

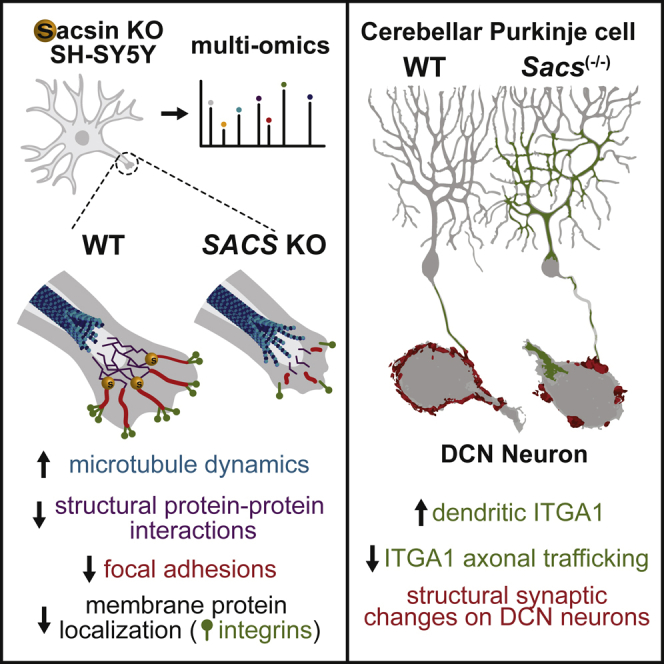
Summary
Autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) is a childhood-onset cerebellar ataxia caused by mutations in SACS, which encodes the protein sacsin. Cellular ARSACS phenotypes include mitochondrial dysfunction, intermediate filament disorganization, and progressive death of cerebellar Purkinje neurons. It is unclear why the loss of sacsin causes these deficits or why they manifest as cerebellar ataxia. Here, we perform multi-omic profiling in sacsin knockout (KO) cells and identify alterations in microtubule dynamics and mislocalization of focal adhesion (FA) proteins, including multiple integrins. Deficits in FA structure, signaling, and function can be rescued by targeting PTEN, a negative regulator of FA signaling. ARSACS mice possess mislocalization of ITGA1 in Purkinje neurons and synaptic disorganization in the deep cerebellar nucleus (DCN). The sacsin interactome reveals that sacsin regulates interactions between cytoskeletal and synaptic adhesion proteins. Our findings suggest that disrupted trafficking of synaptic adhesion proteins is a causal molecular deficit in ARSACS.
Keywords: ARSACS, sacsin, proteomics, cell surface, microtubules, focal adhesions, synaptic adhesion proteins, integrins, Purkinje neurons, synapse
Graphical abstract
Highlights
-
•
Sacsin regulates cytoskeletal organization, microtubule dynamics, and protein trafficking
-
•
Focal adhesion structure, function, and signaling are affected in sacsin knockout cells
-
•
Cerebellar Purkinje neurons have altered integrin localization and synaptic structure
Romano et al. comprehensively characterize the protein landscape of ARSACS model cells, finding that sacsin regulates several core cellular processes including regulation of protein activity, protein trafficking, cell structure, and localization of synaptic adhesion proteins. These findings help explain why the loss of sacsin causes neurodegeneration and cerebellar ataxia.
Introduction
Autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) is a childhood-onset neurological disease characterized by pyramidal spasticity, cerebellar ataxia, and Purkinje cell loss, which is thought to have both neurodegenerative and neurodevelopmental components (Vermeer et al., 1993). ARSACS was initially believed to be restricted to the Charlevoix-Saguenay region of Quebec, Canada, due to a founder effect mutation (Bouchard et al., 1978). However, since the discovery of the causal gene, more than 170 distinct mutations in SACS have been identified worldwide, and ARSACS is now estimated to be the second most common autosomal recessive cerebellar ataxia (Engert et al., 2000; Synofzik et al., 2013).
Sacsin/DNAJC29 expression is ubiquitous but is especially high in large neurons in brain regions associated with motor systems, including layer-V pyramidal neurons in the motor cortex and cerebellar Purkinje cells (Saunders et al., 2018). Sacsin is a large 520 kDa modular protein with domains that implicate it in molecular chaperone and protein quality control systems (Anderson et al., 2010; Parfitt et al., 2009). These include an N-terminal ubiquitin-like domain, regions of homology to the ATPase domain of Hsp90, and a functional J-protein domain, suggesting that sacsin has the ability to modulate Hsp70 chaperone activity. However, the large size of sacsin has hampered biochemical and structural investigations into its function. Patient-derived fibroblasts and sacsin knockout (KO) cell models demonstrate reorganization of the vimentin intermediate filament cytoskeleton, altered mitochondrial network dynamics and trafficking, decreased mitochondrial respiration, and increased mitochondrial stress (Bradshaw et al., 2016; Duncan et al., 2017; Gentil et al., 2019; Girard et al., 2012; Lariviere et al., 2015). Aptamer-based proteomics in sacsin KO SH-SY5Y neuroblastoma cells also found altered expression of proteins involved in synaptogenesis and cell engulfment (Morani et al., 2020). Sacs(−/−) mice recapitulate the motor deficits and cerebellar atrophy observed in ARSACS patients, and undergo progressive age-dependent loss of cerebellar Purkinje neurons, abnormal bundling of non-phosphorylated neurofilament (Lariviere et al., 2015, 2019), and changes to the structure of Purkinje neuron synapses in the deep cerebellar nucleus (DCN) (Ady et al., 2018).
While these cellular phenotypes may affect neuron function and survival, their precise relationship to neurodegeneration in ARSACS is unclear. For example, diverse neurodegenerative diseases exhibit altered mitochondrial dynamics and intermediate filament phenotypes (Didonna and Opal, 2019; Stanga et al., 2020), although whether these phenotypes are causal or merely components of a conserved neurodegenerative cascade is an important unanswered question (Gan et al., 2018). Here, we take a multi-omic approach to determine how the loss of sacsin causes these phenotypes and why this disease manifests as a cerebellar ataxia. Our data suggest that altered trafficking of synaptic adhesion proteins is a causal molecular deficit in ARSACS.
Results
Comprehensive proteomic characterization of sacsin KO cells
To understand the molecular deficiencies in ARSACS, we generated a sacsin KO human SH-SY5Y cell line (Figure S1A), which is widely used to model neurodegenerative diseases (Xicoy et al., 2017). Consistent with ARSACS patient fibroblasts (Duncan et al., 2017) and Sacs(−/−) mice (Lariviere et al., 2015), KO cells had abnormal bundling and asymmetric partitioning of multiple intermediate filaments, including vimentin (Figures 1A and S1B), neurofilament heavy, and peripherin (Figures S1C–S1E). As phosphorylation is a key post-translational modification controlling intermediate filament assembly and disassembly (Snider and Omary, 2014), we performed quantitative proteomic and phosphoproteomic profiling of sacsin KO cells (Table S1). We identified decreased abundance of several proteins previously described in ARSACS patient fibroblasts, including vimentin, the mitochondrial protein ATP5J, and the autophagy-regulated scaffold SQSMT1/p62 (Duncan et al., 2017) (Figure 1B and Table S1). Among the overabundant proteins were the tau-tubulin kinase 1 (TTBK1) and microtubule-associated protein tau (MAPT) (Figures 1B and S1F–S1I), which was hyperphosphorylated at several sites (Figures S1J–S1L). To assess the functional significance of each phosphosite, we analyzed our data in light of a recent machine-learning approach that estimated the effects of individual phosphosites on organism fitness (Ochoa et al., 2020). This analysis identified several highly functional hypophosphorylated residues in vimentin and the nuclear lamina intermediate filaments LMNA/LMNB2 (Figure 1C), which is intriguing considering that ARSACS neurons have altered nuclear shape and positioning (Duncan et al., 2017). Other hypophosphorylated proteins included the focal adhesion (FA) protein zyxin (ZYX) and ataxin 2-like protein (ATXN2L). In addition to tau, several other microtubule-regulating proteins were hyperphosphorylated, including the primary cilia protein ARL3 (Zhou et al., 2006), and the scaffold stathmin (STMN1), which promotes microtubule assembly in a pS16-dependent fashion (Di Paolo et al., 1997). When analyzing changes in phosphorylation corrected for changes in total protein levels, the most hypophosphorylated proteins were RPS6, NLM1, and ATXN2L, which have been implicated in neuronal autophagy and likely reflect increased autophagy in sacsin KO cells (Figure S1M) (Duncan et al., 2017; Key et al., 2020; Klionsky et al., 2021; Tang et al., 2021). The most hyperphosphorylated residues were again in microtubule-related proteins, such as HN1/JPT1 and ARL3. In all, these results suggest that altered phosphorylation may be a contributing factor to cellular ARSACS phenotypes.
Figure 1.

Proteomic profiling of sacsin KO cells
(A) Representative confocal images of control (WT) and sacsin KO SH-SY5Y neuroblastoma cells immunostained for the intermediate filament protein vimentin. Scale bars, 10 μm.
(B) Global proteomic profiling of sacsin KO SH-SY5Y cells. Significance cutoffs: p < 0.05 and log2 fold change (f.c.) ±0.4, denoted by black outline.
(C) Functional analysis of altered phosphosites in sacsin KO cells. y axis is the functional score assigned by Ochoa et al. (2020), with higher scores reflecting increased effects on fitness. Dot color and size reflect log2 f.c. Black outlines label phosphosites with p < 0.05 and log2 f.c. ±0.4.
(D) Phylogenetic tree of the kinome in sacsin KO cells. Color indicates log2 f.c. of kinase abundance, size indicates −log10 p value. Underlined abbreviations refer to phylogenetically related kinase families.
(E) Protein map of tau isoform 2 (2N4R). Phosphosites identified in phosphoproteomic profiling are labeled above diagram. Tau kinases identified in the kinome profiling are listed below, indicating validated phosphosites. Colored circles correlate with log2 f.c. of differentially expressed phosphosites or kinases.
(F–H) Western blot and quantification for BRSK2, and the BRSK2 target residue pTAU S262. n = 3, SEM, Student’s t test, ∗∗∗p < 0.001.
Kinases are attractive drug targets (Krahn et al., 2020) but are typically lowly expressed and difficult to detect with standard proteomics. Therefore, we enriched for kinases using multiplexed kinase inhibitor beads and performed quantitative mass spectrometry (Cooper et al., 2013). The kinome was broadly altered in sacsin KO cells (Figures S1N and S1O; Table S1). Interestingly, specific families were generally misexpressed in similar directions. For example, the tyrosine kinase family (TK) members were generally downregulated, while CMGC family members were generally upregulated (Figure 1D). Strikingly, we identified ten overexpressed kinases which directly phosphorylate tau at residues that were hyperphosphorylated in sacsin KO cells (Figures 1E and S1N). The most overabundant kinase, BRSK2, and additional CAMK family members MARK1/2/3, all phosphorylate Ser262 in the microtubule-binding domain of tau (Ando et al., 2016; Kishi et al., 2005) (Figures 1E–1H). Phosphorylation of tau Thr231 by DYRK1A is also associated with the detachment of tau from microtubules (Coutadeur et al., 2015; Sengupta et al., 1998). In pathological settings, tau overabundance and hyperphosphorylation can cause the aggregation of insoluble tau and the formation of neurofibrillary tangles. However, we did not find evidence of increased tau aggregation in either undifferentiated or neuronally differentiated sacsin KO cells (Figure S1P). Yet, independent of aggregation, tau phosphorylation can affect microtubule stability, interfere with motor protein function, and disrupt axonal trafficking (Dixit et al., 2008; Ikezu et al., 2020; Stoothoff and Johnson, 2005). Combined with the altered phosphorylation of other microtubule-related proteins, these data suggest that microtubule structure or function may be altered in sacsin KO cells.
Microtubule organization and dynamics are altered in sacsin KO cells
We next sought to determine whether microtubule structure and function are affected in sacsin KO cells. We found that cage-like vimentin bundles form around γ-tubulin, a marker of the microtubule organizing center (MTOC), which is a central hub for microtubule nucleation and cargo transport (Martin and Akhmanova, 2018) (Figures 2A and S2A). Acetylated α-tubulin, a microtubule-stabilizing post-translational modification, was increased in sacsin KO cells without affecting total α-tubulin distribution or level (Figures 2B–2D). To assess microtubule dynamics, we treated cells with the microtubule destabilizer nocodazole and found enhanced microtubule polymerization following nocodazole washout (Figures 2E and 2F). Sacsin KO cells also demonstrated increased microtubule polymerization and disordered movements as assessed by live cell imaging of the microtubule plus-end binding protein EB1:GFP (Figures 2G and 2H; Videos S1 and S2).
Figure 2.

Altered microtubule structure and dynamics in sacsin KO cells
(A) Confocal immunofluorescent images of sacsin WT/KO cells stained for vimentin, and the MTOC marker γ-tubulin. Arrowheads point to the most intense signal in each cell, showing that vimentin bundles surround the MTOC in sacsin KO cells. Scale bar, 10 μm.
(B) Confocal images of immunostaining for α-tubulin, neurofilament heavy, and acetylated tubulin in WT and sacsin KO cells. Arrowheads mark coincidence of acetylated tubulin and neurofilament bundles, suggesting that acetylated tubulin structures are found in proximity to neurofilament bundles, but also localize throughout the cell. Scale bar, 10 μm.
(C and D) Quantification of images in (B). n = 3, SEM, Student’s t test.
(E) Confocal images of WT/KO cells treated with nocodazole (NDZ) labeled for α-tubulin and acetylated tubulin at indicated time points following nocodazole washout. Scale bar, 10 μm.
(F) Quantification of (E). n = 3 coverslips, SEM, one-way ANOVA with Tukey’s post test.
(G) Representative TIRF microscopy images from WT and sacsin KO cells expressing EB1-GFP. Microtubule growth tracks are color coded marking their position over time. Insets show the enlargement of outlined regions and movement of individual comets over time (circles). Numbers refer to seconds.
(H) Quantification of microtubule polymerization velocity marked by EB1-GFP movement in WT/KO cells (F and Video S1). n = 34 WT and n = 25 sacsin KO cells from three independent experiments, Student’s t test, ∗∗p < 0.01.
Mitochondrial trafficking in neurons is dependent on microtubules (Melkov and Abdu, 2018), and tau overexpression and hyperphosphorylation can cause decreased mitochondrial trafficking (Ando et al., 2016; Lopes et al., 2017; Reddy, 2011), buildup of mitochondria around the MTOC (Ebneth et al., 1998), and DRP1 mislocalization and reduced mitochondrial fission (DuBoff et al., 2012; Manczak and Reddy, 2012). In ARSACS, mitochondria also accumulate around proximal dendrites (Girard et al., 2012) and exhibit reduced DRP1-dependent fission (Bradshaw et al., 2016). We observed occlusion of mitochondria around vimentin bundles (Figure S2B) with no alterations in the actin cytoskeleton (Duncan et al., 2017) (Figure S2C). To assess how these alterations affect mitochondria in neurons, we performed neuronal differentiation of SH-SY5Y cells (Shipley et al., 2016). While wild-type (WT) and sacsin KO cells expressed indistinguishable levels of neuronal markers, neurites were fewer and shorter in sacsin KO cells (Figures S2D–S2G), contained fewer mitochondria (Figure S2H), and had diminished mitochondrial movement (Figure S2I and Video S3). Our proteomics data also identified several hyperphosphorylated kinesin proteins, which shuttle mitochondria along microtubule tracts (Frederick and Shaw, 2007) (Table S1). In all, these results demonstrate that the loss of sacsin affects microtubule structure, dynamics, and function, in agreement with recent findings demonstrating that sacsin directly interacts with microtubules (Francis et al., 2022).
