

Abstract
Microporous annealed particle (MAP) scaffolds are generated from assembled hydrogel microparticles (microgels). It has been previously demonstrated that MAP scaffold are porous, biocompatible, and recruit neural progenitor cells (NPCs) to the stroke cavity after injection into the stroke core. Here, the goal is to study NPC fate inside MAP scaffolds in vitro. To create plain microgels that can later be converted to contain different types of bioactivities, the inverse electron-demand Diels–Alder reaction between tetrazine and norbornene is utilized, which allows the post-modification of plain microgels stoichiometrically. As a result of adhesive peptide attachment, NPC spreading leads to contractile force generation which can be recorded by tracking microgel displacement. Alternatively, non-adhesive peptide integration results in neurosphere formation that grows within the void space of MAP scaffolds. Although the formed neurospheres do not impose a contractile force on the scaffolds, they are seen to continuously transverse the scaffolds. It is concluded that MAP scaffolds can be engineered to either promote neurogenesis or enhance stemness depending on the chosen post-modifications of the microgels, which can be key in modulating their phenotypes in various applications in vivo.
Keywords: hydrogels, microgel/microparticles, microporous annealed particles, neural progenitor cells, neural stem cells, norbornene, tetrazine
1. Introduction
Tissue regeneration relies on endogenous depots of progenitor cells. In the adult brain, the progenitor cells, which are called NPCs, reside in the subgranular zone and the subventricular zone (SVZ).[1] Because NPCs can self-renew or differentiate into the cell types of the central nervous system,[2] they are an ideal target for brain repair after injury.[3] Post-injury, such as traumatic brain injury or stroke, NPCs travel toward the damaged site, however, they mainly remain in the surrounding region only.[4] We have previously shown that injecting MAP scaffolds in the stroke cavity increased the recruitment of NPCs toward the stroke site and resulted in NPC infiltration into the stroke core.[5,6] This provides an opportunity to modulate NPC fate and lead to improved endogenous regeneration by tailoring the microgels in our MAP scaffold.
Hydrogels have historically been used to culture[7] and/or transplant[8] cells in vitro and in vivo, respectively. Their use as extra-cellular matrix mimics allows for studying various chemical and physical properties that are necessary to derive desired cellular interactions and phenotypes.[7] The most utilized hydrogel systems for NPC renewal or differentiation are nonporous hydrogels formed through crosslinking biocompatible polymers decorated with laminin-derived peptides.[9] These hydrogels can easily be produced with tailored properties, for example varying degrees of degradability and stiffnesses, and NPCs can be seeded inside or on top of the gels. However, conflicting results arise when investigating NPC seeding within or on top of hydrogel substrates. Stiffness was historically thought to be key in modulating NPC differentiation[9] and was significantly investigated when NPCs were seeded atop of a polylysine/polyacrylamide-IKVAV bifunctional hydrogel.[10] Farrukh et al. found that the optimal gel elasticity for promoting neurogenesis was dependent on if the NPCs were embryonic or adult NPCs. However, more recent work from Madel et al. found that when seeded within a nonporous hydrogel, material degradability (rather than stiffness) was necessary for adult NPC self-renewal and differentiation.[11] It was determined that in nonporous elastin-like protein hydrogels containing only RGD adhesion ligands, the hydrogel’s degradability maintained the NPC stemness and self-renewal regardless of hydrogel stiffness.[11] Additionally, they demonstrated that hydrogels that were more degradable, or allowed more time for cell remodeling prior to the induction of differentiation, significantly increased NPC differentiation. While degradation and stiffness have both been found to be critical in modulating either NPC renewal or differentiation, it seems to be highly dependent on cell seeding, that is, within or atop, regarding the hydrogel substrate. The granular and porous nature of MAP scaffolds makes it inherently different from bulk nonporous hydrogels,[12] thus prior findings may not be directly applicable. To our knowledge, the culture of NPCs in MAP scaffolds has not been previously reported, and we aimed to understand how to design microgels to promote either NPC renewal or differentiation within MAP scaffodls.
MAP scaffolds are formed by packing microgels and linking them to one another through covalent or non-covalent interactions (hydrophobic, electrostatic, guest–host, cell–cell/cell–material, etc).[13] When microgels are injected into a tissue, they jam together and support cell infiltration through the porous structure of the scaffold. We covalently link microgels to each other after injection to form stable MAP scaffolds.[12] Microgels can be produced from a variety of methods including emulsion, microfluidics, and electrospraying, which generate microgels with different size distributions.[14] Most microgels are generated in a water-in-oil emulsion, from a polymer macromer that is crosslinked using either protease degradable peptides,[12] polymers,[15] or small molecules;[16,17] microfluidic platforms generate the narrowest particle size distributions and have been increasingly used for microgel production.
Besides the method of fabrication, the polymer backbone and crosslinking chemistry used for creating microgels are two of the most important aspects to be considered. Click-chemistry has been a popular avenue for hydrogel synthesis and modification due to its fast, versatile, and selective reactivity.[18,19] The Diels–Alder reaction is one example of click-chemistry and was first developed in 1928.[20] It has since been widely used for stereoselective synthesis of various complex molecules. However, it was not until 30 years later that the inverse-electron-demand Diels–Alder click reaction between tetrazine and electron-rich dienophiles was reported.[21] The tetrazine–norbornene reaction was first applied in bioconjugation reactions in 2008,[22] and it has since become a widely used reaction in biomaterials.[23] We and others have previously reported the use of tetrazine–norbornene modification of microgels and linked them to form a MAP scaffold.[16,17] However, this reaction has not been previously demonstrated to stoichiometrically modify microgel surfaces post-fabrication. Herein, we report for the first time the stoichiometric modification of microgels via tetrazine–norbornene chemistry to introduce bioactivity, and the use of these microgels to form NPC-loaded MAP scaffolds to assess NPC renewal or differentiation.
2. Results and Discussion
2.1. One-Pot Microfluidic Synthesis of Microgels
The physical and biochemical properties of microgels dictate the microstructure and biochemical properties of MAP scaffolds. Thus, our ability to generate microgels with narrow polydispersity in their diameter and shape is important for our ability to predict the scaffold microstructure. Previously our lab has used an emulsion batch system and a click-by-click approach to generate microgels via a light-activated thiol–ene reaction, and subsequently annealed them to form a MAP scaffold (via tetrazine–norbornene cycloaddition).[17] However, this method of batch production also led to a greater distribution of microgels, ranging from 50–250 μm, such that the microgels had to be filtered through various size sieves to selectively control microgel diameters used for different scaffold designs (i.e., in vitro and/or in vivo experimentation). To narrow the size distribution, we implemented a flow-focusing microfluidic device with one inlet for the precursor and crosslinker mixture, and another inlet for the oil. The photoinitiation for crosslinking the microgels was designed to occur downstream off chip after droplet formation (Figure 1a).[24] The simplification of the device also decreased microgel production times compared to previous microfluidic methods[5] and allowed for the use of a single syringe pump capable of handling two (i.e., gel and oil) syringes simultaneously, which eliminated non-synchronous oscillatory fluctuations across multiple syringe pumps.[25] Overall, this resulted in lower size variability of micrs (Figure 1a).
Figure 1.
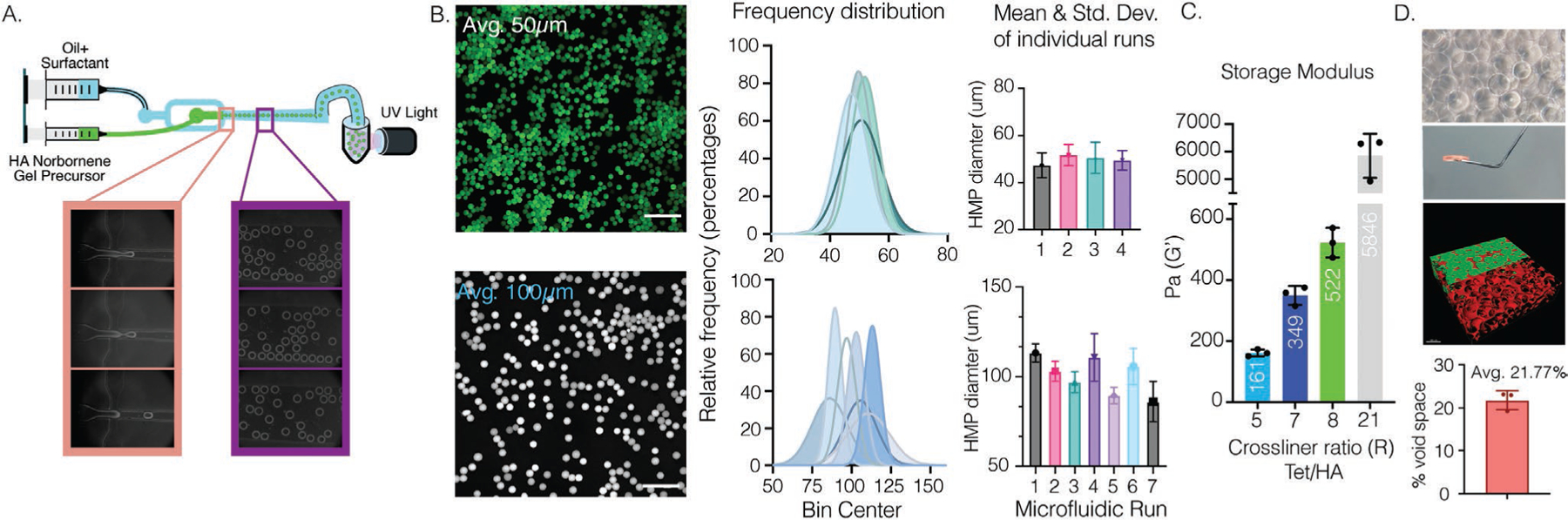
A) Overview of flow-focusing microfluidic design for creating HA–NB microgels. B) Fluorescence imaging of labeled microgels. Histograms of various independent runs demonstrate control over microgel size generation of either an average of 100 or 50 μm depending on the microfluidic design used. Mean and standard deviation as error bars are plotted for each individual run. Scale bar: 500 μm. C) Microgels can be crosslinked with PEG–Tet. The bulk storage modulus of the MAP scaffold can be tailored by annealing of microgels with PEG–Tet at various ratios. Microgels with an average 100 μm were used. D) Representative images of MAP scaffolds and average calculated percent void space.
