

Abstract
The [FeFe] hydrogenase catalyzes the redox interconversion of protons and H2 with a Fe-S “H-cluster” employing CO, CN, and azadithiolate ligands to two Fe centers. The biosynthesis of the H-cluster is a highly interesting reaction carried out by a set of Fe-S maturase enzymes called HydE, HydF, and HydG. HydG, a member of the radical S-adenosylmethione (rSAM) family, converts tyrosine, cysteine, and Fe(II) into an organometallic Fe(II)(CO)2(CN)cysteine “synthon”, which serves as the substrate for HydE. Although key aspects of the HydG mechanism have been experimentally determined via isotope sensitive spectroscopic methods, other important mechanistic questions have eluded experimental determination. Here we use computational quantum chemistry to refine the mechanism of the HydG catalytic reaction. We utilize QM/MM molecular dynamics simulations to investigate the reactions at the canonical Fe-S cluster, where a radical cleavage of the tyrosine substrate takes place and proceeds through a relay of radical intermediates to form HCN and a COO•− radical anion. We then carry out a broken symmetry DFT study of the reactions at the unusual five-iron auxiliary Fe-S cluster, where two equivalents of CN− and COOH• coordinate to the fifth “dangler iron” in a series of substitution and redox reactions that yield the synthon as the final product for further processing by HydE.
Graphical Abstract
1. Introduction
Hydrogenases are fascinating metalloenzymes that catalyze the reversible interconversion of H2 and H+/e–. They are categorized into [FeFe], [NiFe] and [Fe] subtypes according to the metal composition of their active site cofactors.1,2 The [FeFe] hydrogenase is highly active in H2 production, with rates up to 104/s, making it of great interest to the renewable energy community.3 This high activity is rendered by the unique catalytic center of [FeFe] hydrogenases, a six-Fe “H-cluster” consisting of a [4Fe–4S]H cluster linked through a cysteine S to a [2Fe]H cluster in which the two iron centers are coordinated by diatomic CO and CN− ligands, as well as an unusual azadithiolate (adt, NH(CH2S–)2) bridging ligand (Scheme 1). The unique structure and activity of the H-cluster thus raise the intriguing question as to its biosynthesis; a multi-component, step-by-step assembly made challenging by toxic ligands, oxygen sensitivity, and the inherent chemical instability of the adt moiety.
Scheme 1:
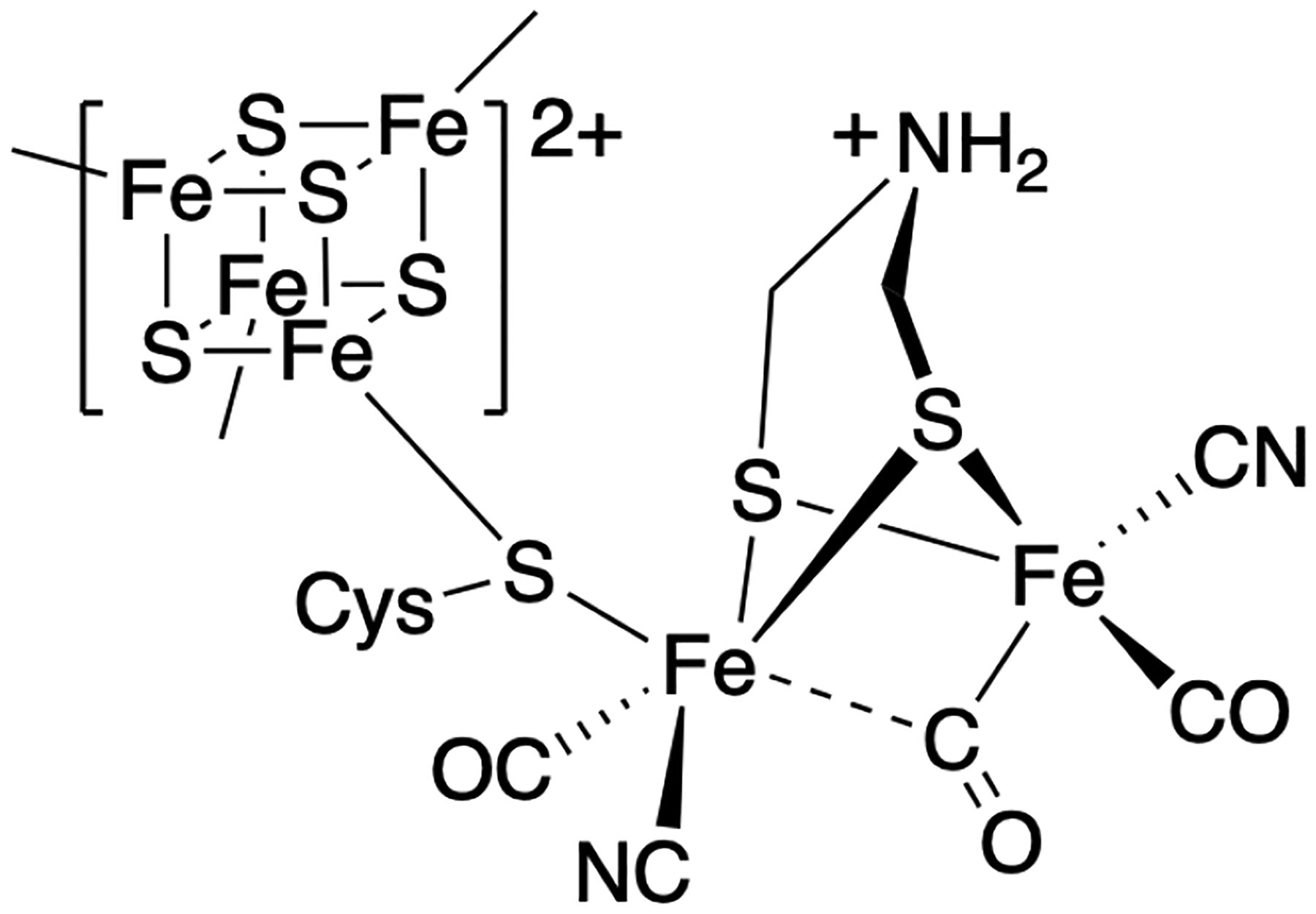
The H-cluster in the active site of [FeFe] hydrogenases.
Genetic and biochemical studies have shown that three Fe-S cluster proteins, HydE, HydF and HydG, are essential to the biosynthesis of the H-cluster.4,5 In particular, it is demonstrated that the radical S-adenosyl-L-methionine (rSAM) enzyme HydG is responsible for the biogenesis of the toxic CO and CN− ligands and their passivation by the formation of a [Fe(CO)2(CN)(cysteinate)] organometallic synthon.6–9 Recent work shows that this HydG product synthon serves as the substrate for another rSAM enzyme, HydE, which in turn generates [FeI(CO)2(CN)S]-containing intermediates that are proposed to undergo dimerization to form the core of the [2Fe]H subcluster.10 It was also recently shown that the NH(CH2)2 component of the adt ligand is sourced from serine.11 Once assembled, presumably on the HydF protein,12 the [2Fe]H cluster is delivered to an apo-hydrogenase that harbors only the [4Fe-4S]H subcluster, allowing the completion of the fully active H-cluster.13
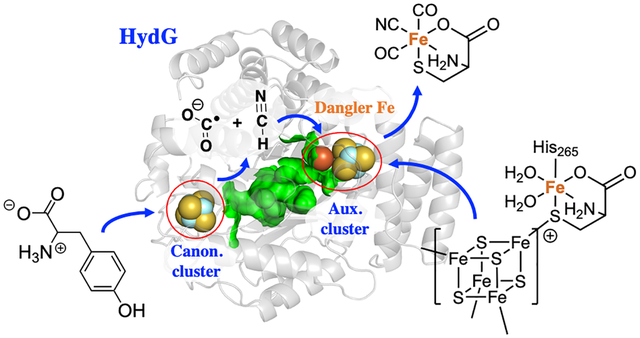
Despite these recent advances, questions remain regarding the detailed steps of the bioassembly pathway, even those involving the best understood maturase, HydG. Specifically, the molecular mechanism of CO and CN− formation is only partially defined by experiments. HydG contains two Fe-S clusters at either end of a 24 Å hydrophobic TIM barrel. These two clusters fulfill two distinct functions.14 As shown in Figure 1, the N-terminal cluster, a rSAM 4Fe–4S cluster such as found in all radical SAM enzymes, initiates the rSAM chemistry to generate the 5′-dAdo• radical which abstracts an amino hydrogen atom from tyrosine and induces Cα-Cβ homolysis to form a 4-hydroxylbenzyl radical (4-OB•) as detected by EPR spectroscopy, along with a proposed dehydroglycine (DHG) molecule.15 It is thought that the DHG is then converted into a CO and CN− pair inside HydG; these diatomic ligands are delivered to the C-terminal [4Fe-4S]-[Fe(cysteinate)] auxiliary cluster, where they bind to the unique fifth “dangler Fe” to form a [4Fe-4S]-[Fe(CO)(CN)(cysteinate)] intermediate revealed by stop-flow FTIR and EPR spectroscopy.16? ,17 A second pair of CO/CN− generated from Tyr further convert this intermediate to the [Fe(CO)2(CN)(cysteinate)] synthon product and a [4Fe-4S]-CN cluster that is subsequently reconverted to the resting state configuration with a fresh cysteine ligand replacing the CN− and then binding a new Fe2+.16
Figure 1:
Summary of reactivity at the HydG rSAM Fe-S cluster (left) and auxiliary cluster (right) leading to the production of the Fe(CO)2CN synthon. The cycles on the left and right represent the reactions that occur at the canonical and auxiliary iron-sulfur clusters, respectively. Herein, the red arrows indicate the controversial steps in HydG that we focus on in the study.
