


Abstract
Peptide nucleic acids (PNAs) are promising therapeutic molecules for gene modulation; however, they suffer from poor cell uptake. Delivery of PNAs into cells requires conjugation of the PNA to another large molecule, typically a cell-penetrating peptide or nanoparticle. In this study, we describe a new PNA-based molecule with cyclic tetrahydrofuran (THF) backbone modifications that in some cases considerably improve cell uptake. We refer to these THF-PNA oligomers as thyclotides. With THF groups at every position of the oligomer, the cell uptake of thyclotides targeted to miR-21 is enhanced compared with the corresponding unmodified PNA based on an aminoethylglycine backbone. An optimized thyclotide can efficiently enter cells without the use of cell-penetrating peptides, bind miR-21, its designated microRNA target, decrease expression of miR-21 and increase expression of three downstream targets (PTEN, Cdc25a and KRIT1). Using a plasmid with the PTEN-3′UTR coupled with luciferase, we further confirmed that a miR-21-targeted thyclotide prevents miR-21 from binding to its target RNA. Additionally, the thyclotide shows no cytotoxicity when administered at 200 times its active concentration. We propose that thyclotides be further explored as therapeutic candidates to modulate miRNA levels.
INTRODUCTION
Cancer is a leading cause of death worldwide, and millions of new cases are diagnosed each year (1). Among the many therapeutic strategies being investigated to reduce the burden of cancer, modulating micro-RNA (miRNA) expression is a very attractive strategy (2–4). miRNAs are non-coding RNA molecules that bind to complementary mRNA or RNA binding proteins (5,6) and are expressed in many tissues (7). They are involved in many processes, including cell differentiation, proliferation and survival (8). By binding to a mRNA target, miRNAs can inhibit translation or promote mRNA degradation, thereby regulating gene expression (9). Dysregulation of miRNAs is related to the development of diseases such as cancer, which is why they are considered promising targets for both diagnostic detection and therapeutic intervention (2,10). Therapeutics based on RNA are emerging, and the first siRNA drug, Patisiran, was approved by the FDA in 2018. Other miRNA candidate drugs are in clinical development for medical intervention, or to help in reducing miRNA-induced chemoresistance (11,12). Although antisense oligonucleotides are one possible means to inhibit miRNAs (13–16), unmodified oligonucleotides have a limited ability to effectively target miRNAs due to low binding affinity, sensitivity to nuclease degradation, and limited cell penetration (17). Some modified antisense oligonucleotides have been used to inhibit miRNAs. For example, locked nucleic acids (LNAs), which contain a rigidifying 2'-O, 4'-C methylene bridge bond in their ribose ring, have been used to inhibit miRNAs, both in vitro using transfection to deliver the LNA into cells, and in vivo by injection (18–20). Similarly, cholesterol-conjugated single-stranded short RNA molecules, or antagomiRs, have also been used to silence miRNAs (21–24). Phosphorodiamidate morpholino oligomers (PMOs) are a class of non-ionic DNA analogues (25–27) that are resistant to enzymatic degradation and bind with high affinity to RNA targets. PMOs are powerful antisense agents (28), and they have also been used as miRNA inhibitors (29–32).
PNAs are DNA analogues in which the phosphate deoxyribose backbone is replaced by uncharged N-(2-aminoethyl)glycine (aeg) units (33). As with the nonionic backbone of PMOs, the absence of charged phosphate groups on PNA eliminates the electrostatic repulsion that occurs in nucleic acid duplexes, thereby enabling PNA to bind with high affinity to complementary DNA and RNA sequences through Watson–Crick and Hoogsteen base pairing mechanisms (34–36). Additionally, PNAs are resistant to degradation by proteases and nucleases as they are not recognized substrates of these enzymes (36). These two features make PNAs very attractive biomedical tools, and several studies have examined PNAs as antisense and antigene agents (37–43). Without modifications, PNAs suffer from poor cell uptake, which is why numerous methods to improve their delivery into cells are being investigated to enhance their utility in biomedical applications (44–49).
One of the most successful methods to enhance the cell uptake of PNAs is conjugation with cell-penetrating peptides (CPPs) (49–51). The CPPs typically contain several positively charged amino acids such as arginine and lysine that interact with cell surface phospholipids and glycoproteins (52). Antisense activity of CPP-PNA conjugates directed toward miRNAs has been reported, demonstrating the therapeutic potential of PNAs for several diseases, including cancer (53–55). Four lysines residues were sufficient to promote the cell uptake of PNAs targeting miR-122 (56,57) and miR-155 (58). There have been other types of carriers used successfully to deliver PNAs with antisense activity into cells. For instance, hybridization of PNAs with overlapping DNA oligonucleotides enables the association of cationic lipids to the DNA strand, allowing the PNA strand to be passively imported into the cell (59). The introduction of phosphonate moieties (60) or the conjugation of cholesterol or cholic acid to the PNA (61) also were successfully used in vitro with lipofectamine transfection. Nanoparticle-based delivery is another attractive strategy for the therapeutic application of PNAs, and has been developed for antisense applications, gene editing, splice correction, RNA detection and mRNA imaging (37). Mesoporous silica nanoparticles have also been used in vitro to deliver PNAs into HeLa cells to silence BCL-2 (62). Nanoparticle efficiency in PNA delivery was also demonstrated in vivo in a miR-155-dependent mouse model of lymphoma (63). PNAs were also successfully conjugated to ligands to target specific receptors on the cell surface. Bhingardeve et al. have shown that PNAs conjugated to N-acetyl galactosamine were recognized by the asialoglycoprotein receptor in HepG2 cells which promoted an active uptake into these cells (64). Further, PNA cell uptake was improved through the use of a nucleocapsid protein derived from the SV40 virus. The pseudovirions carrying PNAs were able to efficiently enter KB-8–5 carcinoma cells and decrease MDR1 expression (65). Finally, as an alternative to conjugation or chemical modification of the PNA, it was recently shown that a tetraargininocalix[4]arene could transport PNAs into U251 glioma cells, with minimal toxicity of the vehicle and relatively good efficiency. Indeed, 2 μM of PNAs simply mixed with 2.5 μM of calixarene was able to reduce by half the expression levels of miR-221, which was similar to the inhibition levels obtained with the same PNA conjugated to an oligoarginine peptide (66). Modifying the backbone of PNAs to enhance their cell uptake and anti-microRNA activity against miR-210 has also been achieved using PNAs containing arginine sidechains incorporated into the backbone (67).
While there are several approaches to deliver PNAs into cells, problems due to cytotoxicity (68,69), endosome entrapment (70) or the high concentration required to fully inhibit a target sequence are often associated with the existing methods. In this manuscript, we report the development and validation of a new type of PNA-based molecule that we call a thyclotide. In contrast to the prior strategies that rely on highly charged CPPs, transfection reagents, or non-covalent association with macromolecular structures to facilitate cell uptake of the PNA, the thyclotide modification simply incorporates uncharged THF groups into the peptidic backbone to achieve prominent uptake across a range of cells and potent inhibition of a miRNA target.
MATERIALS AND METHODS
Reagents and materials
All 9-fluorenylmethoxycarbonyl (Fmoc)-PNA monomers were purchased from PolyOrg, Inc. (Leominster, MA, USA). Acetonitrile, Fmoc-Lys(Boc)-OH, acetic anhydride (Ac2O), pyridine, dichloromethane (DCM), 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU), N,N-diisopropylethylamine (DIPEA), diethyl ether (Et2O), N,N-dimethylformamide (DMF), Kaiser test reagents, m-cresol, N-methyl-2-pyrrolidinone (NMP), piperidine, and trifluoroacetic acid (TFA) were purchased from Sigma-Aldrich (St Louis, MO, USA). High purity water (18 MΩ) was generated from a Millipore (Billerica, MA, USA) Milli-Q water system. Rink Amide ProTide resin (LL, 100–200 mesh, 0.19 mmol/g) was purchased from CEM corporation (Matthews, NC, USA). NHS-Fluorescein (5/6-carboxyfluorescein succinimidyl ester, mixed isomer) was purchased from ThermoFisher Scientific (Waltham, MA, USA). Fmoc-8-amino-3,6-dioxaoctanoic acid was purchased from Peptides International, Inc. (Louisville, KY, USA). All Fmoc-protected thyclotide monomers were prepared using commercially available starting material from Entrechem (Spain) using protocols previously reported by our group (71).
General method for PNA and thyclotide synthesis
The detailed synthetic route for the preparation of thyclotide monomers is available in the Supporting Information (Supplementary Figure S1). Nuclear Magnetic Resonance (NMR) spectra for new molecules S3–S5 are reported in Supplementary Figures S2–S27. Thyclotides and PNAs were prepared on 5 μmol scale using Fmoc-solid phase peptide synthesis protocols on an Intavis Multipep CF with HATU as the amide-forming reagent. All thyclotides and PNAs were synthesized on Rink Amide ProTide resin. The resin, in a peptide synthesis vessel, was first washed with 20% piperidine in DMF (2 × 5 ml for 5 min) to deprotect the Fmoc group. The progress of the deprotection was followed by a qualitative Kaiser test. If the test was negative, the resin was resubmitted to additional deprotection. After a positive result for primary amines, the resin was drained and treated with cleavage cocktail (2 ml, 5% m-cresol in TFA) for 1 h. The cleavage mixture was collected in a glass vial using N2 pressure to drain the vessel. The resin was resubmitted to a fresh cleavage cocktail and cleaved for 1 hour, then drained into the first cleavage fraction. The volatiles were removed by flowing dry N2 over the solution to produce a yellow-brown oil. Approximately 10 ml of Et2O was added to the cleavage oil to create a suspended white precipitate. The suspension was partitioned into five 2 ml microcentrifuge tubes and chilled over dry ice for 10 minutes. The tubes were centrifuged at 13 800 rcf for 40 s to produce a white pellet. Et2O was carefully decanted, leaving the white crude PNA solid. Further washing was performed by adding about 1.6 ml of Et2O to each tube, mixing to resuspend the precipitate, then chilling on dry ice for 5 min. Following centrifugation and decanting, the washes were repeated twice without dry ice. After the final wash, the white precipitate was dried by carefully passing a stream of dry N2 over the crude PNAs and thyclotides.
Labeling of PNAs (or thyclotide) with NHS-Fluorescein
The resin was first washed in a peptide synthesis vessel with 20% piperidine in DMF (2 × 5 ml for 5 min) to deprotect the Fmoc group. The progress of the deprotection was followed by a qualitative Kaiser test. If the test was negative, the resin was resubmitted to additional deprotection. After a positive test for primary amines, the resin was drained and ready for the next step. To the resin was added a solution of NHS-Fluorescein (47 mg, 100 μmol) in anhydrous DMF (0.4 ml) and DIPEA (30 μl) at room temperature. The suspension was stirred at room temperature overnight. The next day, the resin was washed thoroughly with DMF (4 × 5.0 ml) followed by PNA (or thyclotide) cleavage from solid support using standard procedures.
