

Abstract
Dysregulation of Sonic hedgehog (SHH) signaling drives the growth of distinct cancer subtypes, including medulloblastoma (MB). Such cancers have been treated in the clinic with a number of clinically relevant SHH inhibitors, the majority of which target the upstream SHH regulator, Smoothened (SMO). Despite considerable efficacy, many of these patients develop resistance to these drugs, primarily due to mutations in SMO. Therefore, it is essential to identify druggable, signaling components downstream of SMO to target in SMO inhibitor resistant cancers. We utilized an integrated functional genomics approach to identify epigenetic regulators of SHH- signaling and identified a novel complex of Ubiquitin-like with PHD and RING finger domains 1 (UHRF1), DNA methyltransferase 1 (DNMT1), and GLI proteins. We show that this complex is distinct from previously described UHRF1/DNMT1 complexes, suggesting that it works in concert to regulate GLI activity in SHH driven tumors. Importantly, we show that UHRF1/DNMT1/GLI complex stability is targeted by a repurposed FDA-approved therapy, with a subsequent reduction in the growth of SHH-dependent MB ex vivo and in vivo.
Keywords: GLI, UHRF1, DNMT1, medulloblastoma, SHH
INTRODUCTION
Medulloblastoma is the most common malignant, pediatric brain tumor [1]. Large-scale genomic analyses have been used to stratify MB patients, based on their molecular drivers, into four major molecular subgroups WNT, SHH, group 3, and group 4 [1–3], and numerous subtypes within each subgroup [4]. SHH subgroup MB (SHH-MB) accounts for ~30% of MB cases and is characterized by hyperactivation of SHH signaling [5]. Although SHH-MB patients have an approximately 75% 5-year survival rate under standard of care treatment (surgery, radiation, and cytotoxic chemotherapy) (reviewed in [6]), many of them exhibit treatment-induced long-term sequelae, including neurocognitive difficulties, endocrinopathies and secondary malignancies [7]. Further, SHH-MB comprises four molecular subtypes (α, β, γ and δ) with distinct prognosis [4]. Of these subtypes, SHH α and β are associated with the worst outcome (5-year survival rates below 70%), likely due to their higher rate of TP53 mutation (SHH α) or increased metastases (SHH β) [4]. These clinical challenges highlight a significant need for targeted therapies that reduce the toxicity and long-term treatment induced sequelae of SHH-MB patients and improves the survival of subsets of this cohort with significantly poorer outcomes. Targeted therapeutics for SHH-MB patients have been undergoing evaluation in the clinic, including those capable of directly blocking SHH signaling [8–11]. Although these SHH inhibitors showed significant efficacy in SHH-MB patients, rapid drug resistance was also observed [8, 10]. Such work highlighted both the promise and challenges of inhibiting SHH signaling in MB patients.
SHH binding to its receptor, the main component of which is the multi-transmembrane protein Patched 1 (PTCH1), initiates its signaling [12, 13] (and reviewed in [14]) . PTCH1 functions as a negative regulator of the SHH pathway [15, 16], consistent with its role as a common tumor suppressor in basal cell carcinoma, MB, and rhabdomyosarcoma [17]. Ligand-bound PTCH1 releases its inhibition on the pivotal positive regulator of the SHH signaling pathway, SMO [18], a seven transmembrane protein that also functions as the target for three FDA approved small-molecule inhibitors [19–21]. SMO ultimately acts to regulate the activity, localization, and stability of the GLI family of transcription factors (GLI1-3) [14]. The three GLI transcription factors function as the ultimate effector, among which GLI1 and GLI2 are the dominant transcriptional activators, to regulate the expression of GLI target genes, including GLI1 [14]. In the absence of SHH, activation of GLI is prevented via their association with the negative regulator SUFU [22, 23]. GLI proteins also function in conjunction with various other transcriptional co-activator and co-repressor proteins [24]. Such proteins include Histone deacetylase 1 (HDAC1) and Histone deacetylase 2 (HDAC2), which enhance GLI transcriptional activity by removing their acetylate modifications [25]; Bromodomain containing 4 (BRD4), which coactivates the transcription of GLI-target genes [26, 27]; and Polycomb repressive complex 2 (PRC2) or lysine demethylase 6B (KDM6B/JMJD3), which interacts with GLI proteins to epigenetically mediate inhibition or activation of GLI-target genes respectively [28].
Given that rapid resistance to SMO inhibitors is driven mostly by mutations in SMO itself [29, 30], we set out to identify novel, druggable components of the SHH pathway that act downstream of SMO. We performed an epigenetic regulator focused siRNA-based screen and identified UHRF1 and DNMT1 as potential positive GLI regulators. Meta-analyses of MB genomic data showed that increased expression of either gene correlated with poor SHH-MB patient outcome. We also showed that both gene-products do indeed regulate SHH signaling downstream of SMO. Using SHH-MB sphere cultures, we provide evidence that UHRF1 and DNMT1 exist in complexes with GLI proteins, where they act to regulate the GLI activity that drives SHH-MB cell growth. Importantly, we also show that a clinically relevant DNMT1 inhibitor is able to inhibit the progression of MB in a SHH-MB orthotopic mouse model.
METHODS
Cell culture and constructs
Light2 (RRID:CVCL_2721), an NIH-3T3-derived cell line stably expressing a GLI-driven Firefly luciferase and a constitutive TK-driven Renilla luciferase reporter gene, was a gift of Dr. Philip A. Beachy (Stanford University, CA, USA) and was cultured in DMEM with 10% newborn calf serum (NCS), 1% penicillin/streptomycin, 0.2 μg/μl G418, and 0.1 μg/μl Zeocin [31]. Light2 cells were functionally authenticated for hedgehog responsiveness, using the SMO agonist SAG and a known SMO inhibitor (Vismodegib), but not molecularly authenticated. Immortalized Sufu−/− mouse embryonic fibroblasts (iMEFs) were a gift of Dr. Rune Tofgard (Karolinska Institute, Stockholm, Sweden) and cultured in DMEM with 10% FBS [32]. All immortalized cell lines were used for experiments between passage 20 and 30. MB4 and MB2 tumors were isolated from a spontaneous MB in Ptch1+/− (MB4 tumor) or Ptch1+/−;Trp53+/− (MB2 tumor) mice respectively [33, 34]. These tumors were further enriched in the flank of CD1-Foxn1nu mice and were never cultured ex vivo. MB sphere cultures (MSC4) (Ptch1−/−) and MSC2 (Ptch1−/−;Trp53−/−) were isolated from MB4 and MB2 tumors and cultured ex vivo (in DMEM/F12 with 2% B27, and 1% penicillin/streptomycin) as described before [35, 36]. MB propagating cells (MPC4) (Ptch1−/−) and MPC2 (Ptch1−/−;Trp53−/−) were similarly isolated from MB4 and MB2 tumors respectively, and selected in MPC enrichment culture (Neurobasal serum-free medium with 2% B27, 1% Glutamax, 1% N2, 25 ng/ml epidermal growth factor, 25 ng/ml fibroblast growth factor and 1% penicillin/streptomycin) as described before [37, 38]. MSCs and MPCs were genotyped by Transnetyx (Cordova, TN), and maintained in culture for less than 10 passages. The granular cell precursors (GCPs) were isolated from P4-6 C57BL/6 mice and cultured as described before [36]. The lab uses a mycoplasma sentinel monitoring system, in which a different lab member tests their cells for mycoplasma monthly using a MycoAlert detection kit (Lonza).
FLAG-tagged GLI1 and GLI2 plasmids were purchased from Addgene (Cat# 84922, RRID:Addgene_84922; Cat# 84920, RRID:Addgene_84920). Smart-pool siRNA targeting specific genes (Uhrf1, Smc1a, Dnmt1, Hdac7, Ddx39a, Gli2, Brd4) were purchased (Dharmacon). Individual siRNA targeting Uhrf1 were purchased from Dharmacon (#13: A-055507-13-0005; #14: A-055507-14-0005; #15: A-055507-15-0005; #16: A-055507-16-0005). All lentiviral shRNA constructs were purchased from Dharmacon as premade viral particles or as a bacterial glycerol stock expressing the relevant packaging plasmid and lentivirus generated as per manufacturer’s instructions (shUhrf1: V3SM11241-233453003; shDnmt1: #34:V3SM11241-234560879, #49:V3SM11241-236301813, #83:V3SM11241-234084147).
Transfection, transduction, and drug treatment
For the siRNA screen, 5 x 103 Light2 cells/well were seeded on 96-well plates. 100 nM pooled siRNA picked from a Mouse Druggable Genome siRNA Library v1.0 (Qiagen) were transfected the next day using Lipofectamine 2000 (Invitrogen) as per the manufacturer’s instructions. Three days later, 100 nM of the SMO agonist SAG (Selleckchem), was added to the cells in the presence of 0.5 % new born calf serum for 1 day. In all other experiments, Light2 cells and Sufu−/− iMEFs, were seeded at <40% confluency and then transfected using Lipofectamine 2000 (Invitrogen) or Lipofectamine RNAiMAX (Invitrogen), for plasmids or siRNA respectively, following the manufacturer’s instructions. For SAG treatment, in all experiments other than the siRNA screen, 100 nM SAG treatment was performed 48 h post-transfection in the presence of 0.5 % serum for 24 h. For MPC4 and MPC2, Lipofectamine 2000 (Invitrogen) and siGENOME smartpool siRNAs (Dharmacon) were used for gene knockdown. Briefly, spheres were digested in Accutase (Gibco) for 3 min and then 2 μL Lipofectamine 2000 was used to transfect siRNA, at a final concentration of 50 nM, into 3 x 105 cells. For MSC4 and MSC2, Accell smartpool siRNAs (Dharmacon) were used for gene knockdown. Spheres were similarly digested in Accutase (Gibco), and 4 x 105 cells resuspended in the mixture of Accell siRNA Delivery Media (Dharmacon) and siRNA at the final siRNA concentration at 1 μM. Cells were supplemented with B27 (Gibco) 24 h later, and additional complete media added 48 h later. Transduction for ex vivo experiments was initiated by infecting MSC4 cells via centrifugation in the presence of the lentivirus (MOI=1) for 2 h at 2,000 rpm, at room temperature. The infected cells were selected in 200 ng/mL puromycin (Invitrogen) for 3 days. For ex vivo drug treatments, 5-aza (Selleckchem), 5-aza-dCR (Selleckchem), GSK3484862 (MedChemExpress), cycloheximide (MilliporeSigma), and MG132 (Selleckchem) were used.
