

Abstract
Microenvironmental changes in the early metastatic niche may be exploited to identify therapeutic targets to inhibit secondary tumor formation and improve disease outcomes. We dissected the developing lung metastatic niche in a model of metastatic, triple-negative breast cancer using single-cell RNA-sequencing. Lungs were extracted from mice at 7-, 14-, or 21 days after tumor inoculation corresponding to the pre-metastatic, micro-metastatic, and metastatic niche, respectively. The progression of the metastatic niche was marked by an increase in neutrophil infiltration (5% of cells at day 0 to 81% of cells at day 21) and signaling pathways corresponding to the hallmarks of cancer. Importantly, the pre-metastatic and early metastatic niche were composed of immune cells with an anti-cancer phenotype not traditionally associated with metastatic disease. As expected, the metastatic niche exhibited pro-cancer phenotypes. The transition from anti-cancer to pro-cancer phenotypes was directly associated with neutrophil and monocyte behaviors at these time points. Predicted metabolic, transcription factor, and receptor-ligand signaling suggested that changes in the neutrophils likely induced the transitions in the other immune cells. Conditioned medium generated by cells extracted from the pre-metastatic niche successfully inhibited tumor cell proliferation and migration in vitro and the in vivo depletion of pre-metastatic neutrophils and monocytes worsened survival outcomes, thus validating the anti-cancer phenotype of the developing niche. Genes associated with the early anti-cancer response could act as biomarkers that could serve as targets for the treatment of early metastatic disease. Such therapies have the potential to revolutionize clinical outcomes in metastatic breast cancer.
Keywords: Breast cancer, Single-cell sequencing, Metastasis, Cancer immunology, Anti-cancer signaling
Introduction
As one in eight women are diagnosed with breast cancer in their lifetime, metastatic breast cancer is a leading cause of death worldwide [1]. Metastasis, the colonization of tumor cells at distal tissues, leads to the development of secondary tumors that compromise the function of vital organs, such as the lungs, liver, and brain [2]. In many instances, cancer remains undetected until the patient experiences physical symptoms associated with metastasis (e.g., respiratory distress, blurred vision) [3]. Current studies on metastatic cancer often focus on late-stage disease; yet, recent advances offer new opportunities to identify disease at the pre- or micro-metastatic stages [4–7]. An understanding of the local microenvironment may be utilized to identify novel biomarkers and therapeutic targets that could inhibit secondary tumor formation. Such treatments could be particularly effective in breast cancer, where localized disease has a 99% 5 year survival rate that drops to a mere 27% in cases of metastasis [1].
Research based on Stephen Paget’s “seed-and-soil” hypothesis has shown that specific distal tissues are primed for the arrival of metastatic cells by tumor-secreted factors and tumor-conditioned immune cells [8–10]. Specific cell types (e.g., myeloid derived suppressor cells (MDSCs)), signaling molecules (e.g., S100A8/9), and structural changes (e.g., extracellular matrix (ECM) remodeling) have been identified that support this hypothesis and enable the formation of the pre-metastatic niche [11]. While changes in cell populations at metastatic sites are well established [12–14], the emergence of single-cell sequencing provides an avenue to monitor the phenotypic evolution of cells as the tissue evolves from its initial healthy state to a pre-metastatic niche, and ultimately to a mature metastatic niche with a macroscopic tumor. Characterization of the microenvironment throughout this transition establishes potential to facilitate early detection and subsequently improve the treatment of metastatic disease.
Herein, we detail the temporal evolution of the lung metastatic niche in a mouse model of metastatic, triple-negative breast cancer. The cell types and phenotypic progression at the lung from healthy to metastatic disease were analyzed using single-cell RNA-sequencing [15, 16]. This technology has been applied extensively to investigate cancer cell clonal evolution and stemness, chemotherapeutic response, and rare populations [17–19]. We have utilized these methods to analyze the dynamic metastatic microenvironment. We illustrate that cells in the pre-metastatic niche maintain an anti-cancer phenotype that transitions to pro-metastatic as the disease progresses. Monocytes and neutrophils were identified as the primary contributors to this phenotypic shift. These data were supported through gene and protein expression, predicted signaling pathways, and functional assays. This interrogation of the localized microenvironment at a metastatic niche can be employed to develop a staging of metastatic disease, and identify novel therapeutic targets based on these stages.
Materials and methods
All materials and reagents were purchased from Thermo Fisher Scientific (Waltham, MA) unless otherwise stated.
Animal models
All animal studies were conducted with approval from and in accordance with the University of Michigan Institutional Animal Care and Use Committee guidelines and protocols. Female BALB/c mice were purchased from Jackson Laboratory (Bar Harbor, ME). At 7–8 weeks old, mice received an orthotopic injection of 4T1-tdTomato-luc2 cells (Perkin Elmer; Waltham, MA) (2 million cells/50 μl phosphate buffered saline (PBS)) in the fourth right mammary fat pad [4, 20–22]. The 4T1 murine cell line is a reproducible model of triple-negative breast cancer with spontaneous metastases to the lung and bone marrow [23].
For the Gr1 cell depletion studies, mice received 100 μg intraperitoneal injections (1 mg/ml in PBS) of InVivoMab anti-Gr1 antibody (clone RB6–8C5; Bio X Cell; Lebanon, NH) according to one of three different time courses: (1) days 1 and 3, (2) days 7 and 10, or (3) days 14 and 17 [22]. Extent of CD45 + Gr1 + cell depletion was measured 24 h after the second dose through flow cytometry with a ZE5 Cell Analyzer (Bio-Rad; Hercules, CA). The body condition of the mice was monitored daily throughout the survival study and mice were euthanized if tumor diameter > 2 cm, ulceration encompassed > 50% tumor surface area, tumor interference caused partial paralysis, or the mouse was found moribund [21].
Tissue processing
Lungs were extracted from mice at 7-, 14-, or 21 days post-inoculation and from their healthy counterparts (day 0). Samples from three diseased mice or two healthy mice were pooled together at each time point to account for biological variability. Tissues were finely diced with a scalpel and incubated in RPMI containing 0.2 U/ml Liberase TL (Roche; Basel, Switzerland) and 150 U/ml DNase I (Sigma-Aldrich; St. Louis, MO) at 37 °C for 20 min [20, 22, 24]. The lungs were filtered through a 70 μm mesh to create a single cell suspension. Cells were washed with PBS containing 30% (w/v) bovine serum albumin (BSA) and 0.5 M ethylenediaminetetraacetic acid (EDTA). Erythrocyte lysis was conducted by sequential exposure to hypotonic (0.2% w/v) and hypertonic (1.6% w/v) solutions of sodium hydroxide. Cells were washed twice before they were counted with the Countess™ Automated Cell Counter. Cells were resuspended to a final concentration of 100,000 cells/ml in 0.01% (w/v) BSA in PBS.
Library preparation and sequencing
Droplet generation oil, cells, and oligo-labeled beads (Chemgenes; Wilmington, MA) were passed through a Drop-Seq device (FlowJEM; Toronto, CA) as described by Macosko, et al. [25]. The device was allowed to flow for 10 min and the resulting cell-bead pairs (STAMPs) were collected and the oil was removed. Approximately 1000 STAMPs underwent reverse transcription, Exonuclease I treatment, and universal PCR. cDNA concentrations and quality were measured with a Bioanalyzer DNA high sensitivity chip (Agilent Technologies; Santa Clara, CA). Tagmentation of the cDNA was conducted with Tn5 transposase as described by Picelli et al. [26]. Samples were then loading into Illumina HiSeq sequencers and approximately 30,000 reads/cell were generated with asymmetric reads.
Post–processing and analysis
The Drop-Seq pipeline (v1.2) was used to map raw Illumina reads to the mouse genome (mm10), determine cell barcodes and gene counts, and create the digital gene expression (DGE) file [25]. DGE files were inputted into Seurat v2 designed by the Satija research group for cell type identification and analysis [27, 28]. Sequencing data from the healthy controls from each time point were combined to establish a single healthy control (day 0) (Fig. S1). A single Seurat object was compiled that included all samples. Cells were classified as having 200 to 5000 RNA fragments and less than 25% percent mitochondrial genes. One cluster of dead cells remained in the resulting object and was removed computationally. At least 1000 cells from each time point were included in the final object.
Gene set enrichment analysis (GSEA)
Differentially expressed genes were converted to their human orthologs using biomaRt [29]. Pre-ranked gene lists were assembled for significant genes (adjusted p-value < 0.05) using the fold change between conditions as the ranking variable. The corresponding human gene sets underwent GSEA using a pre-ranked gene list to identify significantly enriched pathways [30]. Gene sets were sampled from the hallmark, BIOCARTA, KEGG, REACTOME, PID, and gene ontology (GO) databases obtained from the Molecular Signatures Database (MSigDB) collections [30].
Temporal changes in gene expression and pseudotime
Time dependency of gene expression and pseudotime calculations were conducted using Monocle3 established by the Trapnell lab [31]. Seurat objects were converted to Monocle3 objects using the established vignette. Changes in gene expression were analyzed with regression models over real time. Genes were considered to significantly change over time when q < 0.05. The pseudotime roots were automatically identified, and they closely matched the true roots at day 0. Pseudotime values for each cell were compared to the true time points and cell subsets to assess correlation. Cells that were excluded from the analysis had reported pseudotimes of infinity.
