

Abstract
The endoplasmic reticulum (ER) is an intracellular organelle that fosters the correct folding of linear polypeptides and proteins, a process tightly governed by the ER-resident enzymes and chaperones. Failure to shape the proper 3-dimensional architecture of proteins culminates in the accumulation of misfolded or unfolded proteins within the ER, disturbs ER homeostasis, and leads to canonically defined ER stress. Recent studies have elucidated that cellular perturbations, such as lipotoxicity, can also lead to ER stress. In response to ER stress, the unfolded protein response (UPR) is activated to reestablish ER homeostasis (“adaptive UPR”) or conversely, to provoke cell death when ER stress is overwhelmed and sustained (“maladaptive UPR”). It is well documented that ER stress contributes to the onset and progression of multiple hepatic pathologies including non-alcoholic fatty liver disease (NAFLD), alcoholic liver disease (ALD), viral hepatitis, liver ischemia, drug toxicity, and liver cancers. Here, we review key studies dealing with the emerging role of ER stress and the UPR in the pathophysiology of liver diseases from cellular, murine, and human models. Specifically, we will summarize current available knowledge on pharmacological and non-pharmacological interventions that may be employed to target maladaptive UPR for the treatment of non-malignant liver diseases.
Keywords: ER stress, UPR, Liver disease, Liver toxicity, Liver viral infections, Liver cancer
Graphical Abstract
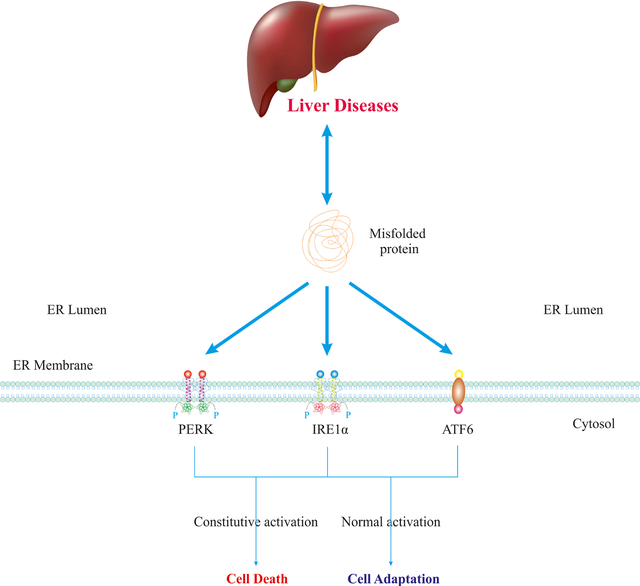
1. Introduction: ER Stress and Canonical UPR Activation
The endoplasmic reticulum (ER) constitutes one of the largest subcellular compartments and mediates a cadre of biological functions including cytosolic co-translational folding of proteins, Ca2+ homeostasis, and biosynthesis of steroids/lipids (1). The ER serves as a network for vesicular transport including trafficking of endosomes (2). ER function is tightly orchestrated by essential regulatory factors such as protein oxidoreductase chaperones, proteolysis/glycosylation/sulfation enzymes, Ca2+ transporters, and channels. Both physiological and pathological stimuli, such as B cell maturation, glucose-induced insulin secretion, nutrient deprivation, oxidative stress, hypoxia, and ER-relevant gene mutations may all perturb ER homeostasis, resulting in ER stress (3). In response to ER stress, eukaryotic cells have adapted the ER-nucleus signaling system, commonly known as the “unfolded protein response” (UPR) to resolve ER stress or execute cell death. Upon mild to moderate ER stress, UPR is initiated to remove unfolded/misfolded proteins and restore ER homeostasis. This type of UPR is referred to as “adaptive/cytoprotective” UPR. However, upon severe or persistent ER stress, UPR is overwhelmingly hyperactivated, leading to the activation of intrinsic apoptotic machinery (4). This type of UPR is referred as “maladaptive/unchecked/terminal” UPR (5). Recent studies have demonstrated that UPR sensors can be turned on by stimuli that do not directly affect protein folding, e.g., saturated free fatty acids, leading to the evolution of the term “canonical UPR” which refers to the activation of the three UPR sensors upon buildup of unfolded/misfolded proteins (6). Accumulating evidence indicates that severe ER stress and maladaptive UPR contribute to the pathogenesis of hepatic pathologies (7), including dysmetabolism, acute and chronic viral infection, drug toxicity, and cancer (8). Hence, a better comprehension of UPR mechanisms in the pathogenesis of these conditions may help in the identification of novel therapeutic strategies. This review discusses some of the recent insights into ER stress and UPR signaling in hepatic pathophysiology.
2. Mechanisms of UPR Signaling and Activation
UPR signaling is initiated by three ER transmembrane proteins commonly referred to as UPR branches/sensors including PERK (encoded by EIF2AK3), IRE1α (encoded by ERN1), and ATF6α (Fig. 1, 2). Under physiological conditions, the luminal domains of UPR sensors interact with and bind to the ER-resident chaperones, in particular BiP/GRP78 (encoded by HSPA5). When unfolded/misfolded proteins accumulate within the ER lumen, they bind to, and saturate BiP, thus competitively dissociating BiP from luminal domains of UPR sensors, leading to their subsequent oligomerization and activation. Several lines of evidence have also suggested a direct binding of unfolded/misfolded proteins with the luminal domains of UPR sensors that result in their activation and oligomerization. In contrast, under lipotoxic conditions, IRE1α and PERK can be activated via their transmembrane domains (9).
Fig. 1. Mild ER Stress And Adaptive UPR.
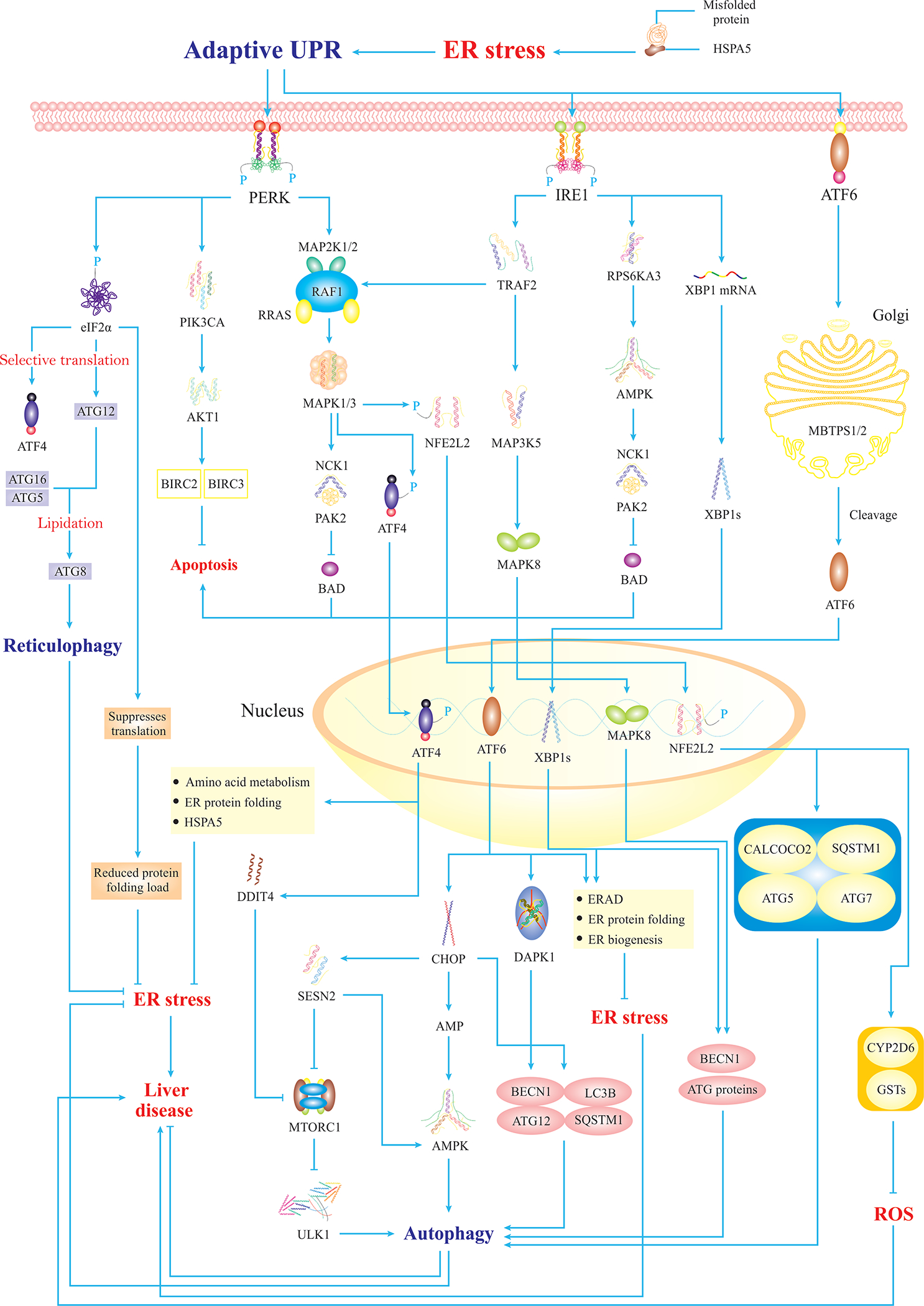
Mild ER stress triggers adaptive UPR signalings that consist of three main branches including PERK, IRE1, and ATF6α. PERK mediates phosphorylation of eIF2α that participates in autophagy of the ER (reticulophagy) through a selective translation of ATG12. Also, PERK mediates selective translation of ATF4 that translocates to the nucleus and transactivates UPR-target genes associated with autophagy induction and alleviation of ER stress. Besides, PERK activates the PIK3CA-AKT1 axis to suppress apoptosis. Moreover, PERK turns on MAPK signaling, which inhibits BAD-mediated apoptosis, and activates NFE2L2 that translocates to the nucleus and transactivates genes associated with autophagy and ROS inhibition. Similarly, IRE1 triggers pathways to block BAD-mediated apoptosis. Importantly, IRE1 induces activation of MAPK8 that translocates to the nucleus and upregulates autophagy genes. Likewise, IRE1-activated XBP1s translocates to the nucleus and transactivates autophagy and ERAD-related genes. Finally, ATF6α is cleaved and activated in the Golgi then translocates to the nucleus to mildly upregulate CHOP, resulting in mild autophagy. It is noteworthy that ULK1 forms an autophagy initiation complex, which is regulated by MTORC1, as ULK1 phosphorylation renders ULK1 inactivation and autophagy inhibition (5).
Fig. 2. Severe ER Stress Triggers Maladaptive UPR.
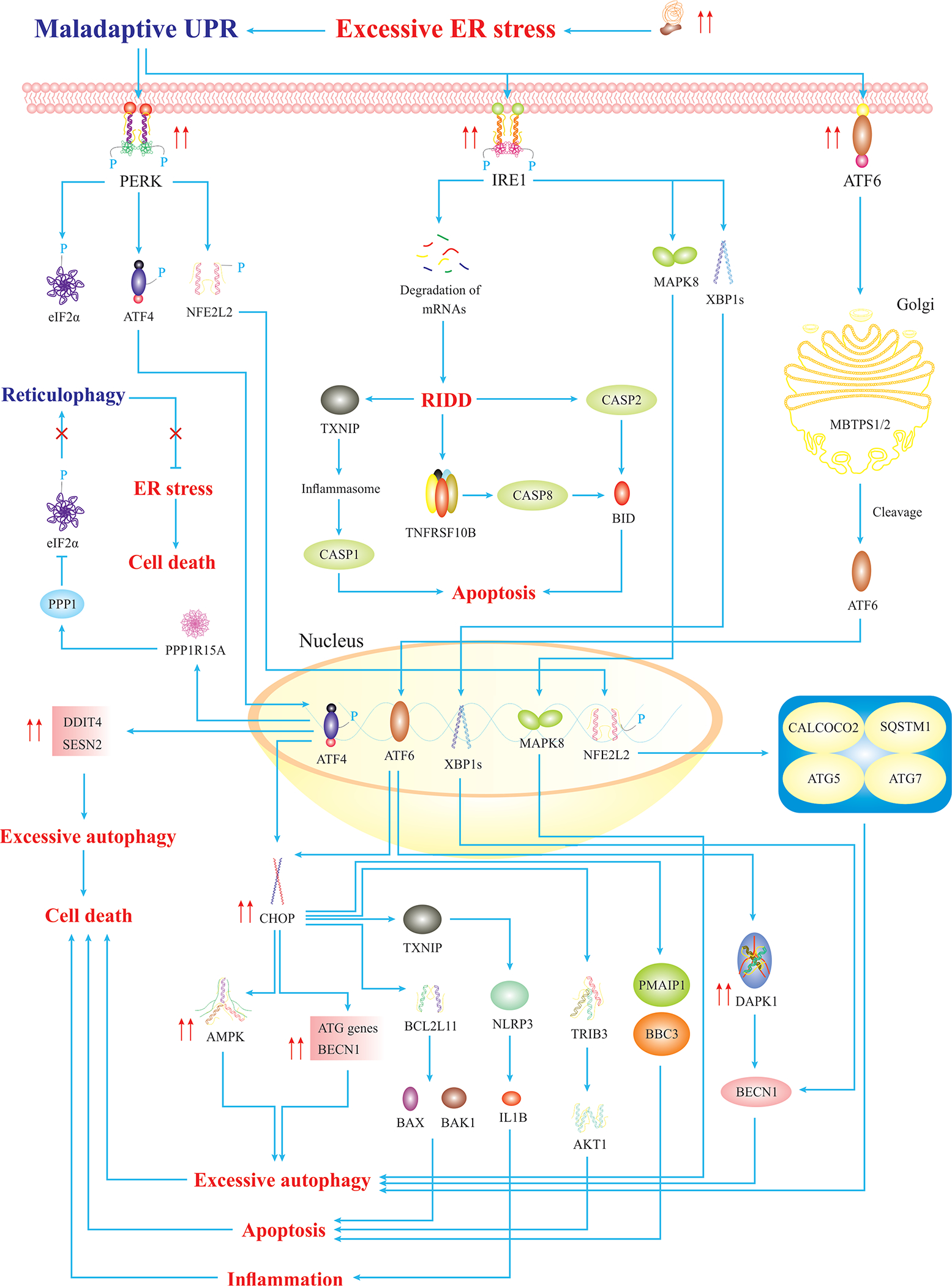
Severe ER stress triggers maladaptive UPR, leading to activation of cell death. Hyperactivated PERK triggers overexpression of NFE2L2-/ATF4-target genes leading to excessive autophagy, cell death, and CHOP-mediated apoptosis. Also, hyperactivation of IRE1 induces apoptosis via CASP2 activation. Hyperactivated MAPK8 and XBP1s induce overexpression of autophagy genes, resulting in excessive autophagy. Similarly, hyperactivation of ATF6α induces overexpression of proapoptotic factors and autophagy genes, ultimately, leading to cell death (5).