It remains elusive how DHG undergoes C-C bond cleavage to form CO and CN−; the simple hydrolysis reaction would yield ammonium and glyoxylate instead. It is also unclear whether and how the auxiliary cluster is involved in the decomposition of DHG, although this relevance is inferred from the observation that the auxiliary cluster knock-out mutant of HydG generates only CN− at a much slower rate without any detectable CO,9 and that the H265N mutant, which abolishes the dangler Fe in the auxiliary cluster, generates cyanide and formate only, again without CO.18 These results seem to imply, as alleged in the latter study, that the formation of CN− may occur at the rSAM site, whereas the formation of CO requires the dangler Fe site in the auxiliary cluster. This proposal is yet incomplete however, in that the detailed reaction mechanism for both CO and CN− formation needs to be clarified, and also given that the ligand environment and the electronic structure of the dangler Fe is dramatically altered upon binding of the first CO/CN− pair (high-spin, S = 2 to low-spin, S = 0),19 so the chemistry leading to the second CO ligand must differ from that of the first CO and CN− addition. Despite these previous efforts, it is challenging to address these remaining issues by purely experimental approaches. Tracking the fate of DHG in the active site pocket and the protein channel is difficult to achieve since relevant spectroscopic markers are currently lacking.
Here we investigate the reactions in HydG using computational quantum chemistry in order to gain insights into the experimentally inaccessible portions of the catalytic mechanism and guide further experimental design. The study involves two main parts: the first part is a series of hybrid QM/MM molecular dynamics calculations of the reactions at the canonical Fe-S cluster. Here, the 5’-deoxyadenosyl radical (5’dAdo•) initiator cleaves the tyrosine substrate, the products of which proceed through a relay of radical intermediates ending in HCN and a COO•− radical anion. The second part is a broken symmetry DFT study of the reactions at the auxiliary Fe-S cluster where two equivalents of CN− and COOH• coordinate to the dangler Fe in a series of substitution and redox reactions that yield the synthon as the final product. The presented mechanistic hypothesis is supported by computational data and consistent with experimental results, and reveals important and previously hidden features of the catalytic mechanism of HydG.
2. Computational Methods
Herein, the crystal structure of HydG (pdb ID: 4WCX) was used as a starting point to study the catalytic mechanism.14,16,20 The reactions that occur around the canonical [4Fe-4S] cluster were simulated using a hybrid density functional theory QM/MM umbrella sampling approach with the help of the Q-Chem/AMBER interface.21–23 The reactions occurring at the auxiliary [4Fe-4S] cluster were studied using a cluster model of the active site due to elevated computational cost, with calculations performed by the TeraChem software package.24–26 EPR properties for selected structures were comptued using the ORCA software package.27,28 Further details of the calculations including structural modeling, the level of QM theory,29,30 basis set,31–34 and the choice of QM region and force fields35–38 used in the QM/MM calculations are provided in the Supporting Information.
3. Results and Discussion
Tyrosine Radical Formation.
It has been reported by Britt, et al. that the HydG catalytic reaction is initiated by electron transfer from the reduced rSAM Fe-S cluster to its bound SAM cofactor, leading to the formation of a 5’-deoxyadenosyl radical (5’dAdo•) that initiates tyrosine homolytic cleavage.39,40 Because the Fe-S cluster induced decomposition of SAM is a well-known and well-studied radical reaction in biological systems, it is not a focal point of this study, and here we only aim to demonstrate consistency with experiments. By driving the defined reaction coordinate RC1 = d(C…S) – d(Fe…S) (defined in Figure S4), we found that the activation energy of this reaction is approximately 26.3 kcal/mol (Figure S4) which is comparable with the HydG experimental kinetics studies (about 23 kcal/mol).41 Following this step, Dinis et al. proposed that the 5’-dAdo• radical abstracts a hydrogen atom from the tyrosine amino group,14 by analogy to the tryptophan lyase NosL, where X-ray structural analysis and computations indicated that H-atom abstraction occurs at a tryptophan amino group.42 As shown in Figure 2, the key structures of tyrosine radical generation (Figure 2(A)) and the corresponding diagrams (Figure 2(B) and 2(C)) were depicted. The activation free energy of H• abstraction from the tyrosine amino group was calculated as 15 kcal/mol, demonstrating that this H-atom abstraction is feasible at room temperature and is exothermic, releasing more than 6 kcal/mol of energy. This exothermic nature of the reaction is attributed to the increased electronegativity of the tyrosyl N relative to the 5’-dAdo• C1 atom and by the rapid delocalization of the radical over the aromatic tyrosyl side chain, which we observed by monitoring the spin density along the reaction coordinate. We also considered an alternative pathway involving H-atom abstraction from the tyrosine α carbon in Figure S5, and ruled it out due to the activation barrier being higher by ≈5 kcal/mol.
Figure 2:
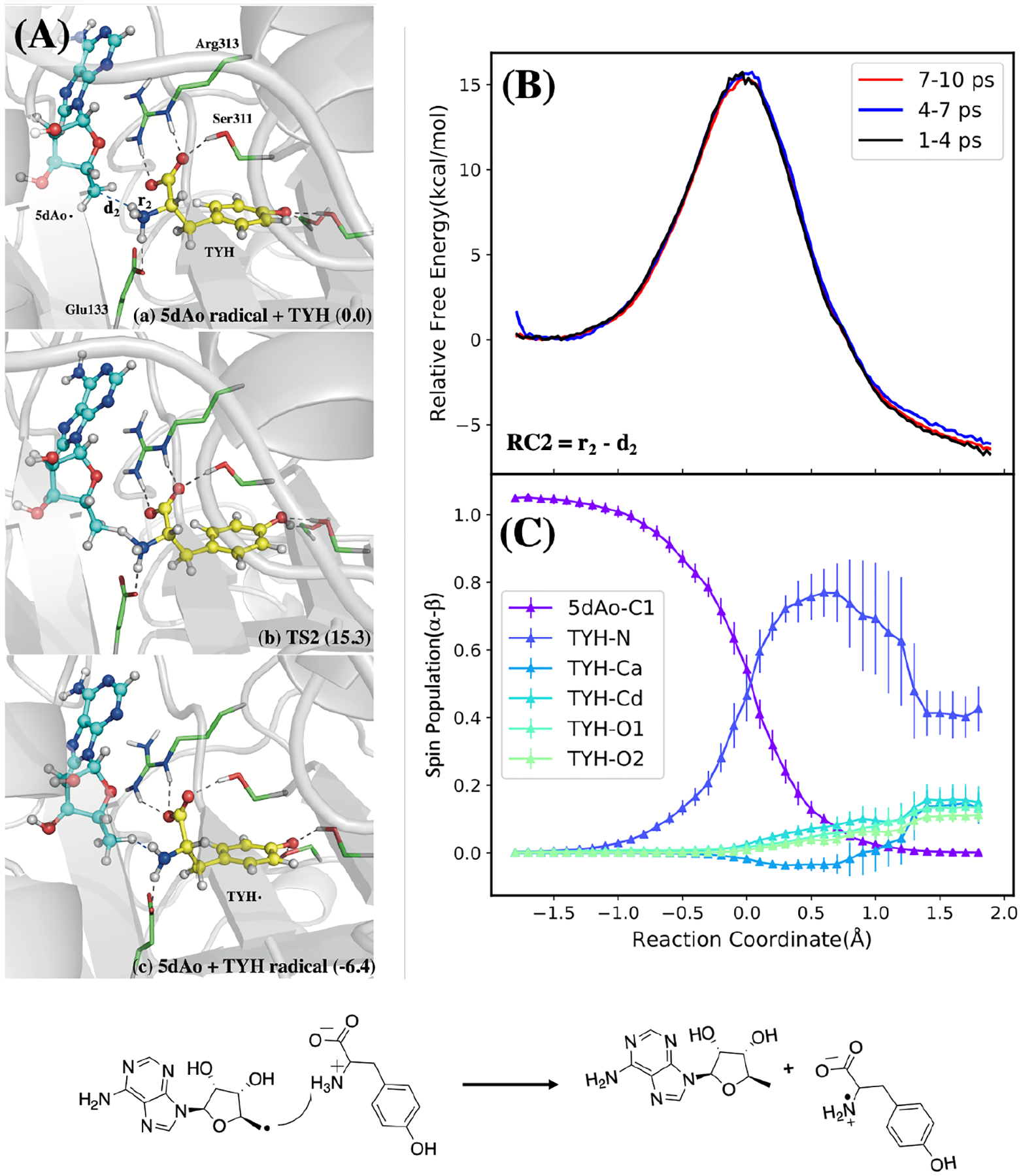
Radical transfer from 5-Ado radical to tyrosine substrate. (A): Structures of reactant, transition state, and product. (B): QM/MM free energy profile calculated using different QM/MM trajectory segments with the defined reaction coordinate (RC). (C): Mulliken spin populations along reaction coordinate with error bars taken from the standard deviations of the QM/MM MD simulations. Bottom: Reaction scheme.
Tyrosine Decomposition.
As mentioned above, HydG and NosL share many similarities both in terms of protein sequence and functionality.14,20 However, there is one significant difference in the catalytic mechanism between NosL and HydG: it is reported that NosL catalyzes cleavage of the tryptophan Cα—C(=O) bond which generates a COO•− radical instead of a radical localized on the indole side chain.42 In HydG, it is believed that the DHG is a key intermediate, which indicates that the cleavage of Cα—Cβ is required instead. We calculated the activation energies starting from different protonation states of the Tyr carboxyl group and found that it plays an important role in directing which Cα—C bond undergoes homogenous cleavage. As shown in Figure S6, the relative activation energies of the two different C—C cleavage mechanisms depends strongly on the protonation state of the model, and that the zwitterionic TYH model tends to undergo Cα—Cβ cleavage while the neutral TYY model tends to undergo Cα—C(=O) cleavage. The analysis of frontier orbitals in Figure S6 supports this result; the β LUMO, which is singly occupied, possesses σ-bonding character between Cα—Cβ in TYH that is absent in TYY, and this is consistent with TYH favoring Cα—Cβ cleavage. All of these results suggest that TYH is the appropriate protonation state in HydG, and differences in protonation state may contribute to the distinct mechanisms of NosL and HydG, along with the intrinsic difference between Tyr and Trp side chains.
