. Context‐dependent induction of different antiapoptotic BCL‐2 family proteins, such as MCL‐1 might be an explanation. Recently, scRNA sequencing of prostate cancer cells from Pten pc−/− and Pten pc−/−;Timp1 −/− mouse models showed Mcl‐1 induction upon senescence. Consistently, the MCL‐1 inhibitors S63845, UMI77 and AZD5991 efficiently eliminated those senescent prostate cancer cells [208].
Quercetin, fisetin and other flavonoids
Senescent cells activate anti‐apoptotic and pro‐survival pathways. Quercetin was identified as a senolytic drug chosen from selected candidates regulating anti‐apoptotic and pro‐survival pathways induced during senescence [227]. In that study, it was shown that the combination of the tyrosine kinase inhibitor (TKI) dasatinib and quercetin (D + Q) was promising in the clearance of senescent cells. Interestingly, D + Q effectively cleared senescent cells of old, progeroid Ercc 1−/Δ mice and irradiated mice to increase their cardiovascular function, healthspan and physical endurance [227]. Following the discovery of quercetin as a senolytic, a panel of flavonoids was screened to test their capacity to kill senescent mice and human fibroblasts [228]. Among the flavonoids tested, fisetin demonstrated senolytic activity on Ercc1 −/− mouse embryonic fibroblasts (MEFs) and etoposide‐treated IMR‐90 fibroblasts and reduced senescent cell burden of p16 +/Luc;Ercc1 −/Δ progeroid mice, aged mice, and in human explants. Fisetin eliminates senescent human umbilical vein endothelial cells (HUVECs), but not senescent IMR‐90 cells or primary human adipocytes [229]. Procyanidin C1, another flavonoid, has also been recently shown to display senolytic properties [230].
While flavonoids are commonly consumed in human diet, none of these compounds are currently approved for medical use, despite numerous publications on beneficial effects to treat cancer or cardiovascular disease. Recently, first‐in‐human studies have suggested the efficacy of the senolytic combination dasatinib + quercetin in the improvement of the condition of patients with pulmonary fibrosis and chronic kidney disease [231, 232]. Several clinical trials are currently in progress to evaluate the senolytic activity of quercetin, fisetin (NCT05276895; NCT04815902; NCT04770064) and dasatinib + quercetin (NCT04063124; NCT04685590, NCT02848131; NCT04313634) in patients.
FOXO4 peptidomimetics
FOXO4 is upregulated in senescent cells and contributes to maintain their viability. FOXO4 resides in PML bodies and colocalises with DNA segments with chromatin alterations reinforcing senescence (DNA‐SCARS) where it interacts and sequesters p53 in the nucleus, to prevent apoptosis. Thus, FOXO4‐DRI peptides [D‐Retro Inverso (DRI)‐isoform] compete with endogenous FOXO4, releasing p53 and causing apoptosis of senescent cells [233]. This strategy was successfully validated on irradiated IMR‐90 fibroblasts and eliminates senescent cells in premature ageing mouse models and mice treated with doxorubicin [233]. More recently, other FOXO4 peptidomimetics have been developed [217]. These new peptides showed 3–7 times more effective senolytic activity on doxorubicin‐treated A375 melanoma cells and orthotopic grafts.
Galacto‐conjugated senolytics
Senescent cells display an increase in lysosomal mass that results in higher activity of lysosomal β‐galactosidase [31]. As a result, SA‐β‐Gal activity is one of the most frequently used markers for senescence. Several strategies have taken advantage of the higher β‐galactosidase activity of senescent cells to design drugs with enhanced selectivity towards senescent cells.
This rationale was first tested by designing galacto‐oligosaccharide‐capped nanoparticles, which preferentially released their cargo on senescent cells [234]. This strategy could be combined with imaging agents to identify senescent cells. Encapsulation limited the cytotoxic effects of doxorubicin and transformed it into a senolytic drug. In addition, nano‐encapsulated navitoclax Gal‐NP(Nav) inhibited the tumour growth and the emergence of metastases, while limiting the whole‐body toxicity of navitoclax [108].
Galactose conjugation was taken a step further by using galactose‐derived pro‐drugs rather than encapsulating drugs in galacto‐oligosaccharide‐capped nanoparticles. A galactose derivative prodrug of navitoclax (Nav‐Gal) showed a higher specificity toward cisplatin‐induced senescent A549 cancer cells compared to unconjugated navitoclax. In vivo, the concomitant treatment with cisplatin and Nav‐Gal cleared the senescent cancer cells and reduced the growth of cancer xenografts, sparing platelets and not causing thrombocytopenia [213]. Other studies converted cytotoxic drugs into senolytics by generating galactose‐conjugated pro‐drugs. For example, galactose prodrug derivatives of the alkylating antibiotic duocarmycin displayed senolytic activity both in culture and in vivo [214]. Similarly, SSK1, a galactose prodrug derived from gemcitabine targeted senescent cells, attenuated inflammation and restore the physical activity of old mice [235].
Cardiac glycosides
Unbiased phenotypic screens have also served to identify senolytic drugs. Taking these approaches, two groups independently identified the senolytic potential of cardiac glycosides [209, 211]. Cardiac glycosides (CGs), such as ouabain, digoxin and digitoxin, are natural compounds used in cardiology as inhibitors of Na+/K+‐ATPase. Interestingly, there is an increase in the concentration of intracellular cations in senescent cells, and treatment with CGs decrease K+ levels more prominently in senescent cells, what eventually cause induction of the pro‐apoptotic NOXA gene. Thus, the senolytic activity of cardiac glycosides might be explained by inhibition of the Na+/K+‐ATPase and depolarization of the plasma membrane. In vivo, CGs were able to eliminate different senescent cells including preneoplastic hepatocytes and naturally arising senescent cells in old mice [209]. Moreover, CGs synergise with gemcitabine or doxorubicin to eliminate cancer xenografts [211]. Interestingly, another inhibitor of ATPase pumps, curcumin, and derivatives such as o‐vanillin and EF24 also displays senolytic activity [209, 236, 237].
Other small compounds with senolytic activity
Drug screens have served to identify other drugs with senolytic properties. HSP90 chaperone inhibitors such as geldanamycin, and its derivates 17‐AAG (tanespimycin) and 17‐DMAG (alvespimycin) downregulate the pro‐survival PI3K/AKT pathway, which is associated with a decreased number of senescent cells and an increased in healthspan of Ercc1−/Δ progeroid mice [238].
Another drug screen identified the BET (Bromodomain and Extraterminal domain) family protein degrader (BETd) as a senolytic drug and validated the ARV825 compound as the BET inhibitor with the strongest senolytic activity [215]. ARV825 is a PROTAC (proteolysis targeting chimera), consisting in two active domains, one able to engage an E3 ligase to allow the proteolytic degradation of the chosen protein targeting by the second active domain. This PROTAC eliminates senescent cells in a model of obesity‐induced hepatocellular carcinoma (HCC), restraining tumour development. Other drugs, such as the HDAC inhibitor panobinostat have been shown to have senolytic activity. In A549 non‐small cell lung cancer (NSCLC) and FaDu head and neck squamous cell carcinoma (HNSCC) cells, taxol and panobinostat cotreatment showed a synergistic killing of cancer cells [216]. This apoptosis of the cancer cells was attributed to the induction of senescence by taxol followed by panobinostat‐induced senolysis.
Exploiting the immune system to eliminate senescent cells
Beside small compounds, senescent cells can be targeted taking advantage of the immune system. For example, antibodies against senescent‐specific antigens such as DPP4 can be used to target senescent cells by potentiating antibody‐dependent cell‐mediated cytotoxicity (ADCC) [239]. An antibody against B2M, an extracellular epitope identified as a membrane marker of senescence, conjugated to duocarmycin was able to kill doxorubicin‐induced senescent HCT‐116 colorectal cancer cells [240]. Recently, the therapeutic concept of chimeric antigen receptor (CAR) T cells was applied to senotherapy. The urokinase plasminogen activator receptor (uPAR) is a cell surface protein upregulated in senescent cells. The authors thus designed CAR T cells directed against uPAR that successfully eliminate senescent cells in vitro and in vivo upon TIS in Kras G12D;Trp53 −/− lung adenocarcinoma mouse model, what resulted in extended survival [241].
Other senotherapies
Besides the elimination of senescent cells by senolytics, senotherapies include senomorphic drugs that modulate the SASP to ensure the immune‐mediated senescent cell clearance. For example, the combination of MEK and CDK4/6 inhibitors induces senescence in Kras‐driven models of lung cancer and pancreatic ductal adenocarcinoma (PDAC). In lung cancer, that is sufficient to promote NK‐mediated regression [106], while in PDAC, the SASP increases tumour vascularization, facilitating chemotherapy delivery and efficacy, and increasing the recruitment of CD8+ T immune cells, thus sensitising these “cold” tumours to immunotherapy [242]. These studies suggest that immune response against senescent cancer cells could be further stimulated by immune checkpoint inhibitors (ICI).
Alternatively, senomorphic drugs altering the SASP phenotype without killing the senescent cells could be used to prevent the adverse effects of the remaining senescent cells or favour senescence immunosurveillance. Senomorphic drugs include IκB kinase (IKK) and NF‐κB kinase inhibitors [243], JAK/STAT signalling pathway inhibitors [244] or mTOR inhibitors [12, 13]. Interestingly, the antidiabetic drug metformin improves the healthspan and lifespan in mice, notably via the AMP‐activated protein kinase (AMPK) activation resulting in decreased oxidative damage and chronic inflammation [245, 246]. While metformin has pleiotropic effects, it also inhibits SASP production by preventing NF‐kB activation [247, 248]. Rapamycin, a selective inhibitor of mTOR, also inhibits the proinflammatory SASP [12, 13]. Everolimus, a second‐generation rapamycin derivate, is currently approved to treat certain types of breast [249], pancreatic [250], gastrointestinal [251], lung [252], kidney cancers [253, 254] and astrocytoma [255]. However, whether everolimus (or other senomorphic drugs) behave as a senomorphic drug in cancer patients has not been evaluated to date.
The “one‐two punch” approach: combining cancer therapies with senolytics
The extension of lifespan and healthspan observed upon genetic ablation of senescent cells in the INK‐ATTAC mice [198] fuelled the interest in developing senolytic drugs, initially aimed to treat an array of age‐related diseases. The potential that eliminating senescent cells have as an anticancer therapy became evident already in these early studies, as the expansion of lifespan observed in the INK‐ATTAC mice is mostly explained by a delay in cancer‐related deaths. While senescent cells influence many aspects of tumour progression, a way to deploy senotherapeutics for cancer treatment is the so‐called “one‐two punch” approach [256]. The rationale of “one‐two punch” therapies is that many cancer therapies induce senescence and using senolytics (as a second punch) would therefore target a newly exposed vulnerability in the cancer cells (Fig. 1).
