

ABSTRACT
Macrophages are one of the first innate immune infiltrates in the cornea of mice following ocular infection with herpes simplex virus 1 (HSV-1). Using gamma interferon (IFN-γ) and interleukin-4 (IL-4) injections to polarize macrophages into M1 and M2, respectively, and in M1 and M2 conditional knockout mice, we have shown that M1 macrophages play an important role in suppressing both virus replication in the eye and eye disease in HSV-1-infected mice. Autophagy is also important in controlling HSV infection and integrity of infected cells. To determine if blocking autophagy in M1 and M2 macrophages affects HSV-1 infectivity and eye disease, we generated two transgenic mouse strains expressing the HSV-1 γ34.5 autophagy gene under the M1 promoter (M1-γ34.5) or the M2 promoter (M2-γ34.5). We found that blocking autophagy in M1 macrophages increased both virus replication in the eyes and eye disease in comparison to blocking autophagy in M2 macrophages or wild-type (WT) control mice, but blocked autophagy did not affect latency-reactivation. However, blocking autophagy affected fertility in both M1 and M2 transgenic mice. Analysis of 62 autophagy genes and 32 cytokines/chemokines from infected bone marrow-derived macrophages from M1-γ34.5, M2-γ34.5, and WT mice suggested that upregulation of autophagy-blocking genes (i.e., Hif1a, Mtmr14, mTOR, Mtmr3, Stk11, and ULK2) and the inflammatory tumor necrosis factor alpha (TNF-α) gene in M1-γ34.5 transgenic mice correlated with increased pathogenicity, while upregulation of proautophagy genes (Nrbf2 and Rb1cc1) in M2-γ34.5 macrophages correlated with reduced pathogenicity. The in vivo and in vitro responses of M1-γ34.5 and M2-γ34.5 transgenic mice to HSV-1 infection were independent of the presence of the γ34.5 gene in wild-type HSV-1. Our results suggest that M1 macrophages, but not M2 macrophages, play an important role in autophagy relative to primary virus replication in the eye and eye disease in infected mice.
IMPORTANCE Autophagy plays a critical role in clearing, disassembling, and recycling damaged cells, thus limiting inflammation. The HSV-1 γ34.5 gene is involved in neurovirulence and immune evasion by blocking autophagy in infected cells. We found that blocking autophagy in M1 macrophages enhances HSV-1 virus replication in the eye and eye disease in ocularly infected transgenic mice. Our results also show the suppressive effects of γ34.5 on immune responses to infection, suggesting the importance of intact autophagy in M1 but not M2 macrophages in controlling primary infection and eye disease.
KEYWORDS: autophagy, eye disease, HSV-1, latency, M1-M2 macrophage, reactivation, virus replication
INTRODUCTION
Although protecting corneal transparency from infection is fundamental to safeguarding vision, herpes simplex virus 1 (HSV-1) infection remains the primary contributing factor to viral eye infections and virus-induced blindness in the United States (1, 2). Cellular infiltrates detected in the corneas of mice ocularly infected with HSV-1 have been implicated in this disease process (3–9). Specifically, a combination of innate and adaptive immune responses, including neutrophils (10), macrophages (11), dendritic cells (12), NKT cells (13), and T cells (14, 15), is associated with HSV-1-induced eye disease. Macrophages are widely distributed throughout the body (16, 17) and play key roles in the immune defense system, including central roles in innate and natural immunity (18–21). Macrophage infiltrates appear to play different roles in the disease process depending on the milieu, including abundance and characteristics of the various infiltrates, infection dose, and mouse model (strain of mouse, strain of virus, and whether infection is conducted with or without corneal scarification) (7, 22–24). Macrophages are mononuclear phagocytes that are important for tissue homeostasis and resolution of inflammation and have a crucial role in eliciting potent antiviral immune responses (25, 26). We have shown previously that macrophages form an early and predominant infiltrate in the corneas of mice ocularly infected with HSV-1 (22).
One of the most important characteristics of macrophages is their plasticity and associated adaptive heterogeneity. As different macrophage subsets are likely to mediate distinct biological sequelae (27, 28), it is important to understand the regulatory mechanisms that control differentiation of macrophage subsets and their relative abundance. In the previously suggested M1/M2 macrophage polarization paradigm (29), M1 (classically activated) macrophages produce significant amounts of proinflammatory cytokines after gamma interferon (IFN-γ) induction (27, 28), while M2 (alternatively activated) macrophages are induced by exposure to interleukin-4 (IL-4), IL-13, IL-10, or glucocorticoids and produce low levels of proinflammatory cytokines and increased levels of anti-inflammatory cytokines (28, 30). In the specific context of ocular HSV-1 infection in mice, we reported previously that injection of mice with macrophage colony-stimulating factor (M-CSF) pushes macrophages toward an M2 response, whereas injection of mice with IFN-γ enhances an M1 response (31). We have found that enhancing the M2 macrophage subpopulation by injecting colony-stimulating factor 1 (CSF-1) or infecting with a recombinant HSV-1 expressing IL-4 (HSV-IL-4) reduced virus replication in the eye and eye disease. In contrast, activation of the M1 subpopulation by injection of IFN-γ or infection with a recombinant HSV-1 expressing IFN-γ (HSV-IFN-γ) was proinflammatory and exacerbated eye disease (31, 32). To evaluate the role of M1 and M2 macrophages in controlling HSV-1 infection in vivo, we used M1 and M2 conditional knockout mice. Our results demonstrated that a proper balance between M1 and M2 macrophages must be maintained to achieve an effective immune response against ocular HSV-1 infection (33) and that M1 macrophages are crucial elements of innate immunity to control viral replication in the eye of infected mice and resulting eye disease (34).
The HSV-1 ICP34.5 gene, also known as γ34.5, is a neurovirulence gene that targets several pathways to counteract the host IFN response, and viruses that lack the γ34.5 gene are avirulent in vivo (35–40). Pathogens, including HSV-1, can inhibit autophagy, thereby preventing the destruction of infected cells (41, 42). The ability of HSV-1 to block autophagy is controlled by the γ34.5 gene, which encodes ICP34.5 protein that binds beclin-1 (41–43). Macrophages are one of the most dominant infiltrates in the eyes after ocular HSV-1 infection (22), yet the role of autophagy in the context of functional M1 and M2 activities is unknown. To determine what role, if any, blocking autophagy in M1 and M2 macrophages may play in HSV-1 infectivity in vitro and in vivo, we constructed two transgenic mouse lines that specifically express the HSV-1 autophagy gene γ34.5 in M1 and M2 macrophages. These transgenic mice were used to investigate the effects of blocking autophagy on M1 and M2 macrophage function in vitro and in vivo. Here, we report for the first time that (i) blocking autophagy in M1 but not M2 macrophages increased virus replication in the eye and eye disease in ocularly infected mice, while blocking autophagy in M1 or M2 macrophages had no effect on latency-reactivation, and (ii) the presence of the γ34.5 gene in M1-γ34.5 transgenic mice but not in M2-γ34.5 transgenic mice correlated with upregulation of the antiautophagy genes Hif1a, Mtmr14, mTOR, Mtmr3, Stk11, and ULK2 as well as the inflammatory cytokine tumor necrosis factor alpha (TNF-α).
RESULTS
Construction and validation of M1-γ34.5 and M2-γ34.5 transgenic mice.
We have made two transgenic mouse lines expressing the HSV-1 antiautophagy γ34.5 gene under the control of the M1 (nitric oxide synthase 2 [NOS2]) or M2 (ARG1) promoters. To generate M1-γ34.5 transgenic mice, the full-length γ34.5 gene coding sequence with three FLAG tag sequences before the stop codon followed by the internal ribosome entry site (IRES) sequence was commercially synthesized and inserted into pGL2-NOS2 (44, 45), in correct orientation (Fig. 1, left panel). To generate M2-γ34.5 transgenic mice, the full-length γ34.5 gene coding sequence (described above) was inserted into pGL3-mARG1 (46) (Fig. 1, right panel). M1-γ34.5 and M2-γ34.5 transgenic mice were both created using a standard DNA microinjection procedure. DNA from tail, cornea, trigeminal ganglion (TG), brain, spleen, and liver of M1-γ34.5 and M2-γ34.5 transgenic mice was isolated, and total DNA from these tissues was analyzed by PCR using IRES primers as described in Materials and Methods. The presence of a 428-bp IRES DNA in the tail, cornea, TG, brain, spleen, and liver of M1-γ34.5 and M2-γ34.5 transgenic mice was confirmed by TaqMan PCR (Fig. 2). Tissues isolated from both transgenic mouse groups expressed the 428-bp IRES DNA (Fig. 2). To verify the expression of inserted γ34.5 mRNA in tail, cornea, TG, brain, spleen, and liver of M1-γ34.5 and M2-γ34.5 mice, total RNA was extracted from each tissue and quantitative real-time PCR (qRT-PCR) was used to detect γ34.5 mRNA expression in each tissue. Expression of γ34.5 mRNA was detected in tail, cornea, TG, brain, spleen, and liver of M1-γ34.5 transgenic mice but was not statistically different among these tissues (Fig. 3A; P > 0.05 in all tissues), whereas in M2-γ34.5 transgenic mice, the expression of γ34.5 mRNA was similar in tail, cornea, TG, brain, and spleen but was significantly higher in liver (P < 0.05) (Fig. 3B), confirming that both transgenic mouse lines express the γ34.5 gene in these tissues.
FIG 1.

Construction of transgenic mice. Transgenic M1-γ34.5 and M2-γ34.5 mice were constructed by inserting the HSV-1 γ34.5 gene into the BglII site of the pGL2-NOS2 expression vector containing the mouse nitric oxide synthase (NOS2) promoter and the XhoI site of the mouse arginase 1 (ARG1) promoter from pGL3-mARG1. Abbreviations: ORF, open reading frame; SV40, simian virus 40; EBV, Epstein-Barr virus; NLS, nuclear localization signal.
FIG 2.
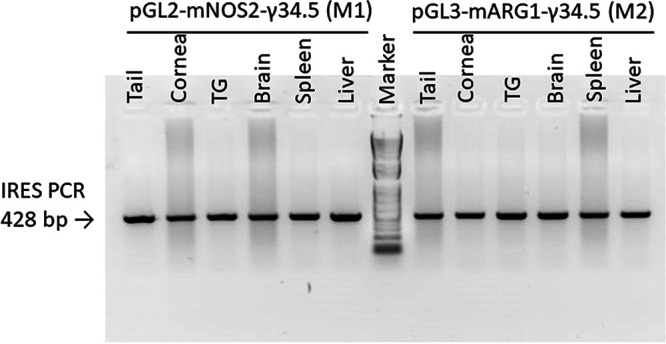
Detection of IRES in different M1 and M2 transgenic mouse tissues. Total DNA was isolated from tail, cornea, trigeminal ganglion (TG), brain, spleen, and liver of transgenic M1-γ34.5 and M2-γ34.5 mice, and quantitative PCR (qPCR) was performed using IRES primers.
FIG 3.

Detection of γ34.5 mRNA in isolated transgenic mouse tissues. Tail, cornea, trigeminal ganglion (TG), brain, spleen, and liver from M1-γ34.5 and M2-γ34.5 mice were harvested, total RNA was isolated, qRT-PCR was performed on isolated RNA, and the γ34.5 gene copy number was determined. (A) γ34.5 copy number in M1-γ34.5 mice; (B) γ34.5 copy number in M2-γ34.5 mice. Each bar represents the mean ± standard error of the mean (SEM) from two independent experiments (n = 6).
