

Abstract
Carbohydrates, one of the three primary macromolecules of living organisms, play significant roles in various biological processes such as intercellular communication, cell recognition, and immune activity. While the majority of established methods for the installation of carbohydrates through the anomeric carbon rely on nucleophilic displacement, anomeric radicals represent an attractive alternative because of their functional group compatibility and high anomeric selectivities. Herein, we demonstrate that anomeric nucleophiles such as C1 stannanes can be converted into anomeric radicals by merging Cu(I) catalysis with blue light irradiation to achieve highly stereoselective C(sp3)–S cross-coupling reactions. Mechanistic studies and DFT calculations revealed that the C–S bond-forming step occurs via the transfer of the anomeric radical directly to a sulfur electrophile bound to Cu(II) species. This pathway complements a radical chain observed for photochemical metal-free conditions where a disulfide initiator can be activated by a Lewis base additive. Both strategies utilize anomeric nucleophiles as efficient radical donors and achieve a switch from an ionic to a radical pathway. Taken together, the stability of glycosyl nucleophiles, a broad substrate scope, and high anomeric selectivities observed for the thermal and photochemical protocols make this novel C–S cross coupling a practical tool for late-stage glycodiversification of bioactive natural products and drug candidates.
Graphical Abstract
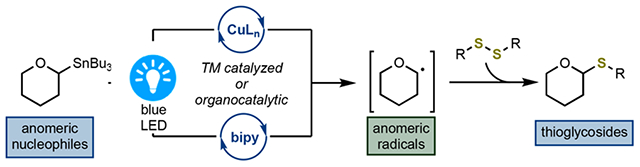
INTRODUCTION
As one of the main classes of biopolymers, carbohydrates are a structural element frequently found in bioactive natural products.1 While the O-linked saccharides constitute the bulk of known carbohydrates, modifications at the anomeric position such as C-, N-, and S-glycosylations represent an important class of glycoconjugates (Scheme 1A).2 In expanding the synthetic toolbox of preparative carbohydrate chemistry to access these valuable materials, chemical methodologies that capitalize on a (formal) nucleophilic displacement at the anomeric carbon have dominated the mechanistic thinking.3 Certain limitations of these methodologies such as suboptimal chemo- and regioselectivities still require general and broadly applicable solutions. In this regard, reactions that proceed through radical intermediates are an unexplored class of transformations. Anomeric radicals 6 are an established species, and their reactivity has been studied mostly in the context of C-glycosylations.4 Methods for generating anomeric radicals include photochemical activation of anomeric halides,5 nitro compounds,6 sulfones,7 and selenoglycosides8 (Scheme 1B). “Inverse” anomeric radicals located at the C5 position in pyranoses can be derived from the Hantzsch9,10 and redox-active esters11–14 or through decarbonylation of alkoxyacyl tellurides 8.15 Radicals generated with these methods have been utilized in the synthesis of complex targets such as sesquiterpenoids16 and diterpenoids,17,18 demonstrating their synthetic utility. However, these strategies are not without limitations, and the need to prepare “inverse” radical donors, the use of inherently unstable anomeric halides, and acyl 1,2-migrations19 remain as several of the key obstacles reducing their practical value.
Scheme 1.
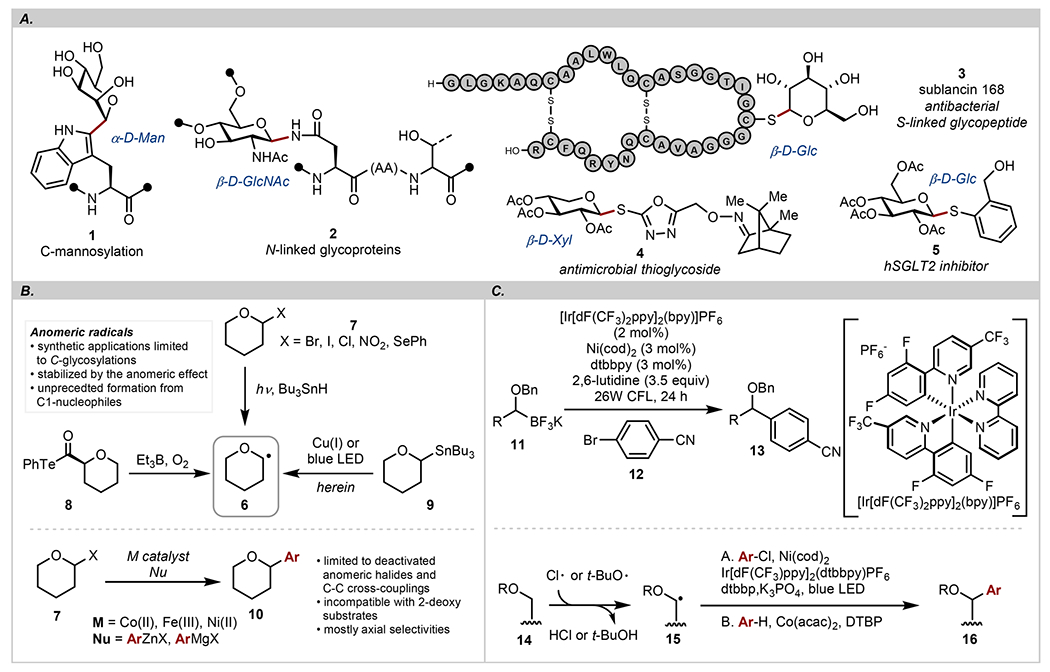
Considering the potential utility of anomeric radicals as synthetic intermediates, the merger of transition-metal-based catalysis and photochemical activation represents a fruitful strategy.20 However, despite their great potential, very little is known about transition-metal-mediated transformations that proceed through anomeric radicals. Compared to known strategies that utilize transition metals and photocatalysis,11,21–42 this represents a significant knowledge gap, and only recent studies have outlined the promise of this approach.43,44 Cross-coupling reactions with anomeric bromides/chlorides 5 and aryl organometallic reagents (ArZnX, ArMgX) using Co,45 Fe,46 and Ni9,10,47–49 catalysts indicate that the anomeric selectivities are dictated by the anomeric preference of the transition-metal center provided that the C—C bond-forming step occurs through a stereoretentive reductive elimination. Molander reported that under the photoredox conditions, the Hantzsch esters derived from C6 aldehydes form acyl C-glycosides.9,10 The authors postulated that the putative anomeric radicals undergo recombination with a Ni(0) center followed by reductive elimination, leading to a new C—C bond. Collectively, these studies show that anomeric control can be achieved by invoking kinetic anomeric effects of C1 radicals, representing a productive strategy to attach saccharides to various C(sp2) acceptors.
We recently described carbohydrate C1–stannanes, a class of anomeric nucleophiles, as competent partners in C–C,50 C–S(e),51,52 and C–O53 cross-coupling reactions. These processes proceed with high levels of stereoretention across a broad range of substrates, including saccharides with free hydroxyl groups. Inspired by the prior work with α-alkoxy boronates 11 that can generate heteroatom-substituted carbon-centered radicals54 and direct arylation reactions involving alkoxy radical intermediates 15 produced through a C–H cleavage reported with Co55 and Ni26,34 catalysts (Scheme 1C), we wondered if anomeric nucleophiles 9 could also function as a source of C1 radicals. This conceptually novel approach requires the generation of radicals from C1 nucleophiles with mildly oxidizing reagents, which could be realized with late transition metals56 or under light irradiation. The first method calls for a set of conditions that can weaken the putative C1–metal bond and generate a radical species as opposed to formation of oxocarbenium intermediates. Alternatively, the C–Sn bond in anomeric nucleophiles can be cleaved homolytically with a suitable radical initiator such as thiyl radicals.57,58 Here, we show that anomeric stannanes are a convenient source of anomeric radicals under thermal conditions using Cu(I) precatalyst or generated directly through light-mediated cleavage of a disulfide. Both pathways display high anomeric selectivities and are operational for a broad collection of saccharides, resulting in the synthesis of thioglycosides. Mechanistic and computational studies revealed that the transfer of a glycosyl group proceeds from a Cu(II) intermediate through a radical rebound pathway. In addition, we show that bipyridine additives facilitate the disulfide cleavage step by a previously unknown mechanism of chalcogen bonding, therefore representing a novel approach for merging organo-catalysis with light-mediated activation.59 Because stable anomeric nucleophiles of all common saccharides are available or can be readily prepared from commercial reagents, this method establishes a platform for direct incorporation of glycosides into various carbon- and heteroatom-based acceptors.
RESULTS AND DISCUSSION
Reaction Development with Cu(I) Catalysts.
