

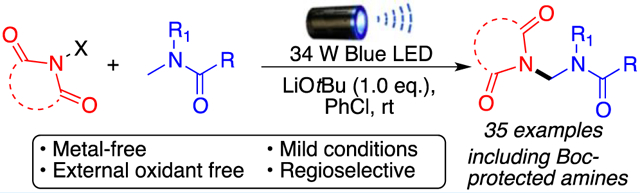
Abstract
A metal-free regioselective C(sp3)–H amination of amides using N-haloimides in the presence of lithium tert-butoxide and visible light is presented herein. This photoexcited approach is straightforward, and it aminates a wide variety of amides under mild conditions without the use of photocatalysts, external radical initiators, or oxidants. A halogen-bonded intermediate between the tert-butoxide base and the N-haloimide is proposed to be responsible for the increased photoreactivity. Calculations show that the formation of this electron donor–acceptor complex presents an exergonic energy profile.
Graphical Abstract
Amines and amides are key intermediates in many chemical syntheses, as both groups are present in biologically active natural products, synthetic intermediates, and pharmaceutical agents.1 In particular, the amido aminal functional group is found in various bioactive compounds such as metolazone, fluspirilene, lymecycline, and primidone (Scheme 1A).
Scheme 1.
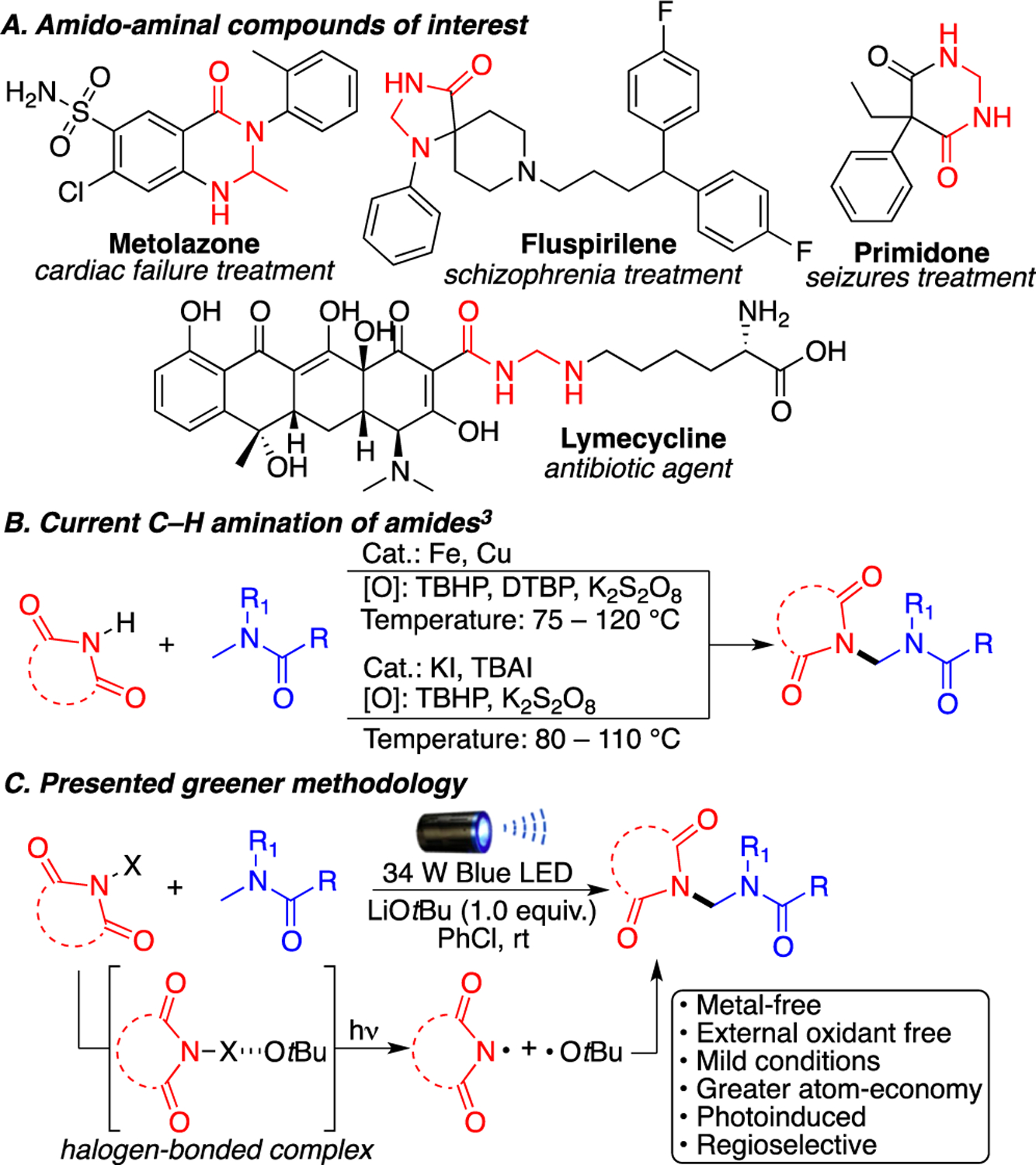
(A) Examples of FDA-Approved Drugs Containing the Amido Aminal Group, (B) Current Methods for Accessing the Amido Aminal Functional Group That Require the Use of Catalysts and Strong Oxidants at High Temperatures, and (C) Our Photoexcited C–H Activation under Mild Reaction Conditions
Traditionally, these difficult to access compounds have been generated through condensation reactions with formaldehyde.2 Due to the importance of this amido aminal framework, the development of C–H amination methodologies that selectively target the amino carbon of amides has attracted some attention in the organic community.3 Currently, two general approaches have been employed to achieve this (Scheme 1B): (i) transition metal-catalyzed C–H aminations3a–d and (ii) metal-free C–H aminations.3e–h Unfortunately, both of these approaches require the use of stoichiometric strong oxidants or peroxides at high temperatures, increasing the risk of an accident and the waste footprint. Consequently, the development of metal-free and atom-economical strategies is particularly important.