Purification and characterization of crude PNA and thyclotides
Purification was performed on an Agilent (Santa Clara, CA, USA) 1260 Series RP-HPLC with automatic fraction collection using ultraviolet detection at 260 nm. A Waters (Milford, MA, USA) XBridge C18 (10 × 250 mm, 5 μm) column was used in conjunction with Solvents A and B for purification at 45 °C. Another Waters (Milford, MA, USA) XBridge C18 (4.6 × 250 mm, 5 μm) column was used in conjunction with Solvents A and B to check purity at room temperature. Solvent A was 0.05% TFA in water and Solvent B consisted of 90% acetonitrile in water. PNA and thyclotide HPLC isolates were characterized using electrospray ionization-mass spectrometry on a Waters/Micromass LCT Premier time-of-flight mass spectrometer. The instrument was operated in W-mode at a nominal resolution of 10 000. The electrospray capillary voltage was 2 kV and the sample cone voltage was 60 V. The desolvation temperature was 275 °C and the desolvation gas was N2 with a flow of 300 l/ h. Accurate masses were obtained using the internal reference standard method. The sample was introduced into the mass spectrometer via the direct loop injection method. Deconvolution of multiply charged ions was performed with MaxEnt I. All PNA and thyclotide oligomers yielded molecular ions consistent with the calculated theoretical product values. PNA and thyclotide sequences are shown in Table 1.
Table 1.
List and mass characterization data of thyclotides and aegPNA oligomers. Tetrahydrofuran residues are represented by the symbol * in the sequences. Tetrahydrofuran stereochemistry is (R,R). The data for 14-mer PNAs or thyclotides in this table correspond to the triply charged ion [M + 3H]3+ and the data for the 20-mer PNAs or thyclotides in this table correspond to the quadruply charged ion [M + 4H]4+. FAM = 5/6-fluorescein, AEEA = 2-(2-aminoethoxy)ethoxyacetyl group
| Entry | PNA or thyclotide | Calculated | Observed |
|---|---|---|---|
| 1antimiR-21 14-nucleobase PNA | NH2-AGTCTGATAAGCTA-AEEA-CONH2 | 1327.3 | 1327.5 |
| 2antimiR-21 14-nucleobase thyclotide | NH2-A*G*T*C*T*G*A*T*A*A*G*C*T*A*-AEEA-CONH2 | 1523.3 | 1523.6 |
| 3antimiR-21 14-nucleobase partial thyclotide | NH2-AGTCTGA*T*AAGCTA-AEEA-CONH2 | 1355.2 | 1355.2 |
| 4FAM-antimiR-21 14-nucleobase PNA | FAM-(6)5-AGTCTGATAAGCTA-AEEA-CONH2 | 1543.3 | 1543.6 |
| 5FAM-antimiR-21 14-nucleobase thyclotide | FAM-(6)5-A*G*T*C*T*G*A*T*A*A* G*C*T*A*-AEEA-CONH2 | 1739.7 | 1739.6 |
| 6FAM-antimiR-21 20-nucleobase PNA | FAM-AEEA-AEEA-AACATCAGTCTGAT AAGCTA-AEEA-CONH2 | 1556.4 | 1556.6 |
| 7FAM-antimiR-21 20-nucleobase thyclotide | FAM-AEEA-AEEA-A*A*C*A*T*C*A*G*T*C*T*G*A* T*A*A*G*C*T*A*-AEEA-CONH2 | 1766.7 | 1766.7 |
| 8scrambled 20-nucleobase thyclotide | NH2-G*T*A*C*A*T*T*A*C*G*A*C*G* A*T*A*C*A*A*T*-AEEA-CONH2 | 1604.4 | 1604.6 |
| 9antimiR-21 20-nucleobase PNA | NH2-AACATCAGTCTGATAAGCTA-AEEA-CONH2 | 1394.6 | 1394.6 |
| 10antimiR-21 20-nucleobase thyclotide | NH2-A*A*C*A*T*C*A*G*T*C*T*G*A* T*A*A*G*C*T*A*-AEEA-CONH2 | 1604.4 | 1604.6 |
| 11antimiR-21 20-nucleobase Lys-PNA | NH2-Lys-AACATCAGTCTGATAAGCTA-Lys-CONH2 | 1422.1 | 1422.3 |
| 12antimiR-21 20-nucleobase Lys-thyclotide | NH2-Lys-A*A*C*A*T*C*A*G*T*C*T*G*A* T*A*A*G*C*T*A*-Lys-CONH2 | 1632.6 | 1632.4 |
| 13scrambled 20-nucleobase Lys-thyclotide | NH2-Lys-G*T*A*C*A*T*T*A*C*G*A*C*G* A*T*A*C*A*A*T*-Lys-CONH2 | 1632.6 | 1632.4 |
| 14FAM-antimiR-21 20-nucleobase Lys-PNA | FAM-AEEA-AEEA-Lys-AACATCAGTC TGATAAGCTA-Lys-CONH2 | 1584.1 | 1584.4 |
| 15FAM-antimiR-21 20-nucleobase Lys-thyclotide | FAM-AEEA-AEEA-Lys-A*A*C*A*T*C*A*G* T*C*T*G*A*T*A*A*G*C*T*A*-Lys-CONH2 | 1794.4 | 1794.5 |
| 16FAM-scrambled 20-nucleobase Lys-thyclotide | FAM-AEEA-AEEA-Lys-G*T*A*C*A*T*T*A*C*G* A*C*G*A*T*A*C*A*A*T*-Lys-CONH2 | 1794.4 | 1794.7 |
HPLC analysis of thyclotide and PNA oligomers
For HPLC, the column was equilibrated with 100% A (0.05% TFA in water) and 0% B (90% acetonitrile in water) for 15 min prior to run, 0% B (100% A) for 2 min, a linear gradient from 0% B (100% A) to 100% B (0% A) over 18 min, 100% B (0% A) for 4 min, and a linear gradient to 0% B (100% A) over 1 min. The flow rate was 1.2 ml/min. HPLC chromatograms and mass spectra of thyclotide and PNA oligomers are shown in Supplementary Figures S28–S59.
UV melting experiments
Oligonucleotides were ordered from IDT (Coralville, IA, USA). High purity water (18 MΩ) was generated from a Millipore (Billerica, MA, USA) Milli-Q water system. 1× PBS buffer (154 mM NaCl, 1.55 mM KH2PO4, 5.12 mM Na2HPO4, pH 7.2) was purchased from KD Medical (Columbia, MD, USA). The ultraviolet concentration was determined by adding 4 μl of DNA, RNA, and PNA (or thyclotide) solution to 196 μl milli-Q water. If the signal was too intense, the concentration was diluted by adding 198 μl of water to 2 μl of the original DNA, RNA, and PNA (or thyclotide) solution. For the background, a blank measurement was taken with water at 90 °C on an Agilent 8453 UV/Vis spectrometer equipped with an Agilent 89090A Peltier temperature controller and a computer interface. The unknown solution was added to the quartz cell (Helma) and vigorously shaken, replaced in the spectrophotometer and the absorbance was read at 260 nm. The mixing and reading were repeated three times. Values were converted to concentration based on the average absorbance. After initial measurement by ultraviolet, the concentration was determined based on appropriate ϵ260 nm (calculated on nearest neighbour approximation for PNA (or thyclotide) or provided by IDT or Thermo Scientific for oligonucleotides) and then used from that point forward for additional experiments. Thermal melting experiments were performed by preparing 1 μM PNA (or thyclotide) solution and 1 μM DNA (5′-TAGCTTATCAGACT-3′) or 1 μM RNA (matched RNA: 5′-UAGCUUAUCAGACU-3′; TU mismatched RNA: 5′-UAGCUUUUCAGACU-3′; TC mismatched RNA: 5′-UAGCUUCUCAGACU-3′; TG mismatched RNA: 5′-UAGCUUGUCAGACU-3′) solution in 1× PBS buffer (pH 7.2). Experimental temperatures ranged from 90 to 15 °C and back to 95 °C at 1 °C intervals while monitoring at 260 nm. An equilibration of 60 seconds at each temperature measurement step was performed before each reading. Cooling and heating profiles were generated for each sample in triplicate. The Tm (melting temperature) for duplexes was determined using the maximum first derivative of the heating curves, then taken as an average of three runs. The absorbances at 260 nm for the aegPNA–DNA duplex were normalized by defining the smallest absorbance in the data as 0% and the largest absorbance as 100%. The melting temperature transition curve for the thyclotide–DNA duplex is not complete. Therefore, the absorbance value representing dissociation of the thyclotide–DNA duplex was estimated by adding the value by which the absorbance increases for the aegPNA–DNA duplex to the smallest absorbance of the thyclotide–DNA duplex. Then, the absorbances at 260 nm for the thyclotide–DNA duplex were normalized by defining the smallest absorbance as 0% and the largest expected absorbance as 100%.
Cell lines and cell culture
SKHEP1, HepG2, RKO and MCF7 cells were ordered from American Type Culture Collection (ATCC, Manassas, VA, USA). All cells were cultured at 37°C with 5% CO2 in a humidified incubator. All cells were cultured in EMEM (ATCC, Manassas, VA, USA) supplemented with 10% FBS. For cell treatments, PNAs and thyclotides were diluted in the complete cell culture medium.
Electrophoretic mobility shift assay (EMSA)
100 nM of FAM-conjugated hsa-miR-21-5p (5′-/FAM/rUrArG rCrUrU rArUrC rArGrA rCrUrG rArUrG rUrUrG rArCrU-3′) was incubated in 5 mM HEPES pH 7.3 with 12 mM NaCl at 50 °C with increasing concentrations of antimiR-21 aegPNA 11 (25, 50, 75, 100, 200 and 300 nM) or thyclotide 12 (25, 50, 75, 100 and 200 nM). The solutions were loaded onto a 14% acrylamide:bis-acrylamide (29:1) gel in incubation buffer supplemented with 10% glycerol along with a low molecular weight DNA ladder (New England Biolabs, Ipswich, MA, USA) and electrophoresis was done for 3 h at 200 V at room temperature in 1× Tris borate EDTA. The gel was imaged with the Typhoon FLA9500 (GE Healthcare Life Sciences, Chicago, IL, USA). Following fluorescence imaging, gel was stained with GelRed (Millipore Sigma, Burlington, MA, USA) to reveal the ladder. A ladder image was then merged with the fluorescence image of the EMSA.
Fluorescence-activated cell sorting (FACS)
Cells were seeded in 6-well plates and the next day were either non-treated or treated with fluorescein-labelled aegPNA or thyclotide diluted in the culture medium. Three hours after the treatment (SKHEP1, RKO and MCF7 cells) or 16 h after the treatment (HepG2 cells), the PNA-containing culture medium was removed, and the cells were washed three times with PBS. Cells were detached with trypsin-EDTA and the enzyme was stopped with total growth medium. Cellular uptake was quantified with fluorescence analyzed by flow cytometry using a BD FACSCanto II flow cytometer (BD Biosciences, San Jose, CA, USA) and the FlowJo software v10 (BD Biosciences, San Jose, CA, USA).