Assays
For analyses of gene expression, total RNA was extracted from cells using RNeasy Plus mini kit (Qiagene). RNA was converted into cDNA (Applied Biosystems) and analyzed using real-time qPCR (RT-qPCR) and TaqMan probes (Invitrogen). Gapdh expression was used for normalization.
Reduction of 3-(4,5-dimethyl-2-thiazolyl) 2,5-diphenyl-2H-tetrazolium bromide (MTT) to formazan [36] or CellTiter-Glo (Promega) was used to determine cell viability. EdU incorporation assays were performed using the Click-iT EdU Cell Proliferation Kit (Thermo Fisher Scientific), as per the manufacturer’s instructions.
For proximity ligation assays (PLA), sphere cultures were digested in Accutase (Gibco) for 3 min and 5 x 105 cells subsequently seeded on poly-L-Lysine pre-coated coverslips (Corning), in a 24-well plate, by centrifugation at 2000 rpm for 5min. PLA was then performed on these cells using a Duolink Proximity Ligation Assay (MilliporeSigma) as described by the manufacturer. The following antibodies were used for PLA: UHRF1 (Cat# sc-373750, Santa Cruz Biotechnology, RRID:AB_10947236), DNMT1 (Cat# 5032, Cell Signaling Technology, RRID:AB_10548197), GLI1 (prepared as described before [39]), GLI2 (Cat# AF3635, R&D Systems, RRID:AB_2111902), rabbit IgG (Cat# 12370, MilliporeSigma), mouse IgG (Cat# sc2025, Santa Cruz Biotechnology). DAPI co-stain was done using ProLong Gold Antifade Mountant with DAPI (Invitrogen)
Biochemistry
Cellular protein was extracted using RIPA buffer (Thermo Fisher Scientific) containing protease inhibitors cocktail (Thermo Fisher) and subjected to immunoblotting with the following antibodies: GLI1 (Cat# 2643, Cell Signaling Technology, RRID:AB_2294746), GLI2 (Cat# AF3635, R&D Systems, RRID:AB_2111902), UHRF1 (Cat# sc-373750, Santa Cruz Biotechnology, RRID:AB_10947236), DNMT1 (Cat# 5032, Cell Signaling Technology, RRID:AB_10548197), GAPDH (Cat# 5174, Cell Signaling Technology, RRID:AB_10622025), PCNA (Cat# 2586, Cell Signaling Technology, RRID:AB_2160343), cleaved Caspase 3 (Cat# 9661, Cell Signaling Technology, RRID:AB_2341188), HDAC1 (Cat# 59581, Cell Signaling Technology, RRID:AB_2799568). Protein levels were quantified using ImageJ (RRID:SCR_003070). Reporter activity of Light2 cells was determined using the Dual-Luciferase Reporter Assay kit (Promega).
Subcellular fractions for direct immunoblot or immunoprecipitation were prepared essentially as described before [40]. Briefly, cells were lysed in buffer A (10 mM HEPES pH 7.5, 1.5 mM MgCl2, 10 mM KCl, 0.4% NP-40, 1X protease inhibitor cocktail (Thermo Fisher) for 2 min on ice. These lysates were centrifuged for 10 min at 2000 g at 4 °C, and the supernatant saved as a cytoplasmic enriched fraction. The resultant pellets were washed twice, resuspended in buffer B (50 mM Tris-HCl pH 8.0, 300 mM NaCl, 1 mM EDTA, 1% NP-40, 1X protease inhibitor cocktail (Thermo Fisher) for 10 min on ice. These extracts were centrifuged for 10 min at 10,000g at 4°C, and the supernatant saved as a nuclear enriched fraction. For immunoprecipitations from the subcellular fractions, the cytoplasmic and nuclear enriched fractions were equilibrated in an equal volume of buffer B and buffer A, respectively. The fractions were subsequently pre-cleared in 20 μl Protein A/G magnetic beads (Thermo Fisher) and incubated with 2 μg of antibodies or IgG for 2 h at 4 °C. 20 μl Protein A/G magnetic beads (Thermo Fisher) were then added and incubated overnight at 4 °C. The beads were subsequently isolated and washed three times for 10 min, at 4 °C. The resulting beads were resuspended in Laemmli sample buffer (Biorad), boiled for 5 min, and subjected to immunoblotting. For immunoprecipitations from whole cell lysates, cells were directly lysed in buffer B (50 mM Tris-HCl pH 8.0, 300 mM NaCl, 1 mM EDTA, 1% NP-40, 1X protease inhibitor cocktail (Thermo Fisher), then subjected to the same procedure. The following antibodies were used for immunoprecipitation: GLI1 (prepared as described before [39]), GLI2 (Cat# AF3526, R&D Systems, RRID:AB_2279108), rabbit IgG (Cat# 12370, MilliporeSigma), sheep IgG (Cat# 5-001-A, R&D Systems).
For the size-exclusion chromatography, a Superose 6 Increase 10/300 size-exclusion chromatography column (Cytiva) was calibrated with a high molecular weight calibration kit (Cytiva) in 150 mM NaCl, 40 mM Tris·HCl pH 7.4, 1 mM EDTA, 0.5 mM DTT, 1% glycerol, 0.001% NP-40, using an AKTA Purifier FPLC (GE Healthsciences). Nuclear enriched fractions of MSC4 cells were prepared as described before [41] and 500 μL of sample at 2 mg/mL (adjusted using the column buffer) was loaded onto the column. The various fractions were subsequently analyzed by immunoblotting.
Immunohistochemical staining
Immunohistochemical analysis of the patient-derived MB TMA (#BC17012c, Biomax) and mouse tumor sections used the UHRF1 antibody (Cat# sc-373750, Santa Cruz Biotechnology, RRID:AB_10947236) or GLI1 antibody (Cat# MA5-26638, Thermo Fisher Scientific, RRID:AB_2724371) and the previously described procedure [36]. Blinded quantification of the Immunohistochemical TMA staining was performed by Dr. Nadji, (University of Miami) who is board-certificated in Anatomic and Clinical Pathology, using the following scoring system: 0 (negative), 1+ (< 10% of cells), 2+ (11-50% of cells), 3+ (>50% of cells).
Mouse experiments
All mouse experiments were conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee at the University of Miami and Medical University of South Carolina. Ptch1+/− (Ptch1tm1Mps/J, RRID:IMSR_JAX:003081)[33] and Trp53+/− (B6.129S2-Trp53tm1Tyj/J, RRID:IMSR_JAX:002101)[34] mice (Jackson laboratory) were mated to generate breeding colonies. Spontaneous tumors from the Ptch1+/− mice or the Ptch1+/−;Trp53+/− mice were digested using Accutase (Gibco) for 15 minutes, and single cell suspensions obtained by serial passages through a 100 μm and a 70 μm mesh. The resulting cells were further expanded in vivo as allografts using 6-week-old CD1-Foxn1nu mice (Charles River Laboratories), or ex vivo as MSC and MPC cultures. A patient derived orthotopic xenograft, from a patient with medulloblastoma possessing a TP53 germline missense mutation (A138P) and MYCN amplification, SJSHHMB-14-7196, was a kind gift of Dr. Roussel before publication (St Jude Children Research Hospital (Memphis)) [36] and was maintained in CD1-Foxn1nu mice. Orthotopic implantation methods and tissue processing were performed as described before [37].
For the orthotopic MSC4 implantation experiment, MSC4 cells were infected with Firefly luciferase expressing lentivirus (Cellomics Technology) and lentivirus carrying either a control shRNA or a Uhrf1 targeting shRNA via centrifugation in the presence of lentivirus (MOI=1) particles for 2 h at 2,000 rpm, at room temperature. The infected cells were allowed to recover overnight. 4 x 104 transduced cells were subsequently implanted into the cerebella of CD1-Foxn1nu mice, as described before [37]. D-Luciferin (Perkin Elmer) was administrated intraperitoneally at a concentration of 150 mg/kg 10 minutes before imaging. IVIS imaging was performed using a Caliper/Xenogen IVIS SPECTRUM. Bioluminescence intensity was quantified using a Living Image Advanced In Vivo Imaging Software (Perkin Elmer).
For the 5-Azacytidine (5-aza) treatment experiment, 2 x 106 MB4 tumor derived cells (which were never cultured ex vivo) were subcutaneously implanted into the flanks of CD1-Foxn1nu mice. When tumors reached ~100 mm3, mice were treated with 5 mg/kg 5-aza (Selleckchem) or vehicle (PBS) every other day through IV injections for a total of 4 doses. Tumors were collected 6 h after their last treatment. For the 5-aza treatment experiment in an orthotopic tumor model, 3 x 105 MB4 tumor derived cells were implanted orthotopically into 6-week-old CD1-Foxn1nu mice (Charles River Laboratories). Fourteen days after implantation, these mice were treated with 5 mg/kg 5-aza (Selleckchem) or vehicle (PBS) every three days through IV injections for a total of 6 doses. The mice were sacrificed 6 h after their last treatment and their brain tissues isolated and analyzed.
Bioinformatics and statistics
The gene dependence data of patient-derived cancer cell lines retrieved from Depmap (depmap.org/portal) were analyzed using GraphPad Prism (RRID:SCR_002798). The survival and expression data were retrieved from previous studies [4, 42–45], and analyzed using the GlioVis platform (gliovis.bioinfo.cnio.es) [46] and GraphPad Prism.
Unless otherwise indicated, results from ex vivo analyses show the mean of at least 3 independent experiments ± SEM. For EdU staining, 4 fields per condition from 3 independent experiments were quantified. For tumor size studies, the results shown represent the mean ± SEM of 7 mice for the shUHRF1 experiment or the mean ± SEM of 4 mice for the 5-aza experiment per experimental condition. The number of positive cells for IHC staining was quantitated from 4 fields for each mouse and summarized in bar graphs as the mean ± SEM of 4 mice per experimental condition. Statistical significance was determined by Student’s t test for 2-sample analyses, or Chi-square test for correlation, or log-rank Mantel-Cox test for analyzing Kaplan-Meier survival curves, or two-way ANOVA for 2-group analyses. p < 0.05 was considered as statistically significant and labelled with one asterisk.
Data availability
Data were generated by the authors are available within the paper and its supplementary files or from the authors on request. The bioinformatics data analyzed in this study was obtained as described in the “Bioinformatics analysis” section above.