2. 1. IRE1 Branch
UPR signaling through IRE1 is highly conserved in eukaryotic cells (10). The mammalian genome encodes two isoforms of IRE1; IRE1α (ERN1) and IRE1β (ERN2). IRE1α and IRE1β possess different functions owing to distinct RNase domains and substrate specificities. IRE1α is ubiquitously expressed and mediates downstream signaling through its kinase and endoribonuclease cytosolic domains. The role of IRE1α is well-established in UPR signaling and will be our focus in the subsequent sections. IRE1β is uniquely expressed in intestinal epithelial cells and it is assumed that IRE1β-mediated RNA cleavage participates in digestive tissue-specific UPR (11, 12). Under normal protein load in the ER, IRE1α activation is constrained by BiP as described above. Upon the dissocation of BiP, IRE1α undergoes oligomerization and autophosphorylation (13). IRE1α autophosphorylation activates its endoribonuclease domain to excise a 26 base intron from XBP1 mRNA, leading to XBP1 splicing into the shorter XBP1 mRNA, mediated by RtcB RNA ligase (14). As a result, translation of shorter XBP1 mRNA yields active spliced XBP1 protein (XBP1s) (Fig. 1, 2), which traffics to the nucleus to transactivate ER chaperones and ER secretory genes (15). Moreover, XBP1s transactivates genes involved in ER-associated degradation (ERAD), which boosts the transport of unfolded/misfolded proteins out of ER for degradation by proteasomes (16). In addition, the IRE1α endoribonuclease can degrade nonessential microRNAs and mRNA in a process known as “regulated IRE1α dependent decay” (RIDD) (17–19). RIDD is increasingly associated with cell survival and cell death as well as metabolic anomalies in the liver (discussed in subsequent sections) (17–19).
2. 2. PERK Branch
PERK is a transmembrane protein with its cytosolic kinase domain activated by oligomerization upon ER stress. The major substrate of PERK is the α subunit of the heterotrimeric eukaryotic translation initiation factor 2 (eIF2α), which may also be phosphorylated by three additional eIF2α kinases, HRI, GCN2, and PKR, which respond to different stress stimuli. This regulation couples the rate of protein synthesis to environmental factors such as viral infection (PKR), amino acid starvation (GCN2), and oxidative stress (HRI). PERK-mediated eIF2α phosphorylation leads to the general suppression of translation while allowing selective translation of certain mRNAs, such as ATF4 mRNA (20). ATF4 traffics to the nucleus to transactivate UPR target genes, leading to either cell survival or cell death. CHOP is considered one of the most critical and essential proapoptotic factors downstream of eIF2α and ATF4 and contributes to the pathogenesis of liver diseases (21, 22) (Fig. 1, 2).
2. 3. ATF6 Branch
ATF6 is a transmembrane protein with a cytosolic bZIP transcription factor dimerization domain. Two mammalian paralogues, ATF6α and ATF6β, exist and contain distinct transcriptional activation domains. It is suggested that ATF6β may function as a repressor to regulate the duration and extent of ATF6α-mediated gene modulation in UPR (23). Upon ER stress, BiP releases ATF6α from the ER, enabling ATF6α to traffic to the cis-Golgi (24). There, full-length ATF6α is cleaved into ATF6p50 by two Golgi-resident processing enzymes, namely site-1 protease (S1P)/MBTPS1 and site-2 protease (S2P)/MBTPS2, the same enzymes required for processing the liver-specific regulators of lipid homeostasis, SREBP1/3, and SREBP2. Following proteolytic cleavage, the active form of ATF6α (ATF6p50), traffics to the nucleus and transactivates the UPR target genes. Moreover, ATF6p50 can heterodimerize with XBP1s, favoring upregulation of ERAD-associated genes (24) (Fig. 1, 2).
2. 4. Crosstalk among UPR Branches
A possible crosstalk among IRE1α, PERK, and ATF6 in UPR activation is rather intricate. It was reported that ablation of ATF6 evoked uncontrolled activity of IRE1α and upregulated XBP1 splicing (25). Also, cells with ATF6 ablation exhibited temporary upregulation of IRE1 and IRE1 protein levels upon ER stress, indicating a unique accelerated role for IRE1α signaling in the abscense of ATF6. Vice versa, ATF6 overexpression reversed the enhanced activity and levels of IRE1α (25). Moreover, activation of the PERK-eIF2α-ATF4 axis jacked up ATF6 and its target genes. Indeed, the PERK signaling axis fostered the synthesis and translocation of ATF6 to the Golgi for ultimate activation (26). Interestingly, a novel crosstalk between XBP1-ATF6 and PERK branches was identified in a recent study whereby PERK-mediated activation and phosphorylation of eIF2α suppressed certain subset of genes pertaining to XBP1-ATF6 signalings thus, governing cell fate and survival (27). Also, it is postulated that ER stress-induced apoptosis is characterized by concerted timing of various UPR branches rather than input from a single branch or a switch between branches (28).
3. ER Stress in Nonalcoholic Fatty Liver Disease and Steatohepatitis
Nonalcoholic fatty liver disease (NAFLD) is characterized by the accumulation of triglycerides in the liver as often seen in overweight or obese individuals. A subset of NAFLD patients develop liver injury, inflammation, and fibrosis, collectively termed as nonalcoholic steatohepatitis (NASH) (29, 30). ER stress and UPR sensors are involved in steatosis, inflammation, and fibrosis in the context of NAFLD. Initial findings noted a close tie between obesity-induced ER stress and insulin resistance (31). These initial observations were later supported by several studies elucidating the role of ER stress and UPR sensors in hepatic steatosis and individual liver cell types (cited in later sections). We discuss these further in the context of each of the UPR sensors.
3. 1. IRE1α in NAFLD/NASH
IRE1α signaling via XBP1 generation and RIDD are both implicated in NAFLD (32). Several observations support a harmful role for IRE1α activation in NASH. For example, the ER-resident protein BI-1 constrians IRE1α activation. When BI-1 is deleted mice are sesnsitived to HFD-induced NASH along with increased activation of IRE1α and XBP1. Upregulation of BI-1 inhibits IRE1α activity, leading to reduced levels of XBP1s in animal models of tunicamycin-induced ER stress (33). Activated IRE1α promotes transcription of serine palmitoyltransferase genes via XBP1s, resulting in ceramide biosynthesis and release of extracellular vesicles (EVs) from hepatocytes (33). In mice with diet-induced NASH, EVs recruit macrophages to the liver to accentuate inflammation and injury in diet-mediated steatohepatitis. Levels of XBP1, serine palmitoyltransferase, and EVs are all elevated in liver tissues from patients with NASH (33). In contrast, S-nitrosylation of IRE1α resulted in reduced endoribonuclease activity of hepatic IRE1α and thereby, reduced XBP1 splicing in HFD-fed and ob/ob mice (34, 35). Nonetheless, reinitiation of hepatic IRE1α expression by establishing IRE1α resistant to s-nitrosylation culminated in increased XBP1 levels, thus, augmenting glucose homeostasis (34). Additional parameters relevant to NAFLD were not reported in this study. However, in a separate study, Wang and colleagues revealed an increase in liver triglyceride levels owing to s-nitrosylation of IRE1α and damaged endoribonuclease activity, which inhibited IRE1α-induced degradation of miR-34 and miR-200, key microRNAs which regulate hepatic lipid metabolism (36). Moreover, this group revealed that liver-specific knockout of IRE1a, resulted in enhanced liver fibrosis and inflammation in mice under HFD, suggesting the protective role of IRE1α against NASH (35, 36). These differences in the role of IRE1α may be partly explained by context dependent signaling, such that a balance of XBP1 splicing and RIDD determines the eventual outcome of IRE1α activation. The possibility of additional regulatory molecules, similar to BI-1, cannot be excluded either.
3. 2. PERK Pathway in NAFLD/NASH
The PERK-eIF2α pathway leads to the upregulation of UPR target genes and induces the proapoptotic protein CHOP (Fig. 1). Novoa and colleagues revealed that GADD34 binds to PP1 catalytic subunit (PPIc), leading to eIF2α dephosphorylation and attenuated levels of DDIT3 and ATF4, thus, suppressing the UPR (37). Li and colleagues established high-fat diet-induced obesity in C57BL/6 mice and administered salubrinal (indirectly blocks eIF2α through inhibiting its α-subunit dephosphorylation) subcutaneously, prior to evaluation of autophagy and ER stress in vitro and in vivo. Histological analyses revealed that salubrinal alleviated obesity in association with reduction in hepatic steatosis and fibrosis. High-fat diet did not induce eIF2α phosphorylation, while a rise in ATF4 was paradoxically associated with dampened DDIT3 expression in this model, suggesting an alternative mechanism of action. Indeed, eIF2α phosphorylation induced autophagy, which might be the likely mechanism for hepato-protective effects of salubrinal (38). However, ER stress-induced cell death is attenuated in CHOP deficient hepatocytes. The role of CHOP in NASH is more complex, as CHOP-dependent macrophage apoptosis was found to protect mice from NASH (22).
3. 3. ATF6α in NAFLD/NASH
Decreased ER membrane fluidity can selectively activate ATF6α and cause NASH (39). Recently, ATF6α was shown to be activated by sphingolipids such as dihydrosphingosine and dihydroceramide through a mechanism distinct from protein misfolding. Sphingolipid activation does not induce UPR genes, but rather turns on ER lipid biosynthetic genes (40). ATF6p50 may indirectly suppress lipogenic genes through inducing CHOP via its dominant-negative inhibition of C/EBPα, ultimately reducing fatty acid oxidation, lipoprotein secretion, or BiP, the overexpression of which inhibits SREBP1 proteolytic cleavage (41, 42).
3. 4. Secondary UPR Activation in NAFLD/NASH
In addition to the above described roles of each UPR sensor in NAFLD, several studies have placed ER stress activation downstream of perturbations which modulate lipotoxicity and NAFLD. For example, mice deficient in the lysosomal membrane protein SID1 transmembrane family member 2 (SIDT2) developed liver injury and altered lipid metabolism, causing hepatic steatosis, and inflammatory infiltration in lobular and portal areas. SIDT2 deficiency caused significant elevation of SREBP1 protein and SREBF1 mRNA levels, as well as upregulation of genes implicated in fatty acid biosynthesis. Importantly, SIDT2 deficiency induced unchecked ER stress and UPR activation accompanied by ER damage in the liver (43). Kim and colleagues demonstrated that ER stress-induced upregulation of CASP2 led to the accumulation of TG and free cholesterol in hepatocytes by activating SREBP1/2 transcriptional factors in a diet-induced model of NASH (44). The underlying mechanism behind SREBP1/2 activation involves colocalization of CASP2 with MBTPS1 prior to cleavage of MBTPS1 into a soluble active form, which in turn, truncates the ER loop in SREBP1/2 (44). Consequently, SREBP1 upregulates genes associated with fatty acid synthesis, and SREBP2 upregulates genes in cholesterol synthesis, leading to NASH progression. In addition, pharmacological inhibition or genetic ablation of CASP2 reversed ER stress-induced NASH (44). Excess cholesterol activated ER stress in macrophages plays a role in atherosclerosis (45). Similarly, in a dietary NASH model with high cholesterol (2%), accumulation of cholesterol was associated with ER stress and inflammasome activation (46). Though the high cholesterol content limits wider generalization, these observations suggest that lipotoxicity may mediate activation of the ER stress response in models which interfere with lipid metabolism and lead to the accumulation of bioactive lipid species.
Nutrient-dependent metabolic regulators may link gene expression with ER stress. In this context, Shin and coworkers noted that SIRT7 modulated gene expression, leading to reduced ER stress and NAFLD pathology in obese mice. The possible underlying mechanism was that induction of ER stress elicits SIRT7 activation, which directly inhibits MYC transcriptional factor to silence gene expression and relieve ER stress (47). These authors indicated that MYC is likely to stabilize SIRT7 at the promoters of ribosomal proteins to mediate chromatin remodeling and gene repression, leading to suppression of both ER stress-associated genes and ER stress. In addition, SIRT7 deficiency potentiated chronic hepatosteatosis, while pharmacological inhibition of ER stress or MYC inactivation mitigated ER stress and murine fatty liver (47). This finding suggests that therapeutic targeting of the SIRT7-MYC axis could revert ER stress and murine fatty liver.
Overall, ER stress and the UPR play a cardinal role in the pathogenesis of NAFLD/NASH. Mild ER stress triggers an adaptive UPR pathway that alleviates the disease pathology, while, severe and sustained ER stress leads to overwhelmed UPR activation, resulting in exacerbation of disease pathology through inflammation, cell death, and EV secretion.
4. ER Stress and Alcoholic Liver Disease
4. 1. PERK in Alcoholic Liver Disease
Of the three UPR sensors, the PERK signaling pathway is most well-studied in the context of ALD. Alcohol feeding in mice fostered PERK activation and ATF4-dependent upregulation of NNMT, a key cytosolic methyltransferase enzyme, in association with upregulation of lipogenesis genes and steatosis in ALD. Upregulation of NNMT was dependent on ATF4 as ATF4 knockout reverted NNMT upregulation. (48). This study suggests that the PERK-ATF4-NNMT axis upregulates de novo lipogenesis in the liver in association with alcohol consumption, leading to steatosis. Nonetheless, it remains to be determined whether ATF4-triggered amino acid synthesis and transport also contribute to the onset of steatosis in ALD.
4. 2. Secondary ER Stress in ALD
ER stress can occur secondary to primary hepatic pathological stress. Mouse hepatocyte-specific knockout of DGAT1, a rate-limiting enzyme of triglyceride synthesis, worsened steatosis and hepatocellular injury in ALD by promoting FFA build-up in the liver, thus triggering ER stress in concert with downregulation of LAMP2 levels and defective autophagy. Mechanistically, FFAs-induced ER stress activated the ATF4 branch to mediate LAMP2 downregulation and subsequent inhibition of LAMP2-dependent autophagic flux. Moreover, lowering liver FFAs by PPARA activation reversed ER stress, replenished LAMP2, and reactivated autophagy (49). This finding suggests inhibitory modulation of secondary ER stress (e,g., ATF4 or its upstream signals) may constitute a potential strategy for the treatment of hepatocellular injury and steatosis in ALD.
