

Abstract
Dinoflagellates from the family Symbiodiniaceae are phototrophic marine protists that engage in symbiosis with diverse hosts. Their large and distinct genomes are characterized by pervasive gene duplication and large-scale retroposition events. However, little is known about the role and scale of horizontal gene transfer (HGT) in the evolution of this algal family. In other dinoflagellates, high levels of HGTs have been observed, linked to major genomic transitions, such as the appearance of a viral-acquired nucleoprotein that originated via HGT from a large DNA algal virus. Previous work showed that Symbiodiniaceae from different hosts are actively infected by viral groups, such as giant DNA viruses and ssRNA viruses, that may play an important role in coral health. Latent viral infections may also occur, whereby viruses could persist in the cytoplasm or integrate into the host genome as a provirus. This hypothesis received experimental support; however, the cellular localization of putative latent viruses and their taxonomic affiliation are still unknown. In addition, despite the finding of viral sequences in some genomes of Symbiodiniaceae, viral origin, taxonomic breadth, and metabolic potential have not been explored. To address these questions, we searched for putative viral-derived proteins in thirteen Symbiodiniaceae genomes. We found fifty-nine candidate viral-derived HGTs that gave rise to twelve phylogenies across ten genomes. We also describe the taxonomic affiliation of these virus-related sequences, their structure, and their genomic context. These results lead us to propose a model to explain the origin and fate of Symbiodiniaceae viral acquisitions.
Keywords: Symbiodiniaceae evolution, virus horizontal gene transfer, dinoflagellate virus, Symbiodinium virus, algal virus
1. Introduction
Dinoflagellates from the family Symbiodiniaceae are phototrophic marine protists that engage in symbiosis with diverse hosts such as foraminifera, ciliates, radiolarians, mollusks, and cnidarians such as anemones and, most notably, corals (LaJeunesse et al. 2018). Their large and distinct genomes are characterized by pervasive gene duplications (Aranda et al. 2016; Nand et al. 2021), expansion of lineage-specific gene families with unknown functions (González-Pech et al. 2021), and large-scale retroposition events (Song et al. 2017). However, little is known about the role of horizontal gene transfer (HGT) in Symbiodiniaceae evolution and the scale of gene acquisition events (González-Pech et al. 2021). In Fugacium kawagutii and Brevioloum minutum (formerly Symbiodinium Clades F and B, respectively), ∼0.7 per cent of their genes (246 and 244 sequences) have putatively originated from prokaryote-derived HGT events (Fan et al. 2020).
Higher levels of HGTs have been observed in other dinoflagellates and appear to be linked to major genomic transitions (Wisecaver, Brosnahan, and Hackett 2013; Janouškovec et al. 2017). Remarkably, all sequenced dinoflagellate genomes contain a family of viral-acquired nucleoproteins known as the dinoflagellate/viral nucleoproteins (DVNPs), which have a putative role in chromatin regulation (Irwin et al. 2018). In B. minutum, nineteen divergent DVNPs have been identified, and these may be involved in regulating chromosome structure (Shoguchi et al. 2013). Furthermore, it has been proposed that the DVNP protein family originated in the ancestor of dinoflagellates via HGT from a large DNA algal virus, possibly from the family Phycodnaviridae (Gornik et al. 2012). Acquisition of the DVNP gene family coincides with massive genomic expansion in this group and may have conferred immunity to infection by the same group of exogenous viruses (Gornik et al. 2019).
Symbiodiniaceae isolated from corals are actively infected by several viral groups, ranging from giant DNA viruses to ssRNA viruses (Correa et al. 2013; Weynberg et al. 2014; Wood-Charlson et al. 2015; Correa et al. 2016; Levin et al. 2017; Messyasz et al. 2020). Most importantly, the presence and abundance of viral sequences were found to be associated with bleached and diseased corals, suggesting that viruses may play a role in coral health and bleaching events by infecting (subsequently killing or disrupting the function of) Symbiodiniaceae cells in the host (Thurber and Correa 2011). Recently, Grupstra et al. (2022) demonstrated the rapid formation of diverse viral major capsid protein (MCP) ‘aminotypes’ (unique amino acid sequences) from a Symbiodiniaceae-infecting dinoflagellate RNA virus in coral colonies exposed to high temperatures (30.3°C) that are known to cause coral bleaching. This result suggests that the switch from a persistent to a productive viral infection may be triggered by thermal stress.
Early observations linking symbiotic dinoflagellates from anemones with viruses, stresses, and coral diseases (Wilson et al. 2001) led to the hypothesis that the algal cells harbored latent proviruses. In this type of infection, a virus persists in the cytoplasm as an episome (an extrachromosomal molecule) or becomes integrated into the host genome, replicating as a (latent) provirus, synchronized with the host cell (Hyman and Abedon 2012). After exposure to stress, the latent provirus enters a lytic cycle, lysing the host cell and invading other susceptible hosts. This hypothesis was partially corroborated with multiple Symbiodiniaceae isolates from various cnidarian hosts, showing the production of viral-like particles (VLPs) in thermally and ultraviolet (UV) light-stressed algal cells (Wilson et al. 2005; Davy et al. 2006; Lohr, Munn, and Wilson 2007; Lawrence et al. 2014, 2017; Weynberg et al. 2017; Benites et al. 2018). However, the cellular localization of putative latent viruses (i.e. cytoplasmic or genomic) and their precise taxonomic affiliations are still unknown.
In other algal groups, such as the brown algae (Phaeophyceae), double-stranded DNA viruses from Phycodnaviridae can persist as integrated proviruses with fragments of the viral genome interspersed as repeats and pseudogenes throughout the host genome. The viral genome remains latent until it becomes active in reproductive cells after light and temperature stimulation (Bräutigam et al. 1995; Müller, Kapp, and Knippers 1998; Delaroque and Boland 2008; McKeown et al. 2017). In Ectocarpus (strain Ec 32), the genome of a virus was found to be integrated into the host genome; however, the viral genes were not expressed and viral particles were not produced (Cock et al. 2010). This is similar to observations in Feldmannia, which has viral fragments in its genome but does not show evidence of viral production (Lee, Ivey, and Meints 1998). In the red seaweed Chondrus crispus (Rhodophyta), multiple copies of a partial RNA viral genome are expressed, including a copy of the capsid gene, but there is no apparent virus production or host symptoms of infection (Rousvoal et al. 2016). In green algae (Chlorophyta), although some of their genomes contain numerous giant ‘endogenous viral elements’ (EVEs) with spliceosomal introns and segmental gene duplications (Moniruzzaman et al. 2020), the only evidence thus far of viral latency is from the chlorophyte Cylindrocapsa geminella. This species produces VLPs, and a lytic infection is observed after heat-shock treatment (Hoffman and Stanker 1976).
Recently, Irwin et al. (2022) reported that at least twenty-three genes in Symbiodiniaceae (Fkaw, Sgor, and Smic) show strong support for HGTs from virus donors such as Nucleocytoviricota giant viruses (although this analysis was not focused on Symbiodiniaceae). These HGT-derived genes have possible roles in the catalysis of ATP-dependent conversion of ribonucleotides and HNH endonuclease, among other functions. In addition, despite the fact that viral sequences account for <3 per cent of the Symbiodinium microadriaticum genome (Aranda et al. 2016) and ∼0.1 per cent of the Cladocopium goreaui genome (Liu et al. 2018), their origin, taxonomic breath, and metabolic potential have not been explored in-depth, and no evidence exists of viral genome integration into Symbiodiniaceae nuclear DNA. The latter finding would support the proviral hypothesis (Wilson et al. 2001).
Therefore, to address questions about the occurrence of integrated viral sequences in Symbiodiniaceae genomes and to uncover their possible origins and functions, we searched predicted proteins from the thirteen available Symbiodiniaceae genomes (Shoguchi et al. 2013, 2018; Lin et al. 2015; Liu et al. 2018; Chen et al. 2020; González-Pech et al. 2021) for sequences of putative viral origin. We identified fifty-nine putative viral HGTs that form twelve phylogenies, from across ten of the genomes. We describe, for each of these sequences, their taxonomic affiliation with viruses and the composition, structure, and genomic context of their associated genes. Moreover, we searched for corroborative evidence that would support the hypothesis that these sequences are HGT-derived and may have arisen from viral genome integrations.
2. Methods
2.1. Screening for viral HGT events in Symbiodiniaceae protein datasets
Each predicted protein from the thirteen Symbiodiniaceae genomes (445,306 total) was searched against the RefSeq viral database using DIAMOND (v0.9.22; BLASTp, sensitive mode) (Buchfink, Xie, and Huson 2015) with an e-value cutoff of <1e−5, retaining just the best viral hits per query sequence. To exclude false positive hits, we performed another DIAMOND (BLASTp) search against the complete GenBank non-redundant database, using only the Symbiodiniaceae protein queries with hits to the RefSeq viral database. For each query, the top 400 hits were retained and filtered using an e-value of <1e−5. For each hit, the full taxonomy of the subject sequence was retrieved for the top fifty filtered hits using a custom perl script (available from https://github.com/LFelipe-B/Symbiodiniaceae_vHGT_scripts), which uses the subject accession number to fetch the whole taxonomic tree from the GenBank taxonomy suite. Query sequences that had at least one of their top fifty hits to a viral sequence were retained for downstream analysis (Supplementary Table S1 and flowchart of methods in Supplementary Fig. S1). The genomes are abbreviated as follows: Brevioloum minutum (Bmin); Cladocopium sp. C92 (CC92); Cladocopium goreaui (Cgor); Fugacium kawagutii (Fkaw); Symbiodinium linucheae CCMP2456 (Slin_CCMP2456); S. microadriaticum (Smic); S. microadriaticum 04-503SCI.03 (Smic_04-503SCI.03); S. microadriaticum CassKB8 (Smic_CassKB8); S. natans CCMP2548 (Snat_CCMP2548); S. necroappetens CCMP2469 (Snec_CCMP2469); S. pilosum CCMP2461 (Spil_CCMP2461); S. tridacnidorum (Stri); and S. tridacnidorum CCMP2592 (Stri_CCMP2592).
2.2. Inferring pre-candidates for viral HGTs
To determine the scale of viral HGT in Symbiodiniaceae genomes, we collected sequences that showed significant similarity to viral sequences by calculating four independent metrics using custom Python scripts and the full taxonomic annotations retrieved using the subject accession numbers from the DIAMOND outputs. Each hit to the complete GenBank non-redundant database was categorized as ‘non-target’ if the subject sequence was from a virus and as ‘target’ if the subject sequence was from a eukaryote; hits to Symbiodiniaceae sequences were omitted from the subsequent calculations to avoid hits to sequences from the same or similar species already submitted to GenBank from biasing the analysis. The first metric calculated was the Alien index (AI) (Gladyshev, Meselson, and Arkhipova 2008; Rancurel, Legrand, and Danchin 2017), which assesses how many orders of magnitude there are between the e-values of the top target and non-target hits; query sequences were retained if non-target sequences scored AI > 30. The second metric calculated was the HGT index (hU) (Boschetti et al. 2012), defined as the difference between the bit scores of the top non-target and target hits; query sequences with a Hu ≥ 30 were retained. The third metric was calculated by counting the total number of target and non-target hits (collapsing repeated taxa) associated with each query; query sequences were retained if the sum of non-target hits was >50 per cent of the total number of hits. The fourth metric was calculated by taking the sum of the non-target hit bit scores and comparing it with the sum of the target hit bit scores, retaining query sequences that had higher non-target bit score totals compared to their target bit score totals. Query sequences with scores above the cutoffs associated with each of the four metrics were considered pre-candidates for viral HGTs (pre-vHGTs).