RESULTS
Identification of GLI regulators
To screen for novel GLI regulators we selected siRNA targeting 577 transcriptional modulators from a siRNA library (Supplemental Table S1) and determined their ability to reduce GLI activity using a well-established GLI-dependent reporter cell line (Light2 cells) [31]. These cells were transfected with the individual siRNA and subsequently treated with the SMO agonist SAG to induce GLI activity (Fig. 1A and B). Focusing on those candidates whose knockdown reduced activity by greater than 50% relative to a control siRNA (Fig. 1C and Supplemental Fig. S1), we identified 83 potential regulators of GLI signaling (Supplemental Table S2). Amongst these candidates, 8 were previously reported to positively modulate SHH signaling (Supplemental Table S2) [28, 47–52], providing internal validation of our screen. We next used a bioinformatic analysis to further stratify these 83 candidates [4], and identified 8 whose expression correlates exclusively with SHH-MB patient outcome: UHRF1, Structural maintenance of chromosomes 1A (SMC1A), DNMT1, HDAC7, LIM homeobox 5 (LHX5), RNA polymerase II subunit D (POLR2D), RNA polymerase II subunit J (POLR2J) and DExD-box helicase 39A (DDX39A) (Table 1, Fig. 1D and E). This linkage again implicates them as potential regulators of GLI activity.
Figure 1. A focused siRNA screen identifies novel regulators of SHH signaling.
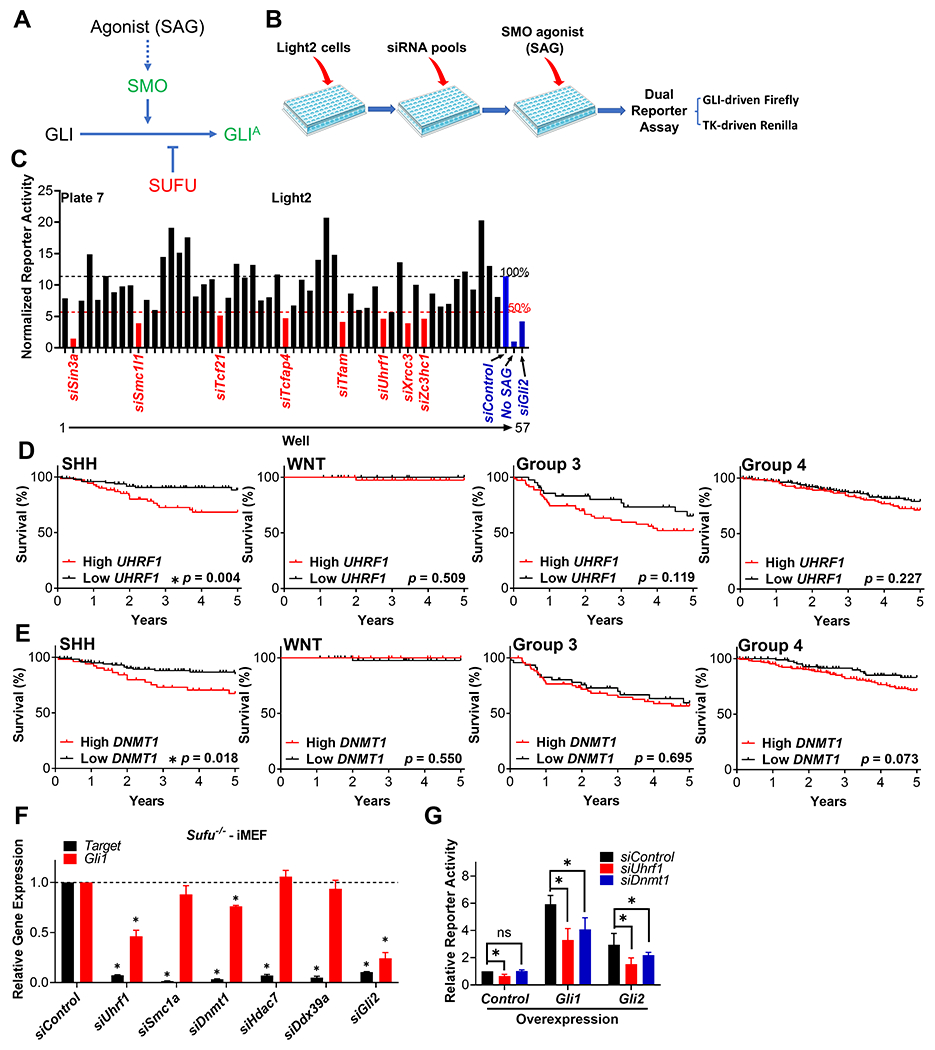
(A) A simplified schematic of the SHH signaling pathway, with positive regulators labeled in green and negative regulators labeled in red. GLIA: GLI activator form. (B) A schematic outlining the design of our siRNA-based screen. Light2 cells were seeded in 96-well plates, followed by transfection of pooled siRNAs (a mix of four distinct siRNA) from a siRNA library for 72h. The cells were then treated with the SMO agonist SAG in 0.5% serum for another 24h. Cells were then collected and GLI-driven Firefly luciferase activity determined and normalized to that of TK-driven Renilla luciferase. (C) Data from a representative plate in the siRNA screen. The normalized luciferase activity of each sample was compared to that of a scramble siRNA control (siControl) treated sample. The siRNA targets whose reduction in expression attenuated luciferase activity more than 50% were identified as positive hits and labeled in red. siRNA targeting Gli2 was included as a positive control. Cells that were treated with control siRNA (siControl) without SAG induction (No SAG) were included as an additional control. Using the data from Cavalli et al [4], the expression level of (D)UHFR1 and (E)DNMT1 in patient derived MB tissue was assigned to a high or low expression group based on the median values of UHRF1 expression, and each group’s 5-year survival determined across the four major MB subgroups. (F) Sufu−/− iMEFs were transfected with pooled siRNA targeting the indicated prioritized genes for 96 h. The expression of the siRNA-target (Target) and the SHH-dependent target gene Gli1 were determined. These data were normalized to that from siControl treated cells and presented as relative gene expression. LHX5 was not detected in MEFs and two candidates encoding subunits of RNA polymerase II (Polr2d and Polr2j) were not pursued further. (G) Light2 cells were transfected with the indicated pooled siRNA for 2 days, followed by transfection of the indicated plasmids for another 2 days. Cells were then collected and GLI-driven Firefly luciferase activity determined and normalized to TK-driven Renilla luciferase activity. Normalized reporter activity was then compared to the control samples and presented as relative reporter activity.
Table 1. The expression of eight GLI regulator candidates is associated with poor SHH-MB patient outcome.
Using outcome data from Cavalli et al [4], logrank tests of the 83 GLI regulator candidates identified in our siRNA-based screen were performed to determine if the expression level of each candidate was associated with patient outcome (5-year survival curve) across the four major subgroups of MB. Significant p values (<0.05) are highlighted in green. Only candidate genes whose expression was significantly associated with poor SHH-MB patient outcome are shown.
| Genes | p values (Logrank test) | |||
|---|---|---|---|---|
| SHH | WNT | Group 3 | Group 4 | |
| UHRF1 | 0.004 | 0.509 | 0.119 | 0.227 |
| SMC1A | 0.010 | 0.302 | 0.144 | 0.779 |
| DNMT1 | 0.020 | 0.545 | 0.854 | 0.074 |
| HDAC7 | 0.020 | 0.812 | 0.449 | 0.778 |
| LHX5 | 0.024 | 0.317 | 0.635 | 0.123 |
| POLR2D | 0.034 | 0.283 | 0.082 | 0.209 |
| POLR2J | 0.044 | 0.197 | 0.764 | 0.806 |
| DDX39A | 0.049 | 0.163 | 0.573 | 0.385 |
We next utilized Sufu−/− iMEFs to further prioritize these candidates based on their ability to act downstream of SMO (Fig. 1F), as GLI signaling is constitutively activated independently of SMO in such cells [32] (Fig. 1A). Among the remaining candidates only knockdown of Uhrf1 or Dnmt1 attenuated the expression of the GLI target gene Gli1, consistent with their regulation of GLI activity functioning downstream of SMO (Fig. 1F). Furthermore, knockdown of Uhrf1 or Dnmt1 also attenuated GLI activity in Light2 cells in which SHH signaling was induced by transfecting cells with exogenous Gli1 or Gli2 (Fig. 1G). Together, these results implicated UHRF1 and DNMT1 as potential regulators of GLI signaling downstream of SMO.
UHRF1 is overexpressed in SHH-MB
Given that UHRF1 was our top candidate in these initial analyses (Table 1 and Fig. 1D–G) and is a novel GLI regulator that has not been previously described (Supplemental Table S2), we focused on characterization of its role in SHH-MB. Meta-analysis across four published MB patient derived microarray datasets [42–45] showed that UHRF1 expression is significantly higher in MB tissue than normal brain tissue (Fig. 2A). We also utilized the RNAi/shRNA screening datasets from the DepMap project at the Broad Institute [53] to analyze UHRF1 dependency scores across a panel of 712 cancer cell lines. These data showed that MB cell lines are more dependent on UHRF1 than cell lines from other types of cancer (Fig. 2B), implicating UHRF1 in patient-derived MB cell growth. Moreover, our follow-up meta-analysis showed that UHRF1 expression is significantly higher in the tumors derived from the two subtypes of SHH-MB (α and β) that have the worst patient outcome in SHH-MB [4] (Fig. 2C). Consistent with our gene expression-based pan-MB patient survival analysis (Table 1 and Fig. 1D), these data indicated that UHRF1 overexpression may contribute to the deterioration of SHH-subgroup MB patients. Consistent with our findings, UHRF1 protein was previously reported to enriched in patient derived SHH-MB tissues [54]. We next determined UHRF1 protein levels in SHH-MB mouse models harboring mutations in known MB drivers, loss of Ptch1 (Fig. 2D), increased expression of a constitutively active Smo mutant (Fig. 2E), and a patient derived MB model characterized by a MYCN amplification and TP53 mutation [36] (Fig. 2F). SHH-MB tissue from all three of these mouse SHH-MB models exhibited high levels of UHRF1 staining, localized predominantly in the nucleus, relative to adjacent normal cerebellar tissues. We also determined UHRF1 and GLI1 levels in a patient-derived MB tissue microarray (Supplemental Fig. S2 A and B). We consistently observed increased levels of UHRF1 in MB tissues (Supplemental Fig. S2A and C). Moreover, we show for the first time that MB tissues with high levels of GLI1 also have higher levels of UHRF1 (Supplemental Fig. S2D), suggesting that UHRF1 is overexpressed in SHH-MB in a manner that may regulate GLI activity.
