

SUMMARY
Codon usage of each genome is closely correlated with the abundance of tRNA isoacceptors. How codon usage bias is resolved by tRNA post-transcriptional modifications is largely unknown. Here we demonstrate that the N1-methylation of guanosine at position 37 (m1G37) on the 3′-side of the anticodon, while not directly responsible for reading of codons, is a neutralizer that resolves differential decoding of proline codons. A genome-wide suppressor screen of a non-viable Escherichia coli strain, lacking m1G37, identifies proS suppressor mutations, indicating a coupling of methylation with tRNA prolyl-aminoacylation that sets the limit of cell viability. Using these suppressors, where prolyl-aminoacylation is decoupled from tRNA methylation, we show that m1G37 neutralizes differential translation of proline codons by the major isoacceptor. Lack of m1G37 inactivates this neutralization and exposes the need for a minor isoacceptor for cell viability. This work has medical implications for bacterial species that exclusively use the major isoacceptor for survival.
Graphical Abstract
In brief
Masuda et al. show that loss of m1G37 from the 3′ side of the tRNA anticodon renders a modified wobble nucleotide of the anticodon insufficient to decode a set of rare codons, providing a functional underpinning for the “modification circuit” between position 37 and the wobble position of the tRNA anticodon.
INTRODUCTION
In the degeneracy of the genetic code, proteins can be coded in multiple ways using different sets of synonymous codons. Each codon choice makes a specific demand for the supply of the tRNA isoacceptors with matching anticodons. This supply-to-demand balance is broadly observed throughout the domains of life. The impact of this balance is manifested in multiple aspects of a cellular life. However, the quality of a codon-anticodon pair is determined not only by the abundance of each isoacceptor but also by the epigenetic modifications to the anticodon of the isoacceptor (de Crecy-Lagard and Jaroch, 2021). The evolution of epigenetic modifications is an important mechanism to explore the wobble rule of the genetic code, where the wobble nucleotide of the anticodon at position 34 of tRNA can form non-canonical base pairing with the third nucleotide of a codon, enabling one isoacceptor to translate multiple codons (Agris et al., 2018). Indeed, many genomes in all three domains of life encode just one isoacceptor for all synonymous codons. How a single isoacceptor translates all synonymous codons with the speed and quality necessary to support life is poorly understood.
The wobble nucleotide hosts a diverse and high density of post-transcriptional modifications. Several wobble modifications can expand the anticodon-codon pairing beyond a canonical W-C structure to affect translation of multiple tRNA families. For example, the position-34 modification of adenosine to inosine (A34 to I34), expanding base pairing from A34 with U to I34 with C/U/A (Torres et al., 2014), is conserved in Eukarya in eight tRNA families (Ala, Arg, Ile, Leu, Pro, Ser, Thr, and Val). This expansion is most critical in organisms that use NNC as the preferred codons, but lack the corresponding tRNA(GNN) isoacceptors, implying that the I34-modified tRNA(ANN) isoacceptors are solely responsible for translation of NNC codons. Indeed, knockout (KO) of the A34-to-I34 conversion enzymes is lethal to cells, while I34 deficiency (KD) re-programs expression of NNC-enriched genes (Lyu et al., 2020). Similarly, several other position-34 modifications also expand the capacity of decoding (Agris et al., 2018). Deficiency of these modifications also produces changes of gene expression (Begley et al., 2007; Chionh et al., 2016; Zinshteyn and Gilbert, 2013). However, not all wobble base pairings are created equal (e.g., Figure S1), raising the question of whether some may be deficient in thermo-dynamic stability and thus would be discriminated against during the high demand for protein synthesis to support life.
An intriguing question is whether deficiency of a wobble base pairing can be compensated by other post-transcriptional modifications in the tRNA. Of interest is position 37 on the 3′ side of the anticodon, which is populated in the density and diversity of chemical modifications similarly to position 34. Importantly, while the position-37 modification is not within the anticodon triplet and is not directly involved in the anticodon-codon pairing, it is often conserved with the position-34 modification or other anticodon-loop modifications in a “modification circuit” (Han and Phizicky, 2018). These modification circuits are thought to enhance enzyme modification specificity, such that the first modification in the circuit is a recognition element that facilitates the second modification (Han and Phizicky, 2018). While this notion rationalizes the ordered enzymatic synthesis of the modification circuit and is supported experimentally (Han and Phizicky, 2018; Masuda et al., 2018), the question of why the circuit is formed is unanswered. Particularly, whether the position-37 modification compensates deficiency of the position-34 modification to improve the quality of anticodon-codon pairing at the level necessary for cell viability is unknown.
We focus on the m1G37 methylation that is invariably associated with tRNA families of Pro, Leu, and Arg. Deficiency of m1G37 stalls ribosomes at most of the associated codons (Masuda et al., 2021), indicating a role of the methylation in regulating the quality of anticodon-codon pairing. However, which anticodon-codon pairing is most critically dependent on m1G37 is unknown. Additionally, the biology of m1G37 is complex. It is required for accuracy of the translational reading frame (Gamper et al., 2015, 2021a, 2021b); loss of m1G37 results in +1 frameshifting and premature termination of protein synthesis, leading to cell death (Bjork et al., 1989; Masuda et al., 2019). It is also required for efficient and specific aminoacylation of the associated tRNA (Masuda et al., 2021; Perret et al., 1990); loss of m1G37 results in accumulation of uncharged tRNA during protein synthesis (Masuda et al., 2021). Despite this complexity, the essentiality of m1G37 for cell viability provides a way to explore the aspect of its biology that is most relevant to cellular life.
Here we describe a systematic and unbiased approach to determine whether m1G37 can regulate the quality of anti-codon-codon pairing and resolve codon usage bias at the limit of cell viability. We removed the methylation to “expose” the deficiency in anticodon-codon pairing that caused cell death. We then used a genome-wide screen to isolate suppressors that support cell growth. We found that, while m1G37 is associated with multiple tRNAs and multiple functions, all suppressors in our screen are mapped to the single gene: proS for prolyl-tRNA synthetase (ProRS) responsible for prolyl-aminoacylation of tRNA. This exclusiveness indicates that it is the coupling of methylation with prolyl-aminoacylation that distinguishes life from death. This coupling is disrupted, however, in proS suppressors, thus generating prolyl tRNAs that are now charged but not methylated for analysis of the deficiency of anticodon-codon pairings. We show that a minor prolyl isoacceptor, which is normally dispensable when m1G37 is present, has become indispensable when m1G37 is absent, revealing a deficiency of the anticodon-codon pairing upon removal of the methylation. This deficiency results from the insufficiency of the major prolyl isoacceptor to use wobble pairings to decode a subset of codons. Thus, m1G37 is the determinant that resolves codon-reading bias of the major prolyl isoacceptor. We find that many bacterial pathogens of serious threat to human health do not encode the minor prolyl isoacceptor, suggesting that their cell fitness and viability would be severely compromised in m1G37 deficiency.
RESULTS
A genome-wide suppressor screen identifies proS mutations in m1G37 deficiency
We used Escherichia coli as a genetic model, where the seven isoacceptors that are associated with m1G37 (Pro(CGG, GGG, and UGG), Leu(CAG, GAG, and UAG), and Arg(CCG)) are well defined and experimentally tractable (Bjork and Hagervall, 2014). E. coli encodes a complete set of all essential isoacceptors, as well as an additional set of non-essential tRNAs, which together assemble into a more-than-sufficient ribosome-tRNA apparatus that readily translates all sense codons. In bacteria, m1G37 is synthesized by the conserved TrmD methyl transferase that is also essential for cell viability (Bystrom and Bjork, 1982) (Figure 1A). This essentiality suggests that, to generate an m1G37-deficient condition, a simple trmD-KO cannot be made. We therefore created a modified E. coli trmD-KO strain, in which the chromosomal trmD was replaced by the antibiotic Kan marker, while cell survival was maintained by expression of the gene under the control of an arabinose (Ara)-inducible promoter from a plasmid that had a temperature-sensitive (ts) origin of replication (Figure 1B). This strain was grown overnight in the presence of Ara at the permissive temperature 30°C and was then diluted into an Ara-free medium at the non-permissive temperature 43°C for multiple cycles of growth and dilution. This cycling was designed to irradicate the trmD-carrying ts plasmid and to eliminate residuals of the TrmD enzyme and its m1G37-tRNA products. After three cycles of growth and dilution at 43°C, when cell count was no longer visible, an aliquot of the culture was streaked on an Ara-free plate and incubated at 43°C to screen for suppressors. We chose not to use a mutagen for the screen to avoid mutagen-induced cell stress. We also minimized a potential bias of the screen, due to different genetic background of the host, by parallel analysis of three well-characterized strains, each constructed and grown in the same trmD-KO condition. We isolated five colonies from MG1655, four colonies from BW25113, and three colonies from XAC. Each isolate was streaked purified, confirmed for loss of the ts plasmid (Figure S2A), and subjected to whole-genome sequencing.
Figure 1. E. coli trmD-KO suppressors are mapped to proS.

(A) Synthesis of m1G37 (red) by TrmD on the 3′ side of the anticodon at positions 34–36 (blue).
(B) E. coli trmD-KO in MG1655, BW25113, and XAC for isolation of suppressors.
(C) Exclusive mapping of all suppressor mutations to proS (Table S1).
(D) Clusters of suppressor mutations in the crystal structure of the Enterococcus faecalis proS enzyme with a prolyl analog (PDB: 2J3L). Mutations are mapped to residues in red in monomer #1, while the prolyl analog is in white.
(E) (Top) Suppressor mutations (yellow triangles) in a linear diagram of E. coli proS structural domains. (Bottom) A multi-sequence alignment of the enzyme, showing residues with mutations (yellow), those conserved (boxed in blue), and those in structural similarity (blue letters). An expanded alignment is in Figure S2C.
To our surprise, all but one clones mapped their single-nucleotide substitutions to the gene proS, totaling nine unique mutations (Figure 1C). The exception was a single clone from BW25113 that did not yield a clear whole-genome sequencing result. The exclusive mapping of all suppressor mutations to proS, occurring across the three strains, indicates a connection between m1G37 methylation and prolyl-aminoacylation of tRNA that is required for cell viability. In this connection, m1G37 would promote efficient prolyl-aminoacylation to generate Pro-charged tRNA isoacceptors to support ribosome translation of Pro codons. This connection would be consistent with our recent work showing that loss of m1G37 reduces prolyl-aminoacylation, causing ribosome stalling at Pro codons and arresting cell growth (Masuda et al., 2021). Thus, while m1G37 is associated with multiple tRNAs in multiple aspects of translation, it is most intimately associated with prolyl-aminoacylation of tRNA at the nexus that determines cell death and survival.
Mapping of each unique suppressor mutation to a known crystal structure of the proS enzyme identified three clusters (Yaremchuk et al., 2000) (Figures 1D and 1E). Cluster I mutations (D98N, G107C, S123R) are between catalytic motif 1 and motif 2, which are formed at the monomer-monomer interface of the obligate dimer enzyme (Figure S2B). Cluster II mutations (T199I, T199S, and Q211L) are between the catalytic motif 2 and an insertion domain (i.e., the editing domain), the latter of which performs proof-reading to remove mis-acylated tRNAPro (Vargas-Rodriguez and Musier-Forsyth, 2013). Cluster III mutations (A231E, N232S, and A236E) are near the start of the insertion domain. Some mutations in clusters II and III are also adjacent to the Pro-binding pocket of the enzyme (Figure S2B). Thus, all mutations are mapped to functionally important regions of proS. Additionally, all mutations are mapped to highly conserved positions across a wide range of bacterial proS enzymes (Figures 1E and S2C), indicating that they arose from the necessity to restore cell viability against a strong selective pressure of sequence conservation.
Each unique proS suppressor mutation was reconstructed by genome editing in the native MG1655 strain (Figure 2A). In each edited strain, the chromosomal trmD was then deleted and confirmed for loss of the trmD enzyme (Figure S2D). Note that the plus (+) and minus (−) symbols indicate the presence and absence of trmD, respectively. Importantly, all reconstructed suppressors supported cell viability (Figure 2B), whereas a trmD-KO expressing the wild-type (WT) proS in an m1G37-deficient condition did not (bottom two rows). Thus, each proS mutant is a bona fide suppressor of trmD-KO. However, these suppressors exhibited growth deficiency compared with the WT, with the highest deficiency in the proS-T199S and proS-S123R suppressors (Figure 2C). Even the two most robust suppressors, proS-D98N and proS-A231E (Figures 2D and S2E), showed reduced fitness over time in a growth competition with the WT. This reduced fitness was correlated with an increased frequency of ribosomal +1 frameshifting in a reporter assay that monitored expression of the nLuc gene containing a frameshift-prone insertion (Figure 2E). Thus, consistent with the notion that m1G37 ensures accuracy of the translational reading frame, these m1G37-lacking suppressors are prone to +1 frameshifting, providing an explanation for their deficient growth and compromised cell fitness relative to the WT.