Activation of innate immune system is an essential component of ALD. Ethanol-induced secondary ER stress activated the STING1-IRF3 signaling cascade. As a result, activated IRF3 interacted with and activated proapoptotic factor BAX, leading to hepatocyte apoptosis and ALD. STING1 deficiency caused IRF3 inactivation and therefore, inhibition of hepatocyte apoptosis. Of note, these studies noted that IRF3-mediated pathogenic action did not rely on the presence of type I interferons or inflammation (50). Taken together, the current literature denotes that ER stress occurs constitutively in or secondary to the state of ALD, leading to maladaptive UPR activation thus, promoting disease progression. In this regard, inhibition targeting of UPR components seems to be a proper therapeutic approach for the treatment of ALD.
5. ER Stress in Chronic Viral Hepatitis B, C
5. 1. ER Stress in Hepatitis B
Viral replication usurps host’s cellular protein synthesis machinery. In this regard, hepatitis B virus (HBV) and hepatitis C virus (HCV) infection is associated with many components of ER stress signaling. He and coworkers revealed that level of B56γ subunit of PPP2CA (a major cellular serine/threonine phosphatase) was positively correlated with HBV x protein (HBx) levels in HBx-transgenic mice, hepatic cells expressing HBx in vitro and human hepatocytes introduced into humanized chimeric mice (51). Mechanistically, HBx expression in hepatocytes induced ER stress which in turn activated CREB3L3 signaling, leading to activation of JUN and its nuclear translocation, to transactivate gene encoding B56γ subunit. As a result, B56γ induced dephosphorylation of TP53 and consequently, CDKN1A activation, which triggered cell cycle arrest at the G1 phase and hepatocyte apoptosis (51). Therefore, HBx-induced ER stress activates the CREB3L3-JUN-TP53-CDKN1A axis, which triggers cell cycle arrest and cell death, indicating that targeting ER stress could mitigate HBV-induced liver injury.
During HBV infection, several HBV envelope proteins are upregulated in the ER. These envelope proteins include MHBst (truncated middle S protein) and LHBs (large S protein), which regulate gene expression of host hepatocytes, contributing to liver injury. Li and colleagues demonstrated that MHBst expression upregulated IL-6 via activation of the MAPK14 and NF-κB pathways. Besides, they showed that MHBst accumulation in the ER activated ER stress that was perceived to be the underlying mechanism of activation for MAPK14 and NF-κB signaling (52). Therefore, MHBst triggers ER stress-induced MAPK14 and NF-κB activation, which mediates IL-6 production and liver disease progression. Hence, suppressing ER stress and MAPK14 and NF-κB activation may diminish liver damage upon HBV infection. Overall, these observations suggest that constitutive ER stress inflicts maladpative effects and promotes HBV infection. Hence, inhibiting ER stress and maladptive UPR may ameliorate HBV pathology.
5. 2. ER Stress in Hepatitis C
Hepatitis C virus (HCV) is known to induce ER stress and upregulate ER stress-associated genes. In line with this, it was reported that expression and replication of HCV proteins in the human liver Huh7 cell line, which carries complete HCV replicon, triggered ER stress and upregulation of CHOP, resulting in apoptotic cell death. Further analysis revealed that ER stress-induced ATF4 and ATF6α components of UPR were responsible for upregulation and activation of DDIT3/CHOP. Besides, treating HCV replicon cells with H2O2 further increased CHOP upregulation, suggesting that oxidative stress and ER stress can lead to CHOP upregulation and cell death in HCV replicon cells. Furthermore, using small interfering RNA to suppress the DDIT3 gene reversed cell death under oxidative and ER stress (53). Hence, inhibitory targeting of ER stress and oxidative stress may prevent HCV-induced liver injury.
Asselah and associates also revealed features of ER stress and activation of all three UPR branches (IRE1α, ATF6α, and PERK) as well as upregulation of inflammatory and apoptotic genes in infected hepatocytes from patients. Also, these investigators observed that ER stressed-hepatocytes formed clusters scattered across hepatic parenchyma. Besides, HCV managed to suppress UPR-responsive genes from propagating ER stress among hepatocytes via unknown mechanisms (54). This study further pinpoints the potential of ER stress inhibition and activation of UPR-responsive genes in the management of HCV-induced liver damage.
Altogether, current available evidence suggests that ER stress induction under HCB and HCV infection is relatively severe, thus evoking persistent activation of UPR, resulting in the progression of hepatitis disease pathology. Hence, inhibition targeting of ER stress and the UPR branches may be a sensible approach in the management/treatment of HCB and HCV. However, generally, apoptosis is not a frequent feature of HBV of HCV infection as both viruses usurp ER machinery to replicate. Therefore, it is very difficult to conceptualize these limited studies or learn the role of ER stress in HBV or HCV.
6. ER Stress and Liver Cancer: Hepatocellular Carcinoma
6. 1. Implications of the UPR in Hepatocarcinogenesis
HCC is the most prevalent primary liver malignancy. ER stress and UPR activation occur in multiple malignancies including HCC. It is well-known that HBV potentiates HCC and serves as the primary cause of HCC globally. Also, recent data suggest that HCC can result from NASH prior to onset of cirrhosis (55). Shuda and team assessed the expression of ATF6α, BiP, XBP1 in HCC tissue samples and observed augmentation of ATF6α and HSPA5 mRNAs, accumulation of BiP in the cytoplasm, increased nuclear localization of ATF6p50 and XBP1 mRNA splicing, in association with more severe histological scores (56). Hence, ER stress-activated UPR branches such as ATF6α and IRE1α may play a role in hepatocarcinogenesis (56, 57).
Besides, mutation profiling in human HCC identified non-synonymous mutations in numerous ATF6α target genes and a somatic mutation in a gene that prevents ATF6α degradation, thus promoting ATF6α accumulation and activation (58). On the other hand, a single nucleotide polymorphism in ATF6α increased levels of ATF6α mRNA, ATF6α target genes and susceptibility to HCC (59). It is noteworthy that ATF6α loss-of-function mutations possess a mild phenotype in human, resulting in achromatopsia - a form of progressive retinal degeneration (60, 61), suggesting that short-term inhibition of ATF6α may be affordable to treat human NASH and HCC.
Moreover, IRE1α signaling plays a significant role in HCC onset, as hepatic-specific IRE1 deficiency prevented HCC development in mice under diethylnitrosamine treatment (62). IRE1 deficiency was accompanied by STAT3 activation, restrained hepatocyte proliferation, enhanced hepatocyte apoptosis, and attenuated levels of TNF-α and IL-6 (62). It is perceived that UPR activation is an adaptive mechanism in malignant hepatic cells to cope with ER stress under unfavorable environmental conditions and chemotherapeutic drugs. However, severe/constitutive ER stress may induce cell death in HCC cells, thus, exhibiting promises in the prevention of HCC progression. Besides, pharmacological inhibition of IRE1α endoribonuclease activity reduced tumor burden in mice with fibrotic HCC (63). Treatment with the IRE1α inhibitor 4μ8c blocked stellate cell activation, restrained proliferation and migration of tumor cells in 2D and 3D in vitro co-cultures, possible due to reduced levels of intracellular ROS. Hence, IRE1α may play a role in the crosstalk between hepatic stellate cells and tumor cells in liver cancers and possess therapeutic promises as a pharmacological target.
6. 2. Mild ER Stress and Adaptive UPR in HCC
Examing ZNF263 (a transcription factor) in the face of ER stress-mediated drug resistance in HCC cell lines and HCC patients revealed that ZNF263 was significantly upregulated. Besides, level of ZNF263 was found to correlate with ER stress, clinical stage, and shorter survival of HCC patients. Mechanistically, ZNF263 induced ER stress and, subsequently, autophagy that conferred chemoresistance to HCC cells. Thus, ZNF263 knockdown using RNA interference diminished proliferation, reduced chemoresistance, and enhanced apoptosis in HCC (64). This study suggests that ZNF263-induced ER stress provokes adaptive autophagy that reduces the sensitivity of HCC cells to chemotherapeutic drugs. Hence, abrogating autophagy may boost the anticancer effect of chemical drugs.
Liu and associates reported upregulation of ER stress markers including BiP/HSPA5, ATF6α, PERK/EIF2AK3, IRE1α/ERN1 in HCC tissues, which was negatively correlated with the overall patient survival, recruitment of CD68+ macrophages and CD274 expression in HCC tissues. HCC cells exposed to tunicamycin (an ER stress inducer) were found to release abundant EV containing microRNA (MIR)-23a-3p, and MIR-23a-3p may be responsible for the upregulation of CD274 in macrophages both in vivo and in vitro. Mechanistically, MIR-23a-3p regulated CD274 via inhibiting PTEN expression and therefore, enhanced AKT1 phosphorylation (activation), leading to upregulation of CD274. Moreover, coculturing T-cells with EV-treated macrophages induced T-cell apoptosis and reduced CD8+ T-cell levels (65). Hence, this study suggests that ER stressed-HCC cells release EV to confer T cell-immunosuppressive properties to macrophages.
6. 3. Severe/Constitutive ER Stress and Maladaptive UPR in HCC
Severe/constitutive ER stress and unchecked UPR may be useful in the treatment of HCC. For example, Lee and team applied combination of TNFSF10 (a cytokine that induces apoptosis) and NSAIDs (nonsteroidal anti-inflammatory drugs) on TNFSF10-resistant/CD44-expressing HCC cells. They discovered that the NSAID celecoxib and its derivative 2,5-dimethyl celecoxib reversed TNFSF10 resistance in HCC cells. Mechanistically, celecoxib dose-dependently induced ER stress, ATF4 and CHOP expression, PRKAA2 (aka AMPK) activation, and inhibition of AKT1-MTORC1 pathway, leading to facilitated autophagy, as indicated by increased MAP1LC3B levels and decreased p62 levels (66). Altogether, this finding suggests that combination of NSAID and TNFSF10 improves the clinical efficacy of TNFSF10-based HCC therapy likely through induction of profound ER stress and unchecked autophagy.
To investigate the role of ER stress in obesity-induced liver tumorigenesis, Nakagawa and coworkers utilized HFD-fed wild-type and MUP-PLAU mice. In MUP-PLAU transgenic model, hepatocyte-specific overexpression of PLAU causes hepatocyte ER stress at 6 wks of age that is attenuated by 10 wks due to extinction of PLAU expression. When fed an HFD starting at 10wks, MUP-PLAU mice developed symptoms of human NASH and HCC by 9 months. Thus, transient ER stress is sufficient to establish an environment conducive to NASH and HCC development upon subsequent HFD feeding. Indeed, both HFD-fed wild-type and MUP-PLAU mice were insulin resistant, although MUP-PLAU mice displayed more pronounced liver injury, lipogenesis, and immune infiltration, resulting in NASH and steatohepatitis-driven HCC. Mechanistically, hepatocyte ER stress in MUP-PLAU mouse livers stimulated the recruitment of inflammatory macrophages to produce TNF and contribute to the pathogenesis of NASH and HCC (55). These findings consolidate the notion that ER stress is embedded in a molecular cascade leading to hepatic carcinogenesis.
In summary, the role of ER stress and the UPR is a double-edged sword in HCC, as these malignant cells hijack adaptive UPR to suppress mild ER stress and maintain cell growth and proliferation. On the other side of the coin, induction of severe ER stress and constitutive UPR serves as an effective measure to eliminate carcinomaous cells via UPR-dependent cell death.
7. ER Stress and Liver Ischemia/Reperfusion Injury
7. 1. ATF6α in Liver Ischemia/Reperfusion Injury
The contribution of ER stress to ischemia-associated deregulation of innate immunity is largely elusive. Rao and team observed that warm ischemia evoked ER stress and UPR in mice, particularly, ATF6α signaling in Kupffer cells (KC), thus modulating their response to TLRs agonists. As a result, ischemic and ER stressed KC reduced the production of anti-inflammatory cytokines but enhanced that of pro-inflammatory cytokines. Suppression of ER stress by means of the chemical chaperon 4-PBA or silencing ATF6α by small interfering RNAs curbed pro-inflammatory cytokine production and consequently dampened the local activation of immune cells (67). This study suggests that inhibiting ER stress may be beneficial for inflammatory complications of hepatic ischemia.
7. 2. IRE1-α in Liver Ischemia/Reperfusion Injury
Ischemic preconditioning confers partial protection from I/R injury. In an in vivo model of ER stress preconditioning, Liu and coworkers injected rats with tunicamycin prior to liver I/R challenge (68). Meanwhile, these researchers treated L02 and HepG2 cells with tunicamycin and thapsigargin. Liver I/R injury was apparently mitigated by ER stress induction and subsequent activation of IRE1α and BiP signaling. Of note, these authors observed that IRE1α interacted with RACK1, leading to the activation of IRE1-RACK1-PRKAA2 and IRE1-RACK1-BCL2 pathways, culminating in apoptosis inhibition and autophagy induction (68). This study suggests that ER stress preconditioning can alleviate liver I/R damage via activation of the IRE1-RACK1 axis.
7. 3. Secondary ER Stress in Liver Ischemia/Reperfusion Injury
Lu and associates investigated the impact of ER stress on IL-23A production upon I/R liver injury in vivo and in vitro. These authors revealed that ischemia upregulated IL23A and IL12B in liver non-parenchymal cells (NPCs), leading to an immune response against I/R injury and progression of hepatocellular inflammation and injury. It is believed that ER stress induction, autophagy inhibition, and TLR4 activation serve as the main underlying mechanism of IL23A and IL12B upregulation. Therefore, inhibiting TLR4 and alleviating ER stress using rapamycin and 4-PBA, respectively, led to inhibition of IL23A production and liver protection from I/R injury (69). This study denotes a synergistic effect among ER stress induction, autophagy inhibition, and TLR4 activation in I/R-induced liver injury. In conclusion, the current literature uncaps both preventive and promoting role of ER stress and UPR in liver I/R injury, depending on the extent and severity of ER stress and UPR activation.
8. ER Stress and Liver Fibrosis and Cirrhosis
8. 1. IRE1α in Liver Fibrosis
Fibrogenic signals trigger transcription of procollagen I, which then, enters the ER and passes through the ER-Golgi secretory compartment to be released into the extracellular matrix. Maiers and coworkers observed that perturbation of ER export of procollagen I induced ER stress and UPR activation, leading to apoptosis in hepatic stellate cells (HSCs) (70). They also revealed that HSCs-specific depletion of TANGO1 (a protein that participates in collagen I secretion) suppressed collagen I secretion, resulting in ER retention of procollagen I, ER stress, UPR induction, and apoptosis, thereby, alleviating hepatic fibrosis in murine and human tissue samples (70). Further analysis showed that the IRE1α-XBP1s axis could induce TANGO1 upregulation upon TGF-β treatment (70). This study suggests a key role for TANGO1 in hepatic fibrosis through facilitating collagen I secretion. UPR activation induces TANGO1 upregulation, thus, promoting hepatic fibrosis. On the contrary, TANGO1 deficiency triggers UPR-mediated cell death, thereby, slowing down fibrogenesis.
