

Abstract
The mechanisms of how cancer cells are selected and evolve to establish distant metastatic colonies remain unclear. Tumor heterogeneity and lack of biomarkers are some of the most difficult challenges in cancer biology and treatment. Here using mouse models for triple negative breast cancer metastasis, we report heterogeneous expression of DNA methyltransferase 3B (DNMT3B) in both mouse and human primary tumors. High levels of DNMT3B were correlated with poor clinical outcomes in multiple human breast cancer datasets. Mechanistically, clonal cells with high DNMT3B (DNMT3BH) showed higher vimentin (VIM) expression and displayed enhanced epithelial-to-mesenchymal transition (EMT) capacity. Deletion of VIM diminished the metastatic phenotype of DNMT3BH cells. Importantly, in preclinical mouse models in which the primary tumors were surgically removed, perioperative targeting of DNMT3B in combination with chemotherapy markedly suppressed tumor recurrence and metastasis. Our studies identify DNMT3B-mediated transcription regulation as an important mediator of tumor heterogeneity and show that DNMT3B is critical for tumor invasion and metastasis, reinforcing its potential as a target for treating metastatic disease.
Implications:
Our findings of transcriptome changes mediated by DNMT3B provide new mechanistic insight for intra-tumor heterogeneity and chemo-resistance, and therapeutic targeting of DNMT3B in combination with chemotherapy offer additional treatment options for metastatic disease especially for TNBC patients.
Keywords: Metastasis, Tumor heterogeneity, DNA methylation, Epigenetic, Inflammation, therapy
Introduction
Advances in early detection and targeted therapies have significantly improved outcomes for breast cancer patients. However, there are limited therapeutic options to treat metastatic disease, resulting in significant patient mortality (1). Triple-negative breast cancer (TNBC) is an aggressive subtype characterized by extensive intra-tumoral heterogeneity, and frequently develops resistance to chemotherapy (2). Tumor heterogeneity and a lack of biomarkers remain to be some of the most difficult challenges in achieving high therapeutic efficacy (3,4). Aside from the distinct genomic mutations that promote emergence or expansion of therapy-resistant cancer cells (5,6), epigenetic and transcriptomic changes have been recently identified as key factors in tumor heterogeneity and evolution (7–10). In addition, neoadjuvant therapy can promote the development of resistant subpopulations in human breast tumors through transcriptional reprogramming without genomic alterations (11,12). These studies demonstrate that tumor cells are selected for and evolved from a heterogeneous population of primary tumors through acquired transcriptional alterations. Therefore, identification of transcriptional regulatory mechanisms that drive metastatic clonal emergence or expansion is critical to enhance our understanding of the metastatic process and treatment options.
DNA methylation pattern is one of the strongest signatures to classify cancer cell type with its origin in The Cancer Genome Atlas (TCGA) analysis (13). DNA methylation is also one of the key molecular mechanisms that regulates epithelial-mesenchymal transition (EMT), which provides cellular plasticity for metastatic advantage (14,15). Of the three DNA methyltransferases (DNMTs) that catalyze DNA methylation at the 5’ cytosine of CpG sites, DNMT3B mediates de novo DNA methylation and is required to silence genes through promoter hypermethylation during development (16). Aberrant overexpression of DNMT3B is reported in several human cancers, including breast cancer, while its expression levels are low in many adult tissues (17–19). Preclinical studies demonstrated that overexpression of DNMT3B promotes primary tumor progression of melanoma and colon cancer (20–22). Recently, we reported that DNMT3B is induced at metastatic sites and facilitates metastatic colonization (23). However, the intra-tumoral DNMT3B heterogeneity and its mechanism of function remain to be investigated.
Surgical resection of primary tumors or metastatic nodules have been reported to mobilize inflammatory immune cells and to increase inflammatory molecules in circulation (24,25). In addition, neoadjuvant and adjuvant chemotherapy, which are the most common way to treat cancer, also increase the systemic secretion of inflammatory molecules (26,27). It is not clear whether targeting the surgery-or chemotherapy-induced inflammation would be a valid approach to suppress metastatic spread and to minimize therapeutic adverse effects.
In this study, we report intra-tumoral heterogeneity of DNMT3B in breast cancer tissues, and that high DNMT3B levels are correlated with poor patient survival and more aggressive subtypes of breast cancer. In mouse models, tumor cells with high DNMT3B level display enhanced migration and metastatic colonization. Importantly, targeting DNMT3B expression with perioperative application of anti-inflammatory drugs and targeting DNMT3B enzymatic activities potentiated the chemotherapeutic efficacy in suppressing breast cancer metastasis. Our studies reveal a novel mechanism of DNMT3B in metastatic spread and targeted therapies aimed at the specific clonal cells with high DNMT3B expression will likely provide additional treatment options.
Materials and Methods
Cell lines and mice.
The murine mammary tumor cell lines 4T1 and 6DT1 were maintained in 10% FBS DMEM (Life Technologies) in a 37 °C incubator with 5% CO2. All cell lines were obtained from ATCC and were verified to be mycoplasma negative with the MycoAlert Mycoplasma Detection Kit (Lonza). The cells were used within two months after thawing. To establish clonal cell lines with high or low DNMT3B expression, clonal cells were derived from single cell seeding, and the clonal cell lines maintained the high or low DNMT3B levels after three passages were selected. The experiments with the clonal cell lines were performed within two weeks after thawing. The generation of the DNMT3B knockdown and DNMT3B-inducible cell line was described in our previous study (23). Cell line authentication has not been performed. All mice were housed at the National Cancer Institute (NCI) animal facility, and experiments were approved by the NCI Animal Care and Use Committee.
Immunohistochemistry (IHC) and immunofluorescence (IF).
Paraffin sections of mouse mammary tumors and a human breast tumor tissue array (BRM961, US Biomax Inc.) were used. DNMT3B expression was detected with a DNMT3B antibody (1:100, ab2851, Abcam), and hematoxylin was utilized as a nuclear counterstain. For immunofluorescence staining, the frozen lung and tumor sections were fixed with 4% formaldehyde (28906, Thermo Fisher Scientific), then incubated with Vimentin (Proteintech), Ki67 (M7240, Agilent), or Annexin V (11060-1-AP, Proteintech) antibodies overnight at 4°C after blocking with 10% normal serum. Fluorescence-tagged secondary antibodies and DAPI were utilized for visualization. Images were obtained (Carl Zeiss LSM 880), and quantitative analyses of the IHC and IF images were performed by ImageJ.
Human data analysis.
Publicly available human datasets, GEO-GSE6532 (28), Yau (2010) (29), TCGA-BRCA, and METABRIC (30) for patient survival, GEO-GSE12276 (31) and GEO-GSE25065 (32) for metastasis status of patients, TCGA-BRCA for tumor stages and subtypes, were used to investigate the correlation of DNMT3B expression with stages, cancer subtypes, and patient survival. The gene expression associated with promoter methylation and its correlation with breast cancer patient survival was analyzed using the Methylation Expression Index (MEI) as previously reported (33). The MEI for 10 DNMT3B targeted genes from the previous study (23) were determined in the GSE59000 dataset and applied to GEO-GSE6532, Yau (2010), TCGA-BRCA, and METABRIC datasets for patient survival analysis.
Mouse tumor models and treatment.
Tumor cells (4T1 or 6DT1, 2.5 x 105) were injected into the #2 mammary fat pad (MFP) or tail vein (TVI) of eight-week-old female BALB/c, nude, or FVB/N mice (Charles River). In some experiments, TVI was performed in mice that received MFP injection 2 weeks earlier. Mice were sacrificed at given time points as described in each experimental design. Weight of primary tumors and the number of metastatic nodules were evaluated at necropsy, and primary tumors, lungs, and CTCs were collected for further analysis.
For drug treatment experiments, the 4T1 or 6DT1 primary tumors were surgically removed on day 14. The mice were treated with Paclitaxel (6 mg/kg bodyweight, i.v. once a week), Gemcitabine (2 mg/kg bodyweight, i.v. once a week), 5-Aza (1 mg/kg body weight, i.p. three times per week) or Celecoxib (1500 ppm in diet). To target surgery-induced inflammation, Celecoxib was given to the mice perioperatively or long-term, as shown in the experimental design. For preclinical treatment, mice were sacrificed when the recurrent tumor size reached > 2 cm or mice showed labored breathing or bodyweight loss of more than 10%.
Western blotting.
Protein extraction from cultured tumor cells, primary tumors, and metastatic nodules was performed using a RIPA protein extraction buffer containing protease/phosphatase inhibitors (Thermo Fisher Scientific). Antibodies against DNMT3B (ab79822, Abcam), Vimentin (10366-1-AP, Proteintech), β-Actin (sc-69879, Santa Cruz Biotechnology), p-STAT3 (9145, Cell Signaling Technology), STAT3 (12640, Cell Signaling Technology), and p-p65 NFκB, (3033, Cell Signaling Technology) were diluted at 1:500 to 1:1000. Quantification of Western blots density was measured by ImageJ software, each band was normalized by corresponding β-Actin band.
