

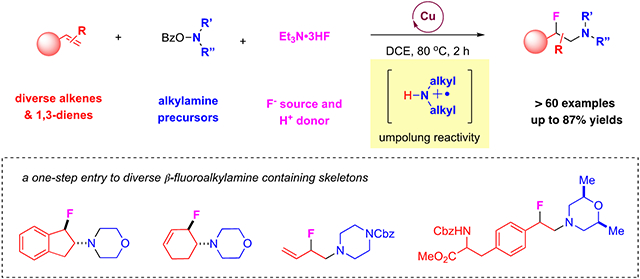
Abstract
Rapid and efficient access to structurally diverse β-fluoroalkylamines is in high demand, with their wide presence and great importance in medicinal chemistry and drug development. Direct 1,2-aminofluorination of alkenes offers an ideal strategy for one-step entry to β-fluorinated amines from readily available starting materials. Yet the synthesis of valuable β-fluorinated alkylamines remains an unsolved challenge, due to the inherent incompatibility between electrophilic fluoride sources and the electron-rich alkylamines. We report an unprecedented, catalytic, three-component aminofluorination of diverse alkenes and 1,3-dienes, which has been achieved by an innovative copper-catalyzed electrophilic amination strategy using O-benzoylhydroxylamines as alkylamine precursors. The use of Et3N•3HF is also critical, not only as a commercially available and inexpensive fluoride source to enable effective fluorination but also as an acid source for the formation of aminyl radical cation for electrophilic amination. Mechanistic experiments suggest the involvement of aminyl radical species and carbon-radical intermediates under reaction conditions. This method features high regioselectivity, good tolerance of diverse functional groups, and provides a practical and direct entry to a broad range of β-fluorinated electron-rich alkylamines. Synthetic applications of this method have also been highlighted by its use for the rapid entry to β-fluoridated amine-containing pharmaceuticals, natural products, and bioactive compounds.
Graphical Abstract
INTRODUCTION
The importance of nitrogen-containing molecules is evident by their ubiquitous presence in bioactive natural products, small-molecule probes, and pharmaceuticals. Among FDA-approved small-molecule drugs, over 80% entail at least one nitrogen atom. Therefore, rapid, efficient synthesis of novel bioactive nitrogen-containing compounds is of the utmost importance in biomedical research and drug discovery. The introduction of a fluorine to amine-containing compounds is a powerful strategy in medicinal chemistry and drug discovery that can productively influence their conformation, pKa, physical, chemical, biological, and pharmacokinetic properties.1-6 Particularly, β-fluorination of an amino functionality can greatly modulate the basicity of the vicinal amino group, offering dramatic enhancements in bioavailability, lipophilicity, and biological activity (Figure 1, A).7, 8 Accordingly, β-fluoroamine motifs are highly valued in drug development and the preparation of β-fluorinated amines is in great demand.
Figure 1. Alkene Aminofluorination for the Synthesis of β-Fluoroamines.

Traditionally, the synthesis of β-fluorinated amines has typically proceeded with a multiple step sequence, e.g., through substitution reactions on aziridines, β-amino alcohols, or β-fluoroalcohols with a fluoride or amine nucleophile.9 Alternatively, direct 1,2-aminofluorination of alkenes offers an ideal strategy allowing rapid access to β-fluorinated amines from readily available starting materials (Figure 1, B).10-12 Extensive efforts have been devoted to alkene aminofluorination in recent years, which elegantly expedite the synthesis of vicinal aminofluorine scaffolds.13, 14 However, the known transformations have been limited to the use of a few specially substituted electro-deficient amines for the preparation of β-fluorosulfonamides,15-21 β-fluorocarbamates,22-24 β-fluorosulfoximines,25 and the use of azides to realize β-fluoroazides.26 Despite the attractiveness of these studies, the amine functionalities introduced in these methods need to be further derivatized to access medicinally valuable entities. In sharp contrast, a one-step entry to β-fluoroalkylamines remains an unsolved synthetic challenge.27 It is fundamentally problematic to develop such a transformation using conventionally nucleophilic alkylamines, given the inherent incompatibility of electron-rich alkylamines with electrophilic fluorination reagents or strong oxidizing conditions.28 To overcome these obstacles, an innovative synthetic strategy is needed to realize such a desired yet challenging transformation that allows for direct access to valuable β-fluoroalkylamines.
We envision that an umpolung electrophilic amination strategy would offer an effective solution for the direct installation of electron-rich alkylamines (Figure 1, C). Such an unconventional strategy engages a different alkene activation pathway through electrophilic amination using heteroatom-substituted alkylamine precursors (LG–NR2), allowing for a subsequent coupling with nucleophilic partners. Our group has recently discovered a novel alkene activation mode initiated by copper-catalyzed N─O bond cleavage of O-acyl N, N-dialkylhydroxylamines and electrophilic amination addition to the alkenes, and achieved a series of copper-catalyzed alkene amino oxygenation reactions.29-31 The Morandi group has also reported elegant examples of iron-catalyzed aminochlorination using O-pivaloyl hydroxylamine triflic acid derivatives, conceivably through the formation of electrophilic protonated aminyl radical species.32, 33 Although this umpolung strategy represents a highly promising new platform for alkene amino functionalization, the incorporation of different nucleophilic partners is not trivial, owing to high reactivities of the transient nitrogen or carbon radicals that can readily undergo undesired degradation and side reactions. Furthermore, the development of the aminofluorination is particularly difficult for this umpolung approach via a protonated aminyl radical species, due to the low nucleophilicity of fluoride ions in protic solvents.
