

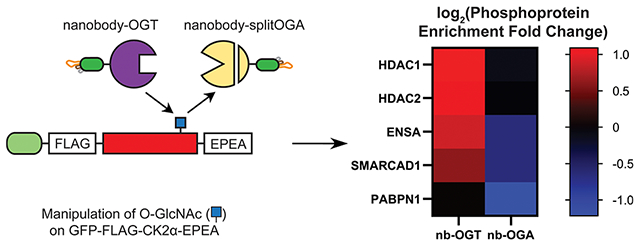
Abstract
O-GlcNAc is an essential carbohydrate modification that intersects with phosphorylation signaling pathways via crosstalk on protein substrates or by direct modification of the kinases that write the phosphate modification. Casein kinase 2 alpha (CK2α), the catalytic subunit of the ubiquitously expressed and constitutively active kinase CK2, is modified by O-GlcNAc, but the effect of this modification on the phosphoproteome in cells is unknown. Here, we apply complementary targeted O-GlcNAc editors, nanobody-OGT and -splitOGA, to selectively write and erase O-GlcNAc from a tagged CK2α to measure the effects on the phosphoproteome in cells. These tools effectively and selectively edit the Ser347 glycosite on CK2α. Using quantitative phosphoproteomics, we report 51 phosphoproteins whose enrichment changes as a function of editing O-GlcNAc on CK2α, including HDAC1, HDAC2, ENSA, SMARCAD1, and PABPN1. Specific phosphosites on HDAC1 Ser393 and HDAC2 Ser394, both reported CK2 substrates, are significantly enhanced by O-GlcNAcylation of CK2α. These data will propel future studies on the crosstalk between O-GlcNAc and phosphorylation.
Graphical Abstract
INTRODUCTION
O-linked N-acetyl glucosamine (O-GlcNAc) is a monomeric glycan modification found at Ser or Thr residues of thousands of nuclear, cytosolic, and mitochondrial proteins within the cell. O-GlcNAcylation of these many substrates is governed by one enzyme pair. The enzyme O-GlcNAc transferase (OGT) installs O-GlcNAc onto proteins, while O-GlcNAcase (OGA) removes it. Both enzymes are essential in development—deletion of the gene encoding OGT in mice leads to embryonic lethality,1 while deletion of the OGA gene results in perinatal death.2 The O-GlcNAc modification often intersects with other post-translational modifications (PTMs) like phosphorylation to tune signaling outcomes.3,4 Crosstalk between O-GlcNAc and phosphorylation has attracted considerable interest as both modifications may occur on individual proteins, where proximal glycosites and phosphosites can regulate each other in a reciprocal manner.5 Alternatively, O-GlcNAc may regulate phosphorylation pathways via direct modification of the kinase, which alters downstream substrate selection and phosphorylation.
To date, O-GlcNAc has been reported on over 100 kinases in the human, mouse, and rat kinomes, spanning all of the kinase families.6 This diverse repertoire indicates the potential for O-GlcNAc to tune kinase activity across signaling pathways to the nutrient availability of the cell through one promiscuous enzyme pair (OGT and OGA) and the glucose-derived cosubstrate UDP-GlcNAc. For example, the calcium/calmodulin-dependent kinase CaMKIV is glycosylated at multiple sites.7 Mutating the Ser189 glycosite to Ala increases CaMKIV kinase activity and Thr200 phosphorylation, indicative of a reciprocal relationship between O-GlcNAc and phosphorylation at these sites. Separately, Akt1 possesses dual glycosites at Thr308 and Ser473 that, when modified by O-GlcNAc, reduce kinase activity and lead to apoptosis in ischemic brain tissue.8 In addition, protein kinase CK2 is a dual-specific kinase consisting of two α or β’ catalytic subunits and two noncatalytic β subunits that is constitutively active and ubiquitously expressed in human cells.9 CK2 mainly targets Ser/Thr residues proximal to negatively charged residues,10 though tyrosine kinase activity has also been reported.11,12 CK2α is O-GlcNAcylated at a conserved Ser347 near the C-terminus.13 This glycosite is proximal to a reported phosphosite at Thr344, which is necessary for stabilizing a protein–protein interaction (PPI) between CK2α and peptidyl–prolyl isomerase Pin1. In 2012, Cole and co-workers discovered that glycosylation of CK2α at Ser347 inhibits phosphorylation at Thr344 and induces a net destabilization of CK2α by downregulating interactions with Pin1.13 They also observed a small subset of phosphoproteins on a microarray whose phosphorylation by CK2 was dependent on the presence or absence of synthetically installed S-GlcNAc on CK2α. Whether addition or removal of O-GlcNAc on CK2α influences substrate selection and the phosphoproteome more broadly in cells has yet to be explored. As CK2 plays a central role in cell cycle progression14 and cell survival15 and regulates differentiation during development16 on hundreds of substrates,17 the effects on the phosphoproteome by O-GlcNAcylation of CK2α may be broadly impactful.
Here, we selectively edit O-GlcNAc on a tagged CK2α in cells with a targeted writer and eraser of O-GlcNAc and measure the resulting changes to the phosphoproteome (Figure 1A). Recently, we reported a method to write and erase O-GlcNAc from a desired target protein in cells using an engineered nanobody-OGT or -splitOGA system.18,19 The nanobody is a small, single-domain protein binder that recruits the fused O-GlcNAc editor to a target protein for selective modulation of O-GlcNAc in a cellular environment, without significantly affecting other glycoproteins. These targeted methods complement methods for global perturbation of O-GlcNAc (e.g., chemical inhibitors of OGT or OGA or genetic knockout)20–22 and the study of specific glycosites by site-directed mutagenesis.23 Mutation of a given glycosite to Ala removes its ability to be modified by OGT, while mutation to Cys may be modified by OGT to produce a stable S-GlcNAc modification that cannot be hydrolyzed by OGA.24 However, point mutagenesis presents challenges to studies of O-GlcNAc and phosphorylation crosstalk, and whether OGT will broadly accept Cys residues at a diversity of Ser/Thr glycosites is unknown. We show that tuning O-GlcNAc on a tagged CK2α is readily promoted by the nanobody-OGT and -splitOGA system, which yields changes to the global phosphoproteome, including direct and indirect substrates of CK2, and demonstrates the cross regulation of phosphorylation by O-GlcNAc.
Figure 1.
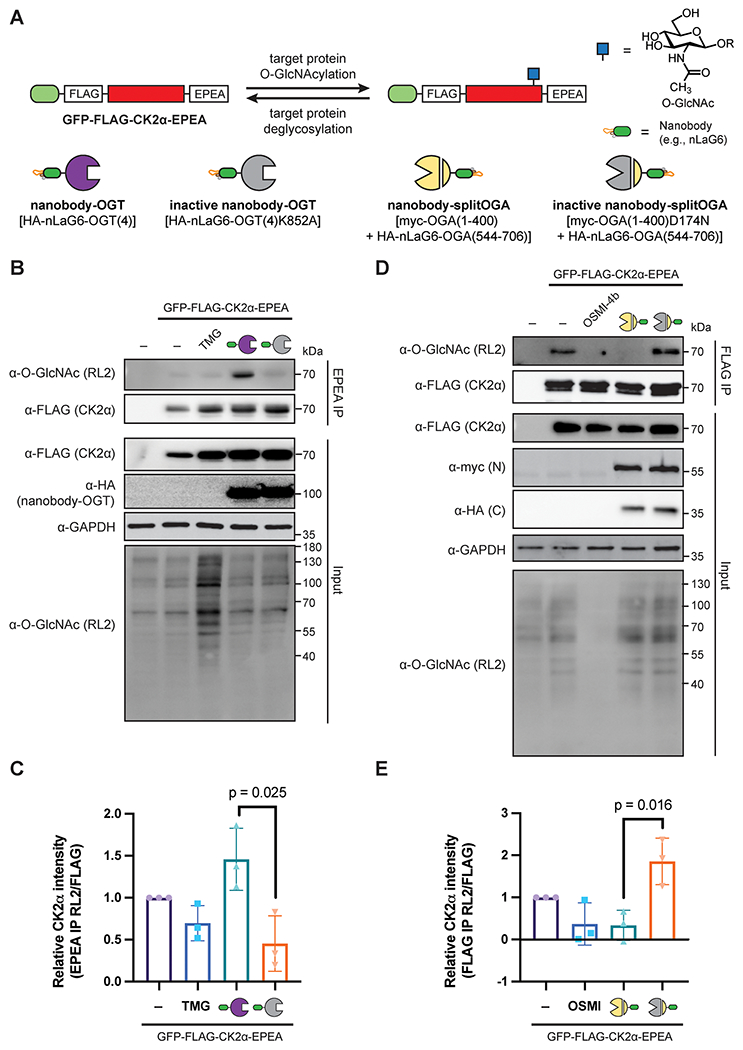
O-GlcNAc levels on the target protein GFP-FLAG-CK2α-EPEA can be manipulated by nanobody-OGT and nanobody-splitOGA. (A) Schematic of the selective writing or erasing of O-GlcNAc from GFP-FLAG-CK2α-EPEA by nanobody-OGT or -splitOGA. (B) Nanobody-OGT selectively increases O-GlcNAc on GFP-FLAG-CK2α-EPEA. HEK293T cells were transfected with GFP-FLAG-CK2α-EPEA with active or inactive nanobody-OGT or treated with Thiamet G (TMG; 100 μM), prior to immunoprecipitation with C-tag resin. (C) Relative O-GlcNAc levels on GFP-FLAG-CK2α-EPEA from part B. (D) Nanobody-splitOGA selectively decreases O-GlcNAc on GFP-FLAG-CK2α-EPEA. HEK293T cells were transfected with GFP-FLAG-CK2α-EPEA with active or inactive nanobody-splitOGA or treated with OSMI-4b (25 μM), prior to immunoprecipitation with anti-FLAG resin. (E) Relative O-GlcNAc levels on GFP-FLAG-CK2α-EPEA from part D. In C and E, error bars represent ± one standard deviation from three independent replicates, and a two-tailed Student’s t-test was used for statistical analysis.