Flow cytometry.
Mouse primary tumors and lungs were chopped into small pieces using sharp blades followed by incubation in tissue dissociation buffer (Collagenase, Dispase, and DNase in DMEM) to generate a single-cell suspension. CTCs were collected from the blood through cardiac puncture. The samples were stained with DAPI to determine cell viability and analyzed by flow cytometry (LSRFortessa, BD Bioscience). The data were analyzed by FlowJo (BD Bioscience).
Cytokine antibody array.
Proteins were extracted from the lungs of control or Celecoxib diet-treated mice and were examined using a cytokine protein antibody array (AAM-CYT-1000-2, Raybiotech) per the manufacturer’s instructions.
Wound healing migration assay.
For Vim knockout in 4T1 cells, the cells were transduced with Pspcas9-2A-puro-px459 constructs containing Vim sgRNAs, followed by 2 μg/ml puromycin selection for 2 days. The knockout of VIM was verified by Western blots. Control or VIM-KO cells were incubated until they reached 100% confluency and were then treated with mitomycin c for 1 hr to inhibit proliferation. 700 - 800 micron-wide wounds were made using an Incucyte®Woundmaker (Essen BioScience, Inc.). Images to monitor cell migration were taken at 0, 6, 12, 18 and 24 hrs by Incucyte S4 (Essen BioScience, Inc.), and the relative wound recovery at 12 hr after wounding was quantitated by Image J with MRI wound healing tool plug in.
Transwell invasion assay.
Transwell inserts of 24-well plate (Millipore Sigma) were coated with matrigel (Millipore Sigma). Cancer cells were seeded into the inserts and incubated for 18 hr. The invaded cells were fixed by 5% glutaraldehyde for 10 min then stained with 0.5% crystal violet solution for 20 min. The matrigel and cells in the inserts were removed by wet cotton swap. The numbers of invaded cells at the bottom of inserts were measured by EVOS cell imaging system.
Statistics.
GraphPad Prism v6.01 and R were used for graphs and statistics. Unless otherwise indicated, data were expressed as mean ± SD. All data were analyzed using the Student’s t-test for comparison of two groups or one-way ANOVA for three groups or more. Differences were considered statistically significant when the p-value was < 0.05.
Results
DNMT3B expression is heterogeneous in primary tumors, with high DNMT3B expression correlating with poor prognosis for breast cancer patients
Our previous study showed that DNMT3B is increased in tumor cells from metastatic nodules compared with those from primary tumors (23). In further examining the primary tumor tissues, we observed heterogeneous levels of DNMT3B; some tumor cells expressed high (DNMT3BH) while others expressed modest or low levels (DNMT3BL). The DNMT3BH and DNMT3BL cell heterogeneity was consistent in both mouse mammary tumors (from 4T1 and 6DT1 mouse models, Fig. 1a and Supplementary Fig.1a and 1b) and primary tumor tissues from breast cancer patients (Fig. 1b). High expression of DNMT3B was not detected in the normal mammary glands (Supplementary Fig 1a). We hypothesize that DNMT3BH tumor cells may exhibit distinct biological behavior in the primary tumor microenvironment. Using multiple clinical datasets containing a large number of breast tumor tissues, we first found that the high DNMT3B levels correlated with decreased metastasis-free survival (GSE6532 and Yau), decreased relapse-free survival (TCGA), or decreased disease-specific survival (METABRIC) (Fig. 1c), which was not observed for DNMT3A and DNMT1 in the same datasets (Supplementary Fig. 1c). In addition, significantly higher DNMT3B levels were found in the primary tumors from patients with metastases compared with those without metastases (Fig. 1d). High levels of DNMT3B but not DNMT3A or DNMT1 were also observed in advanced clinical stages as well as HER2-enriched and basal-like aggressive subtypes (Fig. 1e, Supplementary Fig. 1d). These results suggest that high expression of DNMT3B in primary tumors correlates with more aggressive tumor subtypes and poor prognosis for breast cancer patients.
Fig. 1. Expression of DNMT3B in primary tumors and its clinical correlation with human breast cancers.
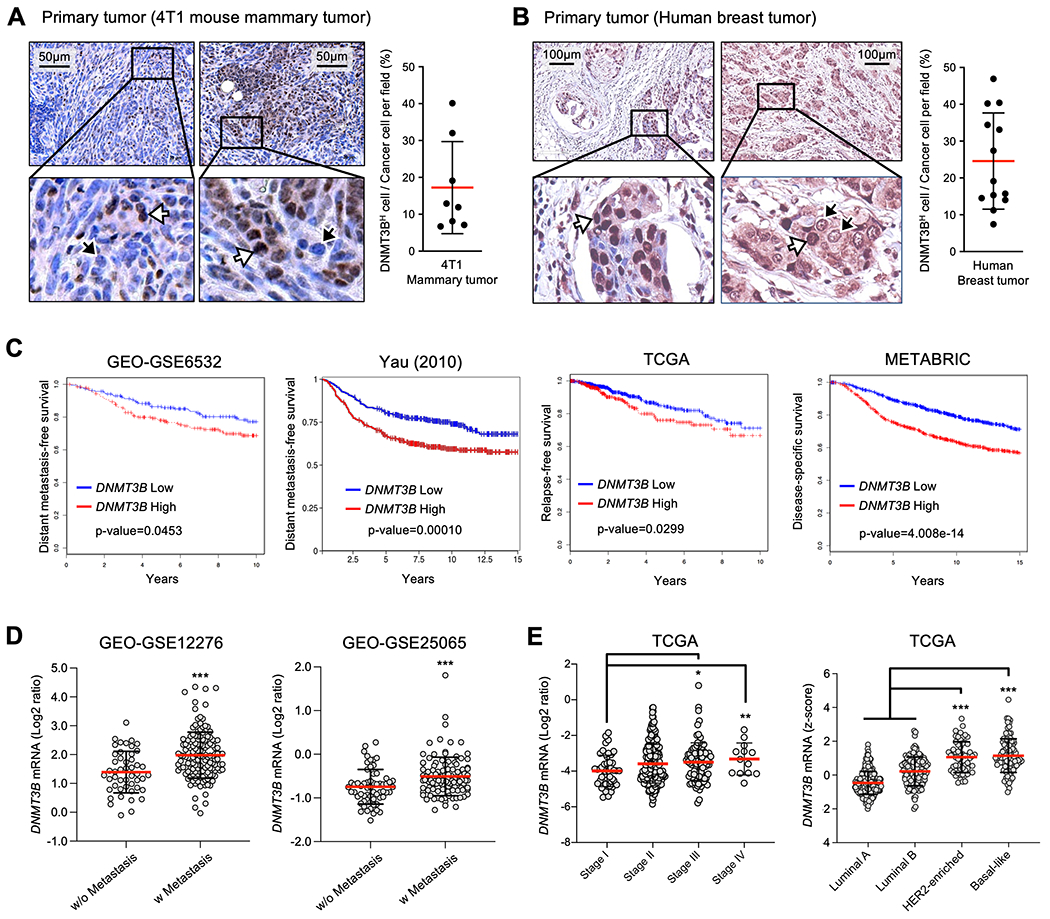
A. DNMT3B IHC of 4T1 mammary tumors and percentage of DNMT3BH cells in all tumor cells. Intensity threshold above 150 in each nucleus was set for DNMT3BH cells by Image J. B. Representative DNMT3B IHC of human breast tumors and percentage of DNMT3BH cells in all tumor cells. Intensity threshold above 150 in each nucleus was set for DNMT3BH cells by Image J. The white arrows indicate tumor cells with high DNMT3B expression, and the black arrows indicate low DNMT3B expression (tissue array, 13 sections of primary tumors examined). C. Kaplan-Meier plots for correlation between DNTM3B expression and patient survival in four different human breast cancer datasets. D. DNMT3B expression levels in breast tumors from patients with or without metastatic diseases (GEO-GSE12276 and GEO-GSE25065). E. DNMT3B expression levels in breast tumors at different stages (left panel) and in different subtypes (right panel). Data are presented as mean ± SD. * p < 0.05, ** p < 0.01, *** p < 0.001.
We have previously reported several genes that are downregulated by DNMT3B-mediated promoter hypermethylation (23). The majority of these genes showed a negative correlation between promoter methylation and gene expression in the human dataset examined here (GSE59000), which has both gene expression and DNA methylation data from same primary tumor samples (Supplementary Table 1). We next selected the 10 top-ranked genes with a Spearman r of less than −0.2 and performed the Methylation Expression Index (MEI) analysis as previously described (33). The lower MEI values indicate lower gene expression with increased promoter methylation of the 10 genes. A scatter plot between gene expression and DNA methylation of CLDN4 was shown as an example (Fig. 2a). The MEI was then applied to determine patient survival rates in multiple human breast cancer datasets. Patients with low MEI, indicating a decreased overall expression of the 10 DNMT3B target genes, showed significantly decreased survival in all datasets examined (Fig. 2b). Together our data support a strong correlation between high DNMT3B expression and aggressive cancer types as well as poor prognosis of breast cancer patients mediated by epigenetic and transcriptomic changes.
