

Abstract
Background
Cytochrome P450 (CYP) 3A contributes to the metabolism of many approved drugs. CYP3A perpetrator drugs can profoundly alter the exposure of CYP3A substrates. However, effects of such drug-drug interactions are usually reported as maximum effects rather than studied as time-dependent processes. Identification of the time course of CYP3A modulation can provide insight into when significant changes to CYP3A activity occurs, help better design drug-drug interaction studies, and manage drug-drug interactions in clinical practice.
Objective
We aimed to quantify the time course and extent of the in vivo modulation of different CYP3A perpetrator drugs on hepatic CYP3A activity and distinguish different modulatory mechanisms by their time of onset, using pharmacologically inactive intravenous microgram doses of the CYP3A-specific substrate midazolam, as a marker of CYP3A activity.
Methods
Twenty-four healthy individuals received an intravenous midazolam bolus followed by a continuous infusion for 10 or 36 h. Individuals were randomized into four arms: within each arm, two individuals served as a placebo control and, 2 h after start of the midazolam infusion, four individuals received the CYP3A perpetrator drug: voriconazole (inhibitor, orally or intravenously), rifampicin (inducer, orally), or efavirenz (activator, orally). After midazolam bolus administration, blood samples were taken every hour (rifampicin arm) or every 15 min (remaining study arms) until the end of midazolam infusion. A total of 1858 concentrations were equally divided between midazolam and its metabolite, 1’-hydroxymidazolam. A nonlinear mixed-effects population pharmacokinetic model of both compounds was developed using NONMEM®. CYP3A activity modulation was quantified over time, as the relative change of midazolam clearance encountered by the perpetrator drug, compared to the corresponding clearance value in the placebo arm.
Results
Time course of CYP3A modulation and magnitude of maximum effect were identified for each perpetrator drug. While efavirenz CYP3A activation was relatively fast and short, reaching a maximum after approximately 2–3 h, the induction effect of rifampicin could only be observed after 22 h, with a maximum after approximately 28–30 h followed by a steep drop to almost baseline within 1–2 h. In contrast, the inhibitory impact of both oral and intravenous voriconazole was prolonged with a steady inhibition of CYP3A activity followed by a gradual increase in the inhibitory effect until the end of sampling at 8 h. Relative maximum clearance changes were +59.1%, +46.7%, −70.6%, and −61.1% for efavirenz, rifampicin, oral voriconazole, and intravenous voriconazole, respectively.
Conclusions
We could distinguish between different mechanisms of CYP3A modulation by the time of onset. Identification of the time at which clearance significantly changes, per perpetrator drug, can guide the design of an optimal sampling schedule for future drug-drug interaction studies. The impact of a short-term combination of different perpetrator drugs on the paradigm CYP3A substrate midazolam was characterized and can define combination intervals in which no relevant interaction is to be expected.
Clinical Trial Registration
The trial was registered at the European Union Drug Regulating Authorities for Clinical Trials (EudraCT-No. 2013-004869-14).
Supplementary Information
The online version contains supplementary material available at 10.1007/s40262-022-01175-6.
Key Points
| Time and magnitude of the in vivo changes in hepatic cytochrome P450 (CYP) 3A activity, in response to CYP3A inhibitors, inducers, and activators were identified using a nonlinear mixed-effects population pharmacokinetic model of the CYP3A substrate midazolam and its metabolite 1’-hydroxymidazolam based on clinical data. |
| Quantification of the changes in CYP3A activity over time identified different modulatory mechanisms by their time of onset. This can contribute to better planning of drug-drug interaction studies and optimize sampling times by intensifying sampling around the time of onset of the interaction. |
| The identified impact of short-term combinations of perpetrator drugs on CYP3A substrates can define combination intervals in which no relevant interaction is to be expected. |
Introduction
Cytochrome P450 (CYP) 3A isozymes participate in the metabolism of over 50% of all marketed drugs. For many of them, this is a rate-determining step in the elimination from plasma. CYP3A, mainly expressed in the liver and small intestine, exhibits large interindividual variability (IIV) in enzyme activity that can further be modulated by co-morbidities and co-medication [1, 2]. Mechanisms of drug interactions involved in the modulation of CYP3A activity are inhibition, activation, induction, or repression, with the latter two taking the longest time to evolve (days) [3–5]. Therefore, knowledge on the impact of CYP3A activity modulation on drug exposure by perpetrator drugs is essential for the safe and effective use of CYP3A substrates whose clearance (CL) largely relies on these isozymes.
The European Medicines Agency, the US Food and Drug Administration, and the Japanese regulatory authority (PMDA) recommend the detection and quantification of metabolic drug-drug interactions (DDIs) as a significant potential cause of change in drug exposure of victim drugs [6–8].
To detect the overall magnitude of the DDI effect, a common approach is to report the fold-change in maximum plasma concentrations or the area under the concentration–time curve obtained from noncompartmental analyses, or to calculate their geometric mean ratios [7]. Recommendations on the use of population pharmacokinetic (PK) analyses, in particular the nonlinear mixed-effects (NLME) approach, are limited to scenarios when noncompartmental analyses may not prove reliable to derive exposure parameters, for example, for drugs with a long half-life [7] or late-stage clinical studies with sparse blood sampling [6]. Therefore, a population PK (NLME) analysis also focuses on the change in systemic exposure. Currently, the recommended endpoints in DDI studies are exposure based and time independent, and mostly report worst-case scenarios, meaning that the dynamic change of effect is usually not measured or reported, i.e., thus ignored.
Surrogate probe substrates, i.e., drugs exclusively metabolized by a single drug-metabolizing enzyme and sensitive to small changes in the enzyme’s activity, are a commonly used tool in DDI trials [3]. Midazolam (MDZ), a short-acting benzodiazepine derivative, is almost exclusively metabolized by CYP3A to its main metabolite 1’-hydroxymidazolam (1’-OH-MDZ) [9]. Its metabolic CL to 1’-OH-MDZ is considered to reliably reflect in vivo CYP3A activity [10, 11]. As a result, both the European Medicines Agency and the US Food and Drug Administration recommend MDZ as a CYP3A probe substrate [6, 12]. Because MDZ pharmacokinetics is linear within a 30,000-fold dose range [13], the use of microdosed MDZ to determine CYP3A activity is advantageous as these very small doses are well tolerated without experiencing any benzodiazepine-related pharmacological effects [12, 14].
The aim of this clinical trial and subsequent analysis was to identify the in vivo time of onset of perpetrator drugs on MDZ pharmacokinetics, as a probe substrate for hepatic CYP3A activity. Hence, a top-down approach (i.e., model building based on measured data) best suited the availability of densely sampled clinical data. By applying a population NLME modeling approach, we aimed to quantify the time course and extent of metabolic DDIs in healthy individuals using a continuous microdosed intravenous (i.v.) MDZ infusion. During the infusion time, we investigated the time-dependent impact of the CYP3A perpetrator drugs voriconazole (inhibitor, oral and i.v.), efavirenz (activator), and rifampicin (inducer).