We next asked if M1 macrophages from M1-γ34.5 transgenic mice have higher levels of γ34.5 gene expression than those from M2-γ34.5 transgenic mice. Bone marrow (BM)-derived macrophages isolated from M1-γ34.5 mice and M2-γ34.5 mice were prepared as described in Materials and Methods. Macrophages from both groups of transgenic mice were harvested on day 6 posttreatment and polarized in vitro for 24 h with no stimulation to generate M0, IL-4 stimulation to generate M2, or IFN-γ stimulation to generate M1 macrophages. Total RNA was isolated, and γ34.5 gene expression under the control of the NOS2 (M1) or ARG1 (M2) promoter was measured by qRT-PCR. As expected, without activation (M0) the level of γ34.5 gene expression was low and was similar between the two groups of transgenic mice (Fig. 4, M0, P = 0.9). Expression of the γ34.5 gene in M1 macrophages from M1-γ34.5 transgenic mice was significantly higher than in M1 macrophages from M2-γ34.5 transgenic mice (Fig. 4, M1, P < 0.001). Conversely, γ34.5 gene expression in M2 macrophages from M2-γ34.5 transgenic mice was significantly higher than in M2 macrophages from M1-γ34.5 transgenic mice (Fig. 4, M2, P < 0.001). These results demonstrate the specificity of γ34.5 expression under the control of the NOS2 or ARG1 promoter.
FIG 4.
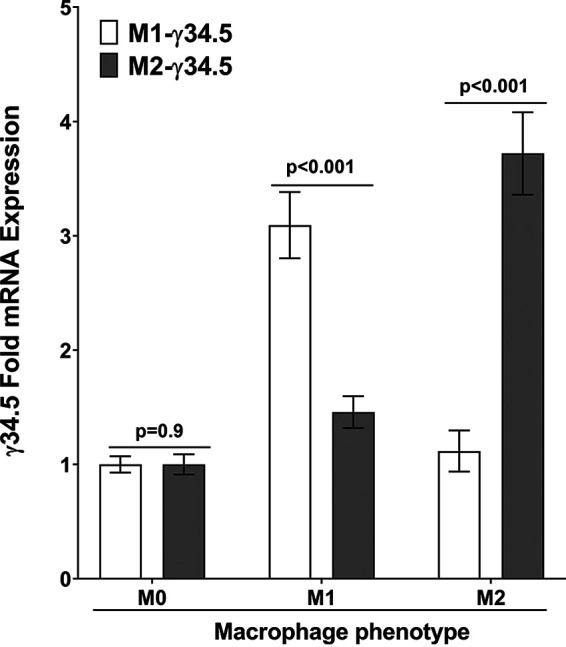
Validation of macrophage phenotype of transgenic mice in vitro. BM-derived macrophages from M1-γ34.5 mice and M2-γ34.5 mice were generated and differentiated into M1 and M2 macrophages as described in Materials and Methods. Cells were then harvested, total RNA was isolated, and TaqMan RT-PCR was performed using γ34.5-specific primers. Expression of γ34.5 mRNA was normalized to that of GAPDH RNA. Each bar represents the mean ± SEM from two independent experiments (n = 6).
Expression of cytokines/chemokines in WT, M1-γ34.5, and M2-γ34.5 mice.
BM-derived macrophages from wild-type (WT), M1-γ34.5, and M2-γ34.5 mice were generated and infected with 10 PFU/cell of HSV-1 strain McKrae. Infected cells were cultured for 24 h, and culture medium was collected from each well to measure cytokine and chemokine expression using a Luminex bead-based multiplex cytokine profiling assay as we described previously (31). Significant differences in proinflammatory cytokine expression were observed among WT, M1-γ34.5, and M2-γ34.5 mice (Table 1). IL-6 expression was significantly higher in M2-γ34.5 macrophages than in M1-γ34.5 or WT macrophages (P = 0.05 and P = 0.03, respectively). In contrast, IL-17 expression was significantly lower in M2-γ34.5 macrophages than in WT macrophages (P = 0.01) but did not differ significantly from that in M1-γ34.5 macrophages (P > 0.05). IL-6 and IL-17 are widely recognized for their proinflammatory nature (47). LIX expression, involved in NF-κB activation and proinflammatory pathways (48), was significantly higher in M2-γ34.5 macrophages than in WT macrophages (P = 0.04) but did not differ significantly from that in M1-γ34.5 macrophages (P > 0.05). Monocyte chemoattractant protein 1 (MCP-1) expression was significantly higher in WT macrophages than in M1-γ34.5 or M2-γ34.5 macrophages (P = 0.02 for both groups). We also saw significantly higher vascular endothelial growth factor (VEGF) expression in M1-γ34.5 and M2-γ34.5 macrophages than in WT macrophages (P = 0.02). Finally, TNF-α expression in M1-γ34.5 macrophages was significantly higher than in M2-γ34.5 and WT macrophages (P = 0.04). However, expression of granulocyte colony-stimulating factor (G-CSF), eotaxin, granulocyte-macrophage colony-stimulating factor (GM-CSF), IFN-γ, IL-1α, IL-1β, IL-2, IL-4, IL-3, IL-5, IL-7, IL-9, IL-10, IL-12(p40), IL-12(p70), leukemia inhibitory factor (LIF), IL-15, interferon-inducible protein 10 (IP-10), KC, macrophage inflammatory protein 1α (MIP-1α), MIP-1β, M-CSF, MIP-2, MIG, and RANTES did not differ among the mouse groups. These results suggest that (i) the γ34.5 gene in M2-γ34.5 mice reversed the anti-inflammatory effects of M2 macrophages and may promote proinflammatory M1 macrophages as reported previously (49), (ii) the γ34.5 gene significantly enhanced the overall inflammatory cytokine secretion cascade by macrophages regardless of M1 or M2 macrophages, and (iii) the γ34.5 gene in WT HSV-1 McKrae has a suppressive effect on the majority of tested cytokines and chemokines. Overall, these results suggest that the absence of autophagy enhances the expression of crucial proinflammatory cytokines and chemokines secreted by macrophages.
TABLE 1.
Cytokine/chemokine expression in BM-derived macrophages from WT and transgenic mice infected with WT McKrae virusa
| Cytokine/chemokine | Level (pg/mL) in infected macrophages: |
P valueb | ||
|---|---|---|---|---|
| M1-γ34.5 | M2-γ34.5 | WT | ||
| G-CSF | 4.9 ± 0.8 | 2.8 ± 0.4 | 2.7 ± 0.5 | >0.05 |
| Eotaxin | 0.7 ± 0.4 | 0.2 ± 0.0 | 0.2 ± 0.0 | >0.05 |
| GM-CSF | 6.2 ± 3.0 | 6.3 ± 3.0 | 3.3 ± 0.07 | >0.05 |
| IFN-γ | 2.0 ± 0.20 | 2.6 ± 0.5 | 1.5 ± 0.2 | >0.05 |
| IL-1α | 9.9 ± 0.9 | 10.1 ± 1.7 | 7.2 ± 0.9 | >0.05 |
| IL-1β | 2.0 ± 0.4 | 2.1 ± 0.1 | 2.4 ± 0.1 | >0.05 |
| IL-2 | 1.3 ± 0.0 | 1.3 ± 0.0 | 1.3 ± 0.0 | >0.05 |
| IL-4 | 0.7 ± 0.1 | 0.7 ± 0.1 | 0.7 ± 0.1 | >0.05 |
| IL-3 | 0.8 ± 0.1 | 0.8 ± 0.1 | 0.8 ± 0.1 | >0.05 |
| IL-5 | 4.5 ± 0.6 | 4.4 ± 0.2 | 5.5 ± 0.4 | >0.05 |
| IL-6 | 1,851.3 ± 163.3 | 2,742.7 ± 219.2 | 1,700.3 ± 195.2 | + |
| IL-7 | 0.6 ± 0.1 | 0.6 ± 0.2 | 0.6 ± 0.1 | >0.05 |
| IL-9 | 33.4 ± 4.7 | 58.3 ± 6.1 | 58.3 ± 6.1 | >0.05 |
| IL-10 | 6.0 ± 0.5 | 5.7 ± 0.1 | 4.4 ± 0.7 | >0.05 |
| IL-12(p40) | 0.7 ± 0.2 | 1.9 ± 0.4 | 1.2 ± 0.5 | >0.05 |
| IL-12(p70) | 0.8 ± 0.2 | 1.0 ± 0.3 | 0.8 ± 0.1 | >0.05 |
| LIF | 0.4 ± 0.2 | 0.4 ± 0.2 | 0.3 ± 0.1 | >0.05 |
| LIX | 182.9 ± 25.9 | 230.0 ± 18.8 | 123.6 ± 18.9 | ++ |
| IL-15 | 4.3 ± 1.2 | 7.5 ± 1.2 | 7.2 ± 2.7 | >0.05 |
| IL-17 | 26.9 ± 1.8 | 19.9 ± 1.5 | 30.7 ± 1.6 | + |
| IP-10 | 13,725.3 ± 1,357.7 | 22,988.3 ± 1,607.6 | 14,730.7 ± 3,254.4 | >0.05 |
| KC | 584.6 ± 69.1 | 565.8 ± 60.4 | 412.4 ± 56.9 | >0.05 |
| MCP-1 | 9,549.3 ± 1,059.4 | 9,494.7 ± 859.0 | 14,393.7 ± 619.3 | +++ |
| MIP-1α | 1,072.9 ± 56.1 | 658.4 ± 105.6 | 2,510.3 ± 263.2 | +++ |
| MIP-1β | 3.2 ± 0.0 | 3.2 ± 0.0 | 3.2 ± 0.0 | >0.05 |
| M-CSF | 6.5 ± 0.5 | 6.0 ± 0.9 | 5.9 ± 0.6 | >0.05 |
| MIP-2 | 4,550.3 ± 817.8 | 5,212.0 ± 790.2 | 2,502.0 ± 346.5 | >0.05 |
| MIG | 14,409.0 ± 500 | 15,187.0 ± 0.0 | 15,187.0 ± 0.0 | >0.05 |
| RANTES | 400.0 ± 0.0 | 400.0 ± 0.0 | 400.0 ± 0.0 | >0.05 |
| VEGF | 6.9 ± 0.4 | 6.0 ± 0.9 | 3.4 ± 0.2 | ++ |
| TNF-α | 1,417.2 ± 332.3 | 779.1 ± 176.7 | 334.1 ± 33.1 | +++ |
Cytokine and chemokine levels in culture media of BM-derived macrophages from M1-γ34.5 and M2-γ34.5 transgenic mice and WT mice were analyzed using mouse 32-plex panels. Experimental procedures are detailed in Materials and Methods. Briefly, cells were infected with 10 PFU/cell of WT McKrae virus for 1 h at 37°C, washed with PBS, and incubated for an additional 24 h in fresh medium. Results indicate mean ± SEM (n = 3).
Symbols: +, significant differences between WT and M2-γ34.5 group (P < 0.05); ++, significant differences between WT and M1-γ34.5 group (P < 0.05); +++, significant differences among all three mouse groups (P < 0.05).
NanoString analysis of autophagy genes expressed in infected BM-derived macrophages.