The results of tyrosine decomposition to DHG and 4-OB• are shown in Figure 3. The mechanism involves the transfer of a proton from the tyrosyl radical H2N• moiety to Glu133, thus a two-dimensional umbrella sampling calculation was carried out to calculate the reaction free energy along the Cα—Cβ bond length and the proton transfer coordinate. According to the free energy map and the key structures in Figure 3, the highest point on the barrier mainly involves C—C bond dissociation and the proton is transferred afterward. After the reaction, the intermediates DHG and 4-OB• are formed, and the radical on the latter is stabilized by the aromatic system.
Figure 3:
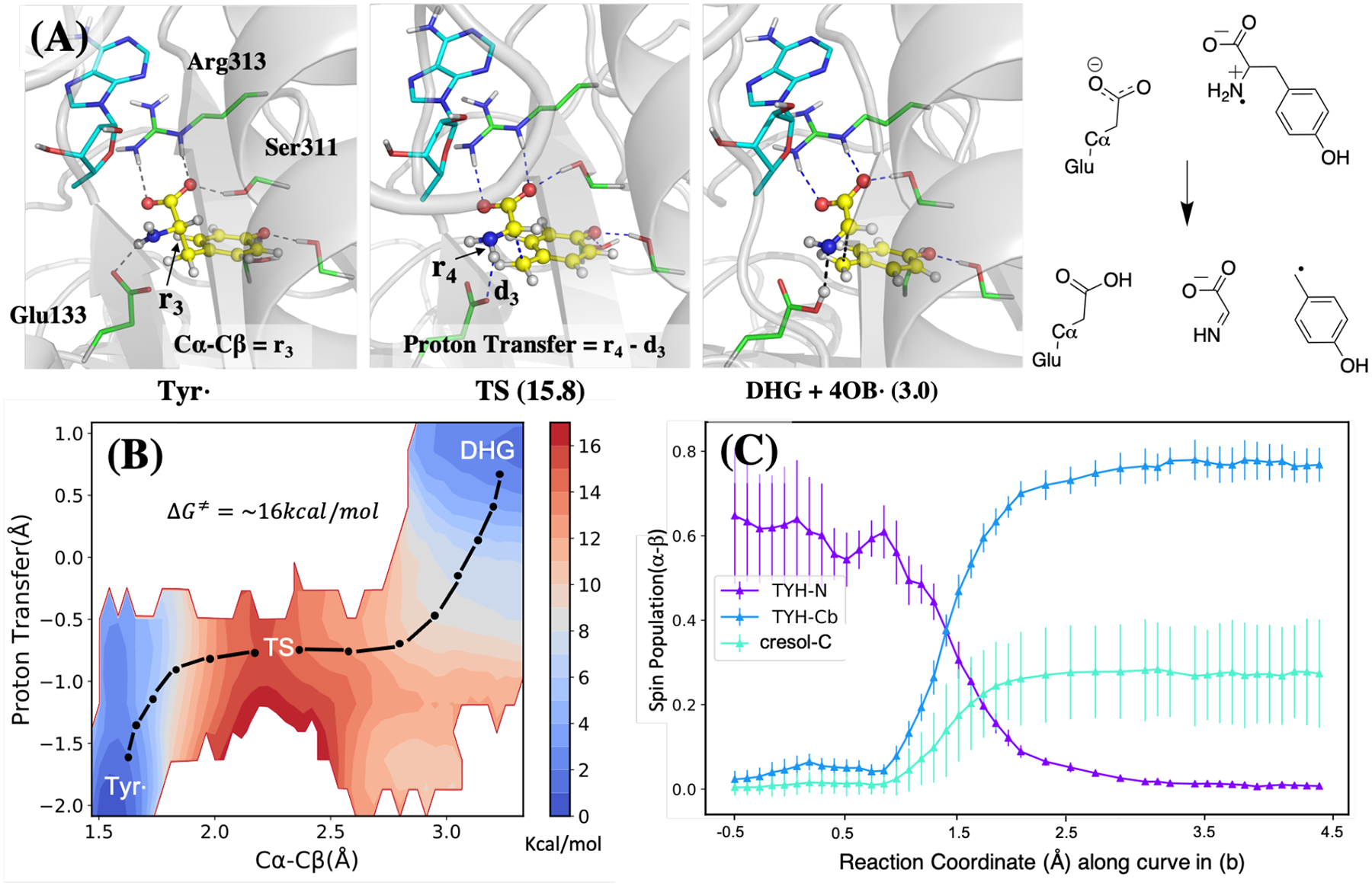
The calculated mechanism of tyrosine radical decomposition and DHG formation. The structures of three key states are shown on top (A), and the free energy map and spin density of different atoms are shown at the bottom left (B) and right (C) respectively. The color bar in (B) indicates the scale of the energy in kcal/mol. The spin population in (C) is plotted along the one-dimensional path indicated using the black dot-dash in the free energy map (B). The 2D scheme of the reaction is shown at the top right.
DHG Decomposition and CN and COOH Generation.
Isotope labeling studies have confirmed that the CO and CN ligands originate from the atoms of tyrosine that belong to the DHG homolysis product. However, our calculations show that directly breaking the C-C bond in DHG to form HCN and CO2 is not feasible at room temperature (Ea > 35 kcal/mol). In addition, CO2 would then need to be reduced to CO, and the highly negative reduction potential of CO2 makes this process even less feasible. Herein, we propose a “radical relay” mechanism where the 4-hydroxybenzyl radical (4-OB•) produced by tyrosine lysis now abstracts an H atom from the DHG nitrogen (Figure 4 left). We calculate that the radical transfer process from the 4-OB• to the DHG is exothermic with ΔG = −4 kcal/mol and ΔG‡ = 18 kcal/mol, and more favorable than an alternative pathway involving H-atom abstraction from carbon (Figure S5). Next, the homolytic cleavage of the C—C bond in this newly formed DHG radical occurs with ΔG = +4 kcal/mol and ΔG‡ = 15 kcal/mol (Figure 4 right), making the sum of these two DHG-centered radical reaction steps thermodynamically neutral. The products of this new fission are HCN and another radical, COO•−. The well-studied radical anion of carbon dioxide has most of the unpaired spin localized on carbon.43 Importantly, this proposed mechanism provides a crucial role for the 4-OB•; rather than being simply quenched to form p-cresol, it plays an active role in fragmenting the co-formed DHG along the proper reaction pathway to produce CO and CN− rather than ammonia and glyoxylate.
Figure 4:
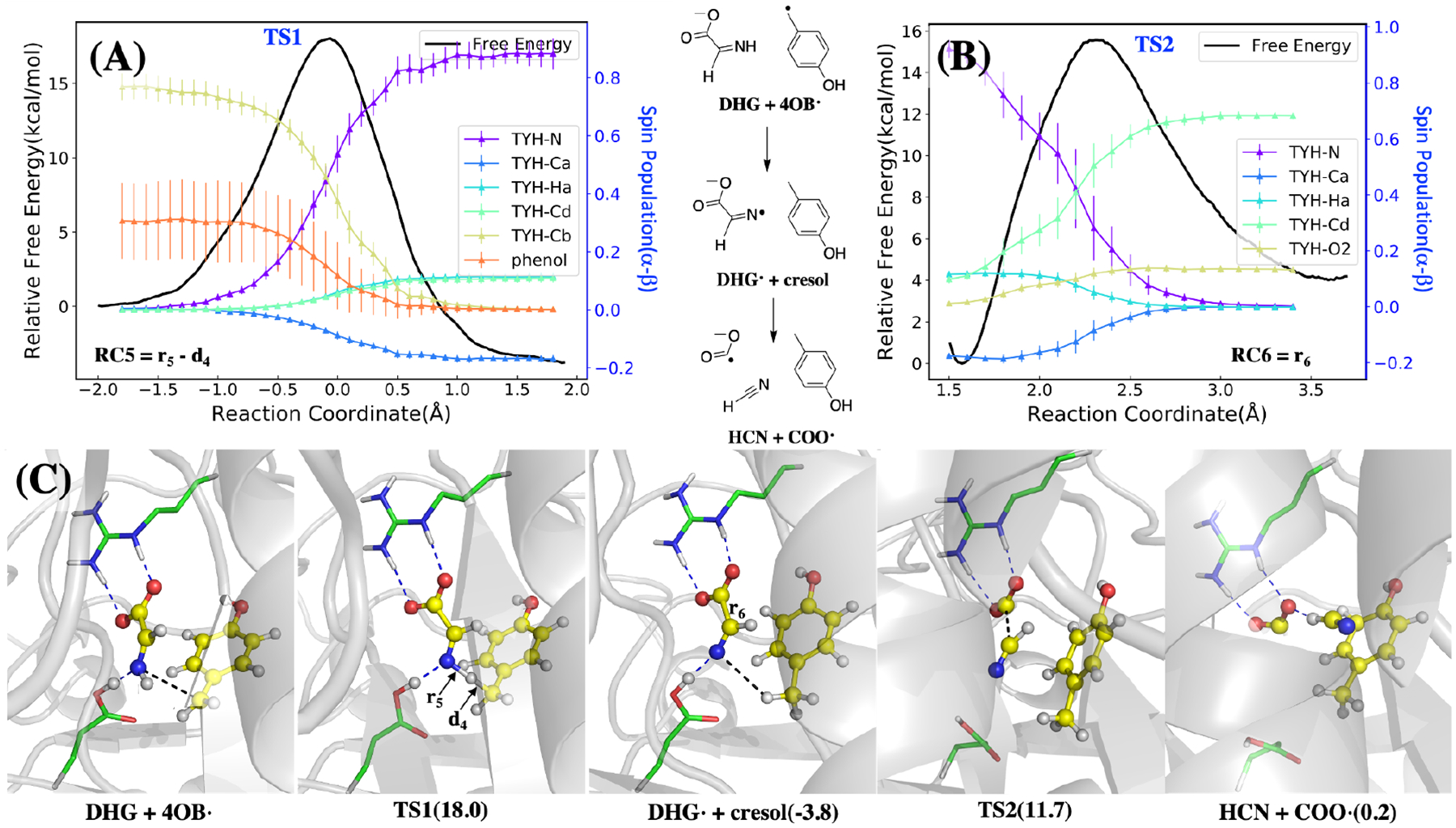
The calculated mechanism of DHG radical formation and decomposition, starting with H• abstraction from the DHG nitrogen, followed by decomposition of DHG radical to COO•− and HCN. Top: Free energy profile (A) and spin populations profiles (B) along the umbrella sampling reaction coordinates. The reaction coordinate of the DHG radical formation (top left) is defined as r5 − d4 where distances are indicated in the bottom left panel, and for the DHG radical decomposition it is defined as r6 shown in the bottom panel. The energy curves are both colored in black with the y-axis on the left, while the spin density profiles are shown for selected atoms in color with the y-axis are shown on the right. Bottom (C): Key structures during these two reactions with hydrogen bonds shown in blue dotted lines and the distances of the reaction coordinate shown in black dotted lines. As the structures shown in the bottom, the radical transfers from the 4-OB• to the nitrogen in the DHG, which helps the decomposition of the DHG into COO• and HCN. The barriers of these two steps are about 18 and 15.5 kcal/mol respectively. The radical transfer is exothermic while the DHG decomposition is endothermic and makes these two reactions overall barely absorb energy.