Metabolic activity analysis
We performed data imputation on the filtered sequencing matrix using Markov Affinity-based Graph Imputation of Cells (MAGIC) to increase the amount of information [32]. The MAGIC-imputed gene expression matrix was normalized using a robust version of the Z-score method [33], where MAD = median absolute deviation (Eq. 1). Gene-wise two-tailed P-values were computed for each cell using the Z-score, with α = 0.05.
| (1) |
Up- and down-regulated metabolic genes intersecting with the iHUMAN metabolic reconstruction [34] were used in Flux Balance Analysis [35]. A modified version of the iMAT algorithm (CFR() in MATLAB) [36] was used to maximize the corresponding metabolic flux, while the probabilistic regulation of metabolism (PROM) down-regulation constraint minimized reaction flux [37]. Parsimonious Flux Balance Analysis was used to assume efficient allocation of metabolic resources and to obtain unique metabolic flux distributions [38]. The fold-change for metabolic flux was computed by taking the value from all cells relative to the mean metabolic flux at the previous time point.
Transcription factor regulatory analysis
Activity of the most variable transcription factors in the monocytes and neutrophils was determined using the DoRothEA (v1.2.1) algorithm for single-cell RNA-sequencing [39–41]. Mouse regulons (grades A–C) were obtained from the DoRothEA R package and Virtual Inference of Protein-activity by Enriched Regulon analysis (VIPER) scores were computed. We extracted the 50 most variable transcription factors over time. The predicted transcription factors were compared to their corresponding gene expression, and those with low real expression (normalized expression < 0.5) were removed from analysis.
Receptor–ligand interactions
Time-dependent neutrophil- and monocyte-specific ligands were determined using NicheNet (v1.0) [42]. Monocytes and neutrophils were used as the “sender” cell types (ligands), while all immune cells were deemed “receivers” (targets). Ligands that were differentially expressed in neutrophils or monocytes relative to the general population were considered cell-specific and used for further analyses. All genes were then converted to their human orthologs [29]. Scores for receptor-ligand pairs were calculated using CellPhoneDB with the following parameters: counts data type = hgnc_symbol, ratio of cells in a cluster expressing a gene = 0.1, p-value = 0.05, precision = 3, and number of statistical iterations = 10 [43]. Interaction scores refer to the product of the mean expression of the receptor gene in one cell type and the mean expression of the ligand gene in the other cell type.
Antibody staining
Neutrophil and monocyte phenotypes were validated through immunofluorescence staining [44]. Upon collection, tissues were flash frozen in isopentane and embedded in optimal cutting temperature compound prior to sectioning. Samples were subsequently fixed with 4% (w/v) paraformaldehyde and permeabilized with 0.1% (v/v) Triton-X, with three 5 min PBS washes after each step. The tissues were blocked with 10% (v/v) normal donkey serum (Sigma-Aldrich) in 0.02% (v/v) Tween-20 (Sigma-Aldrich) for 1 h at room temperature, protected from light. Samples were incubated overnight at 4 °C with the primary antibodies in 1% (v/v) donkey serum in 0.02% (v/v) Tween-20. Antibodies for the following proteins were used: Ly6G (1:100, BV421-conjugated; Biolegend; San Diego, CA), IL1B (1:200, AF647-conjugated; Novus Biologicals; Littleton, CO), MMP9 (1:200, Cy3-conjugated; Bioss Antibodies; Woburn, MA), CCL6 (1:200, FITC-conjugated; R&D Systems; Minneapolis, MN), CSTA (1:100, AF594-conjugated; Bioss Antibodies), S100A8 (1:200, Cy5-conjugated; Bioss Antibodies), Ly6C (1:100, BV421-conjugated; Biolegend), CHI3L3 (1:100, goat, unconjugated; R&D Systems), Collagen I (1:100, AF594-conjugated; Novus Biologicals), and Fibronectin (1:100, FITC-conjugated; Novus Biologicals). The following day, samples containing the CHI3L3 antibody were washed, and incubated with a secondary antibody (1:400, donkey anti-goat, AF647-conjugated; Abcam; Cambridge, UK) for 2 h at room temperature for detection of CHI3L3. After a final washing step, glass coverslips were mounted with Fluoromount and sealed with nail polish. Imaging was conducted on a Zeiss Axio Observer.Z1 inverted microscope and analyzed with the Zen software package (Zeiss; Oberkochen, Germany).
Generation of conditioned medium
Lungs were extracted from three mice at each stage of disease (days 0, 7, 14, and 21) and processed as described above. After erythrocyte lysis, cells were resuspended in phenol red-free RPMI at a concentration of 5 million cells/ml. Cells were cultured for 24 h before the medium and cells were collected and centrifuged at 12,000×g for 5 min. The supernatant was sterile filtered and stored at − 80 °C until use. Protein concentrations of the conditioned medium were determined using a BCA assay and samples were diluted to 300 μg/ml with phenol red-free RPMI.
Scratch assay
4T1 tumor cells were seeded in flat-bottomed 96-well plates at a concentration of 10,000 cells/well and allowed to proliferate until confluence. A 100 μl pipette tip was used to create a scratch down the center of each well [24]. Wells were washed with PBS to remove any non-adherent cells. The wells were replenished with 50% tumor cell medium (RPMI with 10% fetal bovine serum) and 50% conditioned medium [20]. Cells were imaged immediately with a Zeiss Axio Observer.Z1 inverted microscope with two images/well. The locations of the images were stored and re-imaged after 5 h and 24 h. The reduction in area of the scratch was calculated using the ImageJ software.
Tumor cell proliferation assay
4T1-tdTomato-luc2 tumor cells were seeded in flat-bottomed 96-well plates at a concentration of 7500 cells/well in phenol red-free RPMI with 10% fetal bovine serum. A standard curve was established using 4T1 cells in a serial dilution with a maximum concentration of 200,000 cells/well. Conditioned media was added to the cells (50% total volume) with phenol red-free RPMI added to the controls and standard curve. Changes in cell proliferation were determined by reading the 96-well plate in a Biotek Synergy H1 Microplate Reader (Winooski, VT) for tdTomato fluorescence (Ex554/Em581) at t = 0, 24, 48, and 72 h [20]. Cell concentrations were calculated by comparing fluorescent intensity to the standard curve at t = 0 h.
T cell proliferation assay
T cells were collected from the spleens of healthy, female BALB/c mice. The spleens were mechanically processed to a single-cell suspension, the erythrocytes were lysed, and T cells were isolated with the Pan T Cell Isolation Kit II (Miltenyi Biotec; Bergisch Gladbach, Germany). The T cell population was stained with the CellTrace Far Red Cell Proliferation Kit. Stained T cells were seeded in a round-bottom 96-well plate at a density of 150,000 cells/well. The positive controls and samples were treated with Dynabeads™ Mouse T-Activator CD3/CD28 for T-Cell Expansion and Activation (20 μl/ml) and anti-mouse CD28 antibody (3 μg/ml; Biolegend) [45, 46]. The negative and positive controls were supplemented with 50% RPMI while the samples were supplemented with 50% conditioned medium. Cultures were incubated at 37 °C for 72 h. At the end of the culture, the 96-well plate was placed directly in a ZE5 Cell Analyzer. T cell proliferation was assessed by the fluorescent intensity of eFlour 660 (Ex633/Em669) using FlowJo software (BD; Franklin Lakes, NJ).
Lentivirus production
Third-generation lentivirus was produced by co-transfecting lentiviral packaging vectors (pMDL-GagPol, pRSV-Rev, pIVS-VSV-G) and lentiviral vectors using JetPrime (Polyplus; Strasbourg, France) into HEK-293 T cells [47]. Viral supernatant was collected after 48 h in culture, concentrated using PEG-it (Systems Biosciences; Palo Alto, CA), re-suspended in PBS, and stored at − 80 °C until use.
Ex vivo bioluminescence imaging
TRanscriptional activity cell arrays (TRACER) was used to monitor activity of transcription factors in the tumor cells in response to conditioned medium [47, 48]. Unlabeled 4T1 tumor cells were seeded in a black 384-well plate at 10,000 cells/well with activity reporter lentivirus for IRF1, NFκB, or SP1. Cells were exposed to d-luciferin after 24 h of culture. After an additional 24 h, cells were exposed to conditioned medium (50% total volume) and re-exposed to d-luciferin. The activities of the transcription factors were monitored with luciferase readings on an IVIS Spectrum (Perkin Elmer) at 0, 2, 4, 6, and 8 h after the addition of conditioned medium. The luciferase values were internally normalized to t = 0 h and then further normalized to the day 0 conditioned medium-treated samples to calculate the relative changes in transcription factor activity.
Metastatic burden was measured through luciferase on an IVIS Spectrum. Briefly, lungs were explanted 21 days post-tumor inoculation and incubated in 50 μM d-luciferin for 10 min. Imaging was conducted with large binning and automatic exposure to determine the integrated light flux (photons/s).
Statistical analysis
Differential gene expression was determined using default Seurat settings with the Wilcoxon rank-sum test. Significance of signaling pathways was output from the GSEA software. The p-values of pathway scores were determined with Student’s t-tests using the Bonferroni multiple hypothesis correction (α = 0.01). Significance between samples for functional assays was conducted using ANOVA with the Tukey multiple comparison test. In vitro experiments were conducted with n = 3 biological replicates and n = 3 technical replicates. All figures show mean ± SEM with α = 0.05, unless otherwise stated.
Results
Dynamics of lung cell populations in metastatic progression
Lungs were explanted from mice in an orthotopic, syngeneic model of triple-negative metastatic breast cancer (4T1) at 7, 14, or 21 days post-inoculation (Fig. 1a), which correlate to the pre-metastatic, early metastatic (micro-metastatic), and metastatic niche, respectively [4, 21]. Controls, denoted as “day 0”, were obtained from the lungs of healthy mice. Single-cell RNA sequencing was used to investigate temporal dynamics in cell populations and their respective phenotypes. We identified 13 major cell types in the lungs (Figs. 1b, S2). Although we did not identify tumor cells in our data, 4T1-tdTomato-luc2 cells could be detected by imaging in the day 14 and day 21 lungs (Fig. S2). Therefore, the low number of tumor cells were likely contained within other cell clusters. Healthy lungs were predominantly composed of B Cells, T Cells, and alveolar macrophages (19.3%, 14.3%, and 16.0% of all cells, respectively). Macrophages, neutrophils, natural killer (NK) cells, pneumocytes (alveolar type I and type II cells) and stromal cells (primarily fibroblasts) each comprised around 5% of the lung composition.