Figure 2. UHRF1 is overexpressed in SHH-MB.
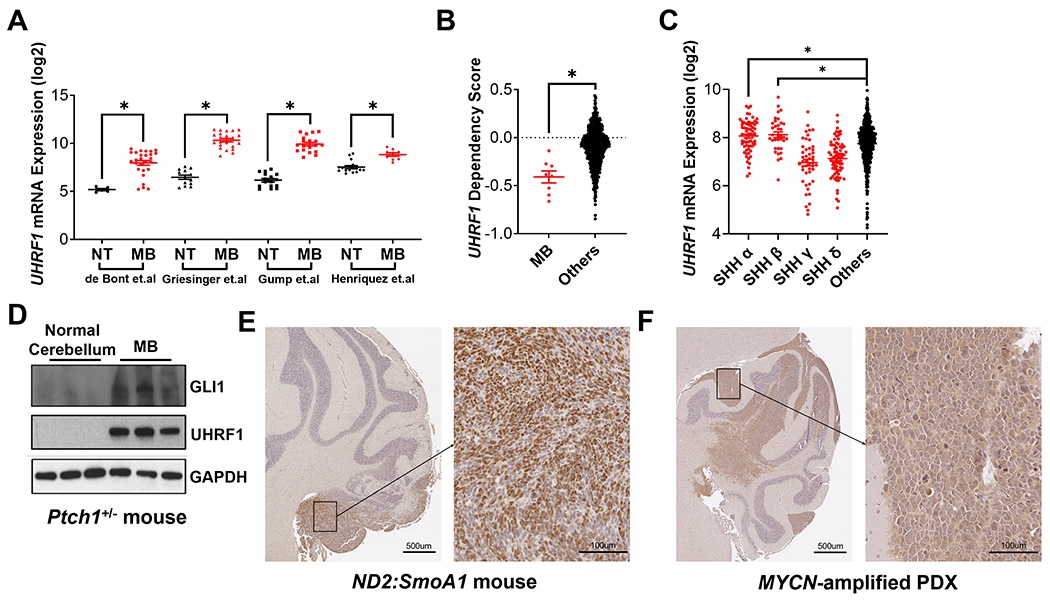
(A) Meta-analysis comparing the expression of UHRF1 between normal brain tissue (NT) and patient derived MB tissue (MB) using the four indicated microarray datasets [42–45]. (B) The dependency on UHRF1 for patient-derived cancer cell line viability was compared between MB cell lines (MB) and non-MB cancer cell lines (Others) using the data from the DepMap project (Broad Institute). (C) Meta-analysis comparing the expression of UHRF1 among the four SHH subtypes (SHH α, β, γ and δ) and other subgroups of MB (Others), using microarray data from Cavalli et al [4]. (D) Normal mouse cerebella or SHH-MB tissue from Ptch1+/− mice were isolated and immunoblotted to determine the levels of GLI1, UHRF1 and GAPDH (n=3 mice per group). MB tissue from (E) ND2:SmoA1 or (F) patient-derived TRP53-mutant, MYCN-amplified MB xenograft (SJSHHMB-14-7196) mice were immunostained for UHRF1. Representative images are shown (n=3).
SHH-MB growth requires UHRF1
We used MB sphere cultures (MSCs) isolated from spontaneous MBs in Ptch1+/− mice (MSC4, Ptch1−/−) or Ptch1+/−;Trp53+/− mice (MSC2, Ptch1−/−;Trp53−/−) to further determine the role of UHRF1 in SHH-MB, all of which are SHH activity dependent [35, 36]. Knockdown of Uhrf1 expression by distinct or pooled siRNA reduced cell viability in MB sphere cultures, regardless of Trp53 status, suggesting that UHRF1 positively regulates SHH-MB growth (Fig. 3A and Supplemental Fig. S3A). We also utilized MB sphere cultures known to be SMO inhibitor-resistant, MPC4 (Ptch1−/−) and MPC2 (Ptch1−/−;Trp53−/−) [37]. Interestingly, Uhrf1 knockdown also inhibited the growth of these SMO inhibitor-resistant MB sphere cultures (Fig. 3B), providing further evidence that UHRF1 acts downstream of SMO. Consistent with UHRF1 acting to regulate GLI signaling in MB sphere cultures, knockdown of Uhrf1 reduced the expression of the GLI target gene Gli1 (Fig. 3C and D) as well as overall GLI1 protein levels (Fig. 3E). We next examined the level of cell proliferation in order to better understand the cause of the decreased cell viability observed subsequent to Uhrf1 knockdown. Knockdown of Uhrf1 reduced the levels of the G1/S cell cycle biomarker Proliferating cell nuclear antigen (PCNA) (Fig. 3E) and the extent of EdU (5-ethynyl-2´-deoxyuridine) incorporation (Supplemental Fig. S4), indicating that UHRF1 promotes MB cell proliferation. Uhrf1 knockdown also increased the cleavage of the effector Caspase, Caspase 3, (Fig. 3E) suggesting that increased apoptosis may also contribute to the observed decrease in cell viability. We next determined the role UHRF1 plays in regulating SHH-MB growth in vivo. MSC4 cells transduced with an shRNA targeting Uhrf1, or a scrambled shRNA control, were implanted orthotopically into the cerebellum of mice. Similar to our ex vivo data, knockdown of Uhrf1 inhibited SHH-MB growth in vivo (Fig. 3F and G). Together, these results suggest that UHRF1 functions to potentiate SHH-MB growth.
Figure 3. SHH-MB growth requires UHRF1.
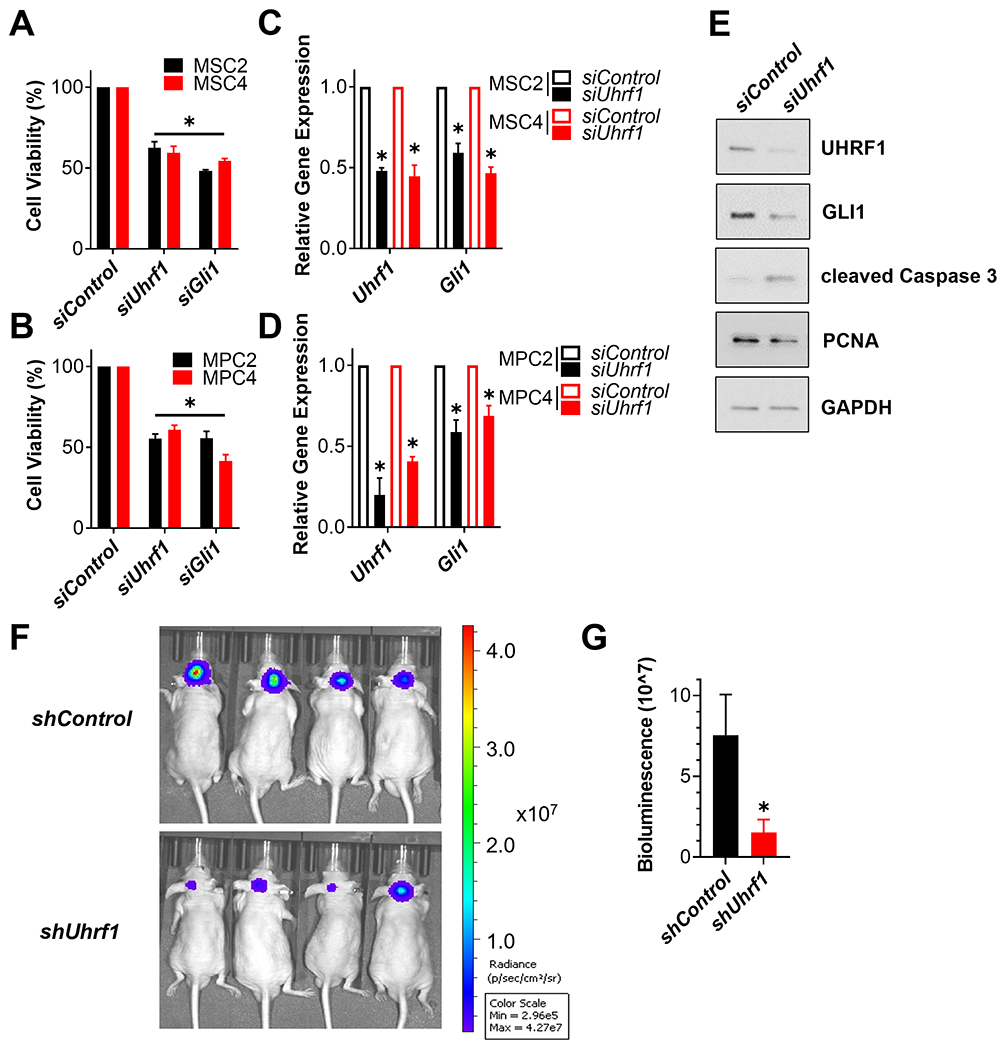
(A and B) The indicated SHH-MB sphere cultures were transfected with a scramble siRNA control (siControl) or pooled siRNA targeting Uhrf1 or Gli1. The number of viable cells were determined 5 days later using an MTT reduction assay and the data normalized to results from siControl transfected cells. (C and D) The indicated SHH-MB sphere cultures were transfected with control or pooled siRNA targeting Uhrf1 for 4 days. The expression of Uhrf1 and Gli1 was then determined by RT-qPCR, initially normalized to the expression of Gapdh and then to the results from siControl transfected cells. (E) MSC4 cells were transfected with the indicated siRNA for 4 days and the resultant cell lysates immunoblotted for the indicated proteins. A representative blot is shown (n=3). (F) MSC4 cells expressing Firefly luciferase were transduced with a scramble shRNA control (shControl) or shRNA targeting Uhrf1 (shUhrf1) overnight. The next day, these transduced MSC4 cells were orthotopically implanted into the cerebella of CD1-Foxn1nu mice and tumor burden imaged 10 days later by IVIS. Representative IVIS imaging is shown (n=7 mice per group). (G) Bioluminescence quantification of all the mice in this cohort is shown (n=7 mice per group).