BM-derived macrophages from WT, M1-γ34.5, and M2-γ34.5 mice were generated and infected with 10 PFU/cell of HSV-1 strain McKrae for 24 h as described above. Twenty-four hours postinfection (p.i.), total RNA from infected macrophages was isolated and analyzed for 62 autophagy genes using NanoString. Gene expression levels in WT, M1-γ34.5, and M2-γ34.5 infected macrophages were normalized to those in mock-infected macrophages (Table 2). Comparison of macrophages from M1-γ34.5 and M2-γ34.5 mice with those from WT mice identified 10 genes whose expression differed significantly between WT and M1-γ34.5 macrophages (Akt2, Atg12, Atg13, Atg3, Bcl2l1, Becn1, Lamp2, Mapk9, Pik3cd, and Ulk2), six genes whose expression differed significantly between WT and M2-γ34.5 macrophages (Atg13, Atg3, Becn1, Bnip3, Cflar, and Prkcd), and seven genes whose expression differed significantly between M1-γ34.5 and M2-γ34.5 macrophages (Atg12, Bnip3, Cflar, Ctsd, Hif1a, Mapk9, and Rb1cc1) (Table 2). Expression of Cflar, which is known to downregulate autophagy by inhibiting caspase 8 expression, was significantly higher in all three macrophage groups than in mock-infected macrophages (50), suggesting that autophagy is inhibited in macrophages from WT, M1-γ34.5, and M2-γ34.5 mice with the maximum effect in M1-γ34.5 mice. Upregulation of Cflar expression in macrophages from M1-γ34.5 and M2-γ34.5 transgenic mice is likely associated with internal expression of γ34.5 as well as its presence in the virus, while the elevation of Cflar expression in WT macrophages is probably associated with viral infection.
TABLE 2.
Expression of autophagy genes in BM-derived infected macrophages from transgenic and WT micea
| Gene | Fold change (mean ± SEM) |
||
|---|---|---|---|
| M1-γ34.5 | M2-γ34.5 | WT | |
| Akt1e | P > 0.05 | P > 0.05 | P > 0.05 |
| Akt2 | P > 0.05 | P > 0.05 | 2.22 ± 0.54*f |
| Akt3e | P > 0.05 | P > 0.05 | P > 0.05 |
| Atg10e | P > 0.05 | P > 0.05 | P > 0.05 |
| Atg101e | P > 0.05 | P > 0.05 | P > 0.05 |
| Atg12d | 1.26 ± 0.01 | 1.48 ± 0.03 | 1.51 ± 0.04 |
| Atg13d | 1.70 ± 0.10 | 1.76 ± 0.03 | 2.11 ± 0.14 |
| Atg3d | 1.64 ± 0.06 | 1.57 ± 0.02 | 1.37 ± 0.05 |
| Atg4ad | 1.18 ± 0.07 | 1.31 ± 0.02 | 1.2 ± 0.05 |
| Atg4bd | 1.58 ± 0.11 | 1.45 ± 0.05 | 1.41 ± 0.09 |
| Atg5e | P > 0.05 | P > 0.05 | P > 0.05 |
| Atg7e | P > 0.05 | P > 0.05 | P > 0.05 |
| Atg9a | 1.88 ± 0.17* | 1.59 ± 0.06* | P > 0.05 |
| Badd | 1.50 ± 0.11 | 1.64 ± 0.09 | 1.70 ± 0.14 |
| BclIIe | P > 0.05 | P > 0.05 | P > 0.05 |
| Bcl2l1d | 1.81 ± 0.07 | 1.63 ± 0.08 | 1.57 ± 0.03 |
| Becn1d | 1.23 ± 0.03 | 1.55 ± 0.05 | 1.40 ± 0.01 |
| Bnip3b | −2.32 ± 0.09 | −1.56 ± 0.10 | −4.72 ± 1.9 |
| Cflarc | 7.22 ± 0.37 | 5.31 ± 0.38 | 4.06 ± 0.26 |
| Ctsdd | −1.89 ± 0.01 | −1.68 ± 0.04 | −1.66 ± 0.03 |
| Ctsle | P > 0.05 | P > 0.05 | 2.06 ± 0.38* |
| Ddit4e | P > 0.05 | P > 0.05 | P > 0.05 |
| Eif2ak3d | −1.32 ± 0.15 | −1.50 ± 0.09 | −1.50 ± 0.09 |
| Ern1d | 1.56 ± 0.05 | 1.66 ± 0.11 | 1.55 ± 0.12 |
| Hif1a | 2.72 ± 0.44* | P > 0.05 | P > 0.05 |
| Hmgb1 | −1.52 ± 0.07 | −2.66 ± 0.6 | −2.95 ± 0.67 |
| Hrasd | 1.34 ± 0.07 | 1.36 ± 0.04 | 1.38 ± 0.05 |
| Krasd | 1.85 ± 0.23 | 1.92 ± 0.19 | 2.20 ± 0.14 |
| Lamp1d | 1.07 ± 0.02 | 1.51 ± 0.03 | 1.28 ± 0.02 |
| Lamp2d | 1.39 ± 0.04 | 1.75 ± 0.03 | 1.69 ± 0.06 |
| Map2k1d | 1.91 ± 0.1 | 1.85 ± 0.09 | 1.63 ± 0.09 |
| Map2k2d | 2.01 ± 0.16 | 1.60 ± 0.17 | 1.58 ± 0.14 |
| Map3k7e | P > 0.05 | P > 0.05 | P > 0.05 |
| Mapk1e | P > 0.05 | P > 0.05 | P > 0.05 |
| Mapk3d | −1.31 ± 0.05 | −1.38 ± 0.14 | −1.26 ± 0.06 |
| Mapk8e | P > 0.05 | P > 0.05 | P > 0.05 |
| Mapk9d | 1.72 ± 0.03 | 1.89 ± 0.06 | 1.53 ± 0.04 |
| Mtmr14 | 1.65 ± 0.09* | P > 0.05 | P > 0.05 |
| Mtmr3 | 1.37 ± 0.08* | P > 0.05 | P > 0.05 |
| mTOR | 1.39 ± 0.18* | P > 0.05 | P > 0.05 |
| Nrase | P > 0.05 | P > 0.05 | P > 0.05 |
| Nrbf2 | P > 0.05 | 1.29 ± 0.12* | P > 0.05 |
| Pik3c3e | P > 0.05 | P > 0.05 | P > 0.05 |
| Pik3cae | P > 0.05 | P > 0.05 | P > 0.05 |
| Pik3cbe | P > 0.05 | P > 0.05 | P > 0.05 |
| Pik3cd | P > 0.05 | P > 0.05 | 4.00 ± 1.13* |
| Pik3r2e | P > 0.05 | P > 0.05 | P > 0.05 |
| Pik3r3e | P > 0.05 | P > 0.05 | P > 0.05 |
| Pik3r4e | P > 0.05 | P > 0.05 | P > 0.05 |
| Prkcd | 1.44 ± 0.05* | 1.37 ± 0.09* | P > 0.05 |
| Prkcqe | P > 0.05 | P > 0.05 | P > 0.05 |
| Ptene | P > 0.05 | P > 0.05 | P > 0.05 |
| Raf1d | 1.84 ± 0.08 | 1.76 ± 0.14 | 1.59 ± 0.13 |
| Rb1cc1 | P > 0.05 | 1.20 ± 0.05* | P > 0.05 |
| Rps6kb1d | 1.30 ± 0.11 | 1.43 ± 0.03 | 1.38 ± 0.04 |
| Rras2 | P > 0.05 | P > 0.05 | −2.63 ± 0.51* |
| Stk11 | 1.25 ± 0.08* | P > 0.05 | P > 0.05 |
| Traf6d | 2.17 ± 0.06 | 1.94 ± 0.27 | 1.87 ± 0.21 |
| Tsc2e | P > 0.05 | P > 0.05 | P > 0.05 |
| Ulk1e | P > 0.05 | P > 0.05 | P > 0.05 |
| Ulk2 | −1.52 ± 0.11* | P > 0.05 | P > 0.05 |
| Wipi1 | −1.68 ± 0.13* | −2.26 ± 0.83* | P > 0.05 |
BM-derived macrophages from M1-γ34.5 and M2-γ34.5 transgenic mice and WT mice were infected with 10 PFU/cell of WT McKrae virus for 1 h at 37°C, washed with PBS, and incubated for an additional 24 h in fresh medium. Expression of 62 autophagy genes in infected cells was analyzed by NanoString, and the level of each gene was normalized to its level in mock-infected BM-derived macrophages. Results are shown as fold increase or decrease relative to mock-infected gene expression. Results indicate mean ± SEM (n = 3).
For Bnip3 gene expression, there were no differences between the WT and M1-γ34.5 groups (P > 0.05), while differences between WT and M2-γ34.5 groups and among WT, M1-γ34.5, and M2-γ34.5 were statistically significant (P < 0.05).
For Cflar gene expression, the three groups of infected macrophages were statistically significantly different from each other (P < 0.05).
The three groups of infected macrophages were not significantly different from each other (P > 0.05), but they were significantly different from their mock-infected counterparts (P < 0.05).
M1-γ34.5, M2-γ34.5, and WT macrophages were not significantly different from each other or their mock-infected counterparts for AKT1, AKT3, AKT10, Atg101, Atg5, Atg7, BclII, Ddit4, Map3k7, Mapk1, Pik3c3, Pik3ca, Pik3cb, Pik3r2, Pik3r3, Pik3r4, Prkcq, Pten, Tsc2, and Ulk1 genes (P > 0.05).
Each gene indicated by an asterisk among the three groups of macrophages was similar to one another and significantly different from their counterparts without the asterisk.
Based on their expression in the three macrophage groups, the 62 autophagy genes were divided into six subsets: (i) significant common genes among all three groups, (ii) uniquely expressed genes in WT macrophages, (iii) uniquely expressed genes in macrophages from M1-γ34.5 mice, (iv) uniquely expressed genes in macrophages from M2-γ34.5 mice, (v) partially common genes expressed among two of the three groups, and (vi) nonsignificant autophagy genes expressed in all three groups with respect to mock groups (Fig. 5 and Table 2). These results identified uniquely expressed anti-inflammatory/repairing genes, e.g., Nrbf2 and Rb1cc1 (3% of 62 autophagy genes), in M2-γ34.5 mice and uniquely expressed proinflammatory genes, e.g., Hif1a, Mtmr14, mTOR, Stk11, Mtmr3, and Ulk2 (10% of 62 autophagy genes), in M1-γ34.5 mice. WT mice expressed Akt2, Ctsl, Pik3cd, and Rras2 genes (6% of the 62 autophagy genes). Only 5% of the genes were expressed in either of two groups, namely, Atg9a, Prkcd, and Wipi1, whereas the majority of autophagy genes (40%) were commonly expressed among all three mouse strains. However, 36% of the 62 genes were grouped as nonsignificant autophagy genes among HSV-1-infected macrophages (Fig. 5). Overall, our data suggest that the γ34.5 gene inhibits autophagy more efficiently in M1 macrophages than in M2 macrophages after HSV-1 infection.
FIG 5.