2.3. Orthogroup clustering of pre-vHGTs and phylogenetic reconstruction
All Symbiodiniaceae proteins were clustered into orthogroups using OrthoFinder v2.3.3 (Emms and Kelly 2019), with groups containing at least one pre-vHGT sequence extracted for further analysis. We downloaded the subject sequences associated with each of the pre-vHGT DIAMOND hits to the RefSeq viral and GenBank non-redundant databases. The Symbiodiniaceae proteins in each pre-vHGT-containing orthogroup were combined with the downloaded subject sequences and complemented with subject hits downloaded from a BLASTp search (https://blast.ncbi.nlm.nih.gov) to retrieve the maximum number of subject hits associated with the pre-vHGTs for each group. The combined set of proteins associated with each group was aligned using MAFFT v7.305b (L-INS-i algorithm:—localpair—maxiterate 1000) (Katoh and Standley 2013). The resulting alignments were trimmed using TrimAI v1.2 in automated mode (-automated1) (Capella-Gutiérrez, Silla-Martínez, and Gabaldón 2009). A Maximum Likelihood phylogenetic tree was inferred from each group alignment using IQ-TREE v2.0.3 (Nguyen et al. 2015), with the best evolutionary model selected by ModelFinder (Kalyaanamoorthy et al. 2017) (-m TEST) and 10,000 ultrafast bootstrap replicates (UFBoot) (Minh, Nguyen, and von Haeseler 2013). Phylogenetic trees were midpoint rooted and screened for topologies in which Symbiodiniaceae query sequences (target) were clustered or nested with viral sequences (non-target) in a clade with ≥90 per cent ultrafast bootstrap support using PhySortR (Stephens et al. 2016) (clade.exclusivity = 0.95, min.prop.target = 0.7). Phylogenies were further processed using the packages ggtree (Yu et al. 2017), phangorn (Schliep 2011), and phytools (Revell 2012) in the R environment (version 3.4.2). A final manual curation step was implemented, discarding orthogroups with ambiguous phylogenetic signals, i.e. cases, where there was an over-representation of non-viral sequences or taxa, with excessively long branch lengths were considered as having weak evidence of viral HGT and discarded. The phylogenetic groups that remained after filtering were subjected one final time to IQ-TREE v2.0.3 analysis using the same parameters, except this time using 100 nonparametric bootstrap (BP) replicates for the calculation of branch support values. The candidate sequences, based on phylogenetic evidence in trees containing Symbiodiniaceae and viruses, that passed all thresholds were designated as vHGTs (Supplementary Tables S2 and S3).
2.4. Functional analysis of Symbiodiniaceae vHGTs
To gain insights into the putative functions of virus-derived sequences, we analyzed protein family membership and gene ontology (GO) terms for the vHGTs by re-annotating individual sequences using the InterProScan online suite (https://www.ebi.ac.uk/interpro/) and QuickGO (https://www.ebi.ac.uk/QuickGO/annotations). In some instances, we also subjected the vHGTs to a BLASTp search against the UniProtKB database (https://www.uniprot.org/uniprot/). Proteins with hypothetical, unnamed, or uncharacterized annotations were subjected to an additional distant homology search using the HHpred webserver (https://toolkit.tuebingen.mpg.de/tools/hhpred) (Söding, Biegert, and Lupas 2005) with default parameters against the Pfam-A_v35 database; HHpred results with a probability value >50 per cent and e-value < 1 were kept for further analysis.
2.5. Compositional and structural analysis of Symbiodiniaceae vHGTs
We retrieved information about the GC-content, length of coding sequences (CDSs), presence of introns, intron length, and location along the scaffold of the gene models associated with the vHGTs. For each of these features, the difference between the mean values calculated for the vHGTs was compared against a background of all Symbiodiniaceae genes using the Welch two-sample t-test in R. The presence of a dinoflagellate spliced leader (DinoSL) sequence upstream of the first exon of the vHGT genes in the genome was assessed to determine if mRNA recycling (Slamovits and Keeling 2008) has played a role in the evolution of these genes. DinoSL and relic DinoSL sequences were identified by an approach similar to that used by Stephens et al. (2020). Briefly, the DinoSL sequence (CCGTAGCCATTTTGGCTCAAG) and relic DinoSL sequences (which are composed of two or more DinoSL sequences joined together at their canonical splice sites) were searched against each Symbiodiniaceae genome using BLASTn v2.10.1 (-max_target_seqs 1000000 -task blastn-short -evalue 1000). Hits were retained if they started ≤5 bp from the 5′-end of the query sequence (which is the position of the conserved canonical splice site) and ≤2 bp from the 3′-end of the query. The proximity of the identified DinoSL and relic DinoSL sequences to vHGT genes within the genome was assessed using bedtools v2.29.2 (‘bedtools closest -s -D a -id’). Only cases where a DinoSL or relic DinoSL was identified on the same strand as the vHGT gene were upstream or overlapping with the vHGT and were <2 kbp from the first (most upstream) exon of the vHGT gene were reported.
2.6. Gene expression analysis and gene model confirmation of Symbiodiniaceae vHGTs
We retrieved RNA-seq data from the Sequence Read Archive (SRA) database (https://www.ncbi.nlm.nih.gov/sra) to study the expression of vHGTs and to validate vHGT gene models (Supplementary Table S4). When possible (with the exception of Snec, which lacks RNA-seq data), we used transcriptome data generated from the same isolate to verify gene modes in the genome. The RNA-seq data were downloaded from NCBI using Fastq-dump v2.9.6 (https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=toolkit#x005F;doc&f=fastq-dump), trimmed using Cutadapt v1.18 (Martin 2011) (quality-cutoff 20 and minimum-length 25), and mapped against the associated reference genome using HISAT2 v2.2.1 (Kim et al. 2019). The resulting alignment files were sorted and indexed using SAMtools v1.11 (Li et al. 2009), before being visualized using the Integrative Genomics Viewer (IGV) v2.12.3 tool (Robinson et al. 2011). The fragments per kilobase million (FPKM) value for each vHGT was calculated by StringTie2 v2.2.1 (Pertea et al. 2015) using the HISAT2-aligned RNA-seq reads.
The gene models associated with the putative sites of Symbiodinium +ssRNA virus integration into the host genome were further analyzed for internal stop codons and frameshift mutations to determine if these sequences had become pseudogenes or were potentially functional. For each viral +ssRNA gene of interest, the genome sequence, starting 2 kbp upstream and ending 2 kbp downstream, was extracted using seqkit v0.15.0 (-u 2000 -d 2000) (Shen et al. 2016). These sequences were used in alignments that included known RNA-dependent RNA polymerase (YP_009337004.1 and YP_009342067.1; top hits using online BLASTP against the viral gene models) and major capsid (AOG17586.1) proteins using Exonerate v2.3.0 (—model protein2genome—exhaustive yes) (Slater and Birney 2005).
3. Results
3.1. Screening Symbiodiniaceae genomes using four different HGT scoring metrics
The four HGT metrics (see Methods) were applied to Symbiodiniaceae proteins with hits to viral sequences in RefSeq to produce the list of viral HGT pre-candidates (pre-vHGTs). We identified additional viral HGT candidates by constructing orthogroups and taking all sequences that were in an orthogroup with a pre-vHGT. The Symbiodiniaceae sequences from each group, or individually, if single sequences, were combined with the sequences retrieved from the top hits of each pre-vHGT against a taxonomically diverse database and expanded to contain the maximum number of subject hits with BLASTp. For each group of combined sequences, a multiple sequence alignment was created, which was then used for phylogeny reconstruction. The resulting phylogenies were filtered, retaining only those in which viruses were overrepresented and in which viral sequences were nested with Symbiodiniaceae sequences in a clade that had UFBoot branch support ≥90 per cent. Our workflow resulted in the identification of fifty-nine vHGT sequences (comprising twelve phylogenies) that are spread across ten Symbiodiniaceae genomes, ranging from n = 14 sequences in Smic to n = 1 in the Cgor and Slin_CCMP2456 genomes (Fig. 1). The majority of vHGTs were identified in the genomes of species from the Symbiodinium genus and were absent from the genomes of CC92, Fkaw, and the free-living Spil_CCMP2461 species (Supplementary Table S1).
Figure 1.
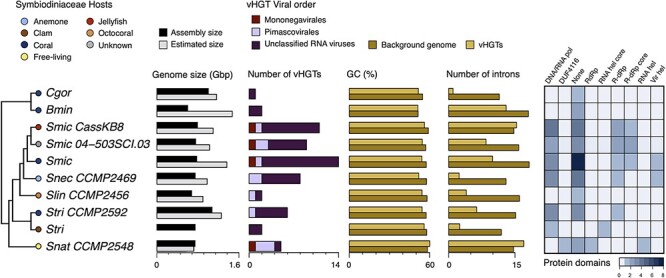
The main characteristics of genes in Symbiodiniaceae genomes that were acquired by viral horizontal gene transfer (vHGT). Cladogram depicting phylogenetic relationships of Symbiodiniaceae genomes (adapted from González-Pech et al. 2021) in which vHGTs were identified (left); the hosts associated with each Symbiodiniaceae species are represented by colored circles (top left key). Assembly and estimated genome sizes are shown in gigabase pairs (Gbp). The total number of vHGTs and their putative taxonomic origins (at the order level; center key), the CDS percent Guanine–Cytosine (GC%) content, the average number of introns, and the total number of associated protein domains are shown for the vHGTs identified in each genome. The CDS GC% content and average number of introns is shown separately for the vHGTs and the background Symbiodiniaceae sequences (i.e. all Symbiodiniaceae CDS or genes [respectively] excluding the vHGTs). Abbreviations: Brevioloum minutum (Bmin); Cladocopium sp. C92 (CC92); Cladocopium goreaui (Cgor); Fugacium kawagutii (Fkaw); Symbiodinium linucheae CCMP2456 (Slin_CCMP2456); S. microadriaticum (Smic); S. microadriaticum 04–503SCI.03 (Smic_04-503SCI.03); S. microadriaticum CassKB8 (Smic_CassKB8); S. natans CCMP2548 (Snat_CCMP2548); S. necroappetens CCMP2469 (Snec_CCMP2469); S. pilosum CCMP2461 (Spil_CCMP2461); S. tridacnidorum (Stri); S. tridacnidorum CCMP2592 (Stri_CCMP2592); DNA/RNA pol (DNA/RNA polymerase superfamily); DUF4116 (Domain of unknown function DUF4116); None (None predicted); RdRp (RNA-dependent RNA polymerase); RNA hel core (RNA virus helicase core domain); R-dRP (RNA-directed RNA polymerase); R-dRp core (RNA-directed RNA polymerase catalytic domain); RNA hel (Viral (Superfamily 1) RNA helicase); Vir hel (Viral helicase1) (Supplementary Tables S2 and S12).