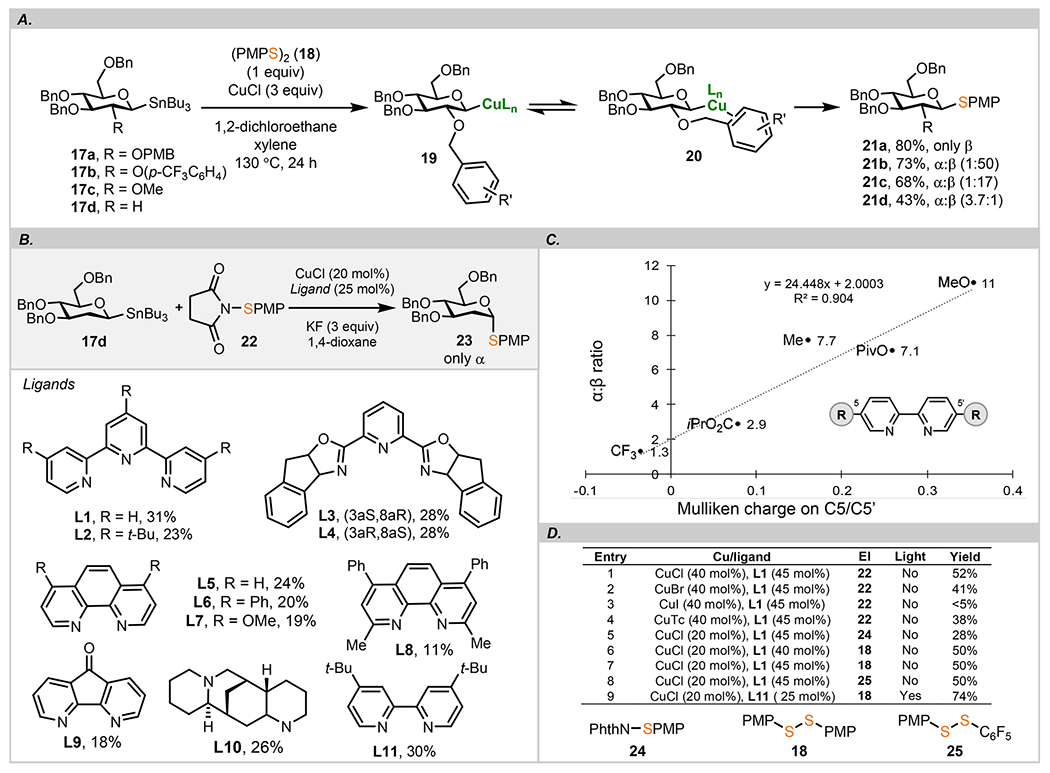
At the outset of our studies, we investigated reactions of anomeric stannanes as the source of carbanionic reactivity using copper(I) catalysts and various bidentate ligands.50 A stereoretentive transfer of glycosyl nucleophiles to Pd/Cu is consistent with the observed retention of configuration in C–C,50,60–62 C–S,51 and C–Se52 bond-forming processes. Selection of C–S cross coupling was dictated by the use of copper as the sole catalyst, therefore minimizing mechanistic analysis. Our investigations on a potential anomerization were prompted by experiments resulting in the erosion of stereochemical integrity correlated with the nature of a substituent at C2 (Scheme 2A). d-Glucose stannane 17a protected as a p-methoxybenzyl (PMB) ether afforded 21a in exclusive β selectivity using symmetrical disulfide (PMP)2S (18) as the sulfur electrophile. The anomeric ratio was slightly compromised when a benzyl ether containing a p-CF3 group was placed at C2 (21b, dr > 50:1). This observation led us to propose that the aromatic ring located in the vicinity of the putative C1 organocopper intermediate can be stabilized by π bonding (20).63,64 The extent of this stabilization and consequently the anomeric selectivity can correlate with the electronic factors of the benzyl group. This proposal is consistent with the observations reported by Molander in Pd-catalyzed cross-coupling reactions of α-alkoxytrifluoroborates.64 Next, we wondered if removal of a coordinating group at C2 could further compromise the selectivity. Indeed, installation of a methyl ether in 17c reduced the α:β ratio to 1:17, and with complete removal of oxygen in 17d, reversal of the anomeric preference was noted (21d). The high anomeric selectivity could be restored through a stereoretentive pathway using a bulky phosphine (JackiePhos)65 as the ligand stabilizing the putative Cu(III) intermediate (for details, see the SI). The results with 17 indicate that, in the absence of an aromatic ether at C2 or in electron-rich 2-deoxy pyranoses, dissociation of the C1–Cu bond may be a reasonably facile process and that the product distribution is likely a result of a competition between the stereoretentive and the stereoconvergent pathways (vide infra).
Scheme 2. Reaction Development with C1–Stannanes.
aReaction conditions: 17d (1.5 equiv), 22 (0.100 mmol, 1.0 equiv), CuCl (20 mol %), ligand (25 mol %), and 1,4-dioxane (2.0 mL) under N2, 130 °C, 24 h. Unless otherwise specified, only the α anomer was detected. Anomeric selectivities determined by 1H NMR analysis of unpurified reaction mixtures. bMulliken charge on C5/C5′ calculated at the B3LYP/6-31G(d) level of theory. cNinety-six hour reaction time. Entry 9: 5 W blue LED lamp was used.
Intrigued by these results, we next wondered if stereoretentive reactions observed in ligand-free C–S cross couplings51 could be diverted into a completely stereoconvergent pathway provided that the putative C1–Cu bond could be weakened to allow for a reversible anomerization prior to the reductive elimination step. This process could be induced by external ligands, and amines that can stabilize Cu(II/III) were selected as the likely candidates based on prior studies.66 A number of asymmetric copper-catalyzed C–C or C–heteroatom bond-forming reactions which proceed via intermediacy of a carbon-centered radical with copper(II) to generate an organocopper-(III) species67–69 were reported to operate with nitrogen-containing ligands. In addition to electronic stabilization of Cu in a high oxidation state, large ligands may (a) enhance dissociation of a C1–Cu bond due to a steric clash between the ligand and the pyranose ring and (b) reduce the rate of oxidative addition across the S–S bond, hence enabling a more facile epimerization.
The reaction of d-glucopyranosylstannane 17d and N-(4-methoxyphenyl)sulfenylsuccinimide 22 was selected to assess the impact of nitrogen-containing ligands on the anomeric selectivity in 23 (Scheme 2B). To minimize the effect of the substituents at C2, we focused on 2-deoxy substrates at this stage of reaction development. Tridentate ligands (L1–L4) in the presence of CuCl (20 mol %) and KF (3 equiv) in 1,4-dioxane at 130 °C were assayed first. Terpyridine L1 and 4,4′,4″-tritert-butyl-2,2′:6′,2″-terpyridine L2 produced the desired thioglycoside 23 in 31% and 23% yield, respectively, with exclusive α selectivity. To test the impact of more congested ligands, both enantiomers of PYBOX L3 and L4 were also evaluated under identical conditions and both led to identical results with a 28% isolated yield of 23 and exclusive α anomer. Other chiral pyridines tested had no effect on the selectivity, indicating that the stereodetermining step is not controlled by the environment around the copper center but likely is a result of the inherent stereochemical preferences of the anomeric radical intermediate.
In order to improve the yield, we next evaluated bidentate nitrogen ligands (L5–L11). Phenanthroline (L5) and its derivatives such as L6–L8 slightly decreased the overall yield while the stereoselectivity remained unaffected likely due to the rigidity of the planar phenanthroline scaffold. 4, 5-Diazafluoren-9-one (L9) and sparteine (L10) were equally less efficient, while 4,4′-di-tert-butylpyridine L11 provided an interesting lead for further optimizations because of its exclusive axial selectivity and a slightly improved yield (31%). Assessment of various bipyridine ligands indicates that the electronic nature of the substituent at C5/C5′ controls the selectivity of the reaction (Scheme 2C). Electron-donating groups such as ethers and esters promote the reaction in high α preference, although a notable erosion of anomeric integrity was noted when compared to other ligands described earlier (e.g., L11). Electron-withdrawing substituents, on the other hand, such as CF3 resulted in almost equimolar amounts of both anomers. The observed α:β ratio correlates well with the calculated Mulliken charges on C5/C5′ (Scheme 2D). This direct relationship between the electronic nature of the group at C5/C5′ and selectivity sharply contrasts with a clean conversion of 17d into the axial anomer 23 with tert-butylbipyridine L11. We note that other modifications of the 2,2′-bipyridine scaffold at C4 and C6 revealed a similar trend, where the electron-donating groups diminished the ratio of the axial anomer (for details, see the SI). Collectively, these results indicate that electron-rich substituents favor high axial selectivity likely because of their propensity to promote cleavage of the equatorial C–Cu bond to generate anomeric radicals rather than a direct stereoretentive reductive elimination from a Cu(III) species to produce the equatorial anomer.