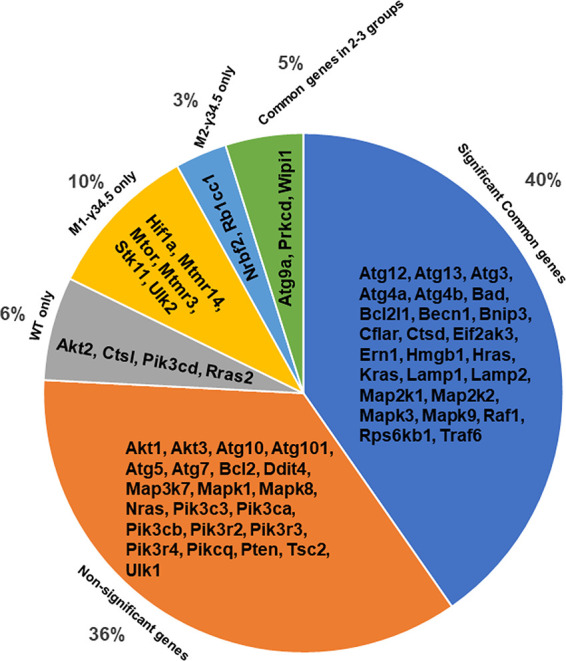
Analysis of expression of 62 autophagy genes in macrophages isolated from transgenic mice. BM cells from WT, M1-γ34.5, and M2-γ34.5 mice were isolated, and macrophages were generated and infected with 10 PFU/cell of HSV-1 strain McKrae. Total RNA was isolated from infected macrophages and used for NanoString analysis of 62 autophagy genes. The autophagy panel of 62 genes is illustrated in a pie chart showing the autophagy genes commonly expressed in WT, M1-γ34.5, and M2-γ34.5 groups (significant common genes), uniquely expressed in M1-γ34.5 macrophages, M2-γ34.5 macrophages, or WT macrophages, or commonly expressed in 2 of 3 groups and autophagy genes whose expression did not differ significantly from mock (nonsignificant genes). Gene expression analysis is based on three replications.
Following NanoString analysis of autophagy genes, BM-derived macrophages from WT, M1-γ34.5, and M2-γ34.5 mice were further analyzed for apoptosis, autoimmune, and myeloid gene panels of over 1,200 genes to detect any abnormality or toxic effects due to constitutive expression of the γ34.5 gene. Our data revealed that apoptosis gene markers were similar in all three groups of mice, suggesting that constitutive expression of the γ34.5 gene had no effect on cell death and did not cause any toxicity in M1-γ34.5 or M2-γ34.5 transgenic mice (see Table S1 in the supplemental material).
Blocking autophagy in M1 macrophages increases virus replication in ocularly infected mice.
Our results described above (Fig. 1 to 4) have shown that M1-γ34.5 transgenic mice express γ34.5 at higher levels in M1 macrophages than in M2 macrophages, while M2-γ34.5 transgenic mice express γ34.5 at higher levels in M2 macrophages than in M1 macrophages. We have previously shown that M1 macrophages play a more important role against ocular HSV-1 infection than do M2 macrophages (31–34). To determine how blocking autophagy in M1 or M2 macrophages affects ocular HSV-1 infection, we infected WT, M1-γ34.5, and M2-γ34.5 mice with 2 × 105 PFU/eye of WT HSV-1 strain McKrae. Tear films from the eyes of infected mice were collected on days 1 to 7 postinfection (p.i.), and viral titers were determined by plaque assay. Among the three infected mouse groups, M1-γ34.5 mice had higher viral titers on days 4 and 5 p.i. that were statistically different from the other two groups (Fig. 6; P < 0.05), and we did not find significant differences between M2-γ34.5 mice and WT mice (Fig. 6; P > 0.05). Virus replication in the eyes of M1-γ34.5 mice was approximately 2-fold higher than that in M2-γ34.5 mice and WT mice. This is in line with our previous studies (11, 12, 24). Thus, blocking autophagy in M1-γ34.5 mice appears to correlate with increased virus replication in the eyes of ocularly infected mice. These results confirm our previous finding that mice lacking M1 macrophages had more virus replication in their eyes than mice lacking M2 macrophages (33, 34).
FIG 6.
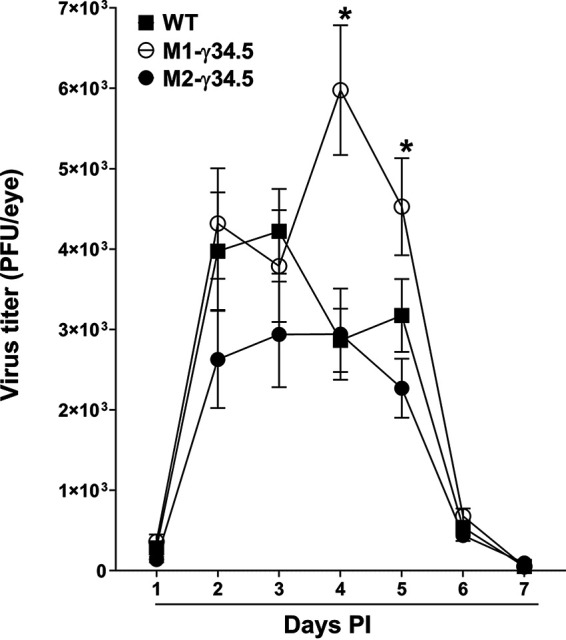
Virus replication in the eye of infected transgenic mice. WT (solid square), M1-γ34.5 (open circle), and M2-γ34.5 (solid circle) mice were ocularly infected with 2 × 105 PFU/eye of McKrae virus. Tear films were collected on days 1 to 7 p.i., and virus titers were determined by standard plaque assay as described in Materials and Methods. Each point represents the mean virus titer ± SEM for 20 eyes per group.
Blocking autophagy in M1 macrophages increases CS in ocularly infected mice.
Corneal scarring (CS) in surviving WT, M1-γ34.5, and M2-γ34.5 mice on day 28 p.i. was monitored, and lesion severity was scored as described in Materials and Methods. The three groups of infected mice showed similar levels of survival (data not shown). However, similar to our virus replication data above (Fig. 6), blocking autophagy in M1-γ34.5 mice was associated with significantly higher levels of eye disease in M1-γ34.5 mice than in M2-γ34.5 mice (Fig. 7; P < 0.01) or WT mice (Fig. 7; P < 0.05). No significant differences in eye disease were detected between M2-γ34.5 mice and WT mice (Fig. 7; P > 0.05). These results are consistent with our previous study in which mice lacking M1 macrophages were shown to be more susceptible to virulent HSV-1 strain McKrae, leading to more eye disease in infected mice (34). Thus, the absence of M1 macrophages (34) and blocking autophagy in M1 macrophages both suggest that M1 macrophages play an indispensable role in controlling virus replication and eye disease in ocularly infected mice.
FIG 7.
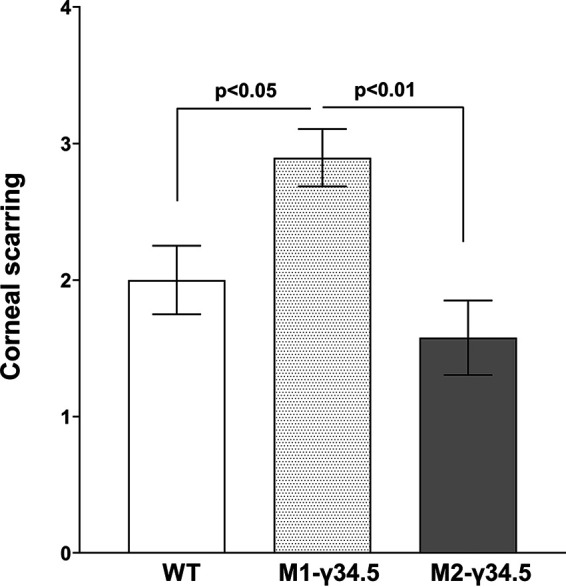
Corneal scarring (CS) in ocularly infected mice. CS on day 28 p.i. was determined in surviving mice as described in Materials and Methods. CS scores represent the mean ± SEM from 20 eyes for each group.
Blocking autophagy in macrophages did not alter latency-reactivation in ocularly infected mice.
To determine how blocking autophagy mediated by the γ34.5 gene affects latency, groups of 10 WT, M1-γ34.5, and M2-γ34.5 mice were ocularly infected with 2 × 105 PFU/eye of HSV-1 strain McKrae. TGs from latently infected mice were harvested on day 28 p.i., and latency levels were determined by qRT-PCR of HSV-1 latency-associated transcript (LAT) expression as described in Materials and Methods. LAT expression did not change in any of the infected mouse groups (Fig. 8A; P > 0.05), suggesting that blocking autophagy by the γ34.5 gene did not affect latency establishment in M1 or M2 groups compared with the WT group.
FIG 8.
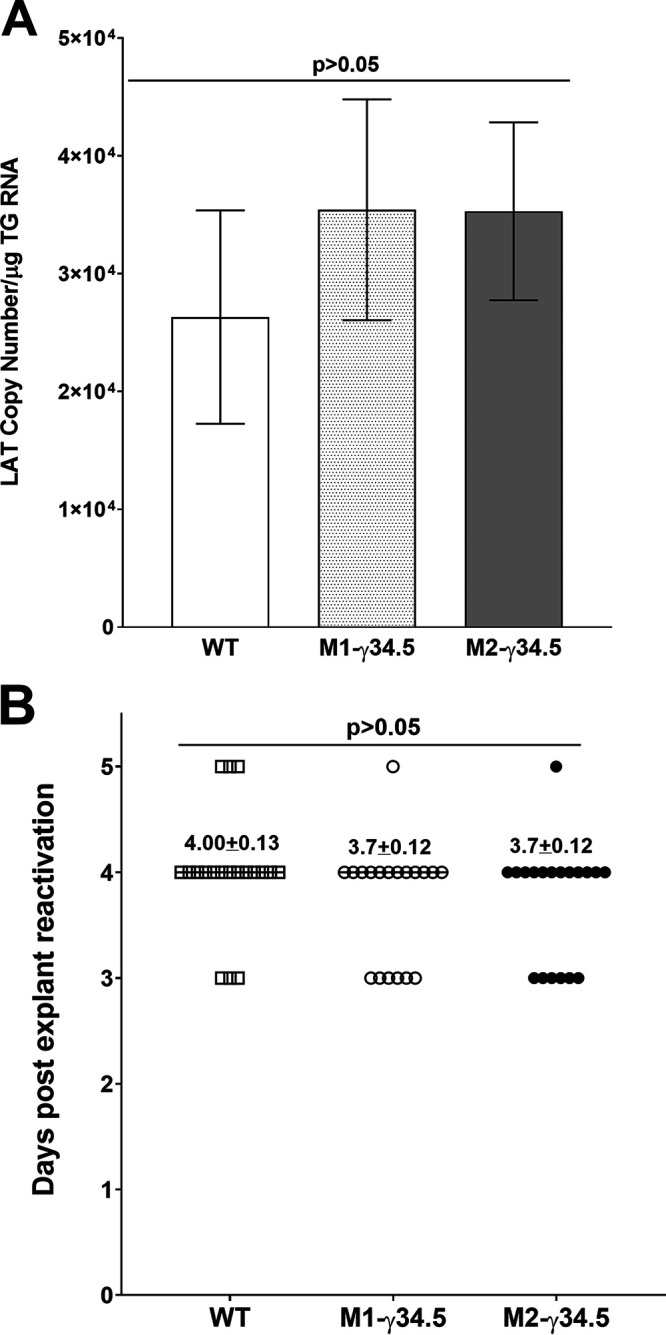
Latency and explant reactivation in TG of latently infected mice. (A) LAT expression in trigeminal ganglion (TG) of infected mice. Latency levels in TG were analyzed by harvesting TG from latently infected WT, M1-γ34.5, and M2-γ34.5 mice on day 28 p.i. and performing qRT-PCR on individual TGs from each mouse. In each experiment, estimated relative LAT copy number was calculated using standard curves generated from pGEM-5317. Briefly, pGEM-5317 DNA, serially diluted 10-fold such that 5 μL contained from 103 to 1011 copies of LAT, was subjected to TaqMan PCR with the same set of LAT primers. Copy number for each reaction was determined by comparing the normalized threshold cycle of each sample to the threshold cycle of the standard. GAPDH expression was used to normalize relative LAT RNA expression in the TG. Twenty TGs were analyzed per mouse group. (B) Explant reactivation in TG of latently infected mice. TGs from latently infected WT, M1-γ34.5, and M2-γ34.5 mice were individually isolated on day 28 p.i. Each individual TG was incubated in 1.5 mL of tissue culture medium at 37°C. Medium aliquots were removed from each culture daily for up to 5 days and plated on indicator RS cells to assess the appearance of reactivated virus. Results are plotted as the number of TGs that reactivated daily. The average time that TGs from each group first showed cytopathic effect ± SEM is shown. Reactivation is based on 20 TGs from 10 mice per group.
