

Abstract
Gain-of-function mutations in isocitrate dehydrogenase (IDH) in human cancers result in the production of d-2-hydroxyglutarate (d-2HG), an oncometabolite that promotes tumorigenesis through epigenetic alterations. The cancer cell–intrinsic effects of d-2HG are well understood, but its tumor cell–nonautonomous roles remain poorly explored. We compared the oncometabolite d-2HG with its enantiomer, l-2HG, and found that tumor-derived d-2HG was taken up by CD8+ T cells and altered their metabolism and antitumor functions in an acute and reversible fashion. We identified the glycolytic enzyme lactate dehydrogenase (LDH) as a molecular target of d-2HG. d-2HG and inhibition of LDH drive a metabolic program and immune CD8+ T cell signature marked by decreased cytotoxicity and impaired interferon-γ signaling that was recapitulated in clinical samples from human patients with IDH1 mutant gliomas.
The discovery of mutations in genes encoding key metabolic enzymes has highlighted a direct link between altered metabolism and disease. In particular, the discovery of mutations in fumarate hydratase, succinate dehydrogenase, and isocitrate dehydrogenase (IDH), all enzymes of the tricarboxylic acid cycle, has uncovered how mitochondrial metabolites can accumulate and act as disease-driving signaling molecules, becoming bona fide oncometabolites (1). Beyond their established intracellular functions, oncometabolites have tumor cell–nonautonomous roles and mediate intercellular cross talk in the tumor microenvironment (TME).
The oncometabolite d-2-hydroxyglutarate (d-2HG) is produced by cancers with gain-of-function mutations in IDH1 and IDH2 and accumulates to levels up to ~30 mM in the surrounding TME (2–8). d-2HG is an inhibitor of many alpha ketoglutarate (αKG)–dependent dioxygenases, leading to slow accumulating alterations in the epigenetic landscape of cells that result in oncogenic transformation (9–12). Despite our understanding of the cancer cell–intrinsic regulation of epigenetics by d-2HG, the tumor cell–nonautonomous effects of d-2HG in the TME still remain poorly understood. IDH-mutant gliomas have fewer tumor-infiltrating lymphocytes, including T cells, compared with their wild-type (WT) counterparts, suggesting that d-2HG promotes the establishment of an immunosuppressive TME (7, 13–16). However, the mechanisms by which d-2HG alters antitumor immunity remain unknown.
We focused on CD8+ T cells, which are a major component of the adaptive immune response and specialize in the direct killing of malignant cells, to investigate the impact of tumor-derived d-2HG on antitumor immunity.
Results
Acute and reversible inhibition of CD8+ T cell proliferation, cytotoxicity, and interferon-γ signaling by d-2HG
To evaluate the tumor cell–nonautonomous mechanisms by which d-2HG influences antitumor immunity, we examined the effects of the oncometabolite on the function of murine CD8+ T cells. We compared d-2HG with its enantiomer, l-2HG, a metabolic by-product of enzyme promiscuity that supports T cell survival and function (17, 18). To determine whether pathophysiological concentrations of d-2HG in the TME of IDH-mutant tumors (2–8) permit cellular uptake by CD8+ T cells, we treated activated CD8+ T cells with various doses of non-cell-permeable d-2HG and l-2HG and monitored uptake by liquid chromatography–mass spectrometry. Both enantiomers were taken up rapidly and in similar quantities when supplied in millimolar concentrations (Fig. 1A and fig. S1A). Next, we quantified the intracellular levels of each enantiomer. When 20 mM d-2HG or l-2HG was added, we estimated that the intracellular concentration of 2HG reached low millimolar levels (Fig. 1B). At this concentration, neither enantiomer had a detrimental effect on cell viability over a 6-day time course (fig. S1B).
Fig. 1. d-2HG impairs CD8+ T cell proliferation, cytotoxicity, and IFN-γ signaling in an acute and reversible fashion.
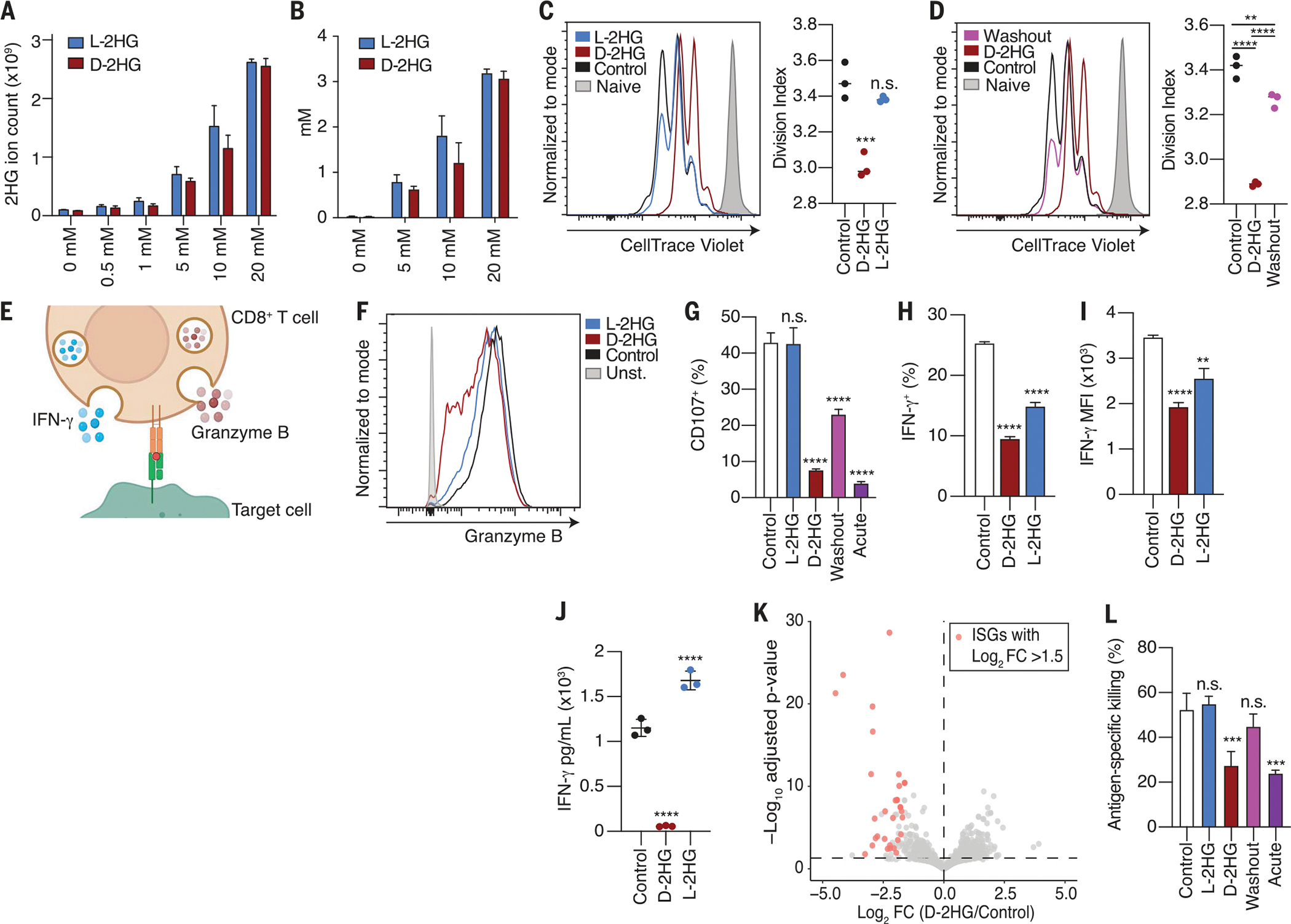
(A) Total 2HG levels in activated CD8+ T cells after 24-hour treatment with increasing concentrations of d-2HG or l-2HG. Total ion count was normalized to cell number (n = 3). (B) Intracellular 2HG concentration determined by assuming a T cell volume of 500 fl (n = 3). (C) Left: CellTrace Violet dilution assay at day 3 of CD8+ T cells activated by CD3/CD28 mAbs in the presence of 20 mM d-2HG, 20 mM l-2HG, or left untreated. Stained, naive cells (shaded gray) that did not proliferate are shown. Right: Division index corresponds to the average number of cell divisions that a cell in the total cell population has undergone (n = 3). (D) CellTrace Violet dilution assay according to experimental design described in fig. S1E to assess reversibility of proliferation phenotype. (E) Schematic of a cytotoxic CD8+ T cell releasing granzyme B and IFN-γ. (F) Intracellular granzyme B levels of CD8+ T cells activated by CD3/CD28 mAbs in the presence of 20 mM d-2HG, 20 mM l-2HG, or left untreated for 3 days. (G) Percentage degranulation as assessed by CD107a/b staining of CD8+ T cells activated by CD3/CD28 mAbs and restimulated by CD3 mAb in the presence of 20 mM d-2HG, 20 mM l-2HG, or left untreated. In the washout condition, CD8+ T cells were activated in the presence of 20 mM d-2HG, and the oncometabolite was subsequently washed out before restimulation. In the acute condition, d-2HG was added solely at the time of restimulation. In all other conditions, the metabolites were kept for the entirety of the assay (n = 3). (H) Percentage of IFN-γ+ CD8+ T cells after intracellular cytokine staining of CD8+ T cells activated with phorbol myristate acetate (PMA) and ionomycin for 4 hours in the presence of 20 mM d-2HG, 20 mM l-2HG, or left untreated (n = 3). (I) Mean fluorescent intensity for IFN-γ after intracellular cytokine staining of CD8+ T cells activated with PMA and ionomycin for 4 hours in the presence of 20 mM d-2HG, 20 mM l-2HG, or left untreated (n = 3). (J) IFN-γ levels in the medium of CD8+ T cells activated with 20 mM d-2HG, 20 mM l-2HG, or left untreated for 24 hours (n = 3). (K) Volcano plot showing distribution of the top down-regulated and up-regulated genes in CD8+ T cells activated in the presence of 20 mM d-2HG or left untreated. Statistically significant ISGs with a fold change >1.5 are marked in pink (n = 3). (L) Antigen-specific killing of B16 ovalbumin-positive tumor cells by OT1 CD8+ T cells that were activated in the presence of 20 mM d-2HG, 20 mM l-2HG, or left untreated. In the washout condition, d-2HG was removed before co-culture seeding of tumor cells and T cells. In the acute condition, d-2HG was added at the time of tumor cell and T cell co-culture. In all other conditions, the metabolites were kept for the entirety of the assay (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 (one-way and two-way ANOVA). Data are representative of at least two independent experiments.