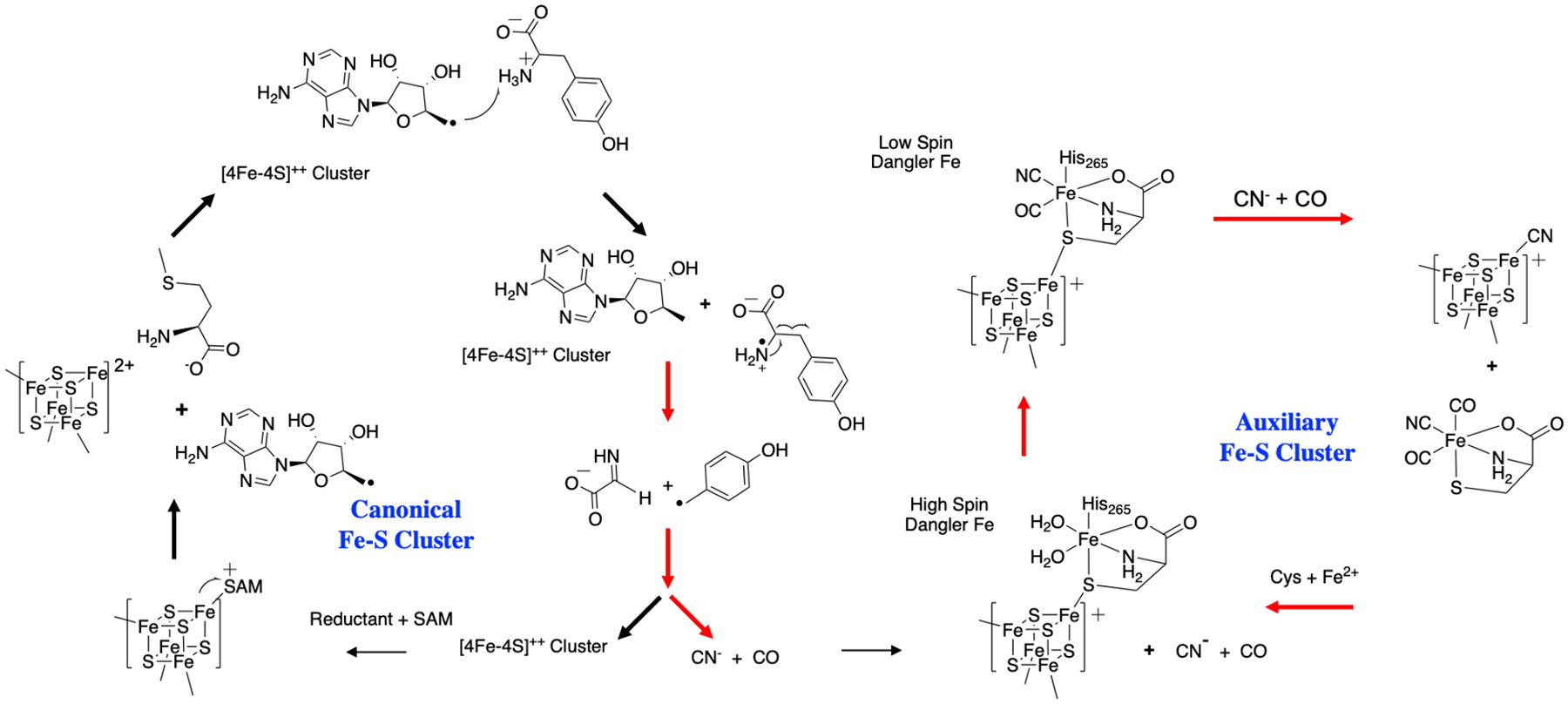
The HCN and COO•− species are structurally similar but not identical to the CN− and CO that ultimately bind to the dangler iron located near the auxiliary cluster. Reduction of COO•− to CO requires a hydrogen atom donor, which we could not find in the vicinity of the rSAM Fe-S cluster; attempts to transfer an H atom from cresol or nearby ionizable residues all resulted in activation energies of 30 kcal/mol or higher (Figure S7). Although it has been reported that COO•− can react with tryptophan side chains42 and disulfide bonds,44 the TIM barrel does not contain any of these structures.14 Due to the lack of other plausible reaction pathways for COO•−, we propose that these species diffuse through the TIM barrel to the dangler iron of the auxiliary cluster. Changes in the protonation state of COO•− and HCN to COOH• and CN− are also thermodynamically accessible, as HCN is weakly acidic, and COO•− is able to accept a proton from Glh133 (which was itself protonated during the prior Tyr decomposition step) with a slightly positive ΔE of 5.0 kcal/mol. Following the diffusion and changes in protonation state, the remaining reaction steps are proposed to take place at the auxiliary cluster, summarized in Figure 5.
Figure 5:
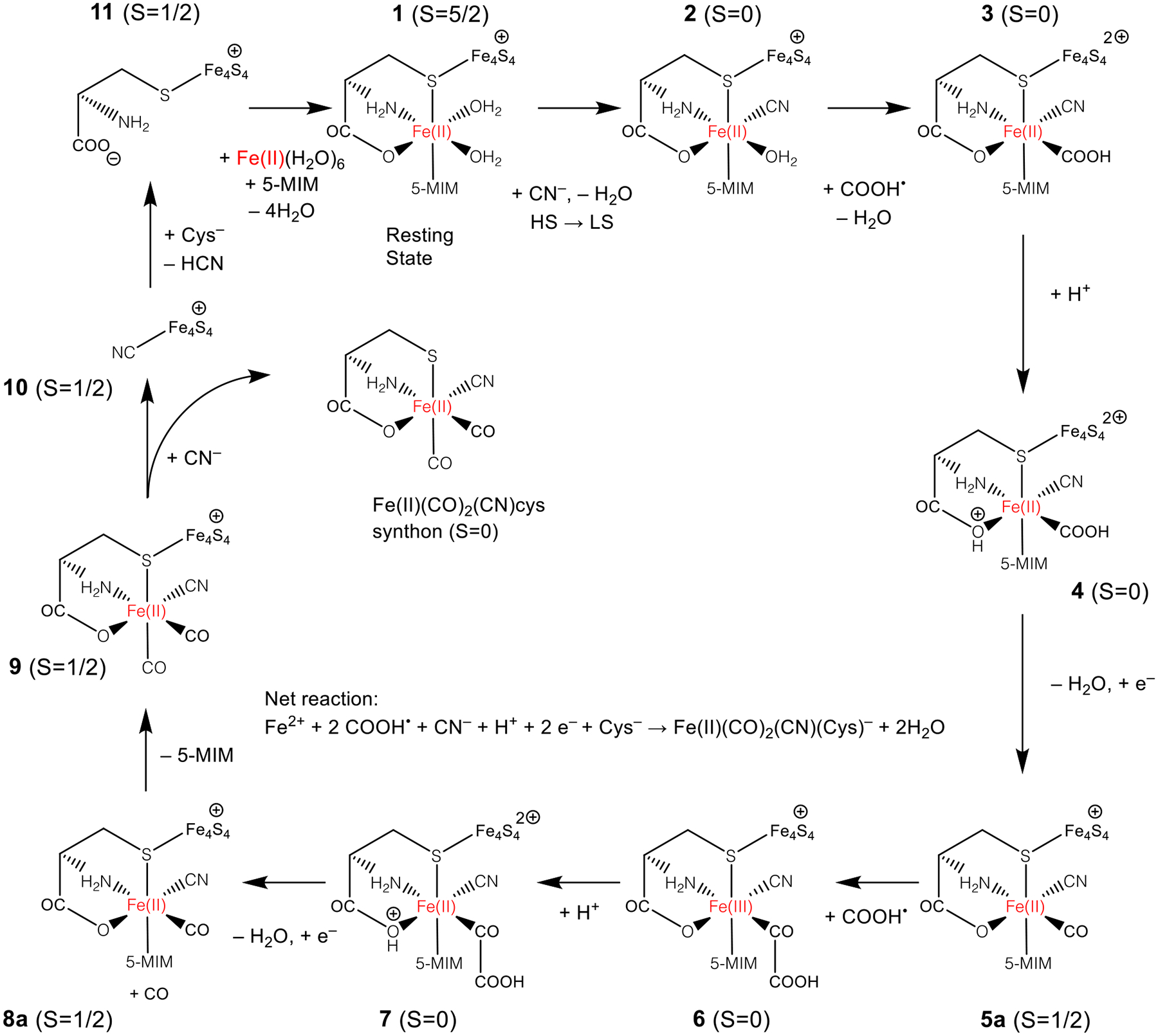
Catalytic cycle for formation of Fe(II) synthon at auxiliary cluster. The total spin is indicated for each species; additional properties are listed in Table S1. The resting state of the catalyst is shown at the top (1). The catalytic cycle involves three ligand substitutions in the coordination sphere of the dangler Fe and the reduction of two COOH• species to CO. The 5-methylimidazole (5-MIm) species models the histidine residue in the protein.