From the initial optimizations, terpyridine L1 emerged as the optimum ligand for subsequent studies. We next focused on fine tuning the catalytic transformation with L1 in the presence of CuCl and a fluoride source in a mixture of m-xylene and 1,4-dioxane at 130 °C. Different fluoride sources were first assayed (LiF, NaF, CsF), but they all produced 23 in yields lower than KF. By increasing the amount of CuCl to 40 mol % (entry 1), 23 could be obtained in 52% yield as a single α anomer. Other common copper salts were also screened (entries 2–4), but again, CuBr and CuTc afforded 23 only in moderate yields with a trace amount of the product found for CuI (entry 3). To further improve the yield, we next evaluated various unsymmetrical and symmetrical sulfides equipped with competent leaving groups (entries 5–8). Our investigation of these electrophiles was based on the hypothesis that a suitable leaving group in the electrophilic component might facilitate C–S cross coupling by accelerating the oxidative addition step.70,71 During these studies, we discovered that phthalimide-containing thiophenol 24 was less effective than succinimide-modified thiophenol 22 and provided 23 in only 28% yield (entry 5). Disulfides such 18 and 25 furnished 23 in a moderate yield (50%, entry 6) but with a very good α selectivity (α:β > 30:1). We reasoned that the competition between terpyridine and thiophenolate bound to a highly oxidized copper center resulted in the observed scrambling of stereochemistry. Curiously, only the α anomer was detected when a small excess (45 mol %) of L1 was employed (entry 7). When testing the reaction with unsymmetrical disulfide 25, 23 was isolated in 36% yield and the competing transfer of the pentafluorophenyl thiolate to C1 position was not detected (entry 8).
When conducting the thioglycosylation reaction for an extended time (4 days), we observed that the reaction stopped at about 70% conversion of the sulfur electrophile. According to the literature report, disulfides can undergo a homolytic cleavage to form thiyl radicals under blue light irradiation.72 We next hypothesized that if two sulfur radicals could combine with Cu(I) to form a Cu(III) intermediate, then the overall efficiency of the reaction could be improved. Indeed, under blue LED irradiation, disulfide 18 and 4,4′-di-tert-butyl-2,2′-dipyridine L11 furnished 23 in 73% yield (entry 9). To our delight, the optimal yield was achieved when the amount of CuCl was kept below 20 mol %.
Scheme 3 lists thioglycosides 27 obtained under the optimized conditions. Common monosaccharides such as d-glucose, d-galactose, and l-fucose could be converted into thioglycosides efficiently. We observed that the anomeric selectivities in reactions with d-glucose are correlated with the size of the protective group at C2. For instance, large groups such as benzyl ethers show only a slight preference toward the axial anomer 27a, while a reduced steric bulk at C2 improved the selectivity as well as yields (27b, 27c). These results can be rationalized by the stabilizing interactions between C1–Cu and the electron-rich group, as depicted in Scheme 2A. Considering the effect of 1,2-cis interactions between the Cu(I)/Cu(III) center, the kinetic ability of anomeric radicals to undergo dissociation has to be taken into account as well. In general, negligible effects of the nature of the protective group on the anomeric selectivities were noted (27d, 27e). Similarly, the identity of the saccharide core (e.g., d-galactose 27f and l-fucose 27g) had little impact on the overall efficiency of the process as long as the positions neighboring the putative radical are a methylene group. Along the same lines, strong axial preferences were observed regardless of the nature of the sulfur electrophile, although electron-rich thiols resulted in better yields (27i–271). This protocol represents a viable solution to the general synthesis of 2-deoxy sugars which cannot be easily prepared from anomeric halides due to a rapid elimination and formation of glycals.46 To further extend the scope of this transformation to other electrophiles, we investigated diselenides, which are known to partake in retentive C–Se cross couplings (Scheme 3B).52 We found, similarly to the C–S bond-forming processes, that these products could be formed in high axial selectivities (27m–27o). However, substantial competition between a stereoretentive manifold and radical anomerization was observed, likely due to a direct substitution mechanism operational for the synthesis of selenoglycosides from anomeric nucleophiles.52
Scheme 3. Scope of Cu(I)-Catalyzed Thio(seleno)glycosylationc.
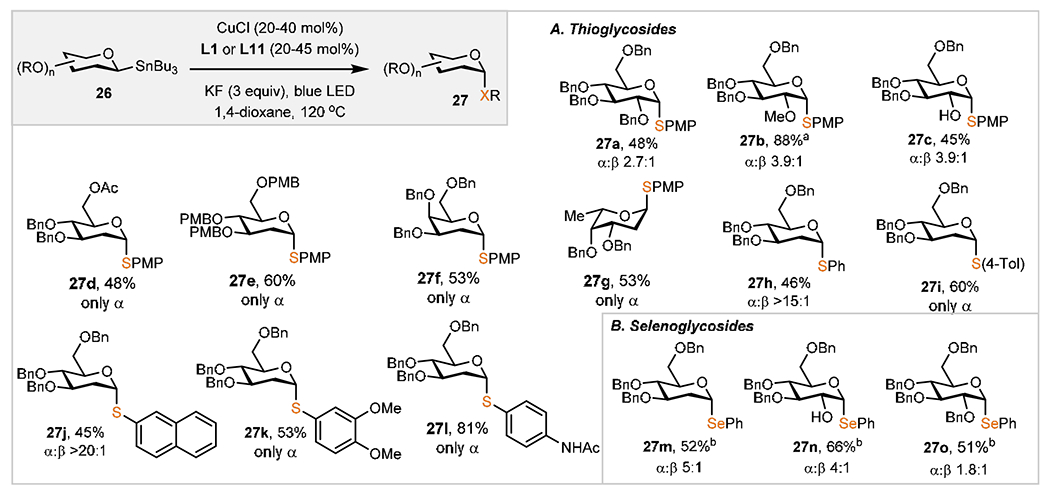
aCuCl (20 mol %), 26 (1.5 equiv), L1 (25 mol %), KF (3 equiv), 1,4-dioxane (2 mL), 130 °C, 96 h. bCuCl (20 mol %), 26 (1.5 equiv), L1 (25 mol %), KF (3 equiv), m-xylene:1,4-dioxane (1:1, 2.00 mL), 130 °C, 96 h. cGeneral reaction conditions: diaryl disulfides or diaryl diselenides (0.100 mmol, 1 equiv), CuCl (20–40 mol %), 26 (1.5 equiv), L11 (25–45 mol %), KF (3 equiv), blue LED (5 W), 1,4-dioxane (2 mL), 120 °C, 96 h; isolated yields. Anomeric selectivities determined by 1H NMR analysis of unpurified reaction mixtures.
Mechanistic Studies on Cu(I)-Catalyzed C–S Cross Coupling.
Consistently high anomeric selectivities led us to undertake mechanistic studies to test the overarching mechanistic hypothesis summarized in Scheme 4A. After the initial transmetalation step of stannanes 9 to Cu(I) complex 28, the organocopper intermediate 29 undergoes oxidative addition with a disulfide or N-thiosuccinimide 30 forming transient Cu(III) species 31. Although these two steps can be, in principle, reversed and transmetalation from organostannane 9 to Cu(III) can be envisioned as a possible pathway, we believe this order of events would likely be characterized by unfavorable thermodynamics due to the necessary ligand dissociation step. Four pathways for the ultimate formation of a new C(sp3)–S bond in 34 and 35 were considered: (i) stereoretentive reductive elimination from 31 that leads to the overall retention at the anomeric carbon (path A), (ii) homolytic cleavage of the C1–Cu bond in 31 resulting in formation of a pair of radicals 8 and 32 that subsequently transfer the sulfur group directly from 32 through a rebound pathway involving 36 (path B), (iii) recombination of radicals 8 and 32 followed by a stereoretentive reductive elimination resulting in the axial anomer 33 followed by reductive elimination (path C), and (iv) direct reaction of disulfides or N-thiosuccinimides 30 with anomeric radicals 8 (path D). All of these pathways account for the observed catalytic nature of the studied process, and the radical chain reactions described for pathway D can be initiated by Cu(I).
Scheme 4.
