

Abstract
Background and Purpose
Misuse of opioids has greatly affected our society. One potential solution is to develop analgesics that act at targets other than opioid receptors. These can be used either as stand‐alone therapeutics or to improve the safety profile of opioid drugs. Previous research showed that activation of Gq/11 proteins by G‐protein coupled receptors has pro‐nociceptive properties, suggesting that blockade of Gq/11 signalling could be beneficial for pain control. The aim of this study was to test this hypothesis pharmacologically by using potent and selective Gq/11 inhibitor YM‐254890.
Experimental Approach
We used a series of behavioural assays to evaluate the acute responses of mice to painful thermal stimulation while administering YM‐254890 alone and in combination with morphine. We then used electrophysiological recordings to evaluate the effects of YM‐254890 on the excitability of dorsal root ganglion (DRG) nociceptor neurons.
Key Results
We found that systemic administration of YM‐254890 produced anti‐nociceptive effects and also augmented morphine analgesia in both hotplate and tail flick paradigms. However, it also caused substantial inhibition of locomotion, which may limit its therapeutic utility. To circumvent these issues, we explored the local administration of YM‐254890. Intrathecal injections of YM‐254890 produced lasting analgesia in a tail flick test and greatly augmented the anti‐nociceptive effects of morphine without any significant effects on locomotor behaviour. Electrophysiological studies showed that YM‐254890 reduced the excitability of DRG nociceptors and augmented their opioid‐induced inhibition.
Conclusion and Implications
These findings indicate that pharmacological inhibition of Gq/11 could be explored as an analgesic strategy.
Keywords: analgesia, G proteins, GPCR, Gq/11 signalling, opioids, pain, YM‐254890
Abbreviations
- DMEM
Dulbecco's modified Eagle's medium
- DRG
dorsal root ganglion
- FR
FR‐900359
- GDNF
glial cell line‐derived neurotrophic factor
- NGF
nerve growth factor
- PLCβ
phospholipase C beta
- YM
YM‐254890
What is already known
Opioids are effective analgesics but have unwanted side effects including addiction, dependence and respiratory depression.
Propagation of μ‐opioid receptor signals may be regulated by other signalling systems.
What does this study add
Gαq/11 blockade enhances the efficacy of systemic morphine administration in mouse models of acute pain.
Gαq/11 inhibition suppresses the activity of nociceptor neurons and synergizes with morphine effects on excitability.
What is the clinical significance
Pharmacological blockade of Gαq/11 may be a viable strategy for pain management.
1. INTRODUCTION
G‐protein coupled receptors (GPCRs) constitute the largest family of cell surface receptors with immense roles in nearly all known physiological processes including nociception, cardiovascular function and inflammation (Pfleger et al., 2019; Salzer et al., 2019; Sun & Ye, 2012). Accordingly, GPCRs are frequently targeted by small‐molecule drugs for therapeutic benefits (Davenport et al., 2020; Hauser et al., 2017). However, the pleiotropic effects associated with their activation or inhibition also often lead to unwanted side effects (Brianso et al., 2011; Sriram & Insel, 2018). Many GPCR actions are mediated by heterotrimeric G proteins (Neves et al., 2002; Oldham & Hamm, 2008). GPCRs activate G proteins by catalysing their nucleotide exchange leading to the release of Gβγ subunits from Gα (Hollmann Markus et al., 2005). Both active Gα‐GTP and free Gβγ transduce signals by interacting with a range of downstream effector molecules to elicit a cellular response (Hubbard & Hepler, 2006). There are 12 Gα subunits in mammals, grouped into five subfamilies (Gαs/olf, Gαi/o, Gαq/11, Gα12/13 and Gα15) and characterized by unique properties and selectivity with which they regulate their effectors (Hilger et al., 2018; Masuho et al., 2015). Studies with knockout mice indicate that individual G‐protein channels selectively contribute to various aspects of GPCR signalling and physiological reactions controlled by them (Cha et al., 2019; van den Bos et al., 2020).
Most GPCRs can activate several different G proteins, and signalling from different GPCRs also converge on the same set of G proteins (Marinissen & Gutkind, 2001). Furthermore, there is also significant evidence for both synergistic and opposing influence of different G proteins in regulating their effector molecules (Gupte et al., 2017; Oduori et al., 2020; Van Eps et al., 2018). Together, this points to the interplay between individual Gα subunits as an important, yet not well understood, process in shaping GPCR signalling and in vivo actions of these receptors. Accordingly, pharmacological targeting of individual G proteins is emerging as an attractive strategy with therapeutic potential (Campbell & Smrcka, 2018).
One particularly prominent pharmacological area of need is the modulation of pain. GPCR signalling is heavily involved in this process with many receptors strategically positioned across both ascending and descending nociceptive circuits and noted involvement in regulating various pain states (Geppetti et al., 2015). A notable example is provided by the μ‐opioid receptor (μ receptor) heavily targeted by opioid drugs to produce powerful analgesia (Gálvez & Pérez, 2012). Notably, several other GPCR systems also produce powerful analgesia (Stone & Molliver, 2009). However, activation of most of these GPCRs also triggers unwanted side effects including dependence, somatic, dysphoria prompting the search for alternative analgesic strategies (Volkow & McLellan, 2016).
In contrast to major anti‐nociceptive effects associated with activation of Gi/o receptors (Galeotti et al., 2002; Yudin & Rohacs, 2018), triggering Gq and Gs signalling generally leads to opposite outcomes, that is, hyperalgesia, sensitization to pain and allodynia (Crain & Shen, 2000; Malin & Molliver, 2010). Interestingly, increased expression of Gq and G11 proteins has been reported in animal models of pain (Belmadani et al., 2021; Saika et al., 2021). Genetic studies in mice indicate that loss of Gq and G11 results in reduced pain hypersensitivity in chronic pain states (Tappe‐Theodor et al., 2012). Moreover, knockout of Gαq/11 modulates properties of nociceptors and reduces basal pain sensitivity as well as pain‐sensitizing effects associated with activation of several Gαq/11‐coupled GPCRs (Wirotanseng et al., 2013). These observations suggest that suppressing Gq/11 may be a promising analgesic strategy.
Recently, related cyclic depsipeptides YM‐254890 (YM) and FR‐900359 (FR) were identified as potent and selective Gαq inhibitors. Their actions have been well characterized mechanistically (Nishimura et al., 2010), and they have been shown to have high chemical and metabolic stability (Schlegel et al., 2021). Their in vivo efficacy has been also established for cardiovascular effects (Meleka et al., 2019; Uemura, Kawasaki, et al., 2006), yet they have not been evaluated for the effects in the nervous system. Here, we present the results of exploring the use of the YM compound as an analgesic using acute pain models in mice. Using different routes of administration and nociceptive tests, we demonstrate in vivo anti‐nociceptive efficacy of Gq inhibition in a stand‐alone regime and in combination with opioids.
2. METHODS
2.1. Ethical statement
All studies were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institute of Health. All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) protocol (#16‐032) at The Scripps Research Institute. Every effort was made to minimize the number of animals used in the following experiments. Animal studies are reported in compliance with the ARRIVE guidelines (Percie du Sert et al., 2020) and with the recommendations made by the British Journal of Pharmacology (Lilley et al., 2020).
2.2. Compliance with requirements for studies using animals
Because mice have been widely used in previous investigations for opioid‐related research (Jirkof, 2017), mice were used to allow comparisons of the current results with the previous literature. Mice were also appropriate because they have been widely used in translational pharmacological research on opioid analgesia and other neurological disorders. Consequently, to allow comparison between subcutaneous and intrathecal studies, mice were used for all experiments. As sex‐specific differences in thermal nociception have been reported following opioid administration, we compared both routes of drug administration in male and female mice. Female mice have been reported to be no more variable than male mice across diverse traits relevant to neuroscience studies (Becker et al., 2016). Thus, both male and female mice were used for all the experiments described in the current study.
2.3. Experimental animals
C57BL/6 mice (6–8 weeks old) of both sexes were used and were bred in the vivarium. Mice were housed in groups of three to five (unless otherwise stated) on a 12‐h light–dark cycle (6:00 am light cycle; 6:00 pm dark cycle) with food (Teklad Global 16% protein rodent diets; Envigo Inc., WI, USA) and water available ad libitum. Animal groups were compiled to ensure minimization of factors (i.e., weight, sex and health). Mice were within 20–28 g in weight at start of all studies. All tested groups contained a control group and consisted of male and female mice. Males and females were tested on the same day but at different times so that they were not in the room at the same time. All the experiments were tested during the light cycle between 8:00 am and 3:00 pm to avoid any long‐term disruption of sleep cycles. Mice were transferred to the behavioural testing room at least 45 min before the first test to acclimatize. Following behavioural evaluation, animals were killed by CO2 inhalation, followed by cervical dislocation and postmortem decapitation.
2.4. Drug treatments
YM was purchased from AdipoGen® Life Sciences (San Diego, CA, USA). The morphine used was sterile and free of preservatives (morphine sulphate, Sigma‐Aldrich, St. Louis, MO, USA). For all in vivo studies, the vehicle used for YM compound is 0.05% dimethyl sulfoxide (DMSO) in 5% dextrose solution unless normal saline (NaCl 0.9%) is indicated as for morphine. To observe the systemic effects of YM administration, YM doses (0.1, 0.3, 0.5 and 1 mg·kg−1) were given subcutaneously. The dose of YM for subcutaneous injection was taken from previous published studies (Meleka et al., 2019). The dose of morphine selected for subcutaneous administration (2.5, 5 and 10 mg·kg−1) is commonly used in mouse analgesia (Kest et al., 2002). For the combined administration of morphine and YM on thermal anti‐nociception, YM was administered 10 min before the morphine. The latency of response was measured before injection of drugs (baseline latency response) and at different time intervals (post‐treatment latency response) after drugs injection.