Figure 2. Each proS suppressor supports cell viability without trmD.

(A) CRISPR-Cas9 reconstruction of each suppressor in MG1655.
(B) A serial dilution of each reconstructed suppressor on an Luria-Bertani (LB) plate at 37°C. WT is the native MG1655, while each suppressor is shown by the proS mutation. The plus (+) and minus (−) symbols indicate the presence and absence of the chromosomal trmD. Growth of MG1655-trmD-KO is shown at the bottom with or without Ara, representing m1G37+ and m1G37− conditions.
(C) Growth of each reconstructed suppressor at 37°C. Data are mean ± SD (n = 3).
(D) A 1:1 mixture of the WT (expressing YFP) and the proS-D98N suppressor (expressing mCherry) monitored for cell fitness at 37°C in LB. Data are mean ± SD (n = 3).
(E) A +1-frameshifting assay for expression of nLuc in MG1655 or in a reconstructed suppressor. Data are mean ± SD (n = 4). **p < 0.05, ***p < 0.01.
Decoupling of m1G37 methylation from tRNA aminoacylation in proS suppressors
We addressed how each suppressor mutation would affect the prolyl-aminoacylation activity of proS. The WT proS enzyme requires the presence of m1G37 for efficient prolyl-aminoacylation of tRNAPro (Masuda et al., 2021), indicating a mechanistic coupling between tRNA methylation and prolyl-aminoacylation. The isolation of proS suppressors that are viable but lack m1G37 suggests that this coupling is disrupted, such that each suppressor restores prolyl-aminoacylation without the need for m1G37. We tested this possibility by measuring the intracellular prolyl-aminoacylation status of each proS suppressor (Figure 3A). Total RNA was isolated from each and the prolyl-aminoacylation status at the time of cell harvest was monitored for the Pro(UGG) isoacceptor using an oligonucleotide probe specific to the tRNA (Figure S3A). This experiment was performed in an acid condition to preserve the charged state, starting with isolation of total RNA to separation of the charged from the uncharged states on an acid denaturing gel. The prolyl-aminoacylation status was calculated by the band intensity of the charged state over the sum of both the charged and uncharged state. The result showed that the prolyl-aminoacylation status is generally high across all suppressors, ranging from 63% for proS-A231E (−) to 38% and 42% for proS-T199S (−) and proS-S123R (−) (Figures 3A and S3B). The high charging status of proS-A231E (−) at 63% is comparable with the status of 73% of the WT (+) that expresses trmD.
Figure 3. Prolyl-aminoacylation of proS suppressors is less dependent on m1G37.

(A) Acid-urea gel of cellular prolyl-aminoacylation status of each reconstructed suppressor. An uncharged and un-modified Pro(UGG) is in lane 1, a pair of control tRNA samples of MG1655-trmD-KO grown in m1G37+ and m1G37− conditions in lanes 2 and 3, and the WT MG1655 sample is in lane 4. Data are mean ± SD (n = 3).
(B–D) Kinetics of prolyl-aminoacylation of (B) Pro(UGG), (C) Pro(GGG), and (D) Pro(CGG), showing loss of kcat/Km of the WT enzyme upon removal of m1G37 relative to proS mutants. Data are mean ± SD (n = 3).
Both a negative and a positive control of total RNA were isolated from a separately constructed trmD-KO strain (Gamper et al., 2015), where the chromosomal trmD was removed, while cell viability was maintained by a plasmid-borne human counterpart trm5 under the control of an Ara-inducible promoter. In this trmD-KO strain, addition of Ara to the medium induced the m1G37+ condition, producing a charging status of Pro at 58%, while depletion of Ara from the medium induced the m1G37− condition, producing a charging status at 6% (Figure 3A). We chose human trm5 as the maintenance gene because the protein product is rapidly degraded upon turning off the Ara-controlled promoter (Christian et al., 2013), thus immediately generating the m1G37− condition. In contrast, a trmD-KO strain maintained by E. coli trmD would take a much longer time to reach an m1G37− condition (Masuda et al., 2021). We found that, relative to these controls, even the low charging status of proS-T199S (−) and proS-S123R (−) (at 38%–42%) is considerably higher than the charging status of the negative control and is within 2-fold of the positive control (Figure 3A). Thus, all of the proS suppressors maintain a high intracellular prolyl-aminoacylation status, despite the absence of m1G37.
We next probed the enzymatic prolyl-aminoacylation activity of selected suppressor mutants, using in vitro kinetic assays to generate intrinsic parameters that define the catalytic efficiency of each enzyme. These kinetic assays served as an independent approach from cell-based assays to verify the decoupling of aminoacylation from m1G37. They also offered the kinetic sensitivity to distinguish among prolyl isoacceptors for differences in the level of decoupling. E. coli expresses a complete set of prolyl isoacceptors, consisting of Pro(UGG), Pro(GGG), and Pro(CGG). To focus on the effect of m1G37, each isoacceptor was prepared in the G37 state and in the m1G37 state, differing in the presence and absence of the methylation. The G37 state was transcribed from the gene of each isoacceptor, lacking m1G37 and any other modification, whereas the m1G37 state was transcribed and was post-transcriptionally modified by TrmD with the single methylation.
We selected D98N, T199I, and N232S for kinetic analysis, each representing a member of cluster I, II, and III, respectively (Figure 1E), and each showing a robust intracellular prolyl charging status (Figure 3A). Although each does not represent members of the entire cluster, the high charging status would give the sensitivity necessary to discriminate among small changes of kinetic parameters. Each mutant enzyme was purified to near homogeneity and was assayed for prolyl-aminoacylation of one of the isoacceptors. The assay was performed in steady-state condition as a function of tRNA concentration, yielding kinetic parameters Km (the concentration of the tRNA that produces half-maximum of the enzyme activity), kcat (the catalytic turnover), and kcat/Km (the catalytic efficiency). For prolyl-aminoacylation of the Pro(UGG) isoacceptor, while the kcat/Km of the WT enzyme decreased by 20.7-fold upon loss of m1G37, it decreased by no more than 3-fold for the three proS suppressor enzymes (Figures 3B and S4A). Specifically, the decrease was by 2.9-fold for the proS-D98N enzyme, by 3.0-fold for the proS-T199I enzyme, and by 2.5-fold for the proS-N232S enzyme. The large decrease of kcat/Km of the WT enzyme upon loss of m1G37 is consistent with our recent result (Masuda et al., 2021), confirming the dependence on the methylation for aminoacylation, whereas the much smaller decrease of kcat/Km of the three suppressor enzymes indicates a loss of the dependence. Thus, based on representative members of the three clusters, prolyl-aminoacylation is no longer tightly couDled to m1G37.
Loss of coupling was also observed for prolyl-aminoacylation of the other two isoacceptors. We focused on the proS-D98N enzyme, due to its high levels of intracellular charging and over-expression. For prolyl-aminoacylation of the Pro(GGG) isoacceptor, loss of m1G37 decreased kcat/Km by 20.8-fold for the WT enzyme but by 4.4-fold for the mutant enzyme (Figures 3C and S4B). Similarly, for prolyl-aminoacylation of the Pro(CGG) isoacceptor, loss of m1G37 decreased kcat/Km by 18.8-fold for the WT enzyme but by 3.4-fold for the mutant enzyme (Figures 3D and S4C). Thus, consistent across all three isoacceptors, while prolyl-aminoacylation by the WT proS enzyme is strongly coupled to the presence of m1G37, this coupling is decreased in suppressor mutants, resulting in synthesis of Pro-tRNAPro despite the loss of m1G37. Notably, while each tested suppressor mutant catalyzes prolyl-aminoacylation, the efficiency is reduced from that of the WT enzyme (Figure S4), supporting the notion that each has compromised an important aspect of the catalytic activity (Figures 1E and S2B). The decrease of the catalytic efficiency of these mutants can also contribute to the loss of cell fitness (Figure 2D).
The Pro(GGG) isoacceptor is required for cell viability of proS suppressors
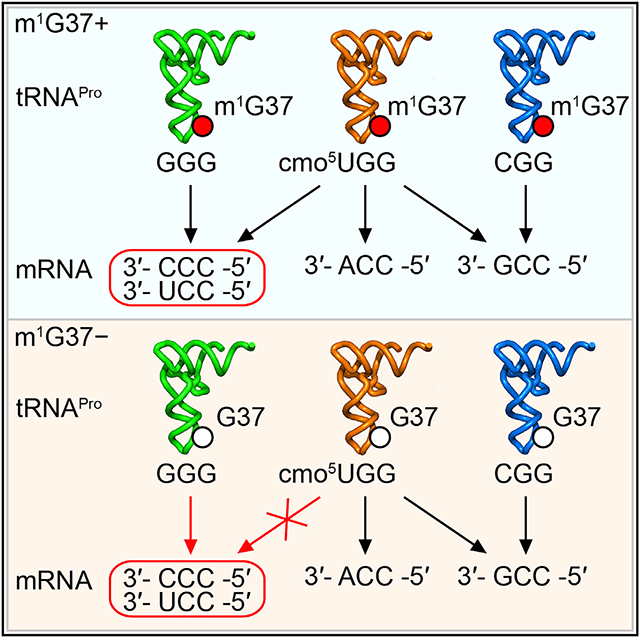
The prolyl-aminoacylation activity of proS suppressors generated Pro-charged tRNAs that were aminoacylated but not methylated with m1G37, thus allowing us to determine whether there was anticodon-codon pairing bias in the absence of the methylation. We addressed this question by removing one of the isoacceptors (and thus one of the anticodons) to uncover deficiency during protein synthesis. In the native E. coli with m1G37, each prolyl isoacceptor has a dedicated role in decoding: Pro(CGG) (encoded by proK) is for reading the CCG codon, Pro(GGG) (encoded by proL) is for reading the CCC and CCU (CC[C/U]) codons, and Pro(UGG) (encoded by proM) is capable of reading all four CCN codons due to the presence of the cmo5U34 (5-carboxymethoxyuridine 34) modification that expands the decoding capacity to all four nucleotides (Nasvall et al., 2004, 2007) (Figures 4A and S1). Of the three, Pro(UGG) is the major isoacceptor (Wei et al., 2019) and is the only one that is essential for viability and cannot be deleted (Nasvall et al., 2004). We therefore deleted the GGG or the CGG isoacceptor from a proS suppressor strain.
Figure 4. Dependence on Pro(GGG) for survival of proS suppressors.

(A) E. coli proK-encoded Pro(CGG) is responsible for reading the CCG codon; the proL-encoded Pro(GGG) is responsible for reading CC[C/U], and the proM-encoded Pro(UGG) is capable of reading CCN due to the cmo5U34 modification.
(B) Construction of a trmD-KO strain with a proS suppressor in MG1655, followed by deletion of proK or proL.
(C) Depletion analysis of Pro(GGG) or Pro(CGG) of the reconstructed trmD-KO strains expressing proS-WT or proS-D98N.
(D) Isoacceptor complementation analysis of reconstructed trmD-KO strains expressing proS-WT or proS-D98N. O/E, over-expression; GGG to UGG, a variant of Pro(GGG) that changed the GGG anticodon to UGG; CAU, tRNAMet(CAU).
Deletion of the GGG or the CGG isoacceptor in cells without m1G37 might be lethal if there was a decoding deficiency that blocked genome-wide protein synthesis. We thus chose the previously constructed trmD-KO strain for this purpose, whose viability can be maintained by the Ara-induced expression of a plasmid-borne human trm5 (Figure 4B) (Gamper et al., 2015). In this trmD-KO strain, we reconstructed a proS mutation to the host genome in the Ara-induced m1G37+ condition and performed deletion of the GGG or the CGG isoacceptor. We evaluated cell viability in the m1G37− condition upon removal of Ara.