Here we disclose the development of an unprecedented copper-catalyzed protocol for aminofluorination of diversely substituted alkenes and 1,3-dienes for the selective formation of a wide range of β-fluoroalkylamines using readily available N, N-dialkylhydroxylamines34, 35 as a precursor of electron-rich alkylamines (Figure 1, D). In this study, we identified Et3N•3HF as the critical nucleophilic partner, which not only enables effective fluorination step as a convenient fluorinating agent but also contributes as an acid source for the formation of aminyl radical species that participates in the electrophilic alkene amination step. This work provides, for the first time, a catalytic aminofluorination transformation that allows for one-step installation of β-fluoro electron-rich tertiary amines onto alkenes and 1,3-dienes in a three-component manner.36 Synthetic values of this method have been highlighted by its application to the rapid synthesis of β-fluoroamine-containing pharmaceuticals, natural products, and bioactive compounds.
RESULTS AND DISCUSSION
ALKENE AMINOFLUORINATION – CONDITION DEVELOPMENT
Our studies began with the aminofluorination of 4-methylstyrene 1a using O-benzoyl-N-hydroxylmorpholine 2a as the alkylamine precursor (Figure 2). First, various fluoride sources were tested including Olah’s reagent, silver fluoride, cesium fluoride, and tetrabutylammonium fluoride and triethylamine trihydrofluoride (Et3N•c–5). Et3N•3HF, a commercially available and inexpensive fluoride source, was found to be effective in promoting the aminofluorination reaction and provide desired product 3a in 40% yield. Observed under this condition was common byproduct 3a’ which resulted from the nucleophilic addition of benzoate leaving group. We also confirmed that 3a’ was not a competent intermediate for the formation of 3a under reaction conditions. Encouragingly, the formation of 3a’ was suppressed by using excess Et3N•3HF (10 equiv) (entry 6). Next, we examined the effect of the leaving groups of the amine precursors in this reaction (Figure 2, B). Selected examples of the leaving group include different benzoates (A–D), acetoxy group (E), trichloroacetoxy (F), a carbonate group (G), a phosphinate group (H), and O-aryl hydroxylamine (I). Although many of them provided desired product 3a, none was more effective in the aminofluorination reaction than 2a bearing a simple benzoate group (A). Thus, we decided to focus on the aminofluorination of 1a using 2a and Et3N•3HF for further optimization (details in SI). Among varied reaction parameters, the choice of copper catalyst showed a significant effect on the reaction (Figure 2, C). Copper(II) hexafluoroacetylacetonate was found to be the most effective among various copper (I) or (II) catalysts. Even with a reduced 2.5 mol % loading, the reaction afforded 3a in 82% yield, which was chosen as the standard conditions.
Figure 2. Condition Development for Alkene Aminofluorination Reaction.

Conditions: 1a (2.0 equiv), 2a (0.2 mmol, 1.0 equiv), [F−] reagent, DCE (1.0 mL), 80 °C, 2 h, in 10-mL sealed FEP tube. Yields were determined by 1H NMR of the crude mixture with dibromomethane as an internal standard. Isolated yields shown in parentheses. aCu(OAc)2 (10 mol %) used. bCu(OAc)2 (10 mol %) Et3N·3HF (10 equiv), hydroxylamine derivative (1.0 equiv). c2a (1.0 equiv), Et3N·3HF (10 equiv).
ALKENE AMINOFLUORINATION – SCOPE OF ALKENES AND AMINES
With the standard conditions established, we investigated the generality of this aminofluorination reaction on different types of alkenes and amines (Table 1). We first examined the variation of the alkene component toward the formation of β-fluoroamines 3 using 2a. Different styrene derivatives all participated successfully in the reactions, delivering β-fluoroamines 3a–3o in good to high yields (45–83%). Note that various substituents were well tolerated, ranging from electron-withdrawing groups (3d–3e), electron-donating groups (3f–3k), sterically encumbered 2,4,6-trimethyl groups (3l), to sensitive functionalities such as benzylic chloride (3m), acetate (3n) and boronic acid pinacol ester (3o). Aminofluorination of other vinyl arenes and heteroarenes, including naphthalene systems (3p–3q), benzothiophene (3r), indole (3s) and pyridine (3t), also produced the corresponding β-fluoroamine products smoothly in 33–66% yields. The reaction of simple alkyl substituted alkenes were also attempted. The desired β-fluoroamine 3u was formed, albeit in only 12% yield, promoting us to seek for alternative route to access such alkyl-substituted β-fluoroamines (see Figure 3A in the following section). In addition to terminal alkenes, internal alkenes were also effective to produce desired products (3v–3z). These reactions occurred regioselectively, with the addition of the fluoride exclusively at the benzylic position. Only modest levels of diastereoselectivity were observed in the reactions of acyclic internal alkenes (3v–3w) while high diastereomeric ratios (up to 20:1) were obtained for cyclic alkenes (3x–3z). Note that the reactions of (E)- and (Z)-1v both led to the formation of 3v in comparable yields with similar diastereomeric ratios. The anti-selectivity observed for cyclic alkenes (3x–3z) probably resulted from the steric influence in the fluorination step where the fluoride was installed from the opposite side of the amino group.
