

ABSTRACT
Foamy macrophages containing abundant intracellular myelin remnants are an important pathological hallmark of multiple sclerosis. Reducing the intracellular lipid burden in foamy macrophages is considered a promising therapeutic strategy to induce a phagocyte phenotype that promotes central nervous system repair. Recent research from our group showed that sustained intracellular accumulation of myelin-derived lipids skews these phagocytes toward a disease-promoting and more inflammatory phenotype. Our data now demonstrate that disturbed lipophagy, a selective form of autophagy that helps with the degradation of lipid droplets, contributes to the induction of this phenotype. Stimulating autophagy using the natural disaccharide trehalose reduced the lipid load and inflammatory phenotype of myelin-laden macrophages. Importantly, trehalose was able to boost remyelination in the ex vivo brain slice model and the in vivo cuprizone-induced demyelination model. In summary, our results provide a molecular rationale for impaired metabolism of myelin-derived lipids in macrophages, and identify lipophagy induction as a promising treatment strategy to promote remyelination.
Abbreviations: Baf: bafilomycin a1; BMDM: bone marrow-derived macrophage; CD68: CD68 antigen; CNS: central nervous system; LD: lipid droplet; LIPE/HSL: lipase, hormone sensitive; LPS: lipopolysaccharide; MAP1LC3/LC3: microtubule-associated protein 1 light chain 3; MBP: myelin basic protein; MGLL: monoglyceride lipase; MS: multiple sclerosis; NO: nitric oxide; NOS2/iNOS: nitric oxide synthase 2, inducible; ORO: oil red o; PNPLA2: patatin-like phospholipase domain containing 2; PLIN2: perilipin 2; TEM: transmission electron microscopy; TFEB: transcription factor EB; TOH: trehalose.
KEYWORDS: Lipid droplets, lipophagy, multiple sclerosis, phagocyte, remyelination
Introduction
Demyelinating disorders such as multiple sclerosis (MS) are characterized by the accumulation of damaged myelin debris and axonal degeneration [1]. Phagocytes, including microglia and infiltrating macrophages, play a dual role in the pathogenesis of these disorders. Classically, these cells were regarded to promote lesion progression by producing inflammatory mediators and degrading the protective myelin sheath of the nerves. However, phagocytes also exert protective functions as they clear damaged myelin, which is essential for remyelination to occur [2]. Moreover, earlier studies reported that uptake of myelin results in a less inflammatory and more reparative phagocyte phenotype [3–5]. Yet, we recently found that, while myelin phagocytosis initially promotes an early-stage wound-healing phenotype, prolonged uptake and accumulation of myelin-derived lipids offsets the reparative properties of phagocytes and increases the expression of inflammatory markers [6]. Moreover, we showed that preventing excessive accumulation of myelin-derived lipids enhanced central nervous system (CNS) repair. While this strategy can prevent lesion progression, it is not able to restore the disease-resolving ability of late-stage lipid-engorged phagocytes, limiting its therapeutic purpose. Hence, targeting lipid overload in myelin-laden phagocytes offers a promising strategy for the treatment of neurological disorders characterized by the presence of foamy phagocytes.
The foamy phenotype of phagocytes is linked to a disrupted ability to process and efflux the accumulating lipids and resembles the appearance of foam cells in atherosclerotic plaques. In atherosclerosis, the gradual increase in lipid accumulation has been associated with failure of autophagy, where early-stage foam cells display higher levels of autophagy compared to late-stage foam cells [7]. Macroautophagy/autophagy, hereafter referred to as autophagy, is a homeostatic cellular recycling process that mediates the lysosomal delivery and clearance of various cellular components and damaged organelles in double-membraned vesicles termed autophagosomes [8]. Autophagy can be selective and direct the degradation of specific cellular targets. One example of selective autophagy targets lipid droplets (LDs), a process termed lipophagy [9]. Induction of autophagy in foam cells was shown to reduce their cholesterol burden, while the inhibition of autophagy results in the excessive accumulation of cholesterol [10]. Further studies aimed at reducing the lipid load in foamy macrophages showed that induction of autophagy lead to reduced lipid accumulation in macrophages [11]. Whether myelin-laden phagocytes show disturbed autophagy and whether boosting autophagy can promote repair in MS remains unknown. Here, we report that sustained accumulation of LDs disrupts lipophagy in myelin-laden phagocytes. Importantly, we show that stimulating autophagy using the natural disaccharide trehalose, reduces intracellular LD accumulation and dampens the inflammatory macrophage phenotype, and improves repair in models of CNS demyelination.
Results
Lipid-laden macrophages in MS lesions show signs of dysfunctional autophagy
Active MS lesions contain a large number of inflammatory phagocytes laden with myelin remnants [12,13] (Figure 1A). To examine whether these lesions display dysfunctional autophagy, we assessed the distribution of the autophagy marker SQSTM1/p62, a receptor involved in the selective autophagy of cellular targets such as protein aggregates. Accumulation of SQSTM1 has previously been used as a marker to demonstrate autophagy dysfunction in foamy macrophages [14,15]. Here, we defined the staining intensity of SQSTM1 in active MS lesions, comparing its spatial abundance in the center, rim, and normal-appearing white matter surrounding the lesions. The active lesion centers showed a high intensity and marked accumulation of SQSTM1 compared to the other two regions (Figure 1B,C), indicating dysfunctional autophagy. Moreover, the density of phagocytes expressing SQSTM1 was increased in the lesion center compared to the normal appearing white matter and the lesion rim (Figure 1D,E). This accumulation of SQSTM1 was closely associated with the intracellular lipid load, as phagocytes in the lesion center showed a higher accumulation of intracellular lipids compared to phagocytes in the rim (Figure 1A). Taken together, these data suggest that autophagy is impaired in active MS lesions, and in particular in myelin-containing macrophages and microglia.
Figure 1.
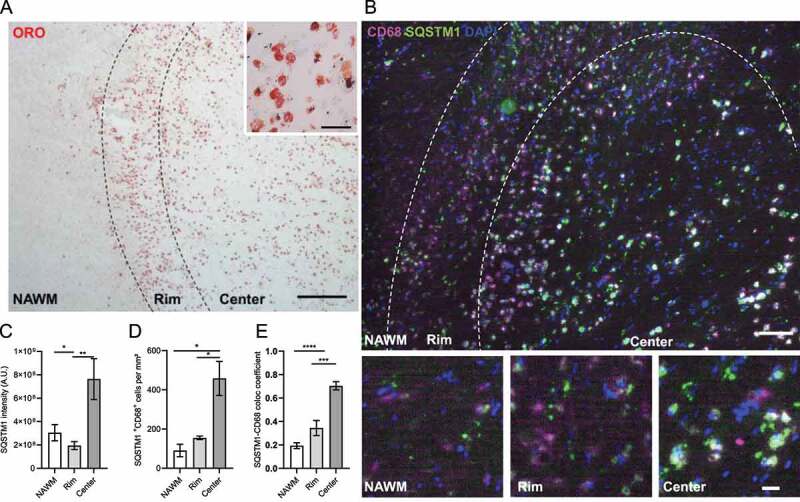
MS lesions show signs of dysfunctional autophagy. (A,B) Representative images of an active MS lesion stained with (A) Oil red O (ORO) or (B) CD68 (magenta) and SQSTM1 (green). (C,D) Quantification of (C) SQSTM1 intensity and (D) the number of phagocytes expressing SQSTM1. (E) Colocalization coefficient of CD68 and SQSTM1. n = 3 lesions from 3 different MS patients. Scale bar: (A, overview) 500 µm, (A, inset) 50 µm, (B, overview) 100 µm, (B, inset) 20 µm. White arrows indicate colocalization. All data are represented as mean ± SEM. *p < 0.05 and **p < 0.01.