8. 2. PERK in Liver Fibrosis
HSCs play a major role in the development of liver fibrosis, a pathological state due to the accumulation of extracellular collagen fibers. Exploration of ER stress in HSCs obtained from patients with HCV infection/fibrosis or mice showed that ER stress evoked activation of HSCs, upregulation of fibrogenic genes, as well as enhanced levels of the pro-fibrotic signaling molecule SMAD2. Targeted delivery of an HSPA5 lentivirus to murine HSCs attenuated hepatic fiber accumulation. Also, SMAD2 knockdown using small interfering RNA attenuated ER stress-induced HSCs activation. Mechanistically, ER stress-induced PERK caused phosphorylation of HNRNPA1 at Thr51, targeting it for proteasomal degradation. As a result of HNRNPA1 degradation, MIR-18A (a suppressor of SMAD2 expression) declines, thus leading to enhanced expression of SMAD2 in murine HSCs. On the contrary, induction of HNRNPA1 expression inhibited liver fibrosis (71). In sum, this study suggests that ER stress in HSCs switches on the PERK-HNRNPA1-MIR-18A-SMAD2 signaling to promote liver fibrosis, offering a new drugable target for liver fibrosis.
Moreover, examination of ER stress in portal myofibroblasts (PMF) using a rat model of bile duct ligation-induced liver fibrosis revealed that PMF upregulated myofibroblastic markers (ACTA2 and COL1A1) and became more proangiogenic, yet dampened their lower migratory and proliferative capabilities. Moreover, fibrotic rat PMF displayed ER stress, as indicated by activation of PERK and its downstream proteins including CHOP, GADD34, and TRB3. The PERK inhibitor GSK2656157 suppressed ER stress, thus attenuating the proangiogenic properties of fibrotic rat PMF and restoring their migratory and proliferative capacity (72). Therefore, inhibitory targeting of the ER stress kinase PERK may alleviate liver fibrosis.
8. 3. Secondary ER Stress in Liver Fibrosis and Cirrhosis
Inflammation plays a significant role in the onset and progression of liver fibrosis. It is well known that IRF3, which is activated by the pro-inflammatory proteins TLR4 and STING1, may trigger hepatocyte apoptosis and favors type I interferon responses. Chronic administration of the liver fibrosis-inducing toxin carbon tetrachloride (CCl4) resulted in the induction of secondary ER stress, IRF3 and type I interferon activation, apoptosis, liver damage, and fibrosis. However, acute/chronic administration of CCl4 to the IRF3- or STING1-deficient mice failed to induce apoptosis and fibrosis in hepatocytes, suggesting an obligatory role for IRF3 and STING in CCl4-induced liver fibrosis. Conversely, mice deficient in type I interferons receptors or TLR4 adaptors (TICAM1 and TRAM1) developed hepatocyte apoptosis and liver fibrosis (73). These findings suggested that ER stress-induced STING1-IRF3 signaling cascade triggers upregulation of type I interferons, resulting in hepatocyte apoptosis and aggravation of liver fibrosis independent of TLR4 signaling.
Zhu and colleagues characterized the effects of salvianolic acid A (SalA, a traditional antioxidant) on liver fibrosis and SIRT1 signaling in an in vitro model of fibrosis (LX2 human hepatic stellate cell line exposed to platelet-derived growth factor-BB homodimer) and in vivo (bile duct ligation) rat models. These researchers found that SalA treatment alleviated ER stress and liver fibrosis via activation of the SIRT1-HSF1 pathway in vivo and in vitro. Mechanistically, SIRT1 activation induced deacetylation and upregulation of HSF1, which stimulated the heat shock response to facilitate proper folding of ER proteins, thus, alleviating ER stress and liver fibrosis (74).
ER stress upstream signaling molecules have been identified as druggable targets for intervention of liver fibrosis. In a rodent model of CCl4-induced liver fibrosis, inhibition of EPHX2 (involved in lipid epoxides metabolism) using TPPU (1-trifluoromethoxyphenyl-3-[1-propionylpiperidin-4-yl] urea) attenuated extracellular collagen deposition and downregulated collagen COL1A2/COL3A1 by 50%. Moreover, CCl4-induced murine liver fibrosis was accompanied by ER stress due to activation of all 3 UPR pathways (IRE1α, PERK, ATF6α), which was alleviated by TPPU treatment (75). Overall, these data suggest that induction of ER stress occurs downstream of EPHX2, which is a druggable target.
Angiotensin II (Ang-II) is a vasoactive molecule in the renin-angiotensin system and plays a key role in liver fibrosis. Mechanistically, Ang-II mediates inflammatory cytokine production, collagen synthesis, proliferation, and mitogenesis in activated HSCs, resulting in development of liver fibrosis (76). Fang and coworkers treated human LX2 cells with Ang II either alone or in combination with MAP3K5 inhibition (using GS-4997) or its silencing (using small interfering RNA) and revealed that Ang II overtly upregulated proinflammatory cytokines (IL1B, IL18, TNF), extracellular matrix proteins (ACTA2, COL1A1, COL3A1), and ER stress markers (HSPA5, EIF2AK3, DDIT3, MAP3K5). However, these effects were abolished by GS-4997 treatment or MAP3K5 silencing. In addition, ER stress-induced MAP3K5 signaling induced EV release, which activated LX2 cells to promote liver fibrosis. Inhibition of EV uptake curtailed activation of LX2 cells and alleviated liver fibrosis in vitro (77). This study suggests that Ang II-induced liver fibrosis involves induction of ER stress, which triggers pathogenic EV release via MAP3K5 signaling.
Finally, intraperitoneal administration of thioacetamide (TAA) to Sprague-Dawley rats induces liver cirrhosis. In this model, Su and associates demonstrated that celecoxib (an inhibitor of PTGS2 for prostaglandin biosynthesis) significantly attenuated serum levels of ALT and AST (markers of liver injury), alleviated ER stress and ER stress markers (BiP, PERK, ATF6α, IRE1), and reduced apoptotic markers (CHOP, CASP12, CASP3), leading to mitigation of liver fibrosis and cirrhosis (78). This study suggests that blocking ER stress-induced apoptosis using celecoxib can effectively prevent cirrhosis, calling for the demand for specific clinical trials.
Taken together, these data conclude that liver fibrosis and cirrhosis are accompanied by severe ER stress, leading to constitutive activation of the UPR branches such as PERK and IRE1α thus, promoting the disease pathology. Hence, therapeutic suppression of the UPR pathways may ameliorate liver fibrosis and cirrhosis.
9. Therapeutic Manipulation of ER Stress for Liver Disease Treatment and Management
9.1. Natural and Pharmaceutical Compounds for ER Stress Manipulation
Given that prolonged ER stress contributes to the pathogenesis of multiple liver diseases, it is important to develop therapeutic strategies to curtail maladaptive UPR. Table.1 lists several natural and pharmaceutical compounds with the potential to suppress maladaptive UPR or alleviate ER stress. Table. 2 lists anticancer agents that induce ER stress-dependent cell death in HCC or other malignant tumors in the liver. Fig. 3 summarizes current knowledge on small therapeutic molecules that specifically elicit or inhibit one of the three UPR branches.
Table. 1.
Bioactive alleviators for hepatic ER stress and UPR in mammalian cells
| Compound | Full Name/Source | Mechanisms of Action | Ref |
|---|---|---|---|
| 4-AAQB | 4-acetylantroquinonol B (natural ubiquinone derivative extracted from mycelia of Antrodia cinnamomea) | Ameliorates ER stress and inflammation in J774A.1 and RAW264.7 cellular model of NAFLD and C57BL/6J mice fed choline/methionine-deficient diet | (87) |
| Berberine | Natural compound found in several herbal plants | Reverses ER stress and ER stress-induced lipogenesis via ATF6α /SREBF1 axis in mice and hepatocyte cell lines, confers resistance to steatosis, and diminishes liver fibrosis, and lipid peroxides | (88) |
| Betaine | Nutrient | Reduces serum homocysteine levels, dampens severe ER stress and UPR in the liver in alcohol-fed mice | (89) |
| Bicyclol | Chemical compound | Ameliorates mouse fatty liver by attenuating ER stress-triggered apoptosis and downregulation of DDIT3, HSPA5, ERN1, and ATF6α | (90) |
| Curcumin | Natural compound derived from Curcuma Longa | Reduces liver ER stress and ER stress markers (BiP, PERK, IRE1, TRAF2, TNF, IL1B, MAPK14, MAP3K5, CEBPB) in diabetic rat liver, suppresses ER stress-mediated apoptosis and boosts liver histopathological and morphological changes and function | (91) |
| Empagliflozin | Medication for type 2 diabetes treatment | Reduces fasting glucose, total triglyceride and cholesterol levels, levels of inflammatory molecules and lipogenic enzymes, ER stress proteins (BiP, IRE1, XBP1, eIF2α, ATF6α, ATF4, CHOP, SQSTM1, HSP90B1), induces adaptive autophagy, and inhibits CASP8 cleavage and apoptosis to relieve NAFLD in HFD-fed mice | (92) |
| Fenofibrate | Medication to reduce blood TG and cholesterol | Ameliorates NAFLD in high-cholesterol diet-fed C57BL/6 mice via suppressing ER stress, inflammation, apoptosis, downregulating ERN1 and XBP1, and reducing MAPK8 phosphorylation | (93) |
| Ginsenoside Mc1 | Compound found in ginseng | Protects from ER stress-induced apoptosis, lipogenesis, and insulin resistance in palmitate-treated HepG2 cells and obese mice | (94) |
| Homocysteine | An amino acid | Mitigates ER stress and replenishes liver S-adenosylmethionine, resulting in alleviation of steatohepatitis in a mouse model of steatohepatitis and cultured hepatocytes. Reduces TNF production from liver macrophages in NASH | (95) |
| Kaempferol | Antioxidation and anti-inflammatory flavonoid | Reduces lethality, increases survival time and alleviates liver injury via inhibition of ER stress-induced apoptosis, downregulation of DDIT3, and upregulation of HSPA5 in mice with acute liver failure and in vitro model | (96) |
| Lactobacillus rhamnosus GG | Friendly bacteria in intestines | Downregulates ER stress and ROS and liver HSPA5 and DDIT3 expression, and normalizes autophagy in acute alcohol-triggered liver damage in C57BL/6 mice | (97) |
| Liraglutide | Analog of glucagon-like peptide-1 | Alleviates ER stress and attenuates chemerin expression in rat livers in HFD-induced insulin resistance | (98) |
| Matrine | Alkaloid obtained from Sophora flavescens | Reverses ER stress, repairs mitochondrial dysfunction, shrinks lipid accumulation and Ca2+ overload by inhibiting ATP2A2, and reduces lipid peroxides and inflammation in the liver of C57BL/6J mice under HFD | (99) |
| Melatonin | Pineal gland released hormone | Suppresses severe UPR activation and ER stress-induced apoptosis, and attenuates expression of DDIT3, HSPA5, HSP90B1, MAPK8, and CASP12 in a rabbit model of fulminant hepatitis ATP2A2 | (100) |
| Disrupts tunicamycin-caused ER stress via downregulation of MIR-23a in primary hepatocytes, alleviates liver steatosis and inflammation | (101) | ||
| Metformin | Medication for type 2 diabetes treatment | Inhibits palmitate-caused ER stress and liver insulin resistance, downregulates HSPA5, DDIT3, ATF6α, eIF2α, and XBP1 in HepG2 cells | (102) |
| Pioglitazone | Anti-diabetic medication | Suppresses ER stress and insulin resistance in diabetic mouse liver | (103) |
| Quercetin | Natural flavonoid | Resolves ER stress, oxidative stress and hepatotoxicity in rat livers exposed to lead. Reduces IRE1 and MAPK8 levels in rat livers | (104) |
| Rapamycin | Macrolide compound | Protects the liver from I/R injury via downregulation of ER stress proteins, inhibits maladaptive UPR in murine and in vitro models of I/R injury | (105) |
| Salvianolic acid B | Natural antioxidant | Suppresses ER stress and hyperactivation of ER stress molecules (CHOP, PERK, IRE1), Refines glucose tolerance, and improves insulin resistance in the liver of ob/ob mice | (106) |
| Schisandra chinensis methanol extract | Chinese herbal medicine | Suppresses ER stress, levels of lipogenic and inflammatory genes, and triglyceride accumulation, alleviates NAFLD in HepG2 cells and obese mice under tunicamycin or palmitate treatment | (107) |
| Silymarin | Chemical compound | Reverses pathologies associated with NAFLD such as ER stress, lipid profile, AST, ALT, HSAP5, and XBP1 levels | (108) |
| Taurine | Amino sulfonic acid | Suppresses ER stress, oxidative stress, CASP3 activation, and apoptosis in H4IIE cells and primary hepatocytes in diet-induced NAFLD models. Counters liver injury, inflammation, lipid accumulation, plasma TG levels | (109) |
| Vitamin C | Vitamin abundantly found in foods | Ameliorates perfluorooctane sulfonate-triggered murine liver steatosis via suppression of maladaptive UPR and ER stress as demonstrated by downregulation of ATF6α, eIF2α, HSPA5, and XBP1, and inhibition of hepatocellular inflammation | (110) |
Table. 2.