Fig. 1.
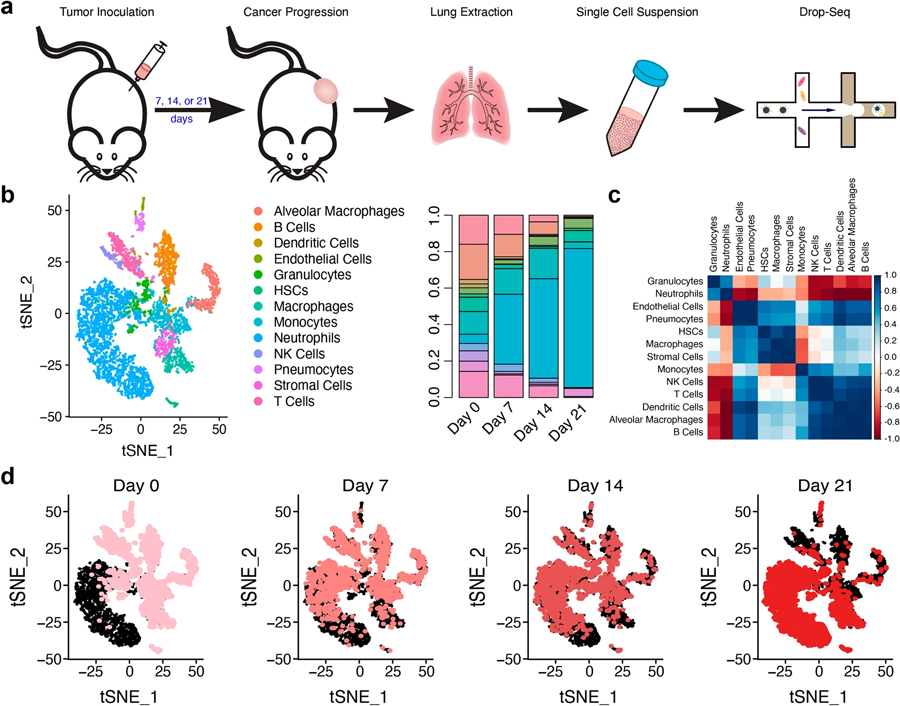
The cellular composition of the lung is dynamic in metastatic progression. a Female Balb/c mice received an orthotopic inoculation of the triple-negative metastatic breast cancer cell line, 4T1. Tumors progressed for either 7, 14, or 21 days, the lungs were explanted, and the tissues were processed into a single-cell suspension. Library preparation was completed using the Drop-Seq platform. b tSNE plot showing the 13 major cell clusters identified in the lung. The proportion of each cell type was time dependent and disease progression was marked by an influx of neutrophils. c Correlation plot of the changes in each cell population over time. Pearson correlations were calculated by comparing the cell proportions at each time point. d tSNE plots over time clearly depict a shift in cell populations. Pink dots (which get darker over time) identify cells at the designated time, while black dots identify the cells at the other time points
After seven days, neutrophil density increased 6.6-fold relative to healthy mice. By day 21, neutrophils (and immature granulocytes) comprised 81.7% of all cells in the lung, consistent with previous reports [49]. The increase in these cells over time corresponded with concordant decreases in the other cell types (Fig. 1b, c). Throughout early-stage disease (day 0 to day 14), the concentration of B cells, dendritic cells, NK cells, and T cells decreased proportionally to the increases in neutrophils, while monocyte concentrations remained relatively constant (12.4–16.7%). Other cell types, including macrophages, hematopoietic stem cells (HSCs), and stromal cells, greatly decreased at day 7, indicating that these cells were highly sensitive to the onset of cancer. The progression of cell types in disease evolution was visualized with t-distributed stochastic neighbor embedding (tSNE) plots (Fig. 1d), primarily defined by an immediate and persistent influx of neutrophils.
The early lung metastatic niche is marked by increases in anti–cancer immune signaling
Overall gene expression was tracked over time to identify phenotypic and signaling transitions in the lung. Regression analysis identified over 2000 genes that significantly changed (q < 0.05) throughout disease progression (Table S1). Top 50 genes that increased over time include S100a6, S100a8, S100a9, Cxcr2, Ifitm1, and Mmp8 (Fig. 2a), which correlated to pathways associated with the inflammatory response, including NK cell activation and T cell differentiation (Table S2). These genes were primarily associated with neutrophils and, to a lesser extent, early granulocytes and monocytes (Fig. S3). Genes that decreased with time correlated to pathways involved in protein and ribosome localization as well as antigen presentation (Table S3).
Fig. 2.
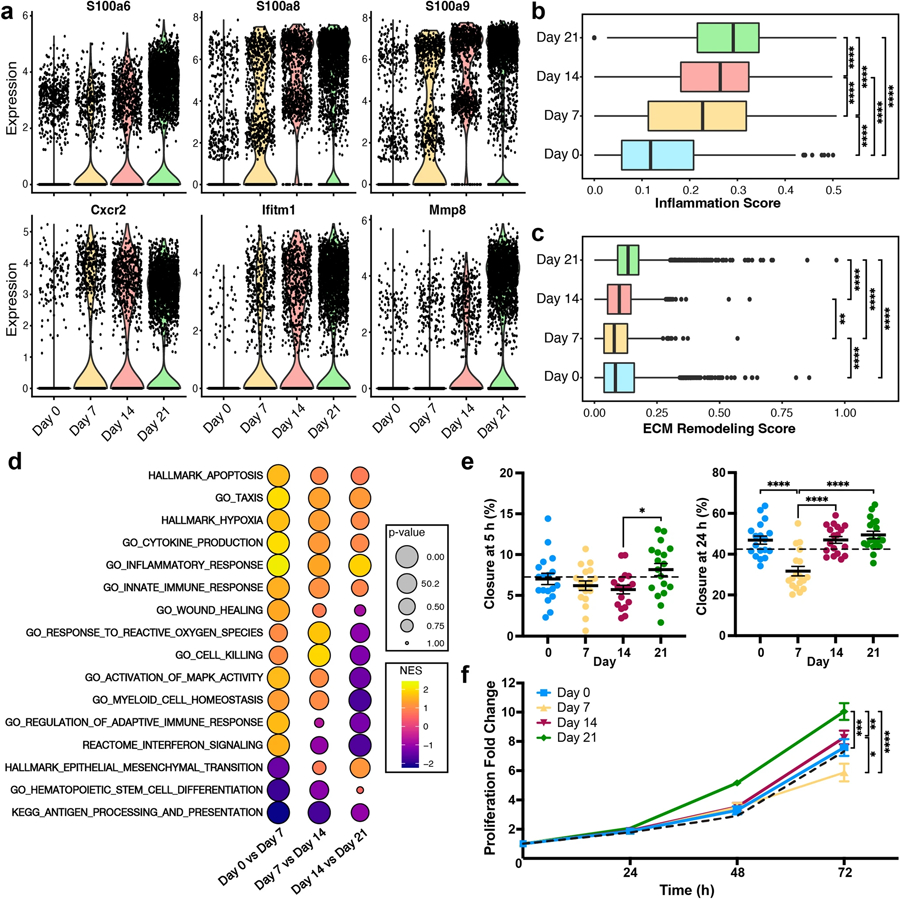
Lung phenotypic shifts in metastatic progression. a Expression of pro-metastatic genes increased as disease progressed. b Inflammation scores were calculated through the average expression of genes in the GO_INFLAMMATORY_RESPONSE pathway. c ECM remodeling scores were calculated through the average expression of genes in the GO_INFLAMMATORY_RESPONSE pathway. Points represent outlying cells as calculated by 1.5*IQR. d Enrichment of signaling pathways (as determined through Gene Set Enrichment Analysis) from differential gene expression between sequential time points, NES normalized enrichment score. e Percent of the scratch that had closed at 5 h and 24 h post-initial scratch when 4T1 tumor cells were exposed to conditioned medium (collected from lungs on days 0, 7, 14, and 21 of culture). The dotted black line represents the RPMI control. f Fold increase in the number of 4T1 cells over 72 h when the cells were exposed to conditioned medium (at t = 0 h) as measured through tdTomato fluorescence, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Gene expression was linked to established hallmarks of cancer [50] by averaging the expression in select MSigDB pathways to calculate corresponding scores for inflammation (Fig. 2b), ECM remodeling (Fig. 2c), hypoxia (Fig. S4), angiogenesis (Fig. S4), genome instability (PARP signaling; Fig. S4), and glycolysis (Fig. S4). Inflammation, hypoxia, and glycolysis scores all increased over time. Specifically, the median inflammation score doubled from day 0 to day 7, consistent with the neutrophil influx between these times, and increased an additional 28% between days 7 and 21. Importantly, this gene set includes both pro- and anti-inflammatory genes, therefore this score reflects the deviation from healthy tissue. Neither genome instability nor matrix remodeling were impacted by early onset disease, yet both scores increased 60–73% from day 7 to day 21. The score for angiogenesis was relatively unaffected by disease progression. These data are consistent with the continued activation of each of these hallmarks of cancer, except angiogenesis, at the metastatic niche.
