

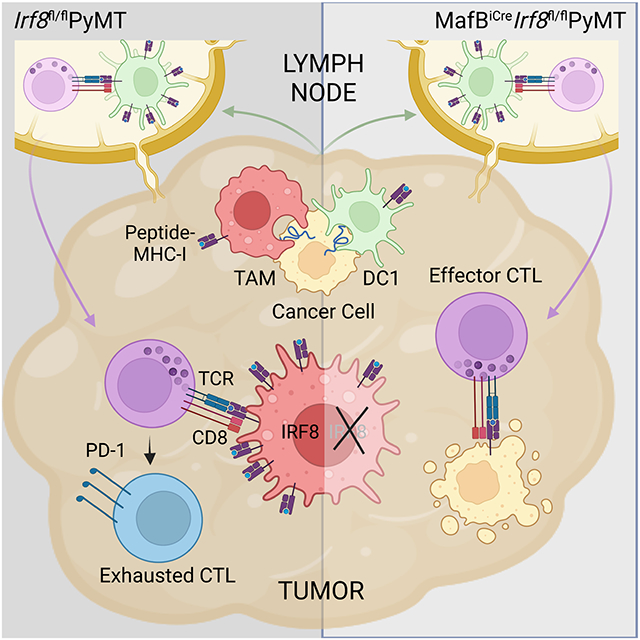
Summary
Tumors are populated by antigen presenting cells (APCs), including macrophage subsets with distinct origins and functions. Here, we examined how cancer impacts mononuclear phagocytic APCs in a murine model of breast cancer. Tumors induced expansion of monocyte-derived tumor-associated macrophages (TAMs) and activation of type 1 dendritic cells (DC1s), both of which expressed and required the transcription factor interferon regulatory factor-8 (IRF8). While DC1s mediated cytotoxic T lymphocyte (CTL) priming in tumor-draining lymph nodes, TAMs promoted CTL exhaustion in the tumor, and IRF8 was required for TAM ability to present cancer cell antigen. TAM-specific IRF8 deletion prevented exhaustion of cancer cell-reactive CTLs and suppressed tumor growth. Tumors from patients with immune-infiltrated renal cell carcinoma had abundant TAMs that expressed IRF8 and were enriched for an IRF8 gene expression signature. Furthermore, the TAM-IRF8 signature co-segregated with CTL exhaustion signatures across multiple cancer types. Thus, CTL exhaustion is promoted by TAMs via IRF8.
Keywords: Tumor-associated macrophage, antigen presentation, IRF8, T cell exhaustion
Graphical Abstract
eTOC blurb:
Macrophages of distinct origin and function populate tumors. Nixon et al. examine how cancer impacts mononuclear phagocytic antigen presenting cells and find that monocyte-derived tumor-associated macrophages (TAMs) present cancer cell antigen and drive cytotoxic T cell exhaustion, promoting tumor growth in a manner dependent on the transcription factor IRF8.
Introduction
Malignancy-associated gene mutations can generate neoantigens to activate cancer cell-reactive CD8+ cytotoxic T lymphocytes (CTLs). However, CTL effector activities are frequently suppressed in the tumor, with CTLs displaying a dysfunctional state of exhaustion characterized by high expression of the inhibitory receptor programmed cell death protein 1 (PD-1) (Chen and Mellman, 2017; McLane et al., 2019). Although PD-1 and its ligand programmed death-ligand 1 (PD-L1) have been successfully targeted for cancer therapy likely by enhancing CTL priming and activation within the tumor-draining lymph nodes (Baumeister et al., 2016; Oh et al., 2020; Sharma and Allison, 2015; Yost et al., 2021), how T cell exhaustion is elicited in the tumor, which is unlikely reversed by anti-PD-1/PD-L1 therapies (Oh et al., 2020), remains poorly understood.
Members of the mononuclear phagocyte system (MPS), including monocytes, macrophages, and dendritic cells (DCs), function as innate immune effectors and regulators as well as antigen presenting cells (APCs), initiating and modulating adaptive T cell responses. DCs, considered a bridge between innate and adaptive immunity (Mellman and Steinman, 2001), capture antigens in tumor tissues and migrate to lymph nodes to activate antigen-specific T cells. There are two classical DC subsets with distinct transcriptional mechanisms of differentiation and APC properties: DC1s, whose differentiation is dependent on the transcription factor interferon regulatory factor-8 (IRF8), are capable of presenting exogenous antigens to CTLs, whereas IRF4-dependent DC2s preferentially regulate CD4+ T cells (Murphy et al., 2016). In several tumor models, ablation of DC1s impairs the priming of cancer cell-reactive CTLs (Hildner et al., 2008; Roberts et al., 2016; Salmon et al., 2016). However, it is unknown whether cancer drives a distinct DC1 activation state compared to infection, and whether this priming predetermines CTL exhaustion.
An alternative model of CTL exhaustion is centered on the hypothesis that it is acquired following CTL migration to the tumor tissue, triggered by distinct tumor tissue-associated APC activities and/or through APC-independent mechanisms. Notably, the monocyte-macrophage lineage of cells constitutes a large proportion of leukocytes infiltrated to tumors and is implicated in suppressing CTL responses in correlation with poor patient outcomes across multiple tumor types (Gentles et al., 2015; Komohara et al., 2014; Zhang et al., 2012). How monocytes and/or macrophages might repress cancer cell-reactive CTLs is not well understood (DeNardo and Ruffell, 2019). They may inhibit CTL proliferation via distinct metabolic activities including depletion of L-arginine and generation of reactive oxygen species (Rodriguez et al., 2004). They also may modulate CTL effector functions through expression of regulatory cytokines such as interleukin-10 (IL-10) and transforming growth factor-β (TGF-β) (Ruffell et al., 2014), or co-inhibitory receptor ligands such as PD-L1 (Lin et al., 2018; Tang et al., 2018). In addition, monocytes and/or macrophages may indirectly suppress CTLs by competing with DCs for cancer cell antigen uptake and dampening the release of immunogenic alarmins from dying cancer cells (Roberts et al., 2017; Zhou et al., 2020). Given that most of these proposed immunosuppressive mechanisms were not assessed for the cancer cell antigen-induced T cell responses, it is unclear to what extent they trigger exhaustion of tumor-reactive CTLs.
A confounding factor in assessing the precise function and mechanism by which the monocyte-macrophage lineage of cells controls CTL responses in tumor is its apparent heterogeneity. Macrophages constitutively populate developing tissues and respond to inflammatory insults or tissue distress signals by in situ proliferation and/or de novo differentiation from monocytes (Ginhoux and Jung, 2014; Wynn et al., 2013). In a transgenic model of murine breast cancer, tumor progression triggers monocyte differentiation to a tumor-associated macrophage (TAM) population phenotypically distinct from mammary tissue macrophages (MTMs), which are the predominant macrophage type in healthy mammary glands (Franklin et al., 2014). Although MTMs can also be monocyte-derived, TAMs and MTMs are ontologically distinct, with Notch signaling specifically promoting TAM differentiation (Franklin et al., 2014). Notably, TAM accumulation is associated with the induction of CTL exhaustion, and defective TAM differentiation in the absence of Notch signaling attenuates CTL exhaustion and suppresses tumor growth (Franklin et al., 2014). Nonetheless, the potential immunosuppressive mechanisms of TAMs remain to be clarified.
In this study, we expanded our analysis of tumor-induced changes to cells of the MPS in the breast cancer model, and further explored their differentiation and function. We found that TAMs represented the most abundant member of the MPS and expressed high IRF8, which was required for their proliferation, maturation, and presentation of cancer cell-associated antigens to CTLs. While CTL priming in tumor-draining lymph nodes was supported by IRF8-dependent DC1s, CTL exhaustion in the tumor was dependent on TAM expression of IRF8, and macrophage-specific ablation of IRF8 suppressed tumor growth in a CTL-dependent manner. To extend these findings to cancer patients, we focused on the highly immune-infiltrated human renal cell carcinoma (RCC) tumors that were abundantly infiltrated with IRF8-expressing TAMs. The TAM IRF8 gene expression signature was highly associated with T cell exhaustion signatures in human RCC, breast cancer, and four other cancer types, including those that are both responsive and non-responsive to immune checkpoint blockade therapy. These findings suggest that CTL exhaustion is promoted by TAMs via IRF8-dependent APC functions, and such an immunoregulatory module may be targeted for novel cancer immunotherapy.
Results
Mononuclear phagocytic cells of the mammary gland are altered in breast cancer
TAMs promote tumor progression in the MMTV-PyMT (PyMT) model of breast cancer, with their terminal differentiation defined by expression of the cell adhesion molecule Vcam1 (Franklin et al., 2014; Lin et al., 2001). Ly6C+ classical inflammatory monocytes give rise to TAMs via a pathway distinct from that of MTMs, which are characterized by high expression of the scavenger receptor Mrc1 (Franklin et al., 2014). Conversely, non-classical monocytes are restricted to vasculature and play roles in tumor metastasis, but not in primary tumor growth (Hanna et al., 2015). To assess how cell transformation broadly impacted the MPS, we characterized Ly6C+ classical inflammatory monocytes, MTMs, and TAMs as well as Xcr1+ DC1s and CD11b+ DC2s in the healthy mammary gland and the tumor (Figure S1A). The proportions of TAMs and MTMs among leukocytes were increased and decreased, respectively, in the tumor (Figure 1A), in line with the notion that mutually exclusive tissue niches support MTM and TAM differentiation. DC2 abundance was unchanged, while DC1s were proportionally underrepresented among tumor-infiltrating leukocytes, although their absolute numbers per gram of tissue increased in the tumor (Figure 1A). Notably, compared to DC1s in the healthy mammary gland, a higher fraction of DC1s in the tumor expressed the integrin molecule CD103 (Figure S1B), a marker associated with maturation and trafficking of tissue DC1s to secondary lymphoid organs (Murphy et al., 2016). Indeed, migratory DC1s, but neither resident DC1s nor DC2s in mammary gland- and tumor-draining lymph nodes, expressed CD103 (Figure S1C). These observations reveal that mammary tissue transformation results in accumulation of tumor-resident TAMs as well as maturation of lymph node-homing DC1s.
Figure 1. Mononuclear phagocytic cells of the mammary gland are altered in breast cancer.
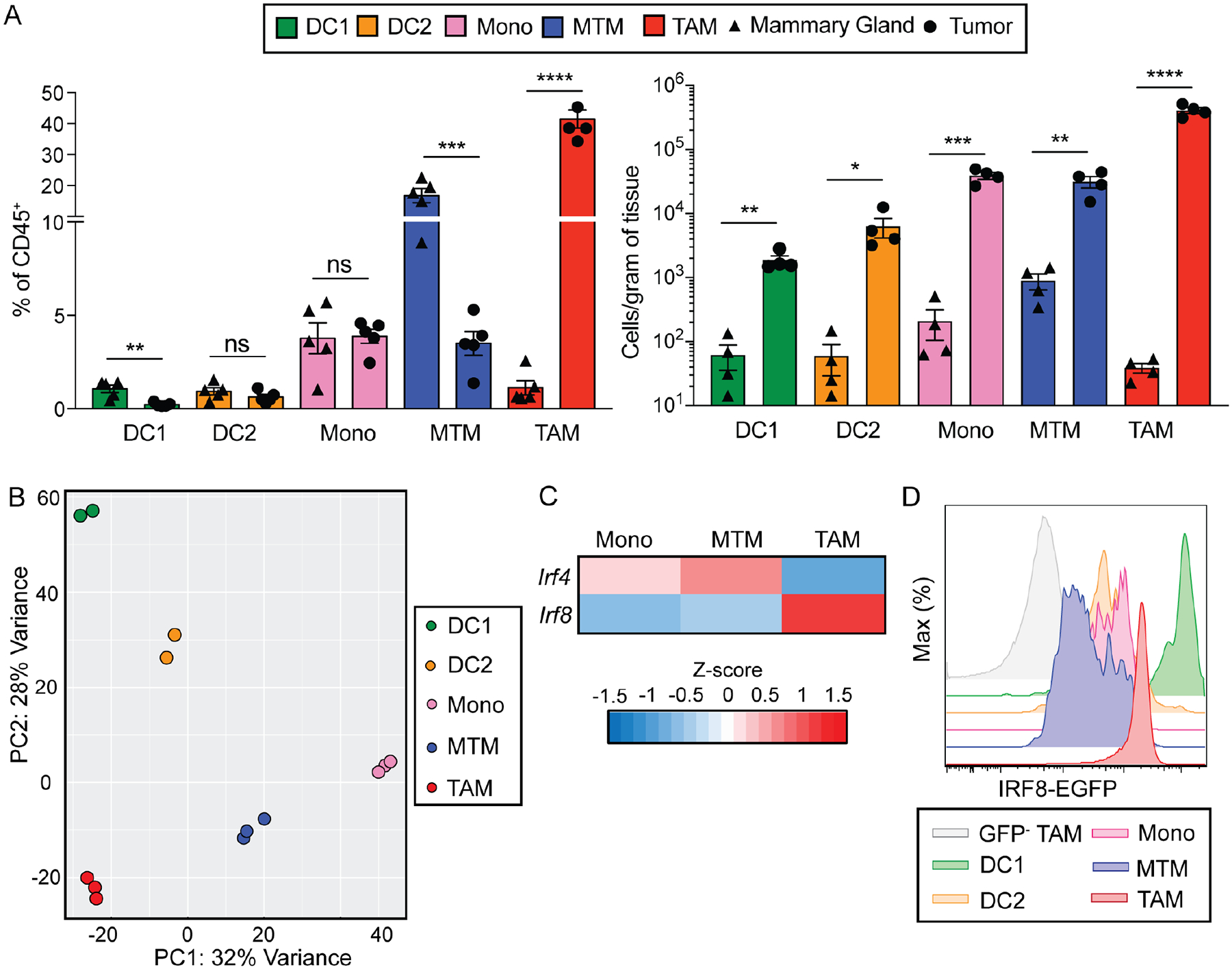
(A) Quantification of DC1s (CD45+Lin−F4/80−Ly6C−CD11c+MHC-II+Xcr1+), DC2s (CD45+Lin-F4/80− Ly6C−CD11c+MHC-II+CD11b+), monocytes (CD45+Lin−F4/80+Ly6C+CD11b+), MTMs (CD45+Lin−F4/80+Ly6C−Mrc1+) and TAMs (CD45+Lin−F4/80+Ly6C−Mrc1−) as a percentage of total immune cells (left) and absolute cell number per gram of tissue (right) in mammary glands from virgin mice (triangle) and in tumors of PyMT mice (circle), n=5 per group (Lin = Ly6G, B220, SiglecF, dead cells).
(B) Principal component analysis on the transcriptome of DC1, DC2, monocyte, MTM and TAM populations isolated from PyMT tumors, n=2 for DC1, DC2, each circle representing pooled tumors from two mice, n=3 for monocyte, MTM and TAM, each circle representing pooled tumors from one mouse.
(C) Expression of two members of the IRF family, IRF4 and IRF8, that are significantly differentially expressed in one or more of the following pairwise comparisons: monocytes versus MTMs, monocytes versus TAMs, and MTMs versus TAMs (base mean expression >100, FDR < 0.05, log2 fold change > 0.5. < −0.5, see also Table S1).
