

Abstract
Sirtuins are NAD+-dependent protein deacylases involved in metabolic regulation and aging-related diseases. Specific activators for the seven human Sirtuin isoforms would be important chemical tools and potential therapeutic drugs. Activators have been described for Sirt1 and act via a unique N-terminal domain of this isoform. For most other Sirtuin isoforms, including mitochondrial Sirt3-5, no potent and specific activators have yet been identified. We describe here the identification and characterization of 1,4-dihydropyridine-based compounds that either act as pan Sirtuin activators or specifically stimulate Sirt3 or Sirt5. The activators bind to the Sirtuin catalytic cores independent of NAD+ and acylated peptide and stimulate turnover of peptide and protein substrates. The compounds also activate Sirt3 or Sirt5 in cellular systems regulating, e.g., apoptosis and electron transport chain. Our results provide a scaffold for potent Sirtuin activation and derivatives specific for Sirt3 and Sirt5 as an excellent basis for further drug development.
Keywords: 1,4-dihydropyridine; Sirtuin activation; aging; drug development; enzyme activator; signaling; deacetylase; deacylase
Graphical Abstract
Introduction
Acetylation and related acylations, such as succinylation, glutarylation, fatty-acylation and others, are post-translational modifications affecting almost all mammalian proteins1, 2. Sirtuins are an evolutionary conserved family of protein deacylases implicated in metabolic regulation, stress responses, and aging processes. They require NAD+ as a cosubstrate, linking the Sirtuin activity to cellular energy levels3, and they contribute to the life- and health-span extending effects of caloric restriction (CR), a severe reduction in calorie intake4, 5. Pharmacological activation of Sirtuins can induce effects similar to CR and is considered a valuable therapeutic approach for several aging-related diseases, such as neurodegeneration, type 2 diabetes, metabolic disorders and cancer6, 7.
Mammals have three mostly nuclear Sirtuin isoforms (Sirt1, −6, −7), one dominantly cytosolic (Sirt2), and three preferentially mitochondrial (Sirt3, −4, −5)8, although additional localizations have been described for several of these isoforms9. Sirt1, −6 and −7 regulate, e.g., DNA homeostasis, and Sirt2 regulates cell cycle progression10. Mitochondrial Sirt3 regulates a wide range of targets through lysine deacetylation, from signaling proteins to metabolic enzymes. It has been implicated in responses to exercise or nutritional regime, in aging-related disorders such as type II diabetes and cardiovascular diseases, and in CR effects11, 12. Sirt5, in contrast, is a weak deacetylase and acts primarily as a protein lysine desuccinylase and deglutarylase13–15. Sirt5 regulates metabolic enzymes and stress response proteins, has been implicated in metabolic adaptations to protein-rich diet, fasting, and long-term CR, and seems to contribute to metabolic dysfunctions and neurodegeneration12. Sirt4 also features weak deacetylase activity but can act as deliponamidase and dehydroxymethylglutarylase, regulates metabolic enzymes, and has been implicated in metabolic disorders16, 17. Pharmacological activation of Sirt3 and Sirt5 could be attractive for treatment of cancer, heart failure and metabolic dysfunctions12. For example, increased Sirt3 activity blocked and reversed cardiac hypertrophy in mice18, reduced β-Amyloid (Aβ levels) in vitro19 and alleviated cognitive deficits of Alzheimer’s disease in transgenic mice20. Sirt5 activation might allow to prevent ischemia/reperfusion-induced oxidative injury21. Also, while the role of Sirt5 in cancer remains controversial, recent findings underline that Sirt5 is a potential suppressor of gastric cancer22, pancreatic cancer23, and possible other cancer types24.
Sirtuins feature a conserved catalytic core, flanked by isoform-specific N- and C-terminal extensions8. The core comprises a Rossmann fold and a Zn2+-binding subdomain, with the catalytic center in a cleft between them25. During catalysis, the oxygen of the lysine-bound acyl group forms an alkylimidate intermediate with ADP-ribose, which is generated from NAD+ through nicotinamide (NAM) release26. Rearrangements lead to a bicyclic intermediate, which is hydrolyzed to yield deacylated polypeptide and 2’-O-acyl-ADP-ribose. Small molecule activation of Sirtuins was first reported for yeast Sir2 and human Sirt1, using resveratrol and other polyphenols27. These activating effects were initially debated25, 28, 29, but Sirt1 activation is now firmly established through the development of potent compounds and biochemical and structural insights in binding site and activation details7, 30–32. Sirt1 activators bind to an N-terminal “Sirt1 activating compound” (STAC) binding domain, with the catalytic core and its bound substrate likely forming the opposite wall of a binding cleft31, 33. The resulting direct activator/substrate interaction explains the influence of the substrate sequence on compound effects30, 34. Sirt2-7 do not feature a Sirt1-like N-terminal activator-binding domain. However, an activating effect and direct interactions with active site-bound substrate were also found for resveratrol and human Sirt5, but most prominently with the artificial “Fluor-de-Lys” (FdL) substrate, whose fluorophore modification mediated a significant compound interaction33. Weak and potentially non-specific activation has also been reported for Sirt3 by the plant metabolite honokiol, and for Sirt6 by fatty acids18, 35. More recently, several synthetic Sirt6 activators32, 36, 37, 38 have been described, revealing the first potent, isoform-specific stimulating effects on isoforms other than Sirt1.
Pharmacological activation of Sirt3 and Sirt5 is considered attractive for treatment of, e.g., cancer and heart failure12. We report here discovery and characterization of potent and specific activators for Sirt3 and Sirt5. The 1,4-dihydropyridine (DHP)-based activators bind to the Sirtuin catalytic core independent of bound substrates and increase turnover. The compounds are selective for Sirt3 or Sirt5 and show cellular activity. Our results establish pharmacological activation for Sirt3 and Sirt5 and provide lead compounds for further drug development.
Results
1,4-DHP as a scaffold for Sirt3 and Sirt5 activation
Based on a structural similarity to the Sirtuin ligands NAM and 2-anilinobenzamide,39, 40 we previously prepared a series of 1,4-dihydropyridine (1,4-DHP) compounds and screened it against Sirt1-3, resulting in Sirt1-specific activators as well as non-specific Sirt1-3 activators41, 42. In particular compound 1 (Figure 1), carrying a benzyl group at the 1,4-DHP N1 position, a phenyl ring at C4, and a double carboethoxy function at the C3/C5 positions, showed Sirt1-3 activating properties in the FdL assay and Sirt1 activation in cellular contexts41. We hypothesized that modifying this scaffold should also enable Sirt3-specific activation and thus selected a panel of internally available derivatives for testing (1–24; Figure 1). It comprised 1, for which Sirt3 data had previously been collected only with the FdL assay, and 5, 6, and 11, which were among the most potent, cellular active Sirt1 activators from a Sirt1-focused follow-up study testing modifications at C2/C6, C3/C5, C4, or N1 and providing no data on Sirt343. For screening these compounds against Sirt3, we used here a coupled deacetylation assay that relies on non-modified acetyl substrates instead of the FdL substrate employed in the previous studies41, 42, which can yield artificial activation signals due to a fluorophore modification26, 33. Assays with 10 or 100 μM compound revealed an ~2-fold activation of Sirt3-dependent deacetylation of a peptide derived from the Sirt3 target acetyl-CoA synthetase 2 (ACS2; Table S1) by 100 μM compound 1 (Figure 2a), similar to its effect in the previous FdL assay41. Compounds 7 and 10, bearing a 2-thiazolyl or 3-quinolinyl ring at C4, and 22 and 24, carrying a 2-naphthoyl or 2-quinoxaloyl group at N1 and a phenyl ring at C4, showed effects on ACS2 peptide deacetylation comparable to the parent compound. Strikingly, compound 11, bearing a benzoyl moiety at N1 and a phenyl ring at C4, showed a significantly stronger effect, activating Sirt3 ~3.5-fold (Figure 2a). Control reactions for compound effects on the coupled enzymes nicotinamidase and glutamate dehydrogenase (GDH) showed negligible effects (Figure S1), confirming that the DHPs act directly on Sirt3 and activate its deacetylase activity.
Figure 1: Chemical structures of the 1,4-DHP compounds studied.
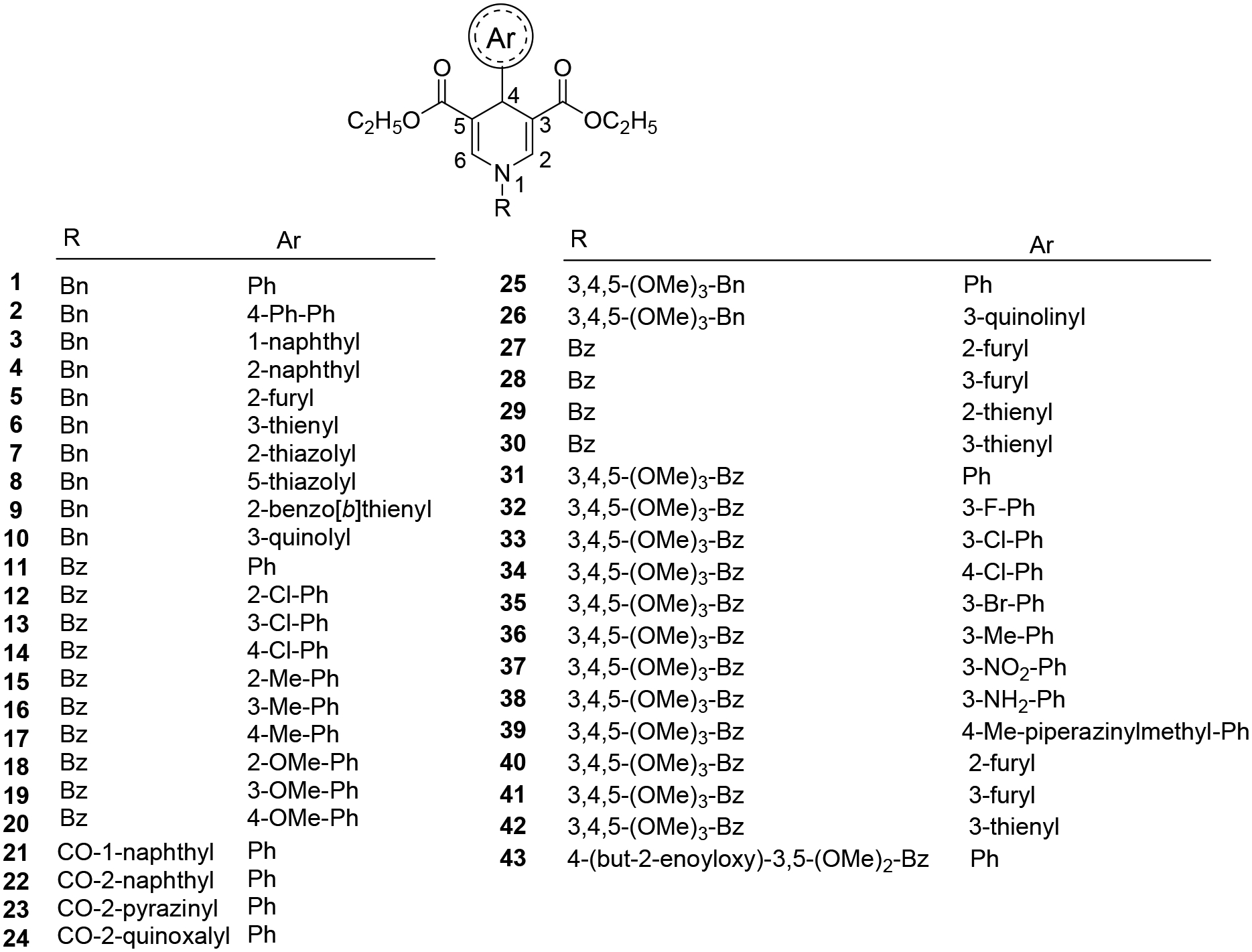
The internal library for testing against Sirtuin isoforms 1, −2, −3 and −5 comprised previously reported (1–24) and newly synthesized (25–43) 1,4-DHP derivatives.
Figure 2: Screening of 1,4-DHP derivatives against recombinant human Sirt1, −2, −3, and −5 at 10 and 100 μM compound concentration.
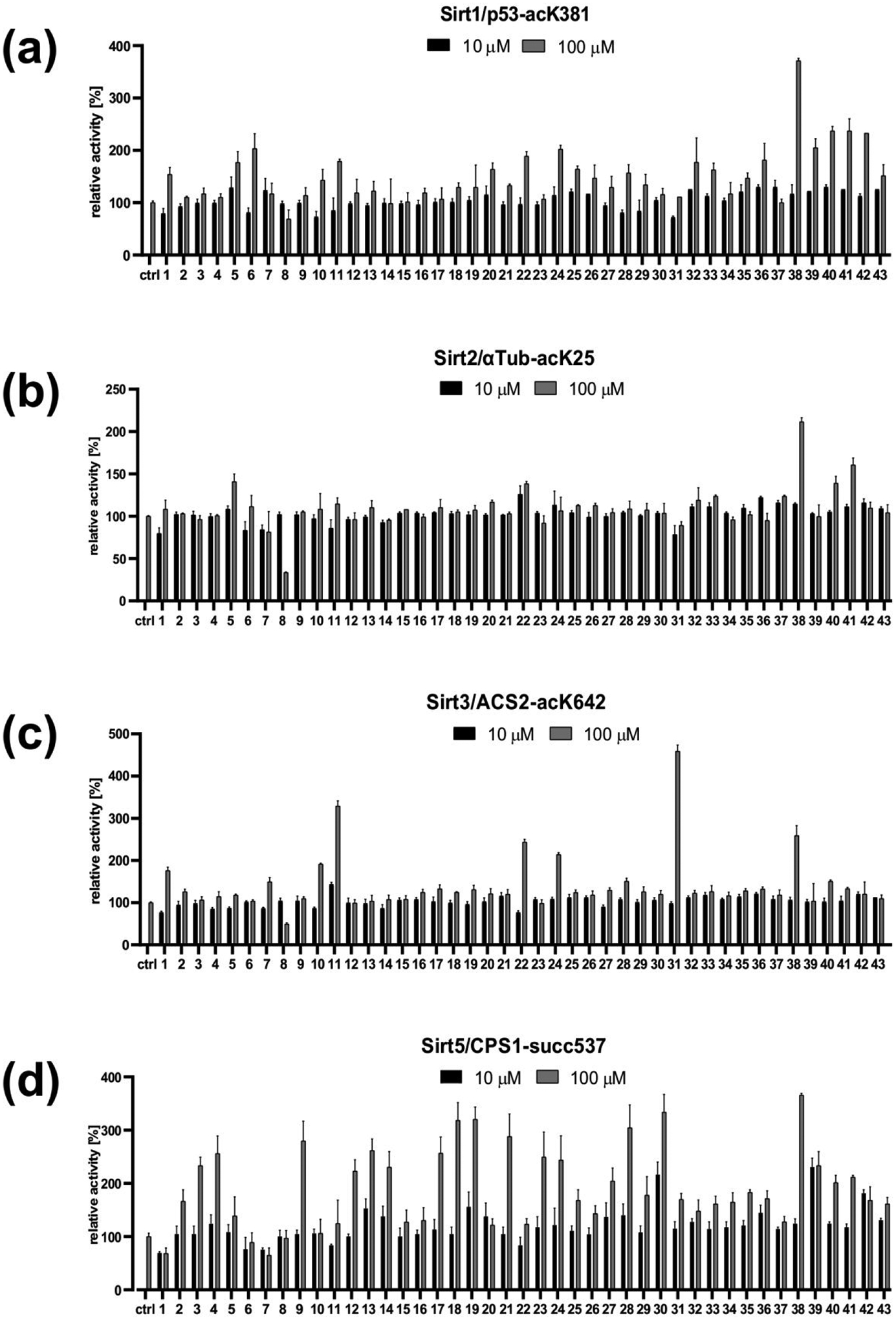
(a) Effects on Sirt1-dependent deacetylation of the p53-acK381 substrate peptide. (b) Effects on Sirt2-dependent deacetylation of the α-tubulin-acK25 substrate peptide. (c) Effects on Sirt3-dependent deacetylation of the ACS2-acK642 substrate peptide. (d) Effects on Sirt5-dependent desuccinylation of the CPS1-succK537 substrate peptide.