Super-resolution microscopy
SKHEP1 cells were seeded in 35 mm Falcon dishes with no. 1.5, 22 × 22 mm glass coverslip kits (MatTek Corporation, Ashland, MA, USA) and treated the next day with fluorescein-labelled aegPNA or thyclotide with CellLight BacMam 2.0 early endosome-RFP marker (Invitrogen, Carlsbad, CA, USA) diluted in the culture medium. The endosome marker transduction was performed at a PPC of 30. Sixteen hours later, cells were washed three times with PBS and then stained with Membrite Fix 640/660 cell membrane marker (Biotium, Fremont, CA, USA). Cells were fixed with 4% PFA (Sigma-Aldrich, St Louis, MO, USA) for 20 min at room temperature. Cells were then stained with 2 μg/ml Hoechst 33342 solution (Invitrogen, Carlsbad, CA, USA) in PBS for 30 min at room temperature and coverslips were mounted on glass slides (Premium plain slides, Fisher scientific, Waltham, MA, USA) with Prolong glass mounting medium (Invitrogen, Carlsbad, CA, USA). Super-resolution images were acquired using a Nikon SoRa spinning disk microscope (Nikon Instrument Inc., Melville, NY, USA) equipped with a 60× oil immersion objective lens (N.A. 1.49) and Photometrics BSI sCMOS camera (Teledyne Photometrics, Tucson, AZ, USA). Image z-stacks were collected using a 0.15 μm z-step size, and were deconvolved in the Nikon Elements software (v. 5.3) using a modified Richardson-Lucy iterative deconvolution algorithm. A volume reconstruction of the deconvolved images was generated using Imaris software (v. 9.3, Bitplane).
RTqPCR
Cells were seeded and treated one day later with scrambled or antimiR-21 thyclotide for 48 h. Cells were washed three times with PBS and collected with Trypsin-EDTA. Total RNA was isolated with the RNeasy Mini Kit (Qiagen, Germantown, MD, USA) and total miRNAs were isolated with the PureLink miRNA kit (Invitrogen, Carlsbad, CA, USA) following manufacturer's protocols. All RNA samples were treated with RNaseOUT recombinant ribonuclease inhibitor (Thermo Fisher Scientific, Waltham, MA, USA) and were stored at –80°C following extraction. Purity was assessed by determining A260/A280 ratio with a NanoDrop One C (Thermo Fisher Scientific, Waltham, MA, USA) and considered good if >1.8. miR-21 quantitation PCR was performed using TaqMan Fast Advanced Master Mix (Thermo Fisher Scientific, Waltham, MA, USA) and TaqMan advanced miRNA Assays, following manufacturer's protocol and reaction conditions in a volume of 20 μl. miR-21 Assay ID: 477975_mir; miR-25 Assay ID: 477994_mir; miR-93 Assay ID: 478210_mir (Thermo Fisher Scientific, Waltham, MA, USA). miR-21 levels were assessed relative to miR-25 and miR-93 levels which were recommended as endogenous controls as they are known to have stable expression across cancer cell lines (72). All PCRs were performed at least in triplicate following MIQE guidelines (73). Expression of miR-21 in aegPNA- and thyclotide-treated cells was normalized to the scrambled control relative to the average of miR-25 and miR-93 endogenous controls expression and calculated using CFX Manager gene expression tool with the ΔΔCq method: the relative quantity of genes of interest is normalized to the relative quantity of the reference genes across samples (BioRad, Hercules, CA, USA). For PTEN PCR, cDNA was prepared using the high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA, USA) and the PCR was performed with the PowerUP SYBR Green Master Mix (Applied Biosystems, Waltham, MA, USA). UDG was activated for 2 minutes at 50°C, then dual lock DNA polymerase was activated for 2 min at 95°C. The PCR was performed for 40 cycles, with 15 s of denaturation at 95°C, 15 s of annealing at 55°C and 45 s of extension at 72°C. All PCRs were performed with a C1000 Touch real-time thermal cycler (BioRad, Hercules, CA, USA). Significance of results was determined with a two-tailed paired t-test using GraphPad Prism software. Primers sequences used during PCRs are shown in Table 2.
Table 2.
Primers sequences (5' to 3') for PTEN, Cdc25a and KRIT1 PCRs
| β-actin (forward) | TGTTTGAGACCTTCAACACC |
| β-actin (reverse) | ATGTCACGCACGATTTCC |
| PTEN (forward) | CGGCAGCATCAAATGTTTCAG |
| PTEN (reverse) | TGGCAGGTAGAAGGCAACTC |
| Cdc25a (forward) | AGAGTCAACTAATCCAGAGAAGG |
| Cdc25a (reverse) | GCAAGTTCACTGCACCCTTG |
| KRIT1 (forward) | ATGCGAGTCTGTAGTGAATCCA |
| KRIT1 (reverse) | TGTGCATGACGTTCATCTAACC |
Western blotting
HepG2 cells were grown in 6-well plates, treated with 25 nM thyclotides for 48 h, then washed with ice-cold PBS, collected with Trypsine-EDTA, washed again with ice-cold PBS before lysis by incubation on ice for 15 min in Pierce RIPA buffer (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 1 mM phenylmethylsulfonyl fluoride, proteases (cOmplete ULTRA Tablets Mini, Millipore Sigma, Burlington, MA, USA) and phosphatases (PhosSTOP, Millipore Sigma, Burlington, MA, USA) inhibitors. Protein concentration of samples was determined with the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). 30 μg of proteins were mixed with Laemmli Sample Buffer (4X) (BioRad Hercules, CA, USA) and 1 mM DTT before being boiled at 95°C for 5 min and loaded in a 4–20% Mini-PROTEAN TGX Precast Protein Gel (BioRad, Hercules, CA, USA). Gels were then transferred to Immun-Blot PVDF membranes (BioRad, Hercules, CA, USA) for 2 h at 52 V in Novex Tris-Glycine transfer buffer (Thermo Fisher Scientific, Waltham, MA, USA). Membranes were blocked in TBS with 0.1% Tween20 and 8% BSA for 45 min at room temperature and then incubated overnight at 4°C with primary antibodies (Santa Cruz Biotechnology, Dallas, TX, USA and Cell Signaling Technology, Danvers, MA, USA) anti PTEN (sc-7974), Cdc25a (sc-7389), and KRIT1 (sc-514371) or GAPDH (CST 97166S). Membranes were then incubated with anti-mouse HRP-conjugated secondary antibody (CST 7076S) and revealed using the ChemiDoc XRS + Imaging System (BioRad, Hercules, CA, USA).
PTEN luminescence assay
Cells were seeded in 12-well plates and co-transfected with pGL3-PTEN-3′UTR (this plasmid was a gift from Joshua Mendell; Addgene plasmid # 21326; http://n2t.net/addgene:21326;RRID:Addgene_21326) and pRL-TK Renilla Luciferase control reporter vector (Promega, Madison, WI, USA) using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA). Six hours after transfection, medium was changed, and cells were treated with 25 nM or 5 μM of either scrambled thyclotide or antimiR-21 thyclotide. Forty-eight hours after transfection, cells were washed three times with PBS, then lysed in wells with passive lysis buffer (Promega, Madison, WI, USA). Luminescence of firefly and renilla luciferase for internal control was then assessed with the dual luciferase assay kit (Promega, Madison, WI, USA) following manufacturer's instructions and using a SpectraMax iD3 plate reader (Molecular Devices, San Jose, CA, USA).
Cytotoxicity assay
Cells were seeded in 96-well plates and treated with DMSO or scrambled thyclotide diluted in complete medium at increasing concentrations for 24, 48, and 72 h. LDH release was used to assess cytotoxicity using the CyQUANT LDH cytotoxicity assay kit (Invitrogen, Carlsbad, CA, USA) and following manufacturer's protocol. Viability was calculated using the following formula:
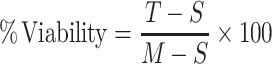 |
Where T is thyclotide-treated LDH activity, S is spontaneous LDH activity, and M is maximum LDH activity.
RESULTS
Thyclotide chemical structure and binding to miR-21
We developed a new molecule to target miR-21 (Figure 1A), an oncomir that is overexpressed in many cancers, and is known to target genes associated with apoptosis, proliferation, migration and invasion such as PTEN, Fas-L, PDCD4, TIMP3 or RECK. We named this new class of molecules thyclotides, designating a class of oligomers with THF groups replacing one or more ethylene diamine units within an aegPNA backbone. Initially, a 14-nucleobase thyclotide that was complementary to miR-21 was studied where THF groups are present at every position in the sequence (thyclotide 2 in Table 1, Figure 1B). We have shown previously that cyclopentane modifications of PNAs displayed increased binding affinity with complementary DNA, resulting in a higher Tm compared with regular aegPNA (71). We therefore compared thermal denaturation and renaturation of both regular aegPNA 1 and thyclotide 2 with the complementary DNA sequence (Figure 1C). The aegPNA-DNA duplex showed a complete melting curve with a Tm of 64°C. However, the Tm of the corresponding thyclotide-DNA duplex could not be accurately calculated as it is likely much higher than 95°C. At a length of 14 nucleobases, a thyclotide with THF groups at every position most likely binds too strongly to complementary oligonucleotide sequences to observe complete denaturation by heating (71). To lower the Tm of the thyclotide + oligonucleotide duplex, we examined the binding of a thyclotide containing two THF groups in the middle of the sequence (thyclotide 3, Table 1) to its complementary RNA sequence. The Tm of the thyclotide 3 + RNA duplex was 77.9°C, which is higher than the Tm of the corresponding aegPNA-RNA duplex (70.6°C, Figure 2A). We then studied the sequence specificity of thyclotides by examining the change of the Tm with RNA containing a single base mismatch in the middle of the sequence. For both the aegPNA 1 (Figure 2B) and thyclotide 3 (Figure 2C), we observed that the Tm of the matched duplex with RNA is significantly higher than the Tm of the mismatched duplex, and that the thyclotide shows sequence specificity slightly better than the corresponding aegPNA (Table 3).
Figure 1.
