

Abstract
Altered synaptic structure and function is a major hallmark of fragile X syndrome (FXS), autism spectrum disorders (ASDs), and other intellectual disabilities (IDs), which are therefore classified as synaptopathies. FXS and ASDs, while clinically and genetically distinct, share significant comorbidity, suggesting that there may be a common molecular and/or cellular basis, presumably at the synapse. In this article, we review brain architecture and synaptic pathways that are dysregulated in FXS and ASDs, including spine architecture, signaling in synaptic plasticity, local protein synthesis, (m)RNA modifications, and degradation. mRNA repression is a powerful mechanism for the regulation of synaptic structure and efficacy. We infer that there is no single pathway that explains most of the etiology and discuss new findings and the implications for future work directed at improving our understanding of the pathogenesis of FXS and related ASDs and the design of therapeutic strategies to ameliorate these disorders.
ASDs and FXS: Causes and Clinical Features
Approximately 1%–3% of the general population is affected by intellectual disabilities (IDs). IDs are neurodevelopmental disorders defined by significant limitations in intellectual functioning and adaptive behaviors and often exhibit comorbidity with autism (Mefford et al., 2012). Autism spectrum disorders (ASDs) are characterized by high phenotypic heterogeneity, comprising deficits in social interaction and communication as well as repetitive and stereotyped behaviors (Bhat et al., 2014; Volkmar and McPartland, 2014). ASDs have a strong and complex genetic component with multiple familial inheritance patterns and are estimated to be linked to mutations in as many as 1,000 genes (see SFARI database). Over the past decade, genome-wide association studies have enabled rapid progress in the identification of risk genes for ASDs (Ramaswami and Geschwind, 2018).
In addition to the genetic factors, environmental and/or immune factors contribute to the risk of autism during early life (Waye and Cheng, 2018). The interaction of environmental exposures with an individual genetic background and particular susceptibility manifests differently in each case, leading to heterogeneous phenotypes and varied comorbid symptoms within the disorder. Among the environmental factors to consider are prenatal infections (maternal immune activation), maternal diabetes, zinc deficiency, prenatal and perinatal stress, and toxins (reviewed in Park et al., 2016). Several reports suggest that exposure to various toxicants, including pesticides, polychlorinated biphenyls, and polybrominated diphenyl ethers (PBDEs), can have detrimental consequences on developmental processes. The negative effects could be exacerbated in genetically susceptible individuals (Mostafalou and Abdollahi, 2017).
Several genetic syndromes are known to have significant associations with autism (Borrie et al., 2017). Study of these autism-related syndromes, which are well-defined genetic disorders, has yielded major insights into ASDs. For example, tuberous sclerosis complex (TSC) is an autosomal dominant genetic disorder characterized by the formation of hamartomas in different organs, including the brain, and it is caused by mutations in the TSC1 or TSC2 genes, encoding hamartin and tuberin, respectively. The TSC protein complex inhibits the mechanistic target of rapamycin (mTOR) signaling pathway (Henske et al., 2016; Jeste et al., 2016). Children and adolescents with TSC display epilepsy and cognitive disorders and 30%–40% of patients have autism (Curatolo et al., 2010).
Another important example of syndromic autism is fragile X syndrome (FXS), the most prevalent neurodevelopmental disorder with a genetic origin characterized by a high incidence of autism. FXS, first described by Martin and Bell in 1943, has a prevalence of approximately 1 in 4,000 males and 1 in 8,000 females. It is an X-linked condition caused, in the majority of cases, by an unstable CGG trinucleotide repeat expansion (200 or more) in the 5′ untranslated region (UTR) of the fragile X mental retardation 1 (FMR1) gene (Bagni et al., 2012), leading to its hypermethylation and transcriptional silencing (Bagni et al., 2012; Berry-Kravis et al., 2018; Nelson et al., 2013). Over the past years, rare FMR1 gene mutations, affecting the coding or non-coding region of the gene and causing FXS, have also been reported (Sitzmann et al., 2018).
Fragile X mental retardation protein (FMRP), the product of the FMR1 gene, is an RNA-binding protein that tightly controls the localization, stability, and translation of a large array of RNAs critical to neuronal development, synaptic plasticity, and dendritic spine architecture (for recent review, see Banerjee et al., 2018; Braat and Kooy, 2015; Contractor et al., 2015; Huber et al., 2015; Richter et al., 2015). The behavioral phenotype of children with FXS can differ from individual to individual, although there are several “core” behaviors seen in the youngest patients. Avoidant eye gaze, attention-deficit/hyperactivity disorder (ADHD), hand flapping, repetitive behaviors, reduced social interactions, anxiety, and speech perseverations have been reported in 60% to 90% of individuals with FXS (Bagni et al., 2012; Roberts et al., 2018). On the more extreme end, some patients with FXS and autism are completely nonverbal and exhibit aggressive or self-injurious, repetitive, and/or stereotyped behaviors and severe intellectual disability. Around 40% to 60% of male and 20% of female patients with FXS meet the criteria for ASDs (Kaufmann et al., 2017). ASDs seem to be a distinctive subphenotype in FXS, characterized by deficits in complex social interaction, with similarities to ASDs in the general population (Hernandez et al., 2009). Individuals with FXS and ASDs also have a lower IQ and more stereotyped and aggressive behaviors, impaired expressive syntax, hyperactivity, emotional lability, attentional deficits, resistance to change, seizures, sleep disorders, hypersensitivity to sensory stimuli, and autonomic dysfunction (Bailey et al., 1998; Hatton et al., 1999; Kaufmann et al., 2017; McDuffie et al., 2010; Thurman et al., 2015). Of note, the observed impaired expressive syntax may contribute to atypical social behaviors and thereby ASD symptoms (Abbeduto et al., 2019).
FXS and ASDs: Common Features in Genetic Makeup and Brain Circuitry
Our brains undergo remarkable changes throughout infancy, childhood, and adolescence. Processes including myelination and synaptogenesis (Remer et al., 2017) occur across the first years of life, and brain remodeling continues into young adulthood. Approximately one-third of the human genes are expressed primarily in the brain and during brain development (Douet et al., 2014). Genes that influence developmental and age-specific brain trajectories play an important role in several critical events in neurodevelopmental disorders such as ASDs (reviewed in Parikshak et al., 2015). The identification of large biological networks of genes affected by rare de novo copy number variants in autism revealed that the genes forming networks are primarily related to synapse development, axon targeting, and cytoskeleton organization (De Rubeis et al., 2014; Gilman et al., 2011), processes that ultimately shape our brain.
Mutations in many genes linked to ASDs and related disorders affect the excitatory/inhibitory balance in neurons. Perturbations in synaptic physiology and neuronal circuitry have recently been reviewed (e.g., Contractor et al., 2015) and will therefore be mentioned only briefly here. Hyperexcitability of excitatory neurons is a recurring topic in FXS (see below). In addition, there is evidence for weakened inhibitory drive. Notable examples are decreased GABAA receptor expression in the hippocampus of FXS patients and Fmr1 knockout (KO) mice (Gantois et al., 2006; Gao et al., 2018; Kang et al., 2017) and a reduction in the frequency and amplitude of miniature and spontaneous inhibitory postsynaptic currents (IPSCs) in the mature amygdala (Olmos-Serrano et al., 2010). Moreover, development of the inhibitory network is delayed. In rodents, GABAA receptors switch on at postnatal day ~7 (~P7) from primarily depolarizing (excitatory) to hyperpolarizing (inhibitory), largely due to a changing Cl− gradient (Cherubini et al., 2011). This switch is delayed in layer IV neurons of the somatosensory cortex of Fmr1 KO mice (He et al., 2014). Similarly, fast-spiking (FS) interneurons in layer IV of neocortex that lack FMRP remain excitatory throughout the critical period (up to P9, Nomura et al., 2017), with important consequences for synaptic and circuit properties in the mature cortex. Of note, an imbalance in the excitatory-to-inhibitory ratio (E/I) is a common theme in FXS and other ASDs (Nelson and Valakh, 2015).
Brain Structure and Functional Connectivity: Lessons from Magnetic Resonance Imaging
While in the past studies in brain relied mainly upon gross anatomical measures of brain structure and morphology, more advanced non-invasive imaging techniques now allow us to identify and quantify functional and microstructural aspects of brain tissue that are more closely related to neurodevelopmental processes (Khundrakpam et al., 2016; Lebel and Deoni, 2018). Specifically, MRI produces detailed three-dimensional anatomical images without the use of damaging radiation. In addition, sophisticated MRI imaging techniques yield invaluable information about brain connectivity. Resting-state functional MRI measures brain functional connectivity (FC). Diffusion tensor imaging (DTI) tractography, and especially its most important readout, the fractional anisotropy (FA), is a measure of axonal integrity. While MRI techniques do not have the spatial and temporal resolution to arrive at the cellular level, they are very powerful because they are in vivo and non-invasive, with a high translational potentiality. Over the past years, neuroimaging studies have linked genetics with brain development from infancy to young adulthood (Douet et al., 2014), providing complementary information to age-specific brain trajectories based on gene expression.
Neuroimaging studies demonstrate that ASDs and other psychiatric disorders are correlated with diffuse alterations in large-scale functional and structural brain networks (Baribeau and Anagnostou, 2013). Likewise, suboptimal levels of FMRP in the developing brain appear to be detrimental for brain wiring (Dennis and Thompson, 2013). Imaging studies in humans with FXS (Swanson et al., 2018) and ASDs (Ha et al., 2015) reveal aberrant structure of multiple brain regions. Specifically, long-range FC is altered in ASD and FXS patients (Hall et al., 2013; Holsen et al., 2008; Yerys et al., 2017). Additionally, axonal integrity and connectivity as measured by DTI is also affected in patients with FXS who exhibit significantly larger or smaller gray matter volume in the bilateral caudate nucleus and other regions relative to controls (Sandoval et al., 2018). The white matter in frontostriatal pathways, as well as in parietal sensory-motor tracts, is also affected (Barnea-Goraly et al., 2003). In addition, microstructural defects as measured by reduction in FA of white matter tracts, predominantly in the corpus callosum, have been found in both FXS individuals and mouse models (Haberl et al., 2015; Swanson et al., 2018).
MRI studies in Fmr1 KO mice identified reduced volume of certain brain areas (Ellegood et al., 2010). Ex vivo DTI of FMR1 KO mice revealed a reduced diffusivity in the cortex of juvenile FXS mice suggesting structural changes in axonal fiber patterns and integrity, indicative of subtle changes in connectivity at early developmental stages (La Fata et al., 2014). In addition, in vivo imaging of FXS mice shows an anatomical hyperconnectivity phenotype in the primary visual cortex but a disproportional low connectivity with other neocortical regions (Haberl et al., 2015; Zerbi et al., 2018). These observations are consistent with findings in FXS human infants at the onset of the critical period, who exhibit an increased density of DTI-reconstructed fibers relative to that of normal children (Haas et al., 2009; Swanson et al., 2018).
Similarly, deletions and/or duplications in ASD-associated chromosomal regions, such as the 16p11.2 and the 15q11.2, also lead to abnormalities in brain FC and morphology. For instance, FC in the prefrontal cortex has been found to be impaired in humans and mice with the 16p11.2 deletion (Bertero et al., 2018). Patients with deletions and duplications in the 16p11.2 chromosomal region also present global and regional differences in brain structure compared to controls (Blackmon et al., 2018; Martin-Brevet et al., 2018; Qureshi et al., 2014). In the 16p11.2 hemideletion mouse, there are pronounced sex-specific changes in males with increased FA in medial fiber tracts, especially in those proximal to the striatum, which correlate with gene expression patterns associated with neurite outgrowth and the mitogen-activated protein kinase (MAPK) pathway (Kumar et al., 2018). Furthermore, patients with deletions and duplications in the 15q11.2 chromosomal region present abnormalities in white matter architecture and brain structure (Silva et al., 2018; Stefansson et al., 2014; Ulfarsson et al., 2017; Vanlerberghe et al., 2015). Finally, genetic variations in the CNTNAP2 gene in humans have been associated with ASDs (Peñagarikano and Geschwind, 2012), and KO of this gene in mice leads to reduced FC and aberrant white matter morphology (Liska et al., 2018; Zerbi et al., 2018). In conclusion, there are consistent deficits detected by MRI in ASD patients and the respective mouse models. Of note, there is heterogeneity in the brain regions affected, which might be helpful for patient stratification (Ellegood et al., 2010).
Spine Structure in Humans and Mice
The maturation and maintenance of the pre- and postsynaptic compartments depends upon the activation status of a synapse that defines its connection strength (Holtmaat and Svoboda, 2009). It is thought that defects in spines can give rise to impaired synaptic maturation and may contribute to cognitive deficits observed in individuals with this debilitating condition. Of note, more than 50% of the ID genes encode for proteins that are part of the pre- or postsynaptic compartments or are implicated in synaptic functions by regulating synapse formation, synaptic actin cytoskeleton assembly or disassembly, or synaptic plasticity (Ropers and Hamel, 2005; Verpelli and Sala, 2012) and form the unifying synapse-based theory for cognitive and behavioral deficits.
Human neuropathological and genetic findings on model systems suggest that FXS and ASDs may be conceptualized as diseases of the synapse because of the aberrant dendritic spine morphology and density (Bourgeron, 2015; Zoghbi and Bear, 2012). Golgi staining of postmortem brain of FXS-affected individuals revealed a deficit in the number, structure, and morphology of the synapses. Specifically, FXS patients exhibit a higher density of dendritic spines on distal segments of apical and basal dendrites in the cingulate, temporal, and visual cortex, with increased incidence of longer (immature) and fewer short (mature) dendritic spines than control subjects (Martínez-Cerdeño, 2017).
Increased spine density has been observed in brain tissues from humans with ASDs, whereas an increase in spine density on apical dendrites of pyramidal neurons in some, but not all, cortical brain areas was detected (Hutsler and Zhang, 2010). Furthermore, reduced developmental spine pruning in layer V pyramidal neurons in postmortem ASD temporal lobe has been shown to correlate with hyperactivated mTOR and impaired autophagy (Tang et al., 2014). The mouse model for FXS also presents an overabundance of dendritic spines with an immature morphology (reviewed in He and Portera-Cailliau, 2013), even though some studies report no detectable spine alteration in FXS. Thus, stimulated emission depletion microscopy (STED) detected only subtle differences in the Fmr1 KO mice that are dependent on age and brain region (Wijetunge et al., 2014). It is possible that the apparent discrepancy is due to a difference in technique, region, or developmental stages (reviewed in He and Portera-Cailliau, 2013). Mouse models of ASDs with specific genetic mutations such as the Cyfip1, Tsc2, Shank3, or Neuroligin-1 genes also present abnormalities in spine morphology and/or number (De Rubeis et al., 2013; Oguro-Ando et al., 2015; Pathania et al., 2014; Peça et al., 2011; Schnell et al., 2014; Varghese et al., 2017).
Molecular Signaling
Another aspect of convergence at the synapse is dysregulation of the mTOR and MAPK/extracellular signal-regulated kinase (ERK) signaling cascades. Molecules in these pathways (Figure 1), such as tuberous sclerosis complex 1 and 2 (TSC1 and TSC2), neurofibromatosis 1 (NF1), phosphatase and tensin homolog (PTEN), MAPK-interacting kinase 1 and 2 (MNK1 and MNK2), and ERK1/2 are dysregulated in FXS, TSC1/2, Angelman syndrome, and Rett syndrome (Borrie et al., 2017; Huber et al., 2015; Mellios et al., 2018; Zoghbi and Bear, 2012). Mutations in TSC1/2, PTEN, MNK1, and MNK2 cause an imbalance in protein synthesis. Of note, the mTOR and MAPK pathways were recently found to be dysregulated in blood from patients with idiopathic ASDs and to possibly constitute a molecular signature for the severity of the disease (Rosina et al., 2019; Figure 1). These observations raise the possibility that dysregulation of mTOR and MAPK may be a common theme in individuals with FXS and a subset of ASDs (Abrahams and Geschwind, 2008; Subramanian et al., 2015).
Figure 1. Simplified Model of the mTOR-ERK1/2-WAVE Signaling Pathways at Synapses.