To characterize the isoform specificity of our compounds, we tested compounds 1–24 against Sirt1, Sirt2, and Sirt5 using the same coupled assay as for Sirt3 and isoform-specific, fluorophore-free substrate peptides (Figure 2b–d). The same concentrations for enzyme, NAD+, and the respective substrate peptide (Sirt1: acetylated p53-K381; Sirt2: acetylated α-tubulin-K25; Sirt5: succinylated CPS1-K537; Table S1) as for Sirt3 were used. Toward Sirt1, several compounds (1, 5, 6, 10, 11, 20–22, 24) furnished 1.5- to 2-fold increased deacetylation (Figure 2b), confirming Sirt1 activating effects previously detected with the FdL substrate41,43. Toward Sirt2, only 5 and 22 displayed slight activation (less than 1.5-fold at 100 μM). The other compounds were unable to activate Sirt2, and 8, bearing a benzyl group at N1 and a 5-thiazolyl ring at C4, even displayed 75% inhibition at 100 μM (Figure 2c), similar to its effect on Sirt3 (60 % inhibition at 100 μM). In contrast, a surprising number of compounds, with various DHP substituents, showed an activating effect on Sirt5 (Figure 1; Figure 2d). Many compounds (2–4, 9, 12–14, 17–19, 21, 23, and 24) were able to activate Sirt5 ~2- to 3-fold at 100 μM, and compounds 4, 13, 14, 19, 20, 23, and 24 exhibited 1.2- to 1.5-fold activation already at 10 μM.
Taken together, we find that 1,4-DHP-based compounds can activate Sirt1, Sirt2, Sirt3, and Sirt5 against peptides representing physiological substrate sites. We find only few, weak activators for Sirt2 but several compounds that activate Sirt3, with higher efficacy than the parent compound albeit no pronounced isoform-specificity, and the same applies to Sirt5 activation, which is activated by a surprisingly large number of derivatives.
Synthesis and screening of novel 1,4-DHP derivatives
Analyzing the potency of the Sirt3 activator 11 in a compound titration yielded a dose-dependent increase in Sirt3 activity, but the decent aqueous solubility of the compound no upper baseline was reached within its solubility limits (Figure 3a). Therefore, the EC50 can only be estimated to be ~100–200 μM. This compound thus shows limited potency and specificity, but since several DHP derivatives showed activating effects against Sirt3 and Sirt5 and differing isoform profiles, we designed, synthesized, and screened additional 1,4-DHP analogues (Figure 1). We prepared compounds 25–43 carrying a 3,4,5-trimethoxybenzyl group at N1 and a phenyl or 3-quinolinyl ring at C4 (25 and 26), or a benzoyl substituent at N1 and heteroaromatic rings at C4 (27–30), as an extension of the previous N1-benzoyl analogues 11–20, or the 3,4,5-trimethoxybenzoyl portion at N1 and variously substituted phenyl, furyl or thienyl rings at C4 (31–42). The substituent at the C4-phenyl ring was inserted mainly at the meta position, one of the favorite ones in the first set of tested compounds. In the last compound 43, the 3,4,5-trimethoxybenzoyl group of 31 was replaced by the 4-crotonyloxy-3,5-dimethoxybenzoyl substituent.
Figure 3: Direct, dose-dependent activation of Sirt3 and Sirt5.
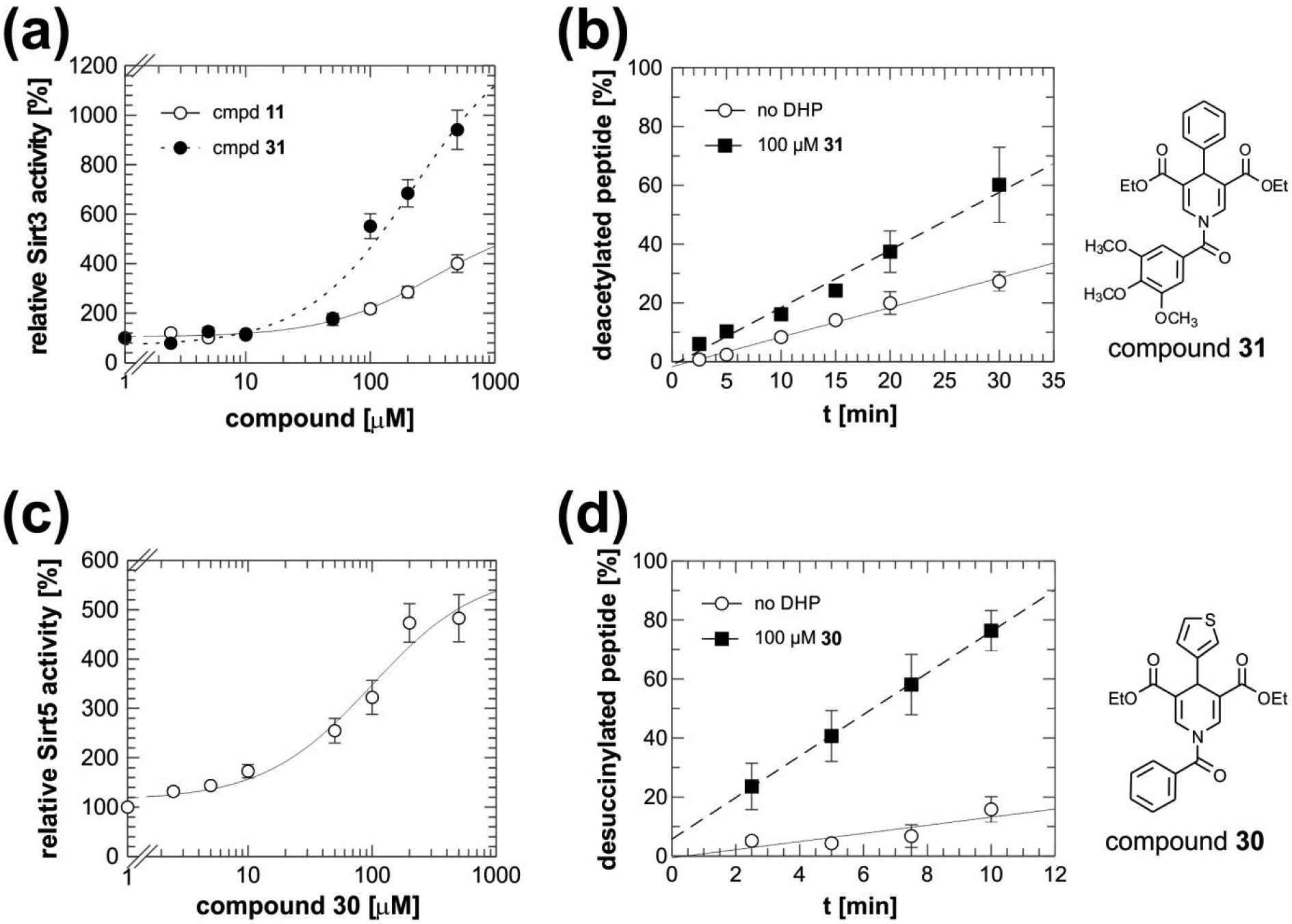
(a) Sirt3 titrations with 11 and 31 result in dose-dependent Sirt3 activation. (b) MS-based deacetylation assay in absence and presence of 31 (100 μM), which confirms direct activation of Sirt3. Identical Sirt3 and substrate concentrations as in the coupled enzymatic assay were used. (c) Sirt5 titration with 30 results in dose-dependent Sirt5 activation. (d) MS-based desuccinylation assay in absence and presence of 30 (100 μM) confirming direct Sirt5 activation. Error bars: Standard errors from at least three independent experiments.
To generate the novel derivatives, we attached the varying aromatic moieties to the diethyl 1,4-DHP-3,5-dicarboxylate scaffold using cyclocondensation chemistry41 43. Multicomponent reaction between ethyl propiolate, benzaldehyde or 3-quinolinecarboxaldehyde and 3,4,5-trimethoxybenzylamine at 80 °C in glacial acetic acid afforded the compounds 25 and 26 (Scheme 1A). For the synthesis of the N-aroyl derivatives 27-43, the key intermediates 44-55 bearing different (hetero)aromatic rings at C4 and hydrogen substituents at N1 had to be prepared through cyclocondensation reaction using properly substituted aldehydes, ethyl propiolate and ammonium acetate (Scheme 1B). Among them, 4444, 4843, 5043, 45, 5143, 45, 5343 and 5445 were already reported by us and/or others. Further N1-acylation of either 44-47 with triethylamine and benzoyl chloride, or 44, 45, 47-55 with triethylamine and 3,4,5-trimethoxybenzoyl chloride, or 44 with triethylamine and 4-crotonyloxy-3,5-dimethoxybenzoyl chloride afforded the final compounds 27-30, 31-37 and 39-42, and 43, respectively (Scheme 1B). The synthesis of 38 was accomplished by reduction of the nitro group of 37 with stannous chloride dihydrate and 37% hydrochloric acid in ethanol (Scheme1B). Elemental analyses for 25-43 are reported in Table 1.
Scheme 1.
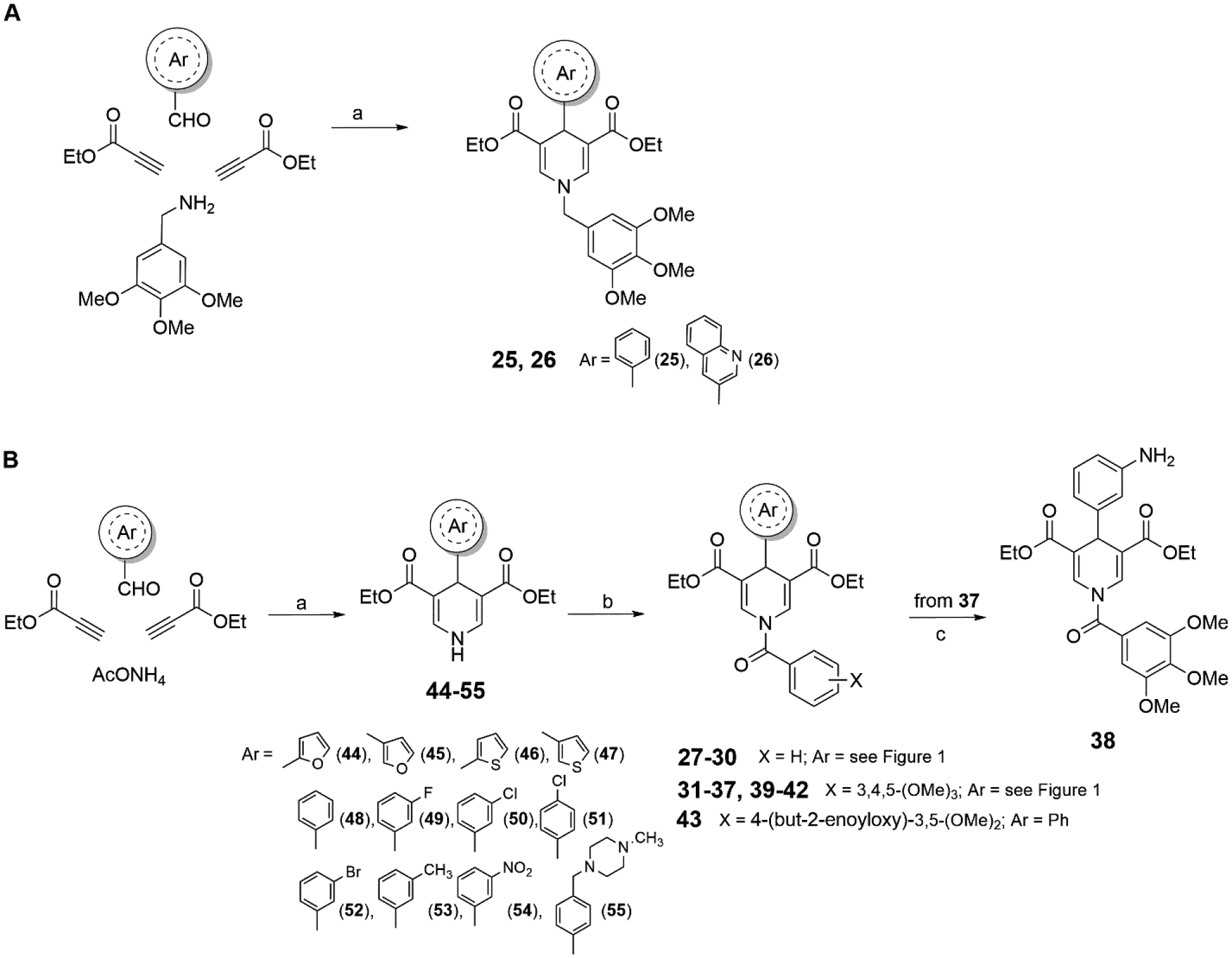
Synthesis of Compounds 25–43a
aReagents and conditions: (a) glacial acetic acid, 80 °C; (b) Et3N, aroyl chloride, dry dichloromethane, rt, overnight; (c) stannous chloride dihydrate, 37% HCl, EtOH, rt, overnight.
Table 1.
Elemental analysis of final compounds 25–43.
| calculated | found | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cpd | C | H | N | O | S | F | Cl | Br | C | H | N | O | S | F | Cl | Br |
| 25 | 67.35 | 6.49 | 2.91 | 23.26 | - | - | - | - | 67.38 | 6.53 | 2.85 | 23.24 | - | - | - | - |
| 26 | 67.66 | 6.06 | 5.26 | 21.03 | - | - | - | - | 67.75 | 5.98 | 5.33 | 20.94 | - | - | - | - |
| 27 | 66.83 | 5.35 | 3.54 | 24.28 | - | - | - | - | 66.91 | 5.28 | 3.49 | 24.32 | - | - | - | - |
| 28 | 66.83 | 5.35 | 3.54 | 24.28 | - | - | - | - | 66.89 | 5.24 | 3.67 | 24.20 | - | - | - | - |
| 29 | 64.22 | 5.14 | 3.40 | 19.44 | 7.79 | - | - | - | 64.13 | 5.22 | 3.51 | 19.33 | 7.81 | - | - | - |
| 30 | 64.22 | 5.14 | 3.40 | 19.44 | 7.79 | - | - | - | 63.99 | 5.11 | 3.52 | 19.55 | 7.83 | - | - | - |
| 31 | 65.44 | 5.90 | 2.83 | 25.83 | - | - | - | - | 65.11 | 5.93 | 2.97 | 25.98 | - | - | - | - |
| 32 | 63.15 | 5.50 | 2.73 | 24.92 | - | 3.70 | - | - | 63.19 | 5.54 | 2.81 | 24.87 | - | 3.59 | - | - |
| 33 | 61.19 | 5.33 | 2.64 | 24.15 | - | - | 6.69 | - | 61.15 | 5.39 | 2.56 | 24.22 | - | - | 6.68 | - |
| 34 | 61.19 | 5.33 | 2.64 | 24.15 | - | - | 6.69 | - | 61.21 | 5.28 | 2.69 | 24.11 | - | - | 6.71 | - |
| 35 | 56.64 | 4.91 | 2.44 | 22.28 | - | - | - | 13.91 | 56.59 | 4.98 | 2.53 | 22.15 | - | - | - | 13.75 |
| 36 | 66.00 | 6.13 | 2.75 | 25.12 | - | - | - | - | 66.07 | 6.02 | 2.88 | 25.03 | - | - | - | - |
| 37 | 60.00 | 5.22 | 5.18 | 29.60 | - | - | - | - | 60.04 | 5.17 | 5.24 | 29.55 | - | - | - | - |
| 38 | 63.52 | 5.92 | 5.49 | 25.07 | - | - | - | - | 63.46 | 5.95 | 5.53 | 25.06 | - | - | - | - |
| 39 | 58.24 | 6.37 | 6.17 | 18.81 | - | - | 10.42 | - | 58.33 | 6.42 | 6.24 | 18.71 | - | - | 10.29 | - |
| 40 | 61.85 | 5.61 | 2.89 | 29.66 | - | - | - | - | 61.78 | 5.69 | 2.85 | 29.68 | - | - | - | - |
| 41 | 61.85 | 5.61 | 2.89 | 29.66 | - | - | - | - | 61.91 | 5.56 | 2.93 | 29.60 | - | - | - | - |
| 42 | 59.87 | 5.43 | 2.79 | 25.52 | 6.39 | - | - | - | 59.82 | 5.57 | 2.72 | 25.41 | 6.48 | - | - | - |
| 43 | 65.57 | 5.69 | 2.55 | 26.20 | - | - | - | - | 65.62 | 5.61 | 2.58 | 26.19 | - | - | - | - |
Compounds 25–43 were then tested against Sirt1, −2, −3, and −5 in the coupled enzymatic assay under the same conditions as compounds 1–24 (Figure 2a–d). Strikingly, the diethyl 4-phenyl-1-(3,4,5-trimethoxybenzoyl)-1,4-dihydropyridine-3,5-dicarboxylate 31 showed a strong, ~5-fold activating effect on Sirt3 and no significant effects in control reactions (Figure S1) and against isoforms Sirt1,2,5 (Figure 2a–d) and Sirt4,6 except for partial Sirt4 inhibition at 100 μM (Figure S2a), identifying it as a Sirt3-specific activator. For additional confirmation of direct Sirt3 activation, we tested 31 effects in a mass spectrometry (MS)-based activity assay that directly analyzes acetylation levels of non-modified substrate, rendering it insensitive to artifacts26, 40. A detailed description of the assay, its sensitivity, and its application for Sirt3 and Sirt5 deacylation assays is provided in40. Compound 31 activated Sirt3-dependent deacetylation of the ACS2 peptide also in the MS-based assay (Figure 3b), confirming that our 1,4-DHP-based compounds are direct Sirt3 activators. Analyzing the potency of 31 against Sirt3 yielded a dose-dependent increase in Sirt3 activity, but again no upper baseline within the compounds’ solubility limits (Figure 3a). Therefore, its EC50 was estimated to be ~100–200 μM, similar to 11. However, while at 100 μM 11 activates Sirt3 to slightly over 300%, 31 already activates to more than 400% (Figure 2c). Compound 31 thus is an activator with fair potency, excellent Sirt3 specificity and increased efficacy, showing that Sirt3 can be activated efficiently and specifically.