FA organization and dynamics are disrupted in sacsin KO cells
To more systematically characterize our proteomic datasets, we performed gene ontology (GO) analysis for the total proteome and phosphoproteome (Figures 3A and 3B; Table S2). The top associated terms in the proteome were related to “focal adhesions,” including “integrin signaling,” “actin filament,” and “regulation of protein localization to plasma membrane.” “Focal adhesion” was also a top term in phosphoproteome, suggesting that FA proteins are affected at both the total protein and post-translational levels. FAs are plasma-membrane-associated macromolecular assemblies that physically link the intracellular cytoskeleton and extracellular matrix (ECM). FAs are composed of integrin receptors bridging the ECM with actin bundles, which interact with microtubules and intermediate filaments to coordinate dynamic regulation of FA structure (Ezratty et al., 2005; Leube et al., 2015; Seetharaman and Etienne-Manneville, 2019). In the brain, FAs are critical for structural remodeling during axon growth, synapse formation, and maintenance (Kilinc, 2018). Immunolabeling for the core FA proteins paxillin and vinculin revealed decreased FA number, area, and aspect ratio in sacsin KO cells (Figures 3C and S3A–S3G) while total levels of these proteins were unaffected (Figure S3H and Table S1). While paxillin is primarily localized at FAs, it also is known to interact with the MTOC (Robertson and Ostergaard, 2011), and we observed perinuclear accumulation of paxillin coinciding with the vimentin bundle (Figure S3A). Microtubules regulate vinculin localization to FAs (Ng et al., 2014), and we found reduced vinculin and vimentin dynamics in sacsin KO cells using fluorescence recovery after photobleaching (FRAP) (Figures 3D, S3I, and S3J). We next removed cell bodies with hypotonic shock, leaving only the structural remnants of cell-ECM interactions, and again found reduced vinculin structures, suggesting that the mislocalization of adhesion proteins also results in decreased cell-ECM interactions (Figures S3K–S3N). These findings were consistent in sacsin KO HEK293 cells, which were generated using an alternative CRISPR-Cas9 genome-editing strategy (Duncan et al., 2017) (Figures S3O–S3S). Our proteomics data also revealed decreased levels of several integrin proteins (Figure S3T). Localization of ITGAV to FAs was diminished in sacsin KO cells (Figure 3E), while ITGA6 was sequestered in the vimentin bundle (Figure S3U). In all, these data suggest that the trafficking, structure, and function of multiple FA proteins is affected in sacsin KO cells.
Figure 3.

FA and integrin localization is altered in sacsin KO cells
(A and B) GO term analysis of differentially expressed proteins (A) and phosphorylation (B) in SH-SY5Y cells (p < 0.05, log2 f.c. cutoff ±0.4).
(C) Confocal images of WT/KO SH-SY5Y cells immunolabeled for vimentin and the FA protein vinculin.
(D) FRAP analysis of the FA protein vinculin in cells expressing tomato-VCL. 2 × 2-μm regions of interest were bleached with a 568-nm laser. Recovery was monitored over 50 cycles of imaging with a 1-s interval. n = 10 cells from each of three independent experiments.
(E) Confocal images of cells immunolabeled for ITGAV. Scale bar, 10 μm.
(F) Scatterplot of statistically significant proteins identified in either SH-SY5Y or primary cortical cultures (p < 0.05, log2 f.c. cutoff ±0.4). Statistical test: Pearson correlation coefficient.
To determine whether levels of adhesion proteins were also affected in neurons, we performed quantitative proteomics of primary cortical neuron cultures derived from embryonic day 15.5 Sacs(−/−) mice (Figure S3V and Table S1). These cultures are composed of ∼75% NeuN+ neurons, but also contain a smattering of other cell types such as astrocytes (Pearson et al., 2016). While no proteins passed statistical cutoffs in both datasets (p < 0.05, log2 fold change ±0.4), comparing levels of proteins which were significantly affected in either dataset revealed a statistically significant relationship, suggesting that a subset of proteins is affected in both cellular contexts (Figure 3F). Notably, these proteins included vimentin and several integrins. The most differentially expressed proteins in cortical cultures included neuron-specific proteins, such as Nrsn1, which binds tubulin and plays a role in vesicular trafficking (Ida et al., 2004; Kiyonaga-Endou et al., 2016), and astrocyte-specific proteins, such as the intermediate filament protein GFAP (Murtinheira et al., 2022). This may suggest that multiple cell types are affected by the loss of sacsin. When we analyzed statistically significant proteins by GO term analysis, we saw similar processes as those identified in SH-SY5Y cells (Figure 3A), including “cell adhesion molecule binding,” “actin filament bundle,” and “focal adhesion” (Figure S3W). As the proteins input into each GO term analysis were completely non-overlapping, this suggests that the loss of sacsin affects cytoskeletal and FA structures independent of cellular context.
Modulating PTEN-FAK signaling rescues cellular deficits in sacsin KO cells
Beyond providing structural support for cells, FAs are enriched with many signaling proteins, which transmit signals from the extracellular milieu to effectors in the cytoplasm and nucleus. A master regulator of FA signaling is the FA kinase (FAK/PTK2) (Sulzmaier et al., 2014). FAK is recruited to integrin adhesion complexes through interactions with paxillin (Brown et al., 1996) and is activated via autophosphorylation at Tyr397 following integrin receptor binding to the ECM (Zhao and Guan, 2011). FAK regulates neuronal outgrowth and synapse formation by phosphorylating multiple downstream effectors of FA signaling (Rico et al., 2004) (Figure 4A). Although total levels of FAK were unaltered in sacsin KO cells, pFAK was significantly reduced, as was its localization to FAs (Figures 4B, 4C, S4A, and S4B). JNK and paxillin, downstream targets of activated pFAK (Zhao and Guan, 2011), were also hypophosphorylated, without corresponding changes in protein levels (Figures 4B and S4C–S4G; Table S1). These data suggest that FAK signaling is suppressed in sacsin KO cells, possibly through disengagement with FAs.
Figure 4.

Targeting upstream FA regulator PTEN rescues FA and vimentin bundling phenotypes in sacsin KO cells
(A) Regulators and effectors of FA signaling.
(B) Western blots for regulators FAK, pFAK-Tyr397, total and phosphorylated JNK, total and phosphorylated PAX, phosphorylated JUN, and PTEN in total cell lysates from sacsin KO and control cells.
(C) Confocal images of cells immunolabeled for pFAK. Scale bar, 10 μm.
(D) Western blots of WT/KO cells treated with siRNAs targeting PTEN or scrambled.
(E–G) Quantification of PTEN (E), pFAK (F), and pPAX (G) levels in WT/KO cells treated with scrambled or PTEN targeting siRNAs. n = 3, SEM, Student’s t test, ∗p < 0.05, ∗∗∗p < 0.001.
(H) Confocal images for cells transfected with siRNAs targeting PTEN or scrambled, and immunolabeled for vimentin and vinculin. Arrowheads indicate cells with prominent FAs, arrows indicate cells with absent or reduced perinuclear accumulations of vimentin. Scale bars, 10 μm.
(I and J) Quantification of the incidence of sacsin KO cells with perinuclear accumulations of vimentin (I) or vinculin-positive FAs (J) 48 h after transfection with siRNAs targeting PTEN or scrambled siRNAs. n = 3 replicates, >100 cells in each replicate, SEM, Student’s t test, ∗p < 0.05, ∗∗∗∗p < 0.0001.
We next considered the mechanism by which FAK signaling is suppressed in sacsin KO cells. The phosphatase PTEN, which dephosphorylates FAK and negatively regulates FAK activity (Tamura et al., 1999), was elevated in sacsin KO cells (Figures 4B and S4H). To investigate whether increased PTEN is a general consequence of intermediate filament disorganization, we treated WT SH-SY5Y cells with simvastin (Trogden et al., 2018), which induced vimentin bundling and perinuclear accumulation but did not affect PTEN levels (Figures S4I–S4K). Conversely, reducing PTEN by small interfering RNA (siRNA)-mediated knockdown to WT levels in sacsin KO cells (Figures 4D and 4E) increased pFAK and pPAX (Figures 4D, 4F, and 4G), reduced the frequency of perinuclear vimentin accumulation, and increased the number of FAs (Figures 4H–4J). FAs also play an important role in the migratory behaviors of cells (De Pascalis and Etienne-Manneville, 2017), and sacsin KO cells exhibited migration deficits in scratch and transwell migration assays (Figures S4L–S4O), which were rescued by PTEN knockdown (Figures S4P and S4Q). Together these results indicate that increased PTEN activity contributes, at least in part, to the intermediate filament and FA phenotypes in sacsin KO SH-SY5Y cells.
Membrane-bound synaptic adhesion molecules are mislocalized in sacsin KO cells
FAs act as signal transduction hubs to integrate information from the outside of the cell to the inside. Some FA proteins, including paxillin and zyxin (Figure 1C), can shuttle to the nucleus and function as transcriptional co-regulators in a phosphorylation-dependent manner (Dong et al., 2009; Suresh Babu et al., 2012). Interestingly, GO term analysis for proteins with altered phosphorylation were highly enriched for terms related to RNA processing, including “RNA binding,” “cytoplasmic stress granules,” “spliceosome,” and “nuclear body” (Figure 3B and Table S2), suggesting that the altered phosphorylation landscape may be affecting the transcriptome. Therefore, we next performed RNA sequencing (RNA-seq) of neuronally differentiated SH-SY5Y cells (Figure S5A and Table S3). We found 876 differentially expressed genes (false discovery rate [FDR] <0.05, log2 fold change ±0.4), suggesting that the loss of sacsin has profound effects on the transcriptome (Figure S5A). Protein interaction mapping revealed altered expression of multiple ECM proteins, integrins, and regulators of integrin activation (Figure S5B). Interestingly, changing the total levels or activity of specific integrins can affect the expression of other integrin subunits, a phenomenon called “integrin crosstalk” (Samarzija et al., 2020). The observation that multiple integrins were affected at both the protein and RNA levels suggests that altered integrin localization may activate regulatory feedback loops which affect the expression of genes that play a role in membrane-based signaling. Indeed, GO term analysis of differentially expressed genes identified terms implicating membrane-related processes, including “postsynaptic membrane,” “axon terminus,” “endomembrane system,” and “cytoplasmic vesicle membrane” (Figure S5C). In all, these data suggest that the altered phosphorylation landscape in sacsin KO cells affects mRNAs encoding for proteins involved in membrane-related processes.
Cell surface proteins are frequently under-represented in proteomics experiments owing to low expression and biochemical properties (Bausch-Fluck et al., 2015). Indeed, while 26% of the genes detected by RNA-seq were detected in the proteome, only 11% of differentially expressed genes (which were enriched for membrane proteins) were detected in the proteome (Figure S5D). Therefore, to better characterize membrane and surface proteins, we incubated live cells with biotin, labeling cellular and exosome membrane/surface proteins, followed by neutravidin purification, and analysis by quantitative mass spectrometry (Nunomura et al., 2005) (Figure 5A and Table S1). This approach identified an additional 870 proteins not in our initial proteomic datasets (Figure S5E). Proteins with altered surface expression in sacsin KO cells included several signaling receptors (FGFR1, LRP4) and GTP-binding proteins involved in signal transduction (GNG2, GNG8) (Figure 5B). Two of the most affected membrane proteins were the synaptic adhesion proteins neuronal cell adhesion molecule (NRCAM) and neurofascin (NFASC), which form a molecular complex and have been linked to movement disorders (Kurolap et al., 2022; Kvarnung et al., 2019; Smigiel et al., 2018) (Figures 5B and 5C). We next compared membrane proteins found in both proteomic and surfaceome datasets, reasoning that conflicting levels between cell surface and total protein levels could reflect improper membrane recycling, precocious membrane localization, or deficits in membrane-bound trafficking. Many proteins with altered surface levels showed no or even opposing change in total protein levels (Figure 5D and Table S1). Among the most mislocalized proteins were synaptic adhesion proteins, including multiple integrins (ITGA1, ITGB1, ITGA3), neuronal cell adhesion molecules (NRCAM, CNTN1, LSAMP), the FA regulator RET/GFRA3 heterodimer, the microtubule-binding protein DCX, and AHNAK, a 700 kDa scaffolding protein with diverse yet poorly understood function (Davis et al., 2015) (Figure 5D).
Figure 5.

The loss of sacsin affects the localization of cell adhesion proteins
(A) Western blot of cell surface protein purification, illustrated by the membrane protein ATP1A1, and the nuclear pore protein NUP98. After purification ATP1A1 is detectable only in conditions that were treated with biotin, and NUP98 is no longer detected, suggesting labeling specificity and enrichment of cell surface proteins.
(B) Mass spectrometry of cell surface proteins in sacsin KO SH-SY5Y cells. Significance cutoffs: p < 0.05 and log2 fold change (f.c.) ±0.4, denoted by black outline. Proteins which pass these cutoffs, but were also detected in biotin negative controls, were not considered for downstream analysis (Table S1).
(C) Western blot of NFASC in total lysate (left), and fractionated cytoplasmic or membrane fractions in WT and sacsin KO cells.
(D) Levels of proteins detected in both cell surface and proteomic datasets. Proteins are colored by the disparity between these two datasets (f.c. surface and f.c. proteome), with red indicating more, and blue less membrane abundance relative to total protein levels. Black outlines are proteins with p < 0.05, log2 f.c. ± 0.4 in the surface dataset.
(E) GO term analysis of proteins differentially localized in membrane of sacsin KO cells (p < 0.05, log2 f.c. ±0.4).
(F and G) Confocal images for fibronectin and vimentin (F) and ER marker KDEL (G) in WT and sacsin KO HEK293 cells. Scale bars, 10 μm.
(H) Disease enrichment analysis with Ingenuity Pathway Analysis of differentially localized surface proteins (p < 0.05, log2 f.c. ±0.4).
GO term analysis of proteins with altered surface levels suggested deficits in processes related to vesicle packaging and transport (Figure 5E). These included eight exosomal Rab proteins, which were increased in the surfaceome and not affected at the total protein level (Figure S5F and Table S1). Rabs are a diverse family of GTPases that coordinate multiple aspects of membrane protein trafficking, including FA turnover, and integrin endo-/exocytosis (Moreno-Layseca et al., 2019). Specific Rabs also regulate trafficking between the Golgi and the endosomal network (RAB8A, RAB10), bidirectional Golgi/endoplasmic reticulum (ER) trafficking (RAB2A, RAB18), and epidermal growth factor receptor (EGFR) internalization (RAB7A) (Bakker et al., 2017; Galea and Simpson, 2015). Kinome profiling also identified multiple regulators of Rab activity and trafficking, including PIK3R4 and PIK3C3, which regulate PTEN activity through localization to vesicles in a microtubule-dependent fashion (Naguib et al., 2015).
To assess trafficking and localization deficits in sacsin KO cells we investigated the localization of the ECM protein fibronectin, which is packaged into vesicles in the ER and Golgi (Kii et al., 2016) and trafficked to the cell periphery along microtubules (Noordstra and Akhmanova, 2017). Fibronectin was not affected in any of our proteomics datasets, allowing us to investigate mislocalization independent of changes in protein level or phosphorylation. In WT HEK293 cells, fibronectin puncta were organized in “chains,” which appear collapsed around the vimentin bundle in sacsin KO cells (Figure 5F). Staining for the ER marker KDEL revealed that fibronectin is retained in the ER in HEK293 and SH-SY5Y sacsin KO cells (Figures 5G and S5G), suggesting that membrane-bound trafficking is affected in sacsin KO cells.
We next used Ingenuity Pathway Analysis to assess whether the misregulated cell surface proteins are associated with any pathological conditions. Resoundingly, the terms were associated with disease traits reminiscent of ARSACS, including “movement disorders,” “neurodegeneration,” and “progressive neurological disorder” (Figure 5H). Notably, three of the most mislocalized proteins, NFASC, NRCAM, and CNTN1, form molecular complexes that are important for axon guidance (Pollerberg et al., 2013), maintenance of synapses by astrocytes (Takano et al., 2020), and interactions between Purkinje neuron axons and glia (Bhat et al., 2001). KO mice or humans which harbor mutations in each of these genes develop cerebellar ataxias with features that resemble ARSACS (see discussion).
Integrin trafficking and synaptic structure are affected in ARSACS mice
Cerebellar atrophy is an early clinical feature of ARSACS (Martin et al., 2007; Synofzik et al., 2013). In the ARSACS mouse model, the progressive death of Purkinje neurons begins around postnatal day 90 (P90) (Lariviere et al., 2015) and is well under way by P120 (Figure 6A). To determine whether any of the proteins that were mislocalized in our sacsin KO cell model were also affected in the brain, we focused on mice at P60, which is when behavioral deficits first emerge but prior to Purkinje neuron death (Lariviere et al., 2015). ITGA1, which was among the most mislocalized proteins in sacsin KO cells (Figure 5D), is normally localized in nuclear Cajal bodies and Purkinje axons in Sacs(+/−) mice (Figures 6B and 6C). However, in Sacs(−/−) mice, we observed striking accumulation of ITGA1 in the soma and dendritic trunk (Figures 6B–6D). Axonal swelling near the Purkinje neuron soma is a consistent feature in Sacs(−/−) mice (Lariviere et al., 2015), and we also observed ITGA1 accumulation in these structures (Figure S6A). In contrast, we observed a decrease of ITGA1 in Purkinje neuron axon tracts (Figures 6E–6G), likely reflecting reduced ITGA1 trafficking.