Fig. 2. Correlation between Methylation Expression Index (MEI) of DNMT3B targeted genes and patient survival.
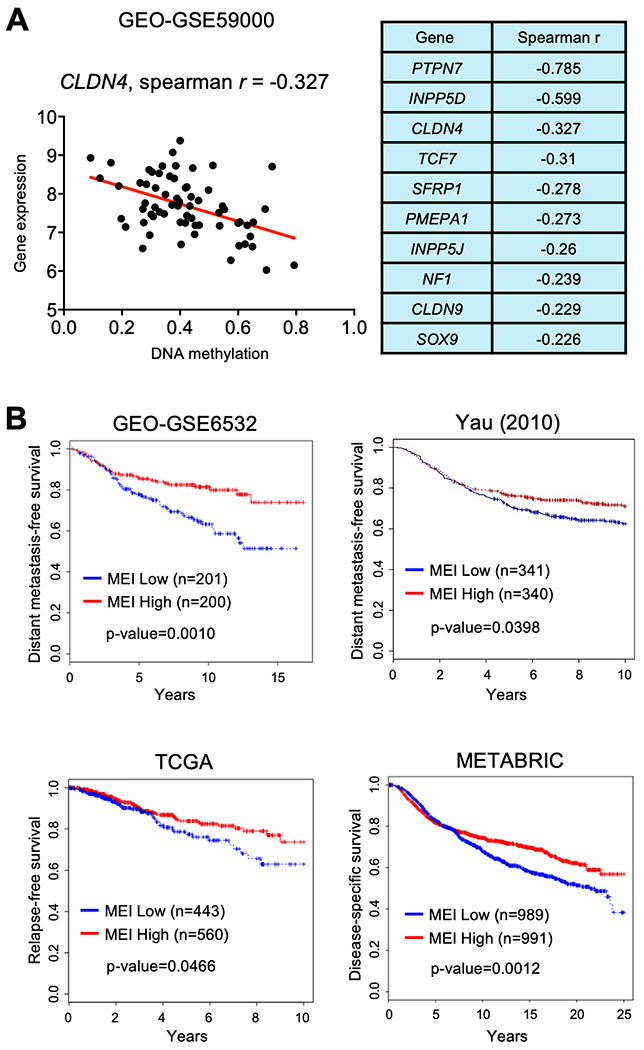
A. Scatter plot of correlation for CLDN4 gene expression and promoter methylation (left panel), and Spearman r values for correlation between gene expression and promoter methylation of 10 DNMT3B-targeted genes (right panel) in a human breast cancer dataset (GSE59000). B. Kaplan-Meier plots for correlation between MEI of the 10 DNMT3B-targeted genes and patient survival from four different human breast cancer datasets (p < 0.05- p < 0.01).
DNMT3BH tumor cells have higher metastatic capacity than DNMT3BL cells
To investigate the biological behavior of DNMT3BH tumor cells in the primary tumor microenvironment, DNMT3BH and DNMT3BL 4T1 clonal cell lines were established from mouse 4T1 cells (Fig. 3a) and labeled with GFP and RFP, respectively. The expression levels of DNMT3B were quantified by measuring DNMT3B band density, with an average 1.26 for DNMT3BH, 0.70 for DNMT3BL cells and 0.68 for normal mammary gland (Fig. 3a and Supplementary Fig. 2a). The protein levels of DNMT1 and DNMT3a were not different between DNMT3BH and DNMT3BL cells (Fig, 3a), and were not affected by DNMT3B knockdown (Supplementary Fig. 2b). Three DNMT3BH or DNMT3BL clonal cell lines were pooled for in vivo experiments to minimize the clonal variation. The pooled cells were injected into mouse mammary fat pads (MFPs). As expected from our previous studies (23), there were more metastases from DNMT3BH cells compared with DNMT3BL cells (Fig. 3b). DNMT3B knockdown in DNMT3BH cells diminished this phenotype (Fig. 3b). To model the coexisting of DNMT3BH and DNMT3BL cells in primary tumors, a clonal competition experiment was performed in which an equal number of DNMT3BH and DNMT3BL cells were co-injected into the MFP (Fig. 3c). The lung nodules from DNMT3BH and DNMT3BL clonal cell lines were counted separately by GFP or RFP label (Fig. 3c). We observed significantly more lung nodules from DNMT3BH cells than DNMT3BL cells (Fig. 3d, left panel). DNMT3B knockdown in DNMT3BH cells markedly decreased the number of lung nodules (Fig. 3d, left panel). This observation was also well-documented by immunofluorescence (IF) imaging of the lungs as DNMT3BH and DNMT3BL cells were labeled with GFP and RFP, respectively (Fig. 3d, right panel). Additional flow cytometry analysis of single-cell suspensions from the lungs of mice that received co-injection showed an increased number of DNMT3BH cells compared with the DNMT3BL cells in the lung (Fig. 3e and Supplementary Fig. 2c).
Fig. 3. Metastatic capacity of DNMT3BH tumor cells compared with DNMT3BL cells.
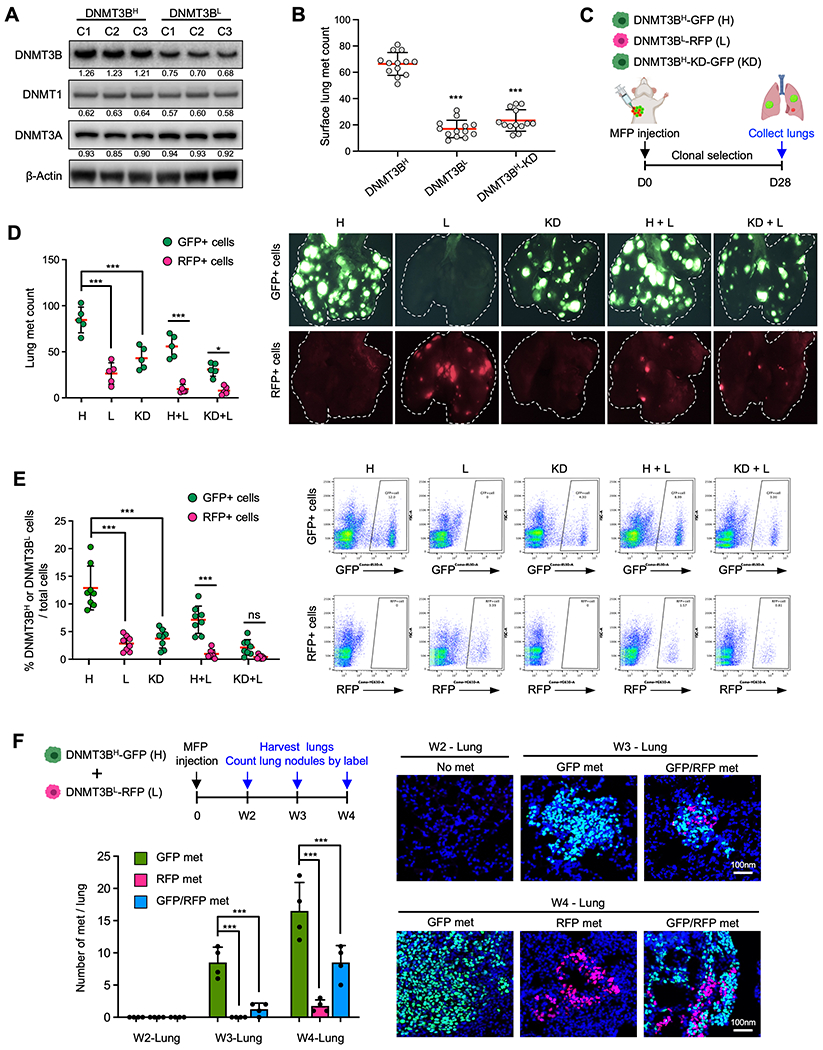
A. DNMT3B, DNMT1, and DNMT4A Western blot for DNMT3BH and DNMT3BL clonal 4T1 cell lines. B. Surface lung metastases (met) counts from mice that received MFP injections of DNMT3BH, DNMT3BL, and DNMT3BH-KD 4T1 cells. C. Schematic experimental design for metastatic potential analysis of DNMT3BH, DNMT3BL, DNMT3BH-KD 4T1 cells. D. Quantitation of DNMT3BH-GFP or DNMT3BL-RFP lung metastases at week 4 from mice that received MFP injection of tumor cells (n=5) (left panel). Representative images are on the right. The white dotted lines indicate the lung boundary. E. Flow cytometry analysis of GFP+ or RFP+ tumor cells at week 4 from the lungs of mice that received MFP injection of tumor cells (left panel), with representative plots of GFP+ or RFP+ tumor cells (right panel). F. DNMT3BH and DNMT3BL metastases in the lungs in a time course experiment (2, 3, and 4 weeks, MFP injection) in which the DNMT3BH and DNMT3BL cells were injected together for clonal competition. Upper left, schematic design; Lower left, quantitative data; Right panels, representative IF images. Quantifications of band density are presented in Western blot. Data are presented as mean ± SD. ** p < 0.01, *** p < 0.001.