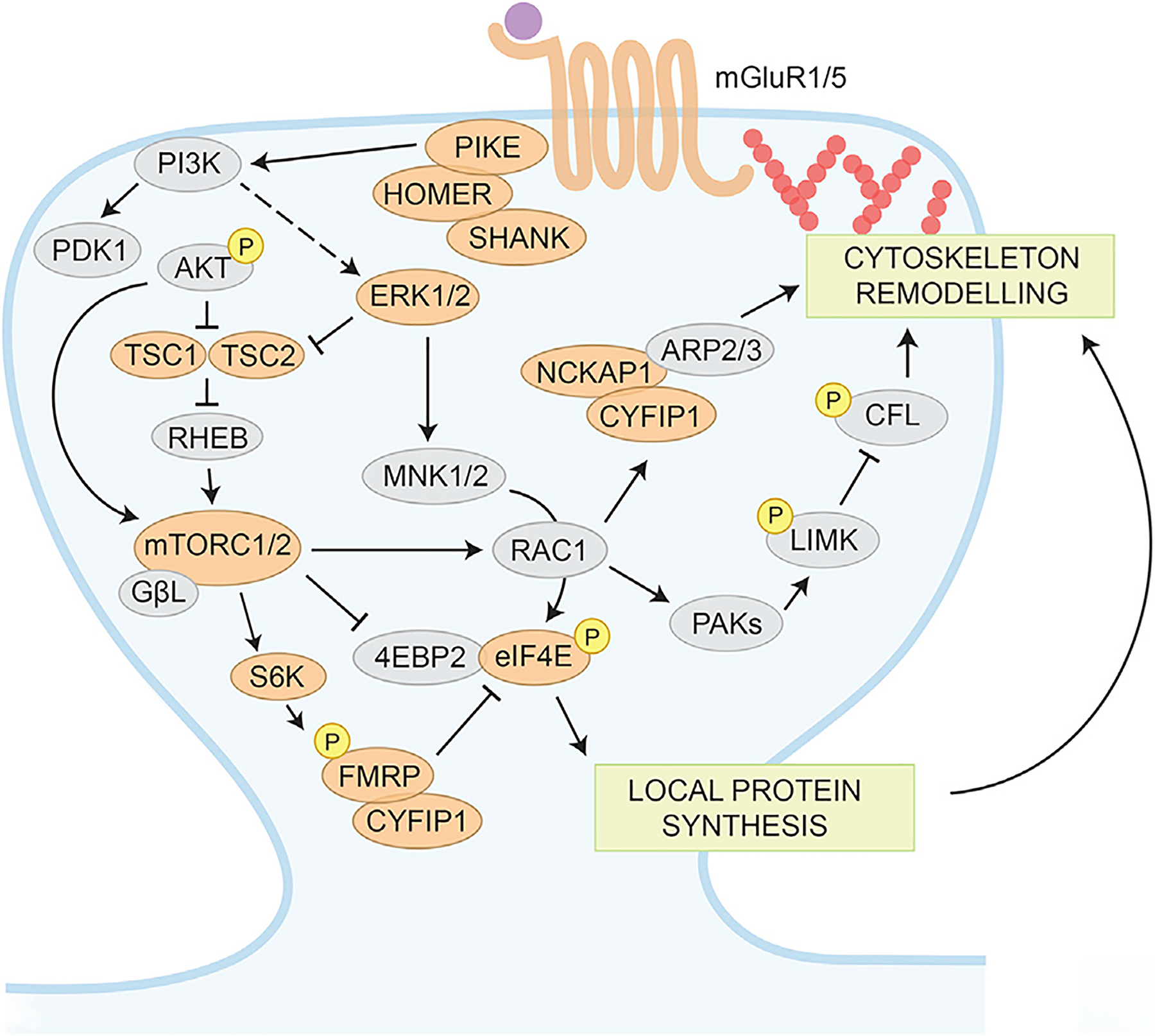
mGluRs mediate activation of PI3K via scaffolding proteins such as PIKE and HOMER, leading to the activation of AKT and inhibition of TSC1-TSC2 complex. TSC2 inhibition of RHEB activates mTORC1, resulting in FMRP phosphorylation by S6 kinase. The phosphorylation status affects its RNA-binding properties as well as its translational regulation. FMRP interacts with the initiation factor eIF4E and regulates translational through the specific eIF4E-binding protein CYFIP1. In Fmr1 KO mice and FXS individuals, PI3K is upregulated, leading to an increased mTOR phosphorylation, culminating in an increased protein synthesis. mTORC2 has a central role in actin remodeling through its actions on RAC1 and LIMK phosphorylation on the actin-depolymerizing factor cofilin (CFL). Proteins encoded by genes associated with ASDs (SFARI database, https://gene.sfari.org) are colored in orange.
Activation of cell surface receptors such as NMDA-type glutamate receptors (NMDARs), metabotropic glutamate receptors (mGluRs), and tyrosine kinase (TRK) receptors activate MAPK via ERK and the mTOR signaling via the phosphoinositide-3 (PI3K) and AKT pathway. The ERK/MAPK pathway plays an important role in several processes, including the transition from pluripotent stem cells to neuronal progenitors, synaptic plasticity, and consolidation of memory, and its dysregulation is implicated in the pathophysiology of fragile X (Zoghbi and Bear, 2012). ERK/MAPK signaling is hyperactivated in brains of both FXS patients and Fmr1 KO mice (Sawicka et al., 2016; Wang et al., 2012). Interestingly, the cleavage product of the amyloid precursor protein (APP), the soluble APPα, is upregulated in Fmr1 KO mice, leading to elevated activation of ERK1/2 (Pasciuto et al., 2015). Furthermore, dysregulated dephosphorylation of ERK in response to mGluR1/5 stimulation was observed in Fmr1 KO cortical synaptoneurosomes (Kim et al., 2008). Of note, the 16p11.2 locus deletion contains the MAPK3 gene and is associated with ASDs (Fasano and Brambilla, 2011). Mice harboring the 16p11.2 deletion show disruption of the MAPK signaling and deficits in behavior. Inhibition of the ERK pathway during development improves the signaling and behavioral deficits observed in these mice, suggesting the MAPK pathway as a potential therapeutic target for 16p11.2 microdeletion syndrome (Pucilowska et al., 2018). Intriguingly, both MAPK and mTOR signaling converge on the regulation of protein synthesis, which plays a central role in synaptic plasticity (see below).
mTOR is a pivotal regulator of cell growth, proliferation, cell cycle, ribosome biogenesis, autophagy, protein synthesis, and the actin cytoskeleton (Costa-Mattioli and Monteggia, 2013; Laplante and Sabatini, 2012). Components of the mTOR signaling pathway localize to synaptic sites where they can modulate synaptic plasticity via regulation of local protein synthesis in dendrites and actin polymerization in spines (Huang et al., 2013; Tang et al., 2002). Whereas mTOR complex 1 (mTORC1) is a master regulator of cap-dependent translation (Darnell and Klann, 2013), mTORC2 is a central regulator of the actin cytoskeleton through its actions on the actin-depolymerizing factor cofilin (Costa-Mattioli and Monteggia, 2013; Laplante and Sabatini, 2012; see also below). Moreover, mTORC2 (but not mTORC1) has been shown to be critical to mGluR-LTD and related behaviors (Zhu et al., 2018).
Fragile X mice exhibit overactivated mTORC1 signaling at hippocampal synapses, which is causally related to an overactivation of p70 S6 kinase 1 (S6K1, Ronesi et al., 2012; Sharma et al., 2010). In addition, crossing Fmr1 KO mice with mice lacking both alleles of the gene encoding S6K1 corrects signaling, spine morphology, and autism-relevant behaviors (Bhattacharya et al., 2016). Although dysregulation of mTORC1 has been documented in fragile X humans (Hoeffer et al., 2012), mice (Sharma et al., 2010), and flies (Gross et al., 2015), a role for mTORC2 is, as yet, unclear. A recent finding that chronic treatment with rapamycin, which inhibits mTOR, had an adverse effect on sleep and social behavior in both control and Fmr1 KO mice (Saré et al., 2018) may simply reflect the not unexpected observation that not only too much, but also too little, mTOR activity is deleterious (Saré et al., 2018). Interestingly, mTOR dysregulation has also been observed in ASD patients with 15q11–13 duplication and in a mouse model overexpressing the Cyfip1, part of the 15q11–13 region (Oguro-Ando et al., 2015).
Lipid Signaling at FXS and ASD Synapses
The brain is particularly enriched in lipids and maintains a highly diverse lipid composition. Overwhelming evidence has identified key roles for signaling lipids in mediating membrane traffic and signal transduction at synapses. For example, phosphoinositides (PIPs) have important pre- and postsynaptic functions (Di Paolo and De Camilli, 2006), including the regulation of exoand endocytosis of synaptic vesicles and postsynaptic receptors, conductance of ion channels, and signaling from activated neuroreceptors such as mGluRs and NMDARs to allow plastic regulation of synaptic function, which is believed to underlie cognitive and behavior features. Furthermore, mechanical deformation of the lipid bilayer is sufficient to modulate the gating properties of several channels (Phillips et al., 2009), suggesting that membrane lipid composition and organization of the spine is different from that of the dendritic shaft, from which these protrusions emerge (Dotti et al., 2014).
In spite of the importance of lipid signaling for regulating synaptic function, its role in synaptopathies remains largely unexplored. Altered lipid signaling is associated with neurodevelopmental diseases. Emerging evidence implicates defective PIP metabolism in IDs and ASDs (Gross, 2017). Loss-of-function mutations in the acid sphingomyelinase (ASM) gene cause Niemann-Pick disease type A (NPA, Schiffmann, 1996), a rare disorder characterized by the accumulation of sphingomyelin (SM) in lysosomes and plasma membrane of neurons. NPA causes severe ID, premature aging, and child death in early age due to the accumulation of SM in lysosomes and plasma membrane of neurons. A contribution to the cause of the disease comes from impaired activity of the plasma membrane calcium ATPase, which increases intracellular calcium and cellular oxidative stress (Pérez-Cañamás et al., 2017).
Therefore, defective lipid signaling seems to be a common theme in the synaptic and neurophysiological deficits underlying ID/ASD and related synaptopathies. In the case of FXS, a retrospective study suggested that total cholesterol, low-density lipoprotein, and high-density lipoprotein levels are lower in males with FXS than in the general population (Berry-Kravis et al., 2015). In the absence of FMRP, diacylglycerol (DAG) levels in the basal state are increased in the cerebellum of patients with FXS as well as in Fmr1 KO neurons. At synapses, DAG molecules are produced through activation of postsynaptic receptors, and DAG signaling triggers dendritic spine growth and stabilization (Kim et al., 2010); excessive DAG would predict the spine properties observed in the Fmr1 KO neurons (Tabet et al., 2016). Of interest, among the list of candidate FMRP targets, there are regulators of the PI3K-based lipid-signaling pathway, mediators of lipid signaling or components that act as downstream effectors of proteins of the PI3K-mTOR-TSC cascade (Ascano et al., 2012; Darnell et al., 2011; Gross et al., 2015; Tabet et al., 2016).
Another important signaling molecule downstream of the PI3K cascade is the glycogen synthase kinase 3 (GSK3), which is inhibited by AKT. This serine/threonine kinase regulates a variety of pathways involved in neurodevelopment, including neurogenesis, migration, axon growth and guidance, and synaptic plasticity (Santos et al., 2014). It is therefore not surprising that dysregulation of GSK3β contributes to a number of diseases such as mood disorders, schizophrenia, and Alzheimer disease (King et al., 2014). Of note, GSK3b activity is elevated in FXS mice (Min et al., 2009) and modulation of GSK3b activity appears to be beneficial in FXS, as it reverses the dendritic spine deficits and the level of protein synthesis (Santos et al., 2014). Interestingly, GSK3b activity is downmodulated by lovastatin (Lin et al., 2016), a statin drug used for lowering cholesterol that also has an effect in inhibiting the Ras-ERK1/2 signaling pathway, exaggerated in FXS (Berry-Kravis et al., 2015; Çaku et al., 2014; Osterweil et al., 2010). This has set the basis for two pilot open-label trials of lovastatin in humans with FXS that were performed and concluded (https://www.clinicaltrials.gov). As lovastatin should actually lower cholesterol and low- and high-density lipoprotein levels, possible problems associated with already low lipid levels in individuals with FXS should be monitored (Berry-Kravis et al., 2015). Although these trials showed potential benefit, a placebo-controlled trial is necessary to ascertain lovastatin efficacy in FXS. Clinical trials with statins have shown mixed results in improving cognition and behavior in individuals with IDs and ASDs such as neurofibromatosis (NF1, van der Vaart et al., 2016), a monogenic form of syndromic autism, although the impaired social and cognitive phenotype in animal models were rescued. Therefore, while the precise mechanisms underlying defective lipid signaling and IDs/ASDs remain to be identified, the availability of FDA-approved drugs targeting lipid signaling might provide a potential avenue for the treatment of IDs/ASDs-related synaptopathies.
Metabotropic Glutamate Receptors
The Gq-coupled, group mGluRs are enriched at excitatory synapses throughout the brain, where they are strategically positioned to regulate glutamatergic signaling (Reiner and Levitz, 2018). They are crucial to synaptogenesis and formation of neural circuitry during brain development (Reiner and Levitz, 2018) and to mGluR-dependent long-term depression (LTD), an activity-dependent, NMDA-independent form of homosynaptic synaptic plasticity at Schaffer collateral to CA1 pyramidal synapses through adulthood (Oliet et al., 1997; Palmer et al., 1997). Importantly, modulation of mGluR5 corrects the bidirectional alteration of synaptic plasticity observed in Tsc2+/− and Fmr1 KO mice, and the synaptic and cognitive defects were restored in the double mutant model (Auerbach et al., 2011). A recent finding shows that β-arrestin2 provides an important link between mGluR5 and local translation in dendrites (Stoppel et al., 2017).
Expression of mGluR-LTD is mediated by persistent internalization of AMPA receptors and in adolescent mice requires de novo protein synthesis (Huber et al., 2002; Waung and Huber, 2009) with a noted requirement for the activity of the immediate early gene Arc encoding the activity-regulated cytoskeletal protein that interacts with dynamin and endophilin to facilitate endocytosis of AMPARs (Chowdhury et al., 2006; Shepherd et al., 2006). Notably, Arc mRNA is an FMRP target at synapses (Zalfa et al., 2003), and dysregulation of Arc activity is implicated in aberrant synaptic plasticity and memory consolidation in Fmr1 KO mice (Park et al., 2008). Specifically, Fmr1 KO mice exhibit deficits in Hebbian forms of LTD and long-term potentiation, altered excitability, and delayed synaptic maturation (Contractor et al., 2015; Huber et al., 2002). Fmr1 KO mice exhibit exaggerated mGluR-LTD at CA1 synapses leading to elevated protein synthesis, a hallmark of FXS (Huber et al., 2002). Genetic reduction of mGluR5 reverses many phenotypes in Fmr1 KO mice, including exaggerated mGluR-LTD (Dölen et al., 2007).
At synapses, association of mGluR5 with the interacting scaffolding protein Homer is compromised in Fmr1 KO mice, a feature that is rescued by genetic deletion of a short, dominant-negative isoform of Homer, H1a (Guo et al., 2016; Ronesi et al., 2012). The mGluR1/5-Homer complex signals to the PI3 kinase enhancer PIKE, a target of FMRP (Darnell et al., 2011), which engages PI3K/AKT/mTOR signaling in response to synaptic stimulation (Figure 1; Gross et al., 2015). It is now well established that PIKE is the upstream effector of overactivated mTOR signaling at hippocampal synapses and provides a link between loss of FMRP and elevated mTOR signaling and exaggerated mGluR-LTD (Gross et al., 2015; Sharma et al., 2010). FMRP itself has also been shown to be part of a complex with the regulatory subunit and a target of the ubiquitin ligase Cdh1-anaphase-promoting complex (Cdh1-APC), which is critical to mGluR-LTD in the brain (Huang et al., 2015).
Given this evidence for a diverse and intricate role for mGluRs in FXS, a panel of mGluR antagonists has been tested not only in mouse models (MPEP, CTEP, Fenobam, AFQ056, STX107, RO491753), but also in individuals diagnosed with FXS (reviewed in Bagni et al., 2012; Hagerman et al., 2017; Richter et al., 2015). However, in clinical trials (https://www.clinicaltrials.gov/), these drugs failed to ameliorate the most severe symptoms in humans. Data generated from several independent laboratories using biological models for the disease are in line with a dysregulated mGluR pathway. The possible discrepancy between the results generated in mice and the clinical trials in humans might suggest that patient stratification together with learning-based outcome measures should be developed (Berry-Kravis et al., 2018)
In addition to fragile X syndrome, dysregulation of mGluRs have been reported in other forms of intellectual disabilities and ASDs (Mahato et al., 2018). Phelan-McDermid syndrome (PMS) is a neurodevelopmental disorder caused by genetic haploinsufficiency of the SHANK3 gene, which is a member of the Shank/ProSAP family of postsynaptic scaffolding proteins (Shank1, Shank2, and Shank3), which form a multiprotein complex assembly in the postsynaptic density of excitatory synapses and have been identified in gene-linkage studies to be associated with autism (Monteiro and Feng, 2017). A mouse model of PMS, the Shank3 KO mouse, exhibits altered mGluR5-mediated signaling due to reduced association of mGluR5 with Homer1b/c and elevated levels of mGluR5 in PSDs, suggesting that mGluR5 function may also be augmented (Wang et al., 2016). Pharmacological manipulation of mGluR5 activity rescued impaired LTD at striatal synapses and elevated neuronal excitability and ameliorated ASD-relevant behaviors (Vicidomini et al., 2017; Wang et al., 2016). Of note, recently a missense variant in the SHANK3 gene revealed a novel function of Shank3 to recruit Abelson interactor 1 (ABI1) and the WAVE complex to the postsynaptic density, an event critical to synapse and dendritic spine development, a function that is independent of its binding to Homer (Wang et al., 2019). Mutations in the human SHANK2 gene (also known as ProSAP1) have been associated with ASDs and intellectual disabilities, and SHANK2 mutant mice carrying a mutation identical to the ASD-associated microdeletion in the human SHANK2 gene exhibit ASD-like behaviors. Those deficits were ameliorated by a positive allosteric modulator of mGluR5 (Won et al., 2012). Furthermore, overexpression of the scaffolding protein of Homer1a in the basal and lateral amygdala impaired auditory fear conditioning and induced autism-like reduced social interactions (Banerjee et al., 2016). These findings are significant in that mGluRs act through scaffolding proteins and downstream signaling to mediate the correct functioning of synapses.
FMRP at Synapses
A recent study has shown that although FMRP is present throughout the brain, its expression varies markedly among different neuronal populations (Zorio et al., 2017). Within a given neuron, FMRP localizes to the soma, developing and mature axons, dendrites, and the base of dendritic spines (Antar et al., 2004, 2006; Ferrari et al., 2007; Zimmer et al., 2017). Because neurons are so highly polarized, transport of mRNAs along the axon and dendrites to undergo local, on-site protein synthesis is key for maintaining proper structural and functional polarity (Bramham, 2008; Cioni et al., 2018; Rangaraju et al., 2017; Tom Dieck et al., 2014). In these subcompartments, FMRP and its homologs, the fragile X-related proteins FXR1P and FXR2P, assemble with other RNA-binding proteins and coding and non-coding RNAs to form large ribonucleoprotein particles or granules that tightly regulate mRNA transport, translation, stability, and degradation (Akins et al., 2012; Korsak et al., 2016; Pasciuto and Bagni, 2014a, 2014b). In axons and dendrites, fragile X granules are transported along microtubules in a translationally-quiescent manner through interactions with kinesin, myosin, and dynein motor proteins (Kanai et al., 2004). Different types of fragile X granules are selectively expressed in specific neuronal circuits where they tightly regulate transport, localization, stability, and translation of target RNAs (Akins et al., 2017; Chyung et al., 2018; Korsak et al., 2016).
High-throughput approaches, such as coimmunoprecipitation followed by microarray analysis (Brown et al., 2001), antibody-positioned-RNA amplification (APRA, Miyashiro et al., 2003) and crosslinking-immunoprecipitation with RNA sequencing (CLIP-seq, Ascano et al., 2012; Darnell et al., 2011), led to the identification of hundreds of putative FMRP target mRNAs (>1,000 in brain and >6,000 in non-neuronal cells, of which ~1,000 were validated by RIP-seq). The impact of FMRP binding to most of these target mRNAs still needs to be evaluated. As yet, only a few have been validated at the protein level (i.e., are known to have altered protein expression in the absence of FMRP), and even fewer have been shown to rescue functional Fmr1 KO phenotypes when their protein abundance is manipulated.