2.5. Intrathecal injections
Intrathecal administration was performed following the method described previously (Hylden & Wilcox, 1980; Li et al., 2019) with slight modifications. All intrathecal injections were performed using a 25‐μl Hamilton syringe with a 30‐gauge needle. The injection volume was 5 μl for each mouse. Data suggest that this is likely to be the upper limit that can be reliably injected into the mouse without any appreciable redistribution of the drugs through the cerebro‐spinal fluid (CSF) to the basal cisterns of the brain (Rieselbach et al., 1962). Each solution was injected without injection cannulae. Intrathecal injections were made into the L5–L6 intervertebral space of unanaesthetized mice. The flick of the tail was considered indicative of a successful intrathecal administration.
2.6. Intracerebral injections
Intracerebroventricular administration was performed following the method described previously with slight modifications (Haley & McCormick, 1957; Narita et al., 2003). On the day of the drug/vehicle injection or on the day of behavioural test, the mouse was secured at the nape of the neck and head by the investigator's thumb and forefinger, and head of the mouse was held against a V‐shaped holder. A 27‐gauge hypodermic needle attached with 25‐μl Hamilton microsyringe was inserted perpendicularly into the unilateral injection site into the hole with the depth of 3.5 mm. The injection volume was 4 μl.
2.7. Behavioural assessment
2.7.1. Hotplate test
Animals were tested for the assessment of anti‐nociception and analgesic effects of morphine using a hotplate set to 52.5°C. The assay was performed as described previously (Bannon & Malmberg, 2007; Wang et al., 2019). Mice were placed in a Plexiglas chamber (16″ tall and 8″ in diameter) on a ceramic plate heated to 52.5°C and the timer started (Ugo Basile, Varese, Italy). Paw licking, paw flicking and jumping were coded as a nociceptive response, upon which the timer was stopped or up to a maximum of 20 s (for YM or vehicle) or 50 s (for morphine). Three trials were made with a 3‐min intertrial interval. The mean paw withdrawal latency from the three trials was used as the baseline latency. The analgesic effect of drug or vehicle was then determined by a single measurement of paw withdrawal or paw flicking or jumping latencies at respective time intervals. On the test day, mice were first tested for the baseline measurement (t = 0, no drug/vehicle given), and the response latency was recorded. The drug (YM or vehicle or morphine) was given after a few minutes, and the nociceptive response was recorded at 10, 20, 30, 60, 90 and 120 min and up to the time it reached baseline. For the combined administration effect of YM with morphine, YM was administered 10 min before the administration of morphine. Time (s) spent on hotplate was graphed as per cent maximum possible effect (MPE): MPE (%) = (test latency − baseline latency)∕(cut‐off time − baseline latency) × 100. Nociceptive responses were monitored on alternate days.
2.7.2. Tail immersion test
Anti‐nociception induced by YM or evaluation of morphine's analgesic effects was determined by tail immersion test. The test was performed as previously described (Wang et al., 2019). Individual mice were transferred to experimentation room and restrained using a well‐ventilated 50‐ml tube with air holes. In order to minimize handling and to facilitate both the drug delivery and testing, each mouse was comfortably positioned in the plastic restrainer tubes with both fore paws and hind paws extending through holes at the bottom of the restrainer. All animals were habituated to restraint 1 h for 3 days prior to behaviour test. After 3 days of training, the mouse usually voluntarily entered the tube‐shaped restrainer during the behavioural testing. No sign of distress was observed in these mice during restraint. Once the animal was immobilized (within 25–30 s), two thirds of the entire tail was dipped in water bath heated to 54°C. Based on our preliminary experiments and reported by others (Bohn et al., 2002; Stone et al., 1997), the tail flick latency is required to be relatively short (2–3 s) for the test to remain valid. To achieve these latencies, the water bath temperature was varied and determined to be 54°C. The tail flick latency was defined as the time from the onset of thermal heat to tail withdrawal. Three trials were made with a 3‐min intertrial interval in between. The mean tail flick latency from the three trials was used as the baseline latency. The analgesic effect of drug or vehicle was then determined by a single measurement of tail flick latencies at respective time intervals. A maximum cut‐off was 10 s (for YM or vehicle) or 20 s (for morphine). The results were then expressed as a percentage of the MPE using the equation described above. On the test day, mice were first tested for the baseline measurement (t = 0, no drug/vehicle given), and the response latency was recorded. The drug (YM or vehicle or morphine) was given after few minutes, and the nociceptive response was recorded at 10, 20, 30, 60, 90 and 120 min and up to the time until baseline was reached. For the effect of combined administration of YM with morphine, YM was administered 10 min before the administration of morphine.
2.7.3. Open‐field test
The open‐field test was used to measure locomotor activity in animals, where both baseline activity and drug‐induced changes can be quantified (Prut & Belzung, 2003). The distance travelled during the test period was recorded as the index of locomotor activity. Locomotor activity and position within the open field were measured for 2 h. The multiple‐unit open‐field maze consisted of four activity chambers. Each chamber was made from white high‐density and non‐porous plastic and measured 50 × 50 × 38 cm. Mice were placed individually in the centre of an open‐field arena with light bottom to contrast with animal colour, and the test started immediately. Light in the chamber was measured to be 40 lx. The arena was cleaned between each test using alcohol 70% to avoid interference from the smell of the previously tested animal. The exploratory activity was analysed within 2 h. Videos were analysed using an EthoVision XT16 system (Noldus Information Technology, Wageningen, The Netherlands) considering two previously defined areas: a central and an outer arena.
2.8. Randomization and blinding
Randomization was accomplished as follows. Animals were first assigned a group designation and weighed. Six different cohorts of C57BL/6J animals were used for the four different behavioural testing as reported in systemic (two cohorts for hotplate and tail immersion test), spinal (one cohort), central (one cohort) and open‐field test (two cohorts for systemic and spinal administration) paradigms. For each cohort, a total of 12 mice (6 male and 6 female) were divided into two different groups (6 animals: 3 males and 3 females per group). Then each mouse was assigned a temporary random number within that group. Then, the cages were randomized within the experimental group. All recordings were blinded prior to scoring. Experimental results that were not blinded were collected by program software. Program software was calibrated (~30 min before starting the experiment). No animals were excluded prior to and during studies because all were healthy.
2.9. Acutely dissociated dorsal root ganglion (DRG) preparation
Male or female mice between 1 and 2 months of age were used for acute DRG isolations. Animals were killed by CO2 inhalation, followed by cervical dislocation and postmortem decapitation. Dissociated DRG cultures were prepared as previously described (Perner & Sokol, 2021). Briefly, DRGs were dissected from 1‐ to 2‐month‐old mice in Hank's buffered salt solution (HBSS) and digested with Collagenase A and Dispase II (Sigma‐Aldrich) for 25 min at 37°, centrifuged at 200× g for 5 min and washed with Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), glutamate, sodium pyruvate, 1% penicillin and 1% streptomycin. DRGs were then triturated in Neurobasal A medium supplemented with 10% FBS, 1% penicillin, 1% streptomycin, 1% GlutaMAX, 2% B27, 25‐ng·L−1 nerve growth factor (NGF) and 2‐ng·L−1 glial cell line‐derived neurotrophic factor (GDNF). After plating on laminin‐coated coverslips, cells were incubated at 37°C overnight. Experiments were performed the day after dissection.
2.10. Whole‐cell patch‐clamp recordings
Nociceptors were identified by physiological classification protocols (Petruska et al., 2000) summarized in Figure S5 and further by responsivity to morphine. Each experimental group consisted of 12–18 DRG recordings from three animals, 1–2 of which were female. Hyperpolarization‐activated current (I h ) and kinetics of resulting transient currents were determined by stepping membrane potential from −60 to −110 mV for 500 ms in 10‐mV increments (Figure S4A). Outward currents were identified by first preconditioning membrane potential at −100 mV for 500 ms and then stepping from −60 to 40 mV in 20‐mV increments for 200 ms (Figure S4D). Inward current dynamics were determined by preconditioning at −80 mV and then stepping from −60 to 40 mV in 10‐mV increments (Figure S4G). A 6‐s, 0‐ to 2‐nA continuous ramp stimulation protocol was used to determine rheobase. Membrane capacitance (C m ), series resistance (R s ) and input resistance (R i ) were tested with a hyperpolarizing 10‐mV pulse of 10‐ms duration initially and at a 30‐s interval throughout the recording period. Cells were recorded only if initial series resistance was ≤20 MΩ and excluded if R s varied >20% while recording. Offline analysis identified clustering of a population n = 30 with uniform morphine responsivity that was selected for further analysis, with basal parameters ± SEM: C m = 27.31 ± 4.72 pF, R i = 492 ± 19.29 MΩ, I h = 7.10 ± 2.45 pA·pF−1, I A tau = 8.39 ± 0.41 ms and action potential (AP) tau = 1.22 ± 0.08 ms. K+ internal was used for all recordings, containing in mM: NaCl 6, NaOH 4, K‐gluconate 130, ethylene glycol‐bis(β‐aminoethyl ether)tetraacetic acid (EGTA) 11, 1 CaCl2, 1 MgCl2, 10 N‐(2‐hydroxyethyl)piperazine‐N′‐(2‐ethanesulphonic acid) (HEPES), Na2ATP 2 and Na2GTP 0.2, adjusted to pH 7.3–7.4 with HCl. External artificial CSF contained in mM: NaCl 125, KCl 3, KH2PO4 1.2, MgSO4 1.2, NaHCO3 25, CaCl2 2 and dextrose 10, adjusted to pH 7.3–7.4 with HCl.