To further understand the metastatic behavior of DNMT3BH tumor cells at different stages of metastatic progression, we performed a time-course experiment in which the DNMT3BH and DNMT3BL cells were co-injected into MFPs, and lungs were evaluated for GFP and RFP metastatic nodules at 2-, 3-, and 4-weeks post-injection (Fig. 3f, upper left panel). At week 2, no DNMT3BH or DNMT3BL cancer cells were detected in the lung (Fig. 3f, lower left and IF). At week 3, there were DNMT3BH-GFP and mixed DNMT3BH-GFP/DNMT3BL-RFP lung nodules (Fig. 3f, lower left and IF). However, no lung nodules contained only DNMT3BL-RFP cells (Fig. 3f, lower left). At week 4, some lung nodules comprised of only DNMT3BL-RFP cells were observed but at lower frequency than DNMT3BH-GFP nodules (Fig. 3f, lower left and IF). In addition, the sizes of DNMT3BL lung nodules at week 4 were smaller than the DNMT3BH lung nodules, as shown in the representative images (Supplementary Fig. 2d). These results suggest that DNMT3BH cells colonize earlier and faster than the DNMT3BL cells.
DNMT3BH cells display an enhanced epithelial-mesenchymal transition (EMT) phenotype
The weights of primary tumors derived from DNMT3BH cells or DNMT3BL cells were not significantly different. DNMT3B knockdown in DNTM3BH cells showed only a modest reduction in tumor weight (Supplementary Fig. 2e). However, when we further examined the clonal composition of the primary tumors from the co-injection of DNMT3BH and DNMT3BL cells, there was higher percentage of DNMT3BH cells than DNTM3BL cells (Supplementary Fig. 2f). This result indicates that DNMT3BH cells have higher capacity for cell proliferation and clonal expansion over DNMT3BL cells in the primary tumors where different clonal cells co-exist and compete. We next determined whether DNMT3BH cells have a better capacity to escape from the primary tumors. We first measured the number of circulating tumor cells (CTCs) at the early time points of tumor cell dissemination (Fig. 4a). We observed a higher number of CTCs in the animals injected with DNMT3BH cells than those injected with DNMT3BL cells, which was even more pronounced at day 17 and day 20 than day 14 (Fig. 4b and Supplementary Fig. 3a). Consistently, in the co-injection of DNMT3BH and DNMT3BL cells, the number of DNMT3BH CTCs was markedly higher than the DNMT3BL CTCs (Fig. 4b). The number of lung metastases from DNMT3BH cells was significantly higher than those from DNMT3BL cells while there was no difference in primary tumor weights between those composed of DNMT3BH or DNMT3BL cells at any of the early time points examined (Supplementary Fig. 3b).
Fig. 4. DNMT3BH tumor cells displayed enhanced dissemination.
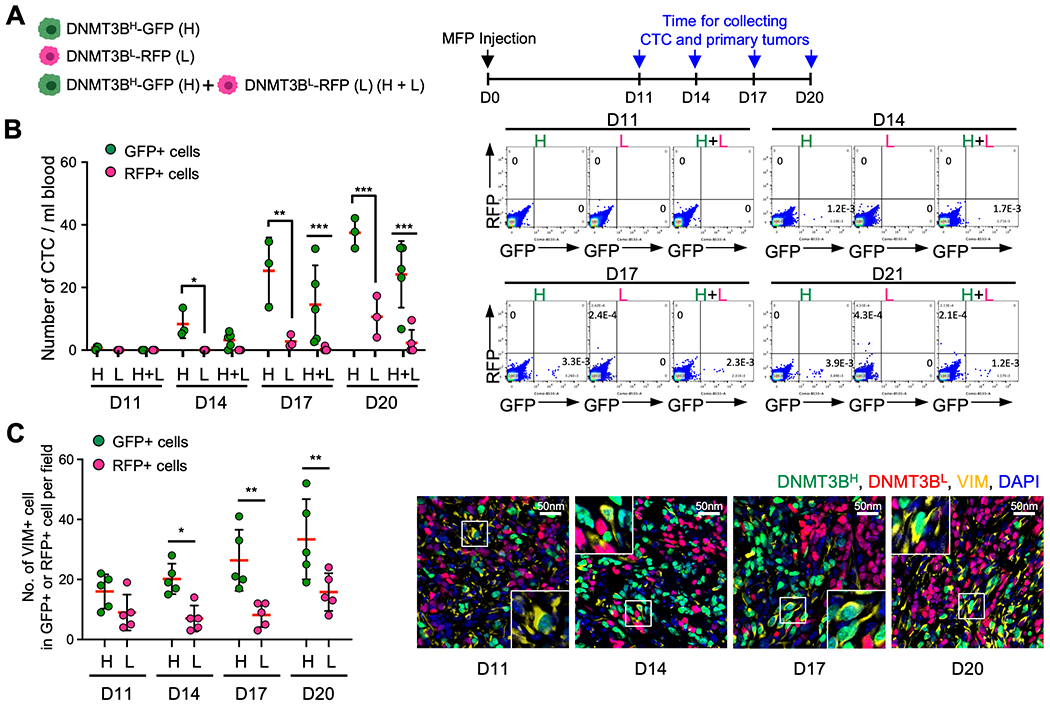
A. Schematic experimental design for a time course of clonal competition of DNMT3BH and DNMT3BL 4T1 tumor cells in early stages of cancer cell dissemination. B. Quantitated data of CTCs from the clonal competition and time course experiment (left panel). Data were collected 11, 14, 17 and 20 days after tumor cell injection. Representative flow cytometry results of CTCs with indication of cell frequencies (right panel). C. Quantitated data of VIM-positive cells among DNMT3BH and DNMT3BL cells in the primary tumors (left panel) from the clonal competition and time course experiment, the same as in 4B. Representative image of primary tumors (right panel). Data are presented as mean ± SD. ** p < 0.01, *** p < 0.001.
To further determine the biological properties that lead to the increased number of the DNMT3BH tumor cells in circulation, we measured proliferation (Ki67), apoptosis (annexin V) and EMT state (vimentin, VIM) of cancer cells in the primary tumors. There was an increase in the number of Ki67-positive DNMT3BH cells at day 20 but not at earlier time points, and no difference in the number of annexin V-positive cells between DNMT3BH and DNMT3BL cells (Supplementary Fig. 3c and 3d). Interestingly, there were clearly more VIM-positive tumor cells in the DNMT3BH population than that in the DNMT3BL population (Fig. 4c). In DNMT3BH cells displaying more mesenchymal-like morphology, almost all were VIM-positive, as shown in immunofluorescence images (Fig. 4c, right panels and Supplementary Fig. 3e). These data revealed that DNMT3BH cells have enhanced EMT phenotype.
High DNMT3B increased VIM expression and the EMT phenotype
We next investigated DNMT3B regulation of VIM and EMT. We observed that DNMT3BH cells expressed higher levels of VIM than DNMT3BL cells (Fig. 5a). DNMT3B knockdown decreased Vim mRNA (Fig. 5b). 5-Aza, an inhibitor targeting DNMT enzymatic activity, also reduced Vim mRNA expression (Fig. 5c). In addition, there were DNMT3B enrichment and high DNA methylation in gene body regions of Vim, as well as Snai1, another well-known EMT marker (Fig. 5d and Supplementary Fig. 4a and 4b). These data suggest a possible regulatory mechanism for Vim and Snai1 expression. Consistently, DNMT3BH cells showed higher levels of Twist2 and Snai1, compared with DNMT3BL cells (Fig. 5e). To further investigate the functional significance of high VIM in DNMT3BH cells, VIM was knocked out in pooled DNMT3BH and DNMT3BL cells (Fig. 5f). VIM knockout diminished the migration of DNMT3BH tumor cells in a wound-healing assay, whereas it had a minimal effect on the DNMT3BL cells (Fig. 5g). VIM knockout also significantly suppressed the invasion of DNMT3BH tumor cells in a matrigel-coated transwell assay, while it did not have effect on DNMT3BL cells (Fig. 5h). Together, these results suggest that the DNMT3BH cells display enhanced EMT through increased expression of Vim, thus intravasate and disseminate from the primary tumors to initiate metastatic colonization.
Fig. 5. DNMT3B increased VIM expression.
