

Abstract
Eukaryotic DNA mismatch repair (MMR) initiates through mispair recognition by the MutS homologs Msh2-Msh6 and Msh2-Msh3 and subsequent recruitment of the MutL homologs Mlh1-Pms1 (human MLH1-PMS2). In bacteria, MutL is recruited by interactions with the connector domain of one MutS subunit and the ATPase and core domains of the other MutS subunit. Analysis of the S. cerevisiae and human homologs have only identified an interaction between the Msh2 connector domain and Mlh1. Here we investigated whether a conserved Msh6 ATPase/core domain-Mlh1 interaction and an Msh2-Msh6 interaction with Pms1 also act in MMR. Mutations in MLH1 affecting interactions with both the Msh2 and Msh6 interfaces caused MMR defects, whereas equivalent pms1 mutations did not cause MMR defects. Mutant Mlh1-Pms1 complexes containing Mlh1 amino acid substitutions were defective for recruitment to mispaired DNA by Msh2-Msh6, did not support MMR in reconstituted Mlh1-Pms1-dependent MMR reactions in vitro, but were proficient in Msh2-Msh6-independent Mlh1-Pms1 endonuclease activity. These results indicate that Mlh1, the common subunit of the Mlh1-Pms1, Mlh1-Mlh2, and Mlh1-Mlh3 complexes, but not Pms1, is recruited by Msh2-Msh6 through interactions with both of its subunits.
Keywords: DNA repair, Msh2-Msh6, Mlh1-Pms1, Mispaired base, Mutagenesis, Protein-protein interactions
1. Introduction
DNA mismatch repair (MMR) prevents the accumulation of mutations by repairing base-base and insertion/deletion mispairs arising mainly through DNA replication errors [1–3]. Defects in MMR in humans are linked to the inherited cancer disposition syndromes Constitutional Mismatch Repair Deficiency and Lynch Syndrome and are also found in many sporadic cancers [4–7]. MMR functions through mispair recognition by MutS homologs, recruitment of MutL homologs, subsequent excision or displacement of the newly synthesized DNA strand eliminating the mispair, and resynthesis by a DNA polymerase [1–3].
The subunits in the complexes formed by MutS and MutL homologs have asymmetric roles in the eukaryotic heterodimers (S. cerevisiae Msh2-Msh6, Msh2-Msh3, Mlh1-Pms1, Mlh1-Mlh2, Mlh1-Mlh3; human MSH2-MSH6, MSH2-MSH3, MLH1-PMS2, MLH1-PMS1, MLH1-MLH3) and even in the bacterial homodimers (MutS-MutS and MutL-MutL). During mispair binding by MutS homologs, only one subunit (a bacterial MutS subunit or the eukaryotic Msh6 and Msh3 subunits of the Msh2-Msh6 and Msh2-Msh3 complexes) recognizes the mispair [8–11]. The endonuclease-proficient MutL homologs are also functionally asymmetric. In the Mlh1-Pms1 complex, the Pms1 subunit (human PMS2) contains most of the endonuclease active site residues and only the Pms1 subunit interacts with PCNA [12–17]. Similarly, in endonuclease-proficient homodimeric bacterial MutL homologs, only one of the two symmetric active sites is predicted to be positioned to mediate DNA cleavage, and this active site needs to be in the subunit that interacts with the bacterial β-clamp [14,18–20].
MutL homolog recruitment by MutS homologs also appears to be mediated by asymmetry that is functional (bacteria) or dictated by different gene products (eukaryotes). The E. coli MutS-MutL structure revealed that MutL interacts with both subunits in the MutS dimer (Fig. 1A,B; Table 1; [21,22]). The two interactions are between the N-terminus of MutL with the connector domain of the first MutS subunit and with the ATPase and core domains of the second MutS subunit. Mutagenesis of E. coli MutL reveals that both interfaces are important for recruitment [21], despite the fact that only the connector-domain interaction was identified by deuterium-exchange mass spectrometry with E. coli MutS and MutL (Fig. 1C; [23]). Mutagenesis of the connector domains in both S. cerevisiae Msh2 and Msh6 revealed that only the Msh2 subunit makes this interaction with Mlh1-Pms1 [23]. This suggests that Msh2-Msh6 can only load a single Mlh1-Pms1 complex at a time. Loading of a single MutL homodimer is also likely true for E. coli MutS homodimers based on single molecule analyses [24], despite the symmetry of the E. coli MutS-MutL recruitment structure, which was trapped by protein crosslinking [21,22]. Similarly, pulldown experiments from nuclear extracts using overexpressed wild-type and mutant human MLH1 proteins have indicated that MLH1 is primarily responsible for MLH1-PMS2 recruitment by MSH2-MSH6 [25,26], and hence MLH1 is likely the subunit equivalent to the crosslinked MutL subunit in the MutS-MutL structure. Despite the fact that mutagenesis studies in E. coli have established the importance of both MutS-MutL interfaces (Fig. 1D), mutagenesis of human MLH1 only probed the MSH2 connector domain interface (Fig. 1E, [26]). Moreover, residual binding observed in pulldowns of mutant MLH1-PMS2 complexes containing amino acid substitutions in MLH1 was attributed to weak interactions with the PMS2 subunit. Here we have probed the recruitment of Mlh1-Pms1 by Msh2-Msh6 in S. cerevisiae using genetic analysis in vivo, by evaluating the functionality of mutant Mlh1-Pms1 complexes in Msh2-Msh6 dependent reconstituted MMR reactions in vitro and Msh2-Msh6 independent Mlh1-Pms1 endonuclease assays, and by evaluating recruitment of mutant Mlh1-Pms1 complexes to mispaired DNA by Msh2-Msh6 to address several outstanding questions. First, can an Msh2-Pms1 interaction equivalent to the established Msh2-connector domain-Mlh1 interaction contribute to MMR in vivo? Second, does recruitment of Mlh1-Pms1 by Msh2-Msh6 solely rely upon the Msh2 connector domain-Mlh1 interaction or does an Msh6 ATPase/core domain-Mlh1 interaction also play a role?
Fig. 1.
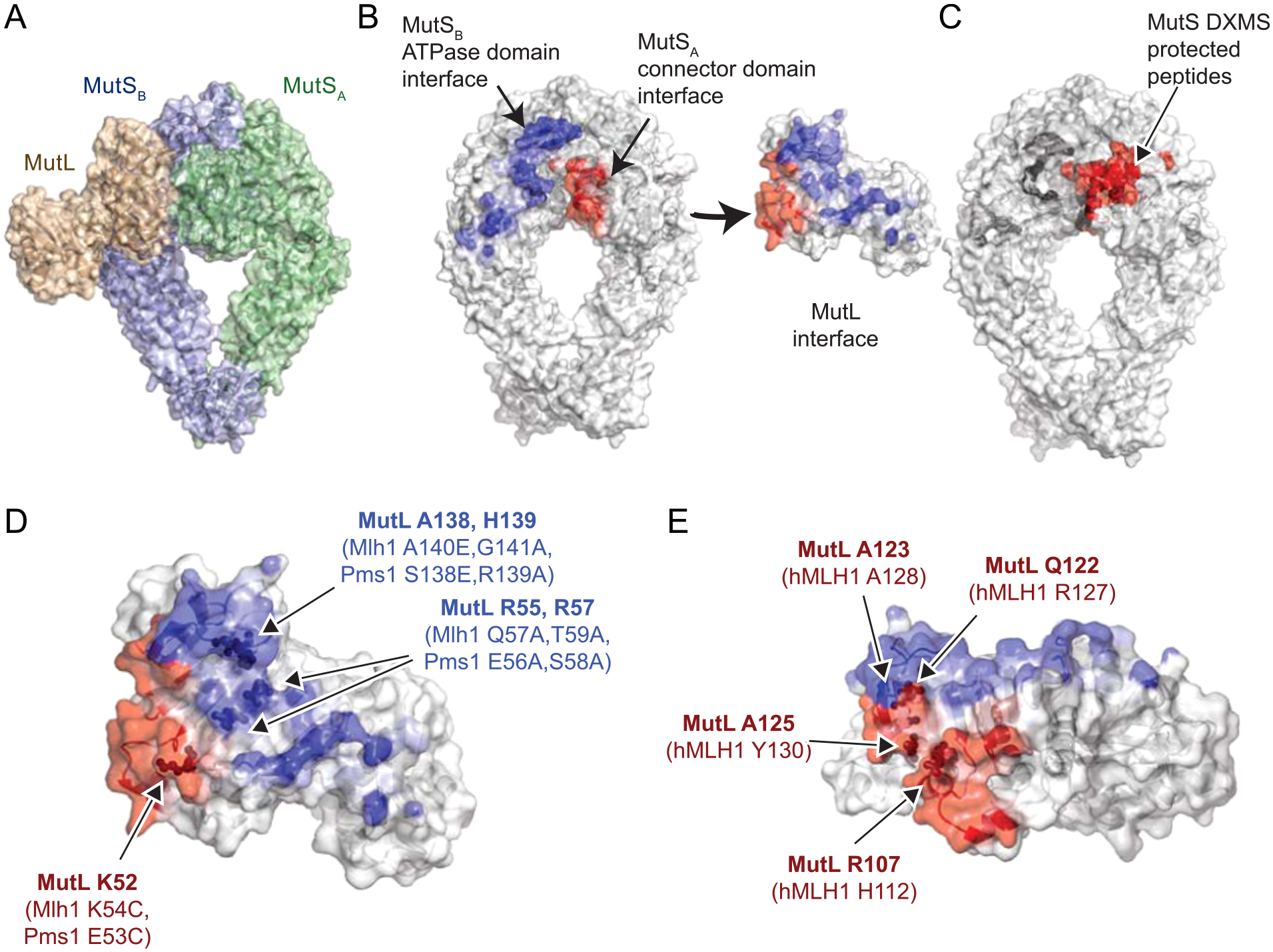
MutL has two interaction surfaces with the MutS dimer. A. In the MutS-MutL recruitment structure [21], the N-terminus of MutL (tan) interacts with both MutS subunits in the MutS dimer via the MutS connector domain in the MutSA subunit (green) and the MutS ATPase domain in the MutSB subunit (blue). B. Residues at the MutL-MutS connector domain interface (red) and the MutL-MutS ATPase domain interface (blue) are depicted on the MutS dimer and the MutL N-terminal monomer. Color saturation for the interface is based on the buried Cx score for each atom (see Methods). C. MutS connector domain peptides protected in DXMS [23] by MutL are colored red. D. Mutagenized residues tested for their effect on E. coli MutL [21] or S. cerevisiae Mlh1-Pms1 recruitment are displayed as spheres relative to the MutL interface surface. E. Residues mutagenized in the hMLH1-hMSH2 connector domain interface found to affect hMLH1-hPMS2 recruitment [26] are shown as spheres.