Tested against Sirt5, all the new 1,4-DHPs, except for the C4 3-nitrophenyl derivate 37, caused an increased desuccinylation of the specific CPS1-succK537 peptide substrate, with 27, 39–41 exhibiting about 2-fold and 28, 30 and 38 about 3- to 3.5-fold activation (Figure 2d). A titration experiment with the N-benzyl-4-(3-thienyl) derivative 30, which showed no significant effects on isoforms Sirt1,2,3 (Figure 2a–d) and only weak, partial inhibition of Sirt4,6 at 100 μM (Figure S2b), yielded a maximum 5-fold activation of Sirt5 beyond 200 μM compound concentration and an EC50 of ~40 μM (Figure 3c). Testing 30 at 100 μM against Sirt5 in the MS-based assay confirmed the activation of Sirt5-dependent desuccinylation of the CPS1-succK537 substrate and thus the direct, isoform-specific activation of Sirt5 (Figure 3d).
When tested against Sirt2, 25–43 were unable to increase the α-tubulin-acK25 deacetylation except for the C4 3-aminophenyl derivative 38, which exhibited almost 2.5-fold activation at 100 μM, and the C4 furyl analogues 40 and 41, which showed smaller effects (Figure 2b). Tested against Sirt1, several compounds (25, 26, 28, 32, 33, and 36) displayed an activating effect similar to the prototype 1, and 38–42 were the most effective, reaching 2.5-(39–42) to 4-fold (38) Sirt1 activation at 100 μM (Figure 2a). Thus, at N1 the 3,4,5-trimethoxybenzoyl, which also allowed Sirt3 activation (by 30 and 38), was better than the 3,4,5-trimethoxybenzyl or the unsubstituted benzoyl group, and at C4 the 3-aminophenyl or the furyl/thienyl rings are preferred by Sirt1. Since the 3,4,5-trimethoxybenzoyl compound 38 was one of the few compounds also showing significant effects on Sirt2, this moiety allows the development of pan Sirtuin activators and, in combination with suitable C4 groups, the development of isoform-specific compounds.
Mechanism of Sirt3 and Sirt5 activation by isoform-specific 1,4-DHPs
Activation of Sirt1 by resveratrol depends on the substrate sequence29, 30, 34. To test whether Sirt3 activation by 31 depends on the ACS2-derived substrate peptide employed in our screen, we analyzed 31’s effects on Sirt3-dependent deacetylation of a panel of eleven acetylated peptides (Figure 4a). As expected, the enzyme converted the substrates with varying deacetylation rates. However, Sirt3-dependent deacetylation of all substrates was stimulated by 31, weakly by 10 μM and significantly by 100 μM compound. Thus, 31 appears to enhance Sirt3-dependent deacetylation independent of the substrate sequence.
Figure 4: Enzymatic characterization of 1,4-DHP-dependent Sirtuin activation.
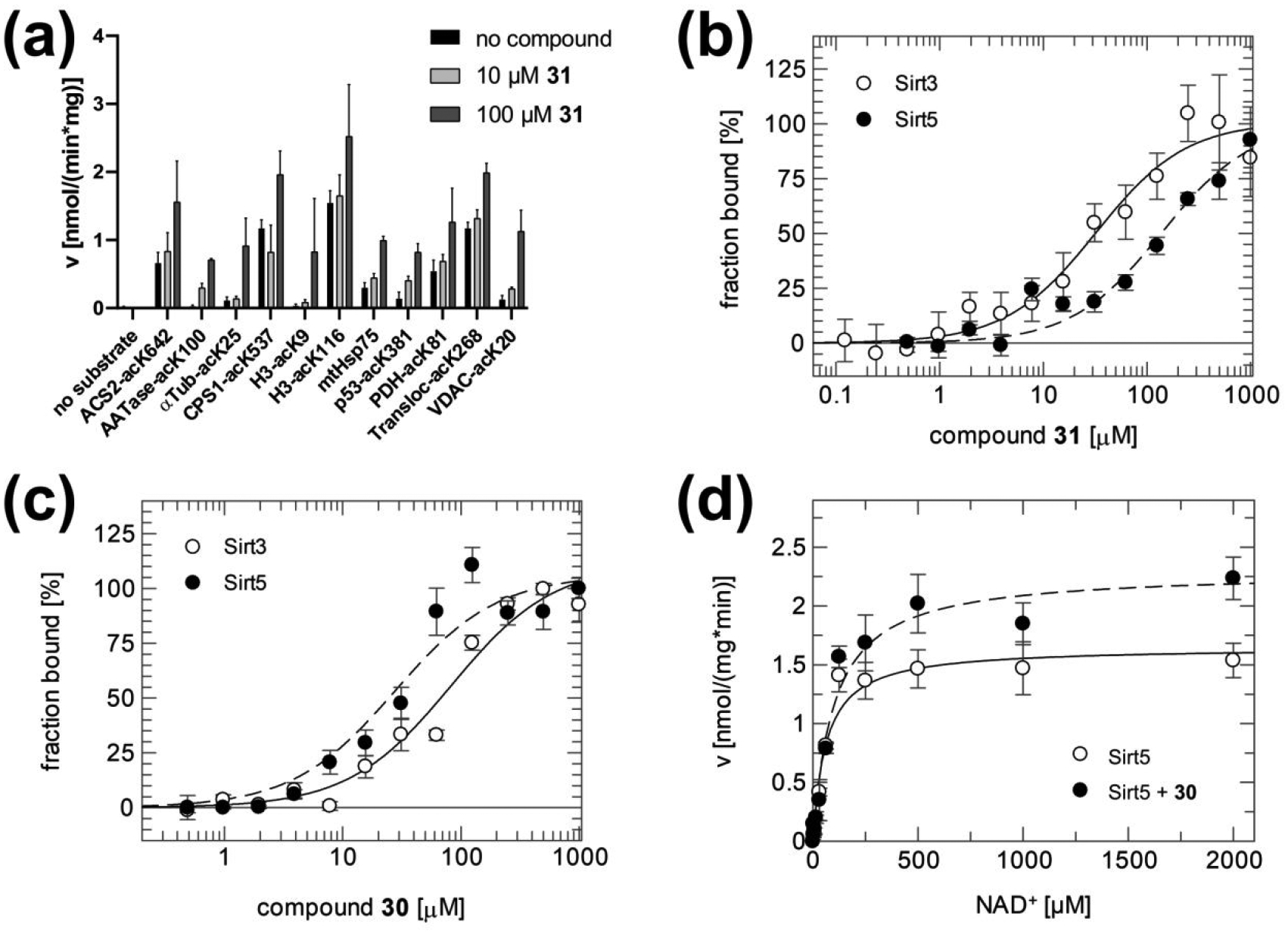
(a) Activation of Sirt3-dependent deacetylation of a variety of acetylated substrate peptides with 31 at 10 and 100 μM. See Table S1 for peptide sequences and origins. (b) Sirt3 and Sirt5 binding titrations with 31 in the absence of any substrate. (c) Sirt3 and Sirt5 binding titrations with 30 in the absence of any substrate. (d) NAD+ titrations of Sirt5 in the presence and absence of 30 (200 μM). Error bars: Standard errors of at least three independent measurements.
To investigate the mechanism of Sirt3 and Sirt5 activation by our 1,4-DHPs further, we performed binding measurements and kinetic studies with 31 (Sirt3 activator) and 30 (Sirt5 activator). Analyzing 31 binding to Sirt3 in absence of any substrate in microscale thermophoresis experiments revealed a Kd of 32 ± 7 μM, confirming that the compound binds directly to the enzyme and that no other ligand is essential for binding (Figure 4b). Consistent with their specificities in activity assays (see above), 31 bound stronger to Sirt3 than to Sirt5, whereas 30 showed higher affinity to Sirt5 than to Sirt3 (Figure 4b,c; Table 2). We next titrated Sirt5 with NAD+ in absence and presence of 200 μM 30 to determine its effect on kinetic constants (Figure 4d; Table 3). An increase in vmax was well defined, while Km was even slightly increased in presence of activator and the effect statistically not significant. Thus, the 1,4-DHP-based activators seem to act by increasing the Sirtuin’s substrate turnover. Furthermore, analyzing the binding affinity of 30 to Sirt5 upon addition of substrate peptide or NAD+ co-substrate revealed no significant effects of these ligands on activator affinity (Table 4). We thus assume that the activators bind remote from the active site and stimulate substrate turnover via a conformational change or an influence on the enzyme’s dynamics. However, conclusions on binding site and molecular mechanism will have to await structural information.
Table 2.
Kd values for binding of compounds 31 and 30 to Sirt3 and Sirt5 in the absence of substrate and cosubstrate.
| Compound | Kd [μM] | |
|---|---|---|
| Sirt3 | Sirt5 | |
| 31 | 32 ± 7 | 83 ± 19 |
| 30 | 138 ± 37 | 28 ± 9 |
Table 3.
Kinetic constants for Sirt5 in the absence and presence of 30.
| 200 μM compound 30 | NAD+ kinetics | ||
|---|---|---|---|
| Km [uM] | vmax [nmol/(mg·min)] | ||
| Sirt5 | − | 62 ± 13 | 1.6 ± 0.1 |
| + | 99 ± 20 | 2.3 ± 0.1 | |
Table 4.
Kd values for binding of compound 30 to Sirt5 in the presence of NAD+ or substrate peptide.
| Substrate present | Kd [μM] |
|---|---|
| Sirt5 / compound 30 | |
| 5 mM NAD+ | 28 ± 7 |
| 0.5 mM CPS1-succK537 peptide | 24 ± 5 |
Activation of Sirt3 and Sirt5 against physiological substrates and in cellular systems
We next tested the identified 1,4-DHP-based Sirt3/Sirt5 activators against physiological substrates and in cellular settings. First, we analyzed compound effects on Sirt3 activity in triple negative breast cancer MDA-MB-231 cells. Treatment with Sirt3 activators 11 and 31 at 50 and 100 μM for 3 h led to a significant increase in FdL deacetylation activity in cell lysates (prepared after the treatment, for assay readout), similar to that observed for MDA-MB-231 cells overexpressing Sirt3 (Figure 5a), indicating that the compounds enable the stimulation of cellular Sirt3 activity. Controls with the Sirt3 inhibitor 3-TYP46 and a Sirt3 knockdown showed the expected decrease of Sirt3 activity. Importantly, Western blot analysis confirmed that compound treatment did not lead to significant changes in Sirt3 protein levels (Figure S3a), showing that the compounds do not affect Sirt3 expression and thus supporting the conclusion that they directly activate cellular Sirt3. We next analyzed compound effects on GDH, a physiological Sirt3 substrate that is activated through deacetylation12, 47, 48. Sirt3 overexpressing MDA-MB-231 cells indeed featured increased GDH activity in cell lysates, and treatment of wild-type MDA-MB-231 cells with 11 or 31 at 50 μM caused a similar increase in GDH activity, indicating Sirt3 activation (Figure 5b). The effect on GDH activity was pronounced already after a 4 h treatment with 31, the most potent in vitro Sirt3 activator, while a second compound addition after 4 h was required for a significant effect of 11. Western blots on GDH immunoprecipitated from MDA-MB-231 cells for overall Lys acetylation - antibodies against specific acetylated GDH lysines are not commercially available – indeed showed decreased acetylation levels upon treatment with compound 11 or 31 (50 μM; Figure S3b). Consistently, GDH acetylation was decreased in Sirt3 overexpressing cells and increased in Sirt3 silenced cells. To further confirm cellular Sirt3 activation by our compounds, we analyzed the deacetylation of MnSOD, another established Sirt3 substrate. Sirt3-dependent deacetylation at Lys68 activates MnSOD and can be monitored with a site-specific acetyl-Lys antibody49. Knocking down Sirt3 in MDA-MB-231 cells caused the expected increase in MnSOD-Lys68 acetylation, and treatment with 50 μM 11 resulted in a decrease in MnSOD-Lys68 acetylation (Figure 5c), indicating that the compound activated Sirt3. A control treatment of Sirt3 silenced cells with 11 for 16 h did not decrease MnSOD-Lys68 acetylation compared to untreated Sirt3 knockdown cells, confirming that the effect of 11 on MnSOD-Lys68 deacetylation is Sirt3-dependent (Figure 5c). Also consistent with activation of cellular Sirt350, treating mouse embryonic fibroblasts (MEFs) with 25 μM of the weaker (but better tolerated by MEFs) Sirt3 activator 10 increased mitochondrial mass about 2.5-fold, and there was no significant compound effect anymore when Sirt3 was knocked-out (Figure 5d). We thus conclude that our DHP-based compounds act as Sirt3 activators against physiological substrates and enable Sirt3 activation in cellular systems.
Figure 5: Activation of Sirt3 and Sirt5 in physiological systems.
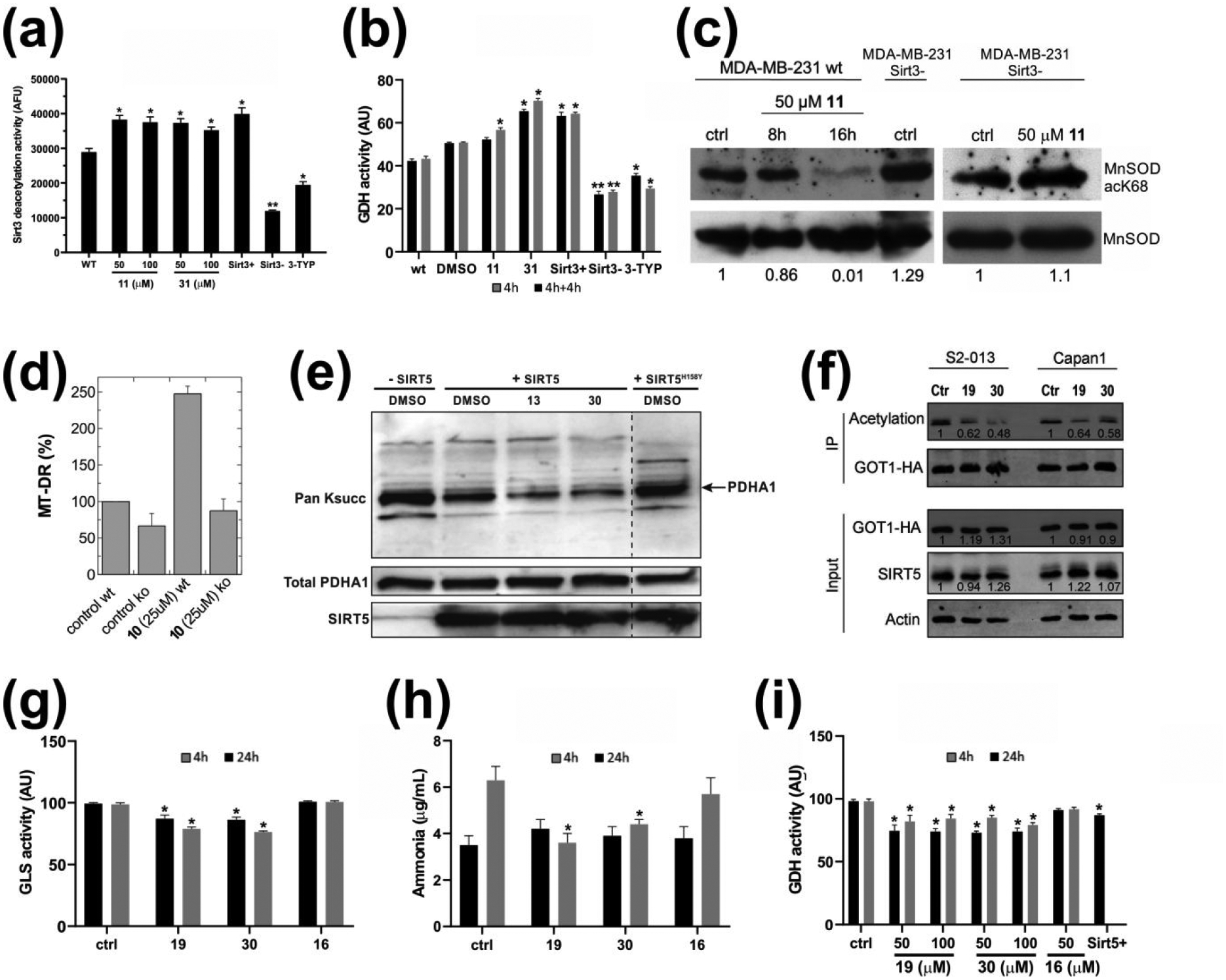
(a) Effects of 11 and 31 on Sirt3 activity in MDA-MB-231 breast cancer cells. Sirt3 activity was determined in cell lysates using the FdL deacetylation assay. For comparison, a treatment with 5 μM Sirt3 inhibitor 3-TYP46 and Sirt3 overexpressing or silencing MDA-MB-231 cells (Sirt3+ and Sirt3−, respectively) are included. (b) Effects of 11 and 31 (50 μM) on GDH activity in MDA-MB-231 cells after 4 h (grey) and after a further 4 h incubation with re-addition of compounds (black). For comparison, GDH activity of Sirt3 overexpressing or silencing MDA-MB-231 cells (Sirt3+ and Sirt3−, respectively) is included. (c) Effects of 11 (50 μM) on acetyl-K68 MnSOD in MDA-MB-231 cells and, as a control, in Sirt3 knock-down MDA-MB-231 cells (Sirt3−). (d) Effects of 10 (25 μM, 24 h) on mitochondrial mass, measured by FACS analysis, of wild-type (wt) and Sirt3 knock-out (ko) MEFs stained with MT-DR for 30 min. (mean ± SD, n = 2) (e) Western-blot showing the effects of 13 or 30 (100 μM) on desuccinylation of Pyruvate dehydrogenase E1 component subunit alpha (PDH-A1; arrow) by recombinant Sirt5. Catalytically inactive Sirt5H158Y is included as a control. (f) Western-blot showing the effects of 19 and 30 (20 μM) on deacetylation of Glutamic-oxaloacetic transaminase 1 (GOT1) in S2–013 and Capan1 cell lines. (g) Effects of 50 μM 19 or 16 (negative control) on GLS activity in MDA-MB-231 cells analyzed after 4 h or 24 h compound treatment. (h) Effects of 50 μM 19 or 16 on ammonia release from MDA-MB-231 cells after 4 h or 24 h compound treatment. (i) Effects of 19 on GDH activity in MDA-MB-231 cell analyzed after 4 h or 24 h compound treatment. Sirt5 overexpressing MDA-MB-231 cells (Sirt5+) are included as a positive control, and compound 16 as a negative control.