Table 1.
Intermolecular Aminofluorination Reactions – the Scope of Alkenes and Amines.
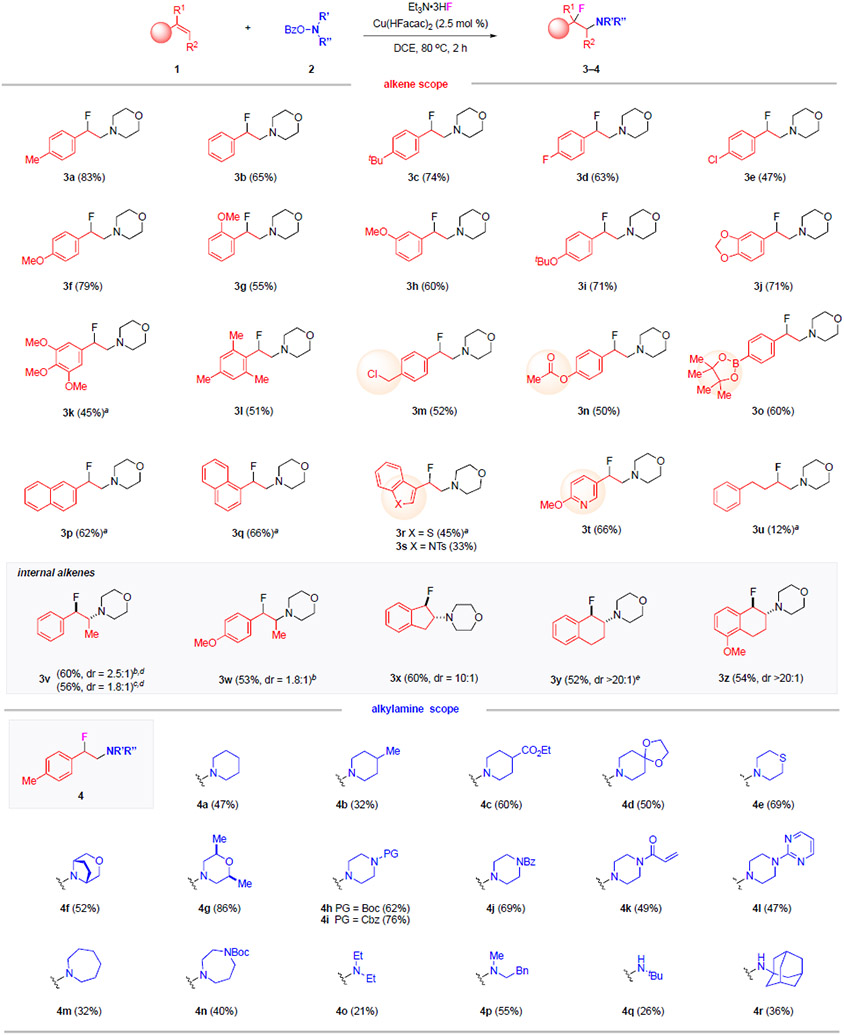
|
Reaction conditions: 1 (2.0 equiv), 2 (1.0 equiv), Et3N•3HF (10 equiv), Cu(HFacac)2 (2.5 mol %), DCE (1.0 mL), 80 °C, 2 h. Isolated yields shown. dr determined by 1H-NMR analysis of the crude mixtures. Major isomer shown. aIPrCuCl (5.0 mol %) used. bFrom E-alkene. cFrom Z-alkene. dRelative stereochemistry of the major diastereomer determined by X-ray analysis. eCH3(CH2)10C(O)O–NR’R” used.
Figure 3. Application toward the Synthesis of β-Fluoroamine-Containing Bioactive Compounds.

(A) Derivatizations of homoallylic β-fluoroalkylamine products. (B) Synthesis of bioactive β-fluoroalkylamine-containing compounds.
The scope of alkylamines in this aminofluorination reaction was found to be extensive, enabling a one-step access to diversely functionalized β-fluoroalkylamines. The reactions of different piperidine-containing aminating reagents provided the desired products 4a–4d. Note that the varied efficiency between 4b (32% yield) and 4c–4d (50–60% yields) revealed a beneficial role of electron-withdrawing substituents of piperidine precursors. Other six-membered cyclic amine precursors readily participated in this reaction, such as thiomorpholine 4e (69% yield), bridged bicyclic morpholine 4f (52% yield) and dimethyl-substituted morpholine 4g (86% yield). A wide range of piperazine-derived precursors bearing different functional groups were all effective, such as N-Boc 4h (62% yield), N-Cbz 4i (76% yield), N-Bz 4j (69% yield), N-acrylamide 4k (49% yield) and pyrimidine–containing piperazine 4l (47% yield). Seven-membered cyclic amine precursors, specifically azepane and N-Boc–protected 1,4-diazepane, successfully delivered 4m and 4n in 32% and 40% yields, respectively. Finally, acyclic hydroxylamine precursors were also found to be effective, as demonstrated in the successful installation of diethylamine (4o), and methylphenethylamine (4p). This protocol was even applicable for the direct formation of secondary amines (4q–4r), thus greatly expanding the types of β-fluoroalkylamines accessible using this method.