Among the “core targets” of FMRP identified by these and other studies are a large number of mRNAs encoding pre- and postsynaptic proteins, including NMDARs, mGluR5, PSD-95, Shank1–3, Homer1, PIKE, neuroligins, neurexins, SNAP-25, AP-2, bassoon, synapsin, and calcium channels (Pasciuto and Bagni, 2014b). Among the FMRP mRNA targets, several hundred (precisely 859) are bona fide axonally and/or dendritically localized mRNAs (Figure 2). A substantial number (271) of these mRNAs are also associated with ASDs (Figure 2). A significant subset of target mRNAs is transported and translated in an activity-dependent manner (Dictenberg et al., 2008; Ferrari et al., 2007). In dendrites (the postsynaptic compartment), FMRP regulates local “on-site” translation of Arc, α-CaMKII, PSD-95, Map1b, Fmr1, and GABAA receptors at dendritic spines (Antar et al., 2004; Dictenberg et al., 2008; Zalfa et al., 2003, 2007). FMRP also promotes stability of specific mRNAs, such as PSD-95 and GABA receptors, thereby preventing their decay in specific brain regions and in specific compartments within a given neuron (Braat and Kooy, 2015; Miyashiro et al., 2003; Zalfa et al., 2007; Zhang et al., 2007).
Figure 2. Shared Genes among FMRP Targets, Axonal/Dendritic Transcriptome, and ASDs.
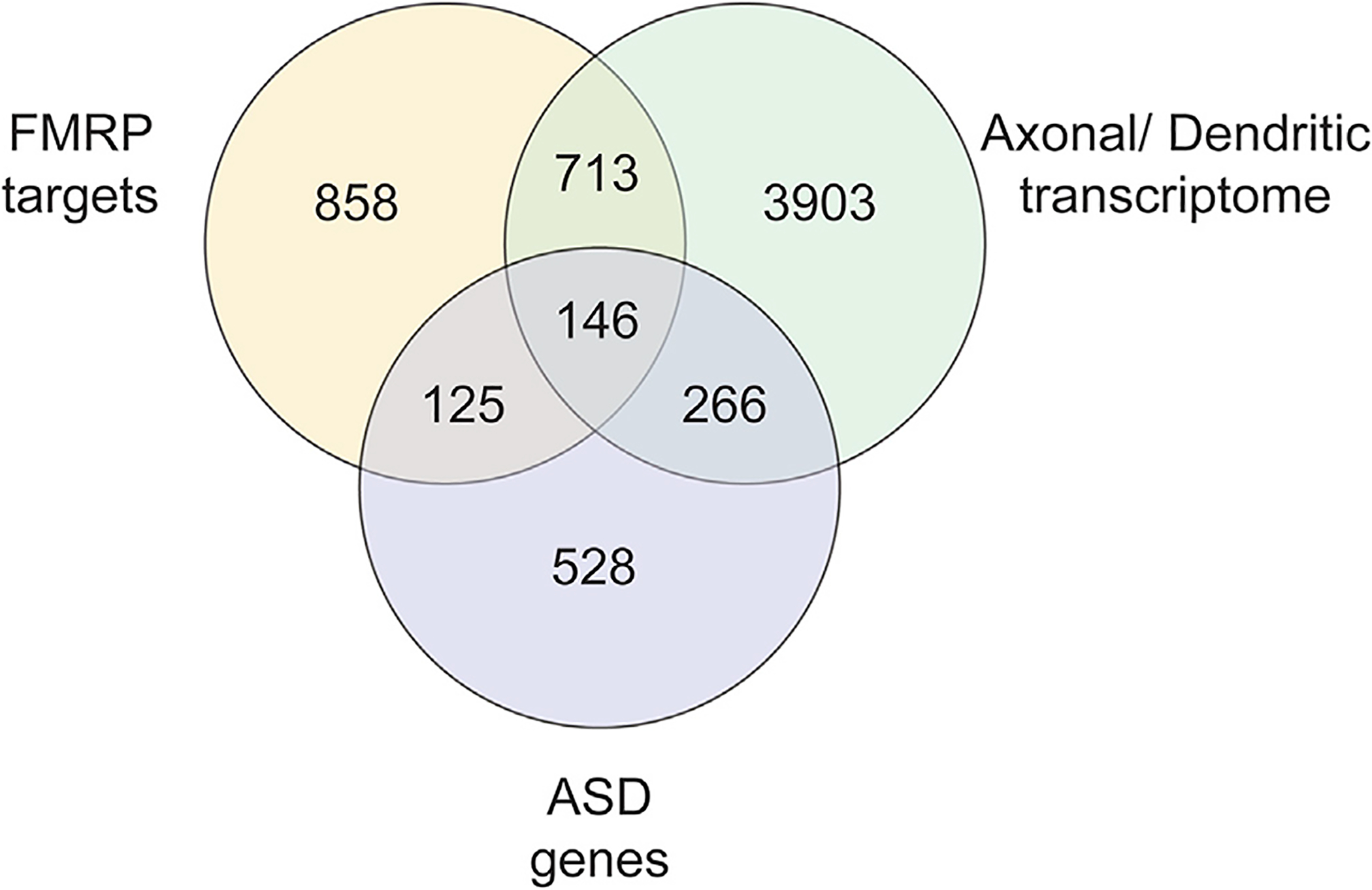
Among the 1,842 identified putative FMRP targets, 271 are in common with ASD genes and 859 are in common with the axonal/dendritic transcriptome. Moreover, 146 genes are shared with all three classes. 1,842 FMRP targets are based on the work from Ascano et al. (2012); Brown et al. (2001); Chen et al. (2003); Darnell et al. (2011); Miyashiro et al. (2003) and the list by Ascano et al. (2012) filtered for brain expression. 1,065 ASD genes are from the SFARI database, AutDB database (Pereanu et al., 2018), and Mahfouz et al. (2015). 5,028 genes from the axonal and dendritic localized transcriptome are based on the work by Cajigas et al. (2012) and Gumy et al. (2011).
It is now well accepted that maintaining a fine balance between the synthesis and degradation of RNAs and correct number of mRNAs and RNA-binding proteins is crucial to the proper structure and function of the synapse and, when dysregulated, result in synaptopathies (Grant, 2012) such as FXS and other ASDs (Bagni et al., 2012; Gandal et al., 2018; Klein et al., 2016; Parras et al., 2018). In normal cells, FMRP tightly regulates mRNA stability, transport, and translation. Some of these post-transcriptional processes are also affected in other ASDs. FMRP appears to fine-tune the proteome to match the changing needs of the cell and/or synapse. Neuronal gene expression is controlled with exquisite spatial and temporal resolution, including activity-dependent transcription in the nucleus and local translation at the synapse. Targets of FMRP include many mRNAs that encode synaptic and other plasticity-related proteins, which, when dysregulated, result in the cognitive impairment and other features in common between FXS and ASDs (Figure 1). Typical of RNA-binding proteins, the targeting specificity of FMRP seems to be determined by a cooperative interplay among several RNA-binding motifs within FMRP itself and/or its binding partners (Achsel and Bagni, 2016).
An additional source of complexity and heterogeneity comes from chemical modification, e.g., methylation and pseudouridylation, of synaptic mRNA and RNA. While enzyme-mediated mRNA modifications are known to regulate cellular mRNA turnover, the role of these modifications in regulating synaptic RNA has been examined only recently. It was shown that the brain noncoding RNA BC1 is differentially 2′-O-methylated in the cell body and at synapses and the change in modification affects its interaction with FMRP (Figure 3) in the different subcellular compartments (Lacoux et al., 2012). Methylation of adenosine in RNA to N6-methyladenosine (m6A), the most common mRNA modification present in about one-quarter of all RNAs (Arguello et al., 2018), appears to be relevant to synaptic pathologies. Recently, the synaptic m6A epitranscriptome was mapped and shown to be functionally enriched in pathways thought to be important in neurodevelopmental and neuropsychiatric diseases (Merkurjev et al., 2018).
Figure 3. FMRP Differently Recognizes Neuronal mRNAs.
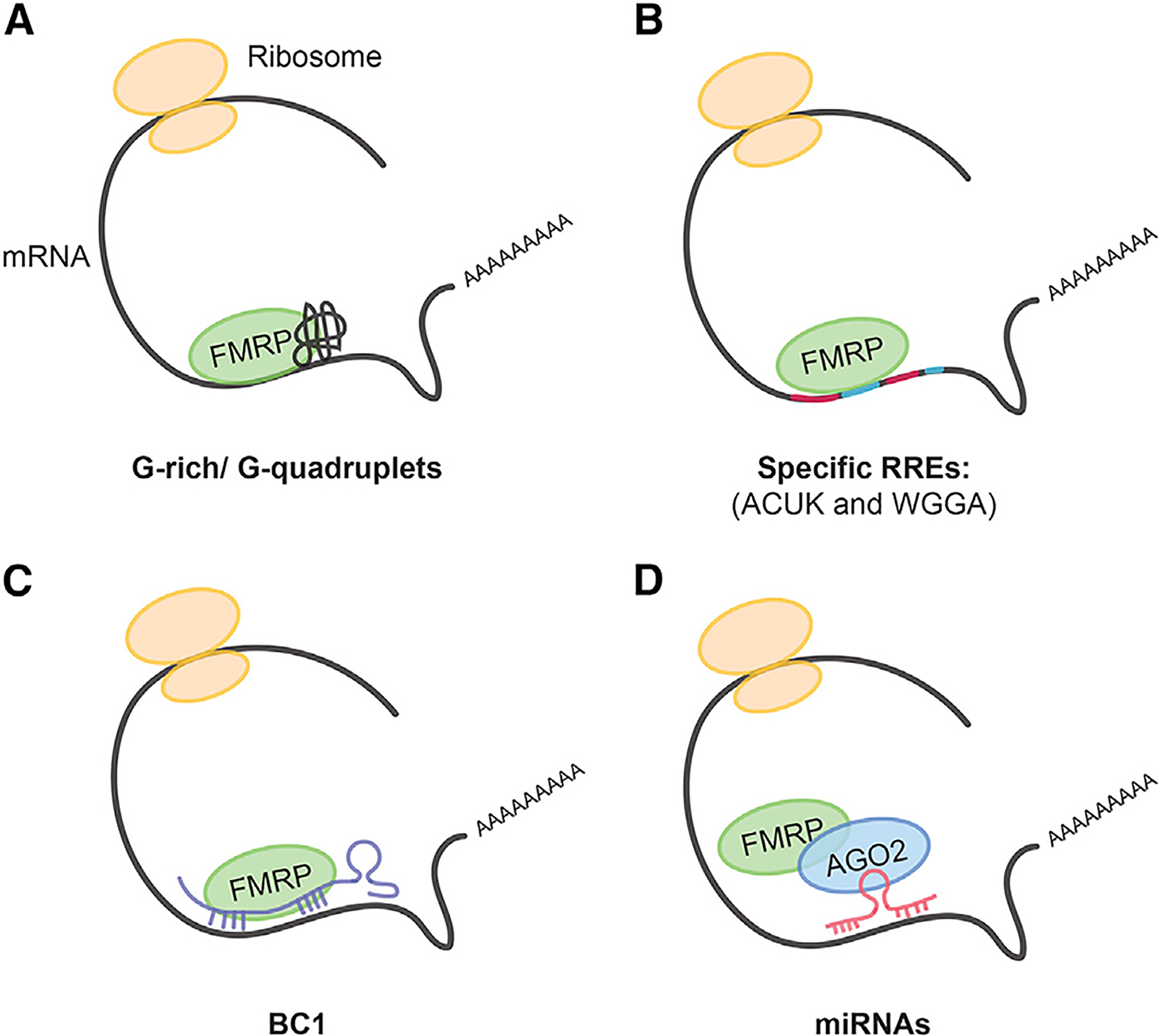
Four principal mechanisms by which FMRP recognizes mRNA targets.
(A) FMRP recognizes G-quartet structure and G-rich regions of the target mRNA.
(B) FMRP binds specific RNA recognition elements (RREs) such as ACUK and WGGA (Ascano et al., 2012).
(C and D) FMRP binds mRNA targets indirectly through either the small non-coding RNA brain cytoplasmic RNA 1 (BC1) (C) or microRNAs (miRNAs) (D). Modified from Bagni and Greenough (2005).
Additional evidence was generated underlining a key role of FMRP in recognition and regulation of modified RNAs. High methylation levels of FMRP target mRNAs suggest that m6A methylation is likely to be used for selective recognition of target mRNAs by FMRP at the synapse (Chang et al., 2017). FMRP binds to the m6A moiety within its mRNA targets and interacts with the m6A reader YTHDF2 in a manner independent of the mRNA. FMRP stabilizes its mRNA targets, whereas YTHDF2 on its own promotes their degradation. These data indicate that FMRP regulates the stability of target mRNAs marked with m6A via its interaction with YTHDF2 (Zhang et al., 2018). Furthermore, transcriptomic analysis of postmortem brains of patients with ASDs and FXS revealed hypoediting in the brains of both, which was shared across brain regions and involved many synaptic genes. These findings are mechanistically explained by the interaction of the fragile X proteins FMRP and FXR1P, which interact with RNA-editing enzymes (ADAR proteins) and modulate A-to-I editing (Arguello et al., 2018; Tran et al., 2019).
FMRP Lost in Synaptic Translation
After more than 18 years of studies addressing the properties of neurons lacking FMRP, excessive translation is still posited to be a major contributor to the synaptic and cognitive deficits associated with fragile X syndrome (Bagni et al., 2012; Banerjee et al., 2018; Darnell and Klann, 2013; Richter et al., 2015). Of note, enhanced mRNA translation was first reported in lymphoblastoid cells derived from humans with FXS (Brown et al., 2001) and at synapses of fragile X mice (Dölen et al., 2007; Greenough et al., 2001; Zalfa et al., 2003). FMRP represses protein synthesis by different mechanisms regulating initiation (Figure 1) and elongation of mRNA translation (Bagni et al., 2012; Richter et al., 2015; Zukin et al., 2009).
In brain and at synapses, several converging lines of evidence suggest that translational repression occurs mainly at the level of initiation (Sossin and Costa-Mattioli, 2018), halting assembly of the pre-initiation complex containing the small ribosomal subunits. The efficiency of mRNA translation is regulated through phosphorylation of translation initiation factors such as eIF4E or the eIF4E-binding proteins (4E-BPs). Phosphorylation of eukaryotic initiation factors eIF4E and/or 4EBPs by mTORC1 or MNK1 releases the brake imposed on translation (Bramham et al., 2016), enabling assembly of eIF4E with eIF4G to form the initiation complex. Mice carrying a genetic mutation in eIF4E-BP2 exhibit increased ratio of E/I, increased translation of neuroligins (NLGNs), as well as autistic behaviors. Modulation of Nlgn1 corrected the defects and worsened autistic phenotypes (Gkogkas et al., 2013; Wang and Doering, 2013). Of note, NLGNs promote synaptic transmission (Varoqueaux et al., 2006) and are regulated by FMRP at synapses (Chmielewska et al., 2018; Darnell et al., 2011; Pasciuto and Bagni, 2014b).
Hypersensitivity to mGluR5 and ERK signaling also contributes to excessive protein synthesis in the hippocampus of Fmr1 KO mice. At synapses, the MAPK pathway regulates local protein synthesis by phosphorylation of TSC2, thus modulating mTOR activity (Lipton and Sahin, 2014). Moreover, phosphorylation of ribosomal protein S6, primarily a downstream target and effector of mTOR in wild-type neurons, is elevated and phosphorylated by ERK via the alternative S6 kinase RSK in neurons lacking FMRP (Sawicka et al., 2016). Finally, MAPK phosphorylates and activates MAP kinase-serine/threonine-protein kinases (MNKs); phosphorylation and activation of eIF4E by MNKs promotes elongation and translation (Panja et al., 2014).
While the above-mentioned mechanisms regulate general mRNA translation, specific mRNA translation is orchestrated by FMRP via multiple mechanisms (Figures 1 and 3). FMRP regulates translational initiation by binding cytoplasmic FMR1-interacting protein 1 (CYFIP1), which subsequently sequesters and inhibits eukaryotic initiation factor (Napoli et al., 2008). In this complex, CYFIP1 acts as a non-canonical 4E-BP, which represses translation of target mRNAs by occluding the eIF4G-binding site on eIF4E (Napoli et al., 2008). The CYFIP1-FMRP complex represses translation of dendritically localized FMRP target mRNAs such as Map1b, αCaMKII, Arc, and APP (Napoli et al., 2008). Under basal conditions, ~30% of neuronal CYFIP1 is contained within the CYFIP1-FMRP-eIF4E complex, and ~70% is contained in the WAVE regulatory complex, consistent with the concept that the same CYFIP1 molecule cannot be part of both complexes at the same time (De Rubeis et al., 2013). Remarkably, synaptic activity in the form of either brain-derived neurotrophic factor (BDNF) or mGluR signaling triggers a conformational change in CYFIP1, from globular (Di Marino et al., 2015) to planar (Chen et al., 2010b). This conformational change is driven in part by the interaction of CYFIP1 with the small Rho GTPase Ras-related C3 botulinum toxin substrate 1 (Rac1) and releases eIF4E from CYFIP1, enabling assembly of eIF4E with eIF4G to form the first translation initiation complex, which can then proceed to position ribosomes on the mRNA (De Rubeis et al., 2013; Napoli et al., 2008). CYFIP1 can be phosphorylated by MNK, releasing translational inhibition and promoting many protein-synthesis forms of synaptic plasticity (Panja et al., 2014). More specifically, it was shown that at synapses of the dentate gyrus early in translation, MNK triggers release of the CYFIP1-FMRP inhibitory repressor complex from the mRNA. During the final steps of protein synthesis, MNK inhibits 4E-BP2, the canonical repressor of cap-dependent protein synthesis, in a synapse- and compartment-specific manner. This step is coupled to MNK-dependent elevated dendritic mRNA translation (Panja et al., 2014). Therefore, the FMRP-CYFIP1-eIF4E inhibitory complex affords quality control such that local translation occurs at just the right rate in response to neuronal activity, which is crucial for activating and/or maintaining long-term synaptic plasticity at glutamatergic synapses (Figure 1).
