

Abstract
A deeper understanding of how hydrogen/deuterium exchange mass spectrometry (HDX-MS) reveals allostery is important because HDX-MS can reveal allostery in systems that are not amenable to nuclear magnetic resonance (NMR) spectroscopy. We were able to study thrombin and its complex with thrombomodulin, an allosteric regulator, by both HDX-MS and NMR. In this Perspective, we compare and contrast the results from both experiments and from molecular dynamics simulations. NMR detects changes in the chemical environment around the protein backbone N–H bond vectors, providing residue-level information about the conformational exchange between distinct states. HDX-MS detects changes in amide proton solvent accessibility and H-bonding. Taking advantage of NMR relaxation dispersion measurements of the time scale of motions, we draw conclusions about the motions reflected in HDX-MS experiments. Both experiments detect allostery, but they reveal different components of the allosteric transition. The insights gained from integrating NMR and HDX-MS into thrombin dynamics enable a clearer interpretation of the evidence for allostery revealed by HDX-MS in larger protein complexes and assemblies that are not amenable to NMR.
Graphical Abstract
Proteins are dynamic molecules that interconvert between conformational substates (a recent review has an analysis similar to what follows1). The rate of such dynamic interconversions depends on the energy barrier between states as depicted in Figure 1. Some substates, such as 1 and 2, which interconvert rapidly (on the order of microseconds) and with a very low energy barrier, are not resolved in most experiments. Instead, the average of these states will be measured. In contrast, states 2 and 3 interconvert more slowly because the energy barrier between them is higher. These states are more likely to be observed as separate populations. The relative population of each state depends on their energies according to Boltzmann’s law. This energy landscape view helps diffuse the perceived conflict between the “induced fit” and “conformational selection” models traditionally used to interpret evidence for allostery.2,3 As one can see from the diagram, both states 2 and 3 exist; however, one may be at such a low concentration due to its higher energy that it will not be observed until a ligand binds, reflecting a conformational relationship better described by “induced fit”. When both states are observed because their energies are not that different, or the experiment is sensitive enough to observe the weakly populated state, “conformational selection” will be invoked as the more informative model of allostery. Clearly, these models describe two limits of the same physical phenomenon: the association of a ligand alters the protein energy landscape to form a new distribution of conformational states.