Figure 6.

Altered localization of membrane proteins and synapses in ARSACS mice
(A) Confocal imaging of Purkinje neurons in littermate controlled P120 SACS(+/−) and SACS(−/−) mice, demonstrating substantial Purkinje cell loss. Purkinje marker calbindin-D28K (CALB1) and neuronal marker (NEUN). Sacs(+/−) mice are phenotypically normal, analogous to unaffected human carriers. Scale bar, 200 μm.
(B) Confocal image of cerebellum in P60 mice, stained for integrin A1 (ITGA1), one of the most mislocalized proteins in KO cells (Figure 5D). Arrowheads denote ITGA1 staining in white matter axonal tracts. Scale bar, 200 μm.
(C) High magnification of ITGA1 staining in the Purkinje neuron layer in P60 mice. Scale bar, 20 μm.
(D) Quantification of ITGA1 accumulation in cerebellar sagittal sections. Abnormal accumulation was defined as mean ITGA1 intensity in dendritic arbor greater than 3 standard deviations above the mean in Sacs(+/−) mice. A replicate is defined as a sex-matched het/KO animal from the same litter. Average of two sections per animal, ∼240 Purkinje counted neurons per section. n = 3 litters, SEM, paired t test (litter as pairing variable).
(E) Confocal image of Purkinje axon tracts through the NEUN+ granule cell layer, and NEUN− white matter tracts in P60 mice. Scale bar, 20 μm.
(F and G) Zoomed-in region from (E), single z-plane. Dashed lines (F) mark white matter axonal tracts for quantification (G). n = 3, SEM, replicates defined as in (D), paired t test.
(H) Representative confocal image of the DCN in P60 mice, demonstrating synaptic changes between Purkinje neuron synaptic termini and NEUN+ DCN neurons. Scale bar, 20 μm.
(I and J) Quantification of images from (H). For each large-diameter DCN neuron we counted the number (I) and size (J) of CALB+ structures immediately adjacent to each DCN neuron using an automated analysis pipeline (see STAR Methods). Replicates defined as in (D), n = 4, paired t test.
(K) Confocal image of DCN neurons, with NEUN channel overexposed to enhance projections. Closed arrowheads mark DCN neuron projections, open arrowhead DCN neuron soma. Scale bar, 20 μm.
(L) Quantification of images from (K). Projections were defined as small (5–15 μm diameter) NEUN+/DAPI− structures. Replicates defined as in (D), n = 4. Quantification in Figure S6F provides quantification of DCN neuron soma.
Purkinje axons synapse onto neurons in the DCN, which in turn project to multiple brain regions. As the primary output hub of the cerebellum (Ito, 2002), alterations in the Purkinje-DCN circuit have substantial effects on both motor and non-motor processes (Baek et al., 2022; Sathyamurthy et al., 2020), and are observed in multiple neurodegenerative ataxias (Barron et al., 2018; Feng et al., 2022; Walter et al., 2006). We observed striking disorganization of Purkinje neuron synapses in the DCN in Sacs(−/−) mice at P60 (Figure 6H) and P120 (Figures S6B and S6C), in agreement with a previous report (Ady et al., 2018). The number of Purkinje synapses on each DCN neuron was reduced in Sacs(−/−) mice (Figure 6I), while the size of Purkinje axon termini apposed to DCN neurons was substantially increased (Figure 6J). We observed accumulation of ITGA1 in large CALB+ structures in Sacs(−/−) mice, suggesting that while ITGA1 trafficking is not altogether abolished in Sacs(−/−) mice, ITGA1 does accumulate in these pathological swellings (Figures S6D and S6E). Interestingly, we also observed increased ITGA1 staining in the cell bodies of DCN neurons (Figures 6K and S6F) and accumulation of ITGA1 in the large-diameter dendrites of DCN neurons (Figures 6K and 6L). This pattern was similar to the dendritic ITGA1 accumulation seen in Purkinje neurons (Figure 6C), suggesting that altered protein localization is not unique to Purkinje neurons. As DCN neurons project throughout the brain, the physical disruption between Purkinje and DCN neurons suggests that cerebellar output to multiple brain regions may be directly affected in ARSACS.
The loss of sacsin disrupts protein-protein interactions
To identify how the loss of sacsin causes abnormal protein trafficking, we performed quantitative label-free mass spectrometry of proteins which co-immunoprecipitate with endogenous sacsin in WT SH-SY5Y cells. KO cells were also used to control for non-specific protein pull-down. Our analysis identified 96 proteins as putative sacsin interactors, including vimentin and vinculin (Table S4). Immunofluorescence revealed sacsin puncta in and around vinculin-positive FAs (Figures S7A and S7B) and in close proximity to vimentin structures, with sacsin often being between them (Figure 7A). Reciprocal co-immunoprecipitation (co-IP) experiments confirmed interactions between sacsin, vimentin, and vinculin, but the interaction between vimentin and vinculin was dramatically reduced in sacsin KO cells (Figure 7B). NFASC has been reported to interact with vimentin (Sistani et al., 2013), leading us to wonder whether NFASC may also interact with FA proteins. Co-IP experiments identified an interaction between NFASC and vinculin, which was dramatically reduced in sacsin KO cells (Figure 7C). These results suggest that sacsin promotes the formation and/or stabilization of adhesion protein interactions.
Figure 7.

The loss of sacsin disrupts protein-protein interactions
(A) Airyscan confocal analysis of sacsin, vimentin, and transfected tdTomato:vinculin staining in WT SH-SY5Y cells, demonstrating sacsin localization along vimentin tracts and FAs. Scale bar, 10 μm.
(B) Vimentin or sacsin were immunoprecipitated from WT and sacsin KO SH-SY5Y cells, and co-immunoprecipitated proteins (sacsin, vinculin, vimentin) were analyzed by western blot.
(C) Co-IP of NFASC and vinculin in WT and sacsin KO cells shows that the interaction between VCL and NFASC is greatly reduced in sacsin KO cells, despite NFASC being substantially overexpressed in SACS KO cells (Figure 5C). n = 3, SEM, Student’s t test, ∗∗∗p < 0.001.
(D) STRING protein interaction map depicting proteins quantified in this study. Lines between proteins indicate high-confidence interactions (interaction score >0.7). We removed proteins with redundant interactions for clarity (for example, most integrins have largely overlapping interactomes). Proteins identified in the sacsin interactome profiling are circled, with the thick circles marking interactors identified in all replicates and the thin circles marking interactors identified in <3 replicates. Proteins are colored by log2 f.c. in proteome (left half) and cell surface proteome (right half). Clusters identified by k-means clustering are marked by gray background.
To identify central proteins which may explain the cellular phenotypes in sacsin KO cells, we performed STRING network analysis (Szklarczyk et al., 2019). We considered all proteins altered in any of our datasets and assessed only high-confidence physical or regulatory interactions. k-Means clustering of network interactions identified three clusters, which highlight complementary pathways by which sacsin contributes to cell structure and signaling (Figure 7D). Central to cluster 1 is the interaction between sacsin and intermediate filament proteins, which interact with a variety of cell surface receptors. Combined with our biochemical experiments, this suggests that the loss of sacsin leads to improper localization of adhesion proteins to the plasma membrane, possibly through decreased protein interactions between intermediate filaments, adaptors, and adhesion proteins. The network also highlighted the microtubule-associated kinase MAST1, which stabilizes PTEN (Valiente et al., 2005) and is protected from proteasomal degradation by the sacsin interactor HSP90B1 (Pan et al., 2019).
Cluster 2 is composed of the interaction between sacsin, chaperone network proteins, and microtubules, which in concert regulate membrane protein processing, trafficking, and localization (McClellan et al., 2007). Multiple heat-shock protein (HSP) chaperones were part of the sacsin interactome (Figure 7D), including the marker of ER stress HSPA5/BIP and several HSP90 proteins, which can stabilize FAK, modulate cell migration (Xiong et al., 2014), and regulate microtubules (Quinta et al., 2011). Recent evidence suggests HSP90 is essential for microtubule acetylation (Wu et al., 2020), suggesting that the loss of sacsin may alter microtubule stability via HSP proteins (Figures 2B–E). HSPs also regulate Rab proteins (Chen and Balch, 2006) (cluster 3), which have diverse roles in vesicular trafficking, including PTEN and EGFR trafficking (Shinde and Maddika, 2016). Rabs are highly enriched in synapses, play key roles in endo- and exocytosis, and are linked to many neurodegenerative diseases (Kiral et al., 2018). The increased surface abundance of multiple Rab proteins without corresponding changes in total Rab levels is consistent with the precocious microtubule stability and dynamics we observe in sacsin KO cells. GO term analysis revealed that 65% of sacsin interacting proteins are involved in exosome-related processes, with additional interactors being implicated in unfolded protein binding (HSPs) and FAs (Figure S7C). In all, these results suggest that sacsin plays a direct role in bridging protein quality control systems, microtubule-dependent vesicular transport, and membrane localization of adhesion proteins.
Discussion
This study identifies sacsin as a central regulator of multiple aspects of cellular structure, including intermediate filament architecture, microtubules, protein trafficking, and FAs. The complex and intertwined relationships between these processes complicates our understanding of their precise pathophysiological relevance, but our results raise some intriguing possibilities. Sacsin possesses a functional J domain, which interacts with HSP70 chaperone proteins (Genest et al., 2019; Parfitt et al., 2009) (Figure 7D). HSPs play a role in ubiquitin-dependent turnover of intermediate filaments (Gavriilidis et al., 2018), and neurofilament bundling in ARSACS neurons can be rescued by HSP expression (Gentil et al., 2019). Sacsin also possesses an ATPase domain with homology to HSP90 proteins. The sacsin interactor HSP90B1 stabilizes FAK (Xiong et al., 2014), suggesting that restoring FAK signaling may rescue intermediate filament structure through HSP activity (Figures 4 and 7D). It is also possible that sacsin transiently interacts with HSP90-regulated kinases, such as FAK (Xiong et al., 2014), and has a more direct role at FAs. HSP70/90 complexes bind to microtubules in an acetylation-dependent fashion (Giustiniani et al., 2009) and interact with hyperphosphorylated tau to increase tau’s interaction with microtubules (Lackie et al., 2017). Since HSPs are known to regulate all of the protein clusters with deficits in sacsin KO cells (Figure 7D), we hypothesize that the interaction between HSPs and sacsin may be an especially critical interaction that is lost in ARSACS. Furthermore, as illustrated by sacsin’s mediation of the interaction between intermediate filaments and FAs, changes in additional as yet uncharacterized protein-protein interactions may explain specific ARSACS phenotypes, such as disrupted autophagy, nuclear morphology, and aberrant localization of mitochondria.
Proper localization of synaptic adhesion proteins is critical for neuronal health and is disrupted in many neurodegenerative diseases (Kiral et al., 2018). As cell adhesion proteins, integrins play key roles in modulating axon outgrowth, dendritic arborization, and regulating synaptic structure and function (Park and Goda, 2016). More specifically, multiple integrins and pFAK are localized to dendritic spines in cultured Purkinje neurons, where they regulate spine remodeling (Heintz et al., 2016). However, little is known about the role of ITGA1 in the brain (Murase and Hayashi, 1998), and the lack of a mechanistic connection between ITGA1 localization and the changes to synaptic structure in ARSACS mice is a limitation of our findings. As multiple levels of data suggest that integrins as a class are affected in sacsin KO cells (proteomics, transcriptomics, and surfaceomics), exploring the localization of additional integrin subunits may shed light on this question. Furthermore, integrins are in general most highly expressed during brain development (Nieuwenhuis et al., 2018). Thus, defining when changes in integrin mislocalization and synaptic structure first emerge may yield important insight into the pathomechanistic origins of ARSACS.
Our data also suggest that restoring FA signaling by reducing PTEN levels may rescue some cellular deficits in sacsin KO SH-SY5Y cells. PTEN is highly enriched in axons, where it regulates neurite outgrowth, organelle trafficking, and synaptic plasticity (Kreis et al., 2014). Reducing PTEN activity with competitive peptides, small molecules, or genetically have shown therapeutic potential in acute models of axonal injury and stroke (Park et al., 2008; Shabanzadeh et al., 2019) as well as a progressive neurodegenerative tauopathy (Benetatos et al., 2020). As PTEN directly regulates multiple pathways, including PI3K/AKT/mTOR, targeting downstream components of PTEN-dependent regulatory cascades may also have therapeutic potential (Jacobi et al., 2022) and bypass concerns over PTEN’s roles in neurodevelopment and tumor suppression (Skelton et al., 2020). However, as we did not detect evidence of increased PTEN levels in our primary cortical culture proteomic data, determining whether PTEN signaling is hyperactive in Purkinje cells in ARSACS requires further investigation. Nevertheless, as the above examples achieve neuroprotective effects by reducing PTEN activity in multiple neurodegenerative contexts, this approach remains an intriguing strategy.
Why do mutations in sacsin, which is expressed throughout the brain, present as a cerebellar ataxia? Proteins whose abundance or localization is altered in sacsin KO cells, and which also cause cerebellar ataxia, could suggest a causal molecular deficiency in ARSACS. The interactions between NFASC, NRCAM, and CNTN1 are critical for brain development, and mutation of each causes phenotypes reminiscent of ARSACS. Cntn1 KO mice have deficits in axon guidance and develop cerebellar ataxia (Berglund et al., 1999). Nrcam KO mice have phenotypes only in lobules 4/5 of the cerebellar vermis (Sakurai et al., 2001), which are also specifically affected in ARSACS (Ady et al., 2018; Lariviere et al., 2015, 2019). Lastly, human mutations in NFASC which selectively remove the 155 kDa glial isoform cause congenital hypotonia, demyelinating neuropathy (as in ARSACS), and severe motor coordination deficits (Smigiel et al., 2018), while mutations of the neuron-specific 186 kDa NFASC isoform cause cerebellar ataxia (Kvarnung et al., 2019). These convergent phenotypes lead us to hypothesize that improper localization of synaptic cell adhesion molecules may be a causal molecular deficiency in ARSACS.
In development, if an axon fails to make productive synaptic connections and receive neurotrophic input from nearby cells, molecular cascades are activated, which cause localized pruning of non-productive axonal branches (Dekkers et al., 2013). This process, which initiates at the synapse and advances back toward the cell body, is referred to as the dying back model, and can cause neuronal death (Raff et al., 2002). Although this is a normal mechanism to ensure proper wiring of the nervous system in the face of stochastic errors in axon guidance, this process is co-opted in many neurodegenerative disorders, including amyotrophic lateral sclerosis (Dadon-Nachum et al., 2011), Alzheimer’s disease (Salvadores et al., 2017), Huntington’s disease (Han et al., 2010), Parkinson’s disease (Dauer and Przedborski, 2003), and hereditary spastic paraplegias (Fink, 2013). A common molecular thread across these diseases is microtubule-based axonal transport (Morfini et al., 2009), and many of the proteins implicated in the aforementioned diseases were also identified in this study (e.g., tau, tau kinases, Rabs, synaptic adhesion proteins). This leads us to speculate that the loss of sacsin alters microtubule function, resulting in improper trafficking of synaptic adhesion proteins, deficits in synaptic structure, activation of axonal degeneration, and ultimately Purkinje cell death. A mechanistic exploration of this hypothesis will be necessary for the development of rationally designed therapeutic strategies aimed at delaying or preventing ARSACS progression by restoring synaptic structure and function.