We identified differentially expressed signaling pathways between each time point (day 0 vs. day 7, day 7 vs. day 14, day 14 vs. day 21) to investigate unsupervised lung phenotypic changes (Table S4). In addition to the increased expression of the cancer-related gene sets, progression of disease was also marked by continual down-regulation of GO_HEMATOPOIETIC_STEM_CELL_DIFFERENTIATION and KEGG_ANTIGEN_PROCESSING_AND_PRESENTATION (Fig. 2d). GO_CELL_KILLING, GO_WOUND_HEALING, and GO_RESPONSE_TO_REACTIVE_OXYGEN_SPECIES were up-regulated at days 7 and 14 but down-regulated at day 21—suggesting early anti-cancer responses. Late-stage immune dysregulation was observed through the down-regulation of GO_REGULATION_OF_ADAPTIVE_IMMUNE_RESPONSE, GO_MYELOID_CELL_HOMEOSTASIS, and REACTOME_INTERFERON_SIGNALING at day 21. The HALLMARK_EPITHELIAL_MESENCHYMAL_TRANSITION gene set was down-regulated at day 7 but up-regulated at days 14 and 21, consistent with tumor cell arrival [4, 21, 22]. These data suggest that the pre- and early metastatic niche may be defined by a pro-inflammatory, anti-cancer microenvironment that transitions to the widely reported pro-cancer environment of the metastatic niche.
The functional impact of this dynamic microenvironment was assessed by treating 4T1 tumor cells with conditioned medium from a 24 h culture of lung cells collected at each time point. Using a scratch assay to investigate cell migration (Fig. 2e), the closure of cells exposed to day 7- and day 14-conditioned medium was slightly less than the RPMI controls five hours after the initial scratch. The closure of cells exposed to day 21-conditioned medium was 32% and 43% higher than the day 7- and day 14-conditioned (p < 0.05) samples, respectively. Twenty-four hours after the initial scratch, the areas of the day 0-, day 14-, and day 21-conditioned samples were essentially identical, while the day 7-conditioned samples exhibited 50% less closure (p < 0.05) than the other samples. In the proliferation assay (Fig. 2f), after 48 h, day 21-conditioned samples proliferated 5.2-fold while all other samples only increased 3.3–3.5-fold. At 72 h, tumor cells proliferated 7.6-fold (day 0), 5.7-fold (day 7), 8.3-fold (day 14), and 10.0-fold (day 21). These findings validate the anti-cancer role of the day 7 microenvironment, the transitional role of the day 14 microenvironment, and pro-cancer role of the day 21 microenvironment.
The immune cells that contribute to inflammatory changes at the lung were identified by comparing gene expression at days 0 (healthy mice) and 21 within each cell type (Fig. S5). For the monocytes, 20 genes were differentially expressed between the two time points with p < 0.05 and a fold change greater than 2.0. Neutrophils had 87 genes with similar characteristics and 22 genes were identified for both the B cells and T cells. The other immune cell populations had a maximum of four genes that met these criteria. Subsequent analyses were conducted on monocytes and neutrophils due to their more substantial numbers, greater changes in gene expression, and higher expression of genes that relate to overall phenotypic shifts in the lung.
Tumor–associated monocytes arrive at day 21 and contribute to ECM remodeling
Five monocyte phenotypes were identified in the lung—classical monocytes, MHCII + monocytes (antigen presenting), non-classical monocytes, monocyte precursors, and Chi3l3 + classical monocytes (Fig. 3a, b). Between days 0 and 14, MHCII +, non-classical, and precursor monocytes decreased 23%, 70%, and 80%, respectively. Over this same time frame, classical monocytes increased over threefold. Chi3l3 + classical monocytes comprised 36% of the day 21 population but were virtually undetected at earlier time points indicating that this population correlated to tumor-associated monocytes. These phenotypic shifts were marked by concomitant decreases in monocyte heterogeneity (Fig. 3c).
Fig. 3.
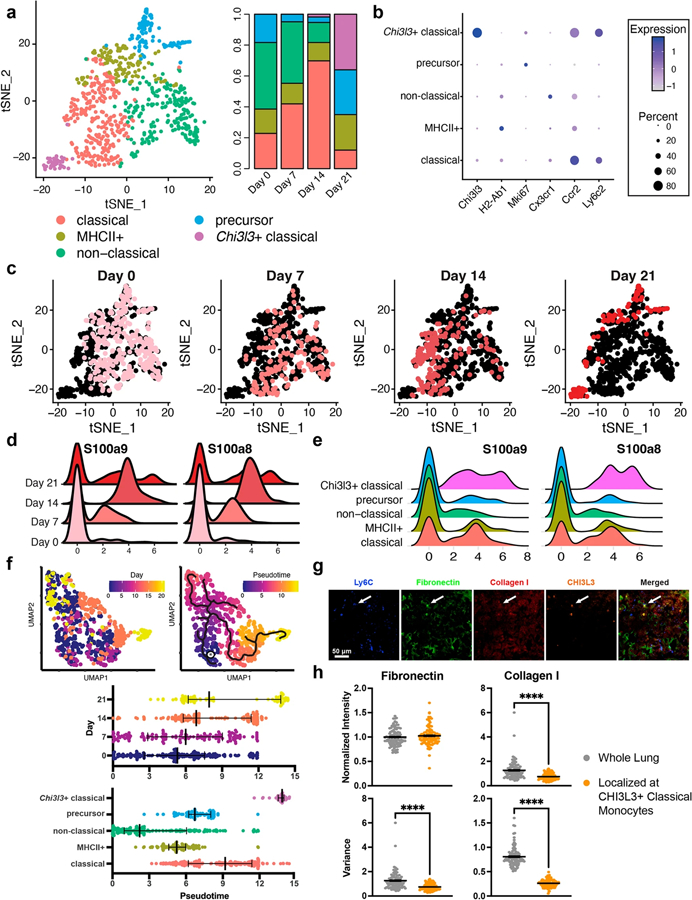
Tumor-associated monocytes in late-stage disease support ECM remodeling. a Five monocyte phenotypes were identified in the lung and their populations were time dependent. b Classical and non-classical (alternatively activated) monocytes were identified through Ccr2 and Cx3cr1 expression. c Monocyte heterogeneity decreased as time progressed. Pink dots identify cells at the designated time point. d Time-dependent responses of S100a8 and S100a9 in the monocytes. e Responses of S100a8 and S100a9 in each of the monocyte subsets. f Comparison of the real-time monocyte progression to pseudotime analysis. The star indicates the pseudotime origin as predicted by the Monocle3 algorithm. g Immunofluorescence staining of monocyte/ECM-related proteins in lung sections at day 21, blue = Ly6C, green = fibronectin, red = collagen I, orange = CHI3L3. Arrows point to CHI3L3 + monocytes. h Quantification of fibronectin and collagen I intensity and variance in the lung and in the area immediately surrounding CHI3L3 + monocytes (refer to Fig. S7 for determination of localized area), ****p < 0.0001
Normalized expression of S100a8/a9, known indicators of metastasis [21, 51], in monocytes steadily increased from day 0 to day 14 (Fig. 3d). On day 21, expression of these genes in Chi3l3 + classical monocytes was 3.5-fold higher than the classical monocytes and 6.0-fold higher than any other subset (Fig. 3e). These findings support the pro-cancer role of Chi3l3 + classical monocytes at day 21. Increases in such genes as Tgfbi, Sell, Lcn2, and S100a4, resulted in the enrichment of the GO_EXTRACELLULAR_MATRIX pathway (among other ECM-related pathways) in the monocytes at day 21 (normalized enrichment score (NES) = 3.17) (Fig. S6). At day 7 and day 14 NES values were − 2.46 (p < 0.001; down-regulation) and 1.18 (p = 0.26), respectively. The non-classical phenotype of early monocytes and the down-regulation of ECM pathways suggests that these cells do not support the foundations for a metastatic niche. Furthermore, lineage tracing of the monocyte pseudotime trajectory paralleled the real time disease (Fig. 3f), suggesting that late-stage monocytes were, at least in part, a result of phenotypic shifts in the localized monocytes. When the pseudotime analysis was conducted on the monocyte clusters, it was evident that monocytes transition from non-classical to classical to Chi3l3 + classical as disease progresses.
The connection between monocytes and ECM proteins in the microenvironment was visualized by immunofluorescence staining. We identified relatively constant levels of fibronectin and collagen I in the lung throughout metastatic progression (Figs. 3g, S7). However, in the areas immediately surrounding (within 50 μm) the CHI3L3 + classical monocytes (white arrows), significant changes were seen in ECM (Fig. S7). Specifically, the variance of fibronectin and collagen I greatly decreased in this region relative to the rest of the lung. The differences in variance correlated to changes in protein conformation near the monocytes. Combined, these data support the hypothesis that the tumor associated, Chi3l3 + classical monocytes contribute to ECM remodeling in metastasis.
Neutrophils in early metastatic disease exhibit anti–cancer phenotypes
Four major neutrophil phenotypes were identified in the lung (Fig. 4a, b). Tumor-associated neutrophils were identified through their expression of Mmp8 [52]. Mmp9 was used to identify pro-cancer neutrophils [53] with Ltf expression delineating immature MDSCs [54]. The anti-cancer neutrophils, which were Mmp9-, were concordantly positive for Il1b. Neutrophils in the healthy lung (day 0) were 87.4% Il1b + mature, while an additional 11.0% clustered with the Il1b + anti-cancer phenotype, indicating that these cells do not inherently support metastatic niche formation. On days 7 and 14, only 20.2% and 32.0% of the neutrophils were classified as either Mmp9 + Ltf- pro-cancer or Mmp9 + Ltf + MDSCs, respectively. Mmp9 + Ltf + MDSCs comprised no more than 2.0% of the day 7 and day 14 populations. In contrast, 37.3% and 27.1% of neutrophils on day 21 were Mmp9 + Ltf- pro-cancer neutrophils and Mmp9 + Ltf + MDSCs, respectively. These data suggest that early neutrophils in metastases may act to combat tumor cell colonization.
