

Abstract
Down syndrome (DS) is typically caused by triplication of chromosome 21. Phenotypically, DS presents with developmental, neurocognitive, and immune features. Epidemiologically, individuals with DS have less frequent viral infection, but when present, these infections lead to more severe disease. The potent antiviral cytokine Type I Interferon (IFN-I) receptor subunits IFNAR1 and IFNAR2 are located on chromosome 21. While increased IFNAR1/2 expression initially caused hyper-sensitivity to IFN-I, it triggered excessive negative feedback. This led to a hypo-response to subsequent IFN-I stimuli and an ensuing viral susceptibility in DS compared to control cells. Upregulation of IFNAR2 expression phenocopied the DS IFN-I dynamics independent of trisomy 21. CD14+ monocytes from individuals with DS exhibited markers of prior IFN-I exposure and had muted responsiveness to ex-vivo IFN-I stimulation. Our findings unveil oscillations of hyper and hypo-response to IFN-I in DS, predisposing to both lower incidence of viral disease and increased infection-related morbidity and mortality.
IN BRIEF
Individuals with Down syndrome have fewer viral infections, but when infected, they suffer from more severe disease. Malle et al. find that triplication of IFNAR1 and IFNAR2, the receptor subunits of the potent antiviral cytokine IFN-I, in DS results in hyper-active IFN-I signaling. The ensuing hyper-induction of IFNAR negative regulators suppresses subsequent IFN-I stimuli and effectively represses further antiviral defenses.
Graphical Abstract
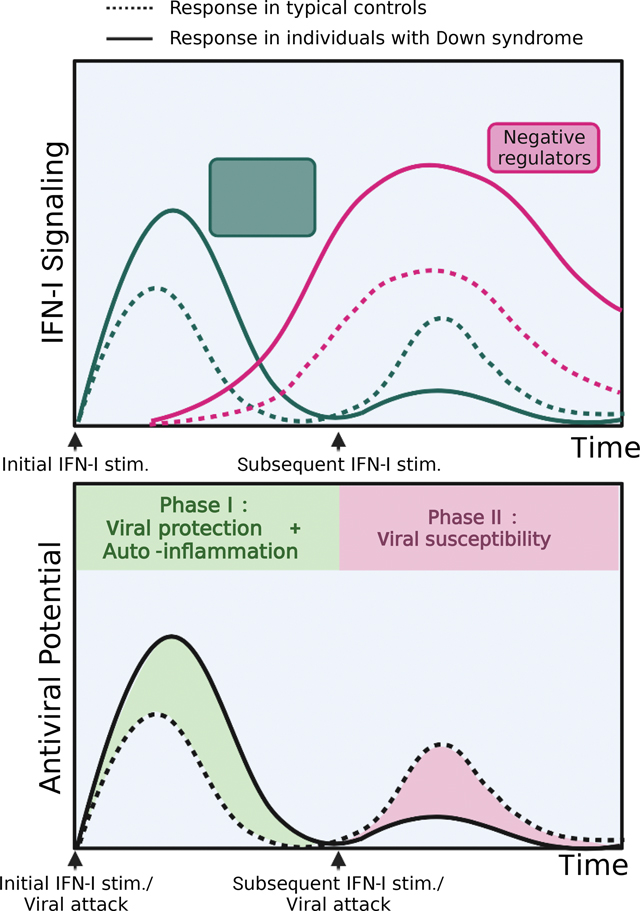
INTRODUCTION
Down syndrome (DS), or trisomy 21, is the most common chromosomal anomaly in the US, affecting 1 in 700 newborns(Mai et al., 2019). This syndrome affects multiple organ systems, causing a heterogenous clinical presentation which includes intellectual disability, developmental delays, congenital heart and gastrointestinal abnormalities, and Alzheimer’s disease in older individuals with DS(Bull, 2020). Recently, it has become clear that abnormal antiviral response is another important feature of DS. A recent study of the largest cohort of individuals with DS to date revealed protection from most infections in DS compared to non-DS controls, including influenza (IAV) (OR:0.62, p<0.0001), unspecified upper respiratory infections (OR:0.37, p<0.0001), mononucleosis (OR:0.32, p=0.0010), Herpes zoster (OR:0.26, p<0.0001), and intestinal infections (OR:0.50, p<0.0001)(Fitzpatrick et al., 2022). Despite this initial protection, there is extensive evidence that once infected, patients with DS are more likely to progress to severe disease including pneumonia (OR: 4.13–6.60), in particular viral pneumonia, acute respiratory distress syndrome, and sepsis(Bruijn et al., 2007; Uppal, Chandran and Potluri, 2015; Santoro et al., 2021; Fitzpatrick et al., 2022). Increased rates of hospitalization have been documented for influenza A virus (IAV), respiratory syncytial virus (RSV), and severe acute respiratory syndrome due to coronavirus (SARS-CoV-2) infections(Pérez-Padilla et al., 2010; Mitra et al., 2018; Malle et al., 2020, 2021). Infection-related mortality accounts for 20–40% of deaths in people with DS, compared to ~4.5% in the general population before the COVID-19 pandemic(Bcheraoui et al., 2018; O’Leary et al., 2018).
While the clinical phenotype of people with DS shows clear signs of immune disturbance, it has yet to be elucidated how a supernumerary chromosome 21 leads to dysregulation of viral defenses. The etiology is likely multi-factorial given the presence of an extra set of ~200 genes but delineating the mechanisms at play is essential to improve the care of people with DS. Studies on the adaptive immune system have shown a profound decrease in B cell numbers and altered B cell subsets, and also suggest that T cells can be over-activated and exhausted(Verstegen and Kusters, 2020). Evidence of innate immune dysfunction in DS has also started to emerge. Recently, studies have shown that the increased copy number of IFNAR1 and IFNAR2, which encode the two units of the type I interferon receptor (IFNAR), results in overactivation of the type I interferon (IFN-I) pathway in DS(Sullivan et al., 2016; Kong et al., 2020).
IFN-Is are potent pro-inflammatory cytokines that are essential to fight against viral infections(Taft and Bogunovic, 2018). To prevent overt inflammation, negative feedback is put in place: IFN-I signaling induces the production of negative regulators (such as USP18) that bind to the receptor, prevent further signaling, and restore homeostasis(François-Newton et al., 2011; Zhang et al., 2015). The hyper-response to IFN-I previously reported in DS is in line with the lower incidence of most viral infections described epidemiologically. However, an important question remains: why are people with DS more susceptible to infection-related morbidity and mortality? In this study, we interrogate how increased copy number of IFNAR1 and IFNAR2 contribute to IFN-I negative regulation, and we examine its effects on viral susceptibility. Our findings add to our understanding of the immune disturbances in DS and help to resolve the clinical paradox of concurrent initial viral protection and increased risk of infection-induced complications.
RESULTS
Increased IFNAR2 expression is sufficient for the hypersensitivity to IFN-I observed in DS
We first confirmed previous findings of increased gene dosage of IFNAR1 and IFNAR2 in DS. As expected, we observed higher expression of IFNAR1 and IFNAR2 in hTERT-immortalized fibroblasts derived from individuals with DS (DS) compared to those of healthy controls (HC), at both the mRNA and protein levels (Supplemental Fig 1A, B). This elevated receptor expression resulted in elevated proximal signaling in response to IFN-I stimulation, demonstrated by higher induction of phospho-STAT1 and phospho-STAT2 in DS compared to HC fibroblasts (Fig 1A), as expected(Sullivan et al., 2016; Kong et al., 2020). Non-canonical signaling, as measured by the induction of STAT3 phosphorylation, was also increased in DS compared to WT controls (Fig 1A).
Figure 1. Hyper-response to IFN-I in Down syndrome, or with increased expression of IFNAR2 alone.
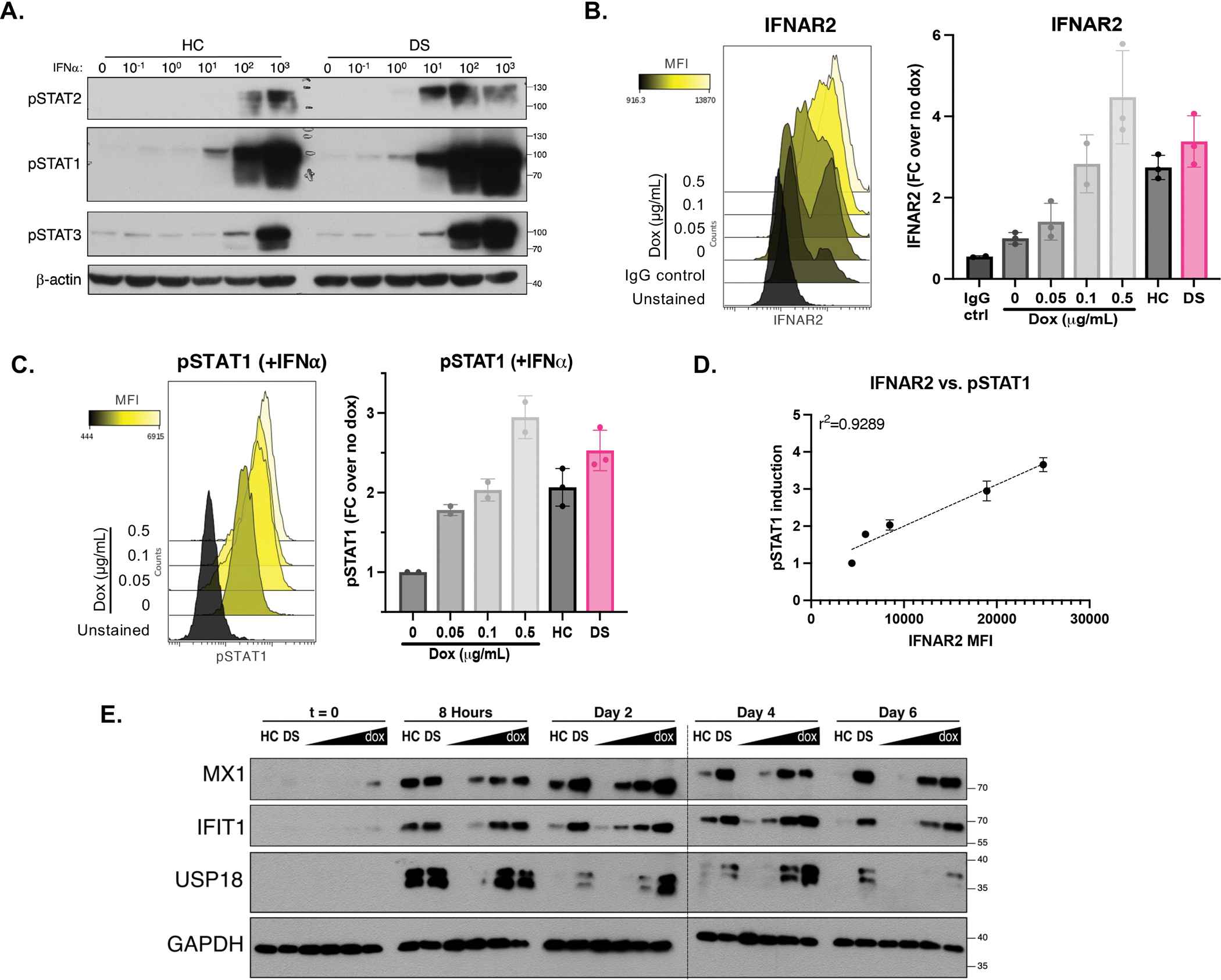
(A) Immunoblotting for STAT1, STAT2 and STAT3 phosphorylation in HC and DS hTERT-immortalized fibroblasts stimulated for 15 min with indicated doses of IFN-α (IU/mL). Result representative of fibroblasts derived from n=3 HCs and n=3 individuals with DS.