Prolonged myelin accumulation leads to increased foaminess and reduced autophagy in macrophages
The accumulation of SQSTM1 in active lesions, together with the abundance of lipid-rich phagocytes and previous evidence linking increased lipid load with autophagy dysfunction, led us to investigate the effect of sustained myelin uptake on autophagy. We have previously shown that long-term exposure to myelin (72 h) renders mouse bone marrow-derived macrophages (BMDMs) more foamy and more inflammatory compared to those exposed to myelin for a short period (24 h), mimicking respectively the late-stage and early-stage foamy phagocyte phenotypes in MS lesions [6]. To test whether sustained myelin uptake by macrophages impairs autophagy, BMDMs were incubated with isolated myelin for 24 h (mye-24) or 72 h (mye-72), or were left untreated. To confirm intracellular accumulation of lipids, the amount of LDs was monitored as a proxy for intracellular lipid load. As expected, the number of BODIPY+ LDs increased with prolonged myelin incubation (Figure 2A,B,D). To evaluate autophagy levels, we first assessed the accumulation of SQSTM1+ puncta. Late-stage mye-macrophages showed a profound accumulation of SQSTM1-bodies, resembling what we observed in active lesions (Figure 2A,C). Next, cells were stained for the autophagosome marker MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3), hereafter called LC3, a commonly used autophagy marker present on autophagosomes. Consistent with the SQSTM1 staining, late-stage mye-macrophages showed a reduced amount of LC3+ puncta compared to early-stage ones, indicating lower autophagy levels upon sustained LD accumulation (Figure 2D,E). The reduced presence of autophagosomes was confirmed using transmission electron microscopy (TEM) analysis (Figure 4B,F, Fig. S5).
Figure 2.
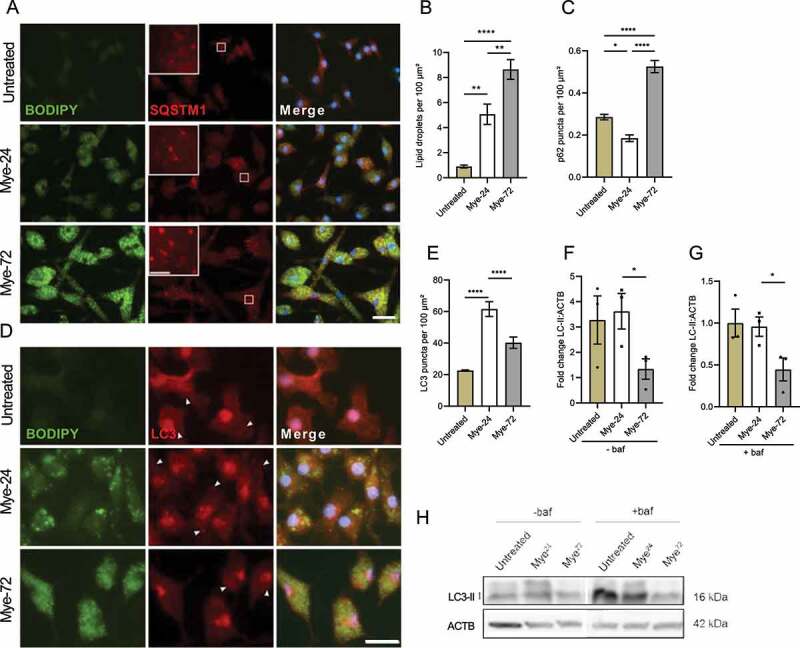
Prolonged myelin accumulation reduces autophagy levels in macrophages. (A-E) Immunofluorescence staining (A,D) and puncta-analysis (C,E) of bone marrow-derived macrophages (BMDMs) treated with myelin for a short (Mye-24) or prolonged (Mye-72) time. BODIPY (green), SQSTM1 (A, red), LC3 (D, red). Scale bar: overview 20 µm, inset 5 µm. (B) Quantification of number of BODIPY+-lipid droplets (LDs) in mye-24- and mye-72-BMDMs. Data originates from 3 independent experiments (50+ cells per condition). (F-H) Western blot analysis of LC3-II in untreated, mye-24- and mye-72-BMDMs stimulated with bafilomycin A1 (baf) 2 h prior to lysing (G) or non-stimulated (F). LC3-II level is calculated from 3 independent experiments and normalized to β-actin levels (loading control). All data are represented as mean ± SEM. *p < 0.05, **p < 0.01 and ****p < 0.0001.
Figure 4.
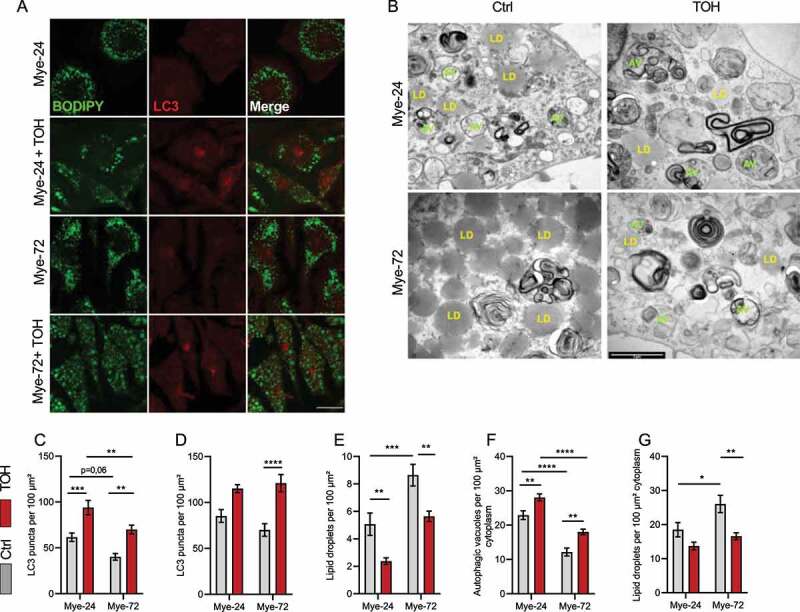
Trehalose increases autophagy in mye-macrophages. Bone marrow-derived macrophages were treated with myelin for a short (Mye-24) or prolonged (Mye-72) time and were additionally stimulated with trehalose (TOH) for the final 6 h of myelin exposure and/or bafilomycin A1 for the final 2 h. (A) Representative pictures of immunofluorescent staining of BODIPY (green) and LC3 (red). Scale bar: 20 µm. (B) Representative transmission electron microscopy images. Scale bar: 2 µm. (C,D) Quantification of LC3 puncta in the absence (C) or presence (D) of bafilomycin A1. (E) Quantification of BODIPY+ lipid droplets (LDs). (F,G) Quantification of the number of (F) autophagic vacuoles (AV, green) and (G) LDs (yellow). Identification was based on ultrastructural morphological characteristics. All quantifications are calculated from 3 independent experiments. All data are represented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001.
Upon induction of autophagy, LC3 is usually converted from the nascent LC3-I form to the lipidated LC3-II form, which correlates with the number of autophagosomes [8]. To determine whether the reduced amount of autophagosomes observed is not due to a high rate of lysosomal fusion and clearance of autophagosomes in late-stage mye-macrophages, we used immunoblotting to detect the levels of LC3-II in myelin-treated macrophages with or without bafilomycin A1 (baf), a vacuolar-type H+-ATPase inhibitor that prevents the fusion of autophagosomes with lysosomes. In line with the microscopy analysis of LC3 puncta, the levels of LC3-II were lower in mye-72-macrophages than in mye-24-ones (Figure 2F,H). Treatment with baf failed to reverse this effect, indicating that the reduced amount of autophagosomes in late-stage mye-macrophages is not due to a higher lysosomal clearance rate (Figure 2G,H). Together, these data show that, alongside increased LD accumulation, macrophages display lower autophagy levels upon prolonged myelin uptake.
Recruitment of LDs to autophagosomes and lysosomes is decreased upon prolonged myelin accumulation
Lipophagy is a specific form of autophagy in which the autophagy machinery targets LDs for degradation [9]. During lipophagy, the autophagy marker LC3 associates with LDs [9,16]. To examine whether lipophagy is impaired upon prolonged myelin uptake, we used super-resolution Airyscan confocal microscopy to measure colocalization of LC3 puncta with LDs. This showed that the percentage of LC3+ LDs was reduced in late-stage compared to early-stage mye-macrophages (Figure 3A,B). Autophagosomes ultimately fuse with lysosomes to degrade their cargo. Moreover, recently, a subtype of lipophagy has been shown to proceed by direct lysosome-based interactions in hepatocytes [17]. Given the latter, we assessed whether LDs in early-stage mye-macrophages associate more with LAMP1+ lysosomes than LDs in late-stage mye-macrophages. We noted a reduction in LAMP1+ LDs after prolonged myelin treatment, similar to the reduction in LC3+ LDs (Figure 3C,D). These results were further confirmed using the LD-marker perilipin 2 (PLIN2) (Fig. S2).
Figure 3.