(D) Flow cytometric analysis of tumor-infiltrating immune cells from IRF8EGFPPyMT tumors, which express EGFP as an indicator of IRF8 expression. DC1s (green), DC2s (orange), monocytes (pink), MTMs (blue), TAMs (red) from IRF8EGFPPyMT tumor and TAMs from control PyMT tumor (GFP−, gray) are shown. Image is representative of five independent experiments.
To delineate gene expression programs in the tumor MPS, we performed RNA sequencing (RNAseq) experiments of DC1s, DC2s, monocytes, MTMs, and TAMs isolated from PyMT tumors. Principal component (PC) analyses showed that PC2 separated DC1s and DC2s from monocytes and macrophages, in support of their distinct origin, whereas PC1 segregated monocytes and MTMs from TAMs, DC1s, and DC2s (Figure 1B), revealing shared characteristics among TAMs and DCs. These observations were in line with the finding that TAM terminal differentiation is dependent on RBPJ (Franklin et al., 2014), a transcriptional coregulator of the Notch signaling pathway that also promotes DC maturation (Murphy et al., 2016). To investigate whether TAM differentiation from Ly6C+ monocytes was additionally controlled by cell-intrinsic transcriptional programs, we performed differentially expressed gene (DEG) analysis of transcription factors among Ly6C+ monocytes, MTMs, and TAMs (Table S1), and focused on the IRF family, which controls various leukocyte fate decisions. Six of the nine IRF family members were differentially expressed among Ly6C+ monocytes, MTMs, and TAMs (Table S1), including IRF3, which is associated with TAMs (Biswas et al., 2006). We focused on two members, IRF4 and IRF8, given their opposing expression patterns in MTMs and TAMs (Figure 1C), which resembled antagonism observed in other immune cell lineages, including DC1s and DC2s (Murphy et al., 2016; Singh et al., 2013). IRF8 functions both as a cell lineage-defining transcription factor, as in DC1s and monocyte precursors (Aliberti et al., 2003; Lanca et al., 2022; Schiavoni et al., 2002; Sichien et al., 2016), and as a mediator of acute responsiveness to extracellular signals, such as to interferon (Langlais et al., 2016). Notably, the interferon target IRF1 was not induced in TAMs relative to monocytes (Table S1), suggesting that IRF8 expression is increased via TAM-intrinsic mechanisms. To validate this pattern of IRF8 expression, we crossed PyMT mice to the Irf8EGFP background in which the enhanced green fluorescent protein (EGFP) is expressed as a fusion protein with IRF8. Indeed, TAMs expressed higher EGFP than monocytes or MTMs, although DC1s expressed EGFP at a still higher level (Figure 1D). Of note, MPS cells were characterized in a related PyMT model, in which the proposed TAM1 and TAM2 populations did not appear to express IRF8 (Broz et al., 2014). It is currently unknown what caused the discrepancy, but MTMs were not taken into consideration for macrophage classification and the purported CD11b+ DC subset appeared to express macrophage markers (Broz et al., 2014).
IRF8 deficiency impairs both dendritic cell and macrophage compartments in the tumor
To interrogate the role of IRF8 in the tumor MPS compartment, we crossed a conditional allele of Irf8 (Irf8fl) with both CD11cCre and PyMT transgenic mice, resulting in IRF8 deletion in CD11c+ cells, including TAMs and both subsets of DCs (Figure S2A). In line with the known specific function for IRF8 in DC1 differentiation in PyMT mice (Broz et al., 2014; de Mingo Pulido et al., 2018), there was an approximate 11-fold reduction of tumor DC1s but no impact on DC2 abundance in CD11cCreIrf8fl/flPyMT mice (Figure 2A). In addition, there was an almost complete loss of DC1s systemically, in both the resident and migratory DC compartments in tumor-draining lymph nodes (Figure S2B). Notably, a 1.5-fold decrease in total F4/80+SiglecF− cells was observed; however, this defect was due neither to Ly6C+ monocytes nor to MTMs, as their abundances remained comparable (Figure 2B). In contrast, there was a 1.9-fold decrease in total TAMs (Figure 2B). Among the TAMs that remained, we observed a 2.2-fold reduction in MHC-II and 1.6-fold reduction in Vcam1 expression levels (Figure 2C), which are markers of TAM terminal differentiation in the PyMT model, revealing that IRF8 is essential for the expansion and maturation of TAMs.
Figure 2. IRF8 deficiency impairs both dendritic cell and macrophage compartments in the tumor.
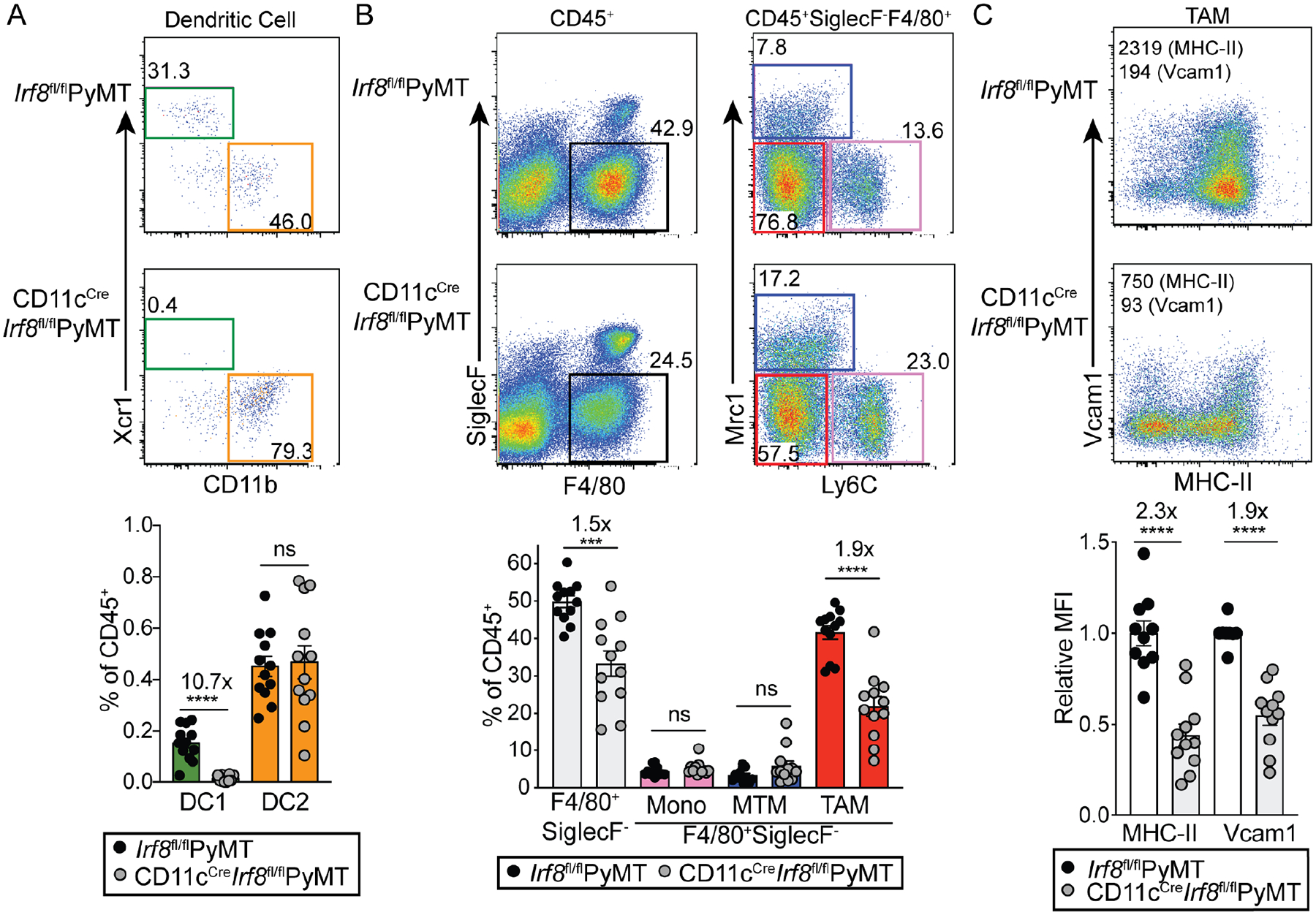
Quantification of tumor-infiltrating myeloid cell populations in Irf8fl/flPyMT and CD11cCreIrf8fl/flPyMT tumors. Representative flow plots (upper) and quantification (lower) of DCs (A) and monocytes and macrophages (B) are shown. Irf8fl/flPyMT (black circles), CD11cCreIrf8fl/flPyMT (gray circles), n=12 per group.
(C) Representative flow plots and quantification of relative geometric mean fluorescence intensity (MFI) of MHC-II and Vcam1 in CD11cCreIrf8fl/fl TAMs (n=11) relative to Irf8fl/flPyMT TAMs (n=10), fold calculated within each experiment. Each circle represents pooled tumors from one mouse; comparably sized tumors were selected between Irf8fl/flPyMT and CD11cCreIrf8fl/fl mice. All data is shown as mean +/− SEM, relative fold change values of means are displayed above significant comparisons (unpaired student’s t test, two-tailed, *** = p<0.001, **** = p<0.0001).
See also Figure S2.
Given the disruption in both DC1 and TAM myeloid compartments, we wondered whether the tumor lymphocyte response was impacted. We observed a dramatic loss of CD8+ T cells in the tumor, with no difference in abundance of either conventional CD4+ cells or Foxp3+ regulatory T (Treg) cells (Figure S2C). Although we observed a CTL defect, CD11cCreIrf8fl/flPyMT mice showed no difference in tumor growth relative to littermate controls (Figure S2D). This confirms that the CTL response in the PyMT model is ineffective in battling the tumor, and removal of the dysfunctional CD8+ T cells does not impact disease outcomes (DeNardo et al., 2009). Given that DC1s are known critical regulators of CTL priming, we were not able to tease apart potential contributions of TAMs to this CTL phenotype, and we aimed to further explore the role IRF8 plays specifically in TAMs.
IRF8 in TAMs drives a broad antigen presenting cell gene expression program
Given that IRF8 regulates TAM differentiation, we investigated the IRF8-dependent gene expression program by performing RNAseq experiments on TAMs isolated from Irf8fl/flPyMT and CD11cCreIrf8fl/flPyMT mice. Expression levels of 118 and 92 genes were significantly decreased and increased, respectively, in CD11cCreIrf8fl/fl TAMs (Figure 3A and S3A, and Table S2). We applied further FPKM filters to ensure for robust gene expression and generated two signatures, which included 58 and 53 genes with decreased and increased expression, respectively (Table S2). The IRF8-dependent genes with decreased and increased expression were most and least enriched, respectively, in TAMs as well as in DC1s and DC2s, while the opposite trend was observed for monocytes and MTMs (Figure 3B and S3B). Additionally, Ingenuity Pathway Analysis revealed that the DC maturation pathway was enriched in Irf8fl/fl TAMs (Figure 3C and Table S3). Notably, when the Irf8fl/fl and CD11cCreIrf8fl/fl TAM pairs were added to the PC plot, the CD11cCreIrf8fl/fl TAMs moved toward MTMs and monocytes but away from Irf8fl/fl TAMs and DC1s along PC1 (Figure S3C), supporting the notion that IRF8 drives DC-like properties in TAMs. Importantly, IRF8 deficiency in TAMs did not alter their PC plot position along PC2 (Figure S3C). Furthermore, both Irf8fl/fl and CD11cCreIrf8fl/fl TAMs had minimal expression of the DC-specific transcription factor Zbtb46 (Table S2), demonstrating that TAMs acquire DC characteristics in an IRF8-dependent manner while still maintaining their macrophage identity.
Figure 3. IRF8 in TAMs drives a broad antigen presenting cell gene expression program.
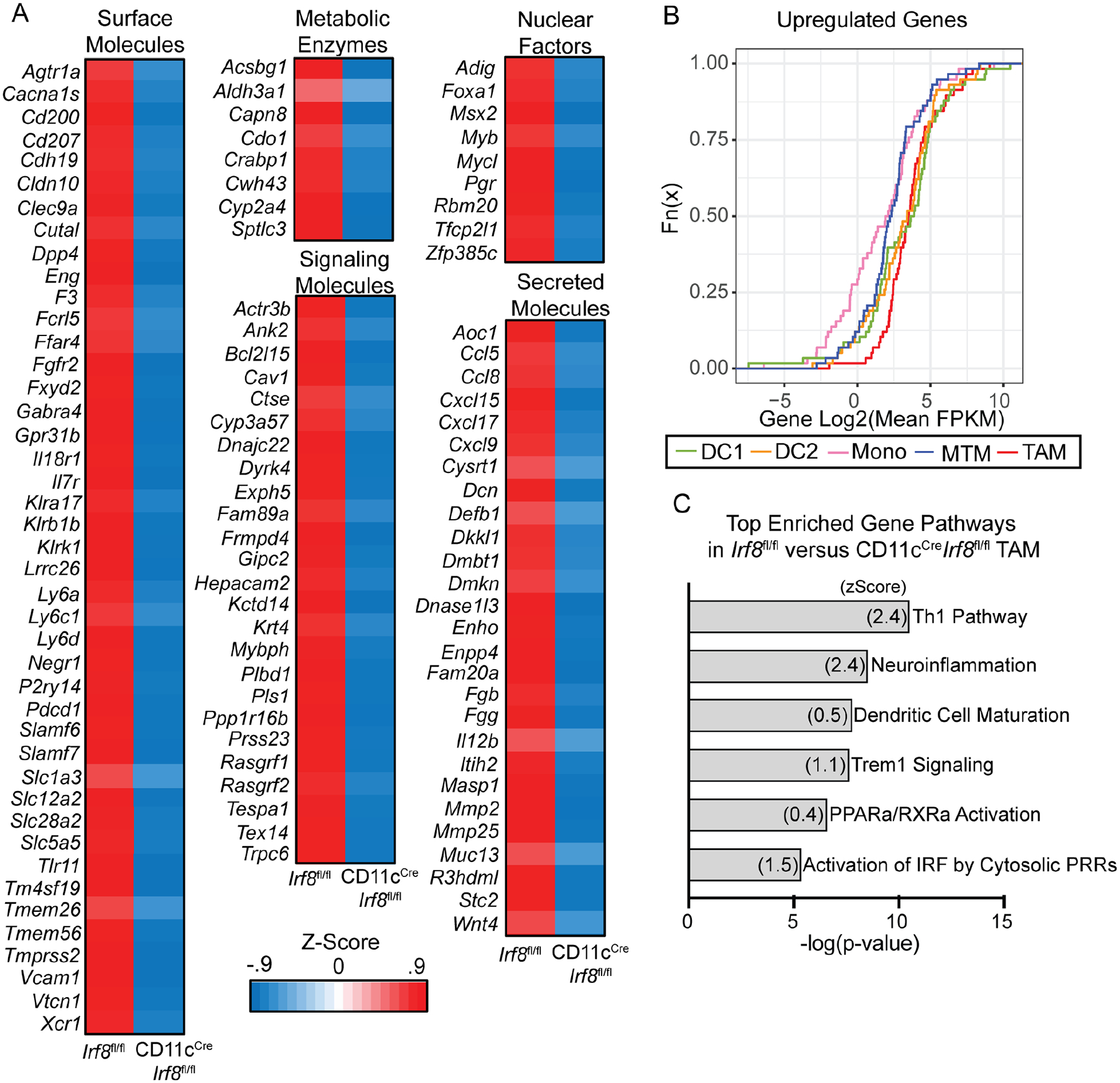
(A) Average z-score of genes significantly downregulated in TAMs sorted from tumors of Irf8fl/flPyMT and CD11cCreIrf8fl/flPyMT mice. RNA was extracted and sequenced. Genes were grouped based on cell localization and function, excluding genes with unknown functions, pseudogenes and noncoding RNAs (BC035044, BC064078, F630111L10Rik, Gm32014, 2610035D17Rik, and A630012P03Rik).