2.2. From Hyaluronic Acid Microgels to MAP Scaffolds
Homogenous HA–NB microgels were synthesized in two steps. First, HA was functionalized with norbornene to allow for use of orthogonal click reactions via thiol–ene or tetrazine–norbornene.[17,26] Functionalization with norbornene was determined via H′ NMR (Figure S1, Supporting Information) and different functionalization degrees were explored, 22%, 33.5%, and 52%. We found that 33.5% resulted in effective microgel generation with sufficient norbornene groups for crosslinking, modification, and annealing of microgels. Interestingly, 52% functionalization led to microgel fabrication issues within our devices, causing gelation to occur at the flow-focusing junction or an increase in overall size distribution. Similarly, modifications around 22% resulted in insufficient leftover norbornene groups for creating scaffolds due to the NB depletion during microgel formation and post-modifications. After HA–NB polymer modification, microgels were formed within our microfluidic device via thiol–ene click chemistry by mixing HA–NB (3.5 wt%) with a dicysteine-containing matrix metalloproteinase degradable peptide (2.8 × 10−3 m) and LAP photo initiator (9.90 × 10−3 m). Previous microgel methods incorporated modification of the HA-polymer with peptides and Alexa Fluor (AF) labels prior to microgel fabrication.[5,12,27] However, our goal was to create a “naked” microgel with a tunable functional handle that would allow for an off-the-shelf material that could be modified post-fabrication for any application. Therefore, all HA–NB microgels were generated without peptides or AF labels.
After microgel production and purification, HA–NB microgels were modified with tetrazine–AF488 to ease the visualization and the characterization of size distribution using a MATLAB code (Figure 1b). Depending on the device used, microgels with an average diameter range of 50 μm and a std of +/−4–6 μm, or microgels with an average 100 μm and a std of +/−5–11 μm, could be fabricated (Figure 1b). Microgels were subsequently crosslinked with 4-arm PEG–Tetrazine to form bulk scaffolds. Microgels were dried using centrifugation and buffer removal before the tetrazine annealing agent was added. Rheological characterization was performed using a plate-to-plate analysis. The storage (G′) and loss modulus (G′′) were constant at frequencies ranging from 0.1 and 10 s−1 and the average G′ and G′′ readings were reported (Figure 1c). As previously reported,[17] changing the moles of Tetrazine to moles of HA ratio (Tet/HA ratio) significantly altered the storage modulus, demonstrating the dynamic range of HA–NB MAP scaffolds from 161 Pa to 5846 Pa (Figure 1c). A Tet/HA ratio of 7, or R7, was chosen for future in vitro experiments with NPCs due to its similarity to the storage modulus of brain.[28] Further characterization was also performed to visualize the interconnected pores of MAP scaffolds by incubating the scaffolds with 70 kDa Dextran Tetramethylrhodamine solution for at least an hour (Figure 1d). The dextran was easily diffused throughout the open pores and did not penetrate the nanoporous microgels, indicating that the macroporous void is interconnected. Confocal images of the scaffolds were analyzed via IMARIS rendering to quantify overall void space in scaffolds with an R7, which led to an average of 21.77% void space (Figure 1d).
2.3. NPCs Isolated from the Sub-Ventricular Zone are GFAP+ and SOX2+
NPCs were isolated from P12 C57/BL6 mice by first dissecting the SVZ niche from the rest of the brain (Figure S2a, Supporting Information). All mouse experiments were performed according to an approved protocol by the Institutional Animal Care and Use Committee at Duke University. The mouse line was purchased from JAX: C57BL/6J (Strain #:000664). The separated tissue was then gently homogenized with a pipette and centrifuged. The supernatant that contained NPCs was then plated on TC-treated 24 well plates in a NSCs proliferation media (also called N5 media) and left to adhere for 1 day. Cells that adhered were cataloged as astrocytes and disposed of, while those remaining suspended within the media were transferred onto a new TC-treated 24 well plate. These cells were considered passage 1 (P1) and left to grow for about a week with adding N5 media every 2 days. Once confluent, cells were passaged and expanded for experiments. P2 and P3 cells were used for all in vitro experiments and allowed to grow for 3 days before use if they were thawed from frozen stocks (Figure S2a, Supporting Information). To determine the efficiency of primary NPCs culture, flow cytometry was performed on P2 cells prior to expansions. Glial fibrillary acidic protein (GFAP) was used as a lineage marker for NPCs from the SVZ,[2,29] and SRY-box 2 (SOX2) was used to confirm that they retained stemness potency. 61.7% of cells in culture were found positive for both GFAP and SOX2 proteins, while 21.8% and 16.2% were found positive for SOX2 only or GFAP only, respectively (Figure S2b, Supporting Information).
2.4. Stoichiometric Modification of Microgels via Norbornene–Tetrazine Cycloaddition
As mentioned earlier, an ideal system for microgels would be a “naked,” off-the-shelf material that can be easily modified with desired constituents of interest. Our previous methods incorporated peptides or labels before microgel synthesis, constraining their formulation to what was synthesized. Historically, norbornene–tetrazine cycloadditions have been shown to be high yielding, selective, and fast in aqueous solutions and therefore used for live cell imaging.[22] As such, we believed that by using norbornene as a functional handle, the microgel can be used in a more versatile off-the-shelf fashion.
While the literature has demonstrated the efficiency of norbornene–tetrazine reactions and maintained bioactivity of molecules after norbornene–tetrazine reactions, we checked whether this held true for the addition of multiple tetrazine-modified molecules onto our microgel system at once. First, we wanted to ensure that we had precise control over post-modification of the microgels using the norbornene–tetrazine click reaction. To verify, three Alexa Fluor colors were all modified to have a tetrazine handle added to them, tetrazine–Alexa Fluors (Tet–AF) 488, 555, and 647. Their predicted color combinations were then created using Adobe Illustrator, taking into account the ratio of the combined Tet–AF. If the reaction remained efficient enough to be stoichiometric, we could expect that the microgels would look like the predicted colors. Microgels were then incubated with these various ratios and confocal images were taken as well as regular photos of the microgels in a 1 mL micro centrifuge tubes (Figure 2a,b). The fluorescent intensity was measured for each color combination and compared to the predicted quantities to determine if the reaction was indeed stoichiometric (Figure 2c,d). Additional images of microgel mixtures were taken to give a glimpse of the possibilities to generate composite and heterogenous scaffolds with these modifiable microgels (Figure 2e). These results indicated that the norbornene–tetrazine chemistry follows a stoichiometric addition of multiple Tet–AF molecules post microgel production, making them ideal for off-the-shelf material that can be tailored for tissue, disease, or patient specific applications.
Figure 2.
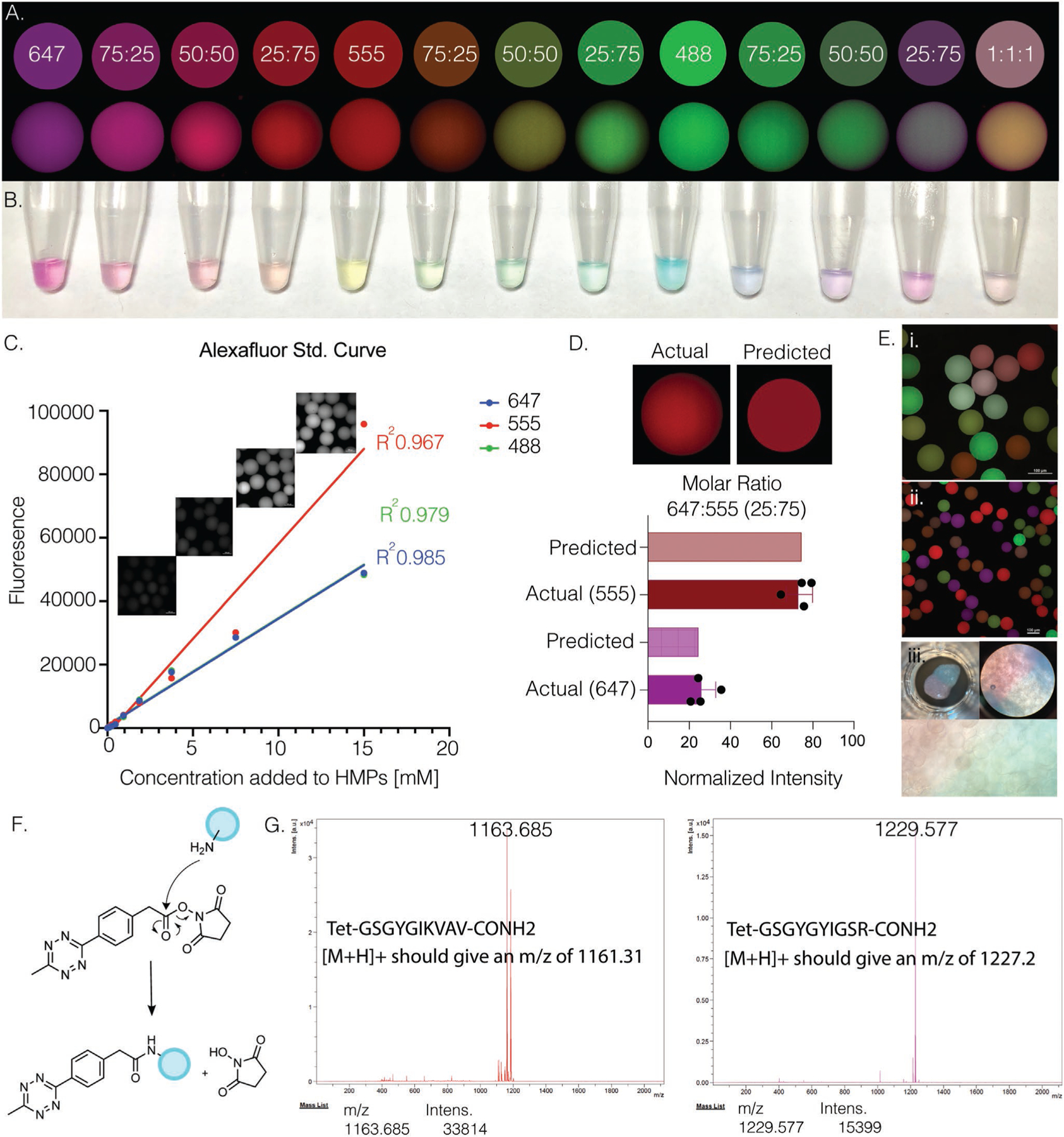
A) Comparison of predicted (top) versus actual (bottom) confocal images of microgels after post-modification with tet–AFs at various mixed ratios. B) Photo of microgels after adding tet–AF. C) Tet–AF standard curve measured via fluorescence and confocal imaging. D) Fluorescence intensity of microgels after post-modification was measured and compared to predicted values. E) i,ii) Confocal images of various modified microgels show heterogeneity and precise control over post-modification via norbornene–tetrazine click reaction. iii) Photos of two different labeled MAP scaffolds adjacent to one another. F) Chemical schematic of attaching tetrazine to desired peptide via aminolysis. G) MALDI analysis of peptides after HPLC purification shows correct m/z of IKVAV and YIGSR tetrazine peptides.