Spin crossover and first CN− substitution.
The assembly of the synthon at the dangler Fe is proposed to begin with coordination of cyanide by displacement of a labile aqua ligand on the ferrous center (1 + CN− → 2 + H2O in Figure 5). Prior to the reaction, the Fe4S4 cluster is in the reduced state with a total charge of +1 and 1 unpaired electron (α), the dangler Fe(II) is high-spin, having 4 unpaired electrons (α) and the COOH• has one unpaired electron (β). The coordination of CN− stabilizes the low spin electronic state, a ubiquitous feature of octahedral ferrous cyanides.45,46 To support the result that the coordination of a single CN− ligand is sufficient to stabilize the low-spin state, we carried out multireference density matrix renormalization group (DMRG) calculations performed on Fe(cys)(5-MIm) in the gas phase, with the results summarized in Table S2 and Figure S8. Consistent with our DFT results, these calculations predict the ground state is high-spin with two H2O ligands, and changes to low spin when one of the ligands is replaced by CO or CN–.
Prior to any ligand substitution, the structure of 1 was optimized in both high-spin and low-spin electronic states as well as the minimum energy crossing point (MECP) between the two. All three optimized structures are highly similar with the largest differences coming from the Fe—S bond lengths, which are 0.16 Å shorter in the LS state. The MECP is energetically slightly uphill from the HS minimum by ΔE = 8.2 kcal/mol, and the LS minimum is only 0.4 kcal/mol lower than the MECP. With the addition of free energy corrections, we computed ΔG(LS-HS) = 11.9 kcal/mol for spin crossover prior to ligand substitution. Starting from the LS energy minimum of 1, the substitution reaction of the H2O ligand by CN− is found to proceed with a modest energy barrier, involving a number of structural intermediates as described in Figure S9. The overall energy parameters of the 1(HS) → 2(LS) reaction are given as ΔE = −7.2; Ea = 17.2; ΔG = −3.9, ΔG‡ = 20.1 (all values in kcal/mol).
First COOH• substitution, decomposition, and reduction.
Starting from the low-spin Fe(II)-CN complex 2, the substitution reaction of the second H2O ligand by COOH• (2 + COOH• → 3 + H2O) was also found to be easily accessible and exothermic, with the energy parameters computed as ΔE = −29.1;Ea = 10.2;ΔG = −25.4;ΔG‡ = 11.4 (all values in kcal/mol). The spin densities on COOH and Fe, respectively, change from (−0.92, −0.02) → (0.04, −1.09) during this reaction step, clearly indicating that the spin density on COOH• moves to Fe, and resulting in an electronic state that we interpret as (+, III). We also found that a second ET step from the auxiliary cluster to the dangler Fe is energetically favorable with ΔE = −8.9 kcal/mol, resulting in a ground state of (2+, II). Based on this finding, we think one possible role for the auxiliary cluster is to act as an electron source for the addition of COOH•, which would otherwise oxidize the dangler Fe.
In order for the COOH ligand to decompose into CO + H2O, an electron and proton must be transferred to the OH group. The electrochemical reduction of M-COOH to CO is well-known in the broad literature on CO2 reduction electrocatalysis.47–52 We investigated two possible pathways of this reaction step, summarized as 3 + H+ + e− → 5a + H2O overall. In the first possible pathway, protonation occurs first, followed by ligand decomposition, then reduction. We modeled this step by adding a proton to one of the displaced H2O molecules and placing both molecules close to the OH group in a hydrogen bonding conformation. After optimization of the minimum energy path, the reactant structure was protonated at the cysteine carboxyl oxygen coordinating to the dangler Fe(II). The free energy of protonation is found to be positive, as ΔG(3 + H+ → 4) = 12.9 kcal/mol. The proton is transferred via a H2O relay to the OH group on COOH with a very small energy barrier of < 1.0 kcal/mol, and the resulting H2O dissociates leaving a CO coordinated to Fe. The free energy diagram of the steps 3 → 4 → 5 are shown as the first red pathway in Figure 6, and the free energy parameters are calculated as ΔG = −22.1;ΔG‡ = 12.9 kcal/mol relative to 3 (electronic energy differences are not reported in steps involving electron and proton transfer). Finally, an electron is added to the system to reduce the cluster from (2+) to (+), represented as 5 → 5a in Figure 6, and the redox potential is computed as −0.95 V, which corresponds to ΔE = 6.7 kcal/mol when using dithionite as the reducing agent, (−0.66 V), or ≈ 12.7 kcal/mol using ferredoxin (≈ −0.40 ± 0.03V) under physiological conditions.53 The g-tensor eigenvalues of 5a are computed as [2.030, 1.916, 1.862] and found to be in reasonable agreement with the experimental measurement of g =[2.058, 1.922, 1.882] (RMS error = 0.020).19 The hyperfine couplings are computed as A(dangler Fe)=[−1.18, −3.04, −1.58] MHz and A(cysteine Cβ)=[−6.32, −0.80, 0.82] MHz. While these values differ significantly from the experimental measurements of A(dangler Fe)=[0.45, 0.30, 0.50] MHz and A(cysteine Cβ)=[1.00, 0.20, 1.00] MHz, they are consistent in their order of magnitude, indicating only small amounts of spin density on the dangler Fe and cysteine Cβ centers.
Figure 6:
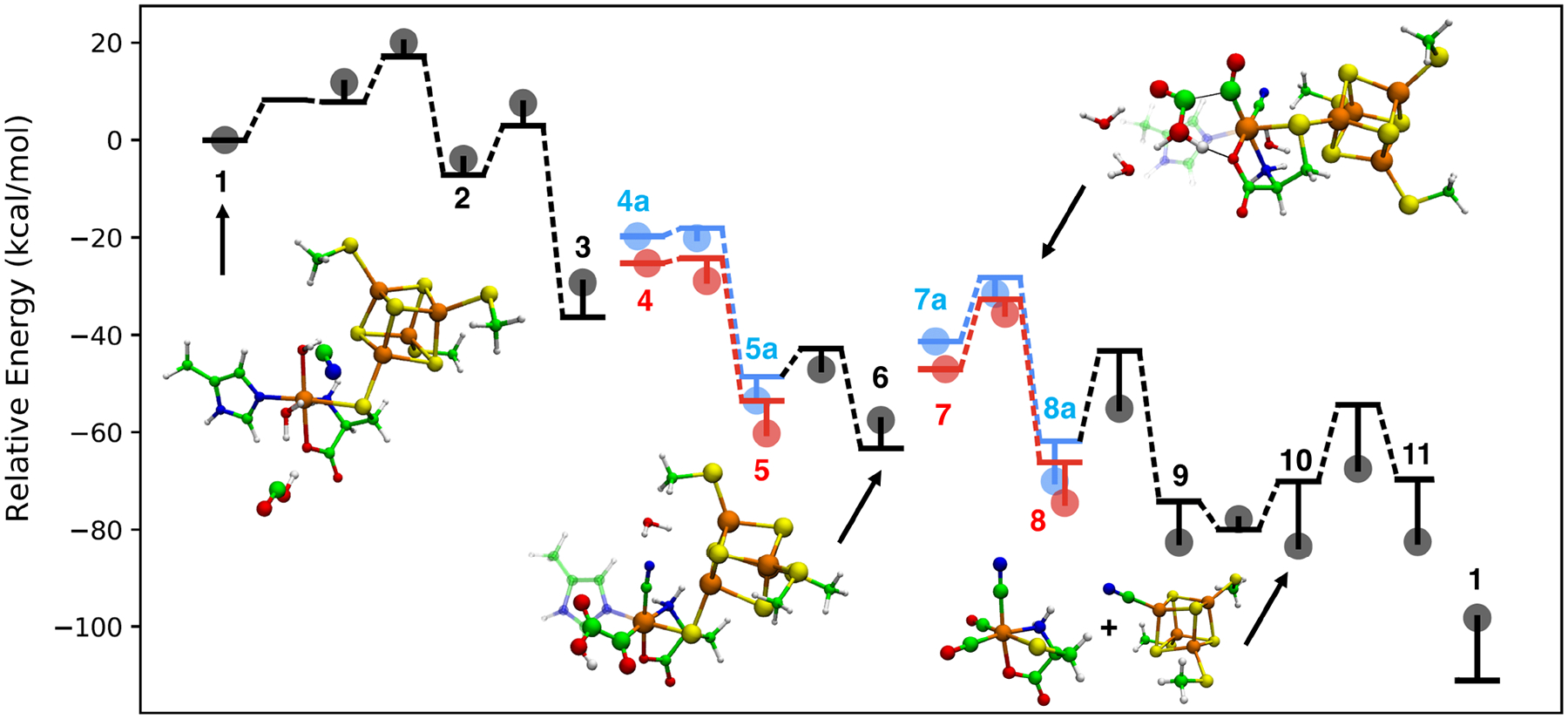
Energy diagram of the catalytic cycle expressed in Fig. 5 for formation of Fe(II) synthon at auxiliary cluster. Bolded numbers correspond to labeled species in Figure 5. Horizontal lines connected by dotted lines represent relative electronic energies that are connected via a minimum energy path. Circles represent relative Gibbs free energies computed using the harmonic oscillator/rigid rotor approximation. In the blue paths, proton-coupled reduction occurs first followed by decomposition. In the red paths, protonation is followed by deprotonation, then followed by reduction.
In the second possible pathway shown as the first blue pathway of Figure 6, reduction of the auxiliary cluster and protonation of the cysteine carbonyl oxygen occur simultaneously, and can be summarized as 3 + H+ + e− → 4a. The redox potential is computed as −1.46 V, which corresponds to ΔG = 18.4 kcal/mol if the reducing agent is dithionite. The proton transfer to the OH group of COOH and the resulting dissociation of H2O (4a → 5a) proceeds in similar fashion to the first pathway, but due to the higher relative free energy of the reduced state, the overall activation free energy is found to be slightly higher (ΔG‡ = 18.4 kcal/mol relative to 3) compared to the first pathway.