Table 1.
Mutation rate analysis.
| Genotype | Strain | hom3-10 reversion rate* | lys2-10A reversion rate* | CanR forward mutation rate* |
|---|---|---|---|---|
| Wild-type | RDKY5964 | 1.77 [0.79–4.32] ×10−9 (1) | 5.94 [1.31–13.6] ×10−9 (1) | 5.26 [3.93–7.62] ×10−8 (1) |
| msh2Δ | RDKY9658 | 4.20 [3.39–7.87] ×10−6 (2373) | 1.36 [1.24–4.65] ×10−4 (22,896) | 2.04 [1.56–4.22] ×10−6 (39) |
| mlh1Δ | RDKY9670 | 4.17 [3.30–6.95] ×10−6 (2356) | 1.42 [1.10–1.59] ×10−4 (23,906) | 2.24 [1.77–5.42] ×10−6 (43) |
| pms1Δ | RDKY9673 | 4.70 [3.14–7.23] ×10−6 (2655) | 9.43 [7.33–11.7] ×10−5 (15,875) | 2.59 [2.35–3.74] ×10−6 (49) |
| mlh1-Q57L,T59L | RDKY9789 | 3.33 [2.29–3.84] ×10−6 (1881) | 9.23 [5.35–13.2] ×10−5 (15,539) | 1.81 [1.27–3.70] ×10−6 (34) |
| mlh1-K5KC | RDKY9792 | 4.89 [4.08–9.28] ×10−8 (28) | 2.88 [2.18–3.89] ×10−6 (485) | 1.24 [0.97–1.98] ×10−7 (2) |
95% confidence intervals in brackets [], fold change over wild-type in parentheses ().
2. Methods
2.1. Identification of interface residues
Interface residues in the crosslinked MutS-MutL recruitment complex [21] were identified through application of the Cx algorithm [27] that identifies surface atoms by calculating the Cx ratio, which is the fraction of a sphere with a radius of 10 Å around each atom which is exposed divided by the fraction of the sphere volume that is taken up by volumes from atoms in the molecule. The Cx ratio ranges from ~15 for exposed atoms to 0 for buried atoms. Residues at interfaces can be identified by calculating the Cx ratios for the subunits and subtracting out the Cx ratio for the complex, as exposed residues that are unchanged have a subtracted value of zero. For example, to identify MutL residues that interact with the MutS dimer, the Cx values of the MutSA-MutSB-MutL trimeric complex were subtracted from Cx values calculated from the MutL monomer to generate MutL Cx interface values.
2.2. Strains
S. cerevisiae strains were grown in YPD (1% yeast extract, 2% bacto peptone and 2% dextrose) or in the appropriate synthetic dropout media (0.67% yeast nitrogen base without amino acids, 2% dextrose, and amino acid dropout mix at the concentration recommended by the manufacturer (US Biological)) at 30 °C [17,28,29]. All transformations with plasmids or PCR-based cassettes were performed using standard lithium acetate transformation protocols [30]. All S. cerevisiae strains used for mutation rate analysis were derived from the S288C strain RDKY5964 (MATa ura3–52 leu2Δ1 trp1Δ63 his3Δ200 hom3–10 lys2∷InsE-A10) [28]. The MLH1 and PMS1 plasmids were derived from pRDK1807 (pRS316 URA3 MLH1) and pRDK1667 (pRS316 URA3 PMS1) [17], respectively, using the GeneArt Site Directed Mutagenesis Kit (Invitrogen) according to the manufacturer’s instructions (Suppl. Table 2). Integrated mutations were constructed by generating a plasmid encoding Cas9 and a suitable guide RNA from pRCC-K [31] by Gibson assembly and cotransforming the resulting plasmid with a template double stranded DNA for introducing the mutations and disrupting the PAM sequence targeting by the gRNA; the oligonucleotide duplexes used for introducing mutations were typically 100 bp long.
2.3. Mutation rate analysis
Mutator phenotypes were evaluated using the hom3–10 and lys2–10A frameshift reversion assays essentially as previously described [28,29]. Qualitative analysis was done by patching colonies onto CSM-Ura plates and replica plating onto CSM-Thr-Ura and CSM-Lys-Ura synthetic dropout media for analysis of papillae growth [29,32]. Mutation rates were determined by fluctuation analysis using a minimum of 2 independently derived strains and 14 or more independent cultures; comparisons of mutation rates were evaluated using 95% confidence intervals or by Mann-Whitney two-tailed tests [28,29].
2.4. Protein purification
MMR proteins were purified according to standard protocols as previously described for Exo1 [33,34], Msh2-Msh6 [35,36], Mlh1-Pms1 [34], PCNA [34,37], DNA polymerase ε [33,38], RFC-Δ1N [39], and RPA [40] and were greater than 95% pure as determined by SDS-PAGE. Multiple protein preparations were used during the course of the experiments presented and many of these preparations were validated in our previously published studies [33,34,41–43]. The expression plasmids encoding the Mlh1-K54C mutant (pRDK1973) and the Mlh1-Q57L, T59L mutant (pRDK1968) were generated from pRDK573 (pRS424/pGAL10-MLH1) [44] using the GeneArt Site Directed Mutagenesis Kit (Invitrogen) according to the manufacturer’s instructions and the sequence of the resulting plasmids was confirmed by Sanger sequencing using a commercial facility.