We next tested whether our Sirt5-specific activators are also functional with physiological Sirt5 substrates and in cellular settings. First, we analyzed the in vitro desuccinylation of the PDH subunit PDHA1, an established Sirt5 substrate51. Sirt5 treatment decreased succinylation of PDHA1 isolated from porcine heart tissue, and the effect was clearly enhanced in presence of 100 μM 13 or 30 (Figure 5e), confirming that our compounds can stimulate Sirt5 activity against a physiological substrate. We next analysed compound effects on GOT1, which we showed very recently to be regulated through Sirt5-dependent deacetylation23. We treated S2–013 and Capan1 pancreatic cancer cell lines for 24 h with compound 30 (20 μM), and for comparison with compound 19 (20 μM), which we recently showed to increase in vitro Sirt5 deacetylase activity and to elicit similar effects to Sirt5 overexpression23. Western blots showed that both compounds decrease GOT1 acetylation, with 30 being even more effective than 19, and effects were negligible when SIRT5 knockout KPC and KPCS cell lines were used (Figure 5f; Figure S4a). We next analysed the activity of glutaminase (GLS), a Sirt5 substrate whose desuccinylation in MDA-MB-231 cells lowers its catalytic activity and the release of ammonia52. Treatment of MDA-MB-231 cells with 50 μM 19 or 30 for 4 and 24 h decreased GLS activity in cell lysates (Figure 5g) and blocked ammonia accumulation (Figure 5h), as expected for increased Sir5 activity, while no such effects were observed with 16 as negative control. For further support, we analyzed GDH activity. Glutaminolysis by GLS leads to glutamate formation, and glutamate is subsequently converted by GDH to α-ketoglutarate. GDH activity thus is expected to decrease upon Sirt5-dependent GLS inhibition due to a lowering of the levels of the GDH substrate glutamate. Indeed, Sirt5-overexpressing MDA-MB-231 cells showed a reduction of GDH activity, and similarly, treatment of wild-type MDA-MB-231 cells with 50 μM Sirt5 activators 19 or 30 for 4 or 24 h decreased GDH activity in cell lysates (Figure 5i). In contrast, treatment of wild-type cells with compound 16 (50 μM) as a negative control yielded no significant changes in GDH activity (Figure 5i). Western blots of treated and untreated MDA-MB-231 cells showed no significant compound effects on Sirt5 expression (Figure S4b), supporting the conclusion that 19 and 30 are cellular active Sirt5 activators. We thus conclude that our DHP-based compounds comprise cellular active, isoform-specific activators for Sirt3 and Sirt5.
Discussion and Conclusions
Initially, a variety of small molecule activators were reported for Sirt130, 32. Our study reveals that potent and specific pharmacological activation of the deacylation of regular, non-modified substrates is also possible for the mitochondrial isoforms Sirt3 and Sirt5. In particular activation of Sirt3, the only known mitochondrial protein deacetylase, might be of therapeutic interest due to the links of mitochondrial protein acetylation and Sirt3 to metabolic disorders, aging-related hearing loss, and heart diseases11, 12, 18. 1,4-DHPs activate several Sirtuin isoforms and thus should employ a binding mode that differs from established Sirt1 activators, which exploit the so-called STAC-binding domain that is unique to Sirt1 for a substrate sequence-specific activation mechanism30, 32, 34. Consistently, 1,4-DHPs effects appear to be largely independent of the substrate sequence and dominated by an increase in substrate turnover, while Sirt1 STACs increase the apparent substrate affinity53. More recently, potent pharmacological activation of Sirt6 confirmed that alternative activation sites exists on the Sirtuin catalytic core, but the known Sirt6 activators again exploit a binding site unique to this isoform36, 54, 55. Our finding that the 1,4-DHP scaffold enables activation of several Sirtuin isoforms, by pan Sirtuin activators as well as by derivatives specific for either Sirt1, Sirt3, or Sirt5, suggests a generic DHP binding site but with isoform-specific features, indicating that 1,4-DHP derivatives specifically activating further Sirtuin isoforms might exist. Candidate sites would be the allosteric sites harboring halistanol sulfate or bromo-resveratrol, respectively, in Sirt356, 57, or the quercetin site found in Sirt254. These sites comprise helices and loops forming active site entrance and cosubstrate binding region and would allow modulation of Sirtuin activity via conformational changes, but definitive conclusions on binding site and mechanism of our activators will have to await structural characterization of Sirtuin/1,4-DHP complexes.
Our internal library of synthetic 1,4-DHPs (1-24), indicated some critical features for Sirtuin activation and isoform specificity. Depending on the aryl substitution at the C4 position, the benzyl moiety at N1 provided mainly Sirt1 and Sirt5 activation, in the first case with a phenyl (1), 2-furyl (5), 3-thienyl (6) or 3-quinoline (10) ring, in the latter with a 4-biphenyl (2), 1- and 2-naphthyl (3 and 4), and 2-benzo[b]thienyl (9) ring at C4. A weak Sirt2 activation was obtained by introducing a 2-furyl moiety (5) at C4, while C4 substitutions with a phenyl (1), 2-thiazolyl (7), and 3-quinolinyl (10) ring afforded Sirt3 activation. The N1-aroyl series mainly resulted in specific Sirt3, specific Sirt5, or mixed Sirt1, −3, −5 activators. By keeping fixed the phenyl ring at C4 and adding a benzoyl (11) or 2-naphthoyl (22) group at N1, a Sirt1 or mainly Sirt3 activation was observed, especially with 11 which gave 3-fold (Sirt3) and less than 2-fold (Sirt1) increased activities. For specific Sirt5 activation, particularly important was the introduction of the benzoyl group at N1 and differently substituted phenyl rings (2-, 3-, 4chloro- (12-14), 4-methyl- (17) and 2- and 3-methoxyphenyl (18, 19)) at C4, or 1-naphthoyl (21) and 2-pyrazinoyl (23) group at N1 and a phenyl ring at C4. The insertion of the 2-quinoxalinoyl (24) moiety at N1 furnished a mixed Sirt1, −3, −5 activator, more potent toward Sirt5 (2.5-fold activation) than toward Sirt1/3 (about 2-fold activation).
Among the newly synthesized compounds (25-43), the N1-benzoyl derivatives bearing furyl/thienyl rings at C4 (27-30) confirmed their capability to specifically activate Sirt5, particularly the 3-furyl (28) and 3-thienyl (30) analogues. The introduction of the 3,4,5-trimethoxy substitution at the N1-benzyl ring of 1 and 10 (Sirt1/3 activators) produced 25 and 26, which lost their effects towards Sirt3. Instead, inserting the 3,4,5-trimethoxy substituent in the N1-benzoyl derivatives afforded 31 (C4 = phenyl), able to strongly (>4-fold) increase Sirt3-catalyzed deacetylations with high selectivity. Compound 38, carrying a 3-aminophenyl ring at C4, displayed a 2.5-fold increase of Sirt3 activity but lacked selectivity, activating also Sirt1 (>3-fold), Sirt2 (2-fold), and Sirt5 (3.5-fold). Introducing the 3,4,5-trimethoxy substitution in 27, 29, 30 furnished compounds 40-42, which displayed improved Sirt1 but lower Sirt5 activation with respect to the unsubstituted analogues. Taken together, our data show that the 1,4-DHP scaffold enables activation of all Sirtuin isoforms tested, by pan activators as well as isoform-specific derivatives, and provide first structure-activity relationship (Figure 6) insights for positions 1 and 4, illustrating potential for improving isoform specificity and potency.
Figure 6.
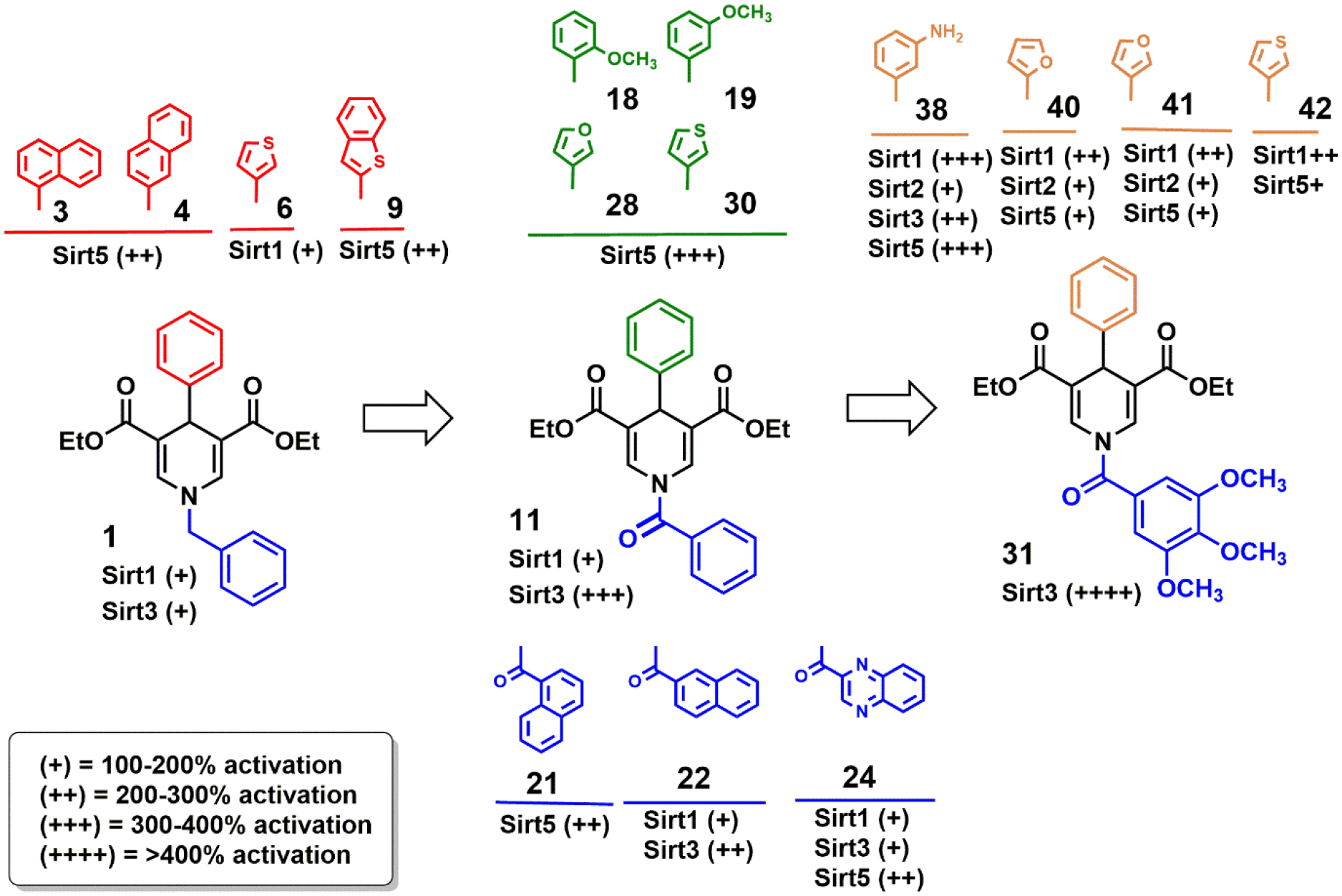
Overview of Sirtuin Isoforms Activation by DHP-based compounds: Structure-Activity Relationship
The biphenyl lignan honokiol, previously reported to weakly activate Sirt318, shows some structural similarity to 4-phenyl-1,4-DHPs. Honokiol might exploit a 1,4-DHP-like mechanism, but it was also reported to stimulate Sirt3 expression and might mainly increase Sirt3 activity through this mechanism.
DHP-based drugs targeting other proteins than Sirtuins have been developed, such as the PDE3 inhibitor Milrinone (a 1,6-DHP) and a variety of calcium channel blockers (1,4-DHPs) such as Nifedipine.58 This compound moiety apparently allows for the development of well acting drugs with good pharmacological properties. Our studies show that the DHP-based compounds enable isoform-specific Sirtuin activation and indeed feature good aqueous solubility (up to ~1 mM) and are active in a cellular environment, and SwissADME (http://www.swissadme.ch/) predicts good ADME (absorption, distribution, metabolism, excretion) properties for them. Fortunately, some of our Sirtuin-activating DHPs were previously tested against PDEs (PDE4) as well as for effects on calcium signaling and yielded no or very weak effects on these targets43, indicating that the pharmacologically favorable DHP fragment enables the development of Sirtuin specific drugs. Although both, our Sirt3-and Sirt5-specific activator scaffolds would require further development improving potency and efficacy, they can serve as first exciting experimental tools with proper controls, and as starting points for drug development. Overall, our findings and compounds are an exciting basis for potent, isoform-specific pharmacological activation of Sirtuin isoforms other than Sirt1 and Sirt6.
Experimental Section
Chemicals
All chemicals were synthesized in the Mai lab or obtained from Sigma (Saint Louis, USA) if not stated differently. HPLC-purified synthetic peptides were from GL Biochem (Shanghai, China). See Table S1 for peptide sequences and the proteins they were derived from.
Protein production and purification
N-terminally his-tagged human Sirtuins were expressed in E. coli using constructs in pET15b (Sirt1) or pET151D/Topo (Sirt3-114-380 and Sirt5-34-302)47, 59. Sirt2-43-356 was expressed from a modified pET19b containing a TEV protease cleavage site and an N-terminal SUMO-tag60. Proteins were expressed and purified as described elsewhere61 47, 59, 60. In brief, proteins were expressed in E. coli, crude extract were cleared by centrifugation, and the proteins were affinity purified using NiNTA beads. After tag removal through proteolytic cleavage and reverse affinity chromatography, Sirtuin proteins were subjected to size exclusion chromatography in 20 mM Tris/HCl pH 7.5 – 8.5 and 150 mM NaCl.
In vitro Sirtuin activity assays
Sirtuin deacylation activities were analyzed by using a coupled enzymatic assay as described in62. For initial modulation screenings, substrate peptide specificity analysis, and compound titrations 1.5 μM Sirtuin was incubated with 50 μM substrate peptide and 100 μM NAD+ in absence or presence of 10 μM, 100 μM or the indicated concentration of 1,4-DHP at a final DMSO concentration of 5%. For kinetic experiments, 1.5 μM Sirtuin was incubated with saturating concentrations of either substrate peptide (0.4 mM) or NAD+ (2 mM), with the second substrate’s concentration being varied in absence or presence of 200 μM 1,4-DHP at a final concentration of 5% DMSO. Potential 1,4-DHP effects on the coupled enzymes GDH and nicotinamidase were tested in control assays containing 50 μM nicotinamide instead of substrate peptide.