Upon entering the TME of IDH-mutant tumors, infiltrating CD8+ T cells undergo local expansion and reactivation in the presence of high amounts of d-2HG. Therefore, we evaluated the effects of d-2HG or l-2HG on the activation and proliferation of CD8+ T cells. After stimulation with anti-CD3/CD28 antibodies, CD8+ T cells fully activated in the presence of either enantiomer, as assessed by cell surface expression of the activating markers CD69 and CD25 (fig. S1C). Although both enantiomers achieved similar intracellular concentrations, d-2HG was a more potent inhibitor of T cell proliferation than l-2HG (Fig. 1C). The inhibitory effects of d-2HG on proliferation were dose dependent and observed only at concentrations sufficient for cellular uptake (fig. S1D). Removing d-2HG from the culture medium alleviated this proliferative block (Fig. 1D and fig. S1E), demonstrating that the effects of d-2HG on proliferation are reversible.
CD8+ T cells can eliminate target cells through exocytosis of preformed lysosomes containing cytotoxic proteins such as granzyme B (Fig. 1E) (19). After T cell activation, intracellular granzyme B levels were decreased in the presence of d-2HG (Fig. 1F). During the process of degranulation, the membrane of granzyme-containing lysosomes fuses with the plasma membrane of activated CD8+ T lymphocytes to allow for cargo release (20). To evaluate degranulation, we assessed the cycling of lysosomal-associated membrane protein 1 and 2 (LAMP-1 and LAMP-2, also known as CD107a and CD107b) to the cell surface in response to T cell receptor signaling (21). CD8+ T cells activated in the presence of d-2HG failed to degranulate upon antigen restimulation in a dose-dependent manner (fig. S1, F, G, and I, and Fig. 1G). The total amounts of intracellular LAMP-1 were not affected by d-2HG treatment (fig. S1H). Given these observations, we then investigated whether the loss of LAMP-1/2 surface expression could be attributed to an impairment in granule formation at the time of ex vivo activation (day 0) or a defect in degranulation at the time of restimulation (day 6). To test this, CD8+ T cells were activated in the presence of d-2HG for 6 days, and then d-2HG was removed at the time of restimulation. Washing away d-2HG at the time of restimulation partially restored the ability of CD8+ T cells to degranulate (fig. S1, F and I, and Fig. 1G, washout condition). Conversely, CD8+ T cells that were activated for 6 days and subsequently exposed to d-2HG only at the time of restimulation failed to degranulate (fig. S1, F and I, and Fig. 1G, acute condition). These data indicate that d-2HG leads to acute defects in CD8+ T cell degranulation by affecting processes involved in the formation of lytic effectors and the release of lytic contents.
Together with granzyme B, CD8+ T cells also secrete interferon-γ (IFN-γ), a cytokine that sensitizes tumor cells to killing by T cells (Fig. 1E) (22). After stimulation, d-2HG–treated CD8+ T cells exhibited a dose-dependent defect in IFN-γ production, as determined by decreased IFN-γ intracellular staining and reduced release of the cytokine into the medium (Fig. 1, H to J, and fig. S1J). IFN-γ controls specific gene expression programs of hundreds of genes (23). Therefore, we performed gene expression profiling by RNA sequencing of CD8+ T cells activated in the presence of d-2HG or l-2HG or left untreated. Gene Ontology (GO) analysis showed that most of the down-regulated genes upon d-2HG treatment belonged to the class of IFN-γ–stimulated genes (ISGs) (Fig. 1K and fig. S1, K and L). Thus, CD8+ T cells exhibit a decrease in the IFN-γ–induced signature, reflecting a decrease in IFN-γ production and secretion after d-2HG treatment.
To assess whether the decreased production of cytotoxic proteins and effector cytokines had functional consequences, we probed the antigen-specific antitumor killing capacities of d-2HG– or l-2HG–treated cells in an in vitro killing assay. d-2HG–treated CD8+ T cells exhibited poor killing of cancer cells both when d-2HG was provided starting at the time of their activation and when it was administered to activated CD8+ T cells only at the time of co-culture with cancer cells (Fig. 1L and fig. S1, M to O). Thus, the effects of d-2HG on cytotoxicity are rapid and independent of upstream events involving T cell activation. Removal of d-2HG at the time of co-culture from CD8+ T cells that had been activated in the presence of d-2HG restored their antitumor killing capacities (Fig. 1L and fig. S1M, washout condition), indicating that d-2HG must be present at the time of tumor cell recognition to impair their killing. Thus, we conclude that d-2HG acutely impairs the antitumor killing properties on CD8+ T cells.
d-2HG alters glycolysis in CD8+ T cells
Because of their acute and reversible nature, the effects of d-2HG on CD8+ T cell proliferation, cytokine production, and cytotoxicity appear to be independent of its established role as an epigenetic regulator. In support of this, acute treatment with d-2HG did not result in epigenetic reprogramming of T cells (fig. S2A). We therefore investigated whether d-2HG had an effect on metabolism. First, we tested whether either enantiomer could be metabolized. The chiral-specific reduction of 2HG to αKG is mediated by d-2HG and l-2HG dehydrogenase (d-2HGDH and l-2HGDH, respectively) (24). In agreement with the literature (25), Western blot analysis demonstrated undetectable amounts of d-2HGDH and l-2HGDH in CD8+ T cells (fig. S2B). Furthermore, stable isotope tracing of 13C5 d-2HG or l-2HG did not show evidence of l-2HG and d-2HG metabolism over a 48-hour time course, despite achieving steady-state isotopic enrichment (fig. S2, C to E). We conclude that d-2HG and l-2HG are not readily metabolized in CD8+ T cells.
Given that d-2HG and l-2HG led to distinct functional effects on CD8+ T cells, we performed steady-state metabolomics of intracellular metabolites and metabolites from cell culture medium after acute 2HG treatment with the goal of identifying metabolic pathways that were differentially altered by the two enantiomers (Fig. 2A). The metabolic pathway most altered upon d-2HG treatment was glycolysis. d-2HG, but not l-2HG, promoted the accumulation of many glycolytic intermediates (Fig. 2B and fig. S2F). Consistent with altered glucose utilization, we also observed increased pyruvate and decreased lactate in the medium of d-2HG–treated CD8+ T cells (Fig. 2C and fig. S2G). l-2HG stabilizes hypoxia-inducible factor-1α (HIF-1α), which is a master transcriptional regulator of glycolysis (17, 26, 27). Therefore, we assessed whether the reported changes in glucose catabolism could be attributed to d-2HG–mediated HIF-1α regulation. We found that d-2HG, unlike its enantiomer l-2HG (28), did not promote HIF-1α stability in T cells (fig. S2H). This result was further confirmed by quantitative polymerase chain reaction analysis, showing no major changes in the expression of genes involved in glucose utilization upon d-2HG treatment (fig. S2I). Thus, we conclude that d-2HG alters glucose catabolism through an HIF-1α–independent mechanism.
Fig. 2. d-2HG alters glycolysis in CD8+ T cells.