REGIOSELECTIVE 1,2 AMINOFLUORINATION OF 1,3-DIENES
We next investigated this copper-catalyzed aminofluorination of 1,3-dienes toward the formation of homoallylic β-fluoroamines, a class of highly valuable and synthetically flexible compounds (Table 2).37-39 Such aminofluorination of 1,3-dienes comes with its own set of challenges and is even more complex than that of alkenes. First, the presence of two olefins may lead to the reactions occurring at two distinct sites. Second, the reaction may allow for either direct 1,2-addition or conjugated 1,4-addition products. Thus, the amino fluorination of 1,3-dienes could potentially lead to a series of isomeric products such as 1,2-addition products 6 and 6’ as well as 1,4-addition products 7 and 7’. Our examination of reaction parameters toward the regioselective 1,2 aminofluorination suggested that IPrCuCl was a more effective catalyst in the reaction of 1,3-dienes (details in SI). Under the modified conditions, a series of 1-aryl-substiuted 1,3-dienes were studied. All selectively delivered 1,2-aminofluorination products, specifically homoallylic β-fluoroamines 6a–6f in synthetically useful yields. The reactions of 1-alkyl-subsituted 1,3-dienes also favored 1,2-aminofluorination over 1,4-addition products. For example, the reaction of 1-hexyl-1,3-butadiene produced 1,2-product 6g selectively, in a 7:1 ratio over 1,4-product 7g. Similarly, the reaction of 1-cyclohexyl-1,3-butadiene favored 6h over 7h in an 8:1 ratio.
Table 2.
Copper-Catalyzed 1,2-Aminofluorination of 1,3-Dienes.
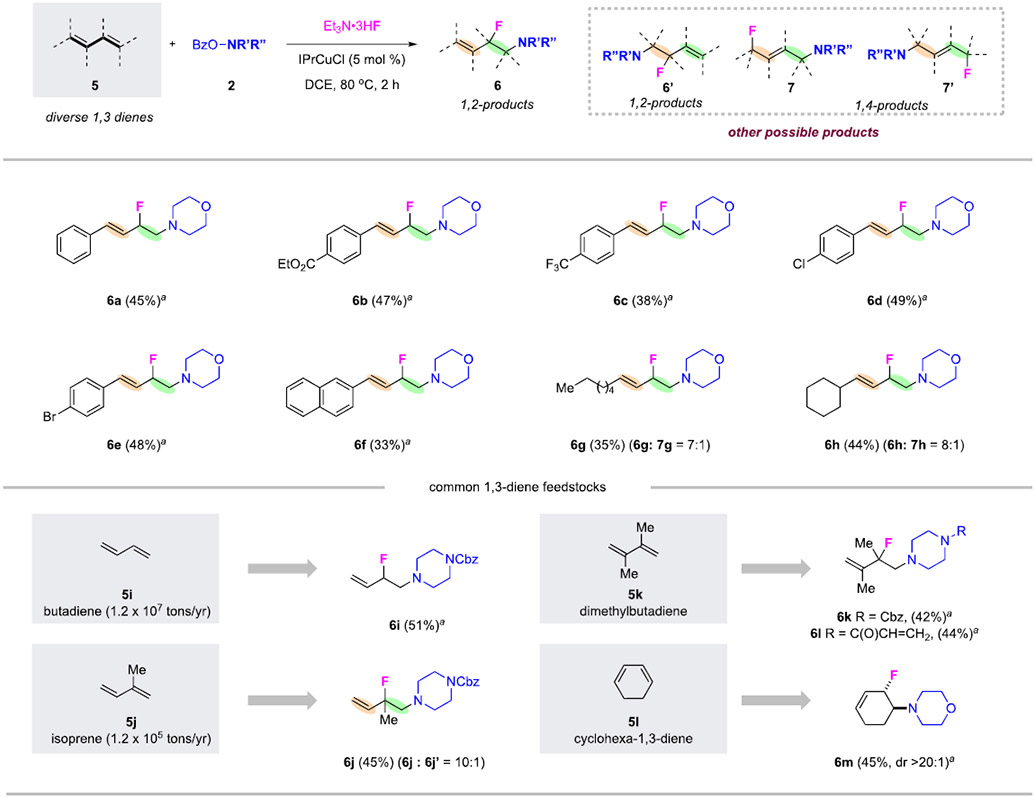
|
Reaction conditions: 5 (2.0 equiv), 2 (0.2 mmol, 1.0 equiv), Et3N•3HF (10 equiv), IPrCuCl (5.0 mol %), DCE (1.0 mL), 80 °C, 2 h. The major products shown. Isolated yields shown. Regioselectivity ratios (rr) were determined by 1H-NMR analysis of the crude reaction mixtures. arr > 20:1.