Figure 1.
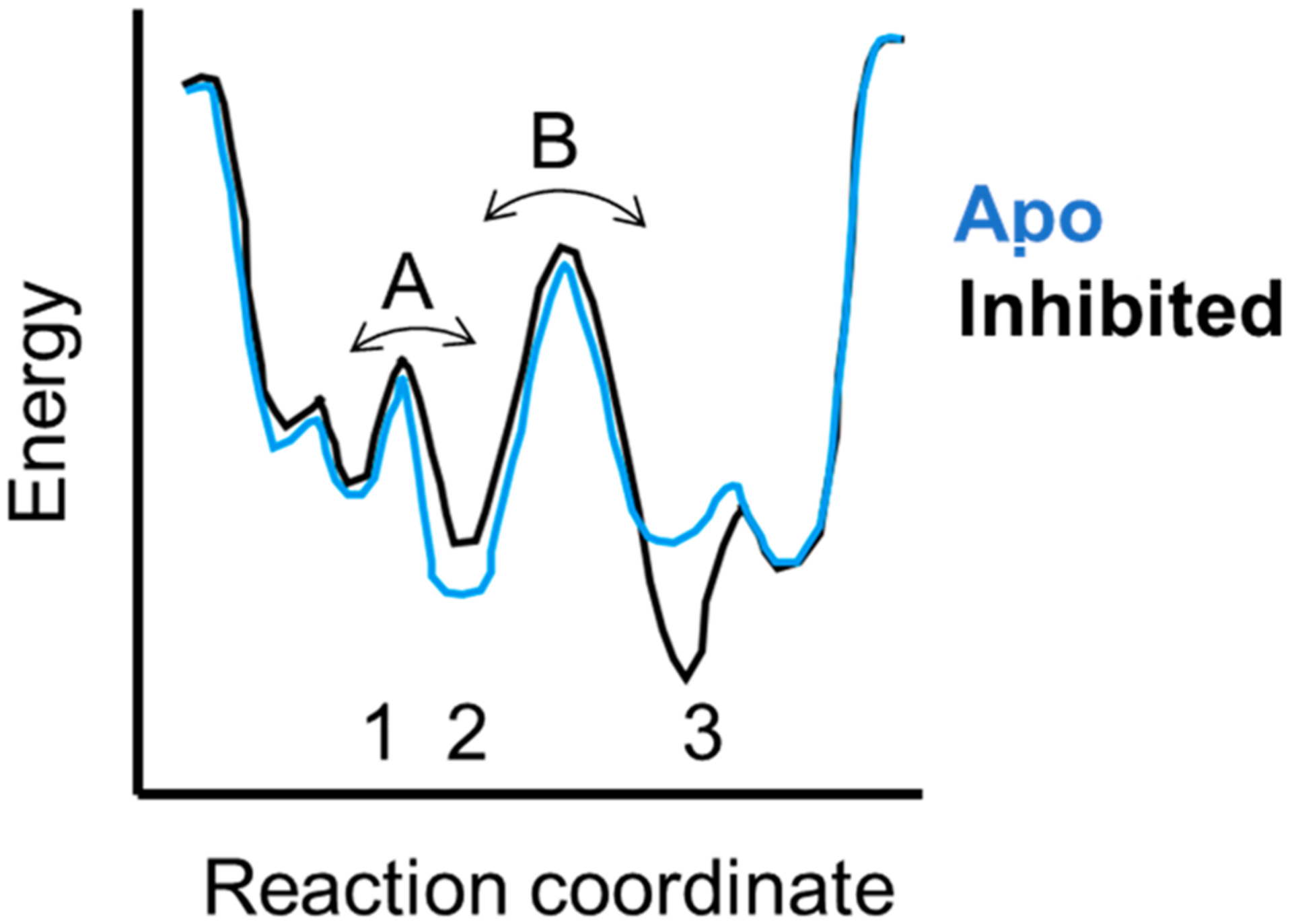
Energy landscape diagram showing three states (1–3) that are separated by two energy barriers (A and B). States 1 and 2 are close in energy, so that most experimental measurements will report the average between them. States 2 and 3 are separated by a larger energy barrier, and their interconversion will occur on the microsecond to millisecond time scale and can be measured by NMR relaxation dispersion or HDX-MS experiments. States such as 2 and 3 are typically stabilized or destabilized by binding of allosteric ligands.
MOTIONAL TIME SCALES FROM ACCELERATED MOLECULAR DYNAMICS (AMD) AND NUCLEAR MAGNETIC RESONANCE (NMR)
Some years ago, we performed conventional NMR relaxation experiments on thrombin to analyze its picosecond to nanosecond time scale dynamics and to compute backbone NH order parameters.4 These experiments can measure only dynamics that occur in a time window shorter than the rotational correlation time of the protein molecule, which in the case of thrombin is 10 ns. Conventional MD simulations run for 20 ns recapitulated the order parameters determined from the picosecond to nanosecond time scale dynamics (Figure 2A).4 Alignment of the amino acid sequence of thrombin with that of the canonical serine protease, chymotrypsin, reveals that thrombin has distinct insertions that form surface loops on the structure. These loops have low order parameters calculated from the picosecond to nanosecond time scale dynamics. NMR-derived order parameters (S2) measure of the degree of motion of the N–H bond and range from 0 (completely disordered) to 1 (completely ordered). The low order parameters of the thrombin loops indicate that these regions are conformationally flexible.4 Analysis of the thrombin structure to determine which side chain interactions in the folded structure were minimally frustrated or highly frustrated5 revealed that these same loops were highly frustrated (Figure 2B,D).6 This makes sense because minimally frustrated regions of proteins have energetically favorable contacts that would be difficult to break, whereas highly frustrated regions would energetically prefer to make interactions different from those captured in the X-ray crystal structure.
Figure 2.