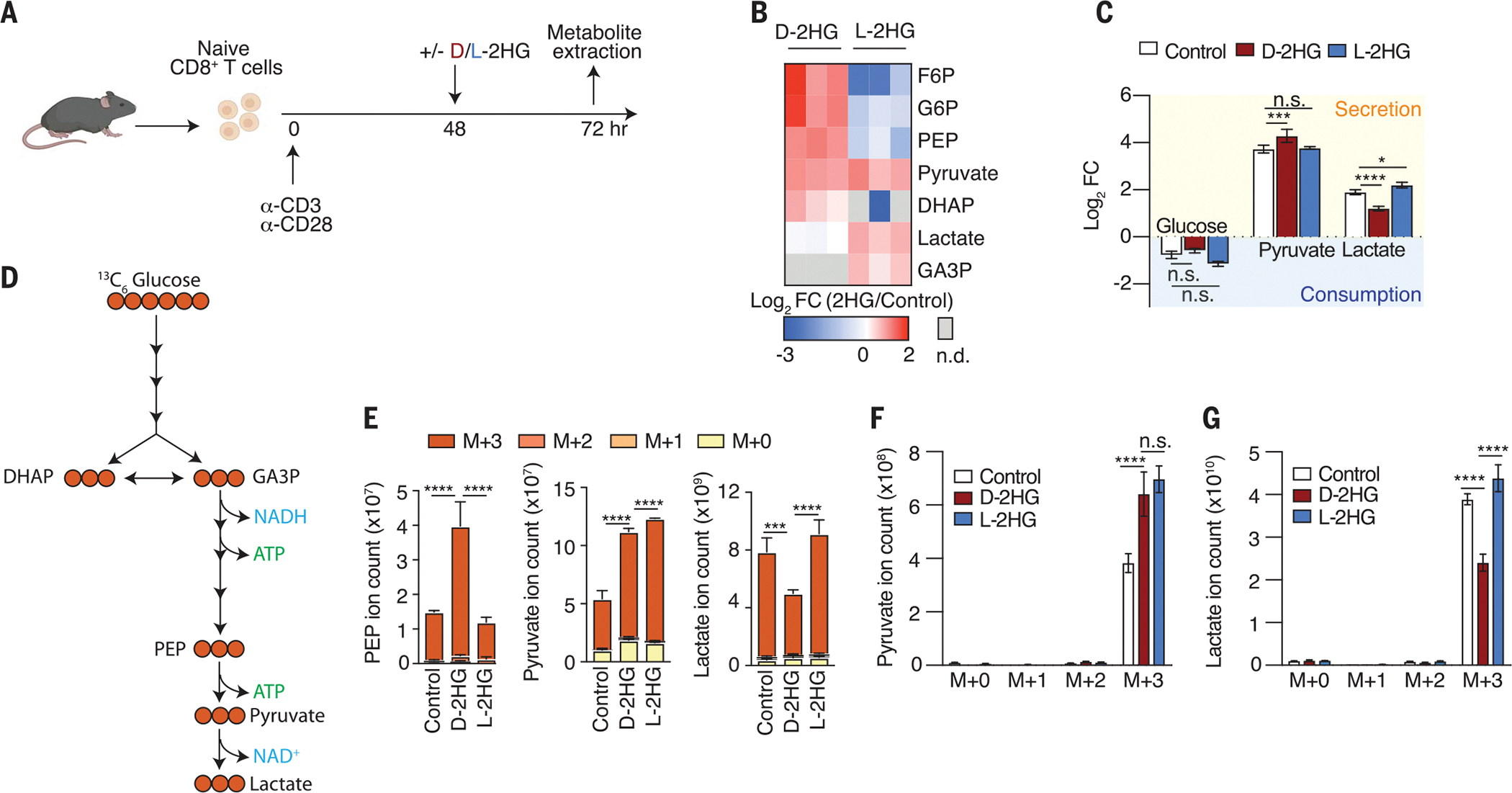
(A) Schematic of experimental design to assess the acute (24 hours) effects of 20 mM d-2HG and 20 mM l-2HG on steady-state metabolite levels in activated CD8+ T cells. (B) Log2 fold changes of glycolytic intermediates in 20 mM d-2HG–treated versus 20 mM l-2HG–treated CD8+ T cells relative to control. F6P, fructose 6-phosphate; G6P, glucose 6-phosphate; DHAP, dihydroxyacetone phosphate; GA3P, glyceraldehyde 3-phosphate; PEP, phosphoenolpyruvic acid (n = 3). (C) Log2 fold changes of key glycolytic metabolites that are secreted into or consumed from the medium of d-2HG–treated, l-2HG–treated, or untreated CD8+ T cells (n = 3). (D) Schematic of expected incorporation of heavy carbons into glycolytic intermediates after 13C6 glucose is provided for 24 hours. (E) Ion intensities of glycolytic intermediates and their respective 13C-isotopologs after 24-hour cotreatment of 13C6 glucose with 20 mM d-2HG, 20 mM l-2HG, or control (n = 3). (F) Levels of secreted pyruvate isotopologs in the medium of 20 mM d-2HG–treated, 20 mM l-2HG–treated, and untreated CD8+ T cells after providing 13C6 glucose for 24 hours (n = 3). (G) Levels of secreted lactate isotopologs in the medium of 20 mM d-2HG–treated, 20 mM l-2HG–treated, and untreated CD8+ T cells after providing 3C6 glucose for 24 hours (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 (two-way ANOVA). Data are representative of at least two independent experiments.
To further investigate how glycolytic flux is altered in the presence of d-2HG and l-2HG, we performed tracing experiments with 13C6 glucose (Fig. 2D). Consistent with our steady-state metabolomic profiles, we observed increased labeling in glycolytic intermediates preceding lactate and a decrease in labeling of lactate (Fig. 2E). These results were confirmed by metabolite analysis in the cell culture medium. We detected increased pyruvate (M+3) accumulation and decreased lactate (M+3) secretion upon d-2HG treatment (Fig. 2, F and G). Collectively, these findings demonstrate that d-2HG, and not l-2HG, strongly inhibits glycolysis in CD8+ T cells.
d-2HG inhibits LDH activity in CD8+ T cells and lowers the NAD+/NADH ratio
Pyruvate to lactate interconversion is catalyzed by the enzyme LDH (Fig. 3A) (29). The dose-dependent decrease in pyruvate to lactate conversion, as measured by the ratio of lactate (M+3) to pyruvate (M+3) upon 13C6 glucose tracing (Fig. 3B and fig. S3A), led us to test whether LDH activity is impaired in the presence of d-2HG. We treated CD8+ T cells with antimycin A, an inhibitor of the electron transport chain (ETC) that limits mitochondria’s ability to generate ATP and increases reliance on glycolysis to support the bioenergetic needs of a cell (Fig. 3C) (30). In response to antimycin A, activated CD8+ T cells induced pyruvate to lactate conversion, as indicated by an increase in the lactate/pyruvate ratio. By contrast, this induction was significantly suppressed in d-2HG–treated CD8+ T cells (Fig. 3D), supporting the model that LDH function is compromised in the presence of d-2HG.
Fig. 3. d-2HG inhibits LDH-A activity in vitro and in CD8+ T cells.
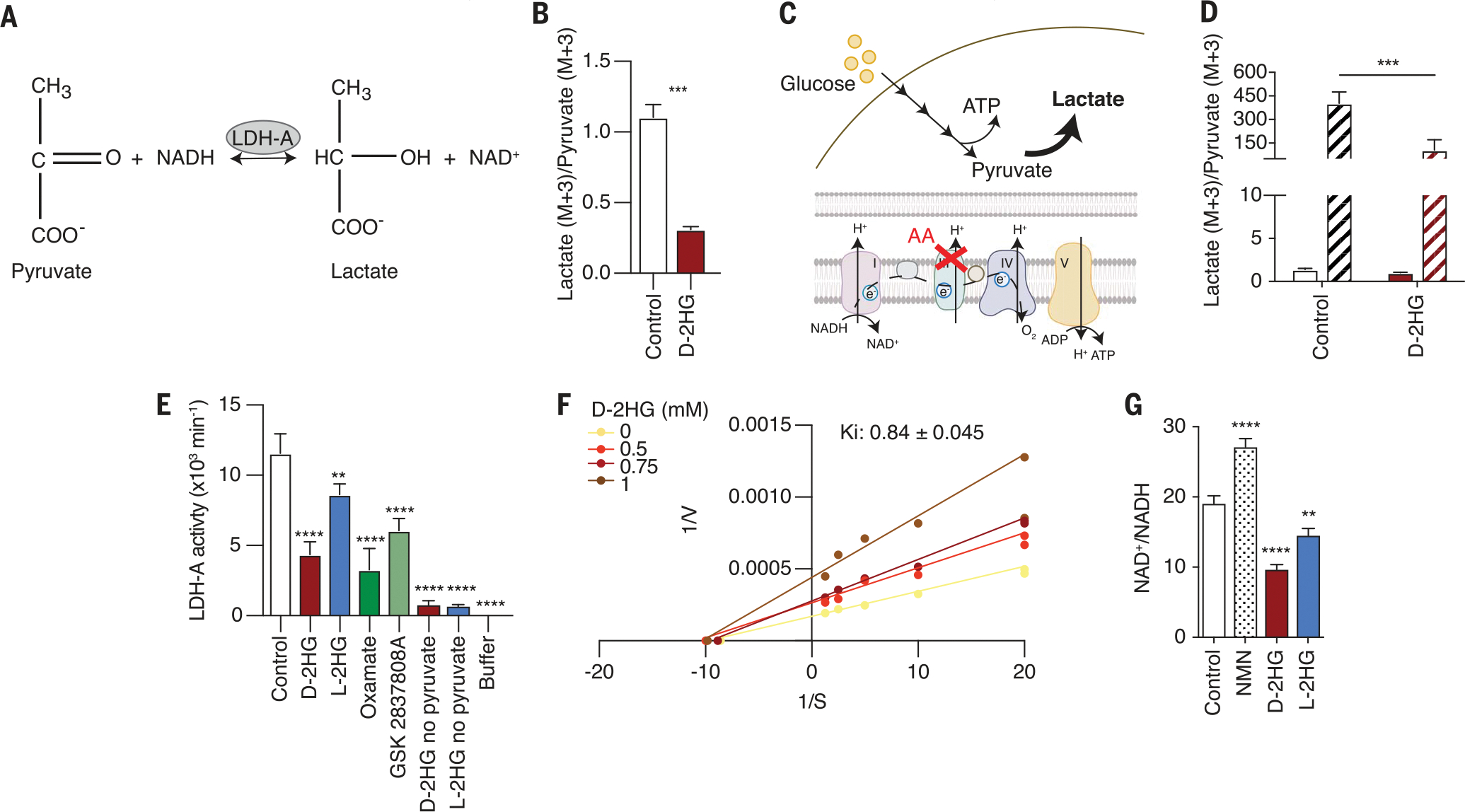
(A) Schematic of LDH reaction. (B) Intracellular lactate (M+3)/pyruvate (M+3) ratio of CD8+ T cells after 24-hour cotreatment of 13C6 glucose with 20 mM d-2HG or control (n = 3). (C) Schematic of the metabolic consequences of antimycin A (AA) treatment on glucose catabolism. (D) Intracellular lactate (M+3)/pyruvate (M+3) ratio after brief 20 mM d-2HG pretreatment, followed by AA and 13C6 glucose cotreatment (n = 3). Solid bars, −AA; striped bars, +AA. (E) In vitro enzymatic assessment of 3 mM l-2HG, 3 mM d-2HG, 80 μM oxamate, and 10 nM GSK2837808A on LDH-A activity. Oxamate and GSK2837808A are known LDH inhibitors and were used as a control (n = 3). (F) Lineweaver-Burk plot for d-2HG. (G) NAD+/NADH ratio of CD8+ T cells treated for 24 hours with control, 1 mM nicotinamide mononucleotide (NMN), 20 mM d-2HG, or 20 mM l-2HG. NMN raises intracellular NAD+ levels and was therefore used as a positive control (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 (Student’s t test, one-way and two-way ANOVA). Data are representative of at least two independent experiments.
