

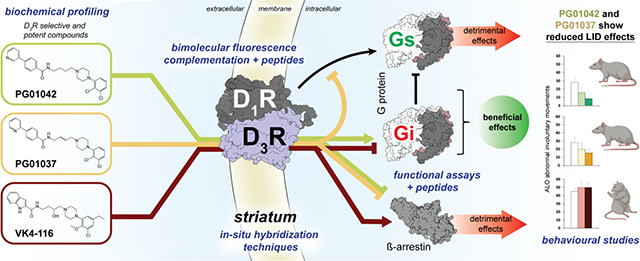
Abstract
A main rationale for the role of G protein-coupled receptor (GPCR) heteromers as targets for drug development is the putative ability of selective ligands for specific GPCRs to change their pharmacological properties upon GPCR heteromerization. The present study provides a proof of concept for this rationale by demonstrating that heteromerization of dopamine D1 and D3 receptors (D1R and D3R) influences the pharmacological properties of three structurally similar selective dopamine D3R ligands, the phenylpiperazine derivatives PG01042, PG01037 and VK4-116. By using D1R-D3R heteromer-disrupting peptides, it could be demonstrated that the three D3R ligands display different D1R-D3R heteromer-dependent pharmacological properties: PG01042, acting as G protein-biased agonist, counteracted D1R-mediated signaling in the D1R-D3R heteromer; PG01037, acting as a D3R antagonist cross-antagonized D1R-mediated signaling in the D1R-D3R heteromer; and VK4-116 specifically acted as a ß-arrestin-biased agonist in the D1R-D3R heteromer. Molecular dynamics simulations predicted potential molecular mechanisms mediating these qualitatively different pharmacological properties of the selective D3R ligands that are dependent on D1R-D3R heteromerization. The results of in vitro experiments were paralleled by qualitatively different pharmacological properties of the D3R ligands in vivo. The results supported the involvement of D1R-D3R heteromers in the locomotor activation by D1R agonists in reserpinized mice and L-DOPA-induced dyskinesia in rats, highlighting the D1R-D3R heteromer as a main pharmacological target for L-DOPA-induced dyskinesia in Parkinson’s disease. More generally, the present study implies that when suspecting its pathogenetic role, a GPCR heteromer, and not its individual GPCR units, should be considered as main target for drug development.
Keywords: G protein-coupled receptor (GPCR) heteromers, dopamine D1 receptor, dopamine D3 receptor, locomotor activation, L-DOPA-induced dyskinesia, mouse, rat
Graphical Abstract
1. Introduction
Dopamine receptors are classically defined as D1-like receptors, which includes D1 and D5 receptors (D1R and D5R), and D2-like receptors, which includes D2, D3 and D4 receptors (D2R, D3R and D4R) [1]. Classically, the psychomotor activity induced by stimulation of the dopaminergic system has been considered secondary to co-activation of segregated D1R and D2R, respectively expressed by the GABAergic striato-nigral and striato-pallidal neurons [2,3]. The discovery of D3R and their predominant localization in striato-nigral neurons of the ventral striatum [4], and the demonstration of functional and molecular interactions between striatal D1R and D3R disclosed a more direct cooperation between D1-like and D2-like receptors, the D1R-D3R heteromer [5–8]. The D1R-D3R heteromer seems to be constituted by D1R and D3R homomers coupled to Gs and Gi proteins, respectively [7,8]. D3R activation in the D1R-D3R heteromer biases signaling away from D1R-Gs-mediated activation of adenylyl cyclase (AC) from a preferential G protein-dependent AC-mediated signaling toward a G protein-independent signaling [7,8]. This switch is associated with a synergistic effect of D1R and D3R agonists on ß-arrestin-dependent signaling [7], which translates into a synergistic locomotor activation [7,8].
Upregulation of otherwise sparsely expressed D3R in the dorsal striatum seems to be a main neurochemical mechanism associated with a side effect of chronic dopamine replacement therapy for Parkinson’s disease known as L-DOPA-induced dyskinesia (LID) [9–14]. It was then suggested that this implies an increase in D1R-D3R heteromers with the concomitant synergistic effect secondary to D1R and D3R co-activation [15]. This hypothesis was strongly supported by results of experiments with a rat model of LID, which consists of analysis of abnormal involuntary movements in the rat with unilateral dopamine denervation and chronic L-DOPA treatment [16]. Hence, LID-like behavior in rats could be elicited by a D1R agonist and by a preferential D3R agonist; co-administration of threshold doses of both agonists induced a synergistic effect [17]. Furthermore, the development of LID-like behavior could be significantly attenuated by the specific genetic suppression of D3R in the D1R-expressing cells of the dorsal striatum while L-DOPA efficacy was preserved [13].
These studies highlight D1R-D3R heteromers in the dorsal striatum as promising pharmacological targets for LID. In fact, we previously demonstrated the efficacy of two similar phenylpiperazine derivatives and very selective D3R ligands, PG01037 and PG01042, in reducing LID-like behavior in rats [18–19]. However, the two compounds demonstrated a very different pharmacological profile in cells transfected with D3R. While PG01037 behaved as an antagonist of D3R agonist-induced AC inhibition and mitogenesis, PG01042 behaved as an agonist at inhibiting AC and as a weak partial agonist at activating mitogenesis [19]. Therefore, the conundrum of how these two selective D3R ligands could be pharmacologically different in vitro and yet promote the same in vivo pharmacological response had to be resolved. Hence, the observation that both PG01037 and PG01042 were able to counteract LID-like behavior in rats can be explained by their ability to counteract D1R-mediated signaling in the D1R-D3R heteromer by means of different allosteric mechanisms. Moreover, the inability of another phenylpiperazine derivative and selective D3R ligand, VK4-116 [20], to counteract and even potentiate LID-like behavior in rats is associated with its inability to counteract D1R signaling and its ability to specifically promote ß-arrestin recruitment by the D1R-D3R heteromer. Computational models of these ligands bound to D3R alone and upon oligomerization with the D1R provide potential molecular mechanisms involved in the D1R-D3R heteromerization-dependent differential pharmacological effects of these three D3R ligands. Therefore, the present study strongly supports D1R-D3R heteromers, and not D1R or D3R, as main therapeutic targets in LID.
2. Materials and Methods
2.1. Expression vectors and fusion proteins
The human cDNAs for D1R and D3R, cloned into pcDNA3.1, were amplified without their stop codons using sense and antisense primers harboring unique EcoRI and BamHI restriction sites. The amplified fragment corresponding to D1R was subcloned to be in-frame with restriction sites of pcDNA3.1-Rluc, pcDNA3.1-cYFP, or pcDNA3.1-nYFP to obtain plasmids that express D1R fused to Renilla luciferase (D1R-Rluc) or to complementary halves of the yellow fluorescence protein (YFP) on the C-terminal end of the receptor (D1R-cYFP, or D1R-nYFP). The amplified fragment corresponding to D3R was subcloned to be in-frame with EcoRI and BamHI restriction sites of pcDNA3.1-YFP, pcDNA3.1-cYFP or pcDNA3.1-nYFP to obtain plasmids that express D3R fused to YFP or to complementary halves of YFP. The receptor-fusion protein expression and function was tested by confocal microscopy and signaling, including extracellular signal-regulated kinases 1 and 2 phosphorylation and cAMP production, as described previously [6,21]. Human β-arrestin-1-Rluc6, cloned in the pcDNA3.1 Rluc6 vector (pRluc-N1 PerkinElmer, Wellesley, MA), was generously given by Dr. Marian Castro from Santiago de Compostela University, Spain.
2.2. Cell culture and transfection
HEK-293T cells were grown in Dulbecco’s modified Eagle’s medium (Gibco, Paisley, Scotland) supplemented with 2 mM L-glutamine, 100 U/ml penicillin/streptomycin, 100 μg/ml sodium pyruvate, minimum essential medium nonessential amino acids solution (1/100), and 5% (v/v) heat-inactivated fetal bovine serum (Invitrogen, Carlsbad, CA) and cells were maintained at 37 °C in an atmosphere of 5% CO2. HEK-239T cells growing in 6-well dishes were transiently transfected with the corresponding fusion protein cDNA by the polyethylenimine method, as described in detail elsewhere [22]. Sample protein concentration was determined using a Bradford assay kit (Bio-Rad, Munich, Germany) and bovine serum albumin dilutions as standards.
2.3. HIV TAT fused-TM peptides
Synthetic peptides with the amino acid sequence of transmembrane domains (TMs) of the D1R were used as heteromer-disrupting tools [7,8,23–25]. Peptides are fused to the cell-penetrating HIV trans-activator of transcription (TAT) peptide (YGRKKRRQRRR) to allow their right orientation when inserted in the plasma membrane [23,24,26]. To obtain the right orientation of the membrane-inserted peptide, the TAT peptide was fused to the C-terminus of peptides with the amino acid sequence of TM 5, and TM 7 of D1R or the D3R (TM5, and TM7 peptides, respectively) or to the N terminus of TM 4, and TM 6 of D1R or the D3R (TM4, and TM6 peptides, respectively). All peptides were synthesized by Genemed Synthesis, Inc. Their sequences were as follows:
YGRKKRRQRRRAAFILISVAWTLSVLISFIPVQLSW for TM4 of D1R,
TYAISSSLISFYIPVAIMIVTYTSIY YGRKKRRQRRR for TM5 of D1R,
YGRKKRRQRRRTLSVIMGVFVCCWLPFFISNCMVPFCG for TM6 of D1R,
FDVFVWFGWANSSLNPIIYAFNADFYGRKKRRQRRR for TM7 of D1R,
YGRKKRRQRRRVALMITAVWVLAFAVSCPLLF for TM4 of D3R,
FVIYSSVVSFYLPFGVTVLVYAYGRKKRRQRRR for TM5 of D3R,
YGRKKRRQRRRMVAIVLGAFIVCWLPFFLTHVL for TM6 of D3R and
ATTWLGYVNSALNPVIYTTFYGRKKRRQRRR for TM7 of D3R.
2.4. Bimolecular fluorescence complementation (BiFC)
HEK-293T cells were transiently co-transfected with the cDNA encoding the corresponding receptor fused to n-YFP and the receptor fused to c-YFP. The amount of transfected cDNA was 4 μg for each construct. After transfection (48 h), cells were treated or not with the indicated TM peptides (4 μM) for 4 h at 37 °C. Reconstituted YFP expression was quantified by distributing the cells (20 μg protein) in 96-well microplates (black plates with a transparent bottom, Porvair, King’s Lynn, UK), and emission fluorescence at 530 nm was read in a Fluo Star Optima Fluorimeter (BMG Labtech) equipped with a high-energy xenon flash lamp, using a 10 nm bandwidth excitation filter at 400 nm reading. Protein fluorescence expression was determined as fluorescence of the sample minus the fluorescence of cells not expressing the fusion proteins (basal). Cells expressing D1R-cYFP and nYFP or D3R-nYFP and cYFP showed similar fluorescence levels to non-transfected cells.
2.5. cAMP formation
To determine cAMP formation, homogeneous time-resolved fluorescence resonance energy transfer (TR-FRET) assays were performed using the Lance Ultra cAMP kit (PerkinElmer, Waltham, MA), based on competitive displacement of a europium chelate-labelled cAMP tracer bound to a specific antibody conjugated to acceptor beads. The optimal cell density for an appropriate fluorescent signal was first established by measuring the TR-FRET signal determined as a function of forskolin concentration using different cell densities. Forskolin dose-response curves were related to the cAMP standard curve to establish a cell density with a response covering most of the dynamic range of the cAMP standard curve. The cAMP formation experiments were performed in HEK-293T cells 48 h after transfection with the indicated amounts of cDNA corresponding to D1R-Rluc and D3R-YFP. Cells were not treated or treated with vehicle or 4 μM of the indicated TM peptides for 4 h at 37 °C in an atmosphere of 5% CO2. Cells were then grown (1000 cells/well) in white ProxiPlate 384-well microplates (PerkinElmer, Waltham, MA) in medium containing 50 μM zardaverine and were pretreated with the antagonists or the corresponding vehicle at 25°C for 20 min and stimulated with agonists for 10 min before adding 0.5 μM forskolin or vehicle and incubated for an additional 15-min period. Fluorescence at 665 nm was analyzed on a PHERAstar Flagship microplate reader equipped with an HTRF optical module (BMG Labtech, Offenburg, Germany).
2.6. β-arrestin recruitment
β-arrestin-1 recruitment was determined by bioluminescence resonance energy transfer (BRET) experiments in HEK-293T cells 48 h after transfection with the indicated amounts of cDNA corresponding to D1R, D3R-YFP, and β-arrestin-1-Rluc. Cells (20 mg total protein from a cell suspension per well in 96-well microplates) were not treated or treated for 10 minutes with the indicated antagonists, and 5 μM coelenterazine H was added before stimulation with the agonist for 7 minutes. BRET readings were collected using a Mithras LB 940 (Berthold Technologies, Oak Ridge, TN) that allows the integration of the signals detected in the short-wavelength filter at 485 nm (440–500 nm) and the long-wavelength filter at 530 nm (510–590 nm). The net BRET is defined as [(long-wavelength emission)/(short-wavelength emission)] - Cf, where Cf corresponds to [(long-wavelength emission)/(short wavelength emission)] for the donor construct expressed alone in the same experiment. BRET is expressed as milli-BRET units (net BRET × 1000). One-way ANOVA followed by Tukey’s post hoc comparisons was used for all the in vitro experiments (see figure legends for statistical results), Prism 9 (GraphPad Software, San Diego CA) was used to carry out all statistical analyses.