The results showed that the trmD-KO strain, when supported by the WT proS, was viable in the m1G37+ condition but was non-viable in the m1G37− condition, consistent with the notion that m1G37 is required for efficient prolyl-aminoacylation to support cell growth (Figure 4C, first row). In contrast, the trmD-KO strain, when supported by the proS-D98N suppressor, was viable in the m1G37− condition, but this viability was observed only when the minor isoacceptor GGG was present (second row). Thus, while proS-D98N was able to support cell viability without m1G37, it failed to do so when the GGG isoacceptor was also lacking. This dependence on the GGG isoacceptor for viability was also observed for proS-T199I and proS-N232S (Figure S5), indicating a shared mechanism. Upon over-expression of a plasmid-borne Pro(GGG), however, cell viability was restored in the m1G37− condition (Figure 4D, second row). This restoration was not observed in the trmD-KO strain supported by the WT proS (first row), consistent with the notion that the native enzyme is insufficient to catalyze prolyl-aminoacylation in the absence of m1G37 to support growth. Neither was restoration observed in the proS-D98N mutant by over-expression of a variant of Pro(GGG) whose anticodon was mutated to UGG (second row, right panel), or by over-expression of a CAU isoacceptor specific for reading the Met AUG codon (second row, right panel). Thus, while the proS-D98N enzyme can catalyze prolyl-aminoacylation to tRNAPro to support cell viability without m1G37, it cannot do so without Pro(GGG). Notably, Pro(GGG) is dedicated for reading of CC[C/U] codons, which in the normal condition is also read by the cmo5U34-modified major isoacceptor Pro(UGG) (Figure 4A). The dependence on Pro(GGG) for viability of proS-D98N implies that decoding of CC[C/U] by the major isoacceptor in the absence of m1G37 is no longer sufficient.
Codon usage bias for expression of cold-shock response genes
We tested the hypothesis that, in the absence of m1G37, translation of CC[C/U] codons would depend on the availability of the minor isoacceptor Pro(GGG). A genome-wide codon usage analysis of E. coli protein-coding genes showed an average usage of CC[C/U] at 0.31 when normalized by the sum of all Pro codons that occur in the same gene (Figure 5A). Notably, 20.4% of E. coli protein-coding genes display an average usage of CC [C/U] higher than 0.5. In Gene Ontology (GO) analysis, these CC [C/U]-enriched genes are most populated in the group involved in organization of cellular components in response to stress with a frequency of 4.2-fold above the background frequency (Figure 5B). Notably, a majority in this group are cold-shock protein (csp) genes that are expressed in response to temperature down-shift (Figure S6A). We tested the possibility that the high enrichment of CC[C/U] codons in csp genes would render the E. coli response to cold-shock dependent on Pro(GGG) when m1G37 is absent. Indeed, the WT strain, when supported by the native proS while expressing trmD and synthesizing m1G37, remained viable upon temperature shift from 37°C to 22°C, whereas several proS suppressors, lacking trmD and unable to synthesize m1G37, did not (Figure 5C). The loss of viability was notable for the proS-D98N mutant even with a milder shift to 30°C (Figure S6B). The high sensitivity of these suppressors to the cold-shock stress is consistent with the notion that, without m1G37, translation of CC [C/U]-enriched csp genes is compromised, likely due to insufficient supply of the Pro(GGG) isoacceptor.
Figure 5. Expression of E. coli csp genes is sensitive to the cellular level of Pro(GGG).

(A) A violin plot of codon usage frequency of CC[C/U] in E. coli protein-coding genes.
(B) GO analysis of genes with an above-average usage of CC[C/U], showing 8 out of 12 genes in the top group in the csp family.
(C) Cold sensitivity assay of WT MG1655 (+) and reconstructed proS-D98N (−) at 37°C (one overnight) or at 22°C (two overnights).
(D) Rescue of cold sensitivity by tRNA expression in proS-D98N.
In E. coli, CC[C/U] codons are rare (average usage at 0.31; Figure 5A) relative to the more abundant CC[A/G] codons for Pro, suggesting that the intrinsic level of the corresponding Pro(GGG) would be similarly low relative to others. This possibility was confirmed by over-expression of Pro(GGG) from a multi-copy plasmid, showing restoration of viability at 22°C (Figures 5D and S6B). In contrast, no restoration of viability was observed by over-expression of the other two isoacceptors. The specific requirement of Pro(GGG), but not others, to restore cell viability at 22°C in m1G37-lacking suppressors supports the notion that the GGG isoacceptor facilitates translation of csp genes that are highly enriched with CC[C/U] codons.
Absence of the Pro(GGG) isoacceptor from bacterial species
The requirement of Pro(GGG) for cell viability in m1G37-lacking suppressors is important for understanding the diversity of bacterial genome structures. While m1G37 is strictly conserved throughout the bacterial domain (Bystrom and Bjork, 1982), not all bacterial species encode Pro(GGG). In such a simplified genome structure, decoding of CC[C/U] could only be achieved by the cmo5U34-modified major isoacceptor Pro(UGG), while decoding of CCG could be achieved by both the major isoacceptor and the other minor isoacceptor Pro(CGG). We thus surveyed the genomic tRNA database (GtRNAdb) to understand expression of all prolyl isoacceptors. We compared the presence and absence of each isoacceptor with a combined 16S and 23S rRNA phylogeny of selected genera in GtRNAdb (Figure 6A). Nearly all examined bacterial genomes encode the major isoacceptor Pro(UGG), supporting its essentiality for cell viability, while a few species lack Pro(UGG), which is likely due to incomplete analysis of the genomes. Some bacterial species also encode a minor Pro(AGG), but the occurrence is extremely rare (12 across the species surveyed) and has no pattern of evolutionary conservation, indicating that it is likely a tRNA pseudo-gene and is not considered further. The resulting phylogenetic tree revealed a bifurcation of bacterial species into phyla with the presence or absence of Pro(GGG). Almost all the clades that are missing Pro(GGG) are also missing Pro(CGG), indicating a co-existence relationship between the two (Figure S7). The absence of both isoacceptors from a genome thus signifies the sole dependence on the major isoacceptor for translation of all Pro codons. This is most notable in the entire clade of Mycoplasma, which are intracellular parasites with minimal sets of isoacceptors (Razin et al., 1998). The only exception to the co-existence rule is in the family Lactobacillaceae, which lacks Pro(GGG) but contains Pro(CGG) (Figure 6A).
Figure 6. Absence of Pro(GGG) from some bacterial species.

(A) A phylogenetic tree showing the presence and absence of tRNAPro genes in representative bacterial genomes at GtRNAdb. Gram-positive (Gram (+)) genera are in bold face, and those of human pathogens are indicated by a red dot. Inner rings show the presence and absence of tRNAPro genes: blue for Pro(CGG), green for Pro(GGG), and yellow for Pro(UGG). The gradient of color darkness indicates the frequency of the species lacking the corresponding tRNA gene in the genus. Darker color means more species lacking the tRNA gene, with the black indicating absence.
(B) Distribution of Gram-positive genera of medical relevance without Pro(GGG).
(C) Distribution of Gram-negative genera of medical relevance without Pro(GGG).
(D) Average frequency of CCC and CCG codons in each protein-coding gene across gram-positive and -negative bacteria with or without Pro(GGG). The p value is for analysis of CCC. KS test, Kolmogorov-Smirnov test. Species names are in STAR Methods.
Many bacterial species lacking Pro(GGG) are human pathogens. In Gram-positive genera, 55 of the 171 species lack Pro(GGG), and 23 of the 55 are human pathogens, including species of the genera Enterococcus and Staphylococcus (Figures 6A and 6B). In Gram-negative genera, 57 of the 424 species lack Pro(GGG), and 25 of the 57 are human pathogens, including species of the genus Acinetobacter (Figures 6A and 6C). Notably, the cited pathogens are members of the ESKAPE family, posing an additional threat to human health, due to their resistance to almost all known antibiotics (De Oliveira et al., 2020).
To explore the relationship between pathogenicity and lack of Pro(GGG), we analyzed codon usage of a broader spectrum of human pathogens. We found that those lacking Pro(GGG) generally have reduced usage of the CCC codon relative to those containing the tRNA (Figure 6D). This pattern is conserved in gram-positive and gram-negative species, but does not correlate with the GC content, indicating that it is a specific requirement of the genome structure. We focused on the CCC codon because its decoding is directly affected by Pro(GGG). In contrast, while the usage of the CCG codon also decreases with the lack of Pro(GGG) and increases with the presence of the tRNA, the trend is correlated with the GC content. Thus, the absence of Pro(GGG) and the reduced codon usage of CCC in pathogens relevant to human health is not random and can have medical implications.
DISCUSSION
Codon usage is well known to be closely correlated with the abundance and composition of the cellular tRNA pool (Rak et al., 2018). Much less known is how codon usage may be controlled or modulated by various tRNA post-transcriptional modifications. Even less known is how codon usage is determined by position-37 modifications, which occur on the 3′ side of the anticodon and are not directly involved in the anticodon-codon pairing. However, elimination of a position-37 modification from a cell causes ribosome stalling and programmatic changes of gene expression (Lamichhane et al., 2016; Masuda et al., 2021; Thiaville et al., 2016), strongly implying that the position-37 modification modulates anticodon-codon pairing. This modulation, however, has not been directly tested. Here we show that the position-37 modification with m1G37 is an important neutralizer that addresses the deficiency in, and bias against, specific anticodon-codon pairings established by the position-34 modification in the major Pro(UGG) isoacceptor. While U34 of the major isoacceptor is typically modified into cmo5U34, which expands decoding to all four Pro codons (Nasvall et al., 2004, 2007), we show that, although this expansion is comprehensive when m1G37 is present, it is insufficient when m1G37 is absent (Figure 4D). Importantly, the insufficiency of the major isoacceptor to decode a subset of Pro codons when m1G37 is absent cannot be solved by increasing cellular levels of the tRNA (Figure 5D), contrary to the conventional thinking that tRNA abundance is the universal solution to resolve codon usage bias. Thus, we present a conceptual framework that tRNA modifications provide a distinct mechanism, separate from tRNA abundance, to resolve codon usage bias.
We suggest that m1G37 is the determinant that expands the decoding capacity of the cmo5U34-modified UGG anticodon, whereas, lacking m1G37, the modified UGG is insufficient to read CC[C/U] and requires the minor isoacceptor Pro(GGG) to assume the responsibility (Figure 7A). Thus, while the minor isoacceptor Pro(GGG) is dispensable in m1G37 abundance, it is essential in m1G37 deficiency, emphasizing the notion that m1G37 resolves the deficiency of the major isoacceptor to read CC[C/U]. Notably, m1G37 is in a conserved modification circuit with the chemical moiety of cmo5U34 in the major isoacceptor Pro(UGG). This includes the methyl addition to the cmo5U34 moiety in E. coli Pro(UGG), generating 5-methoxy-carbonyl-methoxy-uridine 34 (mcmo5U34) in bacteria, and the 5-carbamoyl-methyl-uridine 34 (ncm5U34) modification in eukaryotes, sharing the carbamoyl chemical moiety at the 5 position of the uridine (Figure S1). This broad conservation indicates that the functional compensation of m1G37 for the deficiency of cmo5U34-mediated reading of Pro codons has sustained over the evolution time.
Figure 7. m1G37 methylation of tRNA resolves codon usage bias of the cmo5U34-modified major isoacceptor Pro(UGG) with medical relevance.

(A) (Top) In the presence of m1G37 (a red circle), the cmo5U34-modified Pro(UGG) can read all four Pro codons. (Bottom) In the absence of m1G37 (an open circle), the major isoacceptor is insufficient to read CC[C/U], requiring Pro(GGG) to assume the role.
(B) Upon targeting TrmD by an inhibitor, while cell viability is diminished, a potential resistance mechanism can occur by proS mutations. This resistance would be thwarted if the bacterial species lacks Pro(GGG).
The m1G37 resolution of the deficiency of the cmo5U34-mediated anticodon-codon pairing provides insight into the long-standing question of how the wobble modification expands the pairing capacity to all four nucleotides. In crystal structures, the base pairing of cmo5U34 with C and U is mediated by just one hydrogen (H) bond each, whereas that with A and G is mediated by three H bonds each (Figure S1) (Weixlbaumer et al., 2007). While these structures were obtained with a synthetic anticodon stem-loop (ASL), lacking the position-37 modification, they nonetheless raise the question of how the single H bond can provide the same stability as three H bonds. This question has remained unsolved for decades, due to lack of a tool to probe the question at a biological scale. Here we use a genetic tool to remove m1G37 and, upon removal, we find that the single H bond in the cmo5U34-C/U pairing is indeed insufficient to satisfy the high demand of genome-wide translation required for cell viability. In the major isoacceptor Pro(UGG), stabilization of the cmo5U34-C/U pairing by m1G37 is likely due to the ability of the methylation to remodel the ASL structure, which adopts a disordered form when lacking m1G37 (Maehigashi et al., 2014). This implication is likely far reaching, given that the cmo5U34-mediated anticodon-codon pairing is wide-spread among five other tRNA families. In E. coli, cmo5U34 co-exists with m1G37 in Leu(UAG), with ms2i6A37 in Ser(UGA), with m6A37 in Val(UAC), and with cyclic N6-threonyl-carbamoyl-adenosine 37 (ct6A37) in Thr(UGU). Although cmo5U34 in Ala(UGC) does not co-exist with a position-37 modification, the deficiency of its pairing with C/U may be resolved by other post-transcriptional modifications in the tRNA. Further structural analysis of a cmo5U34-C/U base pairing in the presence of a position-37 modification or accompanied by the full complement of tRNA post-transcriptional modifications will shed light on how the cmo5U34-C/U base pairing is stabilized.