Methods
Study Population, Design, and PK Sampling Schedule
The clinical trial protocol (EudraCT-No. 2013-004869-14) was approved by the competent authority (BfArM, Bonn, Germany) and the responsible Ethics Committee of the Medical Faculty of Heidelberg University (Heidelberg, Germany). The trial was conducted at the Early Clinical Trial Unit of the Department of Clinical Pharmacology and Pharmacoepidemiology (KliPS, certified according to DIN EN ISO 9001:2015, Heidelberg University Hospital, Heidelberg, Germany) in accordance with Good Clinical Practice (ICH-GCP) guidelines and the current version of the Declaration of Helsinki. All individuals gave their written informed consent prior to any study procedures.
The study was an open-label, fixed-sequence, randomized, four-arm, two-period clinical phase I trial. The first period investigated the pharmacokinetics of a single 3 µg i.v. bolus of MDZ in 24 healthy individuals (bolus period). Over 6 h after administration, 14 blood samples were taken and MDZ concentrations were quantified to assess individual drug CL (Table S1 of the Electronic Supplementary Material [ESM]) and calculate an individualized bolus/continuous infusion scheme to achieve a MDZ target concentration of approximately 100 pg/mL in the second period. Afterwards, the individuals were randomized (http://www.randomization.com/) into four arms with different CYP3A perpetrator drugs (perpetrator period). Each arm consisted of six individuals (four were administered the perpetrator drug and two served as a placebo control).
In the second (perpetrator) period, an individualized MDZ dosing scheme was started with a MDZ bolus followed by a continuous i.v. administration at a constant individualized infusion rate (Table S2 of the ESM). Two hours after the start of MDZ administration, the individuals received either a single 400 mg dose of voriconazole (CYP3A inhibitor) either orally or intravenosuly as a 2-hour infusion, a single 400 mg oral dose of efavirenz (CYP3A activator) or 600 mg oral rifampicin (CYP3A inducer) every 24 h for 2 days. Blood samples were taken every 15 min after administration of the MDZ bolus for 10 h (end of MDZ infusion) except for the rifampicin arm, with blood samples collected every hour for 36 h (end of MDZ infusion) [Fig. 1]. For data analysis, four study arms were formed based on the perpetrator drug type and a fifth arm of individuals receiving no perpetrator drug (placebo): oral voriconazole, i.v. voriconazole, rifampicin, efavirenz, and placebo. Adverse events were collected and reported for the different study arms during the clinical trial (ESM).
Fig. 1.

Schematic of the design of the perpetrator drug period of the clinical trial (t = 0 h [h] represents time of perpetrator drug administration). Bold arrows represent drug administration (either midazolam [MDZ] or perpetrator drug); small arrows indicate blood sampling times; horizontal purple rectangle represents 2 h of voriconazole intravenous [i.v.] infusion. VCZ voriconazole (blue: oral, purple: i.v), EFV efavirenz (brass), RIF rifampicin (green), placebo (red)
Bioanalytical Assay
Midazolam and 1’-OH-MDZ were quantified in plasma using a validated liquid chromatography-tandem mass spectrometry assay with a lower limit of quantification of 0.093 pg/mL and 0.255 pg/mL for MDZ and 1’-OH-MDZ, respectively [15, 16].
Parent-Metabolite PK Model Development
To develop the joint PK model for MDZ and 1’-OH-MDZ, the data collected in the perpetrator period of the trial were used. Data collected during the times before perpetrator drug administration (i.e., −2 to 0 h) were considered as part of the placebo arm. The dataset was built using R® version 3.5.3 [4] and Microsoft Excel Version 15.14. The PK population model was developed using Nonlinear Mixed-Effects Modelling program (NONMEM) Version 7.4.3 [5], in combination with Perl-speaks-NONMEM Version 4.8.1 [6] using the ADVAN6 subroutine and the first-order conditional estimation method with interaction.
Descriptive statistics were calculated using R® Version 3.5.3, and exploratory and diagnostic graphics were generated with Xpose4 package Version 4.5.3 [17] and ggplot2 package Version 3.3.1 [7]. R Studio® Version 1.3.1056 [8] and Pirana Version 2.9.4 [9] were used as graphical user interfaces.
PK Base Model for Midazolam and 1’-Hydroxymidazolam
The NLME model was sequentially developed. First, a model for MDZ was developed followed by the incorporation of the 1’-OH-MDZ data. A molar mass correction factor (1.049) was applied to the metabolite compartment, and based on previous knowledge, the fraction of MDZ metabolized by CYP3A was fixed to 0.92 [18]. The structural submodel was developed by analyzing different compartment models based on graphical exploration (one, two, and three compartments), assuming linear elimination. To account for variability between individuals, the IIV was implemented on PK parameters by exponential functions, assuming log-normal distribution of the PK parameters. Different residual variability models (additive, proportional, and combined models) were explored for both MDZ and 1’-OH-MDZ.
Covariate Analysis
Individual Demographics: Based on the identified base MDZ and 1’-OH-MDZ model, demographic characteristics (Table S3 of the ESM) were explored as potential covariates impacting the PK parameters. Because of the correlation between the different covariates (e.g., body mass index, weight, and height), they were tested univariately and the most significant covariate was taken forward. For continuous covariates, linear, power, and exponential relations were investigated. For categorical covariates, the fractional change due to the presence of the different states was estimated. When a parameter-covariate relation was significant in a univariate analysis, correlated covariates were excluded and remaining covariates were tested in multiple forward inclusion steps followed by backward elimination with a drop in the objective function value (OFV) ≥3.84 (≡ 0.05,) or increase in OFV ≥10.8 (≡ 0.001,) for the inclusion and exclusion of a parameter-covariate relationship, respectively.
Perpetrator Drug Effects: The perpetrator drug effect was quantified in each trial arm compared to the pooled placebo arm. Moreover, to describe the dynamics of CYP3A modulation, each concentration-time profile (per study arm) was divided into discrete time intervals and the covariate effect was quantified for each time interval. Initially, the profiles were discretized into 1-hour time intervals (2-hour time intervals for the rifampicin arm) after which, merging of the time intervals was judged after each iteration step. An imprecision (relative standard error [RSE]) >50% for the relative change in MDZ CL parameter was set as a criterion to select a time interval for adjustment. Afterwards, the magnitude of CYP3A modulation of the imprecise time interval was compared to that of the previous and the subsequent time interval to identify the time interval with the smallest difference in CYP3A modulation. The selected imprecise time interval was then merged with the neighboring time interval that had the smallest difference in CYP3A modulation. This avoided a potential bias to the intervals quantifying a stronger modulatory effect. In this way, each arm was described with a tailored time interval strategy to precisely characterize the CYP3A modulatory effect of each perpetrator drug. These effects were explored starting from the time of perpetrator drug administration (i.e., 2 h after the start of MDZ administration). Because of the marginal effect of rifampicin in the first hours, its effect was only quantified starting from the administration of the second dose (i.e., after 24 h). The covariate effect was quantified per unique time interval and study arm, according to Eq. 1.