2.7. Locomotor activity in reserpinized mice
2.7.1. Reserpinization and treatment with dopamine receptor ligands
Male C57BL/6 mice (Charles River Laboratories, Wilmington, MA), experimentally naïve at the start of the study and weighing 20–25 g, were used. All animals were maintained in accordance with the National Institutes of Health Guide for the Care and use of Laboratory Animals. The research project involving the present experiments in mice was first reviewed and approved by the National Institute on Drug Abuse Intramural Program Animal Care and Use Committee (protocol # 15-BNRB-73). Reserpine (Tocris Bio-Techne, Minneapolis, MN) was dissolved in a drop of glacial acetic acid and made up to volume with 5.5% glucose and administered subcutaneously. The dose of reserpine (5 mg/kg) has been previously shown to produce pronounced striatal dopamine depletion in mice (more than 95% depletion) [2]. Mice were reserpinized 20 h prior to the start of the locomotor activity recording. The drugs to be tested were prepared in sterile saline and administered intraperitoneally. The volume of injection was 10 ml/kg for all drugs. The selective D1R agonist SKF81297 and the non-selective D2-like receptor agonist pramipexole were from Tocris Bio-Techne. The selective D3R ligands, PG01037 (N-{4-[4-(2,3-dichlorophenyl)-piperazin-1-yl]-trans-but-2-enyl}−4-pyridine-2-yl)benzamide) [18,27], PG01042 ((N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butyl)-4-(pyridin-3-yl)benzamide HCl) [19], and VK4-116 ([R]-N-[4-(4-[3-chloro-5-ethyl-2-methoxyphenyl]piperazin-1-yl)-3-hydroxybutyl]-1H-indole-2-carboxamide) [20] were synthesized at NIDA IRP. Vehicle was saline for all the drugs except VK4-116 and PG1042, which was 5% DMSO and 5% Tween 80 in sterile water, respectively.
2.7.2. Measurement of locomotor activity
Locomotor activity was recorded immediately after the animals were introduced in the activity chambers with 42.0 × 42.0 cm open fields (Coulbourn Instruments, Allentown, PA), after the administration of SKF81297 and/or pramipexole. The D3R selective compounds were administered 15 min before SKF81297 and/or pramipexole. All values registered per 10 min were transformed (square root) to reduce the asymmetric statistical distribution of the recorded raw data (skewness to the right) and decrease significant differences in the variances between differently treated groups [28]. The average of the results obtained from the 10 min-periods recorded for 1 h was used for calculations. One-way ANOVA followed by Tukey’s post hoc comparisons was used to compare the results of different groups (see figure legends for statistical results), Prism 9 (GraphPad Software) was used to carry out the statistical analyses.
2.8. Expression of dopamine receptor mRNA
RNAscope in situ hybridization was used to image DRD1, DRD2 and DRD3 mRNA. Mouse brains were extracted by rapid decapitation and immediately submerged in 2-methyl-butane to be then stored in −80°C until ready for use. Midbrain coronal sections were collected at 16-μm thickness onto Superfrost Plus slides to be then dehydrated in graduated ethanol (PBS, 50%, 70% and 100% ethanol) and process according to the RNAscope manual assay protocol (ACD Bio-Techne, Nevark, CA). Using RNAscope Reagent Kit (ACD Bio-Techne) the midbrain sections were incubated first in Protease-Pretreat 4 solution (RT, 20 min), rinsed twice in dH20, and then treated with Mm-DRD1 (ACD Biotechne, cat. # 461901-2), Mm-DRD2 (ACD Biotechne; cat. # 406501-C3) and Mm-DRD3 (ACD Biotechne; cat # 447721) probes for 2 h at 40°C. After double rinse in wash buffer, the sections were treated with AMP 1 (30 min, 40°C), AMP 2 (15 min, 40°C), AMP 3 (30 min, 40°C), and AMP 4 Alt A (15 min, 40°C) with double rinse in PB between each amplification step. Next, a drop of DAPI was added to each slide, followed by fluorescent mounting medium (Fluoro-Gel; Electron Microscopy Sciences, Hatfield, PA; cat. # 17985,) and coverslip. Images were taken on a Keyence BZ-X800 fluorescence microscope (Keyence, Iyasca, IL) from 3 sections at 60X magnification. The number of 4′,6-diamidino-2-phenylindole (DAPI) positive cells, expressing D3R and D1R or D3R and D2R were counted at 60X magnification using Image J software (NIH, public domain).
2.9. L-DOPA-induced dyskinesia (LID) in rats
2.9.1. Establishment of LID
Adult male (M) and female (F) Sprague-Dawley rats (16M, 4F) were used throughout the experiment (N = 20). All animals were maintained in accordance with the National Institutes of Health Guide for the Care and use of Laboratory Animals and with the guidelines of the Institutional Animal Care and Use Committee of Binghamton University. All rats underwent a unilateral dopamine neuronal lesion with 6-hydroxydopamine hydrobromide (6-OHDA; Sigma, St Louis, MO, USA) infused in the left medial forebrain bundle (MFB), a procedure previously described [13,29]. In short, rats were administered analgesic Buprenex (buprenorphine HCl; 0.03 mg/kg, i.p.; Reckitt Benckiser Pharmaceuticals Inc, Richmond, VA) prior to surgery and were anesthetized with inhalant isoflurane (2–4%; Sigma) in oxygen (2.5 l/min) and placed in a stereotaxic apparatus (David Kopf Instruments, Tujunga, CA) with the incisor bar positioned at −5.0 mm relative to the interaural line. The targeted site, relative to bregma was: AP, −1.8 mm; ML −2.0 mm; DV, −8.6 mm. A 10 μl Hamilton syringe attached to a 26-gauge needle was lowered into the target through a small hole in the skull and then used to deliver 6-OHDA (3 μg/μl) dissolved in 0.9% NaCl + 0.1% ascorbic acid at a rate of 2 μl/min, for a total volume of 4 μl. The needle was withdrawn 5 min after injection to allow for diffusion from the injection site. To minimize post-surgery pain and discomfort, analgesic Rimadyl (carprofen; Zoetic Inc., Kalamazoo, MI) was administered twice on a 6–12 h interval. After surgery, rats were pair-housed in pre-warmed, clean cages and were monitored for a post-operative period of 10 days in which they received soft food, fruit, and fluid replacement as needed to facilitate recovery. All experiments began 3 weeks post-surgery to allow sufficient recovery time. Three weeks after surgery all lesioned rats (N=16; 12M, 4F) received a daily subcutaneous injection of L-DOPA methyl ester (6 mg/kg; Sigma) + DL-serine 2-(2,3,4-trihydroxybenzyl) hydrazide hydrochloride (benserazide; 15 mg/kg; Sigma), dissolved in vehicle (0.9% NaCl + 0.1% ascorbic acid), for a 2-week priming period to establish stable LID expression [30–32].
2.9.2. Treatments with D3R ligands
Following confirmation of stable LID, dyskinetic rats (n=15; 11M, 4F) entered a counterbalanced within-subjects design where the expression of LID was evaluated following treatment with the D3R-selective compounds PG01037, PG01042 and VK4-116. PG01037 (0, 10, 30 mg/kg; i.p.) and PG01042 (0, 5, 10 mg/kg; i.p.) were prepared in 25% DMSO (VWR, Radnor, PA, USA) in sterile distilled water and administered at 1 ml/kg. To improve solubility, prepared doses of PG01037 and PG01042 were maintained at ~37°C in a water bath until injection immediately prior to L-DOPA (6 mg/kg; s.c.). VK4-116 (0, 10, 30 mg/kg; i.p.) was prepared in 10% DMSO and 15% Tween-80 (Sigma) in sterile distilled water, with the 30 mg/kg dose prepared at a concentration of 15 mg/ml and injected at 2 ml/kg due to solubility constraints. VK4-116 was administered 15 min prior to L-DOPA. Washout periods of at least 2 days interspersed priming day 14 and the first acute challenge and all subsequent acute dose response testing. All rats meeting Abnormal involuntary movements (AIMs) criteria (AIMs >10, N=15) entered a counterbalanced within-subjects design to determine dose response curves for each compound. In the first experiment, rats (n=8; all male) were administered either PG01037 (0, 10, 30 mg/kg; i.p.) or PG01042 (0, 5, 10 mg/kg; i.p) 5 min prior to L-DOPA (6 mg/kg, s.c.). In the second experiment, rats (n=7; 3M, 4F) were administered VK4-116 (0, 10, 30 mg/kg; i.p.) 15 min prior to L-DOPA. Abnormal involuntary movements (AIMs) were rated were quantified on each day of treatment beginning 10 min post-injection of L-DOPA. Washout periods of at least 2 days were interspersed between each test day. Following the completion of all test days, rats were sacrificed off-treatment after a washout period of at least 2 days.
2.9.3. Measurement of abnormal involuntary movements (AIMs)
Rats were monitored for development and expression of rodent dyskinesia using the AIMs rating scale which has been validated pharmacologically through the administration of known anti-dyskinetic compounds [33]. Rats were placed in clear plexiglass cylinders (20 cm in diameter × 25 cm in height) after L-DOPA injection and a trained observer blind to experimental condition rated axial, limb, and orolingual (ALO) behaviors beginning 10 minutes post-injection. ALO behaviors were rated for 60 sec every 10 min for a total duration of 180 min, a procedure previously described [33,34]. Axial AIMs are characterized by dystonic twisting of the trunk contralateral to lesion. Limb AIMs are choreic movement of the fist and forelimb contralateral to lesion. Orolingual AIMs consist of tongue protrusions and jaw tremors. Each ALO behavior is rated on a scale of 0–4: 0, behavior not present; 1, behavior present for <30 sec; 2, behavior present for ≥30sec; 3, behavior present for 60 sec but can be interrupted by a tap on the cylinder; 4, behavior present for 60 sec but cannot be interrupted by an extraneous stimulus. All rats in all experiments went through the same 2-week L-DOPA priming period, and were rated for AIMs on days 1, 8, and 14 of daily L-DOPA treatment to measure the development of LID. On day 14, all animals were required to meet a cumulative ALO sum >10 to continue throughout the study [35].
2.9.4. Measurement of forepaw adjusting steps (FAS)
The FAS test was used as a measure of forepaw akinesia used to evaluate baseline motor impairment consequent to the MFB lesion [16] and was performed as previously described [29,36]. In brief, rats were restrained by a trained experimenter so that both hindlegs and one forelimb were secured. Rats were then moved laterally across a table at a speed of 90 cm/10sec during which an additional experimenter recorded the number of steps taken on both the lesioned and intact paw in both the forehand and backhand direction. FAS serves a proxy for lesion success as rats showing >80% striatal dopamine depletion perform poorly on this test [37]. Forehand percent intact was calculated by dividing the total right forepaw steps by total forepaw steps and multiplying by 100%. Animals showing forehand percent intact values >25% were considered unsuccessfully lesioned and excluded from further study.
2.9.5. Neurochemical analysis
After a washout period of at least 2 days, all animals were rapidly euthanized and brains were collected and flash-frozen on 2-methylbutane kept at approximately −20°C. Brains were cut on a cryostat, and a circular punch of 2 mm diameter × 1.5 mm depth was collected between approximately +0.70 mm and −0.80 mm from bregma from the left and right dorsomedial striatum. Brain tissue punches were stored at −80°C in 1.6 mL microcentrifuge tubes for later analysis. Lesions were confirmed post-mortem in all rats using reverse phase HPLC coupled to electrochemical detection to evaluate striatal dopamine and 3,4-dihydroxyphenylacetic acid (DOPAC) levels as described in prior work [35,36]. The limit of detection was 0.1 nM for monoamines and their metabolites. Tissue homogenate sample oxidation current values were plotted against a standard curve with known concentrations varying from 0.001 to 1.0 nM and were normalized to tissue weight. Monoamine levels are expressed as pg of monoamine per mg of tissue.
2.9.6. Statistical analyses
AIMs data (expressed as median + median absolute deviation; M.A.D.) was analyzed by non-parametric statistics. For all dose-response testing, within-subjects designs were utilized, and the Friedman test was employed with Wilcoxon Match Pairs post-hoc tests. All HPLC monoamine and metabolite data (mean pg/mg of tissue ± S.E.M.) were analyzed using paired t tests. One data point for striatal DOPAC was undetectable and not included. Analyses for all LID experiments in rats were performed with SPSS software (Chicago, IL).