While we emphasize the importance of m1G37 in decoding of prolyl isoacceptors, this does not mean that m1G37 is not important for other tRNAs associated with the methylation (e.g., isoacceptors of Leu and Arg). The distinction is that the importance of m1G37 in prolyl isoacceptors is established at the limit of cell viability, whereas the importance of m1G37 in others is not as critical. Indeed, all mutations in our genome-wide screen of suppressors of trmD-KO are mapped to the single gene proS, indicating a unique dependence on m1G37 for prolyl-aminoacylation that is required for cell viability. In contrast, while we showed previously a similar dependence on m1G37 for aminoacylation of Arg(CCG) and Leu(UAG) (Masuda et al., 2021), no suppressor mutations are mapped to argS or leuS. Additionally, we show a close correlation between proS charging and cell viability. The proS-T199S mutant with the lowest intracellular charging level displays the lowest viability on plates and in liquid culture, while the proS-Q211L mutant with one of the highest charging levels has robust growth (Figures 2B, 2C, and 3A). These results indicate that different structural alterations of the proS enzyme by different mutations have a direct impact on viability. Thus, in the decision of life and death, cells choose to selectively introduce mutations into proS in exchange for viability.
A detailed analysis of some proS suppressor mutants reveals that each has a kinetic deficiency relative to the WT enzyme (Figure S4), resulting in a loss of kcat/Km ranging from 4- to 170-fold. This kinetic deficiency is apparently small enough for the trade-off to continue synthesis of Pro-charged tRNAPro to support protein synthesis and cell viability. Nonetheless, the trade-off does incur a fitness cost. One aspect of the trade-off is a reduced efficiency of prolyl-aminoacylation (Figure 3A), while another is an increased propensity of ribosomal +1 frameshifting (Figure 2D), both of which would compromise cell growth.
The dependence of proS suppressors on the minor isoacceptor Pro(GGG) for cell viability in m1G37 deficiency has medical implications (Figure 7B). TrmD has long been ranked as a high-priority antibacterial target (White and Kell, 2004), due to its essentiality for bacterial viability, conservation across the bacterial domain, fundamental distinction from its human counterpart Trm5 in structure and mechanism (Ahn et al., 2003; Christian et al., 2004, 2010, 2016; Christian and Hou, 2007; Goto-Ito et al., 2008, 2009; Sakaguchi et al., 2014), and possession of a small-molecule binding pocket for the methyl donor that can be targeted by compounds. Notably, the methyl-donor-binding pocket in TrmD is unusual (Ahn et al., 2003), adopting a rare protein topological knot fold that is absent from majority of methyl transferases, indicating the potential of bacteria-specific drug targeting in unexplored chemical space and diversity. The importance of targeting TrmD is also noted by our finding that m1G37-tRNA is required for translation of bacterial membrane-associated genes (Masuda et al., 2019), due to frequent usage of CC[C/U] in these genes. This finding suggests that targeting TrmD has the potential to damage the cell membrane structure to facilitate drug entry and intracellular retention, leading to faster bactericidal action. However, while developing TrmD inhibitors is currently an active pursuit (Hill et al., 2013; Thomas et al., 2020; Whitehouse et al., 2019; Zhong et al., 2019a, 2019b), the enthusiasm is dampened by the discovery that bacteria can find a path via proS mutations to overcome targeting, as shown here and in a related study (Clifton et al., 2021). Nonetheless, this concern is eased somewhat by the discovery that proS suppressors require the minor isoacceptor Pro(GGG) for survival, suggesting that targeting TrmD would be more effective in bacterial species that lack the minor isoacceptor to minimize the potential of resistance arising from proS mutations.
A recent study reported isolation of suppressors of m1G37 deficiency from several E. coli strains (Clifton et al., 2021), each harboring a chromosomal mutation in trmD. Although most of the isolated suppressor mutations are also mapped to proS, with some overlapping with the suppressors we report here, the approach and motivation of the recent study is fundamentally different from ours (Table S1). Specifically, the recent study set out to isolate suppressors of trmD-KD and concluded that proS mutations constitute the dominant mechanism that repairs m1G37 deficiency. In contrast, we used trmD-KO to eliminate m1G37 and to isolate suppressors that would expose differential codon reading by various tRNA isoacceptors that would otherwise have been hidden by the presence of m1G37. From the exposed differential codon reading, we gain insight into how m1G37 resolves codon usage bias. We show that, while m1G37 is normally associated with decoding of Pro, Leu, and Arg, it is most critically associated with resolving codon usage bias against pyrimidine-ending codons of Pro. This discovery has important medical implication for targeting of bacterial pathogens that are enriched with pyrimidine-ending codons of Pro and are also lacking the minor isoacceptor Pro(GGG).
Limitations of the study
The conceptual advance of this work was made solely by studies of E. coli. While E. coli is a model Gram-negative organism, it does not represent all Gram-negative genera, which include species with and without Pro(GGG). Nor can E. coli represent organisms of the gram-positive genera, which also include species with and without Pro(GGG). Nonetheless, this work provides a basis to investigate a broader spectrum of bacterial species, including those relevant to human health.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ya-Ming Hou (ya-ming.hou@jefferson.edu).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact without restriction.
Data and code availability
We deposited our whole-genome sequencing data at EMBL-EBI ArrayExpress with an accession number E-MTAB-12178 (key resources table). These data will be publicly available as of the date of publication. In addition, we used publicly available datasets for codon usage analysis. The accession numbers for the datasets that we used are listed (key resources table). All original codes generated for analyses of publicly available datasets are deposited at GitHub and are publicly available as of the date of publication (key resources table). Any additional information required to reanalyze data reported in this study is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-CysRS antibodies | (Masuda et al., 2021) | N/A |
| Rabbit polyclonal anti-TrmD antibodies | (Li and Bjork, 1999) | N/A |
| Goat polyclonal anti-rabbit IgG antibodies peroxidase conjugate | Sigma-Aldrich | Cat. #A0545; RRID: AB_257896 |
| Bacterial and virus strains | ||
| Escherichia coli strain K-12 substrain MG1655 | ATCC | Cat. #700926 |
| E. coli strain BW25113 | The Coli Genetic Stock Center (CGSC) | CGSC#: 7636 |
| E. coli strain XAC | (Jakowec et al., 1988) | N/A |
| E. coli strain BL21(DE3) | Millipore | Cat. #69450 |
| E. coli strain ArcticExpress (DE3) | Agilent Technologies | Cat. #230191 |
| E. coli strain K-12 substrain MG1655, proS D98N (trmD + or trmD−) | This paper | N/A |
| E. coli strain K-12 substrain MG1655, proS G107C (trmD + or trmD−) | This paper | N/A |
| E. coli strain K-12 substrain MG1655, proS S123R (trmD + or trmD−) | This paper | N/A |
| E. coli strain K-12 substrain MG1655, proS T199I (trmD + or trmD−) | This paper | N/A |
| E. coli strain K-12 substrain MG1655, proS T199S (trmD + or trmD−) | This paper | N/A |
| E. coli strain K-12 substrain MG1655, proS Q211L (trmD + or trmD−) | This paper | N/A |
| E. coli strain K-12 substrain MG1655, proS A231E (trmD + or trmD−) | This paper | N/A |
| E. coli strain K-12 substrain MG1655, proS N232S (trmD + or trmD−) | This paper | N/A |
| E. coli strain K-12 substrain MG1655, proS A236E (trmD + or trmD−) | This paper | N/A |
| E. coli strain K-12 substrain MG1655, proS D98N, trmD−, pACYC-araC-PC-PBAD-human trm5, proK− | This paper | N/A |
| E. coli strain K-12 substrain MG1655, proS D98N, trmD−, pACYC-araC-PC-PBAD-human trm5, proL− | This paper | N/A |
| E. coli strain K-12 substrain MG1655, proS T199I, trmD−, pACYC-araC-PC-PBAD-human trm5, proK− | This paper | N/A |
| E. coli strain K-12 substrain MG1655, proS T199I, trmD−, pACYC-araC-PC-PBAD-human trm5, proL− | This paper | N/A |
| E. coli strain K-12 substrain MG1655, proS N232S, trmD−, pACYC-araC-PC-PBAD-human trm5, proK− | This paper | N/A |
| E. coli strain K-12 substrain MG1655, proS N232S, trmD−, pACYC-araC-PC-PBAD-human trm5, proL− | This paper | N/A |
| Bacteriophage P1vir | CGSC | CGSC#: 12,133 |
| Chemicals, peptides, and recombinant proteins | ||
| L-Arabinose | Gold Biotechnology | Cat. #A-300-250 |
| BamHI | New England BioLabs | Cat. #R0136 |
| EcoRI | New England BioLabs | Cat. #R0101 |
| NdeI | New England BioLabs | Cat. #R0111 |
| PstI | New England BioLabs | Cat. #R0140 |
| XhoI | New England BioLabs | Cat. #R0146 |
| PfuUltraII fusion HS DNA polymerase | Agilent Technologies | Cat. #600670 |
| Phusion™ High-Fidelity DNA Polymerase | ThermoFisher Scientific | Cat. #F530 |
| NEBuilder® HiFi DNA Assembly Cloning Kit | New England BioLabs | Cat. #E5520S |
| T4 DNA ligase | New England Biolabs | Cat. #M0202 |
| T4 polynucleotide kinase | New England Biolabs | Cat. #M0201 |
| Ampicillin | Fisher Scientific | Cat. #BP1760 |
| Chloramphenicol | Gold Biotechnology | Cat. #G-105 |
| Kanamycin | Gemini Bio-products | Cat. #400-114P |
| Anhydrous tetracycline hydrochloride | Cayman Chemicals | Cat. #10009542 |
| [γ-32P]-ATP | PerkinElmer | Cat. #NEG002A |
| [3H]-Proline | PerkinElmer | Cat. #NET483005MC |
| Critical commercial assays | ||
| SuperSignal West Pico Chemiluminescent Substrate | ThermoFisher Scientific | Cat. #34080 |
| ONE-Glo™ Luciferase Assay System | Promega | Cat. #E6120 |
| Deposited data | ||
| Whole-genome sequencing data of E. coli trmD-KO suppressor cells | This paper | EMBL-EBI ArrayExpress: E-MTAB-12178 |
| Codes for codon usage and tRNAPro distribution analysis | This paper | https://doi.org/10.5281/zenodo.7093913 |
| List of annotated genome sequences retrieved from public databases | Table S3 | N/A |
| Experimental models: Organisms/strains | ||
| E. coli strain K-12 substrain MG1655 | ATCC | Cat. #700926 |
| Oligonucleotides | ||
| Oligo DNAs for bacterial strain construction, plasmid construction, CRISPR mutagenesis, and Northern blotting | Table S2 | N/A |
| Recombinant DNA | ||
| pKD46 | CGSC | CGSC #7634 |
| pKD46-EctrmD | This paper | N/A |
| pCP20 | CGSC | CGSC #7629 |
| pACYC-araC-PC-PBAD-human trm5 | (Gamper et al., 2015) | N/A |
| pCas9cr4 | (Reisch and Prather, 2015) | N/A |
| pKDsgRNA | (Reisch and Prather, 2015) | N/A |
| pZS2R-mCherry | (Kelsic et al., 2015) | N/A |
| pZS2R-YFP | (Kelsic et al., 2015) | N/A |
| pKK223-3 E. coli Pro(CGG) (proK) | This paper | N/A |
| pKK223-3 E. coli Pro(GGG) (proL) | (Gamper et al., 2015) | N/A |
| pKK223-3 E. coli Pro(UGG) (proM) | (Masuda et al., 2021) | N/A |
| pKK223-3 E. coli Pro(GGG) (proL) with anticodon UGG | (Masuda et al., 2018) | N/A |
| pKK223-3 E. col Met(CAU) (metV) | (Gamper et al., 2015) | N/A |
| Software and algorithms | ||
| FASTQC v 0.11.5 | Andrews S. (2010) | http://www.bioinformatics.babraham.ac.uk/projects/fastqc |
| Burrows-Wheeler Alignment (BWA) tool v 0.7.15 - r1140 | (Li and Durbin, 2009) | N/A |
| SAMtools v 1.3.1 | (Li et al., 2009) | N/A |
| Bcftools | (Li, 2011) | https://samtools.github.io/bcftools/bcftools.html |
| SnpEff | (Cingolani et al., 2012) | N/A |
| Image Lab v. 6.0 | BIO-RAD | http://www.bio-rad.com/en-us/product/image-lab-software |
| PyMOL v. 2.4.1 | Schrödinger | https://pymol.org/ |
| Clustal X v. 2.1 | (Larkin et al., 2007) | N/A |
| Excel v. 2010 | Microsoft | https://www.microsoft.com/ |
| Kaleidagraph v. 4.5 | Synergy software | https://www.synergy.com/ |
| ImageJ v. 1.51 | National Institutes of Health | https://imagej.nih.gov |
| Python v. 3.7.7 | Python Software Foundation | https://www.python.org/ |
| ETE3 v. 3.1.2 | (Huerta-Cepas et al., 2016) | http://etetoolkit.org/ |
| RefSeq release 211 | (OĽeary et al., 2016) | https://www.ncbi.nlm.nih.gov/refseq/ |
| GenBank release 248 | (Sayers et al., 2022) | https://www.ncbi.nlm.nih.gov/genbank/ |
| FastTree v. 2.1.10 | (Price et al., 2009, 2010) | http://www.microbesonline.org/fasttree/ |
| GtRNAdb release 19 | (Chan and Lowe, 2016) | http://gtrnadb.ucsc.edu/ |
| matplotlib v. 3.5.0 | https://doi.org/10.1109/MCSE.2007.55 | https://matplotlib.org/3.1.1/index.html |
| SINA v. 1.2.11 | (Pruesse et al., 2012) | https://www.arb-silva.de/aligner/ |
| SILVA release 89 | (Quast et al., 2013) | https://www.arb-silva.de/ |
| Scipy v.1.9.0 | (Virtanen et al., 2020) | https://scipy.org |
| Other | ||
| NucleoSpin Gel and PCR Clean-up | Macherey-Nagel | Cat. #740609 |
| Hybond™-N + Nitrocellulose Membrane | GE healthcare | Cat. #RPN203B |
| Nextera XT DNA Library Preparation Kit | Illumina | Cat. #FC-131-1024 |
| Trans-Blot® TurboTM Transfer System | BIO-RAD | Cat. #1704150 |
| MicroPulser Electroporator | BIO-RAD | Cat. #1652100 |
| UV Crosslinker | Fisher Scientific | Cat. #FB-UVXL-1000 |
| Tri-Carb 4910 TR Scintillation Counter | PerkinElmer | N/A |
| Immobilon-P PVDF Membrane | Millipore | IPVH00010 |
| ChemiDoc MP Imaging System | BIO-RAD | Cat. #12003154 |
| Typhoon IP Imaging System | GE Healthcare | N/A |
| Infinite M200 PRO Plate Reader | Tecan | N/A |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
E. coli strains for suppressor screens were MG1655, BW25113, and XAC. All cell-based experiments were done with MG1655 and its genome-edited derivatives. Recombinant proS enzymes were produced from E. coli strains BL21(DE3) and ArcticExpress (DE3).