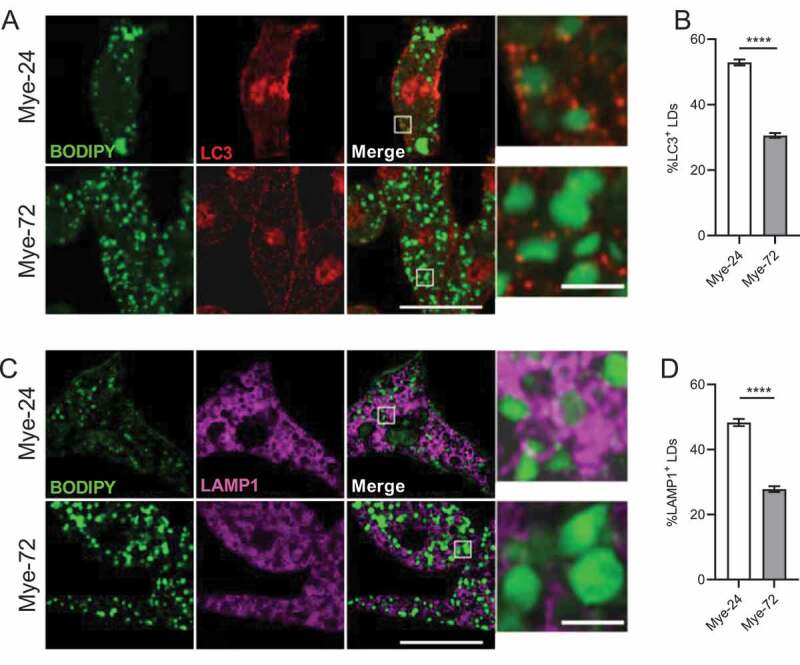
Recruitment of lipid droplets to autophagosomes and lysosomes in macrophages is decreased upon sustained myelin exposure. (A,C) Immunofluorescence staining of bone marrow-derived macrophages treated with myelin for a short (Mye-24) or prolonged (Mye-72) time. BODIPY (green), LC3 (A, red), Lamp1 (C, magenta). Scale bar: overview 20 µm, inset 2 µm. (B,D) Average % of (B) LC3+ and (D) Lamp1+ lipid droplets (LDs) of the total amount of LDs. Quantification is calculated from 3 independent experiments (100+ cells per condition). All data are represented as mean ± SEM. ****p < 0.0001.
As lipophagy shows functional links with lipolysis, a LD degradation process which involves the action of neutral lipases in the cytosol, we aimed to evaluate the effect of prolonged myelin uptake on lipolysis in foamy macrophages. During lipolysis, PNPLA2/ATGL (patatin-like phospholipase domain containing 2) initiates triacylglycerol hydrolysis to diacylglycerol and free fatty acids, followed by LIPE/HSL (lipase, hormone sensitive) and MGLL (monoglyceride lipase)-mediated hydrolysis into monoacylglycerols, glycerol, and free fatty acids [18]. Prolonged myelin treatment increased the expression of Pnpla2 and Mgll, but did not affect Lipe levels (Fig. S3A-C). In agreement, glycerol concentration showed an increasing trend after sustained myelin treatment, which further provides evidence for higher lipolysis levels (Fig. S3D). In summary, these data show that lipophagy-mediated, and not lipolysis-mediated, LD degradation is hampered in macrophages upon sustained myelin accumulation.
Trehalose treatment reduces lipid load and inflammation in foamy macrophages by inducing autophagy
Having established that prolonged lipid accumulation impairs lipophagy in macrophages, we next sought to determine whether stimulating lipophagy can reduce the lipid load and dampen the inflammatory phenotype of these cells. To this end, the disaccharide trehalose, which has been shown to induce autophagy and autophagy-lysosomal biogenesis, was used [11,19]. Notably, the use of trehalose on atherosclerotic macrophages has been shown to reduce their lipid accumulation [11]. BMDMs were treated with myelin for a short or prolonged period and were additionally stimulated with trehalose in the final 6 h of myelin treatment. Here, we show that trehalose boosts autophagy in myelin-laden macrophages, evidenced by microscopy analysis of LC3+ puncta, where late-stage mye-macrophages treated with trehalose showed a significant increase in LC3+ puncta to levels comparable to early-stage macrophages (Figure 4A,C and Fig. S4). This was confirmed using transmission electron microscopy (TEM) analysis which demonstrates an increase in the total amount of autophagosomes after treatment with trehalose (Figure 4B,F). Consistent with these findings, trehalose exposure reduced the accumulation of SQSTM1+ puncta in macrophages upon prolonged myelin uptake (Fig. S1). Collectively, these findings indicate that trehalose promotes autophagy in myelin-containing macrophages.
To assess the impact of trehalose on the lipid load of foamy macrophages, the LD count was determined. For this purpose, we quantified the number of LDs by using TEM and BODIPY staining (Figure 4A,B,E,G). This showed that trehalose reduced the number of LDs in myelin-treated macrophages, which was further confirmed using Oil Red O (ORO)-staining (Figure 5A). As lipophagy is essential for the efficient degradation of LDs prior to lipid efflux, we reasoned that trehalose reduces the lipid burden by stimulating the clearance of LDs. To evaluate this, we assessed the level of cholesterol efflux via ABCA1 (ATP-binding cassette, sub-family A (ABC1), member 1) in mye-macrophages after trehalose treatment. In accordance with our recent findings, prolonged myelin accumulation hampered the cholesterol capacity of macrophages. However, trehalose exposure restored the cholesterol efflux levels of late-stage mye-macrophages, which confirms that trehalose reduces LD load in mye-macrophages by increasing lipid efflux (Figure 5B).
Figure 5.
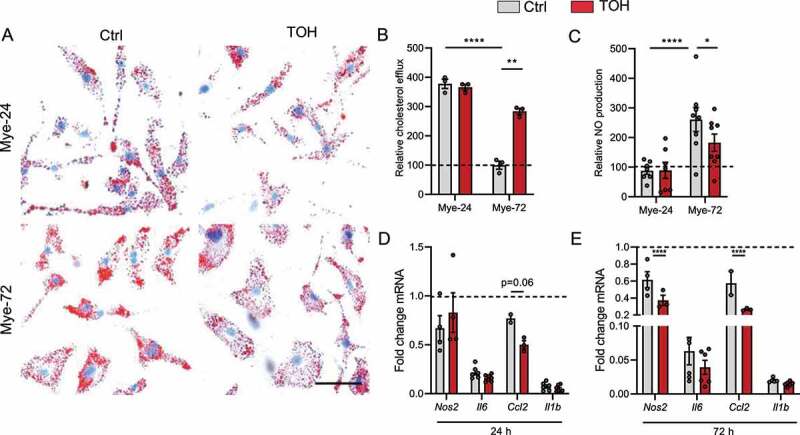
Trehalose stimulates cholesterol efflux and reduces lipid load and inflammation in mye-macrophages. Bone marrow-derived macrophages (BMDMs) were treated with myelin for a short (Mye-24) or prolonged (Mye-72) time and were additionally stimulated with trehalose (TOH) for the final 6 h of myelin-ingestion. (A) Representative images of Oil Red O staining. Scale bar: 50 µm. (n = 4 wells). (B) Capacity of cells to efflux cholesterol and normalized to non-stimulated cells (n = 3). (C) Relative nitric oxide (NO) production by BMDMs stimulated with LPS for 18 h after TOH and myelin treatment (n = 6). (D-E) mRNA expression of pro-inflammatory genes in BMDMs stimulated with LPS for 6 h after TOH and myelin treatment (n = 6). All data are represented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001.
Excessive accumulation of intracellular lipids has previously been linked to the induction of a more inflammatory phenotype in mye-macrophages [12,20]. In line with the enhanced cholesterol efflux and reduced LD load, trehalose switched the macrophage profile of late-stage mye-macrophages to a less-inflammatory one, as evidenced by a reduced nitric oxide (NO) production and a lower mRNA expression of various inflammatory markers (Figure 5C-E). Together, our results prove that trehalose can effectively induce autophagy in myelin-laden phagocytes, thereby increasing lipid efflux and reducing lipid load, which drives them toward a less inflammatory phenotype.
Trehalose reduces lipid load in in lipophagy-defective mye-microglia
Foamy phagocytes inside MS lesions include both myelin-internalizing macrophages as well as microglia. Although these cells have a diverge ontogeny, development, and function, we previously showed that both phagocyte subtypes change their phenotype in a comparable way after myelin internalization [12,21,22]. Consistent with the latter, prolonged myelin treatment reduced the association of LDs with autophagosomes and lysosomes, pointing toward a defect in the lipophagy process (Figure 6A-D). Moreover, additional stimulation with trehalose was able to reduce the LD load in myelin-laden microglia (Figure 6E). Taken together, these results show that late-stage microglia show a similar lipophagy-defective phenotype as macrophages, and that autophagy-induction by trehalose can reduce their lipid burden.