Visible light photoredox processes have recently found many applications in organic synthesis due to the advantages of being readily availability, sustainability, mild reaction conditions, etc.4 Inspired by our previous C–H amination strategies using N-haloimides,5 herein we report a metal-free C(sp3)–H amination of amides in the presence of lithium tert-butoxide using visible light photoexcitation. Our approach does not require a photocatalyst or a peroxide initiator and proceeds under mild reaction conditions, making this methodology among the greenest approaches for selectively generating the amido aminal functional group (Scheme 1C). Mechanistic work proposes that a halogen-bonded electron donor–acceptor complex between tert-butoxide and the N-haloimide is responsible for the observed reactivity.
We started our investigation using N-bromophthalimide (1a) and dimethylacetamide (2a) in the presence of stoichiometric amounts of LiOtBu under blue light (Table 1). Initial solvent screening showed that the desired amination product 3aa could be obtained using PhCF3, PhCl, CH3CN, or benzene (entries 1–4), but chlorobenzene provided the best yields (entry 2). Screening of bases showed that the nature of the counterion seems to play a major role in this chemistry. Indeed, while lithium tert-butoxide generated the product in 67% yield, switching to sodium or potassium tert-butoxide has a major deleterious effect, giving the desired product in only 24% or 2% yield, respectively (entries 5 and 6). The use of bases other than tert-butoxides did not generate the product in synthetically useful yields (see the Supporting Information for a full table of optimization, S4). The stoichiometry of the base was also evaluated (entries 7–10). The absence of LiOtBu in a control reaction did not generate the desired product, and a deviation in the number of equivalents of base from the optimal value of 1 equiv had a deleterious effect on yield. Thermal activation of the reaction mixture in the absence of light showed a sharp decline in the yield (entry 11), further demonstrating that it is light, and not the heat generated by the lamps, that activates the reaction. Also, reducing the number of equivalents of 2a (entries 12 and 13) leads to a reduction in yields of the desired product. Finally, performing the reaction open to the atmosphere leads to a decline in yield (entry 14).
Table 1.
Optimization of the Reaction and Its Conditionsa

| |||
|---|---|---|---|
| entry | base (equiv) | solvent (mL) | yield (%)b |
| 1 | LiOtBu (1.0) | PhCF3 (1.0) | 42 |
| 2 | LiOtBu (1.0) | PhCl (1.0) | 67 (65)c |
| 3 | LiOtBu (1.0) | CH3CN (1.0) | 10 |
| 4 | LiOtBu (1.0) | PhH (1.0) | 49 |
| 5 | NaOtBu (1.0) | PhCl (1.0) | 24 |
| 6 | KOtBu (1.0) | PhCl (1.0) | 2 |
| 7 | – | PhCl (1.0) | – |
| 8 | LiOtBu (0.75) | PhCl (1.0) | 45 |
| 9 | LiOtBu (1.25) | PhCl (1.0) | 63 |
| 10 | LiOtBu (1.5) | PhCl (1.0) | 35 |
| 11d | LiOtBu (1.0) | PhCl (1.0) | 7 |
| 12e | LiOtBu (1.0) | PhCl (1.0) | 44 |
| 13f | LiOtBu (1.0) | PhCl (1.0) | 60 |
| 14g | LiOtBu (1.0) | PhCl (1.0) | 16 |
Reaction conditions: 1a (0.2 mmol, 1 equiv), 2a (1.0 mmol, 5 equiv), base (1 equiv), solvent (1 mL), room temperature around the reaction flask of 35 °C (heating caused by the LED lamp), reaction flask capped, overnight.
1H NMR yields using dibromomethane as the internal standard.
Isolated yield.
Reaction performed at 60 °C without light.
With 2.5 equiv of 2a instead of 5 equiv.
With 4 equiv of 2a intead of 5 equiv.
Reaction performed by adding 20 μL of H2O.
With the optimized conditions in hand, we initiated an amide substrate scope study using 1a as our amine source (Figure 1). As expected, moderate to good yields were obtained. Both secondary and tertiary aliphatic amides generated the desired products 3aa, 3ab, 3ac, and 3af. Interestingly, the use of N-chlorophthalimide leads to an almost complete shutdown of the reaction, providing product 3aa in only 4% yield as opposed to 65% yield when 1a is used. N-Methyl-2-pyrrolidone also generated desired product 3ad in 53% yield with a selective amination of the methylene position over the methyl group. Finally, the presence of an aromatic moiety is well tolerated, and amination of the methyl group is observed predominantly (3ae). Throughout our screens, we did not observe reaction at the α-position of the carboxyl group; polarity-matched hydrogen atom transfer (HAT) processes are known to favor reactions at strong hydridic C–H bonds over weak acidic C–H bonds.4e
Figure 1.