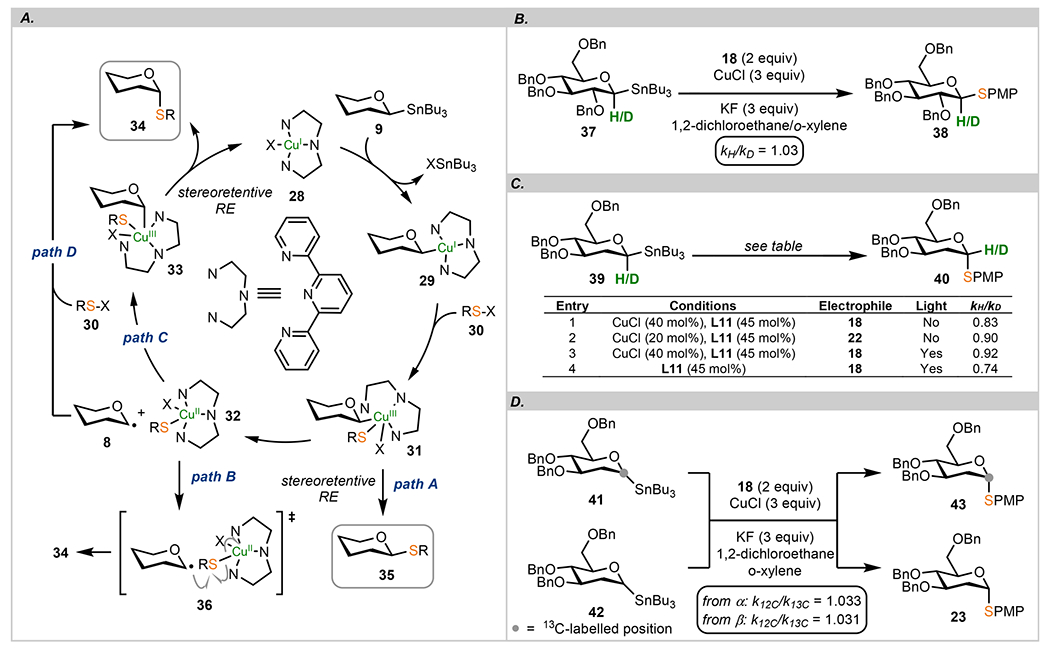
(A) Proposed Mechanism of Cu(I)-Catalyzed Thioetherification; (B) Deuterium Kinetic Isotope Effect Studies of Stereoretentive C(sp3)–S Cross Coupling; (C) Deuterium Kinetic Isotope Effect Studies for Stereoconvergent C(sp3)–S Cross Coupling; (D) 13C Kinetic Isotope Effect Studies of Stereoconvergent C(sp3)–S Cross Coupling
To supplement the mechanistic studies described earlier, we conducted additional investigations as summarized below.
Under the thermal or photochemical conditions, β-linked thioglycosides and glycosyl stannanes do not undergo anomerization to the axial isomers. These observations exclude the possibility that the thermodynamic preferences of the thioglycoside products or stannanes control the selectivity of the C–S cross coupling.
Copper(II) precatalysts such as CuCl2 (20 mol %) with terpyridine L1 do not promote the cross coupling of anomeric stannanes with disulfides and N-sulfenylsuccinimides (1,4-dioxane, 130 °C). Using identical conditions but with blue light irradiation, α anomer 23 was formed in 16% and 52% yield in reactions of 17d with 18 and 22, respectively.
C–S cross couplings can be accomplished with other late transition-metal catalysts; reactions of (PMPS)2, stannane 17d, and terpyridine ligand L1 (45 mol %) with various precatalysts (40%) such as NiCl2(DME), PdCl2(MeCN)2, or AuCl(PPh3)3 afforded only α-thioglycoside 23 in exclusive selectivities but variable yields (16–28%). Interestingly, CoCl2(PPh3)2 completely suppressed the reaction, and 17d was recovered. In the absence of CuCl, no products were found in a reaction of disulfides and N-sulfenylsuccinimide under thermal conditions with 2-deoxy-stannane 17d.
Radical trapping experiments with TEMPO (2 equiv) reduced the yield of thioglycoside 23 to 25% (from 53%) without erosion of anomeric selectivity under the optimized protocol from Scheme 2D. Under the same conditions, TEMPO trapping product was isolated in 49% yield (independently confirmed by synthesis from a disulfide, see the SI for details). Efforts to trap the putative C1 radical with an alkene acceptor such as a 2-O-allyl group afforded d-glucal as the only product (68%).
To distinguish between a stereoretentive and a radical pathway, we performed a CuCl-catalyzed glycosylation of β-stannane 17d and 18 in the presence of an electron-deficient 5,5′-bis(trifluoromethyl)-2,2′-bipyridine (45 mol %) that furnished 23 in a 2.7:1 ratio of α:β anomers. In contrast, identical conditions applied to the α-stannane resulted in exclusive α-selectivity in 23. Furthermore, under identical reaction conditions but without CuCl, both anomers afforded the axial anomer of 23 exclusively. This set of experiments, further confirmed computationally, demonstrates that formation of the β-anomers is a consequence of two competing pathways and the β-anomer is likely a result of a direct reductive elimination from 31.
2-Deoxys sugars and substrates with the C2 position substituted with oxygen show substantially different rates of C(sp3)–S cross coupling. For example, irradiation of (2,3,4,6-tetra-O-benzyl-β-d-glucopyranosyl)tri-n-butyl-stannane and 18 afforded 27a in a trace yield (<5%) when CuCl was absent but a moderate yield when present, and α:β 2.7:1 selectivity was obtained with CuCl under the same conditions (Scheme 3). The disparate reactivity of 2-deoxy sugars in the absence of CuCl indicates that the propensity of anomeric stannanes to form C1 radical is markedly higher when a copper(I) catalyst is absent. We attribute this to a difference in the electronic stabilization of anomeric radicals that are located within a more electron-rich ring.
A summary of the deuterium kinetic isotope effects (DKIEs) is presented in Scheme 4B. For stereoretentive C(sp3)–S cross couplings, normal kH/kD was observed in a reaction with β-anomer 37. This result is consistent with DKIEs for C(sp2)–C(sp3) cross couplings with other configurationally stable C1 nucleophiles.61 Starting from the β-anomer of 2-deoxy-d-glucose 37, inverse DKIEs were observed under thermal conditions with disulfide 18 (entry 1) and succinimide 22 (entry 2) as well as the photochemical conditions with a disulfide (entry 3, Scheme 4C). The magnitude of these effects is dependent on the conditions, and entries 1–3 show similar DKIEs, indicating analogous rate-determining steps for these processes. However, in the absence of CuCl but with blue LED (entry 4), a much lower DKIE was recorded, supporting the proposal that the mechanism of the light-mediated reaction is different from the metal-catalyzed reactions (under identical conditions, N-arylthiosuccinimides were found inert). Under blue LED, formation of the C(sp3)–S bond is the product-limiting step (consistent with the kinetic anomeric effect observed in the reactions of radicals generated from selenoglycosides with alkene acceptors8 or earlier work by Giese5). Secondary DKIEs for Cu(I)-catalyzed reactions indicate that either the oxidative addition step (29 → 31) or radical recombination are the rate-determining steps in the catalytic cycle, whereas reductive elimination is the product-determining step. The magnitude of inverse DKIEs for the reactions with Cu(I) suggests also that the light-mediated reactions are likely to procced by path D (Scheme 4A), and control of the anomeric configuration via the kinetic anomeric effect of C1 radical in this path is different from the reactions in the presence of Cu(I).
To complement DKIE studies, we measured the 13C kinetic isotope effect for stereoconvergent couplings with both anomers of 13C-labeled stannanes 41 and 42 (Scheme 4D). These compounds were easily obtained from commercial 13C1-enriched d-glucose in 3 steps (see the SI for details). At 130 °C in 1,4-dioxane, both anomers showed comparable k12C/k13C values (within experimental error), suggesting that these reactions share similar/identical first irreversible step(s). This observation also implies that the transmetalation steps for both anomers may have similar rates and identical intermediates.
Stereoretentive pathway A is likely operational for ligands that show a weak propensity to stabilize Cu(II) radicals or, in general, for reactions that would be termed as “ligand-free”. Studies with various 2,2′-bipyridine ligands (Scheme 2C) in combination with the reactions of 2-deoxy-α-d-glucose stannane in the presence of 2,2-bipyridine ligands known to lead to scrambling of stereochemistry demonstrated that formation of the β-anomers is a result of the stereoretentive reaction rather than a reaction of anomeric radicals from the equatorial face. Pathway B, where the anomeric radical reacts directly with sulfur bound to Cu(II), cannot be excluded because no difference of anomeric selectivity was found when chiral ligands L3/L4 were used (Scheme 2B). These observations suggest that the steric environment around copper does not impact the stereochemistry of the newly formed bond and C–S bond formation does not proceed from Cu(III) via reductive elimination. Pathway C combines the kinetic anomeric effect and thermodynamic stabilization of anomeric copper intermediates in the axial configuration. Different secondary DKIEs for the metal-catalyzed and metal-free processes support the proposal that pathways B and C have different rate-determining steps, although in the case of Cu-catalyzed reactions, the more appropriate term would be a turnover-limiting step as the reaction with disulfide is a stoichiometric process.
To shed light on pathways B and C for generation of phenyl α- and β-2-deoxy-1-thioglucosides, we resorted to density functional theory (DFT) calculations. The computed free energy profiles and the optimized structures of key transition states are shown in Scheme 5. From the Cu(II) species int1 and the anomeric glycosyl radical int2, the outer-sphere C–S bond formation transition state TS3 requires an 8.7 kcal/mol barrier and leads to the α-2-deoxy-1-thioglucoside product. Alternatively, the facile C–Cu bond formation via TS5 generates the intermediate int6. This α-Cu(III) intermediate int6 has a similar stability as the separated Cu(II) int1 and glycosyl radical int2, indicating the reversible nature of the C–Cu bond formation and a fast equilibrium between the two states. int6 can undergo an inner-sphere C–S bond formation through TS7 to form the same α-2-deoxy-1-thioglucoside product. TS7 is 1.2 kcal/mol less favorable than TS3; thus, generation of α-2-deoxy-1-thioglucoside occurs through the outer-sphere pathway.