2.5. In vitro MMR assays
Reactions were performed as described [33]. 390 fmol of Msh2-Msh6, 390 fmol of Mlh1-Pms1, 290 fmol of RFC-Δ1N, 290 fmol of PCNA, 0.38 fmol of Exo1, 1800 fmol of RPA, and 400 fmol of DNA pol ε were incubated with 100 ng (52 fmol) of CC mispaired substrate with a 3′ single strand nick at the AflIII site 442 bp from the mispair (3′ AflIII CC substrate) in a final volume of 10 μL containing 4 μL of proteins, 1 μL of substrate, and 5 μL of a master reaction buffer mix. The master reaction buffer mix contained 33 mM Tris pH 7.6, 75 mM KCl, 8.3 mM MgCl2, 1 mM MnSO4, 80 ug/mL BSA, 200 μM dNTPs, 1.66 mM glutathione, and 2.5 mM ATP. Reactions were stopped by addition of 0.42 μL of 500 mM EDTA and 20 μL stop solution containing 0.4 mg/mL glycogen (Thermo Scientific) and 360 μg/mL Proteinase K (Sigma Aldrich) to final concentrations of 6.9 mM, 263 μg/mL, and 237 μg/mL, respectively, followed by incubation for 30 min at 55 °C. The reactions were then extracted with phenol, and the DNA substrate was precipitated with ethanol, followed by digestion with PstI and ScaI. Digested DNA substrate was subjected to electrophoresis on a 0.8% agarose gel run in a buffer containing 40 mM Tris, 20 mM acetic acid, 1 mM EDTA at pH 8.3 (TAE; Biorad) with 0.5 mg/mL ethidium bromide for 45 min at 100 V. Quantitation of the relative amounts of the different DNA species in each individual lane was performed with AlphaImager Gel Imaging System (ProteinSimple).
2.6. Mlh1-Pms1 endonuclease assay
Mismatch-independent endonuclease assays were performed as described previously [17]. Briefly, 40 μL reactions containing 7.5 nM PCNA, 30 nM RFC-Δ1N, and 35 nM Mlh1-Pms1, 1 mM MnSO4, 20 mM Tris pH 7.5, 0.5 mM ATP 0.2 mg/mL bovine serum albumin (BSA), 2 mM DTT and 100 ng supercoiled pRS425 plasmid DNA were incubated at 30 °C for 30 min. Reactions were terminated by addition of 10 μL of stop solution containing 0.5% SDS, 70 mM EDTA, 40% glycerol and 2.5 ug/mL proteinase K followed by incubation at 55 °C for 30 min. The samples were then electrophoresed on a 0.8% agarose gel, the gel was stained with 0.5 mg/mL ethidium bromide, and the bands were quantified using an AlphaImager Gel Imaging System (ProteinSimple).
2.7. Surface plasmon resonance
Protein-DNA and protein-protein-DNA interactions were monitored using a Biacore T100 instrument (GE Healthcare) using the conditions described previously [45–47]. The DNA substrates used were 236 bp in length with biotin conjugated at one end, the lacO sequence at the other end, and a centrally located base-base mispair that were constructed as previously described [44,45,48]. Approximately 20 ng (100 ± 5 resonance units (RUs)) of DNA substrates were conjugated to streptavidin-coated Biacore SA chips (GE Healthcare), and an unmodified flow cell was used as a reference surface in each experiment. The DNA ends were blocked by flowing a buffer containing 30 nM LacI, 25 mM Tris, pH 8, 4 mM MgCl2, 110 mM NaCl, 0.01% Igepal, 2 mM DTT, and 2% glycerol over the flow cell. All experiments were performed in the same buffer and also contained 20 nM Msh2-Msh6, the indicated concentrations of Mlh1-Pms1 and 250 μM ATP. Under these conditions, Mlh1-Pms1 does not bind to DNA in the absence of recruitment by Msh2-Msh6 [44]. All experiments were performed at 25 °C at a flow rate of 20 μL/min, and data were collected at a frequency of 10 Hz. The data were analyzed using the BiaEvaluation v3.1 (GE Healthcare).
3. Results
3.1. Mutations of predicted interface residues in Mlh1, but not Pms1, disrupt MMR in vivo
We constructed S. cerevisiae MLH1 and PMS1 plasmids containing mutations affecting predicted interface amino acids based on the mutations that disrupted recruitment of E. coli MutL by E. coli MutS [21]. The alleles created included mlh1-K54C, mlh1-Q57A,T59A, mlh1-Q57L, T59L, mlh1-A140E,G141A, pms1-E53C, pms1-E56A,S58A, pms1-E56L, S58L, and pms1-S138E,R139A (Fig. 1D). These plasmids were then tested for their ability to complement the mutator phenotype of mlh1Δ and pms1Δ single mutant S. cerevisiae strains, respectively, using the hom3–10 and lys2–10A frameshift reversion assays (Fig. 2). The mlh1-K54C single mutation and the mlh1-Q57A,T59A and mlh1-Q57L, T59L double mutations failed to complement the mlh1Δ mutant strain. In contrast, the equivalent pms1-E53C single mutation and the pms1-E56A,S58A and pms1-E56L,S58L double mutations were able to complement the pms1Δ mutant strain. The mlh1-A140E,G141A and pms1-S138E,R139A alleles were unable to complement either the mlh1Δ or pms1Δ mutant strain, respectively.
Fig. 2.
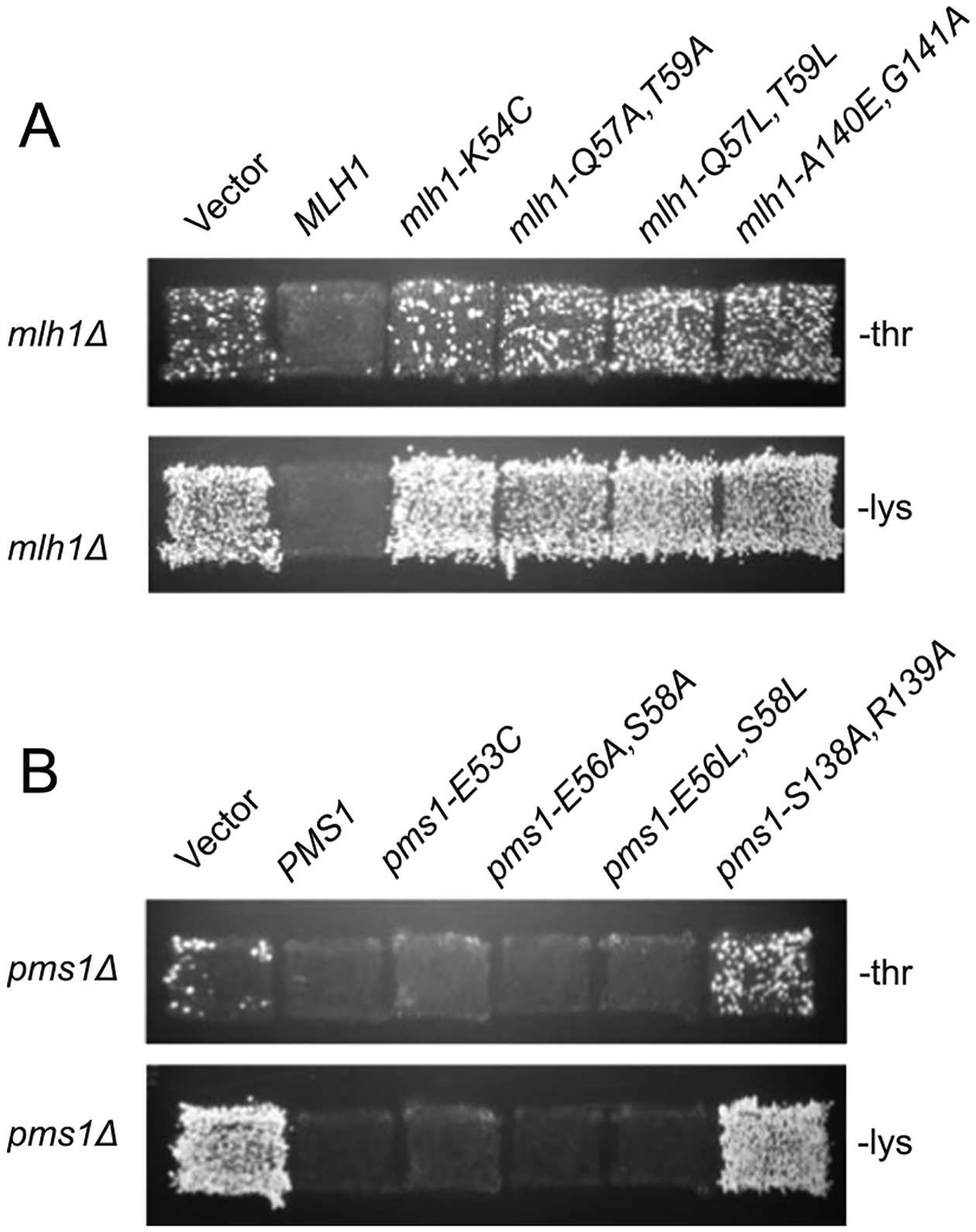
Patch test for MLH1 and PMS1 mutations affecting interface residues. A. Patches of a mlh1Δ strain transformed with a wild-type MLH1 plasmid, an empty plasmid, or a plasmid carrying a mlh1 interface mutation were replica plated onto CSM-Thr and CSM-Lys plates to test for reversion of the hom3–10 and lys2–10A frameshift mutations, respectively. Growth indicates increased mutation rates. B. Patches of a pms1Δ strain transformed with a wild-type PMS1 plasmid, an empty plasmid, or a plasmid carrying various pms1 interface mutations were tested as in panel A above.