For MS-based activity assays, reactions were run with the same components as in the coupled assay but with 50 mM Na-phosphate, pH 7.8, as buffer. To avoid effects of the accumulating product inhibitor nicotinamide, 0.05 μg/μL PNCA was also included. For measuring the activity, product and substrate peptides were then quantified as described in40.
Protein binding analysis
1,4-DHP binding to Sirt3 or Sirt5 in absence or presence of saturating substrate concentrations (0.5 mM peptide or 5 mM NAD+, respectively) at a final DMSO concentration of 10% was analyzed using microscale thermophoresis (MST) on a Monolith NT.115 (NanoTemper Technologies, Munich, Germany). Sirt3 and Sirt5 were fluorescence labelled by incubating the proteins for 2 h at room temperature and protected from light with a twofold molar excess of fluoresceine isothiocyanate (FITC) in 1x PBS pH 7.4. Free FITC was removed using a NAP25 column (GE healthcare). 10 nM of FITC-labelled Sirt3 or Sirt5 were incubated with ligand(s) in 1x PBS for 30 min on ice prior to MST analysis (10% laser power, 25% LED power, 25 °C).
PDC deacylation assays
PDC from porcine heart tissue (Sigma, catalog no. P7032) was concentrated by centrifugation at 135,000 X g for 2 h in 100 mM potassium phosphate, pH 7.5, 0.05% lauryl maltoside, 2.5 mM EDTA, and 30% glycerol as described before51. Desuccinylation reactions were carried out on 7 μg of purified porcine heart PDC, using 2 μg of purified Sirt5 or Sirt5H158Y in a final reaction volume of 20 μL in presence of 25 mM Tris-Cl, pH 8.0, 200 mM NaCl, 5 mM KCl, 1 mM MgCl2, 0.1 % PEG 8000, and 3.125 mM NAD+ at 37 °C for 30 min. During incubation with compounds, tubes were occasionally agitated. Reactions were analyzed by immunoblotting with a succinyl-lysine antibody (PTM Biolabs). After analysis, the membrane was stripped and re-probed with PDHA1 antibody (Abcam) and Sirt5 antibody (Cell Signaling Technology).
MDA-MB-231 cell culture and stable transfection with SIRT3 and SIRT5
The MDA-MB-231 Human Breast Carcinoma cell line (LGC Standards, Milan, Italy) was maintained in 10-cm2 polystyrene dishes (Corning, Oneonta, NY, USA) with RPMI 1640 medium (Mediatech, Herndon, VA, USA), supplemented with 4 mM L-Glutamine, 100 units/mL penicillin, 0.1 mg/mL streptomycin, and 10% heat-inactivated fetal bovine serum. Cells were maintained at 37 °C in a humidified atmosphere of 5% CO2 and 95% air.
MDA-MB-231 cells were stably transfected with a pcDNA3.1 expression vector encoding for human SIRT3-Flag or for SIRT5-Flag (Addgene, Cambridge, MA, USA) using TurboFectin 8.0 (Origene Technologies, Rockville, MD, USA) according to the manufacturer’s guidelines. Briefly, Turbofectin reagents were mixed with serum-free RPMI at room temperature for 5 min, and plasmid DNA was then added, followed by an incubation at room temperature for 30 min. The mixtures were then added to the cells and selection of stable clones was started 24 h later with the addition of 500 μg/mL of Geneticin. Selected clones were isolated and grown individually. SIRT3 or SIRT5 expression was confirmed by western blotting analysis.
Lentiviral transduction
Mission TRC short hairpin RNA (shRNA) lentiviral transduction particles expressing shRNA targeting SIRT3 and lentiviral negative control particles were purchased from Sigma-Aldrich. Stably transduced clones were generated according to the manufacturer’s instructions. Cells were seeded on a 24-well plate and the following day cells were infected. After 24 h medium was changed with fresh RPMI. Selection of stable clones was started 24 h later with the addition of 3 mg/mL of puromycin. SIRT3 silencing was confirmed by western blotting analysis.
Protein extraction, Western Blotting assay (MDA-MB-231 cells)
For whole cell lysates, cells (1 × 106) were pelleted at 1200 rpm (5 min at 4 °C) and lysed with Lysis buffer (50 mM TrisHCl pH 7.4, 5 mM EDTA, 250 mM NaCl, 0.1% Triton, 50 mM NaF, 0.1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl-fluoride and 1X protease inhibitor mixture (P8340, Sigma). After 30 min on ice, lysates were cleared by centrifugation (10 min at 4 °C). Protein concentrations of supernatents were determined using the Bradford assay (Bio-Rad, Hercules, CA, USA). Fifty micrograms of protein for each sample were electrophoresed on SDS-polyacrylamide gels, followed by blotting onto PVDF membranes. After blocking with 5% milk, membranes were incubated with the primary antibody overnight. Finally, proteins expression was analyzed by staining with the appropriate secondary horseradish peroxidase-labeled antibody for 1 h followed by enhanced chemiluminescence detection. The following primary antibodies were used: MnSOD (anti-rabbit; Abcam. Cambridge, UK), MnSOD-acK68 (anti-rabbit; Abcam). Each Western Blot was repeated in triplicate.
GDH immunoprecipitation assay
Proteins were extracted from MDA-MB-231 cells as described above. Protein concentrations were determined using the Bradford assay, and 500 μg lysate protein was brought to a final volume of 1 mL with PBS. Lysates were precleared with 1 μg of appropriate control IgG (Santa Cruz Biotechnology, sc-2763) and 20 μg of protein A/G PLUS-agarose (Santa Cruz Biotechnology, sc-2003) and kept on a rotator for 1 h at 4 °C. Lysates were centrifuged (500 ×g for 5 min at 4 °C) and 2 μg of GDH antibody (Santa Cruz Biotechnology, sc-160382) or corresponding IgG was added to the precleared lysates and kept on ice for 3 to 5 h. Following incubation, 30 μL of protein A/G PLUS-agarose was added to each tube and kept on a rotator overnight at 4 °C. Lysates were then centrifuged (500 ×g for 5 min at 4 °C). The pellet fractions were washed 4 times with PBS and then resuspended in 20 mL of loading buffer. Samples were electrophoresed on an 8% SDS-polyacrylamide gel and immunoblotted with an Acetylated-Lysine Mouse mAb (Ac-K-103) (Cell Signaling) antibody.
Protein extraction, Western Blotting assay (PDAC cells)
For endogenous IP, control and SIRT5-knockdown or knockout (KPCS; using a KPC cell line as a control) PDAC cells were lysed in Pierce IP Lysis Buffer. The IP-grade GOT1 antibody (14886–1-AP, Thermo Fisher) was conjugated to Pierce Protein A/G Magnetic Beads at 4 °C for 4 h before IP experiment. Cell lysates were pre-cleared by Pierce Protein A/G Magnetic Beads and then incubated with GOT1 antibody-Magnetic Beads mixture overnight at 4 °C. The immune complexes were washed with IP Lysis Buffer for five times and subsequently boiled with 2X sample buffer for 10 min, the GOT1-IP samples were then resolved by SDS-PAGE, the endogenous acetylation level of GOT1 was determined by western blotting using an anti-Acetylation antibody (CST, 9441S).
Cellular SIRT3 deacetylase activity assay
SIRT3 deacetylase activity was determined using the SIRT3 Fluorimetric Activity Assay (ENZO Life Technologies Farmingdale, NY) according to manufacturer’s instructions. Briefly, MDA-MB-231 WT, SIRT3+/+ and SIRT3−/− cells were counted and plated on 10-cm2 polystyrene dishes in complete medium and incubated overnight at 37 °C and 5% CO2. Then the medium was substituted with R6504 at pH 7.4 or 6.8 for 2 h. Afterward, mitochondrial extracts of WT, SIRT3+/+ and SIRT3−/− cells (40 μg) were incubated with the “Fluor de Lys substrate” buffer at 37 °C for 40 min followed by incubation with “Fluor de Lys developer” at 37 °C for 10 min. After excitation at 360 nm, light emission was detected at 460 nm using an Infinite 200 microplate fluorometer (Tecan, Mannedorf, Switzerland). The fluorescence intensity of the assay buffer was subtracted from each experimental sample.
GDH Assay
GDH activity in control or treated cells was measured using a GDH activity assay kit (MAK099, Sigma Aldrich) following manufacturer’s instructions. Briefly, 106 cells were lysed in 40 μL of GDH assay buffer and kept 10 min in ice. Afterward, lysates were centrifuged and 10 μL supernatant was added to 40 μL of GDH buffer and 100 μL of a mix containing GDH assay buffer, developer and glutamate. The whole mix was transferred into a 96 well plate and incubated at 37 °C for 3 min. Finally, absorbance was read at 450 nm using a Glomax multi detection system (Promega, Milan, Italy). The absorbance of the assay buffer was subtracted from each experimental sample.
GLS Assay
GLS activity in control or treated cells was measured using a GLS activity assay kit (LSBio, Seattle, WA, USA) following manufacturer’s instructions. Briefly, 106 cells were lysed by freeze and thawing in PBS 4 times. Lysates were centrifuged, and 10 μL supernatant was added to 90 μL of sample diluent and incubated for 2 h. After 1 h incubation with detection reagents A and B, the TMB substrate was added and absorbance read at 450 nm using a Glomax multi detection system (Promega, Milan, Italy). The absorbance of the assay buffer was subtracted from each experimental sample.
Ammonia level assay
Ammonia levels in cell-conditioned medium were determined by using the ammonia assay kit (Sigma-Aldrich) following the manufacturer’s protocol. Briefly, cells were grown for 4 and 24 h. 100 μL of cell medium was mixed with 1 mL of ammonia assay reagent and incubated for 5 min at room temperature. Afterwards, 10 μL of Glutamate dehydrogenase solution was added. After 5 min, absorbance was read at 340 nm in a Glomax multi detection system (Promega). For all measurements ammonia production in the absence of cells was subtracted.
Statistical analysis of assays with MDA-MB-231 and PDAC cells
The results are expressed as means ± S.D. and 95% confidence intervals (95% CI). Student’s t-test was used to determine any significant differences between groups before and after treatment. Significance was set as * p < 0.05, ** p <0.01, and *** p < 0.001. GraphPad Prism 5 and SPSS statistical software package (SPSS Inc., v13.0.1 for Windows, Chicago, IL, USA) were used for all statistical calculations.
Tests on wild-type and Sirt3 knock-out MEFs
To assess mitochondrial mass, cells were challenged with 25 μM 10 for 24 h followed by staining with 10 nM MitoTracker Deep Red (MT-DR) for 30 min at 37 °C. Cells were then washed with PBS, pelleted, and resuspended in PBS with 2% FBS before FACS analysis using FACS Analyzer Canto (10000 cells).
Chemistry
Melting points were determined on a Buchi 530 melting point apparatus and are uncorrected. 1H NMR and 13C NMR spectra were recorded at 400 MHz and 100 MHz, respectively, on a Bruker AC 400 spectrometer; chemical shifts are reported in δ (ppm) units relative to the internal reference tetramethylsilane (Me4Si). EIMS spectra were recorded with a Fisons Trio 1000 spectrometer; only molecular ions (M+) and base peaks are given. All compounds were routinely checked by TLC and 1H NMR. TLC was performed on aluminum-backed silica gel plates (Merck DC, AlufolienKieselgel 60 F254) with spots visualized by UV light. All solvents were reagent grade and, when necessary, were purified and dried by standard methods. Concentration of solutions after reactions and extractions involved the use of a rotary evaporator operating at reduced pressure of ca. 20 Torr. Organic solutions were dried over anhydrous sodium sulfate. Elemental analysis has been used to determine the purity of the described compounds, which is >95%. Analytical results are within ± 0.40% of the theoretical values. All chemicals were purchased from Sigma Aldrich, Milan (Italy), or from Alfa Aesar, Karlsruhe (Germany), and were of the highest purity.
General synthetic procedures and physical and chemical data of the unknown intermediates 46, 47, 49, 52, 55 and of the final compounds 25-43 are reported below (and summarized in Tables S2 and S3), and the elemental analyses for 25-43 are reported in Table 1.
General Procedure A for the Synthesis of Compounds 25, 26.
Ethyl propiolate (18.84 mmol, 1.9 mL), the appropriate aldehyde (benzaldehyde for 25 or quinoline-3-carboxaldehyde for 26) (9.42 mmol, 0.96 mL) and 3,4,5-trimethoxybenzylamine (9.42 mmol, 1.60 mL) in glacial acetic acid (0.5 mL) were heated at 80 °C for 30 min. After cooling, the mixture was poured into water (20 mL) and stirred for 1 h. The solid product was filtered and washed with diethyl ether (3 × 30 mL) to give the pure 25 or 26.
Diethyl 4-Phenyl-1-(3,4,5-trimethoxybenzyl)-1,4-dihydropyridine-3,5-dicarboxylate (25, MC4001, UBCS0357).
Compound 25 was prepared according to general procedure A. Yellow solid; m.p.: 129–131 °C; recrystallization solvent: cyclohexane/toluene; yield: 30 %. 1H NMR (400 MHz, CDCl3) δ 1.11 (t, 6H, −OCH2CH3), 3.79 (s, 3H, −OCH3), 3.81 (s, 6H, −OCH3), 3.95–4.06 (m, 4H, −OCH2CH3), 4.45 (s, 2H, ), 4.85 (s, 1H, ArCH-), 6.41 (s, 2H, aromatic protons), 7.05–7.15 (m, 3H, aromatic protons), 7.21 (s, 2H, dihydropyridine protons), 7.23–7.25 (m, 3H, aromatic protons) ppm. 13C NMR (100 MHz, CDCl3) δ 14.2 (2C), 39.1, 56.2 (2C), 56.8, 61.2, 62.3 (2C), 106.0 (2C), 107.4 (2C), 127.4, 127.8 (2C), 128.4, 129.2, 132.7, 137.8, 139.3 (2C), 140.4, 152.7, 153.1, 166.4 (2C). MS (EI): m/z [M]+: 481.2101.
Diethyl 4-(Quinolin-3-yl)-1-(3,4,5-trimethoxybenzyl)-1,4-dihydropyridine-3,5-dicarboxylate (26, MC4029, UBCS0358).
Compound 26 was prepared according to general procedure A. Off-white solid; m.p.: 158–160 °C; recrystallization solvent: toluene; yield: 20%. 1H NMR (400 MHz, CDCl3) δ 1.19 (t, 6H, −OCH2CH3), 3.88 (s, 6H, −OCH3), 3.91 (s, 3H, −OCH3), 4.04–4.12 (m, 4H, −OCH2CH3), 4.59 (s, 2H, ), 5.15 (s, 1H, ArCH-), 6.53 (s, 2H, aromatic protons), 7.38 (s, 2H, dihydropyridine protons), 7.49–7.53 (m, 1H, quinoline proton), 7.65–7.70 (m, 2H, quinoline protons), 7.95 (s, 1H, quinoline proton), 8.06–8.09 (m, 1H, quinoline proton), 8.97 (d, 1H, quinoline proton) ppm. 13C NMR (100 MHz, CDCl3) δ 14.2 (2C), 36.7, 56.1 (2C), 56.4, 61.2, 62.1 (2C), 105.6 (2C), 107.8 (2C), 127.9, 128.3, 128.7, 129.0, 129.8, 131.2, 132.9, 135.1, 138.2, 139.1 (2C), 146.9, 147.8, 152.6, 152.8, 167.5 (2C). MS (EI): m/z [M]+: 532.2210.
General Procedure B for the Synthesis of the Final Compounds 27–37, 39–43.
The appropriate 1,4-dihydropyridines (44–55) (0.17 mmol), the suitable acyl chloride (0.26 mmol), and triethylamine (0.85 mmol, 0.12 mL) were stirred in dry dichloromethane (7 mL) at room temperature overnight. Afterwards, the reaction was quenched with water (50 mL) and stirred for additional 10 min at room temperature and then extracted with ethyl acetate (4 × 30 mL). The organic layer was washed with brine, dried with sodium sulfate, and evaporated at reduced pressure. The crude was purified via column chromatography on silica using ethyl acetate: n-hexane 1:3, 1:4 or 1:5 as eluting system, and the resulting compounds 27-37, 39-43 were then recrystallized from the appropriate solvent.
Diethyl 1-Benzoyl-4-(furan-2-yl)-1,4-dihydropyridine-3,5-dicarboxylate (27, MC3193, UBCS0347).
Compound 27 was prepared according to general procedure B. Off-white solid; eluting system: ethylacetate: n-hexane 1:3; m.p.: 63–65 °C; recrystallization solvent: cyclohexane; yield: 89%. 1H NMR (400 MHz, CDCl3) δ 1.27 (t, 6H, -OCH2CH3), 4.15–4.23 (m, 4H, −OCH2CH3), 5.15 (s, 1H, ArCH-), 6.21 (d, 1H, furane proton), 6.31–6.32 (m, 1H, furane proton), 7.32 (d, 1H, furane proton), 7.53–7.57 (m, 2H, aromatic protons), 7.62–7.70 (m, 3H, aromatic protons), 8.11 (s, 2H, dihydropyridine protons) ppm. 13C NMR (100 MHz, CDCl3) δ 14.2 (2C), 41.2, 61.6 (2C), 109.0, 110.3, 111.6 (2C), 128.1 (2C), 129.2 (2C), 131.8, 132.6 (2C), 133.7, 142.5, 152.1, 166.4, 167.8 (2C). MS (EI): m/z [M]+: 395.1369.