2.5. Neurogenic Peptide Incorporation via Tetrazine is Biocompatible
To demonstrate post-fabrication modifications of microgels with bioactive molecules, we synthesized peptides with a tetrazine end group. In particular, we chose IKVAV and YIGSR because they are two laminin-derived peptides known to promote NPC growth[30] and migration (Figure 2f,g).[31,32] The same peptides were also synthesized without tetrazine or thiol groups to serve as controls (Figure S3, Supporting Information). MALDI and HPLC were used to check the quality and purify the peptides, respectively. Previous literature has demonstrated the use of IKVAV in hydrogels to improve cell growth and neurogenesis[10,33] while the use of YIGSR in hydrogels has shown some effect on neurons[34] and minimal in others.[35] To investigate the use of these peptides within our MAP scaffolds, microgels were incubated with various concentrations and combinations of the two modified peptides (Figure 2f,g), as well as RGD. Microgels that were previously modified with RGD through a norbornene thiol–ene photoclick reaction, or without RGD, were then modified to contain IKVAV and/or YIGSR. It should be noted, the addition of RGD was not characterized.
Microgels and NPCs were mixed at a concentration of 10 000 cells μL−1 along with the 4arm-PEG–Tet crosslinker, and 10 μL of the mixture was placed within a PDMS-cast cell culture device bound to a cover slip (Figure 3a). The NPC/microgel mixtures were allowed to anneal in TC incubators at 37 °C for 30 min before media was added. The kinetics of the annealing reaction was sufficiently slow[17] to allow adequate mixing of the cells and microgels to ensure even seeding throughout the scaffold (Figure 3a). At day 0, NPCs were evenly distributed throughout the scaffolds and observed to be residing in the void space between microgels. The overall cell viability was assessed with a Live/Dead Kit (Figure 3a) and showed high viability for all the conditions with minimal red signal.
Figure 3.
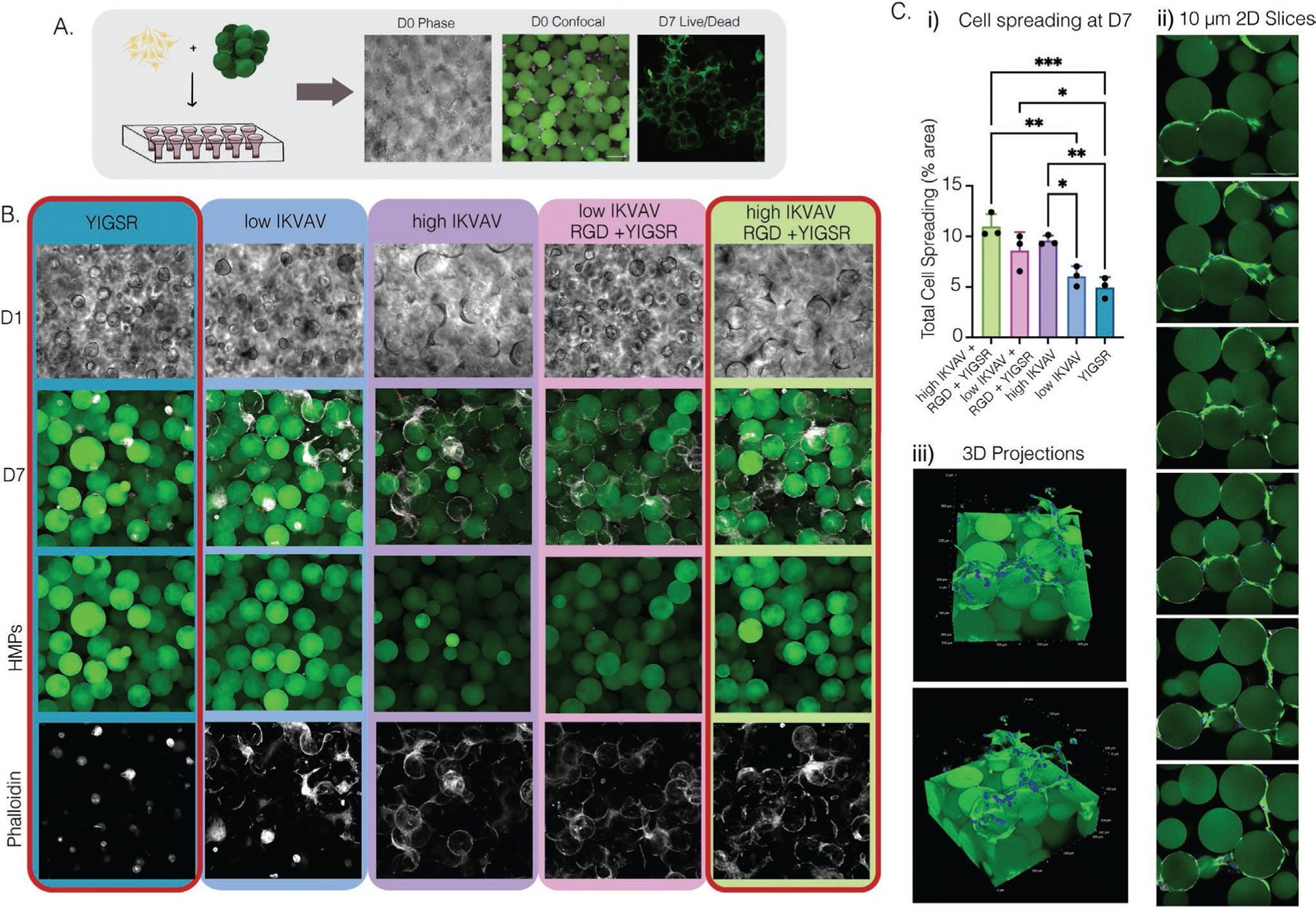
A) Schematic and representative images of NPC seeding within MAP scaffold. D0 in both phase and confocal images show even cell distribution within the scaffold. Confocal image of live/dead stain cell viability at D7. B) 20× phase images of D1 and 20× confocal images at D7 were taken. Microgels were stained (green) and cells were stained for phalloidin (white). C) i) Cell spreading at D7 was qualified using confocal imaging and IMARIS. ii) Slices of z-stack images from the high IKVAV, RGD, + YIGSR condition with NPCs labeled with GFAP (green) and microgels (green). iii) 3D volume renderings of MAP scaffolds containing NPCs from the high IKVAV, RGD, + YIGSR condition, microgels (green), and cell nuclei labeled with DAPI (blue) and NPCs labeled with GFAP (green). Tukey’s multiple comparison test (P < 0.05) (n = 3 unless specified otherwise).
2.6. Peptide Composition Dictates Spreading Versus Neurosphere Formation
Five conditions were investigated; RGD+high[IKVAV]+YIGSR, RGD+low[IKVAV]+YIGSR, high[IKVAV] only, low[IKVAV] only, and YIGSR-only. Significant differences were seen amongst groups and across days in cell spreading and morphology. Most notably was the appearance of neurospheres in conditions lacking IKVAV and significant cell spreading in conditions containing IKVAV with and without RGD+YIGSR. At day 1, we observed via phase contrast imaging that neurospheres were forming within the void space between microgels when there were low concentrations of adhesive peptides. However, low amounts of IKVAV could even salvage some NPC spreading after several days of incubation (Figure 3b). After 7 days of culture, there was significantly more cell spreading in groups that contained high IKVAV or low IKVAV with RGD+YIGSR, compared to YIGSR-only, which maintained only neurospheres (Figure 3c). Additionally, when a 3D projection of z-stack images was created or slices from the image were taken, NPCs were seen to spread attached to one another and in a more linear fashion, as opposed to previous data of human dermal fibroblast grown within our MAP scaffolds (Figure 3d).[17,26] Altogether, this indicated to us that the bioactivity was still intact but highly dependent on peptide combination and concentration within our system, with IKVAV heavily dictating cell spreading over the formation of neurospheres.
2.7. Spreading NPCs Exert a Significant Contractile Force on Microgels
From our initial experiments with the 5 different conditions, the two groups that demonstrated the largest difference in response were RGD+high[IKVAV]+YIGSR, which we also termed ALL, compared to YIGSR-only. As such, these specific groups were chosen for further investigation. Live imaging over the course of 4 days in these two conditions allowed us to visualize the dynamic interaction of NPCs within the scaffold. NPCs seeded in MAP scaffolds composed of microgels modified with ALL peptides spread over time across beads, pulling the microgels with sufficient force that a displacement could be observed (Movie S1, Supporting Information). To quantify the dynamic nature of the NPC/microgel interaction we quantified the videos for the time it takes a cell to spread, and the displacement generated over 2–4 days. Cells began to attach within 2 h (Figure 4a,b), followed by pulling of the microgels and changing the scaffold (Figure 4c,d, Movie S2, Supporting Information). By 12 h, cell spreading was nearly 100%, indicating that the spreading of NPCs occurs rapidly (Figure 4b). Additionally, NPCs were seen to continue their movement and change the scaffold well into the fourth day after seeding in MAP scaffolds (Figure 4c, Movie S2, Supporting Information). We next moved to understand the contractile force that NPCs exert on the scaffold by measuring the displacement of microgels over time and plotting the movement in a cartesian coordinate plane. Though the microgels were covalently annealed to each other, the NPCs appear to exert a force sufficient to move the microgels. The spreading NPCs were constantly pulling on the scaffold, generating an average distance change in microgels of 71 μm over 48 h (Figure 4c,d).
Figure 4.