Scheme 5. DFT-Computed Gibbs Free Energy Diagrams of Proposed Reaction Pathways for Generation of α- and β-Glycosides.
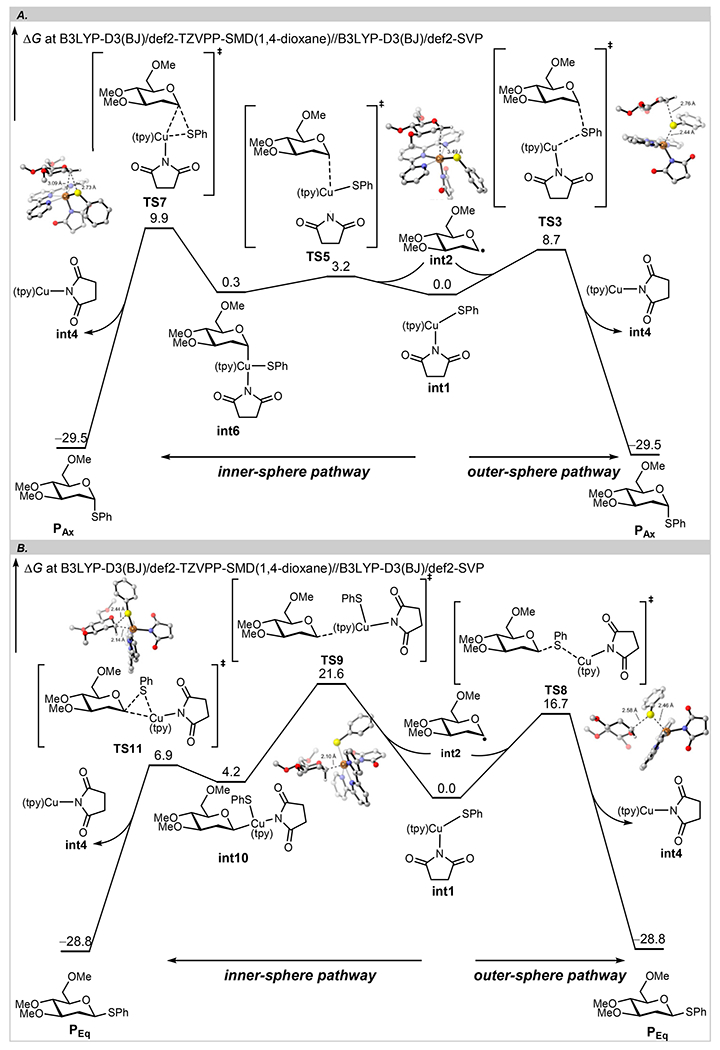
aTrivial hydrogens are omitted for clarity for optimized structures of key transition states.
The same preference for the outer-sphere pathway exists for the β-2-deoxy-1-thioglucoside formation. The outer-sphere C–S bond formation via TS8 is 4.9 kcal/mol more favorable than the C—Cu bond formation via TS9. This suggests that the β-Cu(III) species int10 is not involved in the reaction. It should be emphasized that the intrinsic reductive elimination barrier of int10 is only 2.7 kcal/mol; thus, the stereospecific functionalization of glycosyl is feasible if β-Cu(III) species can be generated from a different approach. Comparing the outer-sphere pathways, formation of α-2-deoxy-1-thioglucoside is 8.0 kcal/mol more favorable than formation of β-2-deoxy-1-thioglucoside (TS3 vs TS8), consistent with our studies on stereoretentive C–S cross coupling. This is in line with the anomeric nature of the glycosyl radical73 and elucidates the molecular basis for the observed stereoconvergent C–S bond formation in a thioglycosylation reaction.
Photochemical Thioglycosylation with Anomeric Stannanes.
Studies of the Cu-catalyzed thioetherifications indicate a likely role of anomeric radicals in the outer-sphere transfer of the glycosyl group. Intrigued by these results, we next investigated reactions promoted solely by light. On the basis of the mechanistic proposal presented in Scheme 4A, direct reaction of a disulfide under blue LED with anomeric radical 8 may generate a thiyl radical, which then can be engaged in a productive chain reaction with anomeric stannanes 9 (path D). To test this hypothesis, we conducted a reaction screen with symmetrical disulfide 18 and 2-deoxy-saccharide 17d, and these studies revealed an interesting trend regarding the role of 2,2′-bipyridine additives on the yield of 23 and the rate of cross coupling (Scheme 6A). By testing a series of 2,2′-bipyridine additives, 4,4′-di-tert-butyl-2,2′-bipyridine L11 emerges as a ligand that improved the reaction yield by approximately 20% (entry 1) with a 2× increase of the reaction rate when compared to ligand-free conditions (entry 3). Other pyridines showed a similar accelerating effect but to a lesser extent (for details, see the SI). Interactions of disulfides with electron-rich alkenes such as styrene were proposed in visible light-mediated cleavage of olefins.74 We extend this mechanistic hypothesis to bipyridine additives, which can engage in chalcogen bonding with a disulfide, effectively weakening the S–S bond in 44 (Scheme 6B).75 We tested this hypothesis by measuring changes in the 1H NMR spectra of mixtures of 18 with L11 and other 2,2′-bipyridine additives. The MeO group in 18 (3.83 ppm, CDCl3) was used as a diagnostic signal indicative of electronic interactions with Lewis basic additives. At a 75:1 ratio of L11 to 18, the MeO signal underwent an upfield shift to 3.81 ppm and then further to 3.59 ppm when the ratio of L11:16 was increased to 1000:1. 4,4′-Dimethoxy-2,2′-bipyridine shows an even more pronounced effect (Δδ 0.03 ppm at 75:1 ratio), whereas the 4-CF3 group exerts a much smaller shift (Δδ 0.01 ppm at 75:1 ratio). These observations are consistent with σ-hole bonding between a disulfide and a Lewis base such as bipyridines. Although these interactions are likely to be weak and the equilibrium between complexed/uncomplexed structures lies heavily on the side of the free substrates, weakening of the S–S bond becomes important in the photochemical initiation step. Alternatively, a charge-transfer complex formed between 2,2′-bipyridine–disulfide can lead to a rapid homolytic cleavage of the S–S bond. This new S–S activation mode is mechanistically similar to a homolytic cleavage of a B–B bond in diboranes by coordination with a Lewis base or through a labile EDA complex.76,77
Scheme 6. Photochemical Thioglycosylation with Anomeric Stannanesd.
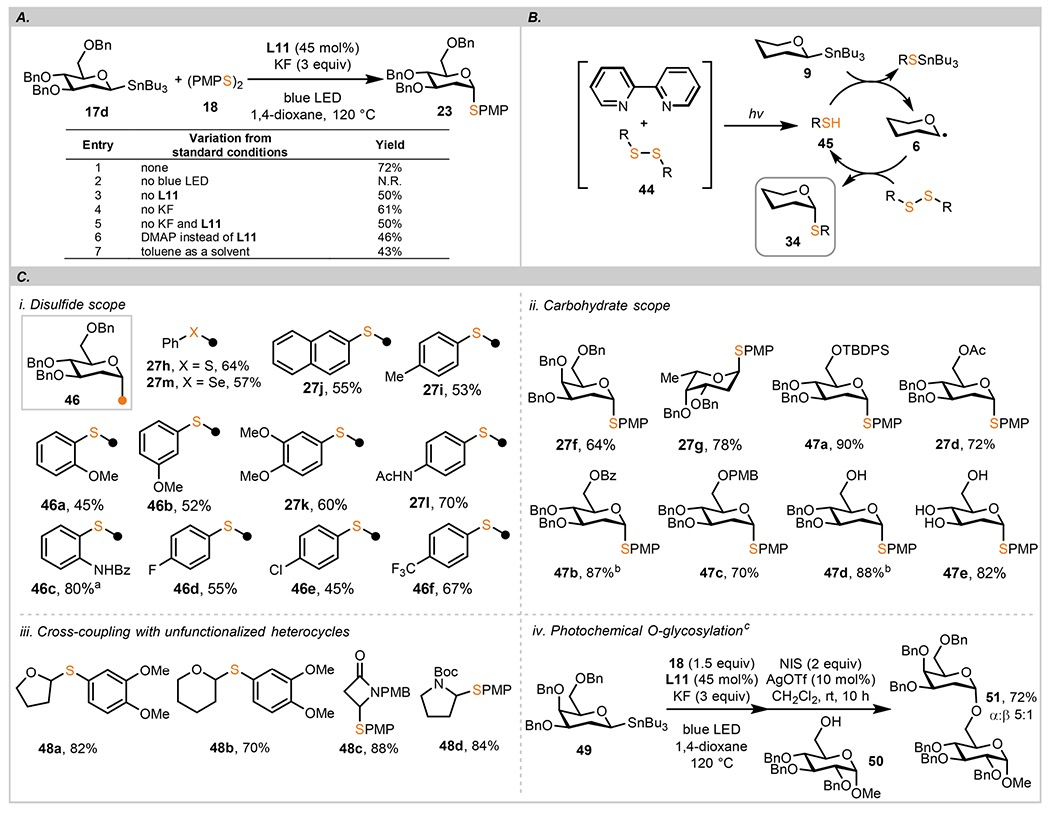
aα:β 10:1. bα-Stannanes were used. c18 (0.150 mmol, 1 equiv), 49 (1.5 equiv), L11 (45 mol %), KF (3 equiv), blue LED (5 W), 1,4-dioxane (3 mL), 120 °C, 48 h, then 2,3,4-tri-O-benzyl-α-d-glucopyranoside 50 (0.100 mmol), NIS (2.0 equiv), AgOTf (10 mol %), CH2Cl2 (5 mL), 23 °C, 10 h. dGeneral reaction conditions: disulfide (0.100 mmol, 1 equiv), anomeric stannanes (1.5 equiv), L11 (45 mol %), KF (3 equiv), blue LED (5 W), 1,4-dioxane (2 mL), 120 °C, 48 h. Unless otherwise specified, only the α anomer was detected. Anomeric selectivities determined by 1H NMR analysis of unpurified reaction mixtures.