Diethyl 1-Benzoyl-4-(furan-3-yl)-1,4-dihydropyridine-3,5-dicarboxylate (28, MC3224, UBCS0349).
Compound 28 was prepared according to general procedure B. Colourless solid; eluting system: ethylacetate: n-hexane 1:3; m.p.: 95–97 °C; recrystallization solvent: cyclohexane; yield: 90%. 1H NMR (400 MHz, CDCl3) δ 1.27 (t, 6H, −OCH2CH3), 4.16–4.24 (m, 4H, −OCH2CH3), 4.95 (s, 1H, ArCH-), 6.36 (s, 1H, furane proton), 7.34–7.35 (m, 2H, furane protons), 7.53–7.57 (m, 2H, aromatic protons), 7.62–7.65 (m, 3H, aromatic protons), 8.07 (s, 2H, dihydropyridine protons) ppm. 13C NMR (100 MHz, CDCl3) δ 14.2 (2C), 32.8, 62.3 (2C), 109.4, 111.0 (2C), 128.0, 128.4 (2C), 128.8, 129.3, 131.8, 134.5, 137.1 (2C), 140.0, 142.8, 166.2, 166.5 (2C). MS (EI): m/z [M]+: 395.1369.
Diethyl 1-Benzoyl-4-(thiophen-2-yl)-1,4-dihydropyridine-3,5-dicarboxylate (29, MC3191, UBCS0348).
Compound 29 was prepared according to general procedure B. Off-white solid; eluting system: ethyl acetate: n-hexane 1:3; m.p.: 108–110 °C; recrystallization solvent: cyclohexane; yield: 87%. 1H NMR (400 MHz, CDCl3) δ 1.27 (t, 6H, −OCH2CH3), 4.15–4.25 (m, 4H, −OCH2CH3), 5.33 (s, 1H, ArCH-), 6.93–6.96 (m, 1H, thiophene proton), 6.99–7.00 (m, 1H, thiophene proton), 7.18 (d, 1H, thiophene proton), 7.55 (t, 2H, aromatic protons), 7.63–7.69 (m, 3H, aromatic protons), 8.09 (s, 2H, dihydropyridine protons) ppm. 13C NMR (100 MHz, CDCl3) δ 14.2 (2C), 42.4, 61.9 (2C), 112.2 (2C), 125.6, 126.4, 127.3, 128.1 (2C), 128.8, 129.5, 131.7, 133.3 (2C), 136.2, 140.2, 166.2, 166.9, 167.0. MS (EI): m/z [M]+: 411.1140.
Diethyl 1-Benzoyl-4-(thiophen-3-yl)-1,4-dihydropyridine-3,5-dicarboxylate (30, MC3215, UBCS0350).
Compound 30 was prepared according to general procedure B. Colourless solid; eluting system: ethylacetate: n-hexane 1:3; m.p.: 92–94 °C; recrystallization solvent: cyclohexane; yield: 84%. 1H NMR (400 MHz, CDCl3) δ 1.15 (t, 6H, −OCH2CH3), 4.01–4.16 (m, 4H, −OCH2CH3), 5.03 (s, 1H, ArCH-), 6.98–6.99 (m, 1H, thiophene proton), 7.04–7.04 (m, 1H, thiophene proton), 7.14–7.16 (m, 1H, thiophene proton), 7.44–7.48 (m, 2H, aromatic protons), 7.53–7.57 (m, 3H, aromatic protons), 8.00 (s, 2H, dihydropyridine protons) ppm. 13C NMR (100 MHz, CDCl3) δ 14.2 (2C), 38.0, 62.2 (2C), 110.7, 111.3, 119.4, 126.4, 126.2, 128.1 (2C), 128.9 (2C), 132.1, 134.2, 134.3, 134.7, 139.6, 165.9.MS(EI): m/z [M]+: 411.1140.
Diethyl 4-Phenyl-1-(3,4,5-trimethoxybenzoyl)-1,4-dihydropyridine-3,5-dicarboxylate (31, MC2789, UBCS0131).
Compound 31 was prepared according to general procedure B. Colourless solid; eluting system: ethylacetate: n-hexane 1:5; m.p.: 174–176 °C; recrystallization solvent: toluene/acetonitrile; yield: 35%. 1H NMR (400 MHz, CDCl3) δ 1.21 (t, 6H, −OCH2CH3), 3.91 (s, 6H, −OCH3), 3.96 (s, 3H, −OCH3), 4.10–4.19 (m, 4H, −OCH2CH3), 4.98 (s, 1H, ArCH-), 6.92 (s, 2H, aromatic protons), 7.23–7.25 (m, 1H, aromatic proton), 7.31–7.38 (m, 4H, aromatic protons), 8.15 (s, 2H, dihydropyridine protons) ppm. 13C NMR (100 MHz, CDCl3) δ 14.2 (2C), 38.9, 55.8 (2C), 61.2, 62.3 (2C), 106.5 (2C), 110.7 (2C), 127.4 (3C), 127.9 (2C), 129.6, 135.4 (2C), 140.3, 143.6, 151.7, 152.0, 165.2, 166.9 (2C). MS (EI): m/z [M]+: 459.1893.
Diethyl 4-(3-Fluorophenyl)-1-(3,4,5-trimethoxybenzoyl)-1,4-dihydropyridine-3,5-dicarboxylate (32, MC4071, UBCS0361).
Compound 32 was prepared according to general procedure B. Colourless solid; eluting system: ethyl acetate: n-hexane 1:4; m.p.: 161–163 °C; recrystallization solvent: toluene; yield: 44%. 1H NMR (400 MHz, CDCl3) δ 1.23 (t, 6H, −OCH2CH3), 3.92 (s, 6H, −OCH3), 3.97 (s, 3H, −OCH3), 4.14–4.23 (m, 4H, −OCH2CH3), 4.99 (s, 1H, ArCH-), 6.91 (s, 2H, aromatic protons), 6.93–6.95 (m, 1H, aromatic proton), 7.04–7.06 (m, 1H, aromatic proton), 7.16–7.20 (m, 3H, aromatic protons), 7.16 (d, 1H, aromatic proton), 7.25–7.30 (m, 1H, aromatic proton), 8.15 (s, 2H, dihydropyridine protons) ppm. 13C NMR (100 MHz, CDCl3-d) δ 14.2 (2C), 38.7, 56.3 (2C), 61.1, 62.3 (2C), 105.8, 106.3, 110.8, 110.9 114.1, 113.8, 124.7, 129.6, 130.0, 135.4 (2C), 139.3, 142.6, 151.7 (2C), 163.7, 165.2, 166.7 (2C). MS (EI): m/z [M]+: 513.1799.
Diethyl 4-(3-Chlorophenyl)-1-(3,4,5-trimethoxybenzoyl)-1,4-dihydropyridine-3,5-dicarboxylate (33, MC4070, UBCS0362).
Compound 33 was prepared according to general procedure B. Colourless solid; eluting system: ethylacetate: n-hexane 1:4; m.p.: 159–161 °C; recrystallization solvent: toluene; yield: 46%. 1H NMR (400 MHz, CDCl3) δ 1.24 (t, 6H, −OCH2CH3), 3.92 (s, 6H, −OCH3), 3.97 (s, 3H, −OCH3), 4.12–4.21 (m, 4H, −OCH2CH3), 4.96 (s, 1H, ArCH-), 6.92 (s, 2H, aromatic protons), 7.21–7.25 (m, 2H, aromatic protons), 7.27–7.30 (m, 2H, aromatic protons), 8.15 (s, 2H, dihydropyridine protons) ppm. 13C NMR (100 MHz, CDCl3) δ 14.2 (2C), 38.8, 55.4 (2C), 60.8, 62.3 (2C), 105.9 (2C), 110.6, 110.7, 127.2, 128.3, 128.9, 129.1, 129.6, 133.5, 136.4 (2C), 140.1, 143.0, 151.7 (2C), 164.8, 166.2, 166.3. MS (EI): m/z [M]+: 529.1503.
Diethyl 4-(4-Chlorophenyl)-1-(3,4,5-trimethoxybenzoyl)-1,4-dihydropyridine-3,5-dicarboxylate (34, MC4069, UBCS0363).
Compound 34 was prepared according to general procedure B. Colourless solid; eluting system: ethylacetate: n-hexane 1:4; m.p.: 216–218 °C; recrystallization solvent: acetonitrile/methanol; yield: 52%. 1H NMR (400 MHz, CDCl3) δ 1.22 (t, 6H, −OCH2CH3), 3.91 (s, 6H, −OCH3), 3.97 (s, 3H, −OCH3), 4.10–4.21 (m, 4H, −OCH2CH3), 4.95 (s, 1H, ArCH-), 6.90 (s, 2H, aromatic protons), 7.27 (d, 2H, aromatic protons), 7.31 (d, 2H, aromatic protons), 8.15 (s, 2H, dihydropyridine protons) ppm. 13C NMR (100 MHz, CDCl3) δ 14.2 (2C), 38.5, 56.2 (2C), 60.8, 62.4 (2C), 105.6 (2C), 110.7 (2C), 126.7 (2C), 129.4, 129.5, 129.6, 132.5, 134.4 (2C), 138.7, 143.0, 150.9 (2C), 165.2, 167.2. (2C). MS (EI): m/z [M]+:529.1503.
Diethyl 4-(3-Bromophenyl)-1-(3,4,5-trimethoxybenzoyl)-1,4-dihydropyridine-3,5-dicarboxylate (35, MC4077, UBCS0364).
Compound 35 was prepared according to general procedure B. Colourless solid; eluting system: ethylacetate: n-hexane 1:5; m.p.: 96–98 °C; recrystallization solvent: cyclohexane; yield: 49%. 1H NMR (400 MHz, CDCl3) δ 1.26 (t, 6H, −OCH2CH3), 3.93 (s, 6H, −OCH3), 3.97 (s, 3H, −OCH3), 4.11–4.22 (m, 4H, −OCH2CH3), 4.95 (s, 1H, ArCH-), 6.92 (s, 2H, aromatic protons), 7.19 (t, 1H, aromatic proton), 7.35 (t, 1H, aromatic proton), 7.45 (s, 1H, aromatic proton), 8.15 (s, 2H, dihydropyridine protons) ppm. 13C NMR (100 MHz, CDCl3) δ 14.2 (2C), 38.6, 56.3 (2C), 60.8, 62.3 (2C), 104.9 (2C), 111.1 (2C), 122.4, 126.7, 129.6, 129.9, 130.1, 131.8, 134.4 (2C), 141.1, 143.1, 152.0 (2C), 165.2, 166.3 (2C). MS (EI): m/z [M]+: 573.0998.
Diethyl 4-(3-Tolyl)-1-(3,4,5-trimethoxybenzoyl)-1,4-dihydropyridine-3,5-dicarboxylate (36, MC4062, UBCS0360).
Compound 36 was prepared according to general procedure B. Colourless solid; eluting system: ethylacetate: n-hexane 1:4; m.p.: 141–143 °C; recrystallization solvent: cyclohexane/toluene; yield: 38%. 1H NMR (400 MHz, CDCl3) δ 1.16 (t, 6H, −OCH2CH3), 2.34 (s, 3H, −CH3), 3.91 (s, 6H, −OCH3), 3.96 (s, 3H, −OCH3), 4.12–4.18 (m, 4H, −OCH2CH3), 4.93 (s, 1H, ArCH-), 6.92 (s, 2H, aromatic protons), 7.03–7.05 (m, 2H, aromatic protons), 7.16–7.20 (m, 3H, aromatic protons), 8.14 (s, 2H, dihydropyridine protons) ppm. 13C NMR (100 MHz, CDCl3) δ 14.2 (2C), 20.8, 38.7, 56.3 (2C), 60.8, 62.3 (2C), 105.9 (2C), 110.8 (2C), 124.7, 126.6, 128.4 (2C), 129.6, 135.3, 136.2, 138.4, 138.8, 143.1, 152.3 (2C), 165.2, 166.9, 168.0. MS (EI): m/z [M]+: 509.2050.
Diethyl 4-(3-Nitrophenyl)-1-(3,4,5-trimethoxybenzoyl)-1,4-dihydropyridine-3,5-dicarboxylate (37, MC4075, UBCS0365).
Compound 37 was prepared according to general procedure B. Colourless solid; eluting system: ethylacetate: n-hexane 1:5; m.p.: 185–187 °C; recrystallization solvent: toluene/acetonitrile; yield: 42%. 1H NMR (400 MHz, CDCl3) δ 1.22 (t, 6H, −OCH2CH3), 3.94 (s, 6H, −OCH3), 3.98 (s, 3H, −OCH3), 4.08–4.22 (m, 4H, −OCH2CH3), 5.11 (s, 1H, ArCH-), 6.95 (s, 2H, aromatic protons), 7.50 (t, 1H, aromatic proton), 7.80 (d, 1H, aromatic proton), 8.11 (d, 1H, aromatic proton), 8.18–8.20 (m, 3H, aromatic and dihydropyridine protons) ppm. 13C NMR (100 MHz, CDCl3) δ 14.2 (2C), 37.8, 55.9 (2C), 60.8, 62.4 (2C), 105.9 (2C), 110.6, 110.9, 121.5, 122.1, 130.0, 130.4, 131.8, 135.4 (2C), 140.3, 143.0, 148.3, 152.1 (2C), 164.8, 167.5 (2C). MS (EI): m/z [M]+: 540.1744.
Diethyl 4-(4-((4-Methylpiperazin-1-yl)methyl)phenyl)-1-(3,4,5-trimethoxybenzoyl)-1,4-dihydropyridine-3,5-dicarboxylate Dihydrochloride Salt (39, MC4003, UBCS0355).
Compound 39 was prepared according to general procedure B. Colourless salt (precipitated from THF with 0.3 mL 4N HCl in dioxane); m.p.: >250 °C; recrystallization solvent: methanol; yield: 88%. 1H NMR (400 MHz, DMSO-d6) δ 1.11 (t, 6H, −OCH2CH3), 2.05 (s, 3H, −CH3), 1.58 (s, 8H, piperazine protons), 3.77 (s, 3H, −OCH3), 3.83 (s, 6H, −OCH3), 3.99–4.11 (m, 4H, −OCH2CH3), 4.77 (s, 1H, ArCH-), 5.02 (s, 2H, ), 7.06 (s, 2H, aromatic protons), 7.28–7.34 (m, 4H, aromatic protons), 7.94 (s, 2H, dihydropyridine protons) ppm. 13C NMR (100 MHz, DMSO-d6) δ 14.2 (2C), 38.9, 44.6, 49.3 (2C), 52.3 (2C), 56.2 (2C), 57.4, 60.8, 61.8 (2C), 106.7 (2C), 109.9 (2C), 127.1 (2C), 128.4, 129.2 (2C), 132.9 (2C), 137.3, 138.3, 143.1, 151.7 (2C), 164.8, 168.2 (2C). MS (EI): m/z [M]+: 679.2427.
Diethyl 4-(Furan-2-yl)-1-(3,4,5-trimethoxybenzoyl)-1,4-dihydropyridine-3,5-dicarboxylate (40, MC4068, UBCS0367).
Compound 40 was prepared according to general procedure B. Colourless solid; eluting system: ethylacetate: n-hexane 1:3; m.p.: 123–125 °C; recrystallization solvent: cyclohexane/toluene; yield: 49%. 1H NMR (400 MHz, CDCl3) δ 1.18 (t, 6H, −OCH2CH3), 3.82 (s, 6H, −OCH3), 3.87 (s, 3H, −OCH3), 4.06–4.19 (m, 4H, −OCH2CH3), 5.08 (s, 1H, ArCH-), 6.13 (d, 1H, furane proton), 6.22–6.24 (m, 1H, furane proton), 6.85 (s, 2H, aromatic protons), 7.19 (d, 1H, furane proton), 8.04 (s, 2H, dihydropyridine protons) ppm. 13C NMR (100 MHz, CDCl3) δ 14.2 (2C), 41.0, 56.3 (2C), 61.2, 61.9 (2C), 106.2 (2C), 109.2, 110.3, 111.8 (2C), 130.2, 132.6 (2C), 142.5, 143.7, 151.6 (2C), 152.0, 164.7, 167.3 (2C). MS (EI): m/z [M]+: 485.1686.
Diethyl 4-(Furan-3-yl)-1-(3,4,5-trimethoxybenzoyl)-1,4-dihydropyridine-3,5-dicarboxylate (41, MC4065, UBCS0368).