(B) CDF plot displaying enrichment of IRF8-activated gene signatures in PyMT tumor DC1 (green), DC2 (orange), monocyte (pink), MTM (blue) and TAM (red). (C) Ingenuity Pathway Analysis among differentially expressed genes between Irf8fl/fl and CD11cCreIrf8fl/fl TAMs. Most significantly enriched pathways in Irf8fl/fl TAMs are shown.
Among the IRF8 target genes with decreased expression in CD11cCreIrf8fl/fl TAMs were those encoding cell surface molecules including scavenger and innate immune receptors Cd207, Clec9a, Tlr11, and Tmprss2 as well as T cell adhesion and regulatory proteins Cd200, Slamf6, Slamf7, Vcam1, and Vtcn1 (Figure 3A). In addition, genes encoding signaling molecules involved in antigen processing such as Ctse as well as an array of targets involved in cytoskeleton and cell mobility control such as Actr3b, Ank2, Pls1, Rasgrf1, and Rasgrf2 were downregulated in CD11cCreIrf8fl/fl TAMs (Figure 3A). Expression of the transcription factor Mycl, a known IRF8 target involved in DC1 priming of T cells (Kc et al., 2014), was also lower in CD11cCreIrf8fl/fl TAMs (Figure 3A). Furthermore, in the absence of IRF8, expression levels of several T cell chemokine genes including Ccl5, Ccl8, Cxcl9, Cxcl15, and Cxcl17 were reduced in TAMs (Figure 3A). Collectively, IRF8 appears to activate a gene expression program in TAMs largely shared with DCs, promoting antigen acquisition and presentation as well as immune cross-talk with T cells.
TAMs present cancer cell antigen and induce PD-1 expression on CTLs via IRF8
IRF8-dependent DC1s excel in antigen cross-presentation to CD8+ CTLs (Murphy et al., 2016). We were intrigued by the finding that IRF8 endows DC-like properties to TAMs and wished to explore whether TAMs could function as APCs and cross-present tumor-associated antigens to CTLs by engineering an in vivo tumor antigen presentation system. Gene expression studies revealed that the S100a8 gene was highly expressed in PyMT cancer cells but not in the untransformed mammary epithelia (data not shown). By crossing S100a8Cre transgenic mice to a PyMT Rosa26LSL-YFP background, where the yellow fluorescent protein (YFP) expression cassette is placed downstream of a floxed “stop” sequence (LSL) in the Rosa26 locus, we confirmed that the Cre recombinase targeted over 80% of cancer cells as well as neutrophils, but not monocytes, MTMs, or TAMs (Figure S4A–B). In order to generate cancer cell-specific antigens, S100a8CrePyMT mice were bred with LSL-USA mice, in which a Universal Self Antigen (USA) expression cassette containing multiple model antigen sequences including the SIINFEKL peptide of ovalbumin is driven by the CAG promoter downstream of an LSL sequence (Zhang et al., 2019). Before tumors grew, S100a8CreLSL-USAPyMT mice were lethally irradiated and reconstituted with congenically mismatched bone marrow cells from C57BL/6 and CD11cCreIrf8fl/fl mice mixed at a 1:1 ratio (Figure 4A). The chimeric mice were aged to allow bone marrow cells to graft and tumors to grow, at which point endogenous neutrophils were replaced by donor bone marrow (data not shown), leading to tumor-restricted antigens. The capabilities of these C57BL/6 and CD11cCreIrf8fl/fl TAMs to cross-present cancer cell-specific antigens picked up in vivo could be compared via coculture with SIINFEKL-reactive CD8+ OT-I T cells in vitro (Figure 4A).
Figure 4. TAMs present cancer cell antigen and induce PD-1 expression on CTLs via IRF8.
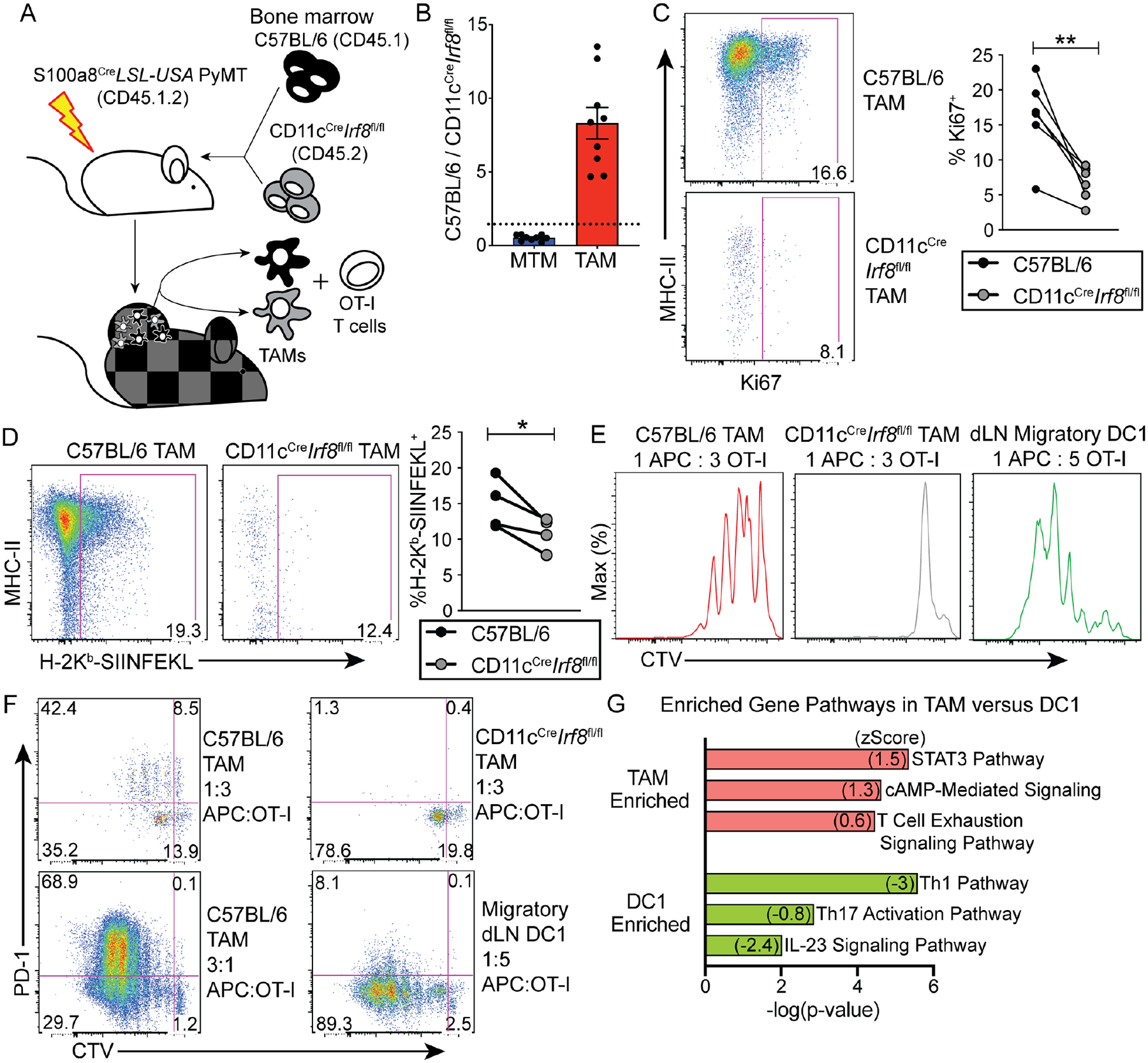
(A) Schematic showings mixed bone marrow chimera experimental setup, with recipient mice (S100a8CreLSL-USAPyMT, CD45.1.2) and bone marrow transfer (1:1) of C57BL/6 CD45.1 (black) and CD11cCreIrf8fl/fl CD45.2 (gray) bone marrow. TAMs were sorted from tumors of chimeric mice and cocultured with naïve OT-I CD8+ T cells.
(B) Reconstitution of TAMs in tumors of chimeric mice. After aging chimeric mice described in (A), and upon sizeable tumor burden, tumor-infiltrating immune cells were quantified. Ratios of C57BL/6 to CD11cCreIrf8fl/fl MTMs and TAMs were calculated, each dot representing pooled tumors from one mouse (n=9), with the dotted line representing a ratio of 1.
Representative flow cytometric gating and quantification of expression of (C) Ki67 and (D) SIINFEKL peptide presented by H-2Kb in C57BL/6 and CD11cCreIrf8fl/fl TAMs from mixed chimeric mice (n=6 for C, n=4 for D), paired student’s t-test, two-tailed, * = p<0.05, ** = p<0.01.
(E) Ability of C57BL/6 and CD11cCreIrf8fl/fl TAMs from tumor and migratory DC1s from tumor-draining lymph nodes (dLN) to trigger proliferation of OT-I T cells. TAMs were sorted from tumors and C57BL/6 migratory DC1 (CD45+MHCIIhighCD11c+Xcr1+) were sorted from dLN of chimeric mice, cocultured with CTV-labeled naïve OT-I T cells at indicated ratios in T cell media at indicated cell ratios. T cells were analyzed by flow cytometry 72 hours later.
(F) PD-1 expression from coculture experiments shown in E as well as a higher TAM concentration condition. Data representative of 3 independent experiments.
(G) Ingenuity Pathway Analysis on differentially expressed genes between TAMs and DC1s from RNAseq dataset in Figure 1. Selected enriched pathways are displayed, TAM-enriched in red, DC1-enriched in green.
Upon initial characterization of the macrophage compartments, CD11cCreIrf8fl/fl bone marrow cells contributed to TAMs approximately 8-fold less than C57BL/6 bone marrow cells, while differentiation into MTMs by CD11cCreIrf8fl/fl cells was enhanced (Figure 4B and S4C). This was coupled with a loss of Ki67 expression in CD11cCreIrf8fl/fl TAMs (Figure 4C). Additionally, CD11cCreIrf8fl/fl TAMs had substantially less surface expression of the H-2Kb-SIINFEKL complex than C57BL/6 TAMs (Figure 4D). These observations corroborate the conclusion that IRF8 promotes the proliferation and differentiation of TAMs, and reveal an important function for IRF8 in support of TAM presentation of cancer cell-associated antigens. TAMs were further isolated from tumors of chimeric mice, cocultured with OT-I T cells labeled with the cell proliferation dye CellTrace Violet (CTV) for 3 days. C57BL/6, but not CD11cCreIrf8fl/fl, TAMs induced substantial OT-I T cell proliferation, although the T cell stimulatory activity of TAMs was less potent than that of C57BL/6 migratory DC1s isolated from the tumor-draining lymph nodes (Figure 4E and S4D). Notably, induction of PD-1 expression was observed in OT-I T cells cocultured with C57BL/6 TAMs at multiple concentrations, leading to varying degrees of proliferation, whereas OT-I T cells cocultured with CD11cCreIrf8fl/fl TAMs or DC1s were much less efficient in gaining PD-1 expression (Figure 4F). To investigate what might account for the differential T cell stimulation activities of TAMs and DC1s, we performed DEG analysis between TAMs and DC1s. Genes more highly expressed in TAMs were enriched for potentially T cell-suppressive pathways including the T cell exhaustion signaling pathway, whereas genes more highly expressed in DC1s were associated with effector T cell stimulatory pathways (Figure 4G and Table S4).
Macrophage-specific deletion of IRF8 attenuates CTL exhaustion in the tumor
In order to further investigate the specific in vivo function of IRF8 in TAMs, we used a strain of MafBiCre transgenic mice that target monocyte lineage of cells (Wu et al., 2016), and generated MafBiCreIrf8fl/flPyMT mice with selective IRF8 depletion in monocytes, MTMs, and TAMs, but not DCs (Figure S4E). These mice displayed intact DC populations both in the tumor-draining lymph nodes and in the tumor (Figure 5A and S5A–B). In contrast, TAMs from MafBiCreIrf8fl/flPyMT mice displayed similar defects as those observed in CD11cCreIrf8fl/flPyMT mice, with reduced abundance as well as defective differentiation and proliferation as determined by the expression of MHC-II, Vcam1, and Ki67 (Figure 5B–C, S5C, and data not shown). This model enabled further assessment of APC defects of IRF8-deficient TAMs with an intact DC compartment. Of note, Irf8fl/fl TAMs expressed the co-stimulatory ligands CD80 and CD86 at comparable levels to that of DCs and MTMs, and their expression were not altered in MafBiCreIrf8fl/fl TAMs (Figure S4F). In addition, Irf8fl/fl TAMs expressed lower levels of the co-inhibitory ligand PD-L1 than DCs, which was not changed in the absence of IRF8 (Figure S4F). These findings suggest that the defective APC activities of MafBiCreIrf8fl/fl TAMs are not due to differential expression of these co-stimulatory or co-inhibitory molecules. As CD11cCreIrf8fl/fl TAMs had lower level of cancer cell antigen presentation than C57BL/6 TAMs (Figure 4D), we wished to determine whether it might account for their defective APC activities. To this end, Irf8fl/fl and MafBiCreIrf8fl/fl TAMs were pulsed directly with the SIINFEKL peptide, bypassing antigen acquisition, processing, and presentation. MafBiCreIrf8fl/fl TAMs were able to induce OT-I T cell proliferation and PD-1 expression to levels comparable to that of Irf8fl/fl TAMs (Figure S4G). Yet, sorted PyMT cancer cells pulsed with SIINFEKL peptide were not able to induce OT-I T cell proliferation or PD-1 expression (Figure S4H). These observations reveal a distinct tolerogenic APC function for TAMs with TAM IRF8 promoting T cell PD-1 expression in part by enabling TAMs to acquire, process, and/or present cancer cell-associated antigens.
Figure 5. Macrophage-specific deletion of IRF8 attenuates CTL exhaustion in the tumor.