To further characterize the mlh1 mutations, we constructed strains in which the mlh1-K54C single mutation or the mlh1-Q57L,T59L double mutation were integrated at the MLH1 genomic locus and used fluctuation analysis to determine the effect of these mutations on mutation rates as assessed using hom3–10 and lys2–10A frameshift reversion assays and the CAN1 forward mutation assay (Table 1). The mlh1-K54C single mutation caused a modest increase in mutation rates that was equivalent to ~2–5% of the increase in mutation rates caused by the msh2Δ, mlh1Δ and pms1Δ single mutations that cause complete MMR defects. In contrast, the mlh1-Q57L,T59L double mutation caused an increase in mutation rates that was not significantly different than that caused by the msh2Δ, mlh1Δ and pms1Δ single mutations. These results along with the results from plasmid complementation studies (Fig. 2) suggest that the Mlh1 K54, Q57, and Q59 side chains are required for recruitment of Mlh1-Pms1, whereas the Pms1 E53, E56, and S58 side chains are not required. Importantly, mlh1-K54C probes the predicted interaction of Mlh1 with the Msh2 connector domain that was previously shown to interact with the Mlh1-Pms1 complex [44], whereas mlh1-Q57A,T59A and mlh1-Q57L,T59L probe the predicted interaction between Mlh1 and the Msh6 ATPase and core domains.
3.2. Mutations affecting the Mlh1 interface cause defects in MMR in vitro
To better understand the defect caused by disrupting the Mlh1-Msh6 interaction, we tested the ability of the Mlh1-Q57L,T59L-Pms1 mutant complex to support Mlh1-Pms1-dependent repair of a mispaired plasmid substrate. The plasmid substrate contains a CC mispair that disrupts a PstI site and a single strand nick at the AflIII site that is 442 bp 3′ to the mispair (Fig. 3A); at the levels of Exo1 used in the experiments presented here [33,34], repair of this substrate depends on the production of 5′ nicks by Mlh1-Pms1 that are substrates for excision by Exo1 [13,14,41]. Repair of the CC mispair was monitored by restoration of the PstI site and formation of diagnostic 1.1 and 1.8 kb product bands upon double digestion with PstI and ScaI. Wild-type Mlh1-Pms1 supported efficient repair of this substrate in reactions that also contained Msh2-Msh6, Exo1, PCNA, RFC-Δ1 N, RPA and DNA polymerase ε, whereas the Mlh1-Q57L,T59L-Pms1 mutant caused a substantial MMR defect (Fig. 3B,C). This defect is consistent with substantial MMR defects caused by the mlh1-Q57L,T59L mutation in vivo.
Fig. 3.
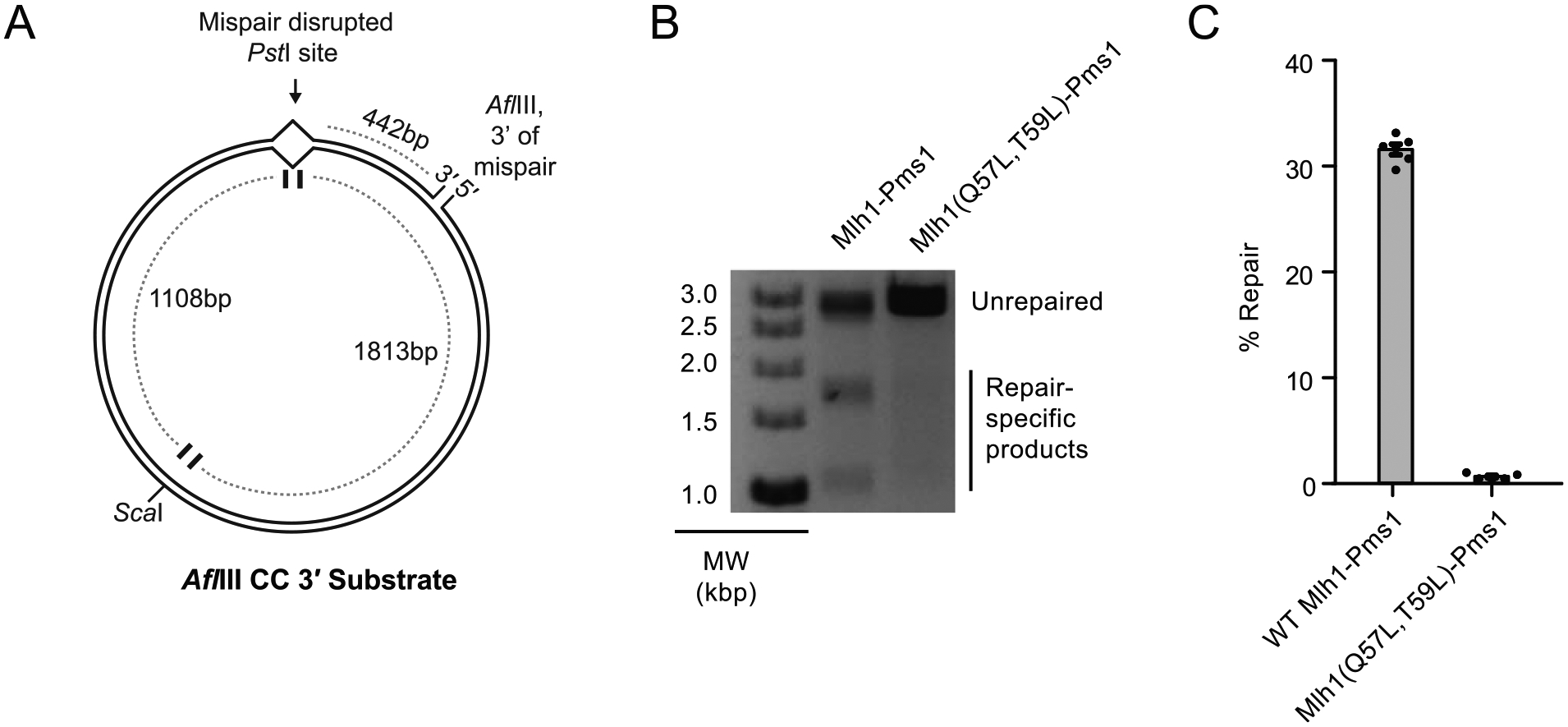
Reconstituted MMR in vitro is not supported by the Mlh1(Q57L,Q59L)-Pms1 mutant. A. The DNA substrate used has a mispair disrupting a PstI site and a nick 442 bp 3′ from the mispair. Repair of this substrate requires Mlh1-Pms1 to make a nick 5′ of the mispair and is monitored by following the restoration of the PstI site using ScaI/PstI double digests. B. Example repair reactions showing robust repair in the presence of wild-type Mlh1-Pms1 complex and substantially reduced repair in the presence of Mlh1-Q57L,Q59L-Pms1 complex. C. Quantification of repair reactions shows that Mlh1(Q57L,Q59L)-Pms1 is unable to support MMR in vitro.
3.3. Mutations affecting the Mlh1 interfaces do not alter endonuclease activity
Because defects in DNA nicking by Mlh1-Pms1 can result in MMR defects, we assayed the mutant Mlh1-K54C-Pms1 and Mlh1-Q57L,T59L-Pms1 complexes for their ability to nick of covalently closed circular supercoiled plasmid DNA in a reaction that is dependent on PCNA and RFC, but is independent of Msh2-Msh6 recruitment and a mispair (Fig. 4) [14,17]. Both mutant complexes both supported efficient conversion of covalently closed circular supercoiled plasmid DNA to nicked circular plasmid DNA and appeared to be at least 80% as active as wild-type Mlh1-Pms1 protein. These results suggest that the MMR defects caused by the mlh1-K54C and mlh1-Q57L,T59L mutations are not due to loss of endonuclease activity.
Fig. 4.