Fig. 4.
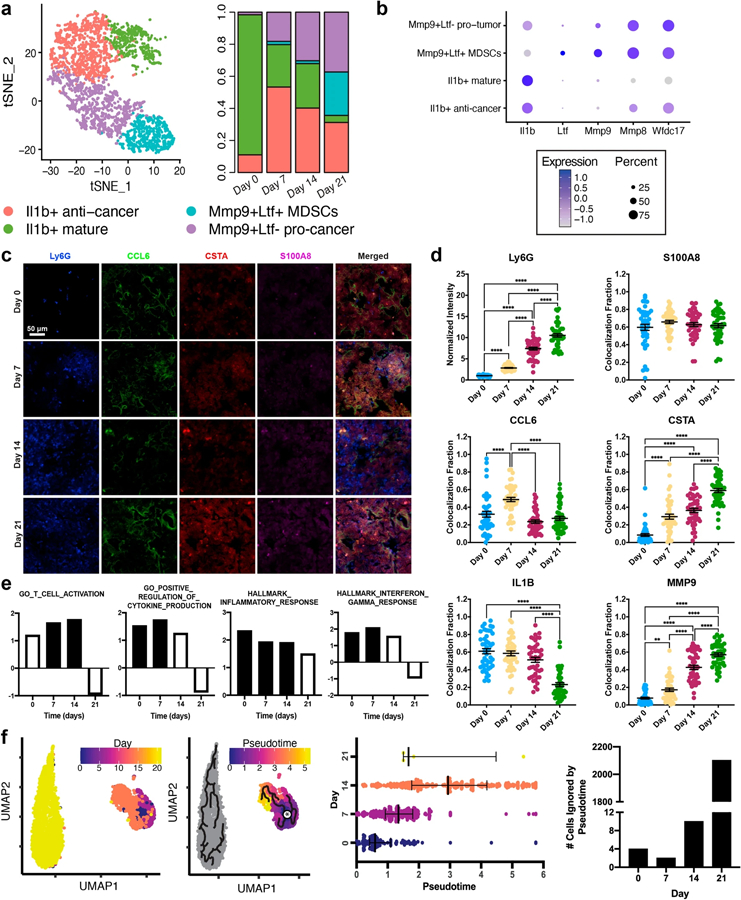
Neutrophils from early metastatic disease have an anti-cancer phenotype. a Four neutrophil phenotypes were identified in the lung and their populations were time dependent. b Pro- and anti-cancer phenotypes were identified through their expression of Mmp9, Ly6g, and Il1b. Cancer-related, but not mature, neutrophils were Wfdc17 +. c Immunofluorescence staining of neutrophil-related proteins in lung sections, blue = Ly6G, green = CCL6, red = CSTA (Stfa2 protein), magenta = S100A8. d Quantification of protein expression. Ly6G intensity denotes the mean fluorescent intensity across each image. Colocalization fraction reports the fraction of BV421 + (Ly6G) pixels that were also positive for the designated protein. e Enrichment of signaling pathways (by Gene Set Enrichment Analysis) from differential gene expression between neutrophils at each time point and all other lung cells, NES normalized enrichment score. Filled bars are significant with p < 0.05. f Comparison of the real-time neutrophil progression to pseudotime analysis. The star indicates the pseudotime origin as predicted by the Monocle3 algorithm. Nearly all the neutrophils collected on day 21 could not be phenotypically linked to those collected between days 0 and 14, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Notably, overall expression of S100a8/a9 increased approximately sixfold from day 0 to day 7 and a further 2.5-fold between days 7 and 21 (Fig. 2a). Interestingly, the normalized expression of these genes in the neutrophils was unchanged over time (Fig. S8). Rather, these data suggest that neutrophil contributions to the S100a8/a9 increase were due to the increased number of cells and independent of phenotypic changes. The protein expression of S100A8 scaled with Ly6G over time (Fig. 4c, d), inferring that this trend is maintained throughout translation.
Markers expressed across the continuum of neutrophil phenotypes and disease stages could provide an avenue to isolate and investigate these early cells. For example, Wfdc17 was widely expressed in all tumor-associated neutrophil populations, but poorly expressed in healthy, mature neutrophils. Furthermore, Ifitm1 and Stfa2 steadily increased throughout disease progression, validated by increased CSTA (Stfa2) protein expression (Figs. 4d, S8). Average Wfdc17, Ifitm1, and Stfa2 expression in the neutrophils was 20-, 40- and 151-fold higher than the average of the other cell types, respectively, indicating strong neutrophil-specificity. Ccl6 and Il1b, although not considered neutrophil-specific, could also be used to monitor neutrophil phenotype (Fig. S8). Gene expression of Ccl6, a pro-metastatic chemokine [55], decreased between day 0 (healthy) and day 14, but increased at day 21. Il1b was highly expressed at days 0 and 7, but markedly decreased at days 14 and 21. Although IL-1 β is often considered pro-tumorigenic, neutrophil-specific IL-1 β can have anti-tumorigenic effects [56]. IL-1 β protein expression was over twofold lower at day 21 relative to the other time points (Figs. 4d, S8), which coincides with the pro-cancer phenotype of neutrophils in late-stage disease. In contrast, MMP9 protein expression increased over time, further corroborating the transition towards pro-cancer. These trends support the hypothesis that neutrophils maintain an anti-cancer phenotype (day 7) at early time points, which transitions (day 14) to pro-cancer (day 21) as disease progresses.
The functional role of neutrophils was investigated by GSEA analysis of the neutrophils at each time point (Fig. 4e). GO_T_CELL_ACTIVATION was significantly up-regulated at days 7 and 14. HALLMARK_INTERFERON_GAMMA_RESPONSE, HALLMARK_INFLAMMATORY_RESPONSE, and GO_POSITIVE_REGULATION_OF_CYTOKINE_PRODUCTION steadily decreased from day 7 to day 21. As each of these pathways are associated with pro-inflammatory, anti-cancer responses, these data are consistent with an anti-cancer neutrophil phenotype at day 7. Furthermore, at day 21, T cell activation, cytokine production, and interferon gamma signaling in the neutrophils were down-regulated, consistent with the predicted strong pro-metastatic phenotype at this late time point.
The pseudotime trajectory of the neutrophils successfully matched real-time disease progression between days 0 and 14 (Fig. 4f). Pseudotime scores increased 1.6-fold from day 0 to day 7 and 2.1-fold from day 7 to day 14 (p < 0.0001 for both), suggesting that neutrophils maintain a consistent lineage at the pre-metastatic site. However, 99.8% of day 21 neutrophils clustered separately from the other neutrophils and were classified as infinite by Monocle3, meaning they could not be connected to the predicted origin. Only 1.5% of neutrophils from days 0 to 14 were part of the infinite cluster. Comparison of the pseudotime analysis with neutrophil clusters demonstrates that a significant portion of each of the tumor-associated neutrophils are a result of recruitment (infinite pseudotime values) rather than differentiation (Fig. S9).
Neutrophil and monocyte signaling regulates metastatic progression
Neutrophil and monocyte signaling was assessed through changes in cell-specific transcription factor and ligand activity, intracellular metabolic pathways, and receptor-ligand signaling with other cell types. Of the 15 transcription factors predicted to be most variable in neutrophils, seven were up-regulated at day 0 through day 14, while the other 8 were up-regulated at day 21 (Fig. 5a). Irf9, Atf4, Cebpb, Stat1, and Stat3 activity were predicted to peak at day 7. The JAK-STAT signaling pathway (Stat1 and Stat3) is known to induce interferon signaling (Irf9), which has strong anti-cancer effects [57]. In contrast, JunD, Jun, and Fos, which increased at day 21, are members of the activator protein-1 (AP-1) that supports tumor progression [58]. In the monocytes, 15 of 18 transcription factors were up-regulated at days 7 and 14 (Fig. S10a). Similar to neutrophils, Stat1, Stat3, Irf1, and Irf9 activity in the monocytes was up-regulated at days 7 and 14 suggesting anti-cancer functions. Oncogenes Sp1, Nfe2l2, and Nfic [59–61] were up-regulated in day 21 monocytes, consistent with pro-tumor signaling and disease progression.
Fig. 5.