(B) Flow cytometry histogram and quantification of IFNAR2 expression in hTERT IFNAR2 knockouts complemented with doxycycline-inducible IFNAR2, treated with indicated amounts of doxycycline, run in triplicates.
(C) Flow cytometry histogram and quantification of STAT1 phosphorylation in hTERTs complemented with doxycycline-inducible IFNAR2, treated with increasing amounts of doxycycline (0, 0.05, 0.1, and 0.5 mg/mL) and stimulated with IFN-α for 15 minutes (1000 IU/mL), run in duplicates.
(D) Correlation of IFNAR2 expression and STAT1 phosphorylation in hTERTs complemented with dox-inducible IFNAR2. (r: Pearson correlation coefficient).
(E) Immunoblotting for ISG induction in hTERTs complemented with dox-inducible IFNAR2 treated with increasing amount of doxycycline followed by stimulation with IFN-α (10 IU/mL) for 8 hours, wash, and rest for indicated times. Result representative of fibroblasts derived from n=3 HCs and n=3 individuals with DS. * indicates samples were run on two separate gels.
For all results in the figure, bars represent the mean ± SD.
Downstream of the IFN-I signaling cascade, induction of IFN-stimulated genes (ISGs) MX1, IFI27, IFIT1, and RSAD2 was correspondingly higher in DS fibroblasts compared to HC fibroblasts (Supplemental Fig 1C, D). Of note, MX1 is located on chromosome 21 therefore its increased induction may not only be due to elevated IFN-I signaling, but also to its increased copy number in DS fibroblasts. While elevation of IFNAR expression and STAT phosphorylation were subtle (<2-fold), ISG induction was more pronounced (up to 6-fold) suggestive of expected signal transduction amplification downstream of the receptor. This further supports the notion that while the gene dosage of IFNAR1 and IFNAR2 is only 1.5x higher in trisomy 21 than in disomic controls, it can result in a profound hyper-response to IFN-I in individuals with DS. Furthermore, the elevation in ISG expression was not only elevated compared to controls at peak induction, but the presence of these ISGs was also sustained for up to a week after IFN-I stimulation compared to only 3 days in controls (Supplemental Fig 1E, F). Combined, these data not only confirm the IFNAR gene dosage effects in DS(Sullivan et al., 2016) but also further suggest that these affect non-canonical signaling and persist over time.
As there are over 200 genes that are triplicated in DS and could play a role in the hyper-response to IFN-I described above, we sought to delineate the impact of increased IFN-I receptor alone to this phenotype. We generated IFNAR2 knockouts in HC fibroblasts and transduced them with a two-lentivirus system: a construct expressing IFNAR2 under a tet-inducible promoter, and a doxycycline-dependent trans-activator (rtTA) (Supplemental Fig2A–D). This resulted in a cell line in which IFNAR2 amounts could be finely titrated with doxycycline treatment to match expression in HC and DS lines (Fig 1B). The intermediate dose of doxycycline (0.1 ug/mL) used corresponded to the HC surface expression, and the DS expression was intermediate between the intermediate (0.1 ug/mL) and high doses (0.5 ug/mL) of doxycycline (Fig 1B).
Increased dosage of IFNAR2 resulted in increased phopho-STAT1 in response to IFN-I (Fig 1C), leading to a strong correlation between IFNAR2 expression and induction of phospho-STAT1 in the presence of IFN-I (Fig 1D). At the ISG level, higher IFNAR2 expression was sufficient to replicate not only the short-term hyper-induction of MX1, IFIT1, and USP18 seen in DS, but also resulted in the long-term persistence of these ISGs at the protein level similar to what we determined in DS hTERTs (Fig 1E). While the induced IFNAR2 expression matched to HC and DS precisely corresponded to the respective levels of STAT1 phosphorylation, the ISGs induced were not perfectly matched which suggests that some other factors may be at play downstream of the receptor. In conclusion, independent of trisomy 21, increased IFNAR2 expression is sufficient to result in hyper-response to IFN-I.
Initial hyper-response to IFN-I underlies heightened IFN-I negative regulation
To prevent the potentially nefarious inflammatory effects of IFN-I, the pathway is tightly regulated by negative feedback mechanisms(Porritt and Hertzog, 2015). In the absence of this physiological process, significant pathology and even death occurs(Meuwissen et al., 2016; Alsohime et al., 2020). This regulatory break of IFN-I signaling is carried out by several proteins, USP18 being one of the most potent of these negative regulators(François-Newton et al., 2011). USP18 is induced by IFN-I and binds to IFNAR2, thereby preventing JAK1 phosphorylation and downstream STAT phosphorylation. As a result, cells “primed” with an initial IFN-I stimulus become refractory to further IFN-I stimulation(François-Newton et al., 2011; Zhang et al., 2015).
Given that naïve DS fibroblasts hyper-respond to IFN-I stimulation, which results in increased production of the negative regulator USP18 (Supplemental Fig 1C–F), we interrogated this negative feedback loop in DS. While naïve DS hTERT hyper-respond to IFN-I compared to control cells, when they were first “primed” with IFN-I, they in fact become hypo-responsive to a second IFN-I stimulus, as evidenced by decreased STAT1 and STAT2 phosphorylation compared to their HC counterparts (Fig 2A). IFNAR1 and IFNAR2 surface expression was not different between primed HC and DS fibroblasts (Supplemental Fig 3A), therefore this hypo-response is not mediated by differential receptor expression. The hypo-response is concomitant with elevated USP18 induction in primed DS cells compared to primed HC cells (Fig 2A). SOCS1, another known IFNAR negative regulator(Blumer et al., 2017), was not differentially induced in primed DS cells (Supplemental Fig 3B), which suggests that it is not a key player in the increased negative regulation we observed in DS. Therefore, increased sensitivity to IFN-I can trigger excessive negative feedback likely via increased USP18 induction, ultimately resulting in an immunosuppressive state.
Figure 2. Initial hyper-response to IFN-I underlies heightened IFN-I negative regulation.
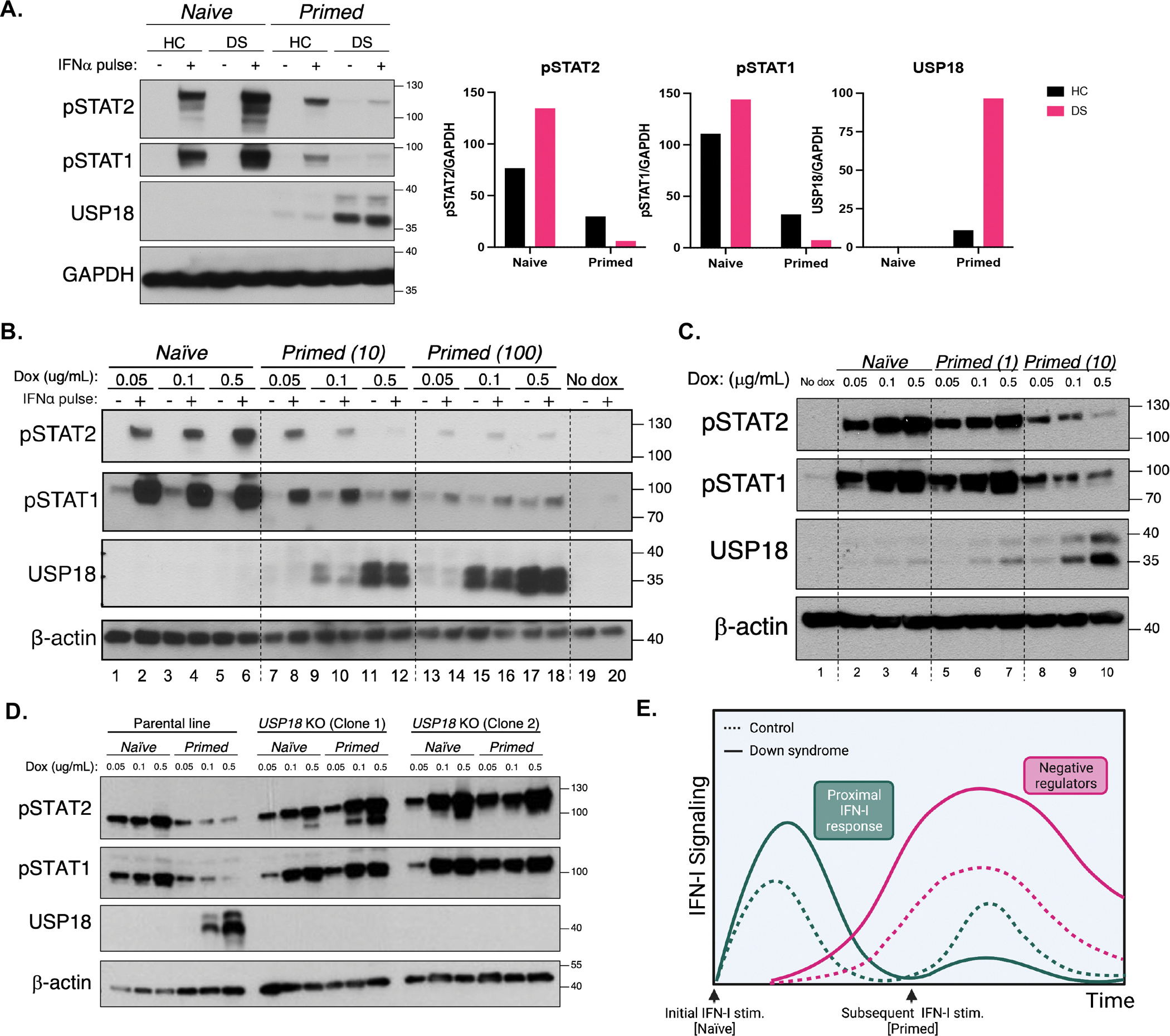
(A) Immunoblotting for STAT phosphorylation after stimulation for 15 min with IFN-α (100 IU/mL) of HC and DS hTERT-immortalized fibroblasts previously stimulated (Primed) or not (Naive) with a primary stimulus of IFN-α (10 IU/mL) for 12 h, washed, and allowed to rest for 36 h. Result representative of fibroblasts derived from n=3 HCs and n=3 individuals with DS.