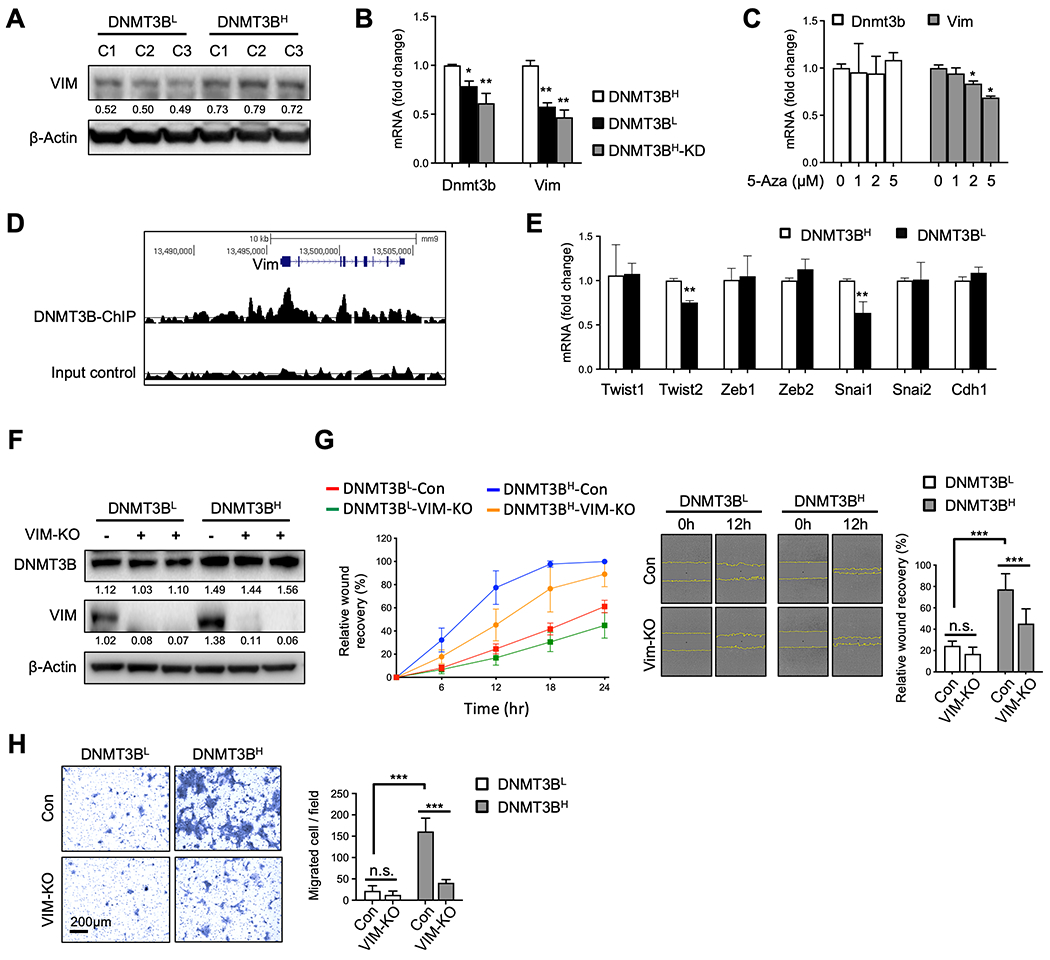
A. VIM Western blot for DNMT3BH and DNMT3BL clonal cell lines. B. mRNA levels of Dnmt3b and Vim in DNMT3BH, DNMT3BL and DNMT3BH-KD cells. C. mRNA levels of Dnmt3b and Vim in DNMT3BH cells with 5-Aza treatment. D. DNMT3B enrichment on Vim from DNMT3B ChIP-seq data (GSE146010). E. mRNA levels of Twist1, Twist2, Zeb1, Zeb2, Snai1, Snai2, and Cdh1 in DNMT3BH and DNMT3BL cells. F. DNMT3B and VIM Western blot for pooled DNMT3BH and DNMT3BL clonal cells with and without VIM knockout. G. Wound healing migration assay for pooled DNMT3BH and DNMT3BL clonal cells with and without VIM knockout. Relative wound recovery in time course (left panel), representative images at 0 hr and 12 hr after scratch (middle panel), and relative wound recovery at 12 hr (right panel). The edge of migrating cells was indicated in yellow. H. Transwell invasion assay for pooled DNMT3BH and DNMT3BL clonal cells with or without VIM knockout. Representative images of invaded cells (left panel) and number of invaded cells per field (right panel). Quantifications of band density are presented under each Western blot. Data are presented as mean ± SD. ** p < 0.01, *** p < 0.001.
DNMT3B is a key target of anti-inflammatory drugs to inhibit metastasis
Our previous studies identified prostaglandin E2 (PGE2) as one of the most significantly increased inflammatory molecules in the metastatic microenvironment that is critical for DNMT3B induction through the STAT3 and NFκB signaling pathways (23). Celecoxib is the most widely prescribed FDA-approved selective inhibitor of COX2, a prostaglandin-endoperoxide synthase also known as cyclooxygenase 2, and it significantly decreases PGE2 production. We thus first investigated the effects of Celecoxib on the levels of various cytokines in the lungs of 4T1 tumor-bearing mice by a cytokine array experiment. Treatment with Celecoxib decreased the levels of pro-tumor cytokines (G-CSF, VEGF, IL-1a, Eotaxin, SCF) and increased anti-tumor cytokines (XCL1, IGFBP6, PF4) (Fig. 6a). Treatment with Celecoxib decreased DNMT3B and inhibited the activation of STAT3 and NFκB in the lung nodules (Fig. 6b). To further examine the cause-effect role of DNMT3B for metastasis suppression by Celecoxib, GFP-labeled 4T1 cells with a doxycycline-inducible overexpression of DNMT3B vector were injected through the tail vein into the mice bearing primary tumors (Fig. 6c). As expected, Celecoxib decreased the number of metastatic nodules (Fig. 6d), while doxycycline-induced overexpression of DNMT3B significantly increased the number of metastatic nodules (Fig. 6d). Importantly, doxycycline induction of DNMT3B in tumor cells rescued the metastasis suppression by Celecoxib treatment (Fig. 6d, left and middle panels), with no difference in primary tumor weight (Fig. 6d, right panel). Thus, DNMT3B is a key target in metastasis suppression by Celecoxib.
Fig. 6. Anti-inflammatory drug targeting of PGE2 in DNMT3B-mediated metastasis.
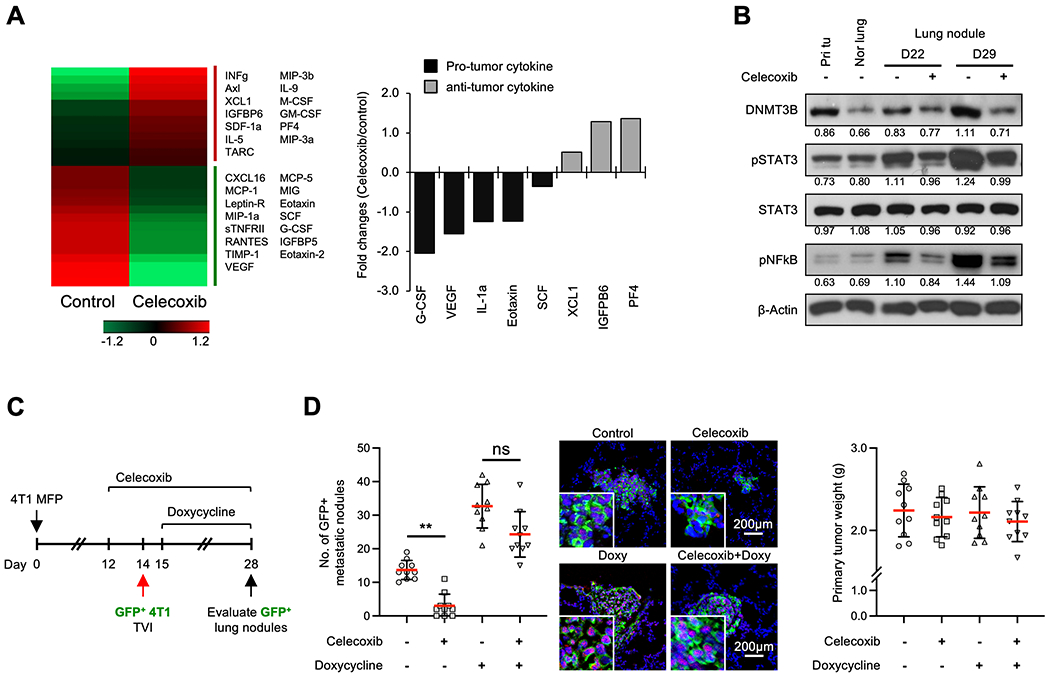
A. Heat map from cytokine array experiments on protein extracts of lungs from 4T1 tumor-bearing mice treated with Celecoxib or control (left panel), with the quantitative data for the fold changes of the pro- and anti-tumor cytokines (right panel). B. DNMT3B, pSTAT3 and pNFκB Western blots of primary tumor, normal lung and metastatic nodules from mice treated with Celecoxib or control. C. Schematic experimental design for Celecoxib treatment and Dox-DNMT3B induction (left panel). Nude mice were injected (TVI) with 4T1-GFP cells (with inducible DNMT3B) 14 days after the initial injection of 4T1 tumor cells in the MFP. The mice were treated with doxycycline (Dox) to induce DNMT3B expression. D. The number of GFP+ lung nodules from mice that received TVI of 4T1-GFP cells and representative images (middle panel), and the weights of primary tumors (right panel). Quantifications of band density are presented in Western blot. Data are presented as mean ± SD. * p < 0.05, ** p < 0.01.