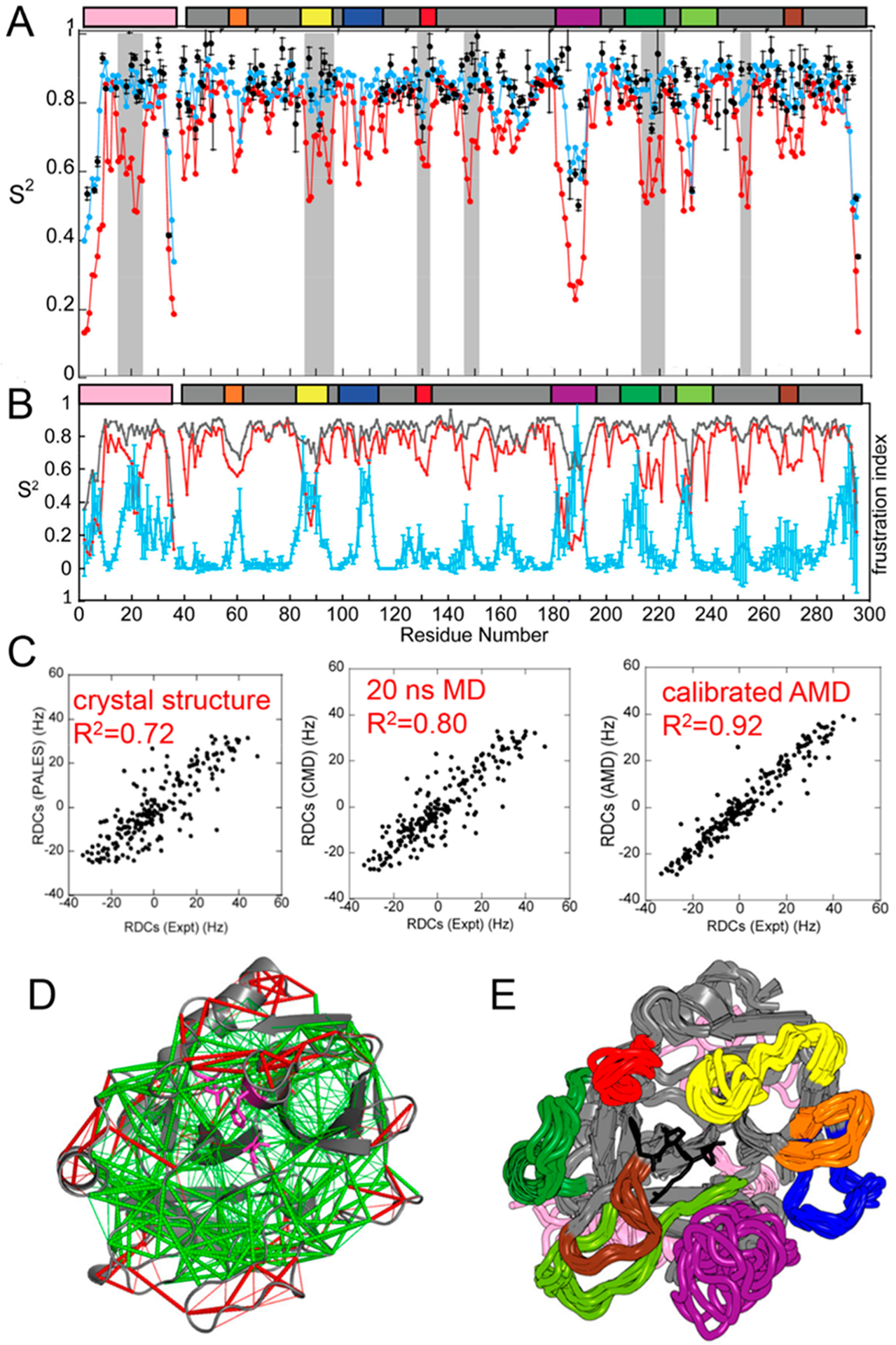
Characterization of the dynamic ensemble of thrombin. (A) Conventional MD (CMD) simulations report on dynamics on the picosecond to nanosecond time scale; measured order parameters (black circles) correlate well with order parameters back-calculated from the CMD ensemble (blue circles), whereas the aMD simulations capture additional motions (red circles). The gray bars represent regions of thrombin for which motions were observed only in the aMD simulations. (B) Order parameters (gray, from CMD simulations; red, from aMD simulations) also correlate with the plot of residual frustration (cyan) because highly frustrated residues are typically found in disordered loops. (C) RDCs were measured from the thrombin crystal structure (Protein Data Bank entry 1PPB) using PALES50 or from the ensemble of structures derived from CMD or aMD simulations and compared to the RDCs measured from the NMR experiments. The RDCs calculated from the aMD ensemble better recapitulate the RDCs. (D) Structure of thrombin with minimally frustrated contacts colored green and highly frustrated contacts colored red. The inhibitor, PPACK (magenta), is located in the active site. (E) Molecular ensemble of thrombin computed from Boltzmann reweighted aMD simulations that were calibrated against the experimental residual dipolar couplings. The various loops are colored in the figure and the same way in the bars above the plots in panels A and B, and the active site inhibitor, PPACK, is colored black.