Most excitingly, this amino fluorination reaction of common, variously substituted 1,3-diene feedstocks readily delivered high-value homoallylic β-fluoroamines. For example, the reaction of the simplest 1,3-butadiene 5i gave 1,2-product 6i in 51% yield exclusively. The reaction of isoprene 5j gave selectively 1,2-aminofluorination products, with 6j and 6j’ formed in a ratio of 10:1, favoring the aminofluorination at the more branched site of terminal olefins. This site selectivity indicated the fluorination preferentially occurred at a more stabilized intermediate. The reactions of 2,3-dimethyl-1,3-butadiene 5k successfully led to regioselective formation of 1,2-aminofluorination products such as 2-fluoropiperazine derivatives 6k and 6l using different alkylamine precursors. Finally, the reaction of cyclohexa-1,3-diene 5l afforded trans-1,2-aminofluorination product 6m in 45% yield, in a highly regio- and diastereoselective manner.
SYNTHETIC APPLICATIONS
To further illustrate the synthetic values of this aminofluorination method, we investigated the derivatizations of homoallylic β-fluoroamines (Figure 3, A). First, the homoallylic amines were successfully transformed into alkyl-substituted β-fluoroamines by Pd-catalyzed hydrogenation. Selected examples include 4-(2-fluoro-4-phenylbutyl)morpholine 3u in 93% yield, N-Cbz (2-fluorobutyl)piperazine 7 in 96% yield, and trans-2-fluorocyclohexyl-morpholine 8 in 92% yield. Such a route to β-fluoroalkylamines offers an effective entry to a wide range of β-fluoroalkylamines, alternative to the reactions using alkyl-substituted olefins. The ozonolysis of 6k readily afforded ketone product 9 in 80% yield. Such a highly functionalized compound may find wide use in chemical synthesis. Furthermore, we have applied this aminofluorination system to the one-step preparation of structurally complex β-fluoroamine-containing bioactive compounds 10 (Figure 3, B). These successful derivatizations of important drugs such as ibuprofen (10a), fenofibrate (10b), indomethacin (10c), D-phenylalanine (10d), estrone (10e) and tryptamine derivative (10f) demonstrated high efficiency of this method and the remarkable tolerance of this catalytic method against different functional groups such as amide, ester, chloride, and ketone. Finally, we applied this method for a rapid synthesis of β-fluoropiperazine [F]-YZ185 (12), as a novel analog of this class of sigma1 receptor ligands that have favorable features for anticocaine actions.40 These representative examples highlight the advantages and potential application of this aminofluorination method in medicinal chemistry.
MECHANISTIC STUDIES
To shed light on the operating mechanism of this catalytic system, we investigated the reaction pathways that are engaged in this alkene aminofluorination reaction. Our previous studies have revealed a novel alkene activation mode initiated by copper-catalyzed amination using O-benzoyl-N-hydroxylamines, which involves the possible generation of nitrogen and carbon radical intermediates. To probe if a radical intermediate is involved in this aminofluorination reaction, control reaction using 1a and 2a was performed in the presence of butylated hydroxytoluene (BHT) as a radical scavenger (Figure 4, A). While only trace amount of the expected β-fluoroamine product 3a was detected, BHT adduct 13a was formed in 54% yield, strongly suggesting the generation of a stabilized carbon-centered benzylic radical intermediate under standard reaction conditions. Neither of the byproducts 13b or 13c was detected, suggesting no absence of long-lived nucleophilic nitrogen radicals. Furthermore, radical clock experiments using cyclopropylstyrenes 14a and 14b were performed (Figure 4, B). Aminofluorination of α-cyclopropylstyrene 14a under the standard conditions produced only trace amount of aminofluorination product 15 and instead afforded the ring opening amination product 16 in 64% yield, which further implicates the involvement of a radical pathway and the presence of a benzylic radical. Such a species bearing the radical α to cyclopropane would be expected to undergo ring opening leading to a terminal radical and facile intramolecular trapping to produce 16. In contrast, the reaction of β-trans-cyclopropylstyrene 14b provided no desired product 17 nor any detectable ring-opening byproducts. This observation is consistent with the above hypothesis: the steric bulk of this 1,2-disubstituted alkene likely inhibits the reaction, even if the benzylic carbon radical is generated β to cyclopropane, no ring opening should be observed either.
Figure 4. Mechanistic Studies on Cu-Catalyzed 1,2 Aminofluorination of Alkenes.

(A) Control experiment in the presence of butylated hydroxytoluene (BHT). (B) Comparative reactions of α- and β-cyclopropylstyrenes. (C) Proposed reaction pathways.
Based on these experimental results and related studies,30, 31, 41 the following reaction pathways are proposed (Figure 4, C). The reaction is initiated by copper-catalyzed N─O bond cleavage of O-benzoylhydroxylamines 2 in the presence of Et3N•3HF, generating a protonated aminyl radical cation (I).32, 33, 42-44 The subsequent electrophilic amination of carbon-carbon double bond would produce a carbon radical (II), which was supported by the formation of 13a and 16 (Figure 4, A and B). Under standard conditions, the resulting radical (II) would undergo copper-mediated fluorination. Et3N•3HF was anticipated to have dual roles in this reaction, by contributing to the formation of aminyl radical cation as an acid source and enabling the effective fluorination step as a nucleophilic fluoride. Considering that the C─N bond formation the alkene electrophilic amination has always been facile and fast in our studies, the C─F bond formation step is likely the rate-determining step in this aminofluorination reaction. It remains inconclusive if the reaction engaged the formation of a possible aziridinium intermediate. Our future studies will be directed to elucidate the nature of the C─F bond formation step.