Fig. 1.
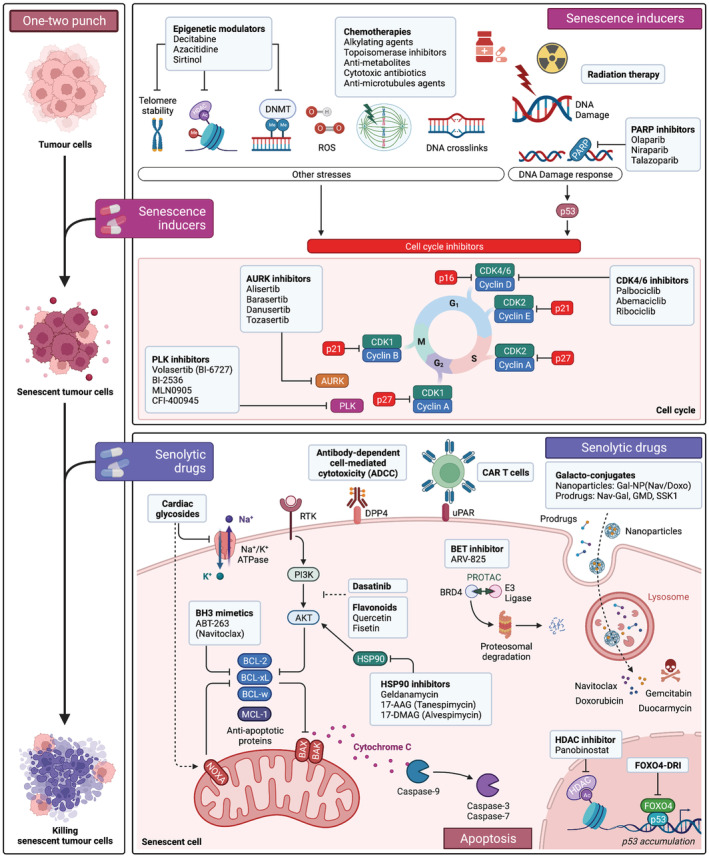
Strategies to target senescence in cancer (“one‐two punch”). Anti‐cancer therapy (first punch) induces the senescence of tumour cells. Conventional chemotherapy, radiotherapy and targeted therapies can be used to induce senescence. Induction of senescence in tumour cells unveils novel targetable vulnerabilities. Senolytic drugs can then be deployed to selectively target and kill senescent tumour cells (second punch). Several senolytic drugs have been described so far and their mechanisms of action are summarised here. Selective killing of senescent cancer cells will improve tumour control and regression. This “one‐two punch” approach to cancer therapy relies on the precise schedule of the administration of the sequential drug treatment to sensitise tumour to senolysis and restore tissue integrity. 17‐AAG, 17‐allylamino‐17‐demethoxygeldanamycin; 17‐DMAG, 17‐dimethylaminoethylamino‐17‐demethoxygeldanamycin; ac, acetylation; ADCC, antibody‐dependent cell‐mediated cytotoxicity; AURK, Aurora kinase; BET, bromodomain and extraterminal domain; CAR‐T cell, chimeric antigen receptor T cell; CDK, cyclin‐dependent kinase; DNMT, DNA methyltransferase; Doxo, doxorubicin; DRI, D‐retro inverso; gal‐NP, galactose‐nanoparticle; GMD, galactose‐modified duocarmycin; HDAC, histone deacetylase; HSP, heat shock protein; Nav, navitoclax; PARP, poly(ADP‐ribose) polymerase; PI3K, phosphatidylinositol tri‐phosphate; PLK, polo‐like kinase; ROS, reactive oxygen species; RTK, receptor tyrosine kinase; uPAR, urokinase plasminogen activator receptor. Created with BioRender.com.
On 36 breast cancer patients who received adjuvant chemotherapy, te Poele et al. [79] found that 41% of the tumours were SA‐β‐galactosidase and p16 positive, where only 10% of the 20 patients who did not receive chemotherapy stained positive, confirming that cytotoxic chemotherapy induces senescence in patients. Another demonstration comes from Roberson et al. [50] who showed that lung tumours from patient treated with neoadjuvant chemotherapy present large area of SA‐β‐galactosidase positive cells compared to tumours from a patient treated with surgery alone. This also illustrates the heterogeneity of tumour responses to therapy. Given that lingering senescent cells can promote tumour growth, metastasis and therapy resistance, strategies that target the elimination of senescent tumour cells post‐therapy can have multiple benefits.
One of the first demonstrations of the “one‐two punch” concept came from cell culture experiments in which treating A549 cancer cells with Aurora kinase inhibitors (such as alisertib or barasetib) were shown to undergo senescence [131]. Subsequent treatment with ABT‐263 killed the senescent cancer cells, suggesting the validity of such a combination. Given that cancer cells with their different origins and genetic composition might present different vulnerabilities, the Bernards' group concentrated in finding pro‐senescence therapies and senolytics tailored to HCC. Inhibition of CDC7, a DNA replication kinase, with XL413 selectively induces the senescence of p53 mutant cells, while inhibitors of mTOR signalling killed the senescent liver cancer cells [147]. Another example of the one‐two punch treatment is the effective combination of the PARPi olaparib and the senolytic drug navitoclax in a mouse model of ovarian cancer [142]. After a successful demonstration in vivo, the authors plan to launch in the mid‐2022 the first phase 1 clinical trial, evaluating the safety and the recommended dose for combined olaparib + navitoclax treatment in ovarian cancer patients, with intermittent delivery of navitoclax to prevent its hematologic toxicity. A future phase 2 study will evaluate the one‐two punch approach in solid tumour, ovarian cancer (NCT05358639).
One‐two punch protocols have been tried with a wide range of other senolytics (Table 3), including cardiac glycosides [211], BRD4 inhibitors [215], and galacto‐coated nanoparticles loaded with doxorubicin or navitoclax [108] or the Gal‐Nav prodrug [213]. There are several clinical trials evaluating the effect of the senolytic Navitoclax in combination with chemotherapy in cancer patients. However, the contribution of senescence and senolysis to the therapeutic effect will not be evaluated on most of those trials (NCT05358639 is an exception, Table 4). In addition, other senolytics, such as D + Q or fisetin, are being evaluated in different trails, including one aiming to improve frailty in adult survivors of childhood cancer (NCT04733534, Table 4). In addition to clinical trials, retrospective analysis is another way to test the potential of drugs repurposed as senolytics. For example, cancer patients treated with the cardiac glycoside digoxin during chemotherapy have a better overall survival [257]. Cardiac glycosides have pleiotropic effects, and the aforementioned study attributed the effect to immunogenic cell death. But given that cardiac glycosides have senolytic properties [209, 211], it would be worthy investigating whether senolysis might explain those results. Finally, a question that remains is what the best timeline for senotherapy is in combination with tumour therapy to achieve maximal benefit.
Table 4.
Examples of clinical trials testing senolytic drugs in the context of cancer.
| Senolytic drug | CT phase | Cancer | Senescence induction | CT number |
|---|---|---|---|---|
| Navitoclax | Phase 1 | Small cell lung cancer | Etoposide, cisplatin | NCT00878449 |
| Navitoclax | Phase 1 | Solid tumours | Erlotinib, irinotecan | NCT01009073 |
| Navitoclax | Phase 1 | Solid tumours | Gemcitabine | NCT00887757 |
| Navitoclax | Phase 1 | Solid tumours | Paclitaxel | NCT00891605 |
| Navitoclax | Phase 1/2 | Solid tumours | Trametinib | NCT02079740 |
| Navitoclax | Phase 1 | Solid tumours | Docetaxel | NCT00888108 |
| Navitoclax | Phase 1/2 | Solid tumours | Dabrafenib, trametinib | NCT01989585 |
| Navitoclax | Phase 1 | Advanced or metastatic non‐small cell lung cancer | Osimertinib | NCT02520778 |
| Navitoclax | Phase 1 | High‐grade serous carcinoma, triple‐negative breast cancer | Olaparib | NCT05358639 |
| Navitoclax | Phase 1 | Myeloid Neoplasms | Decitabine | NCT05455294 |
| Navitoclax | Phase 1 | Refractory acute myeloid leukaemia | Decitabine | NCT05222984 |
| Dasatinib and quercetin / Fisetin | Phase 2 | Childhood cancer survivors | N/A | NCT04733534 |
Effects of senolytics beyond the tumour
Different types of senescent cells are present in the TME during cancer progression and treatment. In addition, cancer treatments are also able to induce senescence in normal tissues. While the use of senolytics as anticancer treatment might be first deployed in the context of “one‐two punch” regimes, senolytics might have benefits that extend beyond killing senescent cancer cells.
OIS, occurring in preneoplastic lesions is a tumour suppressor mechanism that limits tumour progression. Senolytic drugs, such as CGs, have the ability of eliminating preneoplastic senescent cells [209], and therefore might not only target the senescent tumour cells but also limit cancer initiation. Senescent cells (such as fibroblasts) present in the tumour microenvironment are known to promote tumour growth, in part by secreting a pro‐tumorigenic SASP [64]. We and others have shown that strategies that inhibit this pro‐inflammatory SASP decrease tumour growth [12, 13, 258]. Similarly, senolytics such as procyanidin C1 have been proposed to inhibit tumour growth by targeting pro‐tumorigenic fibroblasts in the TME [230]. Moreover, side effects of chemotherapies have been linked to induction of senescence outside the tumour [37]. Using a mouse model for genetic ablation of senescent cells, DeMaria et al. [37] showed how killing senescent cells after chemotherapy reduced bone marrow suppression, cardiac dysfunction, cancer recurrence and improved physical activity and strength. Therefore, an additional advantage of using senolytics for cancer treatment will be limiting the side effects of cancer therapies.
Besides these additional benefits of senolytics we need to consider their side effects. One of the attractions of senolytics is that the original studies suggested an absence of side effects associated with eliminating senescent cells other than delayed wound healing [37, 198, 199]. However, since cancer often affects elderly people, and ageing is associated with slowed wound‐repair and healing abilities [259], delayed wound healing might be problematic. In addition, senolytics such as navitoclax present toxicities associated not with the elimination of senescent cells but with the specific drug target. These drug‐specific toxicities (such as thrombocytopenia caused by navitoclax) also need to be evaluated. In addition, recent studies suggest that, in a way that might depend on the senolytic regime used, elimination of certain senescent cells such as liver sinusoidal endothelial cells (LSECs) [260] can cause additional side effects such as fibrosis. Therefore, the short‐ and long‐term side effects of senolytics remain unknown and need to be considered carefully.