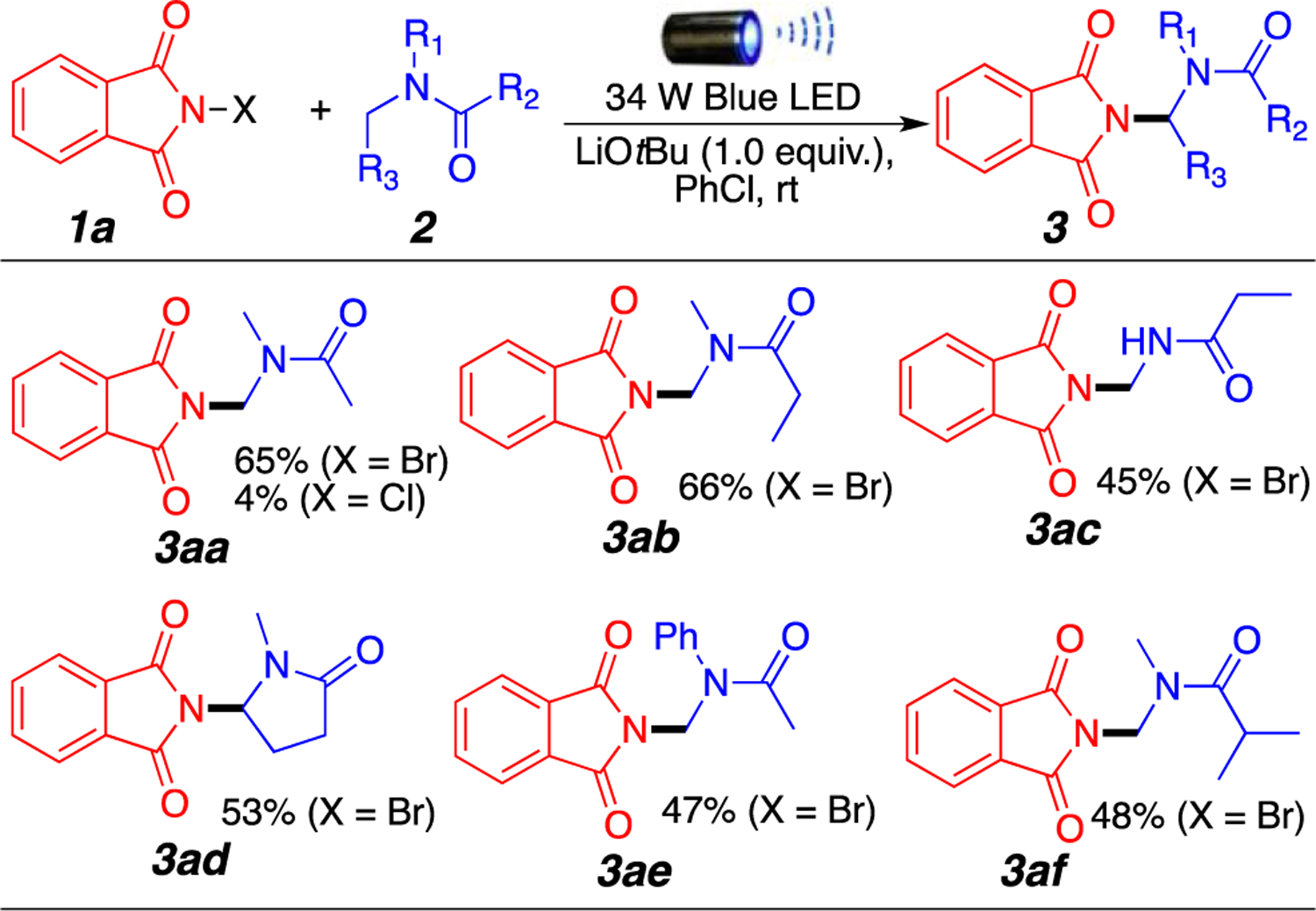
Amination of amides with N-bromophthalimide. Reaction conditions: 1a (0.2 mmol), 2 (1.0 mmol), LiOtBu (0.2 mmol), 1.0 mL of PhCl, room temperature around the reaction flask of 35 °C (heating caused by the LED lamp), overnight. Isolated yields.
We then investigated other possible N-haloimide sources and found that N-halosaccharin 1b was significantly more efficient at performing the desired amination than 1a. As shown in Figure 2, a large variety of amides can be selectively aminated at the amino carbon in moderate to good yields. This is particularly important because literature precedent has shown that N-halosaccharin reacts effectively with aromatic compounds via C(sp2)–H activation.6 Despite our solvent being chlorobenzene, the major product of these reactions remains the formation of the amino aminal product, indicating that the addition of LiOtBu plays a role in inducing chemoselectivity. The main reactivity difference between 1a and 1b comes from the difference in pKa between phthalimide (8.3) and saccharin (1.6). The stronger electron-withdrawing groups in saccharin destabilize the imidyl radical, further increasing its reactivity. Also, stronger electron-withdrawing groups in saccharin will increase the σ-hole of the halogen, thereby favoring stronger halogen bonding interactions with the tert-butoxide base.