(B) Immunoblotting for STAT phosphorylation after stimulation for 15 min with IFN-α (100 IU/mL) of IFNAR2 knockouts complemented with doxycycline-inducible IFNAR2, treated with indicated amounts of doxycycline and previously stimulated (Primed) or not (Naïve) with a primary stimulus of IFN-α (10 or 100 IU/mL). Results representative of n=3 independent experiments.
(C) Immunoblotting for STAT phosphorylation after stimulation for 15 min with IFN-α (100 IU/mL) of IFNAR2 knockouts complemented with doxycycline-inducible IFNAR2, treated with indicated amounts of doxycycline and previously stimulated (Primed) or not (Naïve) with a primary stimulus of IFN-α (10 or 100 IU/mL).
(D) Immunoblotting for STAT phosphorylation after stimulation for 15 min with IFN-α (100 IU/mL) of IFNAR2 knockouts complemented with doxycycline-inducible IFNAR2, treated with increasing concentrations of doxycycline and Primed (with 10 IU/mL IFN-α) or not (Naïve), in the presence (parental line) or absence (USP18 KO Clones 1 and 2) of endogenous USP18.
(E) Diagram of hyper- and hypo-responses to IFN-I over time with subsequent stimulations in HC and DS individuals.
See also Figure S3.
Importantly, IFNAR2 expression was sufficient to replicate this exacerbated refractory state. While IFNAR2 expression positively correlated with STAT1 and STAT2 phosphorylation in naïve cells (Fig 1C, Fig 2B Lanes 1–6), this relationship was inverted in primed cells: increasing IFNAR2 resulted in decreasing of phospho-STATs (Fig 2B Lanes 7–12, 2C Lanes 8–10). Like in DS fibroblasts, this heightened negative feedback is concomitant with the increase in USP18 expression in cells that express more IFNAR2 (Fig 2B, 2C). Furthermore, knocking out USP18 reverts this phenotype so that primed cells remain responsive to a second IFN-I stimulation in an IFNAR2-dependent fashion (Fig 2D). Therefore, USP18 is necessary do induce IFNAR2-dose-dependent refractoriness to IFN-I.
We further explored the dynamics of IFN-induced negative feedback by adjusting the dose of IFN-I priming. The factors that dictate the response to a secondary IFN-I stimulus are not solely the quantity of receptors expressed, but also the quantity of the ligands initially present. Priming with a low dose of IFN-I was not sufficient to induce significant negative regulation, regardless of IFNAR2 expression (Fig 2C Lanes 5–7). At this dose of priming (1 IU/mL), the positive correlation between IFNAR2 expression and STAT phosphorylation remained even at re-stimulation, which was only reversed with priming at a higher dose of IFN-I (10 IU/mL) (Fig 2C Lanes 8–10). On the other end of the spectrum, the threshold of negative regulation was passed regardless of IFNAR2 expression if cells were primed with a high-enough dose of IFN-I (100 IU/mL). This resulted in a similar remarkable hypo-response to the secondary stimulus regardless of the amount of IFNAR2 (Fig 2B Lanes 13–18). In conclusion, the induction of IFN-I negative feedback is a malleable process and depends on both the levels of receptor expressed and the dose of IFN-I priming (Supplemental Fig 3). In our system, we showed that intermediate IFN-I priming opens a window of stronger, temporal, immune suppression. This phenotype could be reversed with a short-term pulse with Tofacitinib, a Jak inhibitor, which suggests that negative regulation is enacted by an IFN-induced protein and that transient immunosuppression can rescue cellular responsiveness to IFN-I (Supplemental Fig 3C).
In all, our results confirm that higher expression of the IFN-I receptor can not only result in IFN-I mediated auto-inflammation in a naïve state, but that it can also predispose cells to become refractory to IFN-I, and thereby prolong the immunosuppressed state.
Myeloid cells from individuals with DS exhibit baseline IFN-I signaling and partial IFN-I desensitization.
While cell lines are useful tools to test cytokines responses in a highly controlled manner, ex vivo studies in blood and peripheral blood mononuclear cells (PBMCs) are perhaps more physiologically reflective of the in vivo phenotype, at least at the time of sampling. We first evaluated whole blood from individuals with DS and age-matched controls by CyTOF (Cytometry by Time-Of-Flight) for two cell surface-expressed ISGs, CD169/SIGLEC-1 and CD64/FcɣRI. Both of these ISGs were elevated in the myeloid cells of individuals with DS in the absence of any exogenous stimulation (Fig 3A, 3B), suggesting the presence of basal IFN-I signaling. Additionally, and in accordance with other studies(Kong et al., 2020), phospho-STAT1 was detected by flow cytometry in the absence of stimulation in the CD14+ monocytes of individuals with DS compared to their HC counterparts (Fig 3C), suggestive of some IFN-I signaling at the steady-state in people with DS.
Figure 3. Basal IFN-I signaling and increased IFN-I negative regulation in monocytes from individuals with Down syndrome ex vivo.
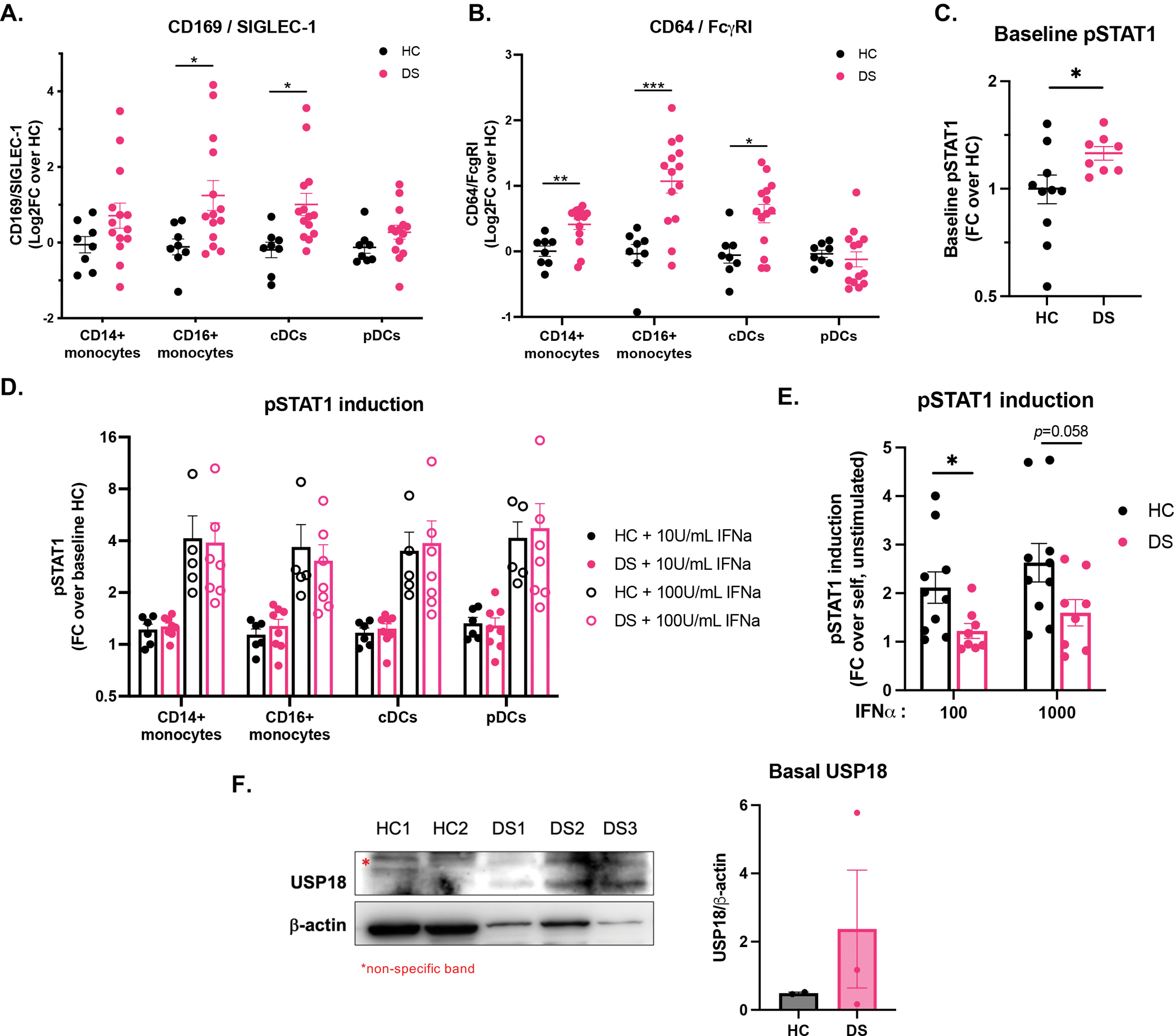
(A-B) CyTOF quantification of (A) CD169/SIGLEC-1 and (B) CD64/FcɣRI at baseline in CD14+ and CD16+ monocytes, and conventional (cDCs) and plasmacytoid dendritic cells (pDCs) from HC (n=8) and DS (n=14) whole blood.
(C) Flow cytometry quantification of baseline STAT1 phosphorylation in CD14+ monocytes from HCs (n=6) and individuals with DS (n=7), expressed as fold-change over baseline healthy control.
(D) CyTOF quantification of STAT1 phosphorylation in CD14+ and CD16+ monocytes, cDCs, and pDCs conventional (cDCs) from HC (n=5) and DS (n=7) whole blood stimulated for 15 minutes with 10 and 100 IU/mL IFN-α, expressed as fold-change over the average baseline of HCs.
(E) Flow cytometry quantification of STAT1 phosphorylation in CD14+ monocytes after stimulation of HC (n=6) and DS (n=7) PBMCs for 15 min with indicated amounts of IFN-α (IU/mL ), expressed as fold-change over each sample unstimulated.
(F) Immunoblotting and quantification for basal USP18 in total PBMCs from individuals with DS (n=3) and age-matched controls (n=2). * denotes a non-specific band.
For all results in the figure, bars represent the mean ± SEM. Significance assessed by unpaired t tests, ns denotes p>0.05, *p ≤ 0.05; **p ≤ 0.005; ***p ≤ 0.0005.