For each time interval ,
| 1 |
where is the individual MDZ CL at time interval , is MDZ population CL of the placebo arm, and is the fractional change in MDZ population CL as a result of perpetrator drug administration per time interval , unique per study arm.
Model Selection and Evaluation
Decisions on model selection during the model development process were based on the change in OFV or Akaike Information Criterion, goodness-of-fit (GOF) plots, and precision and plausibility of parameter estimates. A more complex nested model was accepted if the drop in OFV was ≥3.84 (≡ 0.05, ) or the increase in OFV was ≥10.8 (≡ 0.001, ) for the inclusion and exclusion of a parameter, respectively. Precision was estimated as RSE with a limit of <30% for structural parameters but <50% for covariate effects and variability parameters owing to the low number of individuals per study arm.
Predictive performance of the model was assessed by a visual predictive check stochastically simulating 1000 datasets from the original dataset including IIV and stratifying to either MDZ or 1’-OH-MDZ (eight bins of equal observations in each). The 10th, 50th, and 90th percentiles of the observed data were compared with the 95% confidence interval of the same percentiles of the simulated data [19, 20]. Parameter precision and robustness of the final developed model were evaluated using sampling importance re-sampling including three iterations of 1000, 2000, and 5000 samples and 200, 400, and 1000 resamples [21].
Quantification of the Perpetrator Drug Effect Over Time
For each individual, the magnitude of the estimated CL values as well as the relative change in CL over time following administration of each perpetrator drug were calculated based on the final developed model for each study arm. Moreover, the timepoint was identified, within each study arm, at which CL significantly changed from the estimated population CL of the placebo arm (i.e., deviating beyond the limits of 80–125% as a chosen boundary for clinical relevance).
Results
Data and Study Population Characteristics
A total of 24 healthy individuals (12 were female, 12 were male, demographics in Table S3 of the ESM) were included in the PK model development. Individualized MDZ doses of the 24 individuals in the perpetrator part of the trial are specified in Table S2 of the ESM. The bolus dose ranged from 2.70 to 6.10 µg and the infusion rate ranged from 2.00 to 4.40 µg/h for 36 h (rifampicin arm) or 10 h (all other arms). The dataset included 1870 samples, equally divided between MDZ and 1’-OH-MDZ. Only 12 samples (0.64%) were missing (six in each analyte) summing up to 1858 quantified concentrations with an average of 38.7 concentration/individual/analyte (range 36–40 concentration per individual). No concentrations were below the lower limit of quantification. MDZ concentrations ranged from 28.3 to 187 pg/mL and 1’-OH-MDZ concentrations from 0.550 to 30.7 pg/mL.
PK Model for MDZ 1’-Hydroxymidazolam
The MDZ and 1’-OH-MDZ population PK model was parameterized in terms of CL and volume of distribution (V). The final model structure is depicted in Fig. 2. A one-compartment model with linear elimination described MDZ continuous infusion better than a two-compartment model. A two-compartment model had worse precision of parameter estimates compared to the one-compartment model for both structural parameters (RSE ≤36% vs ≤23% for the base model, rising to ≤41% vs ≤11% in the covariate model) and random effects (RSE ≤466% vs ≤65% in the covariate model) with no improvement in GOF plots. Moreover, MDZ CL estimates, as the parameter of primary interest, were comparable with a relative percent change of −1.02% and −3.19% for the base and covariate model, respectively. The pharmacokinetics of 1’-OH-MDZ was best described with a one-compartment model with linear elimination, assuming non-reversible metabolism.
Fig. 2.
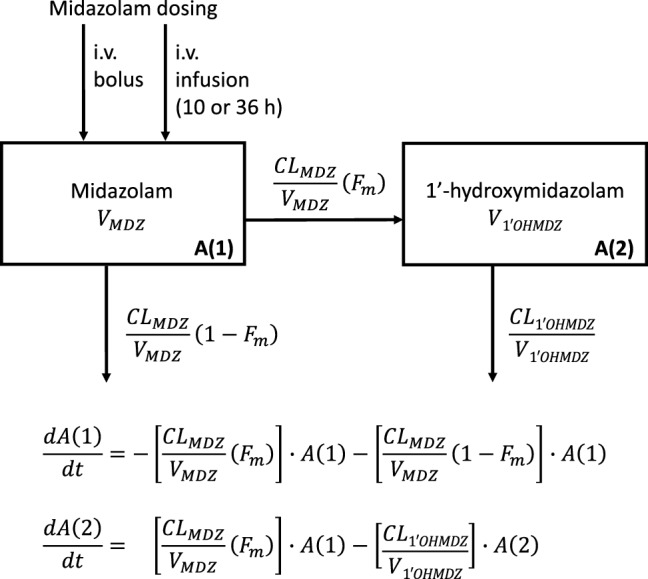
Schematic of joint midazolam (MDZ) and 1’-hydroxymidazolam (1’-OH-MDZ) [parent and metabolite] structural pharmacokinetic model consisting of a one-compartment model for each compound. CLMDZ total MDZ clearance, CL1’OHMDZ 1’-OH-MDZ clearance, Fm fraction of MDZ metabolized, VMDZ MDZ volume of distribution, V1’OHMDZ 1’-OH-MDZ volume of distribution. A(1) and A(2) are MDZ and 1’-OH-MDZ compartments, respectively. h hours, i.v. intravenous
The data supported the inclusion of IIV on all four structural parameter estimates of MDZ and 1’-OH-MDZ compartments (results of IIV ≤93.7 % coefficient of variation). A proportional residual variability model best fitted MDZ and 1’-OH-MDZ data.
As expected, with this homogenous group of individuals, the demographic characteristics of the healthy population were insignificant as covariates on any PK model parameter. The effect of perpetrator drugs was quantified per time interval, starting from the time of their administration until the end of MDZ infusion in the respective study arm. No effect was estimated for the placebo arm and the first 2 h of each perpetrator arm. The final optimal time intervals were the following (note: square brackets include the number; round brackets do not):
For oral voriconazole, [0, 1], (1, 2], (2, 3], (3, 4], (4, 5], (5, 6], (6, 7], and (7, 8] h;
For i.v. voriconazole, [0, 1], (1, 3], (3, 4], (4, 5], (5, 6], (6, 7], and (7, 8] h;
For efavirenz, [0, 2], (2, 3], (3, 4], (4, 5], and (5, 6] h;
For rifampicin, [22, 24], (24, 26], (26, 28], (28, 30], and (30, 34] h.
The last efavirenz time interval (6–8] h and the first rifampicin time interval [0–22) h were considered to not contain enough information to inform the perpetrator drug effect and were thus excluded from the covariate model and considered as part of the placebo arm. Inclusion of the perpetrator drug effects resulted in a reduction of 740 points in the OFV compared with the base model and a relative reduction in the IIV of CLMDZ, VMDZ, and V1’-OH-MDZ of 34.2%, 16.4%, and 57.4%, respectively. Despite the low number of individuals, the final model showed precise estimates. Sampling importance re-sampling was adequately confirmed (Fig. S1 of the ESM) and its reported RSE was between 4.37% and 43.5% (Table 1). Diagnostic GOF plots and plots of residuals for MDZ and 1’-OH-MDZ showed a slight misspecification especially for 1’-OH-MDZ (Fig. S2 of the ESM). The visual predictive check for the final model adequately predicted MDZ and 1’-OH-MDZ, except for the median MDZ concentrations at the later time interval between 8 and 34 h where an underprediction was observed (Fig. S3 of the ESM).