Synaptic plasticity requires actin remodeling and protein synthesis; thus, a balance between these two processes is crucial for brain functioning (De Rubeis et al., 2013). CYFIP1 is also a component of the WAVE regulatory complex (a heteropentamer formed by WAVE1/2/3, CYFIP1/2, ABI1/2, NCKAP1, and HPSC300), which promotes actin remodeling through its interaction with the Arp2/3 complex (Chen et al., 2010b; Eden et al., 2002). CYFIP1 shuttles between the FMRP-eIF4E complex and the Rac1-WAVE regulatory complex, thereby linking protein synthesis to spine dynamics and actin remodeling (De Rubeis et al., 2013). Moreover, treatment of mice with 4EGI-1, an inhibitor of the eIF4E-eIF4G translation initiation complex creates free eIF4E that competes with Rac1 to bind CYFIP1, thus restoring the balance between these two pathways and ultimately correcting deficits in hippocampal-based memory and spine morphology (Santini et al., 2017). These findings are consistent with the concept that dysregulation of protein translation and actin dynamics are critical to the pathophysiology of fragile X.
FMRP can repress mRNA translation at the level of initiation through the action of microRNAs such as miR-125a on PSD-95 mRNA or miR-196a on HOXB8 mRNA (Li et al., 2014; Muddashetty et al., 2011) or microRNA 125b on NR2A mRNA (Edbauer et al., 2010; Figure 3). In this context, modulation of the translational inhibition occurs through FMRP phosphorylation. Neuronal activity and/or activation of mGluR5 promotes protein phosphatase 2A (PP2A)-dependent dephosphorylation of FMRP, which induces release of RISC and stimulation of active translation. Activation of mGluRs also promotes ubiquitination of FMRP, which primes FMRP for ubiquitin-based, proteasomal degradation locally (Hou et al., 2006; Nalavadi et al., 2012), likewise releasing FMRP from the complex. In addition to ubiquitinylation, FMRP can be sumoylated with similar effects: FMRP is rapidly sumoylated in response to neuronal activity and by activation of mGluR5 (Khayachi et al., 2018). The increase in sumoylation promotes homomerization and release of FMRP from dendritic RNA granules, enabling local protein synthesis, which promotes spine elimination and maturation (Khayachi et al., 2018). Additional evidence for a mechanism of FMRP-mediated translational regulation at the level of initiation comes from a study showing that the antidiabetic drug metformin rescues some FXS phenotypes in mice, which coincides with normalization specifically of ERK signaling and eIF4E phosphorylation (Figure 1; Gantois et al., 2017). Of note, metformin is a drug known to activate autophagy, which is impaired in fragile X (section below and References), making metformin a candidate for drug redirection (Tranfaglia et al., 2018).
The role of FMRP, however, is not limited to inhibition of translational initiation. FMRP can also regulate mRNA translation during elongation via stalled polyribosomes (Darnell et al., 2011) or even activate translation of small number of specific mRNAs (Tabet et al., 2016). In the future, it will be interesting to address whether the identity of the target mRNA determines the regulatory mechanism (Achsel and Bagni, 2016) or whether FMRP can switch its mechanistic role depending on the developmental or physiological state of the cell and/or subcellular compartment.
FMRP regulates the expression and activity of a substantial number of voltage-gated ion channels (Contractor et al., 2015; Ferron, 2016; Richter et al., 2015). For example, FMRP controls the translation of Kv4.2, Kv3.1, and HCN1 mRNAs encoding K+ channels, which control neuronal excitability (Brown et al., 2001; Darnell et al., 2011; Miyashiro et al., 2003). In addition to these translation-dependent mechanisms by which FMRP controls channel abundance, FMRP acts via non-canonical (translation-independent) mechanisms to regulate activity and stability of other voltage-gated channels by direct protein-protein interactions, which, in turn, alters cellular excitability in a translation-independent manner. In particular, the N-terminal region of FMRP contains two Tudor motifs that typically recognize methylated amino acids (Maurer-Stroh et al., 2003) in proteins or FMRP RNA-binding partners (Lacoux et al., 2012). FMRP binds and modulates activity of the Slack and BK potassium channels through this domain, while it interacts with calcium channels via its carboxy-terminus. The interaction of FMRP with Slack opens the channel (Brown et al., 2010) and, in the case of BK channels, which are also expressed presynaptically, causes broadening of the action potential (Deng and Klyachko, 2016). The interaction of FMRP with calcium channels targets the channels for ubiquitin-based, proteasomal degradation and modulates N-type calcium channel density and synaptic vesicular release (Ferron et al., 2014).
Synapse Elimination and Spine Pruning
Synaptic activity influences the number and strength of synapses that form between neurons. During early brain development, synapse formation exceeds elimination, resulting in an excess of immature excitatory synapses. Later, synapse destabilization and elimination reduce the number of synapses, thus refining neural circuits that underlie behavior and cognition (Rakic et al., 1986). Synaptogenesis and synapse elimination follow different time lines in anatomically and functionally distinct regions. Excess synapses are eliminated or pruned in a manner that depends on sensory experience and synaptic activity (Berry and Nedivi, 2017; Chen and Sabatini, 2012; Mostany et al., 2013; Südhof, 2018).
An important clue as to the mechanism(s) by which synapse elimination is regulated during brain development came from studies on the myocyte enhancer factor (MEF) 2 family of transcription factors. MEF2 members are highly expressed in brain and tightly regulated in response to Ca2+ signaling and promote expression of a set of genes that restrict the number of synaptic connections in between hippocampal neurons. Three autism genes, namely Fmr1, the ubiquitin E3 ligase murine double minute-2 (Mdm2), and Protocadherin-10 (Pcdh10), a member of the cadherin superfamily of Ca2+-dependent cell adhesion proteins, are required for MEF2-dependent synapse elimination and act downstream of MEF2 (Pfeiffer et al., 2010; Tsai et al., 2012). Upon activation of MEF2, Mdm2 ubiquitinates the synaptic scaffolding protein PSD-95, enabling recognition by PCDH10, which targets PSD-95 to the proteasome for degradation and promotes synapse elimination. FMRP controls expression of eukaryotic elongation factor 1a (EF1a), which, in turn, prevents MEF2-dependent PSD-95 ubiquitination and synapse elimination (Tsai et al., 2017). Moreover, PCDH10 was discovered to serve as a ligand for the WAVE regulatory complex implicated in control of actin cytoskeletal dynamics, linking PCDH10 to cofilin signaling and structural remodeling of spines (Chen et al., 2014). Of note, human genetic studies have implicated PCDH10 in ASDs (Morrow et al., 2008) and the MEF2 family member MEF2C as a convergence point for multiple signaling pathways involved in the pathogenesis of ASDs (Gilissen et al., 2014; Tu et al., 2017). These findings reveal novel roles for multiple autism-linked genes in activity-dependent synapse elimination.
Actin Dynamics at FXS and ASD Synapses
Synaptic strength is largely influenced by changes in synaptic structure that rely on regulation of receptor signaling, local protein synthesis, and structural remodeling of the cytoskeleton (Jędrzejewska-Szmek and Blackwell, 2019; Nakahata and Yasuda, 2018). A major player is actin, the most abundant cytoskeletal protein in spines (Cingolani and Goda, 2008; Hotulainen and Hoogenraad, 2010; Lamprecht, 2014). One of the key regulators of actin dynamics, and hence synaptic plasticity, is the small Rho GTPase “Ras-related C3 botulinum toxin substrate 1” (Rac1, Figure 1). Rac1-GTP binds to CYFIP1, activating WRC-mediated actin polymerization while at the same time disinhibiting local protein synthesis (see above). Next, Rac1-GTP activates the p21-activated kinases (PAK1–PAK3), which activate LimK. Activated LimK1, in turn, phosphorylates and inactivates its target cofilin. Cofilin is a depolymerizing factor and major determinant of dendritic spine structure; cofilin severs the actin filament (F-actin) at its pointed ends (Bamburg and Bernstein, 2010; Mizuno, 2013) and thereby regulates spine morphology, spine motility, and synaptic plasticity (Bosch and Hayashi, 2012; Hotulainen and Hoogenraad, 2010; Lin and Webb, 2009; Pontrello and Ethell, 2009). Growing evidence from studies involving non-neuronal cells indicates that mTORC2 regulates the actin cytoskeleton through Rac1 (Hernández-Negrete et al., 2007; Huang et al., 2013). In addition, cofilin is a direct target of mTORC2 (Tejada-Simon, 2015).
Dysregulation of the synaptic cytoskeleton is a common theme in the pathogenesis of neurodevelopmental disorders such as FXS (Bai et al., 2015; Bourgeron, 2015; De Rubeis et al., 2014; Joensuu et al., 2018; Penzes and Cahill, 2012; Penzes et al., 2011; Pyronneau et al., 2017). Considerable evidence indicates that Rac1 signaling is a point of convergence in ASDs and IDs. Mutations in Rac1 have been detected in ASDs and IDs, and disruption of Rac1 signaling contributes to ASD-like behaviors in animal models (De Rubeis et al., 2013; Dolan et al., 2013; Tian et al., 2018; Zeidán-Chuliá et al., 2013). Aberrant Rac1 activation leads to either abnormally weak or strong glutamatergic synapses (Sadybekov et al., 2017). Of note, Rac1 levels are increased in FXS patients (Fatemi et al., 2013), and baseline activation of the Rac1 pathway is upregulated in the Fmr1 KO mouse model (Santini et al., 2017). Loss of FMRP leads to increased Rac1 signaling, which is mediated by increased interaction between Rac1 and the scaffolding protein CYFIP1. In Fmr1 KO brains, an increased abundance of the initiation complex eIF4E-eIF4G and CYFIP1-Rac1 complexes is observed, thereby blunting synaptic plasticity and learning. Treatment of FXS mice with 4EGI-1, which inhibits interactions between eIF4E and eIF4G, restored the equilibrium between the CYFIP1-Rac1 and protein synthesis and thereby reversed defects in hippocampal-based memory and spine structure (Santini et al., 2017).
Formation and maintenance of dendritic spines is maintained through PAK1, and several PAK isoforms have been implicated in IDs (Asrar et al., 2009; Causeret et al., 2009; de la Torre-Ubieta et al., 2010). Abundance and activity of Rac1 and PAK1 are elevated in the somatosensory cortex of fragile X mice (Bongmba et al., 2011; Castets et al., 2005; Chen et al., 2010a; de Diego-Otero et al., 2009; Dolan et al., 2013), and inhibition of PAK1 rescues, in part, the increased density and length of dendritic spines in Fmr1 KO mice (Dolan et al., 2013; Hayashi et al., 2007; Pyronneau et al., 2017). Pak2 haplo-insufficiency results in markedly decreased density of synapses, impaired long-term potentiation, and autism-relevant behaviors in mice due to reduced activity of the key actin regulators LIMK kinase (LIMK)1/cofilin (Wang et al., 2018). PAK1 and PAK3 are also involved in spine morphogenesis and neural plasticity (Boda et al., 2006). Moreover, PAK3 is associated with non-syndromic X-linked mental retardation (Allen et al., 1998; Bienvenu et al., 2000). PAKs are also the molecular targets of SHANK3, which is also linked to autism-related behaviors (Duffney et al., 2015; see also above).
There is considerable evidence that implicates dysregulation of LIMK/cofilin signaling in the aberrant spine morphology associated with fragile X. Cofilin is dysregulated in the somatosensory cortex of young Fmr1 KO mice and causally related to spine abnormalities (Pyronneau et al., 2017). Constitutively active cofilinS3A, which lacks the conserved LIMK phoshorylation site, rescues spine morphology and density in the somatosensory cortex, implicating elevated cofilin phosphorylation and the high density of immature spines. LIMK-cofilin signaling can also be stimulated by bone morphogenetic protein type II receptor (BMPR2). In humans, the full-length isoform of bone morphogenetic protein type II receptor (BMPR2) is abnormally high and activates LIMK1 (Kashima et al., 2016). Heterozygosity for BMPR2 or pharmacological inhibition of LIMK1 reduced the density of immature spines and restored synaptic function in fragile X mice. It would be interesting to explore whether BMPR2 is the link between FMRP loss and Rac1 activation or whether these represent independent pathways that converge on LIMK1.
mTOR and Autophagy
The correct number of proteins is maintained by a fine balance between protein synthesis and protein degradation. Elevated levels of synaptic proteins can arise as a consequence of elevated translation and/or impaired protein degradation. Autophagy is a process of programmed degradation and recycling of cellular components via the lysosomal pathway that is increased during periods of cellular stress (Harris and Rubinsztein, 2011; Mizushima et al., 2008; Nixon, 2013; Wong and Cuervo, 2010). In neurons, mTORC1 is strategically positioned at pre- and postsynaptic sites where it serves as a brake on autophagy. Under nutrient-rich conditions, mTOR phosphorylates Unc-51-like autophagy-activating kinase 1 (ULK-1) at S757 (Figure 4), a target of mTORC1 and well-established anti-autophagy site (Jung et al., 2009). This, in turn, sequesters ULK-1 away from AMP kinase (AMPK) and halts the initiation of autophagy. By contrast, under conditions of starvation, AMPK phosphorylates and activates ULK-1 at S317. Upon activation, ULK-1 promotes phosphorylation and activation of Beclin-1 at S14, a critical step in the “nucleation phase” of autophagy (Russell et al., 2013). Beclin-1 promotes lipidation of LC3-I to generate its lipidated form LC3-II, enabling elongation of the limiting membrane and formation of autophagosomes (Rubinsztein et al., 2012). Upon lipidation, LC3-II localizes to the phagophore membrane, enabling elongation of the limiting membrane and formation of mature LC3-II (+) autophagosomes, which engulf the cargo (Harris and Rubinsztein, 2011; Mizushima et al., 2008; Nixon, 2013). Autophagosomes act via cargo adaptor proteins, such as p62, to deliver ubiquitinated proteins, protein aggregates, and organelles to lysosomes for degradation.
Figure 4. A Schematic of the Process and Main Regulatory Machinery of the Autophagy.
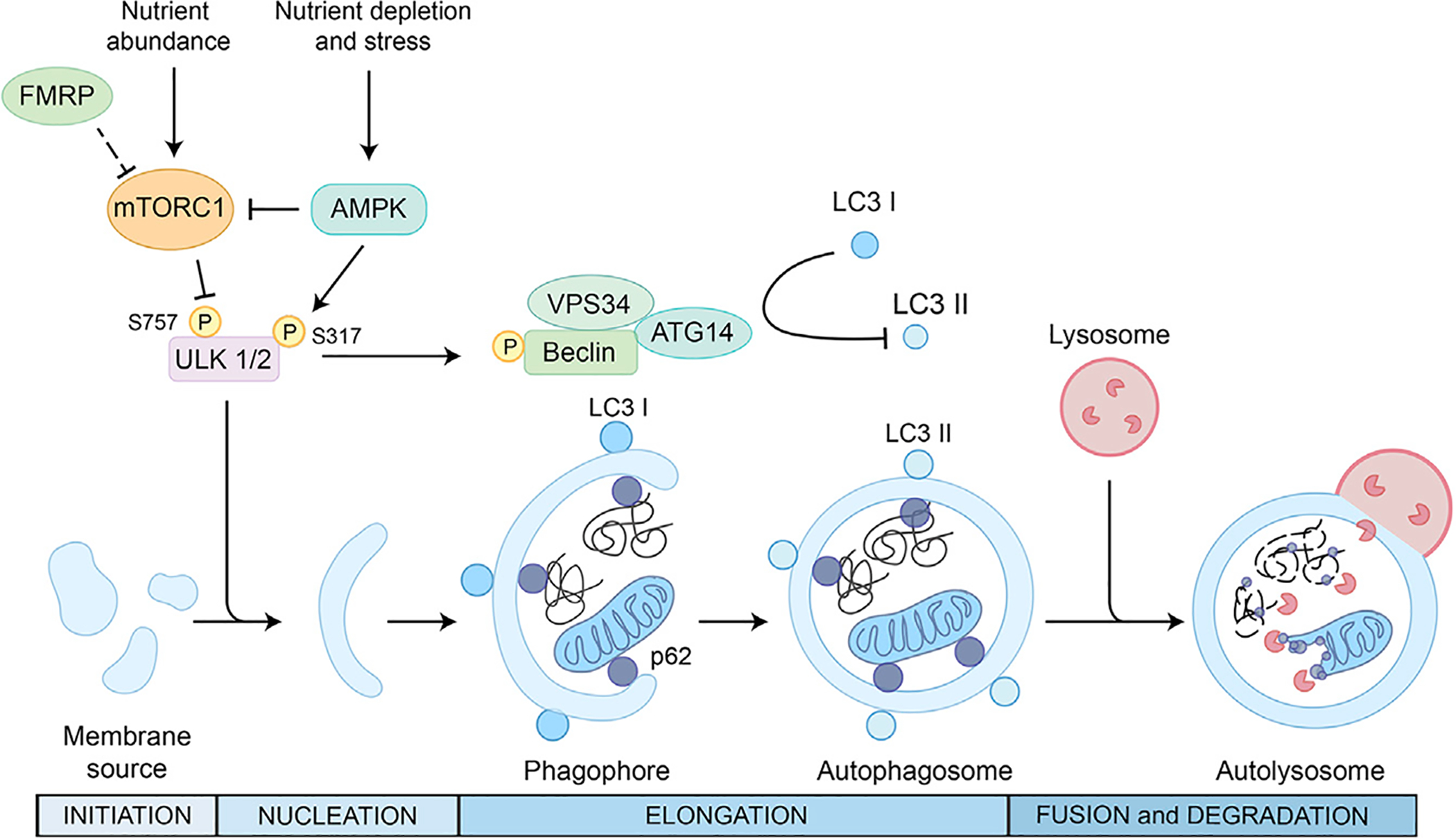
The metabolic sensors mTOR and AMPK are the main regulators of autophagy activation. mTORC1 phosphorylates ULK-1, inhibiting autophagy. In condition of nutrient depletion and cellular stress, AMPK phosphorylates and activates ULK-1, promoting phosphorylation and activation of Beclin-1, a critical step in the nucleation phase of autophagy. Cytoplasmic material is engulfed by double membranes forming a cup-shaped structure called phagophore. Beclin-1 promotes LC3-I lipidation to generate its lipidated form LC3-II, enabling elongation of the limiting membrane of the phagophore and formation of autophagosomes. Autophagosomes require protein adaptors such as p62 to deliver proteins and organelles to lysosomes for degradation in the autolysosome. FMRP inhibits mTORC1, and Fmr1 KO mice show an overactivation of mTORC1 with consequent hyperphosphorylation of ULK1/2 and downregulation of autophagy (Yan et al., 2018).