2.11. Data collection and statistical analysis
Sample sizes appropriate for each type of experiment were estimated on the basis of pilot studies and were calculated on the basis of the equation (Eng, 2003): CI95 = 1.96s/√n, where CI stands for the confidence interval, 1.96 is the corresponding tabulated value for CI95, s is the standard deviation of the mean and n is the sample size. When experiments are novel, it is difficult to perform a priori sample size calculations (Curtis et al., 2018) because the effect size and variance are unknown. Therefore, for animal experiments, estimates of the expected variance and effect size from previous experiments using similar methods were used to estimate appropriate sample sizes a priori through statistical power calculations. Data are presented as mean ± SEM. A D'Agostino–Pearson test and Shapiro–Wilk normality tests were applied to evaluate data normality and homogeneity. Parametric statistics for normally distributed variables included unpaired t test and two‐way analysis of variance (ANOVA). In addition, group differences using two factors or independent variables were evaluated by two‐way ANOVA. Bonferroni's post hoc for multiple comparisons was applied when the main effects of factor were significant in the ANOVA analysis. A non‐parametric test (Spearman rank, R) was used to check correlations when one of the variables was not normally distributed. Kruskal–Wallis non‐parametric test followed by Dunn's multiple comparisons test was applied for the data that were not normally distributed. For the open‐field test, data recorded by EthoVision software were exported and tabulated in Excel. Thereafter, statistical analysis of these data was carried out using GraphPad Prism software. No animals were excluded from the study, and the data were monitored for statistical outliers. For electrophysiological data, measurements were performed with Clampfit Version 10.5 (Molecular Devices, San Jose, CA, USA). All the statistical analysis was performed using GraphPad Prism Version 9.0.0 for Windows (GraphPad Software, San Diego, CA, USA). Post‐hoc tests were run only if F achieved P<0.05 and there was no significant variance inhomogeneity. A P value less than 0.05 (P < 0.05) was considered statistically significant.
2.12. Compliance with design and statistical analysis requirements
The manuscript complies with the design and statistical analysis requirements of the British Journal of Pharmacology (Curtis et al., 2015) and its update (Curtis et al., 2018).
2.13. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander, Christopoulos et al., 2021; Alexander, Fabbro et al., 2021).
3. RESULTS
3.1. Effects of YM administration on central nociception and locomotion
We started by evaluating the effects of YM on pain responses of mice in the hotplate test by performing the dose–response studies administering the drug systemically via subcutaneous injections. A concentration range of 0.1–1 mg·kg−1 that we explored has been reported to be safe for systemic administration (Meleka et al., 2019). We found that treatment with YM at higher doses (0.5 and 1.0 mg·kg−1) produced increase in paw withdrawal latencies reaching maximal analgesic response at 30 min (unpaired t test, P < 0.05) after injection (Figures 1a and S1A). We observed no significant effect of YM at lower doses (0.1 and 0.3 mg·kg−1) (Figures 1a and S1A). To confirm that YM directly affects the central nervous system to produce analgesic effects, the drug was delivered directly into the brain. Indeed, we observed a dose‐dependent increase in both the extent and duration of analgesia upon YM administration (Figure S2).
FIGURE 1.
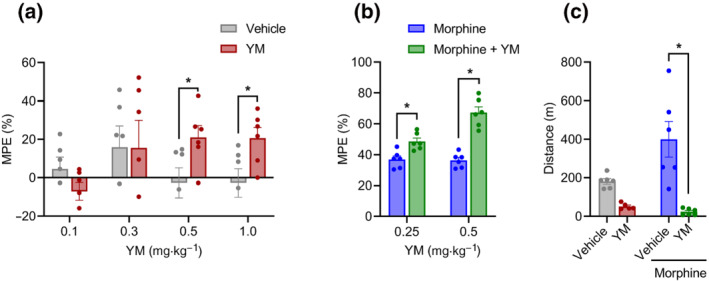
The effect of systemic subcutaneous administration of YM‐254890 (YM) on nociception and locomotion. (a) Dose–response effect of different concentrations of subcutaneous YM (0.1, 0.3, 0.5 and 1 mg·kg−1) and vehicle was tested on the hotplate test after 30 min of administration. Unpaired two‐tailed Student's t test of YM (0.5 mg·kg−1), P < 0.05, and YM (1.0 mg·kg−1), P < 0.05. (b) Effect of combined administration of subcutaneous YM (0.25 and 0.5 mg·kg−1) with a single dose of subcutaneous morphine (5 mg·kg−1) on the hotplate test after 30 min of morphine administration. YM was administered 10 min before the administration of morphine. Treatment: F (1, 10) = 59.06; dose: F (1, 10) = 13.21; and interaction: F (1, 10) = 12.08. Two‐way analysis of variance (ANOVA) with Bonferroni's post hoc test. (c) Changes in cumulative locomotor activity during 120 min of observation in open‐field test by subcutaneous administration of YM (0.5 mg·kg−1), morphine (5 mg·kg−1) and YM (0.5 mg·kg−1, 10 min before morphine) with morphine (5 mg·kg−1) and vehicle‐treated mice. Treatment: F (1, 19) = 26.02; effect of morphine: F (1, 19) = 3.71; and interaction: F (1, 19) = 6.2. Two‐way ANOVA with Bonferroni's post hoc test. In all panels, statistical analysis was performed combining both sexes, and significance was *P < 0.05; data sets (mean ± SEM) as analysed using two‐way ANOVA with Bonferroni's multiple comparisons test. MPE, maximum possible effect.
Next, we evaluated the effect of YM treatment on opioid‐induced analgesia. Administration of subthreshold dose of YM (0.25 mg·kg−1) significantly potentiated anti‐nociceptive effects in the hotplate test (Figures 1b and S1B). Increasing the dose (YM 0.5 mg·kg−1) led to further enhancement of opioid analgesia (Figures 1b and S1B). Interestingly, although YM treatment increased the maximal degree of the effect, it had no effect on either the onset timing or duration of the opioid analgesia with all the animals reaching maximal latencies in ~30 min and recovering to the baseline nociceptive thresholds in ~3 h.
To evaluate effects of YM treatment on locomotion behaviour, mice were tested in the open‐field test. We injected YM at the doses that produce the most significant analgesic effects both alone and in combination with morphine. Analysis of the results indicated that although morphine alone produced well‐known locomotor‐sensitizing effect, co‐treatment with the YM completely suppressed these effects and resulted in substantial decrease in locomotion comparable with the levels seen with the YM treatment alone (Figure 1c).
3.2. Effects of systemic Gαq/11 inhibition on spinal analgesia
Because changes in locomotor activity can confound the interpretation of the hotplate test, which relies on complex body movements, we next used the tail flick test, which relies on spinal reflexes to assess nociception in mice. We found that YM induced a significant anti‐nociceptive response at the highest dose of 1 mg·kg−1 (Figures 2a and S3A). However, no significant difference in nociceptive response was observed with lower doses (Figures 2a and S3A).
FIGURE 2.
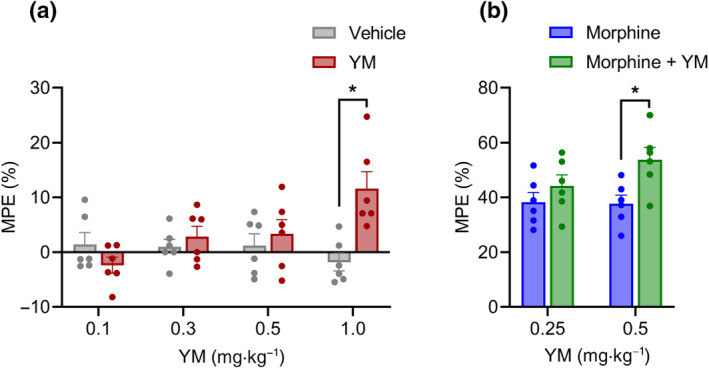
The effect of systemic subcutaneous administration of YM‐254890 (YM) on spinal analgesia. (a) Dose–response effect of different concentrations of subcutaneous YM (0.1, 0.3, 0.5 and 1 mg·kg−1) and vehicle was tested on the tail immersion test after 30 min of administration. Treatment: F (1, 40) = 5.14; dose: F (3, 40) = 2.175; and interaction: F (3, 40) = 5.85. Two‐way analysis of variance (ANOVA) with Bonferroni's post hoc test. (b) Effect of combined administration of subcutaneous YM (0.25 and 0.5 mg·kg−1) with a single dose of subcutaneous morphine (5 mg·kg−1) on the tail immersion test after 30 min of administration. YM was administered 10 min before the administration of morphine. Treatment: F (1, 20) = 8.21; dose: F (1, 20) = 1.38; and interaction: F (1, 20) = 1.75. Two‐way ANOVA with Bonferroni's post hoc test. In all panels, statistical analysis was performed combining both sexes, and significance was *P < 0.05; data sets (mean ± SEM) as analysed using two‐way ANOVA with Bonferroni's post hoc test. MPE, maximum possible effect.
Next, we evaluated the effect of YM treatment at subthreshold doses of morphine analgesia using the same tail immersion test (Figure 2b). We observed significant enhancement of the anti‐nociceptive effects of morphine at 0.5‐mg·kg−1 dose of YM (Figures 2b and S3B). Reducing the dose to 0.25 mg·kg−1 eliminated this effect (Figures 2b and S3B). In summary, these results indicate that when administered systemically, YM has analgesic properties, which are however confounded by effects on locomotor behaviour.
3.3. Analgesic properties of intrathecal YM treatment and its synergy with opioids
To avoid the side effects associated with systemic YM administration, we explored spinal intrathecal delivery, a route routinely used in clinical practice for mitigating unwanted effects especially in the context of opioid treatment (Fairbanks, 2003). When injected intrathecally, YM showed significant anti‐nociceptive properties in the tail immersion test at doses above 1.5 nmol per injection (Figures 3a and S4A). The magnitude of the effect did not change upon increasing the dose to 4.5 nmol suggesting the ceiling effect. The duration of the effect did not differ between the two maximally effective doses (3.0 and 4.5 nmol) with the animals returning to baseline nociception in about 2 h (Figures 3a and S4A).