Table 1.
Final parameters of the joint midazolam and 1’-hydroxymidazolam model (n = 24)
| Parameter | Description | Estimate | Uncertaintya | ||
|---|---|---|---|---|---|
| RSE, % | 95% CI [lower, upper] | ||||
| Structural parameters | |||||
| CLMDZ [L/h] | MDZ CL for placebo, efavirenz (6, 8] h and rifampicin [0, 22) h | 43.9 | 4.37 | 40.0, 47.5 | |
| VMDZ [L] | MDZ volume of distribution | 56.7 | 6.84 | 49.9, 65.1 | |
| CL1’-OH-MDZ [L/h] | 1’-OH-MDZ CL | 264 | 7.68 | 224, 305 | |
| V1’-OH-MDZ [L] | 1’-OH-MDZ volume of distribution | 300 | 8.87 | 250, 353 | |
| Fm | Fraction of MDZ metabolized | 0.92b | n.a. | n.a. | |
| Interindividual variability [%CV] | |||||
| ω2 CLMDZ | IIV of MDZ CL | 21.9 | 27.4 | 16.8, 28.5 | |
| ω2 VMDZ | IIV of MDZ volume of distribution | 29.9 | 32.9 | 22.3, 40.7 | |
| ω2 CL 1’-OH-MDZ | IIV of 1’-OH-MDZ CL | 42.0 | 25.7 | 33.1, 53.2 | |
| ω2 V 1’-OH-MDZ | IIV of 1’-OH-MDZ volume of distribution | 39.9 | 37.5 | 28.5, 56.5 | |
| Residual variability [proportional, %CV] | |||||
| MDZ | Proportional residual variability for MDZ | 12.6 | 4.93 | 12.1, 13.3 | |
| 1’-OH-MDZ | Proportional residual variability for 1’-OH-MDZ | 22.6 | 4.84 | 21.6, 23.8 | |
| Perpetrator drug effects [%]c | |||||
| Oral voriconazole | |||||
| [0, 1] | Oral voriconazole effect on MDZ CL during [0, 1] h | −39.9 | 14.8 | −50.6, −27.4 | |
| (1, 2] | Oral voriconazole effect on MDZ CL during (1, 2] h | −29.0 | 20.1 | −39.2, −16.5 | |
| (2, 3] | Oral voriconazole effect on MDZ CL during (2, 3] h | −23.8 | 25.8 | −34.2, −10.2 | |
| (3, 4] | Oral voriconazole effect on MDZ CL during (3, 4] h | −31.9 | 17.5 | −42.2, −20.7 | |
| (4, 5] | Oral voriconazole effect on MDZ CL during (4, 5] h | −63.9 | 10.1 | −76.4, −51.1 | |
| (5, 6] | Oral voriconazole effect on MDZ CL during (5, 6] h | −54.0 | 8.33 | −63.0, −45.1 | |
| (6, 7] | Oral voriconazole effect on MDZ CL during (6, 7] h | −70.6 | 5.57 | −78.5, −62.5 | |
| (7, 8] | Oral voriconazole effect on MDZ CL during (7, 8] h | −-69.4 | 5.66 | −76.5, −61.1 | |
| Intravenous voriconazole | |||||
| [0, 1] | I.v. voriconazole effect on MDZ CL during [0, 1] h | −16.6 | 37.4 | −28.8, −4.10 | |
| (1, 3] | I.v. voriconazole effect on MDZ CL during (1, 3] h | −11.1 | 38.0 | −19.0, -−2.90 | |
| (3, 4] | I.v. voriconazole effect on MDZ CL during (3, 4] h | −15.2 | 38.8 | −27.0, −4.30 | |
| (4, 5] | I.v. voriconazole effect on MDZ CL during (4, 5] h | −36.4 | 18.3 | −49.1, −23.5 | |
| (5, 6] | I.v. voriconazole effect on MDZ CL during (5, 6] h | −39.3 | 14.7 | −49.7, −27.5 | |
| (6, 7] | I.v. voriconazole effect on MDZ CL during (6, 7] h | −61.1 | 7.70 | −70.0, −51.5 | |
| (7, 8] | I.v. voriconazole effect on MDZ CL during (7, 8] h | −58.3 | 8.13 | −67.0, −48.7 | |
| Efavirenz | |||||
| [0, 2] | Efavirenz effect on MDZ CL during [0, 2] h | 15.0 | 31.3 | 6.20, 24.4 | |
| (2, 3] | Efavirenz effect on MDZ CL during (2, 3] h | 59.1 | 14.4 | 44.1, 77.1 | |
| (3, 4] | Efavirenz effect on MDZ CL during (3, 4] h | 56.0 | 14.5 | 40.3, 72.5 | |
| (4, 5] | Efavirenz effect on MDZ CL during (4, 5] h | 28.5 | 25.9 | 13.1, 41.7 | |
| (5, 6] | Efavirenz effect on MDZ CL during (5, 6] h | 33.3 | 26.0 | 18.1, 52.3 | |
| Rifampicin | |||||
| [22, 24] | Rifampicin effect on MDZ CL during [22, 24] h | 27.9 | 26.4 | 13.1, 42.1 | |
| (24, 26] | Rifampicin effect on MDZ CL during (24, 26] h | 17.6 | 43.5 | 4.00, 33.3 | |
| (26, 28] | Rifampicin effect on MDZ CL during (26, 28] h | 23.7 | 33.6 | 8.50, 40.5 | |
| (28, 30] | Rifampicin effect on MDZ CL during (28, 30] h | 46.7 | 15.6 | 32.1, 61.6 | |
| (30, 34] | Rifampicin effect on MDZ CL during (30, 34] h | 10.2 | 38.6 | 3.30, 17.8 | |
CI confidence interval, CL clearance, CV coefficient of variation, Fm fraction metabolized, h h, IIV interindividual variability, i.v. intravenous, MDZ midazolam, n.a. not available, 1’-OH-MDZ 1’-hydroxymidazolam, RSE relative standard error, V volume of distribution
aObtained from sampling importance resampling
bFixed value[18]
cChange in CL per time interval relative to the estimated population midazolam CL in the placebo arm
Quantification of the Perpetrator Drug Effect Over Time
Figure 3 shows the effect of CYP3A modulation on MDZ CL, as a result of perpetrator drug administration, over time. It also reveals the time at which maximum modulatory effects occur—with inhibitors (oral and i.v. voriconazole), inducers (rifampicin), and activators (efavirenz). For oral voriconazole (Fig. 3), an inhibitory effect of approximately −30% was stable for the first 4 h, after which a two-fold increase in inhibition occurred at 4–6 h, relative to the first 4 h, and continued to reach the maximum inhibitory effect until the end of the sampling time at 8 h (relative change in CL [RelCL] −70.6%, Fig. 4). A similar trajectory to oral voriconazole was observed for i.v. voriconazole (Fig. 3, RelCL −61.1%, Fig. 4). For oral efavirenz, CYP3A activation increased rapidly and gradually, reaching a maximum effect at 2–4 h (Fig. 3, RelCL +59.1%, Fig. 4) after its administration and then started to return to baseline. For oral rifampicin, the induction effect increased and peaked between 28 and 30 h (i.e., 2–4 h after administration of the second dose, Fig. 3, RelCL +46.7%, Fig. 4). Detailed results for the estimated CL values per time interval and study arm are presented in Table S4 of the ESM.