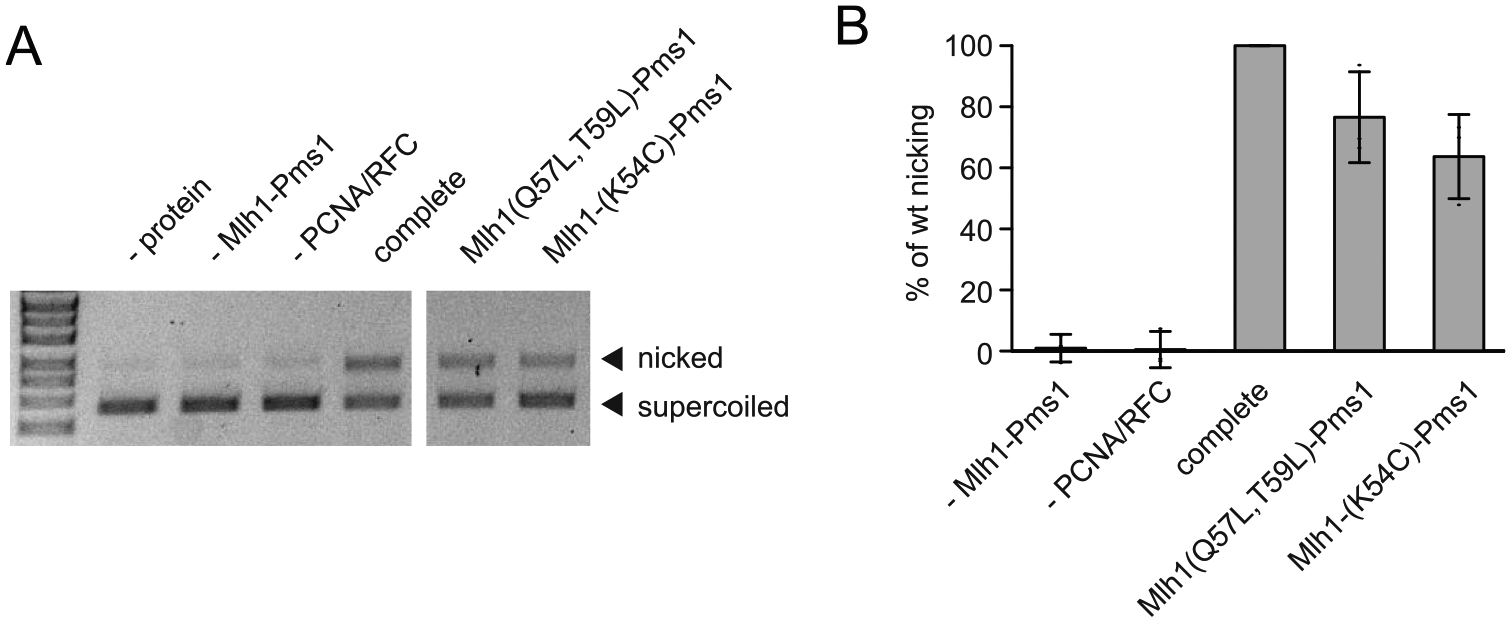
Mlh1-Pms1 interface mutants are proficient for Msh2-Msh6-independent endonuclease function. A. PCNA/RFC-dependent, Msh2-Msh6-independent nicking of a supercoiled plasmid by Mlh1-Pms1 is followed by formation of a more slowly migrating nicked circular plasmid. B. Quantification shows that efficient nicking depends on Mlh1-Pms1 and PCNA and RFC, which activate the Mlh1-Pms1 endonuclease. The Mlh1-Pms1 interface mutants tested were largely proficient for endonuclease activity.
3.4. Mutations affecting both interfaces between Mlh1 and Msh2-Msh6 cause defects in mispair-dependent recruitment
We next used Surface Plasmon Resonance (SPR) to monitor recruitment of Mlh1-Pms1 by Msh2-Msh6 bound to an 236 bp end-blocked DNA substrate containing a GT mispair (Fig. 5). Msh2-Msh6 bound to the GT mispaired substrate, and further protein binding was observed when the bound Msh2-Msh6 was exposed to different concentrations of wild-type Mlh1-Pms1 protein in solutions that also contained the original concentration of Msh2-Msh6. Under these conditions, Mlh1-Pms1 does not bind to DNA in the absence of Msh2-Msh6 [44]. When the Mlh1-Q57L,T59L-Pms1 mutant complex was analyzed, no protein binding above that attributable to Msh2-Msh6 alone was observed, indicating that this mutant was completely defective for recruitment by Msh2-Msh6. In contrast, when the Mlh1-K54C-Pms1 mutant complex was analyzed, some recruitment was observed but at a much lower level than seen with wild-type Mlh1-Pms1. These results indicate that the Mlh1-Q57L,T59L amino acid substitutions eliminates Mlh1-Pms1 recruitment by Msh2-Msh6 and that the Mlh1-K54C amino acid substitution partially eliminates recruitment, and that these recruitment defects likely explain the complete and partial defects of these two mutant complexes, respectively, seen in vivo and in reconstituted MMR reactions in vitro.
Fig. 5.
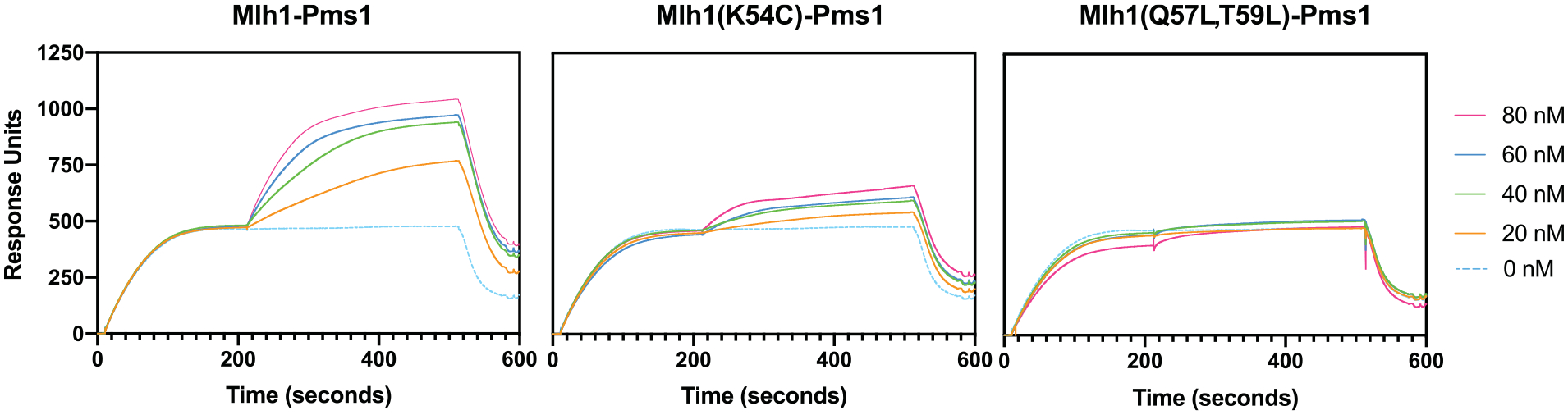
Mlh1-Pms1 interface mutants are defective for Msh2-Msh6-mediated recruitment to mispaired DNA as measured by SPR. Binding of Msh2-Msh6 to an end-blocked DNA containing a mispair was monitored for the first 200 s. After this, flow was switched to buffer containing Msh2-Msh6 and the indicated concentrations of wild-type Mlh1-Pms1 (left), Mlh1-K54C-Pms1 (middle), or Mlh1-Q57L,T59L-Pms1 (right). Robust recruitment of wild-type Mlh1-Pms1 by Msh2-Msh6 is indicated by the rapid, concentration-dependent increase in response units observed. In contrast, the Mlh1-K54C-Pms1 and Mlh1-Q57L,T59L-Pms1 complexes show substantially or completely defective recruitment, respectively.