Future challenges
Senolytics are a relatively novel class of drugs and clinical trials for senolytics are just in their infancy. Senolytics hold immense promise, not just for cancer treatment, but also as therapies for a wide range of age‐related diseases and to treat multimorbidity. However, there are many unknowns and challenges that future research and ongoing trials will help us understand.
There are multiple therapeutics, both novel and clinically approved with senolytic activity and they target different mechanisms. Senolytics include not only small molecules, but also cell and immunotherapies. While this is potentially a very good starting point, it also comes with the added question of which drugs are better suited for which indications. In the case of “one‐two punch” approaches for anticancer therapies, an additional issue is to establish the best regime: when to treat with the senescence‐inducing drug and when with the senolytic? While the rationale argues for sequential treatment (first inducing senescence and then treating with the senolytic), in real life, the heterogeneity of the senescence response in the tumour might need concurrent treatment or a more complicated regime. That of course increases the potential for drug interactions and toxicities that would need to be monitored and managed.
While using senolytics in the context of ‘one‐two punch’ approaches is been postulated as the most clear anticancer therapies, we should not forget about senotherapies that could reduce the pro‐tumorigenic effects associated with the SASP, or ‘reprogram’ the SASP to potentiate anticancer immune surveillance.
Senescence is a cell stress response that is evoked in response to many cancer drugs and in multiple cancer types. While this implies a potentially universal use of senolytics as anticancer therapies, it also opens the question of what are the best cancer types in which start testing the “one‐two punch paradigm”. While pre‐existing data from preclinical models and patients can serve to suggest reasonable starting points, a key element to make senolytic therapies to work is being able to monitor senescence. Identification of senescent cells is one of the issues that has slowed progress in the senescent field for years, even if recent approaches are in development to detect senescence especially in tumours [261]. The issue becomes even more urgent if we want to move senotherapeutics for the clinic. Detection of senescence should be key to enrol and stratify patients and to monitor the efficiency of therapeutic strategies.
To this extent, we ideally need to identify biomarkers that can be routinely surveyed (in plasma or urine) or imaging techniques that could allow a non‐invasive, longitudinal monitoring of senescence in patients. Secreted proteins into biological fluids are routinely used for the surveillance of cancer. Proteins secreted by senescent cells as part of the SASP represent a promising source of biomarkers that could be exploited. For example, a SASP signature has been proposed as an indicator of age and medical risk [262]. Another soluble senescence markers that could be detected in blood include a soluble form of uPAR (suPAR) that is cleaved and secreted by senescent cells and has been used as a biomarker of several senescence‐associated diseases [241]. Interestingly, a study of the lipids present in senescent cells identified the intracellular prostaglandin dh‐15d‐PGJ2 and other prostaglandin D2‐related lipids that accumulate during senescence. Moreover, the dihomo‐prostaglandin is released upon senolysis and thus represents a potential marker of senolysis [263].
The ideal goal would be to monitor the senescence by non‐invasive imaging. While no such an approach is currently used in the clinic, several are in development. For example, [18F]FPyGal is a PET tracer aimed to report β‐galactosidase [264, 265]. Activity of β‐galactosidase is often used as a marker of senescence. The [18F]FPyGal tracer uptake is increased in senescent cells. [18F]FPyGal can reveal the presence of senescence in tumours and was initially evaluated in a patient treated with alisertib, revealing the presence of liver metastasis [264, 265]. Consequently, a clinical trial (NCT04536454) was initiated to further evaluate [18F]FPyGal to image senescence in cancer patients.
Overall senotherapies are a very interesting nascent field. While most of the focus has been placed on using senolytics for age‐related diseases, we anticipate that the next years will also see an increase in the use of senolytics in anticancer therapies. The discovery of novel therapeutics and the establishment of protocols to deploy them effectively, combined with strategies to monitor senescence, will all be necessary steps in this process.
Conflict of interest
JG has acted as a consultant for Unity Biotechnology, Geras Bio, Myricx Pharma and Merck KGaA. Unity Biotechnology and Pfizer have funded research in JG's laboratory. JG owns equity in Geras Bio. JG is a named inventor in MRC and Imperial College patents related to senolytic therapies.
Author contributions
JG and LB discussed the organisation of the manuscript. LB produced a first draft of the manuscript that was revised and modified by JG and LB.
Acknowledgements
We thank A Innes and other members of JG for their comments. This work was supported by a core grant from MRC (MC_U120085810) and a grant from CRUK (C15075/A28647) to JG.
Data accessibility
There is no data associated with this review.
References
- 1. Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, et al. Cellular senescence: defining a path forward. Cell. 2019;179:813–27. [DOI] [PubMed] [Google Scholar]
- 2. Herranz N, Gil J. Mechanisms and functions of cellular senescence. J Clin Invest. 2018;128:1238–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hernandez‐Segura A, Nehme J, Demaria M. Hallmarks of cellular senescence. Trends Cell Biol. 2018;28:436–53. [DOI] [PubMed] [Google Scholar]
- 4. Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, et al. Oncogene‐induced senescence is a DNA damage response triggered by DNA hyper‐replication. Nature. 2006;444:638–42. [DOI] [PubMed] [Google Scholar]
- 5. Freund A, Laberge R‐M, Demaria M, Campisi J. Lamin B1 loss is a senescence‐associated biomarker. Mol Biol Cell. 2012;23:2066–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Faget DV, Ren Q, Stewart SA. Unmasking senescence: context‐dependent effects of SASP in cancer. Nat Rev Cancer. 2019;19:439–53. [DOI] [PubMed] [Google Scholar]
- 7. Birch J, Gil J. Senescence and the SASP: many therapeutic avenues. Genes Dev. 2020;34:1565–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rodier F, Coppé J‐P, Patil CK, Hoeijmakers WAM, Muñoz DP, Raza SR, et al. Persistent DNA damage signalling triggers senescence‐associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11:973–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. 2013;15:978–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. 2017;550:402–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Glück S, Guey B, Gulen MF, Wolter K, Kang T‐W, Schmacke NA, et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol. 2017;19:1061–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, et al. mTOR regulates MAPKAPK2 translation to control the senescence‐associated secretory phenotype. Nat Cell Biol. 2015;17:1205–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Laberge R‐M, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, et al. MTOR regulates the pro‐tumorigenic senescence‐associated secretory phenotype by promoting IL1A translation. Nat Cell Biol. 2015;17:1049–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, et al. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science. 2015;349:aaa5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response‐independent regulator of the senescence‐associated secretory phenotype. EMBO J. 2011;30:1536–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, et al. Control of the senescence‐associated secretory phenotype by NF‐κB promotes senescence and enhances chemosensitivity. Genes Dev. 2011;25:2125–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kuilman T, Michaloglou C, Vredeveld LCW, Douma S, van Doorn R, Desmet CJ, et al. Oncogene‐induced senescence relayed by an interleukin‐dependent inflammatory network. Cell. 2008;133:1019–31. [DOI] [PubMed] [Google Scholar]
- 18. Ohanna M, Giuliano S, Bonet C, Imbert V, Hofman V, Zangari J, et al. Senescent cells develop a PARP‐1 and nuclear factor‐{kappa}B‐associated secretome (PNAS). Genes Dev. 2011;25:1245–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006–18. [DOI] [PubMed] [Google Scholar]
- 20. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. [DOI] [PubMed] [Google Scholar]
- 21. Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345:458–60. [DOI] [PubMed] [Google Scholar]
- 22. d'Adda di Fagagna F, Reaper PM, Clay‐Farrace L, Fiegler H, Carr P, Von Zglinicki T, et al. A DNA damage checkpoint response in telomere‐initiated senescence. Nature. 2003;426:194–8. [DOI] [PubMed] [Google Scholar]
- 23. Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010;24:2463–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. [DOI] [PubMed] [Google Scholar]
- 25. Muñoz‐Espín D, Cañamero M, Maraver A, Gómez‐López G, Contreras J, Murillo‐Cuesta S, et al. Programmed cell senescence during mammalian embryonic development. Cell. 2013;155:1104–18. [DOI] [PubMed] [Google Scholar]
- 26. Storer M, Mas A, Robert‐Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell. 2013;155:1119–30. [DOI] [PubMed] [Google Scholar]
- 27. Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF‐AA. Dev Cell. 2014;31:722–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134:657–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kang T‐W, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, et al. Senescence surveillance of pre‐malignant hepatocytes limits liver cancer development. Nature. 2011;479:547–51. [DOI] [PubMed] [Google Scholar]
- 30. Muñoz‐Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15:482–96. [DOI] [PubMed] [Google Scholar]
- 31. Kurz DJ, Decary S, Hong Y, Erusalimsky JD. Senescence‐associated (beta)‐galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J Cell Sci. 2000;113(Pt 20):3613–22. [DOI] [PubMed] [Google Scholar]
- 32. Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, et al. Senescence in premalignant tumours. Nature. 2005;436:642–2. [DOI] [PubMed] [Google Scholar]