UHRF1/DNMT1/GLI protein complexes
We noted that our other top GLI regulator candidate, DNMT1 (Fig. 1E–G), is a well-described UHRF1 interactor [55]. We thus hypothesized that DNMT1 might function together with UHRF1 to regulate SHH-MB growth. To test this hypothesis, we first determined DNMT1 RNA expression in MB tissue using the meta-analysis described in Fig. 2, and found an enrichment of DNMT1 in MB tissues compared to normal brain tissues (Supplemental Fig. S5A). Although, this analysis indicated that there is in general less DNMT1 RNA expression in SHH-MB tissue than other MB subtypes of MB (Supplemental Fig. S5C), DNMT1 protein levels do appear to enrich in SHH-MB tissue [56]. However, based on analysis using DepMap, DNMT1 appears to be an essential gene across cell lines derived from many types of cancer (Supplemental Fig. S5B). To explore its role in our SHH-MB sphere cultures, we knocked down DNMT1 and noted a reduction in the viability of the SHH-MB cells (Fig. 4A and Supplemental Fig. S3B) and in the expression of the GLI target gene, Gli1 (Fig. 4B and Supplemental Fig. S3C). We also observed a reduction of EdU incorporation in SHH-MB cells upon Dnmt1 knockdown, consistent with DNMT1 regulating the proliferation of SHH-MB cells (Supplemental Fig. S4).
Figure 4. DNMT1 forms complexes with UHRF1 and GLI to regulate the viability of SHH-MB sphere cultures.
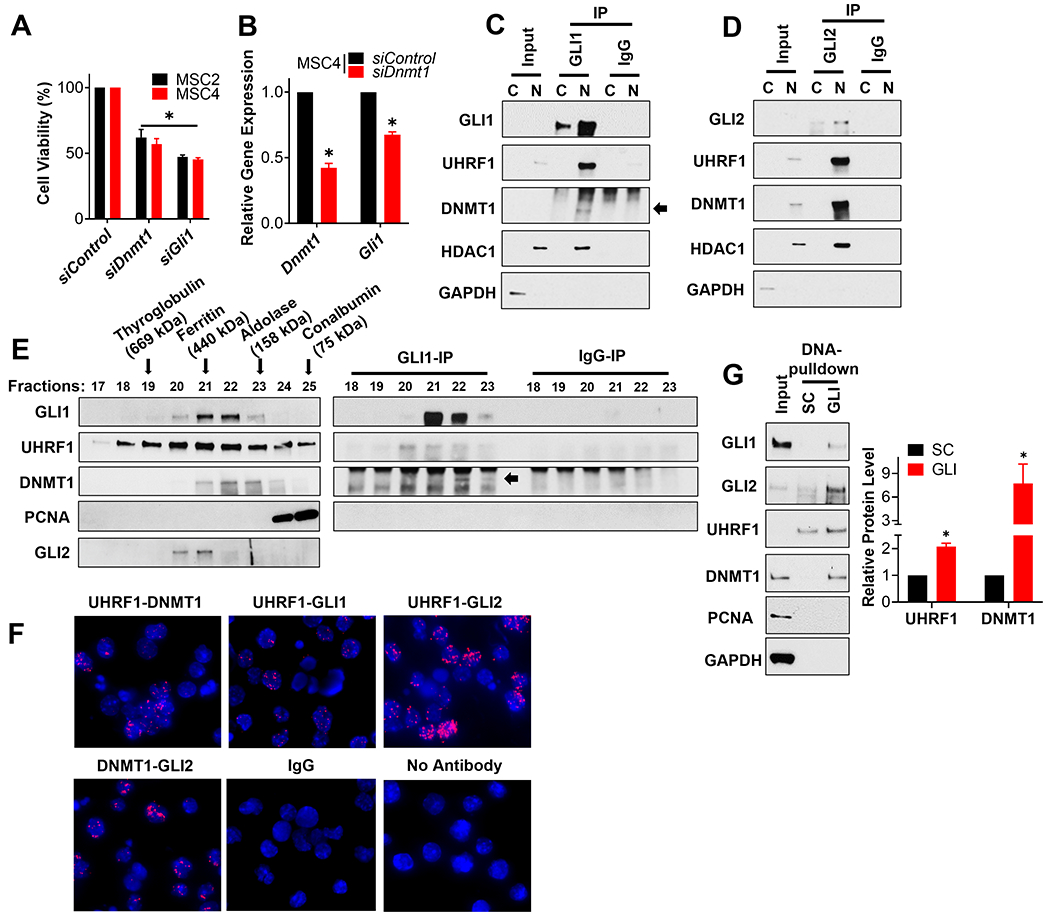
(A) The indicated SHH-MB sphere cultures were transfected with a scramble siRNA control (siControl) or pooled siRNA targeting Dnmt1 or Gli1. The number of viable cells was determined 5 days later using an MTT reduction assay and the data normalized to results from siControl transfected cells. (B) MSC4 cells were transfected with a scramble siRNA control (siControl) or pooled siRNA targeting Dnmt1 for 4 days. The expression of Dnmt1 and Gli1 was then determined by RT-qPCR, initially normalized to the expression of Gapdh and then to the results from siControl transfected cells. MSC4 cells were fractionated into nuclear (N) and cytoplasmic (C) enriched fractions, and GLI1 (C) or GLI2 (D) subsequently immunoprecipitated from such lysates. The immunoprecipitates were analyzed by immunoblotting for the indicated proteins. GAPDH is a cytoplasmic loading control and HDAC1, a known interactor of GLI1 and GLI2 [25], is a nuclear loading control. Representative blots are shown (n=3). (E) The nuclear enriched fraction of MB4 tumor cells was fractionated over a size-exclusion chromatography column. The various fractions were analyzed by immunoblotting for the indicated proteins (Left), or subjected to immunoprecipitation and immunoblotting for the indicated proteins (Right). The elution peaks of the indicated protein standards used to calibrate this gel filtration column are indicated with an arrow above the immunoblot. A representative blot is shown (n=3). (F) MSC4 cells were seeded on cover slips and subsequently subjected to a Proximity Ligation Assay (PLA) to detect potential direct interactions between the indicated proteins (red) with DAPI co-stained (blue). No antibody and IgG controls were included. Representative images are shown (n=3). (G) MSC4 cell lysates were used in a DNA pull-down assay with beads coupled to oligonucleotides containing either a GLI-binding site (GLI) or a scrambled control (SC) sequence. The bead-enriched proteins were then immunoblotted for the indicated proteins. A representative blot is shown (n=3) (Left). The level of UHRF1 or DNMT1 in the GLI-binding site pulldown was quantitated and normalized to that of the control lane (SC) to calculate relative UHRF1 or DNMT1 levels (n=3) (Right).
As UHRF1 and DNMT1 both regulate GLI activity (Fig. 3C–E and 4B; Supplemental Fig. S3C) and SHH-MB cell growth (Fig. 3A–B and 4A; Supplemental Fig. S3A–B), and are known to interact with one another [55], we hypothesized that they interact with GLI proteins to directly regulate GLI function in SHH-MB. To test this model, we immunoprecipitated GLI1 (Fig. 4C) or GLI2 (Fig. 4D) from cytoplasm- or nucleus-enriched subcellular fractions of SHH-MB sphere cultures, followed by immunoblotting for potential interacting proteins. We show that GLI1 and GLI2 associate with UHRF1 and DNMT1 in the nuclear fraction of SHH-MB sphere cultures (Fig. 4C and D). We next used size-exclusion chromatography of a nuclear enriched SHH-MB cell extract (Fig. 4E left) to show that UHRF1 and DNMT1 co-migrate in similar sized fractions (#21-23) to that of GLI proteins. PCNA, another well-known interactor of UHRF1 and DNMT1 [55], did not show substantial overlap with GLI proteins (Fig. 4E left), suggesting that a UHRF1/DNMT1/GLI complex may be distinct from the PCNA/UHRF1/DNMT1 complex that regulates untargeted DNA methylation [57, 58]. Importantly, the presence of a UHRF1/DNMT1/GLI1 complex in these fractions (#22-23) was also validated in GLI1 immunoprecipitates from these fractions (Fig. 4E right). Moreover, we found the interactions between the components of the UHRF1/DNMT1/GLI complex tend to be relatively direct, as they detected by a Proximity ligation assay (PLA) (Fig. 4F) that only recognizes interactions at distances <40 nm [59, 60]. We next used sepharose beads conjugated to scrambled oligonucleotides (SC) or oligonucleotides containing a defined GLI-binding (GACCACCCA) site [61] (GLI) to isolate proteins from SHH-MB sphere culture extracts that enrich on GLI DNA consensus sites (Fig. 4G). GLI-binding site beads enriched a GLI/UHRF1/DNMT1 complex, but not PCNA, relative to SC beads (Fig. 4G). These results provide evidence that GLI proteins can recruit UHRF1 and DNMT1 onto DNA.
A DNMT1 inhibitor attenuates SHH-MB progression
As UHRF1 and DNMT1 appear to function together in a complex to regulate SHH-MB growth, we wanted to disrupt the activity of this complex using UHRF1 or DNMT1 inhibitors. We noted that although several UHRF1 inhibitors have been previously described, none of them are yet clinically relevant [62–64]. However, two DNMT1 inhibitors are already FDA approved (5-Azacytidine (5-aza) and 5-aza-2’ deoxycytidine (5-aza-dCR)) [65]. We therefore evaluated the efficacy of 5-aza and 5-aza-dCR in SHH-MB cells, and showed that both inhibitors attenuate the growth of SHH-MB cells (Fig. 5A). We noted that 5-aza shows a greater efficacy than 5-aza-dCR in attenuating the viability SHH-MB cells (Fig. 5A), similar to observations described in acute myeloid leukemia (AML) cells [66]. Interestingly, a recently developed DNMT1 specific non-covalent inhibitor, GSK3484862 [67], was also able to attenuate the viability of SHH-MB cells (Fig. 5A), supporting the idea that DNMT1 specific inhibition is a reasonable therapeutic strategy for SHH-MB. To evaluate the specificity of DNMT1 inhibition on SHH signaling we utilized granule cell precursors (GCPs), whose proliferation is dependent on SHH activity [68–70]. While the SMO agonist SAG induced the proliferation of GCPs, this activity was significantly inhibited by 5-aza, again suggesting that DNMT1 positively regulates the activity of the SHH signaling pathway (Supplemental Fig. S6). As 5-aza was the most efficacious inhibitor among the three DNMT1 inhibitors we evaluated, and a previous report that 5-aza-dCR treatment alone had no effect on MB development in a SHH-MB mouse model [71], we focused on the evaluation of 5-aza to inhibit SHH-MB growth. We showed that 5-aza attenuated the growth of MSC4 cells with an IC50 of 2.0 μM (Fig. 5B). Importantly, this IC50 is similar to that previously reported to inhibit the growth of AML cells [66], for which 5-aza is FDA approved. Moreover, we confirmed that 5-aza attenuates the viability of SHH-MB cells through DNMT1, as knockdown of DNMT1 rendered these cells resistant to 5-aza treatment (Fig. 5C).