See also Figure S3.
In line with this seemingly “lightly primed” state, there was no notable hyper-response to IFN-I in DS myeloid cells, reflective of the plasticity of the system (Fig 3D). The IFN-I-stimulated induction of phospho-STAT1 over baseline was in fact lower in CD14+ monocytes from people with DS compared to that of HCs (Fig 3E). Further downstream, the induction of ISGs MX1, IFIT1, and IFI27 was also decreased in PBMCs derived from individuals with DS compared to HCs (Supplemental Fig 3E). In accordance with these data, immunoblotting for USP18 revealed that this negative regulator was present at steady state in PBMCs derived from individuals with DS but not that of HCs (Fig 3F), which indicate that these cells may be refractory to further IFN-I stimulation at baseline.
We postulate that basal phospho-STAT1, increased CD169 and CD64 surface expression, and increased amount of USP18 are suggestive of prior exposure to IFN-I, so that these cells are likely in a partial refractory state and thus not hyper-responding to subsequent IFN-I stimulation.
Increased IFN-I negative regulation underlies viral susceptibility.
Individuals with DS are at higher risk of severe viral infections. This has been documented for RSV, IAV, and most recently for SARS-CoV-2(Pérez-Padilla et al., 2010; Mitra et al., 2018; Malle et al., 2020; Clift et al., 2021). This risk is significant (adjusted hazard ratio of 10.39 for death from COVID-19)(Clift et al., 2021), but this viral susceptibility remains less stark than that of patients with complete deficiencies of IFNAR1, IFNAR2, STAT1, and STAT2, which often result in death from various viral insults(Boisson-Dupuis et al., 2012; Hambleton et al., 2013; Duncan et al., 2015; Taft and Bogunovic, 2018; Hernandez et al., 2019). In DS, while anatomical defects and dysregulated adaptive immune responses likely play a significant role in this susceptibility(Verstegen and Kusters, 2020; De Lausnay et al., 2021), we sought to determine if excessive IFN-I inflammation leading to more profound IFN-I-induced desensitization could be a contributing factor in this clinical phenotype.
To assess if refractoriness to IFN-I had an impact of viral susceptibility, we infected HC fibroblasts and HC fibroblasts constitutively over-expressing the IFN-I negative regulator USP18 (HC+USP18) with IAV-GFP. While HC and HC+USP18 cells are infected at similar rates at baseline, stimulating the cells with IFN-I confers sizeable protection to HCs, while HC+USP18 cells only have a modest, ~10% reduction in infection (Fig 4A). Thus, the IFN-I refractory state established by USP18 results in susceptibility to IAV infection.
Figure 4. Increased IFN-I negative regulation underlies viral susceptibility.
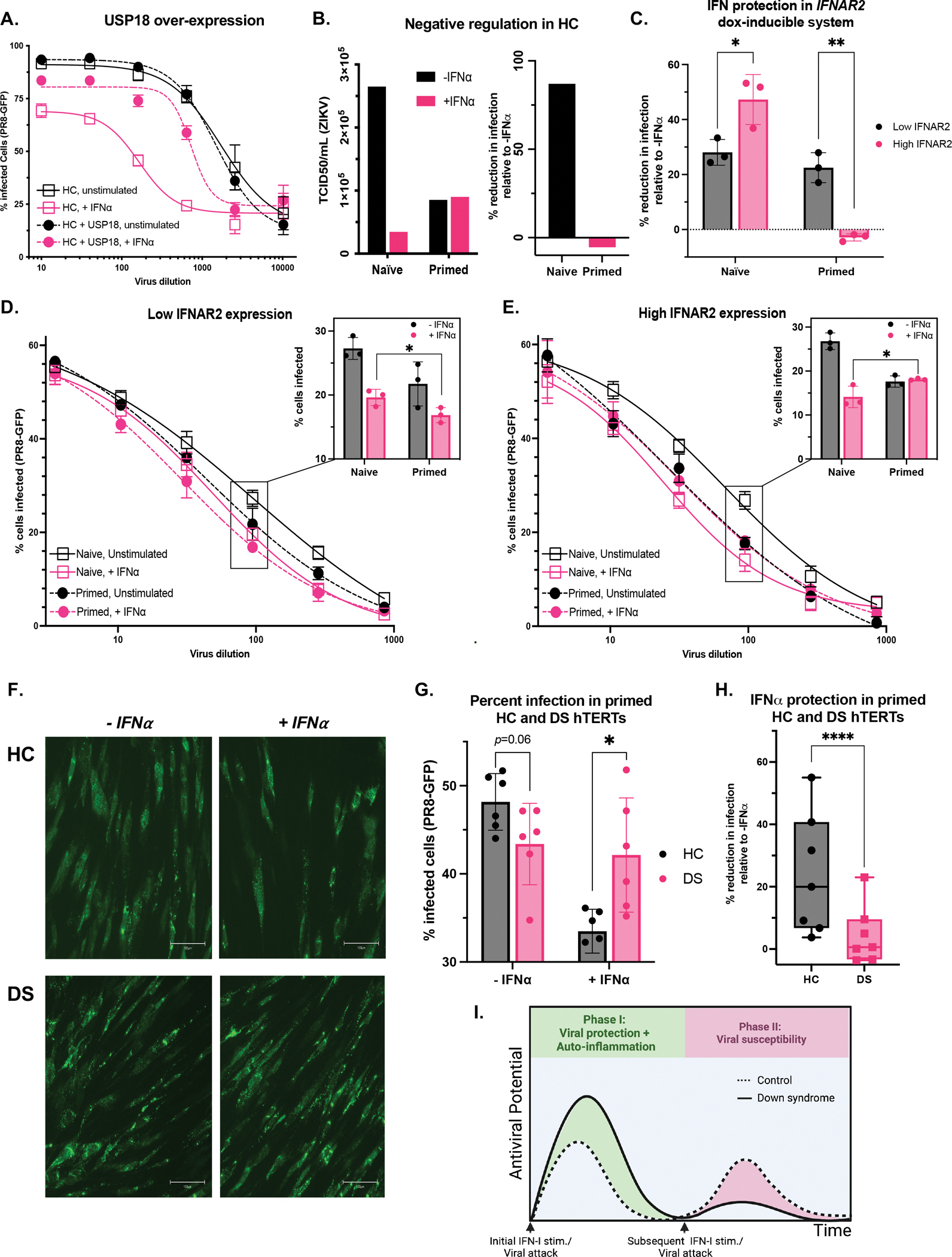
(A) HC hTERTs transduced or not with USP18 were stimulated overnight with IFN-α (10 IU/mL) and infected with PR8-GFP for 8 hours.
(B) ZIKV was titrated on HC HTERTs that were naïve or primed for 8h with IFN-α (100 IU/mL), rested for 2.5 days, and restimulated with IFN-α (100 IU/mL) for 15 min, and infected with ZIKV 8 hours later. To calculate the TCID50, infected wells were considered GFP+ if they displayed a mean intensity above the mean of the un-infected wells.
(C-E) hTERT IFNAR2 knockouts complemented with doxycycline-inducible IFNAR2 were treated with 0.05 (“Low IFNAR2”) or 0.5 (“High IFNAR2”) μg/mL doxycycline. Cells were initially treated (Primed) or not (Naïve) with a primary stimulus of IFN-α (10 IU/mL) for 8 h, allowed to rest for 4.5 days in DMEM+Dox, and restimulated with IFN-α (1000 IU/mL) for 15-min followed by 8-hr rest and overnight infection with PR8-GFP. (C) Percent reduction in infection by IFN-I stimulation at 1:100 viral dilution. Raw percent infection at all viral dilutions tested (curves) and at 1:100 viral dilution (bar graphs) at (D) low and (E) high IFNAR2 levels.
(F-H) HC and DS hTERTs were initially treated with a primary stimulus of IFN-α (10 IU/mL) for 8 h, allowed to rest for 2.5 days, and restimulated with IFN-α (100 IU/mL) for 15-min followed by 8-hr rest and overnight infection with PR8-GFP. (F) PR8-GFP fluorescence captured at 20x (scale bar denotes 100μm) and (G) Raw percent infection at 1:100 viral dilution. (H) Percent reduction in infection by IFN-I stimulation at all viral dilutions tested. Results of n=2 independent experiments.
(I) Diagram of auto-inflammatory and immuno-suppressed states over time with subsequent IFN-α stimulations in HC and DS individuals.
For all results in the figure, bars represent the mean ± SD. Significance assessed by unpaired t tests, ns denotes p>0.05, *p ≤ 0.05; **p ≤ 0.005; ***p ≤ 0.0005.
We then tested whether the endogenous refractory state induced in HC cells, triggered as a part of naturally occurring balance of protection from overt inflammation, can similarly have an impact on viral susceptibility. We infected naïve and primed HC hTERT fibroblasts with ZIKV in absence or presence of an IFN-I stimulus. The IFN-I stimulus appreciably reduced infectivity in naïve cells (86% reduction in infection) (Fig 4B). Primed, unstimulated cells are still protected compared to naïve unstimulated cells, which shows that IFN-I confers some long-term resistance to cells, albeit waning as expected. Importantly, these primed cells do not better control the virus after a second IFN-I stimulus (Fig 4B). Despite the lingering protection from the IFN-I prime, these cells are more susceptible to the virus than naïve, stimulated cells. Thus, IFN-I priming prevents cells from mounting an antiviral state equivalent to that of naïve cells, making them more vulnerable to viral infection.
Next, we took advantage of our inducible system to study whether increased IFNAR2 expression, which we’ve shown results in greater refractoriness to IFN-I, could result in more viral susceptibility. With low IFNAR2 expression, IFN-I treatment elicits a similar amount of viral protection in naïve and primed cells (28% and 22% decrease, respectively, compared to the unstimulated control) (Fig 4C, black boxes). By contrast, with high IFNAR2 expression, while IFN-I stimulation results in greater protection in their naïve state (47% reduction in infection), IFN-I has no additional effect in primed cells (no reduction in infection) (Fig 4C, pink boxes). This reflects the initial hyper-response to IFN-I in naïve cells and subsequent hypo-response in primed cells that we described with increasing IFNAR2 expression (Fig 2B). The heightened negative feedback in these cells thus results in an inability to harness IFN-I to potentiate an antiviral response.