Figure 2.
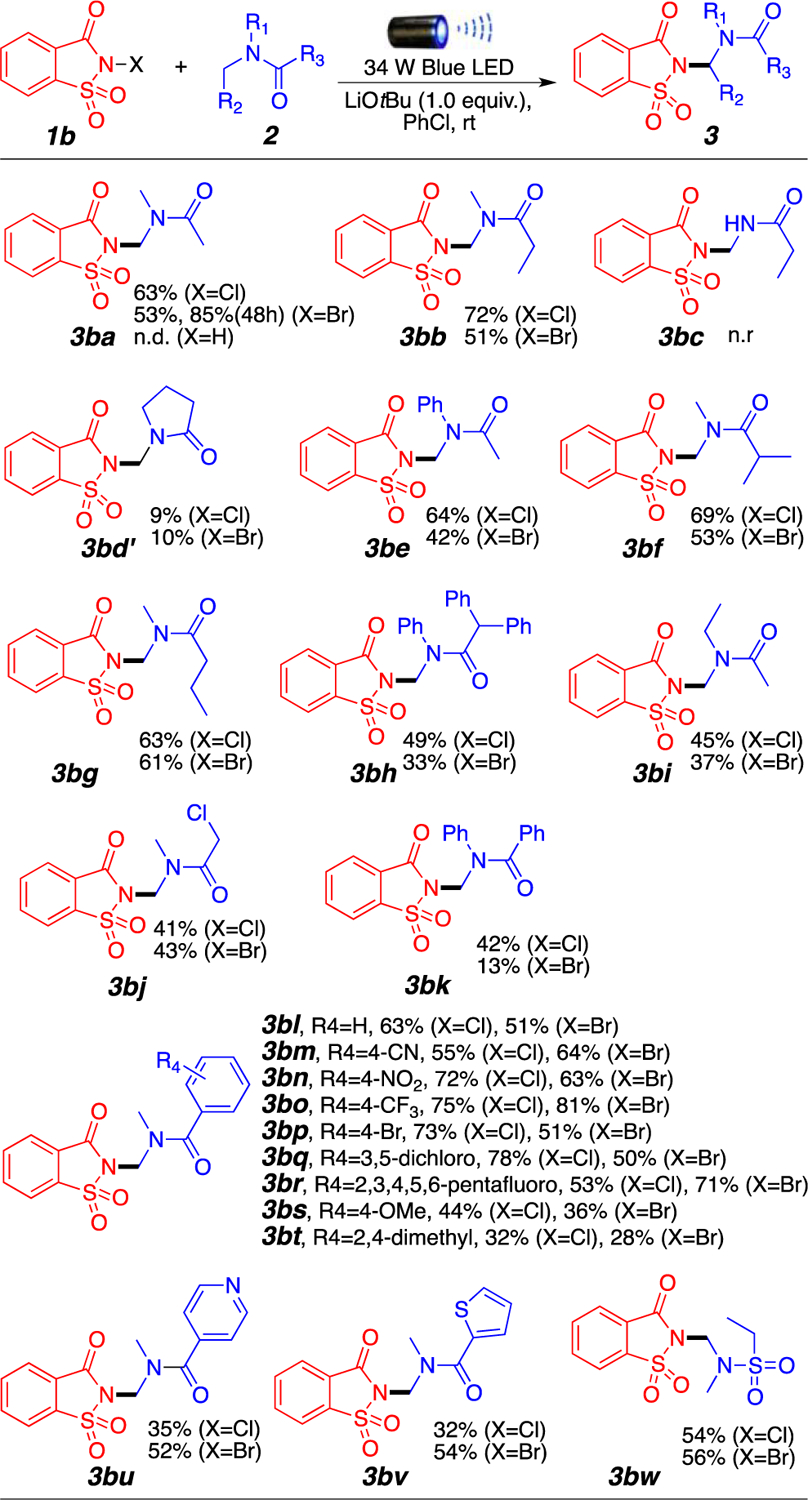
Amination of amides by N-halosaccharins. Reaction conditions: 1b (0.2 mmol), 2 (1.0 mmol), LiOtBu (0.2 mmol), 1.0 mL of PhCl, room temperature around the reaction flask of 35 °C (heating caused by the LED lamp), overnight. Isolated yields.
Aliphatic amide derivatives could be aminated in good to excellent yields using both N-bromo- and N-chlorosaccharin (3ba and 3bb, respectively). As shown for product 3ba, the reaction does not proceed when the halogen is replaced with hydrogen, indicating the necessity of using the N-haloimide motif. An important observation is the complementary reactivity between N-bromophthalimide 1a and N-halosaccharin 1b; while secondary amide 2c and N-methyl-2-pyrrolidone provided the desired product when using 1a, this was not the case with 1b, giving little to no products 3bc and 3bd′. In addition, N-methyl-2-pyrrolidone reactivity seemed to switch from the methylene position to the methyl position (3bd′). Presumably, these observed changes are due to a difference in the stability of the imidyl radicals of phthalimide and saccharin. The more reactive N-centered saccharin radical reacts with the N–H bond of secondary amides, leading to its decomposition (3ac vs 3bc). Similarly, a less reactive phthalimide radical may allow for equilibration to the most stable secondary radical in the amide in 3ad (Figure 1), while the highly reactive saccharin radical presumably reacts with the kinetic amide radical at the methyl position [3bd′ (Figure 2)].