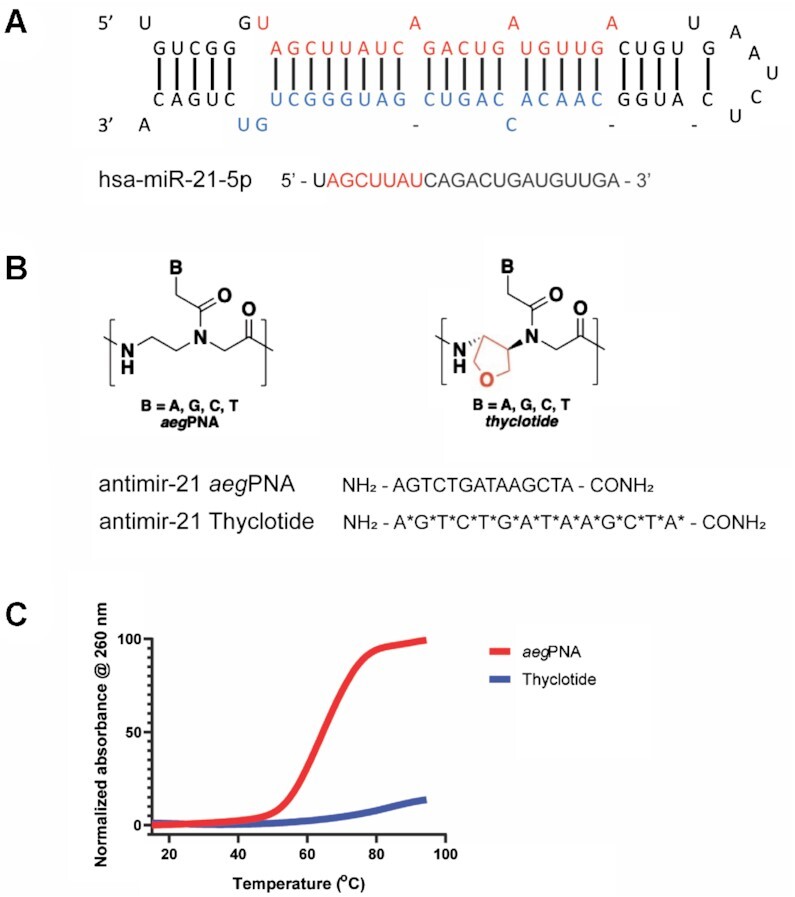
Thyclotide, a miR-21 targeting PNA-based molecule bearing tetrahydrofuran rings in the backbone. (A) Secondary structure of pre-miR-21 (top) and sequence of mature hsa-miR-21-5p (bottom). The seed sequence of mature miR-21 is in red. (B) Structural schematic of the regular aegPNA (left) and thyclotide (right) and the 14-nucleobase sequences representing aegPNA 1 and thyclotide 2. Thyclotides possess tetrahydrofuran rings in the backbone, designated by red in the chemical drawing and by the symbol * in the sequence. (C) Normalized UV melting curves of aegPNA 1 and thyclotide 2 with their complementary DNA. The Tm of the aegPNA-DNA duplex is 64°C. The Tm of the thyclotide-DNA duplex is likely >95°C and could not be accurately calculated.
Figure 2.

UV melting curves of aegPNA or thyclotide with their complementary RNA or single base mismatched RNA. The thyclotide 3 used in these experiments has two tetrahydrofuran residues (Table 1 and Table 3). (A) Comparison of the melting temperature curves of aegPNA 1 and thyclotide 3 with their complementary RNA. (B) UV melting curves for aegPNA-RNA complexes. RNA sequence is either fully matched with the PNA (red) or has a mismatch as indicated in Table 3 (TC: green, TU: purple, TG: blue). (C) UV melting curves for thyclotide 3-RNA complexes. RNA sequence is either fully matched with the thyclotide (red) or has a mismatch as indicated in Table 3 (TC: green, TU: purple, TG: blue).
Table 3.
Discrimination of single base mismatches and improvements in thyclotide. Tetrahydrofuran stereochemistry is (R,R). Tetrahydrofuran residues are represented by the symbol * in the sequences. PNA and thyclotide sequences are written from N- to C-terminal. All temperature values are reported in units of°C. ΔTm represents the difference in melting temperature between the complementary RNA and the RNA with the indicated mismatch. All mismatches were opposite to residue symbolized with T or T*
| Sequence | T m (ΔTm) (°C) | ||
|---|---|---|---|
| TU mismatch | TC mismatch | TG mismatch | |
| NH2-AGTCTGATAAGCTA-AEEA-CONH2 (1) | 62.0 (−8.6) | 60.5 (−10.1) | 63.4 (−7.2) |
| NH2-AGTCTGA*T*AAGCTA-AEEA-CONH2 (3) | 68.2 (−9.7) | 66.8 (−11.1) | 70.3 (−7.6) |
Studies on cell uptake of 14-nucleobase thyclotide
To assess cellular uptake, we treated cultured human cells with fluorescein-labelled aegPNA or fluorescein-labelled thyclotide, with both molecules containing the same 14-nucleobase sequence (Table 1; FAM-antimiR-21 PNA 4 and FAM-antimiR-21 thyclotide 5 respectively), and assessed cell-based fluorescence by flow cytometry. Regular aegPNAs typically do not enter cells efficiently without conjugation to positively charged CPPs, incorporation of positively charged sidechains in the backbone, or incorporation into larger macromolecular structures (such as nanoparticles or viruses). FACS analysis of SKHEP1 cells treated with 5 μM of the 14-nucleobase thyclotide 5 demonstrated successful uptake after 3 hours of treatment (Figure 3A). Similarly, thyclotide 5 was successfully taken into HepG2 cells after 16 hours of treatment (Figure 3B). This change in cellular uptake time could be due to differences in the morphology of the HepG2 cells, which grow in clumps. In both cell lines, the uptake of thyclotide is significantly higher than the uptake of aegPNA (P < 0.0001). Specifically, thyclotide 5 was successfully taken in by 78.8% of SKHEP1 cells and 72.65% of HepG2 cells, with positive cells having fluorescence values higher than untreated cells. In comparison, only 4% of SKHEP1 and 12.7% of HepG2 cells were positive for aegPNA 4. The mean fluorescence intensity (MFI) of aegPNA-treated positive cells was significantly lower compared with the MFI of thyclotide-treated positive cells (SKHEP1: P < 0.0001 and HepG2: P = 0.0009), showing that the quantity of molecules taken up by all fluorescent cells was always higher with thyclotide 5 than aegPNA 4. To more closely examine the cellular uptake of thyclotide, we used super resolution microscopy to visualize SKHEP1 cells that were treated with either aegPNA 4 or thyclotide 5. As expected, no detectable signal was observed with aegPNA 4. Remarkably, green fluorescence was easily observed in the cells treated with thyclotide 5 and good cytoplasmic diffusion was evident. In cases where aegPNAs are conjugated to CPPs to facilitate cell uptake, aegPNA-CPP conjugates are often trapped in endosomes (74). It was therefore important to check whether thyclotide 5 was similarly trapped in endosomes. For this purpose, cells were treated with an early-endosomes marker to check the subcellular localization of thyclotide 5 after entering the cell. No colocalization of thyclotide 5 with endosomes was observed in SKHEP1 cells (Figure 3C), indicating that the thyclotide enters cells by mechanisms that may be different than those of CPP-conjugated aegPNAs.
Figure 3.

Thyclotide 5 is successfully taken up by cells and does not colocalize with endosomes 16 hours after treatment. (A) FACS of SKHEP1 cells either non-treated (blue) or treated with 5 μM of either FAM-aegPNA 4 (orange) or FAM-thyclotide 5 (red). Positive cells were determined by having a fluorescence higher than the non-treated cells. MFI refers to the mean fluorescence intensity. (B) FACS of HepG2 cells either non-treated (blue) or treated with 5 μM of FAM-aegPNA 4 (orange) or FAM-thyclotide 5 (red). Positive cells were determined by having a fluorescence higher than the non-treated cells. MFI refers to the mean fluorescence intensity. (C) 3D volume reconstruction of 0.15 μm z-steps super-resolution microscopy imaging of SKHEP1 cells treated with FAM-thyclotide 5. Cells were stained with Membrite Fix 640/660 cell membrane marker (white), CellLight BacMam 2.0 early endosomes-RFP marker (orange) and Hoechst 33342 nucleus staining solution (blue). The membrane marker channel was removed from the right picture for a better visualization of the cytoplasmic diffusion of FAM-thyclotide 5 (green).
Inhibition of miR-21 with a 20-nucleobase thyclotide
With initial data showing that a thyclotide of 14 nucleobases had good cellular uptake, we next investigated the ability of a thyclotide to directly inhibit the activity of miR-21 in cells. Although the 14-base sequence was useful for development and proof of concept, the short length might lead to off-target effects in cells. Indeed, a BLAST (Basic Local Alignment Search Tool) search of the human genome using the thyclotide 5 sequence as the query revealed that it was too short to achieve sequence specificity within the cell. This sequence could target other regions of the genome in addition to the pre-miR-21, mature miR-21, or chromosome 17q23.2, which is the location of this miRNA (75). Therefore, we synthesized a 20 nucleobase thyclotide that is complementary to most of the mature miR-21 sequence, which is 22 nucleobases long. A BLAST search indicated this longer sequence would be specific for miR-21. Before beginning the studies to examine inhibition of miR-21, we examined the cellular uptake of thyclotide versus aegPNA with the same 20 nucleobase sequence to determine whether the longer length could impact the ability of thyclotide to enter cells. Similar to the previous studies with 14 nucleobase sequences, we synthesized fluorescein-conjugated 20-nucleobase sequences of aegPNA and thyclotide (FAM-antimiR-21 aegPNA 6 and FAM-antimiR-21 thyclotide 7 respectively). Following treatment of cultured human cells with 5 μM of fluorescein-labelled oligomers, FACS analysis showed that the thyclotide 7 enters cells efficiently (Figure 4A and B), with 73% of SKHEP1 and 81% of HepG2 cells positive for thyclotide 7. In contrast, only 5% of SKHEP1 cells and 19.5% of HepG2 cells were positive for the aegPNA 6. Next, we directly tested whether thyclotide 7 inhibited miR-21 in HepG2 cells, which are hepatocellular carcinoma cells that are known to overexpress miR-21. For these experiments, we synthesized a 20-nucleobase scrambled control thyclotide (thyclotide 8) as well as the antimiR-21 aegPNA 9 and thyclotide 10 (all without fluorescein). Cells were treated for 48 hours with 1 μM of scrambled control thyclotide 8, aegPNA 9 or thyclotide 10. After total miRNA isolation, we performed RTqPCR to quantify the levels of miR-21. As expected, aegPNA 9 showed no effect on miR-21 levels relative to the scrambled control thyclotide 8. In contrast, 1 μM of thyclotide 10 reduced miR-21 to undetectable apparent levels in HepG2 cells, confirming the ability of this molecule to enter the cell and bind its miRNA target (Figure 4C). Antisense oligonucleotides, PNAs and thyclotides will bind more strongly to their miRNA target than PCR primers. It is likely that the apparent decrease in miR-21 levels detected by RTqPCR does not mean that the actual levels of miR-21 are decreased, but that antimiR-21 thyclotides act as steric blockers of miR-21, preventing the miRNA to bind its target sequence.
Figure 4.

A 20-nucleobase thyclotide enters cells and inhibits miR-21. (A) FACS of SKHEP1 cells either non-treated (blue) or treated with 5 μM of FAM-aegPNA 6 (orange) or FAM-thyclotide 7 (red). (B) FACS of HepG2 cells either non-treated (blue) or treated with 5 μM of either FAM-aegPNA 6 (orange) or FAM-thyclotide 7 (red). (C) Determination of miR-21 expression by RT-qPCR. HepG2 cells were treated with either 1 μM of scrambled thyclotide 8, antimiR-21 aegPNA 9 or antimiR-21 thyclotide 10, and apparent miR-21 levels were assessed and normalized to the average of miR-25 and miR-93 endogenous controls relative to the control scrambled expression.