FIGURE 3.
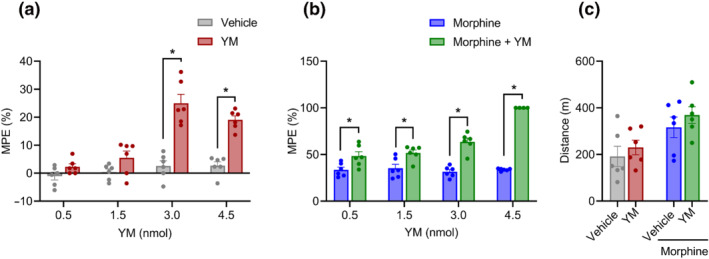
The effect of local intrathecal treatment of YM‐254890 (YM) on spinal analgesia and locomotion. (a) Dose–response effect of different concentrations of intrathecal YM (0.5, 1.5, 3.0 and 4.5 nmol) and vehicle was tested on tail immersion test after 30 min of administration. Treatment: F (1, 40) = 81.67; dose: F (3, 40) = 22.93; and interaction: F (3, 40) = 12.19. Two‐way analysis of variance (ANOVA) with Bonferroni's post hoc test. (b) Effect of combined administration of intrathecal YM (0.5, 1.5, 3.0 and 4.5 nmol) with a single low dose of subcutaneous morphine (2.5 mg·kg−1) on the tail immersion test after 30 min of administration. YM was administered 10 min before the administration of morphine. Treatment: F (1, 38) = 185.2; dose: F (3, 38) = 22.00; and interaction: F (3, 38) = 22.88. Two‐way ANOVA with Bonferroni's post hoc test. (c) Changes in cumulative locomotor activity during 120 min of observation in open‐field test by intrathecal administration of YM (3.0 nmol), subcutaneous morphine (2.5 mg·kg−1) and intrathecal YM (3.0 nmol, 10 min before morphine) with subcutaneous morphine (2.5 mg·kg−1)‐treated mice. Treatment: F (1, 20) = 1.35; effect of morphine: F (1, 20) = 11.36; and interaction: F (1, 20) = 0.032. Two‐way ANOVA with Bonferroni's post hoc test. In all panels, statistical analysis was performed combining both sexes, and significance was *P < 0.05; data sets (mean ± SEM) as analysed using two‐way ANOVA with Bonferroni's multiple comparisons test. MPE, maximum possible effect.
As before, we next tested the effects of intrathecal YM administration on systemic morphine analgesia in the tail immersion test. We observed that the lowest subthreshold dose of 0.5 nmol already significantly enhanced the anti‐nociceptive effects of morphine (Figures 3b and S4B). This effect was increased dose dependently until maximizing at the cut‐off value for the test (Figure S4B). At each of the doses, both the maximal extent and duration of the analgesic effects of morphine were increased. Because inhibition of motor activity was a significant confound of the YM‐induced analgesia upon systemic administration, we further monitored its effects on animal locomotion following intrathecal delivery. As expected from local delivery method, we found no significant changes in motor activity induced by YM when administered either alone or in combination with morphine (Figure 3c). Together, these results indicate effectiveness of YM as an analgesic at the level of the spinal cord and its substantial synergy with opioid‐induced analgesia.
3.4. Gαq/11 inhibition suppresses activity of DRG nociceptors and augments their responsiveness to opioid inhibition
To obtain insights into the mechanisms by which inhibition of Gq/11 produces analgesic effects and enhances morphine action, we examined the impact of YM on electrophysiological properties of DRG nociceptors in the peripheral nervous system. These neurons play a crucial role in nociception and express the direct target of opioid analgesics— the μ receptor (Rau et al., 2005; Ruda, 1986).
DRG nociceptors were identified by multiple electrophysiological characteristics (Figure S5) including presence of a hyperpolarization‐activated current, A‐type current inactivation rate, inward current dynamics and responsiveness to morphine. The effectiveness of YM as a Gq/11 antagonist in this population was verified by its ability to inhibit the effects of substance P, which mediates its effects via canonical Gq/11‐coupled pathway (Figure S6). Consistent with prior observations (Mizuta et al., 2012; Womack & McCleskey, 1995), application of morphine substantially decreased excitability of these DRG neurons as evidenced by a significant increase in rheobase when using ramp stimulation protocol (Figure 4a,b). Treatment with YM alone also caused significant decrease in the excitability of DRG nociceptors (Figure 4a,b). The magnitude of this effect was smaller as compared with morphine, consistent with the lower analgesic efficacy of YM relative to morphine observed in behavioural studies. A similar interaction was observed with resting input resistance (Figure 4c).
FIGURE 4.
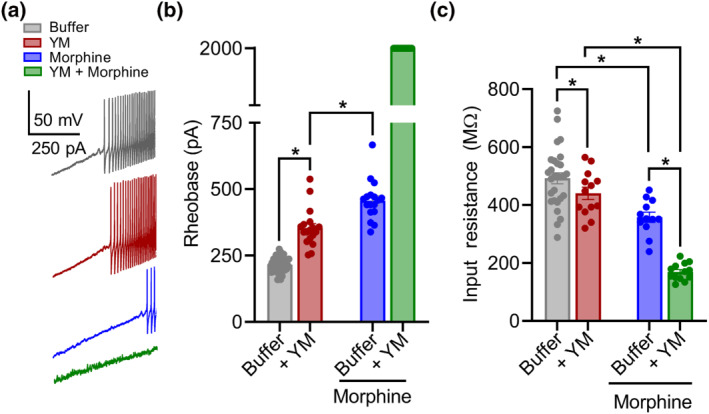
The effect of YM‐254890 (YM) treatment on morphine‐provoked inhibition of dorsal root ganglion (DRG) nociceptors. (a) Representative voltage traces from a continuous 0‐ to 2‐nA ramp stimulation protocol illustrating excitability of a cultured DRG neuron at baseline (black), after bath application of either 1‐μM morphine (blue) or 100‐nM YM (maroon) followed by both 100‐nM YM + 1‐μM morphine (green). (b) Quantification of rheobase from DRG recordings illustrated in (a). Co‐application of YM and morphine prevented action potential firing throughout the 2‐nA ramp protocol in all recordings. YM: F(1, 24) = 6032; morphine: F(1, 24) = 7591; and interaction: F(1, 24) = 4208. Two‐way analysis of variance (ANOVA) with Bonferroni's multiple comparisons test. (c) Quantification of resting input resistance at baseline (black) and after bath application of either 1‐μM morphine (blue) or 100‐nM YM (maroon) followed by both 100‐nM YM + 1‐μM morphine (green). YM: F(1, 24) = 6.636; morphine: F(1, 24) = 172.7; and interaction: F(1, 24) = 57.75. Two‐way ANOVA with Bonferroni's multiple comparisons test. Statistical analysis was performed combining both sexes, and significance was *P < 0.05.
To better understand these effects, we further studied AP dynamics with a voltage‐step protocol (Figure S5G–I). Again, application of either YM or morphine moderately reduced AP amplitude to approximately similar extents (Figure 4c,d). However, co‐application of YM and morphine largely prevented evoked AP responses (Figure 5a,b). This synergistic interaction was also apparent in the modulation of rapidly inactivating A‐type potassium currents (I A ) tested with voltage‐step protocol 2 (Figure S5D–F). Both YM and morphine significantly inhibited I A , whereas their coadministration nearly eliminated I A . These physiological effects demonstrate that YM and morphine interact synergistically to reduce nociceptor excitability as a mechanism for producing analgesic effects.
FIGURE 5.
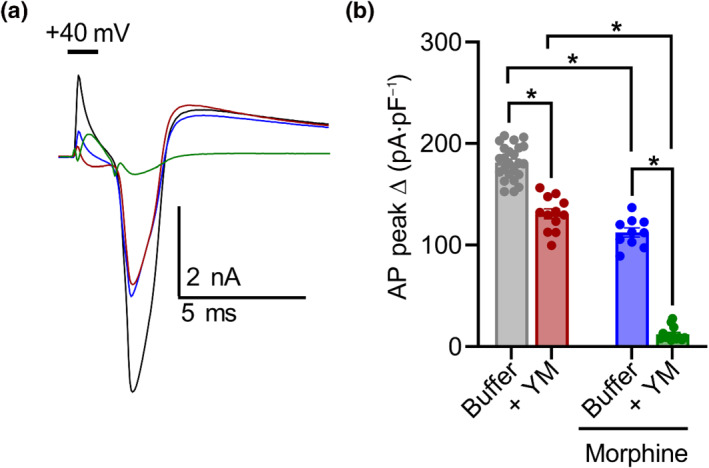
YM‐254890 (YM) and morphine interact to inhibit action potentials (APs) of dorsal root ganglion (DRG) nociceptors. (a) Representative current traces of AP profiles evoked by a 2‐ms, 40‐mV voltage step at baseline (black) and after bath application of either 1‐μM morphine (blue) or 100‐nM YM (maroon) followed by 100‐nM YM + 1‐μM morphine (green). (b) Quantification of peak AP amplitude normalized to capacitance. Co‐application of YM and morphine greatly inhibited evoked depolarization. YM: F(1, 20) = 283.0; morphine: F(1, 20) = 633.3; and interaction: F(1, 20) = 46.29. Two‐way analysis of variance (ANOVA) with Bonferroni's multiple comparisons test. Statistical analysis was performed combining both sexes, and significance was *P < 0.05.