Limitations of the study
The precise molecular function of sacsin remains elusive, in part because of the difficulty of performing biochemical assays with such a large protein. We attempted to shed light on sacsin’s function by identifying interacting proteins, but an important limitation of co-IP experiments is that many of the interactions may be indirect. Determining which proteins interact directly with sacsin may help clarify the mechanism by which sacsin regulates the processes we describe in this study. While we provide multiple lines of evidence that microtubule-dependent trafficking of membrane proteins is affected in sacsin KO SH-SY5Y cells, and some evidence in primary cortical neuron cultures and cerebellar neurons in the brain, a more systematic exploration of this phenotype in Purkinje neurons is warranted. Specifically, analyzing integrin localization at Purkinje synapses, the activity of PTEN/FAK signaling, and the role of vesicular transport and membrane protein turnover will be necessary to assess the physiological relevance of our findings. Furthermore, of the many cellular phenotypes that have been found in ARSACS, teasing apart which are causal and which are merely a part of neurodegenerative processes is necessary to understand the fundamental role of sacsin in the brain. We posit that exploring the neurodevelopmental aspects of this disease, prior to the onset of the neurodegenerative cascade, may help shed light on this question.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| acetylated tubulin | Cell Signaling | #5335; RRID:AB_10544694 |
| Alexa Fluor 488 | Life Technologies | A-11029; RRID:AB_2534088 |
| Alexa Fluor 488 | Life Technologies | A-11008; RRID:AB_143165 |
| Alexa Fluor 568 | Life Technologies | A-11004; RRID:AB_2534072 |
| Alexa Fluor 568 | Life Technologies | A-11011; RRID:AB_143157 |
| Alexa Fluor 633 | Life Technologies | A-21052; RRID:AB_2535719 |
| Alexa Fluor 633 | Life Technologies | A-21070; RRID:AB_2535731 |
| Alexa Fluor 633 Phalloidin | Life Technologies | A22284 |
| alpha-tub | ThermoFisher | MA1-80017; RRID:AB_2210201 |
| anti-rabbit HRP | Fisher | #PI31466 |
| API | SIGMA | D9542 |
| ATP1A1 | Cell Signaling | #3010; RRID:AB_2060983 |
| Beta-Actin | Abcam | ab8226; RRID:AB_306371 |
| BRSK2 | Cell Signaling | 5460S |
| CALB1 | Novus | NBP2-50028 |
| FAK | CST | 71433S; RRID:AB_2799801 |
| Fibronectin | Abcam | ab268020 |
| gamma-tubulin | ThermoFisher | MA1-19421; RRID:AB_1075282 |
| GAPDH | Abcam | ab8245; RRID:AB_2107448 |
| Integrin alpha 6 | Abcam | ab235905 |
| Integrin beta1 | Abcam | ab134179 |
| Integrin alpha V | Abcam | ab179475; RRID:AB_2716738 |
| IRDye®680 RD | Li-Cor | 926–68070 |
| IRDye®680 RD | Li-Cor | 926–68071 |
| IRDye®6800 CW | Li-Cor | 926–32210 |
| IRDye®6800 CW | Li-Cor | 926–32211 |
| ITGA1 | BiCell | 10001 |
| JNK | CST | 9252S; RRID:AB_2250373 |
| KDEL | Enzo Life Sciences | ADI-SPA-827-D |
| MAP2 | Abcam | Ab5392; RRID:AB_2138153 |
| NES | Abcam | Ab22035; RRID:AB_446723 |
| NEUN | Millipore | ABN90P; RRID:AB_2341095 |
| NFH | Abcam | Ab8135; RRID:AB_306298 |
| NUP98 | Cell Signaling | #2598; RRID:AB_2267700 |
| p-FAK (Tyr397) | ThermoFisher | 44–625G; RRID:AB_2533702 |
| p-JNK (Tyr183/185) | CST | 4668S; RRID:AB_823588 |
| p-JUN (Ser 73) | CST | 3270S; RRID:AB_2895041 |
| p-Paxillin (Tyr118) | CST | 69363S; RRID:AB_2174466 |
| Paxillin | CST | 12065S; RRID:AB_2797814 |
| PRKCG | Santa-Cruz | sc-211; RRID:AB_632234 |
| PRPH | Abcam | Ab4666; RRID:AB_449340 |
| PTEN | Santa-Cruz | sc-7974; RRID:AB_628187 |
| Sacsin | Abcam | ab181190 |
| Sacsin | Abcam | Cab80195; RRID:AB_10714400 |
| SYN1 | Abcam | Ab8; RRID:AB_2200097 |
| TAU p262 | ThermoFisher | 44-750-G; RRID:AB_2533743 |
| tau S199 | Abcam | Ab81268; RRID:AB_1641106 |
| TAU5 | Abcam | Ab80579; RRID:AB_1603723 |
| Vimentin | Abcam | ab8978; RRID:AB_306907 |
| Vimentin | Abcam | ab16700; RRID:AB_443435 |
| Vinculin | ThermoFisher | MA5-11690; RRID:AB_10976821 |
| Chemicals, peptides, and recombinant proteins | ||
| Nocodazole | Sigma | #SML1665 |
| all-trans retinoic acid | Sigma | #R2625 |
| Brain-Derived Neurotrophic Factor | Sigma | #B3795 |
| dibutyryl cyclic AMP | Santa Cruz | sc-201567A |
| MitoTracker Green FM | Invitrogen | #M7514 |
| Critical commercial assays | ||
| Tau Aggregation Kit | Cisbio | 6FTAUPEG |
| Minute Plasma Membrane Protein Isolation and Cell Fractionation Kit | Invent | SM-005 |
| Deposited data | ||
| PRIDE | This paper | PXD036906 |
| SRA | This paper | GSE214213 |
| Experimental models: Cell lines | ||
| SH-SY5Y | this study | ATCC CRL2266 |
| HEK293T | Duncan et al. 2017 | ATCC CRL1573 |
| Experimental models: Organisms/strains | ||
| C57BL/6-Sacstm2Bebr/J | The Jackson Laboratory | Strain #:033221 |
| Oligonucleotides/primers | ||
| WT SACS F: GCTGTCAGGGGGAAATCTGATAAAG | The Jackson Laboratory | N/A |
| WT SACS R: GCAGCACCTTTAGACAAAAGATTGC | The Jackson Laboratory | N/A |
| KO SACS F: CAACCTTGGAGAAACTGTGCCTG | The Jackson Laboratory | N/A |
| KO SACS R: CACCGACGCCAATCACAAACAC | The Jackson Laboratory | N/A |
| PTEN siRNA Pool | Horizon | 5728 |
| Recombinant DNA | ||
| hCas9 | Addgene | #41815 |
| pGFP-EB1 | Addgene | #17234 |
| tdTomato:Vinculin | Addgene | #58146 |
| EGFP-Vimentin-7 | Addgene | #56439 |
| Software and algorithms | ||
| ImageJ Fiji | NIH | https://imagej.net/software/fiji/downloads |
| CellProfiler | Broad | www.cellprofiler.org |
| MaxQuant | Max Planck | https://www.maxquant.org |
| Zen Black and Zen Blue | Zeiss | https://www.zeiss.com/microscopy/en/products/software/zeiss-zen.html |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to the lead contact, Justin Wolter (justin_wolter@med.unc.edu).
Materials availability
Cell lines generated in this study are freely available from the lead authors upon request.
Experimental model and subject details
Cell lines
Human female SH-SY5Y neuroblastoma cells were obtained from the American Type Culture Collection and were grown in 1:1 Dulbecco’s Minimum Eagle Medium (DMEM)/Ham’s F12 medium, plus 10% heat-inactivated fetal bovine serum,100 U/mL penicillin and 100 mg/mL streptomycin. Human female HEK293T cell lines were obtained from the American Type Culture Collection and were grown in DMEM, plus 10% heat-inactivated fetal bovine serum,100 U/mL penicillin and 100 mg/mL streptomycin.
Animals
All animal procedures used in this study were approved by the Institutional Animal Care and Use Committee at the University of North Carolina at Chapel Hill. Mice were housed in an AAALAC accredited facility in accordance with the Guide for the Care and Use of Laboratory Animal. All animal procedures were approved by the University of North Carolina at Chapel Hill Institutional Animal Care and Use Committee (IACUC). Sacs(−/−) mice were a kind gift from Dr. Stefan Strack. Sacs(+/−) mice were generated by mating Sacs KO mice with wild type C57BL/6J mice (Jackson Laboratories). Primers for genotyping are as follows: WT allele forward primer: 5’ - GCTGTCAGGGGGAAATCTGATAAAG –3', WT allele reverse primer: 5’ - GCAGCACCTTTAGACAAAAGATTGC –3', Sacs KO allele forward primer: 5’ - CAACCTTGGAGAAACTGTGCCTG – 3', Sacs KO allele reverse primer: 5’ - CACCGACGCCAATCACAAACAC –3'.
Mouse primary cortical cultures
Cortices were isolated from E15.5 mouse pups (Simon et al., 2019). Cortices from each animal were dissociated in papain (Pierce, 88285) and DNase (Sigma, D4513) for 30 min at 37°C, and filtered through a 70 μm filter. Each animal was genotyped during dissection using the above PCR primers, and cortices from either WT or KO animals were pooled independent of sex. Cells were plated into 3 wells of poly-D-lysine coated 6 well plates at 1 × 106 cells per well in Neurobasal medium (Life Technologies) containing 5% fetal bovine serum (Invitrogen), B27 (17504–044, Invitrogen), Antibiotic-Antimycotic (15240–062, Invitrogen) and GlutaMAX (35050–061, Invitrogen). On day 3, we performed a full media change, replacing with the above media with serum omitted, and supplemented with the antimitotic FDU to inhibit proliferation of non-neuronal cell types. We performed 50% media changes every third day. Pooled cultures from each litter was considered a single replicate.
Method details
Antibodies
A detailed list of antibodies and dilutions used in this study is provided in Table S5.
Genome editing to generate sacsin KO SH-SY5Y cells
An SH-SY5Y cell line with the sacsin truncation mutation M783∗ was generated using CRISPR/Cas9. We cloned the SACS targeting guide RNA (gRNA) TTTCATGGCTTAAGATGGTTTGG (PAM sequence underlined) into the p1261_GERETY_U6_BasI_gRNA vector for expression of the gRNA under control of the U6 promoter. The gRNA expression vector was co-transfected with a Cas9 expression vector (hCas9, Addgene # 41815) and a targeting vector with homology arms to introduce the M783∗ mutation along with a puromycin selection cassette (pMCS-SACStrunc-PB:PGKpuroDtk) using Lipofectamine 3000. Puromycin-resistant clones were selected and screened by PCR and sequencing.
Proteomic, phosphoproteomic sample preparation: SH-SY5Y
SH-SY5Y were grown to ∼80% confluency in 13 T75 flasks for harvesting. SH-SY5Y flasks were placed on ice, washed twice with ice cold PBS and harvested by scraping cells in lysis buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 0.5 % Triton X-100, 1mM EDTA, 1mM EGTA, 10mM NaF, 2.5mM Na3VO4, complete protease inhibitor cocktail (Roche), and phosphatase inhibitor cocktail 2 and 3 (Sigma). Lysates were sonicated by pulsing once at 30% for 10 s and then twice at 40% for 10 s with 10 s rest on ice between each pulse (Branson 150 Sonifier). Lysate was transferred to microcentrifuge tube and spun at 14,000 × g for 10 min at 4°C. Lysate was filtered through a 0.2 μm syringe filter and stored at −80°C until all replicates were collected. Protein concentration was quantified using a Bradford assay. Cell lysates (1mg, n = 3) were acetone precipitated overnight and stored at −20°C. Protein pellets were resuspended in 8M urea, then reduced with 5M DTT at 56°C for 30 min and alkylated with 15mM iodoacetamide in the dark at RT for 45 min. Samples were diluted to 1M urea, then digested overnight with trypsin (Promega) at 1:100 trypsin:protein ratio. Samples were acidified then desalted using C18 desalting spin columns (Pierce). A peptide BCA colorimetric assay (Pierce) was performed and 500μg of each sample was individually labeled with TMT6 reagent (Thermo). After labeling efficiency was confirmed, the TMT6 labeled samples were mixed and desalted using C18 desalting spin column (Pierce). A 100μg aliquot was set aside for global proteome analysis, and was fractionated into 4 fractions using a High pH reversed phase fractionation spin column (Pierce). The rest of the sample (∼3mg) was enriched for phosphopeptides using Ti-MAC magnetic beads (ReSyn Biosciences). The Ti-MAC eluate was fractionated into 3 fractions using High pH reversed phase fractionation spin column. Global proteome and phosphoproteome fractions were dried down via vacuum centrifugation and stored at −80°C until LC/MS/MS analysis.
MIB/MS kinome enrichment
Multiplexed inhibitor bead (MIB) kinase enrichment was performed as previously described with a slightly modified bead composition (Arend et al., 2017). Cells were lysed in MIB-MS buffer (50 mm HEPES, pH 7.5, 150 mm NaCl, 0.5% Triton X-100, 1 mm EDTA, 1 mm EGTA, 10 mm NaF, Phosphatase Inhibitor Mixture 2 (Sigma, P5726) and 3 (Sigma P0044), and 2.5 mm NaV04 plus Protease Inhibitor Mixture (Roche). Samples were sonicated and clarified by centrifugation at 14,000g and filtered through a 0.2 μm filter (Corning, #431219). Protein was quantified by Bradford assay. Specifically, each sample was applied to an individual 350 μL Poly-Prep chromatography column (Bio-Rad) containing the following immobilized kinase inhibitors: CTx-0294885, PP58, Purvalanol B, UNC2147A, VI-16832, UNC8088A. Proteins were eluted from columns by boiling in elution buffer (100 mm Tris-HCl, pH 6.8, 0.5% SDS, 1% β-mercaptoethanol) for 15 min. Samples were incubated at room temperature for 30 min in the dark. dl-DTT was added to bring the final concentration to 10 mm and samples were incubated at room temperature for in the dark for 5 min. Samples were concentrated to a final volume of ∼100 μL in 10K Amicon Ultra centrifugal concentrators and proteins were purified by methanol chloroform extraction. Samples were re-suspended in 50 mm HEPES, pH 8.0, and digested with sequencing grade porcine trypsin (Promega) overnight at 37°C. Samples were extracted with ethyl acetate 3 times to remove residual detergent, desalted using Pierce C-18 spin columns, and submitted to the UNC Michael Hooker Proteomics Core for LC/MS/MS analysis.
Primary cortical culture sample prep
On day 10 post plating lysates from primary cortical cultures were acetone precipitated overnight and stored at −20°C. Protein pellets (500 μg, n = 3 per genotype) were resuspended in 8M urea, then reduced with 5M DTT at 56°C for 30 min and alkylated with 15mM iodoacetamide in the dark at RT for 45 min. Samples were diluted to 1M urea, then digested overnight with trypsin (Promega) at 1:100 trypsin:protein ratio. Samples were acidified then desalted using C18 desalting spin columns (Pierce). A peptide BCA colorimetric assay (Pierce) was performed and 300μg of each sample was individually labeled with TMT6 reagent (Thermo). After labeling efficiency was confirmed, the TMT6 labeled samples were mixed and desalted using C18 desalting spin column (Pierce). The TMT sample (3 mg total) was fractionated using high pH reversed phase HPLC (Mertins et al., 2018). Briefly, the peptide samples were separated by offline high pH reverse-phase HPLC (Agilent 126) and fractionated over a 90 min method, into 96 fractions using an Agilent Zorbax 300 Extend-C18 column (3.5-μm, 4.6 × 250 mm). Mobile phase A containing 4.5 mM ammonium formate (pH 10) in 2% (vol/vol) LC-MS grade acetonitrile, and mobile phase B containing 4.5 mM ammonium formate (pH 10) in 90% (vol/vol) LC-MS grade acetonitrile were used for separation. The 96 resulting fractions were then concatenated in a non-continuous manner into 24 fractions, dried down via vacuum centrifugation and stored at −80°C until LC-MS/MS analysis.
Cell surface labeling
2 × 106 SH-SY5Y WT and sacsin KO cells were each plated in nine 10 cm dishes, and cultured until 95% confluent (∼3 days) (n = 3 per cell line). To identify proteins which purify non-specifically, an additional replicate of WT/KO lines were processed as below, but without the addition of Biocytin hydrazide. Cells were lifted using CellStripper Dissociation Reagent (Corning, #25056CI) for 20 min at 37°C and resuspended in 1X PBS (pH 6.5) + 1.6 mM NalO4 and rotated at 4°C for 20 min in the dark. Cells were washed three times then resuspended in 1X PBS (pH 6.5) + 10mM Aniline + 1mM Biocytin hydrazide and incubated at room temperature for 60 min, then at 4°C for 20 min while rotating. After three PBS washes, cell pellets were resuspended in RIPA, rotated at 4°C for 30 min, and sonicated with 1 s pulses at 20% power for 1 min. To enrich for the labeled surface proteins, cells were centrifuged 15,000 rpm for 10 min at 4°C and supernatant was incubated in washed Neutravidin High-Capacity Resin (ThermoFisher #29204) for one hour at 4°C. Resin was added to gravity column and washed with RIPA, 1X PBS (pH 7.4) + 1M NaCl, Ammonium Bicarbonate (ABC) + 2M Urea then resuspended in ABC + 2M Urea +5 mM tris(2-carboxyethyl)phosphine (TCEP) and incubated at room temperature in the dark at 55°C shaking at 300 rpm for 30 min. Iodoacetamide (IAM) was then added to a final concentration of 11 mM and shaken at room temp for 30 min in the dark. Resin was centrifuged at 500g for 5 min and resuspended in 1 mL ABC + 2M urea containing 20 μg trypsin (Fisher #P8101) to fragment peptides at RT overnight. To desalt, samples were acidified to pH < 2 with 10% trifluoroacetic acid (TFA) in C-18 spin column (ThermoFisher #89873), washed, and resuspended in 40% acetonitrile +0.1% formic acid then dried with vacuum centrifugation and stored at −80°C.