We asked next whether blocking autophagy by the γ34.5 gene influences the time to reactivation in transgenic and WT mice. Groups of 10 WT, M1-γ34.5, and M2-γ34.5 mice were ocularly infected with 2 × 105 PFU/eye of HSV-1 strain McKrae as described above. Virus reactivation was analyzed by explanting individual TGs from latently infected mice on day 28 p.i. Consistent with latency levels, time of reactivation did not change among the three infected mouse groups (Fig. 8B; P > 0.05). Thus, these results support our previously published studies suggesting that altered macrophage function does not affect latency-reactivation (33, 34). Taken together, these results and our previous studies with M1 and M2 macrophages suggest that the effect of M1 and M2 macrophage subtype on HSV-1 infection is associated with virus replication and eye disease but not latency-reactivation.
DISCUSSION
Autophagy is a critically important intracellular process by which damaged organelles are cleared, disassembled, and recycled (50–52). There is increasing evidence that autophagy is an adaptive response to infection and plays an important role in limiting inflammation. Defects in autophagy result in an inability to recycle damaged organelles and are also associated with impaired apoptosis and defective clearance of dying cells (51), which can activate the immune system, leading to development of immune-mediated disorders and pathology. Thus, autophagy is crucial for self-maintenance and appropriate regulation of innate and adaptive immune responses. In our previous studies, we demonstrated that macrophages play a critical role in HSV-1 infection (7, 11, 22, 31, 33, 34) and form the dominant cell infiltrates in infected corneas (22). In addition to being a significant part of the innate immune system, macrophages are also involved in maintaining homeostasis, in clearing cellular debris, and in tissue repair and remodeling (53–55). Macrophages are broadly classified as M1 and M2 depending on their polarization effects (27, 28, 30). Due to the importance of macrophages in ocular HSV-1 infection and the importance of autophagy in clearing dying cells, we investigated the effect of blocking autophagy in M1 and M2 macrophages. Since the HSV-1 γ34.5 gene blocks autophagy in infected cells (41–43), we constructed two transgenic mouse lines expressing the HSV-1 γ34.5 gene under the control of the M1 (NOS2) (44, 45) or M2 (ARG1) promoter (46). Both M1-γ34.5 and M2-γ34.5 transgenic mice were created using a standard DNA microinjection procedure, and we verified the specificity of γ34.5 expression under the M1 or M2 promoter.
Following ocular infection of M1-γ34.5 and M2-γ34.5 transgenic mice and WT control mice with HSV-1, M1-γ34.5 transgenic mice exhibited higher levels of virus replication in the eyes and more severe eye disease than did M2-γ34.5 transgenic mice or WT control mice. M1-γ34.5 transgenic mice in particular did not display any other abnormality or side effect of γ34.5 insertion other than higher levels of eye disease and higher viral titers in the eye than those in M2-γ34.5 transgenic mice or WT control mice. Similar to our study, infection of mice with a recombinant HSV-1 lacking the beclin-binding domain (BBD) of the γ34.5 gene was shown to increase the autophagy response and induce a more rapid innate immune response that was associated with increased virus replication and retinal damage (56). Similar to this study with M1-γ34.5 transgenic mice, the absence of autophagy in dendritic cells also enhanced eye disease in` infected mice (57). Furthermore, deletion of multiple autophagy genes in macrophages led to uveitis due to uncontrolled inflammasome activation (58). These results are consistent with our recent report showing the important role of M1 macrophages in protecting against ocular HSV-1 infection (34). We recently showed that M1 macrophages are the main players in controlling viral replication, and in the absence of M1 macrophages, enhanced viral replication led to increased eye disease (34). Similar to this study, in which blocking autophagy in M2 macrophages did not affect virus replication in the eyes or eye disease, with levels of both similar to those in control mice, we have also shown that mice lacking M2 macrophages behave similarly to WT control mice (33). M1-γ34.5 transgenic mice have an enhanced ability to block autophagy, leading to increased virus replication in the eyes of these mice, which is consistent with previous work showing that autophagy provides antiviral functions (59). Autophagy also functions as an antiviral against other viruses (60–62) and plays an important role in maintaining healthy reproductive mechanisms in both male and female mice (63, 64). As in previous studies (63, 64), we found that blocking autophagy by γ34.5 affected fertility in both transgenic M1-γ34.5 and M2-γ34.5 mice despite the absence of obvious developmental issues in the pups. However, blocking autophagy in M1 or M2 macrophages did not affect levels of latency-reactivation compared with WT control mice. This is similar to our previous reports that the absence of M1 or M2 in conditional knockout mice did not affect latency-reactivation compared with WT control mice (33, 34).
We also evaluated the effect of γ34.5 expression in transgenic mice by quantifying the expression of autophagic genes and their fundamental function in response to virus infection. We evaluated expression of 62 autophagy transcripts in vitro after infecting isolated macrophages from M1-γ34.5 transgenic mice, M2-γ34.5 transgenic mice, or WT mice with WT McKrae virus. We found significant upregulation of autophagy-inhibitory genes including Hif1a, Mtmr14, mTOR, Stk11, Mtmr3, and Ulk2 in M1-γ34.5 transgenic mice but not in M2-γ34.5 transgenic mice, suggesting that expression of γ34.5 blocks autophagy in M1 macrophages more efficiently than in M2 macrophages. Hif1a can induce or inhibit autophagy depending on the nature of the disease (65, 66), and it has an inhibitory role in macrophage autophagy (67). In inhibiting autophagy, two other genes, Mtmr14 and Mtmr3, play important roles in dephosphorylating phosphatidylinositol 3-phosphate (PI3P), which is involved in autophagosome formation (68). Mtmr14 and Mtmr3 are expressed significantly only in M1-γ34.5 mice and not in M2-γ34.5 mice, reflecting the inhibitory function of the γ34.5 gene in M1 macrophages only. Stk11 is also known to inhibit autophagy but is required for mouse survival and to control infection burden (69). We found that Stk11 was upregulated in M1-γ34.5 mice but not in M2-γ34.5 mice. Ulk2 has not been widely studied but does not yet have a documented direct role in activating autophagy (70). Nrbf2 and Rb1cc1 genes have anti-inflammatory/repairing functions and were upregulated in macrophages from M2-γ34.5 transgenic mice (71). Rb1cc1 is a component of the ULK1-ATG13-RB1CC1/FIP200 complex, which is inhibited by mTOR and is an essential upstream inducer of autophagy (71, 72). Deletion of the RB1CC1 autophagy inducer leads to age-related degeneration of the retinal pigment epithelium (73). We found that Rb1cc1 was significantly upregulated in M2-γ34.5 mice but not in M1-γ34.5 mice, while at the same time, mTOR was upregulated in M1-γ34.5 mice but not in M2-γ34.5 mice, demonstrating that mTOR suppresses Rb1cc1 in M1 macrophages but not in M2 macrophages. Along with the Rb1cc1 gene in M2-γ34.5 mice, the Nrbf2 gene is also significantly upregulated but is not expressed in M1-γ34.5 mice. Nrbf2 regulates autophagy, is involved in apoptotic cell clearance, and plays a positive role in regulating autophagy in macrophages (74, 75). Of the 62 autophagy genes upregulated in M1-γ34.5 mice, 10% are known to inhibit autophagy, while only 3% of autophagy genes upregulated in M2-γ34.5 mice are known to inhibit autophagy. This demonstrates a novel ability of the γ34.5 gene to inhibit autophagy in M1 macrophages while upregulating autophagy in M2 macrophages. Since the HSV-1 γ34.5 gene blocks autophagy (41–43), these results suggest the importance of intact autophagy in controlling virus replication and eye disease.
Cytokines and chemokines play important roles in protection and disease; thus, we investigated whether blocking autophagy in macrophages would affect cytokine and chemokine secretion after infecting macrophages isolated from M1-γ34.5 transgenic mice, M2-γ34.5 transgenic mice, or WT mice with WT McKrae virus. In our in vitro results using Luminex, we found no significant differences in expression of G-CSF, eotaxin, GM-CSF, IFN-γ, IL-1α, IL-1β, IL-2, IL-4, IL-3, IL-5, IL-7, IL-9, IL-10, IL-12(p40), IL-12(p70), LIF, IL-15, IP-10, KC, MIP-1α, MIP-1β, M-CSF, MIP-2, MIG, and RANTES among WT, M1-γ34.5, or M2-γ34.5 macrophages, while significant differences in IL-6, LIX, IL-17, MCP-1, VEGF, and TNF-α were detected among the three macrophage types. For instance, expression of proinflammatory cytokine TNF-α was significantly higher in M1-γ34.5 macrophages than in other groups. The primary source of TNF-α in virally stimulated cells is macrophages (76, 77), and TNF-α is one of the most important proinflammatory cytokines involved in inflammation, apoptosis, and cancer (78). TNF-α expression is reported to be elevated in activated macrophages following chronic cytomegalovirus (CMV) infection and directly correlates with active virus replication (79). Thus, elevated TNF-α expression in M1-γ34.5 transgenic macrophages may be a significant contributor to increased virus replication in the eyes and eye disease relative to WT or M2-γ34.5 mice. Consistent with our study, ocular infection of mice with a recombinant HSV-1 expressing TNF-α has been shown to increase virus replication, inflammation, and pathology in the eyes of infected mice (80).
In summary, we have presented the results of blocking autophagy in M1 and M2 macrophages on HSV-1 replication in vitro and in vivo. The major findings of this study demonstrated that (i) inhibiting autophagy directly correlates with higher virus replication in the eyes of M1-γ34.5 mice and enhanced eye disease, (ii) absence of autophagy may increase the secretion of certain proinflammatory cytokines/chemokines independent of macrophage phenotype, and (iii) impaired autophagy function correlates with poor reproduction and fertility in both male and female mice.
MATERIALS AND METHODS
Ethics statement.
All animal procedures were performed in strict accordance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research and the Guide for the Care and Use of Laboratory Animals (81). Animal research protocols were approved by the Institutional Animal Care and Use Committee of Cedars-Sinai Medical Center (protocols 5030 and 8837).
Construction of transgenic mice expressing the HSV-1γ34.5 gene under the M1 or M2 promoter.