4. DISCUSSION
In this study, we demonstrate that in vivo pharmacological inhibition of Gq/11 induces anti‐nociception in a mouse model of thermal pain. When administered intrathecally to healthy adult mice, YM produced analgesia in a dose‐dependent manner without noticeable side effects. The efficacy of this analgesia was moderate and reached a ceiling of around 30% of the maximal anti‐nociceptive effect that could be recorded in the test we used. However, local YM administration produced marked enhancement of analgesic effect of morphine. Intrathecal YM at the highest dose we used essentially maximized the otherwise low pain suppressant effect of low dose of morphine producing extremely long‐lasting and potent analgesia. These observations suggest that although tonically active Gαq/11 signalling contributes to setting nociceptive thresholds, its major role is likely in intersecting with the receptor signalling pathways, for example, the μ receptor involved in analgesia.
The involvement of Gαq/11 signalling in pain was suggested by earlier genetic studies in mice targeting the Gq/11 pathway, and the interventions at the level of Gq‐coupled GPCRs and downstream Gq/11 effectors also showed modulation of pain responses (Tappe‐Theodor et al., 2012; Wirotanseng et al., 2013). To the best of our knowledge, the present study is the first to explore direct pharmacological blockade of Gαq/11 in the context of pain and nervous system actions. This was made possible by recent development of compounds targeting Gαq/11—YM and FR. The YM compound used in this study is a selective and efficacious Gq inhibitor isolated from Chromobacterium species (Taniguchi et al., 2003). Although specificity of YM across different Gα subunits has been questioned (Peng et al., 2021), recent thorough investigation unequivocally established its selectivity towards Gαq and Gα11 (Patt et al., 2021).
Previously, the therapeutic potential of YM was demonstrated to promote antithrombotic and vasodilatory effects in mice, rats and monkeys (Kawasaki et al., 2003, 2005; Uemura, Kawasaki, et al., 2006; Uemura, Takamatsu, et al., 2006). In these studies, the Gq/11 inhibitor YM (1–30 μg·kg−1) was administered as a bolus injection directly into the bloodstream and resulted in substantial lowering of the systemic blood pressure. Very few studies examined performed systemic YM administration, where effects were observed with dosing ranging between 0.15 and 7.5 mg·kg−1 (Hitchman et al., 2021; Roszko et al., 2017). This generally agrees with the range of 0.1–1 mg·kg−1 of YM that we delivered subcutaneously to observe analgesic effects. Intrathecal and intracerebroventricular routes were explored in this study for the first time. We think that our observations regarding YM effects on inhibition of locomotor activity of mice are related to its depressant effects on cardiovascular function, which likely limit the utility of the systemic YM administration as an analgesic. We showed that these limitations could be bypassed by local administration of YM into the spinal cord, that produced efficacious analgesia without locomotor/cardiovascular side effects. Indeed, spinal delivery of YM may be achievable in humans, because intrathecal and epidural injections present a clinically viable route for the delivery of analgesics (Bottros & Christo, 2014) and because the spinal cord is the first relay site in the transmission of nociceptive information in the central nervous system (Zhuo et al., 2011).
A particularly intriguing observation of our study is an ability of YM to markedly enhance efficacy of opioid analgesics. This could allow lowering the dose of opioids administered, which will likely be beneficial by mitigating side effects associated with opioid therapies. Our mechanistic studies indicate that major site of this interaction occurs at the level of primary nociceptive neurons in the DRG located in the peripheral nervous system. Electrophysiological recordings revealed that although both YM and morphine can supress excitability of DRG nociceptors, their combinatorial application has a synergistic effect completely supressing their firing, thereby blocking reception of noxious stimuli.
Current therapeutically useful opioids such as morphine produce their analgesic and respiratory depressant side effects through activation of the μ receptor (Matthes et al., 1996). The μ receptor is a GPCR that signals through activation of Gi/o proteins and also via β‐arrestin recruitment (Williams et al., 2013). Differential engagement of effectors downstream from μ receptor activation, so‐called biased signalling or functional selectivity, is thought differentially to contribute to various effects associated with μ receptor engagement including its main therapeutic action: analgesia, and unwanted collateral effects such as respiratory depression, constipation and euphoria (DeWire et al., 2013; Manglik et al., 2016). The main focus in the field has been largely on exploring G protein versus β‐arrestin engagement in the efforts to explain functional selectivity of the μ receptor and exploit it therapeutically for dissociating opioid analgesia from the side effects (Bateman & Levitt, 2021; Pineyro & Nagi, 2021). However, recent evidence questioned the utility of this concept (Gillis et al., 2020), suggesting that other signalling mechanisms may be at play in routing μ receptor signals. One attractive area with significant potential for explaining how routing of μ receptor signals can be biased is its signalling crosstalk with other receptor systems that converge on common effectors to allow differential programming of cellular responses. Several signalling systems impacting processing of μ receptor signals and in vivo opioid actions have been described (Gibula‐Tarlowska & Kotlinska, 2020). Interestingly, studying one of these systems, we found that blockade of the Gq/11‐coupled orphan receptor GPR139 exerted augmentation of opioid signalling via the μ receptor and dissociated analgesia from withdrawal at the behavioural level (Wang et al., 2019). These findings are in line with other elements in the Gq pathway counteracting opioid effects initiated by the μ receptor (Javed et al., 2004; Mathews et al., 2008). Thus, we think that our observations with the YM compound enhancing opioid analgesia falls under the same overall theme as these and is likely explained by lifting the Gq effects on downstream effectors such as adenylyl cyclase (AC) and ion channels (Halls & Cooper, 2017; Stoveken et al., 2020) that oppose Gi/o signalling initiated by the μ receptor. Alternatively, blockade of the Gq/11 may diminish desensitizing regulation of the μ receptor by the phospholipase C beta (PLCβ)–PKC axis (Bailey et al., 2009; Xie et al., 1999), a major effector system of Gq/11 proteins. The exact mechanisms and intersecting points of such crosstalk are of interest to determine and should be the focus of future studies.
Our study also has some limitations that will need to be addressed in future. Our experiments have been restricted to acute pain and nociceptor physiology in a mouse model. Although we could exclude locomotor and acute cardiovascular side effects of YM, future work will need to investigate longer term YM effects on other organ systems and during chronic application and translate these observations to other species. Examining the efficacy of YM and other Gq/11 inhibitors in chronic and neuropathic pain models both alone and in combination with opioids also seems warranted. Nevertheless, we believe that the current study demonstrates the utility of pharmacological inhibition of Gq signalling as an analgesic strategy.
5. CONCLUSION
Our findings provide the evidence that pharmacological inhibition of Gq/11 is anti‐nociceptive and enhances opioid analgesia. We hope that these observations will spur further research in potential application of Gq/11 as a pain management strategy.
CONFLICTS OF INTEREST
K. A. M. is listed as an inventor on a provisional patent application related to the potential commercial utility of Gq/11 blockade as an analgesic strategy. S.M. and C.K. declare no conflicts of interest pertaining to this work.
AUTHOR CONTRIBUTIONS
S. M. carried out all the behavioural experiments, analysed the data and wrote the manuscript. C. K. performed electrophysiological studies and contributed to writing of the manuscript. K. A. M. supervised and conceived the study, analysed the data, wrote and revised the manuscript and provided funding for the study.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, and Animal Experimentation, and as recommended by funding agencies, publishers and other organizations engaged with supporting research.
Supporting information
Figure S1. The effect of systemic subcutaneous administration of YM on nociception.
Figure S2. The effect of intracerebroventricular injection of YM on nociception
Figure S3. The effect of systemic subcutaneous administration of YM on spinal analgesia.
Figure S4: The effect of local intrathecal administration of YM on spinal analgesia.
Figure S5. Physiological parameters of morphine‐responsive DRG neurons
Figure S6. The effect of YM on Substance P induced excitation of DRG nociceptors.
ACKNOWLEDGEMENTS
The authors thank Natalia Martemyanova for technical help with mouse husbandry and the members of Martemyanov Laboratory for helpful discussions. This work was supported by the National Institute on Drug Abuse of the NIH (Grant DA036596) (K. A. M.). The funders had no influence over the content of this publication. The authors have no competing financial interests in relation to the work described.
Marwari, S. , Kowalski, C. , & Martemyanov, K. A. (2022). Exploring pharmacological inhibition of Gq/11 as an analgesic strategy. British Journal of Pharmacology, 179(23), 5196–5208. 10.1111/bph.15935
Funding information National Institute on Drug Abuse, Grant/Award Number: DA036596
DATA AVAILABILITY STATEMENT
All data are presented in the manuscript or supporting information.