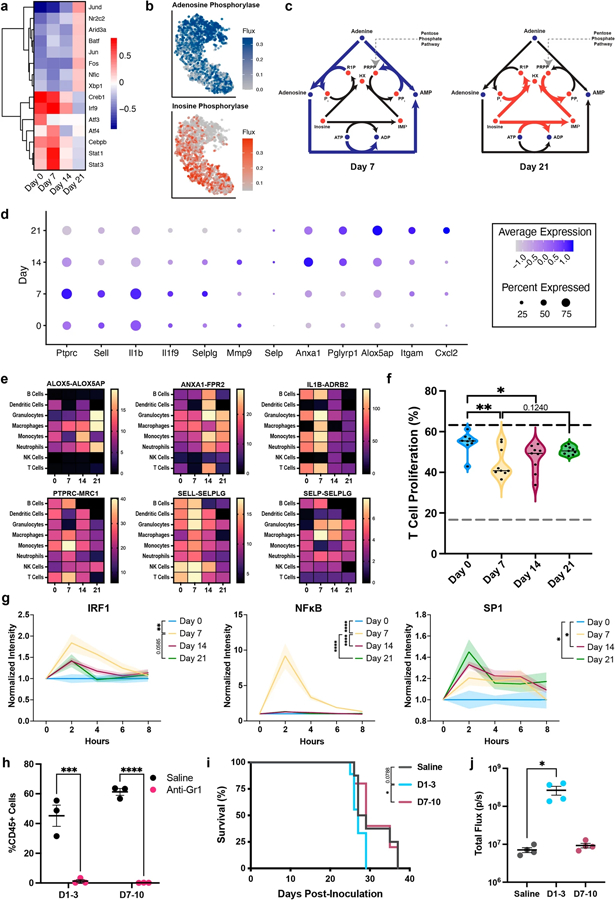
Neutrophil signaling changes throughout metastatic progression. a Heat maps of the most variably active transcription factors in the neutrophils as predicted by DoRothEA. b tSNE plots showing a selection of adenosine- and inosine-related metabolic reactions in the neutrophils. Metabolic events were determined through the gene expression of each cell and independent of time. c Schematics demonstrating the shift in metabolism from adenosine to inosine. Metabolic shifts were assessed using the time course data in Fig. 4. d Dot plot of the twelve time-dependent neutrophil-specific ligands as predicted by NicheNet. e Heat maps of the receptor-ligand interaction values with other immune cell types for select neutrophil ligands. f Percent of T cells that proliferated after 72 h exposure to conditioned medium. Dotted gray line = negative control, dotted black line = positive control. The black line in the violin plots indicate the median of the data set. g Transcription factor activity in 4T1 cells exposed to conditioned medium. h Gr1 + cells (neutrophils and monocytes) were depleted in the lung following two systemic doses of anti-Gr1 antibody. Depletion of early Gr1 + cells i decreased survival outcomes and j increased metastatic burden, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
The impact of the transcriptomic changes on neutrophil and monocyte functional phenotype was also investigated through the prediction of trends in metabolic activity. In neutrophils, nine metabolic reactions were differentially active between consecutive time points, which were primarily related to the TCA cycle and purine metabolism (Table S5). Specifically, we observed a metabolic shift from adenosine metabolism (day 7) to inosine metabolism (day 21) (Figs. 5b, c, S11). Neutrophils use adenosine to promote neutrophil chemotaxis, phagocytosis, cytotoxicity, and pro-inflammatory effector functions [62]. Extracellular inosine inhibits the secretion of pro-inflammatory cytokines required for normal immunological responses by surrounding lymphocytes [63]. No metabolic reactions were predicted to be differentially active in the monocytes between any two time points.
We next looked at receptor-ligand interactions to identify changes in neutrophil and monocyte signaling that could impact other immune cells. Twelve neutrophil-specific ligands significantly changed between consecutive time points (Figs. 5d, S12). Early neutrophils, from days 0 and 7, were enriched for selectins and members of the IL-1 family, consistent with a pro-inflammatory response. Anxa1, which peaks at day 14, has both pro- and anti-inflammatory responses, dependent on its role in the innate or adaptive immune system, respectively [64]. Alox5ap expression, involved in carcinogenesis in multiple types of cancer [65], is highest at day 21. These trends in ligand expression are consistent with the phenotypic shift in neutrophils from anti-cancer to pro-cancer. Neutrophil-specific ligands interacted with their respective receptors on nearly all the immune cell populations showing the widespread potential of neutrophil signaling (Fig. 5e). When an identical analysis was completed on the monocytes, only three ligands were detected (Ceacam1, Tgfb1, Trf) (Fig. S10b, c). The interaction score between Tgfb1 and Tgfbr1 was low (< 4) and relatively independent of time. Combined, these results suggest that although both neutrophils and monocytes are key factors in the phenotypic shift at the metastatic niche, neutrophils are likely the predominant driver of the signaling changes.
The role of phenotypic changes in cell signaling was validated by conditioned medium assays on T cells and tumor cells. First, pan T cells were exposed to conditioned medium, and proliferation was monitored over 72 h (Figs. 5f, S13). Day 7- and day 14-conditioned medium induced significant (p < 0.05) decreases in T cell proliferation relative to day 0, consistent with reports identifying decreases in proliferative T cells in patients responsive to immune checkpoint therapy (an enhanced anti-cancer microenvironment) [66]. Proliferation rates of day 0- and day 21-treated cells only varied by approximately 7%. The similar proliferation rates of the healthy and late-stage disease samples could correlate with decreased immunosurveillance of tumor cells in metastatic cancer. Second, the activities of cancer-associated transcription factors in the tumor cells were monitored in response to conditioned medium (Fig. 5g). Day 7 secreted factors induced increased activity of IRF1 and NFκB in the tumor cells relative to the other conditions. IRF1 is a known tumor suppressor in breast cancer [67]. NFκB is a key player in inflammatory responses associated with cancer [68] and could be induced by the highly pro-inflammatory behavior of the day 7 metastatic niche. The oncogenic transcription factor, SP1 [59], was significantly up-regulated in the day 14- and day 21-treated samples relative to day 0. These data indicate that changes in secreted proteins in the microenvironment, likely induced by the neutrophils, dynamically influence T cell and tumor cell signaling.
The early anti-cancer phenotypes were further validated through the depletion of Gr1 + (Ly6G + /Ly6C +; neutrophils and monocytes, respectively) cells in vivo. Mice were treated with two doses of an anti-Gr1 antibody according to three different schedules: days 1 and 3 post-inoculation (D1–3), days 7 and 10 post-inoculation (D7–10), and days 14 and 17 post-inoculation (D14–17). The anti-Gr1 antibody successfully depleted Gr1 + cells, measured one day after the second dose (Fig. 5h). A single dose of the anti-Gr1 antibody has been reported to deplete systemic neutrophils for up to 5 days in mice [69, 70]. Therefore, the D1–3 mice were likely depleted of Gr1 + cells throughout the pre-metastatic stage and D7–10 mice were likely depleted of Gr1 + cells throughout the early metastatic stage. When treated on D14–17, all mice died within the first 24 h due to the excessive number of Gr1 + cells in the body at this time point (n = 8). Survival of the mice treated at D7–10 was comparable to the saline control, whereas those treated at D1–3 exhibited decreased survival relative to D7–10 (p < 0.05 relative to D7–10, p = 0.0788 relative to saline) (Fig. 5i). Similarly, mice treated with anti-Gr1 at D1–3 exhibited an increased metastatic burden as measured through luminescent flux (Fig. 5j). In summary, these data prove early Gr1 + cells (neutrophils and monocytes) induce an anti-cancer response in the lung pre-metastatic niche, that begin to transition towards a pro-cancer phenotype after day 7.
Discussion
Using a mouse model of triple-negative metastatic breast cancer, we dissected the dynamics of cell types and phenotypes within the lung using single-cell RNA-sequencing along the transition from a healthy lung and into multiple stages of metastatic disease (pre-metastatic, micro-metastatic, and metastatic). These changes have potential as biomarkers for disease progression that could indicate the development of the metastatic niche, prior to the formation of overt tumors, and facilitate early detection. Given the dynamic changes in cell types and phenotypes, personalized therapies could be designed to target the disease processes associated with the stages of metastatic disease. While metastases are currently primarily detected at an advanced stage, emerging technologies, with the potential for earlier detection, highlight clinical opportunities for understanding underlying mechanisms of metastatic progression [6].
We found that the evolution of the lung from healthy tissue to advanced metastatic disease is marked by a gradual transition from anti-cancer phenotypes to pro-cancer phenotypes. This transition was initially predicted in the lung through changes in signaling pathways that indicated a pro-inflammatory microenvironment at the pre-metastatic niche with the development of immune dysregulation in late-stage metastatic disease (Fig. 2). Progressing metastatic disease was also marked by decreases in dendritic cells in the lung (22-fold decrease from day 0 to day 21) and concordant decreases in signaling pathways associated with dendritic cell activity and antigen presentation. This data is consistent with other reports that identify a potent anti-cancer role of conventional dendritic cells in metastasis [71, 72]. The phenotypic shifts in the lung were validated through functional assessment of tumor cells and T cells in response to signaling cues from each microenvironment (Figs. 2, 5). This range of anti-cancer to pro-cancer phenotypes has been previously reported at primary tumors [73, 74], particularly within anti-cancer neutrophils, monocytes, and macrophages. The report herein describes the dynamics of anti-cancer phenotypes for neutrophils and monocytes at metastatic sites and throughout disease progression.
At the primary tumor, monocytes and macrophages promote collagen degradation, ECM remodeling, and tumor growth [75, 76] which are critical to tumor progression. Within the lung metastatic niche, these phenotypes were only observed at day 21. Classical and non-classical monocytes comprise approximately 80% of monocytes in the pre-metastatic niche (day 7), with classical monocytes expanding in abundance by day 14 and non-classical, anti-cancer, monocytes decreasing in abundance. At day 21, the presence of Chi3l3 + classical monocytes at day 21 correlates with increased expression of chitinase 3-like proteins in human tumor-associated macrophages [77]. Monocyte-induced ECM remodeling, as seen in these cells, has been reported in metastatic lung cancer [78]. However, investigations into ECM remodeling in the lung as a metastatic site have been primarily focused on fibroblasts and resident macrophages [11, 79, 80], suggesting a potentially novel role of ECM degradation by tumorassociated monocytes.
These studies support a dual role of neutrophils in metastatic progression. While this phenomenon has been identified at the primary tumor, metastatic lesions are consistently reported as pro-cancer [45, 46, 81–83]. Some studies even identify pro-cancer neutrophils in the pre-metastatic niche, although these experimental designs correlate with our day 14 time point of micro-metastases [46, 82]. In our study, neutrophils had the greatest change in abundance and phenotypes, which were strongly associated with the transition from an anti-cancer to pro-cancer microenvironment. Therefore, neutrophils were implicated as the cell type primarily responsible for progression in cell signaling. Neutrophils, specifically MDSCs, are known to suppress the proliferation of cytotoxic CD8 + T cells in vivo [45, 46]. We identified inhibited proliferation only at day 7 using T cells derived from a healthy spleen, where approximately 70% of T cells have a CD4 + phenotype [84]. This phenomenon is consistent with the up-regulation of interferon signaling at day 7 (Figs. 2d, 4e, and 5g), which can induce apoptosis and inhibit T cell proliferation in naïve CD4 + and Th2 T cells, in part by inhibiting CD28 activation [85–89].