In neurons, autophagy plays an important role in protein homeostasis and is a key regulator of structural remodeling, synaptic plasticity, and memory formation (Nixon, 2013; Shehata et al., 2012). Molecules such as the cargo adaptor protein p62 recognize and bind ubiquitinated proteins and target them for degradation via the lysosomal/autophagy pathway. A possible scenario is that in hippocampal neurons from fragile X mice, overactivated mTOR leads to reduced autophagy and a buildup of a select group of synaptic proteins. It was recently observed that in neurons, inhibition of either ubiquitin-based, proteasomal degradation, or the autophagy/lysosomal pathway lead to the buildup of synaptic proteins such as Arc and PSD-95, consistent with the concept that both pathways are involved in degradation of these and potentially other synaptic proteins under physiological conditions (Yan et al., 2018). Neurons from Fmr1 KO mice exhibited a buildup of ubiquitinated protein aggregates, which colocalized with p62, consistent with the concept that autophagy is impaired in neurons lacking FMRP (Yan et al., 2018). In addition, overactivated mTOR signaling suppresses autophagy in the brains of Tsc1+/− and Tsc2+/− mice during postnatal development, and reduced autophagy is causally linked to impaired pruning of spines in cortical layer V pyramidal neurons. The autism-relevant behaviors in the Tsc mutant mice suggest that impaired autophagy and its link to impaired spine pruning are likely to be more widespread in at least a subset of ASDs (Tang et al., 2014).
Conclusions and Future Perspectives
In this article, we have reviewed dysregulated molecular signaling cascades, receptors, synaptic function and plasticity, and spine architecture that underlie the behavioral and cognitive deficits associated with fragile X syndrome and other forms of syndromic ASDs. We have argued for a partial overlap between the pathogenetic mechanisms that lead to FXS and ASDs. Therefore, advances in our understanding of FXS may also help to inform studies on other ASDs. More importantly, personalized strategies to treat the social endophenotypes of FXS may also be beneficial for other forms of syndromic—and perhaps even idiopathic—ASD.
Several forms of syndromic autism and intellectual disabilities are associated with mutations in genes that regulate protein synthesis and affect structure, transmission, and plasticity of synapses (Zoghbi and Bear, 2012). Failure to maintain the precise number of mRNA molecules and RNA-binding protein levels are crucial entry points of synaptopathies (Grant, 2012). Neuronal gene expression undergoes changes at very fine spatial and temporal scales, including changes induced by activity-dependent transcription in the nucleus and local translation at the synapse. A fascinating frontier for the study of local translation is the question of heterogeneity in the basic translation machinery at synapses and during development. It was recently shown that the ribosome has a very heterogeneous composition, allowing for selective translation of transcripts responsible for different cellular functions such as metabolism, cell cycle, and development (Shi et al., 2017). If and in which context specialized ribosomes are found at axons and dendrites is the new frontier to brain development in physiological and pathological conditions such as FXS and ASD.
FMRP appears to fine-tune the proteome to match changing needs of the cell/synapse. A multitude of mRNAs that are targeted by FMRP belong to various biological pathways that are affected in ASDs (Figure 1). The targeting specificity of FMRP seems to be determined by a cooperative interplay of several RNA-binding activities (Achsel and Bagni, 2016), e.g., FMRP itself, miRNAs, and/or BC1 RNA. The full repertoire of FMRP-containing mRNP complexes is, however, far from elucidated, and new experimental and bioinformatic paradigms are needed to gain a thorough understanding of the molecular functions of FMRP. The identification of FMRP synaptic partners—RNA and proteins—and their cell specificity and expression during a developmental trajectory will shed light into such a complex neurodevelopmental disorder. The multitude of FMRP direct and indirect effects, as well the multitude of genes implicated in ASDs (https://www.sfari.org/), is matched by a consistent heterogeneity in the phenotypes of FXS and ASD patients, indicating that the individual genetic and/or epigenetic background determines which FMRP and or ASD gene mechanism becomes prevalent in the expression of the disease phenotype. This calls for disease models that take individual patients’ backgrounds into account. The heterogeneity of FXS and ASDs in humans that underlies the individual variability may explain, at least in part, the lack of success of clinical trials aiming solely at decreasing protein synthesis. A recent study highlighted a large variability in the levels of protein synthesis in fibroblasts among patients diagnosed with FXS (Jacquemont et al., 2018). These findings suggest that future clinical trials should aim at stratifying the FXS population, define reliable endophenotypes, and then target them in personalized treatment strategies. For such stratification, as well as for earlier identification of ASDs, it is first necessary to identify suitable biomarkers, which have to be selected from the existing lists of genetic and molecular biomarkers, as well as from neuroimaging and cognitive studies.
Although research involving the mouse model has informed our understanding of the pathophysiology of fragile X, including deficits in signaling, synaptic plasticity, and behavior, and several clinical trials have been implemented based on data from the mouse, there is to date no effective treatment for the cognitive and social interaction deficits observed in humans with FXS. Other disease models, such as the Fmr1 KO and other ASD rats, have been developed to better mimic the human phenotype (Berzhanskaya et al., 2017; Hamilton et al., 2014; Harony-Nicolas et al., 2017; Saxena et al., 2018). A recent study involving induced pluripotent stem (iPS) cells derived from patients with fragile X demonstrated rescue of the synaptic deficits associated with fragile X by CRISPR-dependent DNA methylation editing (Liu et al., 2018). This and related studies have propelled a paradigm shift from studies involving the fragile X and ASD rodent models to those involving patient-derived iPS cells (Bhattacharyya and Zhao, 2016; Linda et al., 2018; Sacco et al., 2018). Advantages of iPS cells are several-fold. In particular, iPS cells are derived from fibroblast cells of individual patients and therefore reflect the genomic heterogeneity found in the patient population. inducible-pluripotent stem cell (iPSC)-derived neurons from individuals with FXS have been generated and used to develop a strategy to reestablish FMRP expression by promotor demethylation (Bhattacharyya and Zhao, 2016; Liu et al., 2018). Similar approaches should be used to screen for compounds that reset translational homeostasis, reduce hyperexcitability, and/or accelerate maturation in iPSC-derived neurons from individuals with ASDs and/or FXS. Yet, iPS cells, or even neurons induced from iPS cells, are not a suitable model to study human behavior.
We envision that mechanistic research using animal models and patient-derived stem cells will help researchers to understand the complex etiology of neurodevelopmental disorders such as FXS and ASDs, which will hopefully lead to more effective therapeutic strategies, including personalized pharmacogenomics targets.
ACKNOWLEDGMENTS
The authors are extremely thankful to Vittoria Mariano and Adrian Lo for their help in preparing the figures and Michael Bennet, Tilmann Achsel, Eleonora Rosina, Nuria Domínguez Iturza, and Alexandros Kanellopoulos for critically reading the manuscript and discussions. This work was supported by SNSF 310030-182651 and NCCR Synapsy 51NF40-158776 (Switzerland), Novartis (Switzerland), Italian Fragile X Association (Italy) KU Leuven OTR (Belgium), Telethon Italy (GGP15257) and Department of Defense WW81XWH-15-1-0361 (USA) = to C.B., National Institutes of Health grant MH-092877, a grant from the Simons Foundation and a FRAXA fellowship (USA) to R.S.Z.
We would like to apologize to the colleagues whose work could not be cited due to space limitations.
REFERENCES
- Abbeduto L, Thurman AJ, McDuffie A, Klusek J, Feigles RT, Ted Brown W, Harvey DJ, Adayev T, LaFauci G, Dobkins C, et al. (2019). ASD comorbidity in fragile X syndrome: symptom profile and predictors of symptom severity in adolescent and young adult males. J. Autism Dev. Disord 49, 960–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrahams BS, and Geschwind DH (2008). Advances in autism genetics: on the threshold of a new neurobiology. Nat. Rev. Genet 9, 341–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achsel T, and Bagni C (2016). Cooperativity in RNA-protein interactions: the complex is more than the sum of its partners. Curr. Opin. Neurobiol 39, 146–151. [DOI] [PubMed] [Google Scholar]
- Akins MR, Leblanc HF, Stackpole EE, Chyung E, and Fallon JR (2012). Systematic mapping of fragile X granules in the mouse brain reveals a potential role for presynaptic FMRP in sensorimotor functions. J. Comp. Neurol 520, 3687–3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akins MR, Berk-Rauch HE, Kwan KY, Mitchell ME, Shepard KA, Korsak LI, Stackpole EE, Warner-Schmidt JL, Sestan N, Cameron HA, and Fallon JR (2017). Axonal ribosomes and mRNAs associate with fragile X granules in adult rodent and human brains. Hum. Mol. Genet 26, 192–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen KM, Gleeson JG, Bagrodia S, Partington MW, MacMillan JC, Cerione RA, Mulley JC, and Walsh CA (1998). PAK3 mutation in nonsyndromic X-linked mental retardation. Nat. Genet 20, 25–30. [DOI] [PubMed] [Google Scholar]
- Antar LN, Afroz R, Dictenberg JB, Carroll RC, and Bassell GJ (2004). Metabotropic glutamate receptor activation regulates fragile x mental retardation protein and FMR1 mRNA localization differentially in dendrites and at synapses. J. Neurosci 24, 2648–2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antar LN, Li C, Zhang H, Carroll RC, and Bassell GJ (2006). Local functions for FMRP in axon growth cone motility and activity-dependent regulation of filopodia and spine synapses. Mol. Cell. Neurosci 32, 37–48. [DOI] [PubMed] [Google Scholar]
- Arguello AE, Srikumar T, and Kleiner RE (2018). A photocrosslinking-based RNA chemical proteomics approach to profile m6 A-regulated protein-RNA interactions. Curr. Protoc. Nucleic Acid Chem 75, e69. [DOI] [PubMed] [Google Scholar]
- Ascano M Jr., Mukherjee N, Bandaru P, Miller JB, Nusbaum JD, Corcoran DL, Langlois C, Munschauer M, Dewell S, Hafner M, et al. (2012). FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature 492, 382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asrar S, Meng Y, Zhou Z, Todorovski Z, Huang WW, and Jia Z (2009). Regulation of hippocampal long-term potentiation by p21-activated protein kinase 1 (PAK1). Neuropharmacology 56, 73–80. [DOI] [PubMed] [Google Scholar]
- Auerbach BD, Osterweil EK, and Bear MF (2011). Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature 480, 63–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagni C, and Greenough WT (2005). From mRNP trafficking to spine dysmorphogenesis: the roots of fragile X syndrome. Nat. Rev. Neurosci 6, 376–387. [DOI] [PubMed] [Google Scholar]
- Bagni C, Tassone F, Neri G, and Hagerman R (2012). Fragile X syndrome: causes, diagnosis, mechanisms, and therapeutics. J. Clin. Invest 122, 4314–4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, Xiang X, Liang C, and Shi L (2015). Regulating Rac in the nervous system: molecular function and disease implication of Rac GEFs and GAPs. BioMed Res. Int 2015, 632450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey DB Jr., Mesibov GB, Hatton DD, Clark RD, Roberts JE, and Mayhew L (1998). Autistic behavior in young boys with fragile X syndrome.J. Autism Dev. Disord 28, 499–508. [DOI] [PubMed] [Google Scholar]
- Bamburg JR, and Bernstein BW (2010). Roles of ADF/cofilin in actin polymerization and beyond. F1000 Biol. Rep 2, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A, Luong JA, Ho A, Saib AO, and Ploski JE (2016). Overexpression of Homer1a in the basal and lateral amygdala impairs fear conditioning and induces an autism-like social impairment. Mol. Autism 7, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A, Ifrim MF, Valdez AN, Raj N, and Bassell GJ (2018). Aberrant RNA translation in fragile X syndrome: From FMRP mechanisms to emerging therapeutic strategies. Brain Res. 1693 (Pt A), 24–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baribeau DA, and Anagnostou E (2013). A comparison of neuroimaging findings in childhood onset schizophrenia and autism spectrum disorder: a review of the literature. Front. Psychiatry 4, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnea-Goraly N, Eliez S, Hedeus M, Menon V, White CD, Moseley M, and Reiss AL (2003). White matter tract alterations in fragile X syndrome: preliminary evidence from diffusion tensor imaging. Am. J. Med. Genet. B. Neuropsychiatr. Genet 118B, 81–88. [DOI] [PubMed] [Google Scholar]
- Berry KP, and Nedivi E (2017). Spine dynamics: are they all the same? Neuron 96, 43–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry-Kravis E, Levin R, Shah H, Mathur S, Darnell JC, and Ouyang B (2015). Cholesterol levels in fragile X syndrome. Am. J. Med. Genet. A 167A, 379–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry-Kravis EM, Lindemann L, Jønch AE, Apostol G, Bear MF, Carpenter RL, Crawley JN, Curie A, Des Portes V, Hossain F, et al. (2018). Drug development for neurodevelopmental disorders: lessons learned from fragile X syndrome. Nat. Rev. Drug Discov 17, 280–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertero A, Liska A, Pagani M, Parolisi R, Masferrer ME, Gritti M, Pedrazzoli M, Galbusera A, Sarica A, Cerasa A, et al. (2018). Autism-associated 16p11.2 microdeletion impairs prefrontal functional connectivity in mouse and human. Brain 141, 2055–2065. [DOI] [PubMed] [Google Scholar]
- Berzhanskaya J, Phillips MA, Gorin A, Lai C, Shen J, and Colonnese MT (2017). Disrupted cortical state regulation in a rat model of fragile X syndrome. Cereb. Cortex 27, 1386–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat S, Acharya UR, Adeli H, Bairy GM, and Adeli A (2014). Autism: cause factors, early diagnosis and therapies. Rev. Neurosci 25, 841–850. [DOI] [PubMed] [Google Scholar]
- Bhattacharya A, Mamcarz M, Mullins C, Choudhury A, Boyle RG, Smith DG, Walker DW, and Klann E (2016). Targeting translation control with p70 S6 kinase 1 inhibitors to reverse phenotypes in fragile X syndrome mice. Neuropsychopharmacology 41, 1991–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya A, and Zhao X (2016). Human pluripotent stem cell models of Fragile X syndrome. Mol. Cell. Neurosci 73, 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienvenu T, des Portes V, McDonell N, Carrié A, Zemni R, Couvert P, Ropers HH, Moraine C, van Bokhoven H, Fryns JP, et al. (2000). Missense mutation in PAK3, R67C, causes X-linked nonspecific mental retardation. Am. J. Med. Genet 93, 294–298. [DOI] [PubMed] [Google Scholar]
- Blackmon K, Thesen T, Green S, Ben-Avi E, Wang X, Fuchs B, Kuzniecky R, and Devinsky O (2018). Focal cortical anomalies and language impairment in 16p11.2 deletion and duplication syndrome. Cereb. Cortex 28, 2422–2430. [DOI] [PubMed] [Google Scholar]
- Boda B, Nikonenko I, Alberi S, and Muller D (2006). Central nervous system functions of PAK protein family: from spine morphogenesis to mental retardation. Mol. Neurobiol 34, 67–80. [DOI] [PubMed] [Google Scholar]
- Bongmba OY, Martinez LA, Elhardt ME, Butler K, and Tejada-Simon MV (2011). Modulation of dendritic spines and synaptic function by Rac1: a possible link to Fragile X syndrome pathology. Brain Res. 1399, 79–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrie SC, Brems H, Legius E, and Bagni C (2017). Cognitive dysfunctions in intellectual disabilities: the contributions of the Ras-MAPK and PI3K-AKT-mTOR pathways. Annu. Rev. Genomics Hum. Genet 18, 115–142. [DOI] [PubMed] [Google Scholar]
- Bosch M, and Hayashi Y (2012). Structural plasticity of dendritic spines. Curr. Opin. Neurobiol 22, 383–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeron T (2015). From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat. Rev. Neurosci 16, 551–563. [DOI] [PubMed] [Google Scholar]
- Braat S, and Kooy RF (2015). The GABAA receptor as a therapeutic target for neurodevelopmental disorders. Neuron 86, 1119–1130. [DOI] [PubMed] [Google Scholar]
- Bramham CR (2008). Local protein synthesis, actin dynamics, and LTP consolidation. Curr. Opin. Neurobiol 18, 524–531. [DOI] [PubMed] [Google Scholar]
- Bramham CR, Jensen KB, and Proud CG (2016). Tuning specific translation in cancer metastasis and synaptic memory: control at the MNK-eIF4E axis. Trends Biochem. Sci 41, 847–858. [DOI] [PubMed] [Google Scholar]
- Brown V, Jin P, Ceman S, Darnell JC, O’Donnell WT, Tenenbaum SA, Jin X, Feng Y, Wilkinson KD, Keene JD, et al. (2001). Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell 107, 477–487. [DOI] [PubMed] [Google Scholar]
- Brown MR, Kronengold J, Gazula VR, Chen Y, Strumbos JG, Sigworth FJ, Navaratnam D, and Kaczmarek LK (2010). Fragile X mental retardation protein controls gating of the sodium-activated potassium channel Slack. Nat. Neurosci 13, 819–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cajigas IJ, Tushev G, Will TJ, tom Dieck S, Fuerst N, and Schuman EM (2012). The local transcriptome in the synaptic neuropil revealed by deep sequencing and high-resolution imaging. Neuron 74, 453–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Çaku A, Pellerin D, Bouvier P, Riou E, and Corbin F (2014). Effect of lovastatin on behavior in children and adults with fragile X syndrome: an open-label study. Am. J. Med. Genet. A 164A, 2834–2842. [DOI] [PubMed] [Google Scholar]
- Castets M, Schaeffer C, Bechara E, Schenck A, Khandjian EW, Luche S, Moine H, Rabilloud T, Mandel JL, and Bardoni B (2005). FMRP interferes with the Rac1 pathway and controls actin cytoskeleton dynamics in murine fibroblasts. Hum. Mol. Genet 14, 835–844. [DOI] [PubMed] [Google Scholar]