Bioactive compounds that trigger ER stress-dependent apoptosis in liver cancer
| Compound | Description | Mechanisms of Action | Ref |
|---|---|---|---|
| Annona muricata L. ethanol extract | Plant that contains chemical agents for cancer treatment | Induces ER-stressed apoptosis via enhanced BiP, PERK, HSP90B1, DPYSL5, eIF2α, and CHOP in HepG2 cells | (111) |
| Bacitracin | Cyclic polypeptide antibiotic | Inhibits P4HB, inducing ER stress and apoptosis in HCC in vivo. Boosts anticancer effect of 3-bromopyruvate (inhibitor of HK2) by augmenting ER stress-regulated apoptosis | (112) |
| b-AP15 | Selective inhibitor of USP14 | Induces ER stress and UPR, blocks WNT1-NOTCH1 signaling, reduces cell viability, fosters apoptosis, and perturbs cell cycle in HCC cells | (113) |
| Celastrol | Extracted from the root of Tripterygium regelii and Tripterygium wilfordii | Induces ER stress and activates EIF2A-ATF4 axis, leading to activation of PMAIP1 (pro-apoptotic factor) and apoptosis in combination with ABT-737 drug in HCC cells | (114) |
| Dehydrocostuslactone | Natural sesquiterpene lactone derived from medicinal plants | Blocks HepG2 and PLC/PRF/5 cell proliferation and limits tumor volume by triggering ER stress-induced apoptosis via upregulation of EIF2AK3, ERN1, DDIT3, and increased splicing of XBP1, CASP4 activation | (115) |
| 3,3-Diindolylmethane | Obtained from the digestion of indole-3-carbinol that exists in cruciferous vegetables | Remarkably restrained HCC cell growth, migration, invasion, and proliferation via induction of ER stress-triggered apoptosis in human HCC Huh7 and Hep3B cells | (116) |
| Ginsenoside compound K | 20-O-β-d-glucopyranosyl-20(S)-protopanaxadiol | Induces apoptosis via upregulation of UPR branches and reduces STAT3 levels, blocks colony formation and growth of HCC in human HepG2 and SMMC-7721 cells and human HCC xenografts | (117) |
| Isovitexin | Glycosylflavonoid derived from hulls in rice Oryza sativa | Suppresses hepatic cancer cell growth in vivo by inducing apoptosis through BAX upregulation, enhanced CASP3 cleavage, increased PARP1 and mitochondrial cytochrome c release, induction of prolonged autophagy, and severe ER stress. | (118) |
| Meloxicam | Anti-inflammatory drug and selective inhibitor of PTGS2 | In combination with sorafenib, induces ER stress-mediated apoptosis in human HCC SMMC-7721 cells and in vivo models | (119) |
| N-acetylcysteine | Antioxidant | Abates HCC progression via suppressing ER stress and oxidative stress, UPR, and inflammation in TLR2-knockout mice. Reduces SQSTM1 aggregation in liver | (120) |
| 4-O-carboxymethyl ascochlorin | Synthetic ascochlorin derivative | Triggers ER stress and enhances HSPA5 and DDIT3 expression and autophagy protein levels (BECN1, ATG5, MAP1LC3A), resulting in apoptosis and autophagy-dependent cell death in human HCC HepG2 cells | (121) |
| Piperlongumine | Alkaloid extracted from long pepper fruit | Induces lethal ER stress in HCC in vivo via interacting and inhibiting TXNRD1, resulting in intracellular ROS production | (122) |
| Safranal | Natural compound extracted from saffron | Induces ER stress-caused apoptosis, caspases activation, and DNA-strand breakage in HCC HepG2 cells | (123) |
| Scutebarbatine A | Alkaloid found in Scutellaria barbata D. Don | Inhibits HCC cells growth, induces cell cycle arrest and apoptosis via excessive ER stress and UPR induction (upregulates DDIT3, ATF6α, EIF2AK3) | (124) |
| Sorafenib | Anticancer kinase inhibitor drug | Induces ER stress, leading to apoptosis in human HCC cells, upregulates ERN1. Its combination with chloroquine (autophagy blocker) promotes ER stress-dependent cell death | (125) |
| SNX-2112 | Inhibitor of HSP90AB1 | Induces apoptosis via ER stress induction, inhibition of BiP and all UPR branches in SK-Hep1, Huh7, and HepG2 and murine xenograft models | (126) |
| Sodium demethylcantharidate | Cantharidin derivative | Blocks proliferation of Bel-7402 and SMMC-7721 HCC cells and human HCC xenograft models in a dose- and time-dependent pattern via induction of severe ER stress, leading to apoptosis | (127) |
| V8 | Chemical compound | Induces ER stress-mediated apoptosis via hyperactivation of BiP, PERK, eIF2α, ATF4, and CHOP thus, ceases growth of HCC HepG2 cells in a dose-dependent pattern | (128) |
Fig. 3. Small Molecules Targeting UPR Branches.
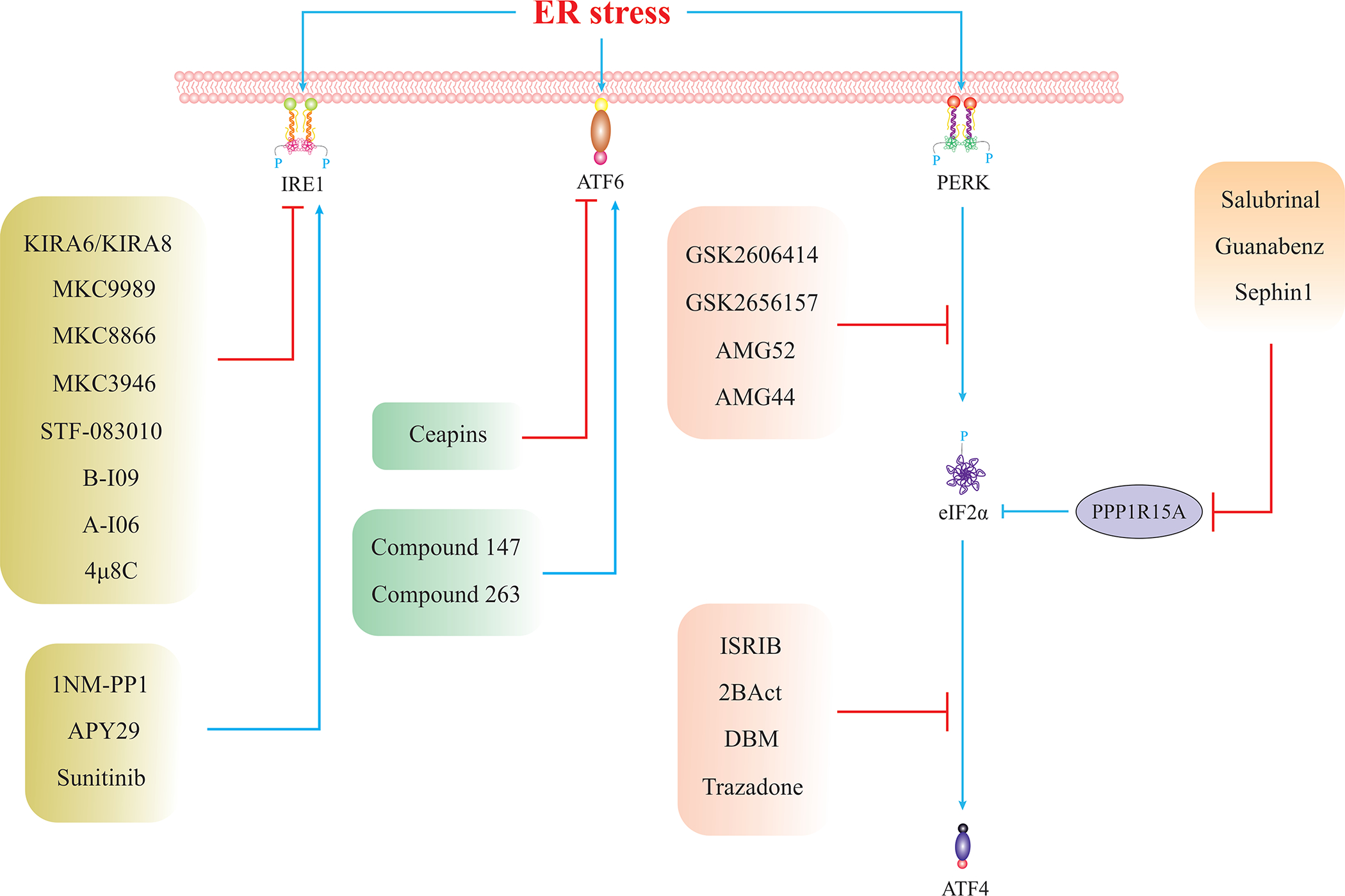
List of small chemicals for specific targeting of UPR components (129, 130). Given that the majority of these compounds have not been examined in the context of liver diseases, future studies are warranted to examine these compounds in the context of liver disease in both pre-clinical and clinical studies.
9. 2. Chemical Chaperones: TUDCA and 4-PBA
Chemical chaperones such as TUDCA and 4-PBA alleviate ER stress by increasing protein-folding capacity. Such molecules might be useful for the management of liver diseases associated with ER stress. For example, TUDCA alleviates the cytotoxicity of polyhexamethyleneguanidine (PHMG), an antimicrobial agent, against HepG2 cells. Moreover, TUDCA ameliorated ER stress-induced apoptosis and mitochondrial depolarization (79). TUDCA boosts multiorgan insulin sensitivity by enhancing protein folding and ameliorating ER stress. In a clinical trial enrolling 20 obese individuals, TUDCA improved insulin sensitivity of the liver and muscle (80). Moreover, in an HFD-induced mouse model of NAFLD, TUDCA was found to suppress ER-associated gut inflammation, insulin resistance, obesity, and liver steatosis (81). Similarly, intraperitoneal injection of 4-PBA into C57BL/6 mice with hepatic I/R injury lowered expression of DDIT3 and eIF2α phosphorylation as well as inhibited ER stress-initiated apoptosis coupled to CASP12 activation. 4-PBA also remarkably attenuated signs of systemic and hepatic inflammation, such as plasma TNF levels and liver MPO, respectively (82). 4-PBA also potently reduces hepatotoxicity of industrial toxicant perfluorooctanoic acid (PFOA). 4-PBA alleviated ER stress and blocked ER stress-induced hyperactivation of autophagy in mouse livers and HepG2 cells exposed to PFOA. 4-PBA also ameliorated other hepatotoxic PFOA-induced effects such as impairment of proteolytic activity, lipid accumulation, and cell cycle arrest (83). Altogether, the current state of the literature suggests that chemical chaperones could be useful for the treatment of ER stress-associated liver diseases.
10. Concluding Remarks and Future Perspectives
Mild ER stress activates adaptive UPR, while severe ER stress triggers maladaptive UPR, leading to cell death. Accumulating clinical and experimental evidence has consolidated the involvement of maladaptive UPR in the pathogenesis of liver diseases. Although maladaptive UPR promotes metabolic diseases, drug toxicity, and viral infections, it can be used as a tool to eliminate cancer cells that emerge in liver malignancies. Therefore, inhibitory targeting of maladaptive UPR could be pursued in non-malignant liver diseases, while inducing severe ER stress could be implemented in the context of liver cancers. In this context, multiple studies have identified and characterized natural and pharmaceutical compounds that induce severe ER stress or, on the contrary, alleviate ER stress in the liver. Chemical chaperones, PE, and CR have a high potential to relieve ER stress and maladaptive UPR in liver diseases. Moreover, it is noteworthy that several intrinsic hepatic hormones and growth factors including fibroblast growth factors (FGFs) display a robust decline in various hepatic injury such as NAFLD and NASH in association with disturbed ER homeostasis (84–86). Although FGFs exhibited promises as novel therapeutic options of hepatic and other metabolic anomalies, possible involvement of ER stress and UPR remains unclear. One burning issue for future research is the identification of novel ER stress modulators that specifically intervene maladaptive UPR in the liver, without affecting adaptive UPR in other organs, or on the contrary, specifically elicit lethal UPR in liver cancer without triggering unwarranted ER stress responses in normal tissues. Moreover, there is a notable lack of studies on IRE1 or ATF6 knockout mouse models in liver disease thus, mandating future research on such models. Also, more effort should be made with regards to the impact of ATF6α or IRE1α inhibitors on liver disease in non-malignant liver diseases.
Fig. 4. Targeting ER Stress In Liver Diseases.
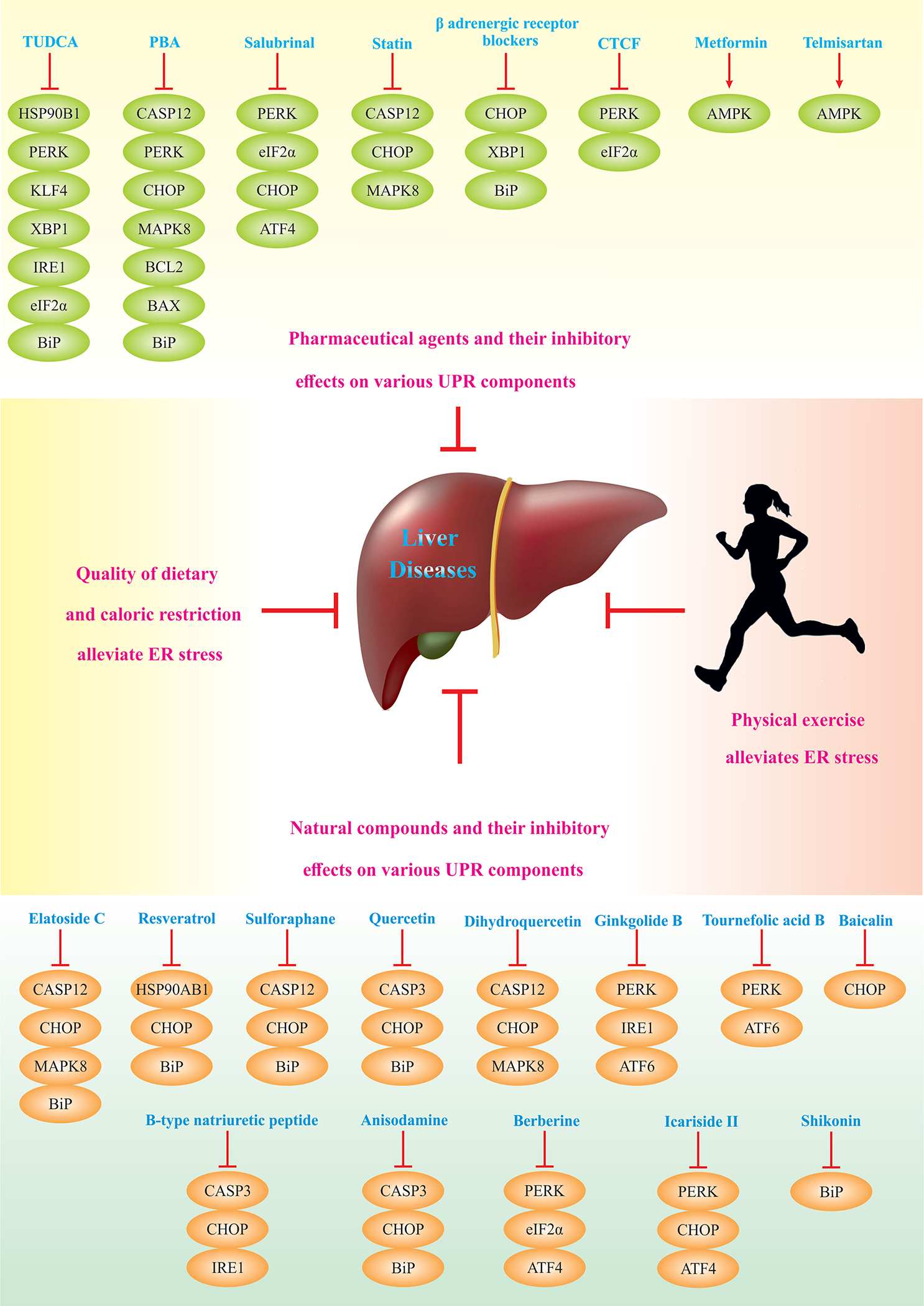
Overall, pharmaceutical and natural agents, physical exercise, and caloric restriction diminish ER stress, and therefore, alleviate non-malignant liver diseases. Pharmaceutical agents suppress UPR components or activate AMPK. Similarly, natural compounds being found abundantly in vegetables, fruits, herbs, spices, and beans, target UPR components. Caloric restriction and physical exercise confer similar effects on ER stress alleviation and liver diseases management.