- 33. Michaloglou C, Vredeveld LCW, Soengas MS, Denoyelle C, Kuilman T, van der Horst CMAM, et al. BRAFE600‐associated senescence‐like cell cycle arrest of human naevi. Nature. 2005;436:720–4. [DOI] [PubMed] [Google Scholar]
- 34. Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AHFM, Schlegelberger B, et al. Oncogene‐induced senescence as an initial barrier in lymphoma development. Nature. 2005;436:660–5. [DOI] [PubMed] [Google Scholar]
- 35. Chen Z, Trotman LC, Shaffer D, Lin H‐K, Dotan ZA, Niki M, et al. Crucial role of p53‐dependent cellular senescence in suppression of Pten‐deficient tumorigenesis. Nature. 2005;436:725–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang L, Lankhorst L, Bernards R. Exploiting senescence for the treatment of cancer. Nat Rev Cancer. 2022;22:340–55. [DOI] [PubMed] [Google Scholar]
- 37. Demaria M, O'Leary MN, Chang J, Shao L, Liu S, Alimirah F, et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 2017;7:165–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fane M, Weeraratna AT. How the ageing microenvironment influences tumour progression. Nat Rev Cancer. 2020;20:89–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ruhland MK, Loza AJ, Capietto A‐H, Luo X, Knolhoff BL, Flanagan KC, et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat Commun. 2016;7:11762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Han L, Long Q, Li S, Xu Q, Zhang B, Dou X, et al. Senescent stromal cells promote cancer resistance through SIRT1 loss‐potentiated overproduction of small extracellular vesicles. Cancer Res. 2020;80:3383–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Burd CE, Sorrentino JA, Clark KS, Darr DB, Krishnamurthy J, Deal AM, et al. Monitoring tumorigenesis and senescence in vivo with a p16INK4a‐luciferase model. Cell. 2013;152:340–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schosserer M, Grillari J, Breitenbach M. The dual role of cellular senescence in developing tumors and their response to cancer therapy. Front Oncol. 2017;7:278. 10.3389/fonc.2017.00278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sturmlechner I, Zhang C, Sine CC, van Deursen E‐J, Jeganathan KB, Hamada N, et al. p21 produces a bioactive secretome that places stressed cells under immunosurveillance. Science. 2021;374:eabb3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE, et al. Non‐cell‐autonomous tumor suppression by p53. Cell. 2013;153:449–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Iannello A, Thompson TW, Ardolino M, Lowe SW, Raulet DH. p53‐dependent chemokine production by senescent tumor cells supports NKG2D‐dependent tumor elimination by natural killer cells. J Exp Med. 2013;210:2057–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dirac AMG, Bernards R. Reversal of senescence in mouse fibroblasts through Lentiviral suppression of p53 * 210. J Biol Chem. 2003;278:11731–4. [DOI] [PubMed] [Google Scholar]
- 48. Zampetidis CP, Galanos P, Angelopoulou A, Zhu Y, Polyzou A, Karamitros T, et al. A recurrent chromosomal inversion suffices for driving escape from oncogene‐induced senescence via subTAD reorganization. Mol Cell. 2021;81:4907–4923.e8. [DOI] [PubMed] [Google Scholar]
- 49. Elmore LW, Di X, Dumur C, Holt SE, Gewirtz DA. Evasion of a single‐step, chemotherapy‐induced senescence in breast cancer cells: implications for treatment response. Clin Cancer Res. 2005;11:2637–43. [DOI] [PubMed] [Google Scholar]
- 50. Roberson RS, Kussick SJ, Vallieres E, Chen S‐YJ, Wu DY. Escape from therapy‐induced accelerated cellular senescence in p53‐null lung cancer cells and in human lung cancers. Cancer Res. 2005;65:2795–803. [DOI] [PubMed] [Google Scholar]
- 51. Milanovic M, Fan DNY, Belenki D, Däbritz JHM, Zhao Z, Yu Y, et al. Senescence‐associated reprogramming promotes cancer stemness. Nature. 2018;553:96–100. [DOI] [PubMed] [Google Scholar]
- 52. Duy C, Li M, Teater M, Meydan C, Garrett‐Bakelman FE, Lee TC, et al. Chemotherapy induces senescence‐like resilient cells capable of initiating AML recurrence. Cancer Discov. 2021;11:1542–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang Q, Wu PC, Dong DZ, Ivanova I, Chu E, Zeliadt S, et al. Polyploidy road to therapy‐induced cellular senescence and escape. Int J Cancer. 2013;132:1505–15. [DOI] [PubMed] [Google Scholar]
- 54. Saleh T, Tyutyunyk‐Massey L, Gewirtz DA. Tumor cell escape from therapy‐induced senescence as a model of disease recurrence after dormancy. Cancer Res. 2019;79:1044–6. [DOI] [PubMed] [Google Scholar]
- 55. Saleh T, Gewirtz DA. Considering therapy‐induced senescence as a mechanism of tumour dormancy contributing to disease recurrence. Br J Cancer. 2022;126:1363–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jackson JG, Pant V, Li Q, Chang LL, Quintás‐Cardama A, Garza D, et al. p53‐mediated senescence impairs the apoptotic response to chemotherapy and clinical outcome in breast cancer. Cancer Cell. 2012;21:793–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Coppé J‐P, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, et al. Senescence‐associated secretory phenotypes reveal cell‐nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gonzalez‐Meljem JM, Haston S, Carreno G, Apps JR, Pozzi S, Stache C, et al. Stem cell senescence drives age‐attenuated induction of pituitary tumours in mouse models of paediatric craniopharyngioma. Nat Commun. 2017;8:1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Prise KM, O'Sullivan JM. Radiation‐induced bystander signalling in cancer therapy. Nat Rev Cancer. 2009;9:351–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Davalos AR, Coppe J‐P, Campisi J, Desprez P‐Y. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev. 2010;29:273–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Laberge R‐M, Awad P, Campisi J, Desprez P‐Y. Epithelial‐mesenchymal transition induced by senescent fibroblasts. Cancer Microenviron. 2011;5:39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Eggert T, Wolter K, Ji J, Ma C, Yevsa T, Klotz S, et al. Distinct functions of senescence‐associated immune responses in liver tumor surveillance and tumor progression. Cancer Cell. 2016;30:533–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Di Mitri D, Toso A, Chen JJ, Sarti M, Pinton S, Jost TR, et al. Tumour‐infiltrating gr‐1+ myeloid cells antagonize senescence in cancer. Nature. 2014;515:134–7. [DOI] [PubMed] [Google Scholar]
- 64. Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci USA. 2001;98:12072–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pazolli E, Luo X, Brehm S, Carbery K, Chung J‐J, Prior JL, et al. Senescent stromal‐derived osteopontin promotes preneoplastic cell growth. Cancer Res. 2009;69:1230–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Alspach E, Fu Y, Stewart SA. Senescence and the pro‐tumorigenic stroma. Crit Rev Oncog. 2013;18:549–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pantsulaia I, Ciszewski WM, Niewiarowska J. Senescent endothelial cells: potential modulators of immunosenescence and ageing. Ageing Res Rev. 2016;29:13–25. [DOI] [PubMed] [Google Scholar]
- 68. Hwang HJ, Lee Y‐R, Kang D, Lee HC, Seo HR, Ryu J‐K, et al. Endothelial cells under therapy‐induced senescence secrete CXCL11, which increases aggressiveness of breast cancer cells. Cancer Lett. 2020;490:100–10. [DOI] [PubMed] [Google Scholar]
- 69. Muñoz DP, Yannone SM, Daemen A, Sun Y, Vakar‐Lopez F, Kawahara M, et al. Targetable mechanisms driving immunoevasion of persistent senescent cells link chemotherapy‐resistant cancer to aging. JCI Insight. 2019;5:124716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chang BD, Broude EV, Dokmanovic M, Zhu H, Ruth A, Xuan Y, et al. A senescence‐like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res. 1999;59:3761–7. [PubMed] [Google Scholar]
- 71. Sun X, Shi B, Zheng H, Min L, Yang J, Li X, et al. Senescence‐associated secretory factors induced by cisplatin in melanoma cells promote non‐senescent melanoma cell growth through activation of the ERK1/2‐RSK1 pathway. Cell Death Dis. 2018;9:260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wang X, Wong SCH, Pan J, Tsao SW, Fung KHY, Kwong DLW, et al. Evidence of cisplatin‐induced senescent‐like growth arrest in nasopharyngeal carcinoma Cells1. Cancer Res. 1998;58:5019–22. [PubMed] [Google Scholar]
- 73. Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM, et al. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell. 2002;109:335–46. [DOI] [PubMed] [Google Scholar]
- 74. Günther W, Pawlak E, Damasceno R, Arnold H, Terzis AJ. Temozolomide induces apoptosis and senescence in glioma cells cultured as multicellular spheroids. Br J Cancer. 2003;88:463–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hirose Y, Katayama M, Mirzoeva OK, Berger MS, Pieper RO. Akt activation suppresses Chk2‐mediated, methylating agent–induced G2 arrest and protects from temozolomide‐induced mitotic catastrophe and cellular senescence. Cancer Res. 2005;65:4861–9. [DOI] [PubMed] [Google Scholar]
- 76. Aasland D, Götzinger L, Hauck L, Berte N, Meyer J, Effenberger M, et al. Temozolomide induces senescence and repression of DNA repair pathways in glioblastoma cells via activation of ATR–CHK1, p21, and NF‐κB. Cancer Res. 2019;79:99–113. [DOI] [PubMed] [Google Scholar]
- 77. Knizhnik AV, Roos WP, Nikolova T, Quiros S, Tomaszowski K‐H, Christmann M, et al. Survival and death strategies in glioma cells: autophagy, senescence and apoptosis triggered by a single type of Temozolomide‐induced DNA damage. PLoS ONE. 2013;8:e55665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mhaidat NM, Zhang XD, Allen J, Avery‐Kiejda KA, Scott RJ, Hersey P. Temozolomide induces senescence but not apoptosis in human melanoma cells. Br J Cancer. 2007;97:1225–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. te Poele RH, Okorokov AL, Jardine L, Cummings J, Joel SP. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res. 2002;62:1876–83. [PubMed] [Google Scholar]
- 80. Elmore LW, Rehder CW, Di X, McChesney PA, Jackson‐Cook CK, Gewirtz DA, et al. Adriamycin‐induced senescence in breast tumor cells involves functional p53 and telomere dysfunction. J Biol Chem. 2002;277:35509–15. [DOI] [PubMed] [Google Scholar]