Figure 5. The DNMT1 inhibitor, 5-aza, attenuates the viability of SHH-MB cells via degradation of DNMT1.
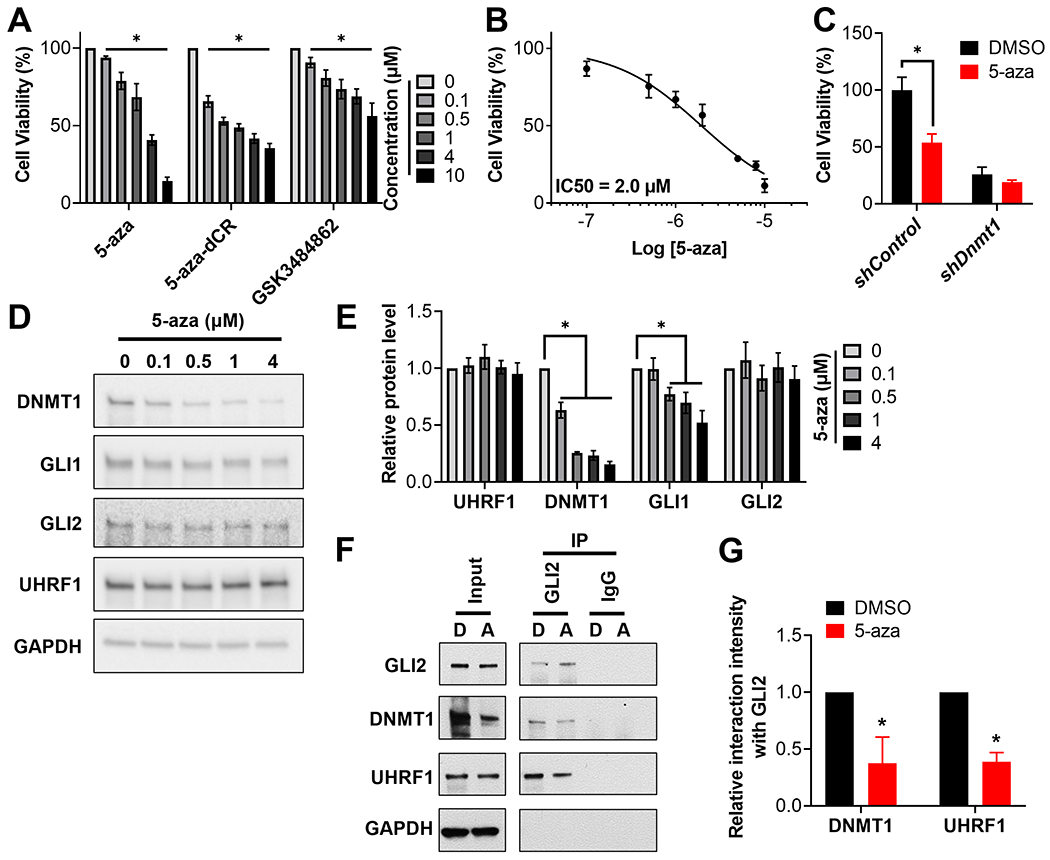
(A) MSC4 cells were treated with 5-aza, 5-aza-dCR or GSK3484862 at the indicated concentrations for 72h. The number of viable cells was then determined using a CellTiter-Glo assay and normalized to results from DMSO treated cells. (B) MSC4 cells were treated with the indicated concentrations of 5-aza for 48h. The number of viable cells was determined using an MTT reduction assay. (C) MSC4 cells were transduced with a control shRNA (shControl) or shRNA targeting Dnmt1 (shDnmt1) and 3 days later selected in puromycin for 4 days. The transduced MSCs was subsequently treated with DMSO or 2 μM 5-aza for 48h. Cellular viability was then determined using a CellTiter-Glo assay and normalized to results from DMSO treated shControl cells. (D) MSC4 cells were treated with the indicated concentrations of 5-aza for 24h followed by immunoblotting of the resultant lysates for the indicated proteins. A representative blot is shown (n=6). (E) The level of the indicated proteins in (D) was quantitated and normalized to that of GAPDH and then to results from DMSO treated samples to calculate relative protein levels (n=6). (F) MSC4 cells were treated with DMSO (D) or 200 nM 5-aza (A) for 24h, and GLI2 subsequently immunoprecipitated from the resultant lysates. A representative blot is shown (n=3). (G) The level of DNMT1 and UHRF1 in the GLI2-immunoprecipatation of the 5-aza lane (A) was quantitated and normalized to that of GLI2, and then normalized to results from the DMSO lane (D) to calculate the relative interaction intensity with GLI2 (n=3).
5-aza was able to reduce the levels of its target protein DNMT1 in a dose-dependent manner (Fig. 5D and E), as previously described [72]. Consistent with DNMT1 potentiating GLI activity, its inhibition by 5-aza also reduced the levels of the GLI target gene product GLI1 (Fig. 5D and E). 5-aza mediated degradation of DNMTs is thought to occur indirectly, incorporating into DNA and subsequently forming irreversible covalent complexes with DNMTs that are subsequently degraded by the proteasome [73]. Thus, it remained possible that 5-aza treatment inhibits GLI activity via proteasomal degradation of the UHRF1/DNMT1/GLI complex. However, while 5-aza significantly reduced DNMT1 and GLI1 levels (Fig. 5D and E), it did not result in GLI2 and UHRF1 degradation (Fig. 5D and E). Further, while 5-aza was able to reduce the half-life of DNMT1 protein in a proteasome dependent manner, it had minimal effect on the half-life of GLI1 (in an 8-hour treatment) and UHRF1 (Supplemental Fig. S7). 5-aza also significantly reduced the stability of the UHRF1/DNMT1/GLI2 complex (Fig. 5F and G). Together, these findings suggest that 5-aza does not result in the proteasomal degradation of the entire UHRF1/DNMT1/GLI complex, but acts rather by decreasing the levels of DNMT1 able to incorporate into and potentially scaffold this GLI complex. Moreover, the evidence that 5-aza is able to disrupt the UHRF1/DNMT1/GLI complex (Fig. 5F and G), supports the model in which the UHRF1/DNMT1/GLI complex potentiates SHH signaling and SHH-MB growth.
To extend the translational relevance of our findings we next evaluated the ability of 5-aza to act on-target in MB tissue in vivo. Cells from a Ptch1+/−; Trp53+/+ driven mouse MB tumor were subcutaneously implanted in mice and subsequently treated with 5-aza once those tumors reached ~100 mm3. Consistent with our ex vivo result (Fig. 5D and E), 5-aza effectively reduced the levels of GLI1 and DNMT1 in these MB tumors (Fig. 6A and B). To examine the ability of 5-aza to attenuate SHH-MB growth in vivo, similar SHH-MB cells were orthotopically implanted into the cerebellum of mice and those mice subsequently treated with 5-aza 14 days after implantation. 5-aza treatment significantly attenuated MB growth in these mice (Fig. 6C and D). DNMT1 inhibition also reduced cell proliferation and induced apoptosis in these tumors, as determined by Ki67 immunostaining and cleaved Caspase 3 levels, respectively (Fig. 6E and F). Thus, we conclude that 5-aza effectively restrains SHH-MB growth in vivo, and does so in an on-target manner. Collectively, these results support the existence of a druggable UHRF1/DNMT1/GLI complex that regulates SHH-MB growth, and can be targeted using a clinically relevant DNMT1 inhibitor.
Figure 6. 5-aza attenuates SHH-MB progression in vivo.
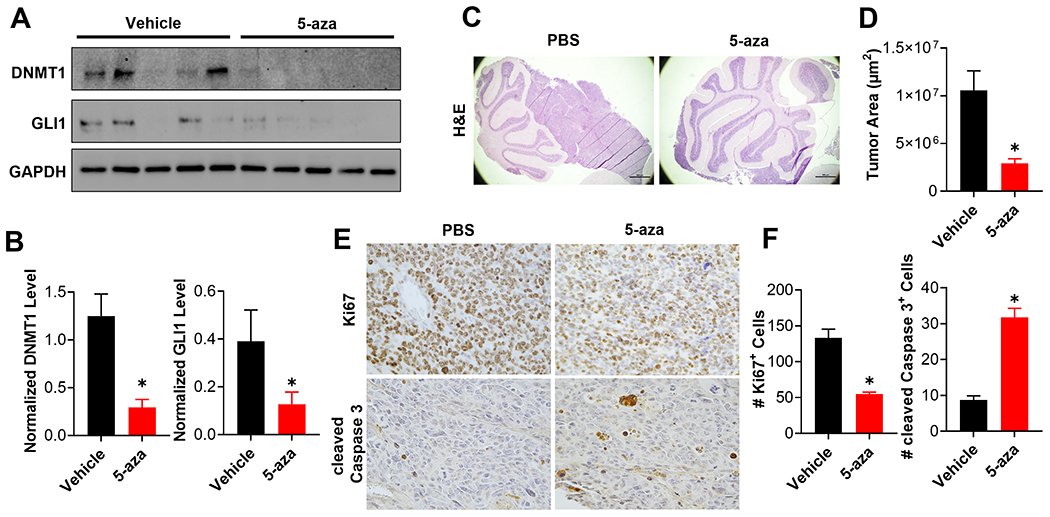
(A) Murine MB cells, derived from a primary MB that had never been cultured ex vivo, were subcutaneously implanted into the flank of 10 CD1-Foxn1nu mice. When tumors reached ~ 100 mm3, these mice (n=5 mice per group) were treated with vehicle or 5-aza (5 mg/kg IV in PBS) every other day for up to 8 days. Residual tumor tissues were harvested 6 hours after the last injection, and subjected to immunoblotting of the resultant tumors for the indicated proteins (n=5 mice per group). (B) The level of DNMT1 (Left) or GLI1 (Right) was quantitated and normalized to that of GAPDH to calculate normalized protein level in each treatment group (n=5 mice per group). (C) Same MB cells as used in (A) were orthotopically implanted into the cerebella of CD1-Foxn1nu mice. These mice were treated 14 days after implantation with vehicle or 5-aza (5 mg/kg IV in PBS) every three days for 18 days. Representative H&E staining of the cerebella from these mice is shown (n=5 mice per group). (D) Tumor area was calculated from 4 independent mice per group and the results summarized in a bar graph. (E) Brain sections from the same mice were subjected to immunohistochemical staining for Ki67 or cleaved Caspase 3. Representative figures are shown (n=4 mice per group). (F) The number of Ki67 positive cells (Left) or cleaved Caspase 3 positive cells (Right) were quantitated in 4 random high magnification fields from each of the 4 mice in each group and the results summarized in bar graphs.