Addition of terminal lysines improves solubility without altering cell uptake
The 20-nucleobase constructs displayed some issues with solubility that were not evident with the shorter 14-nucleobase oligomers. Specifically, aqueous solutions of aegPNA 8 and thyclotide 10 often appeared cloudy after heating. It is well known that aegPNAs can aggregate in aqueous buffers, which is why such solutions are often heated prior to use in an assay (76). It is possible that the 20-nucleobase antimiR-21 sequence has a tendency to aggregate, which could complicate the interpretation of any inhibition data. Therefore, we decided to add one terminal lysine to each end of the 20-nucleobase antimiR-21 sequence (Figure 5A). Lysine side chains are protonated in aqueous buffer at pH 7, which should result in an increase in polarity and solubility of both aegPNAs and thyclotides. We synthesized 20-nucleobase antimiR-21 Lys-aegPNA 11, Lys-thyclotide 12, (Supplementary Figure S60) and its scrambled control Lys-thyclotide 13. All these molecules exhibited good solubility without cloudiness. Next, we performed an electrophoretic mobility shift assay of a miR-21-aegPNA and miR-21-thyclotide duplex with varying concentrations of aegPNA 11 and thyclotide 12 to determine whether the lysines would affect the binding to miR-21. After incubation of miR-21 with different concentrations of aegPNA 11 for 1 hour, we observed a shifted band, confirming the formation of a fully shifted aegPNA-miR-21 duplex at a [aegPNA]/[miR-21] ratio of 2:1. The shifted band is already visible at a ratio [aegPNA]/[miR-21] of 0.25, but the band corresponding to unbound miR-21 completely disappears at a ratio of 2 or higher (Figure 5B). Interestingly, the RNA duplex with thyclotide 12 is also visible at a [thyclotide]/[miR-21] ratio of 0.25, but the shifted band is much stronger than the one observed at a similar ratio with the aegPNA duplex (Figure 5C). In contrast to aegPNA, the free miR-21 RNA band completely disappeared at a ratio [thyclotide]/[miR-21] of 1. The fact that a complete band shift is clearly visible at a 1:1 ratio of [thyclotide]/[miR-21] also indicates that thyclotide 12 does not form aggregates.
Figure 5.

Thyclotide 12 binds miR-21 at a ratio 1:1. (A) Schematic of Lys-aegPNA and Lys-thyclotide structures. One terminal lysine was added on both ends of the molecules. (B) EMSA for the formation of miR-21-aegPNA 11 duplex. Lane 1, DNA marker; lane 2, FAM-hsa-miR-21–5p; lanes 3–8, miR-21-aegPNA 11 duplex with increasing concentrations of aegPNA 11. (C) EMSA for the formation of miR-21-thyclotide 12 duplex. Lane 1, DNA marker; lane 2, FAM-hsa-miR-21-5p; lanes 3–7, miR-21-thyclotide 12 duplex with increasing concentrations of thyclotide. ‘PNA’ and ‘Thy’ in figure refer to antimiR-21 aegPNA 11 and antimiR-21 thyclotide 12, respectively.
To assess cellular uptake of the lysine-modified thyclotides, we then synthesized fluorescein-labelled Lys-aegPNA 14 and Lys-thyclotide 15. At a concentration of 5 μM diluted in the cell culture medium, thyclotide 15 was successfully taken up by SKHEP1, HepG2, RKO and MCF7cells (Figure 6A). In all four cell lines, the uptake of thyclotide 15 is significantly higher than that of aegPNA 14 (P < 0.0001, P = 0.01, P < 0.0001, P < 0.0001, respectively; Figure 6B). Specifically, thyclotide 15 was taken up by 89.9% of SKHEP1 cells, 76.5% of HepG2 cells, 84.2% of RKO cells and 62.3% of MCF7 cells. In comparison, cells treated with aegPNA 14 always showed a significantly lower uptake: 17.5% of SKHEP1 cells, 16.4% of HepG2 cells, 7.2% of RKO cells and 22.4% of MCF7 cells. The MFI of aegPNA-treated cells was always significantly lower compared with the MFI of thyclotide 15-treated cells, especially in SKHEP1 (P < 0.0001) and HepG2 (P = 0.0059) cells, the latter showing the highest MFIThyclotide/MFIaegPNA ratio among all cell lines (HepG2 MFI ratio thyclotide/aegPNA of 50.5). The weak uptake of aegPNA 14 with one terminal lysine on both ends compared with thyclotide 15 shows that the two terminal lysines do not help aegPNA cross the cell membrane, and that the THF groups of the thyclotide are still important for cell uptake. The cellular uptake of thyclotide 15 in cells was confirmed by super resolution microscopy using SKHEP1 cells, which again showed green fluorescent signals in the middle plane of treated cells. Under the same conditions, cells treated with aegPNA 14 showed no detectable signal (Figure 6C).
Figure 6.

Thyclotide 15 is successfully taken up by several cell lines. (A) FACS of cells either non-treated (blue) or treated with aegPNA 14 (orange) or thyclotide 15 (red) conjugated to fluorescein. (B) Statistics of FACS in 4 cell lines. Positive cells were determined by having a fluorescence higher than the non-treated cells. Mean fluorescence intensity was determined with the FlowJo software v10. ‘aeg’ and ‘Thy’ in the figure refer to aegPNA 14 and thyclotide 15 respectively. (C) Representative picture of thyclotide 15 cell uptake by super-resolution microscopy compared with aegPNA 14 treatment in SKHEP1 cells. The picture represents an image obtained from the mid-plane of the cells.
Lysine-modified thyclotide inhibits miR-21 and affects its downstream targets PTEN, Cdc25a and KRIT1
MiRNA-21 is known to be overexpressed in many cancer cell lines, including in the hepatocellular cancer cell line HepG2 (77) which showed the strongest difference between the uptake of thyclotide 15 and aegPNA 14. To investigate the ability of Lys-thyclotide to inhibit miR-21, HepG2 cells were incubated with either antimiR-21 thyclotide 12 or the scrambled control thyclotide 13, at increasing concentrations. After total miRNA isolation from HepG2 cells, we performed RT-qPCR to analyze the apparent levels of miR-21 expression. Compared with control thyclotide 13, increasing concentrations of thyclotide 12 show a strong decrease in apparent miR-21 levels, even at nanomolar concentrations (Figure 7A). After confirming that thyclotide 12 is able to bind and act as a steric blocker of miR-21 in HepG2 cells, we also wanted to assess the impact on the miR-21 downstream target, PTEN. In this experiment, HepG2 cells were treated with control thyclotide 13 or thyclotide 12 for 48 hours and then total RNA was extracted. PTEN mRNA levels were quantified and compared. Consistent with the ability of thyclotide 12 to bind and reduce apparent miR-21 levels, the treatment of HepG2 cells with this thyclotide also induced a significant increase in PTEN mRNA levels compared with cells treated with control thyclotide 13 (P = 0.0158; Figure 7B). We decided to investigate if the increase in PTEN mRNA would translate in a similar increase in protein levels. We performed a western blot on HepG2 cells treated with either 25 nM of scrambled thyclotide 13 or 25 nM of antimiR-21 thyclotide 12 and we observed that the latter increases the levels of the PTEN protein compared with the scrambled control (Figure 7C). To confirm that the observed increase in PTEN mRNA levels was due to the specific interaction of thyclotide 12 with miR-21, HepG2 cells were transfected with a vector containing the luciferase reporter gene downstream of the PTEN-3′UTR (which contains the seed sequence for miR-21 binding) and the pRL-TK vector for Renilla luciferase as an internal control. Following transfection, cells were treated with either control thyclotide 13 or thyclotide 12 and luminescence was assessed 48 hours later. A specific binding of thyclotide to miR-21 prevents miRNA binding to PTEN-3′UTR, therefore inducing an increase in the observed luminescence signal from luciferase. Indeed, thyclotide 12 induced a significant increase in luminescence compared with control thyclotide 13 (P < 0.0001 for both concentrations; Figure 7D). At 25 nM concentration, thyclotide 12 increases luciferase luminescence by 37%, and by 76% at a concentration of 5 μM, confirming that the effect on the downstream miR-21 target PTEN was due to the specific interaction between thyclotide 12 and miR-21. To further confirm the activity of thyclotide 12 towards miR-21, we tested additional downstream targets of this miRNA, Cdc25a and KRIT1 (78–80). Similar to what was observed with PTEN, thyclotide 12 was able to induce an increase in Cdc25a and KRIT1 mRNA and protein levels (Supplementary Figure S61).
Figure 7.

Thyclotide 12 inhibits miR-21 and affects its downstream target PTEN. (A) RT-qPCR of miR-21. Cells were treated with increasing concentrations of scrambled thyclotide 13 or antimiR-21 thyclotide 12 and miR-21 levels were assessed and normalized to the average of miR-25 and miR-93 endogenous controls relative to the control scrambled expression. (B) RT-qPCR of PTEN mRNA. Cells were treated with 25 nM of scrambled thyclotide 13 or antimiR-21 thyclotide 12. Total RNA was isolated and PTEN mRNA levels were assessed and normalized to β-actin control expression. (C) Western Blot for detection of PTEN protein. Cells were treated with either 25 nM of scrambled thyclotide 13 or 25 nM of antimiR-21 thyclotide 12. Densitometry analysis was performed with the software ImageJ. In the densitometry graphic, ‘scr’ refers to scrambled thyclotide 13 and ‘antimiR’ to antimiR-21 thyclotide 12. (D) Dual luciferase reporter assay for PTEN. Cells were co-transfected with 1 μg pGL3-PTEN-3′UTR vector and 1 μg pRL-TK for Renilla luciferase internal control. Transfected cells were then treated with scrambled thyclotide 13 or antimiR-21 thyclotide 12 at low (25 nM) or high (5 μM) concentration.
Lysine-modified thyclotide shows minimal toxicity at active concentrations
To determine whether thyclotides exhibit non-specific toxicity, cells were incubated with increasing concentrations of control thyclotide 13 for 24 and 48 hours, and the release of lactate dehydrogenase was measured as an indicator of cell death. No cytotoxicity was observed in SKHEP1 and RKO cell lines at concentrations up to 25 μM. MCF7 cells did not show significant toxicity after 24 hours of treatment, but after 48 h of treatment at 20 and 25 μM there was a decrease in viability of about 10%. HepG2 cells showed no toxicity at concentrations up to 15 μM for 24 h, but after 48 hours of treatment, 10 and 15 μM resulted in a slight decrease in viability while higher concentrations of 20 and 25 μM induced more significant cell death with viability decreasing to 80% and 70%, respectively (Figure 8). Depending on the cell line, thyclotide-induced cytotoxicity can be observed at higher concentrations, yet the cytotoxic concentrations are ∼200 times higher than the active concentration in HepG2 cells (Figure 7). Even though the slight cytotoxicity observed in HepG2 and MCF7 cells suggests that thyclotide 13 can enter cells, we wanted to confirm its ability to enter a cell line that showed no loss of viability. To confirm that the control thyclotide 13 still enters cells, we synthesized a fluorescein-labelled version of the scrambled control sequence (Lys-thyclotide 16) and tested the cell uptake in SKHEP1 cells by FACS analysis. The results showed that thyclotide 16 was capable of entering cells, indicating that the scrambled sequence does not alter the ability of the thyclotide to enter cells (Supplementary Figure S62).