Scheme 6C lists products obtained in a reaction of C1 stannanes with symmetric disulfides under blue light irradiation. 2-Deoxy-d-glucose thioglucosides 46 were efficiently formed under these conditions irrespective of the electronic nature of the disulfide electrophile. When comparing the two protocols, the light-mediated conditions complement the Cu-catalyzed transformations, which are more suitable for preparation of substituted monosaccharides, whereas 2-deoxy sugars are equally competent under both conditions.
Common protecting groups employed in carbohydrate chemistry such as Bn, TBDPS, Ac, and Bz are compatible with the optimized protocols, and good to excellent yields were obtained (47a–47c). Saccharides with free hydroxyl groups are competent substrates for this transformation as shown in the preparation of 47d and 47e. We note that this process is complementary to the direct thioglycosylation of anomeric alcohols under the Shoda conditions which produces the 1,2-trans anomers.78,79 However, the new protocol is highly α-selective, complementing the established β-selectivity with the imidazolium salts. These results support the notion that the photochemical thioglycosylation can be applied in a late-stage glycodiversification of complex molecules.80 it is worth noting that thioglycosylations starting from the α-stannane are more efficient than those using the β-anomers. These semiquantitative observations match the measured rates of reactions for both anomers (approximately 2.5 times; for details, see the SI). To further demonstrate the versatility of the protocol, we were delighted to find that photochemical thioetherifications are compatible with oxygen-bearing saturated heterocycles (furanosyl/pyranosyl stannanes; 48a, 48b) and other heterocyclic substrates such as azetidine- and pyrrolidine-derived α-amino-stannanes (48c, 48d). The reaction of furanosyl stannane with (PMPS)2 could be accomplished on a 1 g scale in 67% isolated yield. As an extension of the light-mediated thioglycosylation, we were able to develop a two-step protocol that converted anomeric stannane 49 into O-linked diglycoside 51 in an excellent yield (76%) and good anomeric selectivity (α:β 5:1).
CONCLUSIONS
It is becoming abundantly clear that anomeric nucleophiles are competent reagents in one- and two-electron processes catalyzed by late transition metals or through activation with visible light. These reactions can establish the glycosidic linkages with high anomeric selectivities in orthogonal pathways albeit through a common radical intermediate. While the anionic manifold of C1 stannanes has been demonstrated in a series of cross-coupling reactions, the radical pathway remained largely unexplored prior to our studies and focused mainly on mechanistic studies. Here, we showed that a radical pathway with sulfur electrophiles such as disulfides under photochemical strategies can efficiently induce anomeric radicals, and the strereocontrol at the anomeric carbon can be achieved via the kinetic anomeric effect. The overall efficiency of these thioglycosylation reactions and likely other carbon–carbon and carbon–heteroatom forming processes capitalizes on a facile activation by a copper catalyst with nitrogen-based ligands or a direct homolytic cleavage of a disulfide assisted by nitrogen ligands acting as a Lewis base additive. Both strategies achieve a switch of the reaction pathway from ionic to radical. In a broader sense, these novel activation methods are suitable for a late-stage glycodiversification due to its mildness and generality.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the University of Colorado Boulder, National Science Foundation (CAREER Award No. CHE-1753225), National Natural Science Foundation of China (21702182), Zhejiang University, Chinese “Thousand Youth Talents Plan”, and “Fundamental Research Funds for the Central Universities”. Calculations were performed on the high-performance computing system at the Department of Chemistry, Zhejiang University.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c03298.
Detailed experimental procedures, computational details, and copies of NMR spectra for all new compounds (PDF)
The authors declare no competing financial interest.
Contributor Information
Feng Zhu, Department of Chemistry, University of Colorado, Boulder, Colorado 80309, United States.
Shuo-qing Zhang, Department of Chemistry, Zhejiang University, Hangzhou, Zhejiang 310027, People’s Republic of China.
Zhenhao Chen, Department of Chemistry, University of Colorado, Boulder, Colorado 80309, United States.
Jinyan Rui, Department of Chemistry, University of Colorado, Boulder, Colorado 80309, United States.
Xin Hong, Department of Chemistry, Zhejiang University, Hangzhou, Zhejiang 310027, People’s Republic of China.
Maciej A. Walczak, Department of Chemistry, University of Colorado, Boulder, Colorado 80309, United States.
REFERENCES
- (1).Varki A; Cummings RD; Esko JD; Freeze HH; Stanley P; Bertozzi CR; Hart GW; Etzler ME Essentials of Glycobiology; Cold Spring Harbor Laboratory Press, 2009. [PubMed] [Google Scholar]
- (2).Cecioni S; Imberty A; Vidal S Glycomimetics versus Multivalent Glycoconjugates for the Design of High Affinity Lectin Ligands. Chem. Rev 2015, 115, 525–561. [DOI] [PubMed] [Google Scholar]
- (3).Hagen B; Vorm S; Hansen T; Marel GA; Codée JDC Stereoselective Glycosylations–Additions to Oxocarbenium Ions. In Selective Glycosylations: Synthetic Methods and Catalysts; Bennett CS, Ed.; Wiley, 2017; pp 1–28. [Google Scholar]
- (4).Yang Y; Yu B Recent Advances in the Chemical Synthesis of C-Glycosides. Chem. Rev 2017, 117, 12281–12356. [DOI] [PubMed] [Google Scholar]
- (5).Giese B; Dupuis J DiastereoseIective syntheses of C-glycopyranosides. Angew. Chem., Int. Ed. Engl 1983, 22, 622. [Google Scholar]
- (6).Dupuis J; Giese B; Hartung J; Leising M; Korth HG; Sustmann R Electron transfer from trialkyltin radicals to nitrosugars: the synthesis of C-glycosides with tertiary anomeric carbon atoms. J. Am. Chem. Soc 1985, 107, 4332–4333. [Google Scholar]
- (7).Miquel N; Doisneau G; Beau J-M Reductive Samariation of Anomeric 2-Pyridyl Sulfones with Catalytic Nickel: An Unexpected Improvement in the Synthesis of 1,2-trans-Diequatorial C-Glycosyl Compounds. Angew. Chem. Int. Ed 2000, 39, 4111–4114. [DOI] [PubMed] [Google Scholar]
- (8).Abe H; Shuto S; Matsuda A Highly α- and β-selective radical C-glycosylation reactions using a controlling anomeric effect based on the conformational restriction strategy. A study on the conformation-anomeric effect-stereoselectivity relationship in anomeric radical reactions. J. Am. Chem. Soc 2001, 123, 11870. [DOI] [PubMed] [Google Scholar]
- (9).Dumoulin A; Matsui JK; Gutiérrez-Bonet Á; Molander GA Synthesis of Non-Classical Arylated C-Saccharides through Nickel/Photoredox Dual Catalysis. Angew. Chem., Int. Ed 2018, 57, 6614–6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Badir SO; Dumoulin A; Matsui JK; Molander GA Synthesis of Reversed C-Acyl Glycosides through Ni/Photoredox Dual Catalysis. Angew. Chem., Int. Ed 2018, 57, 6610–6613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Toriyama F; Cornella J; Wimmer L; Chen T-G; Dixon DD; Creech G; Baran PS Redox-Active Esters in Fe-Catalyzed C–C Coupling. J. Am. Chem. Soc 2016, 138, 11132–11135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Ji P; Zhang Y; Wei Y; Huang H; Hu W; Mariano PA; Wang W Visible-Light-Mediated, Chemo- and Stereoselective Radical Process for the Synthesis of C-Glycoamino Acids. Org. Lett 2019, 21, 3086–3092. [DOI] [PubMed] [Google Scholar]