The changes in neutrophil ligand expression and transcription factor activity, which correlate to the local niche microenvironment, could act as indicators of staging in metastatic disease. Signaling genes that could serve as potential markers (up-regulated in the pre-metastatic niche but down-regulated upon the progression of disease) include Stat1, Stat3, Irf9, Il1b, and Selplg. Increases in Il1b gene expression were paralleled by intracellular (Figs. 4d and S8) and extracellular (Fig. S14) IL-1β expression, demonstrating the clinical value of such markers for early detection of metastasis. IL-1β is known to have a dual role in cancer, but is more often considered to support metastatic development [90]. Our analysis indicates that Il1b denoted an anti-cancer phenotype in our system. These inconsistencies could be a result of translation or protein activation that would not be detected in the single-cell data.
Clinical efforts towards identification of early metastatic events have primarily focused on the identification of prognostic serum markers [6]. Extensive research has been conducted into the potential of circulating cell-free tumor DNA, circulating tumor cells, immune cell ratios, and gene signatures to predict metastasis prior to the formation of macroscopic lesions [6]. For example, neutrophil and MDSC infiltration is widely reported in both human and mouse models of breast cancer metastasis as seen in our data (Fig. 1) [81, 91]. While previous studies have reported good correlations between the 4T1 model and human metastatic breast cancer [92], clinical disease does not undergo the 15-fold increase in neutrophils exhibited herein. Furthermore, we identified that pneumocytes comprised only 5% of the healthy lung, although literature suggests this number should be closer to 20% [93]. This discrepancy could be due to method biases resulting from inconsistencies in cell type survival and improved recovery of circulatory cells over adherent cells during tissue dissociation. As the lungs were not perfused prior to explant, the presence of circulatory cells is the more likely cause of the immune cell bias identified in this study. Our findings may also be dampened relative to what occurs at the specific (pre-)metastatic niche since we included cells from the entire lung in our analysis. These limitations must be considered in the evaluation of the results as known deviations from the human in vivo microenvironment.
In summary, we dissected the phenotypic dynamics of the lung as it transitioned from a healthy tissue to a metastatic niche in triple-negative breast cancer using single-cell RNA-sequencing. These analyses provide an unprecedented understanding of the evolution of the metastatic niche prior to the development of macroscopic lesions. This study provides a comprehensive analysis of how immune cell phenotype dynamics progress concordantly with metastatic disease. We have identified a behavioral shift in the niche from anti-cancer to pro-cancer, largely associated with substantial changes in monocyte and neutrophil numbers and phenotypes. These cells work to combat the early arrival of metastatic tumor cells and stave off tumor formation. These early anti-cancer phenotypes could be studied and leveraged towards the development of new targeted therapies that work to inhibit the development of secondary tumors in the early stages of metastatic disease.
Supplementary Material
Acknowledgements
Funds for this research were provided by the National Institutes of Health (NIH) R01 CA243916 (L.D.S and J.S.J.), NIH R01 CA214384 (L.D.S and J.S.J.), NIH R35 GM13779501 (S.C.), and NIH R35 CA197585 (M.S.W.). Further support was provided by the Dynami Foundation, the Breast Cancer Research Foundation (BCRF-18–173), and A Sister’s Hope. S.M.O is funded through the UM Postdoctoral Translational Scholar Program and NRSA F32 CA243421. S.E.C is a recipient of the Advanced Proteome Informatics of Cancer Training Program T32 CA140044. J.T.D. is funded through the NIH 5K01 EB028877. G.G.B is a recipient of the NRSA F31 CA224982.
Footnotes
Supplementary Information The online version contains supplementary material available at https://doi.org/10.1007/s10585-022-10185-4.
Declarations
Competing interest The authors declare no competing interests.
Data availability
All data required to evaluate the findings and conclusions are provided in the main text or supplemental data. Additional data is available upon author request.
References
- 1.Siegel RL, Miller KD (2020) Jemal A (2020) cancer statistics. CA Cancer J Clin 70(1):7–30 [DOI] [PubMed] [Google Scholar]
- 2.Gupta GP, Massague J (2006) Cancer metastasis: building a framework. Cell 127(4):679–695 [DOI] [PubMed] [Google Scholar]
- 3.Irvin W Jr, Muss HB, Mayer DK (2011) Symptom management in metastatic breast cancer. Oncologist 16(9):1203–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Azarin SM et al. (2015) In vivo capture and label-free detection of early metastatic cells. Nat Commun 6:8094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Menezes ME et al. (2016) Detecting tumor metastases: the road to therapy starts here. Adv Cancer Res 132:1–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morris AH et al. (2020) Engineered niches to analyze mechanisms of metastasis and guide precision medicine. Cancer Res 80(18):3786–3794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bushnell GG et al. (2021) Disease-induced immunomodulation at biomaterial scaffolds detects early pancreatic cancer in a spontaneous model. Biomaterials 269:120632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paget S (1889) The distribution of secondary growths in cancer of the breast. Lancet 133:571–573 [PubMed] [Google Scholar]
- 9.Kaplan RN et al. (2005) VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 438(7069):820–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aguado BA et al. (2017) Engineering the pre-metastatic niche. Nat Biomed Eng 10.1038/s41551-017-0077 [DOI] [PMC free article] [PubMed]
- 11.Peinado H et al. (2017) Pre-metastatic niches: organ-specific homes for metastases. Nat Rev Cancer 17(5):302–317 [DOI] [PubMed] [Google Scholar]
- 12.Blomberg OS, Spagnuolo L, de Visser KE (2018) Immune regulation of metastasis: mechanistic insights and therapeutic opportunities. Dis Model Mech 10.1242/dmm.036236 [DOI] [PMC free article] [PubMed]
- 13.Sceneay J, Smyth MJ, Moller A (2013) The pre-metastatic niche: finding common ground. Cancer Metastasis Rev 32(3–4):449–464 [DOI] [PubMed] [Google Scholar]
- 14.Liu Y, Cao X (2016) Characteristics and significance of the pre-metastatic niche. Cancer Cell 30(5):668–681 [DOI] [PubMed] [Google Scholar]
- 15.Hwang B, Lee JH, Bang D (2018) Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp Mol Med 50(8):96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eberwine J et al. (2014) The promise of single-cell sequencing. Nat Methods 11(1):25–37 [DOI] [PubMed] [Google Scholar]
- 17.Navin N et al. (2011) Tumour evolution inferred by single-cell sequencing. Nature 472(7341):90–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim N et al. (2020) Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat Commun 11(1):2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fan J, Slowikowski K, Zhang F (2020) Single-cell transcriptomics in cancer: computational challenges and opportunities. Exp Mol Med 52(9):1452–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bushnell GG et al. (2020) Microporous scaffolds loaded with immunomodulatory lentivirus to study the contribution of immune cell populations to tumor cell recruitment in vivo. Biotechnol Bioeng 117(1):210–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oakes RS et al. (2020) Metastatic conditioning of myeloid cells at a subcutaneous synthetic niche reflects disease progression and predicts therapeutic outcomes. Cancer Res 80(3):602–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rao SS et al. (2016) Enhanced survival with implantable scaffolds that capture metastatic breast cancer cells in vivo. Cancer Res 76(18):5209–5218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pulaski BA, Ostrand-Rosenberg S (2001) Mouse 4T1 breast tumor model. Curr Protoc Immunol 20:20–22 [DOI] [PubMed] [Google Scholar]
- 24.Bushnell GG et al. (2019) Biomaterial scaffolds recruit an aggressive population of metastatic tumor cells in vivo. Cancer Res 79(8):2042–2053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Macosko EZ et al. (2015) Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161(5):1202–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Picelli S et al. (2014) Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res 24(12):2033–2040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Butler A et al. (2018) Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36(5):411–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stuart T et al. (2019) Comprehensive Integration of Single-Cell Data. Cell 177(7):1888–1902.e21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Durinck S et al. (2009) Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc 4(8):1184–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Subramanian A et al. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102(43):15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trapnell C et al. (2014) The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 32(4):381–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Dijk D et al. (2018) Recovering gene interactions from single-cell data using data diffusion. Cell 174(3):716–729.e27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lipinski S et al. (2012) RNAi screening identifies mediators of NOD2 signaling: implications for spatial specificity of MDP recognition. Proc Natl Acad Sci U S A 109(52):21426–21431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robinson JL et al. (2020) An atlas of human metabolism. Sci Signal 10.1126/scisignal.aaz1482 [DOI] [PMC free article] [PubMed]
- 35.Orth JD, Thiele I, Palsson BO (2010) What is flux balance analysis? Nat Biotechnol 28(3):245–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zur H, Ruppin E, Shlomi T (2010) iMAT: an integrative metabolic analysis tool. Bioinformatics 26(24):3140–3142 [DOI] [PubMed] [Google Scholar]