We next performed solution NMR residual dipolar coupling (RDC) experiments to verify the structure of the molecule, which was well-known by X-ray crystallography.7 Surprisingly, the measured RDCs for the active site-inhibited thrombin did not agree very well with the RDCs back-calculated from the crystal structure (Figure 2C). RDCs capture the averaged NH bond vector amplitudes of motion from picoseconds to milliseconds. We showed that only RDCs that were averaged from a Boltzmann reweighted ensemble of structures from aMD simulations that captured ~100 μs of total time could recapitulate the motions reported in RDCs (Figure 2C).4 The ensemble of structures that fully represents thrombin in solution has disordered surface loops (Figure 2E).
NMR spectroscopy provides a range of experiments to measure the conformational dynamics across a wide temporal scale. It is now accepted that measurements of chemical exchange, which reflect microsecond to millisecond time scale motions, are likely to be linked to binding (kon ~ 106 M−1 s−1) and catalysis (kcat ~ 103 s−1).8 Chemical exchange between the ground state and an “invisible state” or a poorly populated state occurs on the microsecond to millisecond time scale and is commonly measured by Carr–Purcell–Meinboom–Gill-type (CPMG or relaxation dispersion) experiments.9 In this experiment, backbone NH groups that are exchanging between states have broadened signals due to slightly different chemical shifts of the NH group in states that are interconverting on the “NMR time scale”. Upon application of refocusing pulses, the rate of interconversion between the two states can be measured. In addition, the populations of each state can be ascertained. Typically, pa ≫ pb, with pb ranging from 0.05% to 5%. Relaxation dispersion experiments are widely applied, so we will point the reader to a few recent reviews.10–12 NMR relaxation dispersion experiments were used to measure the kinetics of visitation to a higher-energy “invisible state” in apo-thrombin and thrombin with the covalent inhibitor, d-Phe-Pro-Arg chloromethyl ketone (PPACK), bound to the active site serine.13 In these experiments, 24 backbone NH groups showed significant chemical exchange to an invisible state, and these groups were scattered throughout the apo-thrombin molecule (Figure 3A). The relaxation dispersion curves could be globally fit using the Carver–Richards equation14 to a single exchange rate (kex = 1770 ± 60 s−1) and a minor state population of 4.1 ± 6%.13 We note that the fact that the curves could be globally fit does not necessarily prove that the motions of these NH groups are correlated; in fact, community analysis of the aMD simulations yielded a multitude of small communities moving in an uncorrelated fashion.6,15 The measured exchange rate, kex, is the sum of the forward and reverse rates for the conformational transitions between the ground state (~95%) and the excited state (~5%), so kex represents a transition rate of ~1/1600 s−1 or a transition time from the excited state to the ground state of ~600 μs and a transition rate of 1/177 s−1 or a transition time from the ground state to the excited state of ~6 ms.13 The active site-inhibited thrombin retained some NH groups that were exchanging on the microsecond to millisecond time scale, although motions in the loops that extend from the C-terminal β-barrel to make up the primary substrate binding pocket were completely damped13 (Figure 3B). The relaxation dispersion curves for these nine remaining NH groups could be globally fit only using the Luz–Meiboom equation,16 which is for fast exchange processes and cannot resolve populations, giving a kex of 2440 ± 50 s−1. Collectively, these NMR experiments define the residue-specific changes in thrombin backbone microsecond to millisecond dynamics throughout the protein that occur with ligand binding at the active site.