LC-MS/MS analysis
Kinome, proteome, and phosphoproteome were analyzed by LC/MS/MS using an Easy nLC 1200 coupled to a QExactive HF mass spectrometer (Thermo Scientific). Samples were injected onto an Easy Spray PepMap C18 column (75 μm id × 25 cm, 2 μm particle size) (Thermo Scientific) using a 120 min method. The gradient for separation consisted of 5–50% mobile phase B at a 250 nL/min flow rate, where mobile phase A was 0.1% formic acid in water and mobile phase B consisted of 0.1% formic acid in 80% ACN. The QExactive HF was operated in data-dependent mode where the 15 most intense precursors were selected for subsequent HCD fragmentation.
For kinome samples, QExactive HF was operated as previously described (Arend et al., 2017). QExactive HF resolution for the precursor scan (m/z 350–1600) was set to 120,000 with a target value of 3 × 106 ions and a maximum injection time of 100 ms. MS/MS scans resolution was set to 60,000 with a target value of 1 × 105 ions and a maximum injection time of 100 ms. The normalized collision energy was set to 27% for HCD with an isolation window of 1.6 m/z. Dynamic exclusion was set to 30 s, peptide match was set to preferred, and precursors with unknown charge or a charge state of 1 and ≥8 were excluded.
For TMT proteome and phosphoproteome samples (each biological replicate analyzed in duplicate), QExactive HF resolution for the precursor scan (m/z 350–1600) was set to 60,000 with a target value of 3 × 106 ions and a maximum injection time of 100 ms. MS/MS scans resolution was set to 60,000 with a target value of 1 × 105 ions and a maximum injection time of 100 ms. Fixed first mass was set to 110 m/z and the normalized collision energy was set to 32% for HCD with an isolation window of 1.2 m/z. Dynamic exclusion was set to 30 s, peptide match was set to preferred, and precursors with unknown charge or a charge state of 1 and ≥8 were excluded.
Cell surface samples and the primary cortical culture samples were analyzed by LC-MS/MS using a Thermo Easy nLC 1200 coupled to a Thermo Fusion Lumos mass spectrometer. Samples were injected onto a Thermo PepMap C18 trap column, washed, then loaded onto an Easy Spray PepMap C18 analytical column (75 μm id × 25 cm, 2 μm particle size) (ThermoFisher). The samples were separated over a 120 min method, where the gradient for separation consisted of 5–45% mobile phase B at a 250 nL/min flow rate; mobile phase A was 0.1% formic acid in water and mobile phase B consisted of 0.1% formic acid in 80% acetonitrile. For the cell surface samples, MS1 orbitrap scans were collected at a resolution of 120,000 and 1e6 AGC target. The MS2 scans were acquired in the Orbitrap at 15,000 resolution, with a 1.25e5 AGC, and 50ms maximum injection using HCD fragmentation with a normalized energy of 30%. Dynamic exclusion was set to 30 s and precursors with unknown charge or a charge state of 1 and ≥8 were excluded. For the primary cortical culture TMT proteome fractions (24 total), the Lumos was operated in SPS-MS3 mode (McAlister et al., 2014), with a 3s cycle time. Resolution for the precursor scan (m/z 400–1500) was set to 120,000 with an AGC target set to standard and a maximum injection time of 50 ms. MS2 scans consisted of CID normalized collision energy (NCE) 32; AGC target set to standard; maximum injection time of 50 ms; isolation window of 0.7 Da. Following MS2 acquisition, MS3 spectra were collected in SPS mode (10 scans per outcome); HCD set to 55; resolution set to 50,000; scan range set to 100–500; AGC target set to 200% with a 100 ms maximum inject time.
Proteomic data analysis
Kinome, proteome and phosphoproteome raw data files were analyzed with MaxQuant version 1.6.1.0 and searched against the reviewed human database (downloaded Feb 2017, containing 20,162 entries), using Andromeda within MaxQuant. Enzyme specificity was set to trypsin, up to two missed cleavage sites were allowed, carbamidomethylation of C was set as a fixed modification and oxidation of M and acetyl of N-term were set as variable modifications. For phosphoproteome samples, phosphorylation of S,T,Y was set as a variable modification. For phosphoproteome samples, phosphorylation of S,T,Y was set as a variable modification. For proteome and phosphoproteome samples, TMT6plex of peptide N-termini & K was set as a fixed modification and the quantitation type was set to reporter ion MS2. For kinome label-free quantitation, match between runs was enabled. A 1% FDR was used to filter all data. For kinome data, a minimum of two peptides was required for label-free quantitation using the LFQ intensities.
Cell surface proteome raw data files were processed using MaxQuant version 1.6.15.0 and searched against the reviewed human database (downloaded Feb 2020, containing 20,350 entries), using Andromeda within MaxQuant. Enzyme specificity was set to trypsin, up to two missed cleavage sites were allowed, carbamidomethylation of C was set as a fixed modification and oxidation of M and acetyl of N-term were set as variable modifications. A 1% FDR was used to filter all data and match between runs was enabled. A minimum of two peptides was required for label-free quantitation using the LFQ intensities.
For all proteomic datasets, proteins with a missing value in one replicate were imputed using the KNN imputation method, proteins with two or more missing values were removed from analysis. Linear Models for Microarray Data (LIMMA) was used to calculate log2 fold change of LFQ intensity and perform statistical analysis (Ritchie et al., 2015). FDR was calculated using Benjamini-Hochberg adjusted p values. For proteins identified in the surfaceome, we annotated them as ‘Membrane” or ‘Exosome’ based on DAVID bioinformatics database. Proteins which were identified in unlabeled controls (no biotin) were removed from further analysis. Proteins with p < 0.05 and log2 fold change of KO/WT ±>0.4 were included in downstream analyses. The Kinome tree was generated on CORAL (Metz et al., 2018).
Tau aggregation assay
The Homogenous Time Resolved Fluorescence (HTRF) (Degorce et al., 2009) Tau aggregation kit (Cisbio, MA) was used to determine tau aggregate levels in undifferentiated and differentiated SH-SY5Y lysates using Fluorescence Resonance Energy Transfer (FRET). Cells were scraped in ice-cold PBS and pelleted followed by lysis using buffer provided in the kit. Protein lysates were quantified using Bradford assay. Serially diluted cell lysates were tested in duplicate in a 96 well to determinate the optimal concentration of protein lysates. Anti Tau-d2 and anti tau-Tb conjugates were diluted 1:50 from stock to a final volume, which was calculated based on the total number of samples. The two conjugates were diluted separately, mixed at equal ratios and vortexed before 10μL were added to 10μL (10μg total concentration) of the protein lysates in each well. Lysates and conjugates were then incubated for 2h at room temperature and the plate was read on the CLARIOstar plate reader using the HTRF filter cube which allows for sequential detection of Donor and Acceptor fluorescence. Signal was measured as the peak ratio of 655nm (acceptor fluorescence) to 620nm (donor fluorescence). The results of the two emission signals were plotted as HTRF Ratio or DeltaF% values.
GO term analysis
For GO term graphs, the list of significant genes for each proteomic experiment were input into geneontology.com with human genome as the background, Fisher’s exact test with FDR correction (Gene Ontology, 2021). Graphs include terms in all categories (biological processes, molecular function, cellular component). Due to the hierarchical nature of GO terms in Panther (i.e. groups of terms have a nested nature to assign relationships between them) we only considered the most proximal term in each hierarchy to ensure terms were specific and directly comparable. These proximal terms are listed as “PARENT” in Table S2. All terms underneath each parent are also listed for reference. Terms were ranked by FDR value and the top ten non-redundant top terms were included in each figure. All terms, including graphs, were generated using R scripts from bio-protocol.org (https://doi.org/10.21769/BioProtoc.3429).
Western blots
Cells were lysed with RIPA buffer and 20 ug protein was loaded per lane and on a Novex 4–12% (4–20% for BRSK2) Tris-Glycine gel (ThermoFisher). Protein was transferred to PVDF and blocked with 1X Blocker BSA (ThermoFisher). Blots were washed and incubated with primary antibodies followed by HRP secondary antibodies (ThermoFisher). Protein was quantified using ImageJ software. Each lane was normalized to the relative density of GAPDH/ACTB.
Cell surface protein isolation (Figure 5A) was validated through western blot by collecting 250 μL resin (bound to cell surface proteins) from the wash column and centrifuging at 2000xg for 5 min to pellet resin and remove residual 50mM ABC + 2M Urea. Resin was then resuspended in 250 μL 2X Laemmli buffer with 5% 2-mercaptoethanol. The control sample contained 10 μL of total cell lysates (collected prior to resin addition) from SH-SY5Y WT and sacsin KO cells. The samples were run on a 4–15% TGX Bio-Rad pre-stained gel (Bio-Rad #4568094) and transferred to a PVDF membrane, which was blocked in 5% milk in 1X TBST. Primary antibodies were diluted 1:1000 in 5% BSA in 1X TBST. Goat anti-rabbit HRP secondary antibody was diluted 1:5000 in 5% milk.
Remaining immunoblotting was performed as described previously (Duncan et al., 2017).
Fractionated western blots
SH-SY5Y WT and sacsin KO cells were fractionated with the Minute Plasma Membrane Protein Isolation and Cell Fractionation Kit (Invent). The cytoplasmic fraction was lysed in Buffer A and the plasma membrane fraction was lysed in RIPA buffer, both fractions contained protease inhibitor (Pierce). Proteins were diluted 1:1 with 2× laemmli buffer with betamercaptoethanol and run on TGX prestained gels (Biorad). Total protein images were obtained before transferring onto a PVDF membrane. Individual protein levels were normalized to total protein image using Image J software for quantification.
Endogenous co-immunoprecipitation
SH-SY5Y cells were subjected to chemical cross-linking
The cleavable, homo-bifunctional cross-linker dithiobis[succinimidylpropionate] (DSP; Pierce, Rockford, IL, USA) was diluted to a final concentration of 1mM in PBS and added to the cultured cells. After incubation for 1h at room temperature, cross-linking was stopped by addition of Tris (pH 7.5) to a final concentration of 20mM. Cells were then washed twice in ice-cold PBS, before the cells were harvested in RIPA buffer (NFASC IP) or 50mM Tris-HCl pH7.4 + 150mM NaCl + 1mM EDTA +0.5% Triton X-100, supplemented with protease inhibitors (Pierce/Roche) and incubated on ice for 5 min (sacsin/VIM IP).
Sacsin and vimentin IP
A small aliquot of supernatant was removed for analysis by immunoblotting (input fraction) the remaining supernatant was incubated with rabbit monoclonal anti-vimentin antibody or rabbit monoclonal anti-sacsin overnight at 4°C on a rotor. After 16 h 50μL of magnetic beads (Sigma, Poole, UK) were washed twice in PBS-Tween 0.1% buffer, before being recovered in a magnetic separator. The beads were then resuspended within the cell lysate already incubated with the antibody for 2h at room temperature and washed.
NFASC IP
Protein concentration was assessed with a Bradford assay and a small aliquot of supernatant was removed for analysis by immunoblotting (input fraction). 300 μg of the sample were used for the immunoprecipitation. First, 20 μL of protein A magnetic beads (73778, CST) washed with 1× Cell Lysis buffer (9803, CST) were added to the samples for 20 min for preclearing. Next, beads were removed and 5 μL of the precipitation antibody, anti-neurofascin (PA5-78668, Invitrogen) or normal rabbit IgG isotype control (2729, CST), were added to the samples and incubated overnight at 4°C on a rotor. After 16 h, fresh prewashed magnetic beads were added to the samples and rotated at room temperature for 20 min before being recovered in a magnetic separator and washed five times with 1× Cell Lysis Buffer.
Cell culture immunostaining image acquisitions
All cell culture samples, with the exception of samples prepared for microtubule staining, were fixed with 4% PFA and permeabilized with 0.1 to 0.3% Triton X-100. For microtubule staining, samples were fixed with 100% methanol for 20 min at −20°C. Non-specific binding was reduced by blocking in 5% Normal donkey serum or 1% bovine serum albumin (BSA)+10% normal goat serum. Cells were incubated with primary antibodies for a minimum of 1 h at RT, followed by incubation with labelled secondary antibodies, Phalloidin, and Hoechst for 1 h at RT. Samples were mounted either in Fluoro-Gel mounting medium (Electron Microscopy Sciences, Cat. # 17985–30) or ProLong Diamond Antifade Mountant (Invitrogen, Cat. #P36961). Widefield images were captured with GE IN Cell 2200 high content imaging system equipped with a Plan Fluor 20×/0.75 NA air objective. Confocal images were acquired using an inverted Olympus FV3000RS. Plan Apo 60×/1.4 NA oil (Olympus) or Plan Apo 30×/1.05 NA silicon oil (Olympus) objectives were used. SIM images were acquired and reconstructed in 3D-SIM mode using a Nikon N-SIM system equipped with a Plan Apo TIRF 100×/1.49 NA oil objective.
For FA immunolabelling, coverslips were coated with 10μg/mL fibronectin solutions overnight. Confocal microscopy was performed using a LSM880 (Zeiss) with a 63× objective and an AiryScan module. Quantification of incidence of cells with perinuclear vimentin accumulation and incidence of FA was performed blind to experimental status. Imaging processing was carried out with Zen Blue software (Zeiss). FA isolation from cells was performed by hypotonic shock to remove cells while leaving FAs intact, as described previously (Kuo et al., 2012). Isolated FAs were then immunolabelled to detect vinculin and analysed using confocal microscope with quantification as described previously. All image processing steps were carried out using ImageJ software.
Focal adhesion
EB1-GFP imaging, tracking and quantification
For Total Internal Reflection Fluorescence (TIRF) imaging, SH-SY5Y cells were cultured on 35 mm glass bottom dishes (Mattek) and transfected with plasmid expressing EB1-GFP using FuGene transfection reagent (Promega). pGFP-EB1 was a gift from Lynne Cassimeris (Addgene plasmid #17234). TIRF live cell imaging was carried out 24 h after transfection on an inverted Nikon Eclipse Ti2 equipped with a Plan Apo TIRF 100×/1.49 NA oil (Nikon) objective. TIRF images were captured at single z-plane, every second for a period of 1 min. Tracking of EB1-GFP comet was performed using the FIJI plugin TrackMate with the following analysis settings: Laplacian of Gaussian detector with an estimated spot diameter of 0.16 μm, subpixel localization enabled, simple LAP tracker, minimum number of spots on track >9, and maximum number of spots on track <35. Each trajectory was visually inspected to confirm tracking accuracy. Mean track velocities were plotted and statistical analyses were performed using GraphPad Prism 9.0.
Nocodazole treatment experiment
SH-SY5Y cells were treated with 10 μM nocodazole (Sigma, Cat. #SML1665) or DMSO control for 3 h at 37°C. Washout of nocodazole was performed by one wash with ice old PBS and one wash with pre-warmed media. Cells were fixed with 100% methanol at indicated timepoints. Measurement of microtubule staining in untreated cells and repolymerization after nocodazole washout were performed on images constructed with maximum intensity projections. Areas occupied by microtubules and cells were thresholded and quantified using a custom CellProfiler pipeline. Microtubule density was quantified by thresholding and normalizing area occupied by microtubules to cell area.
SH-SY5Y neuronal differentiation
SH-SY5Y cells were differentiated using a modified version of a previously described method (Shipley et al., 2016). Cells were grown to near confluence under normal maintenance conditions prior to the start of differentiation. 1 × 105 cells/well were seeded in a 6 well plate. After 24 h media was changed to DMEM containing 2% Fetal Bovine Serum (FBS: Gibco, #A3840001) and10 μM of all-trans retinoic acid (ATRA: Sigma, #R2625). At day 4 cells were passaged and resuspended in neurobasal media (Gibco, #21103049) supplemented with B27 (Gibco, #17504044) 50 ng/mL Brain-Derived Neurotrophic Factor (BDNF: Sigma, #B3795), and 10 μM RA. At day 12 media was supplemented with 2 mM dibutyryl cyclic AMP (dc-AMP: Santa Cruz, sc-201567A). Media was changed every other day with fresh aliquots of RA, BDNF and dcAMP. Cells were harvested at day 15 for downstream analysis.