RESULTS AND DISCUSSION
Writing and Erasing O-GlcNAc from CK2α.
We first evaluated whether O-GlcNAc alters CK2α stability in HEK293T cells using a cycloheximide chase experiment and found a minor destabilization of CK2α on Thiamet G treatment, but not OSMI-4b treatment, in line with prior work in HeLa cells (Supplementary Figure 1).13 We next evaluated whether the nanobody-OGT and -splitOGA systems were readily amenable to writing and erasing O-GlcNAc from GFP-FLAG-CK2α-EPEA (Figure 1A). To enhance selectivity for the target protein through the nanobody, we used an engineered OGT(4) bearing a truncated tetratricopeptide repeat (TPR) domain for introduction of O-GlcNAc18 and an engineered splitOGA system for removal of O-GlcNAc.19 We additionally used the GFP-binding nanobody nLaG6 for recruitment of the engineered OGT and splitOGA to the target protein through GFP due to its superior efficiency in writing and erasing O-GlcNAc from a desired target protein.18,25
To evaluate the selective introduction of O-GlcNAc to CK2α, GFP-FLAG-CK2α-EPEA was coexpressed with nanobody-OGT or a catalytically inactive nanobody-OGT [nLaG6-OGT(4)K852A]. We additionally compared the target protein O-GlcNAcylation to global OGA inhibition by transfection of GFP-FLAG-CK2α-EPEA to cells with or without Thiamet G treatment. After 24 h, O-GlcNAc levels of immunoprecipitated CK2α and the broader proteome were examined by Western blot with the α-O-GlcNAc antibody RL2 (Figure 1B). Interestingly, GFP-FLAG-CK2α-EPEA was moderately O-GlcNAcylated, and these levels were not perturbed by Thiamet G treatment, although global elevation of O-GlcNAc on the broader proteome was observed. By contrast, coexpression of nanobody-OGT, but not the inactive nanobody-OGT, selectively and significantly increased glycosylation of GFP-FLAG-CK2α-EPEA (Figure 1C).
We next evaluated if the proximity-directed nanobody-splitOGA system19 would effectively deglycosylate CK2α in a similar manner (Figure 1D,E). The nanobody-splitOGA was expressed as a myc-tagged N-terminal fragment containing residues 1–400 with nLaG6-fused to a HA-tagged C-terminal fragment containing residues 544–706. The catalytically inactive version of splitOGA contains a D174N mutation in the N-terminal fragment [myc-OGA(1–400)D174N]. The targeted nanobody-splitOGA system was additionally compared to global inhibition of OGT by OSMI-4b treatment. To enhance detection of the basal levels of O-GlcNAc on GFP-FLAG-CK2α-EPEA, we increased the cellular input for immunopre-cipitation. We observed a decrease in glycosylation of GFP-FLAG-CK2α-EPEA cotransfected with the nanobody-splitOGA comparable to OSMI-4b treatment that occurred selectively on the target protein without affecting global O-GlcNAc levels (Figure 1D). These observations were reproducible and statistically significant across replicates (Figure 1E). Taken together, the nanobody-OGT and nanobody-splitOGA can write and erase O-GlcNAc from GFP-FLAG-CK2α-EPEA, without broad perturbations to the global glycoproteome.
CK2α is O-GlcNAcylated at Ser347 in Human Cells.
After confirming the function of the nanobody-OGT and -splitOGA system on CK2α, we sought to confirm that the O-GlcNAc modification on GFP-FLAG-CK2α-EPEA aligned with the previously reported Ser347 glycosite from the bovine brain13 and that this glycosite was installed by nanobody-OGT. Using site-directed mutagenesis, we examined Ser347 and the neighboring Ser348 as potential glycosites by mutating one or both to alanine and blotting the immunoprecipitated CK2α for O-GlcNAc (Figure 2A). Nanobody-OGT specifically installed O-GlcNAc to Ser347, and O-GlcNAc levels were readily reduced in mutants carrying S347A (Figure 2B). These data show that Ser347 is the location of the glycosite on CK2α, and it is installed by both endogenous OGT and nanobody-OGT [HA-nLaG6-OGT(4)].
Figure 2.
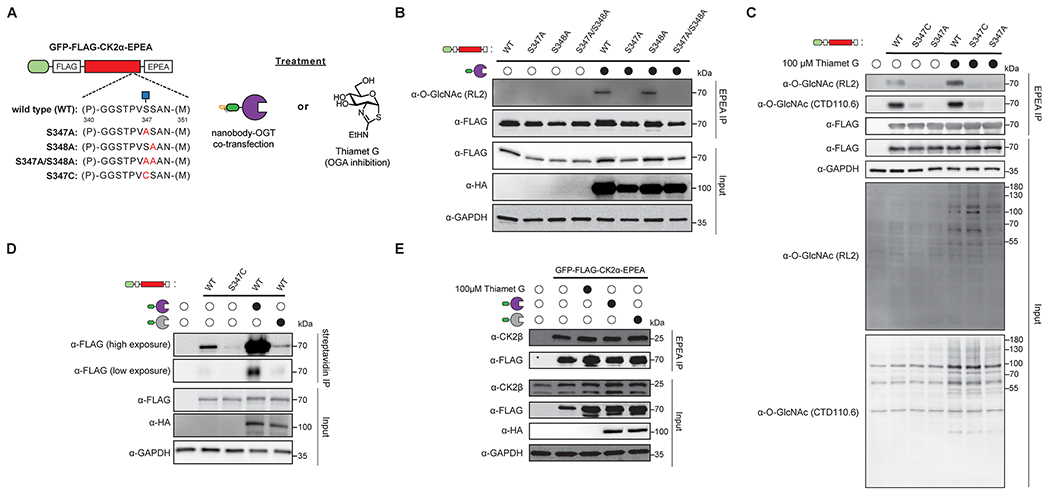
GFP-FLAG-CK2α-EPEA is O-GlcNAcylated at Ser347 and binds to endogenous CK2β. (A) Experimental design to map the glycosite(s) of GFP-FLAG-CK2α-EPEA. GFP-FLAG-CK2α-EPEA was point mutated at positions Ser347 and Ser348 to Ala or Cys and evaluated in in the presence or absence of nanobody-OGT or Thiamet G treatment. (B) GFP-FLAG-CK2α-EPEA is glycosylated at Ser347 by nanobody-OGT. HEK293T cells were transfected with the indicated CK2α construct with or without nanobody-OGT, prior to immunoprecipitation with C-tag resin. (C) Endogenous OGT glycosylates GFP-FLAG-CK2α-EPEA at Ser347. HEK293T cells were transfected with the indicated CK2α construct with or without Thiamet G (100 μM) treatment, prior to immunoprecipitation with C-tag resin. (D) Visualization of the GFP-FLAG-CK2α-EPEA glycosite Ser347 by chemoenzymatic labeling. HEK293T cells were transfected with the indicated CK2α construct with or without nanobody-OGT, prior to chemoenzymatic labeling of O-GlcNAc with GalNAz by GalT1(Y289L). Labeled glycosites were tagged with biotin-PEG(4)-alkyne prior to immunoprecipitation with streptavidin resin. (E) GFP-FLAG-CK2α-EPEA binds to endogenous CK2β. HEK293T cells were transfected with the indicated CK2α construct with or without Thiamet G (100 μM) treatment, prior to nondenaturing lysis, and coimmunoprecipitation with C-tag resin. All Western blot data are representative of three independent replicates.
As Ser347 is the primary glycosite on the tagged CK2α construct, we investigated whether stable introduction of O-GlcNAc to the target protein could also be achieved by point mutagenesis, where mutation to Cys would potentially generate an S-GlcNAc site that is not hydrolyzed by OGA.24 We evaluated these point mutants for responsiveness to Thiamet G treatment with two O-GlcNAc antibodies, RL2 and CTD110.6 (Figure 2C). Curiously, the RL2 antibody appeared more sensitive to O-GlcNAc levels on CK2α in response to Thiamet G treatment than the CTD110.6 antibody. However, both the RL2 and CTD110.6 antibodies showed poor glycosylation of the CK2α S347C mutant.