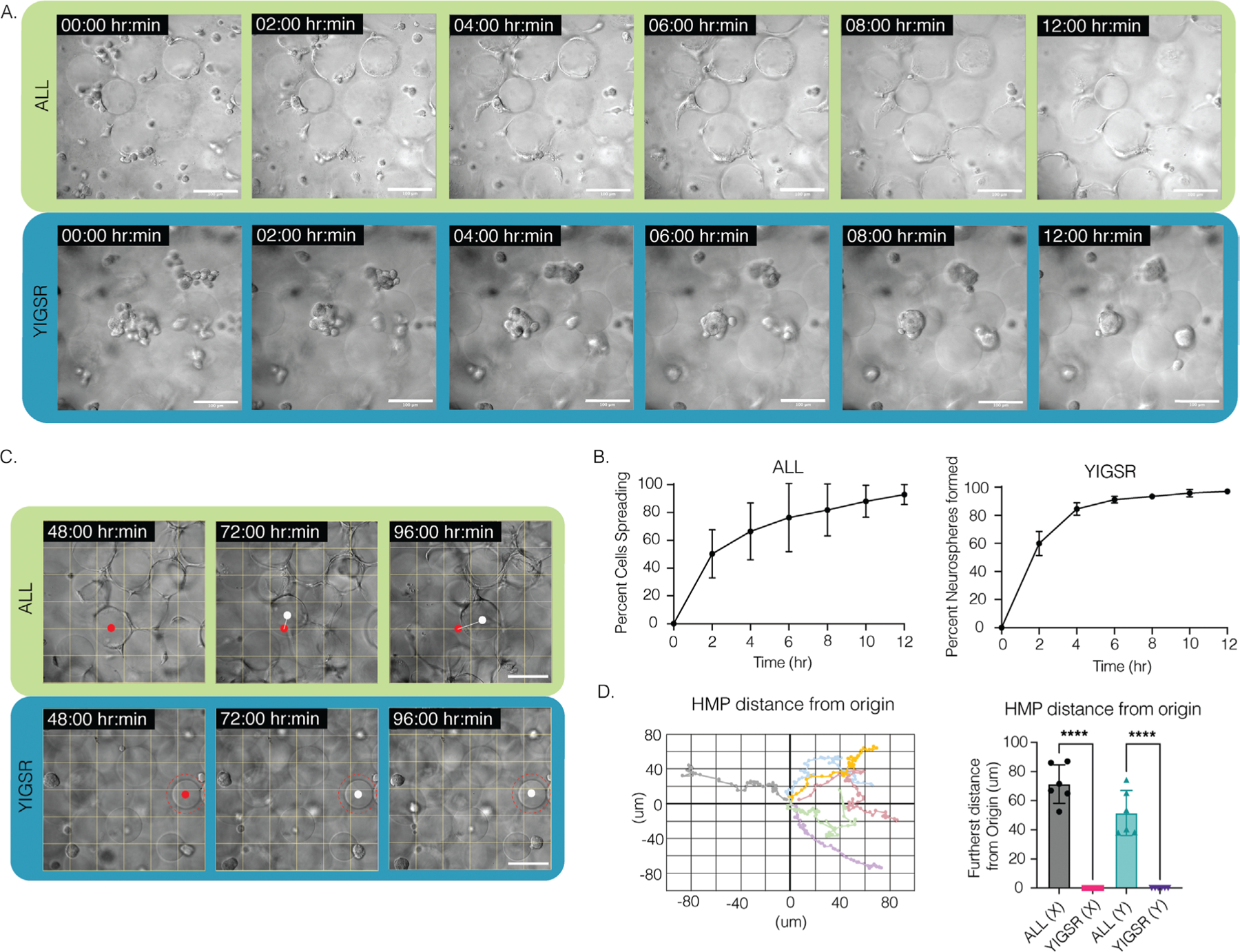
A) Time lapse of NPC spreading in ALL condition and neurosphere formation in YIGSR condition; scale bar: 100 μm. B) Percentage of cell spreading over 12 h in ALL condition versus percentage of neurospheres formed in YIGSR only condition. NPCs take around 12 h to fully spread in MAP scaffolds, whereas neurosphere formation takes ≈6 h. The error bars represent the standard deviation. C) NPCs can be seen attaching and contracting and moving the microgels within the MAP scaffold for up to 4 days after seeding. Meanwhile, neurospheres can be seen reaching for other NPCs but without perturbing or attaching to microgels within the scaffolds. Scale bar: 100 μm. D) Plot of microgels displacement from origin from day 2 to day 4 in ALL condition and bar graph comparing microgel displacement between YIGSR only and ALL conditions, n = 6. Tukey’s multiple comparison test (P < 0.05) (n = 3 unless otherwise specified).
2.8. Neurosphere Formation Does Not Exert a Contractile Force on Microgels
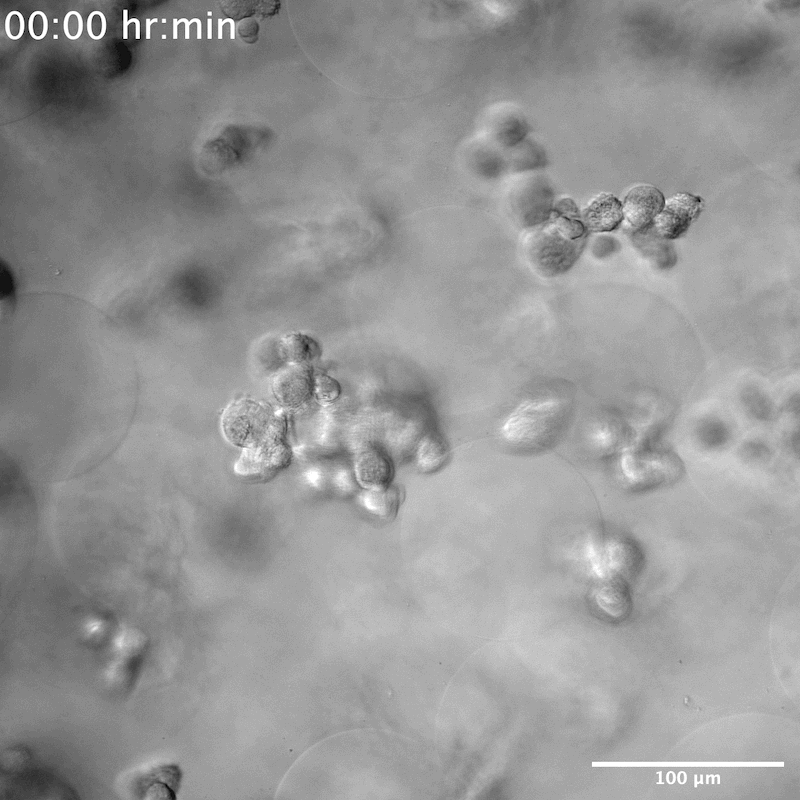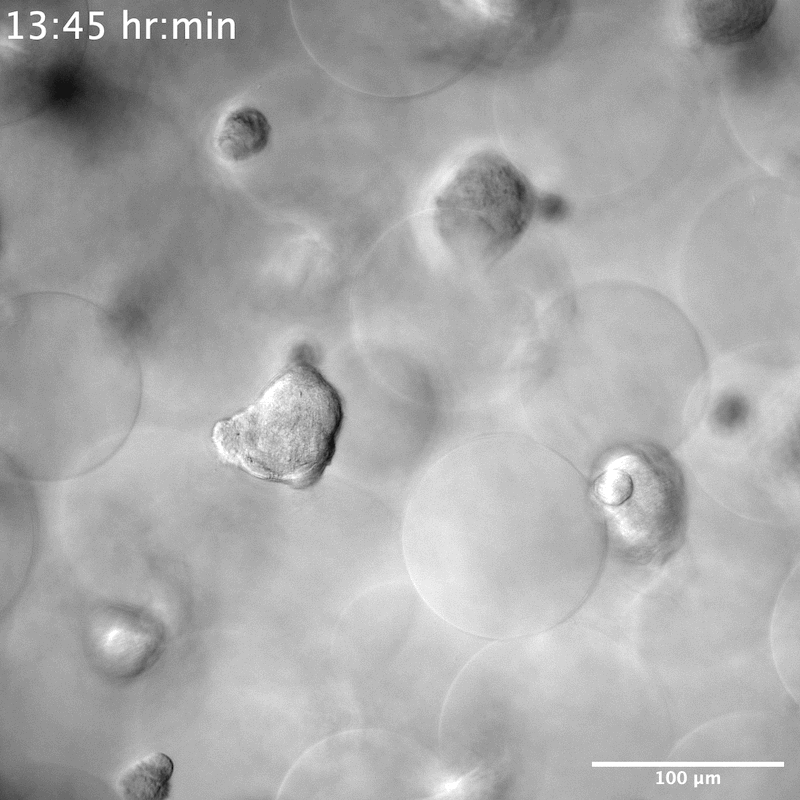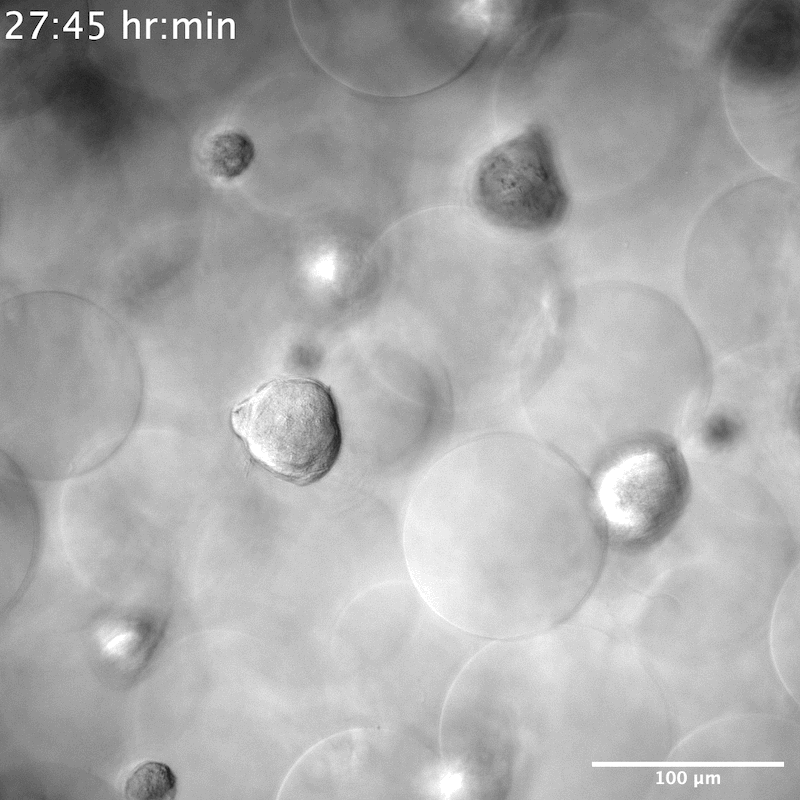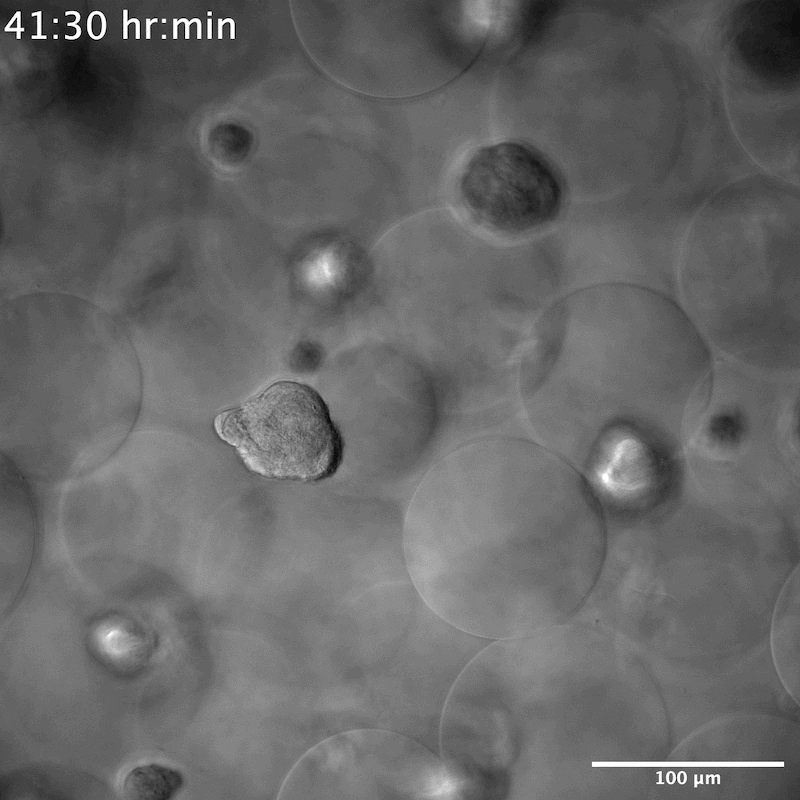
NPCs seeded in the YIGSR-only modified microgels group were seen to stretch and search for something to grab onto. When single cells encountered one another, they formed neurospheres within 50 μm of their reach (Movies S3 and S4, Supporting Information). On average it took 6 h until the neurospheres were completely formed, that is, few new additions or departures of single NPCs and a decrease in movement within the scaffold (Figure 4a,b). This was different than the spreading observed in the ALL scaffolds that required 12 h to reach maximal cell spreading (Figure 4a,b). We next moved to understand the contractile force that neurospheres exert on the scaffold by measuring the displacement of microgels over time. Although neurospheres were seen to traverse the scaffold in both the XY and Z directions (Figure S4, Movies S5 and S6, Supporting Information), they did not adhere to or displace any of the microgels. Despite the scaffold remaining entirely still, we observed the cells moving constantly within the scaffold (Figure 4c,d). This indicated that neurospheres exerted no contractile forces on the scaffold. Historically, neurosphere or brain organoids are developed either through prolonged culture times, within 2D wells or atop of a 2D hydrogel, or on a chip.[36,37] To our knowledge, neurosphere development within a 3D hydrogel system has not been reported. However, there was report of a PEG hydrogel microcell array, similar in structure to chip methods, used to grow neurospheres.[38] The microcell size averaged 35 μm in height and 95 μm in diameter, producing neurospheres similar in size to what we observed, around 40 to 50 μm (Figure S5, Supporting Information), which may infer structural similarities to the void spaces that neurospheres are seen to grow within our system.