Figure 6.
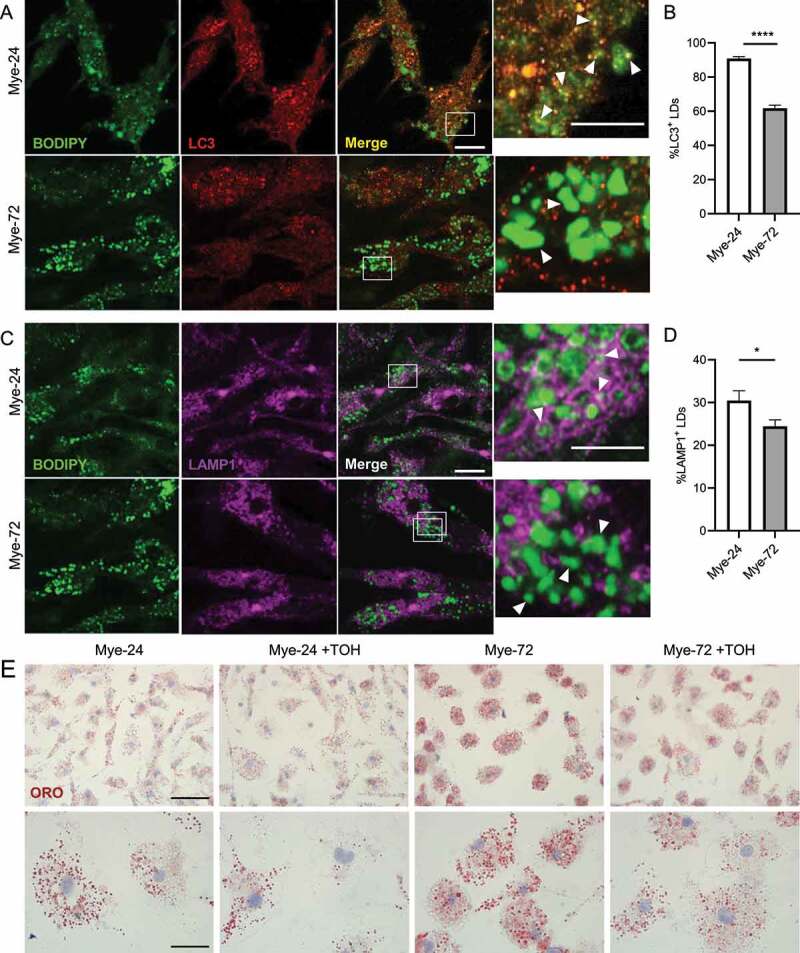
Trehalose reduces LD accumulation in late-stage microglia. (A,C) Immunofluorescence staining of primary mouse microglia treated with myelin for a short (Mye-24) or prolonged (Mye-72) time. BODIPY (green), LC3 (A, red), LAMP1 (C, magenta). Scale bar: overview 10 µm, inset 5 µm. (B,D) Average % of (B) LC3+ and (D) LAMP1+ lipid droplets (LDs) of the total amount of LDs. White arrows indicate colocalization. Quantification is calculated from 2 independent experiments (50+ cells per condition). (E) Representative images of Oil Red O staining of primary mouse microglia which were treated with myelin for a short (Mye-24) or prolonged (Mye-72) time and were additionally stimulated with trehalose (TOH) for the final 6 h of myelin-treatment. Scale bar: overview 50 µm, inset 20 µm. (n = 3 wells). All data are represented as mean ± SEM. *p < 0.05, ****p < 0.0001.
Trehalose stimulates remyelination and reduces foam cell formation in an ex vivo cerebellar brain slice model
We and others demonstrated that increased inflammation impedes remyelination [12,20,23–26]. Since our results show that trehalose-mediated autophagy induction promotes a less-inflammatory macrophage phenotype, we assessed whether trehalose can promote remyelination. To this end, we used the ex vivo cerebellar brain slice model where demyelination is induced using lysolecithin, followed by a period remyelination (Figure 7A). Similar to our in vitro findings, we show that trehalose treatment reduced lipid accumulation in brain slices (Figure 7B,C). Accordingly, there was a lower expression of various pro-inflammatory genes and reduced presence of NO in the supernatant of trehalose-treated brain slices (Figure 7D,E). To examine whether the positive impact of trehalose extends to remyelination, the amount of myelination was evaluated by measuring the colocalization of myelin (myelin basic protein, MBP) on axons (neurofilament, NF). Trehalose-treated slices showed improved remyelination, which was confirmed with three-dimensional reconstructions of the sections (Figure 7F,G). In summary, these findings show that trehalose reduces lipid accumulation, suppresses inflammation, and promotes remyelination in the brain slice model.
Figure 7.
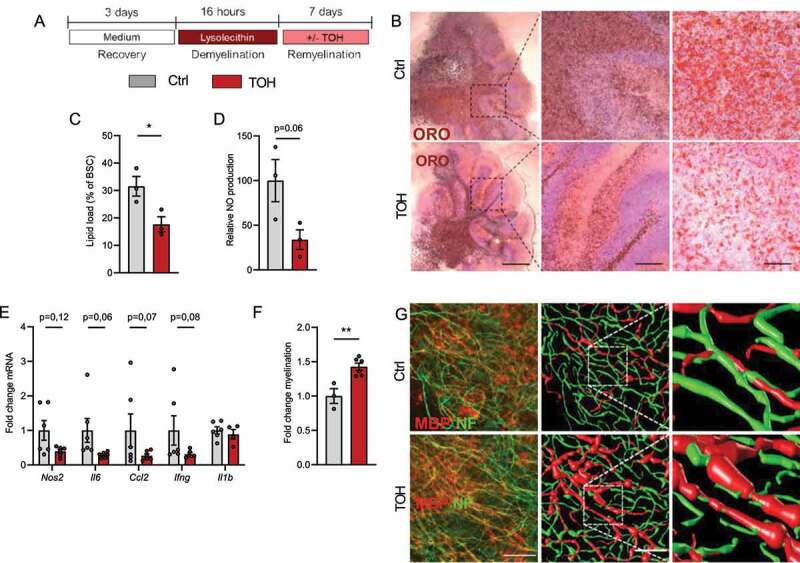
Trehalose promotes remyelination in an ex vivo cerebellar brain slice model. (A) Brain slices were demyelinated with lysolecithin and afterward treated daily with vehicle (Ctrl) or trehalose (TOH) for 1 week. (B,C) Representative images and quantification (lipid load defined as percent area covered in lipid droplets of the total brain slice area) of Oil Red O (ORO) staining of cerebellar brain slices (n = 3 slices). Scale bars: 500 μm (left), 100 μm (middle), 50 µm (right). (D) Relative nitric oxide (NO) concentration measured in supernatant of the brain slice cultures (BSC) (n = 3 wells). (E) mRNA expression of inflammatory mediators in brain slices (n = 6 slices). (F) Fold change MBP+NF+ axons of total NF+ axons (n = 3–6 slices). (G) Representative immunofluorescence images of brain slices stained for neurofilament (NF, green) and myelin basic protein (MBP, red). Scale bar: 50 µm (overview), 150 µm (inset), orthogonal and three-dimensional reconstruction (n = 3–6 slices). All data are represented as mean ± SEM. *p < 0.05 and **p < 0.01.
Trehalose improves remyelination and reduces foam cell formation in vivo
To validate the significance of our findings in vivo, we used the cuprizone-induced de- and remyelination model. Cuprizone feeding leads to demyelination in the corpus callosum and cessation of cuprizone administration after 6 weeks causes spontaneous remyelination. Animals were intraperitoneally injected with a clinically relevant dose of trehalose (2 g/kg) or vehicle (PBS) three times per week and were pathologically characterized both after demyelination (6 weeks, 6 w) and during spontaneous remyelination (7 weeks, 7 w) (Figure 8A). Interestingly, trehalose-treated animals showed an increased g-ratio (the ratio of the inner axonal diameter to the total outer diameter) after 6 weeks of cuprizone feedings and one week after cessation of the cuprizone diet (Figure 8B-E). We further confirm our in vitro and ex vivo findings and show that trehalose reduced the lipid accumulation after demyelination and during remyelination (Figure 8F,G). Moreover, trehalose treatment reduced NOS2/iNOS immunoreactivity and ADGRE1/F4/80+ macrophages (Figure 8F,I), and TMEM119+ microglia (Fig. S5B, E) infiltration after demyelination, but not during remyelination (Figure 8F,H,I). Similarly, there was a lower expression of various pro-inflammatory genes in trehalose-treated animals during demyelination (Figure 8J,K). Of note, trehalose treatment effectively increased autophagosome formation in ADGRE1+ macrophages in the corpus callosum (Fig. S6). Collectively, our results show that trehalose reduces macrophage lipid load and inflammation in vivo by increasing autophagy, thereby enhancing remyelination. In conclusion, we identify trehalose as a novel therapeutic compound to promote remyelination.