We sought to confirm the responsiveness of the O-GlcNAc site on CK2α to nanobody-OGT using chemoenzymatic labeling as an orthogonal O-GlcNAc detection method. In brief, HEK293T cell lysates after transfection were chemo-enzymatically labeled with GalNAz using GalT1 (Y289L).26 The azide on GalNAz is subsequently tagged to a biotin-PEG(4)-alkyne by copper-catalyzed azide–alkyne cycloaddition (CuAAC) to enable enrichment of O-GlcNAcylated proteins with streptavidin resin. Signal detected by α-FLAG should be proportional to the amount of glycosylated GFP-FLAG-CK2α-EPEA. Using this chemoenzymatic enrichment method, significantly more CK2α was pulled down when GFP-FLAG-CK2α-EPEA was cotransfected with active nanobody-OGT as compared to the inactive nanobody-OGT (Figure 2D). In addition, enrichment of the S347C mutant was poor compared to the wild-type CK2α, despite equivalent expression of both proteins. Therefore, the S347C mutant is not readily glycosylated by endogenous OGT, and the modification does not build up as a result.
While CK2α alone is catalytically active, many of the protein targets of the CK2 complex are dependent on association of CK2α with CK2β.27,28 We therefore verified that GFP-FLAG-CK2α-EPEA forms a complex with this important component of the CK2 holoenzyme (Figure 2E). We observed that endogenous CK2β is coimmunoprecipitated when GFP-FLAG-CK2α-EPEA is overexpressed, and the amount of coimmunoprecipitated CK2β is not affected by Thiamet G treatment or nanobody-OGT cotransfection. This implies that O-GlcNAcylation of the C-terminal domain of CK2α does not affect CK2β binding, which aligns with prior reports as the CK2α residues involved in CK2β binding are between residues 36 and 108,29 whereas the Ser347 glycosite is distal to these residues.
O-GlcNAc on CK2α Influences the Phosphoproteome.
As CK2 is a horizontal regulator of multiple cellular processes and has hundreds of reported substrates,13,17,30,31 if O-GlcNAc on CK2α modulates the phosphoproteome, we would expect to observe changes to phosphosites that are directly and indirectly regulated by CK2 within central pathways, such as cell cycle progression, chromatin structure, and apoptosis. To investigate changes to the phosphoproteome, HEK293T cells were cotransfected with GFP-FLAG-CK2α-EPEA and catalytically active or inactive forms of nanobody-OGT or nanobody-splitOGA for 24 h in triplicate (Supplementary Figure 2). The cells were lysed, the lysates were trypsinized, and phosphopeptides were enriched by TiO2 affinity chromatography. The enriched phosphopeptides were reacted with TMT-10plex tags for quantitative analysis by mass spectrometry using Higher-energy C-trap Dissociation (HCD) fragmentation. Phosphopeptides were assigned by database searching using Sequest HT in Proteome Discoverer software.
The phosphoproteomics data were initially analyzed at the protein level (Figure 3A,B, Supplementary Table 1). High confidence [1% False Discovery Rate (FDR)] phosphoproteins were evaluated based on their fold change (FC) in enrichment between the active and inactive nanobody-OGT and -splitOGA conditions, and their associated p value. Specifically, a |log2(FC)| of at least 0.5, a p value less than or equal to 0.05, and at least two peptide-spectrum matches (PSMs) were required for inclusion at the protein level. We considered a |log2(FC)| between 0.5 and 1 a moderate enrichment and |log2(FC)| ≥ 1 a major enrichment. Principal component analyses and normalization box plots for each level of analysis are presented in Supplementary Figures 3–10. This statistical analysis indicates stronger independent clustering between the control and treatment samples in the nanobody-OGT condition compared to the nanobody-splitOGA condition.
Figure 3.
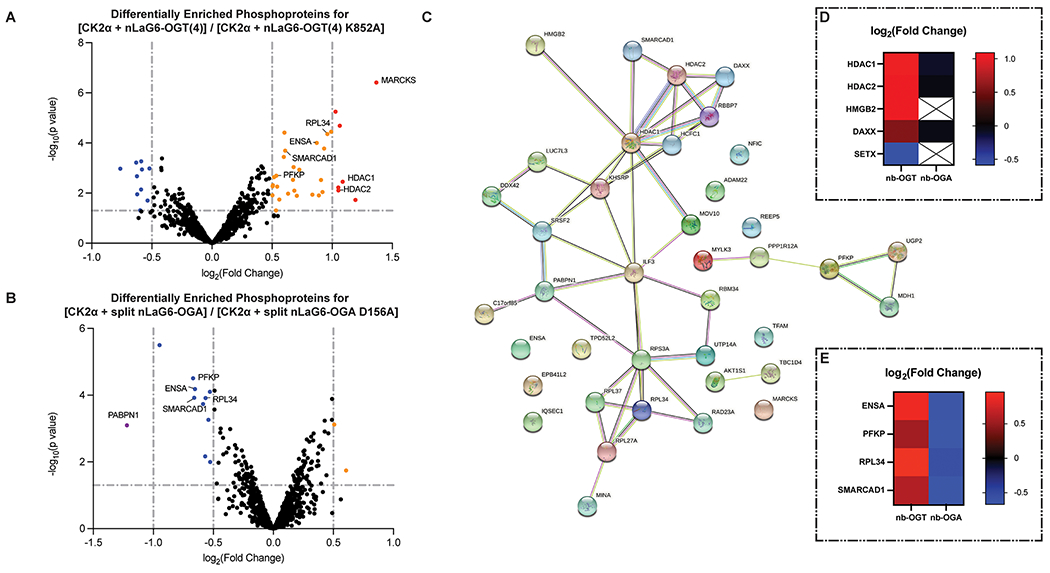
O-GlcNAc on GFP-FLAG-CK2α-EPEA affects the phosphoproteome. (A) Volcano plot of the fold change in phosphoproteins enriched from cell lysates transfected with GFP-FLAG-CK2α-EPEA and the active or inactive nanobody-OGT. (B) Volcano plot of the fold change in phosphoproteins enriched from cell lysates transfected with GFP-FLAG-CK2α-EPEA and the active or inactive nanobody-splitOGA. In A and B, data points were colored purple, blue, orange, or red corresponding to their log2(FC) (≤−1, −1 to −0.5, 0.5 to 1, and ≥1, respectively) if they met the p-value threshold (p ≤ 0.05). (C) STRING network of phosphoproteins with a moderate/major increase in the nanobody-OGT condition or moderate/major decrease in the nanobody-splitOGA condition. (D) Heatmap demonstrating the log2(FC) of reported CK2 substrates HDAC1, HDAC2, HMGB2, DAXX, and SETX. (E) Heatmap demonstrating the similar trend in log2(FC) of ENSA, SMARCAD1, PFKP, and RPL34 in both phosphoproteomics data sets.
In the nanobody-OGT condition, increased O-GlcNAc on CK2α may promote phosphorylation of some substrates while reducing phosphorylation of others, such as substrates that are dependent on interaction with unmodified Ser347 or phosphorylated Thr344 on the C-terminus of CK2α. Seven phosphoproteins were heavily enriched in a nanobody-OGT activity-dependent manner (|log2(FC)| ≥ 1, p value ≤0.05, Figure 3A). Of these phosphoproteins, HDAC1 and HDAC2 are previously reported substrates of CK2.32 An additional 25 phosphoproteins show a moderate increase in enrichment, and 10 phosphoproteins show a moderate decrease in enrichment (0.5 ≤ |log2(FC)| < 1, p value ≤0.05). The majority of other phosphoproteins detected do not change phosphorylation significantly, suggesting that they are unaffected by increased O-GlcNAc on CK2α.
Conversely, phosphoproteins that are dependent on O-GlcNAc on CK2α should decrease in enrichment after treatment with nanobody-splitOGA, while those that are inhibited by O-GlcNAc on CK2α should increase. Only one protein, PABPN1, was extremely down-regulated based on nanobody-splitOGA activity (|log2(FC)| ≥ 1, Figure 3B). PABPN1 is a phosphorylated RNA-binding protein that is a substrate of ATM1, a known CK2 substrate.33 PABPN1 also appears in the nanobody-OGT data set, but its FC is insignificant based on p value. In the moderate FC threshold (0.5 ≤ |log2(FC)| < 1, p value ≤ 0.05), two proteins increase, and 10 proteins decrease in enrichment. The more limited changes in the phosphoproteome due to nanobody-splitOGA treatment may be a result of the low level of basal O-GlcNAc on CK2α observed by Western blot (Figure 1D). Reducing O-GlcNAc on CK2α with nanobody-splitOGA even further may not produce as large an effect on the phosphoproteome as we observed with increasing O-GlcNAc with nanobody-OGT.