Figure 3.
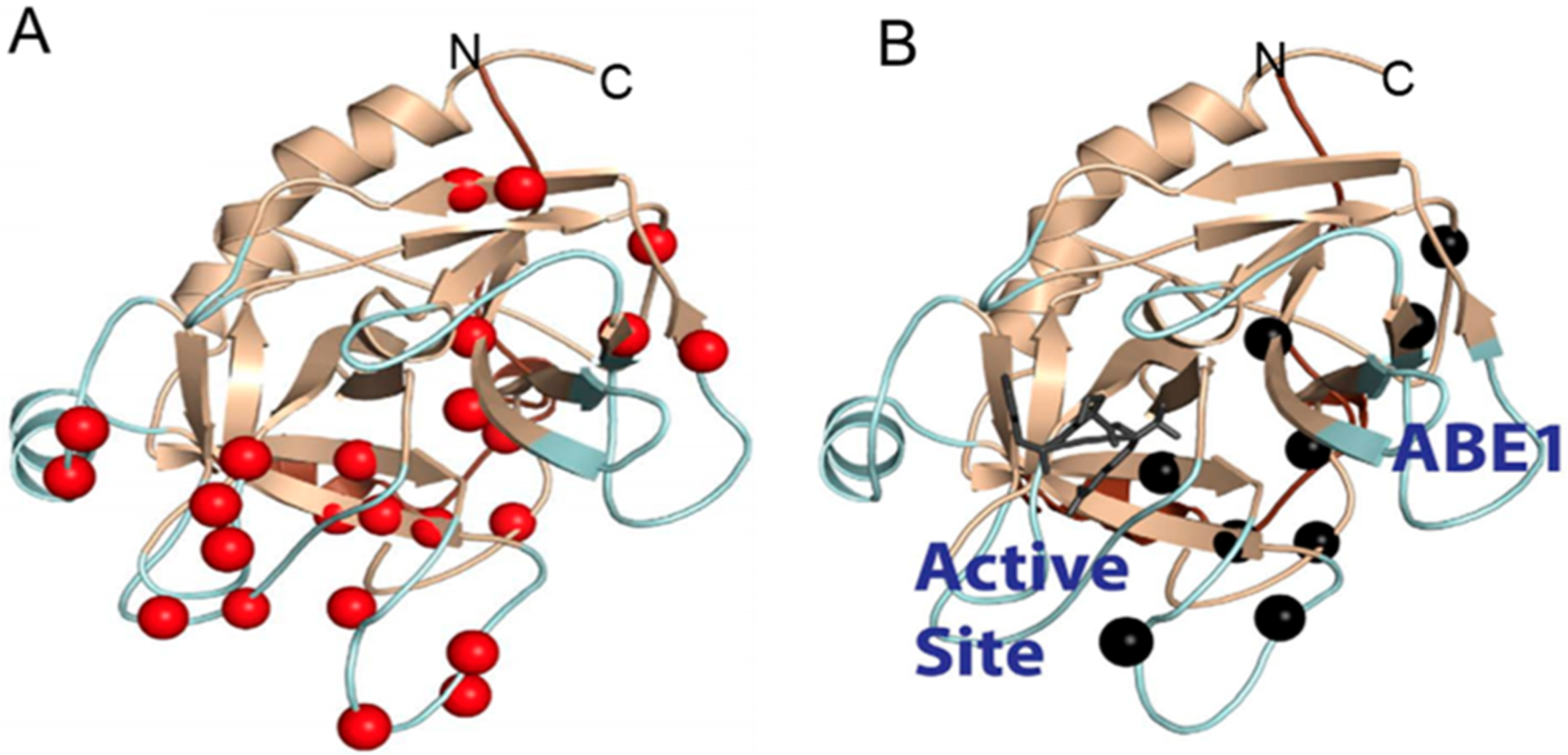
(A) Relaxation dispersion experiments revealed many backbone NH groups throughout the structure of thrombin were moving on the microsecond to millisecond time scale (red spheres). (B) When the covalent active site inhibitor, PPACK, is bound, motions in the C-terminal β-barrel are damped but motions in the N-terminal β-barrel remain and form a pathway of the remaining dynamics from the allosteric site (anion binding exosite 1) to the active site (marked by the PPACK inhibitor in dark gray).