2.9. IKVAV Peptide Presentation Leads to NPC Differentiation
We next wanted to assess the differentiation state of NPCs cultured in the ALL group compared to the YIGSR-only group. At day 7 of culture, MAP scaffolds were fixed, and cells were stained with either neurogenic (Tuj1), astrocytic differentiation (S100beta), or self-renewal marker (SOX2) markers in combination with lineage marker (GFAP) (Figure 5a,b). It is important to note that no differentiation media was used, and NPCs were kept in N5 stem cell maintenance (i.e., proliferation) media only during the entire cell culture period. Upon imaging, the ALL condition was not positive for S100b (a marker for established astrocytes) or SOX2 (a marker for maintained stemness), however, the NPCs were positive for GFAP and Tuj1 (a marker for early stage of neural differentiation). While we did not explore this explicitly, we believe that these results in combination with other reported data may indicate that this differentiation may be driven by mechanisms such as mechanosensing or through the beta1-integrin receptor interaction of the NPCs with the IKVAV peptides. IKVAV is known to directly bind to the transmembrane receptor beta1-integrin.[30,39] The beta1-integrin has also been shown to restrict astrocytic differentiation in adult NPCs.[40,41] Additionally, IKVAV has been reported in multiple hydrogel systems to lead to neuronal differentiation, migration, and neurite extension.[42] Our 3D MAP scaffold is softer than has been described in the literature[10] for inducing NPC differentiation and did not require any degradation.[11] To investigate how RGD alone may affect cell differentiation, we grew NPCs in an RGD only condition. Interestingly, while they maintained their spread morphology, they did not express Tuj1 and instead were positive for SOX2 (Figure S6, Supporting Information). This indicates that in the presence of IKVAV, the NPC’s spread morphology led to neuronal differentiation. Similarly, in the ALL condition, the NPCs are seen attached to one another primarily in a chain configuration, rather than in a fully spread-out morphology (Figure 3d). This is not unlike their typical migratory mechanism along the rostral migratory stream (RMS) that is observed in vivo when NPCs travel from the SVZ to the olfactory bulb (OB).[1,43,44] They are also seen to migrate in this serial, non-invasive fashion, within the brain toward lesions such as strokes or tumors, and are typically seen to differentiate into neurons.[45]
Figure 5.
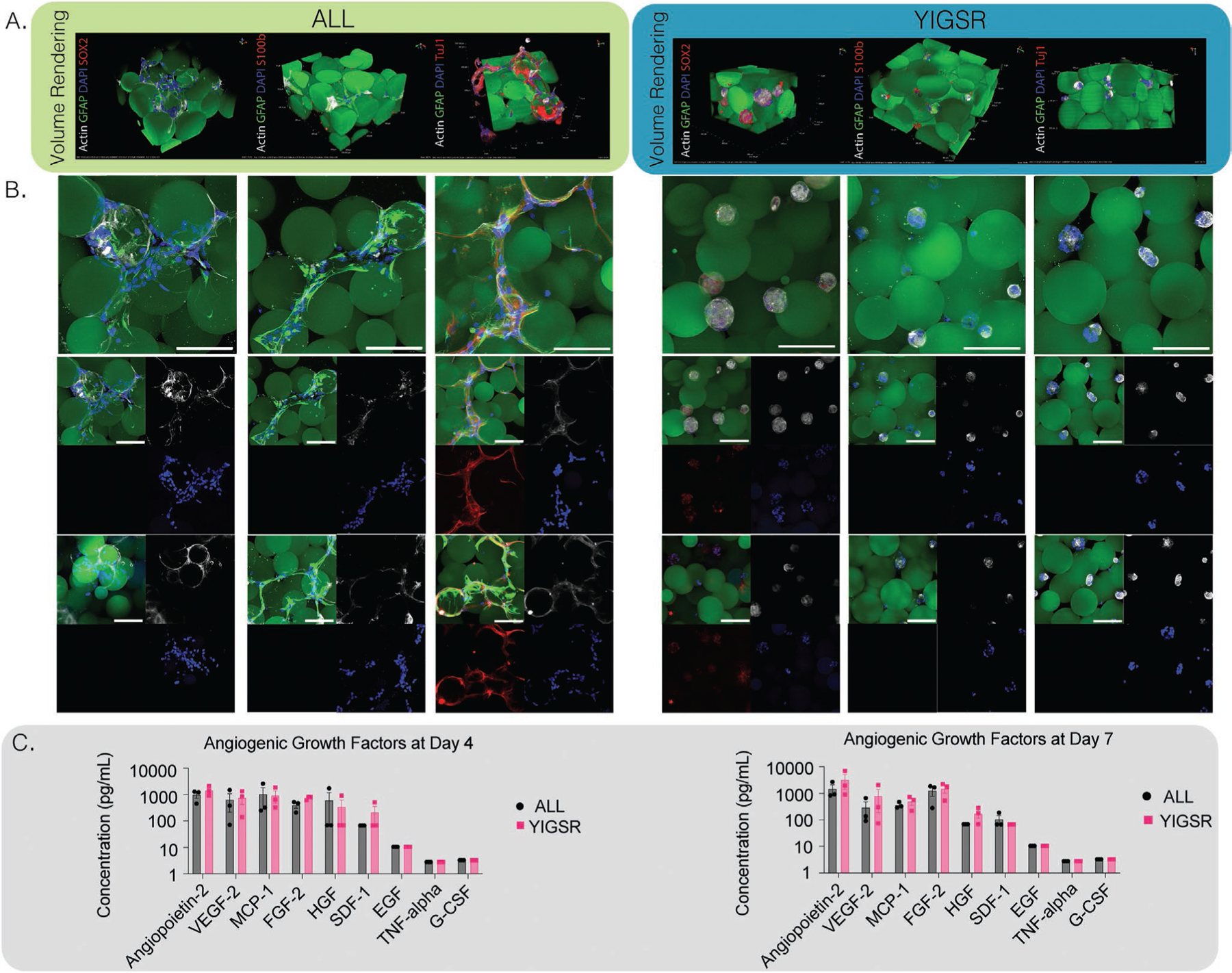
A) NPCs were cultured in vivo for 4 days. Confocal images were taken at 40× and volumed renderings or max intensity projections (MIPs) were displayed for ALL and YIGSR conditions. Scaffolds were stained for SOX2 for stemness, Tuj1 for neurogenesis, or S100b for astrogliosis, scale bar is 100 μm. B) Representative images of ALL and YIGSR conditions stained for differentiation markers, scale bar is 100 μm. C) Luminex multiplex immunoassay for angiogenic markers at day 4 and day 7 for ALL or YIGSR conditions, no statistical difference was observed between days.
2.10. YIGSR Maintains NPC Stemness and Neurospheres within Specific MAP Scaffold Pockets
As we reported earlier, NPCs in microgels decorated with YIGSR peptide only displayed neurosphere morphology across all 7 days. Staining of these scaffolds at D7 showed a lack of Tuj1 and S100b proteins. However, cells were positive for SOX2, indicating no differentiation in the NPCs occurs within this condition and they maintained stemness (Figure 5a). Unlike previously reported literature,[11] no degradation of the material is necessary to maintain the stemness of NPCs. The size of the neurospheres also appeared consistent once fully formed, averaging 45 μm in diameter (Figure S5, Supporting Information). Conditions containing a low amount of IKVAV also showed initial neurosphere formation at D1, but by D5 and D7 NPCs were seen to spread and have morphologies more consistent with microgels decorated with higher concentrations of IKVAV, or in combination with RGD (Figure S7, Supporting Information). To investigate whether YIGSR was necessary at all for neurosphere formation, two additional conditions were performed—a high YIGSR, containing 0.1 × 10−3 m of YIGSR and a no peptide condition (Figure S6, Supporting Information). While neurosphere formation was still observed in the no peptide condition, these NPCs were no longer positive for SOX2. This indicates a necessity of YIGSR in NPC stem maintenance, but not for neurosphere formation within our scaffolds.