As previously seen for ZIKV protection in HCs, in our IFNAR2 system priming the cells with IFN-I results in long-lasting protection, decreasing the cellular infectivity compared to baseline (Fig 4D, 4E, solid black lines compared to dashed black lines). The ability to respond to a second stimulus, however, dictates whether this priming puts cells at higher risk of infection than naïve cells. Given their ability to remain responsive to subsequent IFN-I stimuli, low-IFNAR2 cells mount the greatest antiviral resistance with the combined effects of the prime and the restimulation (Fig 4D, pink dashed line). By contrast, with high amounts of IFNAR2 (DS-matched), primed cells that received an additional IFN-I stimulus (Fig 4E, pink dashed line) are more susceptible to IAV than naïve cells stimulated with IFN-I (Fig 4E, pink solid line and inlay, p=0.04). Therefore, increasing IFNAR2 expression results in heightened viral susceptibility in the context of repeated IFN-I stimuli or a continuous IFN-I inflammatory state, likely present in the clinical setting.
Finally, we turned to primed HC and DS hTERTs to test if the increased negative regulation observed in DS (Fig 2A) results in increased viral susceptibility. Although DS hTERTs are initially more sensitive to IFN-I, upon priming these cells exhibit resistance against IAV that is comparable to HC although it did trend higher (p=0.06) (Fig 4F, 4G). Perhaps more importantly, these DS primed cells cannot mount a second response to IFN-I, resulting in equal infectivity of IAV of about 43% of cells on DS background whether primed, or primed and additionally IFN-I-stimulated (Fig 4G). In contrast, HC cell lines were protected via priming alone which resulted in infectivity of IAV of about 48%, but when additional IFN-I was administered IAV infectivity was further reduced to about 33% (Fig 4G). Additionally, at all IAV dilutions tested, IFN-I stimulation confers additional protection to primed HC hTERTs (24% reduction in infection on average compared to unstimulated cells) while DS hTERTs have a muted response to this second IFN-I stimulation (4% further reduction in infection) (Fig 4H). In conclusion, in the presence of additional IFN-I, primed HC cells are better protected from IAV infection than their DS counterparts (p=0.01). Therefore, the IFN-I desensitization triggered in DS results in increased viral susceptibility compared to HCs.
DISCUSSION
Given the inborn nature of trisomy 21, its clinical immune presentation, and the mounting molecular evidence of genetic causation of immune pathology, DS belongs in the group of diseases known as inborn errors of immunity (IEIs). Its main distinguishing feature from previously described IEIs is that it is not monogenic but rather monochromosomal. Furthermore, DS encompasses both autoinflammation and immune suppression.
The Mendelian IEIs provide valuable insight to frame our understanding of disturbances in the IFN-I pathway. IEIs causing chronic IFN-I signaling, termed Type I Interferonopathies (IFNopathies), cause a severe phenotype characterized by neurological disturbances and inflammatory features typically affecting the skin and/or the lungs(Rodero and Crow, 2016; Lee-Kirsch, 2017), and in some cases provide protection from viral infections. Previous studies have provided evidence that increased IFNAR gene dosage in DS result in inflammatory features that are comparable to a mild Type I IFNopathy, and recent epidemiologic data demonstrates that people with DS are protected from viral infections. However, DS also overlaps clinically with IEIs caused by defects in the IFN-I pathway, as individuals with DS are more susceptible to complications due to viral disease. Indeed, loss-of-function (LOF) mutations in IFNAR1, IFNAR2, IRF7, IRF3, and STAT1 all result in severe viral susceptibility(Andersen et al., 2015; Duncan et al., 2015; Michael J. Ciancanelli, Sarah X. L. Huang, Priya Luthra, Hannah Garner, Yuval Itan, Stefano Volpi, Fabien G. Lafaille, Céline Trouillet, Mirco Schmolke, Randy A. Albrecht, Elisabeth Israelsson, Hye Kyung Lim, Melina Casadio, Tamar Hermesh, Lazaro Lorenzo, Christopher F. Basler and Shen-Ying Zhang, Hans-Willem Snoeck, 2015; Hernandez et al., 2019; Zhang et al., 2020). DS therefore corresponds to a mild form of both IFN-I GOF and LOF phenotypes.
Evidence stemming from these monogenic IEI suggests that both excessive and insufficient IFN-I are pathogenic, and that perturbations in duration and regulation of IFN-I signaling can also cause disease. The increase in IFNAR expression in DS puts individuals at risk of the negative effects of IFN-I at both ends of the spectrum. We show that the hyper-active IFN-I signaling cascade causes increased induction of the IFNAR negative regulator USP18, in turn hyper-suppressing any subsequent response to IFN-I and effectively repressing further antiviral responses. Together with known defects in adaptive immunity in DS in both B and T cell compartments(Verstegen and Kusters, 2020) as well as anatomical anomalies of the respiratory tract such as laryngomalacia and tracheomalacia(De Lausnay et al., 2021), which all likely contribute to the susceptibility to infections in DS, the dampening of the innate antiviral response we describe here may explain why people with DS are more prone to severe viral infections. DS is therefore, in part, an inflammation-induced error of immunity.
While the mechanisms at play are still largely unknown, there is evidence in the general population that excessive and ill-timed IFN-I signaling can worsen infectious disease. Administration of recombinant IFN-I as a therapeutic in viral infections has repeatedly proven ineffective if administered at the inappropriate time or dose, including for HIV, HCV, and SARS-CoV-2 among others (Asmuth et al., 2010; Teijaro, 2016; Kalil et al., 2021). In a cohort of patients with SARS, circulating IFN-I and ISG levels correlated with disease severity(Cameron et al., 2007). Murine studies demonstrate that IFN-I is protective early in infection but later becomes pathogenic(Channappanavar et al., 2016, 2019; Park and Iwasaki, 2020), which was also recently reported in humans in the treatment of SARS-CoV-2, as IFN-β improved recovery time if administered early in the course of disease but proved ineffective or even deleterious when given at advanced stages of disease(Monk et al., 2021; WHO Solidarity Trial Consortium, 2021). These findings are in accordance with the notion that timing of IFN-I is key as it can be protective against viral infection but IFN-I stimulation in “primed” cells is detrimental to viral control. For instance, administration of recombinant IFN-I before viral peak is protective in a murine model of Middle East Respiratory Syndrome (MERS), while late treatment results in nefarious inflammation and lethal pneumonia(Channappanavar et al., 2019). The role that IFN-I negative feedback plays in these observations remains to be investigated.
As both aspects of the typical physiologic IFN-I response are amplified in DS, an intermittent short-circuiting of IFNAR signaling via JAK inhibition or IFNAR blocking may help restore homeostasis. Put another way, transient immunosuppression may actually restore the ability of individuals with DS to fight off viral disease, while ameliorating their hyperinflammation. Our experimental data supports this notion, which warrants further investigation ex vivo and in vivo. So far, JAK inhibitor treatment is promising in curbing inflammatory symptoms in DS(Rachubinski et al., 2019; Pham et al., 2021), but it remains to be studied if this treatment improves individuals with DS’ ability to fend off viruses.
LIMITATIONS OF THE STUDY
We acknowledge that important additional information surrounding IFN-I signaling in DS could be obtained from tissue biopsies at steady state, as well as by further ex vivo studies testing the response to IFN-I in virally infected individuals with DS. Furthermore, follow-up evaluations of the IFN-I response in the adaptive arm of the immune system and its impact on cell and antibody-mediated immunity are needed to fully explain epidemiological findings of infection-related morbidity and mortality in DS.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact.
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dusan Bogunovic (dusan.bogunovic@mssm.edu).
Materials availability.
All plasmids and cell lines used in this study are available from the lead contact.
Data and code availability.
The data that support the findings of this study are available in the article and supplementary materials.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human samples
The study was approved under the IRB protocol at Mount Sinai Health System (MSHS) (IRB-18–00638/ STUDY-18–00627). Patients were approached either in person at the hospital or via email obtained through the NIH’s DS-Connect ® national registry (dsconnect.nih.gov). Recruited patients were 60% male, 40% female, 15.3 years mean age (range: 1.5–38 years). All patient data were de-identified. Written informed consent for all individuals in this study was provided in compliance with an institutional review board protocol. All uninfected patient samples were drawn in the context of an outpatient routine visit and patients exhibited no signs of infection (fever, runny nose, cough, sore throat). From each patient, blood was drawn into a Cell Preparation Tube with sodium heparin (BD Vacutainer). PBMCs were isolated from using Ficoll separation and stored at −80°C until use. Whole blood was stimulated or not with cytokine and fixed using Proteomic Stabilizer PROT1 (SmartTube) and frozen at −80°C.
Cell lines
DS- and HC-derived primary dermal fibroblasts were obtained from ATCC (CCL54, CRL2088) and Coriell (AG08942, GM04616, GM03440, GM08447) and immortalized with human telomerase (hTERT) (2 male and 1 female line for both HC and DS). Constitutively-expressing USP18 cells were previously generated in the lab (Marta NEJM). hTERT fibroblasts, HEK293Ts, and A549 cells were cultured in DMEM containing 10% fetal bovine serum (FBS) (Invitrogen), 1% GlutaMAX (GIBCO), and 1% penicillin/streptomycin (GIBCO). All cells were cultured at 37°C and 10% CO2. All cell lines were routinely tested for mycoplasma contamination with the MycoAlert PLUS Mycoplasma Detection Kit (Lonza) according to the manufacturer’s instructions.
METHOD DETAILS
Cloning
IFNAR2-pMET7 (generously provided by Sandra Pellegrini) was subcloned with In-Fusion (Takara) into a lentiviral compatible vector with a TETon 3G (TRE3G) promoter and BFP expressed from a separate constitutive promoter (generously provided by Ben Tenoever). The TETon system was combined with a separate lentiviral compatible vector containing the reverse tetracycline-controlled transactivator (rtTA) (Addgene: 66810).
Lentiviral particles were generated by co-transfection of TETon-IFNAR2 or rtTA and pCAGGS-VSV-G, pCMV-Gag/Pol by CaCl2 transfection of HEK293Ts. Supernatants were collected 48 h later, purified, and transferred to target cells with polybrene. Cells were selected with Hygromycin (200 μg/ml) and sorted for BFP expression.