REFERENCES
- Alexander, S. P. , Fabbro, D. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , Armstrong, J. F. , Faccenda, E. , Harding, S. D. , Pawson, A. J. , Southan, C. , Davies, J. A. , Boison, D. , Burns, K. E. , Dessauer, C. , Gertsch, J. , Helsby, N. A. , Izzo, A. A. , Koesling, D. , … Wong, S. S. (2021). THE CONCISE GUIDE TO PHARMACOLOGY 2021/22: Enzymes. British Journal of Pharmacology, 178(S1), S313–S411. 10.1111/bph.15542 [DOI] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , Armstrong, J. F. , Faccenda, E. , Harding, S. D. , Pawson, A. J. , Southan, C. , Davies, J. A. , Abbracchio, M. P. , Alexander, W. , Al‐Hosaini, K. , Bäck, M. , Barnes, N. M. , Bathgate, R. , … Ye, R. D. (2021). The Concise Guide to PHARMACOLOGY 2021/22: G protein‐coupled receptors. British Journal of Pharmacology, 178, S27–S156. [DOI] [PubMed] [Google Scholar]
- Bailey, C. P. , Llorente, J. , Gabra, B. H. , Smith, F. L. , Dewey, W. L. , Kelly, E. , & Henderson, G. (2009). Role of protein kinase C and μ‐opioid receptor (MOPr) desensitization in tolerance to morphine in rat locus coeruleus neurons. The European Journal of Neuroscience, 29, 307–318. 10.1111/j.1460-9568.2008.06573.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannon, A. W. , & Malmberg, A. B. (2007). Models of nociception: Hot‐plate, tail‐flick, and formalin tests in rodents. In Current protocols in neuroscience, Chapter 8: Unit 8.9. Wiley. [DOI] [PubMed] [Google Scholar]
- Bateman, J. T. , & Levitt, E. S. (2021). Evaluation of G protein bias and β‐arrestin 2 signaling in opioid‐induced respiratory depression. American Journal of Physiology‐Cell Physiology, 321, C681–C683. 10.1152/ajpcell.00259.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker, J. B. , Prendergast, B. J. , & Liang, J. W. (2016). Female rats are not more variable than male rats: A meta‐analysis of neuroscience studies. Biology of Sex Differences, 7, 34. 10.1186/s13293-016-0087-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmadani, A. , Jayaraj, N. D. , George, D. S. , Ren, D. , Rathwell, C. , Miller, R. J. , & Menichella, D. M. (2021). Activation of keratinocyte Gq‐linked G‐protein coupled receptors regulates degeneration of cutaneous nerves. The Journal of Pain, 22, 581. 10.1016/j.jpain.2021.03.016 [DOI] [Google Scholar]
- Bohn, L. M. , Lefkowitz, R. J. , & Caron, M. G. (2002). Differential mechanisms of morphine antinociceptive tolerance revealed in βarrestin‐2 knock‐out mice. The Journal of Neuroscience, 22, 10494–10500. 10.1523/JNEUROSCI.22-23-10494.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottros, M. M. , & Christo, P. J. (2014). Current perspectives on intrathecal drug delivery. Journal of Pain Research, 7, 615–626. 10.2147/JPR.S37591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brianso, F. , Carrascosa, M. C. , Oprea, T. I. , & Mestres, J. (2011). Cross‐pharmacology analysis of G protein‐coupled receptors. Current Topics in Medicinal Chemistry, 11, 1956–1963. 10.2174/156802611796391285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, A. P. , & Smrcka, A. V. (2018). Targeting G protein‐coupled receptor signalling by blocking G proteins. Nature Reviews. Drug Discovery, 17, 789–803. 10.1038/nrd.2018.135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha, H. L. , Choi, J.‐M. , Oh, H.‐H. , Bashyal, N. , Kim, S.‐S. , Birnbaumer, L. , & Suh‐Kim, H. (2019). Deletion of the α subunit of the heterotrimeric Go protein impairs cerebellar cortical development in mice. Molecular Brain, 12, 57. 10.1186/s13041-019-0477-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crain, S. M. , & Shen, K. F. (2000). Antagonists of excitatory opioid receptor functions enhance morphine's analgesic potency and attenuate opioid tolerance/dependence liability. Pain, 84, 121–131. 10.1016/S0304-3959(99)00223-7 [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , Hoyer, D. , Insel, P. A. , Izzo, A. A. , Ji, Y. , MacEwan, D. J. , Sobey, C. G. , Stanford, S. C. , Teixeira, M. M. , Wonnacott, S. , & Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, M. J. , Bond, R. A. , Spina, D. , Ahluwalia, A. , Alexander, S. P. , Giembycz, M. A. , Gilchrist, A. , Hoyer, D. , Insel, P. A. , Izzo, A. A. , & Lawrence, A. J. (2015). Experimental design and analysis and their reporting: New guidance for publication in BJP. British Journal of Pharmacology, 172, 3461–3471. 10.1111/bph.12856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport, A. P. , Scully, C. C. G. , de Graaf, C. , Brown, A. J. H. , & Maguire, J. J. (2020). Advances in therapeutic peptides targeting G protein‐coupled receptors. Nature Reviews Drug Discovery, 19, 389–413. 10.1038/s41573-020-0062-z [DOI] [PubMed] [Google Scholar]
- DeWire, S. M. , Yamashita, D. S. , Rominger, D. H. , Liu, G. , Cowan, C. L. , Graczyk, T. M. , Chen, X. T. , Pitis, P. M. , Gotchev, D. , Yuan, C. , Koblish, M. , Lark, M. W. , & Violin, J. D. (2013). A G protein‐biased ligand at the μ‐opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. Journal of Pharmacology and Experimental Therapeutics, 344, 708–717. 10.1124/jpet.112.201616 [DOI] [PubMed] [Google Scholar]
- Eng, J. (2003). Sample size estimation: How many individuals should be studied? Radiology, 227, 309–313. 10.1148/radiol.2272012051 [DOI] [PubMed] [Google Scholar]
- Fairbanks, C. A. (2003). Spinal delivery of analgesics in experimental models of pain and analgesia. Advanced Drug Delivery Reviews, 55, 1007–1041. 10.1016/S0169-409X(03)00101-7 [DOI] [PubMed] [Google Scholar]
- Galeotti, N. , Ghelardini, C. , & Bartolini, A. (2002). Antihistamine antinociception is mediated by Gi‐protein activation. Neuroscience, 109, 811–818. 10.1016/S0306-4522(01)00537-1 [DOI] [PubMed] [Google Scholar]
- Gálvez, R. , & Pérez, C. (2012). Is morphine still the best reference opioid? Pain Manag, 2, 33–45. 10.2217/pmt.11.78 [DOI] [PubMed] [Google Scholar]
- Geppetti, P. , Veldhuis Nicholas, A. , Lieu, T. , & Bunnett Nigel, W. (2015). G protein‐coupled receptors: Dynamic machines for signaling pain and itch. Neuron, 88, 635–649. 10.1016/j.neuron.2015.11.001 [DOI] [PubMed] [Google Scholar]
- Gibula‐Tarlowska, E. , & Kotlinska, J. H. (2020). Crosstalk between opioid and anti‐opioid systems: An overview and its possible therapeutic significance. Biomolecules, 10. 10.3390/biom10101376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillis, A. , Kliewer, A. , Kelly, E. , Henderson, G. , Christie, M. J. , Schulz, S. , & Canals, M. (2020). Critical assessment of G protein‐biased agonism at the μ‐opioid receptor. Trends in Pharmacological Sciences, 41, 947–959. 10.1016/j.tips.2020.09.009 [DOI] [PubMed] [Google Scholar]
- Gupte, T. M. , Malik, R. U. , Sommese, R. F. , Ritt, M. , & Sivaramakrishnan, S. (2017). Priming GPCR signaling through the synergistic effect of two G proteins. Proceedings of the National Academy of Sciences, 114, 3756–3761. 10.1073/pnas.1617232114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haley, T. J. , & McCormick, W. G. (1957). Pharmacological effects produced by intracerebral injection of drugs in the conscious mouse. British Journal of Pharmacology and Chemotherapy, 12, 12–15. 10.1111/j.1476-5381.1957.tb01354.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halls, M. L. , & Cooper, D. M. F. (2017). Adenylyl cyclase signalling complexes—Pharmacological challenges and opportunities. Pharmacology & Therapeutics, 172, 171–180. 10.1016/j.pharmthera.2017.01.001 [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , Gray, A. J. G. , Bruce, L. , Alexander, S. P. H. , Anderton, S. , Bryant, C. , Davenport, A. P. , Doerig, C. , Fabbro, D. , Levi‐Schaffer, F. , Spedding, M. , Davies, J. A. , & NC‐IUPHAR . (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser, A. S. , Attwood, M. M. , Rask‐Andersen, M. , Schiöth, H. B. , & Gloriam, D. E. (2017). Trends in GPCR drug discovery: New agents, targets and indications. Nature Reviews. Drug Discovery, 16, 829–842. 10.1038/nrd.2017.178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilger, D. , Masureel, M. , & Kobilka, B. K. (2018). Structure and dynamics of GPCR signaling complexes. Nature Structural & Molecular Biology, 25, 4–12. 10.1038/s41594-017-0011-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitchman, T. D. , Bayshtok, G. , Ceraudo, E. , Moore, A. R. , Lee, C. , Jia, R. , Wang, N. , Pachai, M. R. , Shoushtari, A. N. , Francis, J. H. , Guan, Y. , Chen, J. , Chang, M. T. , Taylor, B. S. , Sakmar, T. P. , Huber, T. , Chi, P. , & Chen, Y. (2021). Combined inhibition of Gαq and MEK enhances therapeutic efficacy in uveal melanoma. Clinical Cancer Research, 27, 1476–1490. 10.1158/1078-0432.CCR-20-2860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollmann Markus, W. , Strumper, D. , Herroeder, S. , Durieux Marcel, E. , & Warltier David, C. (2005). Receptors, G proteins, and their interactions. Anesthesiology, 103, 1066–1078. 10.1097/00000542-200511000-00022 [DOI] [PubMed] [Google Scholar]