3.2. Taxonomic distribution of vHGTs sequences in Symbiodiniaceae
The taxonomic distribution of the vHGTs was assessed at all classification levels using the top viral hit associated with each sequence and associated taxonomic information in the NCBI database. The number of viral sequences grouped at the order level in each of the genomes is shown in Fig. 1. vHGTs were derived from both DNA and RNA viruses; however, the majority were annotated as Unclassified RNA viruses (NCBI Taxonomy ID: 1922348; n = 44 sequences). The genome with the largest number of Unclassified RNA viruses vHGTs was Symbiodinium microadriaticum (n = 14), followed by the isolates S. microadriaticum KB8 (n = 11) and S. microadriaticum 04–503SCI.03 (n = 6). The second most abundant group of vHGTs (n = 11) was similar to giant viruses from the order Pimascovirales (Phylum Nucleocytoviricota; formerly NCLDVs). The third group of vHGTs (n = 4) was annotated to negative-strand RNA viruses from the Mononegavirales (Supplementary Table S2).
3.3. Functional annotation and expression of vHGTs sequences in Symbiodiniaceae
We found that the most abundant predicted functional annotation associated with the vHGTs is the DNA/RNA polymerase superfamily (n = 12), followed by RNA-directed RNA polymerase (n = 11) and viral RNA helicase (n = 3). Eleven vHGTs had RNA-directed RNA polymerase annotations (GO:0003968), followed by ATP binding (GO:0005524), viral RNA genome replication (GO:0039694), and mRNA capping (GO:0006370) as the most abundant GO terms. There were no protein family membership or GO terms predicted for ten of the vHGTs (Fig. 1) (Supplementary Table S2).
We also noted six vHGTs occurring in Smic, Smic_04-503SCI.03, Smic_CassKB8, and Stri_CCMP2592 that have two predicted domains (Supplementary Table S5), one of viral origin and the other of eukaryotic origin (BLASTp analysis supported this result), suggesting these may be chimeric genes (Méheust et al. 2018) (Supplementary Table S1). At the 5ʹ-end of these proteins is a predicted Clavaminate synthase-like or a Winged helix DNA-binding domain, and at the 3ʹ-end is a DNA/RNA polymerase domain. Aligned RNA-seq reads were visualized, compared manually against all the predicted vHGT gene models, and then used to calculate FPKM values for each vHGT to determine if these sequences are being expressed. This analysis showed that 23/51 vHGTs (eight sequences could not be verified given the lack of Snec RNA-seq data) had FPKM values >0, providing evidence of their expression, whereas the remainder lacked read mapping. The latter may be explained by the selective expression of vHGTs under specific (possibly stress) conditions that were not used to generate the available RNA-seq data (Supplementary Table S2 column FPKM). Alternatively, these sequences may not be expressed because they are non-functional. Regarding the putative viral-Symbiodiniaceae chimeric domain proteins, we found that 5/6 had evidence (by having an FPKM > 0) for their expression; however, without additional experimental evidence, we cannot determine if these sequences are true chimeras or artifacts of gene prediction (Supplementary Table S5).
3.4. Phylogenetic profile of vHGTs sequences in Symbiodiniaceae
3.4.1. Unclassified RNA viruses
The majority of vHGTs were grouped into nine phylogenies (n = 44 sequences); these sequences are all putatively related to Unclassified RNA viruses (Shi et al. 2016) (Fig. 2A-I). The taxonomy of top viral hits contained the Wenzhou weivirus-like virus (n = 15), Beihai sobemo-like virus (n = 7), Beihai weivirus-like virus (n = 7), Beihai narna-like virus (n = 4), and Hubei Beny-like virus (n = 3). Re-annotation of these sequences with BLASTp provided evidence of putative homology with hallmark RNA viral genes such as the RNA-dependent RNA polymerase (RdRp; n = 23 sequences), putative MCP (n = 4), viral RNA helicase (n = 4), and polyproteins encoding replicases, including an RNA-dependent RNA polymerase region (n = 5) (Supplementary Table S2). Moreover, these vHGT sequences had hits to the previously characterized Symbiodinium +ssRNA virus (accession KX538960—KX787934) (Levin et al. 2017).
Figure 2.
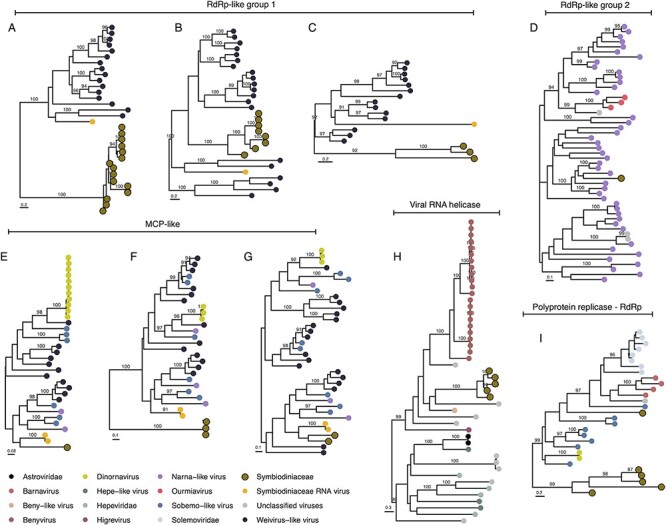
Phylogenetic profile of vHGT’s grouped by the category ‘Unclassified RNA viruses’: RNA-dependent RNA polymerase (RdRps-like group 1 [A, B, C] and RdRp-like group 2 [D]), Major capsid protein (MCPs-like [E, F, G]), viral RNA helicase (H), and polyprotein coding for replicases including RNA-dependent RNA polymerase region (I). Sequence names are not shown in the trees to enhance readability. The taxonomic affiliation of each sequence in the trees is represented by a colored circle, with the colors described in the legend at the bottom of the figure. Symbiodiniaceae vHGTs are highlighted with thicker borders. Only bootstrap support values ≥90 per cent are shown. Complete names and branch support in Supplementary Fig. S5.
3.4.1.1. RdRp-like phylogenies.
The Symbiodiniaceae vHGT-derived RdRps-like sequences were grouped into four phylogenies that matched two different viral groups, indicating that multiple independent vHGTs had occurred. The first viral group (RdRp-like group 1), comprising the sequences from phylogeny A (n = 15 sequences), B (n = 8), and C (n = 3) (Fig. 2 and Supplementary Fig. S5), have hits to Weivirus-like viruses and the Symbiodinium +ssRNA virus. In the phylogenetic tree associated with each group, all Symbiodiniaceae vHGTs are clustered into a separate clade or within Weivirus-like viral sequences, distant from the Symbiodinium +ssRNA virus. Weivirus-like viruses were first identified in the transcriptomes of mollusks and are related to alveolate EVEs (Shi et al. 2016), and were later found in the Porifera Halichondria panacea (breadcrumb sponge) (Waldron, Stone, and Obbard 2018).
The second viral group (RdRp-like group 2) comprised the phylogeny D (n = 1) and was a single Symbiodiniaceae sequence from the Bmin genome. This sequence has hits with RdRps from narna-like viruses and is located in a clade containing crustacean and Porifera-associated viruses (Beihai and Wenling narna-like viruses and Barns Ness breadcrumb sponge narna-like virus) (Shi et al. 2016). Narnavirus genomes often contain a single open reading frame encoding RdRp. This virus replicates in the cytoplasm of the host and may be transmitted vertically through cell division or horizontally during the mating cycle of its fungal host (Dinan et al. 2020). Narnaviruses are associated with trypanosomatids (in which a genomic narnavirus-like element was described; Lye et al. 2016), diatoms (Charon, Murray, and Holmes 2021), and red and brown algae (Dinan et al. 2020). Hits were also observed with plant viruses (Ourmiavirus), which have putative origins via a chimeric fusion between a fungal narnavirus and other plant viruses (Rastgou et al. 2009).
3.4.1.2. MCP-like phylogenies.
The MCP-like vHGT sequences formed three phylogenies: E (n = 1), F (n = 3), and G (n = 2) (Fig. 2 and Supplementary Fig. S5). These sequences matched MCPs from the dinoflagellate Heterocapsa circularisquama RNA virus (HcRNAV) Dinornavirus, the invertebrate-associated Weivirus-like virus, Sobemo-like virus, narna-like virus, and MCPs from the Symbiodinium +ssRNA virus. In the E and G phylogenies, vHGTs are clustered with Symbiodinium +ssRNA virus; the sequences from phylogeny F are clustered at the base of the tree. These patterns also suggest multiple independent vHGTs, with a high divergence of Symbiodiniaceae MCPs-like sequences, given that the viral sequence hits and the tree topology are approximately the same in all three phylogenies.
The remaining vHGTs with unclassified viral hits, from phylogenies H (n = 6) and I (n = 5), were re-annotated as viral RNA helicases and ‘polyprotein coding for replicases including RNA-dependent RNA polymerase region’ (respectively). In the H phylogeny, all Symbiodiniaceae vHGT-derived RNA helicase-like sequences clustered with a fungal virus, Agaricus bisporus virus 8, forming a bigger clade that includes the aggressive plant disease-causing viruses from the genus Benyvirus (Gilmer and Ratti 2017) and Beny-like viruses. In the polyprotein-like vHGTs phylogeny (I), one vHGT sequence (Stri.gene18597) is in a clade with fungal and plant Barnaviruses (Nibert et al. 2018) and Solemoviruses from plant hosts (Sõmera et al. 2021), whereas the other vHGT sequences are at the base of the tree. Interestingly, one sequence in this tree is from the plant Poinsettia, which is not only a latent virus but is also suggested to be a hybrid between polerovirus and sobemovirus (Aus Dem Siepen et al. 2005).
3.4.2. Pimascovirales
The second group of viruses with similarities to vHGTs (n = 11) were all contained within one phylogeny that is presented in Fig. 3 (n = 11 sequences; Supplementary Fig. S5). These vHGTs had best hits with restriction endonucleases encoded by large and giant viruses from Nucleocytoviricota (NCLDVs) as Brazilian marseillevirus (n = 2), Noumeavirus (n = 2), Cannes 8 virus (n = 3), and Kurlavirus BKC-1 (n = 2); all of these taxa are from the Marseilleviridae that infect Acanthamoeba protists (Aherfi et al. 2014). In addition, we searched for putative homologs of these sequences in giant viral metagenome-assembled genomes (GVMAGs) (Schulz et al. 2020) using DIAMOND BLASTp (as described earlier), in order to augment the phylogenetic diversity and resolution of the tree. This analysis added n = 42 additional metagenome-derived viral sequences to our dataset (Supplementary Table S1).
Figure 3.
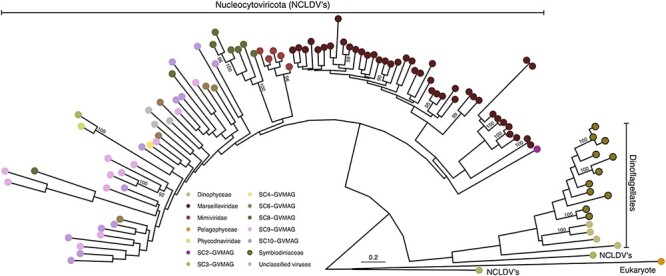
Phylogenetic profile of vHGT’s grouped by the category ‘Pimascovirales’: Symbiodiniaceae vHGT restriction endonuclease-like, viral restriction endonucleases and GVMAG homologs. Sequence names are not shown in the trees to enhance readability. Taxonomic affiliation of each sequence in the tree is represented by a colored circle, with the colors described in the legend at the bottom of the figure. Symbiodiniaceae vHGTs are highlighted with thicker borders. Only bootstrap support values ≥90 per cent are shown. Complete names and branch support in Supplementary Fig. S5.