HDX-MS AND PROTEIN SURFACE PROTECTION
In an HDX-MS experiment, proteins are diluted into deuterated buffer (here 2H2O will be designated D2O) and the exchange of amide hydrogens with deuterium is monitored over time after digestion with an acid-stable protease using mass spectrometry. HDX-MS experiments monitor whether an NH group can exchange its proton. Proton exchange is very different than chemical exchange between nuclear environments, which is monitored by relaxation dispersion NMR experiments. As such, NMR and HDX-MS give complementary information about protein conformational dynamics. Often, large proteins and/or protein complexes are difficult to study by NMR, whereas HDX-MS does not require isotopic labeling; it is now possible to study very large proteins and protein complexes by HDX-MS.17,18
Changes in protein conformational dynamics are elicited upon ligand binding. For both NMR and HDX-MS, it is necessary to deconvolute changes in chemical shifts or changes H/D exchange due to chemical interactions with the ligand. For NMR, this is relatively straightforward because the chemical environment changes for those atoms in direct physical contact with the ligand. For H/D exchange, it was not clear whether the ligand could block surface H/D exchange because ligands mostly do not change the H-bonding of the protein. In 1998, we showed that when a ligand binds to a protein there can be local decreases in amide exchange that are due to the decreased solvent accessibility of surface amides (which may be participating in H-bonds to other protein groups).19 Although this result was surprising at the time, it has now become widely accepted that ligand binding causes decreased surface solvent accessibility and this approach is even used in high-throughput drug screening.20 However, both increases and decreases in H/D exchange may be observed farther from the ligand binding site that are due to changes in the protein ensemble of states and interconversion among these states.
In HDX-MS analysis, the kinetics are traditionally described by eq 1 in which kch is the chemical exchange rate for the NH in a completely unfolded peptide. The values for kch have been tabulated21 and can be obtained from the protein sequence by SPHERE (https://protocol.fccc.edu/research/labs/roder/sphere/).22 For a protein–ligand interaction, two equilibria must be considered: the protein–ligand equilibrium and the equilibrium between conformations that do not exchange (traditionally denoted as Pcl for closed) and conformations that do exchange (denoted as Pop for open conformations) (eq2).23
| (1) |
| (2) |
To observe surface solvent accessibility changes, the conditions of the HDX-MS experiment must be such that the ligand is essentially always bound. When the ligand is at an appropriate concentration to achieve >99% bound, then eq 2 reduces to eq 1 and only the LPcl-H state need be considered in the HDX reaction. The fraction of bound protein depends on the KD of the ligand and can be calculated from the quadratic binding equation.24 This is not trivial because HDX-MS can be measured on relatively low concentrations of protein for which even a ligand KD of 5 nM will result in only 90% of the protein being bound if the L:P ratio is 1:1. At a 2:1 ratio of L:P, the protein will be 99% bound, but over time, the ligand and protein will dissociate and deuteration of the unbound protein will occur, resulting in a substantial amount (~50%) of the unbound protein within 10 min. As an example, the rate of association of thrombomodulin (TM) with thrombin (ka) is 2 × 107 M−1 s−1, and the dissociation rate (kd) is 0.037 s−1, resulting in a KD of 2 nM.25 HDX-MS revealed that TM caused decreased exchange at anion binding exosite 1, the known allosteric regulatory binding site on thrombin.24,26 This protection disappears after 10 min if a1.5:1 TM:thrombin ratio is used.27 Therefore, studies of protein surface protection should be carried out for short deuteration times, and interface protection will occur immediately, at the earliest time point, and will not build up over time. These features of surface protection result in an informative time window for deuterium exchange between 0 and 10 min.
HDX-MS REVEALS PROTEIN CONFORMATIONAL DYNAMICS
Often, conformationally dynamic regions of a protein will show H/D exchange within the first few minutes of exchange. These rapidly H/D exchanging NH groups are frequently not observed in the NMR spectra because the NH group is sampling multiple chemical environments and the signal is broadened beyond visibility. HDX-MS very often (too often to completely reference here) reveals changes in amide exchange far from the known ligand binding site and has been said to reveal allostery.28 These observations are consistent with the definition of allostery as ligand-mediated reconfiguration of the protein ensemble of conformational states.12,29–32 It remains confusing, however, how to relate the rate of amide exchange to the time scales of allostery, which are so readily accessible from NMR experiments.