Highly acidic α-positions such as those in product 3bh (Figure 2) still provided the desired product in moderate yield, and unsymmetrical N-ethyl-N-methyl amides showed regioselectivity toward the methyl group over the methylene carbon (3bi). Aromatic amides bearing electron-withdrawing groups (-CN, -NO2, and -CF3) (3bm–3bo) presented better yields compared with those of electron-donating groups (-OMe and -Me) (3bl–3bt). It is also noteworthy that aromatic halogens (F, Cl, and Br) were well tolerated and provided the desired products in good yields (3bp–3br), enabling further manipulations of the products through cross-coupling transformations. Heteroaromatic amides were also aminated in moderate yields (3bu and 3bv). Importantly, unlike in previously reported aminations using 1b,8 our approach did not seem to lead to significant reaction with the aromatic moieties of the amides, and only small amounts of aminated chlorobenzene were observed. Finally, as expected, sulfonamides tolerated the reaction conditions well, furnishing desired product 3bw in 56% yield (Figure 2).
An additional screen important to the synthetic organic chemistry community is the possibility of selectively aminate-protected amines in the form of carbamates (Figure 3). Indeed, we hypothesized that carbamates and amides would react similarly under our reaction conditions but would provide a facile deprotection strategy. Much to our delight, tert-butyloxycarbonyl (Boc)-protected amines tolerated our mild reaction conditions and provided the desired products in moderate to good yields (Figure 3). Boc-protected dimethyl-amine and N-ethyl-N-methyl amine provided desired products 5ba and 5bb in excellent and good yields, respectively, when using N-halosaccharin. Boc-protected N-methylaniline also generated desired product 5bc, albeit in lower yields. Of particular interest is the ability to selectively aminate Boc-protected methamphetamine derivatives in low to moderate yields, to give rise to potentially new bioactive compounds such as 5bd and 5be. As expected, the use of unsymmetrical amides favors the formation of the aminated product in the methyl position, even in the presence of a more stable benzylic carbon (5bb, 5bd, and 5be). This further emphasizes the possible role of steric hindrance as the driving force for the regioselectivity of the reaction. Finally, N-halophthalimide can also be used to aminate Boc-proteccted amines in lower yields, as shown for product 5aa (Figure 3).
Figure 3.
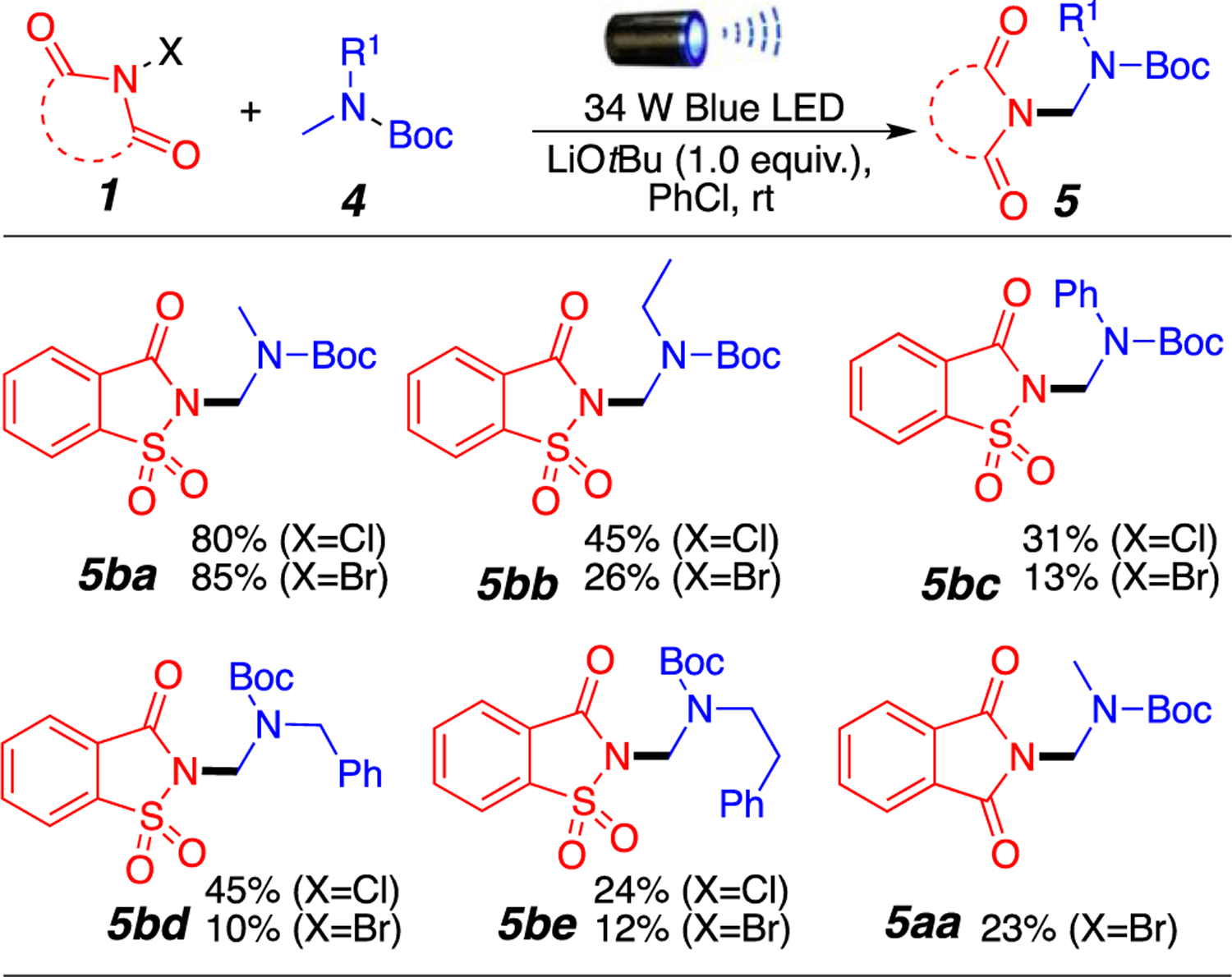
Amination of Boc-protected amines. Reaction conditions: 1 (0.2 mmol), 4 (1.0 mmol), LiOtBu (0.2 mmol), 1.0 mL of PhCl, room temperature around the reaction flask of 35 °C (heating caused by the LED lamp), overnight. Isolated yields.