CRISPR editing
CRISPR editing was performed with Alt-® S.p. Cas9 Nuclease V3 (IDT 1081058) and Alt-R crRNA guide RNAs (IDT) transfected into hTERTs with a 4D-Nucleofector (Lonza) using the manufacturer’s optimized protocol for primary cells (P3 solution). Single-cell clones from CRISPR-generated KOs were isolated via limiting dilution on a feeder layer of irradiated hTERTs. KOs were screened by Flow cytometry and Western blot for the absence of IFNAR2 and USP18 (Fig 1E, Supplemental Fig 2B) and competence of signaling in response to IFNa2b (Supplemental Fig 2C).
| Targeted gene | Guide sequence |
|---|---|
| IFNAR2 | ATTTCCGGTCCATCTTATCA |
| USP18 | CATTACGAACACCTGAATCA |
RNA isolation and quantitative PCR
All cytokine stimulations were performed as indicated with IFN-α 2b (Intron-A) in complete DMEM. RNA was extracted from hTERT-immortalized fibroblasts (Qiagen RNeasy) complete PBMCs (Direct-zol RNA Microprep) and reverse-transcribed (ABI High-Capacity Reverse Transcription). The expression of ISGs (MX1, IFIT1, RSAD2, IFI27, and USP18), relative to the 18S or GAPDH housekeeper gene, was analyzed by TaqMan quantitative real-time PCR (TaqMan Universal Master Mix II with uracil-DNA glycosylases) on a Roche LightCycler 480 II. The relative levels of ISG expression were calculated by the ΔΔCT method, relative to the mean values for the mock-treated controls.
IFNAR negative regulation
To test IFN refractoriness, hTERTs were seeded overnight at 50,000 cells/mL in 12-well plates and stimulated (Primed) or not (Naïve) with 10 IU/mL IFNα2b for 8–16 hours, washed 3 times with PBS, and allowed to rest in DMEM for 36 hours. For Jak inhibition (Primed + TOFA), cells were treated with overnight IFNα2b and Tofacitinib (50mM) was added to the wells 30 minutes after the initiation of the treatment. Cells were then washed 3 times with PBS and allowed to rest in DMEM for 36 hours. Cells were then re-stimulated with 10 or 100 IU/mL IFNα2b as indicated for 15 minutes followed by immunoblotting. In the dox-inducible IFNAR2 system, cells were cultured in DMEM+doxycycline for 48 hours before the prime, and the prime and rest were also performed in DMEM+doxycycline. All experiments were performed with n=3 biological replicates.
Protein assays
Whole-cell extracts for immunoblotting were prepared by incubating cells for 20 min in RIPA lysis buffer (Thermo Fisher Scientific) with 50 mM dithiothreitol and Protease/Phosphatase inhibitor cocktail (Cell Signaling Technology). Protein was quantified with Micro BCA Protein Assay (Thermo Fisher Scientific). Immunoblotting was performed using the Bio-Rad Western blot workflow. Membranes were blocked in 5% BSA for primary antibodies or 5% nonfat dry milk for secondary antibodies. Antibodies used were STAT1 (Santa Cruz Biotechnology), STAT2 (Millipore), phospho-Tyr 701 STAT1 (Cell Signaling Technology), phospho-Tyr 689 STAT2 (Cell Signaling Technology), USP18 (Cell Signaling Technology), IFIT1 (Cell Signaling Technology), MX1 (Abcam), SOCS1 (Thermo Fisher Scientific), β-actin (ABclonal), and GAPDH (Millipore). Signal was detected with enhanced chemiluminescence detection reagent (ECL or SuperSignal West pico, Thermo Fisher Scientific) by film development or capturing on ImageQuant 800 Fluor imaging system (Cytiva). For all representative immunoblots shown, n=3 separate experiments were carried out.
Flow cytometry
Phospho-STAT1 staining: Whole blood from individuals with DS and healthy controls were subject to Ficoll gradient to collect the mononuclear cell layer (PBMC) and frozen. For direct stimulation, thawed PBMCs were incubated in complete RPMI overnight, washed, stained with live-dead in PBS for 10 min at 37°C, and finally were stimulated for 15 min with indicated amounts of IFNα2b. For staining, cells were washed 2 times with 0.5% BSA and surface-stained on ice for 30 minutes (CD16-AF647, CD14-AF488, CD3-BV510, CD19-AF700, CD56-BV711, all from Biolegend) followed by fixation/permeabilization in 90% ice-cold methanol and staining with PE anti-phospho-STAT1 Y701 (1:25, BD).
IFNAR1 and IFNAR2 staining: hTERT-immortalized fibroblasts were scraped off in 5mM EDTA, washed, and stained with live-dead in PBS for 30 min on ice. They were then washed and stained with anti-IFNAR1 (clone AA3, courtesy of Sandra Pellegrini in Supplemental Fig 1A and clone MARI-5A3 from Millipore Sigma for Supplemental Fig 3A) or anti-IFNAR2 (PBL) for 2 hours on ice. The cells were washed and stained with biotin-conjugated rat anti-mouse IgG (H+L) (Thermo Fisher) for 40 min on ice, followed by PE-conjugated Streptavidin for 10 min on ice.
Flow cytometry was acquired on a Cytek Aurora or BD LSR Fortessa, and data were analyzed with Cytobank (https://www.cytobank.org/).
Mass cytometry
Whole blood from the patient and healthy controls were stimulated for 15 min with as indicated with IFN-α2b (Intron-A) in RPMI or mock-treated, fixed using Proteomic Stabilizer PROT1 (SmartTube) and frozen at −80°C. Fixed whole blood was stained and analyzed by mass cytometry (CyTOF) at the Human Immune Monitoring Center of the Icahn School of Medicine at Mt. Sinai. Samples were barcoded then stained together with antibodies against selected surface markers for 30 min on ice. Cells were then washed and fixed, resuspended in diH2O containing EQ Four Element Calibration Beads (Fluidigm), and acquired on a CyTOF2 Mass Cytometer (Fluidigm). Data files were normalized by using a bead-based normalization algorithm (CyTOF software, Fluidigm) and debarcoded using CD45 gating. The gated populations were visualized in lower dimensions using viSNE in Cytobank and manually gated based on the previously described gating scheme(Geanon et al., 2020).
Viral challenges
Influenza A Virus challenge: Cells were exposed to PR8-GFP (Influenza A/PR/8/34 (PR8) virus (H1N1), generously provided by Adolfo Garcia-Sastre) overnight at indicated dilutions from stock. Following the incubation period, cells were fixed and permeabilized with the BD Fixation/Permeabilization kit (BD). Cells were fixed for 20 min at 4°C and stained with DAPI (1:10,000 in 1XPerm/Wash buffer) for 20 min at RT. Cells were then washed and left in PBS. DAPI and GFP signals were acquired on a Celigo Imaging Cytometer (Nexcelom) and quantified in CellProfiler (McQuin et al., 2018). Experiments were carried out with n=3 biological replicates. Zika Virus challenge: Cells were exposed to ZIKV (strain PRVABC59, GenBank: KU501215.1) for one hour, washed three times, and allowed to rest overnight. Supernatants from these cells were then harvested and diluted at 1:100 dilution followed by 1:3 serial dilutions and placed on previously plated Veros (ATCC). After 7 days, Veros were fixed and permeabilized with the BD Fixation/Permeabilization kit (BD). Cells were fixed for 20 min at 4°C and stained with anti-Flavivirus Group Antigen 4G2, 1:1000 (Millipore) for 1 hour at RT. Cells were then washed and stained with secondary (1:10,000) and DAPI for 45 minutes at RT. Cells were then washed and DAPI and AF647 signals were acquired on a Celigo Imaging Cytometer (Nexcelom).
INCLUSION AND DIVERSITY
We worked to ensure gender balance in the recruitment of human subjects. We worked to ensure ethnic or other types of diversity in the recruitment of human subjects. We worked to ensure that the study questionnaires were prepared in an inclusive way. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list. The author list of this paper includes contributors from the location where the research was conducted who participated in the data collection, design, analysis, and/or interpretation of the work.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-STAT1 Clone C-111 | Santa Cruz Biotechnology | Cat No. sc417 |
| Rabbit anti-STAT2 | Millipore Sigma | Cat No. 06502 |
| Rabbit anti-phospho-Tyr 701-STAT1 Clone 58D6 | Cell Signaling Technology | Cat No. 9167 |
| Rabbit anti-phospho-Tyr-689-STAT2 Clone D3P2P | Cell Signaling Technology | Cat No. 88410 |
| Rabbit anti-USP18 Clone D4E7 | Cell Signaling Technology | Cat No. 4813 |
| Rabbit anti-β-actin Clone AC026 | ABclonal | Cat No. AC026 |
| Mouse anti-GAPDH Clone 6C5 | Millipore | Cat No. MAB374 |
| Rabbit anti-IFIT1 Clone D2X9Z | Cell Signaling Technology | Cat No. 14769 |
| Rabbit polyclonal anti-MX1 | Abcam | Cat No. ab95926 |
| Rabbit polyclonal anti-SOCS1 | Thermo Fisher Scientific | Cat No. PA5-27239 |