On the basis of the protein-level analysis, we obtained a list of 51 statistically significant phosphoproteins that change by the activity of nanobody-OGT or nanobody-splitOGA [|log2(FC)| ≥ 0.5, p value ≤0.05]. Of these 51 proteins, 39 increased in enrichment in the nanobody-OGT condition and/or decreased in enrichment in the nanobody-splitOGA condition. This list of phosphoproteins whose regulation correlated with O-GlcNAc on CK2α was imported into STRING, an online database of protein functional interactions (Figure 3C).34 Interactome analysis of these 39 phosphoproteins reveals a nexus of proteins involved in chromatin regulation, including histone deacetylases HDAC1 and HDAC2 and the heavily O-GlcNAcylated cell cycle regulator HCFC1. HDAC1, HDAC2, HMGB2, and DAXX are all reported CK2 substrates (Figure 3D), which may provide the point of entry for CK2 to affect phosphorylation of other connected proteins in a glycosylation-dependent manner. We also observe clusters of phosphoproteins involved in metabolism (PFKP, MDH1, UGP2) and the ribosome (RPS3A, RPL27A, RPL34, RPL36). The STRING network of phosphoproteins that increased under the nanobody-splitOGA condition or decreased under the nanobody-OGT condition contained the remaining 12 proteins, and only two of them were functionally associated (Supplementary Figure 11).
A subset of phosphoproteins (ENSA/ARPP-19, SMARCAD1, PFKP, and RPL34) met the moderate threshold for enrichment under both conditions. Enrichment of phosphopeptides from these proteins increased with active nanobody-OGT cotransfection and decreased with active nanobody-splitOGA cotransfection (Figure 3E). Although these are not reported CK2 substrates, and none of the identified phosphosites possess the consensus sequence typically recognized by CK2,10 these phosphoproteins appear to be dependent on O-GlcNAc on CK2α.
Integration of these indirect phosphorylation changes with direct CK2 substrates points to possible connections for how cellular pathways may be regulated by O-GlcNAc on CK2α (Figure 3C). ENSA/ARPP-19 is a 19 kDa regulator of PP2A. The Ser67 phosphosite detected in our data is installed by the kinase Gwl.35 Phosphorylation of ENSA at Ser67 results in inhibition of PP2A and an increase in Cdk1/Cyclin B activity, a kinase complex known to modify CK2α near its glycosite. This interaction network represents a possible mechanism for glycosylated CK2α to upregulate Cdk1/Cyclin B activity and regulate M phase progression.
Once in the M phase, the cell simultaneously replicates the genome and maintains the epigenetic markers associated with each gene. SMARCAD1 plays a central role in this process by directing deacetylation of H3 and H4, promoting H3 Lys9 methylation, and establishing heterochromatin during the cell cycle.36 SMARCAD1 also interacts with HDAC1 and HDAC2, two CK2 target proteins that similarly deacetylate histones and have phosphosites whose occupancy increases with CK2α glycosylation. Together, these results indicate that these chromatin regulating complexes may be regulated by O-GlcNAc on CK2α.
Phosphorylation of PFKP and RPL34 was also moderately altered in a manner dependent on CK2α glycosylation. PFKP is one of three phosphofructokinase isoforms, all of which catalyze the first step of glycolysis. While PFKP is not directly regulated by CK2, the observed PFKP phosphosite at Ser386 is installed by Akt1, a CK2 target protein.37 This phosphorylation event stabilizes PFKP by blocking its interaction with an E3 ubiquitin ligase.37 It is also upregulated under amino acid deprivation and leads to ATG4B phosphorylation at Ser34 and initiation of autophagic processes in HEK293T cells.38 RPL34 is a protein component of the 60S ribosome. The observed phosphosite at Ser12 has been assigned in other phosphoproteomics studies,39,40 but the specific function of phosphorylation at Ser12 is not known. If the nearby Thr15 on RPL34 is phosphorylated by another kinase, this negative charge three residues downstream of Ser12 creates a favorable environment for CK2 to phosphorylate Ser12; however, no phosphorylated Ser12/Thr15 peptides from RPL34 were assigned in our analysis.
O-GlcNAc on CK2α Enhances Phosphorylation on Direct and Indirect Substrates.
To gain higher resolution to the changes in phosphorylation at specific sites, the phosphoproteomics data were also analyzed at the phosphopeptide level, where the fold change of individual phosphopeptides was examined (Figure 4, Supplementary Table 2). We identified one CK2α phosphosite at S294 in the nanobody-splitOGA condition, although the site occupancy did not change significantly with nanobody-splitOGA activity. To allow for analysis of only the strongest shifts, only the major threshold for phosphopeptide enrichment (|log2(FC)| ≥ 1, p value ≤0.05) was analyzed. Twelve distinct phosphosites from 13 phosphopeptides met these criteria. Four of the 12 phosphosites, HDAC1 Ser393, HDAC2 Ser394, and MDC1 Ser329 and Thr404, are reported sites installed by CK2.32 A peptide derived from HMGB2, another CK2 substrate, also appears in this data set, but the Thr179 phosphosite identified is unlikely to be installed by CK2 based on the consensus sequence.
Figure 4.
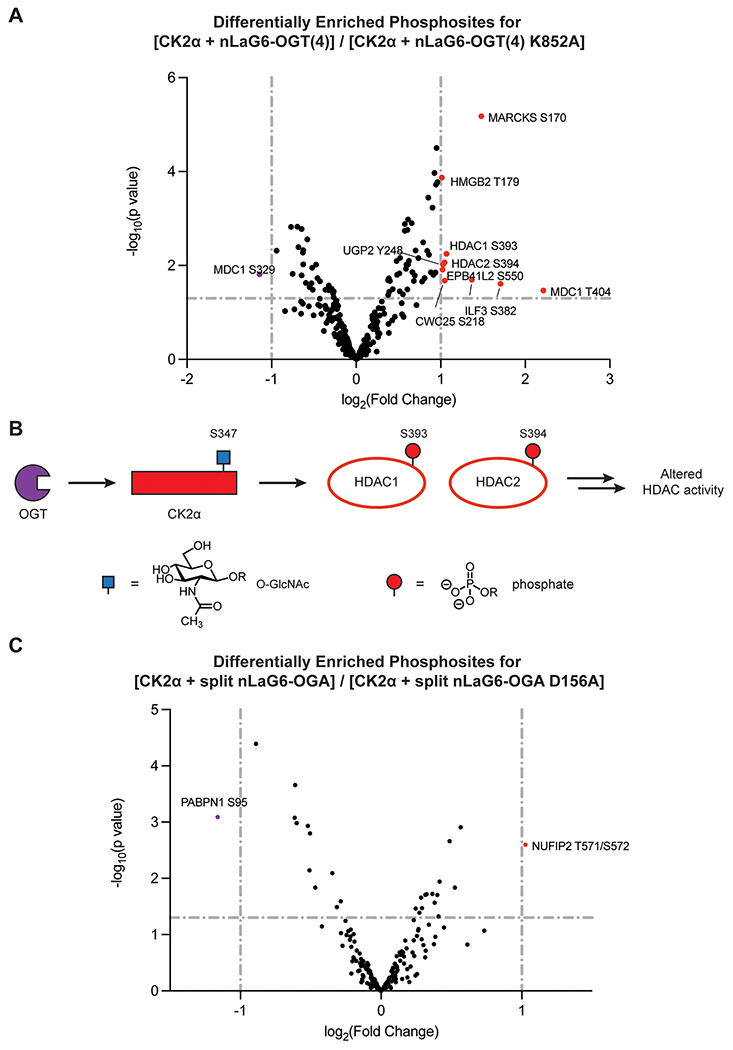
O-GlcNAc on GFP-FLAG-CK2α-EPEA affects specific phosphosites. (A) Volcano plot of the fold change in phosphopeptides enriched from cell lysates transfected with GFP-FLAG-CK2α-EPEA and the active or inactive nanobody-OGT. (B) Proposed pathway representation of the connection between O-GlcNAcylation of CK2α and phosphorylation of HDAC1 at Ser393 and HDAC2 at Ser394. (C) Volcano plot of the fold change in phosphopeptides enriched from cell lysates transfected with GFP-FLAG-CK2α-EPEA and the active or inactive nanobody-splitOGA. Each annotated data point represents a specific phosphopeptide that is increased or decreased in a major way (|log FC| ≥ 1, p value ≤0.5) after enrichment.
Ten of the 12 significantly changing phosphosites were derived from the nanobody-OGT condition (Figure 4A). The highest log2(FC) hit, MARCKS, is a substrate of PKC and plays a role in cross-linking of filamentous actin.41 Inactive MARCKS is normally sequestered to the cytoplasmic side of the plasma membrane. Phosphorylation at multiple sites by PKC, including Ser170, induces its release into the cytoplasm. In mouse melanoma cells, increased phosphorylated MARCKS correlates with increased cell motility,42 which may correspond to increased metastatic potential in cancer. PKC and CK2 function in an antagonistic manner on p53,43 and multiple examples of PKC regulating CK2 have been reported.44,45 Currently, no evidence of direct PKC regulation by CK2 exists.
Enrichment of HDAC1 Ser393 and HDAC2 Ser394 were both increased under the nanobody-OGT condition. HDAC1 and HDAC2 are each catalytic components of protein complexes involved in modulating gene expression and cell cycle progression through chromatin regulation. The role of phosphorylation at these two CK2-regulated sites is to promote dissociation of HDAC1 and HDAC2 heterodimers, leading to formation of other regulatory complexes, including homodimers, during mitosis.32 This likely promotes differential substrate targeting, as HDAC1 and HDAC2 possess both distinct and overlapping substrates (Figure 4B).46
HMGB2 is also a reported CK2 substrate. CK2 is known to phosphorylate HMGB2 at the C-terminus, but the specific location of this phosphosite is not known.47 While Thr179 does not resemble a canonical CK2 phosphosite, especially compared to nearby site Ser168, this finding still warrants further investigation.