The reaction was further probed with other imide and amide derivatives (see the Supporting Information for the full substrate scope, S5); however, the yields obtained were often low. The N-haloimide reagents require the presence of an aromatic moiety to avoid decomposition of the imidyl radical. Additionally, we presume that the aromatic moiety interacts with the solvent (chlorobenzene) via π-stacking, which stabilizes the reactive intermediate.
To provide some insight into the reaction mechanism, a series of control experiments were carried out as shown in Scheme 2. As expected, addition of a radical scavenger such as TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy) drastically hampered the reaction. Importantly, through this experiment, we observed the generation of compound 6 via GC-MS, which indicates the formation of the amide radical at the amino carbon (Scheme 2A). Additionally, throughout our reactions, we can also observe the amination of the solvent (chlorobenzene) to give compound 7 observed via GC-MS as a mixture of regioisomers. This is similar to previously published transformations,6 indicating the formation of the imidyl radical. We presume that homolytic cleavage of the N-haloimide prior to activation by LiOtBu is in part responsible for this side reactivity,8 which is observed as the primary product (7) in the absence of LiOtBu. Also as previously mentioned in the table of optimization, in the absence of LiOtBu or light no product is generated, and replacing N-haloimides with the nonhalogenated imide (such as phthalimide) does not generate any observable product (Scheme 2B).
Scheme 2.
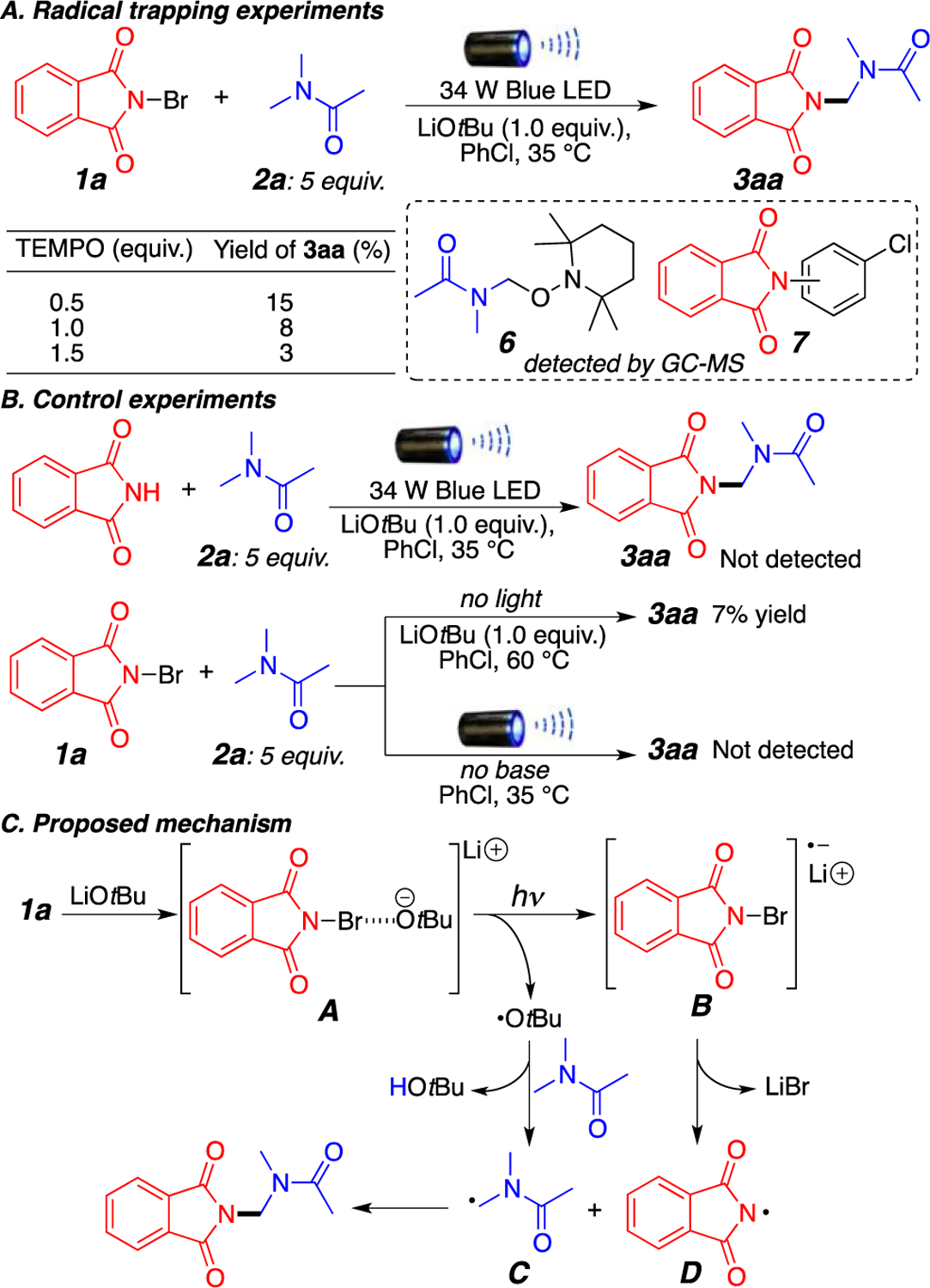
(A) Radical Trapping Experiments, (B) Control Experiments, and (C) Proposed Reaction Mechanism
To further understand the role played by LiOtBu, we performed a series of UV–vis spectroscopic measurements on various combinations of 1a, 1b, and lithium tert-butoxide in PhCl (see the Supporting Information, S6). The results indicate that LiOtBu is interacting with the N-haloimide, possibly via halogen bonding,7 and generates a halogen-bonded adduct A capable of absorbing blue light to initiate the radical reaction.