Figure 8.

Cytotoxicity assay of thyclotide in several cell lines. HepG2, SKHEP1, RKO and MCF7 cells were treated with increasing concentration of scrambled thyclotide 13, and cell viability was assessed by the measurement of LDH release at 24 and 48 hours after the treatment.
Thyclotide nucleobase sequence determines the efficiency of cellular uptake
Thyclotides designed to inhibit miR-21 enter the cytoplasm of different cell lines more efficiently than the corresponding aegPNAs with the same sequence. To examine whether thyclotides perform similarly with a different sequence and in a different biological context, we studied a thyclotide designed to interfere with the intron splicing of a pre-mRNA target. Using CPP-conjugated aegPNA or acridine-conjugated aegPNA in combination with transfection, Shiraishi et al. designed several PNAs that modulate the pre-mRNA splicing of MDM2 (81). For our study, we synthesized the thyclotide and aegPNA versions of one of the published sequences that was reported to affect the splicing of intron 2 of MDM2, and we also prepared a scrambled version of the thyclotide (Supplementary Table S1, Supplementary Figures S63–S72). Using FACS analysis, we compared the cell uptake of aegPNA 17 and thyclotide 18 (which are both fluorescein-labeled) in SKHEP1 and HepG2 cells. Interestingly, thyclotide 18 was unable to enter cells any better than aegPNA 17 in SKHEP1 cells, and only a minor increase in uptake was observed in HepG2 cells (Supplementary Figure S73A). Using the procedures reported by Shiraishi et al (81), we examined whether there was any effect of aegPNA 19 and thyclotides 20 and 21 on the splicing of the MDM2 intron in SKHEP1 and HepG2 cells. Consistent with the cell uptake results, no increase in the specific MDM2 splicing variant was observed following treatment with aegPNA or the thyclotides (Supplementary Figure S73B). To directly evaluate the differences in cell uptake between thyclotides of different sequences, thyclotide 18 (15 nucleobases) and antimiR-21 thyclotide 5 (14 nucleobases) were examined at the same time for cell uptake in SKHEP1 and HepG2 cells. Consistent with previous experiments, antimiR-21 thyclotide 5 displayed good cell uptake while thyclotide 18 did not show good cell uptake (Supplementary Figure S73C).
DISCUSSION
Since miRNA was discovered by Lee et al in 1993 (82), multiple studies have shown that miRNA impacts many areas of cell biology. Specifically, miRNA can play a role in disease development, tumor growth, cell proliferation, apoptosis (83) and even responses to anti-cancer treatments (84). Dysregulation of miRNA is linked to the development of specific cancer phenotypes, which makes miRNA an attractive therapeutic target (85). Antisense-based modulation of miRNA levels has been studied with the use of several tools, such as antagomiRs (15,86,87), and also aegPNAs (55,88,89). Despite their stability and high binding affinity to complementary DNA or RNA targets, aegPNAs are not readily taken into cells. In this study, we developed thyclotide molecules that have THF groups grafted onto the aegPNA backbone. The introduction of the THF groups seems to confer very promising cell uptake properties to an antimiR-21 thyclotide. The introduction of N- and C-terminal lysines on antimiR-21 thyclotides improved aqueous solubility without altering the ability of the thyclotide to enter cells. Clearly, the presence of the THF groups on the thyclotide improves the cellular uptake properties compared to aegPNA. It has been shown that several lysines conjugated to the ends of an aegPNA act like CPPs to promote cell uptake of the PNA and therefore efficient miRNA inhibition. Fabani et al. and Torres et al. used PNAs conjugated to one N-terminal lysine residue and three C-terminal lysine residues to inhibit miR-122 and miR-155 (57,58). It is important to note that these PNAs also contained an N-terminal cysteine, and they seemed to be captured by endosomes. In this regard, we have shown that a single lysine attached to each end of an aegPNA cannot overcome the inability of the aegPNA to enter cells but still improve its solubility. It is remarkable that in our case, the addition of THF groups into the backbone of aegPNA promotes efficient delivery of a thyclotide targeting miR-21 into cells, with only two terminal lysines residues, and without the need to use any transfection reagents, CPPs, or nanoparticles.
There are currently many approaches to deliver aegPNAs into cells, and one of the most successful methods is to conjugate CPPs with aegPNA. However, CPPs have limitations. While the inherent toxicity of amphipathic CPPs can be moderated by using arginine-rich CPP sequences to deliver cargo (such as aegPNA) into cells, dose-dependent cytotoxicity can still be problematic. The delivery efficacy and cytotoxicity of CPP-conjugates can also vary considerably depending on the type and size of the cargo molecule attached to the CPP (68,69). Furthermore, CPP conjugates enter cells via endocytosis that traps the molecule in an endosome. The molecule must subsequently escape the endosome to exert a biological effect inside a cell. Overcoming endosome entrapment can be challenging for CPP-PNA conjugates, and the use of endosome-disruption reagents (90) or the conjugation of endosomolytic-inducing chemical modifications (70,74,91) may be needed to enhance the biological activity of a CPP-PNA conjugate.
The thyclotide we designed to target miR-21 presented in this manuscript seems to overcome many of the limitations of CPP-PNA conjugates. Without using CPPs or any other type of reagent to promote cell uptake, we were able to show that an antimiR-21 thyclotide enters several different types of cells, bind miR-21 in HepG2 cells, and block the binding of this miRNA to PTEN-3′-UTR at nanomolar concentrations. The colocalization of fluorescein-labelled thyclotide 5 with endosomes was not observed by super-resolution microscopy, suggesting that thyclotides do not enter cells by endocytosis. It is possible that cellular uptake would vary depending on the properties of the cell membrane it crosses. Indeed, differences in uptake were observed among the 4 cell lines studied. Even though the cellular uptake of the antimiR-21 thyclotide was always superior compared to aegPNA, thyclotide uptake by MCF7 cells was noticeably lower compared to the other cell lines. It is known that cellular membrane composition can vary depending on the growth phase, which has been shown in HepG2 cells (92), and that cholesterol levels in cell membrane vary between cell types (93), which affects the local rigidity of membranes (94). The nucleobase sequence of the thyclotide also determines the degree of cell uptake. Thyclotides targeting miR-21 successfully enter several cell lines compared to the corresponding aegPNA, but a thyclotide aimed at interfering with the splicing of MDM2 pre-mRNA did not show any difference in cell uptake relative to the aegPNA of same sequence. A brief analysis of the thyclotide sequences does not immediately indicate why one sequence would have better cellular uptake than the other. The percentage of GC bases in the antimiR-21 thyclotide sequences is around 35% while the GC content of the thyclotide sequence to alter the splicing of MDM2 is 47%. At the present time, we speculate that a higher GC content in thyclotides may alter the physical properties of the molecule to inhibit cellular uptake. Future work will focus on understanding the links between thyclotide sequence, membrane composition and its cellular uptake.
DATA AVAILABILITY
All flow cytometry datasets are available in FlowRepository (https://flowrepository.org/). Repository IDs: FR-FCM-Z575 and FR-FCM-Z5XV.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Sharlyn J. Mazur and George Leiman for editing the manuscript.
Contributor Information
Victor Clausse, Synthetic Bioactive Molecules Section, Laboratory of Bioorganic Chemistry, NIDDK, National Institutes of Health, Bethesda, MD 20892, USA.
Hongchao Zheng, Synthetic Bioactive Molecules Section, Laboratory of Bioorganic Chemistry, NIDDK, National Institutes of Health, Bethesda, MD 20892, USA.
Harsha Amarasekara, Synthetic Bioactive Molecules Section, Laboratory of Bioorganic Chemistry, NIDDK, National Institutes of Health, Bethesda, MD 20892, USA.
Michael Kruhlak, Microscopy Core Facility, Center for Cancer Research, NCI, National Institutes of Health, Bethesda, MD 20892, USA.
Daniel H Appella, Synthetic Bioactive Molecules Section, Laboratory of Bioorganic Chemistry, NIDDK, National Institutes of Health, Bethesda, MD 20892, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health [DK031143-15]. Funding for open access charge: National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health.
Conflict of interest statement. Thyclotides are patented (https://patents.google.com/patent/WO2021211786A1/en). Inventors: Daniel H. Appella, Hongchao Zheng, Harsha C. Amarasekara, Victor Clausse, George A. Kubi.