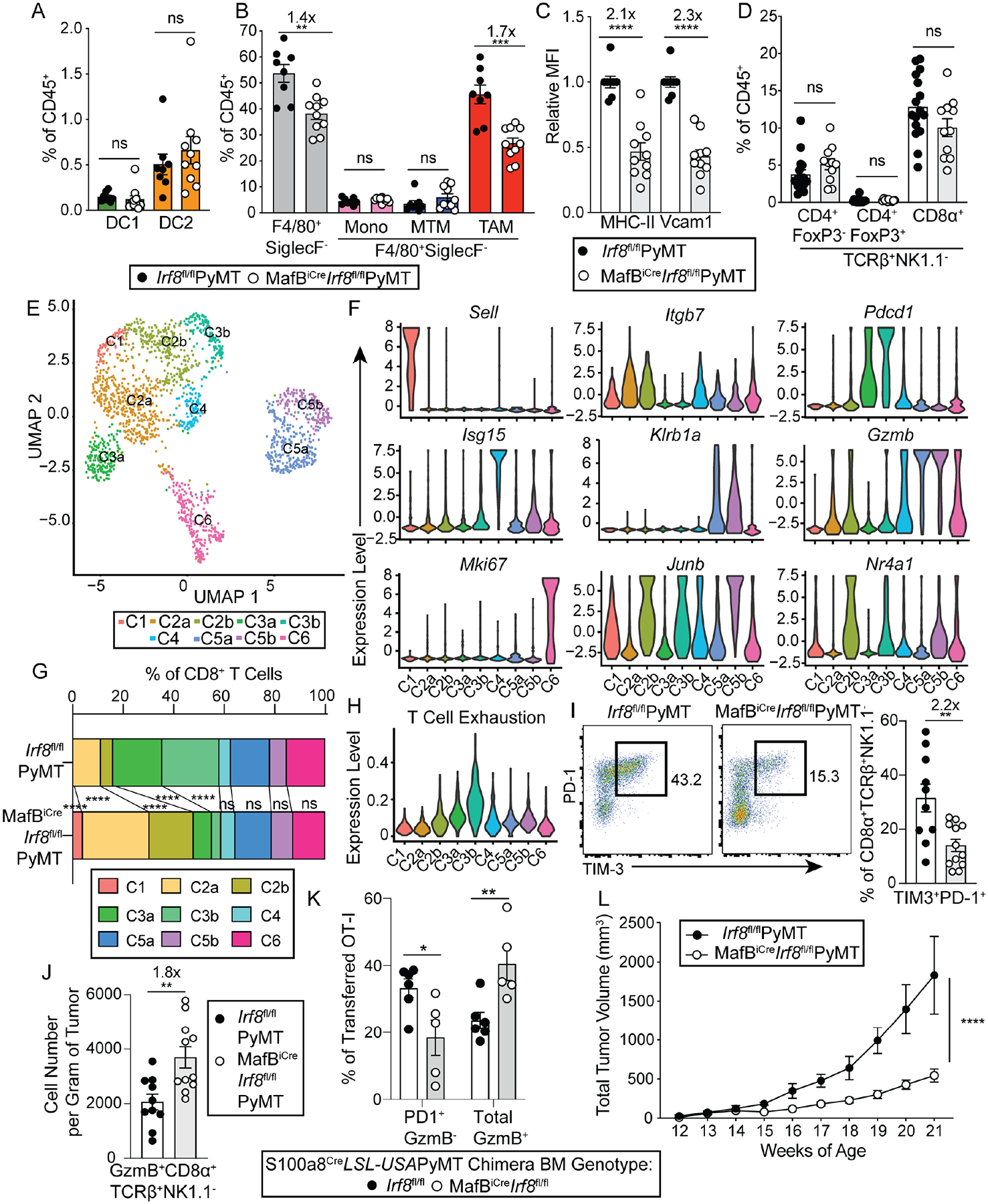
(A) Quantification of tumor-infiltrating dendritic cells in Irf8fl/flPyMT (black circles, n=8) and MafBiCreIrf8fl/flPyMT (white circles, n=10) mice. DC1 (green), and DC2 (orange) are shown.
(B) Quantification of tumor-infiltrating monocyte/macrophage lineage of cells in Irf8fl/flPyMT (black circles, n=8) and MafBiCreIrf8fl/flPyMT (white circles, n=10) mice. Total F4/80+SiglecF− cells (gray), monocytes (pink), MTMs (blue), and TAMs (red) are shown.
(C) Quantification of relative geometric mean fluorescence intensity (MFI) of MHC-II and Vcam1 relative to Irf8fl/flPyMT TAMs, MFI indicated on representative plot (Irf8fl/flPyMT n=8, MafBiCreIrf8fl/flPyMT n=10).
(D) Quantification of tumor-infiltrating conventional and regulatory CD4+ and conventional CD8+ T cells in Irf8fl/flPyMT (n=16) and MafBiCreIrf8fl/flPyMT (n=11) mouse tumors.
(E) UMAP projection of single-cell RNA sequencing (scRNAseq) clusters of TCRβ+CD8α+ cells sorted from multiple tumors of one Irf8fl/flPyMT and one MafBiCreIrf8fl/flPyMT mouse.
(F) Violin plots showing normalized expression of select genes across all 9 clusters from scRNAseq dataset.
(G) Abundance of each cluster as a proportion among total cells within Irf8fl/flPyMT or MafBiCreIrf8fl/flPyMT tumors.
(H) Violin plot showing normalized expression of a T cell exhaustion gene signature across all 9 clusters.
(I) Representative plots and quantification of flow cytometric analysis of co-expression of PD-1 and TIM-3 in TCRβ+NK1.1−CD8α+ T cells from tumors of Irf8fl/flPyMT (n=10) and MafBiCreIrf8fl/flPyMT (n=12) mice.
(J) Quantification of flow cytometric analysis of number of GzmB+TCRβ+NK1.1−CD8α+ cells per gram of tumor in Irf8fl/flPyMT (black line, circles) (n=10) and MafBiCreIrf8fl/flPyMT (gray line, circles) (n=11) mice.
(K) Quantification of flow cytometric analysis of PD-1 and GzmB expression in OT-I T cells two weeks after transfer to tumor-bearing S100a8CreLSL-USAPyMT chimeric mice reconstituted with either Irf8fl/fl (black circles, n=6) or MafBiCreIrf8fl/fl (white circles, n=5) bone marrow.
(L) Tumor measurements from MafBiCreIrf8fl/flPyMT and littermate and cagemate Irf8fl/flPyMT controls (n=7 each). All data are shown as mean +/− SEM, fold change of means are displayed above significant comparisons. (A, B, C, D, I, J, K) Each circle represents pooled tumors from one mouse; comparably sized tumors were selected between mice, unpaired student’s t-test, two-tailed, ** = p<0.01, *** = p<0.001, **** = p<0.0001. G: Fisher’s Exact Test, **** = p<0.0001 J: 2-way ANOVA **** = p<0.0001.
See also Figures S5–S6 and Table S5.
To determine the IRF8-mediated CTL regulatory function of TAMs in vivo, we phenotyped tumor-infiltrating T cells in MafBiCreIrf8fl/flPyMT mice. In contrast to CD11cCreIrf8fl/flPyMT mice, the abundance of CD8+ T cells as well as conventional CD4+ cells and Treg cells in MafBiCreIrf8fl/flPyMT mice were comparable to that of control Irf8fl/flPyMT mice (Figure 5D and S5D–E). These observations suggest that DC1s are critical regulators of the CTL response in the tumor, presumably through priming in the tumor-draining lymph node (Hildner et al., 2008; Roberts et al., 2016; Salmon et al., 2016), whereas TAM IRF8 is largely dispensable for modulating the magnitude of the CTL response.
While we did not observe differences in overall CTL abundance, we investigated whether TAM IRF8 influences CTL phenotypes. To this end, we performed single-cell RNA sequencing (scRNAseq) experiments of tumor-infiltrating CD8+ T cells from Irf8fl/flPyMT and MafBiCreIrf8fl/flPyMT mice, which revealed 9 clusters and 6 broad phenotypes (Figure 5E and S6). Naïve-like T cells, exemplified by expression of Sell, were represented by C1. Early-activated and effector CD8+ T cells, which expressed high levels of Itgb7 and intermediate levels of Gzmb, were found in C2a and CD2b, while exhausted cells, identified by high expression of Pdcd1 as well as Tox, Tigit, and Lag3 but low expression of Gzmb, were found in C3a and C3b (Figure 5F and S6). Other clusters included cells expressing interferon-induced genes such as Isg15 in C4, innate-like T cells (ILTCs) expressing high levels of Klrb1a (encoding NK1.1) and Gzmb as previously reported (Chou et al., 2022; Dadi et al., 2016) in C5a and C5b, and proliferating cells expressing Mki67 in C6 (Figure 5F and S6). Multiple phenotypic categories were broken into two clusters, differentiated by expression of immediate-early genes including Junb and Nr4a1 (C2b, C3b, and C5b) (Figure 5F and Table S5).
While CD8+ T cells from Irf8fl/flPyMT and MafBiCreIrf8fl/flPyMT mice were represented by the same clusters, the relative abundances of specific clusters were altered in MafBiCreIrf8fl/flPyMT mice, with enrichment for naïve-like and early-activated/effector T cells (C1, C2a, C2b) and diminished exhausted clusters (C3a, C3b) (Figure 5G). Of note, C3a-b were most enriched for a T cell exhaustion signature, supporting their identity as exhausted CTLs (Figure 5H). Flow cytometric analysis confirmed the altered abundances observed in scRNAseq. There were fewer PD-1+TIM-3+ exhaustion phenotype cells and an increase in proportion and absolute number of granzyme B-expressing cells among NK1.1− conventional tumor-infiltrating CTLs in MafBiCreIrf8fl/flPyMT mice, including among the antigen-experienced PD-1+ subset (Figure 5I–J and S5F). To investigate whether this altered CTL phenotype could occur in tumor antigen-specific T cells, S100a8CreLSL-USAPyMT mice, which expressed the SIINFEKL OT-I T cell peptide antigen in cancer cells, were reconstituted with congenically mismatched Irf8fl/fl or MafBiCreIrf8fl/fl bone marrow, aged, and upon palpable tumors, received OT-I T cell transfers. Indeed, two weeks post-transfer, OT-I T cells in the tumor of mice with MafBiCreIrf8fl/fl bone marrow expressed less PD-1 among GzmB− cells and more GzmB overall than OT-I T cells from tumors of mice with Irf8fl/fl bone marrow (Figure 5K and S5G–H). Notably, this altered CTL compartment was associated with tumor protection, as MafBiCreIrf8fl/flPyMT mice exhibited slower tumor growth than littermate controls (Figure 5L). This difference in tumor growth was dependent on CTLs, since after CD8+ T cell depletion, tumor growth was comparable between Irf8fl/flPyMT and MafBiCreIrf8fl/flPyMT mice (Figure S5I–J). These observations suggest that in the PyMT model, while DC1s are required for initial priming of tumor-specific CD8+ T cells, re-encountering antigen on IRF8-expressing TAMs in the tumor promotes T cell exhaustion, thus attenuating CD8+ T cell cancer immunosurveillance.
TAMs from human clear cell RCC patients express IRF8 and IRF8-activated genes
We wished to determine whether a similar TAM-dependent immune regulatory module occurred in human solid tumors. We chose to investigate clear cell renal cell carcinoma (ccRCC), as surgery is the first line therapy and naturally evolved tumor-elicited immune responses can be observed in drug treatment-naïve cancer patients. Surgically removed fresh tumor tissues underwent flow cytometric analysis to investigate tumor-associated MPS cells, including DC1s, DC2s, classical monocytes, and TAMs, defined as HLADR+CD14−CD141+, HLADR+CD14−CD1c+, CD14+HLADRlow, and CD14+HLADRhigh leukocytes, respectively (Figure 6A). As in mice, DC1s and DC2s were very rare, making up less than one percent of the immune infiltrate, whereas TAMs were 31-fold more abundant than DC1s and 19-fold more abundant than DC2s (Figure 6A). The IRF8 expression pattern across the MPS compartment was similar to that observed in mice, with DC1s expressing the highest level and TAMs expressing the second highest level (Figure 1D and 6B). Additionally, in most patient samples, we observed higher IRF8 expression in TAMs than in monocytes, again matching what we observed in our mouse tumor model (Figure 1D and 6C).
Figure 6. TAMs from human clear cell RCC patients express IRF8 and IRF8-activated genes.
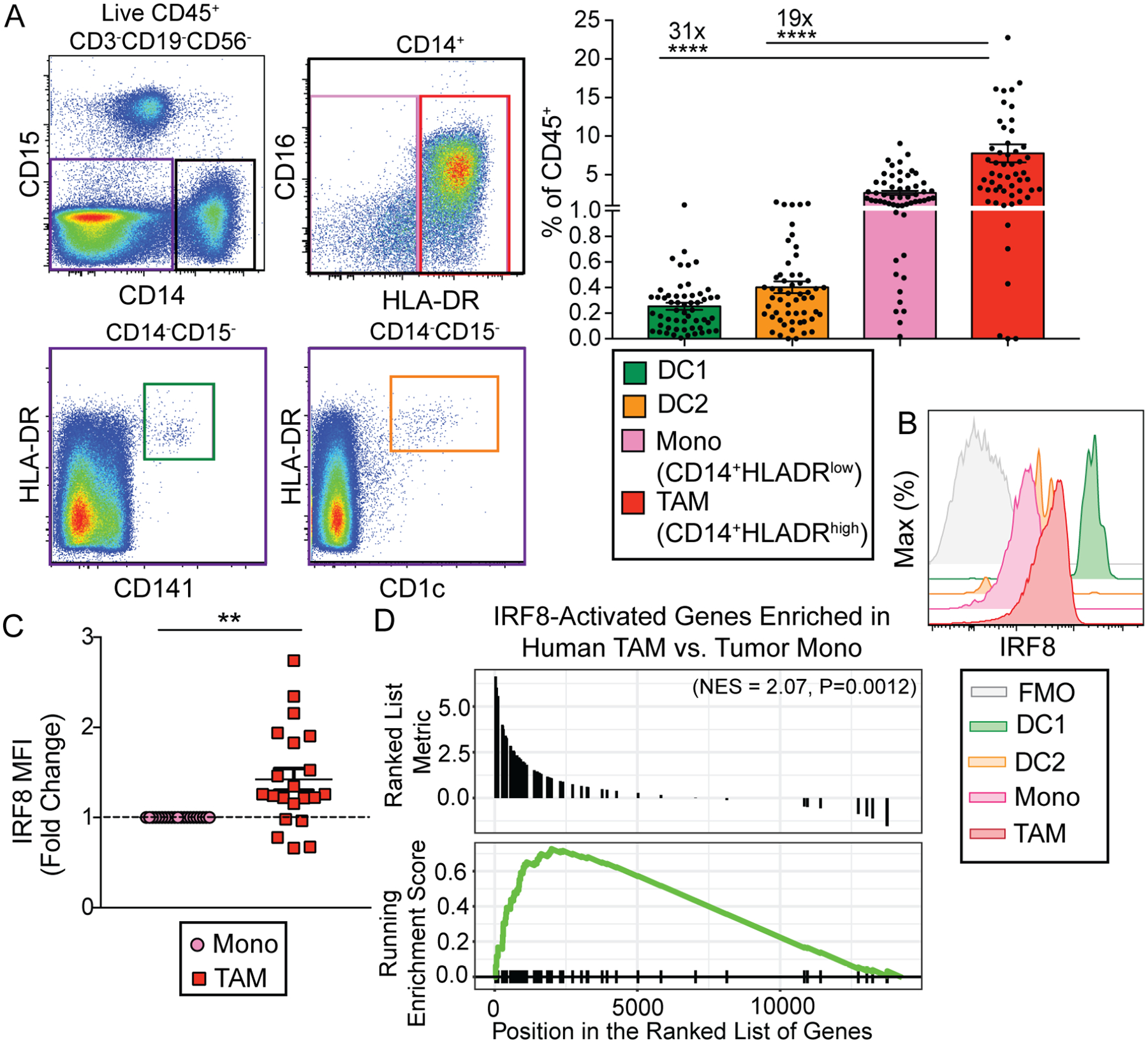
(A) Representative flow plots and quantification of tumor-infiltrating DC1s (green, CD45+Lin−CD14−HLA-DR+CD141+), DC2s (orange, CD45+Lin−CD14−HLA-DR+CD1c+), classical monocytes (pink, CD45+Lin−CD14+HLA-DRlow), and TAMs (red, CD45+Lin−CD14+HLA-DRhigh) in human clear cell RCC tumor samples as determined by flow cytometric analysis (Lin = CD15, CD19, CD3, CD56, dead cells). Fold differences in abundance between TAMs and DC1s, TAMs and DC2s are shown. Each circle represents one patient tumor sample, data shown as mean +/− SEM, n=56 (one-way ANOVA with multiple comparisons, **** = p<0.0001).