| Goat anti-mouse IgG HRP-conjugated | Southern Biotech | Cat No. 101005 |
| Goat anti-rabbit IgG HRP-conjugated | Southern Biotech | Cat No. 403005 |
| anti-CD16 AF647-conjugated Clone 3G8 | Biolegend | Cat No. 302023 |
| anti-CD56 BV711-conjugated Clone 5.1H11 | Biolegend | Cat No. 362541 |
| anti-CD19 AF700-conjugated Clone HIB19 | Biolegend | Cat No. 302225 |
| anti-CD14 AF488-conjugated Clone M5E2 | Biolegend | Cat No. 301811 |
| anti-CD3 BV510-conjugated Clone OKT3 | Biolegend | Cat No. 317331 |
| anti-Stat1 (pY701) PE-conjugated, Clone 4A | BD | Cat No. 612564 |
| anti-IFNAR1, Clone MARI-5A3 | Millipore Sigma | Cat. No. 04-151 |
| Anti-IFNAR1, Clone AA3 | Laboratory of Sandra Pellegrini | N/A |
| anti-IFNAR2, Clone MMHAR-2 | PBL | Cat No. 21385-1 |
| rat anti-mouse IgG (H+L), biotin conjugated | Thermo Fisher | Cat No. 13-4013-85 |
| Streptavidin, PE-conjugated | Thermo Fisher | Cat No. S866 |
| anti-CD45 89Y-conjugated Clone HI30 | Fluidigm | Cat No.3089003B |
| anti-CD57 113In-conjugated Clone HCD57 | Biolegend | Cat No.322302 |
| anti-CD11c 115In-conjugated Clone Bu15 | Biolegend | Cat No.337202 |
| anti-IgD 141Pr-conjugated Clone IA6-02 | Biolegend | Cat No.348202 |
| anti-CD19 142Nd-conjugated Clone HIB19 | Biolegend | Cat No.302202 |
| anti-CD45RA 143Nd-conjugated Clone HI100 | Biolegend | Cat No.304102 |
| anti-CD141 144Nd-conjugated Clone M80 | Biolegend | Cat No.344102 |
| anti-CD4 145Nd-conjugated Clone RPA-T4 | Biolegend | Cat No.300502 |
| anti-CD8 146Nd-conjugated Clone RPA-T8 | Biolegend | Cat No.301002 |
| anti-CD20 147Sm-conjugated Clone 2H7 | Biolegend | Cat No.302302 |
| anti-CD16 148Nd-conjugated Clone 3G8 | Biolegend | Cat No.302014 |
| anti-CD127 149Sm-conjugated Clone A019D5 | Fluidigm | Cat No.3149011B |
| anti-CD1c 150Nd-conjugated Clone L161 | Biolegend | Cat No.331502 |
| anti-CD123 151Eu-conjugated Clone 6H6 | Biolegend | Cat No.306002 |
| anti-CD66b 152Sm-conjugated Clone G10F5 | Biolegend | Cat No.305102 |
| anti-CD86 154Sm-conjugated Clone IT2.2 | Biolegend | Cat No.305410 |
| anti-CD27 155Gd-conjugated Clone O323 | Biolegend | Cat No.302802 |
| anti-CD33 158Gd-conjugated Clone WM53 | Biolegend | Cat No.303402 |
| anti-CD24 159Tb-conjugated Clone ML5 | Biolegend | Cat No.311102 |
| anti-CD14 160Gd-conjugated Clone M5E2 | Biolegend | Cat No.301810 |
| anti-CD56 161Dy-conjugated Clone B159 | BD Biosciences | Cat No.555513 |
| anti-CD169 162Dy-conjugated Clone 7-239 | Biolegend | Cat No.346002 |
| anti-CD69 164Dy-conjugated Clone FN50 | Biolegend | Cat No.310902 |
| anti-CD64 165Ho-conjugated Clone 10.1 | Biolegend | Cat No.305047 |
| anti-CD3 168Er-conjugated Clone UCHT1 | Biolegend | Cat No.300402 |
| anti-CD38 170Er-conjugated Clone HB-7 | Biolegend | Cat No.356602 |
| anti-CD161 171Yb-conjugated Clone HP-3G10 | Biolegend | Cat No.339902 |
| anti-HLADR 174Yb-conjugated Clone L243 | Biolegend | Cat No.307602 |
| anti-pSTAT5 147 Sm-conjugated Clone 47 | Fluidigm | Cat No.3147012A |
| anti-pSTAT6 149 Sm-conjugated Clone 18/P-Stat6 | Fluidigm | Cat No.3149004A |
| anti-pSTAT1 153 Eu-conjugated Clone 4a | Fluidigm | Cat No.3153005A |
| anti-pp38 156 Gd-conjugated Clone D3F9 | Fluidigm | Cat No.3156002A |
| anti-pSTAT3 158 Gd-conjugated Clone 4/P-Stat3 | Fluidigm | Cat No.3158005A |
| anti-pMAPKAP2 159 Tb-conjugated Clone 27B7 | Fluidigm | Cat No.3159010A |
| anti-STAT3 165 Ho-conjugated Clone 124H6 | Fluidigm | Cat No.3173003A |
| anti-STAT1 169 Tm-conjugated Clone 10C4B40 | Biolegend | Cat No.661002 |
| anti-pERK 171 Yb-conjugated Clone D13.14.4E | Fluidigm | Cat No.3171010A |
| anti-pS6 175 Lu-conjugated Clone N7-548 | Fluidigm | Cat No.3175009A |
| anti-Flavivirus Group Antigen, Clone D1-4G2-4-15 | Millipore | MAB10216 |
| Goat anti Mouse IgG (H+L) Secondary Antibody, Alexa Fluor 647 | Thermo Fisher | Cat No. A21235 |
| Bacterial and Virus Strains | ||
| DH5-Alpha Competent E. Coli | Molecular Cloning Laboratories | Cat No. DA-196 |
| PR8-GFP, Influenza A/PR/8/34 (PR8) virus (H1N1) | Laboratory of Adolfo Garcia-Sastre | N/A |
| ZIKV, strain PRVABC59, GenBank: KU501215.1 | Laboratory of Matthew Evans | N/A |
| Biological Samples | ||
| Human whole blood samples | Various institutions | N/A |
| HC-derived primary dermal fibroblasts | ATCC | Cat No. CRL2088 |
| HC-derived primary dermal fibroblasts | Coriell institute | Cat No. GM03440, GM08447 |
| DS-derived primary dermal fibroblasts | ATCC | Cat No. CCL54 |
| DS-derived primary dermal fibroblasts | Coriell institute | Cat No. AG08942, GM04616 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Intron-A Recombinant Interferon Alpha-2b | Merck Pharmaceuticals | Cat No. NDC0085057102 |
| Proteomic Stabilizer Prot1 | SMART TUBE Inc | Cat No. 501351691 |
| Heparin | Sigma | Cat No. 201060 |
| Osmium tetroxide (99.9%) | ACROS organics | Cat No. 191180010 |
| Discovery Ultra antibody block | Roche | Cat No. 760-4204 |
| Pierce ECL Western Blotting Substrate | Thermo Fisher Scientific | Cat No. 32106 |
| Pierce SuperSignal West Pico PLUS Chemiluminescent Substrate | Thermo Fisher Scientific | Cat No. 34580 |
| RIPA Lysis and Extraction Buffer | Thermo Fisher Scientific | Cat No. 89901 |
| Protease/Phosphatase Inhibitor Cocktail | Cell Signaling Technologies | Cat No. 5872 |
| Macherey-Nagel RNA Isolation, RA1 Lysis Buffer | Thermo Fisher Scientific | Cat No. 10335832 |
| Cytofix/Cytoperm Fixation/Permeabilization Solution | BD | Cat No. 554714 |
| Doxycycline hyclate | Sigma-Aldrich | Cat No. D9891 |
| Alt-R S.p. Cas9 Nuclease V3 | IDT | Cat No. 1081058 |
| Histopaque 1077 | Millipore Sigma | Cat No. 10771-500 |
| Critical Commercial Assays | ||
| Direct-zol RNA Microprep | Zymo Research | Cat No. R2060 |
| RNeasy RNA Isolation Kit | Qiagen | Cat No. 74106 |
| Applied Biosystems High-Capacity cDNA Reverse Transcription Kit | Thermo Fisher Scientific | Cat No. 4368814 |
| TaqMan Universal Master Mix II with UNG | Thermo Fisher Scientific | Cat No. 4440039 |
| Infusion HD | Takara Bio | Cat No. 638909 |
| CloneAmp HiFi PCR Premix | Takara Bio | Cat No. 639298 |
| MycoAlert PLUS Mycoplasma Detection Kit | Lonza | Cat No. LT07-703 |
| Invitrogen Taq polymerase | Thermo Fisher | Cat No. 10342020 |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | ATCC CRL-3216 |
| HC-derived primary dermal fibroblasts | ATCC | Cat No. CRL2088 |
| HC-derived primary dermal fibroblasts | Coriell institute | Cat No. GM03440, GM08447 |
| DS-derived primary dermal fibroblasts | ATCC | Cat No. CCL54 |
| DS-derived primary dermal fibroblasts | Coriell institute | Cat No. AG08942, GM04616 |
| Oligonucleotides | ||
| MX1 mRNA TaqMan FAM Hs200895608_m1 | Thermo Fisher | Cat No. 4331182 |
| IFI27 mRNA TaqMan FAM Hs01086373_g1 | Thermo Fisher | Cat No. 4351370 |
| IFIT1 mRNA TaqMan FAM Hs03027069_s1 | Thermo Fisher | Cat No. 4331182 |
| USP18 mRNA TaqMan FAM Hs00276441_m1 | Thermo Fisher | Cat No. 4331182 |
| RSAD2 mRNA TaqMan FAM Hs00369813_m1 | Thermo Fisher | Cat No. 4351370 |
| IFNAR2 CRISPR gRNA ATTTCCGGTCCATCTTATCA | IDT | N/A |
| USP18 CRISPR gRNA CATTACGAACACCTGAATCA | IDT | N/A |
| Recombinant DNA | ||
| pMET7-IFNAR2 | Laboratory of Sandra Pellegrini | N/A |
| IFNAR2_TETonBFP_TRE3G | This paper | N/A |
| Reverse Tetracycline-Controlled Transactivator (rtTA) | Addgene | Plasmid No. 66810 |
| pCAGGS-VSV-G | This paper | N/A |
| pCMV-Gag/Pol | This paper | N/A |
| Software and Algorithms | ||
| Cytobank | Beckman Coulter | N/A |
| FlowJo | Becton Dickinson Company | N/A |
| Fiji | ImageJ | N/A |
| Cell Profiler | Broad Institute | N/A |
| GraphPad Prism 9 | GraphPad Software | N/A |
HIGHLIGHTS.
Triplication of IFNAR1 and IFNAR2 in DS leads to hyper-active IFN-I response in vitro
Excessive IFN-I signaling triggers negative feedback and ensuing viral susceptibility
Increased IFNAR2 expression alone is sufficient to replicate DS IFN-I phenotype
Monocytes from individuals with DS have basal IFN-I activation and muted IFN-I response
ACKNOWLDEGEMENTS
We thank all individuals and their families for their participation. We thank Adeeb Rahman, Daniel Geanon, and Geoffrey Kelly from the Human Immune Monitoring Center at the Icahn School of Medicine for their technical assistance. We also thank Sandra Pellegrini for sharing reagents. This study was funded by the National Institute of Allergy and Infectious Diseases Grants R01AI150300, R01AI150300–01S1. LM was supported by T32 training grant T32HD075735.