To determine whether d-2HG is a direct inhibitor of LDH activity, we used purified recombinant LDH-A protein, the LDH isozyme expressed by T cells, and measured its activity in the presence or absence of d-2HG or l-2HG. Using the intracellular 2HG concentration previously determined (3 mM; Fig. 1B), we observed that d-2HG strongly inhibited LDH-A activity in vitro (Fig. 3E and fig. S3, B and C). LDH-B was also inhibited by d-2HG, suggesting that the inhibitory properties of d-2HG are not isozyme specific (fig. S3D). Although our tracing studies did not provide strong evidence for l-2HG–mediated inhibition of LDH, our in vitro kinetic assay revealed that l-2HG also inhibits, albeit less effectively, LDH-A activity (Fig. 3E and fig. S3, E and F). We determined d-2HG and l-2HG to be noncompetitive inhibitors of LDH-mediated pyruvate reduction, with inhibition constant (Ki) values of 840 ± 0.045 μM and 3.58 ± 0.206 mM, respectively (Fig. 3F and fig. S3G). Given that the reported Ki value for d-2HG is below the estimated intracellular d-2HG concentration, we inhibit LDH activity in CD8+ T cells.
A critical function of the LDH reaction is the regeneration of cytosolic NAD+ to support the continued functioning of glycolysis (31). d-2HG treatment lowered the NAD+/NADH ratio of the cell (Fig. 3G). Thus, our data indicate that d-2HG inhibits LDH activity in CD8+ T cells, resulting in altered glycolytic flux and NAD(H) balance.
d-2HG treatment induces CD8+ T cells to maximize mitochondrial respiration
Upon activation, T cells undergo a switch from oxidative phosphorylation to glycolysis to support their proliferative and effector functions (32). We sought to investigate how d-2HG affects this metabolic switch. We tested the source (mitochondrial or glycolytic) of cellular ATP in d-2HG–treated CD8+ T cells using the Seahorse XF Real-Time ATP rate assay. Consistent with our model of d-2HG–mediated LDH inhibition, d-2HG–treated CD8+ T cells relied less on glycolysis and more on oxidative phosphorylation for ATP production than untreated cells (Fig. 4A). Furthermore, we observed an increased oxygen consumption rate and decreased extracellular acidification rate upon d-2HG treatment (Fig. 4B and fig. S4, A to C).
Fig. 4. Cytosolic NAD(H) imbalance drives mitochondrial membrane hyperpolarization in d-2HG–treated CD8+ T cells.
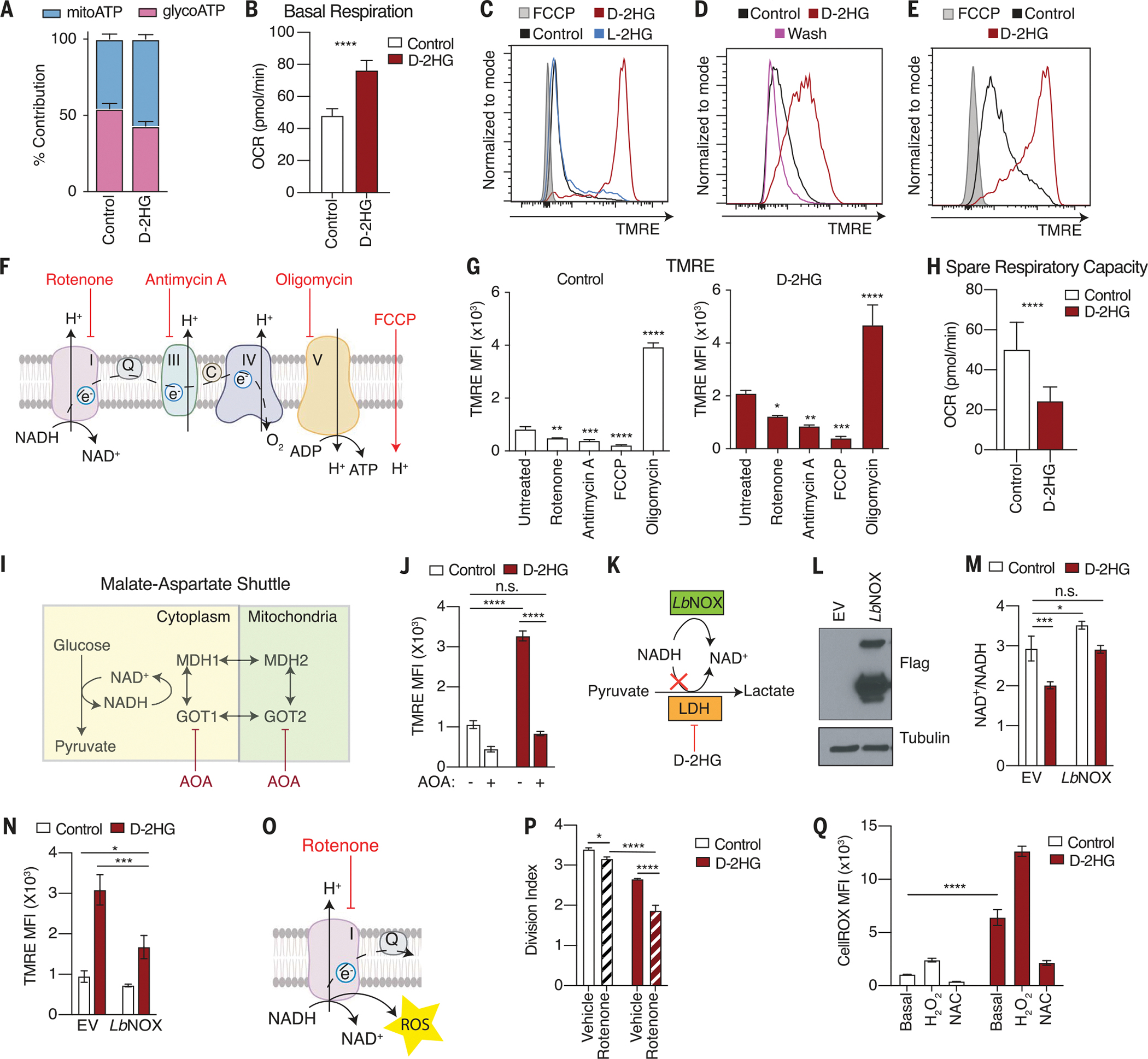
(A) Relative contribution of mitochondrial oxidative phosphorylation and glycolysis to ATP production in response to d-2HG after 24 hours of treatment (n = 10). (B) Quantification of basal respiration in response to 20 mM d-2HG after 24 hours of treatment (n = 10). (C) Mitochondrial membrane potential as assessed by tetramethylrhodamine ethyl ester (TMRE) fluorescence in CD8+ T cells treated with 20 mM d-2HG, 20 mM l-2HG, or control for 3 days. (D) TMRE assay according to experimental design described in fig. S4F. (E) Mitochondrial membrane potential as assessed by TMRE fluorescence in CD8+ T cells treated with 20 mM d-2HG, 20 mM l-2HG, or control for 2 hours. (F) Schematic of targets of rotenone, AA, oligomycin, and FCCP. (G) Effects of inhibition of ETC complexes on mitochondrial membrane potential in control and d-2HG–treated CD8+ T cells (n = 3). (H) Quantification of spare respiratory capacity in response to 20 mM d-2HG after 24 hours of treatment (n = 10). (I) Schematic of targets of AOA. (J) Mitochondrial membrane potential as assessed by TMRE fluorescence in CD8+ T cells treated with 20 mM d-2HG or control for 24 hours in the presence or absence of 1 mM AOA (n = 3). (K) Schematic of LbNOX mechanism of action. (L) Western blot of CD8+ T cells overexpressing empty vector (EV) or FLAG-tagged cytosolic LbNOX. (M) NAD+/NADH ratio of CD8+ T cells overexpressing EV or cytosolic LbNOX after a 24-hour-long treatment with 20 mM d-2HG or control (n = 3). (N) TMRE staining of EV or LbNOX-overexpressing CD8+ T cells treated with 20 mM d-2HG or left untreated for 24 hours (n = 3). (O) Schematic of the target of rotenone. (P) Quantification of division index of CD8+ T cells activated in the presence or absence of 20 mM d-2HG for 3 days and cotreated with 1 nM rotenone (n = 3). (Q) Quantification of intracellular ROS levels as assessed by CellROX staining in CD8+ T cells treated with 20 mM d-2HG or left untreated for 1 day. Hydrogen peroxide (H2O2, 100 mM) and 10 mM NAC were used as positive and negative controls, respectively (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 (Student’s t test, one-way and two-way ANOVA). Data are representative of at least two independent experiments.