MitoTracker green FM live imaging
Light sheet fluorescence microscopy was used to image mitochondrial movement. Cells were incubated with 100 nM of MitoTracker Green FM (Invitrogen, Cat. #M7514) for 20 min at 37°C and then washed with prewarmed media prior to imaging. Mitochondrial movements along the neurites were captured every at 0.5 μm z-steps, 10 s intervals, for 3 min on a ASI’s RAMM frame (ASI), equipped with Mizar TILT light sheet illumination module (Mizar Imaging LLC), Prime 95B sCMOS (Photometrics) camera, and a Plan Apo TIRF 100×/1.45 NA oil (Nikon) objective. Prior to fluorescence imaging, a brightfield image was captured to confirm presence of neurites. Kymographs of mitochondrial movement along neurites were generated using the line tools and resliced command in FIJI.
Plasmids and siRNAs
Plasmids encoding tdTomato:Vinculin (Addgene #58146) or vimentin-EGFP (Addgene #56439) were transfected with Lipofectamine 3000 in Opti-Mem (ThermoFisher) according to the manufacturer’s instructions. To induce vimentin bundle cells were treated for 4 or 24 h with 10 μm simvastatin, in their standard culture medium. For PTEN knockdown a combination of four siRNAs targeting PTEN were used (ON-TARGETplus Human PTEN [5728] siRNA - SMARTpool: Target sequence1#: Sense GAUCAGCAUACACAAAUUA, Antisense: UAAUUUGUGUAUGCUGAUC; Target sequence2#: Sense GACUUAGACUUGACCUAUA, Antisense: UAUAGGUCAAGUCUAAGUC; Target sequence3#: Sense GAUCUUGACCAAUGGCUAA, Antisense: UUAGCCAUUGGUCAAGAUC; Target sequence4#: Sense CGAUAGCAUUUGCAGUAUA, Antisense: UAUACUGCAAAUGCUAUCG). These siRNAs were at a concentration of 10 nm each and were transfected in combination using Lipofectamine 3000 (ThermoFisher), according to the manufacturer’s instructions. A negative control siRNA with no significant sequence similarity to human gene sequences was used as a control.
Fluorescence recovery after photobleaching (FRAP)
Cells were transfected with plasmid encoding vimentin-EGFP or tdTomato:Vinculin. 48 h post transfection FRAP experiments were conducted as described previously using an LSM880 microscope (Zeiss) (Girard et al., 2012). 2μm x 2μm regions of interest in cells expressing vimentin-EGFP or tdTomato:Vinculin were excited with the 488 nm or 568nm laser lines respectively. Following photobleaching regions of interest were imaged a minimum of 50 cycles with a 1 s interval. Image and data acquisition were performed using Zen Black and Zen Blue software (Zeiss). Fluorescence intensity in the photo-bleached regions at each time point were quantified as a percentage of fluorescence intensity before photobleaching.
Transwell migration assays
Cell migration and invasion abilities were assessed using Transwell cell culture inserts (BD Biosciences). For the cell migration assay, 2.5 ×104 cells in 500 μL in serum-free medium were seeded directly into the wells of Transwell chambers with 8 μm-pore membranes. Medium containing 10% FBS, was added into the lower chamber. After 24h, cells were fixed and stained with 2% Giemsa blue stain (Sigma). Cells adhering to the upper surface of the membrane were removed using a cotton applicator. Cells on the lower side of the membrane were counted. Five fields were randomly selected per cell line and the mean number of cells quantified.
Scratch assay
Cells were detached from the tissue culture plate using 0.25% Trypsin-EDTA solution and plated at the appropriate number of cells in a 6-well plate for 100% confluence in 24 h. In a sterile environment the monolayer was scratch with a pipette tip forming a 1-2mm scratch from one edge of the well to the other. The media was removed and replaced with 2mL of fresh media. Following the generation and inspection of the wound the plate was placed in an incubator set at 37°C and 5% CO2. Pictures were taken at different time points. A time-lapse microscope with a controlled temperature at 37°C and 5% CO2, was also used in parallel experiments.
Tissue collection and immunostaining
Mice were anesthetized using pentobarbital and perfused with 4% PFA in PBS via transcardiac perfusion. Tissue was dissected and drop-fixed in 4% PFA for 24 h at 4°C, followed by a 48-h incubation in 30% sucrose at 4°C. Tissue was mounted in M1 Embedding matrix (ThermoFisher) and stored at −80°C. Tissue was sectioned on a CryoStar NX50 (ThermoFisher) in 40μm sections. Slices were transferred into 200μL of permeabilization buffer (5% NDS, 0.3% Triton X-100, 2% DMSO, 0.01% Sodium Azide, 1X PBS) for 60 min on a shaker at room temperature. Following the incubation, buffer was removed and 200μL of primary antibodies diluted in staining buffer were added for 24–48 h with gentle rocking at room temperature. Sections were washed with PBST (0.3% Triton X-100/1X PBS) three times for 10 min, followed by 2 h room temperature incubation with gentle rocking in the dark in 100 μL staining buffer with secondary antibodies. The secondary antibody was removed and DAPI diluted to 1:1000 was added for 5 min before 3 washes in PBST (0.1% Triton X-100/1X PBS). Slices were mounted on Superfrost Plus slides (ThermoFisher), dried, and ∼200μL mounting medium (Sigma, Polyvinyl alcohol mounting medium with DABCO®, antifading) was added prior to placing a cover slip. Slides were dried overnight prior to imaging. Images were acquired on the Nikon 710 confocal microscope.
Image quantification
Raw image Z-stacks were visualized in FIJI. To quantify Calb+ synapses onto DCN Neun+ neurons (Figures 6I and 6J), we selected individual DCN neurons that were ∼20–25 um in diameter using only the NEUN channel (being blind to CALB1 staining). For each DCN neuron the single Z-stack at the widest diameter was manually isolated. Each DCN neuron was then analyzed using a custom CellProfiler pipeline. In brief, we measured mean intensity and size of Calb+ objects within 1 μm of each DCN neuron. n = 4 animals for each genotype, two sections per animal, for a total of 137 DCN neurons. Images in Figures 6L and S6F were quantified using a custom CellProfiler pipeline. In brief, soma and axons were isolated based on their sizes, and mean intensity was calculated for each object.
Sacsin interactome analysis
Proteins were extracted from 8.5 × 106 cells of KO and WT SH-SY5Y cell lines using 500 μL of the lysis buffer (RIPA with phosphatase and protease inhibitors (EDTA free, Merk). The lysates were cleared by centrifugation (13000g) for 15 min at 4°C and then incubated with 1/50 dilution of anti-sacsin antibody (Abcam, ab181190) overnight at 4°C with slow rotation. Protein A Dynabeads (Merk) were equilibrated with the ice-cold lysis buffer and incubated with each cell lysate/antibody mix for 2 h at 4°C with slow rotation. The beads were then washed with the ice-cold lysis buffer and the bound proteins were eluted with 40μL of 2xLaemmli buffer. The samples were then heated at 95°C for 5 min, centrifuged and loaded onto the NuPAGE™ 4 to 12%, Bis-Tris, 1.5 mm, Mini Protein Gel (ThermoFisher). The gels were resolved in 1× MOPC for 2 h at 4°C and then stained with SimplyBlue SafeStain (ThermoFisher) according to the manufacturer’s protocol. Each gel lane was then cut into 10 fragments and the proteins were extracted from the gel using trypsin digest protocol as previously described (Patel et al., 2009). Digests were analyzed using a Waters NanoAcquity Ultra-Performance Liquid Chromatography system and data processed using PLGS v3.0.2 (Waters, UK). For protein interactome analysis we only focused on proteins which were completely absent from all KO lysates, as these are the most stringent, high-confidence interactors. However, several proteins were identified in both WT/KO lysates, but had substantially reduced intensity values in KO cells. These proteins are also included in Table S4.
Quantification and statistical analysis
For each experiment, the statistical test used, sample size, definition of a replicate, and precision measures are defined in the corresponding figure legend. Center points in all figures are mean of all replicates. Statistical tests were performed using either R or Excel.
Acknowledgments
We express our gratitude to Sonia Gobeil and the ARSACS patient community. This work was supported by grants to J.P.C. and J.M.W. from the Fondation de l’Ataxie Charlevoix-Saguenay. We thank Thomas Sterns, Karim Gilbert, Natalie Barker, and Dennis Goldfarb for technical assistance. We thank Stefan Strack for generously providing ARSACS mice. We also thank Mark Zylka, Jason Stein, and Dave Morris for their support and advice. J.P.C. was supported by the BBSRC (BB/R003335/1) and Ataxia UK. J.M.W. was supported by grants from the NICHD T32HD040127 and a Pfizer-NCBiotech postdoctoral fellowship in gene therapy. L.M.G. is supported by NIH R01 GM138520. L.X. is supported by a grant from the USDOD, W81XWH-19-1-0402. The UNC Catalyst for Rare Diseases gratefully acknowledges the support of the Eshelman Institute for Innovation. This research is based in part upon work conducted using the UNC Proteomics Core Facility, which is supported in part by NCI Center Core support grant (2P30CA016086-45) to the UNC Lineberger Comprehensive Cancer Center. Confocal microscopy was performed using a microscope funded by Barts Charity (MGU0293) and at the UNC Neuroscience Microscopy Core (RRID: SCR_019060) supported, in part, by funding from the NIH-NINDS Neuroscience Center support grant P30 NS045892 and the NIH-NICHD Intellectual and Developmental Disabilities Research Center support grant U54 HD079124.
Author contributions
J.M.W. and J.P.C. conceived of the study. J.M.W., J.P.C., and L.E.L.R. analyzed data and prepared the manuscript. S.N., T.M.H., and J.M.W. created sacsin KO neuroblastoma lines. L.M.G. aided in experimental design and provided reagents for proteomics experiments. A.J.H. and K.T. provided reagents and experimental oversight. W.Y.A., K.M.H., B.T.-B., L.X., and J.M.W. performed histology. S.H., T.M.H., and W.Y.A. managed the mouse colony. K.M.H., T.M.H., S.H., J.B., L.E.H., and J.M.W. performed quantitative proteomics experiments. T.V.N. and C.L.H. performed RNA-seq. K.M.H. and J.M.W. analyzed the data. T.V.N. and C.L.H. performed the sacsin interactome experiments. A.H. and K.T. performed co-IP MS experiments. L.E.L.R., W.Y.A., K.M.H., T.V.N., T.M.H., S.H., L.P., J.M.W., and G.C.A. performed cell biology experiments.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: November 1, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111580.
Contributor Information
J. Paul Chapple, Email: j.p.chapple@qmul.ac.uk.
Justin M. Wolter, Email: justin_wolter@med.unc.edu.
Supplemental information
Data and code availability
-
•
A list of all cell lines used in each figure are provided in Table S1. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2022) partner repository with the dataset identifier PXD036906. The RNA-seq dataset generated during the current study is available in the Gene Expression Omnibus (GEO) repository under project identifier GSE214213.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Ady V., Toscano-Marquez B., Nath M., Chang P.K., Hui J., Cook A., Charron F., Lariviere R., Brais B., McKinney R.A., Watt A.J. Altered synaptic and firing properties of cerebellar Purkinje cells in a mouse model of ARSACS. J. Physiol. 2018;596:4253–4267. doi: 10.1113/JP275902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J.F., Siller E., Barral J.M. The sacsin repeating region (SRR): a novel Hsp90-related supra-domain associated with neurodegeneration. JMB (J. Mol. Biol.) 2010;400:665–674. doi: 10.1016/j.jmb.2010.05.023. [DOI] [PubMed] [Google Scholar]
- Ando K., Maruko-Otake A., Ohtake Y., Hayashishita M., Sekiya M., Iijima K.M. Stabilization of microtubule-unbound tau via tau phosphorylation at Ser262/356 by par-1/MARK contributes to augmentation of AD-related phosphorylation and abeta42-induced tau toxicity. PLoS Genet. 2016;12:e1005917. doi: 10.1371/journal.pgen.1005917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arend K.C., Lenarcic E.M., Vincent H.A., Rashid N., Lazear E., McDonald I.M., Gilbert T.S., East M.P., Herring L.E., Johnson G.L., et al. Kinome profiling identifies druggable targets for novel human cytomegalovirus (HCMV) antivirals. Mol. Cell. Proteomics. 2017:S263–S276. doi: 10.1074/mcp.M116.065375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek S.J., Park J.S., Kim J., Yamamoto Y., Tanaka-Yamamoto K. VTA-projecting cerebellar neurons mediate stress-dependent depression-like behaviors. Elife. 2022;11:e72981. doi: 10.7554/eLife.72981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker J., Spits M., Neefjes J., Berlin I. The EGFR odyssey - from activation to destruction in space and time. J. Cell Sci. 2017;130:4087–4096. doi: 10.1242/jcs.209197. [DOI] [PubMed] [Google Scholar]
- Barron T., Saifetiarova J., Bhat M.A., Kim J.H. Myelination of Purkinje axons is critical for resilient synaptic transmission in the deep cerebellar nucleus. Sci. Rep. 2018;8:1022. doi: 10.1038/s41598-018-19314-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bausch-Fluck D., Hofmann A., Bock T., Frei A.P., Cerciello F., Jacobs A., Moest H., Omasits U., Gundry R.L., Yoon C., et al. A mass spectrometric-derived cell surface protein atlas. PLoS One. 2015;10:e0121314. doi: 10.1371/journal.pone.0121314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benetatos J., Bennett R.E., Evans H.T., Ellis S.A., Hyman B.T., Bodea L.G., Götz J. PTEN activation contributes to neuronal and synaptic engulfment by microglia in tauopathy. Acta Neuropathol. 2020;140:7–24. doi: 10.1007/s00401-020-02151-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund E.O., Murai K.K., Fredette B., Sekerková G., Marturano B., Weber L., Mugnaini E., Ranscht B. Ataxia and abnormal cerebellar microorganization in mice with ablated contactin gene expression. Neuron. 1999;24:739–750. doi: 10.1016/s0896-6273(00)81126-5. [DOI] [PubMed] [Google Scholar]
- Bhat M.A., Rios J.C., Lu Y., Garcia-Fresco G.P., Ching W., St Martin M., Li J., Einheber S., Chesler M., Rosenbluth J., et al. Axon-glia interactions and the domain organization of myelinated axons requires neurexin IV/Caspr/Paranodin. Neuron. 2001;30:369–383. doi: 10.1016/s0896-6273(01)00294-x. [DOI] [PubMed] [Google Scholar]
- Bouchard J.P., Barbeau A., Bouchard R., Bouchard R.W. Autosomal recessive spastic ataxia of Charlevoix-Saguenay. Can. J. Neurol. Sci. 1978;5:61–69. [PubMed] [Google Scholar]
- Bradshaw T.Y., Romano L.E.L., Duncan E.J., Nethisinghe S., Abeti R., Michael G.J., Giunti P., Vermeer S., Chapple J.P. A reduction in Drp1-mediated fission compromises mitochondrial health in autosomal recessive spastic ataxia of Charlevoix Saguenay. Hum. Mol. Genet. 2016;25:3232–3244. doi: 10.1093/hmg/ddw173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M.C., Perrotta J.A., Turner C.E. Identification of LIM3 as the principal determinant of paxillin focal adhesion localization and characterization of a novel motif on paxillin directing vinculin and focal adhesion kinase binding. J. Cell Biol. 1996;135:1109–1123. doi: 10.1083/jcb.135.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.Y., Balch W.E. The Hsp90 chaperone complex regulates GDI-dependent Rab recycling. Mol. Biol. Cell. 2006;17:3494–3507. doi: 10.1091/mbc.e05-12-1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper M.J., Cox N.J., Zimmerman E.I., Dewar B.J., Duncan J.S., Whittle M.C., Nguyen T.A., Jones L.S., Ghose Roy S., Smalley D.M., et al. Application of multiplexed kinase inhibitor beads to study kinome adaptations in drug-resistant leukemia. PLoS One. 2013;8:e66755. doi: 10.1371/journal.pone.0066755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutadeur S., Benyamine H., Delalonde L., de Oliveira C., Leblond B., Foucourt A., Besson T., Casagrande A.S., Taverne T., Girard A., et al. A novel DYRK1A (dual specificity tyrosine phosphorylation-regulated kinase 1A) inhibitor for the treatment of Alzheimer's disease: effect on Tau and amyloid pathologies in vitro. J. Neurochem. 2015;133:440–451. doi: 10.1111/jnc.13018. [DOI] [PubMed] [Google Scholar]