Figure 8.
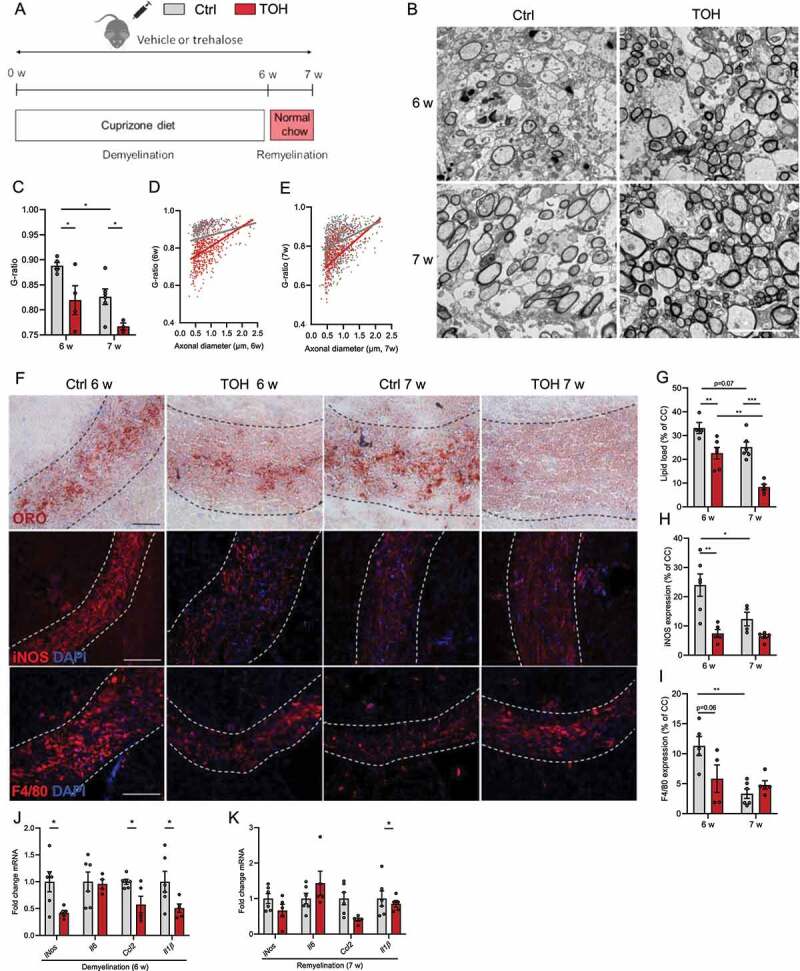
Trehalose stimulates remyelination in the cuprizone model. (A) Experimental setup of cuprizone-induced demyelination in vivo model. (B) Representative images of transmission electron microscopy analysis of corpus callosum (cc) from vehicle (Ctrl)- and trehalose (TOH)-treated animals after demyelination (6 w) and during remyelination (7 w). Scale bar: 100 µm. (C-E) Analysis of the g-ratio (the ratio of the inner axonal diameter to the total outer diameter) and g-ratio as a function of axon diameter in cc. (F) Representative images of Oil Red O (ORO) staining (scale bar: 50 µm), immunofluorescence NOS2 staining (scale bar: 100 µm), and immunofluorescence ADGRE1 staining (scale bar: 100 µm). (G) Quantification of ORO staining (lipid load defined as percent area covered in lipid droplets of the total cc area). (H,I) Relative NOS2+ area and ADGRE1+ area of cc. (J,K) mRNA expression of inflammatory mediators in CC. n = 4–7. All data are represented as mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001.
Discussion
Foamy phagocytes containing substantial amounts of myelin-derived lipid remnants are abundantly present in MS lesions. We and others demonstrated that the presence of these foamy phagocytes is associated with an increased neuroinflammatory burden and reduced CNS repair [12,20]. In this study, we provide convincing evidence showing that impaired lipophagy, a selective form of autophagy responsible for the degradation of LDs, underlies the disease-promoting phenotype shift associated with late-stage myelin-laden macrophages. Furthermore, we show that trehalose restores autophagy, thereby reducing the intracellular lipid load and inflammatory phenotype of macrophages. Importantly, by using experimental models for remyelination, we demonstrate that trehalose enhances remyelination ex vivo and in vivo. Thus, our findings show that lipophagy induction holds great promise to improve remyelination in MS.
Autophagy is a homeostatic cellular recycling process that mediates the lysosomal delivery and clearance of various cellular components and damaged organelles in double-membraned vesicles termed autophagosomes [27]. Here, we demonstrate that the center of MS lesions, where foamy phagocytes are very abundant, is characterized by autophagy dysfunction, as this region showed an accumulation of SQSTM1. SQSTM1 is a chaperone-protein that shuttles selective substrates into autophagosomes. Since SQSTM1 is degraded together with its cargo upon autophagy occurrence, its level is inversely correlated to the level of autophagy [8]. Moreover, prolonged myelin treatment of macrophages led to an increased LD load associated with an accumulation of SQSTM1 aggregates. Besides, late-stage mye-macrophages showed less LC3, a protein that coats autophagosomes and is involved in its maturation. TEM analysis confirmed this and demonstrated a reduced presence of double-membrane-delimited autophagosomes. Importantly, we prove that the accumulation of LDs in late-stage mye-macrophages was associated with reduced lipophagy as well, as the number of LC3+ and LAMP1+ LDs was decreased in these cells.
Lipophagy was previously demonstrated to represent an essential mechanism for cells to dispose LDs [9]. It mediates the delivery of LDs to lysosomes, where lysosomal acidic lipases hydrolyze the LD-associated cholesterol esters to generate free cholesterol. This degradation of LDs is crucial to facilitate ABCA1- and ABCG1 (ATP binding cassette subfamily G member 1)-mediated efflux to respectively apolipoprotein A1 and high-density lipoprotein, for which not esterified cholesterol but free cholesterol is the substrate [7]. Autophagy dysfunction has been shown to be a pathological hallmark in other diseases characterized by excessive lipid accumulation as well. Similar to our findings, in vitro models of atherosclerosis show that early-stage foam cells display higher levels of autophagy than late-stage foam cells. Moreover, a clear correlation between plaque progression and reduced lipophagy in macrophages is reported in vivo in atherosclerosis [11]. This dysfunctional autophagy contributes to increased lipid accumulation, apoptosis, and inflammasome hyperactivation [10,14,28]. Furthermore, there is evidence for defective hepatic autophagy upon high-fat diet feeding as well [29–31].
Until the discovery of lipophagy, degradation of LDs by neutral cytosolic lipases was considered the primary mechanism by which LDs are degraded. Now, increasing evidence suggests a synergistic relationship between lipolysis and lipophagy [32]. Lipolysis is proposed to be responsible for the degradation of bigger LDs, resulting in smaller LDs which can afterward be broken down by lipophagy [33]. Interestingly, a link between LC3 and PNPLA2, the primary cytosolic lipase, has been demonstrated already, providing a molecular rationale for a link between autophagy and lipolysis. LC3 can interact with PNPLA2, which is crucial for the translocation of PNPLA2 toward the LDs [18]. Furthermore, it was recently shown in hepatocytes that LDs can be directly degraded by lysosomes without the interference of autophagosomes [17]. Given this vital role for lipolysis in regulating intracellular lipid stores, it is of interest to elucidate the role of lipolysis in foam cell formation as well. Interestingly, our findings indicate that late-stage mye-macrophages showed an increase in average LD size (data not shown). As bigger LDs are more likely to be degraded by lipolysis, this finding suggests that, besides lipophagy, lipolysis is likely to be dysfunctional in these cells as well. Yet, whether a disturbance in this lipid catabolic process is involved in skewing phagocytes toward a foamier phenotype remains to be further elucidated.