ACKNOWLEDGEMENT
We wish to thank Dr. Yaguang Bi from Zhongshan Hospital Fudan University for helpful discussion. The authors wish to sincerely apologize to those authors whose important work cannot be included due to space limitation. JR is partially supported by National Key R&D Program of China (2017YFA0506000) and National Natural Science Foundation of China (82130011). GK is supported by the Ligue contre le Cancer (équipe labellisée); Agence National de la Recherche (ANR) – Projets blancs; AMMICa US23/CNRS UMS3655; Association pour la recherche sur le cancer (ARC); Association “Ruban Rose”; Cancéropôle Ile-de-France; Fondation pour la Recherche Médicale (FRM); a donation by Elior; Equipex Onco-Pheno-Screen; European Joint Programme on Rare Diseases (EJPRD); Gustave Roussy Odyssea, the European Union Horizon 2020 Projects Oncobiome and Crimson; Fondation Carrefour; Institut National du Cancer (INCa); Inserm (HTE); Institut Universitaire de France; LabEx Immuno-Oncology (ANR-18-IDEX-0001); the Leducq Foundation; a Cancer Research ASPIRE Award from the Mark Foundation;, the RHU Torino Lumière; Seerave Foundation; SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); and SIRIC Cancer Research and Personalized Medicine (CARPEM). This study contributes to the IdEx Université de Paris ANR-18-IDEX-0001. HM is partly supported by NIH grant R01 DK111378.
Abbreviations:
- ACTA2
(actin α2, smooth muscle)
- AKT1
(AKT serine/threonine kinase 1)
- ALT
(alanine transaminase)
- AMP
(adenosine monophosphate)
- AST
(aspartate transaminase)
- ATF4
(activating transcription factor 4)
- ATF6
(activating transcription factor 6)
- ATG5
(autophagy related 5)
- ATG8
(autophagy related 8)
- ATG12
(autophagy related 12)
- ATG16
(autophagy related 16)
- ATP2A2
(ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 2)
- BAD
(BCL2 associated agonist of cell death)
- BAX
(BCL2 associated X, apoptosis regulator)
- BCL2
(BCL2 apoptosis regulator)
- BECN1
(beclin 1)
- BI-1
(Bax inhibitor-1)
- CASP2
(caspase 2)
- CASP3
(caspase 3)
- CASP4
(caspase 4)
- CASP8
(caspase 8)
- CASP9
(caspase 9)
- CASP12
(caspase 12)
- CD274
(cluster of differentiation 274)
- CD68
(cluster of differentiation 68)
- CEBPB
(CCAAT enhancer binding protein β)
- COL3A1
(collagen type III α1 chain)
- COL1A1
(collagen type I α1 chain)
- COL1A2
(collagen type I α2 chain)
- CREB3L3
(cAMP responsive element binding protein 3 like 3)
- CTCF
(cellular CCCTC-binding factor)
- DDIT3/CHOP/GADD153
(DNA damage inducible transcript 3/ C/EBP homologous protein/ growth arrest- and DNA damage-inducible gene 153)
- DGAT1
(diacylglycerol O-acyltransferase 1)
- DPYSL5
(dihydropyrimidinase like 5)
- EIF2AK3/PERK
(eukaryotic translation initiation factor 2 alpha kinase 3/protein kinase R-like endoplasmic reticulum kinase)
- EPHX2
(epoxide hydrolase 2)
- ERN1/IRE1
(endoplasmic reticulum to nucleus signaling 1/inositol-requiring enzyme 1)
- FGF
(fibroblast growth factor)
- GCN2
(general control of nitrogen metabolism)
- GPAM
(glycerol-3-phosphate acyltransferase mitochondrial)
- HK2
(hexokinase 2)
- HNRNPA1
(Heterogeneous Nuclear Ribonucleoprotein A1)
- HRI
(heme regulated inhibitor of mRNA translation)
- HSPA2/HSP70
(heat shock protein family A [Hsp70] member 2)
- HSPA5/BiP/GRP78
(heat shock protein family A [Hsp70] member 5/ binding immunoglobulin protein/ 78-kDa glucose regulated protein)
- HSP90AB1
(heat shock protein 90 α family class B member 1)
- HSP90B1/GRP94
(heat shock protein 90 β family member 1)
- IL1B
(interleukin 1β)
- IL2
(interleukin 2)
- IL12B
(interleukin 12B)
- IL18
(interleukin 18)
- IL23A
(interleukin 23 subunit α)
- IL-6
(interleukin 6)
- IRF3
(interferon regulatory factor 3)
- JUN
(Jun proto-oncogene AP-1 transcription factor subunit)
- LAMP2
(lysosomal associated membrane protein 2)
- MAP1LC3A/LC3A
(microtubule associated protein 1 light chain 3 α)
- MAP1LC3B/LC3B
(microtubule associated protein 1 light chain 3β)
- MAP2K1/2
(mitogen-activated protein kinase kinase 1/2)
- MAPK1
(mitogen-activated protein kinase 1)
- MAPK3
(mitogen-activated protein kinase 3)
- MAPK8/JNK1
(mitogen-activated protein kinase 8/ c-jun NH2-terminal kinase 1)
- MAPK14
(mitogen-activated protein kinase 14)
- MAP3K5
(mitogen-activated protein kinase kinase kinase 5)
- MBTPS1/S1P
(membrane bound transcription factor peptidase, site 1)
- MBTPS2/S2P
(membrane bound transcription factor peptidase, site 2)
- MTORC1
(mechanistic target of rapamycin kinase)
- MYC
(MYC proto-oncogene, BHLH transcription factor)
- NFE2L2
(nuclear factor, erythroid 2 like 2)
- NF-κB
(nuclear factor kappa B subunit 1)
- NNMT
(nicotinamide N-methyltransferase)
- NOTCH1
(notch receptor 1)
- PARP1
(poly[ADP-ribose] polymerase 1)
- PBA
(phenylboronic acid)
- P4HB
(prolyl 4-hydroxylase subunit β)
- PKR
(double stranded RNA activated protein kinase)
- PIK3CA
(phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit α)
- PLAU
(plasminogen activator, urokinase)
- PPARA
(peroxisome proliferator activated receptor α)
- PP1
(protein phosphatase 1)
- PPP1R15A/GADD34
(protein phosphatase 1 regulatory subunit 15A/ growth arrest and DNA damage inducible protein 34)
- PPP2CA
(protein phosphatase 2 catalytic subunit α)
- PRKAA2/AMPK
(protein kinase AMP-activated catalytic subunit α2)
- PTEN
(phosphatase and tensin homolog)
- PTGS2
(prostaglandin-endoperoxide synthase 2)
- RACK1
(receptor for activated C kinase 1)
- RAF1
(Raf-1 proto-oncogene, serine/threonine kinase)
- RRAS
(RAS related)
- RtcB
(RNA 2’,3’-cyclic phosphate and 5’-OH ligase)
- SIDT2
(SID1 transmembrane family member 2)
- SIRT1
(sirtuin 1)
- SIRT7
(sirtuin 7)
- SMAD2
(SMAD family member 2)
- SQSTM1/P62
(sequestosome 1)
- SREBF1/2
(sterol regulatory element binding transcription factor 1/2)
- SREBP1/2/3
(sterol regulatory element binding protein 1/2/3)
- STAT3
(signal transducer and TANGO1 (transport and Golgi organization 1)
- TICAM1
(toll like receptor adaptor molecule 1)
- TLRs
(toll-like receptors)
- TLR2
(toll like receptor 2)
- TLR4
(toll like receptor 4)
- TNF-α
(tumor necrosis factor)
- TNFSF10
(TNF superfamily member 10)
- TP53
(tumor protein p53)
- TRAF2
(TNF receptor associated factor 2)
- TRAM1
(translocation associated membrane protein 1)
- TXNRD1
(thioredoxin reductase 1)
- ULK1
(Unc-51 like autophagy activating kinase 1)
- USP14
(ubiquitin specific peptidase 14)
- XBP1
(X-box binding protein 1)
- WNT1
(Wnt family member 1)
- ZNF263
(zinc finger protein 263)
Footnotes
CONFLICT OF INTEREST
GK holds research contracts with Daiichi Sankyo, Eleor, Kaleido, Lytix Pharma, PharmaMar, Samsara, Sanofi, Sotio, Vascage and Vasculox/Tioma. GK is on the Board of Directors of the Bristol Myers Squibb Foundation France. GK is a scientific co-founder of everImmune, Samsara Therapeutics and Therafast Bio. GK is the inventor of patents covering therapeutic targeting of aging, cancer, cystic fibrosis and metabolic disorders.
REFERENCES
- 1.Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016;529:326–335. [DOI] [PubMed] [Google Scholar]
- 2.Schwaninger R, Plutner H, Bokoch GM, Balch WE. Multiple GTP-binding proteins regulate vesicular transport from the ER to Golgi membranes. The Journal of cell biology 1992;119:1077–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ajoolabady A, Wang S, Kroemer G, Klionsky DJ, Uversky VN, Sowers JR, Aslkhodapasandhokmabad H, et al. ER stress in cardiometabolic diseases: from molecular mechanisms to therapeutics. Endocrine Reviews 2021. [DOI] [PubMed] [Google Scholar]
- 4.Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO reports 2006;7:880–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ren J, Bi Y, Sowers JR, Hetz C, Zhang Y. Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nature Reviews Cardiology 2021;18:499–521. [DOI] [PubMed] [Google Scholar]
- 6.Wei Y, Wang D, Topczewski F, Pagliassotti MJ. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. American Journal of Physiology-Endocrinology and Metabolism 2006;291:E275–E281. [DOI] [PubMed] [Google Scholar]
- 7.Cao SS, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxidants & redox signaling 2014;21:396–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dara L, Ji C, Kaplowitz N. The contribution of endoplasmic reticulum stress to liver diseases. Hepatology 2011;53:1752–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zheng Z, Shang Y, Tao J, Zhang J, Sha B. Endoplasmic reticulum stress signaling pathways: Activation and diseases. Current Protein and Peptide Science 2019;20:935–943. [DOI] [PubMed] [Google Scholar]
- 10.Tirasophon W, Welihinda AA, Kaufman RJ. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes & development 1998;12:1812–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grey MJ, Cloots E, Simpson MS, LeDuc N, Serebrenik YV, De Luca H, De Sutter D, et al. IRE1β negatively regulates IRE1α signaling in response to endoplasmic reticulum stress. Journal of Cell Biology 2020;219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Imagawa Y, Hosoda A, Sasaka S-i, Tsuru A, Kohno K. RNase domains determine the functional difference between IRE1α and IRE1β. FEBS letters 2008;582:656–660. [DOI] [PubMed] [Google Scholar]
- 13.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nature cell biology 2000;2:326–332. [DOI] [PubMed] [Google Scholar]
- 14.Jurkin J, Henkel T, Nielsen AF, Minnich M, Popow J, Kaufmann T, Heindl K, et al. The mammalian tRNA ligase complex mediates splicing of XBP1 mRNA and controls antibody secretion in plasma cells. The EMBO journal 2014;33:2922–2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002;415:92–96. [DOI] [PubMed] [Google Scholar]
- 16.Oda Y, Okada T, Yoshida H, Kaufman RJ, Nagata K, Mori K. Derlin-2 and Derlin-3 are regulated by the mammalian unfolded protein response and are required for ER-associated degradation. The Journal of cell biology 2006;172:383–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han D, Lerner AG, Walle LV, Upton J-P, Xu W, Hagen A, Backes BJ, et al. IRE1α kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 2009;138:562–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 2006;313:104–107. [DOI] [PubMed] [Google Scholar]
- 19.Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. Journal of Cell Biology 2009;186:323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vattem KM, Wek RC. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proceedings of the National Academy of Sciences 2004;101:11269–11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yong J, Parekh VS, Reilly SM, Nayak J, Chen Z, Lebeaupin C, Jang I, et al. Chop/Ddit3 depletion in β cells alleviates ER stress and corrects hepatic steatosis in mice. Science Translational Medicine 2021;13:eaba9796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malhi H, Kropp EM, Clavo VF, Kobrossi CR, Han J, Mauer AS, Yong J, et al. C/EBP homologous protein-induced macrophage apoptosis protects mice from steatohepatitis. Journal of Biological Chemistry 2013;288:18624–18642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thuerauf DJ, Morrison L, Glembotski CC. Opposing roles for ATF6α and ATF6β in endoplasmic reticulum stress response gene induction. Journal of Biological Chemistry 2004;279:21078–21084. [DOI] [PubMed] [Google Scholar]
- 24.Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, Harada A, et al. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6α and XBP1. Developmental cell 2007;13:365–376. [DOI] [PubMed] [Google Scholar]
- 25.Walter F, O’Brien A, Concannon CG, Düssmann H, Prehn JH. ER stress signaling has an activating transcription factor 6α (ATF6)-dependent “off-switch”. Journal of Biological Chemistry 2018;293:18270–18284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Teske BF, Wek SA, Bunpo P, Cundiff JK, McClintick JN, Anthony TG, Wek RC. The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Mol Biol Cell 2011;22:4390–4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gonen N, Sabath N, Burge CB, Shalgi R. Widespread PERK-dependent repression of ER targets in response to ER stress. Sci Rep 2019;9:4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walter F, Schmid J, Düssmann H, Concannon CG, Prehn JH. Imaging of single cell responses to ER stress indicates that the relative dynamics of IRE1/XBP1 and PERK/ATF4 signalling rather than a switch between signalling branches determine cell survival. Cell Death Differ 2015;22:1502–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Angulo P Nonalcoholic fatty liver disease. New England Journal of Medicine 2002;346:1221–1231. [DOI] [PubMed] [Google Scholar]
- 30.Sheth SG, Gordon FD, Chopra S. Nonalcoholic steatohepatitis. Annals of internal medicine 1997;126:137–145. [DOI] [PubMed] [Google Scholar]