METHOD DETAILS
Genome-wide screen of trmD-KO suppressors
Because trmD is essential for viability, a simple KO cannot be made. A maintenance plasmid carrying E. coli trmD was made with a ts origin of replication. The gene together with a partial sequence of PBAD was isolated from pACYC-araC-PC-PBAD-EctrmD (Gamper et al., 2015; Masuda et al., 2019) by excision with BamHI and XhoI (NEB) and gel purified by NucleoSpin Gel and PCR Clean-up (Macherey-Nagel). The ts origin of replication of pKD46 (Datsenko and Wanner, 2000) was amplified with PfuUltraII fusion HS DNA polymerase (Agilent Technologies) between the BamHI and an XhoI sites, excluding the λRed recombinase coding sequence. The PCR product was digested by BamHI and XhoI, then gel-purified by NucleoSpin Gel and PCR Clean-up (Macherey-Nagel). The trmD gene and the amplified region of pKD46 were ligated by T4 DNA ligase (NEB) and verified by Sanger sequencing.
The three E. coli strains in this work were MG1655 (Hayashi et al., 2006), BW25113 (Grenier et al., 2014), and XAC (Dincbas et al., 1999). After transformation with pKD46-EctrmD, each strain was made into trmD-KO by P1 transduction to replace trmD with a Kan marker using the P1 lysate of the trmD-KO strain (Gamper et al., 2015; Masuda et al., 2019). Each strain was grown overnight at 30°C in LB with the presence of Ara 0.2% (w/v), ampicillin (Amp) (100 μg/mL), and Kan (50 μg/mL), allowing stable replication and expression of pKD46-EctrmD from the pBAD promoter. Cells were diluted at 1:100 to fresh LB + Kan and grown at 43°C until OD600 = 1.0 for the first cycle, during which expression of trmD was turned off, replication of the ts-plasmid was inhibited, and cellular TrmD and m1G37-tRNA was reduced with each cell division. Cell cultures at OD600 = 1.0 were diluted (1:20) and passed through two more rounds of growth and dilution to pre-warmed LB + Kan (at 1:20 each) until cell counts were not visible (~4 h). An aliquot (10 μL) of each culture was spread on an LB + Kan plate and incubated at 43°C until distinct colonies were formed. In total, 5 colonies of MG1655, 4 colonies of BW25113, and 3 colonies of XAC were isolated, streak-purified at 43°C, verified for the absence of pKD46-EctrmD, and subjected to whole-genome sequencing.
Whole-genome sequencing and identification of variants
For whole-genome sequencing analysis, we chose two suppressor colonies from each strain. After streak-purification, each was grown in 3 mL LB + Kan at 37°C overnight and cells were harvested and resuspended in 500 μL of 50 mM NaCl and 25 mM EDTA. Cell lysis was performed with the addition of lysozyme (1 mg/mL) at 37°C for 30 min, and further by the addition of 1.5% sarcosyl for 10 min at 37°C. Cell lysates were extracted by phenol twice, phenol-chloroform-isoamyl alcohol once, and ethanol precipitated. Chromosomal DNA was re-suspended in 200 μL TE buffer (pH = 8.0) and was used to generate a genomic library using Nextera XT DNA Library Prep Kit (Illumina). A DNA library from each WT was also prepared as a reference. Each library was paired-end sequenced by MiSeq at 2 x 150 bp, yielding 80x coverage of reads relative to the entire genome.
Sequencing reads were pre-processed using FASTQC (Version 0.11.5) (Andrews, 2010), and mapped to the E. coli BW25113 genome sequence (Grenier et al., 2014) using BWA (Version 0.7.15 - r1140) (Li and Durbin, 2009) and SAMtools (Version 1.3.1) (Li et al., 2009). Single nucleotide polymorphisms (SNPs) in each sample were retrieved as Variant Call Format (VCF) files. A comparative analysis of SNPs between all samples with the BW25113 genome was then performed using bcftools (Li, 2011). Further annotation of SNPs for each comparison was performed using SnpEff (Cingolani et al., 2012). Finally, SNPs with missense and frameshift variants were considered to generate final Excel files. The SNPs only present in the suppressor genome but not in the parental WT genome were identified as variants.
Of the 6 suppressor colonies subjected to whole-genome sequencing, 5 contained a single nucleotide substitution that was mapped to proS, while one was unclear in sequence analysis. We therefore focused on the proS gene, PCR-amplified it from all suppressor clones, and performed Sanger sequence analysis of the amplified gene. We confirmed that all suppressor clones contained a single nucleotide substitution in proS, noting that G107C and N232S were identified independently from both MG1655 and BW25113.
Structural mapping and amino acid sequence alignment
Each suppressor mutation was mapped to the crystal structure of E. faecalis ProRS (PDB: 2J3L) (Crepin et al., 2006) using PyMOL (Schrödinger). The amino acid sequence of the enzyme from bacterial species was retrieved from KEGG database (https://www.genome.jp/kegg/). Multiple-sequence alignment was performed by Clustal X 2.1 (Larkin et al., 2007).
CRISPR reconstruction of each proS suppressor
Each proS suppressor mutation was reconstructed in the WT MG1655 using the scar-less CRISPR-Cas9 technology (Reisch and Prather, 2017). The specific sgRNA for each mutation was expressed from the pKDsgRNA plasmid, which was constructed by round-the-horn PCR using Phusion High-Fidelity DNA Polymerase (ThermoFischer Scientific) and self-ligation using T4 DNA ligase (NEB) (primers listed in Table S2). The sgRNA plasmid was introduced to the WT MG1655 strain that already contained the plasmid pCas9cr4 encoding a Cas9 protein. CRISPR-Cas9 system was induced on an LB plate containing anhydrous tetracycline (aTc) (100 ng/mL). Each proS mutation was assessed by PCR using allele-specific primers, and verified by Sanger sequencing. Subsequently, each strain was made into trmD-KO by P1 transduction without any maintenance plasmid, followed by removal of the Kan marker by FLP recombination with pCP20.
All reconstructed suppressors were confirmed for viability without the support of trmD. Early-log phase cultures were serially diluted, spotted on an LB plate, and grown overnight at 37°C. As a control, the E. coli trmD-KO was depleted of pre-existing Trm5 and m1G37-tRNA by growing in Ara-free LB medium for 5 h, and serial dilution was spotted on an LB plate, with or without 0.2% Ara. The growth curve analysis in a liquid culture was done by inoculating overnight-grown cells to fresh LB media at 1:100, followed by measurement of OD600 at 37°C, over the time course. To confirm the absence of TrmD from each suppressor, cell lysate proteins (15 μg) from a culture of 3 mL LB and harvested at OD = 0.4 were run on a 12% SDS-PAGE gel for Western blot analysis using an anti-TrmD primary antibody (Li and Bjork, 1999), an anti-CysRS primary antibody for a loading control, and an anti-rabbit IgG secondary antibody (Sigma-Aldrich).
Cellular prolyl-aminoacylation status by acid-urea gel analysis
Total RNA was extracted in an acidic condition to maintain the aminoacylation status of tRNAs (Masuda et al., 2021). Each reconstructed proS suppressor lacking trmD was grown in LB at 37°C to OD = 0.4, upon which cells were mixed with 10% (w/v) trichloro-acetic acid (TCA) at an equal volume and incubated on ice for 10 min. Cells were harvested and resuspended in the extraction buffer (0.3 M NaOAc, pH 4.5 and 10 mM EDTA), and extracted with equal volume of ice-cold phenol-chloroform-isoamyl alcohol (25:24:1), pH 4.5. The mixtures were vortexed for 3 cycles of 1 min vortex and 1 min rest on ice, and spun at 12,000 rpm for 10 min at 4°C. The RNA-containing aqueous phase was stored, while a second extraction of the organic phase with an equal volume of fresh extraction buffer was performed. The combined aqueous phase from the two extractions was mixed with an equal volume of cold isopropanol and incubated at −20°C for precipitation. Precipitated RNA was collected at 14,000 rpm for 20 min at 4°C, and rinsed with wash buffer (70% ethanol and 30% 10 mM NaOAc, pH 4.5). Pellets were dried and dissolved in the buffer containing 10 mM NaOAc, pH 4.5, and 1 mM EDTA.
From each isolated total RNA sample, 10% was estimated to represent total tRNA (Masuda et al., 2021). Approximately 700 ng of total tRNA of each sample was mixed with a 2X loading buffer (0.1 M NaOAc, pH 5.0, 9 M Urea, 0.05% bromophenol blue (BPB), and 0.05% xylene cyanol (XC)) and separated on a 6.5%PAGE/7M urea (14 × 17 cm) in 0.1 M NaOAc, pH 5.0 at 250 V for 3 h 45 min at 4°C. Subsequently, the gel region between XC and BPB was excised, washed in 1X Tris-borate pH 8.0 and EDTA (TBE) buffer, transferred to a wet Hybond-N+ nitrocellulose membrane (GE healthcare) in 1X TBE using Trans-Blot TurboTM Transfer System (BIO-RAD) at constant 25 V for 20 min. The membranes were briefly air-dried, crosslinked with the bound RNA in a UV cross-linker (FB-UVXL-1000, Fisher Scientific), and then probed with a [32P]-labeled DNA oligonucleotide targeting positions 18 to 36 of E. coli Pro(UGG) in a Northern blot. The membrane was imaged by a phosphorimager (Typhoon IP, GE) and bands corresponding to the charged and uncharged tRNA were quantified by ImageJ (NIH). The prolyl-aminoacylation status was calculated as the band corresponding to the charged tRNA in the sum of the bands for both charged and uncharged tRNAs. To validate the probe specificity to Pro(UGG), 15 μg of T7 transcripts of E. coli Pro(UGG), Pro(CGG), and Pro(GGG) were run on a denaturing 12% PAGE/7M urea gel, transferred to a nitrocellulose membrane, then reacted with the [32P]-labeled probe.