(B) Representative plot showing expression of IRF8 in tumor-infiltrating DC1s (green), DC2s (orange), monocytes (pink), TAMs (red), and fluorescence minus one (FMO) TAM sample (gray), as determined by flow cytometric analysis.
(C) IRF8 MFI fold change in TAMs relative to tumor-infiltrating monocytes. IRF8 MFI is relative to monocyte MFI (n=21), data shown as mean +/− SEM (paired student’s t test, two-tailed, ** = p<0.01).
(D) Enrichment of human orthologs of IRF8-activated mouse TAM genes among human TAM-specific genes relative to human monocytes. Tumor monocytes (n=4) and TAMs (n=5) were sorted from human RCC samples, underwent RNA sequencing and differential gene expression analysis. The IRF8-activated genes with significantly higher expression identified in mouse TAMs were converted to their human orthologs, and the enrichment of these IRF8-activated genes was tested among human TAM-specific genes.
To define the genetic program that may control TAM differentiation from monocytes, we performed RNAseq experiments on purified monocyte and TAM populations from RCC patient tumors. Significant enrichment of the human orthologs of mouse IRF8-activated target genes was observed in human TAMs over monocytes (Figure 3A and 6D), suggesting that IRF8 induces a similar gene expression program in human TAMs. Thus, we were able to identify TAMs in human solid tumors that expressed both IRF8 and an IRF8-dependent gene program that mirrored what we observed in mouse.
A TAM IRF8 gene signature predicts CTL exhaustion across multiple human cancers
We aimed to generate a human IRF8-TAM gene expression signature using the genomics data we had generated from both mouse and human TAMs. After converting the mouse IRF8-activated genes to their human orthologues, we applied further FPKM filters to ensure robust expression in the human TAM RNAseq dataset. The resulting IRF8-TAM signature consisted of 18 genes (Figure 7A). To explore the specificity of this signature, we looked for its enrichment in RNAseq datasets generated from a number of purified myeloid and lymphoid cell populations from RCC tumors, demonstrating that the signature was most enriched in TAMs, DC1s, and DC2s (Figure 7B). This pattern of enrichment suggests this signature is shared among multiple mature APC compartments in human RCC tumors, similar to what we observed in mice (Figure 3B).
Figure 7. A TAM IRF8 gene signature predicts CTL exhaustion across multiple human cancers.

(A) Human IRF8 signature was generated by using additional FPKM filters (FPKM mean >1, FPKM variance > 3) on human orthologs of IRF8-activated mouse TAM genes.
(B) Enrichment of Human IRF8 Signature across various immune populations sorted from tumor and peripheral blood of RCC patients, including DC1s, DC2s, monocytes, TAMs, neutrophils (CD15+CD16+), B cells (CD19+), conventional CD4+ T cells (CD3+CD4+CD25−), Treg cells (CD3+CD4+CD25+), CD49a+CD103+ CD8+ T cells (CD3+CD8α+CD49a+CD103+), CD49a−CD103− CD8+ T cells (CD3+CD8α+CD49a−CD103−) and NK cells (CD3−CD56dimCD16+) (within a cell type: n=4–21, each sample from a different patient).
Bulk RNAseq samples from patients of six different cancer types in the TCGA database were stratified into IRF8high and IRF8low groups based on IRF8 signature enrichment. Enrichment of (C) a macrophage signature (Mac_CSF1 signature) and (D) a CD8+ T cell exhaustion signature among IRF8high versus IRF8low patients is shown, with both macrophage and exhaustion signatures being significantly enriched in IRF8high patients for all six tumor types (KIRC = kidney renal clear cell carcinoma, BRCA = breast invasive carcinoma, LUSC = lung squamous cell carcinoma, BLCA = urothelial bladder carcinoma, SKCM = skin cutaneous melanoma, LIHC = liver hepatocellular carcinoma).
To investigate whether the IRF8 signature was associated with phenotypically distinct CTL responses in tumor, we used the patient bulk RNAseq datasets for six cancer types from the TCGA database. We included three cancer types known to respond well to checkpoint blockade therapy, including kidney renal clear cell carcinoma (KIRC), lung squamous cell carcinoma (LUSC), and skin cutaneous melanoma (SKCM), as well as three cancer types that do not respond well, namely breast invasive carcinoma (BRCA), bladder urothelial carcinoma (BLCA), and liver hepatocellular carcinoma (LIHC). We subsetted patients based on enrichment for the IRF8 signature into IRF8high and IRF8low groups. The bulk RNAseq from tumors among the IRF8high group was enriched for a macrophage signature (Beck et al., 2009; Bindea et al., 2013) (Figure 7C), confirming that TAMs manifest an IRF8-dependent gene expression program and are the major APC subset in the tumor not only in KIRC but also in other cancer types. In addition, the IRF8high group had significant enrichment for T cell exhaustion signature (Chung et al., 2017) (Figure 7D). These observations suggest that the IRF8-dependent TAM gene program generally promotes CD8+ T cell exhaustion in mouse and human malignancies (Figure S7).
Discussion
Tumor progression is associated with overstimulation of cancer cell-reactive CTLs, resulting in a dysfunctional state of exhaustion. Using murine models of myeloid cell-specific ablation of IRF8, our studies revealed a critical role for TAMs in induction of CTL exhaustion via their ability to present cancer cell-associated antigens to CD8+ T cells. While APC-independent mechanisms of suppression in the tumor microenvironment have been proposed, we did not observe IRF8-dependent expression of genes implicated in such pathways in TAMs. Given that there is no difference in tumor growth in CD11cCreIrf8fl/flPyMT mice, where DC1-primed T cells are absent, any potential CTL-independent pro-tumor TAM activities are not dependent on IRF8 and/or do not contribute in this model. Our findings confirm that IRF8-dependent TAM APC activity is a key regulator of CTL responses in tumor.
Both DC1s and TAMs can present cancer cell antigens to CD8+ T cells with notable differences in APC functions. We observed distinct outcomes in the coculture system, namely induction of high PD-1 expression on T cells by TAMs, robust activation and proliferation of T cells at a low APC to T cell ratio by DC1s, and no proliferation induced by cancer cells. The inability of cancer cells pulsed with antigen to stimulate T cells suggests presentation of antigen alone is not sufficient to induce T cell exhaustion, as cancer cells are generally lack of expression of co-stimulatory molecules. The different APC ratios used in our study are in line with different abundances of TAMs and DC1s in vivo, and the observation supports the notion that DC1s are more potent activators of CTLs than TAMs. Compared to TAMs, DC1s exhibit a lower level of phagocytic activity, but can better preserve antigens as a likely consequence of diminished lysosomal degradation (Broz et al., 2014; Delamarre et al., 2005). How these opposing processes affect net antigen presentation on DC1s and TAMs remains to be determined. IRF8 specifies DC1 identity and is required for DC1-associated functional programs, including cross-presentation (Lanca et al., 2022). In our study, we show IRF8 is required for TAM presentation of tumor antigen and is also required for expression of SLAMF7, a molecule that has been shown to promote CTL exhaustion (O’Connell et al., 2021). Similar to DC1s, IRF8 may influence TAM identity and functional programs, promoting TAM antigen presentation and interactions with CD8 T cells. DC1s and TAMs express similar levels of costimulatory molecules CD80 and CD86, suggesting that co-stimulation may not differentiate TAM and DC1 APC functions. DEG analysis revealed that genes associated with effector T cell stimulatory pathways are more highly expressed in DC1s. Notably, previous studies have shown that DC1s express higher levels of the p40 subunit of interleukin-12 (IL-12) and IL-15 than TAMs (Broz et al., 2014; Kansler et al., 2022; Ruffell et al., 2014), which we confirmed in our TAM versus DC1 RNAseq experiment (data not shown). Future studies will determine if differential expression of these effector CTL differentiation cytokines contributes to the distinct T cell phenotypes observed in DC1 and TAM coculture.
In addition to differential expression of T cell stimulatory factors, distinct in vivo dynamics of DC1s and TAMs may also affect their APC functions. In PyMT mice, tumor-associated DC1s express higher levels of the DC maturation marker CD103, and are underrepresented compared to DC1s in healthy mammary glands. Thus, following maturation, DC1s may readily migrate to tumor-draining lymph nodes, and prime cancer cell antigen-reactive CD8+ T cells that are subsequently recruited to the tumor. Do DC1s play a role locally in the tumor microenvironment to restimulate the antigen-experienced CD8+ T cells? Given their dynamics and critical role in priming T cell responses, this can be difficult to address. Of note, inducible ablation of DC1s following T cell priming does not substantially affect CTL responses (Diao et al., 2018), suggesting against a local role for DC1s in the tumor. In contrast to DC1s, TAMs are tumor-resident, and robustly expand as the tumor grows. Therefore, abundant interactions between TAMs and the antigen-primed CD8+ T cells would occur and persist chronically, which may be the root cause for the induction of T cell exhaustion in tumor. TAMs and exhausted CTLs are often enriched in the same late-stage tumors (Braun et al., 2021; Wagner et al., 2019). These two cell types can be colocalized in the tumor (Braun et al., 2021), and a recent study demonstrated that prolonged TAM-CTL antigen-dependent interactions occur and TAMs can contribute to T cell exhaustion (Kersten et al., 2022). While both TAMs and cancer cells can present antigens to CD8+ T cells, our results suggest this CTL-cancer cell interaction is not sufficient to drive exhaustion. TAMs are more specialized APCs, expressing costimulatory molecules and chemokines, such as Cxcl9, that can attract T cells. However, further studies are required to fully explain the difference between TAM-CTL and cancer cell-CTL interactions. In contrast to conventional T cells, ILTCs (Dadi et al., 2016) require neither DC1s nor TAMs for their activity in the tumor, and instead can directly respond to cancer cells (Chou et al., 2022). Paired TCRαβ sequencing reveals ILTC TCRs have broad self-reactivity, whereas PD-1+ T cell TCRs are not shared with and do not respond to cancer cells from other PyMT mice, and thus presumably respond to neo-antigens (Chou et al., 2022). Thus, distinct APC compartments control different types of T cell responses in the tumor to impact their complementary cancer immunosurveillance functions.
Our work uncovered an important role for the transcription factor IRF8 in TAM APC function. The finding that IRF8 and IRF4 are selectively enriched in TAMs and MTMs raises the possibility that analogous to DC1s and DC2s, a division of labor exists among macrophage populations. In some murine cancer models, tissue-resident macrophages derived from embryonic progenitors have been implicated in tumor regulation (Loyher et al., 2018; Zhu et al., 2017). Resident macrophages in multiple organs, including microglia of the brain and macrophages of the kidney and liver express IRF8, and IRF8 plays a critical function in macrophage maturation and survival (Hagemeyer et al., 2016). It will be interesting to determine whether embryonically-derived macrophages also engage the IRF8-dependent program to induce CTL exhaustion in cancer or other conditions of chronic antigen stimulation, as there is evidence that Kupffer cells in the liver can present hepatocyte antigens to CTLs during infection (Ebrahimkhani et al., 2011). Compared to monocytes, TAMs isolated from human ccRCC patients express higher levels of IRF8, and an IRF8-TAM gene signature predicts T cell exhaustion. Nonetheless, whether IRF8-expressing TAMs acquire cancer cell antigens to present to CTLs in human tumors remain to be determined. High expression of IRF8 in ccRCC TAMs has also been reported in a recent study (Muhitch et al., 2019), which predicted beneficial patient outcome. In contrast, we found that ccRCC patients with abundant CD8+ T cell infiltration trended to have worse survival if they expressed higher levels of the IRF8-TAM gene signature, which predicted T cell exhaustion (data not shown). The reasons underlying this discrepancy are unknown but may be related to the use of different predictors, namely manually scored TAM IRF8 expression versus the IRF8-TAM gene expression signature.
How is IRF8 regulated in TAMs? As opposed to what has been shown for macrophages activated in vitro (Langlais et al., 2016; Xu et al., 2012), TAM expression of IRF8 in the PyMT model does not require Notch or interferon-γ (IFN-γ) (data not shown), in line with the lack of induction of the IFN-γ target gene IRF1 in TAMs. Thus, TAM IRF8 expression appears to be developmentally wired, but unlike DC1s, is independent of the transcription factor Batf3 (data not shown). Future studies will unravel the exact mechanisms by which IRF8 is induced in TAMs. How does IRF8 regulate TAM function? A major defect of IRF8-deficient TAMs is their inability to present cancer cell antigens, which largely accounts for their inability to induce PD-1 expression in vitro. Such a defect may be due to the loss of antigen cross-presentation activities of TAMs following their engulfment of cancer cell constituents. This may occur at the step of antigen acquisition, as some scavenger receptors are downregulated in IRF8-deficient TAMs. Antigen cross-presentation itself may also be affected, but we did not observe defective expression of the canonical cross-presentation genes in IRF8-deficient TAMs. In addition, it is possible that IRF8 may promote TAMs to employ “cross-dressing” by capturing pre-formed peptide-MHC-I complexes from cancer cells (Campana et al., 2015), a process that is critical for DC1 priming of CTLs in some cancer models (MacNabb et al., 2022). The relative contribution of cross-presentation and cross-dressing in TAM presentation of cancer cell antigens warrants further investigation and can be differentiated by animal models with selective ablation of MHC-I antigen presentation genes in TAMs or cancer cells. What’s more, IRF8-deficient TAMs are defective in proliferation, and express low levels of chemokine genes including its direct target Cxcl9 (Langlais et al., 2016), and Cxcl9 may attract CTLs to interact with TAMs in the tumor microenvironment (Dangaj et al., 2019). These defects may collectively contribute to the loss of TAM-induced T cell exhaustion in vivo.
Therapies targeting the PD-1/PD-L1 pathway have shown great success in the clinic for a subset of patients. While initially thought to reinvigorate exhausted T cells in the tumor microenvironment, increasing evidence suggests these therapies target peripheral more stem-like T cell populations, potentially in the tumor-draining lymph node during priming, where DCs are the relevant source of PD-L1 (Dolina et al., 2021; Oh et al., 2020). These therapies can activate new waves of T cells to proliferate and infiltrate the tumor, but these cells can still become exhausted once in the tumor microenvironment (Dolina et al., 2021; Oh et al., 2020). Treatment can also lead to altered TAM phenotypes in responding patients (Bi et al., 2021). It will be interesting to investigate whether the IRF8-regualted TAMs play a role in the induction of T cell exhaustion under these conditions, and thus open additional avenues for novel therapeutic intervention.