- Causeret F, Terao M, Jacobs T, Nishimura YV, Yanagawa Y, Obata K, Hoshino M, and Nikolic M (2009). The p21-activated kinase is required for neuronal migration in the cerebral cortex. Cereb. Cortex 19, 861–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M, Lv H, Zhang W, Ma C, He X, Zhao S, Zhang ZW, Zeng YX, Song S, Niu Y, and Tong WM (2017). Region-specific RNA m6A methylation represents a new layer of control in the gene regulatory network in the mouse brain. Open Biol 7, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, and Sabatini BL (2012). Signaling in dendritic spines and spine microdomains. Curr. Opin. Neurobiol 22, 389–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Yun SW, Seto J, Liu W, and Toth M (2003). The fragile X mental retardation protein binds and regulates a novel class of mRNAs containing U rich target sequences. Neuroscience 120, 1005–1017. [DOI] [PubMed] [Google Scholar]
- Chen LY, Rex CS, Babayan AH, Kramár EA, Lynch G, Gall CM, and Lauterborn JC (2010a). Physiological activation of synaptic Rac>PAK (p-21 activated kinase) signaling is defective in a mouse model of fragile X syndrome. J. Neurosci 30, 10977–10984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Borek D, Padrick SB, Gomez TS, Metlagel Z, Ismail AM, Umetani J, Billadeau DD, Otwinowski Z, and Rosen MK (2010b). Structure and control of the actin regulatory WAVE complex. Nature 468, 533–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Brinkmann K, Chen Z, Pak CW, Liao Y, Shi S, Henry L, Grishin NV, Bogdan S, and Rosen MK (2014). The WAVE regulatory complex links diverse receptors to the actin cytoskeleton. Cell 156, 195–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherubini E, Griguoli M, Safiulina V, and Lagostena L (2011). The depolarizing action of GABA controls early network activity in the developing hippocampus. Mol. Neurobiol 43, 97–106. [DOI] [PubMed] [Google Scholar]
- Chmielewska JJ, Kuzniewska B, Milek J, Urbanska K, and Dziembowska M (2018). Neuroligin 1, 2, and 3 regulation at the synapse: FMRP-dependent translation and activity-induced proteolytic cleavage. Mol. Neurobiol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury S, Shepherd JD, Okuno H, Lyford G, Petralia RS, Plath N, Kuhl D, Huganir RL, and Worley PF (2006). Arc/Arg3.1 interacts with the endocytic machinery to regulate AMPA receptor trafficking. Neuron 52, 445–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chyung E, LeBlanc HF, Fallon JR, and Akins MR (2018). Fragile X granules are a family of axonal ribonucleoprotein particles with circuit-dependent protein composition and mRNA cargos. J. Comp. Neurol 526, 96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani LA, and Goda Y (2008). Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat. Rev. Neurosci 9, 344–356. [DOI] [PubMed] [Google Scholar]
- Cioni JM, Wong HH, Bressan D, Kodama L, Harris WA, and Holt CE (2018). Axon-axon interactions regulate topographic optic tract sorting via CYFIP2-dependent WAVE complex function. Neuron 97, 1078–1093.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contractor A, Klyachko VA, and Portera-Cailliau C (2015). Altered neuronal and circuit excitability in fragile X syndrome. Neuron 87, 699–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, and Monteggia LM (2013). mTOR complexes in neurodevelopmental and neuropsychiatric disorders. Nat. Neurosci 16, 1537–1543. [DOI] [PubMed] [Google Scholar]
- Curatolo P, Napolioni V, and Moavero R (2010). Autism spectrum disorders in tuberous sclerosis: pathogenetic pathways and implications for treatment. J. Child Neurol 25, 873–880. [DOI] [PubMed] [Google Scholar]
- Darnell JC, and Klann E (2013). The translation of translational control by FMRP: therapeutic targets for FXS. Nat. Neurosci 16, 1530–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, et al. (2011). FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Diego-Otero Y, Romero-Zerbo Y, el Bekay R, Decara J, Sanchez L, Rodriguez-de Fonseca F, and del Arco-Herrera I (2009). Alpha-tocopherol protects against oxidative stress in the fragile X knockout mouse: an experimental therapeutic approach for the Fmr1 deficiency. Neuropsychopharmacology 34, 1011–1026. [DOI] [PubMed] [Google Scholar]
- de la Torre-Ubieta L, Gaudillière B, Yang Y, Ikeuchi Y, Yamada T, Di-Bacco S, Stegmüller J, Schüller U, Salih DA, Rowitch D, et al. (2010). A FOXO-Pak1 transcriptional pathway controls neuronal polarity. Genes Dev. 24, 799–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rubeis S, Pasciuto E, Li KW, Fernández E, Di Marino D, Buzzi A, Ostroff LE, Klann E, Zwartkruis FJ, Komiyama NH, et al. (2013). CYFIP1 coordinates mRNA translation and cytoskeleton remodeling to ensure proper dendritic spine formation. Neuron 79, 1169–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, et al. ; DDD Study; Homozygosity Mapping Collaborative for Autism; UK10K Consortium (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng PY, and Klyachko VA (2016). Genetic upregulation of BK channel activity normalizes multiple synaptic and circuit defects in a mouse model of fragile X syndrome. J. Physiol 594, 83–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis EL, and Thompson PM (2013). Typical and atypical brain development: a review of neuroimaging studies. Dialogues Clin. Neurosci 15, 359–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marino D, Chillemi G, De Rubeis S, Tramontano A, Achsel T, and Bagni C (2015). MD and docking studies reveal that the functional switch of CYFIP1 is mediated by a butterfly-like motion. J. Chem. Theory Comput 11, 3401–3410. [DOI] [PubMed] [Google Scholar]
- Di Paolo G, and De Camilli P (2006). Phosphoinositides in cell regulation and membrane dynamics. Nature 443, 651–657. [DOI] [PubMed] [Google Scholar]
- Dictenberg JB, Swanger SA, Antar LN, Singer RH, and Bassell GJ (2008). A direct role for FMRP in activity-dependent dendritic mRNA transport links filopodial-spine morphogenesis to fragile X syndrome. Dev. Cell 14, 926–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolan BM, Duron SG, Campbell DA, Vollrath B, Shankaranarayana Rao BS, Ko HY, Lin GG, Govindarajan A, Choi SY, and Tonegawa S (2013). Rescue of fragile X syndrome phenotypes in Fmr1 KO mice by the small-molecule PAK inhibitor FRAX486. Proc. Natl. Acad. Sci. USA 110, 5671–5676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dölen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, and Bear MF (2007). Correction of fragile X syndrome in mice. Neuron 56, 955–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotti CG, Esteban JA, and Ledesma MD (2014). Lipid dynamics at dendritic spines. Front. Neuroanat 8, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douet V, Chang L, Cloak C, and Ernst T (2014). Genetic influences on brain developmental trajectories on neuroimaging studies: from infancy to young adulthood. Brain Imaging Behav. 8, 234–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffney LJ, Zhong P, Wei J, Matas E, Cheng J, Qin L, Ma K, Dietz DM, Kajiwara Y, Buxbaum JD, and Yan Z (2015). Autism-like deficits in Shank3-deficient mice are rescued by targeting actin regulators. Cell Rep. 11, 1400–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edbauer D, Neilson JR, Foster KA, Wang CF, Seeburg DP, Batterton MN, Tada T, Dolan BM, Sharp PA, and Sheng M (2010). Regulation of synaptic structure and function by FMRP-associated microRNAs miR-125b and miR-132. Neuron 65, 373–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden S, Rohatgi R, Podtelejnikov AV, Mann M, and Kirschner MW (2002). Mechanism of regulation of WAVE1-induced actin nucleation by Rac1 and Nck. Nature 418, 790–793. [DOI] [PubMed] [Google Scholar]
- Ellegood J, Pacey LK, Hampson DR, Lerch JP, and Henkelman RM (2010). Anatomical phenotyping in a mouse model of fragile X syndrome with magnetic resonance imaging. Neuroimage 53, 1023–1029. [DOI] [PubMed] [Google Scholar]
- Fasano S, and Brambilla R (2011). Ras-ERK signaling in behavior: old questions and new perspectives. Front. Behav. Neurosci 5, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, Folsom TD, Kneeland RE, Yousefi MK, Liesch SB, and Thuras PD (2013). Impairment of fragile X mental retardation protein-metabotropic glutamate receptor 5 signaling and its downstream cognates ras-related C3 botulinum toxin substrate 1, amyloid beta A4 precursor protein, striatal-enriched protein tyrosine phosphatase, and homer 1, in autism: a postmortem study in cerebellar vermis and superior frontal cortex. Mol. Autism 4, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari F, Mercaldo V, Piccoli G, Sala C, Cannata S, Achsel T, and Bagni C (2007). The fragile X mental retardation protein-RNP granules show an mGluR-dependent localization in the post-synaptic spines. Mol. Cell. Neurosci 34, 343–354. [DOI] [PubMed] [Google Scholar]
- Ferron L (2016). Fragile X mental retardation protein controls ion channel expression and activity. J. Physiol 594, 5861–5867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferron L, Nieto-Rostro M, Cassidy JS, and Dolphin AC (2014). Fragile X mental retardation protein controls synaptic vesicle exocytosis by modulating N-type calcium channel density. Nat. Commun 5, 3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandal MJ, Zhang P, Hadjimichael E, Walker RL, Chen C, Liu S, Won H, van Bakel H, Varghese M, Wang Y, et al. ; PsychENCODE Consortium (2018). Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 362, 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantois I, Vandesompele J, Speleman F, Reyniers E, D’Hooge R, Severijnen LA, Willemsen R, Tassone F, and Kooy RF (2006). Expression profiling suggests underexpression of the GABA(A) receptor subunit delta in the fragile X knockout mouse model. Neurobiol. Dis 21, 346–357. [DOI] [PubMed] [Google Scholar]
- Gantois I, Khoutorsky A, Popic J, Aguilar-Valles A, Freemantle E, Cao R, Sharma V, Pooters T, Nagpal A, Skalecka A, et al. (2017). Metformin ameliorates core deficits in a mouse model of fragile X syndrome. Nat. Med 23, 674–677. [DOI] [PubMed] [Google Scholar]
- Gao F, Qi L, Yang Z, Yang T, Zhang Y, Xu H, and Zhao H (2018). Impaired GABA neural circuits are critical for fragile X syndrome. Neural Plast. 2018, 8423420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilissen C, Hehir-Kwa JY, Thung DT, van de Vorst M, van Bon BW, Willemsen MH, Kwint M, Janssen IM, Hoischen A, Schenck A, et al. (2014). Genome sequencing identifies major causes of severe intellectual disability. Nature 511, 344–347. [DOI] [PubMed] [Google Scholar]
- Gilman SR, Iossifov I, Levy D, Ronemus M, Wigler M, and Vitkup D (2011). Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron 70, 898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gkogkas CG, Khoutorsky A, Ran I, Rampakakis E, Nevarko T, Weatherill DB, Vasuta C, Yee S, Truitt M, Dallaire P, et al. (2013). Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature 493, 371–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant SG (2012). Synaptopathies: diseases of the synaptome. Curr. Opin. Neurobiol 22, 522–529. [DOI] [PubMed] [Google Scholar]
- Greenough WT, Klintsova AY, Irwin SA, Galvez R, Bates KE, and Weiler IJ (2001). Synaptic regulation of protein synthesis and the fragile X protein. Proc. Natl. Acad. Sci. USA 98, 7101–7106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C (2017). Defective phosphoinositide metabolism in autism. J. Neurosci. Res 95, 1161–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C, Chang CW, Kelly SM, Bhattacharya A, McBride SM, Danielson SW, Jiang MQ, Chan CB, Ye K, Gibson JR, et al. (2015). Increased expression of the PI3K enhancer PIKE mediates deficits in synaptic plasticity and behavior in fragile X syndrome. Cell Rep. 11, 727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumy LF, Yeo GS, Tung YC, Zivraj KH, Willis D, Coppola G, Lam BY, Twiss JL, Holt CE, and Fawcett JW (2011). Transcriptome analysis of embryonic and adult sensory axons reveals changes in mRNA repertoire localization. RNA 17, 85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Molinaro G, Collins KA, Hays SA, Paylor R, Worley PF, Szumlinski KK, and Huber KM (2016). Selective disruption of metabotropic glutamate receptor 5-Homer interactions mimics phenotypes of fragile X syndrome in mice. J. Neurosci 36, 2131–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha S, Sohn IJ, Kim N, Sim HJ, and Cheon KA (2015). Characteristics of brains in autism spectrum disorder: structure, function and connectivity across the lifespan. Exp. Neurobiol 24, 273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BW, Barnea-Goraly N, Lightbody AA, Patnaik SS, Hoeft F, Hazlett H, Piven J, and Reiss AL (2009). Early white-matter abnormalities of the ventral frontostriatal pathway in fragile X syndrome. Dev. Med. Child Neurol 51, 593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberl MG, Zerbi V, Veltien A, Ginger M, Heerschap A, and Frick A (2015). Structural-functional connectivity deficits of neocortical circuits in the Fmr1 (-/y) mouse model of autism. Sci. Adv 1, e1500775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman RJ, Berry-Kravis E, Hazlett HC, Bailey DB Jr., Moine H, Kooy RF, Tassone F, Gantois I, Sonenberg N, Mandel JL, and Hagerman PJ (2017). Fragile X syndrome. Nat. Rev. Dis. Primers 3, 17065. [DOI] [PubMed] [Google Scholar]
- Hall SS, Jiang H, Reiss AL, and Greicius MD (2013). Identifying large-scale brain networks in fragile X syndrome. JAMA Psychiatry 70, 1215–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton SM, Green JR, Veeraragavan S, Yuva L, McCoy A, Wu Y, Warren J, Little L, Ji D, Cui X, et al. (2014). Fmr1 and Nlgn3 knockout rats: novel tools for investigating autism spectrum disorders. Behav. Neurosci 128, 103–109. [DOI] [PubMed] [Google Scholar]
- Harony-Nicolas H, Kay M, Hoffmann JD, Klein ME, Bozdagi-Gunal O, Riad M, Daskalakis NP, Sonar S, Castillo PE, Hof PR, et al. (2017). Oxytocin improves behavioral and electrophysiological deficits in a novel Shank3-deficient rat. eLife 6, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris H, and Rubinsztein DC (2011). Control of autophagy as a therapy for neurodegenerative disease. Nat. Rev. Neurol 8, 108–117. [DOI] [PubMed] [Google Scholar]
- Hatton DD, Bailey DB Jr., Hargett-Beck MQ, Skinner M, and Clark RD (1999). Behavioral style of young boys with fragile X syndrome. Dev. Med. Child Neurol 41, 625–632. [DOI] [PubMed] [Google Scholar]
- Hayashi ML, Rao BS, Seo JS, Choi HS, Dolan BM, Choi SY, Chattarji S, and Tonegawa S (2007). Inhibition of p21-activated kinase rescues symptoms of fragile X syndrome in mice. Proc. Natl. Acad. Sci. USA 104, 11489–11494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He CX, and Portera-Cailliau C (2013). The trouble with spines in fragile X syndrome: density, maturity and plasticity. Neuroscience 251, 120–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Q, Nomura T, Xu J, and Contractor A (2014). The developmental switch in GABA polarity is delayed in fragile X mice. J. Neurosci 34, 446–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henske EP, Jóźwiak S, Kingswood JC, Sampson JR, and Thiele EA (2016). Tuberous sclerosis complex. Nat. Rev. Dis. Primers 2, 16035. [DOI] [PubMed] [Google Scholar]
- Hernandez RN, Feinberg RL, Vaurio R, Passanante NM, Thompson RE, and Kaufmann WE (2009). Autism spectrum disorder in fragile X syndrome: a longitudinal evaluation. Am. J. Med. Genet. A 149A, 1125–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Negrete I, Carretero-Ortega J, Rosenfeldt H, Hernández-García R, Calderón-Salinas JV, Reyes-Cruz G, Gutkind JS, and Vázquez-Prado J (2007). P-Rex1 links mammalian target of rapamycin signaling to Rac activation and cell migration. J. Biol. Chem 282, 23708–23715. [DOI] [PubMed] [Google Scholar]
- Hoeffer CA, Sanchez E, Hagerman RJ, Mu Y, Nguyen DV, Wong H, Whelan AM, Zukin RS, Klann E, and Tassone F (2012). Altered mTOR signaling and enhanced CYFIP2 expression levels in subjects with fragile X syndrome. Genes Brain Behav. 11, 332–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holsen LM, Dalton KM, Johnstone T, and Davidson RJ (2008). Prefrontal social cognition network dysfunction underlying face encoding and social anxiety in fragile X syndrome. Neuroimage 43, 592–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat A, and Svoboda K (2009). Experience-dependent structural synaptic plasticity in the mammalian brain. Nat. Rev. Neurosci 10, 647–658. [DOI] [PubMed] [Google Scholar]
- Hotulainen P, and Hoogenraad CC (2010). Actin in dendritic spines: connecting dynamics to function. J. Cell Biol 189, 619–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou L, Antion MD, Hu D, Spencer CM, Paylor R, and Klann E (2006). Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron 51, 441–454. [DOI] [PubMed] [Google Scholar]
- Huang W, Zhu PJ, Zhang S, Zhou H, Stoica L, Galiano M, Krnjević K, Roman G, and Costa-Mattioli M (2013). mTORC2 controls actin polymerization required for consolidation of long-term memory. Nat. Neurosci 16, 441–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Ikeuchi Y, Malumbres M, and Bonni A (2015). A Cdh1-APC/FMRP ubiquitin signaling link drives mGluR-dependent synaptic plasticity in the mammalian brain. Neuron 86, 726–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, and Bear MF (2002). Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. USA 99, 7746–7750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Klann E, Costa-Mattioli M, and Zukin RS (2015). Dysregulation of mammalian target of rapamycin signaling in mouse models of autism. J. Neurosci 35, 13836–13842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutsler JJ, and Zhang H (2010). Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 1309, 83–94. [DOI] [PubMed] [Google Scholar]