4. Discussion
Here, we mutagenized amino acids of S. cerevisiae Mlh1 and Pms1 that would be predicted to interact with Msh2-Msh6 based on the E. coli MutS-MutL recruitment structure and E. coli MutL mutations that prevented MutL recruitment to mispaired DNA [21,22]. The mlh1-K54C, mlh1-Q57A,T59A, and mlh1-Q57L,T59L mutations caused partial or complete MMR defects in vivo, whereas the pms1-E53C, pms1-E56A, S58A, and pms1-E56L,S58L did not cause MMR defects in vivo. The mlh1-Q57L,T59L mutation caused a complete defect in recruitment of Mlh1-Pms1 to mispaired DNA by Msh2-Msh6, and the mlh1-K54C mutation caused a significant but partial recruitment defect. These results are consistent with results indicating that the human MLH1 subunit mediates MSH2-MSH6 interaction as shown by pulldowns from nuclear extracts (summarized in Table 2; [25]). As would be predicted based on the partial recruitment defect of the Mlh1-K54C-Pms1 complex and the full recruitment defect of the Mlh1-Q57L,T59L-Pms1 complex, Mlh1-Q57L,T59L-Pms1 was essentially completely defective for supporting Msh2-Msh6-dependent MMR repair in reconstituted MMR reactions in vitro, but was not defective for supporting PCNA/RFC-dependent, Msh2-Msh6-independent nicking of supercoiled DNA. Previous mutagenesis studies (summarized in Table 2) of E. coli MutL indicated that MutL interacted with the MutS connector and ATPase/core domains [21] and that human MLH1 interacted with the MSH2 connector domain [26], consistent with our previous finding that Mlh1-Pms1 interacts with the Msh2 connector domain [23]. In combination with the results of these studies, our results indicate that Mlh1, the common subunit of the eukaryotic MutL homolog complexes, is recruited to mispaired DNA through interactions with both the Msh2 connector domain and the Msh6 ATPase/core domains (Table 2); this latter interaction has been demonstrated for the first time by our studies.
Table 2.
Effect of mutations on recruitment of Mlh1 or MutL.
| Mutation | ScMlh1 residue | EcMutL residue | hMLH1 residue | Recruitment defect | Notes | Reference |
|---|---|---|---|---|---|---|
| EcMutL K52C | K54 | K52 | K57 | Yes | Connector domain interface | [21] |
| ScMlh1 K54C | K54 | K52 | K57 | Yes | Connector domain interface | This study |
| EcMutL R55D, R57D | Q57, T59 | R55, R57 | Q60, Q62 | Yes | ATPase/core domain interface | [21] |
| ScMlh1 Q57A, T59A | Q57, T59 | R55, R57 | Q60, Q62 | Yes | ATPase/core domain interface | This study |
| EcMutL R55D, R57D, A138E, H139A | Q57, T59, A140, G141 | R55, R57, A138, H139 | Q60, Q62, A143, G144 | Yes | Both interfaces | [21] |
| hMLH1 N64S | N61 | N59 | N64 | No | Next to ATPase/core domain interface; Lynch syndrome | [26] |
| hMLH1 H112A | R109 | R107 | H112 | Yes | Connector domain interface | [26] |
| hMLH1 H112D | R109 | R107 | H112 | Yes | Connector domain interface | [26] |
| hMLH1 T117M | T114 | S112 | T117 | No | Mostly buried; Lynch syndrome | [26] |
| hMLH1 R127E | R124 | Q122 | R127 | Yes | Connector domain interface | [26] |
| hMLH1 A128P | V125 | A123 | A128 | Yes | Connector domain interface | [26] |
| hMLH1 Y130A | Y127 | A125 | Y130 | Yes | Connector domain interface | [26] |
| hMLH1 D132H | E129 | G127 | D132 | No | Loop between connector domain interface residues; disordered in co-crystal structure | [26] |
| EcMutL A138E | A140 | A138 | A143 | Yes | ATPase/core domain interface | [21] |
| EcMutL A138E, H139A | A140, G141 | A138, H139 | A143, G144 | Yes | ATPase/core domain interface | [21] |
| ScMlh1 A140E, G141A | A140, G141 | A138, H139 | A143, G144 | n.d. | ATPase/core domain interface; MMR defective in both ScMLH1 and ScPMS1 | This study |
| hMLH1 R265C | R265 | R261 | R265 | No | Opposite face of molecule; Lynch syndrome | [26] |
DXMS studies of E. coli MutS and MutL only detected the MutS connector domain interaction with MutL (the Msh2-Mlh1 interaction); a second region that was protected from deuterium exchange was caused by the movement of the connector domain [21,23]. The fact that the MutL interface with the MutS ATPase/core domains was not detected suggests either that this interaction primarily affects side chain environments, which are not directly monitored by DXMS [49], or that the two surfaces have different properties, with interactions with the connector domain being more stable during the time course of the DXMS experiment. It is possible that stable interactions constrain which amino acids are at the interfaces and that this could be detected as increased conservation of stable interface residues. We therefore examined the conservation of residues at the predicted interfaces between Mlh1 and Msh2, Msh6, and Msh3 for 744, 747, 652, and 657 fungal Msh2, Msh6, Msh3, and Mlh1 proteins, respectively (Suppl. Figs. 1,2). For the Msh2 connector domain interface, Msh2 residues predicted to be buried upon complex formation based on the E. coli structure were highly conserved (Suppl. Fig. 1A). Remarkably, the Msh6 and Msh3 residues equivalent to the MutS residues at the ATPase/core domain interface show the opposite pattern; the residues that are the most buried upon MutL recruitment tended to be the least conserved (Suppl. Fig. 1B). Similar conservation patterns were observed at both interfaces for predicted Mlh1 interface residues, although the differences were less pronounced than for the Msh2 and Msh6/Msh3 residues (Suppl. Fig. 2A,B). Increased stability of the connector domain interface relative to the ATPase/core domain interface would be consistent with the fact that the connector domain interface, but not the ATPase/core domain interface, is specific to the recruitment conformation [8,9,21,50]. Thus, licensing of Msh2-Msh6 to recruit Mlh1-Pms1, which requires prior binding to mispaired DNA and ATP [22,44,51,52], may also depend upon a relatively stable interaction through the connector domain as well as a less stable interaction with the ATPase/core interface.
These results fit within a larger picture of the duplication and specialization of the core MMR proteins in eukaryotes. The functional asymmetry present in the symmetric bacterial MutS and MutL homodimers has led to the diversification of function after gene duplication in eukaryotes, which fits the long-standing paradigm proposed by Ohno [53]. Eukaryotic MutS homologs that act in MMR have evolved into Msh2, the common subunit that does not recognize mispairs but interacts with Mlh1 through its connector domain, and the mispair-recognizing subunit Msh6 (and most likely Msh3), which interact with Mlh1 through their ATPase/core domains. These mispair recognition subunits have further diversified their roles. Msh6 preferentially binds single base mispairs and short insertion/deletion mispairs and retains an MutS-like recognition by base flipping and pi-stacking by a conserved phenylalanine side chain [10]. In contrast, Msh3 preferentially binds insertion/deletion mispairs and a subset a single base mispairs and has evolved a mechanism involving insertion of a tyrosine side chain into the DNA and distortion of the insertion/deletion loop that flips the unpaired bases out of the DNA helix [11,54,55]. Eukaryotic MutL homologs that act in MMR have evolved into Mlh1, the common subunit that does not possess an endonuclease active site but is recruited by Msh2 complexes, and a variety of partners, of which S. cerevisiae Pms1 and Mlh3 possess endonuclease activity [14,17,56,57]. The roles for these Mlh1-binding partners are diverse: Mlh1-Pms1 plays a major role in MMR, Mlh1-Mlh3 plays a minor role in MMR but a major role in meiotic crossing over, and Mlh1-Mlh2, which is not an endonuclease, plays only a minor role in MMR that is only apparent at reduced Pms1 levels [57–62]. The asymmetry of the interaction of Mlh1 with Msh2 complexes also likely plays a role during recruitment to DNA [21]; Mlh1 is stabilized through interactions with Msh2-Msh6/Msh2-Msh3, whereas binding of the Pms1/Mlh2/Mlh3 N-terminal domain by the Mlh1 N-terminal domain likely requires interactions between Pms1 and Mlh1 and between the Pms1 N-terminal DNA binding region and DNA. This asymmetry may allow for functional divergence in DNA binding by subunits within Mlh1 heterodimers, and the initial tethering may explain the significance of DNA binding despite the fact that isolated MutL homologs only bind DNA at very low ionic strength [24,63–65]. Remarkably, the asymmetric interaction between the shared Msh2 and Mlh1 subunits, which were revealed in this and previous studies [23,25,26], may have been the lynchpin that allowed for the diversification of the other subunits by ensuring the ability of these diversified heterodimers to still mediate recruitment or be recruited to DNA substrates.
Supplementary Material
Acknowledgements
This study was supported by NIH grant R01 GM50006.
Footnotes
CRediT authorship contribution statement
MLD, CDP, and RDK conceived and designed the project. MLD, TP, and FAC performed the experiments. CDP performed the bioinformatic and structural analyses. RDK and CDP wrote the initial draft of the manuscript, and all authors discussed and commented on the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest with the contents of this article.