- Dadon-Nachum M., Melamed E., Offen D. The “dying-back” phenomenon of motor neurons in ALS. J. Mol. Neurosci. 2011;43:470–477. doi: 10.1007/s12031-010-9467-1. [DOI] [PubMed] [Google Scholar]
- Dauer W., Przedborski S. Parkinson's disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Davis T.A., Loos B., Engelbrecht A.M. Corrigendum to AHNAK: the giant jack of all trades. Cell. Signal. 2015;27:187–188. doi: 10.1016/j.cellsig.2014.10.004. [DOI] [PubMed] [Google Scholar]
- De Pascalis C., Etienne-Manneville S. Single and collective cell migration: the mechanics of adhesions. Mol. Biol. Cell. 2017;28:1833–1846. doi: 10.1091/mbc.E17-03-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degorce F., Card A., Soh S., Trinquet E., Knapik G.P., Xie B. HTRF: A technology tailored for drug discovery - a review of theoretical aspects and recent applications. Curr. Chem. Genomics. 2009:22–32. doi: 10.2174/1875397300903010022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekkers M.P.J., Nikoletopoulou V., Barde Y.A. Cell biology in neuroscience: death of developing neurons: new insights and implications for connectivity. J. Cell Biol. 2013;203:385–393. doi: 10.1083/jcb.201306136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo G., Antonsson B., Kassel D., Riederer B.M., Grenningloh G. Phosphorylation regulates the microtubule-destabilizing activity of stathmin and its interaction with tubulin. FEBS Lett. 1997;416:149–152. doi: 10.1016/s0014-5793(97)01188-5. [DOI] [PubMed] [Google Scholar]
- Didonna A., Opal P. The role of neurofilament aggregation in neurodegeneration: lessons from rare inherited neurological disorders. Mol. Neurodegener. 2019;14:19. doi: 10.1186/s13024-019-0318-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit R., Ross J.L., Goldman Y.E., Holzbaur E.L.F. Differential regulation of dynein and kinesin motor proteins by tau. Science. 2008;319:1086–1089. doi: 10.1126/science.1152993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J.M., Lau L.S., Ng Y.W., Lim L., Manser E. Paxillin nuclear-cytoplasmic localization is regulated by phosphorylation of the LD4 motif: evidence that nuclear paxillin promotes cell proliferation. Biochem. J. 2009;418:173–184. doi: 10.1042/BJ20080170. [DOI] [PubMed] [Google Scholar]
- DuBoff B., Götz J., Feany M.B. Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron. 2012;75:618–632. doi: 10.1016/j.neuron.2012.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan E.J., Larivière R., Bradshaw T.Y., Longo F., Sgarioto N., Hayes M.J., Romano L.E.L., Nethisinghe S., Giunti P., Bruntraeger M.B., et al. Altered organisation of the intermediate filament cytoskeleton and relocalisation of proteostasis modulators in cells lacking the ataxia protein sacsin. Hum. Mol. Genet. 2017;26:3130–3143. doi: 10.1093/hmg/ddx197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebneth A., Godemann R., Stamer K., Illenberger S., Trinczek B., Mandelkow E. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer's disease. J. Cell Biol. 1998;143:777–794. doi: 10.1083/jcb.143.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engert J.C., Bérubé P., Mercier J., Doré C., Lepage P., Ge B., Bouchard J.P., Mathieu J., Melançon S.B., Schalling M., et al. ARSACS, a spastic ataxia common in northeastern Quebec, is caused by mutations in a new gene encoding an 11.5-kb ORF. Nat. Genet. 2000;24:120–125. doi: 10.1038/72769. [DOI] [PubMed] [Google Scholar]
- Ezratty E.J., Partridge M.A., Gundersen G.G. Microtubule-induced focal adhesion disassembly is mediated by dynamin and focal adhesion kinase. Nat. Cell Biol. 2005;7:581–590. doi: 10.1038/ncb1262. [DOI] [PubMed] [Google Scholar]
- Feng T., Luan L., Katz I.I., Ullah M., Van Deerlin V.M., Trojanowski J.Q., Lee E.B., Hu F. TMEM106B deficiency impairs cerebellar myelination and synaptic integrity with Purkinje cell loss. Acta Neuropathol. Commun. 2022;10:33. doi: 10.1186/s40478-022-01334-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink J.K. Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol. 2013;126:307–328. doi: 10.1007/s00401-013-1115-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis V., Alshafie W., Kumar R., Girard M., Brais B., McPherson P.S. The ARSACS disease protein sacsin controls lysosomal positioning and reformation by regulating microtubule dynamics. J. Biol. Chem. 2022;298:102320. doi: 10.1016/j.jbc.2022.102320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederick R.L., Shaw J.M. Moving mitochondria: establishing distribution of an essential organelle. Traffic. 2007;8:1668–1675. doi: 10.1111/j.1600-0854.2007.00644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galea G., Simpson J.C. High-content analysis of Rab protein function at the ER-Golgi interface. BioArchitecture. 2015;5:44–53. doi: 10.1080/19490992.2015.1102826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan L., Cookson M.R., Petrucelli L., La Spada A.R. Converging pathways in neurodegeneration, from genetics to mechanisms. Nat. Neurosci. 2018;21:1300–1309. doi: 10.1038/s41593-018-0237-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavriilidis C., Laredj L., Solinhac R., Messaddeq N., Viaud J., Laporte J., Sumara I., Hnia K. The MTM1-UBQLN2-HSP complex mediates degradation of misfolded intermediate filaments in skeletal muscle. Nat. Cell Biol. 2018;20:198–210. doi: 10.1038/s41556-017-0024-9. [DOI] [PubMed] [Google Scholar]
- Gene Ontology Consortium The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res. 2021:D325–D334. doi: 10.1093/nar/gkaa1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genest O., Wickner S., Doyle S.M. Hsp90 and Hsp70 chaperones: collaborators in protein remodeling. J. Biol. Chem. 2019;294:2109–2120. doi: 10.1074/jbc.REV118.002806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentil B.J., Lai G.T., Menade M., Larivière R., Minotti S., Gehring K., Chapple J.P., Brais B., Durham H.D. Sacsin, mutated in the ataxia ARSACS, regulates intermediate filament assembly and dynamics. Faseb. J.: official publication of the Federation of American Societies for Experimental Biology. 2019;33:2982–2994. doi: 10.1096/fj.201801556R. [DOI] [PubMed] [Google Scholar]
- Girard M., Larivière R., Parfitt D.A., Deane E.C., Gaudet R., Nossova N., Blondeau F., Prenosil G., Vermeulen E.G.M., Duchen M.R., et al. Mitochondrial dysfunction and Purkinje cell loss in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) Proc. Natl. Acad. Sci. USA. 2012;109:1661–1666. doi: 10.1073/pnas.1113166109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giustiniani J., Daire V., Cantaloube I., Durand G., Poüs C., Perdiz D., Baillet A. Tubulin acetylation favors Hsp90 recruitment to microtubules and stimulates the signaling function of the Hsp90 clients Akt/PKB and p53. Cell. Signal. 2009;21:529–539. doi: 10.1016/j.cellsig.2008.12.004. [DOI] [PubMed] [Google Scholar]
- Han I., You Y., Kordower J.H., Brady S.T., Morfini G.A. Differential vulnerability of neurons in Huntington's disease: the role of cell type-specific features. J. Neurochem. 2010;113:1073–1091. doi: 10.1111/j.1471-4159.2010.06672.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintz T.G., Eva R., Fawcett J.W. Regional regulation of Purkinje cell dendritic spines by integrins and eph/ephrins. PLoS One. 2016;11:e0158558. doi: 10.1371/journal.pone.0158558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ida M., Suzuki H., Mori N., Taketani S., Araki M. Neuro-p24 plays an essential role in neurite extension: antisense oligonucleotide inhibition of neurite extension in cultured DRG neurons and neuroblastoma cells. Neurosci. Res. 2004;50:199–208. doi: 10.1016/j.neures.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Ikezu S., Ingraham Dixie K.L., Koro L., Watanabe T., Kaibuchi K., Ikezu T. Tau-tubulin kinase 1 and amyloid-beta peptide induce phosphorylation of collapsin response mediator protein-2 and enhance neurite degeneration in Alzheimer disease mouse models. Acta Neuropathol. Commun. 2020;8:12. doi: 10.1186/s40478-020-0890-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M. Historical review of the significance of the cerebellum and the role of Purkinje cells in motor learning. Ann. N. Y. Acad. Sci. 2002;978:273–288. doi: 10.1111/j.1749-6632.2002.tb07574.x. [DOI] [PubMed] [Google Scholar]
- Jacobi A., Tran N.M., Yan W., Benhar I., Tian F., Schaffer R., He Z., Sanes J.R. Overlapping transcriptional programs promote survival and axonal regeneration of injured retinal ganglion cells. Neuron. 2022;110:2625–2645.e7. doi: 10.1016/j.neuron.2022.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Key J., Harter P.N., Sen N.E., Gradhand E., Auburger G., Gispert S. Mid-gestation lethality of atxn2l-ablated mice. Int. J. Mol. Sci. 2020;21:E5124. doi: 10.3390/ijms21145124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kii I., Nishiyama T., Kudo A. Periostin promotes secretion of fibronectin from the endoplasmic reticulum. Biochem. Biophys. Res. Commun. 2016;470:888–893. doi: 10.1016/j.bbrc.2016.01.139. [DOI] [PubMed] [Google Scholar]
- Kilinc D. The emerging role of mechanics in synapse formation and plasticity. Front. Cell. Neurosci. 2018;12:483. doi: 10.3389/fncel.2018.00483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiral F.R., Kohrs F.E., Jin E.J., Hiesinger P.R. Rab GTPases and membrane trafficking in neurodegeneration. Curr. Biol. 2018;28:R471–R486. doi: 10.1016/j.cub.2018.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishi M., Pan Y.A., Crump J.G., Sanes J.R. Mammalian SAD kinases are required for neuronal polarization. Science. 2005;307:929–932. doi: 10.1126/science.1107403. [DOI] [PubMed] [Google Scholar]
- Kiyonaga-Endou K., Oshima M., Sugimoto K., Thomas M., Taketani S., Araki M. Localization of Neurensin1 in cerebellar Purkinje cells of the developing chick and its possible function in dendrite formation. Brain Res. 2016;1635:113–120. doi: 10.1016/j.brainres.2016.01.038. [DOI] [PubMed] [Google Scholar]
- Klionsky, D.J., Abdel-Aziz, A.K., Abdelfatah, S., Abdellatif, M., Abdoli, A., Abel, S., Abeliovich, H., Abildgaard, M.H., Abudu, Y.P., Acevedo-Arozena, A., et al. (2021). Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition1. 17, 1-382. https://10.1080/15548627.2020.1797280. [DOI] [PMC free article] [PubMed]
- Krahn A.I., Wells C., Drewry D.H., Beitel L.K., Durcan T.M., Axtman A.D. Defining the neural kinome: strategies and opportunities for small molecule drug discovery to target neurodegenerative diseases. ACS Chem. Neurosci. 2020;11:1871–1886. doi: 10.1021/acschemneuro.0c00176. [DOI] [PubMed] [Google Scholar]
- Kreis P., Leondaritis G., Lieberam I., Eickholt B.J. Subcellular targeting and dynamic regulation of PTEN: implications for neuronal cells and neurological disorders. Front. Mol. Neurosci. 2014;7:23. doi: 10.3389/fnmol.2014.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo J.C., Han X., Yates J.R., Waterman C.M. Isolation of focal adhesion proteins for biochemical and proteomic analysis. Methods Mol. Biol. 2012:297–323. doi: 10.1007/978-1-61779-166-6_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurolap A., Kreuder F., Gonzaga-Jauregui C., Duvdevani M.P., Harel T., Tammer L., Xin B., Bakhtiari S., Rice J., van Eyk C.L., et al. Bi-allelic variants in neuronal cell adhesion molecule cause a neurodevelopmental disorder characterized by developmental delay, hypotonia, neuropathy/spasticity. Am. J. Hum. Genet. 2022;109:518–532. doi: 10.1016/j.ajhg.2022.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvarnung M., Shahsavani M., Taylan F., Moslem M., Breeuwsma N., Laan L., Schuster J., Jin Z., Nilsson D., Lieden A., et al. Ataxia in patients with Bi-allelic NFASC mutations and absence of full-length NF186. Front. Genet. 2019;10:896. doi: 10.3389/fgene.2019.00896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackie R.E., Maciejewski A., Ostapchenko V.G., Marques-Lopes J., Choy W.Y., Duennwald M.L., Prado V.F., Prado M.A.M. The hsp70/hsp90 chaperone machinery in neurodegenerative diseases. Front. Neurosci. 2017;11:254. doi: 10.3389/fnins.2017.00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larivière R., Gaudet R., Gentil B.J., Girard M., Conte T.C., Minotti S., Leclerc-Desaulniers K., Gehring K., McKinney R.A., Shoubridge E.A., et al. Sacs knockout mice present pathophysiological defects underlying autosomal recessive spastic ataxia of Charlevoix-Saguenay. Hum. Mol. Genet. 2015;24:727–739. doi: 10.1093/hmg/ddu491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larivière R., Sgarioto N., Márquez B.T., Gaudet R., Choquet K., McKinney R.A., Watt A.J., Brais B. Sacs R272C missense homozygous mice develop an ataxia phenotype. Mol. Brain. 2019;12:19. doi: 10.1186/s13041-019-0438-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leube R.E., Moch M., Windoffer R. Intermediate filaments and the regulation of focal adhesion. Curr. Opin. Cell Biol. 2015;32:13–20. doi: 10.1016/j.ceb.2014.09.011. [DOI] [PubMed] [Google Scholar]
- Lopes S., Teplytska L., Vaz-Silva J., Dioli C., Trindade R., Morais M., Webhofer C., Maccarrone G., Almeida O.F.X., Turck C.W., et al. Tau deletion prevents stress-induced dendritic atrophy in prefrontal cortex: role of synaptic mitochondria. Cereb. Cortex. 2017;27:2580–2591. doi: 10.1093/cercor/bhw057. [DOI] [PubMed] [Google Scholar]
- Manczak M., Reddy P.H. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer's disease neurons: implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 2012;21:2538–2547. doi: 10.1093/hmg/dds072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M., Akhmanova A. Coming into focus: mechanisms of microtubule minus-end organization. Trends Cell Biol. 2018;28:574–588. doi: 10.1016/j.tcb.2018.02.011. [DOI] [PubMed] [Google Scholar]
- Martin M.H., Bouchard J.P., Sylvain M., St-Onge O., Truchon S. Autosomal recessive spastic ataxia of Charlevoix-Saguenay: a report of MR imaging in 5 patients. AJNR. Am. J. Neuroradiol. 2007;28:1606–1608. doi: 10.3174/ajnr.A0603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister G.C., Nusinow D.P., Jedrychowski M.P., Wühr M., Huttlin E.L., Erickson B.K., Rad R., Haas W., Gygi S.P. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem. 2014;86:7150–7158. doi: 10.1021/ac502040v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClellan A.J., Xia Y., Deutschbauer A.M., Davis R.W., Gerstein M., Frydman J. Diverse cellular functions of the Hsp90 molecular chaperone uncovered using systems approaches. Cell. 2007;131:121–135. doi: 10.1016/j.cell.2007.07.036. [DOI] [PubMed] [Google Scholar]