C57BL/6 WT mice (6 to 8 weeks old) were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). M1-γ34.5 transgenic mice were constructed by inserting the full-length γ34.5 gene sequence (GenBank identifier [ID] JX142173.1) (82) with three FLAG tag sequences before the stop codon followed by an IRES sequence and flanked by two BglII restriction sites added to the 5′ and 3′ ends of the insert for cloning purposes. This construct, synthesized by GenScript (Piscataway, NJ), was inserted into the BglII site of the pGL2-NOS2 mammalian expression vector containing the mouse nitric oxide synthase promoter sequence (NOS2) (Addgene, Cambridge, MA; plasmid no. 19296) (45) such that γ34.5 gene expression is controlled by the NOS2 promoter (Fig. 1). Similarly, M2-γ34.5 transgenic mice were constructed using the pGL3-mARG1 mammalian expression vector containing the mouse arginase 1 (ARG1) promoter (Addgene; plasmid no. 34571) (46). An insert identical to that used to make M1-γ34.5 transgenic mice was constructed except that two XhoI restriction sites were added to the 5′ and 3′ ends of the insert for cloning instead of the BglII sites used above. In this construct, γ34.5 gene expression is controlled by the ARG1 promoter (Fig. 1). The Cedars-Sinai Rodent Genetics Core created both the M1-γ34.5 and M2-γ34.5 transgenic mice using standard DNA microinjection procedures as we described previously (80). All mice were bred and maintained in the Cedars-Sinai Medical Center pathogen-free animal facility. Although these mice had reproduction and fertility issues, the homozygous pups appeared healthy and were of normal size and body weight.
Viruses and cells.
Plaque-purified, virulent HSV-1 strain McKrae was used in this study. Rabbit skin (RS) cells were used to prepare virus stocks, culture mouse tear swabs, and determine viral growth kinetics. RS cells were grown in Eagle’s minimal essential medium supplemented with 5% fetal bovine serum as we described previously (83).
Genotyping of transgenic mice by DNA extraction and PCR analysis for the presence of IRES and γ34.5 sequences.
To genotype and confirm the presence of IRES and γ34.5 genes in transgenic mice, tail, cornea, TG, brain, spleen, and liver tissue samples from transgenic mice were collected and lysed at 55°C overnight in 100 μL lysis buffer (100 mM Tris-HCl, pH 8.5, 5 mM EDTA, 0.2% SDS, 200 mM NaCl, and 1 mg/mL proteinase K). After dilution of the lysate 1:10 in distilled water, 1 μL was used as a template for PCR analyses using IRES-specific primers (forward, 5′-AATAAGGCCGGTGTGCGTTTG-3′, and reverse, 5′-TGGGATCTGATCTGGGGCCT-3′) or γ34.5-specific primers (forward, 5′-TCGTCGGACGCGGACTCGGGAACGGTGGAGC-3′, and reverse, 5′-CTCCACGCCCAACTCGGAACCCGCGGTCAG-3′) as we described previously (84). The IRES primer set has an amplicon length of 428 bp, and the γ34.5 primer set has an amplicon length of 138 bp. The γ34.5 DNA copy number was calculated using a standard curve generated using pUC57-γ34.5 DNA as we described previously (85).
Generation of BM-derived macrophages.
Femoral bones were dissected, and all remaining tissue on the bones was removed. Each bone end was cut off, and bone marrow (BM) was expelled. BM-derived macrophages were cultured for 6 days. To differentiate and activate macrophages, 20 ng/mL M-CSF and GM-CSF (Peprotech, Rocky Hill, NJ; catalog no. 315-02 [M-CSF] and 315-03 [GM-CSF]) were added along with the cells to be cultured, as described previously (31, 86). On day 3, new medium containing fresh M-CSF and GM-CSF was replenished and cells were returned to the incubator until day 6. On day 6, medium was removed, and cells were washed three times with phosphate-buffered saline (PBS) to remove floating cells. Macrophages adhering to tissue culture dishes were harvested by scraping and counted for experiments.
Confirmation of phenotype of macrophages isolated from transgenic mice.
BM-derived macrophages were seeded at 2 × 105 cells per well in a 24-well plate. After overnight incubation, medium was replaced with complete Dulbecco’s modified Eagle’s medium (DMEM) containing either 50 ng/mL of murine IFN-γ (Peprotech, Rocky Hill, NJ) and 100 ng/mL of lipopolysaccharide (LPS; Sigma-Aldrich, St. Louis, MO) for M1 activation or 10 ng/mL of murine IL-4 (Peprotech) for M2 activation as we described previously (31, 32). Macrophages were harvested, and cells were counted (Countess cell counting chamber slides; Invitrogen). Cell counts from all three mouse groups were similar with no detectable level of apoptosis/toxicity in response to constitutive expression of the γ34.5 gene. Total RNA was extracted from monolayers of unpolarized (M0) or M1- or M2-polarized macrophages derived from M1-γ34.5 or M2-γ34.5 mice using the RNeasy minikit according to the manufacturer’s protocol (Qiagen, Valencia, CA). qRT-PCR analyses were performed using γ34.5-specific primers, forward, 5′-GGGCTGACCCCTCCCA-3′, and reverse, 5′-TGCTCCGCGGTGACG-3′, and probe, 5′-6-carboxyfluorescein (FAM)-CCCCTCGCGCCCCT-3′ (amplicon length, 83 bp). Copy number of γ34.5 RNA was calculated using a standard curve generated using pUC57-γ34.5 DNA.
Luminex xMAP immunoassay.
BM cells isolated from WT, M1-γ34.5, and M2-γ34.5 mice were cultured and differentiated into macrophages as described above. Differentiated macrophages were infected with 10 PFU/cell of HSV-1 strain McKrae for 24 h. Medium was collected from infected cells, and Luminex assays were performed in the Immune Assessment Core at the University of California, Los Angeles, using mouse 32-plex magnetic cytokine/chemokine kits purchased from EMD Millipore (Billerica, MA) according to the manufacturer’s instructions as we described previously (34). Fluorescence was quantified using a Luminex 200 instrument (Luminex Corp., Austin, TX).
Ocular infection.
Mice were infected ocularly with 2 × 105 PFU per eye of McKrae virus as an eyedrop in 2 μL of tissue culture medium as we described previously (87). Corneal scarification was not performed prior to infection with McKrae virus.
Viral titers from tears of infected mice.
Tear films were collected on days 1 to 7 p.i. from WT, M1-γ34.5, and M2-γ34.5 mouse eyes infected with HSV-1 strain McKrae virus using a Dacron-tipped swab. Each swab was placed in 1 mL of tissue culture medium and squeezed. The amount of virus was determined by standard plaque assay on RS cells as described previously (88).
Monitoring corneal scarring in ocularly infected mice.
The severity of corneal scarring in WT, M1-γ34.5, and M2-γ34.5 mice was examined by slit lamp biomicroscopy on day 28 p.i. The scoring scale was as follows: 0, normal cornea; 1, mild haze; 2, moderate opacity; 3, severe corneal opacity but iris visible; 4, opaque and corneal ulcer; 5, corneal rupture and necrotizing keratitis, as we described previously (14).
In vitro explant reactivation assay.
WT, M1-γ34.5, and M2-γ34.5 infected mice were sacrificed on day 28 p.i., and individual TGs were removed and cultured in tissue culture medium as described previously (85). Medium aliquots were removed from each culture daily and plated on indicator RS cells to detect reactivated virus. As medium from explanted TG cultures was plated daily, we could determine the time at which reactivated virus first appeared in the explanted TG cultures.
RNA extraction, cDNA synthesis, and TaqMan qRT-PCR assay.
To extract RNA from TGs, tissues were collected on day 28 p.i., immersed in TRIzol reagent (Applied Biosystems, Foster City, CA), and stored at −80°C until processing. Total RNA was extracted according to the manufacturer’s protocol. Following RNA extraction, 1 μg of total RNA was reverse transcribed using the High-Capacity cDNA reverse transcription kit (Applied Biosystems, CA) according to the manufacturer’s protocol. mRNA expression levels of the genes of interest were evaluated using TaqMan gene expression assays (Applied Biosystems, CA). Levels of LAT RNA from latent TGs were determined using custom LAT primers and probe as follows: forward, 5′-GGGTGGGCTCGTGTTACAG-3′, and reverse, 5′-GGACGGGTAAGTAACAGAGTCTCTA-3′, and probe, 5′-FAM-ACACCAGCCCGTTCTTT-3′ (amplicon length, 81 bp). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control in all experiments. Quantitative real-time PCR (qRT-PCR) was performed as we described previously (85). LAT RNA copy number was calculated using a standard curve generated using pGEM5317-LAT, as we described previously (85).
NanoString gene expression analysis.
BM-derived macrophages from WT, M1-γ34.5, and M2-γ34.5 mice were generated and infected with 10 PFU/cell of HSV-1 strain McKrae for 24 h, at which time total RNA was purified with RNeasy as we described previously (31). Hybridization was performed using total RNA (20 ng/μL) per well diluted in 15 μL hybridization cocktail (3 μL reporter CodeSet, 5 μL hybridization buffer, 2 μL capture mix, and 5 μL sample). Following hybridization, samples were centrifuged, and NanoString gene expression analysis was performed using gene panels consisting of probes for 62 autophagy-related genes, 1,209 autoimmune and myeloid panel genes, and 20 internal reference genes (8 negatives, 6 positives, and 6 housekeeping) for data normalization. NanoString analysis was performed in a thermocycler at 65°C for 20 h. Samples were placed in a 12-well PCR strip and loaded into the Max/Flex nCounter Prep (NanoString Tech, Seattle, WA) with consumables such as reagent plates and a cartridge for the hybridization. The preparation station ran for 3 h, after which the cartridge was loaded to the digital analyzer (NanoString Tech, Seattle, WA) for imaging analysis. A field of view of 240 was used for the experimental recording. nSolver software 4.0 was used to compute NanoString gene expression values, principal-component analysis of probe counts, and respective fold changes.
Statistical analysis.
For all statistical tests, P values less than or equal to 0.05 were considered statistically significant and are indicated by a single asterisk (*). P values less than or equal to 0.001 are indicated by double asterisks (**). A two-tailed Student t test with unequal variances was used to compare differences between two experimental groups. A one-way analysis of variance (ANOVA) was used to compare differences among three or more experimental groups. All experiments were repeated at least three times to ensure accuracy.
ACKNOWLEDGMENTS
This study was supported by Public Health Service NIH grants RO1EY024649, RO1EY026944, and RO1EY013615.
Footnotes
Supplemental material is available online only.
Contributor Information
Homayon Ghiasi, Email: ghiasih@CSHS.org.