Limitations of the study
This study shows TAM acquisition and presentation of cancer cell antigen and induction of CTL exhaustion require IRF8 expression. Additional studies are needed to further explore which IRF8-dependent genes control which aspects of TAM antigen-presenting cell activity. Additionally, while we observed correlation between TAMs expressing the IRF8-dependent gene program and exhausted CTLs in human cancers, additional studies are needed to directly demonstrate that human TAMs can present antigen and induce CTL exhaustion through chronic antigen stimulation.
STAR Methods
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Ming Li (lim@mskcc.org).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The raw sequencing data for bulk RNA sequencing on myeloid lineage cells from the tumor in mouse and tumor immune populations from RCC patients and for single cell RNA sequencing on CD8+ T cells from Irf8fl/flPyMT and MafBiCreIrf8fl/flPyMT tumors have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse CD103 | BD Biosciences | M290 |
| Anti-mouse CD11b | BD Biosciences | M1/70 |
| Anti-mouse CD11c | BD Biosciences | N418 |
| Anti-mouse CD4 | BD Biosciences | RM4-5 |
| Anti-mouse CD45 | BD Biosciences | 30-F11 |
| Anti-mouse Ki67 | BD Biosciences | B56 |
| Anti-mouse Ly6C | BD Biosciences | AL-21 |
| Anti-mouse MHC-II I-A/I-E | BD Biosciences | M5/114.15.2 |
| Anti-mouse SiglecF | BD Biosciences | E50-2440 |
| Anti-human CD141 | BD Biosciences | 1A4 |
| Anti-human CD1c | BD Biosciences | F10/21A3 |
| Anti-mouse CD206 | BioLegend | C068C2 |
| Anti-mouse H-2Kb bound to SIINFEKL | BioLegend | 25-D1.16 |
| Anti-mouse XCR1 | BioLegend | ZET |
| Anti-human CD15 | BioLegend | W6D3 |
| Anti-human CD16 | BioLegend | 3G8 |
| Anti-human CD19 | BioLegend | HIB19 |
| Anti-mouse CD29 | BioLegend | HmB1-1 |
| Anti-mouse CD326 (EpCAM) | BioLegend | G8.8 |
| Anti-human CD56 | BioLegend | HCD56 |
| Anti-human HLA-DR | BioLegend | L243 |
| Anti-mouse CD24 | eBioscience/Thermo | M1/69 |
| Anti-mouse CD80 | eBioscience/Thermo | 16-10A1 |
| Anti-mouse CD86 | eBioscience/Thermo | GL1 |
| Anti-mouse FoxP3 | eBioscience/Thermo | FJK-16s |
| Anti-mouse NK1.1 | eBioscience/Thermo | PK136 |
| Anti-mouse PD-L1 | eBioscience/Thermo | MIH5 |
| Anti-mouse PD-1 | eBioscience/Thermo | RMP1-30 |
| Anti-mouse TIM-3 | eBioscience/Thermo | 8B.2C12 |
| Anti-mouse Vcam1 | eBioscience/Thermo | 429 |
| Anti-mouse/human IRF8 | eBioscience/Thermo | V3GYWCH |
| Anti-human CD14 | eBioscience/Thermo | 61D3 |
| Anti-human/mouse granzyme B | Invitrogen | GB11 |
| Anti-human CD3d | Invitrogen | 7D6 |
| CellTrace Violet | Invitrogen | |
| Anti-mouse B220 | Tonbo Biosciences | RA3-6B2 |
| Anti-mouse CD8a | Tonbo Biosciences | 53-6.7 |
| Anti-mouse Ly6G | Tonbo Biosciences | 1A8 |
| Anti-mouse TCRB | Tonbo Biosciences | H57-597 |
| Anti-human CD45 | Tonbo Biosciences | 2D1 |
| Ghost Dye | Tonbo Biosciences | 13-0865-T500 13-0863-T500 13-0870-T500 |
| CD8 depleting antibody | Bio × Cell | Clone 2.43 |
| FcR Block (2.4G2 mAB) | Bio × Cell | 25 mg (custom) |
| Human TruStain FcX | BioLegend | 422302 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Collagenase Type 3 | Worthington Biochemical | LS004183 |
| Dnase I | Sigma-Aldrich | |
| Percoll | Sigma-Aldrich | P1644-1L |
| Trizol LS | Invitrogen | |
| OVA(257–264) | Invivogen | |
| Critical Commercial Assays | ||
| Transcription factor Fix/Perm Kit | Tonbo Biosciences | TNB-0607-KIT |
| Naïve CD8 T cell Negative Selection Kit | EasySep, Stem Cell Technologies | 19858 |
| Deposited Data | ||
| Murine bulk RNAseq/scRNAseq sequencing files | GEO | GSE212643 |
| Human TAM monocyte bulk RNAseq sequencing files | EGA | Dataset ID: EGAD00001009393 Study ID: EGAS00001006593 |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6.Cg-Tg(Itgax-cre)1-1Reiz/J (CD11cCre) (Received from Dr. Boris Reizis Lab) | Jackson Laboratories | Stock: 008068 |
| Mouse: B6.SJL-PtprcaPepcb/BoyJ (CD45.1) | Jackson Laboratories | Stock: 002014 |
| Mouse: B6(Cg)-Irf8tm1.1Hm/J (Irf8flox) | Jackson Laboratories | Stock: 014175 |
| Mouse: B6(Cg)-Irf8tm2.1Hm/J(Irf8EGFP) (Received from Dr. Herbert C. Morse III Lab) | Jackson Laboratories | Stock: 027084 |
| Mouse: B6N(129S4)-Mafbtm1.1(cre)Kmm/J (MafBiCre) (Received from Dr. Kenneth Murphy Lab) | Jackson Laboratories | Stock: 029664 |
| Mouse: C57BL/6-Tg(TcraTcrb)1100Mjb/J (OT-I) | Jackson Laboratories | Stock: 003831 |
| Mouse: B6.FVB-Tg(MMTV-PyVT)634Mul/LellJ (PyMT) | Jackson Laboratories | Stock: 022974 |
| Mouse: B6.Cg-Tg(S100A8-Cre,-EGFP) 1Ilw/J (S100a8Cre) | Jackson Laboratories | Stock: 021614 |
| USA-LSL (“Universal Self Antigen”, Received from Dr. James J. Moon Lab) | Zhang Z. et al, 2019 Eur J Immunol. | N/A |
| Mouse: B6.129X1-Gt(ROSA)26Sortm1(EYFP)Cos/J (Rosa26LSL-YFP) | Jackson Laboratories | Stock: 006148 |
| Software and Algorithms | ||
| GraphPad Prism V.7-9 | GraphPad | |
| FlowJo V.9, V.10 | TreeStar | |
| FacsDIVA | BD Biosciences | |
| R software | ||
| Other | ||
Experimental Model and Subject Details
Animal experimental models
CD45.1+, Irf8fl/fl, OT-I, and S100a8Cre mice were purchased from Jackson Laboratory. IRF8EGFP and MafBiCre mice were generated as previously reported (Wang et al., 2014; Wu et al., 2016). LSL-USA mice were generated in the laboratory of Dr. James Moon (Zhang et al., 2019). MMTV-PyMT, CD11cCre and Rosa26LSL-YFP were in the lab as previously reported (Franklin et al., 2014). All mice used in these studies were on the C57BL/6 background and were female given the focus on the mammary gland and PyMT breast cancer. All mice were maintained under specific pathogen-free conditions, and animal experimentation was conducted in accordance with procedures approved by the Institutional Animal Care and Use Committee of MSKCC.
Human experimental models
Informed consent was obtained from ccRCC patients prior to surgery utilizing MSKCC IRB protocol 12–237. Tissue was processed on the same day as surgery. The cohort of ccRCC patients from which tissues were analyzed were 91.6% male, mean age 58.3, median age 58.
Method Details
Tumor measurements.
Beginning at 12 weeks of age, tumors were measured weekly with a caliper. Tumor volume was calculated using the equation [(LxW2) × (π/6)], in which L denotes length and W denotes width. Individual tumor volumes were added together to calculate total tumor burden, with an endpoint when total volume reached 3,000 mm3 or one tumor reached 2,000 mm3. Researchers were blinded to genotypes of mice during measurements.
CD8+ T cell depletion.
Upon palpable tumor appearance, mice were injected intraperitoneally with 300 μg CD8-depleting antibody (clone 2.43, Bio × Cell) every 3 to 4 days for 5 weeks. Tumors were measured every 7 days. Depletion of CD8 T cells was confirmed by checking the blood by flow cytometry.
Immune cell isolation from tissues.
Immune cells and cancer cells were isolated from mouse mammary gland, mouse tumors and human tumors as described previously (Dadi et al., 2016). Briefly, tissues were prepared by mechanical disruption with a razor blade followed by incubation in 280 U/mL Collagenase Type 3 and 4 μg/mL DNase I in HBSS at 37°C for 1 h with periodic vortex. Digested tissues were passed through 70 μm filters and pelleted. Cells were resuspended in 44% Percoll and layered above 66% Percoll. Sample was centrifuged at 1,900 g for 30 min without brake. Cells at interface were collected, stained with antibodies and analyzed by flow cytometry or used for FACS. To isolate cells from tumor-draining lymph nodes, tissues were mechanically disrupted with a razor blade, then incubated in 300 U/mL Collagenase Type 3 in RPMI supplemented with 10% FBS for 35 min at 37°C. EDTA was added for final concentration of 0.01 M, incubated for 5 min at 37°C, then 5 min on ice, then cells were passed through 70 μm filter for single cells suspension and downstream flow cytometric analysis.
Flow cytometry.
Cells were preincubated with FcBlock to block non-specific FcγR binding and Ghost Dye to detect dead cells. They were stained with antibodies targeting surface proteins for 30 min on ice. When needed, cells were then fixed and permeabilized for 30 min on ice and then stained with intracellular antibodies for 30 min on ice. All samples were acquired and analyzed with LSRII flow cytometer (Becton Dickson) and FlowJo software (TreeStar).
Cell sorting.
After gating on morphology and singlets, CD45+Dead− cells were gated as follows: Related to Figure 1: DC1 (Lin−F4/80−Ly6C−MHC-II+CD11c+Xcr1+CD11b−), DC2 (Lin−F4/80−Ly6C−MHC-II+CD11c+Xcr1−CD11b+), monocytes (Lin−F4/80+Ly6C+Xcr1−CD11b+), TAMs (Lin−F4/80+ Ly6C−MHC-II+Xcr1− excluding CD11bhigh), MTMs (Lin−F4/80+Ly6C−MHC-II+Xcr1-CD11bhigh) (Lin = SiglecF, B220, Ly6G, TCRβ, NK1.1). Each biological replicate was pooled tumors from PyMT mice, either two mice for DCs, or one mouse for monocytes, TAMs and MTMs, n = 2 for DC1, DC2, n = 3 for monocytes, MTMs and TAMs. Related to Figure 3: TAMs (F4/80+SiglecF−B220− Ly6G−Ly6C−TCRβ−NK1.1−Xcr1− excluding CD11bhigh [MTMs]) were sorted from tumors of Irf8fl/flPyMT and CD11cCreIrf8fl/flPyMT mice. Each biological replicate was pooled tumors of one mouse, n = 2 for each group. Related to Figure 5: TCRβ+CD8α+ cells were sorted from tumors of Irf8fl/flPyMT and MafBiCreIrf8fl/flPyMT mice (n=1 per genotype). Related to Figure S4: Cancer cells (Live CD45−CD24+CD29+EpCAM+) were sorted from tumors of PyMT mice. Related to Figure 6: Monocytes (Lin−CD14+HLA-DRlow to neg) and TAMs (Lin−CD14+HLA-DRhigh) were sorted from human RCC tumors (Lin = CD3, CD19, C56, CD15). Cell sorting was conducted on Aria II (Becton Dickson).
Bone marrow chimeras.
Recipient mice (CD45.1.2+ or CD45.1.1+ S100a8CreRosa26LSL-USAPyMT) were lethally irradiated with 900 Gyz at 8–12 weeks of age. Eighteen hours after irradiation, bone marrow was harvested from donor mice and injected intravenously into irradiated recipients. For mixed bone marrow chimeras related to Figure 4 and S4, bone marrow was harvested from CD45.1+ C57Bl/6 and CD45.2+ CD11cCreIrf8fl/fl mice, mixed 1:1 and co-transferred into the same recipient. For bone marrow chimeras related to Figure 5 and S5, bone marrow was harvested from CD45.2+ Irf8fl/fl and CD45.2+ MafBiCreIrf8fl/fl donors and transferred into separate recipients. Mice were maintained on sulfatrim antibiotic diet and aged to allow bone marrow to graft and PyMT tumors to grow. Upon large tumor burden (2000–3000 mm3), mice were euthanized and immune cells from tumor and draining lymph node were harvested for downstream analyses.
Antigen-specific T cell response.
Lymph nodes were harvested from OT-I TCR-transgenic mice. Single-cell suspension of lymph nodes was obtained by homogenization between frosted ends of two histological slides and passed through 70 μm filters. Naïve T cells were isolated using negative selection with magnetic beads, as per kit’s instructions. OT-I T cells were transferred into recipient mice, and tumor-infiltrating OT-I T cells were analyzed by flow cytometry two weeks later (Figure 5K and S5G–H). For in vitro experiments, OT-I T cells were stained with CellTrace Violet and plated in 96 U-bottom wells (15,000 T cells/well), cocultured with various ratios of TAMs sorted from tumors or migratory DC1 sorted from tumor-draining lymph nodes of chimeric mice in T cell media (RPMI supplemented with 10% FCS, 1 mM sodium pyruvate, non-essential amino acids [Gibco], 10 mM Hepes, 55 μM 2-Mercaptoethanol, 100 U/mL Penicillin G and 0.1 mg/ml Streptomycin) for 72 h at 37°C. Cells were then analyzed by flow cytometry (Figure 4).
Related to Figure S4G–H, OT-I T cells were isolated and labeled with CTV as described above. TAMs or cancer cells were sorted from tumors of Irf8fl/flPyMT and MafBiCreIrf8fl/flPyMT mice, and were pulsed by culturing in T cell media supplemented with the SIINFEKL peptide at 2 ng/mL for 1 h at 37°C. Cells were washed twice with T cell media, and OT-I T cells were added. TAMs or cancer cells were cultured with OT-I T cells for 72 h at 37°C, and then analyzed by flow cytometry.
RNA sequencing.
For bulk RNA sequencing, immune populations were sorted into Trizol LS and flash frozen. RNA was extracted with chloroform. Isopropanol and linear acrylamide were added, and the RNA was precipitated with 75% ethanol. 0.649–1 ng total RNA with RNA integrity numbers 6.8–10 underwent amplification using SMART-Seq v4 Ultra Low Input RNA Kit (Clontech #63488). 15 ng amplified cDNA was used to prepare libraries with KAPA Hyper Prep Kit (Kapa Biosystems KK8504) using 8 cycles of PCR. Samples were barcoded and run on a HiSeq 2500 in 50bp/50bp paired end run using HiSeq3000/4000 SBS Kit (Illumina) for 30–40 million paired reads. The raw sequencing FASTQ files were aligned against the hg19 and mm10 assembly by STAR for human and mouse samples respectively. FASTQ files of TCGA ccRCC cohort (KIRC) were downloaded from GDC and processed by the same procedures. Gene level count values were then computed by the summarizeOverlaps function from the R package “GenomicAlignments” with UCSC hg19 or mm10 KnownGene as the base gene model for human and mouse samples respectively. The Union counting mode was used and only mapped paired reads were considered. FPKM (Fragments Per Kilobase Million) values were then computed from gene level counts by using fpkm function from the R package “DESeq2”.