- 81. Su D, Zhu S, Han X, Feng Y, Huang H, Ren G, et al. BMP4‐Smad signaling pathway mediates Adriamycin‐induced premature senescence in lung cancer cells. J Biol Chem. 2009;284:12153–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Schwarze SR, Fu VX, Desotelle JA, Kenowski ML, Jarrard DF. The identification of senescence‐specific genes during the induction of senescence in prostate cancer cells. Neoplasia. 2005;7:816–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Mansilla S, Piña B, Portugal J. Daunorubicin‐induced variations in gene transcription: commitment to proliferation arrest, senescence and apoptosis. Biochem J. 2003;372:703–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Nagano T, Nakano M, Nakashima A, Onishi K, Yamao S, Enari M, et al. Identification of cellular senescence‐specific genes by comparative transcriptomics. Sci Rep. 2016;6:31758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Rudolf E, John S, Cervinka M. Irinotecan induces senescence and apoptosis in colonic cells in vitro. Toxicol Lett. 2012;214:1–8. [DOI] [PubMed] [Google Scholar]
- 86. Jonchère B, Vétillard A, Toutain B, Lam D, Bernard AC, Henry C, et al. Irinotecan treatment and senescence failure promote the emergence of more transformed and invasive cells that depend on anti‐apoptotic mcl‐1. Oncotarget. 2014;6:409–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Vétillard A, Jonchère B, Moreau M, Toutain B, Henry C, Fontanel S, et al. Akt inhibition improves irinotecan treatment and prevents cell emergence by switching the senescence response to apoptosis. Oncotarget. 2015;6:43342–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Toso A, Revandkar A, Di Mitri D, Guccini I, Proietti M, Sarti M, et al. Enhancing chemotherapy efficacy in Pten‐deficient prostate tumors by activating the senescence‐associated antitumor immunity. Cell Rep. 2014;9:75–89. [DOI] [PubMed] [Google Scholar]
- 89. Hu W, Sung T, Jessen BA, Thibault S, Finkelstein MB, Khan NK, et al. Mechanistic investigation of bone marrow suppression associated with Palbociclib and its differentiation from cytotoxic chemotherapies. Clin Cancer Res. 2016;22:2000–8. [DOI] [PubMed] [Google Scholar]
- 90. Bojko A, Czarnecka‐Herok J, Charzynska A, Dabrowski M, Sikora E. Diversity of the senescence phenotype of cancer cells treated with chemotherapeutic agents. Cell. 2019;8:1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Modrak DE, Leon E, Goldenberg DM, Gold DV. Ceramide regulates gemcitabine‐induced senescence and apoptosis in human pancreatic cancer cell lines. Mol Cancer Res. 2009;7:890–6. [DOI] [PubMed] [Google Scholar]
- 92. Song Y, Baba T, Mukaida N. Gemcitabine induces cell senescence in human pancreatic cancer cell lines. Biochem Biophys Res Commun. 2016;477:515–9. [DOI] [PubMed] [Google Scholar]
- 93. Hattangadi DK, DeMasters GA, Walker TD, Jones KR, Di X, Newsham IF, et al. Influence of p53 and caspase 3 activity on cell death and senescence in response to methotrexate in the breast tumor cell. Biochem Pharmacol. 2004;68:1699–708. [DOI] [PubMed] [Google Scholar]
- 94. Qiu T, Tian Y, Gao Y, Ma M, Li H, Liu X, et al. PTEN loss regulates alveolar epithelial cell senescence in pulmonary fibrosis depending on Akt activation. Aging (Albany NY). 2019;11:7492–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Aoshiba K, Tsuji T, Nagai A. Bleomycin induces cellular senescence in alveolar epithelial cells. Eur Respir J. 2003;22:436–43. [DOI] [PubMed] [Google Scholar]
- 96. Jones KR, Elmore LW, Jackson‐Cook C, Demasters G, Povirk LF, Holt SE, et al. p53‐dependent accelerated senescence induced by ionizing radiation in breast tumour cells. Int J Radiat Biol. 2005;81:445–58. [DOI] [PubMed] [Google Scholar]
- 97. Quick QA, Gewirtz DA. An accelerated senescence response to radiation in wild‐type p53 glioblastoma multiforme cells. J Neurosurg. 2006;105:111–8. [DOI] [PubMed] [Google Scholar]
- 98. Anders L, Ke N, Hydbring P, Choi YJ, Widlund HR, Chick JM, et al. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell. 2011;20:620–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Vijayaraghavan S, Karakas C, Doostan I, Chen X, Bui T, Yi M, et al. CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers. Nat Commun. 2017;8:15916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Valenzuela CA, Vargas L, Martinez V, Bravo S, Brown NE. Palbociclib‐induced autophagy and senescence in gastric cancer cells. Exp Cell Res. 2017;360:390–6. [DOI] [PubMed] [Google Scholar]
- 101. Michaud K, Solomon DA, Oermann E, Kim J‐S, Zhong W‐Z, Prados MD, et al. Pharmacologic inhibition of cdk4/6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res. 2010;70:3228–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Gadsden NJ, Fulcher CD, Li D, Shrivastava N, Thomas C, Segall JE, et al. Palbociclib renders human papilloma virus–negative head and neck squamous cell carcinoma vulnerable to the senolytic agent navitoclax. Mol Cancer Res. 2021;19:862–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Bollard J, Miguela V, de Galarreta MR, Venkatesh A, Bian CB, Roberto MP, et al. Palbociclib (PD‐0332991), a selective CDK4/6 inhibitor, restricts tumour growth in preclinical models of hepatocellular carcinoma. Gut. 2017;66:1286–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Kovatcheva M, Liu DD, Dickson MA, Klein ME, O'Connor R, Wilder FO, et al. MDM2 turnover and expression of ATRX determine the choice between quiescence and senescence in response to CDK4 inhibition. Oncotarget. 2015;6:8226–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Klein ME, Dickson MA, Antonescu C, Qin L‐X, Dooley SJ, Barlas A, et al. PDLIM7 and CDH18 regulate the turnover of MDM2 during CDK4/6 inhibitor therapy‐induced senescence. Oncogene. 2018;37:5066–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Ruscetti M, Leibold J, Bott MJ, Fennell M, Kulick A, Salgado NR, et al. NK cell‐mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science. 2018;362:1416–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Puyol M, Martín A, Dubus P, Mulero F, Pizcueta P, Khan G, et al. A synthetic lethal interaction between K‐Ras oncogenes and Cdk4 unveils a therapeutic strategy for non‐small cell lung carcinoma. Cancer Cell. 2010;18:63–73. [DOI] [PubMed] [Google Scholar]
- 108. Muñoz‐Espín D, Rovira M, Galiana I, Giménez C, Lozano‐Torres B, Paez‐Ribes M, et al. A versatile drug delivery system targeting senescent cells. EMBO Mol Med. 2018;10:e9355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Leontieva OV, Blagosklonny MV. CDK4/6‐inhibiting drug substitutes for p21 and p16 in senescence: duration of cell cycle arrest and MTOR activity determine geroconversion. Cell Cycle. 2013;12:3063–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Wang T‐H, Chen C‐C, Leu Y‐L, Lee Y‐S, Lian J‐H, Hsieh H‐L, et al. Palbociclib induces DNA damage and inhibits DNA repair to induce cellular senescence and apoptosis in oral squamous cell carcinoma. J Formos Med Assoc. 2021;120:1695–705. [DOI] [PubMed] [Google Scholar]
- 111. Gong X, Litchfield LM, Webster Y, Chio L‐C, Wong SS, Stewart TR, et al. Genomic aberrations that activate D‐type cyclins are associated with enhanced sensitivity to the CDK4 and CDK6 inhibitor Abemaciclib. Cancer Cell. 2017;32:761–776.e6. [DOI] [PubMed] [Google Scholar]
- 112. Goel S, Wang Q, Watt AC, Tolaney SM, Dillon DA, Li W, et al. Overcoming therapeutic resistance in HER2‐positive breast cancers with CDK4/6 inhibitors. Cancer Cell. 2016;29:255–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Goel S, DeCristo MJ, Watt AC, BrinJones H, Sceneay J, Li BB, et al. CDK4/6 inhibition triggers anti‐tumour immunity. Nature. 2017;548:471–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Torres‐Guzmán R, Calsina B, Hermoso A, Baquero C, Alvarez B, Amat J, et al. Preclinical characterization of abemaciclib in hormone receptor positive breast cancer. Oncotarget. 2017;8:69493–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Guenther LM, Dharia NV, Ross L, Conway A, Robichaud AL, Catlett JL II, et al. A combination CDK4/6 and IGF1R inhibitor strategy for Ewing sarcoma. Clin Cancer Res. 2019;25:1343–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Rader J, Russell MR, Hart LS, Nakazawa MS, Belcastro LT, Martinez D, et al. Dual CDK4/CDK6 inhibition induces cell‐cycle arrest and senescence in neuroblastoma. Clin Cancer Res. 2013;19:6173–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Iyengar M, O'Hayer P, Cole A, Sebastian T, Yang K, Coffman L, et al. CDK4/6 inhibition as maintenance and combination therapy for high grade serous ovarian cancer. Oncotarget. 2018;9:15658–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Venturelli S, Berger A, Weiland T, Essmann F, Waibel M, Nuebling T, et al. Differential induction of apoptosis and senescence by the DNA Methyltransferase inhibitors 5‐Azacytidine and 5‐Aza‐2′‐deoxycytidine in solid tumor cells. Mol Cancer Ther. 2013;12:2226–36. [DOI] [PubMed] [Google Scholar]
- 119. Borghesan M, Fusilli C, Rappa F, Panebianco C, Rizzo G, Oben JA, et al. DNA Hypomethylation and histone variant macroH2A1 synergistically attenuate chemotherapy‐induced senescence to promote hepatocellular carcinoma progression. Cancer Res. 2016;76:594–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Grandjenette C, Schnekenburger M, Karius T, Ghelfi J, Gaigneaux A, Henry E, et al. 5‐aza‐2′‐deoxycytidine‐mediated c‐myc Down‐regulation triggers telomere‐dependent senescence by regulating human telomerase reverse transcriptase in chronic myeloid leukemia. Neoplasia. 2014;16:511–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Amatori S, Bagaloni I, Viti D, Fanelli M. Premature senescence induced by DNA demethylating agent (Decitabine) as therapeutic option for malignant pleural mesothelioma. Lung Cancer. 2011;71:113–5. [DOI] [PubMed] [Google Scholar]
- 122. Widodo N, Deocaris CC, Kaur K, Hasan K, Yaguchi T, Yamasaki K, et al. Stress chaperones, Mortalin, and Pex19p mediate 5‐Aza‐2′ deoxycytidine‐induced senescence of cancer cells by DNA methylation‐independent pathway. J Gerontol A Biol Sci Med Sci. 2007;62:246–55. [DOI] [PubMed] [Google Scholar]
- 123. Elknerova K, Myslivcova D, Lacinova Z, Marinov I, Uherkova L, Stöckbauer P. Epigenetic modulation of gene expression of human leukemia cell lines ‐ induction of cell death and senescence. Neoplasma. 2011;58:35–44. [DOI] [PubMed] [Google Scholar]
- 124. Xu W‐S, Perez G, Ngo L, Gui C‐Y, Marks PA. Induction of polyploidy by histone deacetylase inhibitor: a pathway for antitumor effects. Cancer Res. 2005;65:7832–9. [DOI] [PubMed] [Google Scholar]
- 125. Kitadate A, Ikeda S, Teshima K, Ito M, Toyota I, Hasunuma N, et al. MicroRNA‐16 mediates the regulation of a senescence–apoptosis switch in cutaneous T‐cell and other non‐Hodgkin lymphomas. Oncogene. 2016;35:3692–704. [DOI] [PubMed] [Google Scholar]