Jae U. Jung, Lerner Research Institute, Cleveland Clinic
REFERENCES
- 1.Barron BA, Gee L, Hauck WW, Kurinij N, Dawson CR, Jones DB, Wilhelmus KR, Kaufman HE, Sugar J, Hyndiuk RA. 1994. Herpetic eye disease study. A controlled trial of oral acyclovir for herpes simplex stromal keratitis. Ophthalmology 101:1871–1882. 10.1016/s0161-6420(13)31155-5. [DOI] [PubMed] [Google Scholar]
- 2.Wilhelmus KR, Dawson CR, Barron BA, Bacchetti P, Gee L, Jones DB, Kaufman HE, Sugar J, Hyndiuk RA, Laibson PR, Stulting RD, Asbell PA. 1996. Risk factors for herpes simplex virus epithelial keratitis recurring during treatment of stromal keratitis or iridocyclitis. Herpetic Eye Disease Study Group. Br J Ophthalmol 80:969–972. 10.1136/bjo.80.11.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tumpey TM, Chen SH, Oakes JE, Lausch RN. 1996. Neutrophil-mediated suppression of virus replication after herpes simplex virus type 1 infection of the murine cornea. J Virol 70:898–904. 10.1128/JVI.70.2.898-904.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shimeld C, Whiteland JL, Nicholls SM, Easty DL, Hill TJ. 1996. Immune cell infiltration in corneas of mice with recurrent herpes simplex virus disease. J Gen Virol 77:977–985. 10.1099/0022-1317-77-5-977. [DOI] [PubMed] [Google Scholar]
- 5.Hendricks RL, Tumpey TM. 1991. Concurrent regeneration of T lymphocytes and susceptibility to HSV-1 corneal stromal disease. Curr Eye Res 10:47–53. 10.3109/02713689109020357. [DOI] [PubMed] [Google Scholar]
- 6.Newell CK, Martin S, Sendele D, Mercadal CM, Rouse BT. 1989. Herpes simplex virus-induced stromal keratitis: role of T-lymphocyte subsets in immunopathology. J Virol 63:769–775. 10.1128/JVI.63.2.769-775.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ghiasi H, Hofman FM, Wallner K, Cai S, Perng G, Nesburn AB, Wechsler SL. 2000. Corneal macrophage infiltrates following ocular herpes simplex virus type 1 challenge vary in BALB/c mice vaccinated with different vaccines. Vaccine 19:1266–1273. 10.1016/S0264-410X(00)00298-X. [DOI] [PubMed] [Google Scholar]
- 8.Ghiasi H, Wechsler SL, Cai S, Nesburn AB, Hofman FM. 1998. The role of neutralizing antibody and T-helper subtypes in protection and pathogenesis of vaccinated mice following ocular HSV-1 challenge. Immunology 95:352–359. 10.1046/j.1365-2567.1998.00602.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghiasi H, Wechsler SL, Kaiwar R, Nesburn AB, Hofman FM. 1995. Local expression of tumor necrosis factor alpha and interleukin-2 correlates with protection against corneal scarring after ocular challenge of vaccinated mice with herpes simplex virus type 1. J Virol 69:334–340. 10.1128/JVI.69.1.334-340.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gimenez F, Bhela S, Dogra P, Harvey L, Varanasi SK, Jaggi U, Rouse BT. 2016. The inflammasome NLRP3 plays a protective role against a viral immunopathological lesion. J Leukoc Biol 99:647–657. 10.1189/jlb.3HI0715-321R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mott K, Brick DJ, van Rooijen N, Ghiasi H. 2007. Macrophages are important determinants of acute ocular HSV-1 Infection in immunized mice. Invest Ophthalmol Vis Sci 48:5605–5615. 10.1167/iovs.07-0894. [DOI] [PubMed] [Google Scholar]
- 12.Matundan H, Ghiasi H. 2019. Herpes simplex virus 1 ICP22 suppresses CD80 expression by murine dendritic cells. J Virol 93:e01803-18. 10.1128/JVI.01803-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nandakumar S, Woolard SN, Yuan D, Rouse BT, Kumaraguru U. 2008. Natural killer cells as novel helpers in anti-herpes simplex virus immune response. J Virol 82:10820–10831. 10.1128/JVI.00365-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jaggi U, Varanasi SK, Bhela S, Rouse BT. 2018. On the role of retinoic acid in virus induced inflammatory response in cornea. Microbes Infect 20:337–345. 10.1016/j.micinf.2018.04.007. [DOI] [PubMed] [Google Scholar]
- 15.Varanasi SK, Reddy PBJ, Bhela S, Jaggi U, Gimenez F, Rouse BT. 2017. Azacytidine treatment inhibits the progression of herpes stromal keratitis by enhancing regulatory T cell function. J Virol 91:e02367-16. 10.1128/JVI.02367-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wynn TA, Chawla A, Pollard JW. 2013. Macrophage biology in development, homeostasis and disease. Nature 496:445–455. 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, Mazloom AR, Ma’ayan A, Chua W-J, Hansen TH, Turley SJ, Merad M, Randolph GJ, Immunological Genome Consortium. 2012. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol 13:1118–1128. 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Macatonia SE, Hsieh CS, Murphy KM, O’Garra A. 1993. Dendritic cells and macrophages are required for Th1 development of CD4+ T cells from alpha beta TCR transgenic mice: IL-12 substitution for macrophages to stimulate IFN-gamma production is IFN-gamma- dependent. Int Immunol 5:1119–1128. 10.1093/intimm/5.9.1119. [DOI] [PubMed] [Google Scholar]
- 19.Morahan PS, Morse SS, McGeorge MG. 1980. Macrophage extrinsic antiviral activity during herpes simplex virus infection. J Gen Virol 46:291–300. 10.1099/0022-1317-46-2-291. [DOI] [PubMed] [Google Scholar]
- 20.Ramirez MC, Sigal LJ. 2002. Macrophages and dendritic cells use the cytosolic pathway to rapidly cross-present antigen from live, vaccinia-infected cells. J Immunol 169:6733–6742. 10.4049/jimmunol.169.12.6733. [DOI] [PubMed] [Google Scholar]
- 21.Sit MF, Tenney DJ, Rothstein JL, Morahan PS. 1988. Effect of macrophage activation on resistance of mouse peritoneal macrophages to infection with herpes simplex virus types 1 and 2. J Gen Virol 69:1999–2010. 10.1099/0022-1317-69-8-1999. [DOI] [PubMed] [Google Scholar]
- 22.Lee DH, Jaggi U, Ghiasi H. 2019. CCR2+ migratory macrophages with M1 status are the early-responders in the cornea of HSV-1 infected mice. PLoS One 14:e0215727. 10.1371/journal.pone.0215727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mott KR, Underhill D, Wechsler SL, Town T, Ghiasi H. 2009. A role for the JAK-STAT1 pathway in blocking replication of HSV-1 in dendritic cells and macrophages. Virol J 6:56. 10.1186/1743-422X-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mott KR, Gate D, Zandian M, Allen SJ, Rajasagi NK, van Rooijen N, Chen S, Arditi M, Rouse BT, Flavell RA, Town T, Ghiasi H. 2011. Macrophage IL-12p70 signaling prevents HSV-1-induced CNS autoimmunity triggered by autoaggressive CD4+ Tregs. Invest Ophthalmol Vis Sci 52:2321–2333. 10.1167/iovs.10-6536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ginhoux F, Jung S. 2014. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol 14:392–404. 10.1038/nri3671. [DOI] [PubMed] [Google Scholar]
- 26.Kodukula P, Liu T, Rooijen NV, Jager MJ, Hendricks RL. 1999. Macrophage control of herpes simplex virus type 1 replication in the peripheral nervous system. J Immunol 162:2895–2905. [PubMed] [Google Scholar]
- 27.Gordon S, Martinez FO. 2010. Alternative activation of macrophages: mechanism and functions. Immunity 32:593–604. 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 28.Gordon S. 2003. Alternative activation of macrophages. Nat Rev Immunol 3:23–35. 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 29.Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. 2000. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol 164:6166–6173. 10.4049/jimmunol.164.12.6166. [DOI] [PubMed] [Google Scholar]
- 30.Cao Y, Brombacher F, Tunyogi-Csapo M, Glant TT, Finnegan A. 2007. Interleukin-4 regulates proteoglycan-induced arthritis by specifically suppressing the innate immune response. Arthritis Rheum 56:861–870. 10.1002/art.22422. [DOI] [PubMed] [Google Scholar]
- 31.Lee DH, Ghiasi H. 2017. Roles of M1 and M2 macrophages in herpes simplex virus 1 infectivity. J Virol 91:e00578-17. 10.1128/JVI.00578-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee DH, Ghiasi H. 2018. An M2 rather than a TH2 response contributes to better protection against latency reactivation following ocular infection of naive mice with a recombinant herpes simplex virus 1 expressing murine interleukin-4. J Virol 92:e00051-18. 10.1128/JVI.00051-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jaggi U, Yang M, Matundan HH, Hirose S, Shah PK, Sharifi BG, Ghiasi H. 2020. Increased phagocytosis in the presence of enhanced M2-like macrophage responses correlates with increased primary and latent HSV-1 infection. PLoS Pathog 16:e1008971. 10.1371/journal.ppat.1008971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jaggi U, Matundan HH, Yu J, Hirose S, Mueller M, Wormley FL, Ghiasi H, Jr.. 2021. Essential role of M1 macrophages in blocking cytokine storm and pathology associated with murine HSV-1 infection. PLoS Pathog 17:e1009999. 10.1371/journal.ppat.1009999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bolovan CA, Sawtell NM, Thompson RL. 1994. ICP34.5 mutants of herpes simplex virus type 1 strain 17syn+ are attenuated for neurovirulence in mice and for replication in confluent primary mouse embryo cell cultures. J Virol 68:48–55. 10.1128/JVI.68.1.48-55.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brown SM, Harland J, MacLean AR, Podlech J, Clements JB. 1994. Cell type and cell state determine differential in vitro growth of non-neurovirulent ICP34.5-negative herpes simplex virus types 1 and 2. J Gen Virol 75:2367–2377. 10.1099/0022-1317-75-9-2367. [DOI] [PubMed] [Google Scholar]
- 37.Perng GC, Ghiasi H, Slanina SM, Nesburn AB, Wechsler SL. 1996. High-dose ocular infection with a herpes simplex virus type 1 ICP34.5 deletion mutant produces no corneal disease or neurovirulence yet results in wild-type levels of spontaneous reactivation. J Virol 70:2883–2893. 10.1128/jvi.70.5.2883-2893.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chou J, Kern ER, Whitley RJ, Roizman B. 1990. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science 250:1262–1266. 10.1126/science.2173860. [DOI] [PubMed] [Google Scholar]
- 39.Chou J, Roizman B. 1992. The gamma 1(34.5) gene of herpes simplex virus 1 precludes neuroblastoma cells from triggering total shutoff of protein synthesis characteristic of programed cell death in neuronal cells. Proc Natl Acad Sci USA 89:3266–3270. 10.1073/pnas.89.8.3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chou J, Roizman B. 1994. Herpes simplex virus 1 gamma(1)34.5 gene function, which blocks the host response to infection, maps in the homologous domain of the genes expressed during growth arrest and DNA damage. Proc Natl Acad Sci USA 91:5247–5251. 10.1073/pnas.91.12.5247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Orvedahl A, Alexander D, Tallóczy Z, Sun Q, Wei Y, Zhang W, Burns D, Leib DA, Levine B. 2007. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 1:23–35. 10.1016/j.chom.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 42.Gobeil PA, Leib DA. 2012. Herpes simplex virus gamma34.5 interferes with autophagosome maturation and antigen presentation in dendritic cells. mBio 3:e00267-12. 10.1128/mBio.00267-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leib DA, Alexander DE, Cox D, Yin J, Ferguson TA. 2009. Interaction of ICP34.5 with Beclin 1 modulates herpes simplex virus type 1 pathogenesis through control of CD4+ T-cell responses. J Virol 83:12164–12171. 10.1128/JVI.01676-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lorsbach RB, Murphy WJ, Lowenstein CJ, Snyder SH, Russell SW. 1993. Expression of the nitric oxide synthase gene in mouse macrophages activated for tumor cell killing. Molecular basis for the synergy between interferon-gamma and lipopolysaccharide. J Biol Chem 268:1908–1913. 10.1016/S0021-9258(18)53940-5. [DOI] [PubMed] [Google Scholar]