Perioperative application of Celecoxib in combination with 5-Aza or chemotherapeutic drugs inhibits metastasis
Cancer surgery is fundamentally important in the treatment of solid tumors. However, recent studies suggest that surgical removal of primary tumors or metastatic lesions induces systemic inflammatory responses that stimulate metastatic spread and dormant tumor reactivation (24,25). As PGE2 is a key inflammatory mediator (25), we tested whether Celecoxib would decrease cancer metastasis in the 4T1 preclinical mouse model following surgical resection of the primary tumor (Fig. 7a). In addition, considering the adverse cardiovascular events observed for COX-2 inhibitors in previous clinical trials (34), we directly compared the effect of perioperative short-term Celecoxib treatment (7 days, 1500 ppm in chow) and those from long-term treatment (23 days, 1500 ppm in chow) (Fig. 7a). Both short and long-term Celecoxib significantly decreased the number of metastases, with no difference between the short versus the long-term treatment (Fig. 7b, left panel). Of note, long-term but not perioperative Celecoxib treatment decreased recurrent primary tumor weight (Fig. 7b, right panel).
Fig. 7. Perioperative application of Celecoxib in combination with 5-Aza or chemotherapies in metastasis treatment.
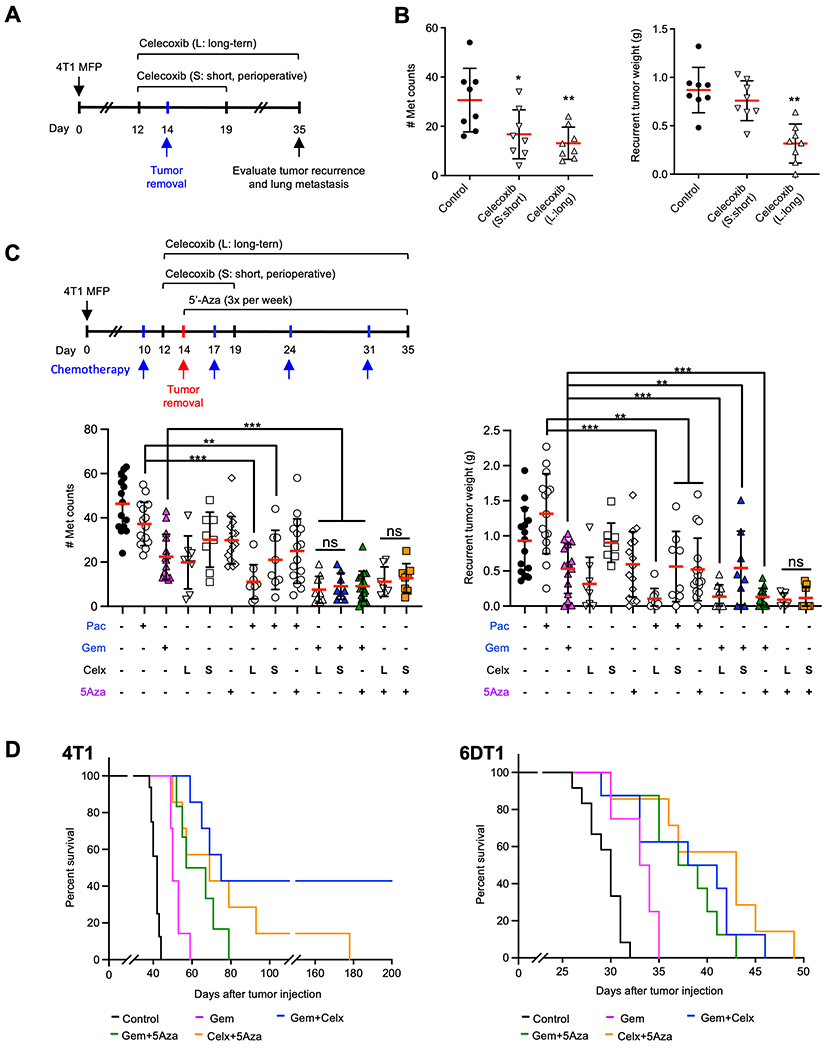
A. Schematic experimental design for surgical removal of primary tumors and perioperative vs long-term Celecoxib treatment. B. Number of metastases in the lungs (middle panel) and the weights of recurrent tumors in mice treated with perioperative vs long-term Celecoxib. C. upper panel: Schematic experimental design for combination of chemotherapeutics with perioperative and long-term Celecoxib treatment; lower panels: number of metastases in the lungs (lower left) and the weights of recurrent tumors (lower right) from mice that received Paclitaxel (Pac), Gemcitabine (Gem), or 5-Aza in combination with perioperative (S, short) or long-term (L, long) Celecoxib (Celx). D. Percent survival of 4T1 (left panel) or 6DT1 (right panel) tumor-bearing mice with single or combined drug treatment with Gemcitabine or 5-Aza alone, the two in combination, or each of the two drugs in combination with perioperative Celecoxib. Data are presented as mean ± SD. * p < 0.05, ** p < 0.01, *** p < 0.001.
The efficacy of perioperative Celecoxib was further investigated in combined treatment with the chemotherapeutic drugs Paclitaxel or Gemcitabine as well as the DNA methyltransferase inhibitor 5-azacytidine (5’-Aza), a drug targeting the enzymatic activity of DNMTs (Fig. 7c, upper left panel). The combinations of perioperative Celecoxib, low-dose 5-Aza, or chemotherapeutic agents decreased the number of metastasis nodules (Fig. 7c, lower left) and tumor recurrence in 4T1 tumor-bearing mice (Fig. 7c, right). Particularly, the perioperative treatment of Celecoxib combined with Gemcitabine or 5-Aza showed similar efficacy against metastasis that were shown as long-term Celecoxib in combination with these two drugs (Fig. 7c). We further evaluated the effect of perioperative Celecoxib with 5-Aza or Gemcitabine on the survival of tumor-bearing mice (Fig. 7d). The perioperative Celecoxib in combination with Gemcitabine significantly increased survival, and 40% of the mice survived 200 days post tumor injection (Fig 7d, left panel), and stayed tumor-free for almost additional one year. In mice bearing 6DT1 tumors, an even more aggressive breast cancer metastasis model than 4T1, the drug combinations of Celecoxib, 5-Aza, and Gemcitabine also significantly increased survival (Fig. 7d, right panel). Our data suggest that perioperative application of Celecoxib, together with low-dose 5-Aza, will likely enhance the efficacy of chemotherapy in metastasis treatment.
Discussion
Our studies show that DNMT3B expression is heterogeneous in primary tumors from both mouse models and human breast cancer patients. In addition, high DNMT3B expression correlated with poor prognosis and was associated with later stages and more aggressive basal-like breast cancers. Mechanistically, we found that the DNMT3BH cells are more mesenchymal-like and have higher migratory capacity to escape from primary tumors when compared with DNMT3BL cells. In therapeutic targeting using preclinical mouse models, we found that perioperative application of Celecoxib in combination with 5-Aza or chemotherapies significantly inhibits metastatic spread. Together with our previous report that DNMT3B is elevated in metastatic cancer cells and facilitates metastatic outgrowth (23), we have identified DNMT3B as a valuable therapeutic target against metastatic disease because of its critical roles in tumor dissemination and metastatic outgrowth, two critical stages of metastatic progression.
Our work supports the key role of transcriptional regulation in tumor heterogeneity. The genetic heterogeneity, such as mutations and copy number variations, cannot fully explain the substantial subclonal complexity of tumors (3,35,36). The mutational landscapes of the metastases are also largely similar to that of the primary tumors (37,38). Rather, histopathology-guided multi-region sampling demonstrates that the heterogeneity in primary tumors is not due to genomic alterations, but due to transcriptional regulation (8,10). Importantly, pre-existing intra-tumor heterogeneity is known to be responsible for acquired resistance during therapy (3,39). Overcoming acquired resistance remains an obstacle to treatment success. Our studies identify DNMT3B as a key regulator in this transcription-mediated tumor heterogeneity and as a non-genetic predictive marker for high metastatic potential.