- 37.Chandrasekaran S, Price ND (2010) Probabilistic integrative modeling of genome-scale metabolic and regulatory networks in Escherichia coli and Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 107(41):17845–17850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lewis NE et al. (2010) Omic data from evolved E. coli are consistent with computed optimal growth from genome-scale models. Mol Syst Biol 6:390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holland CH et al. (2020) Robustness and applicability of transcription factor and pathway analysis tools on single-cell RNA-seq data. Genome Biol 21(1):36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holland CH, Szalai B, Saez-Rodriguez J (1863) Transfer of regulatory knowledge from human to mouse for functional genomics analysis. Biochim Biophys Acta Gene Regul Mech 6:194431. [DOI] [PubMed] [Google Scholar]
- 41.Garcia-Alonso L et al. (2019) Benchmark and integration of resources for the estimation of human transcription factor activities. Genome Res 29(8):1363–1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Browaeys R, Saelens W, Saeys Y (2020) NicheNet: modeling intercellular communication by linking ligands to target genes. Nat Methods 17(2):159–162 [DOI] [PubMed] [Google Scholar]
- 43.Efremova M et al. (2020) Cell PhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat Protoc 15(4):1484–1506 [DOI] [PubMed] [Google Scholar]
- 44.Chen JY et al. (2021) Lentiviral interleukin-10 gene therapy preserves fine motor circuitry and function after a cervical spinal cord injury in male and female mice. Neurotherapeutics 18(1):503–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Coffelt SB et al. (2015) IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature 522(7556):345–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Casbon AJ et al. (2015) Invasive breast cancer reprograms early myeloid differentiation in the bone marrow to generate immunosuppressive neutrophils. Proc Natl Acad Sci U S A 112(6):E566–E575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Decker JT et al. (2020) Cyclin E overexpression confers resistance to trastuzumab through noncanonical phosphorylation of SMAD3 in HER2+ breast cancer. Cancer Biol Ther 21(11):994–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aguado BA et al. (2018) Biomaterial scaffolds as pre-metastatic niche mimics systemically alter the primary tumor and tumor microenvironment. Adv Healthc Mater 7(10):e1700903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bosiljcic M et al. (2019) Targeting myeloid-derived suppressor cells in combination with primary mammary tumor resection reduces metastatic growth in the lungs. Breast Cancer Res 21(1):103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674 [DOI] [PubMed] [Google Scholar]
- 51.Srikrishna G (2012) S100A8 and S100A9: new insights into their roles in malignancy. J Innate Immun 4(1):31–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sirnio P et al. (2018) High-serum MMP-8 levels are associated with decreased survival and systemic inflammation in colorectal cancer. Br J Cancer 119(2):213–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hiratsuka S et al. (2002) MMP9 induction by vascular endothelial growth factor receptor-1 is involved in lung-specific metastasis. Cancer Cell 2(4):289–300 [DOI] [PubMed] [Google Scholar]
- 54.Veglia F et al. (2021) Analysis of classical neutrophils and polymorphonuclear myeloid-derived suppressor cells in cancer patients and tumor-bearing mice. J Exp Med 10.1084/jem.20201803 [DOI] [PMC free article] [PubMed]
- 55.Yi F, Jaffe R, Prochownik EV (2003) The CCL6 chemokine is differentially regulated by c-Myc and L-Myc, and promotes tumorigenesis and metastasis. Cancer Res 63(11):2923–2932 [PubMed] [Google Scholar]
- 56.Baker KJ, Houston A, Brint E (2019) IL-1 family members in cancer; two sides to every story. Front Immunol 10:1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Owen KL, Brockwell NK, Parker BS (2019) JAK-STAT signaling: a double-edged sword of immune regulation and cancer progression. Cancers (Basel) 11(12):2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Langer S et al. (2006) Jun and Fos family protein expression in human breast cancer: correlation of protein expression and clinicopathological parameters. Eur J Gynaecol Oncol 27(4):345–352 [PubMed] [Google Scholar]
- 59.Beishline K, Azizkhan-Clifford J (2015) Sp1 and the “hallmarks of cancer.” FEBS J 282(2):224–258 [DOI] [PubMed] [Google Scholar]
- 60.Fane M et al. (2017) Nuclear factor one transcription factors as epigenetic regulators in cancer. Int J Cancer 140(12):2634–2641 [DOI] [PubMed] [Google Scholar]
- 61.Ju Q et al. (2020) NFE2L2 is a potential prognostic biomarker and is correlated with immune infiltration in brain lower grade glioma: a pan-cancer analysis. Oxid Med Cell Longev 2020:3580719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Barletta KE, Ley K, Mehrad B (2012) Regulation of neutrophil function by adenosine. Arterioscler Thromb Vasc Biol 32(4):856–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marton A et al. (2001) Anti-inflammatory effects of inosine in human monocytes, neutrophils and epithelial cells in vitro. Int J Mol Med 8(6):617–621 [PubMed] [Google Scholar]
- 64.Perretti M, D’Acquisto F (2009) Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat Rev Immunol 9(1):62–70 [DOI] [PubMed] [Google Scholar]
- 65.Wang J, John EM, Ingles SA (2008) 5-lipoxygenase and 5-lipoxygenase-activating protein gene polymorphisms, dietary linoleic acid, and risk for breast cancer. Cancer Epidemiol Biomarkers Prev 17(10):2748–2754 [DOI] [PubMed] [Google Scholar]
- 66.Zhang Y et al. (2021) Single-cell analyses reveal key immune cell subsets associated with response to PD-L1 blockade in triple-negative breast cancer. Cancer Cell 10.1016/j.ccell.2021.09.010 [DOI] [PubMed]
- 67.Bouker KB et al. (2005) Interferon regulatory factor-1 (IRF-1) exhibits tumor suppressor activities in breast cancer associated with caspase activation and induction of apoptosis. Carcinogenesis 26(9):1527–1535 [DOI] [PubMed] [Google Scholar]
- 68.Taniguchi K, Karin M (2018) NF-kappaB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol 18(5):309–324 [DOI] [PubMed] [Google Scholar]
- 69.Weber FC et al. (2015) Neutrophils are required for both the sensitization and elicitation phase of contact hypersensitivity. J Exp Med 212(1):15–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen L, Zhang Z, Sendo F (2000) Neutrophils play a critical role in the pathogenesis of experimental cerebral malaria. Clin Exp Immunol 120(1):125–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bottcher JP, Reise Sousa C (2018) The role of type 1 conventional dendritic cells in cancer immunity. Trends Cancer 4(11):784–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Headley MB et al. (2016) Visualization of immediate immune responses to pioneer metastatic cells in the lung. Nature 531(7595):513–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Singhal S et al. (2019) Human tumor-associated monocytes/macrophages and their regulation of T cell responses in early-stage lung cancer. Sci Transl Med 10.1126/scitranslmed.aat1500 [DOI] [PMC free article] [PubMed]
- 74.Eruslanov EB (2017) Phenotype and function of tumor-associated neutrophils and their subsets in early-stage human lung cancer. Cancer Immunol Immunother 66(8):997–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Afik R et al. (2016) Tumor macrophages are pivotal constructors of tumor collagenous matrix. J Exp Med 213(11):2315–2331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Madsen DH et al. (2017) Tumor-associated macrophages derived from circulating inflammatory monocytes degrade collagen through cellular uptake. Cell Rep 21(13):3662–3671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu T et al. (2018) Tumor-associated macrophages in human breast cancer produce new monocyte attracting and pro-angiogenic factor YKL-39 indicative for increased metastasis after neoadjuvant chemotherapy. Oncoimmunology 7(6):e1436922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Olingy CE, Dinh HQ, Hedrick CC (2019) Monocyte heterogeneity and functions in cancer. J Leukoc Biol 106(2):309–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Paolillo M, Schinelli S (2019) Extracellular matrix alterations in metastatic processes. Int J Mol Sci 10.3390/ijms20194947 [DOI] [PMC free article] [PubMed]
- 80.Erler JT, Weaver VM (2009) Three-dimensional context regulation of metastasis. Clin Exp Metastasis 26(1):35–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jaillon S et al. (2020) Neutrophil diversity and plasticity in tumour progression and therapy. Nat Rev Cancer 20(9):485–503 [DOI] [PubMed] [Google Scholar]
- 82.Wculek SK, Malanchi I (2015) Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature 528(7582):413–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kowanetz M et al. (2010) Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proc Natl Acad Sci U S A 107(50):21248–21255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Grosjean C et al. (2021) Isolation and enrichment of mouse splenic T cells for ex vivo and in vivo T cell receptor stimulation assays. STAR Protoc 2(4):100961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gajewski TF et al. (1989) Regulation of T-cell activation: differences among T-cell subsets. Immunol Rev 111:79–110 [DOI] [PubMed] [Google Scholar]
- 86.Wood KJ, Sawitzki B (2006) Interferon gamma: a crucial role in the function of induced regulatory T cells in vivo. Trends Immunol 27(4):183–187 [DOI] [PubMed] [Google Scholar]
- 87.Wherry EJ, Kurachi M (2015) Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 15(8):486–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tough DF (2012) Modulation of T-cell function by type I interferon. Immunol Cell Biol 90(5):492–497 [DOI] [PubMed] [Google Scholar]
- 89.Konieczny BT et al. (1998) IFN-gamma is critical for long-term allograft survival induced by blocking the CD28 and CD40 ligand T cell costimulation pathways. J Immunol 160(5):2059–2064 [PubMed] [Google Scholar]
- 90.Dinarello CA (2006) The paradox of pro-inflammatory cytokines in cancer. Cancer Metastasis Rev 25(3):307–313 [DOI] [PubMed] [Google Scholar]
- 91.Allen BM et al. (2020) Systemic dysfunction and plasticity of the immune macroenvironment in cancer models. Nat Med 26(7):1125–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schrors B et al. (2020) Multi-omics characterization of the 4T1 murine mammary gland tumor model. Front Oncol 10:1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Stone KC et al. (1992) Allometric relationships of cell numbers and size in the mammalian lung. Am J Respir Cell Mol Biol 6(2):235–243 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data required to evaluate the findings and conclusions are provided in the main text or supplemental data. Additional data is available upon author request.