- (13).Wan ICS; Witte MD; Minnaard AJ From d- to l-Monosaccharide Derivatives via Photodecarboxylation–Alkylation. Org. Lett 2019, 21, 7669–7673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ma Y; Liu S; Xi Y; Li H; Yang K; Cheng Z; Wang W; Zhang Y Highly stereoselective synthesis of aryl/heteroaryl-C-nucleosides via the merger of photoredox and nickel catalysis. Chem. Commun 2019, 55, 14657–14660. [DOI] [PubMed] [Google Scholar]
- (15).Masuda K; Nagatomo M; Inoue M Direct assembly of multiply oxygenated carbon chains by decarbonylative radical–radical coupling reactions. Nat. Chem 2017, 9, 207–212. [DOI] [PubMed] [Google Scholar]
- (16).Kuwana D; Nagatomo M; Inoue M Total Synthesis of 5-epi-Eudesm-4(15)-ene-1β,6β-diol via Decarbonylative Radical Coupling Reaction. Org. Lett 2019, 21, 7619–7623. [DOI] [PubMed] [Google Scholar]
- (17).Nagatomo M; Kamimura D; Matsui Y; Masuda K; Inoue M Et3B-mediated two- and three-component coupling reactions via radical decarbonylation of α-alkoxyacyl tellurides: single-step construction of densely oxygenated carboskeletons. Chem. Sci 2015, 6, 2765–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Hashimoto S; Katoh S.-i.; Kato T; Urabe D; Inoue M Total Synthesis of Resiniferatoxin Enabled by Radical-Mediated Three-Component Coupling and 7-endo Cyclization. J. Am. Chem. Soc 2017, 139, 16420–16429. [DOI] [PubMed] [Google Scholar]
- (19).Giese B; Gröninger KS; Witzel T; Korth H-G; Sustmann R Synthesis of 2-Deoxy Sugars. Angew. Chem., Int. Ed. Engl 1987, 26, 233–234. [Google Scholar]
- (20).Skubi KL; Blum TR; Yoon TP Dual Catalysis Strategies in Photochemical Synthesis. Chem. Rev 2016, 116, 10035–10074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Gong H; Sinisi R; Gagne MR A room temperature Negishi cross-coupling approach to C-alkyl glycosides. J. Am. Chem. Soc 2007, 129, 1908. [DOI] [PubMed] [Google Scholar]
- (22).Andrews RS; Becker JJ; Gagne MR Intermolecular addition of glycosyl halides to alkenes mediated by visible light. Angew. Chem., Int. Ed 2010, 49, 7274. [DOI] [PubMed] [Google Scholar]
- (23).Andrews RS; Becker JJ; Gagne MR A photoflow reactor for the continuous photoredox-mediated synthesis of C-glycoamino acids and C-glycolipids. Angew. Chem., Int. Ed 2012, 51, 4140. [DOI] [PubMed] [Google Scholar]
- (24).Ye Y; Sanford MS Merging visible-light photocatalysis and transition-metal catalysis in the copper-catalyzed trifluoromethylation of boronic acids with CF3I. J. Am. Chem. Soc 2012, 134, 9034–9037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Prier CK; Rankic DA; MacMillan DW Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis. Chem. Rev 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Shields BJ; Doyle AG Direct C(sp3)-H Cross Coupling Enabled by Catalytic Generation ofChlorine Radicals. J. Am. Chem. Soc 2016, 138, 12719–12722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Joe CL; Doyle AG Direct Acylation of C(sp3)-H Bonds Enabled by Nickel and Photoredox Catalysis. Angew. Chem., Int. Ed 2016, 55, 4040–4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Ahneman DT; Doyle AG C-H functionalization of amines with aryl halides by nickel-photoredox catalysis. Chem. Sci 2016, 7, 7002–7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Cornella J; Edwards JT; Qin T; Kawamura S; Wang J; Pan CM; Gianatassio R; Schmidt M; Eastgate MD; Baran PS Practical Ni-Catalyzed Aryl-Alkyl Cross-Coupling of Secondary Redox-Active Esters. J. Am. Chem. Soc 2016, 138, 2174–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Wang J; Qin T; Chen TG; Wimmer L; Edwards JT; Cornella J; Vokits B; Shaw SA; Baran PS Nickel-Catalyzed Cross-Coupling of Redox-Active Esters with Boronic Acids. Angew. Chem., Int. Ed 2016, 55, 9676–9679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Qin T; Cornella J; Li C; Malins LR; Edwards JT; Kawamura S; Maxwell BD; Eastgate MD; Baran PS A general alkyl-alkyl cross-coupling enabled by redox-active esters and alkylzinc reagents. Science 2016, 352, 801–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Kainz QM; Matier CD; Bartoszewicz A; Zultanski SL; Peters JC; Fu GC Asymmetric copper-catalyzed C-N cross-couplings induced by visible light. Science 2016, 351, 681–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Twilton J; Le C; Zhang P; Shaw MH; Evans RW; MacMillan DWC The merger of transition metal and photocatalysis. Nat. Rev. Chem 2017, 1, 0052. [Google Scholar]
- (34).Nielsen MK; Shields BJ; Liu J; Williams MJ; Zacuto MJ; Doyle AG Mild, Redox-Neutral Formylation of Aryl Chlorides through the Photocatalytic Generation of Chlorine Radicals. Angew. Chem., Int. Ed 2017, 56, 7191–7194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Paul A; Smith MD; Vannucci AK Photoredox-Assisted Reductive Cross-Coupling: Mechanistic Insight into Catalytic Aryl-Alkyl Cross-Couplings. J. Org. Chem 2017, 82, 1996–2003. [DOI] [PubMed] [Google Scholar]
- (36).Fu GC Transition-Metal Catalysis of Nucleophilic Substitution Reactions: A Radical Alternative to SN1 and SN2 Processes. ACS Cent. Sci 2017, 3, 692–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Ahn JM; Ratani TS; Hannoun KI; Fu GC; Peters JC Photoinduced, Copper-Catalyzed Alkylation of Amines: A Mechanistic Study of the Cross-Coupling of Carbazole with Alkyl Bromides. J. Am. Chem. Soc 2017, 139, 12716–12723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Ahn JM; Peters JC; Fu GC Design of a Photoredox Catalyst that Enables the Direct Synthesis of Carbamate-Protected Primary Amines via Photoinduced, Copper-Catalyzed N-Alkylation Reactions of Unactivated Secondary Halides. J. Am. Chem. Soc 2017, 139, 18101–18106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Ackerman LKG; Martinez Alvarado JI; Doyle AG Direct C-C Bond Formation from Alkanes Using Ni-Photoredox Catalysis. J. Am. Chem. Soc 2018, 140, 14059–14063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Key RJ; Vannucci AK Nickel Dual Photoredox Catalysis for the Synthesis of Aryl Amines. Organometallics 2018, 37, 1468–1472. [Google Scholar]
- (41).Bartoszewicz A; Matier CD; Fu GC Enantioconvergent Alkylations of Amines by Alkyl Electrophiles: Copper-Catalyzed Nucleophilic Substitutions of Racemic α-Halolactams by Indoles. J. Am. Chem. Soc 2019, 141, 14864–14869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Bour JR; Ferguson DM; McClain EJ; Kampf JW; Sanford MS Connecting Organometallic Ni(III) and Ni(IV): Reactions of Carbon-Centered Radicals with High-Valent Organonickel Complexes. J. Am. Chem. Soc 2019, 141, 8914–8920. [DOI] [PubMed] [Google Scholar]
- (43).Milligan JA; Phelan JP; Badir SO; Molander GA Alkyl Carbon–Carbon Bond Formation by Nickel/Photoredox Cross-Coupling. Angew. Chem., Int. Ed 2019, 58, 6152–6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Wiles RJ; Molander GA Photoredox-Mediated Net-Neutral Radical/Polar Crossover Reactions. Isr. J. Chem 2020, 60, 281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Nicolas L; Angibaud P; Stansfield I; Bonnet P; Meerpoel L; Reymond S; Cossy J Diastereoselective Metal-Catalyzed Synthesis of C-Aryl and C-Vinyl Glycosides. Angew. Chem., Int. Ed 2012, 51, 11101–11104. [DOI] [PubMed] [Google Scholar]
- (46).Adak L; Kawamura S; Toma G; Takenaka T; Isozaki K; Takaya H; Orita A; Li HC; Shing TKM; Nakamura M Synthesis of Aryl C-Glycosides via Iron-Catalyzed Cross Coupling of Halosugars: Stereoselective Anomeric Arylation of Glycosyl Radicals. J. Am. Chem. Soc 2017, 139, 10693–10701. [DOI] [PubMed] [Google Scholar]