DISCUSSION
In this study we use an epigenetic focused siRNA screen to identify 83 potential positive regulators of GLI signaling. UHRF1 and DNMT1 were prioritized as the top candidates from this screen as their expression was correlated with SHH-MB patient outcome, they function downstream of SMO, and have been previously implicated in MB progression [54, 71, 74]. In this work we show for the first time that UHRF1 and DNMT1 promote SHH-MB growth via a novel UHRF1/DNMT/GLI multi-protein complex. Though there are no clinically relevant UHRF1 inhibitors available (reviewed in [75]), we show that an FDA-approved DNMT1 inhibitor, 5-aza, is able to disrupt the stability of the complex and attenuate SHH-MB growth. Taken together, we describe a druggable UHRF1/DNMT1/GLI complex that functions to regulate GLI activity in SHH dependent tumor growth.
Besides functioning in DNA methylation inheritance via association with PCNA [57], UHRF1 and DNMT1 also function as transcriptional repressors that associate with specific transcription factors to mediate target gene methylation and subsequently silencing [58, 76, 77]. Although we do not yet fully understand the mechanism by which UHRF1 and DNMT1 regulate GLI activity, our results do suggest that UHRF1 and DNMT1 function as positive regulators of GLI signaling and that they do so by interacting with and regulating GLI proteins. To account for these findings in the context of UHRF1/DNMT1 acting primarily to silence gene expression, we suggest a model in which UHRF1 and DNMT1 form a corepressor complex with GLI proteins to attenuate expression of a subset of GLI target genes that function as repressors of GLI signaling. Consistent with such a model, the expression of two GLI-target genes that encode repressors of SHH signaling (reviewed in [14]), PTCH1 and HHIP, is transcriptionally repressed by DNMT1 [78] or the UHRF1/DNMT1 complex [79], respectively. However, we cannot exclude the possibility that UHRF1 and DNMT1 function as transcriptional coactivators for GLI proteins, as in certain scenarios DNMT mediated DNA methylation increases the binding of specific transcription factors to increase the transcription of a subset of target genes [80–82].
A growing interest in discovering the oncogenic role of UHRF1 in distinct cancers has stimulated efforts to identify and develop UHRF1 inhibitors (reviewed in [75]). Rather than directly targeting UHRF1 protein, these inhibitors mainly function by reducing UHRF1 levels via alternative mechanisms, such as 17AAG degrading UHRF1 through inhibiting HSP90 [62] and polyphenols attenuating UHRF1 expression through the p53/p21/p16 signaling axis [75]. Unlike these UHRF1 inhibitors, which are not advanced clinically, several DNMT1 specific inhibitors have already been evaluated in clinic trails [65]. Among these inhibitors, 5-aza and 5-aza-dCR are FDA approved for myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML) [65]. Interestingly, one study showed that 5-aza-dCR treatment alone had no effect on MB development in Ptch1+/− mice, however, in combination with the HDAC inhibitor valproic acid (VPA), 5-aza-dCR was able to inhibit MB growth [71]. Though 5-aza and 5-aza-dCR are structurally similar (reviewed in [65]), the two compounds induce different responses in various types of tumor cells [66, 83, 84]. Here, we observed that 5-aza is less potent but more efficacious than 5-aza-dCR in inhibiting the viability of SHH-MB cells, consistent with similar observations in AML cells [66]. Although we did not evaluate 5-aza-dCR in SHH-MB in vivo, we show for the first time that 5-aza monotherapy effectively inhibits tumor growth in a SHH-MB orthotopic model. Moreover, several clinical evaluations have been done to provide the pediatric dosage of 5-aza (NCT02450877, NCT01861002 and NCT02447666), which could benefit the application of this drug in SHH-MB pediatric patients.
Supplementary Material
Implications:
This work describes a novel, druggable UHRF1/DNMT1/GLI complex that regulates SHH-dependent tumor growth, and highlights an FDA-approved drug capable of disrupting this complex to attenuate tumor growth.
Acknowledgements
We thank all members of the Robbins, Rodriguez-Blanco, Capobianco, and Ayad laboratories for providing insights and discussion regarding this work. We thank Dr. Mehrdad Nadji (University of Miami) for scoring the IHC staining of the MB TMA, Dr. Lluis Morey (University of Miami) for providing conceptual advice, and Dr. Steven Y. Cheng (Nanjing Medical University) for providing a critical review of this manuscript. We also thank Sylvester Cancer Center Cancer Modeling Shared Resource for IVIS imaging, the Miami Project for providing the siRNA library, and the Department of Surgery Tissue and Pathology core (University of Miami) for tumor sample processing.
Financial support:
This work was supported by a National Institutes of Health (NIH) Grants (R01NS110591, to D.J. Robbins; R01NS118023, to N.G. Ayad; K01NS119351, to J. Rodriguez-Blanco), FDOH, Live Like Bella Pediatric Cancer Research Initiative (8LA03, to D.J. Robbins), and the Rally Foundation (20CDN46, to J. Rodriguez-Blanco).
Footnotes
Disclosure of Potential Conflicts of Interest: The authors declare no potential conflicts of interest.
REFERENCE
- 1.Louis DN, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. 2016. 131(6): p. 803–820. [DOI] [PubMed] [Google Scholar]
- 2.Taylor MD, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol, 2012. 123(4): p. 465–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thompson MC, et al. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. Journal of clinical oncology, 2006. 24(12): p. 1924–1931. [DOI] [PubMed] [Google Scholar]
- 4.Cavalli FMG, et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell, 2017. 31(6): p. 737–754 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kool M, et al. Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. 2012. 123(4): p. 473–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gajjar AJ and Robinson GW, Medulloblastoma-translating discoveries from the bench to the bedside. Nat Rev Clin Oncol, 2014. 11(12): p. 714–22. [DOI] [PubMed] [Google Scholar]
- 7.Crawford JR, MacDonald TJ, and Packer RJ, Medulloblastoma in childhood: new biological advances. Lancet Neurol, 2007. 6(12): p. 1073–85. [DOI] [PubMed] [Google Scholar]
- 8.Rudin CM, et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med, 2009. 361(12): p. 1173–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gajjar A, et al. Phase I study of vismodegib in children with recurrent or refractory medulloblastoma: a pediatric brain tumor consortium study. Clinical Cancer Research, 2013. 19(22): p. 6305–6312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robinson GW, et al. Vismodegib exerts targeted efficacy against recurrent sonic hedgehog–subgroup medulloblastoma: results from phase II pediatric brain tumor consortium studies PBTC-025B and PBTC-032. Journal of Clinical Oncology, 2015. 33(24): p. 2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kieran MW, et al. Phase I study of oral sonidegib (LDE225) in pediatric brain and solid tumors and a phase II study in children and adults with relapsed medulloblastoma. Neuro-oncology, 2017. 19(11): p. 1542–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stone DM, et al. The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature, 1996. 384(6605): p. 129–134. [DOI] [PubMed] [Google Scholar]
- 13.Marigo V, et al. Biochemical evidence that patched is the Hedgehog receptor. Nature, 1996. 384(6605): p. 176–179. [DOI] [PubMed] [Google Scholar]
- 14.Robbins DJ, Fei DL, and Riobo NA, The Hedgehog signal transduction network. Sci Signal, 2012. 5(246): p. re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Y and Struhl G, Dual roles for patched in sequestering and transducing Hedgehog. Cell, 1996. 87(3): p. 553–563. [DOI] [PubMed] [Google Scholar]
- 16.Ingham P, Taylor A, and Nakano Y, Role of the Drosophila patched gene in positional signalling. Nature, 1991. 353(6340): p. 184–187. [DOI] [PubMed] [Google Scholar]
- 17.Raleigh DR and Reiter JF, Misactivation of Hedgehog signaling causes inherited and sporadic cancers. The Journal of clinical investigation, 2019. 129(2): p. 465–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alcedo J, et al. The Drosophila smoothened gene encodes a seven-pass membrane protein, a putative receptor for the hedgehog signal. Cell, 1996. 86(2): p. 221–232. [DOI] [PubMed] [Google Scholar]
- 19.Norsworthy KJ, et al. FDA approval summary: glasdegib for newly diagnosed acute myeloid leukemia. Clinical Cancer Research, 2019. 25(20): p. 6021–6025. [DOI] [PubMed] [Google Scholar]
- 20.Casey D, et al. FDA approval summary: sonidegib for locally advanced basal cell carcinoma. Clinical Cancer Research, 2017. 23(10): p. 2377–2381. [DOI] [PubMed] [Google Scholar]
- 21.Axelson M, et al. US Food and Drug Administration approval: vismodegib for recurrent, locally advanced, or metastatic basal cell carcinoma. Clinical Cancer Research, 2013. 19(9): p. 2289–2293. [DOI] [PubMed] [Google Scholar]
- 22.Humke EW, et al. The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes & development, 2010. 24(7): p. 670–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tukachinsky H, Lopez LV, and Salic A, A mechanism for vertebrate Hedgehog signaling: recruitment to cilia and dissociation of SuFu–Gli protein complexes. Journal of Cell Biology, 2010. 191(2): p. 415–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Niewiadomski P, et al. Gli proteins: regulation in development and cancer. Cells, 2019. 8(2): p. 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Canettieri G, et al. Histone deacetylase and Cullin3–REN KCTD11 ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nature cell biology, 2010. 12(2): p. 132. [DOI] [PubMed] [Google Scholar]
- 26.Huang Y, et al. Regulation of GLI Underlies a Role for BET Bromodomains in Pancreatic Cancer Growth and the Tumor Microenvironment. Clin Cancer Res, 2016. 22(16): p. 4259–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taylor R, et al. Regulation of GLI1 by cis DNA elements and epigenetic marks. DNA repair, 2019. 79: p. 10–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi X, et al. An epigenetic switch induced by Shh signalling regulates gene activation during development and medulloblastoma growth. Nature communications, 2014. 5: p. 5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yauch RL, et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science, 2009. 326(5952): p. 572–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Atwood SX, et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer cell, 2015. 27(3): p. 342–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taipale J, et al. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature, 2000. 406(6799): p. 1005–9. [DOI] [PubMed] [Google Scholar]