- 126. Ota H, Tokunaga E, Chang K, Hikasa M, Iijima K, Eto M, et al. Sirt1 inhibitor, Sirtinol, induces senescence‐like growth arrest with attenuated Ras‐MAPK signaling in human cancer cells. Oncogene. 2006;25:176–85. [DOI] [PubMed] [Google Scholar]
- 127. Lee SH, Um S‐J, Kim E‐J. CBX8 suppresses Sirtinol‐induced premature senescence in human breast cancer cells via cooperation with SIRT1. Cancer Lett. 2013;335:397–403. [DOI] [PubMed] [Google Scholar]
- 128. Lee SH, Um S‐J, Kim E‐J. CBX8 antagonizes the effect of Sirtinol on premature senescence through the AKT‐RB‐E2F1 pathway in K562 leukemia cells. Biochem Biophys Res Commun. 2016;469:884–90. [DOI] [PubMed] [Google Scholar]
- 129. Huck JJ, Zhang M, McDonald A, Bowman D, Hoar KM, Stringer B, et al. MLN8054, an inhibitor of Aurora a kinase, induces senescence in human tumor cells both in vitro and in vivo. Mol Cancer Res. 2010;8:373–84. [DOI] [PubMed] [Google Scholar]
- 130. Van Brocklyn JR, Wojton J, Meisen WH, Kellough DA, Ecsedy JA, Kaur B, et al. Aurora‐a inhibition offers a novel therapy effective against intracranial glioblastoma. Cancer Res. 2014;74:5364–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Wang L, de Oliveira RL, Wang C, Neto JMF, Mainardi S, Evers B, et al. High‐throughput functional genetic and compound screens identify targets for senescence induction in cancer. Cell Rep. 2017;21:773–83. [DOI] [PubMed] [Google Scholar]
- 132. Zhang Y, Ma Y, Wang Y, Mukhopadhyay D, Bi Y, Ji B. Aurora kinase a inhibitor MLN8237 suppresses pancreatic cancer growth. Pancreatology. 2022;22:619–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Bonet C, Giuliano S, Ohanna M, Bille K, Allegra M, Lacour J‐P, et al. Aurora B is regulated by the mitogen‐activated protein kinase/extracellular signal‐regulated kinase (MAPK/ERK) signaling pathway and is a valuable potential target in melanoma cells *. J Biol Chem. 2012;287:29887–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Cilibrasi C, Guzzi A, Bazzoni R, Riva G, Cadamuro M, Hochegger H, et al. A ploidy increase promotes sensitivity of glioma stem cells to Aurora kinases inhibition. J Oncol. 2019;2019:e9014045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Driscoll DL, Chakravarty A, Bowman D, Shinde V, Lasky K, Shi J, et al. Plk1 inhibition causes post‐mitotic DNA damage and senescence in a range of human tumor cell lines. PLoS ONE. 2014;9:e111060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Kreis N‐N, Louwen F, Zimmer B, Yuan J. Loss of p21Cip1/CDKN1A renders cancer cells susceptible to polo‐like kinase 1 inhibition. Oncotarget. 2014;6:6611–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Van den Bossche J, Deben C, De Pauw I, Lambrechts H, Hermans C, Deschoolmeester V, et al. In vitro study of the polo‐like kinase 1 inhibitor volasertib in non‐small‐cell lung cancer reveals a role for the tumor suppressor p53. Mol Oncol. 2019;13:1196–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Chan CY‐K, Yuen VW‐H, Chiu DK‐C, Goh C‐C, Thu KL, Cescon DW, et al. Polo‐like kinase 4 inhibitor CFI‐400945 suppresses liver cancer through cell cycle perturbation and eliciting antitumor immunity. Hepatology. 2022. 10.1002/hep.32461 Online ahead of print. [DOI] [PubMed] [Google Scholar]
- 139. Tse KY, Chan KT, Yuen V, Zhang MS, Ip PPC, Ngan HYS, et al. Abstract A29: anticancer effects of polo‐like kinase‐4 inhibitor (CFI‐400945) in ovarian cancer. Clin Cancer Res. 2020;26:A29. [Google Scholar]
- 140. Singh CK, Denu RA, Nihal M, Shabbir M, Garvey DR, Huang W, et al. PLK4 is upregulated in prostate cancer and its inhibition reduces centrosome amplification and causes senescence. Prostate. 2022;82:957–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Esposito MT, Zhao L, Fung TK, Rane JK, Wilson A, Martin N, et al. Synthetic lethal targeting of oncogenic transcription factors in acute leukemia by PARP inhibitors. Nat Med. 2015;21:1481–90. [DOI] [PubMed] [Google Scholar]
- 142. Fleury H, Malaquin N, Tu V, Gilbert S, Martinez A, Olivier M‐A, et al. Exploiting interconnected synthetic lethal interactions between PARP inhibition and cancer cell reversible senescence. Nat Commun. 2019;10:2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Wang Z, Gao J, Zhou J, Liu H, Xu C. Olaparib induced senescence under P16 or P53 dependent manner in ovarian cancer. J Gynecol Oncol. 2019;30:e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Lombard AP, Armstrong CM, D'Abronzo LS, Ning S, Leslie AR, Sharifi M, et al. Olaparib‐induced senescence is bypassed through G2–M checkpoint override in Olaparib‐resistant prostate cancer. Mol Cancer Ther. 2022;21:677–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Chatterjee P, Choudhary GS, Sharma A, Singh K, Heston WD, Ciezki J, et al. PARP inhibition sensitizes to low dose‐rate radiation TMPRSS2‐ERG fusion gene‐expressing and PTEN‐deficient prostate cancer cells. PLOS ONE. Public library of. Science. 2013;8:e60408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Efimova EV, Mauceri HJ, Golden DW, Labay E, Bindokas VP, Darga TE, et al. Poly(ADP‐ribose) polymerase inhibitor induces accelerated senescence in irradiated breast cancer cells and tumors. Cancer Res. 2010;70:6277–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Wang C, Vegna S, Jin H, Benedict B, Lieftink C, Ramirez C, et al. Inducing and exploiting vulnerabilities for the treatment of liver cancer. Nature. 2019;574:268–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Lee Y‐H, Kang BS, Bae Y‐S. Premature senescence in human breast cancer and colon cancer cells by tamoxifen‐mediated reactive oxygen species generation. Life Sci. 2014;97:116–22. [DOI] [PubMed] [Google Scholar]
- 149. Tuttle R, Miller KR, Maiorano JN, Termuhlen PM, Gao Y, Berberich SJ. Novel senescence associated gene, YPEL3, is repressed by estrogen in ER+ mammary tumor cells and required for tamoxifen‐induced cellular senescence. Int J Cancer. 2012;130:2291–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Burton DGA, Giribaldi MG, Munoz A, Halvorsen K, Patel A, Jorda M, et al. Androgen deprivation‐induced senescence promotes outgrowth of androgen‐refractory prostate cancer cells. PLoS One. 2013;8:e68003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Ewald JA, Desotelle JA, Church DR, Yang B, Huang W, Laurila TA, et al. Androgen deprivation induces senescence characteristics in prostate cancer cells in vitro and in vivo. Prostate. 2013;73:337–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Carpenter V, Saleh T, Min Lee S, Murray G, Reed J, Souers A, et al. Androgen‐deprivation induced senescence in prostate cancer cells is permissive for the development of castration‐resistance but susceptible to senolytic therapy. Biochem Pharmacol. 2021;193:114765. [DOI] [PubMed] [Google Scholar]
- 153. Däbritz JHM, Yu Y, Milanovic M, Schönlein M, Rosenfeldt MT, Dörr JR, et al. CD20‐targeting immunotherapy promotes cellular senescence in B‐cell lymphoma. Mol Cancer Ther. 2016;15:1074–81. [DOI] [PubMed] [Google Scholar]
- 154. Rosemblit C, Datta J, Lowenfeld L, Xu S, Basu A, Kodumudi K, et al. Oncodriver inhibition and CD4+ Th1 cytokines cooperate through Stat1 activation to induce tumor senescence and apoptosis in HER2+ and triple negative breast cancer: implications for combining immune and targeted therapies. Oncotarget. 2018;9:23058–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Hasan MR, Ho SHY, Owen DA, Tai IT. Inhibition of VEGF induces cellular senescence in colorectal cancer cells. Int J Cancer. 2011;129:2115–23. [DOI] [PubMed] [Google Scholar]
- 156. Foersch S, Sperka T, Lindner C, Taut A, Rudolph KL, Breier G, et al. VEGFR2 signaling prevents colorectal cancer cell senescence to promote tumorigenesis in mice with colitis. Gastroenterology. 2015;149:177–189.e10. [DOI] [PubMed] [Google Scholar]
- 157. Schick U, Kyula J, Barker H, Patel R, Zaidi S, Gregory C, et al. Trametinib radiosensitises RAS‐ and BRAF‐mutated melanoma by perturbing cell cycle and inducing senescence. Radiother Oncol. 2015;117:364–75. [DOI] [PubMed] [Google Scholar]
- 158. Hartman ML, Sztiller‐Sikorska M, Czyz M. Whole‐exome sequencing reveals novel genetic variants associated with diverse phenotypes of melanoma cells. Mol Carcinog. 2019;58:588–602. [DOI] [PubMed] [Google Scholar]
- 159. Haferkamp S, Borst A, Adam C, Becker TM, Motschenbacher S, Windhövel S, et al. Vemurafenib induces senescence features in melanoma cells. J Invest Dermatol. 2013;133:1601–9. [DOI] [PubMed] [Google Scholar]
- 160. Liu J, Zheng X, Li W, Ren L, Li S, Yang Y, et al. Anti‐tumor effects of Skp2 inhibitor AAA‐237 on NSCLC by arresting cell cycle at G0/G1 phase and inducing senescence. Pharmacol Res. 2022;181:106259. [DOI] [PubMed] [Google Scholar]
- 161. Lin H‐K, Chen Z, Wang G, Nardella C, Lee S‐W, Chan C‐H, et al. Skp2 targeting suppresses tumorigenesis by Arf‐p53‐independent cellular senescence. Nature. 2010;464:374–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Chan C‐H, Morrow JK, Li C‐F, Gao Y, Jin G, Moten A, et al. Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell. 2013;154:556–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Ewald JA, Peters N, Desotelle JA, Hoffmann FM, Jarrard DF. A high‐throughput method to identify novel senescence‐inducing compounds. J Biomol Screen. 2009;14:853–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Chang B‐D, Xuan Y, Broude EV, Zhu H, Schott B, Fang J, et al. Role of p53 and p21waf1/cip1 in senescence‐like terminal proliferation arrest induced in human tumor cells by chemotherapeutic drugs. Oncogene. 1999;18:4808–18. [DOI] [PubMed] [Google Scholar]
- 165. Dabrowska M, Mosieniak G, Skierski J, Sikora E, Rode W. Methotrexate‐induced senescence in human adenocarcinoma cells is accompanied by induction of p21waf1/cip1 expression and lack of polyploidy. Cancer Lett. 2009;284:95–101. [DOI] [PubMed] [Google Scholar]
- 166. Suzuki K, Mori I, Nakayama Y, Miyakoda M, Kodama S, Watanabe M. Radiation‐induced senescence‐like growth arrest requires TP53 function but not telomere shortening. Radiat Res. 2001;155:248–53. [DOI] [PubMed] [Google Scholar]