Frameshift reporter assay
The nano-luciferase (nLuc) reporter gene was subcloned into plasmid pKK223-3 using NEBuilder HiFi DNA Assembly Cloning Kit (NEB) and expressed in MG1655 or a proS suppressor. Each strain expresses the reporter harboring an in-frame CCC triplet or a +1-frame CCC-C quadruplet next to the initiation codon AUG (primers in Table S2). The frequency of +1 frameshifting is the ratio of the reporter expressing the CCC-C quadruplet relative to the CCC triplet, each normalized by expression of a pZS2R-mCherry plasmid. An overnight culture in LB + Amp + Kan at 37°C was inoculated to a fresh medium at 1:200 for WT and 1:100 for mutants. Each fresh culture was grown at 37°C for 6 h and measured for the luciferase activity using ONE-Glo Luciferase Assay System (Promega) on an Infinite M200 PRO plate reader (Tecan). The luminescence, normalized by the mCherry fluorescence at excitation 586 nm and emission 617 nm, was used to determine the nLuc activity.
Competition fitness assay
A fitness assay was developed by mixing a reconstructed proS suppressor expressing the plasmid pZS2R-mCherry, and the MG1655 WT strain expressing pZS2R-YFP (Kelsic et al., 2015). Each strain was separately grown in LB + Kan overnight at 37°C, and diluted 10-fold in LB. According to the OD600 of each, the same number of cells of each culture was inoculated to a single culture of 3 mL LB + Kan at ~106 colony-forming unit (CFU)/mL The 1:1 mixture was grown at 37°C shaking, and an aliquot of the mixture was removed over time, diluted, and spread on an LB + Kan plate. After incubation at 37°C overnight, the number of yellow colonies for WT and pink colonies for the mutant was counted. The plates were incubated at 4°C as needed to enhance development of colors. The CFU of each culture at T = 0 was adjusted to 50:50, which was used to normalize the fractional distribution of the two strains over time.
Kinetic assay of prolyl-aminoacylation of tRNAPro in vitro
Each E. coli tRNAPro in the G37-state was synthesized by transcription, while each in the m1G37-state was synthesized by post-transcriptional methylation of the transcript with TrmD (Christian et al., 2004). Recombinant WT enzyme of E. coli proS was affinity-purified from E. coli BL21(DE3) expressing pET22b-Ec ProRS (Masuda et al., 2021). Suppressor enzymes of proS-D98N, proS-T199I, and proS-N232S were purified similarly from strains harboring the designated mutations but expressed in ArcticExpress (DE3) (Agilent). Each tRNA substrate was heat-denatured at 85°C for 3 min and re-annealed at 37°C for 15 min. Aminoacylation in steady-state conditions was performed at 37°C in a 24 μL reaction of 0.2–20 μM tRNA, 0.5 to 40 nM ProRS, and 20 μM [3H]-Proline (PerkinElmer, 7.5 Ci/mmol) in the aminoacylation buffer of 20 mM KCl, 10 mM MgCl2, 4 mM DTT, 0.15 mg/mL BSA, 2 mM ATP (pH 8.0), and 50 mM Tris-HCl, pH 7.5. Reaction aliquots of 4 μL were removed at different time intervals and precipitated with 5% (w/v) TCA on filter pads for 10 min twice. The filter pads were washed with 95% ethanol twice, with ether once, air-dried, and measured for radioactivity in Tri-Carb 4910 TR scintillation counter (PerkinElmer). Counts were converted to pmoles based on the specific activity of the [3H]-Proline after correction for signal quenching by filter pads. Data corresponding to the initial rate of aminoacylation as a function of tRNA concentration were fit to the Michaelis-Menten equation to derive the Km (tRNA), kcat (catalytic turnover of the enzyme), and kcat/Km (tRNA) (the catalytic efficiency of aminoacylation) using Kaleidagraph v. 4.5 (Synergy software).
Evaluation of tRNAPro isoacceptors for cell viability
In E. coli, the Pro(GGG) and Pro(CGG) isoacceptors are non-essential and can be deleted. To test their role in cell viability, several reconstructed proS suppressor strains were transformed to harbor the maintenance plasmid pACYC-araC-PC-PBAD-human trm5 (Gamper et al., 2015; Masuda et al., 2019) to afford analysis of both the m1G37+ and m1G37− conditions. Deletion of one of these isoacceptors was made by λRed recombination (primers in Table S2) on the WT MG1655 strain to replace the respective gene with a Kan marker (proL for Pro(GGG) and proK for Pro(CGG)). P1 lysates of the proL-KO strain (Gamper et al., 2015; Masuda et al., 2019) and of the proK-KO strain were used to transduce the reconstructed proS suppressor strains harboring the trm5-maintenance plasmid. After removal of the Kan marker by FLP recombination with pCP20, each deletion strain was maintained in the presence of Ara until analysis of cell viability in the m1G37− condition.
To test cell viability, each strain was grown overnight at 37°C in LB + chloramphenicol (Cm) (34 μg/mL) + Ara (0.2% (w/v)). Each strain was inoculated 1:100 to an Ara-free medium of LB + Cm and grown at 37°C for 5 h for depletion of the Trm5 protein and m1G37-tRNAs. Cells were then streaked out in the m1G37 + condition (LB + Cm + Ara) or the m1G37− condition (LB + Cm) and grown overnight at 37°C.
Growth complementation analysis was achieved by introducing each isoacceptor in an over-expression pKK223-3 plasmid. The proK gene was synthesized by Sequenase extension and cloned into pKK223-3 (primers in Table S2). The resultant proK-pKK223-3 plasmid, and proL-pKK223-3 and proM-pKK223-3 plasmids (Gamper et al., 2015; Masuda et al., 2021), was separately introduced to reconstructed proS suppressors harboring the trm5 maintenance plasmid. A variant of the proL-pKK223-3 plasmid, carrying the UGG anticodon replacing the GGG anticodon was made by site-direct mutagenesis (Masuda et al., 2018). A control plasmid, over-expressing Met(CAU) (initiator tRNAMet encoded by metV) was chosen. Expression of each tRNA in MG1655 was constitutive, due to lack of the lacI repressor. Growth complementation was tested in both m1G37+ and m1G37− conditions at 37°C.
Codon usage analysis of CC[C/U] codons in E. coli
The coding sequences (CDS) of E. coli str. K-12 substr. MG1655 were obtained from NCBI (GenBank: GCA_000005845.2). The frequencies of each Pro codon were calculated relative to the total number of all Pro codons in each CDS using Python (version 3.7.7). GO analysis was used to predict pathways with enriched codon usage of CC[C/U]. From the 3,876 genes analyzed, those with the frequency of CC[C/U] usage equal to or higher than 0.5 (792 genes, the top 20.4%) were selected and categorized into 4 groups according to their biological processes (http://geneontology.org/).
Codon usage analysis of a broader spectrum of bacterial pathogens was performed with a wide phylogenetic diversity for species with and without Pro(GGG). The species analyzed are Streptococcus pneumoniae (Spn), Staphylococcus aureus (Sau), Enterococcus faecalis (Efa), Clostridium botulinum (Cbo), Bacillus thuringiensis (Bth), Paenibacillus graminis (Pgr), Geobacillus kaustophilus (Gka), Rhodococcus opacus (Rop), Ruminococcus albus (Ral), Nostoc punctiforme (Npu), Vibrio parahaemolyticus (Vpa), Acinetobacter baumannii (Aba), Francisella tularensis (Ftu), Brachyspira pilosicoli (Bpi), Campylobacter jejuni (Cje), Vibrio cholerae (Vch), Escherichia coli (Eco), Pseudomonas aeruginosa (Pae), Treponema pallidum (Tpa), and Helicobacter pylori (Hpy). The CDS of each gene in each genome was retrieved from NCBI RefSeq (O’Leary et al., 2016) annotations and used to count gene-specific codon frequencies with a custom script.
Temperature-sensitivity assay
The E. coli MG1655 WT strain and reconstructed proS suppressor strains were grown in LB overnight, then spotted in serial dilution on an LB plate and grown at various temperatures (37, 30, 22 (room temp), and 18°C) for one to three overnights. Rescue of cold sensitivity was determined by over-expression of individual tRNAPro species from the pKK223-3 plasmid.
Phylogenetic analysis of prolyl isoacceptors across bacterial species
Bacterial tRNA genes were retrieved from the Genomic tRNA Database (GtRNAdb) (Chan and Lowe, 2016). The number of each tRNA gene was counted for each genome. The Biopython Entrez package designed for searching genome information in NCBI was used in a custom Python script to obtain the taxonomy classifications (phylum and genus) of each genome based on the taxonomy IDs (Table S3). Assemblies lacking Pro(UGG) isoacceptors for 4 box tRNAs were removed from analysis, which was likely caused by an incomplete assembly (3 assemblies). Genera with three or more representative species were selected for further analysis. Genus-level frequencies of each prolyl isoacceptor were computed by normalizing the number of species with each isoacceptor by the total number of species in the genus within this curated set. Genera were categorized into Gram-positive and Gram-negative using information obtained from PubMed literature search. The same search was used to highlight genera containing medically significant species.
To present the distribution of prolyl isoacceptors across the phylogenetic tree of bacteria, sequences of the 16S and 23S ribosomal RNA (rRNA) of each genome were retrieved from NCBI Refseq (O’Leary et al., 2016) or GenBank (Sayers et al., 2022). If rRNA annotations of a particular genome were not available, it was excluded from further analysis (3958 retrieved, 86 skipped). Sequences of 16S and 23S rRNAs were aligned using the SINA aligner (Pruesse et al., 2012) with the non-redundant SSU (small subunit of the ribosome) and LSU (large subunit of the ribosome) reference datasets (Ref NR 99) from SILVA (Quast et al., 2013) respectively. The 16S and 23S alignments of each genome were concatenated to a single FASTA entry. A single representative sequence was chosen arbitrarily from each genus selected for frequency analysis of a prolyl tRNA gene. A maximum-likelihood phylogenetic tree was generated with the prepared concatenated 16S and 23S alignments using FastTree (Price et al., 2009, 2010). Tree image was created using matplotlib with the ETE toolkit (Huerta-Cepas et al., 2016). Figure 6D was generated with the matplotlib python module (https://matplotlib.org/stable/index.html).
QUANTIFICATION AND STATISTICAL ANALYSIS
The statistical significance of +1 frameshifting in Figure 2E was tested by Welch’s t test with a p value using Microsoft Excel. The n represents the number of independent biological replicates of the assay and the mean of 4 independent experiments is shown with an SD (see Figure 2 legend). In Figure 6D, the correlation between the distribution of Pro(GGG) and the frequency of the CCC codon for Pro was computed by the Kolmogorov-Smirnov (KS) test, a non-parametric and distribution-free statistical test, using scipy and numpy in Python 3. Similarly, we used the Scipy python module (https://docs.scipy.org/doc/scipy/) to calculate the average of Pro codon frequencies across all protein-coding genes with at least one Pro codon for each species. We then compared this average codon frequency between the set of species with and without Pro(GGG) (n = 5 species from each group as shown in Figure 6D) (see Figure 6 legend and STAR Methods). The analysis was done for Gram-positive and Gram-negative species independently.