2.10. Computational methods
The structure of inactive D3R bound to eticlopride (PDB id 3PBL) [38] was used. The inactive structure of D1R was modeled from the active structure of D1R bound to a non-catechol agonist and Gs (7JOZ) [39] and the inactive structure of the ß1-adrenergic receptor (4BVN) [40] (see Suppl. Fig. 1 for details). Fusion proteins were removed, and stabilizing mutations were mutated to the native sequence. The D1R-D3R heteromer was built from the TM 5/6 dimeric interface observed in the crystal structure of μ-opioid receptor (4DKL) [41], as previously performed for the adenosine A1R-A2AR [42] and μ-opioid-Gal1 receptor heteromers [43]. PG01042, PG01037 and VK4-116 were modeled into the orthosteric binding cavity of D3R using the structure of eticlopride bound to D3R (3PBL) as a template, whereas the spacer and the second pharmacophore were oriented towards the extracellular domain as previously proposed [44,45]. The structures of the D3R protomer and the D1R-D3R heteromer in the apo form, with no ligand bound, and with PG01042, PG01037 and VK4-116, were embedded in a lipid bilayer box, constructed using PACKMOL-memgen [46], containing 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine, water molecules, and monoatomic Na+ and Cl− ions. Molecular dynamic (MD) simulation of these systems were performed with GROMACS 2019 [47] using the protocol previously reported [49]. The analysis of the trajectories was performed using MDAnalysis [49] and GetContacts (https://getcontacts.github.io/).
3. Results
3.1. Effect of PG01042, PG01037 and VK4-116 on G protein-dependent signaling in mammalian cells transfected with D3R and D1R.
We have previously shown that the selective D3R ligands PG01042, PG01037, and VK4-116 have different pharmacological profiles in cells transfected only with D3R [18,19,27]. PG01042 behaved as an agonist at inhibiting AC, whereas PG01037 and VK4-116 behaved as effective antagonists. To analyze the properties of these compounds in the D1R-D3R heteromer, we first measured cAMP production in HEK-293T cells co-transfected with D1R, a Gs-coupled receptor, and D3R, a Gi-coupled receptor. In agreement with our previous results in cells with only D3R, the agonist PG01042 significantly counteracted the effect of forskolin, both at 10 and 100 nM, with the highest concentration (100 nM) being almost as effective as a high dose (30 nM) of the D3R-agonist pramipexole (at 30 nM) (D3Rago, Fig. 1a). On the other hand, PG01037 and VK4-116 were not able to decrease cAMP as anticipated from their antagonist profile (at 10 and 100 nM) (Fig. 1a). We next analyzed possible allosteric interactions in the D1R-D3R heteromer by comparing the effect of simultaneously adding the D1R agonist SKF81297 (at 30 nM) (D1Rago) and PG01037 or VK4-116 in cells transfected only with D1R (Fig. 1b) with those transfected with D1R and D3R (Fig. 1c). As expected, none of the selective D3R ligands had any effect in SKF81297-induced cAMP production in cells transfected with only D1R. In contrast, PG01042 significantly counteracted the SKF81297-mediated increase of cAMP, the canonical Gs-Gi antagonistic interaction at the AC level (type III allosterism, see ref. [50]). Using heteromer disrupting peptides, we previously demonstrated that D1R-D3R heteromerization is necessary to allow the canonical antagonistic Gs-Gi interaction at the AC level, by which a D3R agonist inhibits AC activation by a D1R agonist [8]. We, thus, measured cAMP levels in the presence of TM peptides with the amino acid sequence of TMs TM 6 and TM 7 of the D1R (TM6 and TM7 peptides) fused to an HIV-TAT sequence (see Materials and Methods). As expected, the canonical antagonistic interaction between SKF81297 and the PG01042 agonist was dependent on D1R-D3R heteromerization, since it was absent in cells only transfected with D1R (Fig. 1b) and in D1R-D3R transfected cells in the presence of the disrupting TM6 peptide (Fig. 1d), but not in the presence of the control TM7 peptide (Fig. 1e).
Figure 1. Differential effects of PG01042, PG01037, and VK4-116 on G protein-dependent signaling in HEK-293T cells transfected with D3R and D1R.
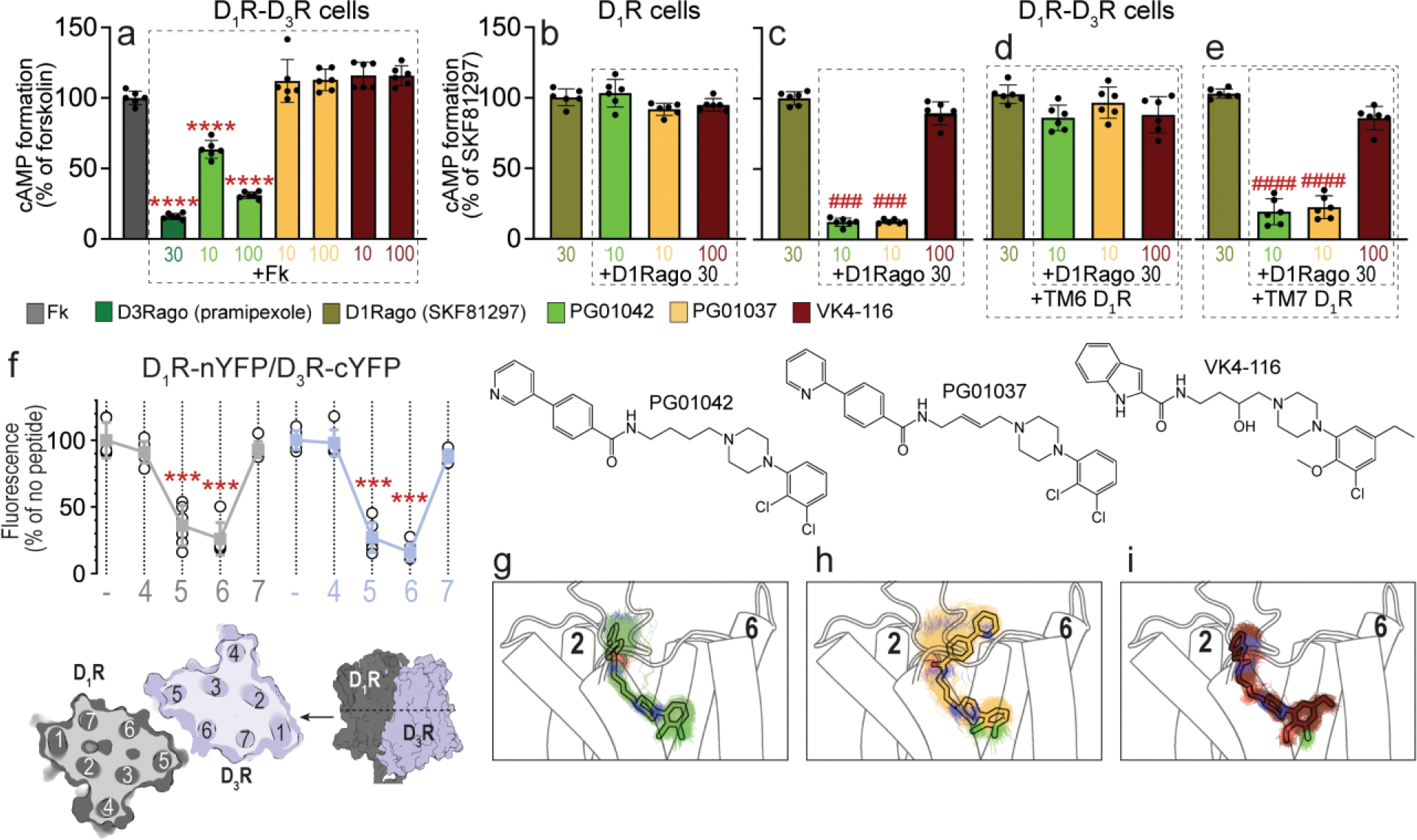
a-e. Results from cAMP formation experiments in HEK-293T cells transfected with D1R-Rluc cDNA (1 μg) with or without D3R-YFP cDNA (1.5 μg) (D1R-D3R cells and D1R cells, respectively). In a, cells are treated with the D2-like receptor agonist pramipexole (D3Rago; 30 nM for 10 min) or the D3R ligands PG01042, PG01037 and VK4-116 (all at 10 or 100 nM for 15 min) before forskolin (Fk, 0.5 μM). In b-e, cells are pre-treated or not with D1R TM6 or TM7 peptides (4 μM for 4 h) and treated with PG01042 (10 nM), PG01037 (10 nM) and VK4-116 (100 nM) for 15 min before the D1R agonist SKF81297 (30 nM; D1Rago). Values of cAMP formation are shown as mean ± S.D. (n = 6) and expressed as percentage of Fk-treated or D1Rago-treated cells in each condition (100% represents 80–100 pmols cAMP/106 cells). ****: p < 0.0001 versus FK; ### and ####: p < 0.001 and p < 0.0001 versus D1Rago, respectively (one-way ANOVA followed by Tukey’s post hoc comparisons). f. Results from BiFC experiments in HEK-293T cells co-transfected with D1R-nYFP and D3R-cYFP in the absence (−) or the presence of the indicated TM peptides (at 4 μM) from D1R (gray symbols and plots) or D3R (blue symbols and plots). Fluorescence values (in means ± S.D.) are expressed as the percentage of the fluorescence in the absence (−) of the indicated TM peptides (n = 6, with triplicates); ***: p < 0.001 versus control values (one-way ANOVA followed by Dunnett’s post hoc comparisons). The schemes illustrate extracellular and parallel to the membrane views of the computational model of the D1R-D3R heteromer built using the TM 5/6 interface (see text). g-i. Representative structures (solid sticks) and evolution (lines) of PG01042 (g, in green), PG01037 (h, in orange) and VK4-116 (i, in purple) in complex with D3R (white cylinders, only the initial structure is shown) as devised from three replicas of unbiased 1μs MD simulations. The second pharmacophore unit of PG01042 and VK4-116 remained stable at the ECD near TM 2, whereas this part of PG01037 favors its interaction with TM 6 (see Suppl. Fig. 2).
Another previously described allosteric property of the D1R-D3R heteromer is cross-antagonism, by which a D3R antagonist blocked D1R-mediated signaling [7] (type I allosterism, [50]). Thus, cross-antagonism requires a direct D1R-D3R interaction since antagonists do not signal on their own. Interestingly, PG01037, but not VK4-116, blocked the SKF81297-mediated increase of cAMP (Fig. 1c). The cross-antagonism of PG01037 was absent in cell expressing only D1R and in the presence of the disruptive TM6 peptide of D1R (Fig. 1d), but not with the TM7 control peptide. We have proposed a mechanism by which cross-antagonism could be explained within a heteromer that discloses a symmetrical TM 5/6 interface [51]. The antagonist of one of the protomers could favor a specific conformation of TM 5 and TM 6 that supports high surface complementarity with TM 5 and TM 6 of the partner receptor. This four-helix bundle interaction previously described in the μ-opioid receptor dimer [41] could block the opening of the intracellular cavity for G protein binding [51]. We previously proposed a symmetrical TM 5/6 interface of the D1R-D3R heteromer, based on the disrupting effects of TM5 and TM6 peptides of the D1R on BiFC and functional experiments [7,8]. We now performed additional BiFC experiments with TM peptides of both D1R and D3R. HEK-293T cells were co-transfected with D1R and D3R separately fused to complementary halves of the yellow fluorescent protein (N-terminal, D3R-nYFP, or C-terminal, D1R-cYFP). Confirming a symmetrical TM 5/6 interface, we observed a statistically significant fluorescence decrease with TM5 and TM6 of both D1R and D3R, but not with TM4 or TM7 peptides (Fig. 1f).
To understand the pharmacological differences among PG01042, PG01037, and VK4-116 at the molecular level, we first performed three replicas of unbiased 1 μs MD simulations (see Methods) of the D3R monomer in the presence of these compounds (Figs. 1g–1i, Suppl. Fig. 2). These phenylpiperazine compounds contain a primary and a second pharmacophore, linked by a spacer, which bind at the orthosteric binding pocket and at a secondary binding site (at the extracellular domain, ECD), respectively. Root-mean-square deviations (rmsd) of the simulations (Suppl. Fig. 2a) show that PG01042 and VK4-116 remained highly stable bound to the orthosteric and at a secondary pocket located at the extracellular part of TM 2 (see probability density plots of key distances in Suppl. Fig. 2b). In contrast, the second pharmacophore unit of PG01037 is unstable near TM 2 and moves toward TM 6 as shown by the large rmsd values and the probability density plots. This change is not due to the differential double bond located at the spacer (see dihedral angles plots at Suppl. Fig. 3) but the hydrogen bond interaction between PG01037 and Asn6.58 in TM 6. These different binding modes prompt a different conformation of the ECD and TM 6, which might therefore explain the respective presence and absence of cross-antagonism with PG01037 and VK4-116 binding.