Fig. 3.

Individual midazolam clearance-time profiles showing the different time intervals and modulatory effects of the different perpetrator drugs at the different time intervals, per study arm. Time −2 to 0 hours (h) represents baseline individual (colored) or population (black) clearance before perturbation. The dashed vertical line indicates the time of perpetrator drug administration, dotted vertical lines indicate the time intervals, and the solid black line indicates midazolam population clearance per study arm. The shaded gray area indicates a significant change in clearance beyond 80–125% (i.e., <35.1 L/h or >54.9 L/h). The following time intervals were estimated as the placebo value: 6–8 h (efavirenz arm), 0–22 h (rifampicin arm). i.v. intravenous
Fig. 4.

Relative change in midazolam clearance over time relative to the placebo arm, as a result of perpetrator drug administration stratified per perpetrator arm. The dashed vertical line indicates the time of perpetrator drug administration. In the plot of the rifampicin arm, the x-axis is truncated for better illustration and only day 2 is shown. Insert: full-time course of the rifampicin arm. h hours, i.v. intravenous
For all perpetrator drugs, a significant change in CL relative to the placebo population CL (43.9 L/h) was defined as below 35.1 L/h or above 54.9 L/h (i.e., beyond the boundary of 80–125%). These significant changes in CL occurred as follows in each arm: for oral voriconazole, immediately after administration; for i.v. voriconazole, 4 h after administration; for efavirenz, 2 h after administration; and for rifampicin, 28 h after administration of the first dose (4 h after the second dose).
Discussion
The time-dependent modulatory effect of CYP3A inhibition (oral and i.v. voriconazole), induction (rifampicin), and activation (efavirenz) on hepatic CYP3A activity was quantified using a joint MDZ and 1’-OH-MDZ-perpetrator population PK model leading to the identification of timepoints at which CL is expected to significantly change. The clinical trial involved four different perpetrator drug study arms and a fifth pooled placebo arm in which all individuals were administered a long-term i.v. infusion of microdosed MDZ to monitor CYP3A activity. Population NLME PK modeling was selected as the approach of choice to best quantify modulatory effects and account for IIV. Moreover, we wanted to expand further the use of population NLME modeling in detecting DDIs and particularly characterizing the dynamic profile rather than an overall exposure.
A one-compartment model for MDZ and 1’-OH-MDZ with compound-specific proportional residual variability best described the data. In contrast to the established two-compartment models for MDZ generally developed for i.v. bolus and oral dosing [14, 22, 23] rather than continuous i.v. infusions [24], the one-compartment model was sufficient to characterize the pharmacokinetics of MDZ and 1’-OH-MDZ in the current trial. This can be explained by the presence of a continuous long-term infusion [25] over the entire observation time, which likely masked the distinct differentiation of distribution, metabolism, and excretion processes. The slight misspecifications observed in the GOF plots were due to the fact that we aimed to precisely estimate the parameter MDZ CL avoiding overparameterization with respect to the low number of individuals. Nevertheless, these slight misspecifications did not impact the model performance, and the very dense sampling and clinical trial design allowed the precise estimation of MDZ CL. Thus, because the scope of this work was assessing the impact of perpetrator drugs on MDZ CL, as a surrogate for CYP3A activity, a one-compartment model was sufficient.
So far, no PK parameters have been reported for a continuous i.v. infusion of microdosed MDZ. In comparison to values derived from oral and i.v. bolus administration, parameter estimates of MDZ were in line with literature values of MDZ CL (estimated: 43.9 L/h, reported: 15.2–45.0 L/h) [22, 23, 26, 27] and V (estimated: 56.7 L, reported: 33.3–180 L/h) [22, 23, 26, 27]. 1’-OH-MDZ CL and V were close to estimates by Wiebe et al. [23] and Tomalik-Scharte et al. [25], respectively (CL estimated: 264 L/h, reported: 186–208 L/h; V estimated: 300 L, reported: 256–876 L) but rather higher than values reported by Hotz et al. for the metabolite in the absence of a parent compound (reported CL: 34.4–56.7 L/h, reported V: 49.7–79.8 L) [27]. This was due to the fact that the rate of 1’-OH-MDZ formation dominates its terminal concentration-time profile (“flip-flop kinetics”) and is the rate-limiting step (our calculated formation rate constant was 0.712 h-1 vs an elimination rate constant of 0.880 h-1) and our PK parameters were calculated from the metabolite concentration after metabolic conversion of MDZ rather than from an independent administration of 1’-OH-MDZ, which does not take the metabolite formation rate into consideration [27].
Although age and sex were previously reported as potential covariates modulating MDZ V [22], it was in line with our expectation that none of the demographic characteristics of the healthy and rather smaller number of individuals in this PK study significantly explained the observed variability. This was because the study population was homogenous and shared similar characteristics. This was also an advantage because it did not significantly influence the model parameters and thus allowed an estimation of the perpetrator drug effect with negligible confounding variables. Perpetrator drug effects were tested only for MDZ CL because the V is not expected to change in the short course of a metabolic DDI trial [28]. We only focused on the CL of the parent drug MDZ because the impact observed on 1’-OH-MDZ is mainly a result of the modulation of MDZ CL. Additionally, as no 1’-OH-MDZ was administered separately, its above-mentioned flip-flop kinetics make the precise analysis on 1’OH-MDZ challenging [27]. Yet, inclusion of the metabolite in the population PK model allowed a more precise estimation of MDZ CL. In developing the model, 1-hour time intervals were initially chosen over the 15-minute time intervals (the planned sampling schedule) to accurately capture effects of smaller magnitude when estimated over a wider time interval and to avoid overparameterizing the model.