2.11. Peptide Presentation Has No Significant Effect on Angiogenic Growth Factor Expression of NPCs within MAP Scaffolds
NPCs have also been known to express angiogenic growth factors that aid in vessel formation or protection through two-way communication with endothelial cells during development and injury.[46,47] Additionally, in vitro[48] and in vivo[49] co-cultures of endothelial cells and NPCs in a macroporous hydrogel showed significant increase in vessel formation through VEGF- and BDNF-mediated cross-talk. As such, we hypothesized that NPCs within our hydrogel system would produce similar angiogenic factors. For that reason, we investigated how changes in NPC morphology and their differentiation may lead to changes in their growth factor expression. Media was collected from either the ALL condition or YIGSR-only condition at days 4 and 7. Initial VEGF ELISA assays showed promising expression in the NPC media (data not shown). Therefore, we opted to run a standard angiogenesis multiplex containing nine different angiogenic factors to measure expression at days 4 and 7 (Figure 5c). While the expression of VEGF-2 was expected, we also observed expression of Angiopoietin-2 (ANG1), MCP-1, and FGF-2 proteins. There was a slight increase in Angiopoietin-2 and FGF-2 proteins from day 4 to day 7, whereas a slight decrease in VEGF-2 and MCP-1 proteins was seen from day 4 to day 7. However, no significant difference was observed in the amount of growth factors that were produced between groups or days (Figure 5c and Figure S8, Supporting Information).
The VEGF-2 and FGF-2 proteins are potent angiogenesis inducers and have previously been reported to be expressed by NPCs. FGF-2 is known to promote angiogenesis through VEGF expression in endothelial cells.[50] Additionally, when the expression of FGF-2 increases in NPCs, the potential for brain repair has been reported to be enhanced.[51] Similarly, NPCs and their differentiated neuronal cells are known to secrete VEGF-2.[52] The VEGF-2 protein is the major mediator of VEGF-A-mediated tropic effects in the vascular system and regulates the proliferation, migration, and survival of endothelial cells.[52] As such, it would be highly beneficial to maintain NPCs in a state that continually secretes these proteins in hopes to increase the revascularization of damaged brain tissue.
MCP-1 has also been recognized as an angiogenic chemokine and found to induce angiogenesis by up-regulating hypoxia-inducible factor 1 alpha (HIF-1 alpha) gene expression.[53] However, this observation was made in human aortic endothelial cells. In relation to NPCs, MCP-1 has mainly been studied for its ability to induce NPC migration in vitro[54,55] and in vivo.[56] Little research has explored NPC production of MCP-1 itself, although one study demonstrated MCP-1 protein production in NPCs when exposed to proinflammatory cytokine tumor necrosis factor alpha.[57] This may signify how NPCs can aid in angiogenesis or recruitment of additional NPCs in the brain by increased MCP-1 expression in disease models.
Similarly to MCP-1, ANG1 is another well-known angiogenic factor. Although its role in angiogenesis is believed to be context-dependent, either acting as an antagonist to angiogenesis or inducing endothelial cell functions,[58] its relation to NPCs has been in mediating differentiation and migration of NPCs in the SVZ after stroke.[59] Other research of ANG1 has also shown that when acting in concert with VEGF, ANG1 may stimulate endothelial cell migration and proliferation.[60,61] Emerging evidence continues to indicate that angiogenesis is coupled with neurogenesis under physiological and pathophysiological conditions, so it is no surprise that NPCs would be seen to express angiogenic factors just as endothelial cells have been reported to express NPC cytokines.[48,49] Further investigation on these two scaffolds in relation to co-cultures with endothelial cells would be required to fully elucidate if the amount of expression of these four growth factors is sufficient to be beneficial in angiogenesis after stroke. However, the changes in peptide presentation do not have significant effect on changing NPC expression of these specific growth factors after 7 days.
3. Conclusion
We have demonstrated the use of norbornene as a functional handle for stoichiometric post-modification of microgels to create an off-the-shelf material that can be easily tailored. We specifically showed this through developing two very different scaffolds containing laminin-derived peptides IKVAV, YIGSR, and RGD (ALL) or YIGSR-only. Scaffolds containing ALL peptides allowed for NPC migration and differentiation without the use of differentiation media or scaffold degredation. NPCs in the ALL condition were seen to fully spread by 12 h and continued to rearrange the scaffold even 4 days after initiation of the cell culture. In this condition, NPCs spread in a linear-like fashion, similar to the traveling of NPCs in vivo. Meanwhile, YIGSR-only scaffolds showed no NPCs spreading but instead formed neurospheres. These neurospheres were formed within 6 h and observed to keep moving through the scaffold but without contracting or adhering to the microgels as observed in the ALL condition. They also maintained their stemness and were similar in size to neurospheres of 45 μm in diameter as reported in literature.[38]
Additionally, the distinct differences in the morphology of NPCs within the scaffolds are correlated with changes in their differentiation, but not in growth factor expression. NPCs within the ALL condition that exhibited spreading were also seen to be positive for Tuj1, which is a structural protein found within neurons. When investigating the differential expression of growth factors in the media of these scaffolds from D4 to D7, these cells were seen to slightly increase in levels of ANG2 and FGF-2 proteins but slightly decrease in levels of VEGF-A and MCP-1 proteins. When their protein expressions are compared to the protein expression from YIGSR-only conditions, no significant differences were noticed between groups. However, the YIGSR NPCs were reported to express SOX2, indicating that YIGSR NPCs are maintaining their stemness potency. These growth factors are key contributors to vascularization,[46,47] supporting that NPCs may aid in re-vascularization of damaged brain tissue if NPCs are promoted to the sites of injury and kept within our scaffolds. While peptide presentation did not affect these growth factors, they should still be kept in consideration if NPC differentiation toward neurogenesis is desired.
4. Experimental Section
Modification of Hyaluronic Acid with Norbornene:
Hyaluronic acid–norbornene (HA–NB) was synthesized through the activation and subsequent functionalization of the HA carboxylic acid group by dissolving 1.0 g of HA (MW 79 000 Da) (Contipro) and 3.1 g 4-(4,6-dimethoxy[1.3.5] triazin-2-yl)-4-methylmorpholinium chloride (DMTMM) (MW: 294.74 Da) (TCI America, Portland, OR, USA) (4 molar equivalents) each in 40 mL of 200 × 10−3 m MES buffer, pH 5.5, combining the solutions and allowing the reaction to stir for 10 min. Then 0.677 mL of 5-norbornene-2-methanamine (a mixture of isomers) (NMA) (TCI America, Portland, OR) (2 molar equivalents) was added drop-wise into the mixture. The reaction was stirred at room temperature overnight and then precipitated in 1 L of 100% Ethanol. All precipitates were collected and dissolved in 2 m brine solution and dialyzed against DI water for 30 min then 1 m brine solution for 30 min. This dialysis process was repeated three times and then dialyzed against DI water for 24 h. The final solution was collected and lyophilized to yield the final product. HA–NB was confirmed by 1H-NMR with 33.5% NB functionalization. 1H NMR shifts of pendant norbornenes in D20, δ6.33 and δ6.02 (vinyl protons, endo), and δ6.26 and δ6.23 ppm (vinyl protons, exo) were compared to the HA methyl group δ2.05 ppm to determine functionalization. All equivalents were based on the moles of the HA repeat unit.
Tetrazine–Peptide Synthesis and Purification:
Peptides were synthesized by microwave-assisted solid-phase synthesis on a CEM Liberty Blue instrument using rink amide ProTide Resin (CEM #R003). Methyltetrazine was conjugated to the N-terminus of peptides by on-resin reaction with methyltetrazine–NHS ester (Click Chemistry Tools #1128) in dimethyl sulfoxide. Peptides were cleaved from resin using a cleavage cocktail containing 95% trifluoroacetic acid, 2.5% water, and 2.5% triisopropylsilane, followed by precipitation in cold diethyl ether. Peptides were purified by reverse-phase HPLC on a C18 column. Peptide identity was confirmed using MALDI mass spectrometry on a Bruker Autoflex Speed LRF instrument withα-cyano-4-hydroxycinnamic acid as the matrix.
Synthesis of Tetrazine Crosslinker and Alexa Fluors:
Tetra-polyethylene glycol–tetrazine (4arm-PEG–Tet) was synthesized as previously described.[17] Briefly, 100 mg of tetra-PEG-SH (MW: 20 000 Da) (NOF America, White Plains, NY, USA) and 15 mg of methyltetrazine–PEG4–maleimide (MW: 514.53 Da) (Kerafast, Boston, MA, USA) were dissolved in 0.5 mL CDCl3(maleimide/SH ratio of 1.05), then adding 1 μL of triethylamine (TEA) (0.5 molar equivalent). The mixture was stirred at room temperature for 4 h. The product was precipitated in 50 mL cold diethyl ether and confirmed by 1H-NMR with 76.5% conversion (Figure S9, Supporting Information).
Alexa Fluor 488/555/647 C2–tetrazine (Alexa488/555/647–Tet) was synthesized through two base-catalyzed thiol-Michael addition reactions in series, as previously described.[17] Briefly, 2.8 mg of HS-PEG-SH (MW: 3500 Da) (JenKem Technology USA, Plano, TX, USA) (1 molar equivalent to Alexa Fluor C2 maleimide) and 0.41 mg of methyltetrazine–PEG4–maleimide (MW: 514.53 Da) (Kerafast, Boston, MA, USA) (1 molar equivalent to Alexa Fluor C2 maleimide) were dissolved in 0.17 mL CDCl3, then adding 0.11 μL of triethylamine (TEA) (0.5 molar equivalent). The mixture was stirred at room temperature overnight. Next, by dissolving 1 mg Alexa Fluor 488 C2 maleimide (MW: 1250 Da) (Thermo Fisher Scientific) in 0.17 mL CDCl3, then adding the reaction mixture and 0.11 μL of triethylamine (TEA) (0.5 molar equivalent). The mixture was stirred at room temperature overnight. The product was precipitated in 10 mL of cold diethyl ether and dried under vacuum overnight. The resulting product was dissolved in dimethylformamide at 1 mg mL−1 and stored at −20 °C.