REFERENCES
- 1. Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., Bray F.. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021; 71:209–249. [DOI] [PubMed] [Google Scholar]
- 2. Ji W., Sun B., Su C.. Targeting micrornas in cancer gene therapy. Genes (Basel). 2017; 8:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ling H., Fabbri M., Calin G.A.. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat. Rev. Drug Discov. 2013; 12:847–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Svoronos A.A., Engelman D.M., Slack F.J.. OncomiR or tumor suppressor? The duplicity of micrornas in cancer. Cancer Res. 2016; 76:3666–3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Garzon R., Marcucci G., Croce C.M.. Targeting microRNAs in cancer: rationale, strategies and challenges. Nat. Rev. Drug Discov. 2010; 9:775–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zealy R.W., Wrenn S.P., Davila S., Min K.W., Yoon J.H.. microRNA-binding proteins: specificity and function. Wiley Interdiscip. Rev. RNA. 2017; 8:e1414. [DOI] [PubMed] [Google Scholar]
- 7. Ludwig N., Leidinger P., Becker K., Backes C., Fehlmann T., Pallasch C., Rheinheimer S., Meder B., Stahler C., Meese E.et al.. Distribution of miRNA expression across human tissues. Nucleic Acids Res. 2016; 44:3865–3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huang Y., Shen X.J., Zou Q., Wang S.P., Tang S.M., Zhang G.Z.. Biological functions of microRNAs: a review. J. Physiol. Biochem. 2011; 67:129–139. [DOI] [PubMed] [Google Scholar]
- 9. Rupaimoole R., Slack F.J.. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017; 16:203–222. [DOI] [PubMed] [Google Scholar]
- 10. Wang J., Chen J.Y., Sen S.. MicroRNA as biomarkers and diagnostics. J. Cell. Physiol. 2016; 231:25–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hanna J., Hossain G.S., Kocerha J.. The potential for microRNA therapeutics and clinical research. Front Genet. 2019; 10:478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Allegra A., Ettari R., Innao V., Bitto A.. Potential role of microRNAs in inducing drug resistance in patients with multiple myeloma. Cells. 2021; 10:448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Boutla A., Delidakis C., Tabler M.. Developmental defects by antisense-mediated inactivation of micro-RNAs 2 and 13 in drosophila and the identification of putative target genes. Nucleic Acids Res. 2003; 31:4973–4980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Esau C.C. Inhibition of microRNA with antisense oligonucleotides. Methods. 2008; 44:55–60. [DOI] [PubMed] [Google Scholar]
- 15. Krutzfeldt J., Rajewsky N., Braich R., Rajeev K.G., Tuschl T., Manoharan M., Stoffel M.. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005; 438:685–689. [DOI] [PubMed] [Google Scholar]
- 16. Nguyen D.D., Chang S.. Development of novel therapeutic agents by inhibition of oncogenic MicroRNAs. Int. J. Mol. Sci. 2017; 19:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li Z., Rana T.M.. Therapeutic targeting of microRNAs: current status and future challenges. Nat. Rev. Drug Discov. 2014; 13:622–638. [DOI] [PubMed] [Google Scholar]
- 18. Di Martino M.T., Gulla A., Gallo Cantafio M.E., Altomare E., Amodio N., Leone E., Morelli E., Lio S.G., Caracciolo D., Rossi M.et al.. In vitro and in vivo activity of a novel locked nucleic acid (LNA)-inhibitor-miR-221 against multiple myeloma cells. PLoS One. 2014; 9:e89659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Javanmard S.H., Vaseghi G., Ghasemi A., Rafiee L., Ferns G.A., Esfahani H.N., Nedaeinia R.. Therapeutic inhibition of microRNA-21 (miR-21) using locked-nucleic acid (LNA)-anti-miR and its effects on the biological behaviors of melanoma cancer cells in preclinical studies. Cancer Cell Int. 2020; 20:384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Obad S., Santos C.O., Petri A., Heidenblad M., Broom O., Ruse C., Fu C., Lindow M., Stenvang J., Straarup E.M.et al.. Silencing of microRNA families by seed-targeting tiny LNAs. Nat. Genet. 2011; 43:371–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Campbell A., Morris G., Heller J.P., Langa E., Brindley E., Worm J., Jensen M.A., Miller M.T., Henshall D.C., Reschke C.R.. Antagomir-mediated suppression of microRNA-134 reduces kainic acid-induced seizures in immature mice. Sci. Rep. 2021; 11:340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Haftmann C., Riedel R., Porstner M., Wittmann J., Chang H.D., Radbruch A., Mashreghi M.F.. Direct uptake of Antagomirs and efficient knockdown of miRNA in primary B and T lymphocytes. J. Immunol. Methods. 2015; 426:128–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maschmeyer P., Petkau G., Siracusa F., Zimmermann J., Zugel F., Kuhl A.A., Lehmann K., Schimmelpfennig S., Weber M., Haftmann C.et al.. Selective targeting of pro-inflammatory Th1 cells by microRNA-148a-specific antagomirs in vivo. J. Autoimmun. 2018; 89:41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Velu C.S., Grimes H.L.. Utilizing antagomiR (antisense microRNA) to knock down microRNA in murine bone marrow cells. Methods Mol. Biol. 2012; 928:185–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Corey D.R., Abrams J.M.. Morpholino antisense oligonucleotides: tools for investigating vertebrate development. Genome Biol. 2001; 2:REVIEWS1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nan Y., Zhang Y.J.. Antisense phosphorodiamidate morpholino oligomers as novel antiviral compounds. Front Microbiol. 2018; 9:750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Summerton J., Weller D. Morpholino antisense oligomers: design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 1997; 7:187–195. [DOI] [PubMed] [Google Scholar]
- 28. Summerton J.E. Peptide Nucleic Acids, Morpholinos and Related Antisense Biomolecules. 2006; Boston, MA: Springer US; 89–113. [Google Scholar]
- 29. Adusumilli L., Facchinello N., Teh C., Busolin G., Le M.T., Yang H., Beffagna G., Campanaro S., Tam W.L., Argenton F.et al.. miR-7 controls the dopaminergic/oligodendroglial fate through Wnt/beta-catenin signaling regulation. Cells. 2020; 9:711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Falcao M.A.P., Walker C.I.B., Disner G.R., Batista J., Soares A.B.S., Balan-Lima L., Lima C., Lopes-Ferreira M.. Knockdown of miR-26a in zebrafish leads to impairment of the anti-inflammatory function of TnP in the control of neutrophilia. Fish Shellfish Immun. 2021; 114:301–310. [DOI] [PubMed] [Google Scholar]
- 31. Huang Y., Huang C.X., Wang W.F., Liu H., Wang H.L.. Zebrafish miR-462-731 is required for digestive organ development. Comp. Biochem. Physiol. Part D Genomics Proteomics. 2020; 34:100679. [DOI] [PubMed] [Google Scholar]
- 32. Li X., Gao Y., Tian F., Du R., Yuan Y., Li P., Liu F., Wang C.. miR-31 promotes neural stem cell proliferation and restores motor function after spinal cord injury. Exp. Biol. Med. (Maywood). 2021; 246:1274–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nielsen P.E., Egholm M., Buchardt O.. Peptide nucleic acid (PNA). A DNA mimic with a peptide backbone. Bioconjug Chem. 1994; 5:3–7. [DOI] [PubMed] [Google Scholar]
- 34. Betts L., Josey J.A., Veal J.M., Jordan S.R.. A nucleic acid triple helix formed by a peptide nucleic acid-DNA complex. Science. 1995; 270:1838–1841. [DOI] [PubMed] [Google Scholar]
- 35. Leijon M., Graslund A., Nielsen P.E., Buchardt O., Norden B., Kristensen S.M., Eriksson M.. Structural characterization of PNA-DNA duplexes by NMR. Evidence for DNA in a B-like conformation. Biochemistry. 1994; 33:9820–9825. [DOI] [PubMed] [Google Scholar]
- 36. Gupta A., Mishra A., Puri N.. Peptide nucleic acids: advanced tools for biomedical applications. J. Biotechnol. 2017; 259:148–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gupta A., Bahal R., Gupta M., Glazer P.M., Saltzman W.M.. Nanotechnology for delivery of peptide nucleic acids (PNAs). J. Control Release. 2016; 240:302–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hanvey J.C., Peffer N.J., Bisi J.E., Thomson S.A., Cadilla R., Josey J.A., Ricca D.J., Hassman C.F., Bonham M.A., Au K.G.et al.. Antisense and antigene properties of peptide nucleic acids. Science. 1992; 258:1481–1485. [DOI] [PubMed] [Google Scholar]
- 39. Brolin C., Lim E.W.K., Grizot S., Olsen C.H., Yavari N., Krag T.O., Nielsen P.E.. Approaches for systemic delivery of dystrophin antisense peptide nucleic acid in the mdx mouse model. Nucleic Acid Ther. 2021; 31:208–219. [DOI] [PubMed] [Google Scholar]
- 40. Yavari N., Goltermann L., Nielsen P.E.. Uptake, stability, and activity of antisense Anti-acpP PNA-Peptide conjugates in Escherichia coli and the role of SbmA. ACS Chem. Biol. 2021; 16:471–479. [DOI] [PubMed] [Google Scholar]
- 41. Hu J., Corey D.R.. Inhibiting gene expression with peptide nucleic acid (PNA)–peptide conjugates that target chromosomal DNA. Biochemistry. 2007; 46:7581–7589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wancewicz E.V., Maier M.A., Siwkowski A.M., Albertshofer K., Winger T.M., Berdeja A., Gaus H., Vickers T.A., Bennett C.F., Monia B.P.et al.. Peptide nucleic acids conjugated to short basic peptides show improved pharmacokinetics and antisense activity in adipose tissue. J. Med. Chem. 2010; 53:3919–3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hu J., Dodd D.W., Hudson R.H., Corey D.R.. Cellular localization and allele-selective inhibition of mutant huntingtin protein by peptide nucleic acid oligomers containing the fluorescent nucleobase (bis-o-(aminoethoxy)phenyl)pyrrolocytosine. Bioorg. Med. Chem. Lett. 2009; 19:6181–6184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Avitabile C., Accardo A., Ringhieri P., Morelli G., Saviano M., Montagner G., Fabbri E., Gallerani E., Gambari R., Romanelli A.. Incorporation of naked peptide nucleic acids into liposomes leads to fast and efficient delivery. Bioconjugate Chem. 2015; 26:1533–1541. [DOI] [PubMed] [Google Scholar]
- 45. Bae Y.M., Kim M.H., Yu G.S., Um B.H., Park H.K., Lee H.I., Lee K.T., Suh Y.D., Choi J.S.. Enhanced splicing correction effect by an oligo-aspartic acid-PNA conjugate and cationic carrier complexes. J. Control Release. 2014; 175:54–62. [DOI] [PubMed] [Google Scholar]
- 46. Llovera L., Berthold P., Nielsen P.E., Shiraishi T.. Cell number and transfection volume dependent peptide nucleic acid antisense activity by cationic delivery methods. Artif. DNA PNA XNA. 2012; 3:22–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McNeer N.A., Anandalingam K., Fields R.J., Caputo C., Kopic S., Gupta A., Quijano E., Polikoff L., Kong Y., Bahal R.et al.. Nanoparticles that deliver triplex-forming peptide nucleic acid molecules correct F508del CFTR in airway epithelium. Nat. Commun. 2015; 6:6952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shen G., Fang H., Song Y., Bielska A.A., Wang Z., Taylor J.S.. Phospholipid conjugate for intracellular delivery of peptide nucleic acids. Bioconjug. Chem. 2009; 20:1729–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shiraishi T., Nielsen P.E.. Peptide nucleic acid (PNA) cell penetrating peptide (CPP) conjugates as carriers for cellular delivery of antisense oligomers. Artif DNA PNA XNA. 2011; 2:90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bendifallah N., Rasmussen F.W., Zachar V., Ebbesen P., Nielsen P.E., Koppelhus U.. Evaluation of cell-penetrating peptides (CPPs) as vehicles for intracellular delivery of antisense peptide nucleic acid (PNA). Bioconjug. Chem. 2006; 17:750–758. [DOI] [PubMed] [Google Scholar]