To study how d-2HG increases reliance on mitochondrial function in response to d-2HG, we assessed mitochondrial function by measuring changes in the mitochondrial membrane potential (MMP). d-2HG caused hyperpolarization of the MMP in a dose-dependent manner (Fig. 4C and fig. S4D). Kinetics studies revealed that the membrane of d-2HG–treated mitochondria remained hyperpolarized over several days after T cell activation (fig. S4E). To determine whether the increase in MMP by d-2HG was reversible, we provided d-2HG to CD8+ T cells for 2 days and then removed it (fig. S4F). Removing d-2HG for as little as 1 hour was sufficient to completely restore the MMP (Fig. 4D). Conversely, a 2-hour exposure to d-2HG was sufficient to promote mitochondrial membrane hyperpolarization (Fig. 4E). The increase in MMP did not result from an increase in mitochondrial number, because d-2HG treatment did not alter the mitochondrial to nuclear DNA ratio or the expression of subunits of the ETC (fig. S4, G and H). Thus, d-2HG hyperpolarizes the mitochondrial membrane and increases mitochondrial respiration in an acute and reversible manner.
The MMP is maintained by the activity of the proton-pumping complexes (I, III, and IV) of the ETC (33). To determine whether the increase in MMP upon d-2HG treatment was due to increased complex activity, we used a series of inhibitors against various ETC complexes (Fig. 4F). In both untreated and d-2HG–treated T cells, inhibition of complexes I and III by rotenone and antimycin A, respectively, lowered the MMP. Similarly, treatment with carbonyl cyanide ptrifluoromethoxyphenylhydrazone (FCCP), an uncoupling agent that collapses the proton gradient, decreased the MMP. Conversely, inhibition of complex V by oligomycin, which prevents proton reentry into the matrix, further increased the MMP (Fig. 4G). Despite respiring at a higher rate, the mitochondria of d-2HG–treated CD8+ T cells exhibited decreased maximal respiration and thus a decreased spare respiratory capacity, a measure of the cells’ ability to respond to increased energy demand or stress (Fig. 4H and fig. S4, I and J). These data indicate that the mitochondria of CD8+ T cells maximize mitochondrial respiration in response to d-2HG.
Cytosolic NAD(H) imbalance supports mitochondrial function in d-2HG–treated CD8+ T cells
LDH inhibition results in cytosolic accumulation of NADH, which can enter mitochondria and provide reducing equivalents for the ETC (34). We tested whether cytosolic NADH might be diverted to the mitochondria for oxidation in response to d-2HG. Because mitochondria are impermeable to NADH, cytosolic NADH transfer is facilitated by a variety of shuttles, one being the malate-aspartate shuttle (35). To prevent shuttling of cytosolic NADH to the mitochondria, we used aminooxyacetate (AOA), which blocks cytosolic and mitochondrial aspartate aminotransferase reactions (Fig. 4I), and measured the state of the MMP. Although d-2HG treatment promoted mitochondrial membrane hyperpolarization, simultaneous treatment with AOA prevented this effect (Fig. 4J).
To test the contribution of cytosolic NAD(H) imbalance to driving the increase in MMP, we overexpressed a cytosol-localized NADH oxidase from Lactobacillus brevis, LbNOX, which generates NAD+ from NADH independently of LDH activity (Fig. 4, K and L) (36). In the presence of LbNOX, the NAD+/NADH ratio of d-2HG–treated CD8+ T cells was increased (Fig. 4M). Cytosolic LbNOX overexpression lowered the MMP in the presence of d-2HG (Fig. 4N and fig. S4, K and L), indicating that cytosolic NAD+/NADH imbalance partially drives the MMP of the cell.
Because NADH is oxidized by complex I of the ETC, we tested whether interventions that limit NAD+ recycling in the mitochondria might exacerbate the effect of d-2HG treatment. We exposed d-2HG–treated CD8+ T cells to rotenone, a complex I inhibitor, and monitored the cells’ ability to proliferate (Fig. 4O). Although rotenone alone had a small effect on proliferation, the combination of rotenone and d-2HG impaired cell proliferation more strongly than d-2HG alone (Fig. 4P). Thus, NADH oxidation by complex I of the ETC appears to be important to sustain the proliferative potential of CD8+ T cells in the presence of d-2HG. Because complex I is the main source of reactive oxygen species (ROS) in the mitochondria (33, 37), we measured ROS and oxidative stress levels in response to d-2HG. d-2HG significantly increased intracellular ROS levels, which were decreased in cells exposed to the antioxidant N-acetyl-cysteine (Fig. 4Q and fig. S4M). Accordingly, d-2HG treatment was accompanied by a significant increase in total glutathione (GSSG) levels, resulting in a lower GSHreduced/GSSGoxidized ratio, which is indicative of cellular redox stress (fig. S4, N to P). Overall, these data support the idea that d-2HG increases dependency on mitochondrial complex I activity, resulting in higher ROS production.
LDH inhibition is sufficient to recapitulate the effects of d-2HG on CD8+ T cell metabolism and proliferation and IFN-γ signaling and cytotoxicity
To assess the contribution of LDH inhibition to the metabolic phenotypes caused by d-2HG, we tested whether LDH inhibition alone was sufficient to phenocopy treatment of cells with d-2HG using two well-established LDH inhibitors, oxamate and GSK-2837808A (abbreviated as GSK). 13C6 glucose tracing in the presence of either LDH inhibitor recapitulated the effects of d-2HG on glucose catabolism. The LDH inhibitors each promoted the accumulation of glycolytic intermediates up to pyruvate, with concomitant loss of lactate labeling (fig. S5, A and B), resulting in a lower lactate (M+3)/pyruvate (M+3) ratio (Fig. 5A). Inhibition of LDH activity by the inhibitors was further validated by metabolite analysis in cell culture medium, in which we observed increased pyruvate and decreased lactate secretion relative to that from untreated cells (fig. S5, C and D). Consistent with LDH inhibition, the NAD+/NADH ratio of the cell was also decreased in response to either of the inhibitors (Fig. 5B). Thus, oxamate and GSK recapitulate the effects of d-2HG on glucose catabolism.
Fig. 5. LDH inhibition recapitulates the effects of d-2HG on CD8+ T cell metabolism, proliferation, cytotoxicity, and IFN-γ signaling.
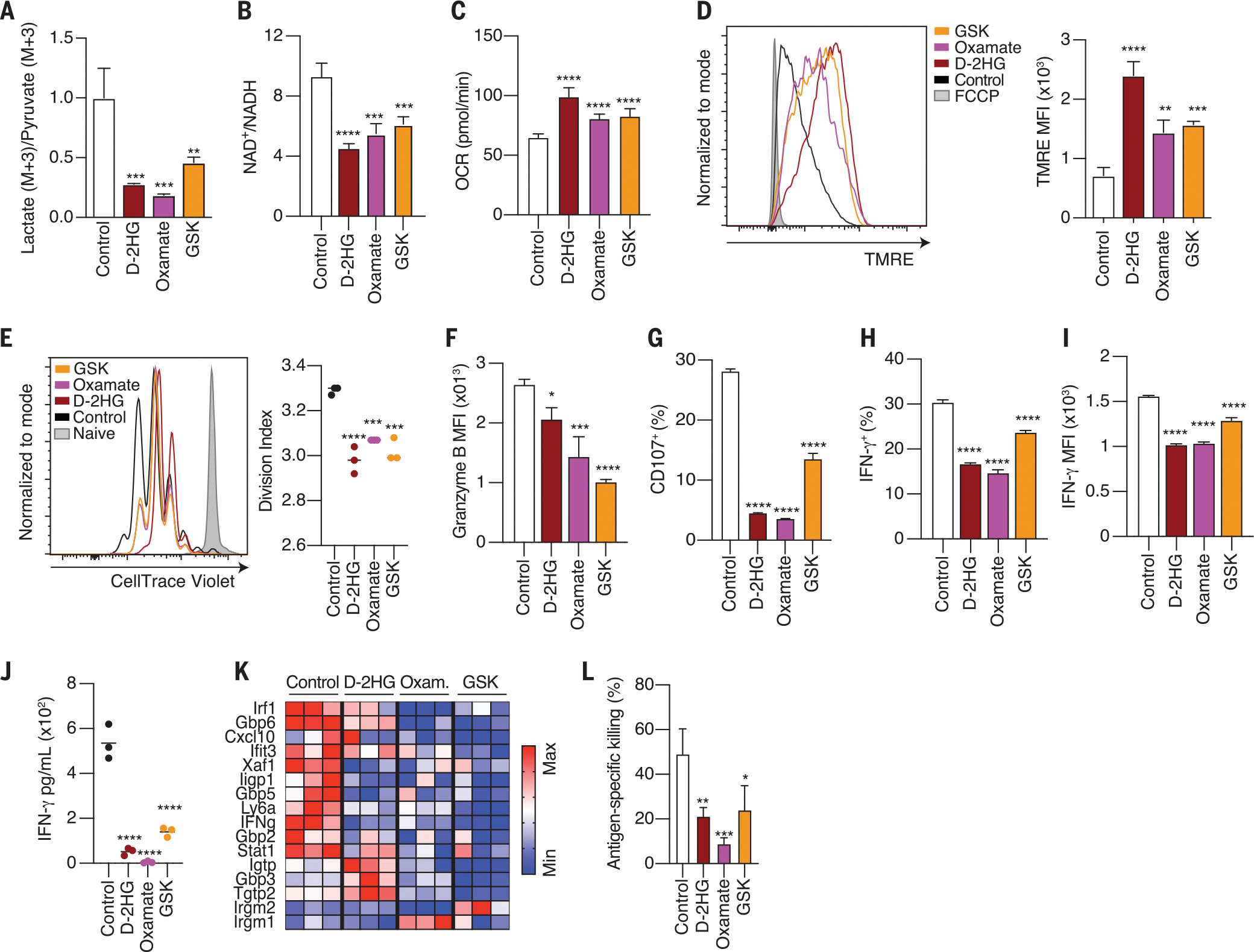
(A) Lactate (M+3)/pyruvate (M+3) ratio in CD8+ T cells treated with 20 mM d-2HG, 20 mM oxamate, 10 μM GSK2837808A, or left untreated for 24 hours (n = 3). (B) NAD+/NADH ratio of CD8+ T cells after a 24-hour-long treatment with 20 mM d-2HG, 20 mM oxamate, 10 μM GSK2837808A, or control (n = 3). (C) Quantification of basal respiration in response to 20 mM d-2HG, 20 mM oxamate, 10 μM GSK2837808A, or control after 24 hours of treatment (n = 10). (D) Left: Mitochondrial membrane potential as assessed by TMRE fluorescence in CD8+ T cells treated with 20 mM d-2HG, 20 mM oxamate, 10 μM GSK2837808A, or control for 24 hours. Right: quantification of mean fluorescence intensity for TMRE (n = 3). (E) Left: CellTrace Violet dilution assay at day 3 of CD8+ T cells activated in the presence of 20 mM d-2HG, 20 mM oxamate, 10 μM GSK2837808A, or left untreated. Right: quantification of division index (n = 3). (F) Quantification of intracellular granzyme B expression in CD8+ T cells activated in the presence of 20 mM d-2HG, 20 mM oxamate, 10 μM GSK2837808A, or control for 3 days (n = 3). (G) Percentage degranulation assessed by CD107a/b staining in CD8+ T cells activated in the presence of 20 mM d-2HG, 20 mM oxamate, 10 μM GSK2837808A, or left untreated (n = 3). (H) Percentage of IFN-γ+ CD8+ T cells after intracellular cytokine staining of CD8+ T cells activated with PMA and ionomycin for 4 hours in the presence of 20 mM d-2HG, 20 mM oxamate, 10 μM GSK2837808A, or left untreated (n = 3). (I) Mean fluorescent intensity for IFN-γ after intracellular cytokine staining of CD8+ T cells activated with PMA and ionomycin for 4 hours in the presence of 20 mM d-2HG, 20 mM oxamate, 10 μM GSK2837808A, or left untreated (n = 3). (J) Cell number–normalized levels of IFN-γ in the cell culture medium of CD8+ T cells activated in the presence of 20 mM d-2HG, 20 mM oxamate, 10 μM GSK2837808A, or control for 2 days (n = 3). (K) Relative mRNA expression of ISGs in CD8+ T cells activated in the presence of 20 mM d-2HG, 20 mM oxamate, 10 μM GSK2837808A, or control for 24 hours (n = 3). (L) Specific killing of B16 ovalbumin-positive tumor cells by OT1 CD8+ T cells activated in the presence of 20 mM d-2HG, 20 mM oxamate, 10 μM GSK2837808A, or control (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 (one-way ANOVA). Data are representative of at least two independent experiments.