- Jacquemont S, Pacini L, Jønch AE, Cencelli G, Rozenberg I, He Y, D’Andrea L, Pedini G, Eldeeb M, Willemsen R, et al. (2018). Protein synthesis levels are increased in a subset of individuals with fragile X syndrome. Hum. Mol. Genet 27, 2039–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jędrzejewska-Szmek J, and Blackwell KT (2019). From membrane receptors to protein synthesis and actin cytoskeleton: mechanisms underlying long lasting forms of synaptic plasticity. Semin. Cell Dev. Biol Published January 11, 2019. 10.1016/j.semcdb.2019.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeste SS, Varcin KJ, Hellemann GS, Gulsrud AC, Bhatt R, Kasari C, Wu JY, Sahin M, and Nelson CA 3rd (2016). Symptom profiles of autism spectrum disorder in tuberous sclerosis complex. Neurology 87, 766–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joensuu M, Lanoue V, and Hotulainen P (2018). Dendritic spine actin cytoskeleton in autism spectrum disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 84 (Pt B), 362–381. [DOI] [PubMed] [Google Scholar]
- Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, and Kim DH (2009). ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 20, 1992–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai Y, Dohmae N, and Hirokawa N (2004). Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron 43, 513–525. [DOI] [PubMed] [Google Scholar]
- Kang JY, Chadchankar J, Vien TN, Mighdoll MI, Hyde TM, Mather RJ, Deeb TZ, Pangalos MN, Brandon NJ, Dunlop J, and Moss SJ (2017). Deficits in the activity of presynaptic g-aminobutyric acid type B receptors contribute to altered neuronal excitability in fragile X syndrome. J. Biol. Chem 292, 6621–6632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashima R, Roy S, Ascano M, Martinez-Cerdeno V, Ariza-Torres J, Kim S, Louie J, Lu Y, Leyton P, Bloch KD, et al. (2016). Augmented noncanonical BMP type II receptor signaling mediates the synaptic abnormality of fragile X syndrome. Sci. Signal 9, ra58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann WE, Kidd SA, Andrews HF, Budimirovic DB, Esler A, Haas-Givler B, Stackhouse T, Riley C, Peacock G, Sherman SL, et al. (2017). Autism spectrum disorder in fragile X syndrome: cooccurring conditions and current treatment. Pediatrics 139 (Suppl 3), S194–S206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khayachi A, Gwizdek C, Poupon G, Alcor D, Chafai M, Cassé F, Maurin T, Prieto M, Folci A, De Graeve F, et al. (2018). Sumoylation regulates FMRP-mediated dendritic spine elimination and maturation. Nat. Commun 9, 757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khundrakpam BS, Lewis JD, Zhao L, Chouinard-Decorte F, and Evans AC (2016). Brain connectivity in normally developing children and adolescents. Neuroimage 134, 192–203. [DOI] [PubMed] [Google Scholar]
- Kim SH, Markham JA, Weiler IJ, and Greenough WT (2008). Aberrant early-phase ERK inactivation impedes neuronal function in fragile X syndrome. Proc. Natl. Acad. Sci. USA 105, 4429–4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Yang J, and Kim E (2010). Diacylglycerol kinases in the regulation of dendritic spines. J. Neurochem 112, 577–587. [DOI] [PubMed] [Google Scholar]
- King MK, Pardo M, Cheng Y, Downey K, Jope RS, and Beurel E (2014). Glycogen synthase kinase-3 inhibitors: rescuers of cognitive impairments. Pharmacol. Ther 141, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein ME, Monday H, and Jordan BA (2016). Proteostasis and RNA binding proteins in synaptic plasticity and in the pathogenesis of neuropsychiatric disorders. Neural Plast. 2016, 3857934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsak LI, Mitchell ME, Shepard KA, and Akins MR (2016). Regulation of neuronal gene expression by local axonal translation. Curr. Genet. Med. Rep 4, 16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar VJ, Grissom NM, McKee SE, Schoch H, Bowman N, Havekes R, Kumar M, Pickup S, Poptani H, Reyes TM, et al. (2018). Linking spatial gene expression patterns to sex-specific brain structural changes on a mouse model of 16p11.2 hemideletion. Transl. Psychiatry 8, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Fata G, Gärtner A, Domínguez-Iturza N, Dresselaers T, Dawitz J, Poorthuis RB, Averna M, Himmelreich U, Meredith RM, Achsel T, et al. (2014). FMRP regulates multipolar to bipolar transition affecting neuronal migration and cortical circuitry. Nat. Neurosci 17, 1693–1700. [DOI] [PubMed] [Google Scholar]
- Lacoux C, Di Marino D, Boyl PP, Zalfa F, Yan B, Ciotti MT, Falconi M, Urlaub H, Achsel T, Mougin A, et al. (2012). BC1-FMRP interaction is modulated by 2′-O-methylation: RNA-binding activity of the tudor domain and translational regulation at synapses. Nucleic Acids Res. 40, 4086–4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamprecht R (2014). The actin cytoskeleton in memory formation. Prog. Neurobiol 117, 1–19. [DOI] [PubMed] [Google Scholar]
- Laplante M, and Sabatini DM (2012). mTOR signaling. Cold Spring Harb. Perspect. Biol 4, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebel C, and Deoni S (2018). The development of brain white matter micro-structure. Neuroimage 182, 207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Tang W, Zhang LR, and Zhang CY (2014). FMRP regulates miR196a-mediated repression of HOXB8 via interaction with the AGO2 MID domain. Mol. Biosyst 10, 1757–1764. [DOI] [PubMed] [Google Scholar]
- Lin WH, and Webb DJ (2009). Actin and actin-binding proteins: masters of dendritic spine formation, morphology, and function. Open Neurosci. J 3, 54–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Lin HI, Chen ML, Lai TT, Cao LP, Farrer MJ, Wu RM, and Chien CT (2016). Lovastatin protects neurite degeneration in LRRK2-G2019S parkinsonism through activating the Akt/Nrf pathway and inhibiting GSK3β activity. Hum. Mol. Genet 25, 1965–1978. [DOI] [PubMed] [Google Scholar]
- Linda K, Fiuza C, and Nadif Kasri N (2018). The promise of induced pluripotent stem cells for neurodevelopmental disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 84 (Pt B), 382–391. [DOI] [PubMed] [Google Scholar]
- Lipton JO, and Sahin M (2014). The neurology of mTOR. Neuron 84, 275–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liska A, Bertero A, Gomolka R, Sabbioni M, Galbusera A, Barsotti N, Panzeri S, Scattoni ML, Pasqualetti M, and Gozzi A (2018). Homozygous loss of autism-risk gene CNTNAP2 results in reduced local and long-range prefrontal functional connectivity. Cereb. Cortex 28, 1141–1153. [DOI] [PubMed] [Google Scholar]
- Liu XS, Wu H, Krzisch M, Wu X, Graef J, Muffat J, Hnisz D, Li CH, Yuan B, Xu C, et al. (2018). Rescue of fragile X syndrome neurons by DNA methylation editing of the FMR1 gene. Cell 172, 979–992.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahato PK, Ramsakha N, Ojha P, Gulia R, Sharma R, and Bhattacharyya S (2018). Group I metabotropic glutamate receptors (mGluRs): ins and outs. Adv. Exp. Med. Biol 1112, 163–175. [DOI] [PubMed] [Google Scholar]
- Mahfouz A, Ziats MN, Rennert OM, Lelieveldt BP, and Reinders MJ (2015). Shared pathways among autism candidate genes determined by co-expression network analysis of the developing human brain transcriptome. J. Mol. Neurosci 57, 580–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Brevet S, Rodríguez-Herreros B, Nielsen JA, Moreau C, Modenato C, Maillard AM, Pain A, Richetin S, Jønch AE, Qureshi AY, et al. ; 16p11.2 European Consortium; Simons Variation in Individuals Project (VIP) Consortium (2018). Quantifying the effects of 16p11.2 copy number variants on brain structure: a multisite genetic-first study. Biol. Psychiatry 84, 253–264. [DOI] [PubMed] [Google Scholar]
- Martínez-Cerdeño V (2017). Dendrite and spine modifications in autism and related neurodevelopmental disorders in patients and animal models. Dev. Neurobiol 77, 393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer-Stroh S, Dickens NJ, Hughes-Davies L, Kouzarides T, Eisenhaber F, and Ponting CP (2003). The Tudor domain ‘Royal Family’: Tudor, plant Agenet, Chromo, PWWP and MBT domains. Trends Biochem. Sci 28, 69–74. [DOI] [PubMed] [Google Scholar]
- McDuffie A, Abbeduto L, Lewis P, Kover S, Kim JS, Weber A, and Brown WT (2010). Autism spectrum disorder in children and adolescents with fragile X syndrome: within-syndrome differences and age-related changes. Am. J. Intellect. Dev. Disabil 115, 307–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mefford HC, Batshaw ML, and Hoffman EP (2012). Genomics, intellectual disability, and autism. N. Engl. J. Med 366, 733–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellios N, Feldman DA, Sheridan SD, Ip JPK, Kwok S, Amoah SK, Rosen B, Rodriguez BA, Crawford B, Swaminathan R, et al. (2018). MeCP2-regulated miRNAs control early human neurogenesis through differential effects on ERK and AKT signaling. Mol. Psychiatry 23, 1051–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkurjev D, Hong WT, Iida K, Oomoto I, Goldie BJ, Yamaguti H, Ohara T, Kawaguchi SY, Hirano T, Martin KC, et al. (2018). Synaptic N6-methyladenosine (m6A) epitranscriptome reveals functional partitioning of localized transcripts. Nat. Neurosci 21, 1004–1014. [DOI] [PubMed] [Google Scholar]
- Min WW, Yuskaitis CJ, Yan Q, Sikorski C, Chen S, Jope RS, and Bauchwitz RP (2009). Elevated glycogen synthase kinase-3 activity in Fragile X mice: key metabolic regulator with evidence for treatment potential. Neuropharmacology 56, 463–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashiro KY, Beckel-Mitchener A, Purk TP, Becker KG, Barret T, Liu L, Carbonetto S, Weiler IJ, Greenough WT, and Eberwine J (2003). RNA cargoes associating with FMRP reveal deficits in cellular functioning in Fmr1 null mice. Neuron 37, 417–431. [DOI] [PubMed] [Google Scholar]
- Mizuno K (2013). Signaling mechanisms and functional roles of cofilin phosphorylation and dephosphorylation. Cell. Signal 25, 457–469. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, and Klionsky DJ (2008). Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro P, and Feng G (2017). SHANK proteins: roles at the synapse and in autism spectrum disorder. Nat. Rev. Neurosci 18, 147–157. [DOI] [PubMed] [Google Scholar]
- Morrow EM, Yoo SY, Flavell SW, Kim TK, Lin Y, Hill RS, Mukaddes NM, Balkhy S, Gascon G, Hashmi A, et al. (2008). Identifying autism loci and genes by tracing recent shared ancestry. Science 321, 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostafalou S, and Abdollahi M (2017). Pesticides: an update of human exposure and toxicity. Arch. Toxicol 91, 549–599. [DOI] [PubMed] [Google Scholar]
- Mostany R, Anstey JE, Crump KL, Maco B, Knott G, and Portera-Cailliau C (2013). Altered synaptic dynamics during normal brain aging. J. Neurosci 33, 4094–4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muddashetty RS, Nalavadi VC, Gross C, Yao X, Xing L, Laur O, Warren ST, and Bassell GJ (2011). Reversible inhibition of PSD-95 mRNA translation by miR-125a, FMRP phosphorylation, and mGluR signaling. Mol. Cell 42, 673–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahata Y, and Yasuda R (2018). Plasticity of spine structure: local signaling, translation and cytoskeletal reorganization. Front. Synaptic Neurosci 10, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalavadi VC, Muddashetty RS, Gross C, and Bassell GJ (2012). Dephosphorylation-induced ubiquitination and degradation of FMRP in dendrites: a role in immediate early mGluR-stimulated translation. J. Neurosci 32, 2582–2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napoli I, Mercaldo V, Boyl PP, Eleuteri B, Zalfa F, De Rubeis S, Di Marino D, Mohr E, Massimi M, Falconi M, et al. (2008). The fragile X syndrome protein represses activity-dependent translation through CYFIP1, a new 4E-BP. Cell 134, 1042–1054. [DOI] [PubMed] [Google Scholar]
- Nelson SB, and Valakh V (2015). Excitatory/inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron 87, 684–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson DL, Orr HT, and Warren ST (2013). The unstable repeats–three evolving faces of neurological disease. Neuron 77, 825–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA (2013). The role of autophagy in neurodegenerative disease. Nat. Med 19, 983–997. [DOI] [PubMed] [Google Scholar]
- Nomura T, Musial TF, Marshall JJ, Zhu Y, Remmers CL, Xu J, Nicholson DA, and Contractor A (2017). Delayed maturation of fast-spiking interneurons is rectified by activation of the TrkB receptor in the mouse model of fragile X syndrome. J. Neurosci 37, 11298–11310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oguro-Ando A, Rosensweig C, Herman E, Nishimura Y, Werling D, Bill BR, Berg JM, Gao F, Coppola G, Abrahams BS, and Geschwind DH (2015). Increased CYFIP1 dosage alters cellular and dendritic morphology and dysregulates mTOR. Mol. Psychiatry 20, 1069–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliet SH, Malenka RC, and Nicoll RA (1997). Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron 18, 969–982. [DOI] [PubMed] [Google Scholar]
- Olmos-Serrano JL, Paluszkiewicz SM, Martin BS, Kaufmann WE, Corbin JG, and Huntsman MM (2010). Defective GABAergic neurotransmission and pharmacological rescue of neuronal hyperexcitability in the amygdala in a mouse model of fragile X syndrome. J. Neurosci 30, 9929–9938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterweil EK, Krueger DD, Reinhold K, and Bear MF (2010). Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J. Neurosci 30, 15616–15627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer MJ, Irving AJ, Seabrook GR, Jane DE, and Collingridge GL (1997). The group I mGlu receptor agonist DHPG induces a novel form of LTD in the CA1 region of the hippocampus. Neuropharmacology 36, 1517–1532. [DOI] [PubMed] [Google Scholar]
- Panja D, Kenney JW, D’Andrea L, Zalfa F, Vedeler A, Wibrand K, Fukunaga R, Bagni C, Proud CG, and Bramham CR (2014). Two-stage translational control of dentate gyrus LTP consolidation is mediated by sustained BDNF-TrkB signaling to MNK. Cell Rep 9, 1430–1445. [DOI] [PubMed] [Google Scholar]
- Parikshak NN, Gandal MJ, and Geschwind DH (2015). Systems biology and gene networks in neurodevelopmental and neurodegenerative disorders. Nat. Rev. Genet 16, 441–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Park JM, Kim S, Kim JA, Shepherd JD, Smith-Hicks CL, Chowdhury S, Kaufmann W, Kuhl D, Ryazanov AG, et al. (2008). Elongation factor 2 and fragile X mental retardation protein control the dynamic translation of Arc/Arg3.1 essential for mGluR-LTD. Neuron 59, 70–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HR, Lee JM, Moon HE, Lee DS, Kim BN, Kim J, Kim DG, and Paek SH (2016). A short review on the current understanding of autism spectrum disorders. Exp. Neurobiol 25, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parras A, Anta H, Santos-Galindo M, Swarup V, Elorza A, Nieto-González JL, Picó S, Hernández IH, Díaz-Hernández JI, Belloc E, et al. (2018). Autism-like phenotype and risk gene mRNA deadenylation by CPEB4 mis-splicing. Nature 560, 441–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasciuto E, and Bagni C (2014a). SnapShot: FMRP interacting proteins. Cell 159, 218–218.e1. [DOI] [PubMed] [Google Scholar]
- Pasciuto E, and Bagni C (2014b). SnapShot: FMRP mRNA targets and diseases. Cell 158, 1446–1446.e1. [DOI] [PubMed] [Google Scholar]
- Pasciuto E, Ahmed T, Wahle T, Gardoni F, D’Andrea L, Pacini L, Jacquemont S, Tassone F, Balschun D, Dotti CG, et al. (2015). Dysregulated ADAM10-mediated processing of APP during a critical time window leads to synaptic deficits in fragile X syndrome. Neuron 87, 382–398. [DOI] [PubMed] [Google Scholar]
- Pathania M, Davenport EC, Muir J, Sheehan DF, López-Doménech G, and Kittler JT (2014). The autism and schizophrenia associated gene CYFIP1 is critical for the maintenance of dendritic complexity and the stabilization of mature spines. Transl. Psychiatry 4, e374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peça J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, Lascola CD, Fu Z, and Feng G (2011). Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 472, 437–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peñagarikano O, and Geschwind DH (2012). What does CNTNAP2 reveal about autism spectrum disorder? Trends Mol. Med 18, 156–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, and Cahill ME (2012). Deconstructing signal transduction pathways that regulate the actin cytoskeleton in dendritic spines. Cytoskeleton (Hoboken) 69, 426–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, and Woolfrey KM (2011). Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci 14, 285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereanu W, Larsen EC, Das I, Estévez MA, Sarkar AA, Spring-Pearson S, Kollu R, Basu SN, and Banerjee-Basu S (2018). AutDB: a platform to decode the genetic architecture of autism. Nucleic Acids Res. 46 (D1), D1049–D1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Cañamás A, Benvegnu S, Rueda CB, Rábano A, Satrústegui J, and Ledesma MD (2017). Sphingomyelin-induced inhibition of the plasma membrane calcium ATPase causes neurodegeneration in type A Niemann-Pick disease. Mol. Psychiatry 22, 711–723. [DOI] [PubMed] [Google Scholar]
- Pfeiffer BE, Zang T, Wilkerson JR, Taniguchi M, Maksimova MA, Smith LN, Cowan CW, and Huber KM (2010). Fragile X mental retardation protein is required for synapse elimination by the activity-dependent transcription factor MEF2. Neuron 66, 191–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips R, Ursell T, Wiggins P, and Sens P (2009). Emerging roles for lipids in shaping membrane-protein function. Nature 459, 379–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontrello CG, and Ethell IM (2009). Accelerators, brakes, and gears of actin dynamics in dendritic spines. Open Neurosci. J 3, 67–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pucilowska J, Vithayathil J, Pagani M, Kelly C, Karlo JC, Robol C, Morella I, Gozzi A, Brambilla R, and Landreth GE (2018). Pharmacological inhibition of ERK signaling rescues pathophysiology and behavioral phenotype associated with 16p11.2 chromosomal deletion in mice. J. Neurosci 38, 6640–6652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyronneau A, He Q, Hwang JY, Porch M, Contractor A, and Zukin RS (2017). Aberrant Rac1-cofilin signaling mediates defects in dendritic spines, synaptic function, and sensory perception in fragile X syndrome. Sci. Signal 10, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi AY, Mueller S, Snyder AZ, Mukherjee P, Berman JI, Roberts TP, Nagarajan SS, Spiro JE, Chung WK, Sherr EH, and Buckner RL; Simons VIP Consortium (2014). Opposing brain differences in 16p11.2 deletion and duplication carriers. J. Neurosci 34, 11199–11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P, Bourgeois JP, Eckenhoff MF, Zecevic N, and Goldman-Rakic PS (1986). Concurrent overproduction of synapses in diverse regions of the primate cerebral cortex. Science 232, 232–235. [DOI] [PubMed] [Google Scholar]