3.2. Effect of PG01042, PG01037 and VK4-116 on β-arrestin in mammalian cells transfected with D3R and D1R.
The ability of PG01042, PG01037, and VK4-116 to promote ß-arrestin recruitment was evaluated in HEK-193T cells transiently transfected only with D3R (Fig. 2a) and co-transfected with both D1R and D3R (Figs. 2b–2c). The full D3R agonist pramipexole significantly recruited ß-arrestin in cell transfected with only D3R, but neither PG01037 or PG01042 (10 nM) or VK4-116 (100 nM) were effective (Fig. 2a). Notably, in cells co-expressing D1R and D3R, a significant ß-arrestin recruitment was selectively obtained with VK4-116, at 10 and 100 nM, which was ~60% the effect of a high concentration (30 nM) of pramipexole (Fig. 2b). Neither PG01037 or PG01042, at 10 or 100 nM promoted a significant ß-arrestin recruitment in D1R-D3R transfected cells. We also compared the effect of the three ligands (at 10 nM) alone and in combination with pramipexole (30 nM) on ß-arrestin recruitment from cells expressing D3R versus cells co-expressing D1R and D3R (Suppl. Fig. 4). PG01042 and PG01037 counteracted the effect of pramipexole in cells expressing D3R or both D1R and D3R, while VK4-116 only significantly counteracted pramipexole in D3R cells (Suppl. Fig. 4). Thus, VK4-116 antagonizes D3R agonist-induced ß-arrestin recruitment in D3R-expressing cells, but it behaves as a ß-arrestin biased agonist in cells co-expressing D1R, inducing ß-arrestin-recruitment when administered alone or combined with pramipexole. The experiments of VK4-116 on ß-arrestin-recruitment in cells co-expressing D1R and D3R were repeated in the presence of TM6 and TM7 peptides of D1R (Fig. 2c). Clearly, the ability of VK4-116 to promote ß-arrestin recruitment was specifically lost in the presence of the disruptive peptide TM6 of D1R, but not in the presence of the control peptide TM7 (Fig. 2c). This indicates that ß-arrestin recruitment induced by VK4-116 is dependent on D1R-D3R heteromerization. Therefore, we can conclude that in the D1R-D3R heteromer, PG01042 prompts a conformation of D3R that permits Gi binding (G protein-biased agonism), PG01037 prompts a conformation of D3R that permits high complementarity with D1R (cross antagonism), and VK4-116 prompts a conformation of D3R that permits ß-arrestin recruitment (ß-arrestin-biased agonist).
Figure 2. Differential effects of PG01042, PG01037, and VK4-116 on β-arrestin recruitment in HEK-293T cells transfected with D3R and D1R.
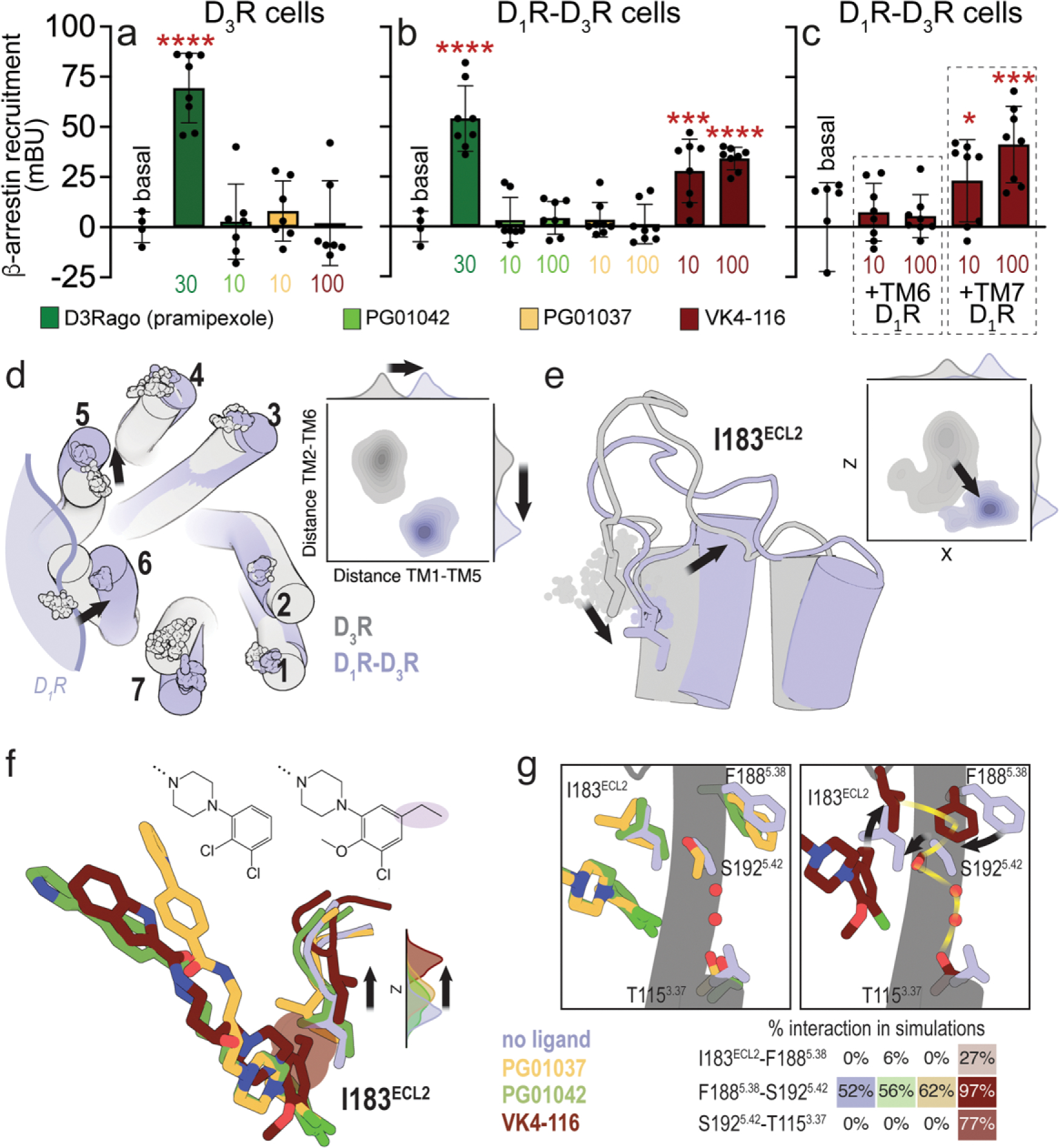
a-c. Results from β-arrestin recruitment experiments in HEK-293T cells transfected with β-arrestin-1-Rluc cDNA (0.5 μg), D3R-YFP cDNA (1 μg cDNA) with or without D1R cDNA (1.5 μg cDNA) (D1R-D3R cells and D3R cells, respectively). In a-b, cells are treated for 10 min with the D2-like receptor agonist pramipexole (D3Rago; 30 nM) or the D3R ligands PG01042, PG01037 and VK4-116 (10 or 100 nM). In c, cells are pre-treated or not with D1R TM6 or TM7 peptides (4 μM for 4 h) and with VK4-116 (10 or100 nM) for 15 min before the D1R agonist SKF81297 (30 nM; D1Rago). Coelenterazine H (5 μM) was added before pramipexole or the selective D3R ligands for 7 minutes and β-arrestin-1 recruitment was measured by BRET (see Material and Methods). Values are mean ± S.D. (n = 8). *, *** and ****: p < 0.05, p < 0.001 and p < 0.0001 versus basal, respectively (one-way ANOVA followed by Dunnett’s post hoc comparisons). d. Evolution of the Cα atoms (spheres) of Y321.35 in TM 1, L892.64 in TM 2, I1013.23 in TM 3, F1704.62 in TM 4, F1885.38 in TM 5, H3546.60 in TM 6, and P3627.32 in TM 7 during three replicas of unbiased 1 μs MD simulations of the D3R monomer (gray) and the D1R-D3R heteromer (blue) with no ligand bound. Contour plots of the distances between Y321.35 in TM 1 and F1885.38 in TM 5 (distance TM1-TM5) and between L892.64 in TM 2 and H3546.60 in TM 6 (distance TM2-TM6) during the MD simulations. Distributions of these distances are shown in the axes. e. Evolution of I183ECL2 (spheres), and contour plots and distributions of X,Z coordinates corresponding to the Cb atom of I183ECL2 during MD simulations. Black arrows represent the movements of D3R in the D1R-D3R heteromer (blue) relative to the D3R protomer (gray). f. Detailed view of I183ECL2 of D3R in the D1R-D3R heteromer during MD simulations with no ligand bound (blue) and PG01042 (green), PG01037 (orange) and VK4-116 (red) bound to D3R. Distributions of the Z coordinate corresponding to the Cα atom of I183ECL2 during MD simulations. The ethyl group of VK4-116 that triggers the upward movement of I183ECL2 is highlighted. g. Detailed views of I183ECL2, F1885.38, S1925.42, and T1153.37 of D3R in the D1R-D3R heteromer during MD simulations with no ligand bound (blue) and PG01042 (green), PG01037 (orange) and VK4-116 (red) bound to D3R. Frequency contacts (%) between side-chain residues, color-coded according to the ligand bound to D3R, as calculated with the GetContacts software. Black arrows represent the movements of these side chains of D3R relative to the unliganded D3R (blue). The xy plane is as defined by the Orientations of Proteins in Membranes (OPM) [73].
The mechanism of agonist-induced receptor activation and G protein binding is accurately characterized [52]. It includes a polar network in the receptor core (NPxxY and DRY motifs and Y5.58) that transmits the signal from the orthosteric ligand binding site to the G protein binding site via the highly conserved PIF motif. On the other hand, the mechanisms of agonist-imduce ß-arrestin recruitment and ß-arrestin-biased agonism are not fully characterized. Significant advances have shown several signatures recently reviewed by Bock & Bermudez [53]. First, bitopic ligands with extended binding modes toward the ECD might modulate binding pocket closure leading to ligand bias [54]. Second, in the analogous dopamine D2 receptor (D2R), the movement of residues Ile184ECL2 [55] and Phe1895.38 [56] has been related to arrestin-biased agonism. Third, the interactions of biased agonists such as formoterol or salmeterol with amino acids in the orthosteric binding site of ß1- or ß2- adrenergic receptors, respectively, are different from the interactions of full agonists [57,58]. And fourth, the polar network in the receptor core can also act as a biased signaling switch, adopting conformations that can activate ß-arrestin but not G protein signaling [59]. Because PG01037, PG01042, and VK4-116 are bitopic ligands interacting with amino acids at the ECD, we explored the conformation of the ECD and orientation of Ile183ECL2 (homologous to Ile184ECL2 in D2R), in MD simulations of the D3R monomer. Our simulations show no significant differences in the conformation of Ile183ECL2 among the ligands (Suppl. Fig. 2c), in agreement with the experimental data indicating that ß-arrestin recruitment is dependent on D1R-D3R heteromerization (Figs. 2a–2c).
Thus, to understand the ability of VK4-116 to recruit ß-arrestin we needed to perform similar MD simulations on the D1R-D3R heteromer that, in this case, requires computational modelling of the D1R/D3R interface (see Suppl. Fig. 1). MD simulations of the D1R-D3R heteromer, modelled via the TM5/6 interface (Fig. 1f), show that D1R significantly influences the conformation of D3R, mainly moving the extracellular part of TMs 5, 6, and 7 (Fig. 2d). Importantly, the movement of TM 5 relocated ECL2 and the position of the key Ile183ECL2 (Fig. 2e). Thus, in contrast to the simulations of the D3R protomer (Suppl. Fig. 2c), the VK4-116 biased agonist influences the conformation of Ile183ECL2 in the D1R-D3R heteromer, relative to the other ligands and apo-D3R (Fig. 2f). The ethyl group of VK4-116 forms a hydrophobic interaction with Ile183ECL2, as also proposed for biased agonists of D2R [55], which is absent in the other ligands. This specific interaction of VK4-116 with Ile183ECL2 moves inward the side chains conformations of Phe1885.38 and Ser1925.42 (Fig. 2g). This causes a polar hydrogen bond network, via specific water molecules, between Ser1925.42 and Thr1153.37, as observed in angiotensin II type 1 receptor in complex with the biased agonist RTV023 [59]. This binding mode of VK4-116 to the D1R-D3R heteromer may underlie its unique pharmacological properties as a biased agonist.
3.3. Behavioral profile of PG01042, PG01037 and VK4-116 in reserpinized mice
Locomotor activation in reserpinized mice is widely accepted as an animal model that allows the pharmacological evaluation of drugs acting directly or indirectly on postsynaptic striatal dopamine receptors, without the interfering influence of endogenous dopamine or presynaptic dopamine receptors [2,3,6]. In previous studies we have reported that the preferential D3R agonist PD128097, at a dose that does not produce locomotor activation in reserpinized mice, strongly potentiates the locomotor activation induced by D1R agonists (SKF38393 and SKF81297) [6,8]. We now reproduce these findings with pramipexole, another agonist with similar preferential affinity for D3R versus D2R than PD128097 (about 10 times higher affinity for D3R than D2R [60]). Pramipexole produced a dose-dependent locomotor activation starting to be significant at 3 mg/kg (Fig. 3a). As previously shown for PD128097, a low dose of pramipexole that was ineffective when administered alone (1 mg/kg) strongly potentiated the locomotor activation induced by SKF81297 (5 mg/kg) (Fig. 3a). It was then assumed that, at 1 mg/kg, pramipexole predominantly activates striatal postsynaptic D3R, while higher doses are necessary to significantly involve the striatal postsynaptic D2R.
Figure 3. Differential effects of PG01042, PG01037, and VK4-116 on the locomotor activation induced by D1R and D2-like receptor agonists in reserpinized mice.