Standard conditions: olefin (2.0 equiv), 2a (0.2 mmol, 1.0 equiv), Cu(HFacac)2 (5.0 mol %), Et3N•3HF (10 equiv), DCE (1.0 mL), 80 °C, 2 h. Isolated yields shown.
CONCLUSIONS
In conclusion, we have developed an unprecedented copper-catalyzed aminofluorination of alkenes and 1,3-dienes using N, N-dialkylhydroxylamines and Et3N•3HF as a direct entry to valuable (hetero)aryl, alkyl and homoallylic β-fluoroalkylamines. The method features high regioselectivity, mild conditions, good tolerance of diverse functional groups and a broad scope of commodity vinyl arenes and 1,3-dienes. Mechanistic experiments suggest a copper-mediated electrophilic amination of alkenes with the sequential formation of amine- and carbon-centered radical intermediates, which undergo nucleophilic fluorination to deliver the desired β-fluoroalkylamine products. The dual role of Et3N•3HF as a fluoride source and an acid is significant and may direct future endeavors in this area of research. Synthetic applications of this method have been highlighted by its use for the rapid entry to β-fluoridated amine-containing pharmaceuticals, natural products, and bioactive compounds. We expect that this novel and practical procedure leading to highly valuable β-fluoroamines will find wide use in synthetic and medicinal chemistry community.
Supplementary Material
ACKNOWLEDGMENT
We acknowledge financial support provided by Duke University and the National Institutes of Health (GM118786). We thank Dr. Peter Silinski (Duke University) for high-resolution mass spectrometry analysis.
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI:
Condition optimizations, experimental procedures, compound characterization, and NMR spectra.
REFERENCES
- (1).van Niel MB; Collins I; Beer MS; Broughton HB; Cheng SKF; Goodacre SC; Heald A; Locker KL; MacLeod AM; Morrison D; et al. Fluorination of 3-(3-(Piperidin-1-yl)propyl)indoles and 3-(3-(Piperazin-1-yl)propyl)indoles Gives Selective Human 5-HT1D Receptor Ligands with Improved Pharmacokinetic Profiles. J. Med. Chem 1999, 42 (12), 2087–2104. DOI: 10.1021/jm981133m. [DOI] [PubMed] [Google Scholar]
- (2).Shah P; Westwell AD The role of fluorine in medicinal chemistry. J. Enzym Inhib. Med. Chem 2007, 22 (5), 527–540. DOI: 10.1080/14756360701425014. [DOI] [PubMed] [Google Scholar]
- (3).Hagmann WK The many roles for fluorine in medicinal chemistry. J. Med. Chem 2008, 51 (15), 4359–4369. [DOI] [PubMed] [Google Scholar]
- (4).Berger R; Resnati G; Metrangolo P; Weber E; Hulliger J Organic fluorine compounds: a great opportunity for enhanced materials properties. Chem. Soc. Rev 2011, 40 (7), 3496–3508, 10.1039/C0CS00221F. DOI: 10.1039/C0CS00221F. [DOI] [PubMed] [Google Scholar]
- (5).Wang J; Sánchez-Roselló M; Aceña JL; del Pozo C; Sorochinsky AE; Fustero S; Soloshonok VA; Liu H Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev 2014, 114 (4), 2432–2506. DOI: 10.1021/cr4002879. [DOI] [PubMed] [Google Scholar]
- (6).Zhou Y; Wang J; Gu Z; Wang S; Zhu W; Aceña JL; Soloshonok VA; Izawa K; Liu H Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II–III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev 2016, 116 (2), 422–518. DOI: 10.1021/acs.chemrev.5b00392. [DOI] [PubMed] [Google Scholar]
- (7).Gillis EP; Eastman KJ; Hill MD; Donnelly DJ; Meanwell NA Applications of Fluorine in Medicinal Chemistry. J. Med. Chem 2015, 58 (21), 8315–8359. [DOI] [PubMed] [Google Scholar]
- (8).Spahn V; Del Vecchio G; Labuz D; Rodriguez-Gaztelumendi A; Massaly N; Temp J; Durmaz V; Sabri P; Reidelbach M; Machelska H; et al. A nontoxic pain killer designed by modeling of pathological receptor conformations. Science 2017, 355 (6328), 966–969. [DOI] [PubMed] [Google Scholar]
- (9).Hunter L b-Fluoroamines (Update 2017). In Science of Synthesis 4.11, 2017 ed.; Christmann M, Paquin JF, Weinreb SM, Carreira EM, Schaumann E Eds.; Science of Synthesis, Georg Thieme Verlag, 2017; pp 413–444. [Google Scholar]
- (10).Stavber S; Zupan M; Poss AJ; Shia GA 1-Fluoro-4-hydroxy-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate) as a new, effective reagent for selective fluorofunctionalisation of alkenes under mild reaction conditions. Tetrahedron Lett. 1995, 36 (37), 6769–6772. DOI: 10.1016/00404-0399(50)1337-H. [DOI] [Google Scholar]