- Ramaswami G, and Geschwind DH (2018). Genetics of autism spectrum disorder. Handb. Clin. Neurol 147, 321–329. [DOI] [PubMed] [Google Scholar]
- Rangaraju V, Tom Dieck S, and Schuman EM (2017). Local translation in neuronal compartments: how local is local? EMBO Rep. 18, 693–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner A, and Levitz J (2018). Glutamatergic signaling in the central nervous system: ionotropic and metabotropic receptors in concert. Neuron 98, 1080–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remer J, Croteau-Chonka E, Dean DC 3rd, D’Arpino S, Dirks H, Whiley D, and Deoni SCL (2017). Quantifying cortical development in typically developing toddlers and young children, 1–6 years of age. Neuroimage 153, 246–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter JD, Bassell GJ, and Klann E (2015). Dysregulation and restoration of translational homeostasis in fragile X syndrome. Nat. Rev. Neurosci 16, 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts JE, Ezell JE, Fairchild AJ, Klusek J, Thurman AJ, McDuffie A, and Abbeduto L (2018). Biobehavioral composite of social aspects of anxiety in young adults with fragile X syndrome contrasted to autism spectrum disorder. Am. J. Med. Genet. B. Neuropsychiatr. Genet 177, 665–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronesi JA, Collins KA, Hays SA, Tsai NP, Guo W, Birnbaum SG, Hu JH, Worley PF, Gibson JR, and Huber KM (2012). Disrupted Homer scaffolds mediate abnormal mGluR5 function in a mouse model of fragile X syndrome. Nat. Neurosci 15, 431–440, S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ropers HH, and Hamel BC (2005). X-linked mental retardation. Nat. Rev. Genet 6, 46–57. [DOI] [PubMed] [Google Scholar]
- Rosina E, Battan B, Siracusano M, Di Criscio L, Hollis F, Pacini L, Curatolo P, and Bagni C (2019). Disruption of mTOR and MAPK pathways correlates with severity in idiopathic autism. Transl. Psychiatry 9, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC, Codogno P, and Levine B (2012). Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov 11, 709–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, and Guan KL (2013). ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol 15, 741–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacco R, Cacci E, and Novarino G (2018). Neural stem cells in neuropsychiatric disorders. Curr. Opin. Neurobiol 48, 131–138. [DOI] [PubMed] [Google Scholar]
- Sadybekov A, Tian C, Arnesano C, Katritch V, and Herring BE (2017). An autism spectrum disorder-related de novo mutation hotspot discovered in the GEF1 domain of Trio. Nat. Commun 8, 601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval GM, Shim S, Hong DS, Garrett AS, Quintin EM, Marzelli MJ, Patnaik S, Lightbody AA, and Reiss AL (2018). Neuroanatomical abnormalities in fragile X syndrome during the adolescent and young adult years. J. Psychiatr. Res 107, 138–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Huynh TN, Longo F, Koo SY, Mojica E, D’Andrea L, Bagni C, and Klann E (2017). Reducing eIF4E-eIF4G interactions restores the balance between protein synthesis and actin dynamics in fragile X syndrome model mice. Sci. Signal 10, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos AR, Kanellopoulos AK, and Bagni C (2014). Learning and behavioral deficits associated with the absence of the fragile X mental retardation protein: what a fly and mouse model can teach us. Learn. Mem 21, 543–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saré RM, Song A, Loutaev I, Cook A, Maita I, Lemons A, Sheeler C, and Smith CB (2018). Negative effects of chronic rapamycin treatment on behavior in a mouse model of fragile X syndrome. Front. Mol. Neurosci 10, 452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawicka K, Pyronneau A, Chao M, Bennett MV, and Zukin RS (2016). Elevated ERK/p90 ribosomal S6 kinase activity underlies audiogenic seizure susceptibility in fragile X mice. Proc. Natl. Acad. Sci. USA 113, E6290–E6297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena K, Webster J, Hallas-Potts A, Mackenzie R, Spooner PA, Thomson D, Kind P, Chatterji S, and Morris RGM (2018). Experiential contributions to social dominance in a rat model of fragile-X syndrome. Proc. Biol. Sci 285, 20180294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffmann R (1996). Niemann-Pick disease type C. From bench to bedside. JAMA 276, 561–564. [PubMed] [Google Scholar]
- Schnell E, Long TH, Bensen AL, Washburn EK, and Westbrook GL (2014). Neuroligin-1 knockdown reduces survival of adult-generated newborn hippocampal neurons. Front. Neurosci 8, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Hoeffer CA, Takayasu Y, Miyawaki T, McBride SM, Klann E, and Zukin RS (2010). Dysregulation of mTOR signaling in fragile X syndrome. J. Neurosci 30, 694–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shehata M, Matsumura H, Okubo-Suzuki R, Ohkawa N, and Inokuchi K (2012). Neuronal stimulation induces autophagy in hippocampal neurons that is involved in AMPA receptor degradation after chemical long-term depression. J. Neurosci 32, 10413–10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, Rumbaugh G, Wu J, Chowdhury S, Plath N, Kuhl D, Huganir RL, and Worley PF (2006). Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron 52, 475–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z, Fujii K, Kovary KM, Genuth NR, Röst HL, Teruel MN, and Barna M (2017). Heterogeneous ribosomes preferentially translate distinct subpools of mRNAs genome-wide. Mol. Cell 67, 71–83.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AI, Ulfarsson MO, Stefansson H, Gustafsson O, Walters GB, Linden DEJ, Wilkinson LS, Drakesmith M, Owen MJ, Hall J, and Stefansson K (2018). Reciprocal white matter changes associated with copy number variation at 15q11.2 BP1-BP2: a diffusion tensor imaging study. Biol. Psychiatry, S0006–3223(18)32009–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitzmann AF, Hagelstrom RT, Tassone F, Hagerman RJ, and Butler MG (2018). Rare FMR1 gene mutations causing fragile X syndrome: a review. Am. J. Med. Genet. A 176, 11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sossin WS, and Costa-Mattioli M (2018). Translational control in the brain in health and disease. Cold Spring Harb. Perspect. Biol. Published online August 6, 2018. 10.1101/cshperspect.a032912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H, Meyer-Lindenberg A, Steinberg S, Magnusdottir B, Morgen K, Arnarsdottir S, Bjornsdottir G, Walters GB, Jonsdottir GA, Doyle OM, et al. (2014). CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature 505, 361–366. [DOI] [PubMed] [Google Scholar]
- Stoppel LJ, Auerbach BD, Senter RK, Preza AR, Lefkowitz RJ, and Bear MF (2017). β-Arrestin2 couples metabotropic glutamate receptor 5 to neuronal protein synthesis and is a potential target to treat fragile X. Cell Rep 18, 2807–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian M, Timmerman CK, Schwartz JL, Pham DL, and Meffert MK (2015). Characterizing autism spectrum disorders by key biochemical pathways. Front. Neurosci 9, 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC (2018). Towards an understanding of synapse formation. Neuron 100, 276–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson MR, Wolff JJ, Shen MD, Styner M, Estes A, Gerig G, McKinstry RC, Botteron KN, Piven J, and Hazlett HC; Infant Brain Imaging Study (IBIS) Network (2018). Development of white matter circuitry in infants with fragile X syndrome. JAMA Psychiatry 75, 505–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabet R, Moutin E, Becker JA, Heintz D, Fouillen L, Flatter E, Krężel W, Alunni V, Koebel P, Dembélé D, et al. (2016). Fragile X mental retardation protein (FMRP) controls diacylglycerol kinase activity in neurons. Proc. Natl. Acad. Sci. USA 113, E3619–E3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, and Schuman EM (2002). A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc. Natl. Acad. Sci. USA 99, 467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Gudsnuk K, Kuo SH, Cotrina ML, Rosoklija G, Sosunov A, Sonders MS, Kanter E, Castagna C, Yamamoto A, et al. (2014). Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83, 1131–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejada-Simon MV (2015). Modulation of actin dynamics by Rac1 to target cognitive function. J. Neurochem 133, 767–779. [DOI] [PubMed] [Google Scholar]
- Thurman AJ, McDuffie A, Kover ST, Hagerman RJ, and Abbeduto L (2015). Autism symptomatology in boys with fragile X syndrome: a cross sectional developmental trajectories comparison with nonsyndromic autism spectrum disorder. J. Autism Dev. Disord 45, 2816–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian C, Kay Y, Sadybekov A, Rao S, Katritch V, and Herring BE (2018). An intellectual disability-related missense mutation in Rac1 prevents LTP induction. Front. Mol. Neurosci 11, 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tom Dieck S, Hanus C, and Schuman EM (2014). SnapShot: local protein translation in dendrites. Neuron 81, 958–958.e1. [DOI] [PubMed] [Google Scholar]
- Tran SS, Jun HI, Bahn JH, Azghadi A, Ramaswami G, Van Nostrand EL, Nguyen TB, Hsiao YE, Lee C, Pratt GA, et al. (2019). Widespread RNA editing dysregulation in brains from autistic individuals. Nat. Neurosci 22, 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tranfaglia MR, Thibodeaux C, Mason DJ, Brown D, Roberts I, Smith R, Guilliams T, and Cogram P (2018). Repurposing available drugs for neurodevelopmental disorders: The fragile X experience. Neuropharmacology. Published online May 4, 2018. 10.1016/j.neuropharm.2018.05.004. [DOI] [PubMed] [Google Scholar]
- Tsai NP, Wilkerson JR, Guo W, Maksimova MA, DeMartino GN, Cowan CW, and Huber KM (2012). Multiple autism-linked genes mediate synapse elimination via proteasomal degradation of a synaptic scaffold PSD-95. Cell 151, 1581–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai NP, Wilkerson JR, Guo W, and Huber KM (2017). FMRP-dependent Mdm2 dephosphorylation is required for MEF2-induced synapse elimination. Hum. Mol. Genet 26, 293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu S, Akhtar MW, Escorihuela RM, Amador-Arjona A, Swarup V, Parker J, Zaremba JD, Holland T, Bansal N, Holohan DR, et al. (2017). NitroSynapsin therapy for a mouse MEF2C haploinsufficiency model of human autism. Nat. Commun 8, 1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulfarsson MO, Walters GB, Gustafsson O, Steinberg S, Silva A, Doyle OM, Brammer M, Gudbjartsson DF, Arnarsdottir S, Jonsdottir GA, et al. (2017). 15q11.2 CNV affects cognitive, structural and functional correlates of dyslexia and dyscalculia. Transl. Psychiatry 7, e1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Vaart T, Rietman AB, Plasschaert E, Legius E, Elgersma Y, and Moll HA; NF1-SIMCODA Study Group (2016). Behavioral and cognitive outcomes for clinical trials in children with neurofibromatosis type 1. Neurology 86, 154–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanlerberghe C, Petit F, Malan V, Vincent-Delorme C, Bouquillon S, Boute O, Holder-Espinasse M, Delobel B, Duban B, Vallee L, et al. (2015). 15q11.2 microdeletion (BP1-BP2) and developmental delay, behaviour issues, epilepsy and congenital heart disease: a series of 52 patients. Eur. J. Med. Genet 58, 140–147. [DOI] [PubMed] [Google Scholar]
- Varghese M, Keshav N, Jacot-Descombes S, Warda T, Wicinski B, Dickstein DL, Harony-Nicolas H, De Rubeis S, Drapeau E, Buxbaum JD, and Hof PR (2017). Autism spectrum disorder: neuropathology and animal models. Acta Neuropathol. 134, 537–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varoqueaux F, Aramuni G, Rawson RL, Mohrmann R, Missler M, Gottmann K, Zhang W, Südhof TC, and Brose N (2006). Neuroligins determine synapse maturation and function. Neuron 51, 741–754. [DOI] [PubMed] [Google Scholar]
- Verpelli C, and Sala C (2012). Molecular and synaptic defects in intellectual disability syndromes. Curr. Opin. Neurobiol 22, 530–536. [DOI] [PubMed] [Google Scholar]
- Vicidomini C, Ponzoni L, Lim D, Schmeisser MJ, Reim D, Morello N, Orellana D, Tozzi A, Durante V, Scalmani P, et al. (2017). Pharmacological enhancement of mGlu5 receptors rescues behavioral deficits in SHANK3 knock-out mice. Mol. Psychiatry 22, 784. [DOI] [PubMed] [Google Scholar]
- Volkmar FR, and McPartland JC (2014). From Kanner to DSM-5: autism as an evolving diagnostic concept. Annu. Rev. Clin. Psychol 10, 193–212. [DOI] [PubMed] [Google Scholar]
- Wang H, and Doering LC (2013). Reversing autism by targeting downstream mTOR signaling. Front. Cell. Neurosci 7, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Snape M, Klann E, Stone JG, Singh A, Petersen RB, Castellani RJ, Casadesus G, Smith MA, and Zhu X (2012). Activation of the extracellular signal-regulated kinase pathway contributes to the behavioral deficit of fragile x-syndrome. J. Neurochem 121, 672–679. [DOI] [PubMed] [Google Scholar]
- Wang X, Bey AL, Katz BM, Badea A, Kim N, David LK, Duffney LJ, Kumar S, Mague SD, Hulbert SW, et al. (2016). Altered mGluR5-Homer scaffolds and corticostriatal connectivity in a Shank3 complete knockout model of autism. Nat. Commun 7, 11459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zeng C, Li J, Zhou Z, Ju X, Xia S, Li Y, Liu A, Teng H, Zhang K, et al. (2018). PAK2 haploinsufficiency results in synaptic cytoskeleton impairment and autism-related behavior. Cell Rep. 24, 2029–2041. [DOI] [PubMed] [Google Scholar]
- Wang L, Pang K, Han K, Adamski CJ, Wang W, He L, Lai JK, Bondar VV, Duman JG, Richman R, et al. (2019). An autism-linked missense mutation in SHANK3 reveals the modularity of Shank3 function. Mol. Psychiatry Published online January 4, 2019. 10.1038/s41380-018-0324-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waung MW, and Huber KM (2009). Protein translation in synaptic plasticity: mGluR-LTD, Fragile X. Curr. Opin. Neurobiol 19, 319–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waye MMY, and Cheng HY (2018). Genetics and epigenetics of autism: a review. Psychiatry Clin. Neurosci 72, 228–244. [DOI] [PubMed] [Google Scholar]
- Wijetunge LS, Angibaud J, Frick A, Kind PC, and Nägerl UV (2014). Stimulated emission depletion (STED) microscopy reveals nanoscale defects in the developmental trajectory of dendritic spine morphogenesis in a mouse model of fragile X syndrome. J. Neurosci 34, 6405–6412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won H, Lee HR, Gee HY, Mah W, Kim JI, Lee J, Ha S, Chung C, Jung ES, Cho YS, et al. (2012). Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 486, 261–265. [DOI] [PubMed] [Google Scholar]
- Wong E, and Cuervo AM (2010). Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci 13, 805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Porch MW, Court-Vazquez B, Bennett MVL, and Zukin RS (2018). Activation of autophagy rescues synaptic and cognitive deficits in fragile X mice. Proc. Natl. Acad. Sci. USA 115, E9707–E9716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yerys BE, Herrington JD, Satterthwaite TD, Guy L, Schultz RT, and Bassett DS (2017). Globally weaker and topologically different: resting-state connectivity in youth with autism. Mol. Autism 8, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalfa F, Giorgi M, Primerano B, Moro A, Di Penta A, Reis S, Oostra B, and Bagni C (2003). The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses. Cell 112, 317–327. [DOI] [PubMed] [Google Scholar]
- Zalfa F, Eleuteri B, Dickson KS, Mercaldo V, De Rubeis S, di Penta A, Tabolacci E, Chiurazzi P, Neri G, Grant SG, and Bagni C (2007). A new function for the fragile X mental retardation protein in regulation of PSD-95 mRNA stability. Nat. Neurosci 10, 578–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeidán-Chuliá F, Rybarczyk-Filho JL, Salmina AB, de Oliveira BH, Noda M, and Moreira JC (2013). Exploring the multifactorial nature of autism through computational systems biology: calcium and the Rho GTPase RAC1 under the spotlight. Neuromolecular Med. 15, 364–383. [DOI] [PubMed] [Google Scholar]
- Zerbi V, Ielacqua GD, Markicevic M, Haberl MG, Ellisman MH, A-Bhaskaran A, Frick A, Rudin M, and Wenderoth N (2018). Dysfunctional autism risk genes cause circuit-specific connectivity deficits with distinct developmental trajectories. Cereb. Cortex 28, 2495–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Wang Q, and Huang Y (2007). Fragile X mental retardation protein FMRP and the RNA export factor NXF2 associate with and destabilize Nxf1 mRNA in neuronal cells. Proc. Natl. Acad. Sci. USA 104, 10057–10062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Kang Y, Wang M, Li Y, Xu T, Yang W, Song H, Wu H, Shu Q, and Jin P (2018). Fragile X mental retardation protein modulates the stability of its m6A-marked messenger RNA targets. Hum. Mol. Genet 27, 3936–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu PJ, Chen CJ, Mays J, Stoica L, and Costa-Mattioli M (2018). mTORC2, but not mTORC1, is required for hippocampal mGluR-LTD and associated behaviors. Nat. Neurosci 21, 799–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer SE, Doll SG, Garcia ADR, and Akins MR (2017). Splice form-dependent regulation of axonal arbor complexity by FMRP. Dev. Neurobiol 77, 738–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY, and Bear MF (2012). Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb. Perspect. Biol 4, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorio DA, Jackson CM, Liu Y, Rubel EW, and Wang Y (2017). Cellular distribution of the fragile X mental retardation protein in the mouse brain. J. Comp. Neurol 525, 818–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zukin RS, Richter JD, and Bagni C (2009). Signals, synapses, and synthesis: how new proteins control plasticity. Front. Neural Circuits 3, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]