An earlier reported semi-physiological model of voriconazole and MDZ suggested a decrease of 75% in hepatic CYP3A activity within 12 h after administration of oral voriconazole [29]. Furthermore, a reduction of 72–89.4% in MDZ CL was observed after 48 h of voriconazole administration [11, 30]. In line with these observations, an ~70% and ~60% decrease in MDZ CL occurred 6–8 h following administration of oral and i.v. voriconazole, respectively. Comparable inhibition results were observed between the two routes of voriconazole administration as intravenously administered MDZ assessed only hepatic CYP3A activity. Thus, the additional impact of oral voriconazole on intestinal CYP3A [31] could not be addressed with this study design. Nevertheless, in the initial time interval, a stronger inhibitory effect of oral voriconazole was observed, compared with i.v. voriconazole. The stronger inhibitory effect observed with oral voriconazole could be attributed to the fact that the whole amount of absorbed voriconazole (oral bioavailability >90% [31]) went directly to the liver in contrast to the same amount that was intravenously infused and distributed over 2 h into the systemic circulation. Moreover, oral voriconazole has initially faster kinetics (first-order kinetics) than the zero-order kinetics of the 2-hour infusion. Thus, voriconazole initially appears at a higher concentration in the liver compared with i.v. voriconazole. The observation most probably would have been different if a bolus i.v. dose of voriconazole was administered or if oral MDZ was used to measure modulation in the CYP3A enzymes of the gut wall. Our estimated efavirenz CYP3A activation was in line with what Mikus et al. reported [32]; where MDZ CL was increased by 27%, 12 h after efavirenz administration. In vitro studies of MDZ and efavirenz also reported an ~1.5-fold increase in metabolite formation rates in pooled human liver microsomes [33]. Rifampicin induction of CYP3A has been reported to gradually increase with repeated dosing [34]. Therefore, within the rifampicin arm, the longer observation time (36 h) was necessary to quantify a strong induction effect. Quantification of the modulatory impact in the first doing interval was negligible and could not be reliably estimated: a 10- to 22-hour time interval or the complete first dosing interval (i.e., 0–22 h) had an estimated relative change in MDZ CL compared to the placebo arm of −1.02% and −1.77% with an RSE of 175% and 203%, respectively. Indeed, this minimal modulation in the first few hours (Fig. S4 of the ESM) affected the overall precise quantification of its effect. This was also supported by Yoshikado et al. [35] who showed an insignificant effect on MDZ area under the concentration-time curve and maximum concentration for the first 12 h after administration of rifampicin. Furthermore, inclusion of the first dosing intervals for rifampicin had a minor impact on the estimates of the subsequent time intervals, i.e., inclusion of a 10- to 22-hour time interval resulted in a percent relative change of −1.43% to −5.20% in the relative change of MDZ CL compared with placebo while inclusion of a 0- to 22-hour time interval resulted in a percent relative change of −5.57% to −17.1%. Therefore, only the second dose effect was retained in the final model. Although our estimated impact of rifampicin on MDZ CL was smaller than what has been simulated before by Yamashita et al. [34], a gradual increase in effect reaching a maximum of 1.5-fold induction with a 2-day dosing was shown. It is expected that with longer sampling times and repeated dosing up to 5 days, an even stronger effect could be observed [36].
With this study, which was designed to capture the time of DDI onset rather than identify its maximum effect, it was possible to distinguish different mechanisms of action of perpetrator drugs (e.g., activator vs inducer), exemplified by efavirenz and rifampicin. Both drugs exhibited different modulatory patterns and times of onset, with a maximum DDI interaction after 2–4 h and 28–30 h, respectively. This was in contrast to voriconazole whose inhibitory effect gradually increased from the time of administration until the end of the sampling time with a maximum effect at 6–8 h.
A limitation of this study was the small number of individuals per arm, also a reason for the misspecification observed in the visual predictive check. Nevertheless, despite the small sample size within each study arm, our approach still proved successful. The covariate model adequately captured the change in MDZ CL, and hence CYP3A activity over time, estimating the magnitude of effect with high precision (sampling importance re-sampling-reported RSE ≤43.5%, Table 1). It is also worth noting that these results are only relevant to the healthy population. It is to be expected that these results can be different for a more heterogeneous group of patients, for example, with comorbidities.
Modeling a continuous perpetrator drug effect was also investigated, for example, using perpetrator concentration or cumulative perpetrator drug area under the concentration-time curve. However, the relationship between the continuous perpetrator drug effect and modulation was implausible because of the dependence of the CYP modulation on the perpetrator drug concentration at the site of action rather than the perpetrator drug plasma concentration alone. Hence, discretization provided the most plausible approach, given the purpose of the clinical study, with best characterization of the dynamics of modulation.
Nevertheless, this modeling approach, especially the discretization of time also presents some limitations. First, weak modulation of CYP3A by a perpetrator drug in a specific time interval may be difficult to precisely quantify; however, this was overcome by merging few time intervals to have a larger magnitude of modulation/more data for this new time interval and consequently a more precise estimation of modulation. Second, the choice of the time intervals is crucial to capture the dynamics of the modulation. The chosen time interval should be as small as possible; the larger it is, the more dynamics is “hidden” and the higher the risk of missing on capturing the maximum modulation. However, the choice of the time intervals is dependent on the informativeness of the data (e.g., number of individuals) for a precise quantification. With larger sample sizes and narrower time intervals, weak modulatory effects can also be precisely captured. On the other hand, this modeling approach has the advantage of accounting for a more detailed dissection of the dynamics of CYP modulation compared to the quantification of a categorical effect over the whole observation period as a dichotomous effect. Thus, measurements of the perpetrator drug concentration are not needed, i.e., a substrate (i.e., MDZ) concentration was sufficient. Microdosed MDZ could successfully assess CYP3A activity and its use as a component of a microdosed cocktail can identify the presence of DDIs. Our results can indeed further inform and validate physiologically based PK models as a complementary approach, irrespective of the availability of clinical data, to detect time-dependent changes in CYP3A activity or in the case of different dosing regimens [6, 37] to provide insights into the time-dependent patterns of CYP3A modulation and prediction of the time of onset. It would be useful to distinguish between different mechanisms of CYP3A modulation by the time of onset and explore whether the modulatory profiles are similar for perpetrator drugs with the same mechanism of CYP3A modulation.
Moreover, identification of the times at which CL significantly changes can serve as suggested monitoring times for a significant modulatory effect and help design an optimal sampling schedule for future DDI studies. It is suggested that a larger sample size along with intensified sampling around the times of expected maximum effects can provide a more informative screening strategy for perpetrator drugs with a pre-hypothesized modulatory impact. Finally, these results could also inform about the impact of a short-term combination of perpetrator drugs on CYP3A substrates and help to define combination intervals in which no relevant interaction is to be expected in clinical practice.
Conclusions
The modulatory effect of three different CYP3A perpetrator drugs over time was successfully quantified by capturing the changes in the CL of the CYP3A substrate MDZ. The developed joint population PK model of MDZ and 1’-OH-MDZ utilized the quantitative change in CL relative to the placebo arm, across the time intervals, to develop a dynamic profile that was specific to each perpetrator drug. This methodology can form a framework for other DDI studies and inform physiologically based PK modeling frameworks.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgments
The authors thank Marlies Stützle‐Schnetz for her excellent assistance during the study performance and Magdalena Longo and Andrea Deschlmayr for their support in the analyses of the samples. Parts of the results were presented as a poster at the 2021 Population Approach Group Europe (PAGE) meeting (www.page-meeting.org/?abstract=9619), 2016 Annual Meeting of the American Society for Clinical Pharmacology and Therapeutics (Hohmann et al. Clin Pharmacol Ther. 2016;99:S40) and 81st Annual Meeting of the Deutsche Gesellschaft für Experimentelle und Klinische Pharmakologie und Toxikologie (Gottwalt et al. Naunyn-Schmiedebergs Arch Pharmacol. 2015;388:S51).