Reduction and decomposition of a second COOH•.
In order to produce the Fe(II) synthon, the 5-methylimidazole ligand must be replaced by CO sourced from a second COOH• ligand produced as a result of a second tyrosine lysis, which we presume follows the same mechanism as the first tyrosine lysis described above. The main question for the second CO formation is how the mechanism is affected by the altered state of the auxiliary cluster, given that the first CO and CN are already bound and the Fe(II) shifted from high spin to low spin. Our attempt to compute this substitution step led to an unexpected result: a transition state for 5-MIm substitution by COOH• was found, but the energy minimization started from the TS led to a reactant structure where COOH• forms a covalent C-C bond with the first CO coordinated to the dangler Fe (6, lower middle structure in Figure. 6). We found that this new intermediate, a glyoxylyl (OC-COOH) ligand to Fe, forms easily from 5 in a pathway where COOH• directly forms a C-C bond with the CO ligand and the energy parameters are ΔE = −14.7, Ea = 5.8, ΔG = −4.0, ΔG‡ = 6.4 kcal/mol; this unusual C—C coupling is reminiscent of CO2 electrolysis experiments at inert electrodes where oxalate is one of the products formed.54 The glyoxylyl intermediate is characterized by a (+, III) oxidation state because the spin density is transferred to the dangler Fe, analogous to the coordination of the first COOH•. The alternate (2+, II) electronic state, in which an electron is transferred from the cluster, is found to be slightly higher in energy ΔG = 1.5 kcal/mol, indicating that the calculations do not significantly favor one state over the other. From this new intermediate, we investigated whether this ligand could be decomposed to CO by an additional reduction step.
We modeled the decomposition of the glyoxylyl ligand by placing a H3O+ cation close to the OH group and searching for a TS involving proton transfer and dissociation of the C-C bond to form H2O and CO, similar to before. In the optimized path, we again found that the reactant structure was protonated at the cysteine carboxyl oxygen coordinating to the Fe atom (7a in figure 6), with ΔG = 10.4 kcal/mol relative to 6. The ground state of the protonated form has nearly zero spin on the dangler Fe indicating a (2+, II) electronic state. Proton transfer results in decomposition of the glyoxylyl ligand to yield CO + H2O + 8, shown as the second red pathway in Figure 6. The free energy barrier of the overall reaction 6 → 8 including the protonation step are the highest in the overall cycle (ΔG = −17.0, ΔG‡ = 21.8 kcal/mol), but are somewhat smaller compared to the highest barrier of 26.3 kcal/mol found for the reactions at the canonical cluster. The Fe4S4 cluster is reduced back to the (+) electronic state (i.e. 8 → 8a with a potential of −0.85 V or ΔG = 4.4 kcal/mol when dithionite is used as the reducing agent, corresponding to ΔG = −12.6 kcal/mol relative to 6.
A second possible pathway was considered in which simultaneous protonation and reduction of 6 occurs first, followed by PT and decomposition of the COOH group. The proton-coupled redox potential was computed as −1.36 V at pH 7, corresponding to an overpotential of 0.70 V, or ΔG = 16.1 kcal/mol. The decomposition reaction then proceeds in analogous fashion, in which the proton is transferred to the glyoxylyl OH group, followed by ligand decomposition to yield CO + H2O + 8a. In the reduced electronic state, the free energy parameters of the reaction 6 + H+ + e− → 7a → 8a were computed as ΔG = −12.6, ΔG‡ = 26.0 kcal/mol, shown as the second blue pathway in Figure 6.
Because the overall free energy barrier is 4.2 kcal/mol higher when proceeding from the reduced state 7a, we think the most likely sequence of reaction steps is protonation of 6 coupled to cluster → dangler electron transfer, decomposition, then reduction; however, the alternate ordering of proton-coupled reduction followed by decomposition is a close alternative possibility. The product includes a free CO ligand which is able to displace the 5-MIm ligand, leading to 9, with energy parameters ΔE = −12.3, Ea = 18.6, ΔG = −12.5, ΔG‡ = 14.9 kcal/mol.
Completion of catalytic cycle.
At this point in the cycle, the coordination sphere of the Fe(II) synthon is completed, and this Fe(II)(CO)2(CN)cysteine complex needs to be released from HydG so it can serve as the substrate for the other rSAM enzyme, HydE.55 We placed the second CN− species close to the auxiliary cluster (about 6Å away from the Fe4S4 cluster) and found that it spontaneously coordinates to the Fe4S4 iron closest to the dangler Fe without a barrier. This coordination mode is highly similar to how the first CN− species coordinates to 1, also seen in titration experiments.19 We note that although we computed the CN− association step after the completion of the coordination sphere of the Fe(II) synthon, it is possible that this coordination may take place at an earlier point in the catalytic cycle, perhaps as early as the delivery of the second COOH• equivalent (6).
The dissociation of the synthon is energetically slightly uphill by ΔE = 9.9 kcal/mol, and the overall reaction of replacing the synthon by CN− has energy parameters given by ΔE, ΔG(9 → 10) = 4.11,−0.85 kcal/mol. The g-tensor eigenvalues of 10 are computed as [2.023, 1.961, 1.931], which can be compared with g =[2.09, 1.94, 1.93] from experiment (RMS error = 0.041).16 The hyperfine tensor on the CN carbon is computed as A =[5.11, 27.39, 27.54] MHz, which is much larger than the experimental measurement of A =[−5.0, −4.0, 0.9] MHz; the overestimation is likely due to the tendency to over-delocalize spin density in DFT.56 To close the cycle, final substitution of this CN− by CH3SH, the side chain analogue of cysteine, is found to be nearly isoenergetic with a moderate barrier (ΔE = +0.4, Ea = 15.7, ΔG = +1.0, ΔG‡ = 16.0, all values in kcal/mol); a proton is transferred from the thiol group to CN− during this step. We computed g =[2.016, 1.967, 1.938], which when compared to the experimental measurement of g =[2.06, 1.90, 1.87], has a relatively large RMS error of 0.061 compared to the other species; this could be due to the use of the Cys side chain analogue in place of the complete amino acid in our calculations. The hyperfine tensor on the CH3SH carbon is computed as A =[5.08, 9.05, 9.64] MHz, which is significantly larger than the experimental A =[0.83, 0.83, 1.09] MHz, a consistent trend with all of the EPR property calculations. The ultimate fate of the HCN species is still experimentally undetermined. Subsequent coordination of Fe(II) to cysteine and 5-MIm completes the catalytic cycle and returns the system to its resting state (ΔE = −40.9, ΔG = −14.8, all values in kcal/mol).
This computed mechanism produces an isomer of the synthon where the CN ligand is opposite to the cysteinyl carboxylate oxygen, whereas a recent experimental study on the crystal structure of HydE with the synthon as its substrate57 assigned the CN ligand as opposite to sulfur based on the proximity of hydrogen bond donating residues. We computed the relative electronic energies of the synthon isomers and found them to be very close in energy (ΔE (opposite S) = 0.00; ΔE (opp. N) = 0.65; ΔE (opp. O) = 1.65, values in kcal/mol). Therefore, we think it is thermodynamically possible for the synthon to undergo isomerization prior to binding to HydE though the isomerization mechanism has yet to be determined.
4. Conclusion.
In this theoretical study, we have been able to elucidate additional mechanistic details concerning the HydG catalytic cycle, building on previous experimental results. We propose that after the initial HydG tyrosine lysis produces a 4-hydroxybenzyl radical (4-OB•) and dehydroglycine (DHG), the nascent 4-OB• radical in turn abstracts an H-atom from the nitrogen of DHG, resulting in a DHG radical. This DHG radical in turns undergoes a spontaneous C-C bond cleavage to form HCN and a new COOH•− anion radical. In the overall formation of the HydG Fe(II)(CO)2(CN)cysteine product this radical cascade occurs twice. However, the specific reactions at the five-Fe auxiliary Fe-S cluster differs between the first and second subcycles. Following the first HydG induced tyrosine lysis and subsequent radical cascade, the fifth “dangler Fe” of the auxiliary cluster is high spin Fe(II) with two aqua ligands. The CN− and COOH•− anion radical can bind in these two positions, substituting for the aqua ligands, and causing a spin crossover of the dangler Fe(II) from high spin to low spin. The COOH is further protonated and decomposes to the CO ligand and an H2O. The auxiliary cluster reaction must differ on the second subcycle since the dangler Fe(II) is now low spin with CO and CN ligands. We model the second cycle with the second COOH• forming a covalent C-C bond at the existing CO Fe ligand, forming a transient glyoxylyl ligand that decomposes upon further reduction. The second CO resulting from this decomposition displaces the histidine ligand to the dangler Fe, while the second CN− attacks the Fe4S4 cluster, releasing the entire HydG Fe(II)(CO)2(CN)cysteine product.
This Fe(II) “synthon” is transferred to HydE where it serves as a substrate for another set of reactions that lead to a highly reactive Fe(I)(CO)2CNS species poised to form the Fe2S2 core of the [2Fe]H subcluster.55,58 The HydG cycle is completed by another cysteine replacing the CN− bound to the Fe4S4 cluster, with this new cysteine and the HydG histidine binding a new Fe(II) to regenerate the HydG resting state. According to the energy barriers of all the reactions above, the rate-limiting step of the HydG catalytic process is either the SAM decomposition, whose energy barrier is 26.3 kcal/mol, or the redox-coupled decomposition of the second COOH• equivalent with a free energy barrier of 21.8 kcal/mol. Both numbers are comparable to experimental kinetics studies (23 kcal/mol). All of the other computed barrier heights for all the steps in this complex reaction are found to be thermodynamically accessible and consistent with the observed (rather slow) timescale of the HydG reaction chemistry, and thus we consider this a plausible overall model for the HydG chemistry, consistent with experimental observables. Looking forward, we think that theoretical studies can also provide insights into the catalytic mechanism of HydE and HydF to complement the experimental data and furnish a complete mechanism for the biosynthesis of the unique [2Fe]H subcluster of the [FeFe] hydrogenase active site.