Compound 41 was prepared according to general procedure B. Off-white solid; eluting system: ethylacetate: n-hexane 1:3; m.p.: 183–185 °C; recrystallization solvent: toluene/acetonitrile; yield: 47%. 1H NMR (400 MHz, CDCl3) δ 1.19 (t, 6H, −OCH2CH3), 3.81 (s, 6H, −OCH3), 3.87 (s, 3H, −OCH3), 4.11–4.20 (m, 4H, −OCH2CH3), 4.87 (s, 1H, ArCH-), 6.25 (s, 1H, furane proton), 6.78 (s, 2H, aromatic protons), 7.23–7.25 (m, 2H, furane protons), 8.00 (s, 2H, dihydropyridine protons) ppm. 13C NMR (100 MHz, CDCl3) δ 14.2 (2C), 33.5, 55.9 (2C), 61.2, 62.5 (2C), 106.2 (2C), 108.7, 111.3 (2C), 127.5, 129.6, 137.ß (2C), 139.9, 142.9, 143.5, 151.6 (2C), 164.8, 167.1 (2C). MS (EI): m/z [M]+: 485.1686.
Diethyl 4-(Thiophen-3-yl)-1-(3,4,5-trimethoxybenzoyl)-1,4-dihydropyridine-3,5-dicarboxylate (42, MC4076, UBCS0369).
Compound 42 was prepared according to general procedure B. Off-white solid; eluting system: ethylacetate: n-hexane 1:4; m.p.: 159–161 °C; recrystallization solvent: toluene; yield: 52%.1H NMR (400 MHz, CDCl3) δ 1.25 (t, 6H, −OCH2CH3), 3.90 (s, 6H, −OCH3), 3.96 (s, 3H, −OCH3), 4.15–4.24 (m, 4H, −OCH2CH3), 5.14 (s, 1H, ArCH-), 6.87 (s, 2H, aromatic protons), 7.06–7.07 (m, 1H, thiophene proton), 7.12–7.12 (m, 1H, thiophene proton), 7.23–7.25 (m, 1H, thiophene proton), 8.11 (s, 2H, dihydropyridine protons) ppm. 13C NMR (100 MHz, CDCl3) δ 14.2 (2C), 37.6, 55.6 (2C), 60.4, 62.3 (2C), 105.7 (2C), 110.6 (2C), 119.4, 125.8, 126.5, 130.2, 135.8 (2C), 139.5, 142.9, 152.0, 152.3, 165.2, 166.8. (2C). MS (EI): m/z [M]+: 501.1457.
Diethyl (E)-1-(4-(But-2-enoyloxy)-3,5-dimethoxybenzoyl)-4-phenyl-1,4-dihydropyridine-3,5-dicarboxylate (43, MC4054, UBCS0359).
Compound 43 was prepared according to general procedure B. Off-white solid; eluting system: ethylacetate: n-hexane 1:4; m.p.: 160–162 °C; recrystallization solvent: toluene; yield: 34%. 1H NMR (400 MHz, CDCl3) δ 1.22 (t, 6H, −OCH2CH3), 2.01 (d, 3H, −CH=CHCH3), 3.87 (s, 6H, −OCH3), 4.10–4.22 (m, 4H, −OCH2CH3), 4.98 (s, 1H, ArCH-), 6.15 (d, 1H, −CH=CHCH3), 7.23–7.32 (m, 6H, aromatic protons and, −CH=CHCH3), 7.36 (d, 2H, aromatic protons), 8.15 (s, 2H, dihydropyridine protons) ppm. 13C NMR (100 MHz, CDCl3) δ 14.2 (2C), 18.2, 38.9, 55.7 (2C), 61.5 (2C), 106.2 (2C), 111.2 (2C), 121.4, 127.4 (2C), 127.9, 127.9 (2C), 130.4, 133.8, 135.4 (2C), 139.6, 145.9, 153.1 (2C), 163.9, 164.7, 166.9 (2C). MS (EI): m/z [M]+: 549.1999.
Procedure for the Synthesis of Diethyl 4-(3-Aminophenyl)-1-(3,4,5-trimethoxybenzoyl)-1,4-dihydropyridine-3,5-dicarboxylate (38, MC4079, UBCS0366).
37% Hydrochloric acid solution (0.15 mL) was slowly added at 0 °C to a mixture of 37 (0.4 mmol, 0.215 g) and stannous chloride dihydrate (2.03 mmol, 0.46 g) in ethanol (5 mL). The reaction was then kept at room temperature overnight. Afterwards, the reaction was quenched at room temperature by 2 N sodium carbonate solution reaching pH = 7 (approx. 20 mL) and the mixture was extracted with ethyl acetate (3 × 30 mL), washed with brine (3 × 30 mL), then dried with anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude solid was recrystallized by toluene to give pure 38 as an off-white solid. M.p.: 160–162 °C; recrystallization solvent: toluene; yield: 70%. 1H NMR (400 MHz, CDCl3) δ 1.24 (t, 6H, −OCH2CH3), 3.63 (bs, 2H, −NH2), 3.91 (s, 6H, −OCH3), 3.96 (s, 3H, -OCH3), 4.12–4.23 (m, 4H, −OCH2CH3), 4.89 (s, 1H, ArCH-), 6.55 (d, 1H, aromatic proton), 6.71–6.75 (m, 2H, aromatic protons), 6.91 (s, 2H, aromatic protons), 7.08 (t, 1H, aromatic proton), 8.13 (s, 2H, dihydropyridine protons) ppm. 13C NMR (100 MHz, CDCl3-d) δ 14.2 (2C), 38.9, 56.0 (2C), 61.2, 62.3 (2C), 105.9 (2C), 111.3 (2C), 114.4, 115.1, 120.9, 129.2, 130.6, 135.4 (2C), 140.0, 142.7, 148.0, 151.7 (2C), 164.9, 166.8 (2C). MS (EI): m/z [M]+: 510.2002.
General Procedure for the Synthesis of Intermediate Compounds 44–55.
Ethyl propiolate (17.84 mmol, 1.82 mL), the appropriate aldehyde (8.92 mmol) and ammonium acetate (8.92 mmol, 0.7 g) were heated to 80 °C for 5 h in glacial acetic acid (5 mL). Back to room temperature, the reaction mixture was quenched with water (50 mL), stirred for additional 10 min at room temperature and then extracted with ethyl acetate (3 × 30 mL). The organic layer was washed with brine, dried with sodium sulfate and evaporated under vacuum. The crude product was purified via column chromatography on silica gel using ethyl acetate/n-hexane 1:2 as eluting system to afford the required diethyl 4-aryl-1,4-dihydropyridine-3,5-dicarboxylates. Compounds 4444, 4843, 5043, 45, 5143, 45, 5343, and 5445 were already described in the literature.
Diethyl 4-(Furan-3-yl)-1,4-dihydropyridine-3,5-dicarboxylate (45).
M.p.: 134–136 °C; recrystallization solvent: cyclohexane; yield: 52%. 1H NMR (400 MHz, CDCl3) δ 1.27 (t, 6H, −OCH2CH3), 4.16–4.24 (m, 4H, −OCH2CH3), 4.91 (s, 1H, ArCH-), 6.23 (bs, 1H, NH), 6.36 (s, 1H, furane proton), 7.34–7.35 (m, 4H, furane protons and dihydropyridine protons) ppm. 13C NMR (100 MHz, CDCl3) δ 14.3 (2C), 32.2, 62.0, 62.2, 100.3 (2C), 109.3, 127.8, 134.2, 134.6, 140.2, 142.9, 166.8, 166.9.
Diethyl 4-(Thiophen-2-yl)-1,4-dihydropyridine-3,5-dicarboxylate (46).
M.p.: 98–100 °C; recrystallization solvent: cyclohexane; yield: 54%. 1H NMR (400 MHz, CDCl3) δ 1.28 (t, 6H, −OCH2CH3), 4.12–4.25 (m, 4H, −OCH2CH3), 5.28 (s, 1H, ArCH-), 6.31 (bs, 1H, NH), 6.88–6.90 (m, 2H, thiophene protons), 7.12 (d, 1H, thiophene proton), 7.35 (d, 2H dihydropyridine protons) ppm. 13C NMR (100 MHz, CDCl3) δ 14.3 (2C), 48.0, 61.9, 62.0, 102.6 (2C), 126.4, 126.5, 126.9, 134.8, 134.8, 141.7, 167.1 (2C).
Diethyl 4-(Thiophen-3-yl)-1,4-dihydropyridine-3,5-dicarboxylate (47).
M.p.: 97–99 °C; recrystallization solvent: cyclohexane; yield: 49%. 1H NMR (400 MHz, CDCl3) δ 1.25 (t, 6H, −OCH2CH3), 4.08–4.23 (m, 4H, −OCH2CH3), 5.08 (s, 1H, ArCH-), 6.21 (bs, 1H, NH), 7.05–7.08 (m, 2H, thiophene protons), 7.16–7.18 (m, 1H, thiophene proton), 7.34 (d, 2H dihydropyridine protons) ppm. 13C NMR (100 MHz, CDCl3) δ 14.3 (2C), 36.3, 61.9, 62.0, 101.7 (2C), 119.2, 125.6, 127.0, 134.2, 134.6, 140.4, 167.0, 167.1.
Diethyl 4-(3-Fluorophenyl)-1,4-dihydropyridine-3,5-dicarboxylate (49).
M.p.: 114–115 °C; recrystallization solvent: cyclohexane; yield: 55%. 1H NMR (400 MHz, CDCl3) δ 1.22 (t, 6H, −OCH2CH3), 4.04–4.19 (m, 4H, −OCH2CH3), 4.94 (s, 1H, ArCH-), 6.25 (bs, 1H, NH), 6.84–6.89 (m, 1H, aromatic proton), 7.04–7.07 (m, 1H, aromatic proton), 7.15–7.25 (m, 2H, aromatic protons), 7.37 (d, 2H dihydropyridine protons) ppm. 13C NMR (100 MHz, CDCl3) δ 14.3 (2C), 36.8, 36.8, 61.9 (2C), 101.1, 101.2, 114.5, 114.7, 114.8, 115.0, 123.8, 130.0, 130.1, 134.4 (2C), 140.3, 162.6, 167.1, 167.3.
Diethyl 4-(3-Bromophenyl)-1,4-dihydropyridine-3,5-dicarboxylate (52).
M.p.: 82–84 °C; recrystallization solvent: cyclohexane; yield: 48%. 1H NMR (400 MHz, CDCl3) δ 1.21 (t, 6H, −OCH2CH3), 4.07–4.21 (m, 4H, −OCH2CH3), 4.93 (s, 1H, ArCH-), 6.21 (bs, 1H, NH), 7.15–7.20 (m, 2H, aromatic protons), 7.26–7.27 (m, 1H, aromatic proton), 7.30–7.31 (m, 1H, aromatic proton), 7.36 (d, 2H dihydropyridine protons) ppm. 13C NMR (100 MHz, CDCl3) δ 14.2 (2C), 36.5, 61.9 (2C), 101.0, 101.1, 122.7, 126.0, 129.8, 129.9, 131.5, 134.4, 134.5, 142.2, 167.1, 167.3.
Diethyl 4-(4-((4-Methylpiperazin-1-yl)methyl)phenyl)-1,4-dihydropyridine-3,5-dicarboxylate (55).
Oil; yield: 36%. 1H NMR (400 MHz, CDCl3) δ 1.23 (t, 6H, −OCH2CH3), 1.58 (s, 8H, piperazine protons), 2.11 (s, 3H, −CH3), 4.05–4.16 (m, 4H, −OCH2CH3), 4.94 (s, 1H, ArCH-), 5.06 (s, 2H, ), 6.21 (bs, 1H, NH), 7.25–7.30 (m, 3H, aromatic protons), 7.35–7.37 (m, 3H aromatic and dihydropyridine protons) ppm. 13C NMR (100 MHz, CDCl3) δ 14.2 (2C), 36.6, 45.3, 52.5 (2C), 53.1 (2C), 62.1 (2C), 100.7 (2C), 127.1, 127.2, 128.3, 128.4, 134.3 (2C), 138.3, 139.2, 167.0, 167.1.
Supplementary Material
Acknowledgments
We thank Dr. Michael Weyand and Susanne Schäfer for technical support. This work was supported by grants from Oberfrankenstiftung (P-Nr. 04115) and Dr. Robert Pfleger Stiftung (mitoSirtuins; to CS), from FCT (PTDC/BIM-MEC/6911/2014), the European Regional Development Fund (ERDF) through Centro 2020 Regional Operational Programme (CENTRO-01-0145-FEDER-000012-HealthyAging2020), the Portugal 2020 - Programme for Competitiveness and Internationalisation, and the Portuguese national funds via FCT (project POCI-01-0145-FEDER-016770; to CP), UID/NEU/04539/2013 (CNC.IBILI Consortium strategic project), from FISR 2019 (n. 00374 – MeDyCa to A.M.), from AIRC 2021 (n. 20172 to S.V.), from FCT PD/BD/114173/2016 to JAA), from NIH (R01GM101171 and R21ES032305) and DoD (CA190267, CA170628, and NF170044; to DL), from NIH (R01CA216853, R01CA210439, R01CA163649 to PKS), and from the Paul F. Glenn Foundation for Medical Research and NIH/NIDDK (R01DK100263; to DAS).
Abbreviations
- ACS2
acetyl-coenzyme A synthetase 2
- CI
confidence interval
- CPS1
carbamoyl phosphate synthetase I
- CR
caloric restriction
- DHP
dihydropyridines
- EIMS
electron ionization mass spectroscopy
- FACS
fluorescent-activated cell sorter
- FBS
fetal bovine serum
- FdL
fluor delys
- FITC
fluoresceine isothiocyanate
- GDH
glutamate dehydrogenase
- GLS
glutaminase
- GOT1
glutamic-oxaloacetic transaminase
- IP
immunoprecipitation
- MEFs
mouse embryonic fibroblasts
- MnSOD
manganese superoxide dismutase
- MST
microscale thermophoresis
- MT-DR
mitotracker deep red
- NAD
nicotinamide adenine dinucleotide
- NAM
nicotinamide
- PDAC
pancreatic ductal adenocarcinoma
- PDC
pyruvate dehydrogenase complex
- PDE3
phosphodiesterase 3
- PDH
pyruvate dehydrogenase
- PDHA1
pyruvate dehydrogenase E1 subunit alpha 1
- PNCA
nicotinamidase
- PVDF
polyvinylidene difluoride
- RNA
ribonucleic acid
- SD
standard deviation
- SDS-PAGE
sodium dodecyl sulphate-polyacrylamide gel electrophoresis
- STAC
sirtuin-activating compounds
- TEV
tobacco etch virus
- TMB
3,3′,5,5′-tetramethylbenzidine
- TRC
the RNAi consortium
- UV
ultraviolet
Footnotes
Supporting Information
Supporting Information contains Figures S1–S4, Tables S1–S3, CSV file of molecular formula strings and associated data.