Declarations
Funding
Open Access funding enabled and organized by Projekt DEAL. This trial and data analysis were funded by internal resources of the Department of Clinical Pharmacology and Pharmacoepidemiology at Heidelberg University Hospital and the Department of Clinical Pharmacy and Biochemistry at the Freie Universitaet Berlin. No medical writing support was used.
Conflicts of Interest/Competing Interests
Yomna M. Nassar, Nicolas Hohmann, Katharina Gottwald, Robin Michelet, Andreas D. Meid, Jürgen Burhenne, Walter E. Haefeli, and Gerd Mikus declare that they have no conflicts of interest that might be relevant to the content of this article. Charlotte Kloft and Wilhelm Huisinga report grants from an industry consortium (AbbVie Deutschland GmbH & Co. KG, AstraZeneca, Boehringer Ingelheim Pharma GmbH & Co.KG, Grünenthal GmbH, F. Hoffmann-La Roche Ltd., Merck KGaA, and Sanofi) for the PharMetrX PhD program. Charlotte Kloft reports a grant from the European Commission for Innovative Medicines Initiative-Joint Undertaking (‘DDMoRe’).
Ethics Approval
All procedures performed in studies involving human participants were in accordance with Good Clinical Practice (ICH-GCP) guidelines and the ethical standards of the current Declaration of Helsinki. The trial protocol (EudraCT 2013-004869-14) was approved by the responsible Ethics Committee of the Medical Faculty of Heidelberg University and the competent authority (BfArM, Bonn, Germany).
Consent to Participate
All included volunteers gave their written informed consent prior to any study procedures.
Consent for Publication
Not applicable.
Availability of Data and Material
The datasets generated and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Code Availability
The NONMEM model code is available in the ESM.
Authors’ Contributions
NH, KG, WEH, and GM designed the clinical study conception and conducted the study; JB performed the midazolam bioanalytical analysis; YMN and ADM conducted the data analyses; YMN, RM, GM, and CK thoroughly discussed the data analysis and were involved in the interpretation of results; RM, WH, and CK provided methodological support; WH, GM, and CK provided supervision; the first version of the manuscript was drafted by YMN; all authors critically reviewed, edited, and commented on previous versions of the manuscript, and approved the final version of the manuscript.
Footnotes
Yomna M. Nassar and Nicolas Hohmann shared first authorship.
Gerd Mikus and Charlotte Kloft shared last authorship.
References
- 1.Dresser GK, Spence JD, Bailey DG. Pharmacokinetic-pharmacodynamic consequences and clinical relevance of cytochrome P450 3A4 inhibition. Clin Pharmacokinet. 2000;38:41–57. doi: 10.2165/00003088-200038010-00003. [DOI] [PubMed] [Google Scholar]
- 2.Paine MF, Shen DD, Kunze KL, Perkins JD, Marsh CL, McVicar JP, et al. First-pass metabolism of midazolam by the human intestine. Clin Pharmacol Ther. 1996;60:14–24. doi: 10.1016/S0009-9236(96)90162-9. [DOI] [PubMed] [Google Scholar]
- 3.Thummel KE, Wilkinson GR. In vitro and in vivo drug interactions involving human CYP3A. Annu Rev Pharmacol Toxicol. 1998;38:389–430. doi: 10.1146/annurev.pharmtox.38.1.389. [DOI] [PubMed] [Google Scholar]
- 4.Nagar S, Argikar UA, Tweedie DJ. Reversible mechanisms of enzyme inhibition and resulting clinical significance. In: Enzyme kinetics in drug metabolism: fundamentals and applications, Vol. 1113. Totowa: Humana Press; 2014; pp 37–56. doi: 10.1007/978-1-62703-758-7_1.
- 5.Eichbaum C, Cortese M, Blank A, Burhenne J, Mikus G. Concentration effect relationship of CYP3A inhibition by ritonavir in humans. Eur J Clin Pharmacol. 2013;69:1795–1800. doi: 10.1007/s00228-013-1530-8. [DOI] [PubMed] [Google Scholar]
- 6.European Medicines Agency. Guideline on the investigation of drug interactions 2010. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-drug-interactions-revision-1_en.pdf. [Accessed 25 May 2021].
- 7.US Food and Drug Administration. Guidance for industry: clinical drug interaction studies—cytochrome P450 enzyme- and transporter-mediated drug interactions. 2020:1–27. Available from: https://www.fda.gov/media/134581/download/. [Accessed 25 May 2021].
- 8.Pharmaceuticals and Medical Devices Agency. Guideline on drug interaction for drug development and appropriate provision of information Title 2019. Available from: https://www.pmda.go.jp/files/000228122.pdf. [Accessed 19 Jul 2022].
- 9.Kronbach T, Mathys D, Umeno M, Gonzalez FJ, Meyer UA. Oxidation of midazolam and triazolam by human liver cytochrome P450IIIA4. Mol Pharmacol. 1989;36:89–96. [PubMed] [Google Scholar]
- 10.Thummel KE, Shen DD, Podoll TD, Kunze KL, Trager WF, Hartwell PS, et al. Use of midazolam as a human cytochrome P450 3A probe: I. In vitro-in vivo correlations in liver transplant patients. J Pharmacol Exp Ther. 1994;271:549–556. [PubMed] [Google Scholar]
- 11.Katzenmaier S, Markert C, Riedel KD, Burhenne J, Haefeli WE, Mikus G. Determining the time course of CYP3A inhibition by potent reversible and irreversible CYP3A inhibitors using a limited sampling strategy. Clin Pharmacol Ther. 2011;90:666–673. doi: 10.1038/clpt.2011.164. [DOI] [PubMed] [Google Scholar]
- 12.Roche. Dormicum Injektionslösung 2019. Available from: https://www.roche.com.sg/content/dam/rochexx/roche-com-sg/documents/product-page/DormicumInjPIApr 2019.pdf. [Accessed 25 May 2021].