We propose that high expression of DNMT3B may confer epigenetic plasticity and allow the cells to readily undergo EMT and metastatic colonization. This notion is supported by our observation that DNMT3BH cells expressed more VIM and Snai1, and knockout of VIM diminished the metastatic advantage of DNMT3BH cells. Our findings suggest that DNMT3BH cells are more mesenchymal-like and have higher capacity to disseminate and initiate the metastatic cascade. While gene silencing by promoter hypermethylation is well-established, DNMT3B can also methylate gene bodies to increase gene expression (40–42). We found DNMT3B enrichment and high DNA methylation in gene body regions of Vim as well as Snai1. In contrast, for methylation-sensitive genes in breast cancer such as Rassf1a, Grhl2 and Sfrp1, DNMT3B enrichment was found in their promoter regions, and DNMT3B knockdown increased expression of these genes (Supplementary Fig. 4c and 4d). We previously reported that DNMT3B is elevated in metastatic cancer cells and facilitates metastatic outgrowth through multiple signaling pathways (23). We now report that in the primary tumors, DNMT3BH cells show an enhanced migratory capacity through elevated VIM expression. Therefore, targeting DNMT3B could inhibit multiple steps of the metastatic cascade.
We explored the therapeutic efficacy of DNMT3B inhibition by using Celecoxib and the DNA methyltransferase inhibitor 5-Aza. Celecoxib, a nonsteroidal anti-inflammatory drug (NSAID) that once attracted huge attention as a potential drug to prevent colorectal adenoma, failed in long-term clinical trials because of an increased risk of severe cardiovascular harm (43). However, a clinical study with arthritis patients showed that moderate doses of Celecoxib are comparable to other nonselective nonsteroidal anti-inflammatory drugs (NSAIDs), such as ibuprofen and naproxen, in cardiovascular safety (34). We found that Celecoxib, when applied perioperatively, showed similar effectiveness as long-term treatment in suppressing metastasis. 5-Aza, a cytosine analog that induces degradation of DNMTs, is an FDA-approved drug for myelodysplastic syndromes and chronic monomyelocytic leukemia. A study with patient-derived xenograft organoids showed anti-cancer activity of 5-Aza against TNBC, which was also highly correlated with DNMT3A and DNMT3B levels (44). However, the clinical benefit of 5-Aza has been inconsistent in trials for the treatment of solid tumors (45–47). In the current study, we showed markedly increased anti-metastatic activity of 5-Aza when combined with Celecoxib or chemotherapies, while treatment with 5-Aza as a single agent was not effective. Our data propose a novel combination of readily available and affordable drugs to target breast cancer metastasis. Drug repurposing is a fast-progressing area that holds the promise of rapid clinical impact at a lower cost than de novo drug development (48), which is particularly useful and urgently needed for TNBC, which currently lacks effective treatment options.
Perioperative Celecoxib in combination with chemotherapy significantly prolonged the survival of mice after tumor removal surgery, and some mice even showed complete response. In a long-term follow up of ER+ breast cancer patients, tumor recurrence and metastasis peaked at 1-2 years after tumor resection, suggesting the relapse risk increased by surgery (49). Surgery has also been found to induce systemic expansion of inflammatory immune cells and caused outgrowth of dormant cells at distant sites (25). Consistent with our observations, perioperative usage of anti-inflammatory drugs improved disease-free survival in colon and breast cancers (50,51). In an animal model of metastatic lung cancer, the preoperative inhibition of PGE2 together with the perioperative resolution of inflammation by resolvins, a family of omega-3 fatty acid derivatives, eliminated micrometastases, with 40% of mice achieving long-term survival without micrometastases (52). Our findings, together with other published evidence, provide a proof of principle for the perioperative usage of anti-inflammatory drugs to treat and to prevent metastatic progression. Altogether, our studies highlight the importance of DNMT3B in tumor dissemination and metastatic outgrowth. Because of its diverse functions at different stages of the metastatic cascade, targeting DNMT3B with Celecoxib or 5-Aza in conjunction with chemotherapy could significantly suppress metastatic progression.
Supplementary Material
Acknowledgements
We thank Dr. Meera Murgai and Dr. Brandi Carofino for critical reading of the manuscript and editorial suggestions. We are grateful to all expert technical supports provided by Animal facility, Flow Cytometry Core and Confocal Microscopy Core, Center for Cancer Research, National Cancer Institute. This work is supported by NCI intramural funding to Dr. Li Yang
Financial support
The work is supported by the NCI intramural funding to Dr. Li Yang.
Footnotes
Conflict of interest statement:
The authors declare no potential conflicts of interest
References
- 1.Steeg PS. Targeting metastasis. Nat Rev Cancer 2016;16(4):201–18 doi 10.1038/nrc.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol 2016;13(11):674–90 doi 10.1038/nrclinonc.2016.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marusyk A, Janiszewska M, Polyak K. Intratumor Heterogeneity: The Rosetta Stone of Therapy Resistance. Cancer Cell 2020;37(4):471–84 doi 10.1016/j.ccell.2020.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McDonald KA, Kawaguchi T, Qi Q, Peng X, Asaoka M, Young J, et al. Tumor Heterogeneity Correlates with Less Immune Response and Worse Survival in Breast Cancer Patients. Ann Surg Oncol 2019;26(7):2191–9 doi 10.1245/s10434-019-07338-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Angus L, Smid M, Wilting SM, van Riet J, Van Hoeck A, Nguyen L, et al. The genomic landscape of metastatic breast cancer highlights changes in mutation and signature frequencies. Nat Genet 2019;51(10):1450–8 doi 10.1038/s41588-019-0507-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yates LR, Knappskog S, Wedge D, Farmery JHR, Gonzalez S, Martincorena I, et al. Genomic Evolution of Breast Cancer Metastasis and Relapse. Cancer Cell 2017;32(2):169–84 e7 doi 10.1016/j.ccell.2017.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sheffield NC, Pierron G, Klughammer J, Datlinger P, Schonegger A, Schuster M, et al. DNA methylation heterogeneity defines a disease spectrum in Ewing sarcoma. Nat Med 2017;23(3):386–95 doi 10.1038/nm.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tavernari D, Battistello E, Dheilly E, Petruzzella AS, Mina M, Sordet-Dessimoz J, et al. Non-genetic evolution drives lung adenocarcinoma spatial heterogeneity and progression. Cancer Discov 2021. doi 10.1158/2159-8290.CD-20-1274. [DOI] [PubMed] [Google Scholar]
- 9.Hinohara K, Polyak K. Intratumoral Heterogeneity: More Than Just Mutations. Trends Cell Biol 2019;29(7):569–79 doi 10.1016/j.tcb.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharma A, Merritt E, Hu X, Cruz A, Jiang C, Sarkodie H, et al. Non-Genetic Intra-Tumor Heterogeneity Is a Major Predictor of Phenotypic Heterogeneity and Ongoing Evolutionary Dynamics in Lung Tumors. Cell Rep 2019;29(8):2164–74 e5 doi 10.1016/j.celrep.2019.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Silwal-Pandit L, Nord S, von der Lippe Gythfeldt H, Moller EK, Fleischer T, Rodland E, et al. The Longitudinal Transcriptional Response to Neoadjuvant Chemotherapy with and without Bevacizumab in Breast Cancer. Clin Cancer Res 2017;23(16):4662–70 doi 10.1158/1078-0432.CCR-17-0160. [DOI] [PubMed] [Google Scholar]
- 12.Kim C, Gao R, Sei E, Brandt R, Hartman J, Hatschek T, et al. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 2018;173(4):879–93 e13 doi 10.1016/j.cell.2018.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018;173(2):291–304 e6 doi 10.1016/j.cell.2018.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer 2013;13(2):97–110 doi 10.1038/nrc3447. [DOI] [PubMed] [Google Scholar]
- 15.Carmona FJ, Davalos V, Vidal E, Gomez A, Heyn H, Hashimoto Y, et al. A comprehensive DNA methylation profile of epithelial-to-mesenchymal transition. Cancer Res 2014;74(19):5608–19 doi 10.1158/0008-5472.CAN-13-3659. [DOI] [PubMed] [Google Scholar]
- 16.Lyko F. The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat Rev Genet 2018;19(2):81–92 doi 10.1038/nrg.2017.80. [DOI] [PubMed] [Google Scholar]
- 17.Rhee I, Bachman KE, Park BH, Jair KW, Yen RW, Schuebel KE, et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature 2002;416(6880):552–6 doi 10.1038/416552a. [DOI] [PubMed] [Google Scholar]
- 18.Girault I, Tozlu S, Lidereau R, Bieche I. Expression analysis of DNA methyltransferases 1, 3A, and 3B in sporadic breast carcinomas. Clin Cancer Res 2003;9(12):4415–22. [PubMed] [Google Scholar]