- 31.Horwath JA, Hurr C, Butler SD, Guruju M, Cassell MD, Mark AL, Davisson RL, et al. Obesity-induced hepatic steatosis is mediated by endoplasmic reticulum stress in the subfornical organ of the brain. JCI insight 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dasgupta D, Nakao Y, Mauer AS, Thompson JM, Sehrawat TS, Liao C-Y, Krishnan A, et al. IRE1A stimulates hepatocyte-derived extracellular vesicles that promote inflammation in mice with steatohepatitis. Gastroenterology 2020;159:1487–1503. e1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lisbona F, Rojas-Rivera D, Thielen P, Zamorano S, Todd D, Martinon F, Glavic A, et al. BAX inhibitor-1 is a negative regulator of the ER stress sensor IRE1α. Molecular cell 2009;33:679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang L, Calay ES, Fan J, Arduini A, Kunz RC, Gygi SP, Yalcin A, et al. S-Nitrosylation links obesity-associated inflammation to endoplasmic reticulum dysfunction. Science 2015;349:500–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maiers JL, Malhi H. Endoplasmic reticulum stress in metabolic liver diseases and hepatic fibrosis. In: Seminars in liver disease; 2019: Thieme Medical Publishers; 2019. p. 235–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang JM, Qiu Y, Yang Z, Kim H, Qian Q, Sun Q, Zhang C, et al. IRE1α prevents hepatic steatosis by processing and promoting the degradation of select microRNAs. Sci Signal 2018;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2α. The Journal of cell biology 2001;153:1011–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li J, Li X, Liu D, Zhang S, Tan N, Yokota H, Zhang P. Phosphorylation of eIF2α signaling pathway attenuates obesity-induced non-alcoholic fatty liver disease in an ER stress and autophagy-dependent manner. Cell Death & Disease 2020;11:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee S, Kim S, Hwang S, Cherrington NJ, Ryu D-Y. Dysregulated expression of proteins associated with ER stress, autophagy and apoptosis in tissues from nonalcoholic fatty liver disease. Oncotarget 2017;8:63370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tam AB, Roberts LS, Chandra V, Rivera IG, Nomura DK, Forbes DJ, Niwa M. The UPR activator ATF6 responds to proteotoxic and lipotoxic stress by distinct mechanisms. Developmental cell 2018;46:327–343. e327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kammoun HL, Chabanon H, Hainault I, Luquet S, Magnan C, Koike T, Ferré P, et al. GRP78 expression inhibits insulin and ER stress–induced SREBP-1c activation and reduces hepatic steatosis in mice. The Journal of clinical investigation 2009;119:1201–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rutkowski DT, Wu J, Back S-H, Callaghan MU, Ferris SP, Iqbal J, Clark R, et al. UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators. Developmental cell 2008;15:829–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gao J, Zhang Y, Yu C, Tan F, Wang L. Spontaneous nonalcoholic fatty liver disease and ER stress in Sidt2 deficiency mice. Biochemical and biophysical research communications 2016;476:326–332. [DOI] [PubMed] [Google Scholar]
- 44.Kim JY, Garcia-Carbonell R, Yamachika S, Zhao P, Dhar D, Loomba R, Kaufman RJ, et al. ER stress drives lipogenesis and steatohepatitis via caspase-2 activation of S1P. Cell 2018;175:133–145. e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feng B, Yao PM, Li Y, Devlin CM, Zhang D, Harding HP, Sweeney M, et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nature cell biology 2003;5:781–792. [DOI] [PubMed] [Google Scholar]
- 46.Bashiri A, Nesan D, Tavallaee G, Sue-Chue-Lam I, Chien K, Maguire GF, Naples M, et al. Cellular cholesterol accumulation modulates high fat high sucrose (HFHS) diet-induced ER stress and hepatic inflammasome activation in the development of non-alcoholic steatohepatitis. Biochimica et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids 2016;1861:594–605. [DOI] [PubMed] [Google Scholar]
- 47.Shin J, He M, Liu Y, Paredes S, Villanova L, Brown K, Qiu X, et al. SIRT7 represses Myc activity to suppress ER stress and prevent fatty liver disease. Cell reports 2013;5:654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Song Q, Chen Y, Wang J, Hao L, Huang C, Griffiths A, Sun Z, et al. ER stress-induced upregulation of NNMT contributes to alcohol-related fatty liver development. Journal of hepatology 2020;73:783–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guo W, Zhong W, Hao L, Dong H, Sun X, Yue R, Li T, et al. Fatty Acids Inhibit LAMP2-Mediated Autophagy Flux via Activating ER Stress Pathway in Alcohol-Related Liver Disease. Cellular and Molecular Gastroenterology and Hepatology 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Petrasek J, Iracheta-Vellve A, Csak T, Satishchandran A, Kodys K, Kurt-Jones EA, Fitzgerald KA, et al. STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proceedings of the National Academy of Sciences 2013;110:16544–16549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.He C, Qiu Y, Han P, Chen Y, Zhang L, Yuan Q, Zhang T, et al. ER stress regulating protein phosphatase 2A-B56γ, targeted by hepatitis B virus X protein, induces cell cycle arrest and apoptosis of hepatocytes. Cell death & disease 2018;9:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li Y-X, Ren Y-L, Fu H-J, Zou L, Yang Y, Chen Z. Hepatitis B virus middle protein enhances IL-6 production via p38 MAPK/NF-κB pathways in an ER stress-dependent manner. PloS one 2016;11:e0159089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ciccaglione A, Marcantonio C, Tritarelli E, Equestre M, Vendittelli F, Costantino A, Geraci A, et al. Activation of the ER stress gene gadd153 by hepatitis C virus sensitizes cells to oxidant injury. Virus research 2007;126:128–138. [DOI] [PubMed] [Google Scholar]
- 54.Asselah T, Bièche I, Mansouri A, Laurendeau I, Cazals-Hatem D, Feldmann G, Bedossa P, et al. In vivo hepatic endoplasmic reticulum stress in patients with chronic hepatitis C. The Journal of Pathology: A Journal of the Pathological Society of Great Britain and Ireland 2010;221:264–274. [DOI] [PubMed] [Google Scholar]
- 55.Nakagawa H, Umemura A, Taniguchi K, Font-Burgada J, Dhar D, Ogata H, Zhong Z, et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer cell 2014;26:331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shuda M, Kondoh N, Imazeki N, Tanaka K, Okada T, Mori K, Hada A, et al. Activation of the ATF6, XBP1 and grp78 genes in human hepatocellular carcinoma: a possible involvement of the ER stress pathway in hepatocarcinogenesis. Journal of hepatology 2003;38:605–614. [DOI] [PubMed] [Google Scholar]
- 57.Vandewynckel Y-P, Laukens D, Bogaerts E, Paridaens A, Van den Bussche A, Verhelst X, Van Steenkiste C, et al. Modulation of the unfolded protein response impedes tumor cell adaptation to proteotoxic stress: a PERK for hepatocellular carcinoma therapy. Hepatology international 2015;9:93–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schulze K, Imbeaud S, Letouzé E, Alexandrov LB, Calderaro J, Rebouissou S, Couchy G, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nature genetics 2015;47:505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu X, Xin Z, Zhang W, Zheng S, Wu J, Chen K, Wang H, et al. A missense polymorphism in ATF6 gene is associated with susceptibility to hepatocellular carcinoma probably by altering ATF6 level. International journal of cancer 2014;135:61–68. [DOI] [PubMed] [Google Scholar]
- 60.Chiang W-C, Chan P, Wissinger B, Vincent A, Skorczyk-Werner A, Krawczyński MR, Kaufman RJ, et al. Achromatopsia mutations target sequential steps of ATF6 activation. Proceedings of the National Academy of Sciences 2017;114:400–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee E-J, Chiang W-CJ, Kroeger H, Bi CX, Chao DL, Skowronska-Krawczyk D, Mastey RR, et al. Multiexon deletion alleles of ATF6 linked to achromatopsia. JCI insight 2020;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wu Y, Shan B, Dai J, Xia Z, Cai J, Chen T, Lv S, et al. Dual role for inositol-requiring enzyme 1α in promoting the development of hepatocellular carcinoma during diet-induced obesity in mice. Hepatology 2018;68:533–546. [DOI] [PubMed] [Google Scholar]
- 63.Pavlović N, Calitz C, Thanapirom K, Mazza G, Rombouts K, Gerwins P, Heindryckx F. Inhibiting IRE1α-endonuclease activity decreases tumor burden in a mouse model for hepatocellular carcinoma. Elife 2020;9:e55865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cui J, Liu J, Fan L, Zhu Y, Zhou B, Wang Y, Hua W, et al. A zinc finger family protein, ZNF263, promotes hepatocellular carcinoma resistance to apoptosis via activation of ER stress-dependent autophagy. Translational Oncology 2020;13:100851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu J, Fan L, Yu H, Zhang J, He Y, Feng D, Wang F, et al. Endoplasmic reticulum stress causes liver cancer cells to release exosomal miR-23a-3p and up-regulate programmed death ligand 1 expression in macrophages. Hepatology 2019;70:241–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee SH, Moon HJ, Lee YS, Kang CD, Kim SH. Potentiation of TRAIL-induced cell death by nonsteroidal anti-inflammatory drug in human hepatocellular carcinoma cells through the ER stress-dependent autophagy pathway. Oncology Reports 2020;44:1136–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rao J, Yue S, Fu Y, Zhu J, Wang X, Busuttil RW, Kupiec-Weglinski JW, et al. ATF6 mediates a pro-inflammatory synergy between ER stress and TLR activation in the pathogenesis of liver ischemia-reperfusion injury. American Journal of Transplantation 2014;14:1552–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu D, Liu X, Zhou T, Yao W, Zhao J, Zheng Z, Jiang W, et al. IRE1–RACK1 axis orchestrates ER stress preconditioning-elicited cytoprotection from ischemia/reperfusion injury in liver. Journal of molecular cell biology 2016;8:144–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lu L, Rao J, Yue S, Li G, Qian Q, Zhang F, Zhai Y, et al. A Tripartite Synergy of TLR4 Activation, ER Stress Induction and Autophagy Inhibition Is Critical for Interleukin 23 Induction in Liver Ischemia and Reperfusion Injury.: Abstract# C1657. Transplantation 2014;98:343. [Google Scholar]
- 70.Maiers JL, Kostallari E, Mushref M, deAssuncao TM, Li H, Jalan-Sakrikar N, Huebert RC, et al. The unfolded protein response mediates fibrogenesis and collagen I secretion through regulating TANGO1 in mice. Hepatology 2017;65:983–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Koo JH, Lee HJ, Kim W, Kim SG. Endoplasmic reticulum stress in hepatic stellate cells promotes liver fibrosis via PERK-mediated degradation of HNRNPA1 and up-regulation of SMAD2. Gastroenterology 2016;150:181–193. e188. [DOI] [PubMed] [Google Scholar]
- 72.Loeuillard E, El Mourabit H, Lei L, Lemoinne S, Housset C, Cadoret A. Endoplasmic reticulum stress induces inverse regulations of major functions in portal myofibroblasts during liver fibrosis progression. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 2018;1864:3688–3696. [DOI] [PubMed] [Google Scholar]
- 73.Iracheta-Vellve A, Petrasek J, Gyongyosi B, Satishchandran A, Lowe P, Kodys K, Catalano D, et al. Endoplasmic reticulum stress-induced hepatocellular death pathways mediate liver injury and fibrosis via stimulator of interferon genes. Journal of Biological Chemistry 2016;291:26794–26805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhu J, Wang R, Xu T, Zhang S, Zhao Y, Li Z, Wang C, et al. Salvianolic acid a attenuates endoplasmic reticulum stress and protects against cholestasis-induced liver fibrosis via the SIRT1/HSF1 pathway. Frontiers in pharmacology 2018;9:1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Harris TR, Bettaieb A, Kodani S, Dong H, Myers R, Chiamvimonvat N, Haj FG, et al. Inhibition of soluble epoxide hydrolase attenuates hepatic fibrosis and endoplasmic reticulum stress induced by carbon tetrachloride in mice. Toxicology and applied pharmacology 2015;286:102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Saber S Angiotensin II: a key mediator in the development of liver fibrosis and cancer. Bulletin of the National Research Centre 2018;42:1–7. [Google Scholar]
- 77.Fang P-p, Pan C-w, Lin W, Li J, Huang S-s, Zhou G-y, Du W-j, et al. ASK1 Enhances Angiotensin II-Induced Liver Fibrosis In Vitro by Mediating Endoplasmic Reticulum Stress-Dependent Exosomes. Mediators of Inflammation 2020;2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Su W, Tai Y, Tang S-H, Ye Y-T, Zhao C, Gao J-H, Tuo B-G, et al. Celecoxib attenuates hepatocyte apoptosis by inhibiting endoplasmic reticulum stress in thioacetamide-induced cirrhotic rats. World Journal of Gastroenterology 2020;26:4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kim SH, Kwon D, Lee S, Ki SH, Jeong HG, Hong JT, Lee Y-H, et al. Polyhexamethyleneguanidine phosphate-induced cytotoxicity in liver cells is alleviated by Tauroursodeoxycholic Acid (TUDCA) via a reduction in endoplasmic reticulum stress. Cells 2019;8:1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kars M, Yang L, Gregor MF, Mohammed BS, Pietka TA, Finck BN, Patterson BW, et al. Tauroursodeoxycholic acid may improve liver and muscle but not adipose tissue insulin sensitivity in obese men and women. Diabetes 2010;59:1899–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang W, Zhao J, Gui W, Sun D, Dai H, Xiao L, Chu H, et al. Tauroursodeoxycholic acid inhibits intestinal inflammation and barrier disruption in mice with non-alcoholic fatty liver disease. British journal of pharmacology 2018;175:469–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vilatoba M, Eckstein C, Bilbao G, Smyth CA, Jenkins S, Thompson JA, Eckhoff DE, et al. Sodium 4-phenylbutyrate protects against liver ischemia reperfusion injury by inhibition of endoplasmic reticulum-stress mediated apoptosis. Surgery 2005;138:342–351. [DOI] [PubMed] [Google Scholar]