- 167. Fumagalli M, Rossiello F, Clerici M, Barozzi S, Cittaro D, Kaplunov JM, et al. Telomeric DNA damage is irreparable and causes persistent DNA‐damage‐response activation. Nat Cell Biol. 2012;14:355–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Ding N‐H, Li JJ, Sun L‐Q. Molecular mechanisms and treatment of radiation‐induced lung fibrosis. Curr Drug Targets. 2013;14:1347–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. He Y, Thummuri D, Zheng G, Okunieff P, Citrin DE, Vujaskovic Z, et al. Cellular senescence and radiation‐induced pulmonary fibrosis. Transl Res. 2019;209:14–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Citrin DE, Shankavaram U, Horton JA, Shield W III, Zhao S, Asano H, et al. Role of type II Pneumocyte senescence in radiation‐induced lung fibrosis. J Natl Cancer Inst. 2013;105:1474–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Su L, Dong Y, Wang Y, Wang Y, Guan B, Lu Y, et al. Potential role of senescent macrophages in radiation‐induced pulmonary fibrosis. Cell Death Dis. 2021;12:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Fouillade C, Curras‐Alonso S, Giuranno L, Quelennec E, Heinrich S, Bonnet‐Boissinot S, et al. FLASH irradiation spares lung progenitor cells and limits the incidence of radio‐induced senescence. Clin Cancer Res. 2020;26:1497–506. [DOI] [PubMed] [Google Scholar]
- 173. O'Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. 2016;13:417–30. [DOI] [PubMed] [Google Scholar]
- 174. Turner NC, Ro J, André F, Loi S, Verma S, Iwata H, et al. Palbociclib in hormone‐receptor‐positive advanced breast cancer. N Engl J Med. 2015;373:209–19. [DOI] [PubMed] [Google Scholar]
- 175. Shapiro G, Rosen LS, Tolcher AW, Goldman JW, Gandhi L, Papadopoulos KP, et al. A first‐in‐human phase I study of the CDK4/6 inhibitor, LY2835219, for patients with advanced cancer. J Clin Oncol. 2013;31:2500.23715580 [Google Scholar]
- 176. Infante JR, Shapiro G, Witteveen P, Gerecitano JF, Ribrag V, Chugh R, et al. A phase I study of the single‐agent CDK4/6 inhibitor LEE011 in pts with advanced solid tumors and lymphomas. J Clin Oncol. 2014;32:2528–8. [Google Scholar]
- 177. Goel S, Bergholz JS, Zhao JJ. Targeting CDK4 and CDK6 in cancer. Nat Rev Cancer. 2022;1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Wagner V, Gil J. Senescence as a therapeutically relevant response to CDK4/6 inhibitors. Oncogene. 2020;39:5165–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Álvarez‐Fernández M, Malumbres M. Mechanisms of sensitivity and resistance to CDK4/6 inhibition. Cancer Cell. 2020;37:514–29. [DOI] [PubMed] [Google Scholar]
- 180. Herrera‐Abreu MT, Palafox M, Asghar U, Rivas MA, Cutts RJ, Garcia‐Murillas I, et al. Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor‐positive breast cancer. Cancer Res. 2016;76:2301–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Taylor‐Harding B, Aspuria P‐J, Agadjanian H, Cheon D‐J, Mizuno T, Greenberg D, et al. Cyclin E1 and RTK/RAS signaling drive CDK inhibitor resistance via activation of E2F and ETS. Oncotarget. 2015;6:696–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Turner NC, Liu Y, Zhu Z, Loi S, Colleoni M, Loibl S, et al. Cyclin E1 expression and Palbociclib efficacy in previously treated hormone receptor‐positive metastatic breast cancer. J Clin Oncol. 2019;37:1169–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Pandey K, Park N, Park K‐S, Hur J, Cho YB, Kang M, et al. Combined CDK2 and CDK4/6 inhibition overcomes Palbociclib resistance in breast cancer by enhancing senescence. Cancers (Basel). 2020;12:E3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Vader G, Lens SMA. The Aurora kinase family in cell division and cancer. Biochim Biophys Acta. 2008;1786:60–72. [DOI] [PubMed] [Google Scholar]
- 185. Tang A, Gao K, Chu L, Zhang R, Yang J, Zheng J. Aurora kinases: novel therapy targets in cancers. Oncotarget. 2017;8:23937–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Carvajal RD, Tse A, Schwartz GK. Aurora kinases: new targets for cancer therapy. Clin Cancer Res. 2006;12:6869–75. [DOI] [PubMed] [Google Scholar]
- 187. Bavetsias V, Linardopoulos S. Aurora kinase inhibitors: current status and outlook. Front Oncol. 2015;5:278. 10.3389/fonc.2015.00278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. de Cárcer G, Manning G, Malumbres M. From Plk1 to Plk5: functional evolution of polo‐like kinases. Cell Cycle. 2011;10:2255–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Gutteridge REA, Ndiaye MA, Liu X, Ahmad N. Plk1 inhibitors in cancer therapy: from laboratory to clinics. Mol Cancer Ther. 2016;15:1427–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Stone A, Cowley MJ, Valdes‐Mora F, McCloy RA, Sergio CM, Gallego‐Ortega D, et al. BCL‐2 Hypermethylation is a potential biomarker of sensitivity to antimitotic chemotherapy in endocrine‐resistant breast cancer. Mol Cancer Ther. 2013;12:1874–85. [DOI] [PubMed] [Google Scholar]
- 191. Prashanth Kumar BN, Rajput S, Bharti R, Parida S, Mandal M. BI2536 – a PLK inhibitor augments paclitaxel efficacy in suppressing tamoxifen induced senescence and resistance in breast cancer cells. Biomed Pharmacother. 2015;74:124–32. [DOI] [PubMed] [Google Scholar]
- 192. Stresemann C, Lyko F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int J Cancer. 2008;123:8–13. [DOI] [PubMed] [Google Scholar]
- 193. Baell JB, Leaver DJ, Hermans SJ, Kelly GL, Brennan MS, Downer NL, et al. Inhibitors of histone acetyltransferases KAT6A/B induce senescence and arrest tumour growth. Nature. 2018;560:253–7. [DOI] [PubMed] [Google Scholar]
- 194. Lord CJ, Ashworth A. PARP inhibitors: synthetic lethality in the clinic. Science. 2017;355:1152–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. González‐Billalabeitia E, Seitzer N, Song SJ, Song MS, Patnaik A, Liu X‐S, et al. Vulnerabilities of PTEN–TP53‐deficient prostate cancers to compound PARP–PI3K inhibition. Cancer Discov. 2014;4:896–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat J‐P, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Nikolaev SI, Rimoldi D, Iseli C, Valsesia A, Robyr D, Gehrig C, et al. Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nat Genet. 2012;44:133–9. [DOI] [PubMed] [Google Scholar]
- 198. Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, et al. Naturally occurring p16Ink4a‐positive cells shorten healthy lifespan. Nature. 2016;530:184–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, et al. Clearance of p16Ink4a‐positive senescent cells delays ageing‐associated disorders. Nature. 2011;479:232–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. 2016;354:472–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Shahbandi A, Rao SG, Anderson AY, Frey WD, Olayiwola JO, Ungerleider NA, et al. BH3 mimetics selectively eliminate chemotherapy‐induced senescent cells and improve response in TP53 wild‐type breast cancer. Cell Death Differ. 2020;27:3097–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Saleh T, Carpenter VJ, Tyutyunyk‐Massey L, Murray G, Leverson JD, Souers AJ, et al. Clearance of therapy‐induced senescent tumor cells by the senolytic ABT‐263 via interference with BCL‐XL ‐BAX interaction. Mol Oncol. 2020;14:2504–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Jochems F, Thijssen B, De Conti G, Jansen R, Pogacar Z, Groot K, et al. The cancer SENESCopedia: a delineation of cancer cell senescence. Cell Rep. 2021;36:109441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Rahman M, Olson I, Mansour M, Carlstrom LP, Sutiwisesak R, Saber R, et al. Selective vulnerability of senescent glioblastoma cells to BCL‐XL inhibition. Mol Cancer Res. 2022;20:938–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Whittle JR, Vaillant F, Surgenor E, Policheni AN, Giner G, Capaldo BD, et al. Dual targeting of CDK4/6 and BCL2 pathways augments tumor response in estrogen receptor–positive breast cancer. Clin Cancer Res. 2020;26:4120–34. [DOI] [PubMed] [Google Scholar]
- 206. Schwarzenbach C, Tatsch L, Brandstetter Vilar J, Rasenberger B, Beltzig L, Kaina B, et al. Targeting c‐IAP1, c‐IAP2, and Bcl‐2 eliminates senescent glioblastoma cells following Temozolomide treatment. Cancers (Basel). 2021;13:3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Lafontaine J, Cardin GB, Malaquin N, Boisvert J‐S, Rodier F, Wong P. Senolytic targeting of Bcl‐2 anti‐apoptotic family increases cell death in irradiated sarcoma cells. Cancers (Basel). 2021;13:386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Troiani M, Colucci M, D'Ambrosio M, Guccini I, Pasquini E, Varesi A, et al. Single‐cell transcriptomics identifies Mcl‐1 as a target for senolytic therapy in cancer. Nat Commun. 2022;13:2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Guerrero A, Herranz N, Sun B, Wagner V, Gallage S, Guiho R, et al. Cardiac glycosides are broad‐spectrum senolytics. Nat Metab. 2019;1:1074–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Deryabin PI, Shatrova AN, Borodkina AV. Apoptosis resistance of senescent cells is an intrinsic barrier for senolysis induced by cardiac glycosides. Cell Mol Life Sci. 2021;78:7757–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Triana‐Martínez F, Picallos‐Rabina P, Da Silva‐Álvarez S, Pietrocola F, Llanos S, Rodilla V, et al. Identification and characterization of cardiac glycosides as senolytic compounds. Nat Commun. 2019;10:4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Galiana I, Lozano‐Torres B, Sancho M, Alfonso M, Bernardos A, Bisbal V, et al. Preclinical antitumor efficacy of senescence‐inducing chemotherapy combined with a nanoSenolytic. J Control Release. 2020;323:624–34. [DOI] [PubMed] [Google Scholar]
- 213. González‐Gualda E, Pàez‐Ribes M, Lozano‐Torres B, Macias D, Wilson JR, González‐López C, et al. Galacto‐conjugation of Navitoclax as an efficient strategy to increase senolytic specificity and reduce platelet toxicity. Aging Cell. 2020;19:e13142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Guerrero A, Guiho R, Herranz N, Uren A, Withers DJ, Martínez‐Barbera JP, et al. Galactose‐modified duocarmycin prodrugs as senolytics. Aging Cell. 2020;19:e13133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Wakita M, Takahashi A, Sano O, Loo TM, Imai Y, Narukawa M, et al. A BET family protein degrader provokes senolysis by targeting NHEJ and autophagy in senescent cells. Nat Commun. 2020;11:1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Samaraweera L, Adomako A, Rodriguez‐Gabin A, McDaid HM. A novel indication for panobinostat as a senolytic drug in NSCLC and HNSCC. Sci Rep. 2017;7:1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Le HH, Cinaroglu SS, Manalo EC, Ors A, Gomes MM, Duan Sahbaz B, et al. Molecular modelling of the FOXO4‐TP53 interaction to design senolytic peptides for the elimination of senescent cancer cells. EBioMedicine. 2021;73:103646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Yosef R, Pilpel N, Tokarsky‐Amiel R, Biran A, Ovadya Y, Cohen S, et al. Directed elimination of senescent cells by inhibition of BCL‐W and BCL‐XL. Nat Commun. 2016;7:11190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An inhibitor of Bcl‐2 family proteins induces regression of solid tumours. Nature. 2005;435:677–81. [DOI] [PubMed] [Google Scholar]