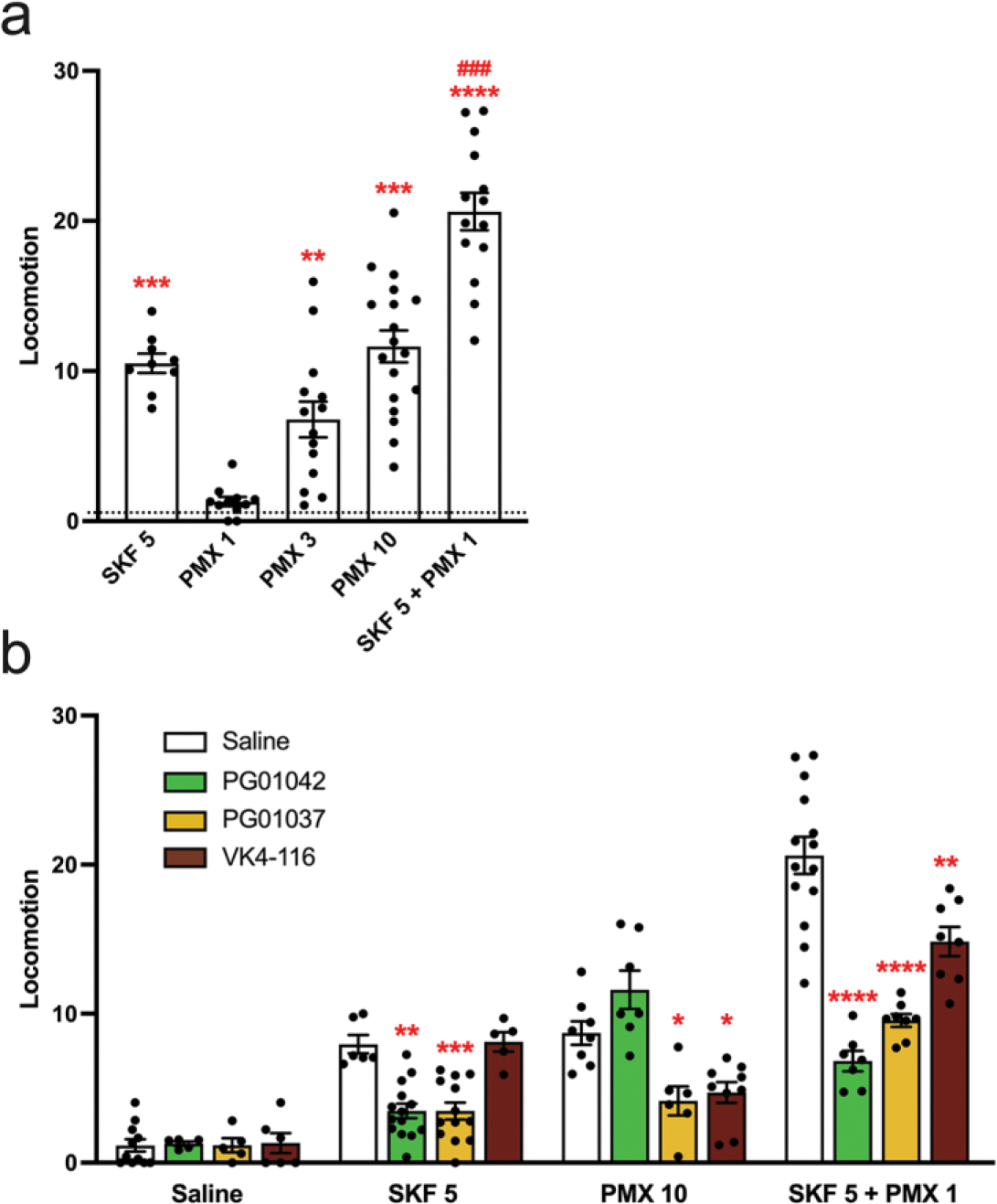
Mice were administered reserpine (5 mg/kg, s.c.) 20 h before administration of the D1R agonist SKF 81297 (SKF; 5 mg/kg, i.p.), the D2-like receptor agonist pramipexole (PMX; 1, 3 or 10 mg/kg, i.p.), or both, without (a) or with (b) previous administration (15 min before) of the D3R ligands PG01042 (10 mg/kg, i.p.), PG01037 (30 mg/kg, i.p.) or VK4-116 (10 mg/kg, i.p.). Values are mean ± S.E.M. (n = 5–18) and are expressed as the average of the transformed counts (squared root) obtained from the 10 min-periods recorded for 1 h. In a, **, *** and ****: p < 0.01, p < 0.001 and p < 0.0001 versus vehicle-treated animals (represented by the dotted line), respectively; ###: p < 0.001 versus SKF (one-way ANOVA followed by Tukey’s post hoc comparisons) (one-way ANOVA followed by Tukey’s post hoc comparisons). In b, *, **, *** and ****: p < 0.05, p < 0.01, p < 0.001 and p < 0.0001 versus respective saline-treated animals, respectively.
PG01042, PG01037 and VK4-116 were then analyzed for their ability to modify the basal locomotor activity in reserpinized mice and the locomotor activation induced by SKF81297 (5 mg/kg), by a high dose of pramipexole (10 mg/kg) and by the co-administration of SKF81297 (5 mg/kg) and the low dose of pramipexole (1 mg/kg) (Fig. 3b). The doses of PG01042 (10 mg/kg), PG01037 (30 mg/kg) and VK4-116 (10 mg/kg) were chosen from results of pilot experiments, based on their minimal doses with a maximal counteracting effect on the locomotor activation induced by SKF81297 (5 mg/kg) or pramipexole (10 mg/kg). In line with their differential biochemical profile, the three D3R ligands behaved in a different qualitative manner. PG01042 counteracted locomotor activation induced by SKF81297 and the synergistic effect of SKF81297 plus the low dose of pramipexole, but not the high dose of pramipexole; PG01037 counteracted SKF81297, by pramipexole and by the synergistic effect of SKF81297 plus the low dose of pramipexole; and VK4-116 counteracted pramipexole and the synergistic effect of SKF81297 plus the low dose of pramipexole, but not SKF81297 (Fig. 3b). Of importance, the effect of PG01037 or PG1042 at counteracting the locomotor activation induced by SKF81297 plus the low dose of pramipexole was more pronounced than the effect of VK4-116 (Fig. 3b).
3.4. Expression of DRD1, DRD2 and DRD3 mRNA in mouse striatum
Previous studies using radioligand-binding or immuno-histochemical techniques in C57BL/6 mice (strain also used in the present study), or genetically modified mice with the same background strain, identified a predominant localization of D3R in the ventral striatum [61]. However, co-localization experiments of D3R with D1R or D2R experiments were only performed in the dorsal striatum, where in fact the low-expressed D3R co-localized with D2R [12]. Nevertheless, in the dorsal striatum of mice rendered dyskinetic upon dopamine denervation and L-DOPA treatment, upregulation of D3R predominantly occurred in D1R-expressing neurons [12]. Using in situ hybridization techniques (RNAscope ISH), we recently obtained similar qualitative results in the dorsal striatum of L-DOPA-induced dyskinetic rats. However, in this case, D3R was found to separately co-localize with D1R and D2R in the dorsal striatum of naïve animals [13]. RNAscope ISH experiments were then performed to simultaneously determine D1R-D3R and D2R-D3R colocalization in the dorsal and ventral striatum of C57BL/6 mice. The qualitative analysis showed that DRD1, DRD2 and DRD3 mRNA are expressed in the dorsal and ventral striatum (Fig. 4). Very little, if any, co-localization of DRD1 and DRD2 mRNA was observed in either the dorsal or ventral striatum. As previously reported [61], DRD3 was significantly more expressed in the ventral than in the dorsal striatum. In the ventral striatum (Fig. 4a), most cells expressing DRD1 also expressed DRD3 mRNA and fewer cells also co-expressed DRD2 and DRD3 mRNA (white arrows). In the dorsal striatum (Fig. 4b), the lower number of cells expressing DRD3 mRNA also preferentially co-expressed DRD1. These results therefore confirm that, in the ventral striatum, D3R are highly co-expressed with D1R and that a less substantial but still evident D2R-D3R co-localization also exists.
Figure 4. D1R, D2R, and D3R gene (DRD1, DRD2 and DRD3) expression in the mouse striatum.

a. RNAscope ISH results in the ventral striatum (framed area in the coronal section) showing strong expression of DRD1, DRD2 and DRD3 mRNA with a strong co-localization of DRD3 and DRD1 and limited expression of DRD3 in DRD2-expressing neurons. b. RNAscope ISH results in the dorsal striatum (framed area in the coronal section) showing a strong expression of DRD1 and DRD2 mRNA and limited expression of DRD3 mRNA, which predominates in DRD1-expressing neurons. Images were taken under 60X magnification. DRD1 in red, DRD2 in blue, DRD3 in green.
3.5. Behavioral profile of PG01042, PG01037 and VK4-116 in L-DOPA-induced dyskinetic rats
Previous work from our laboratory and others has demonstrated that PG01042 and PG01037 reduce established LID in hemi-parkinsonian rats, despite divergent pharmacological properties [18,19]. We now compared their effects to that of VK4-116 [20] on ALO AIMs induced by repeated treatment with L-DOPA in rats with unilateral 6-OHDA-induced lesion of the MFB (see Methods). Lesion efficacy was confirmed using analyses of dopamine and DOPAC content in lesioned vs unmanipulated striata using HPLC. Dopamine (Mlesion = 271.87 pg/mg of tissue, Mun = 15184.30 pg/mg, t14 = 15.02, p < 0.05) and DOPAC (Mlesion = 59.59 pg/mg, Mun = 1528.75 pg/mg, t13 = 15.59, p < 0.05) content in lesioned striata were significantly lower than unmanipulated striata (98.21% and 96.10% reduction, respectively). As shown in Fig. 5, several time-dependent and compound-dependent effects were revealed. With PG01042 (Fig. 5a), rats receiving the low dose (5 mg/kg) demonstrated significantly less ALO AIMs than vehicle-treated rats at the 50 and 60 min timepoints while the high dose (10 mg/kg) attenuated LID expression relative to vehicle in rats at 50, 80 and 90 min-time points. Statistical differences were also found between the low and high doses at 80, 90, and 110 min timepoints. For PG01037 (Fig. 5b), timepoint analyses revealed a significant suppressive effect of the low dose (10 mg/kg) on ALO AIMs 60 min following L-DOPA. The high dose of PG01037 (30 mg/kg) significantly reduced LID expression relative to vehicle treatment at the 60, 80, 90, and 110 min timepoints. Further, differences between AIMs expression in rats receiving the high vs low dose were found at 80 and 110 min such that the high dose significantly alleviated ALO AIMs more than the low dose. Interestingly, VK4-116 did influence severity of AIMs at a single late timepoint (140 min), but in the opposite direction (Fig. 5c). Analysis revealed that the high dose of VK4-116 (30 mg/kg) increased ALO AIMs relative to vehicle (p < 0.05). Although correlational, these behavioral results in dyskinetic rats strongly suggest a central role for ß-arrestin-dependent signaling in LID and as importantly targeting D3R in the D1R-D3R heteromer specifically (see Discussion).
Figure 5. Differential effects of PG01042, PG01037, and VK4-116 on L-DOPA-induced dyskinesia.