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.dnarep.2022.103405.
References
- [1].Fishel R, Mismatch repair, J. Biol. Chem 290 (2015) 26395–26403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Iyer RR, Pluciennik A, Burdett V, Modrich PL, DNA mismatch repair: functions and mechanisms, Chem. Rev 106 (2006) 302–323. [DOI] [PubMed] [Google Scholar]
- [3].Reyes GX, Schmidt TT, Kolodner RD, Hombauer H, New insights into the mechanism of DNA mismatch repair, Chromosoma 124 (2015) 443–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].de la Chapelle A, Genetic predisposition to colorectal cancer, Nat. Rev. Cancer 4 (2004) 769–780. [DOI] [PubMed] [Google Scholar]
- [5].Durno CA, Sherman PM, Aronson M, Malkin D, Hawkins C, Bakry D, Bouffet E, Gallinger S, Pollett A, Campbell B, Tabori U, International BC, Phenotypic and genotypic characterisation of biallelic mismatch repair deficiency (BMMR-D) syndrome, Eur. J. Cancer 51 (2015) 977–983. [DOI] [PubMed] [Google Scholar]
- [6].Lynch HT, Snyder CL, Shaw TG, Heinen CD, Hitchins MP, Milestones of Lynch syndrome: 1895–2015, Nat. Rev. Cancer 15 (2015) 181–194. [DOI] [PubMed] [Google Scholar]
- [7].Post CCB, Stelloo E, Smit V, Ruano D, Tops CM, Vermij L, Rutten TA, Jurgenliemk-Schulz IM, Lutgens L, Jobsen JJ, Nout RA, Crosbie EJ, Powell ME, Mileshkin L, Leary A, Bessette P, Putter H, de Boer SM, Horeweg N, Nielsen M, Wezel TV, Bosse T, Creutzberg CL, Prevalence and prognosis of lynch syndrome and sporadic mismatch repair deficiency in endometrial cancer, J. Natl. Cancer Inst 113 (2021) 1212–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lamers MH, Perrakis A, Enzlin JH, Winterwerp HH, de Wind N, Sixma TK, The crystal structure of DNA mismatch repair protein MutS binding to a G × T mismatch, Nature 407 (2000) 711–717. [DOI] [PubMed] [Google Scholar]
- [9].Obmolova G, Ban C, Hsieh P, Yang W, Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA, Nature 407 (2000) 703–710. [DOI] [PubMed] [Google Scholar]
- [10].Warren JJ, Pohlhaus TJ, Changela A, Iyer RR, Modrich PL, Beese LS, Structure of the human MutSalpha DNA lesion recognition complex, Mol. Cell 26 (2007) 579–592. [DOI] [PubMed] [Google Scholar]
- [11].Gupta S, Gellert M, Yang W, Mechanism of mismatch recognition revealed by human MutSbeta bound to unpaired DNA loops, Nat. Struct. Mol. Biol 19 (2011) 72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Genschel J, Kadyrova LY, Iyer RR, Dahal BK, Kadyrov FA, Modrich P, Interaction of proliferating cell nuclear antigen with PMS2 is required for MutLalpha activation and function in mismatch repair, Proc. Natl. Acad. Sci. USA 114 (2017) 4930–4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kadyrov FA, Dzantiev L, Constantin N, Modrich P, Endonucleolytic function of MutLalpha in human mismatch repair, Cell 126 (2006) 297–308. [DOI] [PubMed] [Google Scholar]
- [14].Kadyrov FA, Holmes SF, Arana ME, Lukianova OA, O’Donnell M, Kunkel TA, Modrich P, Saccharomyces cerevisiae MutLalpha is a mismatch repair endonuclease, J. Biol. Chem 282 (2007) 37181–37190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Dai J, Sanchez A, Adam C, Ranjha L, Reginato G, Chervy P, Tellier-Lebegue C, Andreani J, Guerois R, Ropars V, Le Du MH, Maloisel L, Martini E, Legrand P, Thureau A, Cejka P, Borde V, Charbonnier JB, Molecular basis of the dual role of the Mlh1-Mlh3 endonuclease in MMR and in meiotic crossover formation, Proc. Natl. Acad. Sci. USA 118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gueneau E, Dherin C, Legrand P, Tellier-Lebegue C, Gilquin B, Bonnesoeur P, Londino F, Quemener C, Le MH, Du JA Marquez, M. Moutiez, M. Gondry, S. Boiteux, J.B. Charbonnier, Structure of the MutLalpha C-terminal domain reveals how Mlh1 contributes to Pms1 endonuclease site, Nat. Struct. Mol. Biol 20 (2013) 461–468. [DOI] [PubMed] [Google Scholar]
- [17].Smith CE, Mendillo ML, Bowen N, Hombauer H, Campbell CS, Desai A, Putnam CD, Kolodner RD, Dominant mutations in S cerevisiae PMS1 identify the Mlh1-Pms1 endonuclease active site and an exonuclease 1-independent mismatch repair pathway, PLoS Genet 9 (2013), e1003869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Pillon MC, Lorenowicz JJ, Uckelmann M, Klocko AD, Mitchell RR, Chung YS, Modrich P, Walker GC, Simmons LA, Friedhoff P, Guarne A, Structure of the endonuclease domain of MutL: unlicensed to cut, Mol. Cell 39 (2010) 145–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Putnam CD, Strand discrimination in DNA mismatch repair, DNA Repair (Amst.) 105 (2021), 103161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Liu L, Ortiz Castro MC, Rodriguez Gonzalez J, Pillon MC, Guarne A, The endonuclease domain of Bacillus subtilis MutL is functionally asymmetric, DNA Repair (Amst.) 73 (2019) 1–6. [DOI] [PubMed] [Google Scholar]
- [21].Groothuizen FS, Winkler I, Cristovao M, Fish A, Winterwerp HH, Reumer A, Marx AD, Hermans N, Nicholls RA, Murshudov GN, Lebbink JH, Friedhoff P, Sixma TK, MutS/MutL crystal structure reveals that the MutS sliding clamp loads MutL onto DNA, Elife 4 (2015), e06744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Fernandez-Leiro R, Bhairosing-Kok D, Kunetsky V, Laffeber C, Winterwerp HH, Groothuizen F, Fish A, Lebbink JHG, Friedhoff P, Sixma TK, Lamers MH, The selection process of licensing a DNA mismatch for repair, Nat. Struct. Mol. Biol 28 (2021) 373–381. [DOI] [PubMed] [Google Scholar]
- [23].Mendillo ML, Hargreaves VV, Jamison JW, Mo AO, Li S, Putnam CD, Woods VL Jr., Kolodner RD, A conserved MutS homolog connector domain interface interacts with MutL homologs, Proc. Natl. Acad. Sci. USA 106 (2009) 22223–22228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Liu J, Hanne J, Britton BM, Bennett J, Kim D, Lee JB, Fishel R, Cascading MutS and MutL sliding clamps control DNA diffusion to activate mismatch repair, Nature 539 (2016) 583–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Plotz G, Raedle J, Brieger A, Trojan J, Zeuzem S, N-terminus of hMLH1 confers interaction of hMutLalpha and hMutLbeta with hMutSalpha, Nucleic Acids Res 31 (2003) 3217–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Plotz G, Welsch C, Giron-Monzon L, Friedhoff P, Albrecht M, Piiper A, Biondi RM, Lengauer T, Zeuzem S, Raedle J, Mutations in the MutSalpha interaction interface of MLH1 can abolish DNA mismatch repair, Nucleic Acids Res 34 (2006) 6574–6586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Pintar A, Carugo O, Pongor S, CX, an algorithm that identifies protruding atoms in proteins, Bioinformatics 18 (2002) 980–984. [DOI] [PubMed] [Google Scholar]
- [28].Hombauer H, Campbell CS, Smith CE, Desai A, Kolodner RD, Visualization of eukaryotic DNA mismatch repair reveals distinct recognition and repair intermediates, Cell 147 (2011) 1040–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Amin NS, Nguyen MN, Oh S, Kolodner RD, exo1-Dependent mutator mutations: model system for studying functional interactions in mismatch repair, Mol. Cell Biol 21 (2001) 5142–5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sherman F, Fink GR, Hicks JB, Methods in yeast genetics, Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y, 1986. [Google Scholar]
- [31].Generoso WC, Gottardi M, Oreb M, Boles E, Simplified CRISPR-Cas genome editing for saccharomyces cerevisiae, J. Microbiol Methods 127 (2016) 203–205. [DOI] [PubMed] [Google Scholar]
- [32].Marsischky GT, Filosi N, Kane MF, Kolodner R, Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair, Genes Dev 10 (1996) 407–420. [DOI] [PubMed] [Google Scholar]