MDC1 possesses differentially regulated phosphosites. Two phosphosites on this protein show major changes, but in opposite directions—Thr404 increases, while Ser329 decreases. MDC1 is phosphorylated by CK2 at multiple sites, including these two,48 all of which facilitate its interaction with NBS1. This is a necessary step in the DNA damage response, and these two MDC1 phosphosites serve redundant functions. Further investigation will reveal why MDC1 Thr404 was increased and Ser329 was decreased compared to other CK2 phosphorylation sites on MDC1.
Two of the 12 significantly differentiated phosphosites, NUFIP2 Thr571/Ser572 and PABPN1 Ser95, arose from the nanobody-splitOGA condition (Figure 4C). Both proteins are relatively understudied. NUFIP2 is a 76 kDa RNA-binding protein with important roles in post-transcriptional regulation in nerve and immune cells.49,50 PABPN1 is a heavily phosphorylated RNA-binding protein that facilitates polyA tail elongation.33 Neither are reported CK2 substrates, but PABPN1 Ser95 may be installed by CK2, as Glu98 can facilitate CK2 association.
DISCUSSION
Here, we report the first use of nanobody-OGT and -splitOGA to edit O-GlcNAcylation of a kinase, CK2α, and resultant effects on the phosphoproteome in HEK293T cells. After demonstrating the selective regulation of O-GlcNAc on GFP-FLAG-CK2α-EPEA in cells, we confirmed the glycosite at Ser347 using point mutagenesis. Although point mutagenesis showed that S347A was an effective method to remove O-GlcNAc from CK2α, mutagenesis to S347C as an approach to stably introduce S-GlcNAc24,51 revealed limited introduction of S-GlcNAc to the construct by RL2, CTD110.6, or chemoenzymatic labeling methods for detecting O-GlcNAc. These data imply that while O-GlcNAc may be readily reduced with point mutagenesis to remove the Ser/Thr site, not all O-GlcNAc sites can be turned into S-GlcNAc sites. Additionally, we observed a significant increase in O-GlcNAc on CK2α that was above levels observed by inhibition of OGA, indicating that a trigger to stimulate glycosylation of CK2α may exist. Overall, these data demonstrate that methods for regulating O-GlcNAc at the global, protein, and site levels are useful and needed.
By modulating O-GlcNAc with nanobody-OGT and -splitOGA on a tagged CK2α, we observed a small number of phosphosites over hundreds of PSMs whose enrichment and detection changed in a CK2α glycosylation-dependent manner. By contrast, large changes to the phosphoproteome are observed from global inhibitors of OGT or OGA.52–55 We observed that basal O-GlcNAcylation of overexpressed GFP-FLAG-CK2α-EPEA is relatively low, which can be increased by nanobody-OGT or decreased by nanobody-splitOGA. As the qualitative change of increasing O-GlcNAc on CK2α appears to result in a greater differential than reducing O-GlcNAc on CK2α, this explains the greater differences in the phosphoproteome when modulating O-GlcNAc on CK2α with nanobody-OGT as compared to nanobody-splitOGA. CK2 is different from other kinases in that it is ubiquitously expressed and constitutively active. Because the activity of CK2α is not regulated by a PTM-mediated on or off switch, other mechanisms must lead to differential substrate and phosphosite targeting, including changes in subcellular localization, association with protein complexes, and rate of CK2 holoenzyme assembly.56 We establish here that CK2α glycosylation did not affect the amount of coimmunoprecipitated CK2β, but does alter the phosphoproteome, presumably through altered selectivity or another aforementioned mechanism.
At the protein and phosphosite level, we observed connections between CK2α and three significantly changing phosphoproteins involved in histone deacetylation—SMARCAD1, HDAC1, and HDAC2. With the histone deacetylases, we found that HDAC1 and −2 may relate to the metabolic state of the cell through tuning their association in a CK2α O-GlcNAc-dependent manner, since they are reported CK2 substrates. We found two phosphosites, HDAC1 Ser393 and HDAC2 Ser394, which increased significantly in the nanobody-OGT condition and decreased insignificantly in the nanobody-splitOGA condition. Phosphorylation at these sites is responsible for initiating the dissociation of HDAC1 and HDAC2 from one another, promoting their association with other protein complexes, and differential protein targeting.32 Khan and colleagues also report that HDAC2 is more readily phosphorylated by CK2 than HDAC1. Our data suggest that elevated O-GlcNAc on CK2α may facilitate cell cycle progression by promoting HDAC1/2 dissociation (Figure 4B), where effects on histone acetylation, cell cycle progression, and other processes may be evaluated in the future.
In addition, these data point to new opportunities in the study of the interplay of O-GlcNAc and phosphorylation on the C-terminal tail of CK2α. The main regulator of phosphosites on CK2α proximal to the Ser347 glycosite on its C-terminal tail is Cdk1/Cyclin B.57 Cdk1 is a well-studied mitotic regulator, and prior work established that Thr344 phosphorylation influences CK2α substrate specificity in vitro.13 Therefore, this implies that active Cdk1 redirects CK2 during initiation of mitosis to phosphorylate key substrates. To ascertain what these substrates are and their dependence on CK2α glycosylation, a similar phosphoproteomics experiment with synchronized cells may provide more sensitive quantification of phosphosite differences. In addition, the GFP tag on the CK2α used in this study would facilitate examination of its subcellular localization by microscopy. This would provide insight into how glycosylation and the nanobody system affect CK2α localization.
Although the approach adopted here enabled characterization of the phosphoproteome as a function of O-GlcNAc on CK2α, there are several limitations. Direct measurement of the C-terminal tail of CK2α is challenging with the proteomics methods used here, as this region of CK2α is generally intractable to trypsin or chymotrypsin digestion. Continued expression of endogenous CK2α may reduce the signal-to-noise measured from glycosylation-dependent phosphosites. Because basal protein levels were not quantified, differences in enriched phosphopeptides among samples could arise from changes in phosphosite occupancy or protein expression. The nanobody-OGT and -splitOGA system is also dependent on the fidelity of nLaG6, which may produce off-target effects through the association to tagged CK2α or modification of off-target substrates, which could be better understood through glycoproteomic analysis. Last, our phosphoproteomics data set was obtained after TiO2 enrichment, which could be further expanded using other phosphopeptide enrichment methods, such as Fe-NTA enrichment or Ca2+ precipitation.
The O-GlcNAc modification is associated with nutrient sensing and signaling.58 If the glycosite on CK2α is tuned to the nutrient availability of the cell, then this study implies that pathways like chromatin regulation and cell cycle progression can be tuned by the metabolic state of the cell with OGT as the reporter and CK2α as the messenger. Use of the targeted writer and eraser for O-GlcNAc on overexpressed CK2α enabled this work and will enable the study of additional glycoproteins in the future.
METHODS
Cloning.
The vectors encoding GFP-FLAG-CK2α-EPEA, HA-nLaG6-OGT(4), myc-OGA(1–400), and HA-nLaG6-OGA(544–706) were all generated in a pcDNA3.1 backbone by co-workers in the Woo lab from previously published studies.18,19,59 Subcloning of GFP-FLAG-CK2α-EPEA to generate S347A, S348A, S347A/S348A, and S347C point mutants was achieved using whole-plasmid PCR. Subcloning results were validated via Sanger sequencing.
Cell Culture and Transfection.
HEK293T cells (ATCC) were cultured in Dulbecco’s Modified Eagle’s Medium with 4.5 g L−1 of glucose and l-glutamine (VWR 95042–498) supplemented with penicillin–streptomycin (VWR 12001–692, 50 μg mL−1 each) and fetal bovine serum (Peak Serum PS-FB2, 10% vol/vol). Cultures were maintained at 37 °C in a humidified incubator with 5% CO2 at a passage number no higher than 30.
Cells grown for transfection were seeded in six-well plates (VWR 10062–892, 5 × 105 cells/well), a 10 cm dish (VWR 25382-166, 3.5 × 106 cells/well), or a 15 cm dish (VWR 25383-103, 9 × 106 cells/well) 24 h before transfection. All transfections were performed using TransIT-PRO (Mirus Bio MIR-5740) according to the manufacturer’s instructions for 24 h.
Cells were collected in cold PBS and detached using a cell scraper. Detached cells were moved to microcentrifuge tubes and centrifuged 500g for 3 min. For experiments that required chemoenzymatic labeling of O-GlcNAc with modified GalT1, cells were lysed in PBS + 1% SDS via sonication. For all other experiments, cells were lysed on ice in Cell Signaling Technology lysis buffer (CST #9803) supplemented with a protease/phosphatase inhibitor cocktail (CST #5872) and 100 μM Thiamet G (Selleck Chem S7213).
EPEA/FLAG Immunoprecipitation (CST Lysis Buffer).