- (11).Stavber S; Pecan TS; Papež M; Zupan M Ritter-type fluorofunctionalisation as a new, effective method for conversion of alkenes to vicinal fluoroamides. Chem. Commun 1996, (19), 2247–2248, 10.1039/CC9960002247. DOI: 10.1039/CC9960002247. [DOI] [Google Scholar]
- (12).Stavber G; Zupan M; Jereb M; Stavber S Selective and Effective Fluorination of Organic Compounds in Water Using Selectfluor F-TEDA-BF4. Org. Lett 2004, 6 (26), 4973–4976. DOI: 10.1021/ol047867c. [DOI] [PubMed] [Google Scholar]
- (13).Chen P; Liu G Advancements in Aminofluorination of Alkenes and Alkynes: Convenient Access to β-Fluoroamines. Eur. J. Org. Chem 2015, (20), 4295–4309. DOI: doi: 10.1002/ejoc.201500231. [DOI] [Google Scholar]
- (14).Kong W; Merino E; Nevado C Divergent Reaction Mechanisms in the Aminofluorination of Alkenes. CHIMIA 2014, 68 (6), 430–435. DOI: 10.2533/chimia.2014.430. [DOI] [PubMed] [Google Scholar]
- (15).Wu T; Yin G; Liu G Palladium-Catalyzed Intramolecular Aminofluorination of Unactivated Alkenes. J. Am. Chem. Soc 2009, 131 (45), 16354–16355. DOI: 10.1021/ja9076588. [DOI] [PubMed] [Google Scholar]
- (16).Peng H; Yuan Z; Wang H.-y.; Guo Y.-l.; Liu G Palladium-catalyzed intermolecular fluoroesterification of styrenes: exploration and mechanistic insight. Chem. Sci 2013, 4 (8), 3172–3178, 10.1039/C3SC50690H. DOI: 10.1039/C3SC50690H. [DOI] [Google Scholar]
- (17).Zhang HW; Song YC; Zhao JB; Zhang JP; Zhang Q Regioselective Radical Aminofluorination of Styrenes. Angew. Chem. Int. Ed 2014, 53 (41), 11079–11083. DOI: 10.1002/anie.201406797. [DOI] [PubMed] [Google Scholar]
- (18).Haitao Z; Liu G Palladium-Catalyzed Oxidative Aminofluorination of Styrenes. Acta Chim. Sinica 2012, 70, 2404–2407. [Google Scholar]
- (19).Kong WQ; Feige P; de Haro T; Nevado C Regio- and Enantioselective Aminofluorination of Alkenes. Angew. Chem. Int. Ed 2013, 52 (9), 2469–2473. [DOI] [PubMed] [Google Scholar]
- (20).Mo JN; Yu WL; Chen JQ; Hu XQ; Xu PF Regiospecific Three-Component Aminofluorination of Olefins via Photoredox Catalysis. Org. Lett 2018, 20 (15), 4471–4474. [DOI] [PubMed] [Google Scholar]
- (21).Guo P; Han J-F; Yuan G-C; Chen L; Liao J-B; Ye K-Y Cobalt-Catalyzed Divergent Aminofluorination and Diamination of Styrenes with N-Fluorosulfonamides. Org. Lett 2021, 23 (10), 4067–4071. DOI: 10.1021/acs.orglett.1c01308. [DOI] [PubMed] [Google Scholar]
- (22).Lu D-F; Liu G-S; Zhu C-L; Yuan B; Xu H Iron(II)-Catalyzed Intramolecular Olefin Aminofluorination. Org. Lett 2014, 16 (11), 2912–2915. DOI: 10.1021/ol501051p. [DOI] [PubMed] [Google Scholar]
- (23).Lu D-F; Zhu C-L; Sears JD; Xu H Iron(II)-Catalyzed Intermolecular Aminofluorination of Unfunctionalized Olefins Using Fluoride Ion. J. Am. Chem. Soc 2016, 138 (35), 11360–11367. DOI: 10.1021/jacs.6b07221 PMC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Jiang H; Studer A Amidyl Radicals by Oxidation of alpha-Amido-oxy Acids: Transition-Metal-Free Amidofluorination of Unactivated Alkenes. Angew. Chem. Int. Ed 2018, 57 (33), 10707–10711. [DOI] [PubMed] [Google Scholar]
- (25).Wang C; Tu Y; Ma D; Bolm C Photocatalytic Fluoro Sulfoximidations of Styrenes. Angew. Chem. Int. Ed 2020, 59 (33), 14134–14137. DOI: 10.1002/anie.202005844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Li ZD; Zhang CW; Zhu L; Liu C; Li CZ Transition-metal-free, room-temperature radical azidofluorination of unactivated alkenes in aqueous solution. Org. Chem. Front 2014, 1 (1), 100–104. [Google Scholar]
- (27).Capilato JN; Bume DD; Lee WH; Hoffenberg LES; Jokhai RT; Lectka T Fluorofunctionalization of C═C Bonds with Selectfluor: Synthesis of beta-Fluoropiperazines through a Substrate-Guided Reactivity Switch. J. Org. Chem 2018, 83 (23), 14234–14244. DOI: 10.1021/acs.joc.8b02429. [DOI] [PubMed] [Google Scholar]
- (28).Rozatian N; Hodgson DRW Reactivities of electrophilic N–F fluorinating reagents. Chem. Commun 2021, 57 (6), 683–712, 10.1039/D0CC06339H. DOI: 10.1039/D0CC06339H. [DOI] [PubMed] [Google Scholar]