Supplementary Material
Acknowledgements
We gratefully acknowledge Tom Rauchfuss, Dan Suess, and Lizhi Tao for insightful and thought-provoking discussions. NC and LPW acknowledge financial support from U.S. Army Research Office, award W911NF-17-1-0434. RDB acknowledges support from the National Institutes of Health, award 1R35GM126961-01.
Footnotes
Supporting Information Available
The supporting information contains detailed descriptions of the computational methods and additional details of the reaction mechanisms.
References
- (1).Lubitz W; Ogata H; Rüdiger O; Reijerse E Hydrogenases. Chemical reviews 2014, 114, 4081–4148. [DOI] [PubMed] [Google Scholar]
- (2).Fontecilla-Camps JC; Volbeda A; Cavazza C; Nicolet Y Structure/function relationships of [NiFe]-and [FeFe]-hydrogenases. Chemical reviews 2007, 107, 4273–4303. [DOI] [PubMed] [Google Scholar]
- (3).Pandey K; Islam STA; Happe T; Armstrong FA Frequency and potential dependence of reversible electrocatalytic hydrogen interconversion by FeFe -hydrogenases. Proceedings of the National Academy of Sciences of the United States of America 2017, 114, 3843–3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Posewitz MC; King PW; Smolinski SL; Zhang LP; Seibert M; Ghirardi ML Discovery of two novel radical S-adenosylmethionine proteins required for the assembly of an active Fe hydrogenase. Journal of Biological Chemistry 2004, 279, 25711–25720. [DOI] [PubMed] [Google Scholar]
- (5).Kuchenreuther JM; Britt RD; Swartz JR New Insights into FeFe Hydrogenase Activation and Maturase Function. PLOS ONE 2012, 9, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Driesener RC; Challand MR; McGlynn SE; Shepard EM; Boyd ES; Broderick JB; Peters JW; Roach PL FeFe -Hydrogenase Cyanide Ligands Derived From S-Adenosylmethionine-Dependent Cleavage of Tyrosine. Angewandte Chemie-International Edition 2010, 49, 1687–1690. [DOI] [PubMed] [Google Scholar]
- (7).Shepard EM; McGlynn SE; Bueling AL; Grady-Smith CS; George SJ; Winslow MA; Cramer SP; Peters JW; Broderick JB Synthesis of the 2Fe subcluster of the FeFe -hydrogenase H cluster on the HydF scaffold. Proceedings of the National Academy of Sciences of the United States of America 2010, 107, 10448–10453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Kuchenreuther JM; Myers WK; Suess DLM; Stich TA; Pelmenschikov V; Shiigi SA; Cramer SP; Swartz JR; Britt RD; George SJ The HydG Enzyme Generates an Fe(CO)(2)(CN) Synthon in Assembly of the FeFe Hydrogenase H-Cluster. Science 2014, 343, 424–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Kuchenreuther JM; Myers WK; Stich TA; George SJ; NejatyJahromy Y; Swartz JR; Britt RD A Radical Intermediate in Tyrosine Scission to the CO and CN- Ligands of FeFe Hydrogenase. Science 2013, 342, 472–475. [DOI] [PubMed] [Google Scholar]
- (10).Tao L; Pattenaude SA; Joshi S; Begley TP; Rauchfuss TB; Britt RD Radical SAM Enzyme HydE Generates Adenosylated Fe(I) Intermediates En Route to the [FeFe]-Hydrogenase Catalytic H-Cluster. Journal of the American Chemical Society 2020, 142, 10841–10848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Rao GD; Tao LZ; Britt RD Serine is the molecular source of the NH(CH2)(2) bridgehead moiety of the in vitro assembled FeFe hydrogenase H-cluster. Chemical Science 2020, 11, 1241–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Berggren G; Adamska A; Lambertz C; Simmons TR; Esselborn J; Atta M; Gambarelli S; Mouesca JM; Reijerse E; Lubitz W; Happe T; Artero V; Fonte-cave M Biomimetic assembly and activation of FeFe -hydrogenases. Nature 2013, 499, 66–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Britt RD; Rao G; Tao L Biosynthesis of the catalytic H-cluster of [FeFe] hydrogenase: the roles of the Fe–S maturase proteins HydE, HydF, and HydG. Chemical Science 2020, [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Dinis P; Suess DL; Fox SJ; Harmer JE; Driesener RC; De La Paz L; Swartz JR; Essex JW; Britt RD; Roach PL X-ray crystallographic and EPR spectroscopic analysis of HydG, a maturase in [FeFe]-hydrogenase H-cluster assembly. Proceedings of the National Academy of Sciences 2015, 112, 1362–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Kuchenreuther JM; Myers WK; Stich TA; George SJ; NejatyJahromy Y; Swartz JR; Britt RD A radical intermediate in tyrosine scission to the CO and CN- ligands of FeFe hydrogenase. Science 2013, 342, 472–475. [DOI] [PubMed] [Google Scholar]
- (16).Suess DL; Bürstel I; De La Paz L; Kuchenreuther JM; Pham CC; Cramer SP; Swartz JR; Britt RD Cysteine as a ligand platform in the biosynthesis of the FeFe hydrogenase H cluster. Proceedings of the National Academy of Sciences 2015, 112, 11455–11460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Suess DL; Pham CC; Bürstel I; Swartz JR; Cramer SP; Britt RD The radical SAM enzyme HydG requires cysteine and a dangler iron for generating an organometallic precursor to the [FeFe]-hydrogenase H-cluster. Journal of the American Chemical Society 2016, 138, 1146–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Pagnier A; Martin L; Zeppieri L; Nicolet Y; Fontecilla-Camps JC CO and CN-syntheses by FeFe -hydrogenase maturase HydG are catalytically differentiated events. Proceedings of the National Academy of Sciences of the United States of America 2016, 113, 104–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Rao G; Tao L; Suess DL; Britt RDA [4Fe–4S]-Fe (CO)(CN)-l-cysteine intermediate is the first organometallic precursor in [FeFe] hydrogenase H-cluster bioassembly. Nature chemistry 2018, 10, 555–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Nicolet Y; Zeppieri L; Amara P; Fontecilla-Camps JC Crystal structure of tryptophan lyase (NosL): evidence for radical formation at the amino group of tryptophan. Angewandte Chemie International Edition 2014, 53, 11840–11844. [DOI] [PubMed] [Google Scholar]
- (21).Shao Y; Gan Z; Epifanovsky E; Gilbert AT; Wormit M; Kussmann J; Lange AW; Behn A; Deng J; Feng X; Ghosh D; Goldey M; Horn PR; Jacobson LD; Kaliman I; Khaliullin RZ; Kuś T; Landau A; Liu J; Proynov EI; Rhee YM; Richard RM; Rohrdanz MA; Steele RP; Sundstrom EJ; III HLW; Zimmerman PM; Zuev D; Albrecht B; Alguire E; Austin B; Beran GJO; Bernard YA; Berquist E; Brandhorst K; Bravaya KB; Brown ST; Casanova D; Chang C-M; Chen Y; Chien SH; Closser KD; Crittenden DL; Diedenhofen M Jr.; R. A. D; Do H; Dutoi AD; Edgar RG; Fatehi S; Fusti-Molnar L; Ghysels A; Golubeva-Zadorozhnaya A; Gomes J; Hanson-Heine MW; Harbach PH; Hauser AW; Hohenstein EG; Holden ZC; Jagau T-C; Ji H; Kaduk B; Khistyaev K; Kim J; Kim J; King RA; Klunzinger P; Kosenkov D; Kowalczyk T; Krauter CM; Lao KU; Laurent AD; Lawler KV; Levchenko SV; Lin CY; Liu F; Livshits E; Lochan RC; Luenser A; Manohar P; Manzer SF; Mao S-P; Mardirossian N; Marenich AV; Maurer SA; Mayhall NJ; Neuscamman E; Oana CM; Olivares-Amaya R; O’Neill DP; Parkhill JA; Perrine TM; Peverati R; Prociuk A; Rehn DR; Rosta E; Russ NJ; Sharada SM; Sharma S; Small DW; Sodt A; Stein T; Stück D; Su Y-C; Thom AJ; Tsuchimochi T; Vanovschi V; Vogt L; Vydrov O; Wang T; Watson MA; Wenzel J; White A; Williams CF; Yang J; Yeganeh S; Yost SR; You Z-Q; Zhang IY; Zhang X; Zhao Y; Brooks BR; Chan GK; Chipman DM; Cramer CJ; III WAG; Gordon MS; Hehre WJ; Klamt A; III HFS; Schmidt MW; Sherrill CD; Truhlar DG; Warshel A; Xu X; Aspuru-Guzik A; Baer R; Bell AT; Besley NA; Chai J-D; Dreuw A; Dunietz BD; Furlani TR; Gwaltney SR; Hsu C-P; Jung Y; Kong J; Lambrecht DS; Liang W; Ochsenfeld C; Rassolov VA; Slipchenko LV; Subotnik JE; Voorhis TV; Herbert JM; Krylov AI; Gill PM; Head-Gordon M Advances in molecular quantum chemistry contained in the Q-Chem 4 program package. Molecular Physics 2015, 113, 184–215. [Google Scholar]
- (22).Case D; Belfon K; Ben-Shalom I; Brozell S; Cerutti D; T.E. Cheatham I; Cruzeiro V; Darden T; Duke R; Giambasu G; Gilson M; Gohlke H; Goetz A; Harris R; Izadi S; Izmailov S; Kasavajhala K; Kovalenko A; Krasny R; Kurtzman T; Lee T; LeGrand S; Li P; Lin C; Liu J; Luchko T; Luo R; Man V; Merz K; Miao Y; Mikhailovskii O; Monard G; Nguyen H; Onufriev A; Pan F; Pantano S; Qi R; Roe D; Roitberg A; Sagui C; Schott-Verdugo S; Shen J; Simmerling C; Skrynnikov NR; Smith J; Swails J; Walker R; Wang J; Wilson L; Wolf R; Wu X; Xiong Y; Xue Y; York D; Kollman P AMBER 2020, University of California, San Francisco. [Google Scholar]