To further understand the process in which the reaction takes place, the energetic profile of the mechanism was explored through quantum calculations (see the Supporting Information, S7). The results show that the formation of an electron donor–acceptor (EDA) complex presents an exergonic energy profile (−14.6 kcal/mol), denoting that its formation is favored. Additionally, we also calculated two possible mechanistic pathways for the C–H activation step: (i) a hydrogen atom transfer (HAT) step between •OtBu and the amide and (ii) an electron transfer/proton transfer (ET/PT) step.8 Exploration of both mechanistic pathways reveals that the reaction seems to follow a classic HAT mechanism.
On the basis of the results presented above and previous reports,3e–h,9 we propose a plausible reaction mechanism in Scheme 2C. Reaction between the N-haloimide and LiOtBu forms a halogen bond adduct A that can absorb visible blue light and leads to a single-electron transfer yielding radical anion B and tert-butoxide radical. Regioselective hydrogen abstraction of the amino carbon of the amide by the tBuO• radical generates stable radical intermediate C. Simultaneously, radical anion B decomposes through N–Br bond cleavage to generate phthalimidyl radical D. Fast radical–radical coupling between C and D generates the final product (Scheme 2C). Side reactions between D and the solvent generate aminated byproduct 7, and trapping of intermediate C was shown using TEMPO to give 6.
In summary, the amination of amides was developed via a photochemical sp3 C–H bond functionalization process. This reaction showed good functional group compatibility. As mentioned above, the current methods suffer from external radical initiators, oxidants, heat sources, and a relatively limited substrate scope in many cases. Therefore, this reported process provides a complementary and advantageous approach for accessing amine-containing organic molecules.
Supplementary Material
ACKNOWLEDGMENTS
This publication was made possible, in part, by support from the Indiana Clinical and Translational Sciences Institute funded, in part, by Grant UL1TR002529 from the National Institutes of Health, National Center for Advancing Translational Sciences, Clinical and Translational Sciences Award. Start-up funding from Indiana University-Purdue University Indianapolis (IUPUI) was also used to support this research. National Institute of Dental and Craniofacial Research Grant 1R21DE029156-01.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.orglett.1c00831.