- (29).Hemric BN; Wang Q Copper-catalyzed intermolecular oxyamination of olefins using carboxylic acids and O-benzoylhydroxylamines. Beil. J. Org. Chem 2016, 12, 22–28. DOI: 10.3762/bjoc.12.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Hemric BN; Shen K; Wang Q Copper-Catalyzed Amino Lactonization and Amino Oxygenation of Alkenes Using O-Benzoylhydroxylamines. J. Am. Chem. Soc 2016, 138 (18), 5813–5816. DOI: 10.1021/jacs.6b02840. [DOI] [PubMed] [Google Scholar]
- (31).Hemric BN; Chen AW; Wang Q Copper-Catalyzed Modular Amino Oxygenation of Alkenes: Access to Diverse 1,2-Amino Oxygen-Containing Skeletons. J. Org. Chem 2019, 84 (3), 1468–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Falk E; Makai S; Delcaillau T; Gürtler L; Morandi B Design and Scalable Synthesis of N-Alkylhydroxylamine Reagents for the Direct Iron-Catalyzed Installation of Medicinally Relevant Amines. Angew. Chem. Int. Ed 2020, 59 (47), 21064–21071. DOI: 10.1002/anie.202008247. [DOI] [PubMed] [Google Scholar]
- (33).Legnani L; Prina-Cerai G; Delcaillau T; Willems S; Morandi B Efficient access to unprotected primary amines by iron-catalyzed aminochlorination of alkenes. Science 2018, 362 (6413), 434–439. DOI: doi: 10.1126/science.aat3863. [DOI] [PubMed] [Google Scholar]
- (34).Biloski AJ; Ganem B Improved Oxidation of Amines with Dibenzoyl Peroxide. Synthesis 1983, 1983 (07), 537–538. DOI: 10.1055/s-1983-30410. [DOI] [Google Scholar]
- (35).Berman AM; Johnson JS Copper-Catalyzed Electrophilic Amination of Functionalized Diarylzinc Reagents. J. Org. Chem 2005, 70 (1), 364–366. DOI: 10.1021/jo048168g. [DOI] [PubMed] [Google Scholar]
- (36).During the final preparation of this manuscript, an independently developed AgF-mediated three-component alkene amnofluorination using N-bromodialkylamines was reported: Li Y; Bao J; Zhang Y; Peng X; Yu W; Wang T; Yang D; Liu Q; Zhang Q; Fu J Three-component aminofluorination of alkenes with electronically rich amino sources. Chem 2022, 8 (4), 1147–1163. DOI: 10.1016/j.chempr.2022.02.014. [DOI] [Google Scholar]
- (37).Sorlin AM; Usman FO; English CK; Nguyen HM Advances in Nucleophilic Allylic Fluorination. ACS Catal. 2020, 10 (20), 11980–12010. DOI: 10.1021/acscatal.0c03493. [DOI] [Google Scholar]
- (38).Ovaa H; Stragies R; van der Marel GA; van Boom JH; Blechert S Asymmetric synthesis of indolizidine alkaloids by ring-closing–ring-opening metathesis. Chem. Commun 2000, (16), 1501–1502, 10.1039/B004106H. DOI: 10.1039/B004106H. [DOI] [Google Scholar]
- (39).Ding H; Friestad GK Asymmetric addition of allylic nucleophiles to imino compounds. Synthesis 2005, (17), 2815–2829. DOI: 10.1055/s-2005-875218. [DOI] [Google Scholar]
- (40).Matsumoto RR; Potelleret FH; Mack A; Pouw B; Zhang Y; Bowen WD Structure–activity comparison of YZ-069, a novel σ ligand, and four analogs in receptor binding and behavioral studies. Pharmacology Biochemistry and Behavior 2004, 77 (4), 775–781. DOI: 10.1016/j.pbb.2004.01.014. [DOI] [PubMed] [Google Scholar]
- (41).Hemric BN; Chen AW; Wang Q Copper-Catalyzed 1,2-Amino Oxygenation of 1,3-Dienes: A Chemo-, Regio-, and Site-Selective Three-Component Reaction with O-Acylhydroxylamines and Carboxylic Acids. ACS Catal. 2019, 9 (11), 10070–10076. DOI: 10.1021/acscatal.9b03076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Ruffoni A; Juliá F; Svejstrup TD; McMillan AJ; Douglas JJ; Leonori D Practical and regioselective amination of arenes using alkyl amines. Nat. Chem 2019, 11 (5), 426–433. DOI: 10.1038/s41557-019-0254-5. [DOI] [PubMed] [Google Scholar]
- (43).Ganley JM; Murray PRD; Knowles RR Photocatalytic Generation of Aminium Radical Cations for C─N Bond Formation. ACS Catal. 2020, 10 (20), 11712–11738. DOI: 10.1021/acscatal.0c03567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Pratley C; Fenner S; Murphy JA Nitrogen-Centered Radicals in Functionalization of sp2 Systems: Generation, Reactivity, and Applications in Synthesis. Chem. Rev 2022, 122 (9), 8181–8260. DOI: 10.1021/acs.chemrev.1c00831. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.