- 83.Yan S, Zhang H, Wang J, Zheng F, Dai J. Perfluorooctanoic acid exposure induces endoplasmic reticulum stress in the liver and its effects are ameliorated by 4-phenylbutyrate. Free Radical Biology and Medicine 2015;87:300–311. [DOI] [PubMed] [Google Scholar]
- 84.Song L, Wang L, Hou Y, Zhou J, Chen C, Ye X, Dong W, et al. FGF4 protects the liver from non-alcoholic fatty liver disease by activating the AMPK-Caspase 6 signal axis. Hepatology 2022. [DOI] [PubMed] [Google Scholar]
- 85.Lin Q, Huang Z, Cai G, Fan X, Yan X, Liu Z, Zhao Z, et al. Activating Adenosine Monophosphate–Activated Protein Kinase Mediates Fibroblast Growth Factor 1 Protection From Nonalcoholic Fatty Liver Disease in Mice. Hepatology 2021;73:2206–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ying L, Wang L, Guo K, Hou Y, Li N, Wang S, Liu X, et al. Paracrine FGFs target skeletal muscle to exert potent anti-hyperglycemic effects. Nature communications 2021;12:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yen I-C, Tu Q-W, Chang T-C, Lin P-H, Li Y-F, Lee S-Y. 4-Acetylantroquinonol B ameliorates nonalcoholic steatohepatitis by suppression of ER stress and NLRP3 inflammasome activation. Biomedicine & Pharmacotherapy 2021;138:111504. [DOI] [PubMed] [Google Scholar]
- 88.Zhang Z, Li B, Meng X, Yao S, Jin L, Yang J, Wang J, et al. Berberine prevents progression from hepatic steatosis to steatohepatitis and fibrosis by reducing endoplasmic reticulum stress. Scientific reports 2016;6:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ji C, Kaplowitz N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology 2003;124:1488–1499. [DOI] [PubMed] [Google Scholar]
- 90.Yao X-M, Li Y, Li H-W, Cheng X-Y, Lin A-B, Qu J-G. Bicyclol attenuates tetracycline-induced fatty liver associated with inhibition of hepatic ER stress and apoptosis in mice. Canadian journal of physiology and pharmacology 2016;94:1–8. [DOI] [PubMed] [Google Scholar]
- 91.Afrin R, Arumugam S, Soetikno V, Thandavarayan R, Pitchaimani V, Karuppagounder V, Sreedhar R, et al. Curcumin ameliorates streptozotocin-induced liver damage through modulation of endoplasmic reticulum stress-mediated apoptosis in diabetic rats. Free radical research 2015;49:279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nasiri-Ansari N, Nikolopoulou C, Papoutsi K, Kyrou I, Mantzoros CS, Kyriakopoulos G, Chatzigeorgiou A, et al. Empagliflozin Attenuates Non-Alcoholic Fatty Liver Disease (NAFLD) in High Fat Diet Fed ApoE (−/−) Mice by Activating Autophagy and Reducing ER Stress and Apoptosis. International Journal of Molecular Sciences 2021;22:818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang N, Lu Y, Shen X, Bao Y, Cheng J, Chen L, Li B, et al. Fenofibrate treatment attenuated chronic endoplasmic reticulum stress in the liver of nonalcoholic fatty liver disease mice. Pharmacology 2015;95:173–180. [DOI] [PubMed] [Google Scholar]
- 94.Roh E, Hwang H-J, Kim JW, Hong S-h, Kim JA, Lee Y-B, Choi KM, et al. Ginsenoside Mc1 improves liver steatosis and insulin resistance by attenuating ER stress. Journal of ethnopharmacology 2020;259:112927. [DOI] [PubMed] [Google Scholar]
- 95.Minami S, Miura K, Ishioka M, Morimoto N, Isoda N, Yamamoto H, Iijima K. Homocysteine supplementation ameliorates steatohepatitis induced by a choline-deficient diet in mice. Hepatology Research 2019;49:189–200. [DOI] [PubMed] [Google Scholar]
- 96.Wang H, Chen L, Zhang X, Xu L, Xie B, Shi H, Duan Z, et al. Kaempferol protects mice from d-GalN/LPS-induced acute liver failure by regulating the ER stress-Grp78-CHOP signaling pathway. Biomedicine & Pharmacotherapy 2019;111:468–475. [DOI] [PubMed] [Google Scholar]
- 97.Zhang L, Li H, Kong X, Feng W. Lactobacillus rhamnosus GG supernatant prevents acute alcohol-induced liver steatosis and injury through ER stress and autophagy-mediated signaling pathways. The FASEB Journal 2019;33:680.613–680.613. [Google Scholar]
- 98.Yang J, Ao N, Du J, Wang X, He Y. Protective effect of liraglutide against ER stress in the liver of high-fat diet-induced insulin-resistant rats. Endocrine 2015;49:106–118. [DOI] [PubMed] [Google Scholar]
- 99.Gao X, Guo S, Zhang S, Liu A, Shi L, Zhang Y. Matrine attenuates endoplasmic reticulum stress and mitochondrion dysfunction in nonalcoholic fatty liver disease by regulating SERCA pathway. Journal of translational medicine 2018;16:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tuñón MJ, San-Miguel B, Crespo I, Laliena A, Vallejo D, Álvarez M, Prieto J, et al. Melatonin treatment reduces endoplasmic reticulum stress and modulates the unfolded protein response in rabbits with lethal fulminant hepatitis of viral origin. Journal of pineal research 2013;55:221–228. [DOI] [PubMed] [Google Scholar]
- 101.Kim S-J, Kang HS, Lee J-H, Park J-H, Jung CH, Bae J-H, Oh B-C, et al. Melatonin ameliorates ER stress-mediated hepatic steatosis through miR-23a in the liver. Biochemical and biophysical research communications 2015;458:462–469. [DOI] [PubMed] [Google Scholar]
- 102.Kim D-S, Jeong S-K, Kim H-R, Kim D-S, Chae S-W, Chae H-J. Metformin regulates palmitate-induced apoptosis and ER stress response in HepG2 liver cells. Immunopharmacology and immunotoxicology 2010;32:251–257. [DOI] [PubMed] [Google Scholar]
- 103.Yoshiuchi K, Kaneto H, Matsuoka T-A, Kasami R, Kohno K, Iwawaki T, Nakatani Y, et al. Pioglitazone reduces ER stress in the liver: direct monitoring of in vivo ER stress using ER stress-activated indicator transgenic mice. Endocrine journal 2009:0909180336–0909180336. [DOI] [PubMed] [Google Scholar]
- 104.Liu C-M, Zheng G, Ming Q, Sun J, Cheng C. Protective effect of quercetin on lead-induced oxidative stress and endoplasmic reticulum stress in rat liver via the IRE1/JNK and PI3K/Akt pathway. Free radical research 2013;47:192–201. [DOI] [PubMed] [Google Scholar]
- 105.Zhu J, Hua X, Li D, Zhang J, Xia Q. Rapamycin attenuates mouse liver ischemia and reperfusion injury by inhibiting endoplasmic reticulum stress. In: Transplantation proceedings; 2015: Elsevier; 2015. p. 1646–1652. [DOI] [PubMed] [Google Scholar]
- 106.Shi Y, Pan D, Yan L, Chen H, Zhang X, Yuan J, Mu B. Salvianolic acid B improved insulin resistance through suppression of hepatic ER stress in ob/ob mice. Biochemical and biophysical research communications 2020;526:733–737. [DOI] [PubMed] [Google Scholar]
- 107.Jang M-K, Nam JS, Kim JH, Yun Y-R, Han CW, Kim BJ, Jeong H-S, et al. Schisandra chinensis extract ameliorates nonalcoholic fatty liver via inhibition of endoplasmic reticulum stress. Journal of ethnopharmacology 2016;185:96–104. [DOI] [PubMed] [Google Scholar]
- 108.Sahin E, Bagci R, Bektur Aykanat NE, Kacar S, Sahinturk V. Silymarin attenuated nonalcoholic fatty liver disease through the regulation of endoplasmic reticulum stress proteins GRP78 and XBP-1 in mice. Journal of food biochemistry 2020;44:e13194. [DOI] [PubMed] [Google Scholar]
- 109.Gentile CL, Nivala AM, Gonzales JC, Pfaffenbach KT, Wang D, Wei Y, Jiang H, et al. Experimental evidence for therapeutic potential of taurine in the treatment of nonalcoholic fatty liver disease. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology 2011;301:R1710–R1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Su M, Liang X, Xu X, Wu X, Yang B. Hepatoprotective benefits of vitamin C against perfluorooctane sulfonate-induced liver damage in mice through suppressing inflammatory reaction and ER stress. Environmental toxicology and pharmacology 2019;65:60–65. [DOI] [PubMed] [Google Scholar]
- 111.Liu N, Yang HL, Wang P, Lu YC, Yang YJ, Wang L, Lee SC. Functional proteomic analysis revels that the ethanol extract of Annona muricata L. induces liver cancer cell apoptosis through endoplasmic reticulum stress pathway. Journal of ethnopharmacology 2016;189:210–217. [DOI] [PubMed] [Google Scholar]
- 112.Yu SJ, Yoon J-H, Yang J-I, Cho EJ, Kwak MS, Jang ES, Lee J-H, et al. Enhancement of hexokinase II inhibitor-induced apoptosis in hepatocellular carcinoma cells via augmenting ER stress and anti-angiogenesis by protein disulfide isomerase inhibition. Journal of bioenergetics and biomembranes 2012;44:101–115. [DOI] [PubMed] [Google Scholar]
- 113.Ding Y, Chen X, Wang B, Yu B, Ge J. Deubiquitinase inhibitor b-AP15 activates endoplasmic reticulum (ER) stress and inhibits Wnt/Notch1 signaling pathway leading to the reduction of cell survival in hepatocellular carcinoma cells. European journal of pharmacology 2018;825:10–18. [DOI] [PubMed] [Google Scholar]
- 114.Zhu H, Yang W, He L-j, Ding W-j, Zheng L, Liao S-d, Huang P, et al. Upregulating Noxa by ER stress, celastrol exerts synergistic anti-cancer activity in combination with ABT-737 in human hepatocellular carcinoma cells. PLoS One 2012;7:e52333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hsu Y-L, Wu L-Y, Kuo P-L. Dehydrocostuslactone, a medicinal plant-derived sesquiterpene lactone, induces apoptosis coupled to endoplasmic reticulum stress in liver cancer cells. Journal of Pharmacology and Experimental Therapeutics 2009;329:808–819. [DOI] [PubMed] [Google Scholar]
- 116.Munakarmi S, Shrestha J, Shin H-B, Lee G-H, Jeong Y-J. 3, 3′-Diindolylmethane Suppresses the Growth of Hepatocellular Carcinoma by Regulating Its Invasion, Migration, and ER Stress-Mediated Mitochondrial Apoptosis. Cells 2021;10:1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhang X, Zhang S, Sun Q, Jiao W, Yan Y, Zhang X. Compound K induces endoplasmic reticulum stress and apoptosis in human liver cancer cells by regulating STAT3. Molecules 2018;23:1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lv S-X, Isovitexin Qiao X. (IV) induces apoptosis and autophagy in liver cancer cells through endoplasmic reticulum stress. Biochemical and biophysical research communications 2018;496:1047–1054. [DOI] [PubMed] [Google Scholar]
- 119.Zhong J, Xiu P, Dong X, Wang F, Wei H, Wang X, Xu Z, et al. Meloxicam combined with sorafenib synergistically inhibits tumor growth of human hepatocellular carcinoma cells via ER stress-related apoptosis. Oncology reports 2015;34:2142–2150. [DOI] [PubMed] [Google Scholar]
- 120.Lin H, Liu X-b, Yu J-j, Hua F, Hu Z-w. Antioxidant N-acetylcysteine attenuates hepatocarcinogenesis by inhibiting ROS/ER stress in TLR2 deficient mouse. PLoS One 2013;8:e74130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kang JH, Chang Y-C, Maurizi MR. 4-O-carboxymethyl ascochlorin causes ER stress and induced autophagy in human hepatocellular carcinoma cells. Journal of Biological Chemistry 2012;287:15661–15671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhang Q, Chen W, Lv X, Weng Q, Chen M, Cui R, Liang G, et al. Piperlongumine, a Novel TrxR1 inhibitor, induces apoptosis in hepatocellular carcinoma cells by ROS-mediated ER stress. Frontiers in pharmacology 2019;10:1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chaiboonchoe A, Khraiwesh B, Murali C, Baig B, El-Awady R, Tarazi H, Alzahmi A, et al. Safranal induces DNA double-strand breakage and ER-stress-mediated cell death in hepatocellular carcinoma cells. Scientific reports 2018;8:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pp Feng, Yk Qi, Li N, Fei Hr. Scutebarbatine A induces cytotoxicity in hepatocellular carcinoma via activation of the MAPK and ER stress signaling pathways. Journal of Biochemical and Molecular Toxicology 2021;35:e22731. [DOI] [PubMed] [Google Scholar]
- 125.Shi Y-H, Ding Z-B, Zhou J, Hui B, Shi G-M, Ke A-W, Wang X-Y, et al. Targeting autophagy enhances sorafenib lethality for hepatocellular carcinoma via ER stress-related apoptosis. Autophagy 2011;7:1159–1172. [DOI] [PubMed] [Google Scholar]
- 126.Wang X, Wang S, Liu Y, Ding W, Zheng K, Xiang Y, Liu K, et al. The Hsp90 inhibitor SNX-2112 induces apoptosis of human hepatocellular carcinoma cells: the role of ER stress. Biochemical and biophysical research communications 2014;446:160–166. [DOI] [PubMed] [Google Scholar]
- 127.Ye M, Zhu X, Yan R, Hui J, Zhang J, Liu Y, Li X. Sodium demethylcantharidate induces apoptosis in hepatocellular carcinoma cells via ER stress. American journal of translational research 2019;11:3150. [PMC free article] [PubMed] [Google Scholar]
- 128.Zhang Y, Zhao L, Li X, Wang Y, Yao J, Wang H, Li F, et al. V8, a newly synthetic flavonoid, induces apoptosis through ROS-mediated ER stress pathway in hepatocellular carcinoma. Archives of toxicology 2014;88:97–107. [DOI] [PubMed] [Google Scholar]
- 129.Hetz C, Axten JM, Patterson JB. Pharmacological targeting of the unfolded protein response for disease intervention. Nature chemical biology 2019;15:764–775. [DOI] [PubMed] [Google Scholar]
- 130.Bilekova S, Sachs S, Lickert H. Pharmacological targeting of endoplasmic reticulum stress in pancreatic beta cells. Trends in Pharmacological Sciences 2020. [DOI] [PubMed] [Google Scholar]