For immunoprecipitation of mutant CK2αs by their C-terminal EPEA tag, cell lysates with equal amounts of protein were diluted 5-fold with PBS and incubated with a C-tag affinity matrix (Thermo Scientific, 191307005) overnight at 4 °C with rotation. After washing the resin thrice with excess PBS, the resin was resuspended in 1 × SDS sample buffer and incubated at 95 °C for 5 min. The supernatant was subjected to SDS–PAGE.
For immunoprecipitation of mutant CK2α by the FLAG tag, cell lysates with equal amounts of protein were diluted 5-fold with TBS and incubated with anti-FLAG M2 magnetic beads (Sigma-Aldrich, M8823) overnight at 4 °C with rotation. Beads were washed with excess TBS buffer (50 mM Tris-HCl pH 7.4 and 150 mM NaCl) three times. Enriched proteins were eluted by incubation with 100 ng μL−1 of FLAG peptide in 100 μL of TBS for 1 h at room temperature (RT) with rotation. Eluates were combined with 5 × SDS sample buffer (1 × final concentration) and subjected to SDS–PAGE.
GalT Labeling (1% SDS Lysis Buffer).
Following lysis, protein concentrations were determined by BCA assay. Cell lysates were reduced with 25 mM DTT (Thermo Scientific, 20290) at 95 °C for 5 min and alkylated with 50 mM iodoacetamide (Sigma-Aldrich, I1149) at RT for 1 h. Lysates were precipitated by the addition of excess methanol and were resuspended in 20 mM HEPES at pH 7.9 + 1% SDS at a protein concentration of 5 mg mL−1. Purification of the GalT1 (Y289L) enzyme and labeling of O-GlcNAcylated proteins with GalNAz were performed according to the procedure of Hsieh-Wilson and co-workers.60 For 150 μg of protein, the following components were added to cell lysates in order: water (49 μL), 2.5× GalT labeling buffer (80 μL; final concentrations, 50 mM NaCl, 20 mM HEPES, 2% NP-40, pH 7.9), 100 mM MnCl2 (11 μL), 500 μM UDP-GalNAz (10 μL), and 2 mg mL−1 GalT1 (Y289L; 10 μL). The reaction was gently rotated at 4 °C for at least 24 h, and proteins were precipitated as described above.
Click Chemistry.
As an alternative method for detection of O-GlcNAcylated proteins, copper-catalyzed azide–alkyne cycloaddition (CuAAC) was performed based on the procedure of Woo and co-workers61 following GalT labeling. Lysates were resolubilized in PBS + 1% SDS and were incubated at RT for 1 h with 100 μM THPTA (Sigma-Aldrich, 762342), 0.5 mM CuSO4, 2.5 mM fresh sodium ascorbate, and 100 μM of biotin–PEG4–alkyne (Click Chemistry Tools, TA105) for immunoblotting/enrichment. Reactions were quenched by the addition of excess methanol, and protein pellets were resolubilized in PBS + 1% SDS for downstream applications.
Western Blotting.
SDS–PAGE was performed using 6–12% Tris-Glycine gels in a Mini-PROTEAN BioRad gel system. Following PAGE at 200 V for 40–45 min, proteins were transferred to a nitrocellulose membrane using an iBlot2 system (Thermo Scientific). Membranes were blocked with TBS containing 0.1% Tween-20 (TBST) plus 5% BSA (Sigma-Aldrich, A9647) for 90 min at RT, then incubated with the indicated primary antibodies in TBST + 1% BSA at 4 °C overnight. Primary antibody staining solution was removed, and blots were washed once with TBST for 5 min at RT before the addition of secondary antibodies (diluted 1:10,000) in TBST + 1% BSA and incubation at RT for 1 h. Blots were washed thrice for 5 min in TBST before imaging. Immunoblot images were acquired using an Azure Imager C600 (Azure Biosystems) and analyzed with Fiji ImageJ. All infrared fluorescence Western blot images were converted to grayscale in Fiji ImageJ. Unsaturated exposure images were used for quantification, with the appropriate loading controls used as standards.
Primary antibodies used: α-FLAG (CST #14793), α-GAPDH (CST #97166), α-myc (CST #2276), α-HA (CST #3724), α-CK2α (CST #2656), α-CK2β (abcam ab76025), RL2 (abcam ab2739), and CTD110.6 (CST #9875). All primary antibodies except α-GAPDH were used at a 1:1000 dilution. α-GAPDH was used at a 1:3000 dilution.
Horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Rockland Immunochemicals. IRDye secondary antibodies were purchased from LI-COR Biosciences.
Phosphoproteomics and MS Method.
Cells were lysed by adding 1 mL of lysis buffer (20 mM HEPES pH 7.9, 1% SDS, 1 × protease inhibitors). Protein concentrations were determined by BCA assay. Reduction and alkylation were performed as previously described.62 S-trap digestion was done according to the manufacturer’s instructions resulting in 0.8 mg of tryptic peptides. Samples were desalted on C18 spin columns and evaporated to near dryness in a vacuum concentrator. The enrichment of phosphopeptides was performed using a TiO2 Phosphopeptide Enrichment Kit (cat. no. A32993, ThermoFisher Scientific). Briefly, approximately 0.8 mg of tryptic peptides was resuspended in 150 μL of binding/equilibration buffer. The suspended peptide sample was added to a pre-equilibrated TiO2 spin tip and centrifuged at 1000g for 5 min. The spin tip was then washed with 20 μL of binding/equilibration/wash buffer three times and eluted by 50 μL of elution buffer two times. The eluates were evaporated to near dryness and subjected to the TMT labeling.
For each sample, 10 μL of the corresponding amine-based TMT 10-plex reagents (10 μg μL−1) was added and reacted for 1 h at RT. The reactions were quenched with 2 μL of 5% hydroxylamine solution and combined. The combined mixture was concentrated to dryness. High-pH fractionation (ThermoFisher Scientific) was done according to the manufacturer’s instructions resulting in six fractions.
A Thermo Scientific EASY-nLC 1000 system was coupled to a Thermo Scientific Orbitrap Fusion Tribrid with a nanoelectrospray ion source. Mobile phases A and B were water with 0.1% formic acid (v/v) and acetonitrile with 0.1% formic acid (v/v), respectively. For each fraction, peptides were separated with a linear gradient from 4 to 32% B within 45 min, followed by an increase to 50% B within 10 min and further to 98% B within 10 min and re-equilibration. Peptides were separated using a linear gradient from 4% to 32% B within 50 min, followed by an increase to 50% B within 10 min and further to 98% B within 10 min and re-equilibration. The following instrument parameters were used as previously described.19
Phosphoproteomics Data Analysis.
The raw data were processed using Proteome Discoverer 2.4 (Thermo Fisher Scientific). Data were searched against the UniProt/SwissProt human (Homo sapiens) protein database (19 August 2016; 20 156 total entries) and contaminant proteins using the Sequest HT algorithm. The database was adjusted by adding the sequences of GFP-FLAG-CK2α-EPEA and either HA-nLaG6-OGT(4) or myc-OGA(1–400) + Ha-nLaG6-OGA(544–706). Searches were performed with the following guidelines: spectra with a signal-to-noise ratio greater than 1.5; trypsin as an enzyme; two missed cleavages; variable oxidation on methionine residues (15.995 Da); deamidation on asparagine and glutamine (0.984 Da); phosphorylation on serine, threonine, and tyrosine (79.966 Da); static carbamidomethylation of cysteine residues (57.021 Da); static TMT labeling (229.163 Da) at lysine residues and peptide N-termini; total variable modification max to 3 per peptide; 10 ppm mass error tolerance on precursor ions; and 0.02 Da mass error on fragment ions. Data were filtered with a PSM of 1% FDR using Percolator. The TMT reporter ions were quantified using the Reporter Ions Quantifier with total peptide normalization. For the obtained PSMs, the data were further filtered with the following guidelines: confidence is high; PSM ambiguity is unambiguous; modifications contain phosphorylation; exclude all contaminant proteins. Data were processed using in-house script. After methods are applied for calculating and adjusting missing data in TMT Proteomics data, the file is further filtered with protein FDR confidence is high, unique peptides greater than 2, master proteins only, and no contaminants. Some of the graphs and tables produced include PCA plots, Volcano plots, and tables including all of the statistics presented in the graphs. Applied here is a VSN normalization computed on the imputed matrix using a robust variant of the maximum-likelihood estimator for an additive-multiplicative error model and affine calibration. The model incorporates dependence of the variance on the mean intensity and a variance stabilizing data transformation. A linear model is fitted to the expression data for control and treatment, then t-statistics are computed by empirical Bayes moderation of standard errors toward a common value.
Supplementary Material
ACKNOWLEDGMENTS
We thank D. Ramirez, D. Miyamoto, and B. Budnik for helpful discussions. Mass spectrometry data were collected at the Harvard University Proteomics Facility. Support from the National Science Foundation, CAREER Award (C.M.W.), the Alfred P. Sloan Foundation (C.M.W.), the Camille-Dreyfus Teacher Scholar Award (C.M.W.), and Harvard University is gratefully acknowledged. The MS data were deposited to the ProteomeXchange Consortium via the PRIDE partner repository and are accessible with the identifier PXD030466.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.1c00987.