- Melkov A., Abdu U. Regulation of long-distance transport of mitochondria along microtubules. Cell. Mol. Life Sci. 2018;75:163–176. doi: 10.1007/s00018-017-2590-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertins P., Tang L.C., Krug K., Clark D.J., Gritsenko M.A., Chen L., Clauser K.R., Clauss T.R., Shah P., Gillette M.A., et al. Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography-mass spectrometry. Nat. Protoc. 2018;13:1632–1661. doi: 10.1038/s41596-018-0006-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metz K.S., Deoudes E.M., Berginski M.E., Jimenez-Ruiz I., Aksoy B.A., Hammerbacher J., Gomez S.M., Phanstiel D.H. Coral: Clear and customizable visualization of human kinome data. Cell Syst. 2018;7:347–350.e1. doi: 10.1016/j.cels.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morani F., Doccini S., Chiorino G., Fattori F., Galatolo D., Sciarrillo E., Gemignani F., Züchner S., Bertini E.S., Santorelli F.M. Functional network profiles in ARSACS disclosed by aptamer-based proteomic technology. Front. Neurol. 2020;11:603774. doi: 10.3389/fneur.2020.603774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Layseca P., Icha J., Hamidi H., Ivaska J. Integrin trafficking in cells and tissues. Nat. Cell Biol. 2019;21:122–132. doi: 10.1038/s41556-018-0223-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G.A., Burns M., Binder L.I., Kanaan N.M., LaPointe N., Bosco D.A., Brown R.H., Jr., Brown H., Tiwari A., Hayward L., et al. Axonal transport defects in neurodegenerative diseases. J. Neurosci. 2009;29:12776–12786. doi: 10.1523/JNEUROSCI.3463-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murase S., Hayashi Y. Integrin alpha1 localization in murine central and peripheral nervous system. J. Comp. Neurol. 1998;395:161–176. doi: 10.1002/(sici)1096-9861(19980601)395:2<161::aid-cne2>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Murtinheira F., Migueis M., Letra-Vilela R., Diallo M., Quezada A., Valente C.A., Oliva A., Rodriguez C., Martin V., Herrera F. Sacsin deletion induces aggregation of glial intermediate filaments. Cells. 2022;11 doi: 10.3390/cells11020299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naguib A., Bencze G., Cho H., Zheng W., Tocilj A., Elkayam E., Faehnle C.R., Jaber N., Pratt C.P., Chen M., et al. PTEN functions by recruitment to cytoplasmic vesicles. Mol. Cell. 2015;58:255–268. doi: 10.1016/j.molcel.2015.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng D.H.J., Humphries J.D., Byron A., Millon-Frémillon A., Humphries M.J. Microtubule-dependent modulation of adhesion complex composition. PLoS One. 2014;9:e115213. doi: 10.1371/journal.pone.0115213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwenhuis B., Haenzi B., Andrews M.R., Verhaagen J., Fawcett J.W. Integrins promote axonal regeneration after injury of the nervous system. Biol. Rev. Camb. Philos. Soc. 2018;93:1339–1362. doi: 10.1111/brv.12398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noordstra I., Akhmanova A. Linking cortical microtubule attachment and exocytosis. F1000Res. 2017;6:469. doi: 10.12688/f1000research.10729.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunomura K., Nagano K., Itagaki C., Taoka M., Okamura N., Yamauchi Y., Sugano S., Takahashi N., Izumi T., Isobe T. Cell surface labeling and mass spectrometry reveal diversity of cell surface markers and signaling molecules expressed in undifferentiated mouse embryonic stem cells. Mol. Cell. Proteomics. 2005;4:1968–1976. doi: 10.1074/mcp.M500216-MCP200. [DOI] [PubMed] [Google Scholar]
- Ochoa D., Jarnuczak A.F., Viéitez C., Gehre M., Soucheray M., Mateus A., Kleefeldt A.A., Hill A., Garcia-Alonso L., Stein F., et al. The functional landscape of the human phosphoproteome. Nat. Biotechnol. 2020;38:365–373. doi: 10.1038/s41587-019-0344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan C., Chun J., Li D., Boese A.C., Li J., Kang J., Umano A., Jiang Y., Song L., Magliocca K.R., et al. Hsp90B enhances MAST1-mediated cisplatin resistance by protecting MAST1 from proteosomal degradation. J. Clin. Invest. 2019;129:4110–4123. doi: 10.1172/JCI125963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parfitt D.A., Michael G.J., Vermeulen E.G.M., Prodromou N.V., Webb T.R., Gallo J.M., Cheetham M.E., Nicoll W.S., Blatch G.L., Chapple J.P. The ataxia protein sacsin is a functional co-chaperone that protects against polyglutamine-expanded ataxin-1. Hum. Mol. Genet. 2009;18:1556–1565. doi: 10.1093/hmg/ddp067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K.K., Liu K., Hu Y., Smith P.D., Wang C., Cai B., Xu B., Connolly L., Kramvis I., Sahin M., He Z. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science. 2008;322:963–966. doi: 10.1126/science.1161566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y.K., Goda Y. Integrins in synapse regulation. Nat. Rev. Neurosci. 2016;17:745–756. doi: 10.1038/nrn.2016.138. [DOI] [PubMed] [Google Scholar]
- Patel V.J., Thalassinos K., Slade S.E., Connolly J.B., Crombie A., Murrell J.C., Scrivens J.H. A comparison of labeling and label-free mass spectrometry-based proteomics approaches. J. Proteome Res. 2009:3752–3759. doi: 10.1021/pr900080y. [DOI] [PubMed] [Google Scholar]
- Pearson B.L., Simon J.M., McCoy E.S., Salazar G., Fragola G., Zylka M.J. Identification of chemicals that mimic transcriptional changes associated with autism, brain aging and neurodegeneration. Nat. Commun. 2016;7:11173. doi: 10.1038/ncomms11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Riverol Y., Bai J., Bandla C., García-Seisdedos D., Hewapathirana S., Kamatchinathan S., Kundu D.J., Prakash A., Frericks-Zipper A., Eisenacher M., et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022;50:D543–D552. doi: 10.1093/nar/gkab1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollerberg G.E., Thelen K., Theiss M.O., Hochlehnert B.C. The role of cell adhesion molecules for navigating axons: density matters. Mech. Dev. 2013;130:359–372. doi: 10.1016/j.mod.2012.11.002. [DOI] [PubMed] [Google Scholar]
- Quintá H.R., Galigniana N.M., Erlejman A.G., Lagadari M., Piwien-Pilipuk G., Galigniana M.D. Management of cytoskeleton architecture by molecular chaperones and immunophilins. Cell. Signal. 2011;23:1907–1920. doi: 10.1016/j.cellsig.2011.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff M.C., Whitmore A.V., Finn J.T. Axonal self-destruction and neurodegeneration. Science. 2002;296:868–871. doi: 10.1126/science.1068613. [DOI] [PubMed] [Google Scholar]
- Reddy P.H. Abnormal tau, mitochondrial dysfunction, impaired axonal transport of mitochondria, and synaptic deprivation in Alzheimer's disease. Brain Res. 2011;1415:136–148. doi: 10.1016/j.brainres.2011.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rico B., Beggs H.E., Schahin-Reed D., Kimes N., Schmidt A., Reichardt L.F. Control of axonal branching and synapse formation by focal adhesion kinase. Nat. Neurosci. 2004;7:1059–1069. doi: 10.1038/nn1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson L.K., Ostergaard H.L. Paxillin associates with the microtubule cytoskeleton and the immunological synapse of CTL through its leucine-aspartic acid domains and contributes to microtubule organizing center reorientation. J. Immunol. 2011;187:5824–5833. doi: 10.4049/jimmunol.1003690. [DOI] [PubMed] [Google Scholar]
- Sakurai T., Lustig M., Babiarz J., Furley A.J., Tait S., Brophy P.J., Brown S.A., Brown L.Y., Mason C.A., Grumet M. Overlapping functions of the cell adhesion molecules Nr-CAM and L1 in cerebellar granule cell development. J. Cell Biol. 2001;154:1259–1273. doi: 10.1083/jcb.200104122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvadores N., Sanhueza M., Manque P., Court F.A. Axonal degeneration during aging and its functional role in neurodegenerative disorders. Front. Neurosci. 2017;11:451. doi: 10.3389/fnins.2017.00451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaržija I., Dekanić A., Humphries J.D., Paradžik M., Stojanović N., Humphries M.J., Ambriović-Ristov A. Integrin crosstalk contributes to the complexity of signalling and unpredictable cancer cell fates. Cancers. 2020;12:E1910. doi: 10.3390/cancers12071910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathyamurthy A., Barik A., Dobrott C.I., Matson K.J.E., Stoica S., Pursley R., Chesler A.T., Levine A.J. Cerebellospinal neurons regulate motor performance and motor learning. Cell Rep. 2020;31:107595. doi: 10.1016/j.celrep.2020.107595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders A., Macosko E.Z., Wysoker A., Goldman M., Krienen F.M., de Rivera H., Bien E., Baum M., Bortolin L., Wang S., et al. Molecular diversity and specializations among the cells of the adult mouse brain. Cell. 2018;174:1015–1030.e16. doi: 10.1016/j.cell.2018.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seetharaman S., Etienne-Manneville S. Microtubules at focal adhesions - a double-edged sword. J. Cell Sci. 2019;132:jcs232843. doi: 10.1242/jcs.232843. [DOI] [PubMed] [Google Scholar]
- Sengupta A., Kabat J., Novak M., Wu Q., Grundke-Iqbal I., Iqbal K. Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch. Biochem. Biophys. 1998;357:299–309. doi: 10.1006/abbi.1998.0813. [DOI] [PubMed] [Google Scholar]
- Shabanzadeh A.P., D'Onofrio P.M., Magharious M., Choi K.A.B., Monnier P.P., Koeberle P.D. Modifying PTEN recruitment promotes neuron survival, regeneration, and functional recovery after CNS injury. Cell Death Dis. 2019;10:567. doi: 10.1038/s41419-019-1802-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinde S.R., Maddika S. PTEN modulates EGFR late endocytic trafficking and degradation by dephosphorylating Rab7. Nat. Commun. 2016;7:10689. doi: 10.1038/ncomms10689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipley M.M., Mangold C.A., Szpara M.L. Differentiation of the SH-SY5Y human neuroblastoma cell line. J. Vis. Exp. 2016;2016:53193. doi: 10.3791/53193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon J.M., Paranjape S.R., Wolter J.M., Salazar G., Zylka M.J. High-throughput screening and classification of chemicals and their effects on neuronal gene expression using RASL-seq. Sci. Rep. 2019;9:4529. doi: 10.1038/s41598-019-39016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sistani L., Rodriguez P.Q., Hultenby K., Uhlen M., Betsholtz C., Jalanko H., Tryggvason K., Wernerson A., Patrakka J. Neuronal proteins are novel components of podocyte major processes and their expression in glomerular crescents supports their role in crescent formation. Kidney Int. 2013;83:63–71. doi: 10.1038/ki.2012.321. [DOI] [PubMed] [Google Scholar]
- Skelton P.D., Stan R.V., Luikart B.W. The role of PTEN in neurodevelopment. Mol. Neuropsychiatry. 2020;5:60–71. doi: 10.1159/000504782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smigiel R., Sherman D.L., Rydzanicz M., Walczak A., Mikolajkow D., Krolak-Olejnik B., Kosinska J., Gasperowicz P., Biernacka A., Stawinski P., et al. Homozygous mutation in the Neurofascin gene affecting the glial isoform of Neurofascin causes severe neurodevelopment disorder with hypotonia, amimia and areflexia. Hum. Mol. Genet. 2018;27:3669–3674. doi: 10.1093/hmg/ddy277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider N.T., Omary M.B. Post-translational modifications of intermediate filament proteins: mechanisms and functions. Nat. Rev. Mol. Cell Biol. 2014;15:163–177. doi: 10.1038/nrm3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanga S., Caretto A., Boido M., Vercelli A. Mitochondrial dysfunctions: a red thread across neurodegenerative diseases. Int. J. Mol. Sci. 2020;21:E3719. doi: 10.3390/ijms21103719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoothoff W.H., Johnson G.V.W. Tau phosphorylation: physiological and pathological consequences. Biochim. Biophys. Acta. 2005;1739:280–297. doi: 10.1016/j.bbadis.2004.06.017. [DOI] [PubMed] [Google Scholar]
- Sulzmaier F.J., Jean C., Schlaepfer D.D. FAK in cancer: mechanistic findings and clinical applications. Nat. Rev. Cancer. 2014;14:598–610. doi: 10.1038/nrc3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suresh Babu S., Wojtowicz A., Freichel M., Birnbaumer L., Hecker M., Cattaruzza M. Mechanism of stretch-induced activation of the mechanotransducer zyxin in vascular cells. Sci. Signal. 2012;5:ra91. doi: 10.1126/scisignal.2003173. [DOI] [PubMed] [Google Scholar]
- Synofzik M., Soehn A.S., Gburek-Augustat J., Schicks J., Karle K.N., Schüle R., Haack T.B., Schöning M., Biskup S., Rudnik-Schöneborn S., et al. Autosomal recessive spastic ataxia of Charlevoix Saguenay (ARSACS): expanding the genetic, clinical and imaging spectrum. Orphanet J. Rare Dis. 2013;8:41. doi: 10.1186/1750-1172-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D., Gable A.L., Lyon D., Junge A., Wyder S., Huerta-Cepas J., Simonovic M., Doncheva N.T., Morris J.H., Bork P., et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–D613. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T., Wallace J.T., Baldwin K.T., Purkey A.M., Uezu A., Courtland J.L., Soderblom E.J., Shimogori T., Maness P.F., Eroglu C., Soderling S.H. Chemico-genetic discovery of astrocytic control of inhibition in vivo. Nature. 2020;588:296–302. doi: 10.1038/s41586-020-2926-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura M., Gu J., Takino T., Yamada K.M. Tumor suppressor PTEN inhibition of cell invasion, migration, and growth: differential involvement of focal adhesion kinase and p130Cas. Cancer Res. 1999;59:442–449. [PubMed] [Google Scholar]
- Tang Y., Tao Y., Wang L., Yang L., Jing Y., Jiang X., Lei L., Yang Z., Wang X., Peng M., et al. NPM1 mutant maintains ULK1 protein stability via TRAF6-dependent ubiquitination to promote autophagic cell survival in leukemia. Faseb. J.: official publication of the Federation of American Societies for Experimental Biology. 2021;35:e21192. doi: 10.1096/fj.201903183RRR. [DOI] [PubMed] [Google Scholar]
- Trogden K.P., Battaglia R.A., Kabiraj P., Madden V.J., Herrmann H., Snider N.T. An image-based small-molecule screen identifies vimentin as a pharmacologically relevant target of simvastatin in cancer cells. Faseb. J.: official publication of the Federation of American Societies for Experimental Biology. 2018;32:2841–2854. doi: 10.1096/fj.201700663R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valiente M., Andrés-Pons A., Gomar B., Torres J., Gil A., Tapparel C., Antonarakis S.E., Pulido R. Binding of PTEN to specific PDZ domains contributes to PTEN protein stability and phosphorylation by microtubule-associated serine/threonine kinases. J. Biol. Chem. 2005;280:28936–28943. doi: 10.1074/jbc.M504761200. [DOI] [PubMed] [Google Scholar]
- Vermeer S., van de Warrenburg B.P., Kamsteeg E.J., Brais B., Synofzik M. In: GeneReviews. R, Adam M.P., Ardinger H.H., Pagon R.A., Wallace S.E., Bean L.J.H., Mirzaa G., Amemiya A., editors. 1993. Arsacs. [Google Scholar]
- Walter J.T., Alviña K., Womack M.D., Chevez C., Khodakhah K. Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat. Neurosci. 2006;9:389–397. doi: 10.1038/nn1648. [DOI] [PubMed] [Google Scholar]
- Wu Y., Ding Y., Zheng X., Liao K. The molecular chaperone Hsp90 maintains Golgi organization and vesicular trafficking by regulating microtubule stability. J. Mol. Cell Biol. 2020;12:448–461. doi: 10.1093/jmcb/mjz093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xicoy H., Wieringa B., Martens G.J.M. The SH-SY5Y cell line in Parkinson's disease research: a systematic review. Mol. Neurodegener. 2017;12:10. doi: 10.1186/s13024-017-0149-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X., Wang Y., Liu C., Lu Q., Liu T., Chen G., Rao H., Luo S. Heat shock protein 90beta stabilizes focal adhesion kinase and enhances cell migration and invasion in breast cancer cells. Exp. Cell Res. 2014;326:78–89. doi: 10.1016/j.yexcr.2014.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X., Guan J.L. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv. Drug Deliv. Rev. 2011;63:610–615. doi: 10.1016/j.addr.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C., Cunningham L., Marcus A.I., Li Y., Kahn R.A. Arl2 and Arl3 regulate different microtubule-dependent processes. Mol. Biol. Cell. 2006;17:2476–2487. doi: 10.1091/mbc.e05-10-0929. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
A list of all cell lines used in each figure are provided in Table S1. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2022) partner repository with the dataset identifier PXD036906. The RNA-seq dataset generated during the current study is available in the Gene Expression Omnibus (GEO) repository under project identifier GSE214213.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.