Microgel Production and Purification:
HA–NB microgels were produced using a planar flow-focusing microfluidic device to create uniform particles (Figure 1A). A 1 mL gel precursor solution was made by dissolving HA–NB in 50 × 10−3 m HEPES pH 7.5, di-thiol MMP sensitive linker peptide (Ac-GCRDGPQGIWGQDRCG-NH2, Genscript) (SH/HA ratio of 14), tris(2-carboxyethyl)phosphine (TCEP) (Sigma-Aldrich) (TCEP/SH ratio of 0.25) and 9.90 × 10−3 m lithium phenyl(2,4,6-trimethylbenzoyl)phosphinate photoinitiator (LAP) (TCI America, Portland, OR, USA). The final HA–NB in the precursor solution should be at 3.5% (w/v). The solution was filtered through a 0.22 μm sterile filtered before being transferred to a 1 mL BD Leur-Lok syringe and connected to the inner inlet of the microfluidic device using tubing. A 5 mL BD Leur-Lok syringe was filled with 5% (v/v) Span-80 in heavy mineral oil and attached to the outer inlet of the microfluidic device. A single syringe pump was used to push the differently sized syringes at asymmetric flow rates, as previously described[62] at a flow rate ratio of ≈6.4:1 (oil:aqueous). The precursor solution was pinched at the flow focusing region by the oil and the microgel droplets were crosslinked by consistent exposure to UV light (20 mW cm−2) off chip using an OmniCure LX500 LED Spot UV curing system controller with a OmnniCure LX500 LED MAX head at 365 nm wavelength, 30% power. A 15 mL conical tube wrapped in foil was used to collect the microgel emulsion.
Upon completion of fabrication, the suspension was centrifuged at 5250 rcf for 5 min and the supernatant oil was aspirated off in a sterile hood. Everything following was performed under sterile conditions. The microgels were then washed with sterile filtered HEPES buffer and centrifuged again at the aforementioned conditions. Washing was repeated several times until no more oil was visible in the supernatant. Following washes, endotoxin tests were performed. Endotoxin concentrations were determined with the Pierce LAL Chromogenic Endotoxin Quantitation Kit (Thermo Fisher Scientific) following the manufacturer’s instructions. Particle endotoxin levels were consistently below 0.2 endotoxin U mL−1. Particles were then stored at 4 °C until further use.
μBead Post-Fabrication Modification:
HA–NB μbeads were modified post-fabrication via inverse electron demand Diels–Alder tetrazine–norbornene click reaction, in which excess norbornene groups on the microgels were functionalized with Tet–AF 488, Tet–AF 555, and/or Tet–AF 647 to fluorescently tag the microgels and determine post-modification efficiency. For all in vitro experiments and color combinations (Figure 2c), the microgels were incubated in HEPES containing a final Tet–AF 488, 555, and/or 647 concentration of 0.005 × 10−3 m at 37 °C for 1 h under agitation based on the kinetics from literature.[63] Upon completion of the functionalization, the suspended beads were pelleted by centrifuging at 14 000 rcf for 5 min. The microgels were washed three times with 1× HEPES and recovered with the same centrifugation conditions. Each color combination was imaged on a Nikon Ti Eclipse and compared to the predicted Adobe Illustrator generated color ratios (Figure 2c). For determining increase in intensity over increase in concentration, microgels were incubated as mentioned above but at various × 10−3 m concentrations. The intensity of the labeled μbeads was measured using a Tecan Spark Plate reader for comparison and to measure saturation of post-modification (Figure 2e).
Generation of Scaffolds and Mechanical Testing:
MAP scaffolds were assembled via inverse-electron-demand Diels–Alder tetrazine–norbornene click reaction, in which excess norbornene groups on microgels averaging 100 μm were linked by 4arm-PEG–Tet to form tetrazine-mediated MAP (Tet-MAP) scaffolds. Stiffness of the annealed Tet-MAP scaffolds was measured as the storage modulus (G′) using a plate-to-plate rheometer (Physica MCR, Anton Paar, Ashland, VA, USA). The Tet-MAP scaffolds were formed by combining and mixing microgels with 4arm-PEG–Tet at the desired tetrazine/HA–NB (Tet/HA) ratios of 5, 7, 8, and 21, where an R7 Tet/HA ratio was 28 tet groups/69.8 NB groups or a 0.4 ratio for this specific 33.5% modified HA (Figure 1C). 50 μL of the solution was then placed between two sigma-coated (Sigma-Aldrich) slides with 1 mm spacers on either side and fastened using binder clips and incubated for 2 h at 37 °C unless stated otherwise. This method was used to create triplicate 8 mm by 1 mm height discs of the Tet-MAP scaffolds for rheological testing. The crosslinked gel was transferred into PBS overnight. A frequency sweep was performed on the scaffolds using a strain of 1% with an angular frequency range of 0.5 to 10 rad s−1.
NPCs Isolation:
NPCs were gifted from Dr. Chay Kuo’s lab at Duke University. In brief, SVZ wholemount from P12 C57BL/6J mice was dissected and placed in DMEM/F12 (DF) media plus 100 units mL−1 penicillin, 100 μg mL−1 streptomycin, and 250 ng mL−1 amphotericin B. The dissected tissues were incubated in 0.005% trypsin (15 min, 37 °C, and pH 7.3) and placed overnight in uncoated T75 plastic tissue-culture dishes in N5 medium; containing 35 μg mL−1 bovine pituitary extract, abx, 5% FCS (HyClone), N2 supplements, 40 ng mL−1 EGF, and basic FGF. Unattached cells were collected and subsequently plated again onto a 6 well TC treated plate. These cells were proliferated to confluency in N5 media. Fresh EGF and FGF (20 ng each) were added every other day. The cells were proliferated to 85–95% confluency, until neurospheres formed on the plate, at which point they were trypsinized, collected, and frozen until further use. These cells were marked as passage 2 (P2) and before further use, NPCs were confirmed by checking whether they were positive for GFAP and SOX2 proteins using flow cytometry. Generally, cells were passaged two or three times by using 0.005% trypsin and N5 media, with 20 ng mL−1 EGF/FGF supplementation every other day.
Seeding NPCs in Microgel Scaffolds:
Prior to their use in vitro, microgels were post-modified with RGD (1 × 10−3 m), IKVAV high (0.3 × 10−3 m) or low (0.01 × 10−3 m), and/or YIGSR (0.048 × 10−3 m) (Figure 3c). Additional groups were studied as well, containing high YIGSR (0.1 × 10−3 m) only, no peptides, or RGD (1 × 10−3 m) only (Figure S6, Supporting Information). The RGD peptide (Ac-RGDSPGERCG-NH2) contained a thiol and thus underwent a thiol–ene click reaction using LAP (9.90 × 10−3 m) and UV light for 2 min. Upon completion of the functionalization, the suspended μbeads were pelleted by centrifuging at 14 000 rcf for 5 min. The microgels were washed three times with 1× HEPES to get rid of any excess LAP and recovered with the same centrifugation conditions. IKVAV and YIGSR were synthesized to have a tetrazine group and therefore reacted via tetrazine–norbornene click reaction. For IKVAV and YIGSR post-modifications, μbeads were incubated with the desired concentration and left to react for 2 h at rt before use. RGD-modified μbeads were modified with IKVAV and/or YIGSR after the RGD washes. All microgels were allowed to soak in culture media for a minimum of 2 h prior to centrifuging them again for “dry” microgels before mixing with cells.
Cell Staining and Imaging:
Cell cultures were aspirated of media and fixed at day 7 with 4% PFA for 30 min at room temperature and then washed with 1× PBS 3× for 15 min each wash before staining. Cells were incubated with primary antibodies at the appropriate dilutions in blocking solution 2 h to overnight at 4 °C. After 3 × 5-min washes in 1× PBS, samples were incubated with secondary antibodies at the appropriate dilution in clocking solution for 2 h at room temperature. The samples were then counterstained with nuclear marker DAPI (1:500, Invitrogen) for 15 min at room temperature and then washed and kept in 1× PBS until imaged. Primary antibodies used were as follows: rabbit anti-SOX2 (SOX2, 1:100, Abcam Cambridge, MA, USA, ab97959) for maintained stem cell marker, rabbit anti-beta III Tubulin (Tuj1, 1:100, Abcam Cambridge, MA, USA, ab18207) for neuronal cell marker differentiation, rabbit anti-glial fibrillary acidic protein (GFAP, 1:300, Invitrogen, 13–0300) for NPC lineage marker, or rabbit anti-S100 beta (S100b, 1:100, Abcam Cambridge, MA, USA, ab41548) for astroglia cell marker differentiation. Secondary antibodies conjugated to Alexa Fluor 555 (Donkey anti-rabbit 555, 1:1000, Abcam, Cambridge, MA, USA, ab150074) and Alexa Fluor 647 Phalloidin (F-actin, 1:40, Invitrogen, A22287) for F-actin staining were used. A Nikon Ti Eclipse scanning confocal microscope equipped with a C2 laser with a 20× air objective or 40× water objective was used to take fluorescence images represented as maximum intensity projections. Phase images were taken on a Zeiss Observer Z1 with a 20× air objective for days 0, 3, 5, and 7. Bright-field images for the NPC videos were taken with at the Duke Light Microscopy Core Facility on an Olympus VivaView FL Incubator microscope over the course of 4 days with a 20× air objective.
Imaging Analysis:
Fiji/ImageJ was used to analyze phase or bright-field images. Fiji’s manual tracking was used to output x and y coordinates of moving microgels from live phase images that were taken over the course of 4 days. IMARIS software was used to create surface renderings from confocal images. This data output cell spreading and neurosphere volume and diameter from phalloidin stains, and adjacent microgels based on microgel staining.
Statistical Analysis:
Statistical analysis and plotting were performed using GraphPad Prism 9. Mechanical testing was performed with three technical replicate gels. Statistics assumed that gel samples, which were cast independently, were statistically independent from each other. In vitro experiments were repeated to contain three biological replicates, each with three technical replicates. Each biological replicate contained microgels generated from a different batch and NPCs extracted from a different extraction. Statistical significance was assessed using the biological replicate data, a 95% confidence interval using a one-way ANOVA with Tukey post-hoc test, unless otherwise noted. All error is reported as the standard deviation of error (SD).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
The authors would like to acknowledge the National Institutes of Health and the National Institute of Neurological Disorders and Stroke for funding (R01NS079691). This work was performed in part at the Duke University Shared Materials Instrumentation Facility (SMIF), which was supported by the National Science Foundation (award number ECCS-2025064). This work was performed in part at the Duke University Light Microscopy Core Facility (LMCF) and with help from their staff.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The authors declare no conflict of interest.