In this phylogeny, giant and large viral sequences are overrepresented, such as from the Marseilleviridae and Mimiviridae families, and the inclusion of GVMAG (represented by seven ‘superclades’ [SC]) sequences revealed the presence of only one putative homologous sequence (from SC3) at the base of the Eukaryotic/Dinoflagellate branch, however with low bootstrap support (49 per cent). Symbiodiniaceae vHGTs are clustered in a separate major group with another eukaryote, Pelagomonas calceolata (Pelagophyceae). We also find vHGT homologs in the genomes of the dinoflagellate Polarella glacialis. The distribution of these sequences suggests a more complex scenario involving multiple independent and possibly ancient acquisitions, given that almost all genomes from the Symbiodinium genus, which is the most ancient in relation to the whole family, contained these sequences.
In bacteria, endonucleases together with methyltransferases are part of the restriction/modification (R/M) systems (Wilson and Murray 1991) that confer resistance to foreign DNA integration. In the early stages of Chlorovirus infection (Phycodnaviridae; a large DNA virus infecting green algae), endonucleases play a role in the degradation of host DNA such that it may be recycled, or these endonuclease products may function to prevent viral secondary infections (Agarkova, Dunigan, and Etten 2006). In the amoeba infecting Marseilleviridae, endonucleases are suggested to be hotspots for mutations and the footprints of mobile elements (Doutre et al. 2014; Mueller et al. 2017). These sequences are part of a giant virus R/M system, which may prevent the invasion of co-occurring intracellular parasites and provide protection against genome degradation by DNA methylation of their own DNA (Blanca et al. 2020). This model has been demonstrated, whereby marseilleviruses endonucleases degrade the DNA of other marseilleviruses, reinforcing the role of the R/M system in virus exclusion in cases of multiple infections (Jeudy et al. 2020). It is possible that the ancient acquisition of these sequences also conferred resistance in Symbiodiniaceae against new giant viral integrations (but not for other viral groups) or that a large segment of a giant viral genome was integrated and lost in the Symbiodiniaceae ancestor, and now these sequences are the sole remnants of this past event, with yet-to-be-determined roles. However, the distribution of these sequences in the most ancestral clades, their absence in derived Symbiodiniaceae, and the scarcity of additional vHGTs from giant viruses (using our methods), together with evidence for expression of all these sequences (Supplementary Table S2), suggest some form of biological activity that is yet to be defined.
3.4.3. Mononegavirales
The final group of vHGTs has a similarity to Mononegavirales, an order of ssRNA negative-strand viruses. These vHGTs were contained within two phylogenies (Fig. 4 and Supplementary Fig. S5); four of the vHGTs have top hits in the RNA-directed RNA polymerase catalytic domain (Supplementary Table S2). Whereas the best hits of the vHGTs were affiliated with metazoan viruses (Tacheng Tick Virus 6 [n = 3] and Wuchan romanomermis nematode virus 2 [n = 1]), the majority of sequences in the tree were from Rhabdoviridae or Rhabdo-like viruses, which are complex and diverse viruses that include several plant pathogens such as Strawberry crinkle virus and Citrus leprosis virus, among others (Dietzgen et al. 2017). Whereas in the phylogeny shown in Fig. 4A, the only vHGT present (from Snat) is outside the main group containing plant and metazoan viruses. In the phylogeny shown in Fig. 4B, the vHGTs are positioned at the bottom of the tree, clustered with another eukaryote (the perkinsid Perkinsus olseni). Because we found hits with the same viral sequences that are split into two phylogenies, it is possible that these were two independent HGT events: one involving the S. microadriaticum, Smic, Smic_04-503SCI.03, and Smic_CassKB8 genomes, and the other involving the Snat_CCMP2548 genome.
Figure 4.
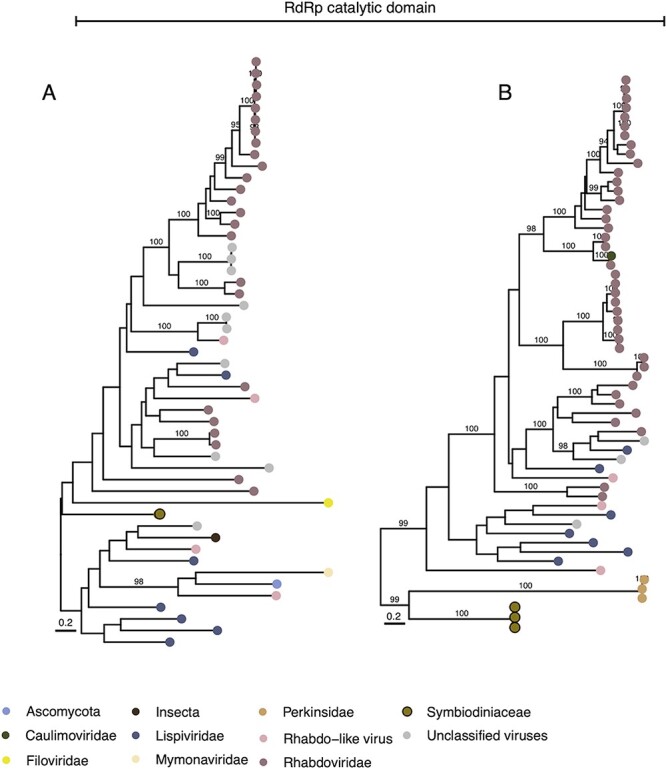
Phylogenetic profile of vHGT’s grouped by the category ‘Mononegavirales’: RNA-directed RNA polymerase catalytic domain (RdRP catalytic domain; A and B). Sequence names are not shown in the trees to enhance readability. The taxonomic affiliation of each sequence in the trees is represented by a colored circle, with the colors described in the legend at the bottom of the figure. Symbiodiniaceae vHGTs are highlighted with thicker borders. Only bootstrap support values ≥90 per cent are shown. Complete names and branch support values are in Supplementary Fig. S5.
3.5. Sequence composition of Symbiodiniaceae vHGTs sequences
We calculated the CDS GC-content for vHGTs and all Symbiodiniaceae CDSs predicted in the genomes that contain vHGTs. That later served as a background for comparative analysis (Fig. 1). Whereas the mean GC-content of the (background) Symbiodiniaceae CDSs ranged from 59.06 per cent (in S. microadriaticum CassKB8) to 51.38 per cent (in B. minutum), the mean GC-content of vHGTs ranged from 60.13 per cent (in S. natans CCMP2548) to 51.29 per cent (in B. minutum). Although the GC-content of the vHGTs was marginally lower than for background CDSs, this difference was statistically significant (P = 3.6E-07). However, when these two sets are compared genome-by-genome (i.e. comparing the vHGTs and background CDSs from the same genome), we only found significant GC-content differences in the Smic_CassKB8 (P = 1.4E−06), Smic (P = 2.4E−05), Snec_CCMP2469 (P = 4.4E−05), and Smic_04-503SCI.03 (P = 3.9E−04) genomes. This comparison was not performed for the Cgor and Slin_CCMP2456 genomes because the number of vHGTs was too small for statistical analysis. There were no significant differences in the length of CDSs between the vHGTs and the background CDSs when comparing all sets together (P = 8.0E−02) or individually, although vHGTs are slightly longer (Supplementary Tables S6 and S7).
3.6. Presence of introns and relic DinoSL motifs in Symbiodiniaceae vHGTs
The vHGTs are predominantly encoded by genes with multiple exons, with only 4/59 present as single-exon genes. The average number of introns per gene in the vHGT gene sets is either roughly equal to (e.g. in S. microadriaticum CassKB8) or lower than (e.g. in S. microadriaticum) the background gene set for each genome (Fig. 1; Supplementary Table S8). The average intron length of the vHGT genes tends to differ from that of background genes in each genome; however, this could be a result of the small number of viral HGT genes being analyzed. Of the fifty-nine vHGT-derived genes, thirteen have a DinoSL sequence that is upstream (within 2 kbp) of the first exon (Supplementary Table S9); there was no evidence of relic DinoSL sequences downstream of the identified DinoSL, in close proximity to the vHGT genes.
3.7. Distribution of vHGT sequences in Symbiodiniaceae genome scaffolds
We evaluated if there was an enrichment of vHGT genes in any of the genome scaffolds in the ten studied taxa. Our reasoning was that multiple vHGTs in a single scaffold from the same taxonomic source may indicate that they were derived from an exogenous virus that was included during sequencing (i.e. a contaminant in the assembly), or it could represent a complete or near-complete viral genome that had integrated into host DNA. The majority of vHGTs (fifty-three out of fifty-nine) were not on the same scaffold as another vHGT (Supplementary Tables S2 and S10). However, in two of the S. microadriaticum genomes, we found cases of two adjacent vHGTs; these genes were in scaffold3501 from Smic_04-503SCI.03 (genes15955 and 15956) and in scaffold792 and scaffold67 from Smic (genes19249−19250 and gene3240–3241, respectively) (Fig. 5). In all three cases, one of the proteins in each pair shares similarity with viral RNA-dependent RNA polymerase proteins and the other with viral major capsid proteins, which are the two proteins that comprise the +ssRNA Symbiodinium virus genome.
Figure 5.
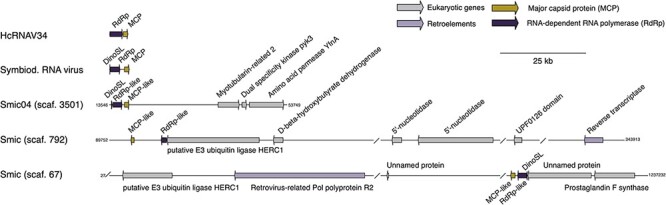
Genome maps comparing the CDSs of the dinoflagellate virus (dinornavirus) HcRNAV34 (Heterocapsa circularisquama RNA virus—accession: AB218608.1) and Symbiodinium RNA virus (Symbiodinium +ssRNA virus—accession KX538960.1) with the putative integrated viral genomes in S. microadriaticum (Smic and Smic04). The putative viral elements (vHGTs) annotated as RdRp- and MCP-like proteins are shown, as are the genes surrounding these elements and the start and end positions of scaffolds. Each feature points in the direction that it is encoded, and a scale bar is shown in the top right corner. Slash bars (/) denote intergenic regions. Abbreviations: scaf. (scaffold); DinoSL (dinoflagellate splice leader; annotated as small arrows); RdRp and RdRp-like (RNA-dependent RNA polymerase); MCP and MCP-like; Retrovirus-related Pol polyprotein R2 (Retrovirus-related Pol polyprotein from type-1 retrotransposable element R2); Amino acid permease YfnA (putative amino acid permease YfnA).