- (47).Gong H; Andrews RS; Zuccarello JL; Lee SJ; Gagné MR Sn-Free Ni-Catalyzed Reductive Coupling of Glycosyl Bromides with Activated Alkenes. Org. Lett 2009, 11, 879–882. [DOI] [PubMed] [Google Scholar]
- (48).Zhao C; Jia X; Wang X; Gong H Ni-Catalyzed Reductive Coupling of Alkyl Acids with Unactivated Tertiary Alkyl and Glycosyl Halides. J. Am. Chem. Soc 2014, 136, 17645–17651. [DOI] [PubMed] [Google Scholar]
- (49).Liu J; Gong H Stereoselective Preparation of α-C-Vinyl/Aryl Glycosides via Nickel-Catalyzed Reductive Coupling of Glycosyl Halides with Vinyl and Aryl Halides. Org. Lett 2018, 20, 7991–7995. [DOI] [PubMed] [Google Scholar]
- (50).Zhu F; Rodriguez J; Yang T; Kevlishvili I; Miller E; Yi D; O’Neill S; Rourke MJ; Liu P; Walczak MA Glycosyl Cross-Coupling of Anomeric Nucleophiles: Scope, Mechanism, and Applications in the Synthesis of Aryl C-Glycosides. J. Am. Chem. Soc 2017, 139, 17908–17922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Zhu F; Miller E; Zhang S-q.; Yi D; O’Neill S; Hong X; Walczak MA Stereoretentive C(sp3)–S Cross-Coupling. J. Am. Chem. Soc 2018, 140, 18140–18150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Zhu F; O’Neill S; Rodriguez J; Walczak MA Stereoretentive Manipulations of Anomeric Nucleophiles: Applications in the Synthesis of Selenoglycosides. Angew. Chem., Int. Ed 2018, 57, 7091–7095. [DOI] [PubMed] [Google Scholar]
- (53).Yang T; Zhu F; Walczak MA Stereoselective oxidative glycosylation of anomeric nucleophiles with alcohols and carboxylic acids. Nat. Commun 2018, 9, 3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Tellis JC; Primer DN; Molander GA Single-electron transmetalation in organoboron cross-coupling by photoredox/nickel dual catalysis. Science 2014, 345, 433–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Li Q; Hu W; Hu R; Lu H; Li G Cobalt-Catalyzed Cross-Dehydrogenative Coupling Reaction between Unactivated C(sp2)–H and C(sp3)–H Bonds. Org. Lett 2017, 19, 4676–4679. [DOI] [PubMed] [Google Scholar]
- (56).Jahn U Radicals in Transition Metal Catalyzed Reactions? Transition Metal Catalyzed Radical Reactions?–A Fruitful Interplay Anyway. In Radicals in Synthesis III; Heinrich M, Gansäuer A, Eds.; Springer Berlin Heidelberg: Berlin, Heidelberg, 2012; pp 191–322. [DOI] [PubMed] [Google Scholar]
- (57).Russell GA; Ngoviwatchai P; Tashtoush H; Hershberger J Reaction of 1-alkenyl and 1-alkynyl derivatives of tin and mercury with hetero-centered radicals. Organometallics 1987, 6, 1414–1419. [Google Scholar]
- (58).Dénès F; Pichowicz M; Povie G; Renaud P Thiyl Radicals in Organic Synthesis. Chem. Rev 2014, 114, 2587–2693. [DOI] [PubMed] [Google Scholar]
- (59).Nicewicz DA; MacMillan DWC Merging Photoredox Catalysis with Organocatalysis: The Direct Asymmetric Alkylation of Aldehydes. Science 2008, 322, 77–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Zhu F; Rourke MJ; Yang T; Rodriguez J; Walczak MA Highly stereospecific cross-coupling reactions of anomeric stannanes for the synthesis of C-aryl glycosides. J. Am. Chem. Soc 2016, 138, 12049–12052. [DOI] [PubMed] [Google Scholar]
- (61).Yi D; Zhu F; Walczak MA Stereoretentive Intramolecular Glycosyl Cross-Coupling: Development, Scope, and Kinetic Isotope Effect Study. Org. Lett 2018, 20, 4627–4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Yi D; Zhu F; Walczak MA Glycosyl Cross-Coupling with Diaryliodonium Salts: Access to Aryl C-Glycosides of Biomedical Relevance. Org. Lett 2018, 20, 1936–1940. [DOI] [PubMed] [Google Scholar]
- (63).Goli M; He A; Falck JR Pd-catalyzed Cross-Coupling of α-(Acyloxy)-tri-n-butylstannanes with Alkenyl, Aryl, and Heteroaryl Electrophiles. Org. Lett 2011, 13, 344–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Molander GA; Wisniewski SR Stereospecific Cross-Coupling of Secondary Organotrifluoroborates: Potassium 1-(Benzyloxy)alkyltrifluoroborates. J. Am. Chem. Soc 2012, 134, 16856–16868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Hicks JD; Hyde AM; Cuezva AM; Buchwald SL Pd-Catalyzed N-Arylation of Secondary Acyclic Amides: Catalyst Development, Scope, and Computational Study. J. Am. Chem. Soc 2009, 131, 16720–16734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Wang F; Chen P; Liu G Copper-Catalyzed Radical Relay for Asymmetric Radical Transformations. Acc. Chem. Res 2018, 51, 2036-2046. [DOI] [PubMed] [Google Scholar]
- (67).Vasilopoulos A; Zultanski SL; Stahl SS Feedstocks to Pharmacophores: Cu-Catalyzed Oxidative Arylation of Inexpensive Alkylarenes Enabling Direct Access to Diarylalkanes. J. Am. Chem. Soc 2017, 139, 7705–7708. [DOI] [PubMed] [Google Scholar]
- (68).Zhang W; Wu L; Chen P; Liu G Enantioselective Arylation of Benzylic C–H Bonds by Copper-Catalyzed Radical Relay. Angew. Chem., Int. Ed 2019, 58, 6425—6429. [DOI] [PubMed] [Google Scholar]
- (69).Hu H; Chen S-J; Mandal M; Pratik SM; Buss JA; Krska SW; Cramer CJ; Stahl SS Copper-catalysed benzylic C–H coupling with alcohols via radical relay enabled by redox buffering. Nat. Catal 2020, 3, 358—367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Ramesh E; Guntreddi T; Sahoo AK AlCl3-Catalyzed Intermolecular Annulation of Thiol Derivatives and Alkynes by 1,2-Sulfur Migration: Construction of 6-Substituted Benzo[b]thiophenes. Eur. J. Org. Chem 2017, 2017, 4405—4413. [Google Scholar]
- (71).Xiong F; Lu L; Sun T-Y; Wu Q; Yan D; Chen Y; Zhang X; Wei W; Lu Y; Sun W-Y; Li JJ; Zhao J A bioinspired and biocompatible ortho-sulfiliminyl phenol synthesis. Nat. Commun 2017, 8, 15912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Wilger DJ; Grandjean J-MM; Lammert TR; Nicewicz DA The direct anti-Markovnikov addition of mineral acids to styrenes. Nat. Chem 2014, 6, 720–726. [DOI] [PubMed] [Google Scholar]
- (73).Giese B; Dupuis J Anomeric effect of radicals. Tetrahedron Lett. 1984, 25, 1349—1352. [Google Scholar]
- (74).Deng Y; Wei X-J; Wang H; Sun Y; Noël T; Wang X Disulfide-Catalyzed Visible-Light-Mediated Oxidative Cleavage of C═C Bonds and Evidence of an Olefin–Disulfide Charge-Transfer Complex. Angew. Chem., Int. Ed 2017, 56, 832—836. [DOI] [PubMed] [Google Scholar]
- (75).Vogel L; Wonner P; Huber SM Chalcogen Bonding: An Overview. Angew. Chem., Int. Ed 2019, 58, 1880–1891. [DOI] [PubMed] [Google Scholar]
- (76).Wang G; Zhang H; Zhao J; Li W; Cao J; Zhu C; Li S Homolytic Cleavage of a B–B Bond by the Cooperative Catalysis of Two Lewis Bases: Computational Design and Experimental Verification. Angew. Chem., Int. Ed 2016, 55, 5985—5989. [DOI] [PubMed] [Google Scholar]
- (77).Wu J; He L; Noble A; Aggarwal VK Photoinduced Deaminative Borylation of Alkylamines. J. Am. Chem. Soc 2018, 140, 10700—10704. [DOI] [PubMed] [Google Scholar]
- (78).Tanaka T; Nagai H; Noguchi M; Kobayashi A; Shoda S.-i. One-step conversion of unprotected sugars to β-glycosyl azides using 2-chloroimidazolinium salt in aqueous solution. Chem. Commun 2009, 3378—3379. [DOI] [PubMed] [Google Scholar]
- (79).Köhling S; Exner MP; Nojoumi S; Schiller J; Budisa N; Rademann J One-Pot Synthesis of Unprotected Anomeric Glycosyl Thiols in Water for Glycan Ligation Reactions with Highly Functionalized Sugars. Angew. Chem., Int. Ed 2016, 55, 15510–15514. [DOI] [PubMed] [Google Scholar]
- (80).Gantt RW; Peltier-Pain P; Thorson JS Enzymatic methods for glyco(diversification/randomization) of drugs and small molecules. Nat. Prod. Rep 2011, 28, 1811–1853. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.