Contributor Information
Katrina L. Wilson, Department of Biomedical Engineering, Duke University, Durham, NC 27708-0281, USA
Sasha Cai Lesher Pérez, Department of Chemical Engineering, University of Michigan, North Campus Research Complex, Building 28, 2800 Plymouth Rd, Ann Arbor, MI 48109-2800, USA.
Moawiah M. Naffaa, Department of Cell Biology, Duke University School of Medicine, Durham, NC 27710, USA; Department of Psychology and Neuroscience, Duke University, Durham, NC 27708, USA
Sean H. Kelly, Department of Biomedical Engineering, Duke University, Durham, NC 27708-0281, USA
Tatiana Segura, Department of Biomedical Engineering, Duke University, Durham, NC 27708-0281, USA; Department of Neurology, Duke University, Durham, NC 27708-0281, USA; Department of Dermatology, Duke University, Durham, NC 27708-0281, USA.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- [1].Obernier K, Alvarez-Buylla A, Development 2019, 4, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Martinez-Cerdeno V, Noctor SC, Front. Neuroanat 2018, 12, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Goldman S, Nat. Biotechnol 2005, 23, 862. [DOI] [PubMed] [Google Scholar]
- [4].Arvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O, Nat. Med 2002, 8, 963. [DOI] [PubMed] [Google Scholar]
- [5].Nih LR, Sideris E, Carmichael ST, Segura T, Adv. Mater 2017, 29, 1606471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sideris E, Yu A, Chen J, Carmichael ST, Segura T, bioRxiv 76829 2019, 10.1101/768291. [DOI] [Google Scholar]
- [7].Caliari SR, Burdick JA, Nat. Methods 2016, 13, 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Oliveira JM, Carvalho L, Silva-Correia J, Vieira S, Majchrzak M, Lukomska B, Stanaszek L, Strymecka P, Malysz-Cymborska I, Golubczyk D, Kalkowski L, Reis RL, Janowski M, Walczak P, npj Regener. Med 2018, 3, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Madhusudanan P, Raju G, Shankarappa S, J. R. Soc., Interface 2020, 17, 20190505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Farrukh A, Ortega F, Fan W, Marichal N, Paez JI, Berninger B, Campo AD, Salierno MJ, Stem Cell Rep 2017, 9, 1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Madl CM, LeSavage BL, Dewi RE, Dinh CB, Stowers RS, Khariton M, Lampe KJ, Nguyen D, Chaudhuri O, Enejder A, Heilshorn SC, Nat. Mater 2017, 16, 1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Griffin DR, Weaver WM, Scumpia PO, Di Carlo D, Segura T, Nat. Mater 2015, 14, 737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Riley L, Schirmer L, Segura T, Curr. Opin. Biotechnol 2019, 60, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Daly AC, Riley L, Segura T, Burdick JA, Nat. Rev. Mater 2020, 5, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Maeda K, Onoe H, Takinoue M, Takeuchi S, Adv. Mater 2012, 24, 1340. [DOI] [PubMed] [Google Scholar]
- [16].Jivan F, Yegappan R, Pearce H, Carrow JK, McShane M, Gaharwar AK, Alge DL, Biomacromolecules 2016, 17, 3516. [DOI] [PubMed] [Google Scholar]
- [17].Darling NJ, Xi W, Sideris E, Anderson AR, Pong C, Carmichael ST, Segura T, Adv. Healthcare Mater 2020, 9, e1901391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Anseth KS, Klok HA, Biomacromolecules 2016, 17, 1. [DOI] [PubMed] [Google Scholar]
- [19].Gopinathan J, Noh I, Tissue Eng. Regener. Med 2018, 15, 531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Alder O. D. a. K., Justus Liebigs Ann. Chem 1928, 460, 98. [Google Scholar]
- [21].Haiber LM, Kufleitner M, Wittmann V, Front. Chem 2021, 9, 654932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Devaraj NK, Weissleder R, Hilderbrand SA, Bioconjugate Chem 2008, 19, 2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Alge DL, Azagarsamy MA, Donohue DF, Anseth KS, Biomacromolecules 2013, 14, 949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Muir VG, Qazi TH, Shan J, Groll J, Burdick JA, ACS Biomater. Sci. Eng 2021, 7, 4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Li Z, Mak SY, Sauret A, Shum HC, Lab Chip 2014, 14, 744. [DOI] [PubMed] [Google Scholar]
- [26].Truong NF, Kurt E, Tahmizyan N, Lesher-Perez SC, Chen M, Darling NJ, Xi W, Segura T, Acta Biomater 2019, 94, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sideris E, Griffin DR, Ding Y, Li S, Weaver WM, Di Carlo D, Hsiai T, Segura T, ACS Biomater. Sci. Eng 2016, 2, 2034. [DOI] [PubMed] [Google Scholar]
- [28].Georges PC, Miller WJ, Meaney DF, Sawyer ES, Janmey PA, Biophys. J 2006, 90, 3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Seri B, Garcia-Verdugo JM, McEwen BS, Alvarez-Buylla A, J. Neurosci 2001, 21, 7153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Agius E, Sagot Y, Duprat AM, Cochard P, Neuroscience 1996, 71, 773. [DOI] [PubMed] [Google Scholar]
- [31].Graf J, Iwamoto Y, Sasaki M, Martin GR, Kleinman HK, Robey FA, Yamada Y, Cell 1987, 48, 989. [DOI] [PubMed] [Google Scholar]
- [32].Graf J, Ogle RC, Robey FA, Sasaki M, Martin GR, Yamada Y, Kleinman HK, Biochemistry 1987, 26, 6896. [DOI] [PubMed] [Google Scholar]
- [33].Li XW, Liu XY, Josey B, Chou CJ, Tan Y, Zhang N, Wen XJ, Stem Cells Transl. Med 2014, 3, 662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Callahan LAS, Xie SB, Barker IA, Zheng JK, Reneker DH, Dove AP, Becker ML, Biomaterials 2013, 34, 9089. [DOI] [PubMed] [Google Scholar]
- [35].Lam J, Carmichael ST, Lowry WE, Segura T, Adv. Healthcare Mater 2015, 4, 534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Park K, Nam Y, Choi Y, In Vitro Cell. Dev. Biol.: Anim 2015, 51, 455. [DOI] [PubMed] [Google Scholar]
- [37].Shou Y, Liang F, Xu S, Li X, Front. Cell Dev. Biol 2020, 8, 579659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Cordey M, Limacher M, Kobel S, Taylor V, Lutolf MP, Stem Cells 2008, 26, 2586. [DOI] [PubMed] [Google Scholar]
- [39].Nomizu M, Kim WH, Yamamura K, Utani A, Song SY, Otaka A, Roller PP, Kleinman HK, Yamada Y, J. Biol. Chem 1995, 270, 20583. [DOI] [PubMed] [Google Scholar]
- [40].Pan L, North HA, Sahni V, Jeong SJ, McGuire TL, Berns EJ, Stupp SI, Kessler JA, PLoS One 2014, 9, e104335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Brooker SM, Bond AM, Peng CY, Kessler JA, Glia 2016, 64, 1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Farrukh A, Zhao SF, del Campo A, Front. Mater 2018, 5, 62. [Google Scholar]
- [43].Lois C, Garcia-Verdugo JM, Alvarez-Buylla A, Science 1996, 271, 978. [DOI] [PubMed] [Google Scholar]
- [44].Sun W, Kim H, Moon Y, Anat. Cell Biol 2010, 43, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lindvall O, Kokaia Z, Cold Spring Harbor Perspect. Biol 2015, 7, a019034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wang J, Chen Y, Yang Y, Xiao X, Chen S, Zhang C, Jacobs B, Zhao B, Bihl J, Chen Y, Mol. Brain 2016, 9, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Tata M, Ruhrberg C, Neuronal Signaling 2018, 2, NS20170139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Li Q, Ford MC, Lavik EB, Madri JA, J. Neurosci. Res 2006, 84, 1656. [DOI] [PubMed] [Google Scholar]
- [49].Ford MC, Bertram JP, Hynes SR, Michaud M, Li Q, Young M, Segal SS, Madri JA, Lavik EB, Proc. Natl. Acad. Sci. USA 2006, 103, 2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Seghezzi G, Patel S, Ren CJ, Gualandris A, Pintucci G, Robbins ES, Shapiro RL, Galloway AC, Rifkin DB, Mignatti P, J. Cell Biol 1998, 141, 1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Dayer AG, Jenny B, Sauvain MO, Potter G, Salmon P, Zgraggen E, Kanemitsu M, Gascon E, Sizonenko S, Trono D, Kiss JZ, Brain 2007, 130, 2962. [DOI] [PubMed] [Google Scholar]
- [52].Wittko-Schneider IM, Schneider FT, Plate KH, Cell. Mol. Life Sci 2013, 70, 1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Hong KH, Ryu J, Han KH, Blood 2005, 105, 1405. [DOI] [PubMed] [Google Scholar]
- [54].Widera D, Holtkamp W, Entschladen F, Niggemann B, Zanker K, Kaltschmidt B, Kaltschmidt C, Eur. J. Cell Biol 2004, 83, 381. [DOI] [PubMed] [Google Scholar]
- [55].Xu Q, Wang S, Jiang X, Zhao Y, Gao M, Zhang Y, Wang X, Tano K, Kanehara M, Zhang W, Ishida T, Clin. Exp. Pharmacol. Physiol 2007, 34, 624. [DOI] [PubMed] [Google Scholar]
- [56].Magge SN, Malik SZ, Royo NC, Chen HI, Yu L, Snyder EY, O’Rourke DM, Watson DJ, J. Neurosci. Res 2009, 87, 1547. [DOI] [PubMed] [Google Scholar]
- [57].Sheng WS, Hu S, Ni HT, Rowen TN, Lokensgard JR, Peterson PK, J. Leukocyte Biol 2005, 78, 1233. [DOI] [PubMed] [Google Scholar]
- [58].Akwii RG, Sajib MS, Zahra FT, Mikelis CM, Cells 2019, 8, 471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Liu XS, Chopp M, Zhang RL, Hozeska-Solgot A, Gregg SC, Buller B, Lu M, Zhang ZG, J. Biol. Chem 2009, 284, 22680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Lobov IB, Brooks PC, Lang RA, Proc. Natl. Acad. Sci. USA 2002, 99, 11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Zhu Y, Lee C, Shen F, Du R, Young WL, Yang GY, Stroke 2005, 36, 1533. [DOI] [PubMed] [Google Scholar]
- [62].Lesher-Perez SC, Weerappuli P, Kim SJ, Zhang C, Takayama S, Micromachines 2014, 5, 1254. [Google Scholar]
- [63].Oliveira BL, Guo Z, Bernardes GJL, Chem. Soc. Rev 2017, 46, 4895. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.