3.8. Symbiodinium +ssRNA virus similarities in Symbiodininiaceae genomes
We found that the majority of vHGTs (n = 34) had similarity with the most well-characterized virus known to infect this group, the Symbiodinium +ssRNA virus (TR74740 c13_g1_i1 and _i2, accession KX538960—KX787934) (Levin et al. 2017; Montalvo-Proaño et al. 2017) that have been implicated in coral holobiont health (Grupstra et al. 2022), and also with the HcRNAV (another dinoflagellate-infecting virus). In addition, in two of the S. microadriaticum genomes, we found adjacent vHGTs re-annotated using BLASTp as the two open reading frames (ORFs) that compose the complete genomes of these two viruses: RNA-dependent RNA polymerase (RdRp) (n = 21) and putative MCP (n = 4) (Supplementary Table S2). Given these findings, we compared the composition and genomic context of the MCPs- and RdRp-like vHGTs with the respective sequences from the +ssRNA Symbiodinium and HcRNAV viruses. We found that the mean GC-content of Symbiodiniaceae vHGTs MCPs-like and RdRps-like genes was higher (52.95 and 53.76 per cent, respectively) than in the putative +ssRNA Symbiodinium virus homologs (48.10 and 43.95 per cent, respectively), lower than the HcRNAV homologs (58.06 and 54.03 per cent, respectively), and lower than in the full Symbiodiniaceae genomes (56.79 per cent). In addition, the mean CDS length of MCPs-like genes was slightly lower than their viral counterparts, whereas the RdRps-like genes were slightly longer (Supplementary Table S11).
Whereas these proteins do not appear to be expressed (i.e. they did not have any RNA-seq reads aligned using HISAT2; Supplementary Figs S2-A, S3-A, and S4-A), they were on scaffolds with other expressed multi-exon genes. Realignment of selected RdRp- and MCP-like proteins against the regions of the host genome that encoded these genes (see Methods) demonstrated that all six of these proteins are significantly degraded (Supplementary Figs. S2-S4). These genes often have similarity to only part of the query viral protein (e.g. Supplementary Fig. S2B), in-frame stop codons, or frameshift mutations (e.g. Supplementary Fig. S3C), which would suggest that they are no longer under selection and have become pseudogenes. Furthermore, two of the putative integrated viral genomes, one from S. microadriaticum (gene3240-3241) and the other from S. microadriaticum 04-503SCI.03 (gene15955–15956) appear to be homologous (Supplementary Figs S2 and S4), potentially having arisen from an infection in the common ancestor of the two isolates. The third putative integrated genome (gene19249−19250) only appears in the S. microadriaticum genome.
3.8. Genomic context of Symbiodiniaceae vHGTs
Through analysis of the position of these sequences in the genome scaffolds, it was observed that the majority of vHGTs encoding MCP- and RdRp-like genes were located at the start of their scaffolds, or in single gene scaffolds. Protein sequences of non-vHGTs up- and downstream of vHGTs (if available) were annotated using the BLASTp online suite, with proteins with hypothetical, uncharacterized, and unnamed domains further analyzed using HHpred to identify distantly related putative homologs. We found that all vHGTs on scaffolds with other genes were located within the majority of non-viral host sequences (Supplementary Table S10). In these scaffolds, we found the presence of protein sequences such as the CCHC-type domain-containing protein (n = 2 sequences in total), which have been shown in other eukaryotes as either an anti- or proviral, targeting several viruses such as RNA viruses (Hajikhezri et al. 2020), which could also be ‘hijacked’ by nuclear-replicating viruses to promote viral production and are also induced after heat-shock stress conditions (Younis et al. 2018). The other genes located close to vHGTs are putative E3 ubiquitin–protein ligases (n = 5), RING finger proteins (n = 3; which have roles in viral evasion of host innate immunity) (Xu et al. 2017), F-box proteins (n = 4; which is a component of E3 ubiquitin–ligase complex that targets proteins for ubiquitination and degradation) (Correa et al. 2013b), and the ankyrin repeat domain-containing proteins (n = 6; which regulate virus–host interactions) (Than et al. 2016). In addition, by using HHpred to annotate the 295 proteins without known domains, we recovered annotations for an additional 112 proteins, of which seventeen remained with the unknown/uncharacterized annotation (Supplementary Table S10). On these, we found instances of predicted viral-like functions such as Adeno_IVa2 Adenovirus IVa2 protein, Phage_integrase Phage integrase family, and also DVNPs. The latter of which is known to be derived from viral HGTs in the dinoflagellate ancestor (Gornik et al. 2012).
Furthermore, some vHGTs were located in close proximity to transposons, retrotransposons, and repetitive sequences, such as the retrovirus-related Pol polyproteins (n = 8), transposon Ty2 Gag-Pol polyprotein (n = 2), pentatricopeptide repeat (n = 2), LINE-1 retrotransposable element ORF2 protein (n = 1), copia protein (n = 5), and reverse transcriptase domain-containing protein (n = 2). More sensitive searches with HHpred using proteins without annotated domains also revealed the presence of transposases (n =11) and additional reverse transcriptase domains (n = 2). Three scaffolds were identified in S. microadriaticum genomes (Fig. 3) in which the MCPs- and RdRps-like genes were located adjacent to each other on the same scaffold (scaffold67 and scaffold792 for Smic and scaffold3501 for Smic 04-503SCI.03), where in Smic sequences were in an orientation that was inverted relative to the +ssRNA Symbiodinium RNA virus genome (i.e. RdRp upstream of MCP). On scaffold67, these sequences are flanked by a downstream unannotated protein and by a Retrovirus-related Pol polyprotein (from a type-1 retrotransposable element R2). Finally, on scaffold792, there is a reverse transcriptase domain-containing protein at the end of this scaffold (Fig. 5).
4. Discussion
In this study, we describe the putative function, genomic features, and taxonomic distribution of fifty-nine high-confidence viral-derived HGT genes present in 10/13 Symbiodiniaceae genomes. These genes were identified using a combination of HGT scoring metrics, phylogeny-based detection, and significant manual curation. Due to the stringent nature of the filtering that was applied and the lack of a comprehensive database consisting of dinoflagellate-infecting viral genomes, the number of vHGTs we found likely represents a lower bound of the actual number of viral HGT events in these genomes. Nevertheless, by describing a more conservative set of viral HGT candidates in Symbiodiniaceae, we open-up the possibility for additional experimental evaluation to be undertaken to advance functional analysis of these genes. There were between 1 and 14 vHGTs identified in each genome, comprising ∼0.2 per cent of the total Symbiodiniaceae gene repertoire. Whereas HGT score metrics, such as AI and HGT indexes, identify sequences that are highly similar to foreign sources, phylogenetics can identify more distantly related sequences. Thus, by combining the two approaches, we have a higher likelihood of identifying genuine viral HGT events (Fan et al. 2020).
Previous reports of viral genes in Symbiodiniaceae genomes showed that in S. microadriaticum, genes with similarity to known viral sequences accounted for <3 per cent of the total gene inventory (Aranda et al. 2016), and in C. goreaui this number was only ∼0.1 per cent (Liu et al. 2018). In the latter case, regions that match viruses known to infect C. goreaui were identified (Weynberg et al. 2017), although the origin of these genomes was not clear. Shoguchi et al. (2018) suggested that DNA methylation in some Symbiodiniaceae genomes (clade SymA and SymC) was related to the presence and expansion of endogenous retroviruses such as retrovirus-related genes. In addition, in the S. microadriaticum genome, recently expanded Ty1-copia-type Long terminal repeats (LTR) retrotransposons are activated under acute temperature increases (Chen et al. 2018).
Our findings expand upon these studies by demonstrating the presence of candidate viral sequences from giant virus sources (Pimascovirales) that are potentially functioning as endonucleases, as well as polymerase sequences originating from RNA viruses that could function in viral replication. Exploration of vHGT sequence composition and structural features showed that their GC-contents are slightly lower than for Symbiodiniaceae background CDSs but higher than homologous viral sequences. The size of the CDSs does not differ from the background Symbiodiniaceae CDS repertoire. This could indicate that some of these transfers were recent in the evolutionary history of this group and are still in the process of being ameliorated into the recipient host genome (Lawrence and Ochman 1997). Moreover, we found that viral sequences are primarily localized to different scaffolds in the genome but tend to be near eukaryotic genes with known roles in virus–host interaction. Finally, we found these viral sequences near mobile genetic elements, such as retrotransposons and transposons, that may act not only as preferential integration sites but also as a mechanism for their integration and expansion.
Currently, the best-characterized virus that infects Symbiodiniaceae is a positive single-stranded RNA virus (+ssRNA) (NCBI:txid1909300) (Nagasaki et al. 2005; Correa et al. 2013a; Montalvo-Proaño et al. 2017; Weynberg et al. 2017), first described in stressed Montastraea cavernosa coral viromes (Correa et al. 2013a), which is related to the dinoflagellate virus Heterocapsa circularisquama (HcRNAV) from the Alvernaviridae family. Its complete genome was described by Levin et al. (2017); they found that thermally stressed Cladocopium (formerly Symbiodinium clade C1) populations experienced an extreme +ssRNA virus infection, suggesting that viral infections may influence Symbiodinium thermal sensitivity and, consequently, coral bleaching. Nevertheless, these authors were unable to amplify MCP sequences from genomic DNA (only from cDNA), suggesting that these sequences were not endogenous in the host genome but rather exogenous. Moreover, they reported the presence of a DinoSL at the 5ʹ-end of the amplified viral transcripts, proposing that this motif was incorporated into the viral genome as a form of ‘molecular mimicry’ to evade the host immune response and drive efficient translation of the viral genome by the host cellular machinery.
It has been suggested that some viral infections in Symbiodiniaceae are latent, given that VLPs and expressed sequences were found in asymptomatic and healthy cultures subjected to stress conditions (Wilson et al. 2001; Lawrence et al. 2014; Brüwer et al. 2017). Lawrence et al. (2017) showed that in Symbiodiniaceae cultures exposed to UV stress, there was upregulation of virus-like genes associated with viral genome replication and protein production, such as ubiquitin-encoding genes and homing endonucleases that could be involved in the excision of the integrated viral genome. This pattern is consistent with latent virus replication and agrees with our results.
Latent viruses are well described in plants and often do not elicit apparent symptoms in wild populations; however, they can cause disease in crops or due to experimentally created stresses such as mechanical wounding (Roossinck and García-Arenal 2015). Mixed viral infections and changes in environmental conditions trigger the switch from latent to active infections and diseases (Takahashi et al. 2019). In plants, the integration of a virus genome is not required for latent viral replication, although partial genome integration and endogenization of entire viral genomes may play a major role in latency (Takahashi et al. 2019). Latent infections and endogenous viral elements (EVEs) present major risks for cultivated crops if they become active (Bertsch et al. 2009). This is the case for the hybrid Musa (Iskra-Caruana et al. 2010), where the endogenous banana streak virus (eBSV), which has multiple copies integrated into the host genome, becomes activated under stress, recombining, producing episomes, viral particles, and disease symptoms such as streaks and necrosis in plant leaves. Recent work using CRISPR/Cas9-mediated editing of eBSV has been successful in preventing activation of these viruses (Tripathi et al. 2019). In contrast, benefits of latent viruses and EVEs are heat and drought tolerance, protection of host plants against virulent viral strains (Agüero et al. 2018), and suppression of heterologous viral infections (Staginnus et al. 2007).