To therapeutically restore lipophagy, we used the natural disaccharide trehalose. Trehalose was previously shown to stimulate macrophage autophagy and autophagy-lysosomal biogenesis in vitro and in vivo. Moreover, trehalose is used as a potent autophagy inducer in neurological disorders such as Alzheimer and Parkinson disease and can cross the blood-brain barrier and enter the CNS [34,35]. We demonstrate that trehalose is a potent inducer of autophagy in mye-macrophages as well, thereby restoring cholesterol efflux and reducing LD accumulation upon prolonged myelin load. Moreover, trehalose was able to alleviate the inflammatory status of late-stage mye-macrophages. To evaluate the therapeutic potential of trehalose in demyelinating disorders, we used the organotypic ex vivo brain slice model and the in vivo cuprizone model. We confirmed our in vitro findings and showed that trehalose was able to reduce macrophage lipid load and inflammation in vivo as well. Interestingly, we provide for the first time a link between lipophagy induction and CNS repair, as trehalose significantly reduced lipid load and increased remyelination in both models. Worth noting, trehalose-treated mice showed higher level of myelination at both 6 and 7 weeks of cuprizone treatment, indicating that trehalose might have a protective influence against demyelination in addition to its remyelination boosting effects. Since an inflammatory environment in the CNS can hamper repair [20,23–26], trehalose likely exerts this disease-resolving effect by its beneficial effects on the macrophage phenotype.
Based on the findings of this project, we propose that autophagy induction by trehalose mediates the observed beneficial effects mainly by reducing macrophage lipid load. Nevertheless, next to regulating the cellular LD load, autophagy also plays a direct role in regulating both anti- and pro-inflammatory processes. On one hand, autophagy can act as a negative feedback regulator of the inflammasome by regulating the degradation of pro-CASP1 and damaged mitochondria [36,37]. On the other hand, a recent study shows that, IL1B/IL-1β is transferred to the intermembrane space of autophagosomes prior to its secretion, suggesting autophagy is essential for IL1B release [38]. Furthermore, specialized lysosomes can secrete both pro- and anti-inflammatory cytokines depending on the inflammatory response of the immune cell [39]. These contradictive actions show that the regulation of inflammation by autophagy likely happens via multiple pathways that have to be regulated appropriately. Yet, it is possible that trehalose reduced the inflammatory burden in our models not only through decreasing LD accumulation but also partly by directly affecting inflammatory signaling pathways. Also, macrophage autophagy was shown to be important for effective phagocytosis [40–42]. Since clearance of myelin debris is crucial for effective remyelination to occur [2], trehalose may have CNS reparative effects via this mechanism as well. For instance, recent research showed that microglial autophagy is essential for myelin phagocytosis and that trehalose administration leads to myelin clearance and disease remission in aged EAE mice [43]. Furthermore, autophagy is involved in antigen presentation as well. More specifically, TFEB (transcription factor EB) activity in dendritic cells increases major histocompatibility complex class II antigen presentation while inhibiting cross-presentation by major histocompatibility complex class I [44,45]. Taken together, autophagy induction by trehalose can have beneficial effects on macrophages phenotype and function and on remyelination independent of reducing lipid accumulation. The mechanism by which trehalose induces autophagy, is still not fully unraveled. It is suggested to induce TFEB nuclear translocation, resulting in the transcriptional activation of autophagy and lysosomal genes. How trehalose causes this translocation remained elusive [11]. However, recently it was shown that trehalose activates TFEB by causing low-grade lysosomal stress [19]. It would be of interest in future studies to explore whether the effects of trehalose on the lysosomes themselves can also impact lipid load and LD degradation in foamy macrophages.
In summary, we provide evidence for lipophagy disturbance in macrophages after sustained accumulation of myelin debris. Using trehalose, we effectively induced autophagy in myelin-loaded macrophages, thereby reducing lipid load and inflammation in vitro and in vivo. Trehalose is already regarded as a potential treatment strategy for different neurological diseases [46,47]. We now show that it is also a promising therapeutic for the treatment of demyelinating disorders characterized by foamy macrophages.
Materials and methods
Antibodies
The following antibodies were used for immunofluorescence: rabbit anti-LC3 (1:500; Sigma-Aldrich, L7543; MBL, PM036), rat anti-LAMP1 (1:500; Abcam, ab25245), rabbit anti-SQSTM1/p62 (1:500; Cell Signaling Technology, 23,214), rat anti-MBP (1:250; Millipore, MAB386), rat anti-ADGRE1/F4/80 (1:100; Bio-Rad, MCA497G) and rabbit anti-NOS2/iNOS (1:100; Abcam, ab15323). The secondary Alexa Fluor antibodies Alexa Fluor 555 (1:500; Life Technologies, A21430) and Alexa Fluor 647 (1:500; Life Technologies, A21247). BODIPY® 493/503 (2 µM; ThermoFisher Scientific, D3922) was used to stain the LDs. The following antibodies were used for western blot: rabbit anti-LC3 (Sigma-Aldrich, L7543) and mouse anti-ACTB/β-actin (1:2000; Santa Cruz Biotechnology, sc-47,778). Appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies for immunoblotting were purchased from Dako (P0260, P0448).
Mice
Wild-type C57BL/6 J mice were purchased from Envigo. Mice were maintained on a 12 h light/dark cycle with free access to water and either a standard chow diet or special formulations as indicated. All studies were conducted in accordance with the institutional guidelines and approved by the Ethical Committee for Animal Experiments of Hasselt University.
Myelin isolation
Myelin was purified from mouse postmortem brain tissue with density-gradient centrifugation, as described previously [48]. Pierce BCA Protein Assay kit (Thermo Fisher Scientific, 23,225) was used to determine myelin protein concentration, according to the manufacturer’s guidelines. Endotoxin content was determined with the Chromogenic Limulus Amebocyte Lysate assay kit (Genscript, L00350). A neglectable amount of endotoxin (<1.8 x 10−3 pg/mg myelin) was present in isolated myelin.
Bone marrow-derived macrophages and cell treatments
Mouse bone marrow-derived macrophages (BMDMs) were isolated and differentiated as described previously [49]. After 7 days in culture, adherent macrophages were harvested and plated at 0.5 × 106 cells/ml to be used for in vitro experiments. BMDMs were treated daily with mouse myelin (100 µg/ml) for 24 h (mye-24-BMDMs) or 72 h (mye-72-BMDMs). To induce autophagy, cells were treated with 100 mM of trehalose (VWR, 6138–23-4) for 6 h before collection. To measure autophagic flux, cells where treated with 10 nM of bafilomycin A1 (InvivoGen, tlrl-baf1) for 2 h before collection.
Transmission electron microscopy
BMDMs cultured on plastic Thermanox® coverslips (Themanox, 72,274) and brain samples were fixed with 2% glutaraldehyde. Afterward, postfixation was performed with 2% osmium tetroxide in 0.05 M sodium cacodylate buffer (pH 7.3) for 1 h at 4°C. Dehydration of the samples was performed by ascending concentrations of acetone. The dehydrated samples were impregnated overnight in a 1:1 mixture of acetone and araldite epoxy resin (Araldite, 20,010,024). Afterward, the samples were embedded in araldite epoxy resin at 60°C. For the BMDMs, the pop-off method was used [50]. Next, the samples were cut in slices of 40–60 nm (BMDMs) or 70 nm (brain tissue), with a Leica EM UC6 microtome (Leica) and transferred to 0.7% formvar-coated copper grids (Aurion, FCF200CU50). Brain tissue was cut perpendicular to the corpus callosum. The samples were contrasted with 0.5% uranyl acetate and lead citrate using a Leica EM AC20 (Leica). Analysis was performed with a Philips EM208 S electron microscope (Philips, Amsterdam, The Netherlands) equipped with a Morada Soft Imaging System camera with iTEM-FEI software (Olympus SIS, Münster, Germany). ImageJ was used to calculate the g-ratio (the ratio of the inner axonal diameter to the total outer diameter), in a blinded fashion using 8 images/animal. Autophagosomes and lipid droplets were identified based on structural characteristics as described previously [51] at 11,000 magnification and normalized to the cytoplasmic area using the imageJ polygon selection tool.
Fluorescence microscopy and image analysis
Mouse BMDMs were cultured on glass cover slides and fixed in ice cold methanol for 10 min. Frozen brain material from active MS lesions was obtained from the Netherlands Brain Bank (NBB, Amsterdam, Netherlands). Cerebellar brain slices were fixed in 4% PFA for 15 min. Cryostat sections (10 µm) were cut using a Leica CM3050 S cryostat (Leica Microsystems, Wetzlar, Germany). Cryosections were fixed in acetone for 10 min and in 70% ethanol for 5 min. Immunostaining and analysis of fixed cells and cryosections were performed as described previously [52]. Cerebellar brain slices were stained by incubating with relevant primary antibody diluted in blocking buffer (1× PBS [Lonza, BE17-516 F] + 1% BSA [Biowest, P6154] + 0.1% Triton X-100 [Sigma-Aldrich, 9036–19-5]). BMDMs and cerebellar brain slices were imaged using LSM 880 Confocal microscope (Zeiss, Oberkochen, Germany) using the Airyscan feature. MS lesions and cuprizone brain tissue were imaged using a Nikon Eclipse 80i microscope (Nikon, Tokyo, Japan). Image quantification and analysis was done using cellprofiler [53] and ImageJ [54] software. Three-dimensional analysis of cerebellar brain slices was performed using the z-stack function on the confocal microscope and images were rendered by the 3D rendering software vaa3d [55]. Pictures indicated in figures are digitally enhanced.