- 220. Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, et al. ABT‐263: a potent and orally bioavailable Bcl‐2 family inhibitor. Cancer Res. 2008;68:3421–8. [DOI] [PubMed] [Google Scholar]
- 221. Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. ABT‐199, a potent and selective BCL‐2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19:202–8. [DOI] [PubMed] [Google Scholar]
- 222. Mijit M, Caracciolo V, Melillo A, Amicarelli F, Giordano A. Role of p53 in the regulation of cellular senescence. Biomolecules. 2020;10:420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Chang J, Wang Y, Shao L, Laberge R‐M, Demaria M, Campisi J, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med. 2016;22:78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Zhu Y, Tchkonia T, Fuhrmann‐Stroissnigg H, Dai HM, Ling YY, Stout MB, et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl‐2 family of anti‐apoptotic factors. Aging Cell. 2016;15:428–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Gandhi L, Camidge DR, Ribeiro de Oliveira M, Bonomi P, Gandara D, Khaira D, et al. Phase I study of Navitoclax (ABT‐263), a novel Bcl‐2 family inhibitor, in patients with small‐cell lung cancer and other solid tumors. J Clin Oncol. 2011;29:909–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Malaquin N, Vancayseele A, Gilbert S, Antenor‐Habazac L, Olivier M‐A, Ait Ali Brahem Z, et al. DNA damage‐ but not enzalutamide‐induced senescence in prostate cancer promotes Senolytic Bcl‐xL inhibitor sensitivity. Cell. 2020;9:1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, et al. The Achilles' heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. 2015;14:644–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Yousefzadeh MJ, Zhu Y, McGowan SJ, Angelini L, Fuhrmann‐Stroissnigg H, Xu M, et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine. 2018;36:18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Zhu Y, Doornebal EJ, Pirtskhalava T, Giorgadze N, Wentworth M, Fuhrmann‐Stroissnigg H, et al. New agents that target senescent cells: the flavone, fisetin, and the BCL‐XL inhibitors, A1331852 and A1155463. Aging (Albany NY). 2017;9:955–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Xu Q, Fu Q, Li Z, Liu H, Wang Y, Lin X, et al. The flavonoid procyanidin C1 has senotherapeutic activity and increases lifespan in mice. Nat Metab. 2021;3:1706–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Justice JN, Nambiar AM, Tchkonia T, LeBrasseur NK, Pascual R, Hashmi SK, et al. Senolytics in idiopathic pulmonary fibrosis: results from a first‐in‐human, open‐label, pilot study. EBioMedicine. 2019;40:554–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Hickson LJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N, Hashmi SK, et al. Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of Dasatinib plus quercetin in individuals with diabetic kidney disease. EBioMedicine. 2019;47:446–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Baar MP, Brandt RMC, Putavet DA, Klein JDD, Derks KWJ, Bourgeois BRM, et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to Chemotoxicity and aging. Cell. 2017;169:132–147.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Agostini A, Mondragón L, Bernardos A, Martínez‐Máñez R, Marcos MD, Sancenón F, et al. Targeted cargo delivery in senescent cells using capped mesoporous silica nanoparticles. Angew Chem Int Ed Engl. 2012;51:10556–60. [DOI] [PubMed] [Google Scholar]
- 235. Cai Y, Zhou H, Zhu Y, Sun Q, Ji Y, Xue A, et al. Elimination of senescent cells by β‐galactosidase‐targeted prodrug attenuates inflammation and restores physical function in aged mice. Cell Res. 2020;30:574–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Cherif H, Bisson DG, Jarzem P, Weber M, Ouellet JA, Haglund L. Curcumin and o‐vanillin exhibit evidence of Senolytic activity in human IVD cells in vitro. J Clin Med. 2019;8:E433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Li W, He Y, Zhang R, Zheng G, Zhou D. The curcumin analog EF24 is a novel senolytic agent. Aging (Albany NY). 2019;11:771–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Fuhrmann‐Stroissnigg H, Ling YY, Zhao J, McGowan SJ, Zhu Y, Brooks RW, et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat Commun. 2017;8:422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Kim KM, Noh JH, Bodogai M, Martindale JL, Yang X, Indig FE, et al. Identification of senescent cell surface targetable protein DPP4. Genes Dev. 2017;31:1529–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Poblocka M, Bassey AL, Smith VM, Falcicchio M, Manso AS, Althubiti M, et al. Targeted clearance of senescent cells using an antibody‐drug conjugate against a specific membrane marker. Sci Rep. 2021;11:20358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Amor C, Feucht J, Leibold J, Ho Y‐J, Zhu C, Alonso‐Curbelo D, et al. Senolytic CAR T cells reverse senescence‐associated pathologies. Nature. 2020;583:127–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Ruscetti M, Morris JP, Mezzadra R, Russell J, Leibold J, Romesser PB, et al. Senescence‐induced vascular remodeling creates therapeutic vulnerabilities in pancreas cancer. Cell. 2020;181:424–441.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Tilstra JS, Robinson AR, Wang J, Gregg SQ, Clauson CL, Reay DP, et al. NF‐κB inhibition delays DNA damage–induced senescence and aging in mice. J Clin Invest. 2012;122:2601–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Xu M, Tchkonia T, Ding H, Ogrodnik M, Lubbers ER, Pirtskhalava T, et al. JAK inhibition alleviates the cellular senescence‐associated secretory phenotype and frailty in old age. Proc Natl Acad Sci USA. 2015;112:E6301–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Martin‐Montalvo A, Mercken EM, Mitchell SJ, Palacios HH, Mote PL, Scheibye‐Knudsen M, et al. Metformin improves healthspan and lifespan in mice. Nat Commun. 2013;4:2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Mohammed I, Hollenberg MD, Ding H, Triggle CR. A critical review of the evidence that metformin is a putative anti‐aging drug that enhances Healthspan and extends lifespan. Front Endocrinol (Lausanne). 2021;12:718942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Moiseeva O, Bourdeau V, Roux A, Deschênes‐Simard X, Ferbeyre G. Mitochondrial dysfunction contributes to oncogene‐induced senescence. Mol Cell Biol. 2009;29:4495–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Hirsch HA, Iliopoulos D, Struhl K. Metformin inhibits the inflammatory response associated with cellular transformation and cancer stem cell growth. Proc Natl Acad Sci USA. 2013;110:972–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Baselga J, Campone M, Piccart M, Burris HA, Rugo HS, Sahmoud T, et al. Everolimus in postmenopausal hormone‐receptor–positive advanced breast cancer. N Engl J Med. 2012;366:520–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Yao JC, Shah MH, Ito T, Bohas CL, Wolin EM, Van Cutsem E, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:514–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Ohtsu A, Ajani JA, Bai Y‐X, Bang Y‐J, Chung H‐C, Pan H‐M, et al. Everolimus for previously treated advanced gastric cancer: results of the randomized, double‐blind, phase III GRANITE‐1 study. J Clin Oncol. 2013;31:3935–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Tarhini A, Kotsakis A, Gooding W, Shuai Y, Petro D, Friedland D, et al. Phase II study of Everolimus (RAD001) in previously treated small cell lung cancer. Clin Cancer Res. 2010;16:5900–7. [DOI] [PubMed] [Google Scholar]
- 253. Choueiri TK, Escudier B, Powles T, Mainwaring PN, Rini BI, Donskov F, et al. Cabozantinib versus Everolimus in advanced renal‐cell carcinoma. N Engl J Med. 2015;373:1814–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus Everolimus in advanced renal‐cell carcinoma. N Engl J Med. 2015;373:1803–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P, et al. Everolimus for subependymal Giant‐cell Astrocytomas in tuberous sclerosis. N Engl J Med. 2010;363:1801–11. [DOI] [PubMed] [Google Scholar]
- 256. Wang L, Bernards R. Taking advantage of drug resistance, a new approach in the war on cancer. Front Med. 2018;12:490–5. [DOI] [PubMed] [Google Scholar]
- 257. Menger L, Vacchelli E, Adjemian S, Martins I, Ma Y, Shen S, et al. Cardiac glycosides exert anticancer effects by inducing immunogenic cell death. Sci Transl Med. 2012;4:143ra99. [DOI] [PubMed] [Google Scholar]
- 258. Georgilis A, Klotz S, Hanley CJ, Herranz N, Weirich B, Morancho B, et al. PTBP1‐mediated alternative splicing regulates the inflammatory Secretome and the pro‐tumorigenic effects of senescent cells. Cancer Cell. 2018;34:85–102.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Gould L, Abadir P, Brem H, Carter M, Conner‐Kerr T, Davidson J, et al. Chronic wound repair and healing in older adults: current status and future research. Wound Repair Regen. 2015;23:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Grosse L, Wagner N, Emelyanov A, Molina C, Lacas‐Gervais S, Wagner K‐D, et al. Defined p16High senescent cell types are indispensable for mouse Healthspan. Cell Metab. 2020;32:87–99.e6. [DOI] [PubMed] [Google Scholar]
- 261. Ou H‐L, Hoffmann R, González‐López C, Doherty GJ, Korkola JE, Muñoz‐Espín D. Cellular senescence in cancer: from mechanisms to detection. Mol Oncol. 2021;15:2634–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Schafer MJ, Zhang X, Kumar A, Atkinson EJ, Zhu Y, Jachim S, et al. The senescence‐associated secretome as an indicator of age and medical risk. JCI Insight. 2020;5:133668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Wiley CD, Sharma R, Davis SS, Lopez‐Dominguez JA, Mitchell KP, Wiley S, et al. Oxylipin biosynthesis reinforces cellular senescence and allows detection of senolysis. Cell Metab. 2021;33:1124–1136.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Krueger MA, Cotton JM, Zhou B, Wolter K, Kuehn A, Schwenck J, et al. Abstract LB‐369: in vivo imaging of tumor senescence with a novel beta‐galactosidase specific PET tracer. Cancer Res. 2018;78:LB‐369. [Google Scholar]
- 265. Krueger MA, Cotton JM, Zhou B, Wolter K, Schwenck J, Kuehn A, et al. Abstract 1146: [18F]FPyGal: a novel ß‐galactosidase specific PET tracer for in vivo imaging of tumor senescence. Cancer Res. 2019;79:1146. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
There is no data associated with this review.