- 19.Nosho K, Shima K, Irahara N, Kure S, Baba Y, Kirkner GJ, et al. DNMT3B expression might contribute to CpG island methylator phenotype in colorectal cancer. Clin Cancer Res 2009;15(11):3663–71 doi 10.1158/1078-0432.CCR-08-2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Linhart HG, Lin H, Yamada Y, Moran E, Steine EJ, Gokhale S, et al. Dnmt3b promotes tumorigenesis in vivo by gene-specific de novo methylation and transcriptional silencing. Genes Dev 2007;21(23):3110–22 doi 10.1101/gad.1594007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Micevic G, Muthusamy V, Damsky W, Theodosakis N, Liu X, Meeth K, et al. DNMT3b Modulates Melanoma Growth by Controlling Levels of mTORC2 Component RICTOR. Cell Rep 2016;14(9):2180–92 doi 10.1016/j.celrep.2016.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ibrahim ML, Klement JD, Lu C, Redd PS, Xiao W, Yang D, et al. Myeloid-Derived Suppressor Cells Produce IL-10 to Elicit DNMT3b-Dependent IRF8 Silencing to Promote Colitis-Associated Colon Tumorigenesis. Cell Rep 2018;25(11):3036–46 e6 doi 10.1016/j.celrep.2018.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.So JY, Skrypek N, Yang HH, Merchant AS, Nelson GW, Chen WD, et al. Induction of DNMT3B by PGE2 and IL6 at Distant Metastatic Sites Promotes Epigenetic Modification and Breast Cancer Colonization. Cancer Res 2020;80(12):2612–27 doi 10.1158/0008-5472.CAN-19-3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tohme S, Simmons RL, Tsung A. Surgery for Cancer: A Trigger for Metastases. Cancer Res 2017;77(7):1548–52 doi 10.1158/0008-5472.CAN-16-1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krall JA, Reinhardt F, Mercury OA, Pattabiraman DR, Brooks MW, Dougan M, et al. The systemic response to surgery triggers the outgrowth of distant immune-controlled tumors in mouse models of dormancy. Sci Transl Med 2018;10(436) doi 10.1126/scitranslmed.aan3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karagiannis GS, Pastoriza JM, Wang Y, Harney AS, Entenberg D, Pignatelli J, et al. Neoadjuvant chemotherapy induces breast cancer metastasis through a TMEM-mediated mechanism. Sci Transl Med 2017;9(397) doi 10.1126/scitranslmed.aan0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keklikoglou I, Cianciaruso C, Guc E, Squadrito ML, Spring LM, Tazzyman S, et al. Chemotherapy elicits pro-metastatic extracellular vesicles in breast cancer models. Nat Cell Biol 2019;21(2):190–202 doi 10.1038/s41556-018-0256-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Loi S, Haibe-Kains B, Desmedt C, Lallemand F, Tutt AM, Gillet C, et al. Definition of clinically distinct molecular subtypes in estrogen receptor-positive breast carcinomas through genomic grade. J Clin Oncol 2007;25(10):1239–46 doi 10.1200/JCO.2006.07.1522. [DOI] [PubMed] [Google Scholar]
- 29.Yau C, Esserman L, Moore DH, Waldman F, Sninsky J, Benz CC. A multigene predictor of metastatic outcome in early stage hormone receptor-negative and triple-negative breast cancer. Breast Cancer Res 2010;12(5):R85 doi 10.1186/bcr2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012;486(7403):346–52 doi 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, et al. Genes that mediate breast cancer metastasis to the brain. Nature 2009;459(7249):1005–9 doi 10.1038/nature08021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hatzis C, Pusztai L, Valero V, Booser DJ, Esserman L, Lluch A, et al. A genomic predictor of response and survival following taxane-anthracycline chemotherapy for invasive breast cancer. JAMA 2011;305(18):1873–81 doi 10.1001/jama.2011.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Figueroa JD, Yang H, Garcia-Closas M, Davis S, Meltzer P, Lissowska J, et al. Integrated analysis of DNA methylation, immunohistochemistry and mRNA expression, data identifies a methylation expression index (MEI) robustly associated with survival of ER-positive breast cancer patients. Breast Cancer Res Treat 2015;150(2):457–66 doi 10.1007/s10549-015-3314-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nissen SE, Yeomans ND, Solomon DH, Luscher TF, Libby P, Husni ME, et al. Cardiovascular Safety of Celecoxib, Naproxen, or Ibuprofen for Arthritis. N Engl J Med 2016;375(26):2519–29 doi 10.1056/NEJMoa1611593. [DOI] [PubMed] [Google Scholar]
- 35.Andor N, Graham TA, Jansen M, Xia LC, Aktipis CA, Petritsch C, et al. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat Med 2016;22(1):105–13 doi 10.1038/nm.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berglund E, Maaskola J, Schultz N, Friedrich S, Marklund M, Bergenstrahle J, et al. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity. Nat Commun 2018;9(1):2419 doi 10.1038/s41467-018-04724-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Priestley P, Baber J, Lolkema MP, Steeghs N, de Bruijn E, Shale C, et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 2019;575(7781):210–6 doi 10.1038/s41586-019-1689-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li C, Bonazzoli E, Bellone S, Choi J, Dong W, Menderes G, et al. Mutational landscape of primary, metastatic, and recurrent ovarian cancer reveals c-MYC gains as potential target for BET inhibitors. Proc Natl Acad Sci U S A 2019;116(2):619–24 doi 10.1073/pnas.1814027116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell 2014;54(5):716–27 doi 10.1016/j.molcel.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang X, Han H, De Carvalho DD, Lay FD, Jones PA, Liang G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 2014;26(4):577–90 doi 10.1016/j.ccr.2014.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duymich CE, Charlet J, Yang X, Jones PA, Liang G. DNMT3B isoforms without catalytic activity stimulate gene body methylation as accessory proteins in somatic cells. Nat Commun 2016;7:11453 doi 10.1038/ncomms11453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baubec T, Colombo DF, Wirbelauer C, Schmidt J, Burger L, Krebs AR, et al. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 2015;520(7546):243–7 doi 10.1038/nature14176. [DOI] [PubMed] [Google Scholar]
- 43.Solomon SD, McMurray JJ, Pfeffer MA, Wittes J, Fowler R, Finn P, et al. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med 2005;352(11):1071–80 doi 10.1056/NEJMoa050405. [DOI] [PubMed] [Google Scholar]
- 44.Yu J, Qin B, Moyer AM, Nowsheen S, Liu T, Qin S, et al. DNA methyltransferase expression in triple-negative breast cancer predicts sensitivity to decitabine. J Clin Invest 2018;128(6):2376–88 doi 10.1172/JCI97924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Appleton K, Mackay HJ, Judson I, Plumb JA, McCormick C, Strathdee G, et al. Phase I and pharmacodynamic trial of the DNA methyltransferase inhibitor decitabine and carboplatin in solid tumors. J Clin Oncol 2007;25(29):4603–9 doi 10.1200/JCO.2007.10.8688. [DOI] [PubMed] [Google Scholar]
- 46.Jones PA, Issa JP, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet 2016;17(10):630–41 doi 10.1038/nrg.2016.93. [DOI] [PubMed] [Google Scholar]
- 47.Oza AM, Matulonis UA, Alvarez Secord A, Nemunaitis J, Roman LD, Blagden SP, et al. A Randomized Phase II Trial of Epigenetic Priming with Guadecitabine and Carboplatin in Platinum-resistant, Recurrent Ovarian Cancer. Clin Cancer Res 2020;26(5):1009–16 doi 10.1158/1078-0432.CCR-19-1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Corsello SM, Bittker JA, Liu Z, Gould J, McCarren P, Hirschman JE, et al. The Drug Repurposing Hub: a next-generation drug library and information resource. Nat Med 2017;23(4):405–8 doi 10.1038/nm.4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Colleoni M, Sun Z, Price KN, Karlsson P, Forbes JF, Thurlimann B, et al. Annual Hazard Rates of Recurrence for Breast Cancer During 24 Years of Follow-Up: Results From the International Breast Cancer Study Group Trials I to V. J Clin Oncol 2016;34(9):927–35 doi 10.1200/JCO.2015.62.3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Forget P, Bentin C, Machiels JP, Berliere M, Coulie PG, De Kock M. Intraoperative use of ketorolac or diclofenac is associated with improved disease-free survival and overall survival in conservative breast cancer surgery. Br J Anaesth 2014;113 Suppl 1:i82–7 doi 10.1093/bja/aet464. [DOI] [PubMed] [Google Scholar]
- 51.Schack A, Fransgaard T, Klein MF, Gogenur I. Perioperative Use of Nonsteroidal Anti-inflammatory Drugs Decreases the Risk of Recurrence of Cancer After Colorectal Resection: A Cohort Study Based on Prospective Data. Ann Surg Oncol 2019;26(12):3826–37 doi 10.1245/s10434-019-07600-8. [DOI] [PubMed] [Google Scholar]
- 52.Panigrahy D, Gartung A, Yang J, Yang H, Gilligan MM, Sulciner ML, et al. Preoperative stimulation of resolution and inflammation blockade eradicates micrometastases. J Clin Invest 2019;129(7):2964–79 doi 10.1172/JCI127282. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.