Materials and methods, endogenous CK2α levels with global OGT/OGA inhibition (Figure S1), phosphoprotoemics workflow (Figure S2), nanobody-OGT and -splitOGA phosphoprotein level normalization and principal component analysis (Figures S3–S6), nanobody-OGT and -splitOGA phosphosite level normalization and principal component analysis (Figures S7–S10), STRING network analysis (Figure S11); supplementary tables of phosphoprotein and phosphosite identifications (Tables S1, S2) (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acschembio.1c00987
The authors declare no competing financial interest.
Contributor Information
Paul A. Schwein, Department of Chemistry and Chemical Biology, Harvard University Cambridge, Massachusetts 02138, United States.
Yun Ge, Department of Chemistry and Chemical Biology, Harvard University, Cambridge, Massachusetts 02138, United States.
Bo Yang, Department of Chemistry and Chemical Biology, Harvard University, Cambridge, Massachusetts 02138, United States.
Alexandria D’Souza, Department of Chemistry and Chemical Biology, Harvard University, Cambridge, Massachusetts 02138, United States.
Alison Mody, Department of Chemistry and Chemical Biology, Harvard University, Cambridge, Massachusetts 02138, United States.
Dacheng Shen, Department of Chemistry and Chemical Biology, Harvard University, Cambridge, Massachusetts 02138, United States.
Christina M. Woo, Department of Chemistry and Chemical Biology, Harvard University, Cambridge, Massachusetts 02138, United States.
REFERENCES
- (1).Shafi R; Iyer SP; Ellies LG; O’Donnell N; Marek KW; Chui D; Hart GW; Marth JD The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proc. Natl. Acad. Sci. 2000, 97 (11), 5735–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Yang YR; Song M; Lee H; Jeon Y; Choi EJ; Jang HJ; Moon HY; Byun HY; Kim EK; Kim DH; et al. O-GlcNAcase is essential for embryonic development and maintenance of genomic stability. Aging cell 2012, 11 (3), 439–48. [DOI] [PubMed] [Google Scholar]
- (3).Wang Z; Udeshi ND; Slawson C; Compton PD; Sakabe K; Cheung WD; Shabanowitz J; Hunt DF; Hart GW Extensive crosstalk between O-GlcNAcylation and phosphorylation regulates cytokinesis. Sci. Signal 2010,3 (104), DOI: 10.1126/scisignal.2000526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Woo CM; Lund PJ; Huang AC; Davis MM; Bertozzi CR; Pitteri SJ Mapping and Quantification of Over 2000 O-linked Glycopeptides in Activated Human T Cells with Isotope-Targeted Glycoproteomics (Isotag). Mol. Cell Proteomics 2018, 17 (4), 764–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Leney AC; El Atmioui D; Wu W; Ovaa H; Heck AJR Elucidating crosstalk mechanisms between phosphorylation and O-GlcNAcylation. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (35), E7255–E7261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Schwein PA; Woo CM The O-GlcNAc Modification on Kinases. ACS Chem. Biol. 2020, 15 (3), 602–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Dias WB; Cheung WD; Wang Z; Hart GW Regulation of calcium/calmodulin-dependent kinase IV by O-GlcNAc modification. J. Biol. Chem. 2009, 284 (32), 21327–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Shi J; Gu JH; Dai CL; Gu J; Jin X; Sun J; Iqbal K; Liu F; Gong CX O-GlcNAcylation regulates ischemia-induced neuronal apoptosis through AKT signaling. Sci. Rep 2015, 5, 14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Borgo C; Ruzzene M Role of protein kinase CK2 in antitumor drug resistance. J. Exp Clin Cancer Res. 2019, 38 (1), 287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Sarno S; Vaglio P; Marin O; Issinger OG; Ruffato K; Pinna LA Mutational analysis of residues implicated in the interaction between protein kinase CK2 and peptide substrates. Biochemistry 1997, 36 (39), 11717–24. [DOI] [PubMed] [Google Scholar]
- (11).Vilk G; Weber JE; Turowec JP; Duncan JS; Wu C; Derksen DR; Zien P; Sarno S; Donella-Deana A; Lajoie G; et al. Protein kinase CK2 catalyzes tyrosine phosphorylation in mammalian cells. Cell Signal 2008, 20 (11), 1942–51. [DOI] [PubMed] [Google Scholar]
- (12).Basnet H; Su XB; Tan Y; Meisenhelder J; Merkurjev D; Ohgi KA; Hunter T; Pillus L; Rosenfeld MG Tyrosine phosphorylation of histone H2A by CK2 regulates transcriptional elongation. Nature 2014, 516 (7530), 267–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Tarrant MK; Rho HS; Xie Z; Jiang YL; Gross C; Culhane JC; Yan G; Qian J; Ichikawa Y; Matsuoka T; et al. Regulation of CK2 by phosphorylation and O-GlcNAcylation revealed by semisynthesis. Nat. Chem. Biol. 2012, 8 (3), 262–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Feng H; Lu J; Song X; Thongkum A; Zhang F; Lou L; Reizes O; Almasan A; Gong Z CK2 kinase-mediated PHF8 phosphorylation controls TopBP1 stability to regulate DNA replication. Nucleic Acids Res. 2021, 49 (4), 2400–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Yamane K; Kinsella TJ CK2 inhibits apoptosis and changes its cellular localization following ionizing radiation. Cancer Res. 2005, 65 (10), 4362–7. [DOI] [PubMed] [Google Scholar]
- (16).Gotz C; Montenarh M Protein kinase CK2 in development and differentiation. Biomed Rep 2017, 6 (2), 127–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Meggio F; Pinna LA One-thousand-and-one substrates of protein kinase CK2? FASEB J. 2003, 17 (3), 349–68. [DOI] [PubMed] [Google Scholar]
- (18).Ramirez DH; Aonbangkhen C; Wu HY; Naftaly JA; Tang S; O’Meara TR; Woo CM Engineering a Proximity-Directed O-GlcNAc Transferase for Selective Protein O-GlcNAcylation in Cells. ACS Chem. Biol. 2020, 15 (4), 1059–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ge Y; Ramirez DH; Yang B; D’Souza AK; Aonbangkhen C; Wong S; Woo CM Target protein deglycosylation in living cells bya nanobody-fused split O-GlcNAcase. Nat. Chem. Biol. 2021, 17 (5), 593–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Martin SES; Tan ZW; Itkonen HM; Duveau DY; Paulo JA; Janetzko J; Boutz PL; Tork L; Moss FA; Thomas CJ; et al. Structure-Based Evolution of Low Nanomolar O-GlcNAc Transferase Inhibitors. J. Am. Chem. Soc. 2018, 140 (42), 13542–13545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Yuzwa SA; Macauley MS; Heinonen JE; Shan X; Dennis RJ; He Y; Whitworth GE; Stubbs KA; McEachern EJ; Davies GJ; et al. A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo. Nat. Chem. Biol. 2008, 4 (8), 483–90. [DOI] [PubMed] [Google Scholar]
- (22).Shen H; Zhao X; Chen J; Qu W; Huang X; Wang M; Shao Z; Shu Q; Li X O-GlcNAc transferase Ogt regulates embryonic neuronal development through modulating Wnt/beta-catenin signaling. Hum. Mol. Genet. 2021, 31, 57. [DOI] [PubMed] [Google Scholar]
- (23).Ge Y; Woo CM Writing and erasing O-GlcNAc from target proteins in cells. Biochem. Soc. Trans. 2021, 49, 2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Gorelik A; Bartual SG; Borodkin VS; Varghese J; Ferenbach AT; van Aalten DMF Genetic recoding to dissect the roles of site-specific protein O-GlcNAcylation. Nat. Struct Mol. Biol. 2019, 26 (11), 1071–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Fridy PC; Li Y; Keegan S; Thompson MK; Nudelman I; Scheid JF; Oeffinger M; Nussenzweig MC; Fenyo D; Chait BT; et al. A robust pipeline for rapid production of versatile nanobody repertoires. Nat. Methods 2014, 11 (12), 1253–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Khidekel N; Arndt S; Lamarre-Vincent N; Lippert A; Poulin-Kerstien KG; Ramakrishnan B; Qasba PK; Hsieh-Wilson LC A chemoenzymatic approach toward the rapid and sensitive detection of O-GlcNAc posttranslational modifications. J. Am. Chem. Soc. 2003,125 (52), 16162–3. [DOI] [PubMed] [Google Scholar]
- (27).Meggio F; Marin O; Boschetti M; Sarno S; Pinna LA HIV-1 Rev transactivator: a beta-subunit directed substrate and effector of protein kinase CK2. Mol. Cell. Biochem. 2001, 227 (1–2), 145–51. [PubMed] [Google Scholar]
- (28).Poletto G; Vilardell J; Marin O; Pagano MA; Cozza G; Sarno S; Falques A; Itarte E; Pinna LA; Meggio F The regulatory beta subunit of protein kinase CK2 contributes to the recognition of the substrate consensus sequence. A study with an eIF2 beta-derived peptide. Biochemistry 2008, 47 (32), 8317–25. [DOI] [PubMed] [Google Scholar]