Western blotting
Cells were lysed in RIPA buffer (150 mM NaCl [VWR, 27,810.295], 50 mM Tris [fisher bioreagents, BD152-5], 1% SDS [Sigma-Aldrich, 75,746], 1% Triton X-100, 0.5% sodium deoxycholate [Sigma-Aldrich, 302–95-4], pH 8) supplemented with protease (Roche, 05892970001) -phosphatase (Roche, 04906837001) inhibitor cocktail. Samples were separated by electrophoresis on a 12% gel and were transferred onto a PVDF membrane. Blots were blocked in 5% milk in PBST (137 mM NaCl [VWR, 27,810.295], 2.7 mM KCl [VWR, 26,764.298, 10 mM Na2HPO4 [VWR, 102,495], 1.8 KH2PO4 [VWR, 26,936.293], 0.5% Tween-20 [Fisher Bioreagents, BP337], pH 7.2) and were incubated with relevant primary antibodies, followed by incubation with the appropriate HRP-conjugated secondary antibody. An enhanced chemiluminescence (ECL) Plus detection kit (ThermoFisher Scientific, 32,132) was used for detection. Densitometry analysis was performed using ImageJ and normalized to actin.
Glycerol assay
Cells were treated with myelin and trehalose and collected in assay buffer. Glycerol assay was performed on the resulting samples according to the manufacturer’s guidelines (Sigma-Aldrich, MAK117).
Oil red O staining
PFA fixed cells, unfixed cryosections, and cerebellar brain slices were stained with 0.3% Oil Red O (ORO; Merck, 1320–06-5) for 10 min (cells and cryosections) or 30 min (brain slices). Counterstaining of cell nuclei was done using hematoxylin incubation. Analysis was carried out using a Leica DM 2000 LED microscope (Leica, Wetzlar, Germany) and ImageJ software.
Cholesterol efflux measurement
Myelin and trehalose-treated BMDMs were exposed to APOA1 (apolipoprotein A-I) which was kindly provided by Alan T. Remaley (National Institute of Health) (50 µg/ml), in phenol- and serum-free medium for 4 h prior to measuring intracellular and extracellular total cholesterol using the AmplexTM Red Cholesterol Assay Kit (Thermo Fisher Scientific, A12216), according to the manufacturer’s instructions. Cholesterol efflux was determined by dividing fluorescence in the supernatants by the total fluorescence in supernatants and cells. Fluorescence was measured at an excitation wavelength of 540 nm and an emission wavelength of 590 nm using the FLUOstar optima microplate reader (BMG Labtech, Ortenberg, Germany).
NO determination
BMDMs seeded in a 48-well plate were treated with myelin and trehalose followed by 18 h of lipopolysaccharide (LPS, 100 ng/µl; Calbiochem, 437,627) stimulation. Afterward, the cell culture supernatant was collected and mixed with the Griess reagent (Sigma-Aldrich, 224,058) at a 1:1 ratio. After a 15-min incubation at room temperature, the absorbance was measured at 540 nm using the FLUOstar optima microplate reader (BMG Labtech, Ortenberg, Germany).
RNA extraction and real-time quantitative PCR
Total RNA from tissue was isolated using Qiazol (Qiagen, 79,306) and the RNeasy mini kit (Qiagen, 74,106), according to the manufacturer’s guidelines. Complementary DNA was synthesized using qScriptTM cDNA SuperMix (Quanta Biosciences, 95,037) according to the manufacturer’s instructions. Real-time quantitative PCR was conducted on a Step One Plus detection system (Applied Biosystems, MA, USA). Cycle conditions were 95°C for 20s, followed by 40 cycles of 95°C for 3 s, and 60°C for 30s. The PCR reaction mixture contained SYBR green master mix (Thermo Fisher Scientific, 4,309,155), 0.3 μM forward and reverse primer (IDT technologies), RNase-free water (Qiagen, 74,106) and 12.5 ng cDNA template. Data were analyzed using the comparative Ct method and normalized to the most stable reference genes, as described previously [56,57]. Primer sequences used for real-time quantitative PCR are available upon request.
Cerebellar slice cultures
Cerebellar slices were obtained from C57BL/6 P9 or P10 mouse pups, as described previously [58,59]. Demyelination was induced by treating the slices with lysolecithin (0.5 mg/ml; Santa Cruz Biotechnology, sc-473611A) for 16 h. Afterward, slices were washed and treated daily with 1 M trehalose for 1 week, followed by histological and biochemical analysis.
Cuprizone-induced demyelination in vivo
Acute demyelination was induced in wild-type C57BL/6 males (10 weeks old) by feeding them ad libitum a diet of 0.3% (w:w) cuprizone (bis[cyclohexanone]oxaldihydrazone; Sigma-Aldrich, C9012) mixed in normal chow for 6 weeks. Upon withdrawal of the cuprizone diet, spontaneous remyelination occurs. Animals were intraperitoneally injected 3 times per week with 2 g/kg of trehalose or PBS as vehicle. After one week of normal chow, the corpus callosum was isolated and snap frozen for real-time quantitative PCR or imbedded for immunohistochemical analysis.
Statistical analysis
Data were statistically analyzed using GraphPad Prism and are reported as mean ± SEM. The D’Agostino and Pearson omnibus normality test was used to test for normal distribution. When datasets were normally distributed, an ANOVA (Tukey’s post hoc analysis) or two-tailed unpaired Student’s t test was used to determine statistical significance between groups. If datasets did not pass normality, the Kruskal–Wallis or Mann–Whitney analysis was applied. P-values <0.05 were considered to indicate a significant difference (*, P < 0.05, **, P < 0.01, and ***, P < 0.001).