- 32.Svard J, et al. Genetic elimination of Suppressor of fused reveals an essential repressor function in the mammalian Hedgehog signaling pathway. Dev Cell, 2006. 10(2): p. 187–97. [DOI] [PubMed] [Google Scholar]
- 33.Goodrich LV, et al. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science, 1997. 277(5329): p. 1109–13. [DOI] [PubMed] [Google Scholar]
- 34.Jacks T, et al. Tumor spectrum analysis in p53-mutant mice. Current biology, 1994. 4(1): p. 1–7. [DOI] [PubMed] [Google Scholar]
- 35.Zhao X, et al. RAS/MAPK Activation Drives Resistance to Smo Inhibition, Metastasis, and Tumor Evolution in Shh Pathway-Dependent Tumors. Cancer Res, 2015. 75(17): p. 3623–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodriguez-Blanco J, et al. A CK1alpha Activator Penetrates the Brain and Shows Efficacy Against Drug-resistant Metastatic Medulloblastoma. Clin Cancer Res, 2019. 25(4): p. 1379–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rodriguez-Blanco J, et al. Inhibition of WNT signaling attenuates self-renewal of SHH-subgroup medulloblastoma. Oncogene, 2017. 36(45): p. 6306–6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang X, et al. Isolation, enrichment, and maintenance of medulloblastoma stem cells. J Vis Exp, 2010(43). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fei DL, et al. Activation of Hedgehog signaling by the environmental toxicant arsenic may contribute to the etiology of arsenic-induced tumors. Cancer research, 2010. 70(5): p. 1981–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Atwood SX, et al. GLI activation by atypical protein kinase C iota/lambda regulates the growth of basal cell carcinomas. Nature, 2013. 494(7438): p. 484–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Robbins DJ, et al. Hedgehog elicits signal transduction by means of a large complex containing the kinesin-related protein costal2. 1997. 90(2): p. 225–234. [DOI] [PubMed] [Google Scholar]
- 42.de Bont JM, et al. Differential expression and prognostic significance of SOX genes in pediatric medulloblastoma and ependymoma identified by microarray analysis. Neuro-oncology, 2008. 10(5): p. 648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Griesinger AM, et al. Characterization of distinct immunophenotypes across pediatric brain tumor types. The Journal of Immunology, 2013. 191(9): p. 4880–4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gump JM, et al. Identification of targets for rational pharmacological therapy in childhood craniopharyngioma. Acta neuropathologica communications, 2015. 3(1): p. 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Henriquez NV, et al. Comparative expression analysis reveals lineage relationships between human and murine gliomas and a dominance of glial signatures during tumor propagation in vitro. Cancer research, 2013. 73(18): p. 5834–5844. [DOI] [PubMed] [Google Scholar]
- 46.Bowman RL, et al. GlioVis data portal for visualization and analysis of brain tumor expression datasets. 2017. 19(1): p. 139–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nakagawa N, et al. APC sets the Wnt tone necessary for cerebral cortical progenitor development. Genes & development, 2017. 31(16): p. 1679–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Coni S, et al. Selective targeting of HDAC1/2 elicits anticancer effects through Gli1 acetylation in preclinical models of SHH Medulloblastoma. Scientific reports, 2017. 7: p. 44079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Y, et al. Oncogenic functions of Gli1 in pancreatic adenocarcinoma are supported by its PRMT1-mediated methylation. Cancer research, 2016. 76(23): p. 7049–7058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jin X, et al. The ID1-CULLIN3 axis regulates intracellular SHH and WNT signaling in glioblastoma stem cells. Cell reports, 2016. 16(6): p. 1629–1641. [DOI] [PubMed] [Google Scholar]
- 51.Laner-Plamberger S, et al. Cooperation between GLI and JUN enhances transcription of JUN and selected GLI target genes. Oncogene, 2009. 28(13): p. 1639–1651. [DOI] [PubMed] [Google Scholar]
- 52.Abe Y, et al. MEP50/PRMT5-mediated methylation activates GLI1 in Hedgehog signalling through inhibition of ubiquitination by the ITCH/NUMB complex. Communications biology, 2019. 2(1): p. 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsherniak A, et al. Defining a cancer dependency map. Cell, 2017. 170(3): p. 564–576. e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang ZY, et al. Clinicopathological analysis of UHRF1 expression in medulloblastoma tissues and its regulation on tumor cell proliferation. Med Oncol, 2016. 33(9): p. 99. [DOI] [PubMed] [Google Scholar]
- 55.Bostick M, et al. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science, 2007. 317(5845): p. 1760–4. [DOI] [PubMed] [Google Scholar]
- 56.Pócza T, et al. High expression of DNA methyltransferases in primary human medulloblastoma. 2016. [DOI] [PubMed] [Google Scholar]
- 57.Smith ZD and Meissner A, DNA methylation: roles in mammalian development. Nat Rev Genet, 2013. 14(3): p. 204–20. [DOI] [PubMed] [Google Scholar]
- 58.Hervouet E, Vallette FM, and Cartron P-F, Dnmt1/transcription factor interactions: an alternative mechanism of DNA methylation inheritance. Genes & cancer, 2010. 1(5): p. 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Söderberg O, et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nature methods, 2006. 3(12): p. 995–1000. [DOI] [PubMed] [Google Scholar]
- 60.Heldin J, et al. Dynamin inhibitors impair platelet-derived growth factor β-receptor dimerization and signaling. Experimental Cell Research, 2019. 380(1): p. 69–79. [DOI] [PubMed] [Google Scholar]
- 61.Wang B and Li Y, Evidence for the direct involvement of βTrCP in Gli3 protein processing. Proceedings of the National Academy of Sciences, 2006. 103(1): p. 33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ding G, et al. Regulation of Ubiquitin-like with Plant Homeodomain and RING Finger Domain 1 (UHRF1) Protein Stability by Heat Shock Protein 90 Chaperone Machinery. J Biol Chem, 2016. 291(38): p. 20125–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Giovinazzo H, et al. A high-throughput screen of pharmacologically active compounds for inhibitors of UHRF1 reveals epigenetic activity of anthracycline derivative chemotherapeutic drugs. Oncotarget, 2019. 10(32): p. 3040–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ibrahim A, et al. Thymoquinone challenges UHRF1 to commit auto-ubiquitination: a key event for apoptosis induction in cancer cells. Oncotarget, 2018. 9(47): p. 28599–28611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ganesan A, et al. The timeline of epigenetic drug discovery: from reality to dreams. Clinical epigenetics, 2019. 11(1): p. 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hollenbach PW, et al. A comparison of azacitidine and decitabine activities in acute myeloid leukemia cell lines. PLoS One, 2010. 5(2): p. e9001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pappalardi MB, et al. Discovery of a first-in-class reversible DNMT1-selective inhibitor with improved tolerability and efficacy in acute myeloid leukemia. Nature cancer, 2021. 2(10): p. 1002–1017. [PMC free article] [PubMed] [Google Scholar]
- 68.Wechsler-Reya RJ and Scott MP, Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron, 1999. 22(1): p. 103–114. [DOI] [PubMed] [Google Scholar]
- 69.Wallace VA, Purkinje-cell-derived Sonic hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Current Biology, 1999. 9(8): p. 445–448. [DOI] [PubMed] [Google Scholar]
- 70.Dahmane N and Ruiz-i-Altaba A, Sonic hedgehog regulates the growth and patterning of the cerebellum. Development, 1999. 126(14): p. 3089–3100. [DOI] [PubMed] [Google Scholar]
- 71.Ecke I, et al. Antitumor effects of a combined 5-aza-2′ deoxycytidine and valproic acid treatment on rhabdomyosarcoma and medulloblastoma in Ptch mutant mice. Cancer research, 2009. 69(3): p. 887–895. [DOI] [PubMed] [Google Scholar]
- 72.Nguyen AN, et al. Azacitidine and decitabine have different mechanisms of action in non-small cell lung cancer cell lines. Lung Cancer: Targets and Therapy, 2010. 1: p. 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gnyszka A, JASTRZĘBSKI Z, and Flis S, DNA methyltransferase inhibitors and their emerging role in epigenetic therapy of cancer. Anticancer research, 2013. 33(8): p. 2989–2996. [PubMed] [Google Scholar]
- 74.Zhang Z-Y, et al. Regulation of UHRF1 by microRNA-378 modulates medulloblastoma cell proliferation and apoptosis. Oncology Reports, 2017. 38(5): p. 3078–3084. [DOI] [PubMed] [Google Scholar]
- 75.Sidhu H and Capalash N, UHRF1: The key regulator of epigenetics and molecular target for cancer therapeutics. Tumour Biol, 2017. 39(2): p. 1010428317692205. [DOI] [PubMed] [Google Scholar]
- 76.Alhosin M, et al. Signalling pathways in UHRF1-dependent regulation of tumor suppressor genes in cancer. J Exp Clin Cancer Res, 2016. 35(1): p. 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hervouet E, et al. Specific or not specific recruitment of DNMTs for DNA methylation, an epigenetic dilemma. Clinical epigenetics, 2018. 10(1): p. 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Berman DM, et al. Medulloblastoma growth inhibition by hedgehog pathway blockade. Science, 2002. 297(5586): p. 1559–1561. [DOI] [PubMed] [Google Scholar]
- 79.Felle M, et al. The USP7/Dnmt1 complex stimulates the DNA methylation activity of Dnmt1 and regulates the stability of UHRF1. 2011. 39(19): p. 8355–8365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhu H, Wang G, and Qian J, Transcription factors as readers and effectors of DNA methylation. Nature Reviews Genetics, 2016. 17(9): p. 551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yin Y, et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science, 2017. 356(6337). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hu S, et al. DNA methylation presents distinct binding sites for human transcription factors. elife, 2013. 2: p. e00726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Qiu X, et al. Equitoxic doses of 5-azacytidine and 5-aza-2′ deoxycytidine induce diverse immediate and overlapping heritable changes in the transcriptome. Plos one, 2010. 5(9): p. e12994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Venturelli S, et al. Differential induction of apoptosis and senescence by the DNA methyltransferase inhibitors 5-azacytidine and 5-aza-2′-deoxycytidine in solid tumor cells. Molecular cancer therapeutics, 2013. 12(10): p. 2226–2236. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data were generated by the authors are available within the paper and its supplementary files or from the authors on request. The bioinformatics data analyzed in this study was obtained as described in the “Bioinformatics analysis” section above.