- (29).Niefind K; Guerra B; Ermakowa I; Issinger OG Crystal structure of human protein kinase CK2: insights into basic properties of the CK2 holoenzyme. EMBO J. 2001, 20 (19), 5320–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Bian Y; Ye M; Wang C; Cheng K; Song C; Dong M; Pan Y; Qin H; Zou H Global screening of CK2 kinase substrates by an integrated phosphoproteomics workflow. Sci. Rep 2013, 3, 3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Rusin SF; Adamo ME; Kettenbach AN Identification of Candidate Casein Kinase 2 Substrates in Mitosis by Quantitative Phosphoproteomics. Front Cell Dev Biol. 2017, 5, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Khan DH; He S; Yu J; Winter S; Cao W; Seiser C; Davie JR Protein kinase CK2 regulates the dimerization of histone deacetylase 1 (HDAC1) and HDAC2 during mitosis. J. Biol. Chem. 2013, 288 (23), 16518–16528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Gavish-Izakson M; Velpula BB; Elkon R; Prados-Carvajal R; Barnabas GD; Ugalde AP; Agami R; Geiger T; Huertas P; Ziv Y; et al. Nuclear poly(A)-binding protein 1 is an ATM target and essential for DNA double-strand break repair. Nucleic Acids Res. 2018, 46 (2), 730–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Szklarczyk D; Gable AL; Lyon D; Junge A; Wyder S; Huerta-Cepas J; Simonovic M; Doncheva NT; Morris JH; Bork P; et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47 (D1), D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Mochida S Regulation of alpha-endosulfine, an inhibitor of protein phosphatase 2A, by multisite phosphorylation. FEBS J. 2014, 281 (4), 1159–69. [DOI] [PubMed] [Google Scholar]
- (36).Rowbotham SP; Barki L; Neves-Costa A; Santos F; Dean W; Hawkes N; Choudhary P; Will WR; Webster J; Oxley D; et al. Maintenance of silent chromatin through replication requires SWI/SNF-like chromatin remodeler SMARCAD1. Mol. Cell 2011, 42 (3), 285–96. [DOI] [PubMed] [Google Scholar]
- (37).Lee J-H; Liu R; Li J; Zhang C; Wang Y; Cai Q; Qian X; Xia Y; Zheng Y; Piao Y; Chen Q; de Groot JF; Jiang T; Lu Z Stabilization of phosphofructokinase 1 platelet isoform by AKT promotes tumorigenesis. Nat. Commun. 2017, 8 (1), 949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Li X; Sun L; Yan G; Yan X PFKP facilitates ATG4B phosphorylation during amino acid deprivation-induced autophagy. Cell Signal 2021, 82, 109956. [DOI] [PubMed] [Google Scholar]
- (39).Mertins P; Qiao JW; Patel J; Udeshi ND; Clauser KR; Mani DR; Burgess MW; Gillette MA; Jaffe JD; Carr SA Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat. Methods 2013, 10 (7), 634–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Dulla K; Daub H; Hornberger R; Nigg EA; Korner R Quantitative site-specific phosphorylation dynamics of human protein kinases during mitotic progression. Mol. Cell Proteomics 2010, 9 (6), 1167–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Uberall F; Giselbrecht S; Hellbert K; Fresser F; Bauer B; Gschwendt M; Grunicke HH; Baier G Conventional PKC-alpha, novel PKC-epsilon and PKC-theta, but not atypical PKC-lambda are MARCKS kinases in intact NIH 3T3 fibroblasts. J. Biol. Chem. 1997, 272 (7), 4072–8. [DOI] [PubMed] [Google Scholar]
- (42).Chen X; Rotenberg SA PhosphoMARCKS drives motility of mouse melanoma cells. Cell Signal 2010, 22 (7), 1097–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Pospisilova S; Brazda V; Kucharikova K; Luciani MG; Hupp TR; Skladal P; Palecek E; Vojtesek B Activation of the DNA-binding ability of latent p53 protein by protein kinase C is abolished by protein kinase CK2. Biochem. J. 2004, 378 (3), 939–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Lee YH; Park JW; Bae YS Regulation of protein kinase CK2 catalytic activity by protein kinase C and phospholipase D2. Biochimie 2016, 121, 131–9. [DOI] [PubMed] [Google Scholar]
- (45).Bren GD; Pennington KN; Paya CV PKC-zeta-associated CK2 participates in the turnover of free IkappaBalpha. J. Mol. Biol. 2000, 297 (5), 1245–58. [DOI] [PubMed] [Google Scholar]
- (46).Jurkin J; Zupkovitz G; Lagger S; Grausenburger R; Hagelkruys A; Kenner L; Seiser C Distinct and redundant functions of histone deacetylases HDAC1 and HDAC2 in proliferation and tumorigenesis. Cell Cycle 2011, 10 (3), 406–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Stemmer C; Schwander A; Bauw G; Fojan P; Grasser KD Protein kinase CK2 differentially phosphorylates maize chromosomal high mobility group B (HMGB) proteins modulating their stability and DNA interactions. J. Biol. Chem. 2002, 277 (2), 1092–8. [DOI] [PubMed] [Google Scholar]
- (48).Wu L; Luo K; Lou Z; Chen J MDC1 regulates intra-S-phase checkpoint by targeting NBS1 to DNA double-strand breaks. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (32), 11200–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Rehage N; Davydova E; Conrad C; Behrens G; Maiser A; Stehklein JE; Brenner S; Klein J; Jeridi A; Hoffmann A; Lee E; Dianzani U; Willemsen R; Feederle R; Reiche K; Hackermuller J; Leonhardt H; Sharma S; Niessing D; Heissmeyer V Binding of NUFIP2 to Roquin promotes recognition and regulation of ICOS mRNA. Nat. Commun. 2018, 9 (1), 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Bardoni B; Castets M; Huot ME; Schenck A; Adinolfi S; Corbin F; Pastore A; Khandjian EW; Mandel JL 82-FIP, a novel FMRP (fragile X mental retardation protein) interacting protein, shows a cell cycle-dependent intracellular localization. Hum. Mol. Genet. 2003, 12 (14), 1689–98. [DOI] [PubMed] [Google Scholar]
- (51).De Leon CA; Levine PM; Craven TW; Pratt MR The Sulfur-Linked Analogue of O-GlcNAc (S-GlcNAc) Is an Enzymatically Stable and Reasonable Structural Surrogate for O-GlcNAc at the Peptide and Protein Levels. Biochemistry 2017, 56 (27), 3507–3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Leney AC; Rafie K; van Aalten DMF; Heck AJR Direct Monitoring of Protein O-GlcNAcylation by High-Resolution Native Mass Spectrometry. ACS Chem. Biol. 2017, 12 (8), 2078–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Hart GW; Slawson C; Ramirez-Correa G; Lagerlof O Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu. Rev. Biochem. 2011, 80, 825–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Zhong J; Martinez M; Sengupta S; Lee A; Wu X; Chaerkady R; Chatterjee A; O’Meally RN; Cole RN; Pandey A; et al. Quantitative phosphoproteomics reveals crosstalk between phosphorylation and O-GlcNAc in the DNA damage response pathway. Proteomics 2015, 15 (2–3), 591–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Tan ZW; Fei G; Paulo JA; Bellaousov S; Martin SES; Duveau DY; Thomas CJ; Gygi SP; Boutz PL; Walker S O-GlcNAc regulates gene expression by controlling detained intron splicing. Nucleic Acids Res. 2020, 48 (10), 5656–5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Litchfield DW Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem. J. 2003, 369 (1), 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Bosc DG; Slominski E; Sichler C; Litchfield DW Phosphorylation of casein kinase II by p34cdc2. Identification of phosphorylation sites using phosphorylation site mutants in vitro. J. Biol. Chem. 1995, 270 (43), 25872–8. [DOI] [PubMed] [Google Scholar]
- (58).Ong Q; Han W; Yang X O-GlcNAc as an Integrator of Signaling Pathways. Front Endocrinol (Lausanne) 2018, 9, 599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Ramirez DH; Ge Y; Woo CM O-GlcNAc Engineering on a Target Protein in Cells with Nanobody-OGT and Nanobody-splitOGA. Curr. Protoc 2021, 1 (5), No. e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Thompson JW; Griffin ME; Hsieh-Wilson LC Methods for the Detection, Study, and Dynamic Profiling of O-GlcNAc Glyco-sylation. Methods Enzymol 2018, 598, 101–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Woo CM; Bertozzi CR Isotope Targeted Glycoproteomics (IsoTaG) to Characterize Intact, Metabolically Labeled Glycopeptides from Complex Proteomes. Curr. Protoc Chem. Biol. 2016, 8 (1), 59–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Yang B; Li R; Liu PN; Geng X; Mooney BP; Chen C; Cheng J; Fritsche KL; Beversdorf DQ; Lee JC; et al. Quantitative Proteomics Reveals Docosahexaenoic Acid-Mediated Neuroprotective Effects in Lipopolysaccharide-Stimulated Microglial Cells. J. Proteome Res. 2020, 19 (6), 2236–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.