- Hubbard, K. B. , & Hepler, J. R. (2006). Cell signalling diversity of the Gqα family of heterotrimeric G proteins. Cellular Signalling, 18, 135–150. 10.1016/j.cellsig.2005.08.004 [DOI] [PubMed] [Google Scholar]
- Hylden, J. L. , & Wilcox, G. L. (1980). Intrathecal morphine in mice: A new technique. European Journal of Pharmacology, 67, 313–316. 10.1016/0014-2999(80)90515-4 [DOI] [PubMed] [Google Scholar]
- Javed, R. R. , Dewey, W. L. , Smith, P. A. , & Smith, F. L. (2004). PKC and PKA inhibitors reverse tolerance to morphine‐induced hypothermia and supraspinal analgesia in mice. European Journal of Pharmacology, 492, 149–157. 10.1016/j.ejphar.2004.03.061 [DOI] [PubMed] [Google Scholar]
- Jirkof, P. (2017). Side effects of pain and analgesia in animal experimentation. Lab Anim (NY), 46, 123–128. 10.1038/laban.1216 [DOI] [PubMed] [Google Scholar]
- Kawasaki, T. , Taniguchi, M. , Moritani, Y. , Hayashi, K. , Saito, T. , Takasaki, J. , Nagai, K. , Inagaki, O. , & Shikama, H. (2003). Antithrombotic and thrombolytic efficacy of YM‐254890, a Gq/11 inhibitor, in a rat model of arterial thrombosis. Thrombosis and Haemostasis, 90, 406–413. [DOI] [PubMed] [Google Scholar]
- Kawasaki, T. , Taniguchi, M. , Moritani, Y. , Uemura, T. , Shigenaga, T. , Takamatsu, H. , Hayashi, K. , Takasaki, J. , Saito, T. , & Nagai, K. (2005). Pharmacological properties of YM‐254890, a specific Gαq/11 inhibitor, on thrombosis and neointima formation in mice. Thrombosis and Haemostasis, 94, 184–192. [DOI] [PubMed] [Google Scholar]
- Kest, B. , Hopkins, E. , Palmese, C. A. , Adler, M. , & Mogil, J. S. (2002). Genetic variation in morphine analgesic tolerance: A survey of 11 inbred mouse strains. Pharmacology Biochemistry and Behavior, 73, 821–828. 10.1016/S0091-3057(02)00908-5 [DOI] [PubMed] [Google Scholar]
- Li, D. , Li, Y. , Tian, Y. , Xu, Z. , & Guo, Y. (2019). Direct intrathecal injection of recombinant adeno‐associated viruses in adult mice. Journal of Visualized Experiments. 10.3791/58565 [DOI] [PubMed] [Google Scholar]
- Lilley, E. , Stanford, S. C. , Kendall, D. E. , Alexander, S. P. H. , Cirino, G. , Docherty, J. R. , George, C. H. , Insel, P. A. , Izzo, A. A. , Ji, Y. , Panettieri, R. A. , Sobey, C. G. , Stefanska, B. , Stephens, G. , Teixeira, M. , & Ahluwalia, A. (2020). ARRIVE 2.0 and the British Journal of Pharmacology: Updated guidance for 2020. British Journal of Pharmacology, 177, 3611–3616. 10.1111/bph.15178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malin, S. A. , & Molliver, D. C. (2010). Gi‐ and Gq‐coupled ADP (P2Y) receptors act in opposition to modulate nociceptive signaling and inflammatory pain behavior. Molecular Pain, 6, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik, A. , Lin, H. , Aryal, D. K. , McCorvy, J. D. , Dengler, D. , Corder, G. , Levit, A. , Kling, R. C. , Bernat, V. , Hübner, H. , Huang, X. P. , Sassano, M. F. , Giguère, P. M. , Löber, S. , Duan, D. , Scherrer, G. , Kobilka, B. K. , Gmeiner, P. , Roth, B. L. , & Shoichet, B. K. (2016). Structure‐based discovery of opioid analgesics with reduced side effects. Nature, 537, 185–190. 10.1038/nature19112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinissen, M. J. , & Gutkind, J. S. (2001). G‐protein‐coupled receptors and signaling networks: Emerging paradigms. Trends in Pharmacological Sciences, 22, 368–376. 10.1016/S0165-6147(00)01678-3 [DOI] [PubMed] [Google Scholar]
- Masuho, I. , Ostrovskaya, O. , Kramer, G. M. , Jones, C. D. , Xie, K. , & Martemyanov, K. A. (2015). Distinct profiles of functional discrimination among G proteins determine the actions of G protein‐coupled receptors. Science Signaling, 8, ra123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews, J. L. , Smrcka, A. V. , & Bidlack, J. M. (2008). A novel Gbetagamma‐subunit inhibitor selectively modulates mu‐opioid‐dependent antinociception and attenuates acute morphine‐induced antinociceptive tolerance and dependence. The Journal of Neuroscience, 28, 12183–12189. 10.1523/JNEUROSCI.2326-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthes, H. W. , Maldonado, R. , Simonin, F. , Valverde, O. , Slowe, S. , Kitchen, I. , Befort, K. , Dierich, A. , le Meur, M. , Dollé, P. , Tzavara, E. , Hanoune, J. , Roques, B. P. , & Kieffer, B. L. (1996). Loss of morphine‐induced analgesia, reward effect and withdrawal symptoms in mice lacking the μ‐opioid‐receptor gene. Nature, 383, 819–823. 10.1038/383819a0 [DOI] [PubMed] [Google Scholar]
- Meleka, M. M. , Edwards, A. J. , Xia, J. , Dahlen, S. A. , Mohanty, I. , Medcalf, M. , Aggarwal, S. , Moeller, K. D. , Mortensen, O. V. , & Osei‐Owusu, P. (2019). Anti‐hypertensive mechanisms of cyclic depsipeptide inhibitor ligands for Gq/11 class G proteins. Pharmacological Research, 141, 264–275. 10.1016/j.phrs.2019.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuta, K. , Fujita, T. , & Kumamoto, E. (2012). Inhibition by morphine and its analogs of action potentials in adult rat dorsal root ganglion neurons. Journal of Neuroscience Research, 90, 1830–1841. 10.1002/jnr.23059 [DOI] [PubMed] [Google Scholar]
- Narita, M. , Imai, S. , Ozaki, S. , Suzuki, M. , Narita, M. , & Suzuki, T. (2003). Reduced expression of a novel μ‐opioid receptor (MOR) subtype MOR‐1B in CXBK mice: Implications of MOR‐1B in the expression of MOR‐mediated responses. The European Journal of Neuroscience, 18, 3193–3198. 10.1111/j.1460-9568.2003.03052.x [DOI] [PubMed] [Google Scholar]
- Neves, S. R. , Ram, P. T. , & Iyengar, R. (2002). G protein pathways. Science, 296, 1636–1639. 10.1126/science.1071550 [DOI] [PubMed] [Google Scholar]
- Nishimura, A. , Kitano, K. , Takasaki, J. , Taniguchi, M. , Mizuno, N. , Tago, K. , Hakoshima, T. , & Itoh, H. (2010). Structural basis for the specific inhibition of heterotrimeric Gq protein by a small molecule. Proceedings of the National Academy of Sciences, 107, 13666–13671. 10.1073/pnas.1003553107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oduori, O. S. , Murao, N. , Shimomura, K. , Takahashi, H. , Zhang, Q. , Dou, H. , Sakai, S. , Minami, K. , Chanclon, B. , Guida, C. , Kothegala, L. , Tolö, J. , Maejima, Y. , Yokoi, N. , Minami, Y. , Miki, T. , Rorsman, P. , & Seino, S. (2020). Gs/Gq signaling switch in β cells defines incretin effectiveness in diabetes. The Journal of Clinical Investigation, 130, 6639–6655. 10.1172/JCI140046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham, W. M. , & Hamm, H. E. (2008). Heterotrimeric G protein activation by G‐protein‐coupled receptors. Nature Reviews. Molecular Cell Biology, 9, 60–71. 10.1038/nrm2299 [DOI] [PubMed] [Google Scholar]
- Patt, J. , Alenfelder, J. , Pfeil, E. M. , Voss, J. H. , Merten, N. , Eryilmaz, F. , Heycke, N. , Rick, U. , Inoue, A. , Kehraus, S. , Deupi, X. , Müller, C. E. , König, G. M. , Crüsemann, M. , & Kostenis, E. (2021). An experimental strategy to probe Gq contribution to signal transduction in living cells. The Journal of Biological Chemistry, 296, 100472. 10.1016/j.jbc.2021.100472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, Q. , Alqahtani, S. , Nasrullah, M. Z. A. , & Shen, J. (2021). Functional evidence for biased inhibition of G protein signaling by YM‐254890 in human coronary artery endothelial cells. European Journal of Pharmacology, 891, 173706. 10.1016/j.ejphar.2020.173706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percie du Sert, N. , Hurst, V. , Ahluwalia, A. , Alam, S. , Avey, M. T. , Baker, M. , Browne, W. J. , Clark, A. , Cuthill, I. C. , Dirnagl, U. , Emerson, M. , Garner, P. , Holgate, S. T. , Howells, D. W. , Karp, N. A. , Lazic, S. E. , Lidster, K. , MacCallum, C. J. , Macleod, M. , … Würbel, H. (2020). The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biology, 18, e3000410. 10.1371/journal.pbio.3000410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perner, C. , & Sokol, C. L. (2021). Protocol for dissection and culture of murine dorsal root ganglia neurons to study neuropeptide release. STAR Protoc, 2, 100333. 10.1016/j.xpro.2021.100333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petruska, J. C. , Napaporn, J. , Johnson, R. D. , Gu, J. G. , & Cooper, B. Y. (2000). Subclassified acutely dissociated cells of rat DRG: Histochemistry and patterns of capsaicin‐, proton‐, and ATP‐activated currents. Journal of Neurophysiology, 84, 2365–2379. 10.1152/jn.2000.84.5.2365 [DOI] [PubMed] [Google Scholar]
- Pfleger, J. , Gresham, K. , & Koch, W. J. (2019). G protein‐coupled receptor kinases as therapeutic targets in the heart. Nature Reviews Cardiology, 16, 612–622. 10.1038/s41569-019-0220-3 [DOI] [PubMed] [Google Scholar]