Impairment of LDH activity by d-2HG caused CD8+ T cells to undergo a metabolic transition toward oxidative phosphorylation. Therefore, we tested whether LDH inhibition was sufficient to cause this effect. LDH inhibition by oxamate or GSK recapitulated the increase in oxygen consumption rate, the decrease in extracellular acidification rate, and the hyperpolarization of the mitochondrial membrane observed in cells treated with d-2HG (Fig. 5, C and D, and fig. S5, E to G). No change in the mitochondrial/nuclear DNA ratio of the cell was detected after LDH inhibition (fig. S5H). Therefore, LDH inhibition is sufficient to cause increased reliance on mitochondrial respiration and hyperpolarization of the mitochondrial membrane in CD8+ T cells.
To test whether LDH inhibition is sufficient to cause the immune phenotypes promoted by d-2HG accumulation, we tested the effects of LDH inhibition alone on viability, antigen-driven proliferation, IFN-γ production, and cytotoxicity. Each of the LDH inhibitors impaired T cell proliferation despite having no effect on cell viability (Fig. 5E and fig. S5I). A d-2HG–mediated decrease in cytotoxicity was also observed in response to each of the LDH inhibitors. Upon LDH inhibition, CD8+ T cells exhibited lower expression of granzyme B and failed to degranulate upon antigen restimulation (Fig. 5, F and G, and fig. S5J). IFN-γ production was also decreased in response to LDH inhibition, resulting in a lower percentage of IFN-γ+ CD8+ T cells, decreased IFN-γ accumulation in the media, and a subsequent decrease in the induction of ISGs (Fig. 5, H to K, and fig. S5K). Ultimately, these cytotoxic defects culminated in impaired killing of tumor cells upon LDH inhibition (Fig. 5L and fig. S5L). Thus, LDH inhibition alone phenocopies the effects of d-2HG on CD8+ T cell proliferation, activation, IFN-γ signaling, and cytotoxicity.
IDH-mutant glioma-infiltrating CD8+ T cells exhibit altered metabolic and cytotoxic programs in patient samples
To determine whether d-2HG impairs antitumor immunity, we used two different syngeneic mouse tumor models. We injected B16 melanoma and MC38 colorectal adenocarcinoma cells overexpressing IDH1WT or IDH1R132H into the flanks of C57BL/6J mice (Fig. 6A). On day 16, when the tumors were comparable in weight, we isolated tumor interstitial fluid and detected low-millimolar levels of d-2HG in tumors harboring the IDH1 mutation (Fig. 6B and fig. S6A). Flow cytometry profiling of cells in the immune infiltrate of these tumors revealed changes in lymphocytes (fig. S6, B and C), including reduced infiltration, proliferation, and cytokine levels of CD8+ T cells in IDH1R132H tumors in both models (Fig. 6, C to E). These results suggest that tumor-derived d-2HG impairs T cell responses in the TME of IDH1R132H tumors.
Fig. 6. Altered metabolic and cytotoxicity signatures in IDH1-mutant relative to IDH1WT cancers.
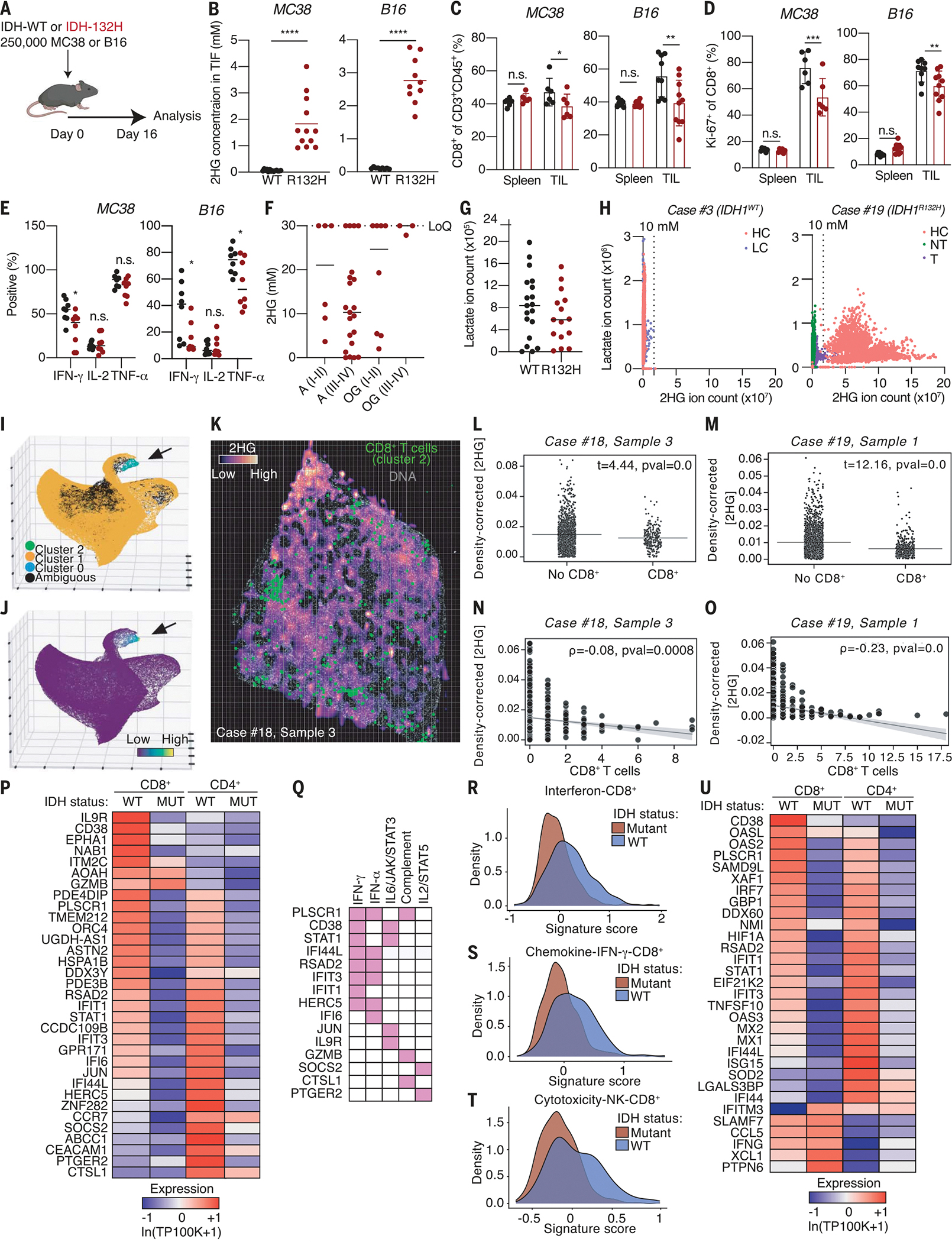
(A) Schematic depicting experimental setup. (B) Quantification of 2HG levels by liquid chromatography–mass spectrometry in the tumor interstitial fluid (TIF) from IDH1WT and IDH1R132H MC38 and B16 tumors (n = 10 to 12). (C) Quantification of the percentage of CD8+ T cells (of CD3+CD45+ cells) in spleen and tumor of IDH1WT and IDH1R132H MC38 and B16 syngeneic tumor mouse models (n = 6 to 10). (D) Quantification of the percentage of Ki67+ CD8+ T cells in spleen and tumor of IDH1WT and IDH1R132H MC38 and B16 syngeneic tumor mouse models (n = 6 to 10). (E) Quantification of the percentage of IFN-γ+, IL-2+, and TNF-α+CD8+ T cells isolated from IDH1WT or IDH1R132H MC38 or B16 tumors on day 16 after inoculation and after ex vivo restimulation with PMA and ionomycin for 4 hours in the presence of 0 or 2.5 mM d-2HG to recapitulate the 2HG levels measured in the TIF (n = 8). (F) MSI-based quantification of 2HG levels in tissue sections of human IDH1R132H gliomas subdivided by grade. The upper limit of 2HG quantification based on the calibration curve was 30 mM; therefore, any value beyond that was collapsed to 30 mM. A, astrocytoma; OD, oligodendroglioma; I-II, low-grade; III-IV, high-grade. (G) Per-section average lactate ion counts across IDH1WT GBM (n = 5) and IDH1R132H (n = 4) astrocytoma, grade IV patients. (H) Per-pixel lactate and 2HG ion counts across all sections from case #3, an IDH1WT GBM patient, and case #19, an IDH1R132H glioma patient. Sections were annotated based on high cellularity (HC), low cellularity (LC), nontumor (NT), and transition (T). (I) Three-dimensional (3D) UMAP embedding of single-cell CyCIF data derived from human brain tumor sections colored by HDBSCAN cluster identifying a population of CD8+ T cells (cluster 2, black arrow). (J) 3D UMAP embedding color mapped according to CD8 signal intensity showing that cluster 2 cells (black arrow) are characterized by high levels of CD8 expression. (K) Overlay of matched MSI and CyCIF images from case #18, sample 3 of an IDH1R132H astrocytoma. Spatial positions of cluster 2 cells are shown in green; Hoechst nuclear dye (DNA) is shown in gray. Grid superimposed on the image shows equal-sized microregions of tissue with x and y intervals corresponding to the pixel size of the original MSI image used to compute statistics in (L) and (M). (L) Welch’s t tests comparing mean tissue density–corrected d-2HG levels in microregions of tissue with and without CD8+ T cells in case #18, sample 3. (M) Welch’s t tests comparing mean tissue density–corrected d-2HG levels in microregions of tissue with and without CD8+ T cells in case #19, sample 1. (N) Spearman’s rank-order correlation showing anticorrelation between mean tissue density-corrected d-2HG levels and CD8+ T cell counts in microregions of tissue in case #18, sample 3. (O) Spearman’s rank-order correlation showing anticorrelation between mean tissue density–corrected d-2HG levels and CD8+ T cell counts in microregions of tissue in case #19, sample 1. (P) Heatmap showing expression of top DE up-regulated genes in CD8+ and CD4+ T cells in IDHWT GBM relative to IDH-mutant gliomas. Gene expression is zero-centered and given in units of ln(TP100K+1). (Q) Grouping of the top up-regulated genes into hallmark gene sets. (R) Density plot of expression of top DE up-regulated genes in the IFN subcluster for CD8+ T cells in IDHWT and IDH-mutant samples. (S) Density plot of expression of the top DE up-regulated CD8+ IDHWT genes in the chemokine–IFN-γ subcluster for T cells in and IDH-mutant samples. (T) Density plot of expression of the top DE up-regulated genes in cytotoxicity-NK subcluster for CD8+ T cells in IDHWT and IDH-mutant samples. (U) Overlay of the top DE ISGs from mouse RNA sequencing onto the top 400 DE up-regulated genes in the CD4+ and CD8+ T cell populations of IDH-mutant and IDHWT tumors showing the relative expression of genes in common between the two datasets. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 (Student’s t test and two-way ANOVA). Data are representative of at least two independent experiments.