- 51. Ghavami M., Shiraishi T., Nielsen P.E.. Cooperative cellular uptake and activity of octaarginine antisense peptide nucleic acid (PNA) conjugates. Biomolecules. 2019; 9:554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Montazersaheb S., Hejazi M.S., Nozad Charoudeh H.. Potential of peptide nucleic acids in future therapeutic applications. Adv Pharm Bull. 2018; 8:551–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Brognara E., Fabbri E., Aimi F., Manicardi A., Bianchi N., Finotti A., Breveglieri G., Borgatti M., Corradini R., Marchelli R.et al.. Peptide nucleic acids targeting miR-221 modulate p27Kip1 expression in breast cancer MDA-MB-231 cells. Int. J. Oncol. 2012; 41:2119–2127. [DOI] [PubMed] [Google Scholar]
- 54. Brognara E., Fabbri E., Bazzoli E., Montagner G., Ghimenton C., Eccher A., Cantu C., Manicardi A., Bianchi N., Finotti A.et al.. Uptake by human glioma cell lines and biological effects of a peptide-nucleic acids targeting miR-221. J. Neurooncol. 2014; 118:19–28. [DOI] [PubMed] [Google Scholar]
- 55. Fabbri E., Tamanini A., Jakova T., Gasparello J., Manicardi A., Corradini R., Sabbioni G., Finotti A., Borgatti M., Lampronti I.et al.. A peptide nucleic acid against MicroRNA miR-145-5p enhances the expression of the cystic fibrosis transmembrane conductance regulator (CFTR) in Calu-3 cells. Molecules. 2017; 23:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fabani M.M., Gait M.J.. miR-122 targeting with LNA/2′-O-methyl oligonucleotide mixmers, peptide nucleic acids (PNA), and PNA-peptide conjugates. RNA. 2008; 14:336–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Torres A.G., Fabani M.M., Vigorito E., Williams D., Al-Obaidi N., Wojciechowski F., Hudson R.H., Seitz O., Gait M.J.. Chemical structure requirements and cellular targeting of microRNA-122 by peptide nucleic acids anti-miRs. Nucleic Acids Res. 2012; 40:2152–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fabani M.M., Abreu-Goodger C., Williams D., Lyons P.A., Torres A.G., Smith K.G., Enright A.J., Gait M.J., Vigorito E.. Efficient inhibition of miR-155 function in vivo by peptide nucleic acids. Nucleic Acids Res. 2010; 38:4466–4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hamilton S.E., Simmons C.G., Kathiriya I.S., Corey D.R.. Cellular delivery of peptide nucleic acids and inhibition of human telomerase. Chem. Biol. 1999; 6:343–351. [DOI] [PubMed] [Google Scholar]
- 60. Shiraishi T., Hamzavi R., Nielsen P.E.. Subnanomolar antisense activity of phosphonate-peptide nucleic acid (PNA) conjugates delivered by cationic lipids to HeLa cells. Nucleic Acids Res. 2008; 36:4424–4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shiraishi T., Nielsen P.E.. Nanomolar cellular antisense activity of peptide nucleic acid (PNA) cholic acid (“umbrella”) and cholesterol conjugates delivered by cationic lipids. Bioconjug Chem. 2012; 23:196–202. [DOI] [PubMed] [Google Scholar]
- 62. Ma X., Devi G., Qu Q., Toh D.F., Chen G., Zhao Y.. Intracellular delivery of antisense peptide nucleic acid by fluorescent mesoporous silica nanoparticles. Bioconjug Chem. 2014; 25:1412–1420. [DOI] [PubMed] [Google Scholar]
- 63. Babar I.A., Cheng C.J., Booth C.J., Liang X., Weidhaas J.B., Saltzman W.M., Slack F.J.. Nanoparticle-based therapy in an in vivo microRNA-155 (miR-155)-dependent mouse model of lymphoma. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:E1695–E1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bhingardeve P., Madhanagopal B.R., Naick H., Jain P., Manoharan M., Ganesh K.. Receptor-Specific delivery of peptide nucleic acids conjugated to three sequentially linked N-Acetyl galactosamine moieties into hepatocytes. J. Org. Chem. 2020; 85:8812–8824. [DOI] [PubMed] [Google Scholar]
- 65. Macadangdang B., Zhang N., Lund P.E., Marple A.H., Okabe M., Gottesman M.M., Appella D.H., Kimchi-Sarfaty C.. Inhibition of multidrug resistance by SV40 pseudovirion delivery of an antigene peptide nucleic acid (PNA) in cultured cells. PLoS One. 2011; 6:e17981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gasparello J., Manicardi A., Casnati A., Corradini R., Gambari R., Finotti A., Sansone F.. Efficient cell penetration and delivery of peptide nucleic acids by an argininocalix(4)arene. Sci. Rep. 2019; 9:3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Manicardi A., Fabbri E., Tedeschi T., Sforza S., Bianchi N., Brognara E., Gambari R., Marchelli R., Corradini R.. Cellular uptakes, biostabilities and anti-miR-210 activities of chiral arginine-PNAs in leukaemic K562 cells. ChemBioChem. 2012; 13:1327–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Cardozo A.K., Buchillier V., Mathieu M., Chen J., Ortis F., Ladriere L., Allaman-Pillet N., Poirot O., Kellenberger S., Beckmann J.S.et al.. Cell-permeable peptides induce dose- and length-dependent cytotoxic effects. Biochim. Biophys. Acta. 2007; 1768:2222–2234. [DOI] [PubMed] [Google Scholar]
- 69. El-Andaloussi S., Jarver P., Johansson H.J., Langel U.. Cargo-dependent cytotoxicity and delivery efficacy of cell-penetrating peptides: a comparative study. Biochem. J. 2007; 407:285–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. LeCher J.C., Nowak S.J., McMurry J.L.. Breaking in and busting out: cell-penetrating peptides and the endosomal escape problem. Biomol. Concepts. 2017; 8:131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zheng H., Botos I., Clausse V., Nikolayevskiy H., Rastede E.E., Fouz M.F., Mazur S.J., Appella D.H.. Conformational constraints of cyclopentane peptide nucleic acids facilitate tunable binding to DNA. Nucleic Acids Res. 2021; 49:713–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Das M.K., Andreassen R., Haugen T.B., Furu K.. Identification of endogenous controls for use in miRNA quantification in human cancer cell lines. Cancer Genomics Proteomics. 2016; 13:63–68. [PubMed] [Google Scholar]
- 73. Bustin S.A., Benes V., Garson J.A., Hellemans J., Huggett J., Kubista M., Mueller R., Nolan T., Pfaffl M.W., Shipley G.L.et al.. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009; 55:611–622. [DOI] [PubMed] [Google Scholar]
- 74. Zhao X.L., Chen B.C., Han J.C., Wei L., Pan X.B.. Delivery of cell-penetrating peptide-peptide nucleic acid conjugates by assembly on an oligonucleotide scaffold. Sci. Rep. 2015; 5:17640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Feng Y.H., Tsao C.J.. Emerging role of microRNA-21 in cancer. Biomed Rep. 2016; 5:395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Braasch D.A., Nulf C.J., Corey D.R.. Synthesis and purification of peptide nucleic acids. Curr. Protoc. Nucleic Acid Chem. 2002; Chapter 4:Unit 4.11. [DOI] [PubMed] [Google Scholar]
- 77. Meng F., Henson R., Wehbe-Janek H., Ghoshal K., Jacob S.T., Patel T.. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007; 133:647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. He Q., Ye A., Ye W., Liao X., Qin G., Xu Y., Yin Y., Luo H., Yi M., Xian L.et al.. Cancer-secreted exosomal miR-21-5p induces angiogenesis and vascular permeability by targeting KRIT1. Cell Death. Dis. 2021; 12:576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Orso F., Balzac F., Marino M., Lembo A., Retta S.F., Taverna D. miR-21 coordinates tumor growth and modulates KRIT1 levels. Biochem. Biophys. Res. Commun. 2013; 438:90–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wang P., Zou F., Zhang X., Li H., Dulak A., Tomko R.J. Jr, Lazo J.S., Wang Z., Zhang L., Yu J. microRNA-21 negatively regulates Cdc25A and cell cycle progression in colon cancer cells. Cancer Res. 2009; 69:8157–8165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Shiraishi T., Eysturskarth J., Nielsen P.E.. Modulation of mdm2 pre-mRNA splicing by 9-aminoacridine-PNA (peptide nucleic acid) conjugates targeting intron-exon junctions. BMC Cancer. 2010; 10:342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lee R.C., Feinbaum R.L., Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993; 75:843–854. [DOI] [PubMed] [Google Scholar]
- 83. Tan W., Liu B., Qu S., Liang G., Luo W., Gong C.. MicroRNAs and cancer: key paradigms in molecular therapy. Oncol. Lett. 2018; 15:2735–2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Si W., Shen J., Zheng H., Fan W.. The role and mechanisms of action of microRNAs in cancer drug resistance. Clin. Epigenetics. 2019; 11:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Shah V., Shah J.. Recent trends in targeting miRNAs for cancer therapy. J. Pharm. Pharmacol. 2020; 72:1732–1749. [DOI] [PubMed] [Google Scholar]
- 86. Cerro-Herreros E., Gonzalez-Martinez I., Moreno-Cervera N., Overby S., Perez-Alonso M., Llamusi B., Artero R.. Therapeutic potential of Antagomir-23b for treating myotonic dystrophy. Mol. Ther. Nucleic Acids. 2020; 21:837–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Cerro-Herreros E., Sabater-Arcis M., Fernandez-Costa J.M., Moreno N., Perez-Alonso M., Llamusi B., Artero R.. miR-23b and miR-218 silencing increase Muscleblind-like expression and alleviate myotonic dystrophy phenotypes in mammalian models. Nat. Commun. 2018; 9:2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Gambari R., Gasparello J., Fabbri E., Borgatti M., Tamanini A., Finotti A.. Peptide nucleic acids for MicroRNA targeting. Methods Mol. Biol. 2020; 2105:199–215. [DOI] [PubMed] [Google Scholar]
- 89. Oh S.Y., Ju Y., Kim S., Park H.. PNA-based antisense oligonucleotides for micrornas inhibition in the absence of a transfection reagent. Oligonucleotides. 2010; 20:225–230. [DOI] [PubMed] [Google Scholar]
- 90. Shiraishi T., Ghavami M., Nielsen P.E.. In vitro cellular delivery of peptide nucleic acid (PNA). Methods Mol. Biol. 2020; 2105:173–185. [DOI] [PubMed] [Google Scholar]
- 91. Shiraishi T., Nielsen P.E.. Enhanced delivery of cell-penetrating peptide-peptide nucleic acid conjugates by endosomal disruption. Nat. Protoc. 2006; 1:633–636. [DOI] [PubMed] [Google Scholar]
- 92. Tsuji T., Morita S.Y., Nakamura Y., Ikeda Y., Kambe T., Terada T.. Alterations in cellular and organellar phospholipid compositions of HepG2 cells during cell growth. Sci. Rep. 2021; 11:2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Casares D., Escriba P.V., Rossello C.A.. Membrane lipid composition: effect on membrane and organelle structure, function and compartmentalization and therapeutic avenues. Int. J. Mol. Sci. 2019; 20:2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Chakraborty S., Doktorova M., Molugu T.R., Heberle F.A., Scott H.L., Dzikovski B., Nagao M., Stingaciu L.R., Standaert R.F., Barrera F.N.et al.. How cholesterol stiffens unsaturated lipid membranes. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:21896–21905. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All flow cytometry datasets are available in FlowRepository (https://flowrepository.org/). Repository IDs: FR-FCM-Z575 and FR-FCM-Z5XV.