- [33].Bowen N, Kolodner RD, Reconstitution of Saccharomyces cerevisiae DNA polymerase epsilon-dependent mismatch repair with purified proteins, Proc. Natl. Acad. Sci. USA 114 (2017) 3607–3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bowen N, Smith CE, Srivatsan A, Willcox S, Griffith JD, Kolodner RD, Reconstitution of long and short patch mismatch repair reactions using Saccharomyces cerevisiae proteins, Proc. Natl. Acad. Sci. USA 110 (2013) 18472–18477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Antony E, Hingorani MM, Mismatch recognition-coupled stabilization of Msh2-Msh6 in an ATP-bound state at the initiation of DNA repair, Biochemistry 42 (2003) 7682–7693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Shell SS, Putnam CD, Kolodner RD, The N terminus of Saccharomyces cerevisiae Msh6 is an unstructured tether to PCNA, Mol. Cell 26 (2007) 565–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Fien K, Stillman B, Identification of replication factor C from Saccharomyces cerevisiae: a component of the leading-strand DNA replication complex, Mol. Cell. Biol 12 (1992) 155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Georgescu RE, Langston L, Yao NY, Yurieva O, Zhang D, Finkelstein J, Agarwal T, O’Donnell ME, Mechanism of asymmetric polymerase assembly at the eukaryotic replication fork, Nat. Struct. Mol. Biol 21 (2014) 664–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Gomes XV, Gary SL, Burgers PM, Overproduction in Escherichia coli and characterization of yeast replication factor C lacking the ligase homology domain, J. Biol. Chem 275 (2000) 14541–14549. [DOI] [PubMed] [Google Scholar]
- [40].Nakagawa T, Flores-Rozas H, Kolodner RD, The MER3 helicase involved in meiotic crossing over is stimulated by single-stranded DNA-binding proteins and unwinds DNA in the 3′ to 5′ direction, J. Biol. Chem 276 (2001) 31487–31493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Smith CE, Bowen N, Jt Graham W, Goellner EM, Srivatsan A, Kolodner RD, Activation of Saccharomyces cerevisiae Mlh1-Pms1 Endonuclease in a Reconstituted Mismatch Repair System, J. Biol. Chem 290 (2015) 21580–21590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Goellner EM, Smith CE, Campbell CS, Hombauer H, Desai A, Putnam CD, Kolodner RD, PCNA and Msh2-Msh6 activate an Mlh1-Pms1 endonuclease pathway required for Exo1-independent mismatch repair, Mol. Cell 55 (2014) 291–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Goellner EM, Putnam CD, Jt Graham W, Rahal CM, Li BZ, Kolodner RD, Identification of Exo1-Msh2 interaction motifs in DNA mismatch repair and new Msh2-binding partners, Nat. Struct. Mol. Biol 25 (2018) 650–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Mendillo ML, Mazur DJ, Kolodner RD, Analysis of the interaction between the Saccharomyces cerevisiae MSH2-MSH6 and MLH1-PMS1 complexes with DNA using a reversible DNA end-blocking system, J. Biol. Chem 280 (2005) 22245–22257. [DOI] [PubMed] [Google Scholar]
- [45].Hargreaves VV, Shell SS, Mazur DJ, Hess MT, Kolodner RD, Interaction between the Msh2 and Msh6 nucleotide-binding sites in the Saccharomyces cerevisiae Msh2-Msh6 complex, J. Biol. Chem 285 (2010) 9301–9310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Jt Graham W, Putnam CD, Kolodner RD, The properties of Msh2-Msh6 ATP binding mutants suggest a signal amplification mechanism in DNA mismatch repair, J. Biol. Chem 293 (2018) 18055–18070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Srivatsan A, Bowen N, Kolodner RD, Mispair-specific recruitment of the Mlh1-Pms1 complex identifies repair substrates of the Saccharomyces cerevisiae Msh2-Msh3 complex, J. Biol. Chem 289 (2014) 9352–9364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Shell SS, Putnam CD, Kolodner RD, Chimeric Saccharomyces cerevisiae Msh6 protein with an Msh3 mispair-binding domain combines properties of both proteins, Proc. Natl. Acad. Sci. USA 104 (2007) 10956–10961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Maier CS, Deinzer ML, Protein conformations, interactions, and H/D exchange, Methods Enzym 402 (2005) 312–360. [DOI] [PubMed] [Google Scholar]
- [50].Mendillo ML, Putnam CD, Mo AO, Jamison JW, Li S, Woods VL Jr., Kolodner RD, Probing DNA- and ATP-mediated conformational changes in the MutS family of mispair recognition proteins using deuterium exchange mass spectrometry, J. Biol. Chem 285 (2010) 13170–13182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Blackwell LJ, Bjornson KP, Allen DJ, Modrich P, Distinct MutS DNA-binding modes that are differentially modulated by ATP binding and hydrolysis, J. Biol. Chem 276 (2001) 34339–34347. [DOI] [PubMed] [Google Scholar]
- [52].Gradia S, Subramanian D, Wilson T, Acharya S, Makhov A, Griffith J, Fishel R, hMSH2-hMSH6 forms a hydrolysis-independent sliding clamp on mismatched DNA, Mol. Cell 3 (1999) 255–261. [DOI] [PubMed] [Google Scholar]
- [53].Ohno S, Evolution by Gene Duplication, Springer-Verlag,, Berlin, 1970. [Google Scholar]
- [54].Dowen JM, Putnam CD, Kolodner RD, Functional studies and homology modeling of Msh2-Msh3 predict that mispair recognition involves DNA bending and strand separation, Mol. Cell Biol 30 (2010) 3321–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Harrington JM, Kolodner RD, Saccharomyces cerevisiae Msh2-Msh3 acts in repair of base-base mispairs, Mol. Cell Biol 27 (2007) 6546–6554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Ranjha L, Anand R, Cejka P, The Saccharomyces cerevisiae Mlh1-Mlh3 heterodimer is an endonuclease that preferentially binds to Holliday junctions, J. Biol. Chem 289 (2014) 5674–5686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Rogacheva MV, Manhart CM, Chen C, Guarne A, Surtees J, Alani E, Mlh1-Mlh3, a meiotic crossover and DNA mismatch repair factor, is a Msh2-Msh3-stimulated endonuclease, J. Biol. Chem 289 (2014) 5664–5673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Campbell CS, Hombauer H, Srivatsan A, Bowen N, Gries K, Desai A, Putnam CD, Kolodner RD, Mlh2 is an accessory factor for DNA mismatch repair in Saccharomyces cerevisiae, PLoS Genet 10 (2014), e1004327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Wang TF, Kleckner N, Hunter N, Functional specificity of MutL homologs in yeast: evidence for three Mlh1-based heterocomplexes with distinct roles during meiosis in recombination and mismatch correction, Proc. Natl. Acad. Sci. USA 96 (1999) 13914–13919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Strand M, Prolla TA, Liskay RM, Petes TD, Destabilization of tracts of simple repetitive DNA in yeast by mutations affecting DNA mismatch repair, Nature 365 (1993) 274–276. [DOI] [PubMed] [Google Scholar]
- [61].Williamson MS, Game JC, Fogel S, Meiotic gene conversion mutants in Saccharomyces cerevisiae. I. Isolation and characterization of pms1-1 and pms1-2, Genetics 110 (1985) 609–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Flores-Rozas H, Kolodner RD, The Saccharomyces cerevisiae MLH3 gene functions in MSH3-dependent suppression of frameshift mutations, Proc. Natl. Acad. Sci. USA 95 (1998) 12404–12409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Ban C, Yang W, Crystal structure and ATPase activity of MutL: implications for DNA repair and mutagenesis, Cell 95 (1998) 541–552. [DOI] [PubMed] [Google Scholar]
- [64].Guarne A, Ramon-Maiques S, Wolff EM, Ghirlando R, Hu X, Miller JH, Yang W, Structure of the MutL C-terminal domain: a model of intact MutL and its roles in mismatch repair, EMBO J 23 (2004) 4134–4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Bende SM, Grafstrom RH, The DNA binding properties of the MutL protein isolated from Escherichia coli, Nucleic Acids Res 19 (1991) 1549–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.