Mouse models of IDH do not fully recapitulate the developmental and morphological features of human IDH-mutant cancers, arguing for characterization of human specimens. Moreover, the reversible and acute nature of the metabolic phenotypes caused by d-2HG makes it important to study these properties in an intact TME. Thus, we leveraged mass spectrometry imaging (MSI) to study the tumor metabolic landscape of five patients with IDH-WT and 17 patients with IDH-mutant gliomas (table S1). Quantification of 2HG levels in tissue sections revealed a range of high-millimolar concentrations of 2HG across all IDH-mutant gliomas regardless of grade (Fig. 6F and fig. S6D). The 2HG signal was restricted to the tumor area and was absent in surrounding healthy tissue, indicating that 2HG levels accumulate at the tumor site (fig. S6E). Comparison of IDH1R132H grade IV astrocytomas with IDH1WT glioblastoma (GBM) revealed trending lower lactate levels in IDH1R132H relative to IDH1WT gliomas (Fig. 6, G and H, and fig. S6, F to H).
Because of the immunosuppressive properties of d-2HG, we hypothesized that tumor regions characterized by high levels of the oncometabolite would contain fewer CD8+ T cells compared with those with lower expression. To test this hypothesis, we coupled cyclic immunofluorescence (CyCIF) of IDH1R132H grade IV astrocytomas with MSI rasters of d-2HG collected on the same tissue sections to study the location of CD8+ T cells with respect to d-2HG expression. Dimensionality reduction [uniform manifold approximation and projection (UMAP)] and clustering [hierarchical density-based clustering (HDBSCAN)] algorithms applied to CyCIF-derived single-cell data identified a population of CD8+ T cells that coexpressed CD8α and CD45RB (Fig. 6, I and J, and fig. S6I). Mapping the coordinates of these cells onto d-2HG MSI rasters, we observed a tendency for these cells to be less present in regions of tissue high in the oncometabolite (Fig. 6K). To quantify this effect, tissues were divided into microregions the size of pixels in the original MSI rasters before computing mean tissue density-corrected d-2HG expression per region. Independent, two-way Welch’s t tests were then used to compare microregions with and without at least one CD8+ T cell (Fig. 6, L and M). Across multiple tissue samples from different patients, this analysis revealed that d-2HG expression was significantly lower in regions of tissue containing CD8+ T cells compared with those that did not. Correlation analysis comparing d-2HG levels and CD8+ T cell numbers using Spearman’s rank-order method established a significant anticorrelation between these variables that was not explained by concurrent anticorrelation between CD8+ T cells and tissue density (Fig. 6, N and O, and fig. S6, K, L, N, and O), suggesting that the avoidance of regions of tissue high in d-2HG by CD8+ T cells was not due to their failure to infiltrate tumor tissue. Together, these studies reveal a dose-dependent relationship between d-2HG and CD8+ T cell density in human gliomas and support the hypothesis that d-2HG may regulate CD8+ T cell proliferation in this context.
To better understand the physiological role of d-2HG accumulation in the microenvironment of IDH-mutant tumors on CD8+ T cell function, we analyzed single-cell RNA-sequencing data from a different set of IDHWT GBM patients (n = 16) and IDH-mutant glioma patients (n = 15) (38) and profiled the gene expression signature of tumor-infiltrating T cells. We identified the top differentially up-regulated genes in CD8+ and CD4+ T cell subpopulations and grouped these into hallmark gene signatures (Fig. 6P and fig. S6P). This showed that several of the top down-regulated genes in CD8+ T cells from IDH-mutant samples fall under the IFN signaling signature (Fig. 6Q).
Previous analysis of this single-cell RNA-sequencing dataset revealed six subclusters for CD8+ T cell subsets [cytotoxicity–natural killer (NK), IFN, effector memory, chemokine–IFN-γ, not determined, and stress cells] and CD4+ T cell subsets (cytotoxicity-chemokine, IFN, effector memory, Treg, memory, and stress cells) (38). Thus, we assessed the gene expression signature of the top 20 up-regulated genes in IDHWT GBM within each of the previously identified subclusters. The top 20 up-regulated genes gave a weaker signature score in the cytotoxic, IFN, and IFN-γ subclusters for CD8+ T cells from IDH-mutant gliomas compared with IDHWT GBM (Fig. 6, R to T). The expression of the top up-regulated genes was not affected in the Tregs and cytotoxicity subclusters for CD4+ T cells, but it was elevated in the CD4+ IFN subcluster for IDHWT compared with IDH-mutant (fig. S6Q). Thus, we concluded that the cytotoxic and IFN programs of effector T cells, in particular effector CD8+ T cells, are lower in IDH-mutant samples compared with IDHWT.
Finally, we validated the relevance of the ex vivo–derived IFN-γ signature from d-2HG–treated mouse CD8+ T cells (fig. S1G) using the human single-cell RNA-sequencing data. We generated a gene list containing the top differentially expressed (DE) ISGs upon d-2HG treatment from the mouse RNA-sequencing experiment and used this gene list to filter the top 400 DE genes in CD4+ and CD8+ T cell populations from IDH-mutant and IDHWT tumors. Overall, both CD8+ and CD4+ T cell populations from IDH-mutant tumors showed decreased expression of the IFN-induced mouse gene signature (Fig. 6U), demonstrating that the human IFN-γ signature of CD8+ T cells derived from IDH-mutant tumors is similar to that of d-2HG–treated mouse CD8+ T cells.
Discussion
Our results suggest that d-2HG may not only affect tumor initiation and growth through tumor cell-intrinsic mechanisms but also by directly affecting the cells in the surrounding TME. In the context of antitumor immunity, IDH-mutant tumors are surveyed by immune cells, which, upon infiltrating, face a newly d-2HG–rich microenvironment. IDH-mutant tumors have fewer tumor-infiltrating T cells compared with IDHWT tumors (7, 13–16), but the factors that mediate the immunosuppressive functions of d-2HG have remained elusive. We propose that d-2HG acts directly on CD8+ T cells in the TME of IDH-mutant tumors by altering their metabolic and cytotoxic signatures.