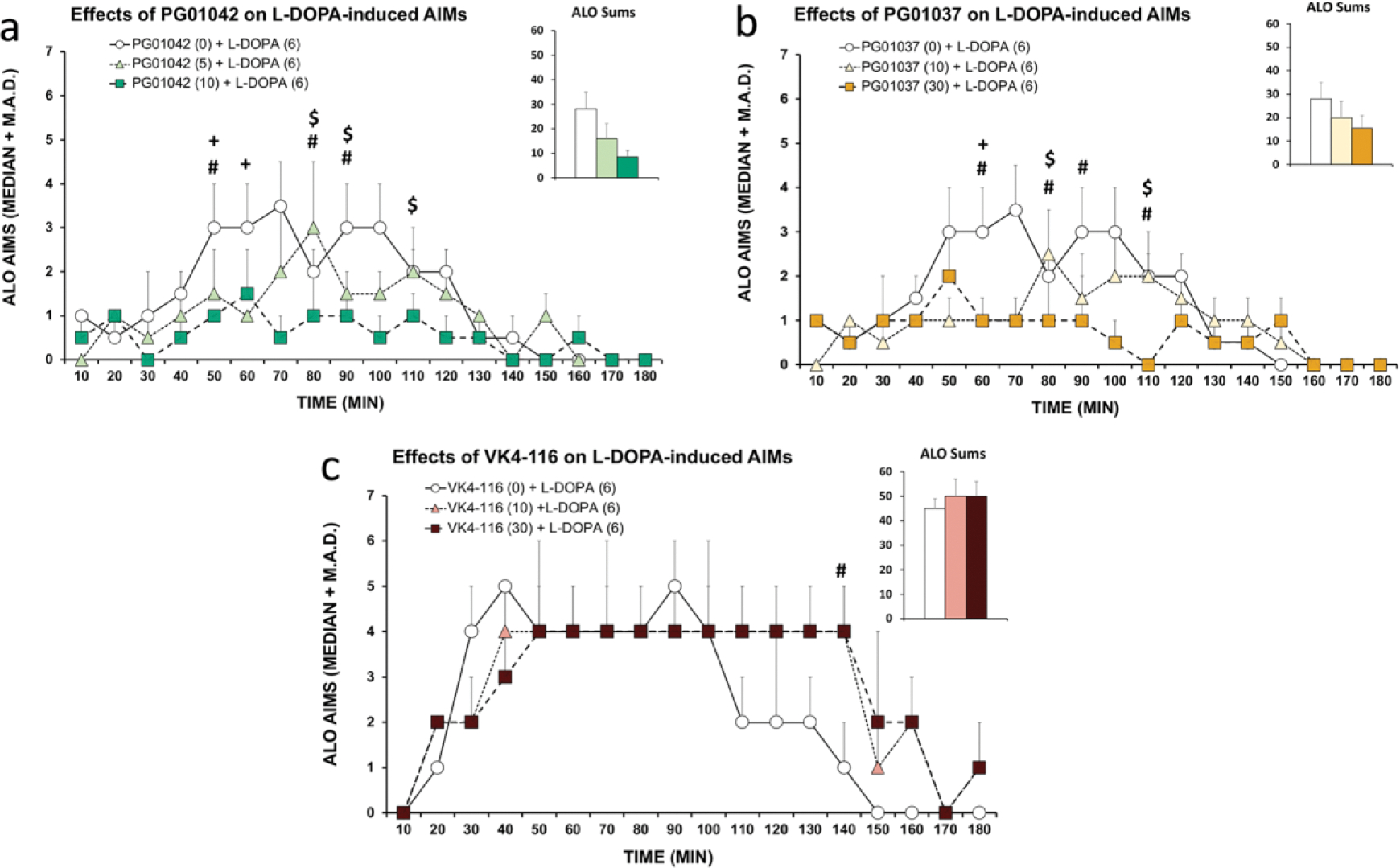
In a within-subjects, counterbalanced design, hemiparkinsonian rats previously rendered dyskinetic were treated with the D3R ligands (a) PG01042 (0, 5, 10 mg/kg; i.p.), (b) PG01037 (0, 10, 30 mg/kg; i.p.), or (c) VK4-116 (VK; 0, 10, 30 mg/kg; i.p.) prior to administration of L-DOPA (6 mg/kg; s.c.). Rats were rated for Axial, Limb and Orolingual (ALO) abnormal involuntary movements (AIMs) every 10 min for 180 min post-injection. AIMs time course and ALO sums are expressed as median + median absolute deviation (M.A.D.). Data were analyzed by non-parametric Friedman ANOVAs with Wilcoxon Match Pairs post-hoc tests. + p < 0.05 for Vehicle versus low dose, # p < 0.05 for vehicle versus high dose, $ p < 0.05 for low dose versus high dose.
4. Discussion
Although GPCR oligomerization represents an increasingly accepted concept [62–64], it is still under considerable debate [65,66]. One of the main concerns is about the localization and functional significance of GPCR heteromers in vivo [65,66]. Nevertheless, in some cases, the simultaneous use of different experimental approaches has provided significant convergent evidence for their functional presence in the experimental animal and for their putative role in pathological conditions and as targets for drug development [62–64]. Particularly valuable has been the use of disrupting synthetic peptides with the amino acid sequence of TMs (TM-peptides) involved in the intermolecular interactions between the GPCR units of the heteromer. The functional and pharmacological properties of GPCR heteromers can in fact be disclosed upon the selective disruption with specific heteromer-disrupting peptides [7,8,24,25,43,51].
Among those properties, GPCR heteromers provide the framework for different emergent allosteric interactions, which have been recently classified in three main types [50]: Type I corresponds to the interactions between orthosteric ligands of the two different GPCRs, by which the ligand of one GPCR changes the properties (affinity or efficacy) of the ligand for the other GPCR; type II corresponds to a ligand-independent interaction, where one of the GPCRs, without ligands, changes the properties of a ligand of the other GPCR; and type III, corresponds to an allosteric interaction through a plasma-membrane effector, such as AC, which forms part of an oligomeric complex that includes the GPCR heteromer. This includes the canonical Gs-Gi antagonistic interaction at the AC level. Using the TM-peptide strategy, we previously demonstrated the existence of type I and III allosterism in the D1R-D3R heteromer, by which a D3R agonist promotes a synergistic effect on D1R agonist-mediated ß-arrestin signaling, while simultaneously inhibiting the D1R agonist-mediated G protein-mediated signaling [7,8]; These simultaneous allosteric interactions result in a significant switch in D1R signaling, from a G protein-AC mediated to a ß-arrestin-mediated signaling [8].
In the present study we provide evidence for the three different types of allosteric interactions in the D1R-D3R heteromer involving three structurally related selective D3R ligands, all of them disclosed by the disruptive TM6 peptide of the D1R. The cross-antagonism of PG01037, its ability to block SKF81297-induced AC activation, represents a type I allosterism. On the other hand, PG01042 promotes the same effect by a canonical Gs-Gi antagonistic interaction at the AC level, which represents a type III allosterism. Finally, the ability of VK4-116 to signal as a ß-arrestin-biased agonist when the D3R heteromerizes with the D1R, represents a ligand-independent type II allosterism. Computational analysis provided possible explanations for the molecular mechanisms of the cross-antagonism of PG01037 and the ß-arrestin functionally selective agonism of VK4-116 in the D1R-D3R heteromer. We found a specific instability of the second pharmacophore unit of PG01037 near TM 2 and its movement toward TM 6 of the D3R, which promotes a different conformation of TM 6, which could favor the four-helix bundle interaction involving TM 5 and TM 6 of both receptors in the D1R-D3R heteromeric interface, previously suggested to provide a mechanism for cross-antagonism in GPCR heteromers [51]. MD simulations of the D1R-D3R heteromer, modelled via the TM5/6 interface, showed that D1R significantly influences the conformation of D3R, mainly by moving the extracellular part of TMs 5, 6, and 7. The movement of TM 5 of the D3R significantly changes the position of a key Ile residue of ECL2 (Ile183ECL2), with which VK4-116, but not PG01037 and PG01042, can specifically interact and promote conformational changes that have been previously associated with biased ß-arrestin agonism (See Results and ref. [59]).
The different biochemical properties disclosed by the three selective D3R ligands, PG01042, PG01037 andVK4-116, and their dependence on D1R-D3R heteromerization, indicated a fundamental involvement of the D1R-D3R heteromer in the locomotor activation induced by D1R agonists in reserpinized mice. Thus, the ability of PG01042 to counteract the locomotor activity induced by the D1R agonist SKF81297 could be explained by its G protein-biased D3R agonism, promoting a negative crosstalk through a D1R-D3R heteromer-dependent canonical antagonistic Gs-Gi interaction at the AC level. PG01037 could also counteract the effect of SKF81297, but through its ability to exert cross-antagonism in the D1R-D3R heteromer. On the other hand, VK4-116 did not show a negative crosstalk or cross-antagonism and, accordingly, did not modify the locomotor activating effect of the D1R agonist.
The ability of PG01042 and PG01037 to strongly counteract the locomotor activation induced by co-administration of D1R and D3R agonists could be explained by their ability to block D1R-mediated G protein-dependent-AC signaling and simultaneously impede the switch to the D1R-mediated ß-arrestin signaling. On the other hand, the ability of VK4-116 to partially counteract the locomotor synergism of the D1R and D3R agonists could be dependent on its selective ability to promote ß-arrestin recruitment by the D1R-D3R heteromer, but without allowing the switch of D1R signaling and consequent synergistic effect. It can also be assumed that the locomotor activation induced by D1R agonists in reserpinized mice generates in the ventral striatum, where D3R are highly expressed and strongly co-localized with D1R in GABAergic striato-nigral neurons (see Introduction). Therefore, a major implication of the present results is that selective D3R ligands can be used to indirectly target ventral striatal D1R-mediated behavioral effects.
The ability of the high dose of pramipexole to elicit locomotor activation in reserpinized mice should depend on its ability to significantly activate postsynaptic D2R, but the counteracting effect of PG01037 and VK4-116 (but not PG01042) indicates that D3R are also involved and exert a facilitatory role. We favor the hypothesis that the effect of the high dose of pramipexole is mediated by a synergistic effect of D2R and D3R activation at D2R-D3R heteromers localized in the GABAergic striato-pallidal neuron of the ventral striatum. Indeed, there is compelling evidence from in vitro experiments for the formation of D2R-D3R heteromers [67–69]. As with D1R-D3R heteromers, D2R-D3R heteromers can modify the properties of D2-like receptor ligands, including an increase in the effect of pramipexole [67]. The results of ISH in the mouse striatum showed that there is a population of striatal cells which co-express both D2R and D3R, preferentially in the ventral striatum (white arrows in Fig. 4).
Locomotor activation induced by pramipexole (or other non-selective D2R-D3R agonists) in reserpinized mice might then provide an in vivo model of D2R-D3R heteromers in the brain. That being the case, biochemical experiments with PG01042, PG01037 and VK4-116 should disclose their pharmacological properties in cells co-transfected with D2R and D3R, which should be able to explain their differential qualitative profile at altering pramipexole-induced locomotor activation in reserpinized mice (experiments in progress). If PG01042, PG01037 and VK4-116 have the same properties at the D3R forming heteromers with D2R than with D1R, the specific inability of PG01042 to counteract pramipexole would suggest that the locomotor activating effect of pramipexole is mostly dependent on G protein-AC-mediated signaling (PG01042 would be substituting for pramipexole by its ability to selectively activate D3R-mediated G protein-dependent signaling). This however would not be supported by other studies that suggest that the locomotor activation mediated by striatal D2R are mostly dependent on ß-arrestin-mediated signaling [70,71].
As briefly reviewed in the Introduction, several studies, including those from our research groups, support the view that D3R upregulation in D1R-expressing neurons of the dorsal striatum is a main mechanism involved in LID. This should lead to the concomitant increase in the population of D1R forming heteromers with D3R. D1R-D3R heteromerization could then explain the sensitization to the effects of L-DOPA, because of its synergistic effect associated with the switch to the ß-arrestin-mediated D1R signaling. The present results add significant pharmacological support to the main role of the D1R-D3R heteromer-mediated ß-arrestin signaling in LID. The results obtained with the different D3R ligands on D1R-mediated locomotion in reserpinized mice paralleled their differential ability to counteract LID-like behavior in rats, which would therefore be mostly mediated by their differential effects on D1R-D3R heteromers in the dorsal striatum. As previously reported, PG01042 and PG01037 displayed an antidyskinetic effect [12,18,19], which was significant at relatively low doses (10 and 5 mg/kg, respectively), while VK4-116 was not only ineffective but it even increased ALO AIMS at the higher dose used (30 mg/kg). The increase occurred during the last period of analysis, when the effect of L-DOPA was wearing off, which could be related to the shared ability of L-DOPA and VK4-116 to promote ß-arrestin-mediated signaling by the D1R-D3R heteromer. This would imply a longer half-life of VK4-116. In fact, previous data demonstrated a high metabolic stability of VK4-116 in rat liver microsomes, suggesting that VK4-116 may undergo slow metabolic clearance in vivo [72].
The in vivo intrastriatal application of heteromer-disrupting peptides could be used to unequivocally demonstrate the key involvement of D1R-D3R heteromers of the ventral striatum and dorsal striatum in the specific pharmacological effects of PG01042, PG01037, and VK4-116 in the respective modulation of locomotor activity in mice and LID in rats. This strategy was recently used in ex vivo experiments in mice, to demonstrate the dependance on D1R-D3R heteromerization in the switch from an AC protein kinase A (PKA)-dependent to a PKA-independent D1R agonist-induced signaling (MAPK activation) upon co-administration of a preferential D3R agonist [8].
The present study constitutes a proof of concept of the significant pharmacological role of GPCR heteromers. We can establish that, to be therapeutically effective in LID, D3R ligands should be either D3R antagonists (such as PG01037) or G protein-biased D3R agonists (such as PG01042), specifically, at the D1R-D3R heteromer. Thus, VK4-116 is an antagonist at the non-heteromerized D3R, but its specific ability to behave as a ß-arrestin-biased agonist at the D1R-D3R heteromer might impede its potential antidyskinetic effect. Hence, when evaluating the role of new D3R ligands as putative antidyskinetic agents, their pharmacological profile in cells expressing D1R-D3R heteromers should be determined. We also provide support for the use of reserpinized mice as a proxy in vivo model to determine the pharmacological properties of D3R ligands at the D1R-D3R heteromers and therefore, their putative efficacy in LID. In general, the present study implies that when suspecting its pathogenetic role, a GPCR heteromer, and not its individual GPCR units, should be considered as a main target for drug development.
Supplementary Material
Acknowledgments
This work was supported by the intramural funds of the National Institute on Drug Abuse (SF); Ministerio de Ciencia e Innovación/Agencia Estatal de Investigación’ (MCIN/AEI) and FEDER, Spain, grant SAF2017-87629-R, and MCIN/AEI 10.13039/501100011033, grant PID2020-113938RB-I00 (E.M, V.C.); MCIN and FEDER, grant PID2019-109240RB-I00 (LP); FPI fellowship, Spain, grant BES-2017-081872 (CLT); Center for Development and Behavioral Neuroscience at Binghamton University and the State University of New York (MC, CB).