- Pineyro, G. , & Nagi, K. (2021). Signaling diversity of mu‐ and delta‐opioid receptor ligands: Re‐evaluating the benefits of β‐arrestin/G protein signaling bias. Cellular Signalling, 80, 109906. 10.1016/j.cellsig.2020.109906 [DOI] [PubMed] [Google Scholar]
- Prut, L. , & Belzung, C. (2003). The open field as a paradigm to measure the effects of drugs on anxiety‐like behaviors: A review. European Journal of Pharmacology, 463, 3–33. 10.1016/S0014-2999(03)01272-X [DOI] [PubMed] [Google Scholar]
- Rau, K. K. , Caudle, R. M. , Cooper, B. Y. , & Johnson, R. D. (2005). Diverse immunocytochemical expression of opioid receptors in electrophysiologically defined cells of rat dorsal root ganglia. Journal of Chemical Neuroanatomy, 29, 255–264. 10.1016/j.jchemneu.2005.02.002 [DOI] [PubMed] [Google Scholar]
- Rieselbach, R. E. , Chiro, G. D. , Freireich, E. J. , & Rall, D. P. (1962). Subarachnoid distribution of drugs after lumbar injection. New England Journal of Medicine, 267, 1273–1278. 10.1056/NEJM196212202672502 [DOI] [PubMed] [Google Scholar]
- Roszko, K. L. , Bi, R. , Gorvin, C. M. , Bräuner‐Osborne, H. , Xiong, X. F. , Inoue, A. , Thakker, R. V. , Strømgaard, K. , Gardella, T. , & Mannstadt, M. (2017). Knockin mouse with mutant Gα11 mimics human inherited hypocalcemia and is rescued by pharmacologic inhibitors. JCI Insight, 2, e91079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruda, M. A. (1986). The pattern and place of nociceptive modulation in the dorsal horn. In Yaksh T. L. (Ed.), Spinal afferent processing (pp. 141–164). Springer US. [Google Scholar]
- Saika, F. , Matsuzaki, S. , Kishioka, S. , & Kiguchi, N. (2021). Chemogenetic activation of CX3CR1‐expressing spinal microglia using Gq‐DREADD elicits mechanical allodynia in male mice. Cell, 10. 10.3390/cells10040874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzer, I. , Ray, S. , Schicker, K. , & Boehm, S. (2019). Nociceptor signalling through ion channel regulation via GPCRs. International Journal of Molecular Sciences, 20. 10.3390/ijms20102488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlegel, J. G. , Tahoun, M. , Seidinger, A. , Voss, J. H. , Kuschak, M. , Kehraus, S. , Schneider, M. , Matthey, M. , Fleischmann, B. K. , König, G. M. , Wenzel, D. , & Müller, C. E. (2021). Macrocyclic Gq protein inhibitors FR900359 and/or YM‐254890—Fit for translation? ACS Pharmacology & Translational Science, 4, 888–897. 10.1021/acsptsci.1c00021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram, K. , & Insel, P. A. (2018). G protein‐coupled receptors as targets for approved drugs: How many targets and how many drugs? Molecular Pharmacology, 93, 251–258. 10.1124/mol.117.111062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone, L. S. , MacMillan, L. B. , Kitto, K. F. , Limbird, L. E. , & Wilcox, G. L. (1997). The α2a adrenergic receptor subtype mediates spinal analgesia evoked by α2 agonists and is necessary for spinal adrenergic‐opioid synergy. The Journal of Neuroscience, 17, 7157–7165. 10.1523/JNEUROSCI.17-18-07157.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone, L. S. , & Molliver, D. C. (2009). In search of analgesia: Emerging poles of GPCRs in pain. Molecular Interventions, 9, 234–251. 10.1124/mi.9.5.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoveken, H. M. , Zucca, S. , Masuho, I. , Grill, B. , & Martemyanov, K. A. (2020). The orphan receptor GPR139 signals via Gq/11 to oppose opioid effects. Journal of Biological Chemistry, 295, 10822–10830. 10.1074/jbc.AC120.014770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, L. , & Ye, R. D. (2012). Role of G protein‐coupled receptors in inflammation. Acta Pharmacologica Sinica, 33, 342–350. 10.1038/aps.2011.200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi, M. , Nagai, K. , Arao, N. , Kawasaki, T. , Saito, T. , Moritani, Y. , Takasaki, J. , Hayashi, K. , Fujita, S. , Suzuki, K. I. , & Tsukamoto, S. I. (2003). YM‐254890, a novel platelet aggregation inhibitor produced by Chromobacterium sp. QS3666. Journal of Antibiotics (Tokyo), 56, 358–363. 10.7164/antibiotics.56.358 [DOI] [PubMed] [Google Scholar]
- Tappe‐Theodor, A. , Constantin, C. E. , Tegeder, I. , Lechner, S. G. , Langeslag, M. , Lepcynzsky, P. , Wirotanseng, R. I. , Kurejova, M. , Agarwal, N. , Nagy, G. , Todd, A. , Wettschureck, N. , Offermanns, S. , Kress, M. , Lewin, G. R. , & Kuner, R. (2012). Gαq/11 signaling tonically modulates nociceptor function and contributes to activity‐dependent sensitization. Pain, 153, 184–196. 10.1016/j.pain.2011.10.014 [DOI] [PubMed] [Google Scholar]
- Uemura, T. , Kawasaki, T. , Taniguchi, M. , Moritani, Y. , Hayashi, K. , Saito, T. , Takasaki, J. , Uchida, W. , & Miyata, K. (2006). Biological properties of a specific Gαq/11 inhibitor, YM‐254890, on platelet functions and thrombus formation under high‐shear stress. British Journal of Pharmacology, 148, 61–69. 10.1038/sj.bjp.0706711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura, T. , Takamatsu, H. , Kawasaki, T. , Taniguchi, M. , Yamamoto, E. , Tomura, Y. , Uchida, W. , & Miyata, K. (2006). Effect of YM‐254890, a specific Gαq/11 inhibitor, on experimental peripheral arterial disease in rats. European Journal of Pharmacology, 536, 154–161. 10.1016/j.ejphar.2006.02.048 [DOI] [PubMed] [Google Scholar]
- van den Bos, E. , Ambrosy, B. , Horsthemke, M. , Walbaum, S. , Bachg, A. C. , Wettschureck, N. , Innamorati, G. , Wilkie, T. M. , & Hanley, P. J. (2020). Knockout mouse models reveal the contributions of G protein subunits to complement C5a receptor–mediated chemotaxis. Journal of Biological Chemistry, 295, 7726–7742. 10.1074/jbc.RA119.011984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Eps, N. , Altenbach, C. , Caro, L. N. , Latorraca, N. R. , Hollingsworth, S. A. , Dror, R. O. , Ernst, O. P. , & Hubbell, W. L. (2018). Gi‐ and Gs‐coupled GPCRs show different modes of G‐protein binding. Proceedings of the National Academy of Sciences, 115, 2383–2388. 10.1073/pnas.1721896115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow, N. D. , & McLellan, A. T. (2016). Opioid abuse in chronic pain—Misconceptions and mitigation strategies. New England Journal of Medicine, 374, 1253–1263. 10.1056/NEJMra1507771 [DOI] [PubMed] [Google Scholar]
- Wang, D. , Stoveken, H. M. , Zucca, S. , Dao, M. , Orlandi, C. , Song, C. , Masuho, I. , Johnston, C. , Opperman, K. J. , Giles, A. C. , Gill, M. S. , Lundquist, E. A. , Grill, B. , & Martemyanov, K. A. (2019). Genetic behavioral screen identifies an orphan anti‐opioid system. Science, 365, 1267–1273. 10.1126/science.aau2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, J. T. , Ingram, S. L. , Henderson, G. , Chavkin, C. , von Zastrow, M. , Schulz, S. , Koch, T. , Evans, C. J. , & Christie, M. D. J. (2013). Regulation of μ‐opioid receptors: Desensitization, phosphorylation, internalization, and tolerance. Pharmacological Reviews, 65, 223–254. 10.1124/pr.112.005942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirotanseng, L. N. , Kuner, R. , & Tappe‐Theodor, A. (2013). Gq rather than G11 preferentially mediates nociceptor sensitization. Molecular Pain, 9, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womack, M. D. , & McCleskey, E. W. (1995). Interaction of opioids and membrane potential to modulate Ca2+ channels in rat dorsal root ganglion neurons. Journal of Neurophysiology, 73, 1793–1798. 10.1152/jn.1995.73.5.1793 [DOI] [PubMed] [Google Scholar]
- Xie, W. , Samoriski, G. M. , McLaughlin, J. P. , Romoser, V. A. , Smrcka, A. , Hinkle, P. M. , Bidlack, J. M. , Gross, R. A. , Jiang, H. , & Wu, D. (1999). Genetic alteration of phospholipase C β3 expression modulates behavioral and cellular responses to μ opioids. Proceedings of the National Academy of Sciences of the United States of America, 96, 10385–10390. 10.1073/pnas.96.18.10385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yudin, Y. , & Rohacs, T. (2018). Inhibitory Gi/O‐coupled receptors in somatosensory neurons: Potential therapeutic targets for novel analgesics. Molecular Pain, 14, 1744806918763646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo, M. , Wu, G. , & Wu, L. J. (2011). Neuronal and microglial mechanisms of neuropathic pain. Molecular Brain, 4, 31. 10.1186/1756-6606-4-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The effect of systemic subcutaneous administration of YM on nociception.
Figure S2. The effect of intracerebroventricular injection of YM on nociception
Figure S3. The effect of systemic subcutaneous administration of YM on spinal analgesia.
Figure S4: The effect of local intrathecal administration of YM on spinal analgesia.
Figure S5. Physiological parameters of morphine‐responsive DRG neurons
Figure S6. The effect of YM on Substance P induced excitation of DRG nociceptors.
Data Availability Statement
All data are presented in the manuscript or supporting information.