4.1. Proposed model and scenarios for vHGT origin in Symbiodiniaceae genomes
Given our findings, we propose a model with two hypothetical scenarios to explain the occurrence of viral sequences in the genomes of Symbiodiniaceae and the underlying mechanisms for their integration (Fig. 6). In this model, whereas several viral groups infect Symbiodiniaceae, independently entering the host cell and subjugating the host machinery to produce viral mRNAs, some virus sequences become EVEs by a series of incidental integration events into Symbiodiniaceae genomes (A). For this to occur, viral mRNAs need to be reverse transcribed before they can be integrated into the host genome. This process might occur via the mRNA recycling mechanism that is prevalent in dinoflagellates (Stephens et al. 2020), whereby spliced-leader-containing transcripts are reverse transcribed (possibly mediated by retrotransposons (Lee et al. 2014; Song et al. 2017) into DNA before being integrated back into the genome through non-homologous recombination (Slamovits and Keeling 2008). The infecting viral elements must escape degradation by host defenses. Because viral mRNAs are highly expressed during an active infection, the host spliceosomal machinery may erroneously add a DinoSL motif onto viral transcripts, making the transcripts appear ‘native’. This may have allowed the viral genome to better evade host defenses and provide the virus with a dinoflagellate TATA-box-like promoter sequence; dinoflagellates appear to use a TTTT motif (which is also present in the DinoSL) in place of the canonical TATA motif in other eukaryotes (Guillebault et al. 2002). This is supported by the presence of a DinoSL in the Symbiodinium RNA virus genome (Levin et al. 2017) and suggests that this motif could have been acquired by the virus from the host either in the common ancestor of these viruses or during each new infection cycle. Whereas convergence is also possible (i.e. independent evolution of a DinoSL-like motif), it is less likely given the high level of sequence similarity between the viral and host DinoSL sequences. Furthermore, the occurrence of DinoSL sequences upstream of thirteen vHGTs, plus the localization of these sequences near retrovirus-related polyproteins, copia proteins, and proteins containing reverse transcriptase domains in the Symbiodiniaceae scaffolds, indirectly supports this mechanism of integration.
Figure 6.
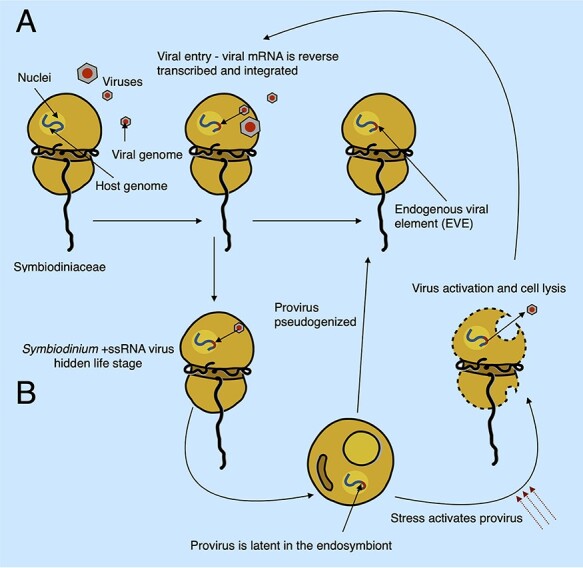
A hypothetical model with two scenarios (A and B) explaining the origin of vHGTs in Symbiodiniaceae genomes. During an acute infection, viral particles enter the host cell, and viral mRNAs accumulate in the cytoplasm to a high abundance. The host machinery can accidentally incorporate viral mRNA into its genome via reverse transcription and non-homologous recombination; this may use the same (or similar) mechanisms as the mRNA recycling process that is prevalent in dinoflagellates. These viral sequences, known as EVEs, would be ‘dead on arrival’ and are expected to decay into pseudogenes (which we would then observe in the genome as vHGTs). In the second scenario (B), integration of the virus genome into the host genome is part of a previously unobserved transient life stage of these viruses (specifically the Symbiodinium +ssRNA virus). The virus infects and is incorporated into the host genome via the same mechanisms as the first scenario (A); however, instead of this process resulting in non-functional EVEs, it produces active proviral elements. These integrated viral genomes can remain silent during times of low host abundance (e.g. during the free-living stage of facultative Symbiodiniaceae lifecycles) and can become activated by environmental cues (such as stress or high Symbiodiniaceae cell density) that result in viral lysis, escape from the host cell, and new infections in susceptible hosts. The proviral elements can also become deactivated, either through random mutation or host-driven process, resulting in EVEs that are expected to decay into pseudogenes, which we would then observe in the genome as vHGTs. The Symbiodiniaceae cell illustration was inspired by drawings by Freudenthal (1962) and the Symbiodinium life cycle illustration was adapted from https://commons.wikimedia.org/wiki/File:Symbiodinium_Life_Cycle.png.
However, given that we found complete viral genomes with similarity to the +ssRNA Symbiodinium virus, we propose a second scenario (B) whereby vHGTs from +ssRNA Symbiodinium viruses result from a previously hidden (cryptic) life stage of these viruses. In this scenario, copies of viral genomes become endogenized and deactivated by the host genome, decaying as pseudogenes. Alternatively, they remain intact, transcriptionally silent (as proviruses), and may eventually become activated in response to stress, shifting from a proviral silent life stage to being infectious. These RNA viruses could actively exploit the dinoflagellate mRNA recycling process to facilitate their integration into host DNA, although this would require the virus to also have a system for permanence (survival) and activation, both of which are unknown at this time. However, given that the genes associated with the three putative integrated Symbiodinium +ssRNA virus genomes, identified in two of the S. microadriaticum isolates, appear to have become pseudogenized, it seems likely that many of the viral genes that we identified in the Symbiodiniaceae genomes are (or are becoming) pseudogenes. This is an expected result given that there is strong selective pressure on the host to inactivate endogenous viruses and that we only expect to see remnants of inactivated endogenous viruses originating from past (failed) infections in the genomes of extant lineages (i.e. active EVEs would likely result in an active viral infection that would cause the lineage to perish). In addition, the two vHGT genes that are part of the putative Symbiodinium +ssRNA viruses identified in the S. microadriaticum (Smic) genome are encoded in the reverse order to what is observed in the available virus reference genome. This may result from the host genome inactivating the invasive viruses by shuffling their gene orders, preventing the formation of infective viral particles. Furthermore, the putative inactivation of these elements by the host suggests that they were active when they were integrated into the genome, lending support to the second scenario (which produces viral elements that are ‘dead on arrival’). Moreover, the multiple, complete and incomplete copies of these viral genomes in Symbiodiniaceae, from different isolates and genera, suggests that viral endogenization is an ongoing process in these organisms, with a potentially rapid turnover of invasion and pseudogenization or activation and infection, similar to Mavirus virophage infection-integration cycles (Hackl et al. 2021).
Under this scenario, endogenization does not need to be an obligatory step in the life cycle of the virus (i.e. it is a transient stage) and does not need to become fixed in all hosts occurring, for instance, only when there is a low number of susceptible hosts. Becoming endogenized when the density of host cells is low is advantageous for the virus, given that a copy of the virus genome is always transmitted to host progeny, effectively preserving the virus until conditions promote the lytic life stage. The virus life cycle may therefore mirror that of the host, remaining inactive during phases where the density of susceptible cells is low, such as during the host coccoid endosymbiont life stage, and becoming active when the density of susceptible cells is high, such as during the host mastigote, free-living stage. From this perspective, the more susceptible stage would be the free-living state with no endogenous provirus in its genome. From the perspective of the Symbiodiniaceae host, it could be argued that having an endogenous provirus would confer superinfection immunity against other exogenous viruses via cross protection and may benefit the cell under certain conditions (Mette et al. 2002). A hidden transient viral life cycle, if confirmed for these viruses, would unify the current knowledge of viruses in Symbiodiniaceae, explaining the observations of: (1) latent viral infections in healthy algal cultures after stress induction, (2) exogenous RNA viruses that infect Symbiodiniaceae, and (3) our findings of endogenous versions of these exogenous Symbiodinium RNA virus genomes.
As previously noted, triggers that cause the switch from latent to active viral infections and diseases in plants and algae (including cultured Symbiodiniaceae) overlap with those that trigger and influence coral bleaching and other coral diseases (Harvell et al. 1999; Lesser and Farrell 2004; Randall and van Woesik 2015; Lawrence et al. 2017). It has been recently proposed that the breakdown of the Symbiodiniaceae-coral symbiosis due to heat stress (coral bleaching) is caused by the increased proliferation of the symbiont (Rädecker et al. 2021). The observed increase in viral particle production under thermal stress may coincide with the increased proliferation of the symbionts during the early stages of coral bleaching. This result suggests that the virus life cycle could be directly linked to that of the host.
Consequently, the main implication of our work is that if EVEs and latent proviruses that are hidden in Symbiodiniaceae genomes could become activated by environmental stressors, they may cause local outbreaks and possibly contribute to the onset of some coral diseases. To conclude, by describing a variety of acquired viral genes originating from multiple HGT events, we provide strong evidence that viruses have invaded and introduced novel genetic material into Symbiodiniaceae genomes. Our work suggests the possibility that endogenous and latent viruses could modulate and participate in the life cycle, evolution, and potentially breakdown of the Symbiodiniaceae-host symbiosis.
Supplementary Material
Acknowledgements
L.F.B. wish to thank Paulo S. Salomon (Universidade Federal do Rio de Janeiro—UFRJ) and coordinators from ‘Programa de Pós-graduação em Biodiversidade e Biologia Evolutiva’ (PPGBBE - UFRJ) for the initial conceptual support in early stage of these ideas; Lílian Caesar for critical reading, discussions, love, and support during all stages of this work; François Bucchini for support in initial codes; GenoToul bioinformatics platform for access to computing cluster facilities during early stages of this work; Girish Beedessee and Cheong Xin Chan for guidance in data retrieval. We thank the anonymous reviewers for their recommendations for manuscript improvement and the editors for handling our article.
Contributor Information
L Felipe Benites, Department of Biochemistry and Microbiology, Rutgers University, 102 Foran Hall, 59 Dudley Road, New Brunswick, NJ 08901-8520, USA.
Timothy G Stephens, Department of Biochemistry and Microbiology, Rutgers University, 102 Foran Hall, 59 Dudley Road, New Brunswick, NJ 08901-8520, USA.
Debashish Bhattacharya, Department of Biochemistry and Microbiology, Rutgers University, 102 Foran Hall, 59 Dudley Road, New Brunswick, NJ 08901-8520, USA.
Data availability
All Symbiodiniaceae sequence data used in this study are available from https://espace.library.uq.edu.au/view/UQ:f1b3a11 and https://espace.library.uq.edu.au/view/UQ:8279c9a. Code to perform the HGT calculations and multifasta protein files with Symbiodiniaceae vHGTs are available from https://github.com/LFelipe-B/Symbiodiniaceae_vHGT_scripts.
Supplementary data
Supplementary data are available at Virus Evolution online.
Funding
This work was supported by an appointment to the NASA Postdoctoral Program at Rutgers University, New Brunswick, administered by Oak Ridge Associated Universities under contract with NASA, to L.F.B. D.B., and T.G.S. were supported by a NASA grant (80NSSC19K0462) to D.B. and a NIFA-USDA Hatch grant (NJ01180) to D.B.
Conflict of interest:
The authors declare no conflict of interest.