Supplementary Material
Funding Statement
The work has been supported by the Flemish Fund for Scientific Research (FWO Vlaanderen; 1141920N, 12U7718N and 1502120N), the Belgian Charcot Foundation (Fondation Charcot 2020-0004), and the special research fund UHasselt (BOF).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here
References
- [1].Alizadeh A, Dyck SM, Karimi-Abdolrezaee S.. Myelin damage and repair in pathologic CNS: challenges and prospects. Front Mol Neurosci. 2015;8:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Miron VE, Boyd A, Zhao J-W, et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci. 2013;16(9):1211–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bogie JFJ, Stinissen P, Hellings N, et al. Myelin-phagocytosing macrophages modulate autoreactive T cell proliferation. J Neuroinflammation. 2011;8:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Boven LA, Van Meurs M, Van Zwam M, et al. Myelin-laden macrophages are anti-inflammatory, consistent with foam cells in multiple sclerosis. Brain. 2006;129(2):517–526. [DOI] [PubMed] [Google Scholar]
- [5].Hikawa N, Takenaka T. Myelin-stimulated macrophages release neurotrophic factors for adult dorsal root ganglion neurons in culture. Cell Mol Neurobiol. 1996;16:517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bogie JFJ, Haidar M, Kooij G, et al. Fatty acid metabolism in the progression and resolution of CNS disorders. Adv Drug Deliv Rev. 2020;159:198–213. [DOI] [PubMed] [Google Scholar]
- [7].Ouimet M, Franklin V, Mak E, et al. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 2011;13(6):655–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature. 2009;458(7242):1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Liu X, Tang Y, Cui Y, et al. Autophagy is associated with cell fate in the process of macrophage-derived foam cells formation and progress. J Biomed Sci. 2016;23:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sergin I, Evans TD, Zhang X, et al. Exploiting macrophage autophagy-lysosomal biogenesis as a therapy for atherosclerosis. Nat Commun. 2017;8(1):15750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bogie JFJ, Grajchen E, Wouters E, et al. Stearoyl-CoA desaturase-1 impairs the reparative properties of macrophages and microglia in the brain. J Exp Med. 2020;217. DOI: 10.1084/jem.20191660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Grajchen E, Hendriks JJA, Bogie JFJ. The physiology of foamy phagocytes in multiple sclerosis. Acta Neuropathol Commun. 2018;6:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Razani B, Feng C, Coleman T, et al. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab. 2012;15(4):534–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sergin I, Bhattacharya S, Emanuel R, et al. Inclusion bodies enriched for p62 and polyubiquitinated proteins in macrophages protect against atherosclerosis. Sci Signal. 2016;9(409):ra2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Miceli C, Roccio F, Penalva-Mousset L, et al. The primary cilium and lipophagy translate mechanical forces to direct metabolic adaptation of kidney epithelial cells. Nat Cell Biol. 2020;22(9):1091–1102. [DOI] [PubMed] [Google Scholar]
- [17].Schulze RJ, Krueger EW, Weller SG, et al. Direct lysosome-based autophagy of lipid droplets in hepatocytes. Proc Natl Acad Sci U S A. 2020;117(51):32443–32452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Martinez-Lopez N, Garcia-Macia M, Sahu S, et al. Autophagy in the CNS and periphery coordinate lipophagy and lipolysis in the brown adipose tissue and liver. Cell Metab. 2016;23(1):113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jeong S-J, Stitham J, Evans TD, et al. Trehalose causes low-grade lysosomal stress to activate TFEB and the autophagy-lysosome biogenesis response. Autophagy. 2021;17(11):3740–3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Cantuti-Castelvetri L, Fitzner D, Bosch-Queralt M, et al. Defective cholesterol clearance limits remyelination in the aged central nervous system. Science. 2018;359(6376):684–688. [DOI] [PubMed] [Google Scholar]
- [21].Goldmann T, Wieghofer P, Jordão MJC, et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol. 2016;17(7):797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chrast R, Saher G, Nave K-A, et al. Lipid metabolism in myelinating glial cells: lessons from human inherited disorders and mouse models. J Lipid Res. 2011;52:419–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Karamita M, Barnum C, Möbius W, et al. Therapeutic inhibition of soluble brain TNF promotes remyelination by increasing myelin phagocytosis by microglia. JCI Insight. 2017;2(8). DOI: 10.1172/jci.insight.87455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lan M, Tang X, Zhang J, et al. Insights in pathogenesis of multiple sclerosis: nitric oxide may induce mitochondrial dysfunction of oligodendrocytes. Rev Neurosci. 2018;29:39–53. [DOI] [PubMed] [Google Scholar]
- [25].Makinodan M, Ikawa D, Miyamoto Y, et al. Social isolation impairs remyelination in mice through modulation of IL-6. FASEB J. 2016;30(12):4267–4274. [DOI] [PubMed] [Google Scholar]
- [26].Vela JM, Molina-Holgado E, Arévalo-Martín A, et al. Interleukin-1 regulates proliferation and differentiation of oligodendrocyte progenitor cells. Mol Cell Neurosci. 2002;20:489–502. [DOI] [PubMed] [Google Scholar]
- [27].Kim KH, Lee M-S. Autophagy—a key player in cellular and body metabolism. Nat Rev Endocrinol. 2014;10(6):322–337. [DOI] [PubMed] [Google Scholar]
- [28].Liao X, Sluimer J, Wang Y, et al. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab. 2012;15(4):545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Yang L, Li P, Fu S, et al. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11:467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Inami Y, Yamashina S, Izumi K, et al. Hepatic steatosis inhibits autophagic proteolysis via impairment of autophagosomal acidification and cathepsin expression. Biochem Biophys Res Commun. 2011;412(4):618–625. [DOI] [PubMed] [Google Scholar]
- [31].Koga H, Kaushik S, Cuervo AM. Altered lipid content inhibits autophagic vesicular fusion. Faseb J. 2010;24:3052–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zechner R, Madeo F, Kratky D. Cytosolic lipolysis and lipophagy: two sides of the same coin. Nat Rev Mol Cell Biol. 2017;18:671–684. [DOI] [PubMed] [Google Scholar]
- [33].Schott MB, Weller SG, Schulze RJ, et al. Lipid droplet size directs lipolysis and lipophagy catabolism in hepatocytes. J Cell Biol. 2019;218(10):3320–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Liu Y, Wang J, Hsiung GR, et al. Trehalose inhibits aβ generation and plaque formation in alzheimer’s disease. Mol Neurobiol. 2020;57:3150–3157. [DOI] [PubMed] [Google Scholar]
- [35].Lu J, Wu M, Autophagy YZ. Parkinson’s disease. Adv Exp Med Biol. 2020;1207:21–51. [DOI] [PubMed] [Google Scholar]
- [36].Shi CS, Shenderov K, Huang NN, et al. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13:255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zhong Z, Umemura A, Sanchez-Lopez E, et al. NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell. 2016;164(5):896–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zhang M, Kenny SJ, Ge L, et al. Translocation of interleukin-1β into a vesicle intermediate in autophagy-mediated secretion. Elife. 2015;4. DOI: 10.7554/eLife.11205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Meer F, Scheltema RA, Mollenkopf H-J, et al. Direct proteomic quantification of the secretome of activated immune cells. Science. 2013;340:475–478. [DOI] [PubMed] [Google Scholar]
- [40].Wong SW, Sil P, Martinez J. Rubicon: LC3-associated phagocytosis and beyond. Febs J. 2018;285:1379–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wu MY, Lu JH. Autophagy and macrophage functions: inflammatory response and phagocytosis. Cells. 2019;9:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Martinez J, Almendinger J, Oberst A, et al. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Nat Acad Sci. 2011;108:17396–17401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Berglund R, Guerreiro-Cacais AO, Adzemovic MZ, et al. Microglial autophagy–associated phagocytosis is essential for recovery from neuroinflammation. Sci Immunol. 2020;5(52):eabb5077. [DOI] [PubMed] [Google Scholar]
- [44].Samie M, Cresswell P. The transcription factor TFEB acts as a molecular switch that regulates exogenous antigen-presentation pathways. Nat Immunol. 2015;16:729–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Crotzer VL, Blum JS. Autophagy and its role in MHC-mediated antigen presentation. J Immunol. 2009;182:3335–3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Rodriguez-Navarro JA, Rodríguez L, Casarejos MJ, et al. Trehalose ameliorates dopaminergic and tau pathology in parkin deleted/tau overexpressing mice through autophagy activation. Neurobiol Dis. 2010;39(3):423–438. [DOI] [PubMed] [Google Scholar]
- [47].Tanaka M, Machida Y, Niu S, et al. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat Med. 2004;10(2):148–154. [DOI] [PubMed] [Google Scholar]
- [48].Norton WT, Poduslo SE. Myelination in rat brain: method of myelin isolation. J Neurochem. 1973;21:749–757. [DOI] [PubMed] [Google Scholar]
- [49].Bogie JF, Timmermans S, Huynh-Thu VA, et al. Myelin-derived lipids modulate macrophage activity by liver X receptor activation. PLoS One. 2012;7:e44998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Bretschneider A, Burns W, Morrison A. “Pop-off” technic. The ultrastructure of paraffin-embedded sections. Am J Clin Pathol. 1981;76:450–453. [DOI] [PubMed] [Google Scholar]
- [51].Ylä-Anttila P, Vihinen H, Jokitalo E, et al. Monitoring autophagy by electron microscopy in Mammalian cells. Methods Enzymol. 2009;452:143–164. [DOI] [PubMed] [Google Scholar]
- [52].Bogie JF, Jorissen W, Mailleux J, et al. Myelin alters the inflammatory phenotype of macrophages by activating PPARs. Acta Neuropathol Commun. 2013;1(1):43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Carpenter AE, Jones TR, Lamprecht MR, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7(10):R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9(7):676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Peng H, Bria A, Zhou Z, et al. Extensible visualization and analysis for multidimensional images using Vaa3D. Nat Protoc. 2014;9:193–208. [DOI] [PubMed] [Google Scholar]
- [56].Nelissen K, Smeets K, Mulder M, et al. Selection of reference genes for gene expression studies in rat oligodendrocytes using quantitative real time PCR. J Neurosci Methods. 2010;187:78–83. [DOI] [PubMed] [Google Scholar]
- [57].Vandesompele J, De Preter K, Pattyn F, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3(7):research0034.0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Meffre D, Massaad C, Grenier J. Lithium chloride stimulates PLP and MBP expression in oligodendrocytes via Wnt/β-catenin and Akt/CREB pathways. Neuroscience. 2015;284:962–971. [DOI] [PubMed] [Google Scholar]
- [59].Hussain R, El-Etr M, Gaci O, et al. Progesterone and nestorone facilitate axon remyelination: a role for progesterone receptors. Endocrinology. 2011;152(10):3820–3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.