For single cell RNA sequencing, around 10,000 FACS sorted live cells (1,000 cells/ul suspension) were suspended in 1X PBS (calcium and magnesium free) containing 0.04% BSA (A2153, SIGMA). Cell suspension was used as input in 10x chromium controller system (10x Genomics Inc., product code 120223) and cell barcoding was performed by gel beads in emulsion (GEMs) in assembly chip. GEM RT reaction was performed in thermocycler (53°C for 45 min, 85°C for 5 min, 4°C hold overnight). SPRIselect dynabeads (B23318, Beckman Coulter) were used for GEM recovery post RT. 2–50ng of DNA was used for target enrichment. cDNA amplification, fragmentation, end repair and A tailing preparation was performed as per manual instructions. High sensitivity DNA chips and Agilent 2100 bioanalyzer (Agilent Technologies) was used for gene expression library profile quality control and quantification at several steps. Quality control was performed twice before sequencing. Two barcoded scRNA samples were pooled together before sequencing. scRNA libraries were sequenced on NovaSeq 6000 S1 with sequencing depth of approximately 300 million reads.
Quantification and Statistical Analysis
Figure 1B and Figure S3C: the regularized log transformation values generated by “DESeq2” of the top 500 varied genes were used for Principal Component Analysis and the first 2 principal components were shown in the plot. Analysis was performed on 17 samples: TAMs (n=3), MTMs (n=3), monocytes (n=3), DC1 (n=2), DC2 (n=2), and two pairs of Irf8fl/fl and CD11cCreIrf8fl/fl TAMs (n=2 each). All groups excluding Irf8fl/fl and CD11cCreIrf8fl/fl TAM pairs are shown in Figure 1B, all 17 samples are shown in Figure S3C.
Related to Figure 1C and Table S1, pairwise differentially expressed gene (DEG) analyses were done among 3 mouse sample groups, including monocytes, MTMs, and TAMs, by the R package DESeq2. Transcription factor genes (as annotated) were included if at least one pairwise comparison passed the following filters: FDR < 0.05, mean expression > 100, log2 fold change > 0.5, < −0.5. Related to Figure 3 and Figure S3, genes were significantly differentially expressed if passing the following filters: FDR < 0.05, mean expression > 50, log2 fold change > 2 or < −2. Mouse IRF8 Irf8fl/fl-UP or IRF8 CD11cCreIrf8fl/fl-UP signatures were built from DEG genes that passed filters listed above as well as mean FPKM > 1 and variance > 3. The mean FPKM value of selected gene was then calculated for each sample group. The log2 transformed mean FPKM values of signature genes were used for the empirical cumulative distribution plot by the R package “ggplot2”. An offset value of 0.001 was added to mean FPKM values prior to log2 transformation to avoid log2(0). DEG results for Irf8fl/fl versus CD11cCreIrf8fl/fl TAMs (Figure S3C) were used in pathway analysis using IPA (QIAGEN Inc.) with the Ingenuity Knowledge Base as the reference set(Kramer et al., 2014). IPA evaluated the expression pattern of genes in canonical pathways and predicted the canonical pathways to be active or inactive.
The mouse DEG genes resulting from comparing Irf8fl/fl to CD11cCreIrf8fl/fl sample group with mean expression > 50, log2 fold change > 2 or < −2, FDR value < 0.05 underwent human homologous gene search. The corresponding human homologous genes were identified by referencing the Mouse Genome Informatics (MGI, http://www.informatics.jax.org/) database and NCBI Orthologs database (ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/gene_orthologs.gz), and any gene without a human homolog was excluded from the human IRF8 signature. Gene Set Enrichment Analysis (GSEA) was done by applying the IRF8-activated human homologous genes against the DEG genes from human TAMs to tumor infiltrated human monocytes comparison through the R package “clusterProfiler” (Figure 6D). To ensure decent expression level in human cells, mouse IRF8-Activated genes were further filtered by having mean FPKM > 1 and variance > 3 among sorted human tumor monocytes and TAMs. These were used to build the human IRF8 signature (Figure 7A). To assess the IRF8 signature enrichment in human immune cell populations, Single-Sample Gene Set Enrichment Analysis (ssGSEA) was done by applying the human IRF8 signatures against the FPKM expression values of sorted human immune cell populations through the R package “GSVA” which estimates the IRF8 signature enrichment score for each of RNASeq samples (Figure 7B).
The cohort sample was classified as IRF8high when the IRF8 signature ssGSEA enrichment score is greater or equal to the median score and as IRF8low when it is smaller than the median score. DEG analysis between IRF8high and low groups was performed and the results were subjected to GSEA analysis against 2 gene signatures, the Mac_CSF1 signature (Beck et al., 2009) (Figure 7C) and the T cell exhaustion/Checkpoint signature including several well-known T cell exhaustion markers and checkpoint markers, such as CTLA4, LAG3, PDCD1, CD274, PDCD1LG2, HAVCR2, BTLA and TIGIT (Figure 7D).
For scRNAseq analysis, the raw reads of single cell RNA sequencing from FASTQ files were aligned against the mm10 mouse genome and the UMI count of individual genes were captured by Cell Ranger v3.0.2 (10X Genomics) with “expected-cells=1000” parameter setting and default value for the rest of required parameters. The R package Seurat 3.1.1(Butler et al., 2018; Stuart et al., 2019) was used for further QC, clustering, plotting and differentially expressed genes analyses. Based on the observation of low mitochondria read percentage distribution and PCA analysis results, cells filtering and batch correction procedure was not performed after merging cells from MafBiCreIrf8fl/flPyMT and Irf8fl/flPyMTmice. The SCTransform() function which implemented the sctransform package (https://github.com/ChristophH/sctransform) was used for UMI count normalization, scaling, and variant gene searching for further dimensionality reduction via calling RunPCA() function. The top 16 principal components (PCs) were chosen and used for Louvain clustering analysis using the FindClusters() function at resolution 0.6 after constructing the Shared Nearest Neighbor (SNN) Graph through calling FindNeighbors() on the selected PCs. A two-dimensional embedding of the data was generated using UMAP with the top 16 PCs as input by the RunUMAP() function. This first run of clustering revealed 11 cell clusters including T cell, innate-like T cell (ILTC), CD68+, possibly degraded T cell, and non-immune cell clusters. After excluding cells from CD68+, possibly degraded T cell, and non-immune cell clusters, a second run of clustering was performed on a total of 375 cells from Irf8fl/flPyMT tumors and 1525 cells from MafBiCreIrf8fl/flPyMT tumors, with top 11 PCs being selected after dimensionality reduction and 9 clusters found, including naïve CD8 (C1: Irf8fl/flPyMT=1 cell, MafBiCreIrf8fl/flPyMT=62 cells), effector/memory CD8 (C2a: Irf8fl/flPyMT=41 cells, MafBiCreIrf8fl/flPyMT=402 cells, C2b: Irf8fl/flPyMT=18 cells, MafBiCreIrf8fl/flPyMT=267 cells), exhaustion CD8 (C3a: Irf8fl/flPyMT=73 cells, MafBiCreIrf8fl/flPyMT=110 cells, C3b: Irf8fl/flPyMT=85 cells, MafBiCreIrf8fl/flPyMT=56 cells), transitioning CD8 (C4: Irf8fl/flPyMT=17 cells, MafBiCreIrf8fl/flPyMT=86 cells), ILTC (C5a: Irf8fl/flPyMT=58 cells, MafBiCreIrf8fl/flPyMT=219 cells, C5b: Irf8fl/flPyMT=25 cells, MafBiCreIrf8fl/flPyMT=130 cells), and proliferating CD8 (C6: Irf8fl/flPyMT=57 cells, MafBiCreIrf8fl/flPyMT=193 cells) clusters. Violin plots of selected genes and gene signature were built by the VlnPlot() function. The Fisher’s Exact test was employed to test for the statistical significance of observed cell abundance difference of each cluster between MafBiCreIrf8fl/flPyMTand Irf8fl/flPyMTgroups. Differential gene expression analysis was conducted through the FindMarkers() function by using the Wilcoxon rank-sum test as the statistical test and a minimum of 10% cell expressing the gene and with FDR P < 0.05. Significantly differentially expressed genes (DEGs) between each set of paired clusters (a & b) were identified and enriched by immediate early genes (IEGs). The top most significant 40 genes between paired clusters were included for building the expression heatmaps by the DoHeatmap() function. Differential gene expression analysis was also conducted for each cluster by comparing the cluster cells to the cells from the other clusters as the reference cells. For those paired clusters, cells from their paired mate cluster were excluded from the reference cell group and the top 20 common DEGs among paired clusters were selected in building the cluster gene expression heatmap along with the top 20 DEGs from singleton clusters.
All data are displayed as mean +/− SEM. For pair-wise comparisons, unpaired or paired student t test, two-tailed was conducted using GraphPad Prism software. For tumor growth, 2-way ANOVA was performed using GraphPad Prism software. The analyzed data is provided in Tables S1–5.
Supplementary Material
Highlights:
IRF8-expressing TAMs present cancer cell antigen, and drive CTL exhaustion
MafBiCreIrf8flPyMT mice lose TAM antigen presentation, gain anti-tumor CTL response
TAMs from human cancer patients express IRF8 and an IRF8-dependent gene program
A TAM-IRF8 gene signature tracks with CTL exhaustion in multiple human malignancies
Acknowledgements
We acknowledge the use of the Integrated Genomics Operation Core, funded by the NCI Cancer Center Support Grant (CCSG, P30 CA08748), Cycle for Survival, and the Marie-Josée and Henry R. Kravis Center for Molecular Oncology. This work was supported by the National Institutes of Health (F31 CA210332 to B.G.N. and R01 CA198280 to M.O.L.), the Howard Hughes Medical Institute (Faculty Scholar Award to M.O.L.), Alan and Sandra Gerry Metastasis and Tumor Ecosystems Center (M.O.L.), the Ludwig Center (A.A.H), the Parker Institute (A.A.H.) and the Weiss Family Foundation (A.A.H).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
All authors declare that they have no competing interests.
Inclusion and Diversity
We support inclusive, diverse, and equitable conduct of research.
References
- Aliberti J, Schulz O, Pennington DJ, Tsujimura H, Reis e Sousa C, Ozato K, and Sher A (2003). Essential role for ICSBP in the in vivo development of murine CD8alpha + dendritic cells. Blood 101, 305–310. [DOI] [PubMed] [Google Scholar]
- Baumeister SH, Freeman GJ, Dranoff G, and Sharpe AH (2016). Coinhibitory Pathways in Immunotherapy for Cancer. Annu Rev Immunol 34, 539–573. [DOI] [PubMed] [Google Scholar]
- Beck AH, Espinosa I, Edris B, Li R, Montgomery K, Zhu S, Varma S, Marinelli RJ, van de Rijn M, and West RB (2009). The macrophage colony-stimulating factor 1 response signature in breast carcinoma. Clin Cancer Res 15, 778–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi K, He MX, Bakouny Z, Kanodia A, Napolitano S, Wu J, Grimaldi G, Braun DA, Cuoco MS, Mayorga A, et al. (2021). Tumor and immune reprogramming during immunotherapy in advanced renal cell carcinoma. Cancer Cell 39, 649–661 e645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, Angell H, Fredriksen T, Lafontaine L, Berger A, et al. (2013). Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 39, 782–795. [DOI] [PubMed] [Google Scholar]
- Biswas SK, Gangi L, Paul S, Schioppa T, Saccani A, Sironi M, Bottazzi B, Doni A, Vincenzo B, Pasqualini F, et al. (2006). A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-kappaB and enhanced IRF-3/STAT1 activation). Blood 107, 2112–2122. [DOI] [PubMed] [Google Scholar]
- Braun DA, Street K, Burke KP, Cookmeyer DL, Denize T, Pedersen CB, Gohil SH, Schindler N, Pomerance L, Hirsch L, et al. (2021). Progressive immune dysfunction with advancing disease stage in renal cell carcinoma. Cancer Cell 39, 632–648 e638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, Barczak A, Rosenblum MD, Daud A, Barber DL, et al. (2014). Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 26, 638–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campana S, De Pasquale C, Carrega P, Ferlazzo G, and Bonaccorsi I (2015). Cross-dressing: an alternative mechanism for antigen presentation. Immunol Lett 168, 349–354. [DOI] [PubMed] [Google Scholar]
- Chen DS, and Mellman I (2017). Elements of cancer immunity and the cancer-immune set point. Nature 541, 321–330. [DOI] [PubMed] [Google Scholar]
- Chou C, Zhang X, Krishna C, Nixon BG, Dadi S, Capistrano KJ, Kansler ER, Steele M, Han J, Shyu A, et al. (2022). Programme of self-reactive innate-like T cell-mediated cancer immunity. Nature 605, 139–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung W, Eum HH, Lee HO, Lee KM, Lee HB, Kim KT, Ryu HS, Kim S, Lee JE, Park YH, et al. (2017). Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat Commun 8, 15081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dadi S, Chhangawala S, Whitlock BM, Franklin RA, Luo CT, Oh SA, Toure A, Pritykin Y, Huse M, Leslie CS, and Li MO (2016). Cancer Immunosurveillance by Tissue-Resident Innate Lymphoid Cells and Innate-like T Cells. Cell 164, 365–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dangaj D, Bruand M, Grimm AJ, Ronet C, Barras D, Duttagupta PA, Lanitis E, Duraiswamy J, Tanyi JL, Benencia F, et al. (2019). Cooperation between Constitutive and Inducible Chemokines Enables T Cell Engraftment and Immune Attack in Solid Tumors. Cancer Cell 35, 885–900 e810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Mingo Pulido A, Gardner A, Hiebler S, Soliman H, Rugo HS, Krummel MF, Coussens LM, and Ruffell B (2018). TIM-3 Regulates CD103(+) Dendritic Cell Function and Response to Chemotherapy in Breast Cancer. Cancer Cell 33, 60–74 e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delamarre L, Pack M, Chang H, Mellman I, and Trombetta ES (2005). Differential lysosomal proteolysis in antigen-presenting cells determines antigen fate. Science 307, 1630–1634. [DOI] [PubMed] [Google Scholar]
- DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, and Coussens LM (2009). CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 16, 91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNardo DG, and Ruffell B (2019). Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol 19, 369–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao J, Gu H, Tang M, Zhao J, and Cattral MS (2018). Tumor Dendritic Cells (DCs) Derived from Precursors of Conventional DCs Are Dispensable for Intratumor CTL Responses. J Immunol 201, 1306–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolina JS, Van Braeckel-Budimir N, Thomas GD, and Salek-Ardakani S (2021). CD8(+) T Cell Exhaustion in Cancer. Front Immunol 12, 715234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimkhani MR, Mohar I, and Crispe IN (2011). Cross-presentation of antigen by diverse subsets of murine liver cells. Hepatology 54, 1379–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, Pamer EG, and Li MO (2014). The cellular and molecular origin of tumor-associated macrophages. Science 344, 921–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD, et al. (2015). The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med 21, 938–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux F, and Jung S (2014). Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol 14, 392–404. [DOI] [PubMed] [Google Scholar]
- Hagemeyer N, Kierdorf K, Frenzel K, Xue J, Ringelhan M, Abdullah Z, Godin I, Wieghofer P, Costa Jordao MJ, Ulas T, et al. (2016). Transcriptome-based profiling of yolk sac-derived macrophages reveals a role for Irf8 in macrophage maturation. EMBO J 35, 1730–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna RN, Cekic C, Sag D, Tacke R, Thomas GD, Nowyhed H, Herrley E, Rasquinha N, McArdle S, Wu R, et al. (2015). Patrolling monocytes control tumor metastasis to the lung. Science 350, 985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, et al. (2008). Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322, 1097–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kansler ER, Dadi S, Krishna C, Nixon BG, Stamatiades EG, Liu M, Kuo F, Zhang J, Zhang X, Capistrano K, et al. (2022). Cytotoxic innate lymphoid cells sense cancer cell-expressed interleukin-15 to suppress human and murine malignancies. Nat Immunol 23, 904–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kc W, Satpathy AT, Rapaport AS, Briseno CG, Wu X, Albring JC, Russler-Germain EV, Kretzer NM, Durai V, Persaud SP, et al. (2014). L-Myc expression by dendritic cells is required for optimal T-cell priming. Nature 507, 243–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersten K, Hu KH, Combes AJ, Samad B, Harwin T, Ray A, Rao AA, Cai E, Marchuk K, Artichoker J, et al. (2022). Spatiotemporal co-dependency between macrophages and exhausted CD8(+) T cells in cancer. Cancer Cell 40, 624–638 e629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komohara Y, Jinushi M, and Takeya M (2014). Clinical significance of macrophage heterogeneity in human malignant tumors. Cancer Sci 105, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer A, Green J, Pollard J Jr., and Tugendreich S (2014). Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanca T, Ungerback J, Da Silva C, Joeris T, Ahmadi F, Vandamme J, Svensson-Frej M, Mowat AM, Kotarsky K, Sigvardsson M, and Agace WW (2022). IRF8 deficiency induces the transcriptional, functional, and epigenetic reprogramming of cDC1 into the cDC2 lineage. Immunity. [DOI] [PubMed] [Google Scholar]
- Langlais D, Barreiro LB, and Gros P (2016). The macrophage IRF8/IRF1 regulome is required for protection against infections and is associated with chronic inflammation. J Exp Med 213, 585–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin EY, Nguyen AV, Russell RG, and Pollard JW (2001). Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med 193, 727–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Wei S, Hurt EM, Green MD, Zhao L, Vatan L, Szeliga W, Herbst R, Harms PW, Fecher LA, et al. (2018). Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade-mediated tumor regression. J Clin Invest 128, 805–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loyher PL, Hamon P, Laviron M, Meghraoui-Kheddar A, Goncalves E, Deng Z, Torstensson S, Bercovici N, Baudesson de Chanville C, Combadiere B, et al. (2018). Macrophages of distinct origins contribute to tumor development in the lung. J Exp Med 215, 2536–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacNabb BW, Tumuluru S, Chen X, Godfrey J, Kasal DN, Yu J, Jongsma MLM, Spaapen RM, Kline DE, and Kline J (2022). Dendritic cells can prime anti-tumor CD8(+) T cell responses through major histocompatibility complex cross-dressing. Immunity 55, 982–997 e988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLane LM, Abdel-Hakeem MS, and Wherry EJ (2019). CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu Rev Immunol 37, 457–495. [DOI] [PubMed] [Google Scholar]
- Mellman I, and Steinman RM (2001). Dendritic cells: specialized and regulated antigen processing machines. Cell 106, 255–258. [DOI] [PubMed] [Google Scholar]
- Muhitch JB, Hoffend NC, Azabdaftari G, Miller A, Bshara W, Morrison CD, Schwaab T, and Abrams SI (2019). Tumor-associated macrophage expression of interferon regulatory Factor-8 (IRF8) is a predictor of progression and patient survival in renal cell carcinoma. J Immunother Cancer 7, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy TL, Grajales-Reyes GE, Wu X, Tussiwand R, Briseno CG, Iwata A, Kretzer NM, Durai V, and Murphy KM (2016). Transcriptional Control of Dendritic Cell Development. Annu Rev Immunol 34, 93–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell P, Hyslop S, Blake MK, Godbehere S, Amalfitano A, and Aldhamen YA (2021). SLAMF7 Signaling Reprograms T Cells toward Exhaustion in the Tumor Microenvironment. J Immunol 206, 193–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SA, Wu DC, Cheung J, Navarro A, Xiong HZ, Cubas R, Totpal K, Chiu H, Wu Y, Comps-Agrar L, et al. (2020). PD-L1 expression by dendritic cells is a key regulator of T-cell immunity in cancer. Nat Cancer 1, 681–+. [DOI] [PubMed] [Google Scholar]
- Roberts AW, Lee BL, Deguine J, John S, Shlomchik MJ, and Barton GM (2017). Tissue-Resident Macrophages Are Locally Programmed for Silent Clearance of Apoptotic Cells. Immunity 47, 913–927 e916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, Kaisho T, Bogunovic D, Bhardwaj N, and Krummel MF (2016). Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 30, 324–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, Delgado A, Correa P, Brayer J, Sotomayor EM, et al. (2004). Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res 64, 5839–5849. [DOI] [PubMed] [Google Scholar]
- Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CM, Pryer N, Daniel D, Hwang ES, Rugo HS, and Coussens LM (2014). Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 26, 623–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, Casanova-Acebes M, Khudoynazarova M, Agudo J, Tung N, et al. (2016). Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 44, 924–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavoni G, Mattei F, Sestili P, Borghi P, Venditti M, Morse HC 3rd, Belardelli F, and Gabriele L (2002). ICSBP is essential for the development of mouse type I interferon-producing cells and for the generation and activation of CD8alpha(+) dendritic cells. J Exp Med 196, 1415–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, and Allison JP (2015). Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 161, 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sichien D, Scott CL, Martens L, Vanderkerken M, Van Gassen S, Plantinga M, Joeris T, De Prijck S, Vanhoutte L, Vanheerswynghels M, et al. (2016). IRF8 Transcription Factor Controls Survival and Function of Terminally Differentiated Conventional and Plasmacytoid Dendritic Cells, Respectively. Immunity 45, 626–640. [DOI] [PubMed] [Google Scholar]
- Singh H, Glasmacher E, Chang AB, and Vander Lugt B (2013). The molecular choreography of IRF4 and IRF8 with immune system partners. Cold Spring Harb Symp Quant Biol 78, 101–104. [DOI] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Liang Y, Anders RA, Taube JM, Qiu X, Mulgaonkar A, Liu X, Harrington SM, Guo J, Xin Y, et al. (2018). PD-L1 on host cells is essential for PD-L1 blockade-mediated tumor regression. J Clin Invest 128, 580–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner J, Rapsomaniki MA, Chevrier S, Anzeneder T, Langwieder C, Dykgers A, Rees M, Ramaswamy A, Muenst S, Soysal SD, et al. (2019). A Single-Cell Atlas of the Tumor and Immune Ecosystem of Human Breast Cancer. Cell 177, 1330–1345 e1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yan M, Sun J, Jain S, Yoshimi R, Abolfath SM, Ozato K, Coleman WG Jr., Ng AP, Metcalf D, et al. (2014). A reporter mouse reveals lineage-specific and heterogeneous expression of IRF8 during lymphoid and myeloid cell differentiation. J Immunol 193, 1766–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Briseno CG, Durai V, Albring JC, Haldar M, Bagadia P, Kim KW, Randolph GJ, Murphy TL, and Murphy KM (2016). Mafb lineage tracing to distinguish macrophages from other immune lineages reveals dual identity of Langerhans cells. J Exp Med 213, 2553–2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA, Chawla A, and Pollard JW (2013). Macrophage biology in development, homeostasis and disease. Nature 496, 445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Zhu J, Smith S, Foldi J, Zhao B, Chung AY, Outtz H, Kitajewski J, Shi C, Weber S, et al. (2012). Notch-RBP-J signaling regulates the transcription factor IRF8 to promote inflammatory macrophage polarization. Nat Immunol 13, 642–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yost KE, Chang HY, and Satpathy AT (2021). Recruiting T cells in cancer immunotherapy. Science 372, 130–131. [DOI] [PubMed] [Google Scholar]
- Zhang QW, Liu L, Gong CY, Shi HS, Zeng YH, Wang XZ, Zhao YW, and Wei YQ (2012). Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PLoS One 7, e50946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Legoux FP, Vaughan SW, and Moon JJ (2019). Opposing peripheral fates of tissue-restricted self antigen-specific conventional and regulatory CD4(+) T cells. Eur J Immunol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Fei M, Zhang G, Liang WC, Lin W, Wu Y, Piskol R, Ridgway J, McNamara E, Huang H, et al. (2020). Blockade of the Phagocytic Receptor MerTK on Tumor-Associated Macrophages Enhances P2X7R-Dependent STING Activation by Tumor-Derived cGAMP. Immunity 52, 357–373 e359. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Herndon JM, Sojka DK, Kim KW, Knolhoff BL, Zuo C, Cullinan DR, Luo J, Bearden AR, Lavine KJ, et al. (2017). Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 47, 597. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw sequencing data for bulk RNA sequencing on myeloid lineage cells from the tumor in mouse and tumor immune populations from RCC patients and for single cell RNA sequencing on CD8+ T cells from Irf8fl/flPyMT and MafBiCreIrf8fl/flPyMT tumors have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse CD103 | BD Biosciences | M290 |
| Anti-mouse CD11b | BD Biosciences | M1/70 |
| Anti-mouse CD11c | BD Biosciences | N418 |
| Anti-mouse CD4 | BD Biosciences | RM4-5 |
| Anti-mouse CD45 | BD Biosciences | 30-F11 |
| Anti-mouse Ki67 | BD Biosciences | B56 |
| Anti-mouse Ly6C | BD Biosciences | AL-21 |
| Anti-mouse MHC-II I-A/I-E | BD Biosciences | M5/114.15.2 |
| Anti-mouse SiglecF | BD Biosciences | E50-2440 |
| Anti-human CD141 | BD Biosciences | 1A4 |
| Anti-human CD1c | BD Biosciences | F10/21A3 |
| Anti-mouse CD206 | BioLegend | C068C2 |
| Anti-mouse H-2Kb bound to SIINFEKL | BioLegend | 25-D1.16 |
| Anti-mouse XCR1 | BioLegend | ZET |
| Anti-human CD15 | BioLegend | W6D3 |
| Anti-human CD16 | BioLegend | 3G8 |
| Anti-human CD19 | BioLegend | HIB19 |
| Anti-mouse CD29 | BioLegend | HmB1-1 |
| Anti-mouse CD326 (EpCAM) | BioLegend | G8.8 |
| Anti-human CD56 | BioLegend | HCD56 |
| Anti-human HLA-DR | BioLegend | L243 |
| Anti-mouse CD24 | eBioscience/Thermo | M1/69 |
| Anti-mouse CD80 | eBioscience/Thermo | 16-10A1 |
| Anti-mouse CD86 | eBioscience/Thermo | GL1 |
| Anti-mouse FoxP3 | eBioscience/Thermo | FJK-16s |
| Anti-mouse NK1.1 | eBioscience/Thermo | PK136 |
| Anti-mouse PD-L1 | eBioscience/Thermo | MIH5 |
| Anti-mouse PD-1 | eBioscience/Thermo | RMP1-30 |
| Anti-mouse TIM-3 | eBioscience/Thermo | 8B.2C12 |
| Anti-mouse Vcam1 | eBioscience/Thermo | 429 |
| Anti-mouse/human IRF8 | eBioscience/Thermo | V3GYWCH |
| Anti-human CD14 | eBioscience/Thermo | 61D3 |
| Anti-human/mouse granzyme B | Invitrogen | GB11 |
| Anti-human CD3d | Invitrogen | 7D6 |
| CellTrace Violet | Invitrogen | |
| Anti-mouse B220 | Tonbo Biosciences | RA3-6B2 |
| Anti-mouse CD8a | Tonbo Biosciences | 53-6.7 |
| Anti-mouse Ly6G | Tonbo Biosciences | 1A8 |
| Anti-mouse TCRB | Tonbo Biosciences | H57-597 |
| Anti-human CD45 | Tonbo Biosciences | 2D1 |
| Ghost Dye | Tonbo Biosciences | 13-0865-T500 13-0863-T500 13-0870-T500 |
| CD8 depleting antibody | Bio × Cell | Clone 2.43 |
| FcR Block (2.4G2 mAB) | Bio × Cell | 25 mg (custom) |
| Human TruStain FcX | BioLegend | 422302 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Collagenase Type 3 | Worthington Biochemical | LS004183 |
| Dnase I | Sigma-Aldrich | |
| Percoll | Sigma-Aldrich | P1644-1L |
| Trizol LS | Invitrogen | |
| OVA(257–264) | Invivogen | |
| Critical Commercial Assays | ||
| Transcription factor Fix/Perm Kit | Tonbo Biosciences | TNB-0607-KIT |
| Naïve CD8 T cell Negative Selection Kit | EasySep, Stem Cell Technologies | 19858 |
| Deposited Data | ||
| Murine bulk RNAseq/scRNAseq sequencing files | GEO | GSE212643 |
| Human TAM monocyte bulk RNAseq sequencing files | EGA | Dataset ID: EGAD00001009393 Study ID: EGAS00001006593 |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6.Cg-Tg(Itgax-cre)1-1Reiz/J (CD11cCre) (Received from Dr. Boris Reizis Lab) | Jackson Laboratories | Stock: 008068 |
| Mouse: B6.SJL-PtprcaPepcb/BoyJ (CD45.1) | Jackson Laboratories | Stock: 002014 |
| Mouse: B6(Cg)-Irf8tm1.1Hm/J (Irf8flox) | Jackson Laboratories | Stock: 014175 |
| Mouse: B6(Cg)-Irf8tm2.1Hm/J(Irf8EGFP) (Received from Dr. Herbert C. Morse III Lab) | Jackson Laboratories | Stock: 027084 |
| Mouse: B6N(129S4)-Mafbtm1.1(cre)Kmm/J (MafBiCre) (Received from Dr. Kenneth Murphy Lab) | Jackson Laboratories | Stock: 029664 |
| Mouse: C57BL/6-Tg(TcraTcrb)1100Mjb/J (OT-I) | Jackson Laboratories | Stock: 003831 |
| Mouse: B6.FVB-Tg(MMTV-PyVT)634Mul/LellJ (PyMT) | Jackson Laboratories | Stock: 022974 |
| Mouse: B6.Cg-Tg(S100A8-Cre,-EGFP) 1Ilw/J (S100a8Cre) | Jackson Laboratories | Stock: 021614 |
| USA-LSL (“Universal Self Antigen”, Received from Dr. James J. Moon Lab) | Zhang Z. et al, 2019 Eur J Immunol. | N/A |
| Mouse: B6.129X1-Gt(ROSA)26Sortm1(EYFP)Cos/J (Rosa26LSL-YFP) | Jackson Laboratories | Stock: 006148 |
| Software and Algorithms | ||
| GraphPad Prism V.7-9 | GraphPad | |
| FlowJo V.9, V.10 | TreeStar | |
| FacsDIVA | BD Biosciences | |
| R software | ||
| Other | ||