References
- Agarkova I. V., Dunigan D. D. and Etten J. L. V. (2006) ‘Virion-Associated Restriction Endonucleases of Chloroviruses’, Journal of Virology, 80: 8114–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agüero J. et al. (2018) ‘Stable and Broad Spectrum Cross-Protection against Pepino Mosaic Virus Attained by Mixed Infection’, Frontiers in Plant Science, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aherfi S. et al. (2014) ‘The Expanding Family Marseilleviridae’, Virology, 466–467: 27–37. [DOI] [PubMed] [Google Scholar]
- Aranda M. et al. (2016) ‘Genomes of Coral Dinoflagellate Symbionts Highlight Evolutionary Adaptations Conducive to a Symbiotic Lifestyle’, Scientific Reports, 6: 39734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aus Dem Siepen M. et al. (2005) ‘Poinsettia Latent Virus Is Not a Cryptic Virus, but a Natural Polerovirus–Sobemovirus Hybrid’, Virology, 336: 240–50. [DOI] [PubMed] [Google Scholar]
- Benites L. F. et al. (2018) ‘Megaviridae-like Particles Associated with Symbiodinium spp. from the Endemic Coral Mussismilia braziliensis’, Symbiosis, 76: 303–11. [Google Scholar]
- Bertsch C. et al. (2009) ‘Retention of the Virus-derived Sequences in the Nuclear Genome of Grapevine as a Potential Pathway to Virus Resistance’, Biology Direct, 4: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanca L. et al. (2020) ‘Comparative Analysis of the Circular and Highly Asymmetrical Marseilleviridae Genomes’, Viruses, 12: 1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boschetti C. et al. (2012) ‘Biochemical Diversification through Foreign Gene Expression in Bdelloid Rotifers’, PLoS Genetics, 8: e1003035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bräutigam M. et al. (1995) ‘Inheritance and Meiotic Elimination of a Virus Genome in the Host Ectocarpus siliculosus (Phaeophyceae)’, Journal of Phycology, 31: 823–7. [Google Scholar]
- Brüwer J. D. et al. (2017) ‘Association of Coral Algal Symbionts with a Diverse Viral Community Responsive to Heat Shock’, BMC Microbiology, 17: 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B., Xie C., and Huson D. H. (2015) ‘Fast and Sensitive Protein Alignment Using DIAMOND’, Nature Methods, 12: 59–60. [DOI] [PubMed] [Google Scholar]
- Capella-Gutiérrez S., Silla-Martínez J. M., and Gabaldón T. (2009) ‘trimAl: A Tool for Automated Alignment Trimming in Large-scale Phylogenetic Analyses’, Bioinformatics, 25: 1972–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charon J., Murray S. and Holmes E. C. (2021) ‘Revealing RNA Virus Diversity and Evolution in Unicellular Algae Transcriptomes’, Virus Evolution, 7: veab070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. E. et al. (2018) ‘Recent Expansion of Heat-activated Retrotransposons in the Coral Symbiont Symbiodinium microadriaticum’, The ISME Journal, 12: 639–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. et al. (2020) ‘Evidence that Inconsistent Gene Prediction Can Mislead Analysis of Dinoflagellate Genomes’, Journal of Phycology, 56: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cock J. M. et al. (2010) ‘The Ectocarpus Genome and the Independent Evolution of Multicellularity in Brown Algae’, Nature, 465: 617–21. [DOI] [PubMed] [Google Scholar]
- Correa A. M. S., Welsh R. M., and Vega Thurber R. L. (2013a) ‘Unique Nucleocytoplasmic dsDNA and +ssrna Viruses Are Associated with the Dinoflagellate Endosymbionts of Corals’, The ISME Journal, 7: 13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa R. L. et al. (2013b) ‘The Role of F-Box Proteins during Viral Infection’, International Journal of Molecular Sciences, 14: 4030–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa A. M. S. et al. (2016) ‘Viral Outbreak in Corals Associated with an In Situ Bleaching Event: Atypical Herpes-Like Viruses and a New Megavirus Infecting Symbiodinium’, Frontiers in Microbiology, 7: 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davy S. K. et al. (2006) ‘Viruses: Agents of Coral Disease?’, Diseases of Aquatic Organisms, 69: 101–10. [DOI] [PubMed] [Google Scholar]
- Delaroque N., and Boland W. (2008) ‘The Genome of the Brown Alga Ectocarpus siliculosus Contains a Series of Viral DNA Pieces, Suggesting an Ancient Association with Large dsDNA Viruses’, BMC Evolutionary Biology, 8: 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietzgen R. G. et al. (2017) ‘The Family Rhabdoviridae: Mono- and Bipartite Negative-sense RNA Viruses with Diverse Genome Organization and Common Evolutionary Origins’, Virus Research, 227: 158–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinan A. M. et al. (2020) ‘A Case for a Negative-strand Coding Sequence in a Group of Positive-sense RNA Viruses’, Virus Evolution, 6: veaa007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doutre G. et al. (2014) ‘Genome Analysis of the First Marseilleviridae Representative from Australia Indicates that Most of Its Genes Contribute to Virus Fitness’, Journal of Virology, 88: 14340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emms D. M., and Kelly S. (2019) ‘OrthoFinder: Phylogenetic Orthology Inference for Comparative Genomics’, Genome Biology, 20: 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X. et al. (2020) ‘Phytoplankton Pangenome Reveals Extensive Prokaryotic Horizontal Gene Transfer of Diverse Functions’, Science Advances, 6: eaba0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freudenthal H. D. (1962) ‘Symbiodinium Gen. Nov. and Symbiodinium microadriaticum Sp. Nov., A Zooxanthella: Taxonomy, Life Cycle, and Morphology.*’, The Journal of Protozoology, 9: 45–52. [Google Scholar]
- Gilmer D., Ratti C., and ICTV Report ConsortiumYR (2017) ‘ICTV Virus Taxonomy Profile: Benyviridae’, Journal of General Virology, 98: 1571–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladyshev E. A., Meselson M., and Arkhipova I. R. (2008) ‘Massive Horizontal Gene Transfer in Bdelloid Rotifers’, Science, 320: 1210–3. [DOI] [PubMed] [Google Scholar]
- González-Pech R. A. et al. (2021) ‘Comparison of 15 Dinoflagellate Genomes Reveals Extensive Sequence and Structural Divergence in Family Symbiodiniaceae and Genus Symbiodinium’, BMC Biology, 19: 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gornik S. G. et al. (2012) ‘Loss of Nucleosomal DNA Condensation Coincides with Appearance of a Novel Nuclear Protein in Dinoflagellates’, Current Biology Cb, 22: 2303–12. [DOI] [PubMed] [Google Scholar]
- ——— et al. (2019) ‘The Biochemistry and Evolution of the Dinoflagellate Nucleus’, Microorganisms, 7: 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grupstra C. G. B. et al. (2022) ‘Thermal Stress Triggers Productive Viral Infection of a Key Coral Reef Symbiont’, The ISME Journal 16: 1430–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillebault D. et al. (2022) ‘A new class of transcription initiation factors, intermediate between TATA box-binding proteins (TBPs) and TBP-like factors (TLFs), is present in the marine unicellular organism, the dinoflagellate Crypthecodinium cohnii’, J Biol Chem. 277: 40881–6. [DOI] [PubMed] [Google Scholar]
- Hackl T. et al. (2021) ‘Virophages and Retrotransposons Colonize the Genomes of a Heterotrophic Flagellate’, eLife, 10: e72674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajikhezri Z. et al. (2020) ‘Role of CCCH-Type Zinc Finger Proteins in Human Adenovirus Infections’, Viruses, 12: 1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvell C. D. et al. (1999) ‘Emerging Marine Diseases—Climate Links and Anthropogenic Factors’, Science, 285: 1505–10. [DOI] [PubMed] [Google Scholar]
- Hoffman L. R., and Stanker L. H. (1976) ‘Virus-like Particles in the Green Alga Cylindrocapsa’, Canadian Journal of Botany. Journal Canadien de Botanique, 54: 2827–41. [Google Scholar]
- Hyman P., and Abedon S. T. (2012) ‘Smaller Fleas: Viruses of Microorganisms’, Scientifica, 2012: e734023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin N. A. T. et al. (2018) ‘Viral Proteins as a Potential Driver of Histone Depletion in Dinoflagellates’, Nature Communications, 9: 1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2022) ‘Systematic Evaluation of Horizontal Gene Transfer between Eukaryotes and Viruses’, Nature Microbiology, 7: 327–36. [DOI] [PubMed] [Google Scholar]
- Iskra-Caruana M.-L. et al. (2010) ‘A Four-partner Plant–Virus Interaction: Enemies Can Also Come from Within’, Molecular Plant-Microbe Interactions, 23: 1394–402. [DOI] [PubMed] [Google Scholar]
- Janouškovec J. et al. (2017) ‘Major Transitions in Dinoflagellate Evolution Unveiled by Phylotranscriptomics’, Proceedings of the National Academy of Sciences of the USA, 114: E171–E180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeudy S. et al. (2020) ‘The DNA Methylation Landscape of Giant Viruses’, Nature Communications, 11: 2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyaanamoorthy S. et al. (2017) ‘ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates’, Nature Methods, 14: 587–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., and Standley D. M. (2013) ‘MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability’, Molecular Biology and Evolution, 30: 772–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. et al. (2019) ‘Graph-based Genome Alignment and Genotyping with HISAT2 and HISAT-genotype’, Nature Biotechnology, 37: 907–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaJeunesse T. C. et al. (2018) ‘Systematic Revision of Symbiodiniaceae Highlights the Antiquity and Diversity of Coral Endosymbionts’, Current Biology CB, 28: 25707–80.e6. [DOI] [PubMed] [Google Scholar]
- Lawrence S. A. et al. (2014) ‘Latent Virus-like Infections are Present in a Diverse Range of Symbiodinium spp. (Dinophyta)’, Journal of Phycology, 50: 984–97. [DOI] [PubMed] [Google Scholar]
- ——— et al. (2017) ‘Exploratory Analysis of Symbiodinium Transcriptomes Reveals Potential Latent Infection by Large dsDNA Viruses’, Environmental Microbiology, 19: 3909–19. [DOI] [PubMed] [Google Scholar]
- Lawrence J. G., and Ochman H. (1997) ‘Amelioration of Bacterial Genomes: Rates of Change and Exchange’, Journal of Molecular Evolution, 44: 383–97. [DOI] [PubMed] [Google Scholar]
- Lee R. et al. (2014) ‘Analysis of EST Data of the Marine Protist Oxyrrhis marina, an Emerging Model for Alveolate Biology and Evolution’, BMC Genomics, 15: 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A. M., Ivey R. G., and Meints R. H. (1998) ‘Repetitive DNA Insertion in a Protein Kinase ORF of a Latent FSV (Feldmannia sp. Virus) Genome’, Virology, 248: 35–45. [DOI] [PubMed] [Google Scholar]
- Lesser M. P., and Farrell J. H. (2004) ‘Exposure to Solar Radiation Increases Damage to Both Host Tissues and Algal Symbionts of Corals during Thermal Stress’, Coral Reefs, 23: 367–77. [Google Scholar]
- Levin R. A. et al. (2017) ‘Evidence for a Role of Viruses in the Thermal Sensitivity of Coral Photosymbionts’, The ISME Journal, 11: 808–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. et al. (2009) ‘1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map Format and SAMtools’, Bioinformatics, 25: 2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S. et al. (2015) ‘The Symbiodinium kawagutii Genome Illuminates Dinoflagellate Gene Expression and Coral Symbiosis’, Science, 350: 691–4. [DOI] [PubMed] [Google Scholar]