1H and 13C{1H} NMR spectra for all aminated products, GC-MS spectra, and the substrate scope and its limitations (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.orglett.1c00831
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Fischer J; Ganellin CR Analogue-based Drug Discovery; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006; p 457. [Google Scholar]; (b) Van Epen JH Experience with Fluspirilene (R 6218), a Long Acting Neuroleptic. Psychiatr., Neurol., Neurochir 1970, 73, 277–284. [PubMed] [Google Scholar]; (c) Taylor-Robinson D; Furr PM Observations on the Antibiotic Treatment of Experimentally Induced Mycoplasmal Infections in Mice. J. Antimicrob. Chemother 2000, 45, 903–907. [DOI] [PubMed] [Google Scholar]; (d) Pellock JM Treatment of Epilepsy in the New Millennium. Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy 2000, 20, 129S–138S. [DOI] [PubMed] [Google Scholar]
- (2).(a) Anisimova NA; Belavin IY; Orlova NA; Sergeev VN; Shipov AG; Baukov YI Silyl Variation of N-Amidoalkylation. Zhurnal Obshchei Khimii 1983, 53, 1198–1199. [Google Scholar]; (b) Hellmann H; Lösehmann I; Lingens F Über N-Mannich-Basen, II. Mitteil.: Synthesen mit Dialkylamino-methyl-phthalimiden und deren quartären Salzen. Chem. Ber 1954, 87, 1690–1699. [Google Scholar]
- (3).For the only known examples of C–H amination to generate amido aminals, see:; (a) Sun M; Zhang T; Bao W FeCl3 Catalyzed sp3 C–H Amination: Synthesis of Aminals with Arylamines and Amides. Tetrahedron Lett. 2014, 55, 893–896. [Google Scholar]; (b) Xia Q; Chen W Iron-Catalyzed N-Alkylation of Azoles via Cleavage of an sp3 C–H Bond Adjacent to a Nitrogen Atom. J. Org. Chem 2012, 77, 9366–9373. [DOI] [PubMed] [Google Scholar]; (c) Saidulu G; Kumar RA; Reddy KR Iron-Catalyzed C–N Bond Formation via Oxidative Csp3–H Bond Functionalization Adjacent to Nitrogen in Amides and Anilines: Synthesis of N-Alkyl and N-Benzyl Azoles. Tetrahedron Lett. 2015, 56, 4200–4203. [Google Scholar]; (d) Deng X; Lei X; Nie G; Jia L; Li Y; Chen Y Copper Catalyzed Cross-Dehydrogenative N2-Coupling of NH-1,2,3-Triazoles with N,N-Dialkylamides: N-Amidoalkylation of NH-1,2,3-Triazoles. J. Org. Chem 2017, 82, 6163–6171. [DOI] [PubMed] [Google Scholar]; (e) Xu S; Luo Z; Jiang Z; Lin D Transition-Metal-Free N9-Amidoalkylation of Purines with N,N-Dialkylamides. Synlett 2017, 28, 868–872. [Google Scholar]; (f) Aruri H; Singh U; Kumar M; Sharma S; Aithagani SK; Gupta VK; Mignani S; Vishwakarma RA; Singh PP Metal-free Cross-Dehydrogenative Coupling of HN-Azoles with α-C(sp3)-H Amides via C-H Activation and Its Mechanistic and Application Studies. J. Org. Chem 2017, 82, 1000–1012. [DOI] [PubMed] [Google Scholar]; (g) Zhu Z; Wang Y; Yang M; Huang L; Gong J; Guo S; Cai H A Metal-Free Cross-Dehydrogenative Coupling Reaction of Amides to Access N-Alkylazoles. Synlett 2016, 27, 2705–2708. [Google Scholar]; (h) Lao Z-Q; Zhong W-H; Lou Q-H; Li Z-J; Meng X-B KI-Catalyzed Imidation of sp3 C–H Bond Adjacent to Amide Nitrogen Atom. Org. Biomol. Chem 2012, 10, 7869–7871. [DOI] [PubMed] [Google Scholar]; (i) Wan Z; Wang D; Yang Z; Zhang H; Wang S; Lei A Electrochemical Oxidative C(sp3)-H Azolation of Lactams Under Mild Conditions. Green Chem. 2020, 22, 3742–3747. [Google Scholar]
- (4).For selected reviews of photochemistry, see:; (a) Ghosh I; Marzo L; Das A; Shaikh R; König B Visible Light Mediated Photoredox Catalytic Arylation Reactions. Acc. Chem. Res 2016, 49, 1566–1577. [DOI] [PubMed] [Google Scholar]; (b) Tucker JW; Stephenson CRJ Shining Light on Photoredox Catalysis: Theory and Synthetic Applications. J. Org. Chem 2012, 77, 1617–1622. [DOI] [PubMed] [Google Scholar]; (c) Prier CK; Rankic DA; MacMillan DWC Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Skubi KL; Blum TR; Yoon TP Dual Catalysis Strategies in Photochemical Synthesis. Chem. Rev 2016, 116, 10035–10074. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Le C; Liang Y; Evans RW; Li X; MacMillan DWC Selective sp3 C–H alkylation via polarity-match based cross-coupling. Nature 2017, 547, 79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Gasonoo M; Thom ZW; Laulhé S Regioselective α-Amination of Ethers Using Stable N-Chloroimides and Lithium tert-Butoxide. J. Org. Chem 2019, 84, 8710–8716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Song L; Zhang L; Luo S; Cheng J Visible-Light Promoted Catalyst-Free Imidation of Arenes and Heteroarenes. Chem. - Eur. J 2014, 20, 14231–14234. [DOI] [PubMed] [Google Scholar]
- (7).Weinberger C; Hines R; Zeller M; Rosokha SV Continuum of Covalent to Intermolecular Bonding in the Halogen Bonded Complexes of 1,4-Diazabicyclo [2.2.2]octane with Bromine Containing Electrophiles. Chem. Commun 2018, 54, 8060–8063. [DOI] [PubMed] [Google Scholar]
- (8).Hancock AN; Tanko JM Radical cation/anion and neutral radicals: a comparison. In Encyclopedia of Radicals in Chemistry, Biology and Materials; John Wiley & Sons: Chichester, U.K., 2012. [Google Scholar]
- (9).(a) Barham JP; Coulthard G; Emery KJ; Doni E; Cumine F; Nocera G; John MP; Berlouis LEA; McGuire T; Tuttle T; Murphy JA KOtBu: A Privileged Reagent for Electron Transfer Reactions? J. Am. Chem. Soc 2016, 138, 7402–7410. [DOI] [PubMed] [Google Scholar]; (b) Liu X; Zhao X; Liang F; Ren B T-BuONa-Mediated Direct C–H Halogenation of Electron-Deficient (Hetero)Arenes. Org. Biomol. Chem 2018, 16, 886–890. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.