Supplementary Material
Highlights.
m1G37 methylation on the 3′ side of the tRNA anticodon is essential for cell survival
Suppressors of loss of m1G37 are found to harbor mutations in prolyl-tRNA synthetase
These suppressors uncouple the dependence on m1G37 for prolyl-aminoacylation of tRNA
Suppressors lacking m1G37 require Pro(GGG) for decoding of proline codons CC[C/U]
ACKNOWLEDGMENTS
We thank Sean Moore for advice, Roy Kishony for plasmid pZS2R, Howard Gamper for Pro(UGG) tRNA, and the BioComputing lab of Bar-Ilan U for discussion. This work is supported by NIH grants R35 GM134931 to Y.-M.H. and R01 HG006753 to T.M.L.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111539.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Agris PF, Eruysal ER, Narendran A, Väre VYP, Vangaveti S, and Ranganathan SV (2018). Celebrating wobble decoding: half a century and still much is new. RNA Biol. 15, 537–553. 10.1080/15476286.2017.1356562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn HJ, Kim HW, Yoon HJ, Lee BI, Suh SW, and Yang JK (2003). Crystal structure of tRNA(m1G37)methyltransferase: insights into tRNA recognition. EMBO J. 22, 2593–2603. 10.1093/emboj/cdg269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begley U, Dyavaiah M, Patil A, Rooney JP, DiRenzo D, Young CM, Conklin DS, Zitomer RS, and Begley TJ (2007). Trm9-catalyzed tRNA modifications link translation to the DNA damage response. Mol. Cell 28, 860–870. 10.1016/j.molcel.2007.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björk GR, and Hagervall TG (2014). Transfer RNA modification: presence, synthesis, and function. EcoSal Plus 6. 10.1128/ecosalplus.ESP-0007-2013. [DOI] [PubMed] [Google Scholar]
- Björk GR, Wikström PM, and Byström AS (1989). Prevention of translational frameshifting by the modified nucleoside 1-methylguanosine. Science 244, 986–989. [DOI] [PubMed] [Google Scholar]
- Byström AS, and Björk GR (1982). The structural gene (trmD) for the tRNA(m1G)methyltransferase is part of a four polypeptide operon in Escherichia coli K-12. Mol. Gen. Genet 188, 447–454. [DOI] [PubMed] [Google Scholar]
- Chan PP, and Lowe TM (2016). GtRNAdb 2.0: an expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 44, D184–D189. 10.1093/nar/gkv1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chionh YH, McBee M, Babu IR, Hia F, Lin W, Zhao W, Cao J, Dziergowska A, Malkiewicz A, Begley TJ, et al. (2016). tRNA-mediated codon-biased translation in mycobacterial hypoxic persistence. Nat. Commun 7, 13302. 10.1038/ncomms13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian T, Evilia C, Williams S, and Hou YM (2004). Distinct origins of tRNA(m1G37) methyltransferase. J. Mol. Biol 339, 707–719. 10.1016/j.jmb.2004.04.025. [DOI] [PubMed] [Google Scholar]
- Christian T, Gamper H, and Hou YM (2013). Conservation of structure and mechanism by Trm5 enzymes. RNA 19, 1192–1199. 10.1261/rna.039503.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian T, and Hou YM (2007). Distinct determinants of tRNA recognition by the TrmD and Trm5 methyl transferases. J. Mol. Biol 373, 623–632. 10.1016/j.jmb.2007.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian T, Lahoud G, Liu C, and Hou YM (2010). Control of catalytic cycle by a pair of analogous tRNA modification enzymes. J. Mol. Biol 400, 204–217. 10.1016/j.jmb.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian T, Sakaguchi R, Perlinska AP, Lahoud G, Ito T, Taylor EA, Yokoyama S, Sulkowska JI, and Hou YM (2016). Methyl transfer by substrate signaling from a knotted protein fold. Nat. Struct. Mol. Biol 23,941–948. 10.1038/nsmb.3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, and Ruden DM (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92. 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifton BE, Fariz MA, Uechi GI, and Laurino P (2021). Evolutionary repair reveals an unexpected role of the tRNA modification m1G37 in aminoacylation. Nucleic Acids Res. 49, 12467–12485. 10.1093/nar/gkab1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crepin T, Yaremchuk A, Tukalo M, and Cusack S (2006). Structures of two bacterial prolyl-tRNA synthetases with and without a cis-editing domain. Structure 14, 1511–1525. 10.1016/j.str.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, and Wanner BL (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 97, 6640–6645. 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Crécy-Lagard V, and Jaroch M (2021). Functions of bacterial tRNA modifications: from ubiquity to diversity. Trends Microbiol. 29, 41–53. 10.1016/j.tim.2020.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Oliveira DMP, Forde BM, Kidd TJ, Harris PNA, Schembri MA, Beatson SA, Paterson DL, and Walker MJ (2020). Antimicrobial resistance in ESKAPE pathogens. Clin. Microbiol. Rev 33, 001811–e219. 10.1128/CMR.00181-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinçbas V, Heurgué-Hamard V, Buckingham RH, Karimi R, and Ehrenberg M (1999). Shutdown in protein synthesis due to the expression of mini-genes in bacteria. J. Mol. Biol 291, 745–759. 10.1006/jmbi.1999.3028 [DOI] [PubMed] [Google Scholar]
- Gamper H, Li H, Masuda I, Miklos Robkis D, Christian T, Conn AB, Blaha G, Petersson EJ, Gonzalez RL Jr., and Hou YM (2021a). Insights into genome recoding from the mechanism of a classic +1-frameshifting tRNA. Nat. Commun 12, 328. 10.1038/s41467-020-20373-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper H, Mao Y, Masuda I, McGuigan H, Blaha G, Wang Y, Xu S, and Hou YM (2021b). Twice exploration of tRNA +1 frameshifting in an elongation cycle of protein synthesis. Nucleic Acids Res. 49, 10046–10060. 10.1093/nar/gkab734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper HB, Masuda I, Frenkel-Morgenstern M, and Hou YM (2015). Maintenance of protein synthesis reading frame by EF-P and m(1)G37-tRNA. Nat. Commun 6, 7226. 10.1038/ncomms8226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto-Ito S, Ito T, Ishii R, Muto Y, Bessho Y, and Yokoyama S (2008). Crystal structure of archaeal tRNA(m(1)G37)methyltransferase aTrm5. Proteins 72, 1274–1289. 10.1002/prot.22019. [DOI] [PubMed] [Google Scholar]
- Goto-Ito S, Ito T, Kuratani M, Bessho Y, and Yokoyama S (2009). Tertiary structure checkpoint at anticodon loop modification in tRNA functional maturation. Nat. Struct. Mol. Biol 16, 1109–1115. 10.1038/nsmb.1653. [DOI] [PubMed] [Google Scholar]
- Grenier F, Matteau D, Baby V, and Rodrigue S (2014). Complete genome sequence of Escherichia coli BW25113. Genome Announc. 2, 010388–e1114. 10.1128/genomeA.01038-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L, and Phizicky EM(2018).A rationale for tRNA modification circuits in the anticodon loop. RNA 24, 1277–1284. 10.1261/rna.067736.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K, Morooka N, Yamamoto Y, Fujita K, Isono K, Choi S, Ohtsubo E, Baba T, Wanner BL, Mori H, and Horiuchi T (2006). Highly accurate genome sequences of Escherichia coli K-12 strains MG1655 and W3110. Mol. Syst. Biol 2, 2006.0007. 10.1038/msb4100049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill PJ,Abibi A,Albert R, Andrews B, Gagnon MM,Gao N, Grebe T, Hajec LI, Huang J, Livchak S, et al. (2013). Selective inhibitors of bacterial t-RNA-(N(1)G37) methyltransferase (TrmD) that demonstrate novel ordering of the lid domain. J. Med. Chem 56, 7278–7288. 10.1021/jm400718n [DOI] [PubMed] [Google Scholar]
- Huerta-Cepas J, Serra F, and Bork P (2016). Ete 3: reconstruction, analysis, and visualization of phylogenomic data. Mol. Biol. Evol 33, 1635–1638. 10.1093/molbev/msw046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakowec M, Prentki P, Chandler M, and Galas DJ (1988). Mutational analysis of the open reading frames in the transposable element IS1. Genetics 120, 47–55. 10.1093/genetics/120.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsic ED, Zhao J, Vetsigian K, and Kishony R (2015). Counteraction of antibiotic production and degradation stabilizes microbial communities. Nature 521, 516–519. 10.1038/nature14485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamichhane TN, Arimbasseri AG, Rijal K, Iben JR, Wei FY, Tomizawa K, and Maraia RJ (2016). Lack of tRNA-i6A modification causes mitochondrial-like metabolic deficiency in S. pombe by limiting activity of cytosolic tRNA-Tyr, not mito-tRNA. RNA 22, 583–596. 10.1261/rna.054064.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. (2007). Clustal W and clustal X version 2.0. Bioinformatics 23, 2947–2948. 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Li H (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993. 10.1093/bio-informatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JN, and Björk GR (1999). Structural alterations of the tRNA(m1G37) methyltransferase from Salmonella typhimurium affect tRNA substrate specificity. RNA 5, 395–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu X, Yang Q, Li L, Dang Y, Zhou Z, Chen S, and Liu Y (2020). Adaptation of codon usage to tRNA I34 modification controls translation kinetics and proteome landscape. PLoS Genet. 16, e1008836. 10.1371/journal.pgen.1008836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maehigashi T, Dunkle JA, Miles SJ, and Dunham CM (2014). Structural insights into +1 frameshifting promoted by expanded or modification-deficient anticodon stem loops. Proc. Natl. Acad. Sci. USA 111, 12740–12745. 10.1073/pnas.1409436111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda I, Hwang JY, Christian T, Maharjan S, Mohammad F, Gamper H, Buskirk AR, and Hou YM (2021). Loss of N(1)-methylation of G37 in tRNA induces ribosome stalling and reprograms gene expression. Elife 10, e70619. 10.7554/eLife.70619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda I, Matsubara R, Christian T, Rojas ER, Yadavalli SS, Zhang L, Goulian M, Foster LJ, Huang KC, and Hou YM (2019). tRNA methylation is a global determinant of bacterial multi-drug resistance. Cell Syst. 8, 302–314.e8. 10.1016/j.cels.2019.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda I, Takase R, Matsubara R, Paulines MJ, Gamper H, Limbach PA, and Hou YM (2018). Selective terminal methylation of a tRNA wobble base. Nucleic Acids Res. 46, e37. 10.1093/nar/gky013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasvall SJ, Chen P, and Bjork GR (2004). The modified wobble nucleoside uridine-5-oxyacetic acid in tRNAPro(cmo5UGG) promotes reading of all four proline codons in vivo. RNA 10, 1662–1673. 10.1261/rna.7106404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Näsvall SJ, Chen P, and Björk GR (2007). The wobble hypothesis revisited: uridine-5-oxyacetic acid is critical for reading of G-ending codons. RNA 13, 2151–2164. 10.1261/rna.731007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D, et al. (2016). Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745. 10.1093/nar/gkv1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perret V, Garcia A, Grosjean H, Ebel JP, Florentz C, and Giegé R (1990). Relaxation of a transfer RNA specificity by removal of modified nucleotides. Nature 344, 787–789. 10.1038/344787a0. [DOI] [PubMed] [Google Scholar]
- Price MN, Dehal PS, and Arkin AP (2009). FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol 26, 1641–1650. 10.1093/molbev/msp077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Dehal PS, and Arkin AP (2010). FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS One 5, e9490. 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruesse E, Peplies J, and Glöckner FO (2012). SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28, 1823–1829. 10.1093/bioinformatics/bts252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, and Glöckner FO (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rak R, Dahan O, and Pilpel Y (2018). Repertoires of tRNAs: the couplers of genomics and proteomics. Annu. Rev. Cell Dev. Biol 34, 239–264. 10.1146/annurev-cellbio-100617-062754. [DOI] [PubMed] [Google Scholar]