- Liu H. et al. (2018) ‘Symbiodinium Genomes Reveal Adaptive Evolution of Functions Related to Coral-dinoflagellate Symbiosis’, Communications Biology, 1: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohr J., Munn C. B., and Wilson W. H. (2007) ‘Characterization of a Latent Virus-Like Infection of Symbiotic Zooxanthellae’, Applied and Environmental Microbiology, 73: 2976–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lye L.-F. et al. (2016) ‘A Narnavirus-Like Element from the Trypanosomatid Protozoan Parasite Leptomonas seymouri’, Genome Announcements, 4: e00713–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. (2011) ‘Cutadapt Removes Adapter Sequences from High-throughput Sequencing Reads’, EMBnet.Journal, 17: 10–2. [Google Scholar]
- McKeown D. A. et al. (2017) ‘Phaeoviruses Discovered in Kelp (Laminariales)’, The ISME Journal, 11: 2869–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méheust R. et al. (2018) ‘Formation of Chimeric Genes with Essential Functions at the Origin of Eukaryotes’, BMC Biology, 16: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messyasz A. et al. (2020) ‘Coral Bleaching Phenotypes Associated with Differential Abundances of Nucleocytoplasmic Large DNA Viruses’, Frontiers in Marine Science, 7: 789. [Google Scholar]
- Mette M. F. et al. (2002) ‘Endogenous Viral Sequences and Their Potential Contribution to Heritable Virus Resistance in Plants’, The EMBO Journal, 21: 461–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minh B. Q., Nguyen M. A. T., and von Haeseler A. (2013) ‘Ultrafast Approximation for Phylogenetic Bootstrap’, Molecular Biology and Evolution, 30: 1188–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moniruzzaman M. et al. (2020) ‘Widespread Endogenization of Giant Viruses Shapes Genomes of Green Algae’, Nature, 588: 141–5. [DOI] [PubMed] [Google Scholar]
- Montalvo-Proaño J. et al. (2017) ‘A PCR-Based Assay Targeting the Major Capsid Protein Gene of a Dinorna-Like ssRNA Virus that Infects Coral Photosymbionts’, Frontiers in Microbiology, 8: 1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller L. et al. (2017) ‘One Year Genome Evolution of Lausannevirus in Allopatric versus Sympatric Conditions’, Genome Biology and Evolution, 9: 1432–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller D. G., Kapp M., and Knippers R. (1998) ‘Viruses in Marine Brown Algae’. In: Maramorosch K., Murphy F. A., and Shatkin A. J. (eds) Advances in Virus Research, Vol. 50, pp. 49–67. Academic Press. [DOI] [PubMed] [Google Scholar]
- Nagasaki K. et al. (2005) ‘Comparison of Genome Sequences of Single-stranded RNA Viruses Infecting the Bivalve-killing Dinoflagellate Heterocapsa circularisquama’, Applied and Environmental Microbiology, 71: 8888–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nand A. et al. (2021) ‘Genetic and Spatial Organization of the Unusual Chromosomes of the Dinoflagellate Symbiodinium microadriaticum’, Nature Genetics, 53: 618–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L.-T. et al. (2015) ‘IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies’, Molecular Biology and Evolution, 32: 268–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nibert M. L. et al. (2018) ‘A Barnavirus Sequence Mined from a Transcriptome of the Antarctic Pearlwort Colobanthus quitensis’, Archives of Virology, 163: 1921–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M. et al. (2015) ‘StringTie Enables Improved Reconstruction of a Transcriptome from RNA-seq Reads’, Nature Biotechnology, 33: 290–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rädecker N. et al. (2021) ‘Heat Stress Destabilizes Symbiotic Nutrient Cycling in Corals’, Proceedings of the National Academy of Sciences of the USA, 118: e2022653118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rancurel C., Legrand L. and Danchin E. G. J. (2017) ‘Alienness: Rapid Detection of Candidate Horizontal Gene Transfers across the Tree of Life’, Genes, 8: 248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall C. J., and van Woesik R. (2015) ‘Contemporary White-band Disease in Caribbean Corals Driven by Climate Change’, Nature Climate Change, 5: 375–9. [Google Scholar]
- Rastgou M. et al. (2009) ‘Molecular Characterization of the Plant Virus Genus Ourmiavirus and Evidence of Inter-kingdom Reassortment of Viral Genome Segments as Its Possible Route of Origin’, The Journal of General Virology, 90: 2525–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revell L. J. (2012) ‘Phytools: An R Package for Phylogenetic Comparative Biology (and Other Things)’, Methods in Ecology and Evolution, 3: 217–23. [Google Scholar]
- Robinson J. T. et al. (2011) ‘Integrative Genomics Viewer’, Nature Biotechnology, 29: 24–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roossinck M. J., and García-Arenal F. (2015) ‘Ecosystem Simplification, Biodiversity Loss and Plant Virus Emergence’, Current Opinion in Virology, 10: 56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousvoal S. et al. (2016) ‘Mutant Swarms of a Totivirus-like Entities are Present in the Red Macroalga Chondrus crispus and Have Been Partially Transferred to the Nuclear Genome’, Journal of Phycology, 52: 493–504. [DOI] [PubMed] [Google Scholar]
- Schliep K. P. (2011) ‘Phangorn: Phylogenetic Analysis in R’, Bioinformatics, 27: 592–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz F. et al. (2020) ‘Giant Virus Diversity and Host Interactions through Global Metagenomics’, Nature, 578: 432–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W. et al. (2016) ‘SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation’, PLOS ONE, 11: e0163962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi M. et al. (2016) ‘Redefining the Invertebrate RNA Virosphere’, Nature, 540: 539–43. [DOI] [PubMed] [Google Scholar]
- Shoguchi E. et al. (2013) ‘Draft Assembly of the Symbiodinium minutum Nuclear Genome Reveals Dinoflagellate Gene Structure’, Current Biology: CB, 23: 1399–408. [DOI] [PubMed] [Google Scholar]
- ——— et al. (2018) ‘Two Divergent Symbiodinium Genomes Reveal Conservation of a Gene Cluster for Sunscreen Biosynthesis and Recently Lost Genes’, BMC Genomics, 19: 458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slamovits C., and Keeling P. (2008) ‘Widespread Recycling of Processed cDNAs in Dinoflagellates’, Current Biology CB, 18: R550–2. [DOI] [PubMed] [Google Scholar]
- Slater G. S. C., and Birney E. (2005) ‘Automated Generation of Heuristics for Biological Sequence Comparison’, BMC Bioinformatics, 6: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söding J., Biegert A., and Lupas A. N. (2005) ‘The HHpred Interactive Server for Protein Homology Detection and Structure Prediction’, Nucleic Acids Research, 33: W244–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sõmera M. et al. (2021) ‘ICTV Virus Taxonomy Profile: Solemoviridae 2021’, The Journal of General Virology, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song B. et al. (2017) ‘Comparative Genomics Reveals Two Major Bouts of Gene Retroposition Coinciding with Crucial Periods of Symbiodinium Evolution’, Genome Biology and Evolution, 9: 2037–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staginnus C. et al. (2007) ‘Endogenous Pararetroviral Sequences in Tomato (Solanum lycopersicum) and Related Species’, BMC Plant Biology, 7: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens T. G. et al. (2016) ‘PhySortR: A Fast, Flexible Tool for Sorting Phylogenetic Trees in R’, PeerJ, 4: e2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2020) ‘Genomes of the Dinoflagellate Polarella glacialis Encode Tandemly Repeated Single-exon Genes with Adaptive Functions’, BMC Biology, 18: 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H. et al. (2019) ‘Virus Latency and the Impact on Plants’, Frontiers in Microbiology, 10: 2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Than T. T. et al. (2016) ‘Ankyrin Repeat Domain 1 Is Up-regulated during Hepatitis C Virus Infection and Regulates Hepatitis C Virus Entry’, Scientific Reports, 6: 20819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurber R. L. V., and Correa A. M. S. (2011) ‘Viruses of Reef-building Scleractinian Corals’, Journal of Experimental Marine Biology and Ecology, 408: 102–13. [Google Scholar]
- Tripathi J. N. et al. (2019) ‘CRISPR/Cas9 Editing of Endogenous Banana Streak Virus in the B Genome of Musa spp. Overcomes a Major Challenge in Banana Breeding’, Communications Biology, 2: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldron F. M., Stone G. N., and Obbard D. J. (2018) ‘Metagenomic Sequencing Suggests a Diversity of RNA Interference-like Responses to Viruses across Multicellular Eukaryotes’, PLoS Genetics, 14: e1007533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weynberg K. D. et al. (2014) ‘Generating Viral Metagenomes from the Coral Holobiont’, Frontiers in Microbiology, 5: 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2017) ‘Prevalent and Persistent Viral Infection in Cultures of the Coral Algal Endosymbiont Symbiodinium’, Coral Reefs, 36: 773–84. [Google Scholar]
- Wilson W. H. et al. (2001) ‘Temperature Induction of Viruses in Symbiotic Dinoflagellates’, Aquatic Microbial Ecology : International Journal, 25: 99–102. [Google Scholar]
- ——— et al. (2005) ‘An Enemy Within? Observations of Virus-like Particles in Reef Corals’, Coral Reefs, 24: 145–8. [Google Scholar]
- Wilson G. G., and Murray N. E. (1991) ‘Restriction and Modification Systems’, Annual Review of Genetics, 25: 585–627. [DOI] [PubMed] [Google Scholar]
- Wisecaver J. H., Brosnahan M. L., and Hackett J. D. (2013) ‘Horizontal Gene Transfer Is a Significant Driver of Gene Innovation in Dinoflagellates’, Genome Biology and Evolution, 5: 2368–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood-Charlson E. M. et al. (2015) ‘Metagenomic Characterization of Viral Communities in Corals: Mining Biological Signal from Methodological Noise’, Environmental Microbiology, 17: 3440–9. [DOI] [PubMed] [Google Scholar]
- Xu Q. et al. (2017) ‘E3 Ubiquitin Ligase Nedd4 Promotes Japanese Encephalitis Virus Replication by Suppressing Autophagy in Human Neuroblastoma Cells’, Scientific Reports, 7: 45375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younis S. et al. (2018) ‘Multiple Nuclear-replicating Viruses Require the Stress-induced Protein ZC3H11A for Efficient Growth’, Proceedings of the National Academy of Sciences of the USA, 115: E3808–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G. et al. (2017) ‘Ggtree: An R Package for Visualization and Annotation of Phylogenetic Trees with Their Covariates and Other Associated Data’, Methods in Ecology and Evolution, 8: 28–36. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All Symbiodiniaceae sequence data used in this study are available from https://espace.library.uq.edu.au/view/UQ:f1b3a11 and https://espace.library.uq.edu.au/view/UQ:8279c9a. Code to perform the HGT calculations and multifasta protein files with Symbiodiniaceae vHGTs are available from https://github.com/LFelipe-B/Symbiodiniaceae_vHGT_scripts.