Footnotes
DECLARATIONS OF INTEREST
D. Bogunovic is the founder of Lab11 Therapeutics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Alsohime F et al. (2020) ‘JAK Inhibitor Therapy in a Child with Inherited USP18 Deficiency’, New England Journal of Medicine. Massachusetts Medical Society, 382(3), pp. 256–265. doi: 10.1056/NEJMoa1905633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen LL et al. (2015) ‘Functional IRF3 deficiency in a patient with herpes simplex encephalitis’, The Journal of Experimental Medicine. The Rockefeller University Press, 212(9), p. 1371. doi: 10.1084/JEM.20142274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asmuth DM et al. (2010) ‘Safely, tolerability, and mechanisms of antiretroviral activity of pegylated interferon alfa-2a in HIV-1monoinfected participants: A phase II clinical trial’, Journal of Infectious Diseases. Oxford University Press, 201(11), pp. 1686–1696. doi: 10.1086/652420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bcheraoui C el et al. (2018) ‘Trends and Patterns of Differences in Infectious Disease Mortality Among US Counties, 1980–2014’, JAMA. American Medical Association, 319(12), pp. 1248–1260. doi: 10.1001/JAMA.2018.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumer T et al. (2017) ‘SOCS1 is an inducible negative regulator of interferon λ (IFN-λ)–induced gene expression in vivo’, The Journal of Biological Chemistry. American Society for Biochemistry and Molecular Biology, 292(43), p. 17928. doi: 10.1074/JBC.M117.788877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisson-Dupuis S et al. (2012) ‘Inborn errors of human STAT1: allelic heterogeneity governs the diversity of immunological and infectious phenotypes’, Current Opinion in Immunology. Elsevier, 24(4), p. 364. doi: 10.1016/J.COI.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruijn M et al. (2007) ‘High incidence of acute lung injury in children with Down syndrome’, Intensive Care Medicine. Springer, 33(12), pp. 2179–2182. doi: 10.1007/s00134-007-0803-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull MJ (2020) ‘Down syndrome’, New England Journal of Medicine. Edited by Ropper AH. Massachusetts Medical Society, 382(24), pp. 2344–2352. doi: 10.1056/NEJMra1706537. [DOI] [PubMed] [Google Scholar]
- Cameron MJ et al. (2007) ‘Interferon-mediated immunopathological events are associated with atypical innate and adaptive immune responses in patients with severe acute respiratory syndrome’, Journal of virology. J Virol, 81(16), pp. 8692–8706. doi: 10.1128/JVI.00527-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Channappanavar R et al. (2016) ‘Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice’, Cell Host and Microbe. Cell Press, 19(2), pp. 181–193. doi: 10.1016/j.chom.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Channappanavar R et al. (2019) ‘IFN-I response timing relative to virus replication determines MERS coronavirus infection outcomes’, Journal of Clinical Investigation. American Society for Clinical Investigation, 129(9), pp. 3625–3639. doi: 10.1172/JCI126363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clift AK et al. (2021) ‘COVID-19 Mortality Risk in Down Syndrome: Results From a Cohort Study of 8 Million Adults’, Annals of Internal Medicine. American College of Physicians, pp. M20–4986. doi: 10.7326/M20-4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan CJA et al. (2015) ‘Human IFNAR2 deficiency: Lessons for antiviral immunity’, Science translational medicine. Sci Transl Med, 7(307). doi: 10.1126/scitranslmed.aac4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick V et al. (2022) ‘Prevalence of Infectious Diseases Among 6078 Individuals With Prevalence of Infectious Diseases Among 6078 Individuals With Down Syndrome in the United States’, Journal of Patient-Centered Research and Reviews. Environmental Public Health Commons, 9(1), pp. 1–17. doi: 10.17294/2330-0698.1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- François-Newton V et al. (2011) ‘USP18-Based Negative Feedback Control Is Induced by Type I and Type III Interferons and Specifically Inactivates Interferon α Response’, PLoS ONE. Edited by Mossman KL. Public Library of Science, 6(7). doi: 10.1371/journal.pone.0022200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geanon D et al. (2020) ‘A Streamlined CyTOF Workflow To Facilitate Standardized Multi-Site Immune Profiling of COVID-19 Patients’, medRxiv. Cold Spring Harbor Laboratory Preprints. doi: 10.1101/2020.06.26.20141341. [DOI] [Google Scholar]
- Hambleton S et al. (2013) ‘STAT2 deficiency and susceptibility to viral illness in humans’, PNAS. Proc Natl Acad Sci, 110(8), pp. 3053–3058. doi: 10.1073/pnas.1220098110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez N et al. (2019) ‘Inherited IFNAR1 deficiency in otherwise healthy patients with adverse reaction to measles and yellow fever live vaccines’, The Journal of experimental medicine. J Exp Med, 18(9), p. 21. doi: 10.1084/jem.20182295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalil AC et al. (2021) ‘Efficacy of interferon beta-1a plus remdesivir compared with remdesivir alone in hospitalised adults with COVID-19: a double-bind, randomised, placebo-controlled, phase 3 trial’, The Lancet Respiratory Medicine. Elsevier Ltd, 9(12), pp. 1365–1376. doi: 10.1016/S2213-2600(21)00384-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong X-F et al. (2020) ‘Three Copies of Four Interferon Receptor Genes Underlie a Mild Type I Interferonopathy in Down Syndrome’, Journal of Clinical Immunology. doi: 10.1007/s10875-020-00803-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lausnay M et al. (2021) ‘Pulmonary complications in children with Down syndrome: a scoping review’, Paediatric Respiratory Reviews. Elsevier BV. doi: 10.1016/j.prrv.2021.04.006. [DOI] [PubMed] [Google Scholar]
- Lee-Kirsch MA (2017) ‘The Type I Interferonopathies’, Annu Rev Med, (68), pp. 297–315. doi: 10.1146/annurev-med-050715-104506. [DOI] [PubMed] [Google Scholar]
- Mai CT et al. (2019) ‘National population-based estimates for major birth defects, 2010–2014’, Birth Defects Research. John Wiley and Sons Inc., 111(18), pp. 1420–1435. doi: 10.1002/bdr2.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malle L et al. (2020) ‘Individuals with Down syndrome hospitalized with COVID-19 have more severe disease’, Genetics in Medicine. doi: 10.1038/s41436-020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malle L et al. (2021) ‘Atypical Inflammatory Syndrome Triggered by SARS-CoV-2 in Infants with Down Syndrome’, Journal of clinical immunology. J Clin Immunol, 41(7), pp. 1457–1462. doi: 10.1007/S10875-021-01078-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen MEC et al. (2016) ‘Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome.’, The Journal of experimental medicine. Rockefeller University Press, 213(7), pp. 1163–74. doi: 10.1084/jem.20151529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciancanelli Michael J., Huang Sarah X. L., Luthra Priya, Garner Hannah, Itan Yuval, Volpi Stefano, Lafaille Fabien G., Trouillet Céline, Schmolke Mirco, Albrecht Randy A., Israelsson Elisabeth, Hye Kyung Lim Melina Casadio, Hermesh Tamar, Lazaro Lorenzo A, Basler Christopher F., F. G. and Zhang Shen-Ying, Snoeck Hans-Willem, J.-L. C. (2015) ‘Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency’, Science. doi: 10.1126/science.aaa3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra S et al. (2018) ‘Hospitalization for Respiratory Syncytial Virus in Children with Down Syndrome Less than 2 Years of Age: A Systematic Review and Meta-Analysis’, The Journal of pediatrics. J Pediatr, 203, pp. 92–100.e3. doi: 10.1016/J.JPEDS.2018.08.006. [DOI] [PubMed] [Google Scholar]
- Monk PD et al. (2021) ‘Safety and efficacy of inhaled nebulised interferon beta-1a (SNG001) for treatment of SARS-CoV-2 infection: a randomised, double-blind, placebo-controlled, phase 2 trial’, The Lancet Respiratory Medicine. Lancet Publishing Group, 9(2), pp. 196–206. doi: 10.1016/S2213-2600(20)30511-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary L et al. (2018) ‘Early death and causes of death of people with Down syndrome: A systematic review’, Journal of Applied Research in Intellectual Disabilities, 31(5), pp. 687–708. doi: 10.1111/jar.12446. [DOI] [PubMed] [Google Scholar]
- Park A and Iwasaki A (2020) ‘Type I and Type III Interferons - Induction, Signaling, Evasion, and Application to Combat COVID-19’, Cell Host and Microbe. doi: 10.1016/j.chom.2020.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Padilla R et al. (2010) ‘Pandemic (H1N1) 2009 Virus and Down syndrome patients’, Emerging Infectious Diseases, 16(8), pp. 1312–1314. doi: 10.3201/eid1608.091931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham AT et al. (2021) ‘JAK inhibition for treatment of psoriatic arthritis in Down syndrome’, Rheumatology. doi: 10.1093/rheumatology/keab203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porritt RA and Hertzog PJ (2015) ‘Dynamic control of type I IFN signalling by an integrated network of negative regulators’, Trends in Immunology. Elsevier Ltd, 36(3), pp. 150–160. doi: 10.1016/j.it.2015.02.002. [DOI] [PubMed] [Google Scholar]
- Rachubinski AL et al. (2019) ‘CASE REPORT Janus kinase inhibition in Down syndrome: 2 cases of therapeutic benefit for alopecia areata’, JAAD Case Reports, 5, pp. 365–367. doi: 10.1016/j.jdcr.2019.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodero MP and Crow YJ (2016) ‘Type I interferon-mediated monogenic autoinflammation: The type I interferonopathies, a conceptual overview’, Journal of Experimental Medicine. Rockefeller University Press, 213(12), pp. 2527–2538. doi: 10.1084/jem.20161596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro SL et al. (2021) ‘Pneumonia and respiratory infections in Down syndrome: A scoping review of the literature’, American Journal of Medical Genetics, Part A. John Wiley and Sons Inc, 185(1), pp. 286–299. doi: 10.1002/AJMG.A.61924. [DOI] [PubMed] [Google Scholar]
- Sullivan KD et al. (2016) ‘Trisomy 21 consistently activates the interferon response’, eLife. doi: 10.7554/eLife.16220.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taft J and Bogunovic D (2018) ‘The Goldilocks Zone of Type I IFNs: Lessons from Human Genetics’, The Journal of Immunology. The American Association of Immunologists, 201(12), pp. 3479–3485. doi: 10.4049/jimmunol.1800764. [DOI] [PubMed] [Google Scholar]
- Teijaro JR (2016) ‘Type I interferons in viral control and immune regulation’, Current Opinion in Virology. Elsevier, 16, p. 31. doi: 10.1016/J.COVIRO.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uppal H, Chandran S and Potluri R (2015) ‘Risk factors for mortality in Down syndrome’, Journal of Intellectual Disability Research. Blackwell Publishing Ltd, 59(9), pp. 873–881. doi: 10.1111/jir.12196. [DOI] [PubMed] [Google Scholar]
- Verstegen RHJ and Kusters MAA (2020) ‘Inborn Errors of Adaptive Immunity in Down Syndrome’, Journal of Clinical Immunology. Springer, pp. 791–806. doi: 10.1007/s10875-020-00805-7. [DOI] [PubMed] [Google Scholar]
- WHO Solidarity Trial Consortium (2021) ‘Repurposed Antiviral Drugs for Covid-19 — Interim WHO Solidarity Trial Results’, New England Journal of Medicine. Massachusetts Medical Society, 384(6), pp. 497–511. doi: 10.1056/nejmoa2023184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q et al. (2020) ‘Inborn errors of type I IFN immunity in patients with life-threatening COVID-19’, Science. American Association for the Advancement of Science, 370(6515). doi: 10.1126/science.abd4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X et al. (2015) ‘Human intracellular ISG15 prevents interferon-α/β over-amplification and auto-inflammation’, Nature. Nature Publishing Group, 517(7532), pp. 89–93. doi: 10.1038/nature13801. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available in the article and supplementary materials.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.