- Razin S, Yogev D, and Naot Y (1998). Molecular biology and pathogenicity of mycoplasmas. Microbiol. Mol. Biol. Rev 62, 1094–1156. 10.1128/MMBR.62.4.1094-1156.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisch CR, and Prather KLJ (2015). The no-SCAR (Scarless Cas9 Assisted Recombineering) system for genome editing in Escherichia coli. Sci. Rep 5, 15096. 10.1038/srep15096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisch CR, and Prather KLJ (2017). Scarless Cas9 assisted recombineering (no-SCAR) in Escherichia coli, an easy-to-use system for genome editing. Curr. Protoc. Mol. Biol 117, 31.8.1–31.8.20. 10.1002/cpmb.29. [DOI] [PubMed] [Google Scholar]
- Sakaguchi R, Lahoud G, Christian T, Gamper H, and Hou YM (2014). A divalent metal ion-dependent N(1)-methyl transfer to G37-tRNA. Chem. Biol 21, 1351–1360. 10.1016/j.chembiol.2014.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayers EW, Cavanaugh M, Clark K, Pruitt KD, Schoch CL, Sherry ST, and Karsch-Mizrachi I (2022). Nucleic Acids Res. 50, D161–D164. 10.1093/nar/gkab1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiaville PC, Legendre R, Rojas-Benítez D, Baudin-Baillieu A, Hatin I, Chalancon G, Glavic A, Namy O, and de Crécy-Lagard V (2016). Global translational impacts of the loss of the tRNA modification t(6)A in yeast. Microb. Cell 3, 29–45. 10.15698/mic2016.01.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas SE, Whitehouse AJ, Brown K, Burbaud S, Belardinelli JM, Sangen J, Lahiri R, Libardo MDJ, Gupta P, Malhotra S, et al. (2020). Fragment-based discovery of a new class of inhibitors targeting mycobacterial tRNA modification. Nucleic Acids Res. 48, 8099–8112. 10.1093/nar/gkaa539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres AG, Piñeyro D, Filonava L, Stracker TH, Batlle E, and Ribas de Pouplana L (2014). A-to-I editing on tRNAs: biochemical, biological and evolutionary implications. FEBS Lett. 588, 4279–4286. 10.1016/j.febslet.2014.09.025. [DOI] [PubMed] [Google Scholar]
- Vargas-Rodriguez O, and Musier-Forsyth K (2013). Exclusive use of trans-editing domains prevents proline mistranslation. J. Biol. Chem 288, 14391–14399. 10.1074/jbc.M113.467795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virtanen MA, Uvarov P, Hubner CA, and Kaila K (2020). NKCC1, an elusive molecular target in brain development: making sense of the existing data. Cells 9. 10.3390/cells9122607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Silke JR, and Xia X (2019). An improved estimation of tRNA expression to better elucidate the coevolution between tRNA abundance and codon usage in bacteria. Sci. Rep 9, 3184. 10.1038/s41598-019-39369-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weixlbaumer A, Murphy FV 4th, Dziergowska A, Malkiewicz A, Vendeix FAP, Agris PF, and Ramakrishnan V (2007). Mechanism for expanding the decoding capacity of transfer RNAs by modification of uridines. Nat. Struct. Mol. Biol 14, 498–502. 10.1038/nsmb1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White TA, and Kell DB (2004). Comparative genomic assessment of novel broad-spectrum targets for antibacterial drugs. Comp. Funct. Genomics 5, 304–327. 10.1002/cfg.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehouse AJ, Thomas SE, Brown KP, Fanourakis A, Chan DSH, Libardo MDJ, Mendes V, Boshoff HIM, Floto RA, Abell C, et al. (2019). Development of inhibitors against Mycobacterium abscessus tRNA (m(1)G37) methyltransferase (TrmD) using fragment-based approaches. J. Med. Chem 62, 7210–7232. 10.1021/acs.jmedchem.9b00809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaremchuk A, Cusack S, and Tukalo M (2000). Crystal structure of a eukaryote/archaeon-like protyl-tRNA synthetase and its complex with tRNAP-ro(CGG). EMBO J. 19, 4745–4758. 10.1093/emboj/19.17.4745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong W, Koay A, Ngo A, Li Y, Nah Q, Wong YH, Chionh YH, Ng HQ, Koh-Stenta X, Poulsen A, et al. (2019a). Targeting the bacterial epi-transcriptome for antibiotic development: discovery of novel tRNA-(N(1)G37) methyltransferase (TrmD) inhibitors. ACS Infect. Dis 5, 326–335. 10.1021/acsinfecdis.8b00275. [DOI] [PubMed] [Google Scholar]
- Zhong W, Pasunooti KK, Balamkundu S, Wong YH, Nah Q, Gadi V, Gnanakalai S, Chionh YH, McBee ME, Gopal P, et al. (2019b). Thieno-pyrimidinone derivatives that inhibit bacterial tRNA (Guanine37-N(1))-methyl-transferase (TrmD) by restructuring the active site with a tyrosine-flipping mechanism. J. Med. Chem 62, 7788–7805. 10.1021/acs.jmed-chem.9b00582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinshteyn B, and Gilbert WV (2013). Loss of a conserved tRNA anticodon modification perturbs cellular signaling. PLoS Genet. 9, e1003675. 10.1371/journal.pgen.1003675. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We deposited our whole-genome sequencing data at EMBL-EBI ArrayExpress with an accession number E-MTAB-12178 (key resources table). These data will be publicly available as of the date of publication. In addition, we used publicly available datasets for codon usage analysis. The accession numbers for the datasets that we used are listed (key resources table). All original codes generated for analyses of publicly available datasets are deposited at GitHub and are publicly available as of the date of publication (key resources table). Any additional information required to reanalyze data reported in this study is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-CysRS antibodies | (Masuda et al., 2021) | N/A |
| Rabbit polyclonal anti-TrmD antibodies | (Li and Bjork, 1999) | N/A |
| Goat polyclonal anti-rabbit IgG antibodies peroxidase conjugate | Sigma-Aldrich | Cat. #A0545; RRID: AB_257896 |
| Bacterial and virus strains | ||
| Escherichia coli strain K-12 substrain MG1655 | ATCC | Cat. #700926 |
| E. coli strain BW25113 | The Coli Genetic Stock Center (CGSC) | CGSC#: 7636 |
| E. coli strain XAC | (Jakowec et al., 1988) | N/A |
| E. coli strain BL21(DE3) | Millipore | Cat. #69450 |
| E. coli strain ArcticExpress (DE3) | Agilent Technologies | Cat. #230191 |
| E. coli strain K-12 substrain MG1655, proS D98N (trmD + or trmD−) | This paper | N/A |
| E. coli strain K-12 substrain MG1655, proS G107C (trmD + or trmD−) | This paper | N/A |
| E. coli strain K-12 substrain MG1655, proS S123R (trmD + or trmD−) | This paper | N/A |
| E. coli strain K-12 substrain MG1655, proS T199I (trmD + or trmD−) | This paper | N/A |
| E. coli strain K-12 substrain MG1655, proS T199S (trmD + or trmD−) | This paper | N/A |
| E. coli strain K-12 substrain MG1655, proS Q211L (trmD + or trmD−) | This paper | N/A |
| E. coli strain K-12 substrain MG1655, proS A231E (trmD + or trmD−) | This paper | N/A |
| E. coli strain K-12 substrain MG1655, proS N232S (trmD + or trmD−) | This paper | N/A |
| E. coli strain K-12 substrain MG1655, proS A236E (trmD + or trmD−) | This paper | N/A |
| E. coli strain K-12 substrain MG1655, proS D98N, trmD−, pACYC-araC-PC-PBAD-human trm5, proK− | This paper | N/A |
| E. coli strain K-12 substrain MG1655, proS D98N, trmD−, pACYC-araC-PC-PBAD-human trm5, proL− | This paper | N/A |
| E. coli strain K-12 substrain MG1655, proS T199I, trmD−, pACYC-araC-PC-PBAD-human trm5, proK− | This paper | N/A |
| E. coli strain K-12 substrain MG1655, proS T199I, trmD−, pACYC-araC-PC-PBAD-human trm5, proL− | This paper | N/A |
| E. coli strain K-12 substrain MG1655, proS N232S, trmD−, pACYC-araC-PC-PBAD-human trm5, proK− | This paper | N/A |
| E. coli strain K-12 substrain MG1655, proS N232S, trmD−, pACYC-araC-PC-PBAD-human trm5, proL− | This paper | N/A |
| Bacteriophage P1vir | CGSC | CGSC#: 12,133 |
| Chemicals, peptides, and recombinant proteins | ||
| L-Arabinose | Gold Biotechnology | Cat. #A-300-250 |
| BamHI | New England BioLabs | Cat. #R0136 |
| EcoRI | New England BioLabs | Cat. #R0101 |
| NdeI | New England BioLabs | Cat. #R0111 |
| PstI | New England BioLabs | Cat. #R0140 |
| XhoI | New England BioLabs | Cat. #R0146 |
| PfuUltraII fusion HS DNA polymerase | Agilent Technologies | Cat. #600670 |
| Phusion™ High-Fidelity DNA Polymerase | ThermoFisher Scientific | Cat. #F530 |
| NEBuilder® HiFi DNA Assembly Cloning Kit | New England BioLabs | Cat. #E5520S |
| T4 DNA ligase | New England Biolabs | Cat. #M0202 |
| T4 polynucleotide kinase | New England Biolabs | Cat. #M0201 |
| Ampicillin | Fisher Scientific | Cat. #BP1760 |
| Chloramphenicol | Gold Biotechnology | Cat. #G-105 |
| Kanamycin | Gemini Bio-products | Cat. #400-114P |
| Anhydrous tetracycline hydrochloride | Cayman Chemicals | Cat. #10009542 |
| [γ-32P]-ATP | PerkinElmer | Cat. #NEG002A |
| [3H]-Proline | PerkinElmer | Cat. #NET483005MC |
| Critical commercial assays | ||
| SuperSignal West Pico Chemiluminescent Substrate | ThermoFisher Scientific | Cat. #34080 |
| ONE-Glo™ Luciferase Assay System | Promega | Cat. #E6120 |
| Deposited data | ||
| Whole-genome sequencing data of E. coli trmD-KO suppressor cells | This paper | EMBL-EBI ArrayExpress: E-MTAB-12178 |
| Codes for codon usage and tRNAPro distribution analysis | This paper | https://doi.org/10.5281/zenodo.7093913 |
| List of annotated genome sequences retrieved from public databases | Table S3 | N/A |
| Experimental models: Organisms/strains | ||
| E. coli strain K-12 substrain MG1655 | ATCC | Cat. #700926 |
| Oligonucleotides | ||
| Oligo DNAs for bacterial strain construction, plasmid construction, CRISPR mutagenesis, and Northern blotting | Table S2 | N/A |
| Recombinant DNA | ||
| pKD46 | CGSC | CGSC #7634 |
| pKD46-EctrmD | This paper | N/A |
| pCP20 | CGSC | CGSC #7629 |
| pACYC-araC-PC-PBAD-human trm5 | (Gamper et al., 2015) | N/A |
| pCas9cr4 | (Reisch and Prather, 2015) | N/A |
| pKDsgRNA | (Reisch and Prather, 2015) | N/A |
| pZS2R-mCherry | (Kelsic et al., 2015) | N/A |
| pZS2R-YFP | (Kelsic et al., 2015) | N/A |
| pKK223-3 E. coli Pro(CGG) (proK) | This paper | N/A |
| pKK223-3 E. coli Pro(GGG) (proL) | (Gamper et al., 2015) | N/A |
| pKK223-3 E. coli Pro(UGG) (proM) | (Masuda et al., 2021) | N/A |
| pKK223-3 E. coli Pro(GGG) (proL) with anticodon UGG | (Masuda et al., 2018) | N/A |
| pKK223-3 E. col Met(CAU) (metV) | (Gamper et al., 2015) | N/A |
| Software and algorithms | ||
| FASTQC v 0.11.5 | Andrews S. (2010) | http://www.bioinformatics.babraham.ac.uk/projects/fastqc |
| Burrows-Wheeler Alignment (BWA) tool v 0.7.15 - r1140 | (Li and Durbin, 2009) | N/A |
| SAMtools v 1.3.1 | (Li et al., 2009) | N/A |
| Bcftools | (Li, 2011) | https://samtools.github.io/bcftools/bcftools.html |
| SnpEff | (Cingolani et al., 2012) | N/A |
| Image Lab v. 6.0 | BIO-RAD | http://www.bio-rad.com/en-us/product/image-lab-software |
| PyMOL v. 2.4.1 | Schrödinger | https://pymol.org/ |
| Clustal X v. 2.1 | (Larkin et al., 2007) | N/A |
| Excel v. 2010 | Microsoft | https://www.microsoft.com/ |
| Kaleidagraph v. 4.5 | Synergy software | https://www.synergy.com/ |
| ImageJ v. 1.51 | National Institutes of Health | https://imagej.nih.gov |
| Python v. 3.7.7 | Python Software Foundation | https://www.python.org/ |
| ETE3 v. 3.1.2 | (Huerta-Cepas et al., 2016) | http://etetoolkit.org/ |
| RefSeq release 211 | (OĽeary et al., 2016) | https://www.ncbi.nlm.nih.gov/refseq/ |
| GenBank release 248 | (Sayers et al., 2022) | https://www.ncbi.nlm.nih.gov/genbank/ |
| FastTree v. 2.1.10 | (Price et al., 2009, 2010) | http://www.microbesonline.org/fasttree/ |
| GtRNAdb release 19 | (Chan and Lowe, 2016) | http://gtrnadb.ucsc.edu/ |
| matplotlib v. 3.5.0 | https://doi.org/10.1109/MCSE.2007.55 | https://matplotlib.org/3.1.1/index.html |
| SINA v. 1.2.11 | (Pruesse et al., 2012) | https://www.arb-silva.de/aligner/ |
| SILVA release 89 | (Quast et al., 2013) | https://www.arb-silva.de/ |
| Scipy v.1.9.0 | (Virtanen et al., 2020) | https://scipy.org |
| Other | ||
| NucleoSpin Gel and PCR Clean-up | Macherey-Nagel | Cat. #740609 |
| Hybond™-N + Nitrocellulose Membrane | GE healthcare | Cat. #RPN203B |
| Nextera XT DNA Library Preparation Kit | Illumina | Cat. #FC-131-1024 |
| Trans-Blot® TurboTM Transfer System | BIO-RAD | Cat. #1704150 |
| MicroPulser Electroporator | BIO-RAD | Cat. #1652100 |
| UV Crosslinker | Fisher Scientific | Cat. #FB-UVXL-1000 |
| Tri-Carb 4910 TR Scintillation Counter | PerkinElmer | N/A |
| Immobilon-P PVDF Membrane | Millipore | IPVH00010 |
| ChemiDoc MP Imaging System | BIO-RAD | Cat. #12003154 |
| Typhoon IP Imaging System | GE Healthcare | N/A |
| Infinite M200 PRO Plate Reader | Tecan | N/A |