- 13.Halama B, Hohmann N, Burhenne J, Weiss J, Mikus G, Haefeli WE. A nanogram dose of the CYP3A probe substrate midazolam to evaluate drug interactions. Clin Pharmacol Ther. 2013;93:564–571. doi: 10.1038/clpt.2013.27. [DOI] [PubMed] [Google Scholar]
- 14.Heizmann P, Eckert M, Ziegler W. Pharmacokinetics and bioavailability of midazolam in man. Br J Clin Pharmacol. 1983;16:43S–S49. doi: 10.1111/j.1365-2125.1983.tb02270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burhenne J, Halama B, Maurer M, Riedel KD, Hohmann N, Mikus G, et al. Quantification of femtomolar concentrations of the CYP3A substrate midazolam and its main metabolite 1′-hydroxymidazolam in human plasma using ultra performance liquid chromatography coupled to tandem mass spectrometry. Anal Bioanal Chem. 2012;402:2439–2450. doi: 10.1007/s00216-011-5675-y. [DOI] [PubMed] [Google Scholar]
- 16.Hohmann N, Kocheise F, Carls A, Burhenne J, Weiss J, Haefeli WE, et al. Dose-dependent bioavailability and CYP3A inhibition contribute to non-linear pharmacokinetics of voriconazole. Clin Pharmacokinet. 2016;55:1535–1545. doi: 10.1007/s40262-016-0416-1. [DOI] [PubMed] [Google Scholar]
- 17.Jonsson EN, Karlsson MO. Xpose - An S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Methods Programs Biomed. 1998;58:51–64. doi: 10.1016/S0169-2607(98)00067-4. [DOI] [PubMed] [Google Scholar]
- 18.Ohno Y, Hisaka A, Ueno M, Suzuki H. General framework for the prediction of oral drug interactions caused by CYP3A4 induction from in vivo information. Clin Pharmacokinet. 2008;47:669–680. doi: 10.2165/00003088-200847100-00004. [DOI] [PubMed] [Google Scholar]
- 19.Karlsson MO, Holford N. A tutorial on visual predictive checks [abstract 1434]. Abstract. Annual Meeting Population Approach Group 17, Marseille, France. 2008: p.17. Available from: www.page-meeting.org/?abstract=1434. [Accessed 31 Aug 2022].
- 20.Owen JS, Fiedler-Kelly J. Introduction to model evaluation. Introduction to population Pharmacokinetics/pharmacodynamics. Anal Nonlinear Mix Eff Model. 2014;2:212–31. doi: 10.1002/9781118784860.ch8. [DOI] [Google Scholar]
- 21.Dosne AG, Bergstrand M, Harling K, Karlsson MO. Improving the estimation of parameter uncertainty distributions in nonlinear mixed effects models using sampling importance resampling. J Pharmacokinet Pharmacodyn. 2016;43:583–596. doi: 10.1007/s10928-016-9487-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang J, Patel M, Nikanjam M, Capparelli EV, Tsunoda SM, Greenberg HE, et al. Midazolam single time point concentrations to estimate exposure and cytochrome P450 (CYP) 3A constitutive activity utilizing limited sampling strategy with a population pharmacokinetic approach. J Clin Pharmacol. 2018;58:1205–1213. doi: 10.1002/jcph.1125. [DOI] [PubMed] [Google Scholar]
- 23.Wiebe ST, Meid AD, Mikus G. Composite midazolam and 1′-OH midazolam population pharmacokinetic model for constitutive, inhibited and induced CYP3A activity. J Pharmacokinet Pharmacodyn. 2020;47:527–542. doi: 10.1007/s10928-020-09704-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maitre PO, Funk B, Crevoisier C, Ha HR. Pharmacokinetics of midazolam in patients recovering from cardiac surgery. Eur J Clin Pharmacol. 1989;37:161–166. doi: 10.1007/BF00558225. [DOI] [PubMed] [Google Scholar]
- 25.Tomalik-Scharte D, Suleiman AA, Frechen S, Kraus D, Kerkweg U, Rokitta D, et al. Population pharmacokinetic analysis of circadian rhythms in hepatic CYP3A activity using midazolam. J Clin Pharmacol. 2014;54:1162–1169. doi: 10.1002/jcph.318. [DOI] [PubMed] [Google Scholar]
- 26.Van Rongen A, Vaughns JD, Moorthy GS, Barrett JS, Knibbe CAJ, Van Den Anker JN. Population pharmacokinetics of midazolam and its metabolites in overweight and obese adolescents. Br J Clin Pharmacol. 2015;80:1185–1196. doi: 10.1111/bcp.12693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hotz MA, Ritz R, Linder L, Scollo-Lavizzari G, Haefeli WE. Auditory and electroencephalographic effects of midazolam and α-hydroxy-midazolam in healthy subjects. Br J Clin Pharmacol. 2000;49:72–79. doi: 10.1046/j.1365-2125.2000.00104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sodhi JK, Benet LZ. A simple methodology to differentiate changes in bioavailability from changes in clearance following oral dosing of metabolized drugs. Clin Pharmacol Ther. 2020;108:306–315. doi: 10.1002/cpt.1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frechen S, Junge L, Saari TI, Suleiman AA, Rokitta D, Neuvonen PJ, et al. A semiphysiological population pharmacokinetic model for dynamic inhibition of liver and gut wall cytochrome P450 3A by voriconazole. Clin Pharmacokinet. 2013;52:763–781. doi: 10.1007/s40262-013-0070-9. [DOI] [PubMed] [Google Scholar]
- 30.Saari TI, Laine K, Leino K, Valtonen M, Neuvonen PJ, Olkkola KT. Effect of voriconazole on the pharmacokinetics and pharmacodynamics of intravenous and oral midazolam. Clin Pharmacol Ther. 2006;79:362–370. doi: 10.1016/j.clpt.2005.12.305. [DOI] [PubMed] [Google Scholar]
- 31.Schulz J, Kluwe F, Mikus G, Michelet R, Kloft C. Novel insights into the complex pharmacokinetics of voriconazole: a review of its metabolism. Drug Metab Rev. 2019;51:247–265. doi: 10.1080/03602532.2019.1632888. [DOI] [PubMed] [Google Scholar]
- 32.Mikus G, Heinrich T, Bödigheimer J, Röder C, Matthee AK, Weiss J, et al. Semisimultaneous midazolam administration to evaluate the time course of CYP3A activation by a single oral dose of efavirenz. J Clin Pharmacol. 2017;57:899–905. doi: 10.1002/jcph.879. [DOI] [PubMed] [Google Scholar]
- 33.Keubler A, Weiss J, Haefeli WE, Mikus G, Burhenne J. Drug interaction of efavirenz and midazolam: efavirenz activates the CYP3A-mediated midazolam 1′-hydroxylation in vitro. Drug Metab Dispos. 2012;40:1178–1182. doi: 10.1124/dmd.111.043844. [DOI] [PubMed] [Google Scholar]
- 34.Yamashita F, Sasa Y, Yoshida S, Hisaka A, Asai Y, Kitano H, et al. Modeling of rifampicin-induced CYP3A4 activation dynamics for the prediction of clinical drug-drug interactions from in vitro data. PLoS ONE. 2013;8:e70330. doi: 10.1371/journal.pone.0070330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoshikado T, Maeda K, Furihata S, Terashima H, Nakayama T, Ishigame K, et al. A clinical cassette dosing study for evaluating the contribution of hepatic OATPs and CYP3A to drug-drug interactions. Pharm Res. 2017;34:1570–1583. doi: 10.1007/s11095-017-2168-5. [DOI] [PubMed] [Google Scholar]
- 36.Backman JT, Olkkola KT, Neuvonen PJ. Rifampin drastically reduces plasma concentrations and effects of oral midazolam. Clin Pharmacol Ther. 1996;59:7–13. doi: 10.1016/S0009-9236(96)90018-1. [DOI] [PubMed] [Google Scholar]
- 37.Einolf HJ. Comparison of different approaches to predict metabolic drug–drug interactions. Xenobiotica. 2007;37:1257–1294. doi: 10.3109/00498250701620700. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.