Glucose metabolism has a central role in regulating cytotoxic T cell responses. As effector CD8+ T cells undergo extensive proliferation in response to antigen stimulation, maintaining a high glycolytic flux is needed to support the bioenergetic needs, as well as to provide the building blocks for cellular biomass (32). Furthermore, glucose metabolism has also been tightly linked to IFN-γ signaling and T cell cytotoxicity (30, 39–46). Our findings show that d-2HG alters glucose metabolism in CD8+ T cells, resulting in impaired proliferation, cytokine production, and cytotoxicity. Specifically, we show that decreased IFN-γ production in d-2HG–treated CD8+ T cells results in a suppressed autocrine IFN-γ gene expression signature, which has been shown to be important for effector function (47). Moreover, although a link between glucose metabolism and the expression of lytic molecules has previously been made (40), our work has uncovered a previously unknown role for LDH and d-2HG in regulating the process of degranulation. Thus, our studies expand upon previous work demonstrating the role of glycolytic metabolism in regulating T cell effector function and proliferation.
Analysis of the metabolic landscape of human gliomas has revealed altered lactate levels and decreased IFN-γ signatures in glioma samples from IDH-mutant patients. Mechanistically, we propose that d-2HG alters CD8+ T cell metabolism through direct LDH inhibition. Decreased LDH-A activity caused by epigenetic silencing in IDH-mutant tumors had previously been reported (48–51), but our work shows that d-2HG can additionally have a new, direct and immediate effect on LDH enzymatic activity in CD8+ T cells. Future studies are required to investigate the in vivo immunotherapeutic relevance of the proposed mechanism and determine whether d-2HG inhibition of LDH activity expands beyond CD8+ T cells in vivo, affecting other cells within the TME.
Finally, we found that many of the effects caused by d-2HG are reversible upon d-2HG removal. In the future, it will be interesting to determine whether therapeutic interventions involving US Food & Drug Administration–approved, mutation-specific IDH inhibitors potentiate immune responses by removing immunosuppressive d-2HG from the TME, in addition to having a direct role in the inhibition of tumor growth. In support of this idea, recent studies have shown that CD8+ T cell infiltration is restored in the presence of inhibitors of mutant IDH (52).
Supplementary Material
ACKNOWLEDGMENTS
We thank Agios Pharmaceuticals, Inc., for providing 13C5 d-2HG and l-2HG; members of the Haigis laboratory for productive discussions; C. Park and lab mates A. Ringel and J. van der Reest for helping with enzyme biochemistry; J. van der Reest for feedback on the manuscript; C. Benoist for facilitating the mouse RNA-sequencing experiment and providing feedback on the manuscript; W. Kaelin Jr. for providing reagents and feedback on the manuscript; and the HMS System Biology FACS facility, HMS Immunology Flow Core, and the Broad Genetic Platform for access to equipment. Schematics were generated in Adobe Illustrator with the support of ChemDraw (PerkinElmer Informatics) and Biorender (https://biorender.com).
Funding:
G.N. was supported by the National Science Foundation (NSF) Graduate Research Fellowship Program (grant DGE1745303). I.E. was supported by the European Molecular Biology Organization (grant ALTF-107802017). K.K. was supported by the Life Sciences Research Foundation. M.H. was supported by National Institutes of Health (NIH) grant R01CA213062. M.H. and P.S. were supported by the Ludwig Center at Harvard Medical School. M.H., P.S., and A.H.S. were supported by NIH grant U54-CA225088. K.W.W., M.L.S., and A.H.S. were supported by NIH grant 1P01CA236749. E.M.P. was supported by the NSF Graduate Research Fellowship Program (fellowship DGE1745303). N.Y.R.A. was supported by NIH grants U54-CA210180 and P41-EB028741 and by a Capital Award from the Massachusetts Life Sciences Center. S.A.S. was supported by NIH grant T32EB025823. G.B. was supported by Marie Skłodowska-Curie grant 713679, by European Union’s Horizon 2020, and by Universitat Rovira i Virgili.
Footnotes
Competing interests: M.H. received research funding from Agilent Technologies and Roche Pharmaceuticals and is on advisory boards for Alixia and Minovia. M.L.S. and K.W.W. are equity holders, scientific cofounders, and advisory board members of Immunitas Therapeutics. K.W.W. serves on the scientific advisory board of TCR2 Therapeutics, T-Scan Therapeutics, SQZ Biotech, and Nextechinvest and receives sponsored research funding from Novartis. N.Y.R.A. is key opinion leader for Bruker Daltonics, scientific adviser to Invicro, and receives support from Thermo Finnegan and EMD Serono. A.H.S. has patents and/or pending royalties on the PD-1 pathway from Roche and Novartis. A.H.S. is on advisory boards for Surface Oncology, SQZ Biotechnologies, Elpiscience, Selecta, Bicara, Monopteros, Fibrogen, Alixia, GlaxoSmithKline, and Janssen. A.H.S. has received research funding from Merck, Vertex, Moderna, Quark/Iome, and AbbVie unrelated to this project. The remaining authors declare no competing interests.
Data and materials availability:
Bulk mouse RNA-sequencing datasets have been deposited to the Gene Expression Omnibus (GEO) website with identifier GSE209757. All other data are available in the main manuscript or supplementary materials.
REFERENCES AND NOTES
- 1.Ryan DG et al. , Nat. Metab 1, 16–33 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Choi C et al. , Nat. Med 18, 624–629 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dang L et al. , Nature 462, 739–744 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Losman JA, Kaelin WG Jr.., Genes Dev 27, 836–852 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andronesi OC et al. , Sci. Transl. Med 4, 116ra4 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andronesi OC et al. , J. Clin. Invest 123, 3659–3663 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bunse L et al. , Nat. Med 24, 1192–1203 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Linninger A et al. , Neuro Oncol 20, 1197–1206 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chowdhury R et al. , EMBO Rep 12, 463–469 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Figueroa ME et al. , Cancer Cell 18, 553–567 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turcan S et al. , Nature 483, 479–483 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu W et al. , Cancer Cell 19, 17–30 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berghoff AS et al. , Neuro Oncol 19, 1460–1468 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klemm F et al. , Cell 181, 1643–1660.e17 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kohanbash G et al. , J. Clin. Invest 127, 1425–1437 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weenink B et al. , Sci. Rep 9, 14643 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tyrakis PA et al. , Nature 540, 236–241 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Intlekofer AM et al. , Nat. Chem. Biol 13, 494–500 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsukumo SI, Yasutomo K, Front. Immunol 9, 101 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barry M, Bleackley RC, Nat. Rev. Immunol 2, 401–409 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Betts MR et al. , J. Immunol. Methods 281, 65–78 (2003). [DOI] [PubMed] [Google Scholar]
- 22.McKenzie MD et al. , Int. Immunol 18, 837–846 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Waddell SJ et al. , PLOS ONE 5, e9753 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Engqvist MK, Eßer C, Maier A, Lercher MJ, Maurino VG, Mitochondrion 19, 275–281 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Struys EA et al. , J. Inherit. Metab. Dis 28, 921–930 (2005). [DOI] [PubMed] [Google Scholar]
- 26.Burr SP et al. , Cell Metab 24, 740–752 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nadtochiy SM et al. , J. Biol. Chem 291, 20188–20197 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koivunen P et al. , Nature 483, 484–488 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Farhana A, Lappin SL, “Biochemistry, lactate dehydrogenase,” in StatPearls [Internet]. Treasure Island, FL: StatPearls Publishing; (2022). [PubMed] [Google Scholar]
- 30.Chang CH et al. , Cell 153, 1239–1251 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lunt SY, Vander Heiden MG, Annu. Rev. Cell Dev. Biol 27, 441–464 (2011). [DOI] [PubMed] [Google Scholar]
- 32.van der Windt GJW, Pearce EL, Immunol. Rev 249, 27–42 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zorova LD et al. , Anal. Biochem 552, 50–59 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu SC, Biochim. Biophys. Acta 1830, 3143–3153 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spinelli JB, Haigis MC, Nat. Cell Biol 20, 745–754 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Titov DV et al. , Science 352, 231–235 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weinberg SE, Chandel NS, Nat. Chem. Biol 11, 9–15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mathewson ND et al. , Cell 184, 1281–1298.e26 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cao Y, Rathmell JC, Macintyre AN, PLOS ONE 9, e104104 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cham CM, Driessens G, O’Keefe JP, Gajewski TF, Eur. J. Immunol 38, 2438–2450 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cham CM, Gajewski TF, J. Immunol 174, 4670–4677 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Gemta LF et al. , Sci. Immunol. 4, eaap9520 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Siska PJ et al. , JCI Insight 2, e93411 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ho PC et al. , Cell 162, 1217–1228 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Quinn WJ 3rd et al. , Cell Rep 33, 108500 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peng M et al. , Science 354, 481–484 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Curtsinger JM, Agarwal P, Lins DC, Mescher MF, J. Immunol 189, 659–668 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chesnelong C et al. , Neuro Oncol 16, 686–695 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dekker LJM et al. , FASEB J 34, 3646–3657 (2020). [DOI] [PubMed] [Google Scholar]
- 50.Chaumeil MM et al. , Neuroimage Clin 12, 180–189 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Balss J et al. , Leukemia 30, 782–788 (2016). [DOI] [PubMed] [Google Scholar]
- 52.Lu M et al. , Cancer Res 80 (Supplement), 2046–2046 (2020). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Bulk mouse RNA-sequencing datasets have been deposited to the Gene Expression Omnibus (GEO) website with identifier GSE209757. All other data are available in the main manuscript or supplementary materials.