References
- (1).Choudhary C; Kumar C; Gnad F; Nielsen ML; Rehman M; Walther TC; Olsen JV; Mann M Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [DOI] [PubMed] [Google Scholar]
- (2).Rauh D; Fischer F; Gertz M; Lakshminarasimhan M; Bergbrede T; Aladini F; Kambach C; Becker CFW; Zerweck J; Schutkowski M; Steegborn C An acetylome peptide microarray reveals specificities and deacetylation substrates for all human sirtuin isoforms. Nat. Commun 2013, 4, 2327. [DOI] [PubMed] [Google Scholar]
- (3).Sauve AA; Wolberger C; Schramm VL; Boeke JD The biochemistry of sirtuins. Annu. Rev. Biochem 2006, 75, 435–465. [DOI] [PubMed] [Google Scholar]
- (4).Canto C; Auwerx J Caloric restriction, SIRT1 and longevity. Trends Endocrinol. Metab 2009, 20, 325–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Baur JA; Chen D; Chini EN; Chua K; Cohen HY; de Cabo R; Deng C; Dimmeler S; Gius D; Guarente LP; Helfand SL; Imai S; Itoh H; Kadowaki T; Koya D; Leeuwenburgh C; McBurney M; Nabeshima Y; Neri C; Oberdoerffer P; Pestell RG; Rogina B; Sadoshima J; Sartorelli V; Serrano M; Sinclair DA; Steegborn C; Tatar M; Tissenbaum HA; Tong Q; Tsubota K; Vaquero A; Verdin E Dietary restriction: standing up for sirtuins. Science 2010, 329, 1012–1013; author reply 1013–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Baur JA; Ungvari Z; Minor RK; Le Couteur DG; de Cabo R Are sirtuins viable targets for improving healthspan and lifespan? Nat. Rev. Drug. Discov 2012, 11, 443–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Hubbard BP; Sinclair DA Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol. Sci 2014, 35, 146–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Michan S; Sinclair D Sirtuins in mammals: insights into their biological function. Biochem. J 2007, 404, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Zwergel C; Tomaselli D; Mai A Sirtuins. In Encyclopedia of Molecular Pharmacology, Offermanns S, Rosenthal W Eds.; Springer International Publishing, 2020; pp 1–15. [Google Scholar]
- (10).Morris BJ Seven sirtuins for seven deadly diseases of aging. Free Radic. Biol. Med 2013, 56, 133–171. [DOI] [PubMed] [Google Scholar]
- (11).Someya S; Yu W; Hallows WC; Xu J; Vann JM; Leeuwenburgh C; Tanokura M; Denu JM; Prolla TA Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 2010, 143, 802–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Gertz M; Steegborn C Using mitochondrial sirtuins as drug targets: disease implications and available compounds. Cell Mol. Life Sci 2016, 73, 2871–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Du J; Zhou Y; Su X; Yu JJ; Khan S; Jiang H; Kim J; Woo J; Kim JH; Choi BH; He B; Chen W; Zhang S; Cerione RA; Auwerx J; Hao Q; Lin H Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 2011, 334, 806–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Tan M; Peng C; Anderson KA; Chhoy P; Xie Z; Dai L; Park J; Chen Y; Huang H; Zhang Y; Ro J; Wagner GR; Green MF; Madsen AS; Schmiesing J; Peterson BS; Xu G; Ilkayeva OR; Muehlbauer MJ; Braulke T; Muhlhausen C; Backos DS; Olsen CA; McGuire PJ; Pletcher SD; Lombard DB; Hirschey MD; Zhao Y Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab 2014, 19, 605–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Roessler C; Nowak T; Pannek M; Gertz M; Nguyen GT; Scharfe M; Born I; Sippl W; Steegborn C; Schutkowski M Chemical probing of the human sirtuin 5 active site reveals its substrate acyl specificity and peptide-based inhibitors. Angew. Chem. Int. Ed. Engl 2014, 53, 10728–10732. [DOI] [PubMed] [Google Scholar]
- (16).Laurent G; German NJ; Saha AK; de Boer VC; Davies M; Koves TR; Dephoure N; Fischer F; Boanca G; Vaitheesvaran B; Lovitch SB; Sharpe AH; Kurland IJ; Steegborn C; Gygi SP; Muoio DM; Ruderman NB; Haigis MC SIRT4 coordinates the balance between lipid synthesis and catabolism by repressing malonyl CoA decarboxylase. Mol. Cell 2013, 50, 686–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Pannek M; Simic Z; Fuszard M; Meleshin M; Rotili D; Mai A; Schutkowski M; Steegborn C Crystal structures of the mitochondrial deacylase Sirtuin 4 reveal isoform-specific acyl recognition and regulation features. Nat. Commun 2017, 8, 1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Pillai VB; Samant S; Sundaresan NR; Raghuraman H; Kim G; Bonner MY; Arbiser JL; Walker DI; Jones DP; Gius D; Gupta MP Honokiol blocks and reverses cardiac hypertrophy in mice by activating mitochondrial Sirt3. Nat. Commun 2015, 6, 6656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ramesh S; Govindarajulu M; Lynd T; Briggs G; Adamek D; Jones E; Heiner J; Majrashi M; Moore T; Amin R; Suppiramaniam V; Dhanasekaran M SIRT3 activator honokiol attenuates beta-amyloid by modulating amyloidogenic pathway. PLoS One 2018, 13, e0190350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Li H; Jia J; Wang W; Hou T; Tian Y; Wu Q; Xu L; Wei Y; Wang X Honokiol alleviates cognitive deficits of Alzheimer’s disease (PS1V97L) transgenic mice by activating mitochondrial SIRT3. J. Alzheimers Dis 2018, 64, 291–302. [DOI] [PubMed] [Google Scholar]
- (21).Liu L; Wang Q; Zhao B; Wu Q; Wang P Exogenous nicotinamide adenine dinucleotide administration alleviates ischemia/reperfusion-induced oxidative injury in isolated rat hearts via Sirt5-SDH-succinate pathway. Eur. J. Pharmacol 2019, 858, 172520. [DOI] [PubMed] [Google Scholar]
- (22).Tang Z; Li L; Tang Y; Xie D; Wu K; Wei W; Xiao Q CDK2 positively regulates aerobic glycolysis by suppressing SIRT5 in gastric cancer. Cancer Sci 2018, 109, 2590–2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Hu T; Shukla SK; Vernucci E; He C; Wang D; King RJ; Jha K; Siddhanta K; Mullen NJ; Attri KS; Murthy D; Chaika NV; Thakur R; Mulder SE; Pacheco CG; Fu X; High RR; Yu F; Lazenby A; Steegborn C; Lan P; Mehla K; Rotili D; Chaudhary S; Valente S; Tafani M; Mai A; Auwerx J; Verdin E; Tuveson D; Singh PK Metabolic rewiring by loss of Sirt5 promotes Kras-induced pancreatic cancer progression. Gastroenterology 2021, 161, 1584–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Li F; He X; Ye D; Lin Y; Yu H; Yao C; Huang L; Zhang J; Wang F; Xu S; Wu X; Liu L; Yang C; Shi J; Liu J; Qu Y; Guo F; Zhao J; Xu W; Zhao S NADP(+)-IDH mutations promote hypersuccinylation that impairs mitochondria respiration and induces apoptosis resistance. Mol. Cell 2015, 60, 661–675. [DOI] [PubMed] [Google Scholar]
- (25).Moniot S; Weyand M; Steegborn C Structures, substrates, and regulators of Mammalian sirtuins - opportunities and challenges for drug development. Front. Pharmacol 2012, 3, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Schutkowski M; Fischer F; Roessler C; Steegborn C New assays and approaches for discovery and design of Sirtuin modulators. Exp. Opin. Drug Disc 2014, 9, 183–99. [DOI] [PubMed] [Google Scholar]
- (27).Howitz KT; Bitterman KJ; Cohen HY; Lamming DW; Lavu S; Wood JG; Zipkin RE; Chung P; Kisielewski A; Zhang LL; Scherer B; Sinclair DA Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [DOI] [PubMed] [Google Scholar]
- (28).Pacholec M; Bleasdale JE; Chrunyk B; Cunningham D; Flynn D; Garofalo RS; Griffith D; Griffor M; Loulakis P; Pabst B; Qiu X; Stockman B; Thanabal V; Varghese A; Ward J; Withka J; Ahn K SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J. Biol. Chem 2010, 285, 8340–8351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Dai H; Kustigian L; Carney D; Case A; Considine T; Hubbard BP; Perni RB; Riera TV; Szczepankiewicz B; Vlasuk GP; Stein RL SIRT1 activation by small molecules: kinetic and biophysical evidence for direct interaction of enzyme and activator. J. Biol. Chem 2010, 285, 32695–32703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Hubbard BP; Gomes AP; Dai H; Li J; Case AW; Considine T; Riera TV; Lee JE; E SY; Lamming DW; Pentelute BL; Schuman ER; Stevens LA; Ling AJ; Armour SM; Michan S; Zhao H; Jiang Y; Sweitzer SM; Blum CA; Disch JS; Ng PY; Howitz KT; Rolo AP; Hamuro Y; Moss J; Perni RB; Ellis JL; Vlasuk GP; Sinclair DA Evidence for a common mechanism of SIRT1 regulation by allosteric activators. Science 2013, 339, 1216–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Dai H; Case AW; Riera TV; Considine T; Lee JE; Hamuro Y; Zhao H; Jiang Y; Sweitzer SM; Pietrak B; Schwartz B; Blum CA; Disch JS; Caldwell R; Szczepankiewicz B; Oalmann C; Yee Ng P; White BH; Casaubon R; Narayan R; Koppetsch K: Bourbonais F: Wu B; Wang J; Qian D; Jiang F; Mao C; Wang M; Hu E; Wu JC; Perni RB; Vlasuk GP; Ellis JL Crystallographic structure of a small molecule SIRT1 activator-enzyme complex. Nat. Commun 2015, 6, 7645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Dai H; Sinclair DA; Ellis JL; Steegborn C Sirtuin activators and inhibitors: Promises, achievements, and challenges. Pharmacol. & Ther 2018, 188, 140–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Gertz M; Nguyen GT; Fischer F; Suenkel B; Schlicker C; Franzel B; Tomaschewski J; Aladini F; Becker C; Wolters D; Steegborn C A molecular mechanism for direct sirtuin activation by resveratrol. PLoS One 2012, 7, e49761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Lakshminarasimhan M; Rauth D; Schutkowski M; Steegborn C Sirt1 activation by resveratrol is substrate sequence-selective. Aging (Albany NY) 2013, 5, 151–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Feldman JL; Baeza J; Denu JM Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by mammalian sirtuins. J. Biol. Chem 2013, 288, 31350–31356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).You W; Rotili D; Li TM; Kambach C; Meleshin M; Schutkowski M; Chua KF; Mai A; Steegborn C Structural basis of Sirtuin 6 activation by synthetic small molecules. Angew. Chem. Int. Ed. Engl 2017, 56, 1007–1011. [DOI] [PubMed] [Google Scholar]
- (37).Huang Z; Zhao J; Deng W; Chen Y; Shang J; Song K; Zhang L; Wang C; Lu S; Yang X; He B; Min J; Hu H; Tan M; Xu J; Zhang Q; Zhong J; Sun X; Mao Z; Lin H; Xiao M; Chin YE; Jiang H; Xu Y; Chen G; Zhang J Identification of a cellularly active SIRT6 allosteric activator. Nat. Chem. Biol 2018, 14, 1118–1126. [DOI] [PubMed] [Google Scholar]
- (38).Klein MA; Liu C; Kuznetsov VI; Feltenberger JB; Tang W; Denu JM Mechanism of activation for the sirtuin 6 protein deacylase. J. Biol. Chem 2020, 295, 1385–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Suzuki T; Imai K; Imai E; Iida S; Ueda R; Tsumoto H; Nakagawa H; Miyata N Design, synthesis, enzyme inhibition, and tumor cell growth inhibition of 2-anilinobenzamide derivatives as SIRT1 inhibitors. Bioorg. Med. Chem 2009, 17, 5900–5905. [DOI] [PubMed] [Google Scholar]
- (40).Fischer F; Gertz M; Suenkel B; Lakshminarasimhan M; Schutkowski M; Steegborn C Sirt5 deacylation activities show differential sensitivities to nicotinamide inhibition. PLoS One 2012, 7, e45098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Mai A; Valente S; Meade S; Carafa V; Tardugno M; Nebbioso A; Galmozzi A; Mitro N; De Fabiani E; Altucci L; Kazantsev A Study of 1,4-dihydropyridine structural scaffold: discovery of novel sirtuin activators and inhibitors. J. Med. Chem 2009, 52, 5496–5504. [DOI] [PubMed] [Google Scholar]
- (42).Spallotta F; Cencioni C; Straino S; Nanni S; Rosati J; Artuso S; Manni I; Colussi C; Piaggio G; Martelli F; Valente S; Mai A; Capogrossi MC; Farsetti A; Gaetano C A nitric oxide-dependent cross-talk between class I and III histone deacetylases accelerates skin repair. J. Biol. Chem 2013, 288, 11004–11012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Valente S; Mellini P; Spallotta F; Carafa V; Nebbioso A; Polletta L; Carnevale I; Saladini S; Trisciuoglio D; Gabellini C; Tardugno M; Zwergel C; Cencioni C; Atlante S; Moniot S; Steegborn C; Budriesi R; Tafani M; Del Bufalo D; Altucci L; Gaetano C; Mai A 1,4-Dihydropyridines active on the SIRT1/AMPK pathway ameliorate skin repair and mitochondrial function and exhibit inhibition of proliferation in cancer cells. J. Med. Chem 2016, 59, 1471–1491. [DOI] [PubMed] [Google Scholar]
- (44).Ghorbani-Choghamarani A; Zolfigol MA; Salehi P; Ghaemi E; Madrakian E; Nasr-Isfahani H; Shahamirian M An efficient procedure for the synthesis of Hantzsch 1, 4-dihydropyridines under mild conditions. Acta Chim. Slov 2008, 55, 644–647. [Google Scholar]
- (45).Sueki S; Takei R; Zaitsu Y; Abe J; Fukuda A; Seto K; Furukawa Y; Shimizu I Synthesis of 1,4-dihydropyridines and their fluorescence properties. Eur. J. Org. Chem 2014, 2014, 5281–5301. [Google Scholar]
- (46).Galli U; Mesenzani O; Coppo C; Sorba G; Canonico PL; Tron GC; Genazzani AA Identification of a sirtuin 3 inhibitor that displays selectivity over sirtuin 1 and 2. Eur. J. Med. Chem 2012, 55, 58–66. [DOI] [PubMed] [Google Scholar]
- (47).Schlicker C; Gertz M; Papatheodorou P; Kachholz B; Becker CF; Steegborn C Substrates and regulation mechanisms for the human mitochondrial sirtuins Sirt3 and Sirt5. J. Mol. Biol 2008, 382, 790–801. [DOI] [PubMed] [Google Scholar]
- (48).Lombard DB; Alt FW; Cheng HL; Bunkenborg J; Streeper RS; Mostoslavsky R; Kim J; Yancopoulos G; Valenzuela D; Murphy A; Yang Y; Chen Y; Hirschey MD; Bronson RT; Haigis M; Guarente LP; Farese RV Jr.; Weissman S; Verdin E; Schwer B Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell Biol 2007, 27, 8807–8814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Chen Y; Zhang J; Lin Y; Lei Q; Guan KL; Zhao S; Xiong Y Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep 2011, 12, 534–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Xin T; Lu C SirT3 activates AMPK-related mitochondrial biogenesis and ameliorates sepsis-induced myocardial injury. Aging (Albany NY) 2020, 12, 16224–16237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Park J; Chen Y; Tishkoff DX; Peng C; Tan M; Dai L; Xie Z; Zhang Y; Zwaans BM; Skinner ME; Lombard DB; Zhao Y SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol. Cell 2013, 50, 919–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Polletta L; Vernucci E; Carnevale I; Arcangeli T; Rotili D; Palmerio S; Steegborn C; Nowak T; Schutkowski M; Pellegrini L; Sansone L; Villanova L; Runci A; Pucci B; Morgante E; Fini M; Mai A; Russo MA; Tafani M SIRT5 regulation of ammonia-induced autophagy and mitophagy. Autophagy 2015, 11, 253–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Milne JC; Lambert PD; Schenk S; Carney DP; Smith JJ; Gagne DJ; Jin L; Boss O; Perni RB; Vu CB; Bemis JE; Xie R; Disch JS; Ng PY; Nunes JJ; Lynch AV; Yang H; Galonek H; Israelian K; Choy W; Iffland A; Lavu S; Medvedik O; Sinclair DA; Olefsky JM; Jirousek MR; Elliott PJ; Westphal CH Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 2007, 450, 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).You W; Riemer S; Zheng W; Chua KF; Steegborn C Structural basis for the activation and inhibition of Sirtuin 6 by quercetin and its derivatives. Sci. Rep 2019, 9, 19176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).You W; Steegborn C Binding site for activator MDL-801 on SIRT6. Nat. Chem. Biol 2021, 17, 519–521. [DOI] [PubMed] [Google Scholar]
- (56).Nakamura F; Kudo N; Tomachi Y; Nakata A; Takemoto M; Ito A; Tabei H; Arai D; de Voogd N; Yoshida M; Nakao Y; Fusetani N Halistanol sulfates I and J, new SIRT1–3 inhibitory steroid sulfates from a marine sponge of the genus Halichondria. J. Antibiot 2018, 71, 273–278. [DOI] [PubMed] [Google Scholar]
- (57).Nguyen GT; Gertz M; Steegborn C Crystal structures of sirt3 complexes with 4’-bromo-resveratrol reveal binding sites and inhibition mechanism. Chem. Biol 2013, 20, 1375–1385. [DOI] [PubMed] [Google Scholar]
- (58).Khedkar SA; Auti PB 1, 4-Dihydropyridines: a class of pharmacologically important molecules. Mini Rev. Med. Chem 2014, 14, 282–290. [PubMed] [Google Scholar]
- (59).Lakshminarasimhan M; Curth U; Moniot S; Mosalaganti S; Raunser S; Steegborn C Molecular architecture of the human protein deacetylase Sirt1 and its regulation by AROS and resveratrol. Biosci. Rep 2013, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Moniot S; Forgione M; Lucidi A; Hailu GS; Nebbioso A; Carafa V; Baratta F; Altucci L; Giacche N; Passeri D; Pellicciari R; Mai A; Steegborn C; Rotili D Development of 1,2,4-oxadiazoles as potent and selective inhibitors of the human deacetylase Sirtuin 2: structure-activity relationship, x-ray crystal structure, and anticancer activity. J. Med. Chem 2017, 60, 2344–2360. [DOI] [PubMed] [Google Scholar]
- (61).Suenkel B; Steegborn C Recombinant preparation, biochemical analysis, and structure determination of Sirtuin family histone/protein deacylases. Methods Enzymol 2016, 573, 183–208. [DOI] [PubMed] [Google Scholar]
- (62).Smith BC; Hallows WC; Denu JM A continuous microplate assay for sirtuins and nicotinamide-producing enzymes. Anal. Biochem 2009, 394, 101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.