- (23).Zhou Y; Wang S; Li Y; Zhang Y Methods in enzymology; Elsevier, 2016; Vol. 577; pp 105–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Seritan S; Bannwarth C; Fales BS; Hohenstein EG; Kokkila-Schumacher SIL; Luehr N; Snyder JW; Song C; Titov AV; Ufimtsev IS; Martínez TJ TeraChem: Accelerating electronic structure and ab initio molecular dynamics with graphical processing units. The Journal of Chemical Physics 2020, 152, 224110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Seritan S; Bannwarth C; Fales BS; Hohenstein EG; Isborn CM; Kokkila-Schumacher SIL; Li X; Liu F; Luehr N; Snyder JW Jr.; Song C; Titov AV; Ufimtsev IS; Wang L-P; Martínez TJ TeraChem: A graphical processing unit-accelerated electronic structure package for large-scale ab initio molecular dynamics. WIREs Computational Molecular Science n/a, e1494. [Google Scholar]
- (26).Wang L-P; Song C Geometry optimization made simple with translation and rotation coordinates. The Journal of Chemical Physics 2016, 144, 214108. [DOI] [PubMed] [Google Scholar]
- (27).Neese F Prediction of electron paramagnetic resonance g values using coupled perturbed Hartree–Fock and Kohn–Sham theory. The Journal of Chemical Physics 2001, 115, 11080–11096. [Google Scholar]
- (28).Neese F; Wennmohs F; Becker U; Riplinger C The ORCA quantum chemistry program package. The Journal of Chemical Physics 2020, 152, 224108. [DOI] [PubMed] [Google Scholar]
- (29).Salomon O; Reiher M; Hess BA Assertion and validation of the performance of the B3LYP* functional for the first transition metal row and the G2 test set. The Journal of Chemical Physics 2002, 117, 4729–4737. [Google Scholar]
- (30).Szilagyi RK; Winslow MA On the accuracy of density functional theory for iron—sulfur clusters. Journal of Computational Chemistry 2006, 27, 1385–1397. [DOI] [PubMed] [Google Scholar]
- (31).Weigend F; Ahlrichs R Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys 2005, 7, 3297–3305. [DOI] [PubMed] [Google Scholar]
- (32).Roy LE; Hay PJ; Martin RL Revised Basis Sets for the LANL Effective Core Potentials. Journal of Chemical Theory and Computation 2008, 4, 1029–1031. [DOI] [PubMed] [Google Scholar]
- (33).Zheng J; Xu X; Truhlar DG Minimally augmented Karlsruhe basis sets. Theoretical Chemistry Accounts 2011, 128, 295–305. [Google Scholar]
- (34).Jang H; Qiu Y; Hutchings ME; Nguyen M; Berben LA; Wang L-P Quantum chemical studies of redox properties and conformational changes of a four-center iron CO2 reduction electrocatalyst. Chem. Sci 2018, 9, 2645–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Wang J; Wolf RM; Caldwell JW; Kollman PA; Case DA Development and testing of a general amber force field. Journal of Computational Chemistry 2004, 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- (36).Chang CH; Kim K Density functional theory calculation of bonding and charge parameters for molecular dynamics studies on [FeFe] hydrogenases. Journal of chemical theory and computation 2009, 5, 1137–1145. [DOI] [PubMed] [Google Scholar]
- (37).Wang L-P; Martinez TJ; Pande VS Building Force Fields: An Automatic, Systematic, and Reproducible Approach. The Journal of Physical Chemistry Letters 2014, 5, 1885–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Wang L-P; McKiernan KA; Gomes J; Beauchamp KA; Head-Gordon T; Rice JE; Swope WC; Martínez TJ; Pande VS Building a more predictive protein force field: a systematic and reproducible route to AMBER-FB15. The Journal of Physical Chemistry B 2017, 121, 4023–4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Sayler RI; Stich TA; Joshi S; Cooper N; Shaw JT; Begley TP; Tantillo DJ; Britt RD Trapping and electron paramagnetic resonance characterization of the 5-dAdo radical in a radical S-adenosyl methionine enzyme reaction with a non-native substrate. ACS central science 2019, 5, 1777–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Sayler RI; Stich TA; Joshi S; Cooper N; Shaw JT; Begley TP; Tantillo DJ; Britt RD Trapping and Electron Paramagnetic Resonance Characterization of the 5-dAdo Radical in a Radical S-Adenosyl Methionine Enzyme Reaction with a Non-Native Substrate. ACS Central Science 2019, 5, 1777–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Driesener RC; Duffus BR; Shepard EM; Bruzas IR; Duschene KS; Coleman NJ-R; Marrison AP; Salvadori E; Kay CW; Peters JW, et al. Biochemical and kinetic characterization of radical S-adenosyl-L-methionine enzyme HydG. Biochemistry 2013, 52, 8696–8707. [DOI] [PubMed] [Google Scholar]
- (42).Amara P; Mouesca J-M; Bella M; Martin L; Saragaglia C; Gambarelli S; Nicolet Y Radical S-Adenosyl-l-methionine Tryptophan Lyase (NosL): How the Protein Controls the Carboxyl Radical• CO2–Migration. Journal of the American Chemical Society 2018, 140, 16661–16668. [DOI] [PubMed] [Google Scholar]
- (43).Villamena FA; Locigno EJ; Rockenbauer A; Hadad CM; Zweier JL Theoretical and experimental studies of the spin trapping of inorganic radicals by 5, 5-dimethyl-1-pyrroline N-oxide (DMPO). 1. Carbon dioxide radical anion. The Journal of Physical Chemistry A 2006, 110, 13253–13258. [DOI] [PubMed] [Google Scholar]
- (44).Favaudon V; Tourbez H; Houee-Levin C; Lhoste JM Carboxyl radical induced cleavage of disulfide bonds in proteins. A .gamma.-ray and pulse radiolysis mechanistic investigation. Biochemistry 1990, 29, 10978–10989. [DOI] [PubMed] [Google Scholar]
- (45).Lord RL; Baik M-H Why Does Cyanide Pretend to be a Weak Field Ligand in [Cr(CN)5]3-? Inorganic Chemistry 2008, 47, 4413–4420. [DOI] [PubMed] [Google Scholar]
- (46).Nakamura M Is Cyanide Really a Strong-Field Ligand? Angewandte Chemie International Edition 2009, 48, 2638–2640. [DOI] [PubMed] [Google Scholar]
- (47).Simón-Manso E; Kubiak CP Dinuclear Nickel Complexes as Catalysts for Electrochemical Reduction of Carbon Dioxide. Organometallics 2005, 24, 96–102, Publisher: American Chemical Society. [Google Scholar]
- (48).Appel AM; Bercaw JE; Bocarsly AB; Dobbek H; DuBois DL; Dupuis M; Ferry JG; Fujita E; Hille R; Kenis PJA; Kerfeld CA; Morris RH; Peden CHF; Portis AR; Ragsdale SW; Rauchfuss TB; Reek JNH; Seefeldt LC; Thauer RK; Waldrop GL Frontiers, Opportunities, and Challenges in Biochemical and Chemical Catalysis of CO2 Fixation. Chemical Reviews 2013, 113, 6621–6658, Publisher: American Chemical Society. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Costentin C; Robert M; Savéant J-M Catalysis of the electrochemical reduction of carbon dioxide. Chemical Society Reviews 2013, 42, 2423–2436. [DOI] [PubMed] [Google Scholar]
- (50).Shen J; Kortlever R; Kas R; Birdja YY; Diaz-Morales O; Kwon Y; Ledezma-Yanez I; Schouten KJP; Mul G; Koper MTM Electrocatalytic reduction of carbon dioxide to carbon monoxide and methane at an immobilized cobalt protoporphyrin. Nature Communications 2015, 6, 8177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Lin S; Diercks CS; Zhang Y-B; Kornienko N; Nichols EM; Zhao Y; Paris AR; Kim D; Yang P; Yaghi OM; Chang CJ Covalent organic frameworks comprising cobalt porphyrins for catalytic CO2 reduction in water. Science 2015, 349, 1208–1213. [DOI] [PubMed] [Google Scholar]
- (52).Gao S; Sun Z; Liu W; Jiao X; Zu X; Hu Q; Sun Y; Yao T; Zhang W; Wei S; Xie Y Atomic layer confined vacancies for atomic-level insights into carbon dioxide electroreduction. Nature Communications 2017, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Maiocco SJ; Arcinas AJ; Booker SJ; Elliott SJ Parsing redox potentials of five ferredoxins found within Thermotoga maritima. Protein Science 2019, 28, 257–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Gennaro A; Isse AA; Severin M-G; Vianello E; Bhugun I; Savéant J-M Mechanism of the electrochemical reduction of carbon dioxide at inert electrodes in media of low proton availability. J. Chem. Soc., Faraday Trans 1996, 92, 3963–3968. [Google Scholar]
- (55).Tao L; Pattenaude SA; Joshi S; Begley TP; Rauchfuss TB; Britt RD The radical SAM enzyme HydE generates adenosylated Fe (I) intermediates en route to the [FeFe]-hydrogenase catalytic H-cluster. Journal of the American Chemical Society 2020, [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Artiukhin DG; Neugebauer J Frozen-density embedding as a quasi-diabatization tool: Charge-localized states for spin-density calculations. The Journal of Chemical Physics 2018, 148, 214104. [DOI] [PubMed] [Google Scholar]
- (57).Rohac R; Martin L; Liu L; Basu D; Tao L; Britt RD; Rauchfuss TB; Nicolet Y Crystal Structure of the [FeFe]-Hydrogenase Maturase HydE Bound to Complex-B. Journal of the American Chemical Society 2021, 143, 8499–8508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Britt RD; Rao G; Tao L Biosynthesis of the catalytic H-cluster of [FeFe] hydrogenase: the roles of the Fe–S maturase proteins HydE, HydF, and HydG. Chemical Science 2020, 11, 10313–10323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.