Abbreviations:
- AIMs
abnormal involuntary movements
- AC
adenylyl cyclase
- ANOVA
analysis of variance
- ALO
axial, limb, and orolingual
- BiFC
bimolecular fluorescence complementation
- BRET
bioluminescence resonance energy transfer
- D1R, D2R, D3R
dopamine D1, D2 and D3 receptors
- DAPI
4′,6-diamidino-2-phenylindole
- DOPAC
3,4-dihydroxyphenylacetic acid
- ECD
extracellular domain
- ECL2
extracellular loop 2
- FAS
forepaw adjusting steps
- GPCR
G protein-coupled receptor
- HEK-293T cells
human embryonic kidney-293T cells
- HIV TAT peptide
HIV trans-activator of transcription peptide
- 6-OD-DA
6-hydroxydopamine
- LID
L-DOPA-induced dyskinesia
- MD
molecular dynamic
- M.A.D.
median absolute deviation
- MFB
medial forebrain bundle
- PKA
protein kinasxe A
- Rluc
Renilla luciferase
- TM
transmembrane domain
- TR-FRET
time-resolved fluorescence resonance energy transfer
- YFP
yellow fluorescence protein
Footnotes
Declaration of Competing Interest
The authors declare no competing interests.
Appendix A. Supplementary material
Supplementary figures associated with this article can be found in the online version at….
Chemical compounds:
PG01037 (PubChem CID: 11477180)
PG01042 (PubChem CID: 11443078)
VK4-116 (PubChem CID: 130431318)
SKF81297 (PubChem CID: 1218)
Pramipexole (PubChem CID: 119570)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. Dopamine receptors: from structure to function. Physiol Rev. 1998;78:189–225. doi: 10.1152/physrev.1998.78.1.189 [DOI] [PubMed] [Google Scholar]
- [2].Starr BS, Starr MS, Kilpatrick IC. Behavioural role of dopamine D1 receptors in the reserpine-treated mouse. Neuroscience. 1987;22:179–188. doi: 10.1016/0306-4522(87)90208-9 [DOI] [PubMed] [Google Scholar]
- [3].Ferré S, Giménez-Llort L, Artigas F, Martínez E. Motor activation in short- and long-term reserpinized mice: role of N-methyl-D-aspartate, dopamine D1 and dopamine D2 receptors. Eur J Pharmacol. 1994;255:203–213. doi: 10.1016/0014-2999(94)90099-x [DOI] [PubMed] [Google Scholar]
- [4].Schwartz JC, Diaz J, Bordet R, Griffon N, Perachon S, Pilon C, Ridray S, Sokoloff P. Functional implications of multiple dopamine receptor subtypes: the D1/D3 receptor coexistence. Brain Res Brain Res Rev. 1998;26:236–242. doi: 10.1016/s0165-0173(97)00046-5 [DOI] [PubMed] [Google Scholar]
- [5].Fiorentini C, Busi C, Gorruso E, Gotti C, Spano P, Missale C. Reciprocal regulation of dopamine D1 and D3 receptor function and trafficking by heterodimerization. Mol Pharmacol. 2008;74:59–69. doi: 10.1124/mol.107.043885 [DOI] [PubMed] [Google Scholar]
- [6].Marcellino D, Ferré S, Casadó V, Cortés A, Le Foll B, Mazzola C, Drago F, Saur O, Stark H, Soriano A, Barnes C, Goldberg SR, Lluis C, Fuxe K, Franco R. Identification of dopamine D1-D3 receptor heteromers. Indications for a role of synergistic D1-D3 receptor interactions in the striatum. J Biol Chem. 2008;283:26016–26025. doi: 10.1074/jbc.M710349200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Guitart X, Navarro G, Moreno E, Yano H, Cai NS, Sánchez-Soto M, Kumar-Barodia S, Naidu YT, Mallol J, Cortés A, Lluís C, Canela EI, Casadó V, McCormick PJ, Ferré S. Functional selectivity of allosteric interactions within G protein-coupled receptor oligomers: the dopamine D1-D3 receptor heterotetramer. Mol Pharmacol. 2014;86:417–429. doi: 10.1124/mol.114.093096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Guitart X, Moreno E, Rea W, Sánchez-Soto M, Cai NS, Quiroz C, Kumar V, Bourque L, Cortés A, Canela EI, Bishop C, Newman AH, Casadó V, Ferré S. Biased G Protein-Independent Signaling of Dopamine D1-D3 Receptor Heteromers in the Nucleus Accumbens. Mol Neurobiol. 2019;56:6756–6769. doi: 10.1007/s12035-019-1564-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bordet R, Ridray S, Carboni S, Diaz J, Sokoloff P, Schwartz JC. Induction of dopamine D3 receptor expression as a mechanism of behavioral sensitization to levodopa. Proc Natl Acad Sci USA. 1997;94(7):3363–3367. doi: 10.1073/pnas.94.7.3363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Cote SR, Chitravanshi VC, Bleickardt C, Sapru HN, Kuzhikandathil EV. Overexpression of the dopamine D3 receptor in the rat dorsal striatum induces dyskinetic behaviors. Behav Brain Res. 2014;263:46–50. doi: 10.1016/j.bbr.2014.01.011 [DOI] [PubMed] [Google Scholar]
- [11].Cote SR, Kuzhikandathil EV. Chronic levodopa treatment alters expression and function of dopamine D3 receptor in the MPTP/p mouse model of Parkinson’s disease. Neurosci Lett. 2015;585:33–37. doi: 10.1016/j.neulet.2014.11.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Solís O, Garcia-Montes JR, González-Granillo A, Xu M, Moratalla R. Dopamine D3 Receptor Modulates l-DOPA-Induced Dyskinesia by Targeting D1 Receptor-Mediated Striatal Signaling. Cereb Cortex. 2017;27:435–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lanza K, Centner A, Coyle M, Del Priore I, Manfredsson FP, Bishop C. Genetic suppression of the dopamine D3 receptor in striatal D1 cells reduces the development of L-DOPA-induced dyskinesia. Exp Neurol. 2021;336:113534. doi: 10.1016/j.expneurol.2020.113534 [DOI] [PubMed] [Google Scholar]
- [14].Farré D, Muñoz A, Moreno E, Reyes-Resina I, Canet-Pons J, Dopeso-Reyes IG, Rico AJ, Lluís C, Mallol J, Navarro G, Canela EI, Cortés A, Labandeira-García JL, Casadó V, Lanciego JL, Franco R. Stronger Dopamine D1 Receptor-Mediated Neurotransmission in Dyskinesia. Mol Neurobiol. 2015;52:1408–1420. doi: 10.1007/s12035-014-8936-x [DOI] [PubMed] [Google Scholar]
- [15].Ferré S, Navarro G, Casadó V, Cortés A, Mallol J, Canela EI, Lluís C, Franco R. G protein-coupled receptor heteromers as new targets for drug development. Prog Mol Biol Transl Sci. 2010;91:41–52. doi: 10.1016/S1877-1173(10)91002-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lundblad M, Andersson M, Winkler C, Kirik D, Wierup N, Cenci MA. Pharmacological validation of behavioural measures of akinesia and dyskinesia in a rat model of Parkinson’s disease. Eur J Neurosci. 2002;15:120–32. doi: 10.1046/j.0953-816x.2001.01843.x [DOI] [PubMed] [Google Scholar]
- [17].Lanza K, Meadows SM, Chambers NE, Nuss E, Deak MM, Ferré S, Bishop C. Behavioral and cellular dopamine D1 and D3 receptor-mediated synergy: Implications for L-DOPA-induced dyskinesia. Neuropharmacology. 2018;138:304–314. doi: 10.1016/j.neuropharm.2018.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kumar R, Riddle LR, Griffin SA, Chu W, Vangveravong S, Neisewander J, Mach RH, Luedtke RR. Evaluation of D2 and D3 dopamine receptor selective compounds on L-dopa-dependent abnormal involuntary movements in rats. Neuropharmacology. 2009;56:956–969. doi: 10.1016/j.neuropharm.2009.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Riddle LR, Kumar R, Griffin SA, Grundt P, Newman AH, Luedtke RR. Evaluation of the D3 dopamine receptor selective agonist/partial agonist PG01042 on L-dopa dependent animal involuntary movements in rats. Neuropharmacology. 2011;60:284–294. doi: 10.1016/j.neuropharm.2010.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kumar V, Bonifazi A, Ellenberger MP, Keck TM, Pommier E, Rais R, Slusher BS, Gardner E, You ZB, Xi ZX, Newman AH. Highly Selective Dopamine D3 Receptor (D3R) Antagonists and Partial Agonists Based on Eticlopride and the D3R Crystal Structure: New Leads for Opioid Dependence Treatment. J Med Chem. 2016;59(16):7634–7650. doi: 10.1021/acs.jmedchem.6b00860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ferrada C, Moreno E, Casadó V, Bongers G, Cortés A, Mallol J, Canela EI, Leurs R, Ferré S, Lluís C, Franco R. Marked changes in signal transduction upon heteromerization of dopamine D1 and histamine H3 receptors. Br J Pharmacol. 2009;157:64–75. doi: 10.1111/j.1476-5381.2009.00152.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Carriba P, Navarro G, Ciruela F, Ferré S, Casadó V, Agnati L, Cortés A, Mallol J, Fuxe K, Canela EI, Lluís C, Franco R. Detection of heteromerization of more than two proteins bysequential BRET-FRET. Nat Methods. 2008;5:727–733. doi: 10.1038/nmeth.1229 [DOI] [PubMed] [Google Scholar]
- [23].He SQ, Zhang ZN, Guan JS, Liu HR, Zhao B, Wang HB, Li Q, Yang H, Luo J, Li ZY, Wang Q, Lu YJ, Bao L, Zhang X. Facilitation of μ-opioid receptor activity by preventing δ-opioid receptor-mediated codegradation. Neuron. 2011;69:120–131. doi: 10.1016/j.neuron.2010.12.001 [DOI] [PubMed] [Google Scholar]
- [24].Navarro G, Cordomí A, Casadó-Anguera V, Moreno E, Cai NS, Cortés A, Canela EI, Dessauer CW, Casadó V, Pardo L, Lluís C, Ferré S. Evidence for functional pre-coupled complexes of receptor heteromers and adenylyl cyclase. Nat Commun. 2018;9(1):1242. doi: 10.1038/s41467-018-03522-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Rivera-Oliver M, Moreno E, Álvarez-Bagnarol Y, Ayala-Santiago C, Cruz-Reyes N, Molina-Castro GC, Clemens S, Canela EI, Ferré S, Casadó V, Díaz-Ríos M. Adenosine A1-Dopamine D1 Receptor Heteromers Control the Excitability of the Spinal Motoneuron. Mol Neurobiol. 2019;56:797–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285(5433):1569–1572 [DOI] [PubMed] [Google Scholar]
- [27].Grundt P, Carlson EE, Cao J, Bennett CJ, McElveen E, Taylor M, Luedtke RR, Newman AH. Novel heterocyclic trans olefin analogues of N-{4-[4-(2,3-dichlorophenyl)piperazin-1-yl]butyl}arylcarboxamides as selective probes with high affinity for the dopamine D3 receptor. J Med Chem. 2005;48(3):839–848. doi: 10.1021/jm049465g [DOI] [PubMed] [Google Scholar]
- [28].Andén NE, Grabowska-Andén M. Stimulation of D1 dopamine receptors reveals direct effects of the preferential dopamine autoreceptor agonist B-HT 920 on postsynaptic dopamine receptors. Acta Physiol Scand. 1988;134(2):285–290. doi: 10.1111/j.1748-1716.1988.tb08490.x [DOI] [PubMed] [Google Scholar]
- [29].Meadows SM, Chambers NE, Conti MM, Bossert SC, Tasber C, Sheena E, Varney M, Newman-Tancredi A, Bishop C. Characterizing the differential roles of striatal 5-HT1A auto- and hetero-receptors in the reduction of l-DOPA-induced dyskinesia. Exp Neurol. 2017;292:168–178. doi: 10.1016/j.expneurol.2017.03.013 [DOI] [PubMed] [Google Scholar]
- [30].Taylor JL, Bishop C, Walker PD. Dopamine D1 and D2 receptor contributions to L-DOPA-induced dyskinesia in the dopamine-depleted rat. Pharmacol Biochem Behav. 2005;81(4):887–893. doi: 10.1016/j.pbb.2005.06.013 [DOI] [PubMed] [Google Scholar]
- [31].Lindgren HS, Rylander D, Ohlin KE, Lundblad M, Cenci MA. The “motor complication syndrome” in rats with 6-OHDA lesions treated chronically with L-DOPA: relation to dose and route of administration. Behav Brain Res. 2007;177(1):150–159. doi: 10.1016/j.bbr.2006.09.019 [DOI] [PubMed] [Google Scholar]
- [32].Putterman DB, Munhall AC, Kozell LB, Belknap JK, Johnson SW. Evaluation of levodopa dose and magnitude of dopamine depletion as risk factors for levodopa-induced dyskinesia in a rat model of Parkinson’s disease. J Pharmacol Exp Ther. 2007;323(1):277–284. doi: 10.1124/jpet.107.126219 [DOI] [PubMed] [Google Scholar]
- [33].Dekundy A, Lundblad M, Danysz W, Cenci MA. Modulation of L-DOPA-induced abnormal involuntary movements by clinically tested compounds: further validation of the rat dyskinesia model. Behav Brain Res. 2007;179(1):76–89. doi: 10.1016/j.bbr.2007.01.013 [DOI] [PubMed] [Google Scholar]