- 45.Lowenstein CJ, Alley EW, Raval P, Snowman AM, Snyder SH, Russell SW, Murphy WJ. 1993. Macrophage nitric oxide synthase gene: two upstream regions mediate induction by interferon gamma and lipopolysaccharide. Proc Natl Acad Sci USA 90:9730–9734. 10.1073/pnas.90.20.9730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pauleau AL, Rutschman R, Lang R, Pernis A, Watowich SS, Murray PJ. 2004. Enhancer-mediated control of macrophage-specific arginase I expression. J Immunol 172:7565–7573. 10.4049/jimmunol.172.12.7565. [DOI] [PubMed] [Google Scholar]
- 47.Miossec P, Kolls JK. 2012. Targeting IL-17 and TH17 cells in chronic inflammation. Nat Rev Drug Discov 11:763–776. 10.1038/nrd3794. [DOI] [PubMed] [Google Scholar]
- 48.Chandrasekar B, Melby PC, Sarau HM, Raveendran M, Perla RP, Marelli-Berg FM, Dulin NO, Singh IS. 2003. Chemokine-cytokine cross-talk. The ELR+ CXC chemokine LIX (CXCL5) amplifies a proinflammatory cytokine response via a phosphatidylinositol 3-kinase-NF-kappa B pathway. J Biol Chem 278:4675–4686. 10.1074/jbc.M207006200. [DOI] [PubMed] [Google Scholar]
- 49.Liu K, Zhao E, Ilyas G, Lalazar G, Lin Y, Haseeb M, Tanaka KE, Czaja MJ. 2015. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy 11:271–284. 10.1080/15548627.2015.1009787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.He MX, He YW. 2013. CFLAR/c-FLIPL: a star in the autophagy, apoptosis and necroptosis alliance. Autophagy 9:791–793. 10.4161/auto.23785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ryter SW, Nakahira K, Haspel JA, Choi AM. 2012. Autophagy in pulmonary diseases. Annu Rev Physiol 74:377–401. 10.1146/annurev-physiol-020911-153348. [DOI] [PubMed] [Google Scholar]
- 52.Deretic V, Saitoh T, Akira S. 2013. Autophagy in infection, inflammation and immunity. Nat Rev Immunol 13:722–737. 10.1038/nri3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saeki N, Imai Y. 2020. Reprogramming of synovial macrophage metabolism by synovial fibroblasts under inflammatory conditions. Cell Commun Signal 18:188. 10.1186/s12964-020-00678-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Herzog C, Pons Garcia L, Keatinge M, Greenald D, Moritz C, Peri F, Herrgen L. 2019. Rapid clearance of cellular debris by microglia limits secondary neuronal cell death after brain injury in vivo. Development 146:dev174698. 10.1242/dev.174698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bosurgi L, Cao YG, Cabeza-Cabrerizo M, Tucci A, Hughes LD, Kong Y, Weinstein JS, Licona-Limon P, Schmid ET, Pelorosso F, Gagliani N, Craft JE, Flavell RA, Ghosh S, Rothlin CV. 2017. Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science 356:1072–1076. 10.1126/science.aai8132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang M, Covar J, Zhang NY, Chen W, Marshall B, Mo J, Atherton SS. 2013. Virus spread and immune response following anterior chamber inoculation of HSV-1 lacking the Beclin-binding domain (BBD). J Neuroimmunol 260:82–91. 10.1016/j.jneuroim.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jiang Y, Yin X, Stuart PM, Leib DA. 2015. Dendritic cell autophagy contributes to herpes simplex virus-driven stromal keratitis and immunopathology. mBio 6:e01426-15. 10.1128/mBio.01426-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saitoh T, Akira S. 2016. Regulation of inflammasomes by autophagy. J Allergy Clin Immunol 138:28–36. 10.1016/j.jaci.2016.05.009. [DOI] [PubMed] [Google Scholar]
- 59.Yordy B, Iwasaki A. 2013. Cell type-dependent requirement of autophagy in HSV-1 antiviral defense. Autophagy 9:236–238. 10.4161/auto.22506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, Herman B, Levine B. 1998. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol 72:8586–8596. 10.1128/JVI.72.11.8586-8596.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu Y, Schiff M, Czymmek K, Talloczy Z, Levine B, Dinesh-Kumar SP. 2005. Autophagy regulates programmed cell death during the plant innate immune response. Cell 121:567–577. 10.1016/j.cell.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 62.Nakashima A, Tanaka N, Tamai K, Kyuuma M, Ishikawa Y, Sato H, Yoshimori T, Saito S, Sugamura K. 2006. Survival of parvovirus B19-infected cells by cellular autophagy. Virology 349:254–263. 10.1016/j.virol.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 63.Lv C, Wang X, Guo Y, Yuan S. 2020. Role of selective autophagy in spermatogenesis and male fertility. Cells 9:2523. 10.3390/cells9112523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gawriluk TR, Hale AN, Flaws JA, Dillon CP, Green DR, Rucker EB, III.. 2011. Autophagy is a cell survival program for female germ cells in the murine ovary. Reproduction 141:759–765. 10.1530/REP-10-0489. [DOI] [PubMed] [Google Scholar]
- 65.Ortiz-Masiá D, Cosín-Roger J, Calatayud S, Hernández C, Alós R, Hinojosa J, Apostolova N, Alvarez A, Barrachina MD. 2014. Hypoxic macrophages impair autophagy in epithelial cells through Wnt1: relevance in IBD. Mucosal Immunol 7:929–938. 10.1038/mi.2013.108. [DOI] [PubMed] [Google Scholar]
- 66.Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, Semenza GL. 2008. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem 283:10892–10903. 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 67.Wang X, de Carvalho Ribeiro M, Iracheta-Vellve A, Lowe P, Ambade A, Satishchandran A, Bukong T, Catalano D, Kodys K, Szabo G. 2019. Macrophage-specific hypoxia-inducible factor-1alpha contributes to impaired autophagic flux in nonalcoholic steatohepatitis. Hepatology 69:545–563. 10.1002/hep.30215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vergne I, Deretic V. 2010. The role of PI3P phosphatases in the regulation of autophagy. FEBS Lett 584:1313–1318. 10.1016/j.febslet.2010.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Khayati K, Bhatt V, Hu ZS, Fahumy S, Luo X, Guo JY. 2020. Autophagy compensates for Lkb1 loss to maintain adult mice homeostasis and survival. Elife 9:62377. 10.7554/eLife.62377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fuqua JD, Mere CP, Kronemberger A, Blomme J, Bae D, Turner KD, Harris MP, Scudese E, Edwards M, Ebert SM, de Sousa LGO, Bodine SC, Yang L, Adams CM, Lira VA. 2019. ULK2 is essential for degradation of ubiquitinated protein aggregates and homeostasis in skeletal muscle. FASEB J 33:11735–11745. 10.1096/fj.201900766R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X. 2009. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 284:12297–12305. 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhou Z, Liu J, Fu T, Wu P, Peng C, Gong X, Wang Y, Zhang M, Li Y, Wang Y, Xu X, Li M, Pan L. 2021. Phosphorylation regulates the binding of autophagy receptors to FIP200 Claw domain for selective autophagy initiation. Nat Commun 12:1570. 10.1038/s41467-021-21874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yao J, Jia L, Khan N, Lin C, Mitter SK, Boulton ME, Dunaief JL, Klionsky DJ, Guan J-L, Thompson DA, Zacks DN. 2015. Deletion of autophagy inducer RB1CC1 results in degeneration of the retinal pigment epithelium. Autophagy 11:939–953. 10.1080/15548627.2015.1041699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lu J, He L, Behrends C, Araki M, Araki K, Jun Wang Q, Catanzaro JM, Friedman SL, Zong W-X, Fiel MI, Li M, Yue Z. 2014. NRBF2 regulates autophagy and prevents liver injury by modulating Atg14L-linked phosphatidylinositol-3 kinase III activity. Nat Commun 5:3920. 10.1038/ncomms4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wu M-Y, Liu L, Wang E-J, Xiao H-T, Cai C-Z, Wang J, Su H, Wang Y, Tan J, Zhang Z, Wang J, Yao M, Ouyang D-F, Yue Z, Li M, Chen Y, Bian Z-X, Lu J-H. 2021. PI3KC3 complex subunit NRBF2 is required for apoptotic cell clearance to restrict intestinal inflammation. Autophagy 17:1096–1111. 10.1080/15548627.2020.1741332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Carpenter EA, Ruby J, Ramshaw IA. 1994. IFN-gamma, TNF, and IL-6 production by vaccinia virus immune spleen cells. An in vitro study. J Immunol 152:2652–2659. [PubMed] [Google Scholar]
- 77.Fields M, Zheng M, Zhang M, Atherton SS. 2006. Tumor necrosis factor alpha and macrophages in the brain of herpes simplex virus type 1-infected BALB/c mice. J Neurovirol 12:443–455. 10.1080/13550280601039030. [DOI] [PubMed] [Google Scholar]
- 78.Zelova H, Hosek J. 2013. TNF-alpha signalling and inflammation: interactions between old acquaintances. Inflamm Res 62:641–651. 10.1007/s00011-013-0633-0. [DOI] [PubMed] [Google Scholar]
- 79.Cousins SW, Espinosa-Heidmann DG, Miller DM, Pereira-Simon S, Hernandez EP, Chien H, Meier-Jewett C, Dix RD. 2012. Macrophage activation associated with chronic murine cytomegalovirus infection results in more severe experimental choroidal neovascularization. PLoS Pathog 8:e1002671. 10.1371/journal.ppat.1002671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tormanen K, Wang S, Jaggi U, Ghiasi H. 2020. Restoring herpesvirus entry mediator (HVEM) immune function in HVEM−/− mice rescues herpes simplex virus 1 latency and reactivation independently of binding to glycoprotein D. J Virol 94:e00700. 10.1128/JVI.00700-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.National Research Council. 1996. Guide for the care and use of laboratory animals. National Academies Press, Washington, DC. [Google Scholar]
- 82.Macdonald SJ, Mostafa HH, Morrison LA, Davido DJ. 2012. Genome sequence of herpes simplex virus 1 strain McKrae. J Virol 86:9540–9541. 10.1128/JVI.01469-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ghiasi H, Slanina S, Nesburn AB, Wechsler SL. 1994. Characterization of baculovirus-expressed herpes simplex virus type 1 glycoprotein K. J Virol 68:2347–2354. 10.1128/jvi.68.4.2347-2354.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yang M, Song L, Wang L, Yukht A, Ruther H, Li F, Qin M, Ghiasi H, Sharifi BG, Shah PK. 2018. Deficiency of GATA3-positive macrophages improves cardiac function following myocardial infarction or pressure overload hypertrophy. J Am Coll Cardiol 72:885–904. 10.1016/j.jacc.2018.05.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jaggi U, Matundan HH, Tormanen K, Wang S, Yu J, Mott KR, Ghiasi H. 2020. Expression of murine CD80 by HSV-1 in place of LAT can compensate for latency-reactivation and antiapoptotic functions of LAT. J Virol 94:e01798-19. 10.1128/JVI.01798-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Allen SJ, Hamrah P, Gate D, Mott KR, Mantopoulos D, Zheng L, Town T, Jones C, von Andrian UH, Freeman GJ, Sharpe AH, BenMohamed L, Ahmed R, Wechsler SL, Ghiasi H. 2011. The role of LAT in increased CD8+ T cell exhaustion in trigeminal ganglia of mice latently infected with herpes simplex virus type 1. J Virol 85:4184–4197. 10.1128/JVI.02290-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ghiasi H, Kaiwar R, Nesburn AB, Slanina S, Wechsler SL. 1994. Expression of seven herpes simplex virus type 1 glycoproteins (gB, gC, gD, gE, gG, gH, and gI): comparative protection against lethal challenge in mice. J Virol 68:2118–2126. 10.1128/JVI.68.4.2118-2126.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ghiasi H, Bahri S, Nesburn AB, Wechsler SL. 1995. Protection against herpes simplex virus-induced eye disease after vaccination with seven individually expressed herpes simplex virus 1 glycoproteins. Invest Ophthalmol Vis Sci 36:1352–1360. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Download jvi.01401-22-s0001.xlsx, XLSX file, 0.1 MB (55.4KB, xlsx)