- [34].Bishop C, George JA, Buchta W, Goldenberg AA, Mohamed M, Dickinson SO, Eissa S, Eskow Jaunarajs KL. Serotonin transporter inhibition attenuates l-DOPA-induced dyskinesia without compromising l-DOPA efficacy in hemi-parkinsonian rats. Eur J Neurosci. 2012;36(6):2839–2848. doi: 10.1111/j.1460-9568.2012.08202.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Chambers NE, Meadows SM, Taylor A, Sheena E, Lanza K, Conti MM, Bishop C. Effects of Muscarinic Acetylcholine m1 and m4 Receptor Blockade on Dyskinesia in the Hemi-Parkinsonian Rat. Neuroscience. 2019;409:180–194. doi: 10.1016/j.neuroscience.2019.04.008 [DOI] [PubMed] [Google Scholar]
- [36].Lanza K, Chemakin K, Lefkowitz S, Saito C, Chambers N, Bishop C. Reciprocal cross-sensitization of D1 and D3 receptors following pharmacological stimulation in the hemiparkinsonian rat. Psychopharmacology. 2020;237(1):155–165. doi: 10.1007/s00213-019-05353-6 [DOI] [PubMed] [Google Scholar]
- [37].Chang JW, Wachtel SR, Young D, Kang UJ. Biochemical and anatomical characterization of forepaw adjusting steps in rat models of Parkinson’s disease: studies on medial forebrain bundle and striatal lesions. Neuroscience. 1999;88(2):617–628. doi: 10.1016/s0306-4522(98)00217-6 [DOI] [PubMed] [Google Scholar]
- [38].Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, Stevens RC. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. 2010;330(6007):1091–1095. doi: 10.1126/science.1197410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sun B, Feng D, Chu ML, Fish I, Lovera S, Sands ZA, Kelm S, Valade A, Wood M, Ceska T, Kobilka TS, Lebon F, Kobilka BK. Crystal structure of dopamine D1 receptor in complex with G protein and a non-catechol agonist. Nat Commun. 2021;12(1):3305. doi: 10.1038/s41467-021-23519-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Miller-Gallacher JL, Nehmé R, Warne T, Edwards PC, Schertler GF, Leslie AG, Tate CG. The 2.1 Å resolution structure of cyanopindolol-bound β1-adrenoceptor identifies an intramembrane Na+ ion that stabilises the ligand-free receptor. PLoS One. 2014;9(3):e92727. doi: 10.1371/journal.pone.0092727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S. Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature. 2012;485(7398):321–326. doi: 10.1038/nature10954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Navarro G, Cordomí A, Brugarolas M, Moreno E, Aguinaga D, Pérez-Benito L, Ferre S, Cortés A, Casadó V, Mallol J, Canela EI, Lluís C, Pardo L, McCormick PJ, Franco R. Cross-communication between Gi and Gs in a G-protein-coupled receptor heterotetramer guided by a receptor C-terminal domain. BMC Biol. 2018;16(1):24. doi: 10.1186/s12915-018-0491-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].De Oliveira PA, Moreno E, Casajuana-Martin N, Casadó-Anguera V, Cai NS, Camacho-Hernandez GA, Zhu H, Bonifazi A, Hall MD, Weinshenker D, Newman AH, Logothetis DE, Casadó V, Plant LD, Pardo L, Ferré S. Preferential Gs protein coupling of the galanin Gal1 receptor in the μ-opioid-Gal1 receptor heterotetramer. Pharmacol Res. 2022;182:106322. doi: 10.1016/j.phrs.2022.106322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lane JR, Donthamsetti P, Shonberg J, Draper-Joyce CJ, Dentry S, Michino M, Shi L, López L, Scammells PJ, Capuano B, Sexton PM, Javitch JA, Christopoulos A. A new mechanism of allostery in a G protein-coupled receptor dimer. Nat Chem Biol. 2014;10(9):745–752. doi: 10.1038/nchembio.1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Moritz AE, Bonifazi A, Guerrero AM, Kumar V, Free RB, Lane JR, Verma RK, Shi L, Newman AH, Sibley DR. Evidence for a Stereoselective Mechanism for Bitopic Activity by Extended-Length Antagonists of the D3 Dopamine Receptor. ACS Chem Neurosci. 2020;11(20):3309–3320. doi: 10.1021/acschemneuro.0c00425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Schott-Verdugo S, Gohlke H. PACKMOL-Memgen: A Simple-To-Use, Generalized Workflow for Membrane-Protein-Lipid-Bilayer System Building. J Chem Inf Model. 2019;59(6):2522–2528. doi: 10.1021/acs.jcim.9b00269 [DOI] [PubMed] [Google Scholar]
- [47].Abraham MJ, Murtola T, Schulz R, Pall S, Smith JC, Hess B, Lindahl E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1–2: 19–25. doi: org/ 10.1016/j.softx.2015.06.001 [DOI] [Google Scholar]
- [48].Navarro G, Gonzalez A, Campanacci S, Rivas-Santisteban R, Reyes-Resina I, Casajuana-Martin N, Cordomí A, Pardo L, Franco R. Experimental and computational analysis of biased agonism on full-length and a C-terminally truncated adenosine A2A receptor. Comput Struct Biotechnol J. 2020;18:2723–2732. doi: 10.1016/j.csbj.2020.09.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Michaud-Agrawal N, Denning EJ, Woolf TB, Beckstein O. MDAnalysis: a toolkit for the analysis of molecular dynamics simulations. J Comput Chem. 2011;32(10):2319–2327. doi: 10.1002/jcc.21787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ferré S, Ciruela F, Dessauer CW, González-Maeso J, Hébert TE, Jockers R, Logothetis DE, Pardo L. G protein-coupled receptor-effector macromolecular membrane assemblies (GEMMAs). Pharmacol Ther. 2022;231:107977. doi: 10.1016/j.pharmthera.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Franco R, Cordomí A, Llinas Del Torrent C, Lillo A, Serrano-Marín J, Navarro G, Pardo L. Structure and function of adenosine receptor heteromers. Cell Mol Life Sci. 2021;78(8):3957–3968. doi: 10.1007/s00018-021-03761-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Weis WI, Kobilka BK. The Molecular Basis of G Protein-Coupled Receptor Activation. Annu Rev Biochem. 2018;87:897–919. doi: 10.1146/annurev-biochem-060614-033910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Bock A, Bermudez M. Allosteric coupling and biased agonism in G protein-coupled receptors. FEBS J. 2021;288(8):2513–2528. doi: 10.1111/febs.15783 [DOI] [PubMed] [Google Scholar]
- [54].Bermudez M, Bock A. Does Divergent Binding Pocket Closure Drive Ligand Bias for Class A GPCRs? Trends Pharmacol Sci. 2019;40(4):236–239. doi: 10.1016/j.tips.2019.02.005 [DOI] [PubMed] [Google Scholar]
- [55].McCorvy JD, Butler KV, Kelly B, Rechsteiner K, Karpiak J, Betz RM, Kormos BL, Shoichet BK, Dror RO, Jin J, Roth BL. Structure-inspired design of β-arrestin-biased ligands for aminergic GPCRs. Nat Chem Biol. 2018;14(2):126–134. doi: 10.1038/nchembio.2527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Sanchez-Soto M, Verma RK, Willette BKA, Gonye EC, Moore AM, Moritz AE, Boateng CA, Yano H, Free RB, Shi L, Sibley DR. A structural basis for how ligand binding site changes can allosterically regulate GPCR signaling and engender functional selectivity. Sci Signal. 2020;13(617):eaaw5885. doi: 10.1126/scisignal.aaw5885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Lee Y, Warne T, Nehmé R, Pandey S, Dwivedi-Agnihotri H, Chaturvedi M, Edwards PC, García-Nafría J, Leslie AGW, Shukla AK, Tate CG. Molecular basis of β-arrestin coupling to formoterol-bound β1-adrenoceptor. Nature. 2020;583(7818):862–866. doi: 10.1038/s41586-020-2419-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Masureel M, Zou Y, Picard LP, van der Westhuizen E, Mahoney JP, Rodrigues JPGLM, Mildorf TJ, Dror RO, Shaw DE, Bouvier M, Pardon E, Steyaert J, Sunahara RK, Weis WI, Zhang C, Kobilka BK. Structural insights into binding specificity, efficacy and bias of a β2AR partial agonist. Nat Chem Biol. 2018;14(11):1059–1066. doi: 10.1038/s41589-018-0145-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Wingler LM, Skiba MA, McMahon C, Staus DP, Kleinhenz ALW, Suomivuori CM, Latorraca NR, Dror RO, Lefkowitz RJ, Kruse AC. Angiotensin and biased analogs induce structurally distinct active conformations within a GPCR. Science. 2020;367(6480):888–892. doi: 10.1126/science.aay9813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Battiti FO, Cemaj SL, Guerrero AM, Shaik AB, Lam J, Rais R, Slusher BS, Deschamps JR, Imler GH, Newman AH, Bonifazi A. The Significance of Chirality in Drug Design and Synthesis of Bitopic Ligands as D3 Receptor (D3R) Selective Agonists. J Med Chem. 2019;62(13):6287–6314. doi: 10.1021/acs.jmedchem.9b00702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Xu M, Koeltzow TE, Santiago GT, Moratalla R, Cooper DC, Hu XT, White NM, Graybiel AM, White FJ, Tonegawa S. Dopamine D3 receptor mutant mice exhibit increased behavioral sensitivity to concurrent stimulation of D1 and D2 receptors. Neuron. 1997;19(4):837–848 [DOI] [PubMed] [Google Scholar]
- [62].Ferré S, Casadó V, Devi LA, Filizola M, Jockers R, Lohse MJ, Milligan G, Pin JP, Guitart X. G protein-coupled receptor oligomerization revisited: functional and pharmacological perspectives. Pharmacol Rev. 2014;66(2):413–434. doi: 10.1124/pr.113.008052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Gomes I, Ayoub MA, Fujita W, Jaeger WC, Pfleger KD, Devi LA. G Protein-Coupled Receptor Heteromers. Annu Rev Pharmacol Toxicol. 2016;56:403–425. doi: 10.1146/annurev-pharmtox-011613-135952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Gaitonde SA, González-Maeso J. Contribution of heteromerization to G protein-coupled receptor function. Curr Opin Pharmacol. 2017;32:23–31. doi: 10.1016/j.coph.2016.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Asher WB, Mathiasen S, Holsey MD, Grinnell SG, Lambert NA, Javitch JA. Extreme vetting of dopamine receptor oligomerization, in: Herrick-Davis K, Milligan G, Di Giovanni G (Eds.), G-Protein-Coupled Receptor Dimers, Springer, Switzerland, 2017, pp. 99–127. [Google Scholar]
- [66].Ferré S, Ciruela F, Casadó V, Pardo L. Oligomerization of G protein-coupled receptors: Still doubted? Prog Mol Biol Transl Sci 2020;169:297–321. doi: 10.1016/bs.pmbts.2019.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Maggio R, Millan MJ. Dopamine D2-D3 receptor heteromers: pharmacological properties and therapeutic significance. Curr Opin Pharmacol. 2010;10(1):100–107. doi: 10.1016/j.coph.2009.10.001 [DOI] [PubMed] [Google Scholar]
- [68].Pou C, Mannoury la Cour C, Stoddart LA, Millan MJ, Milligan G. Functional homomers and heteromers of dopamine D2L and D3 receptors co-exist at the cell surface. J Biol Chem. 2012;287(12):8864–8878. doi: 10.1074/jbc.M111.326678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Scarselli M, Novi F, Schallmach E, Lin R, Baragli A, Colzi A, Griffon N, Corsini GU, Sokoloff P, Levenson R, Vogel Z, Maggio R. D2/D3 dopamine receptor heterodimers exhibit unique functional properties. J Biol Chem. 2001;276(32):30308–30314. doi: 10.1074/jbc.M102297200 [DOI] [PubMed] [Google Scholar]
- [70].Urs NM, Bido S, Peterson SM, Daigle TL, Bass CE, Gainetdinov RR, Bezard E, Caron MG. Targeting β-arrestin2 in the treatment of L-DOPA-induced dyskinesia in Parkinson’s disease. Proc Natl Acad Sci USA. 2015;112(19):E2517–E2526. doi: 10.1073/pnas.1502740112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Donthamsetti P, Gallo EF, Buck DC, Stahl EL, Zhu Y, Lane JR, Bohn LM, Neve KA, Kellendonk C, Javitch JA. Arrestin recruitment to dopamine D2 receptor mediates locomotion but not incentive motivation. Mol Psychiatry. 2020;25(9):2086–2100. doi: 10.1038/s41380-018-0212-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].You ZB, Bi GH, Galaj E, Kumar V, Cao J, Gadiano A, Rais R, Slusher BS, Gardner EL, Xi ZX, Newman AH. Dopamine D3R antagonist VK4-116 attenuates oxycodone self-administration and reinstatement without compromising its antinociceptive effects. Neuropsychopharmacology. 2019;44(8):1415–1424. doi: 10.1038/s41386-018-0284-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Lomize MA, Pogozheva ID, Joo H, Mosberg HI, Lomize AL. OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 2012;40(Database issue):D370–D376. doi: 10.1093/nar/gkr703 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.