Comparing and contrasting the HDX-MS and NMR data from thrombin helped us to understand the time scales of motion that are reported by H/D exchange. NMR spectroscopists understand that ligand binding shifts the equilibrium between protein conformational states that are energetically accessible (cf. Figure 1), that is states that are ~2–10 kcal/mol higher in energy than the ground state.10 It is now widely accepted that relaxation dispersion experiments reveal allostery, because they reveal the “hidden” states that are higher in energy but still can be observed through chemical exchange. Can the rate of exchange to a populated “invisible” state be related to the rate of amide exchange? We realized that when ligand binding reveals a protein conformational state with differentially exchanging amides, that state must be energetically accessible. Using the NMR data we had for thrombin, we made the assumption that the “open” state in the HDX-MS experiment is the same energetically accessible state that was also observed by NMR. This allows us to assign the opening and closing rates in eq 1, which we determined from NMR, to the rate of accessing the “invisible state” and the rate of return to the more populated state13 (cf. eq 3). This is a reasonable assumption because the NMR and HDX-MS experiments were performed under the same solution conditions at 25 °C and pH 6.5. We then used the SPHERE Program (https://protocol.fccc.edu/research/labs/roder/sphere/) to calculate the kch values for the amides in thrombin. These are the rates at which the amide would exchange if it were not in any structured context, and they vary from 0.2 to 10 s−1.22 By rearranging eq 3 to solve for the observed exchange rate in the HDX-MS experiment, kex (cf. eq 4), we could then solve for kex for the range of kch values to obtain the range of kex values we can expect to observe in the HDX experiment. For the range of kch from 0.2 to 10 s−1, kex ranges from 0.02 to 1 s−1. These exchange rates in minutes are 1.2–60 min−1. Translating these exchange rates into half-life yields 0.58–0.11 min, indicating that half of the amides will exchange within 0.1–0.5 min and five half-lives will occur within 0.5–3 min. In fact, we observed most of the differences in thrombin HDX due to TM binding when the HDX was measured from 30 s to 5 min.33 Note that the equation for kex is dominated by the ratio of the opening and closing rates and therefore by the ground state populations of the closed and open states.
| (3) |
| (4) |
Allostery requires an energetically accessible higher-energy state, but one that is not so readily accessible as to be highly populated even in the absence of a ligand. The rate of exchange between states is thus expected to occur within the microsecond to millisecond time regime, exactly the time regime that is measured by relaxation dispersion, and corresponding to an energy barrier of ~2–10 kcal/mol. The microsecond to millisecond time regime of protein conformational rearrangements that would render an amide exchangeable or not corresponds to the 0–5 min time frame for amide exchange. Although an exhaustive search is impossible, many reports of allostery observed by HDX-MS have differences in exchange between the ligand-bound state and the apo state in the first 5 min of deuterium exchange.34–40
We performed a stringent test of the hypothesis that the microsecond to millisecond time regime of protein conformational rearrangements corresponds to an H/D exchange window of 0–10 min. We performed aMD simulations of thrombin and a single-site mutant of thrombin, Trp215Ala. This single-point mutant resulted in changed H/D exchange both near and far from the mutation. We asked whether we could meaningfully extract H/D exchange information from an exhaustive aMD simulation and whether the simulations could capture the subtle differences between the wild-type and mutant thrombin. The enhanced sampling approach of aMD allows much more extensive exploration of the energy landscape to states that are separated by larger barriers such as states 2 and 3 in Figure 1. The simulations that accounted for approximately 1 ms of protein conformational dynamics were analyzed for H-bonding and solvent accessibility, and then differences in these parameters were determined for the wild-type thrombin and the Trp215Ala mutant. The experimental HDX-MS differences and the simulation results accurately predicted the allosteric changes upon mutation when both H-bonding and solvent accessibility were accounted for (Figure 4).41
Figure 4.
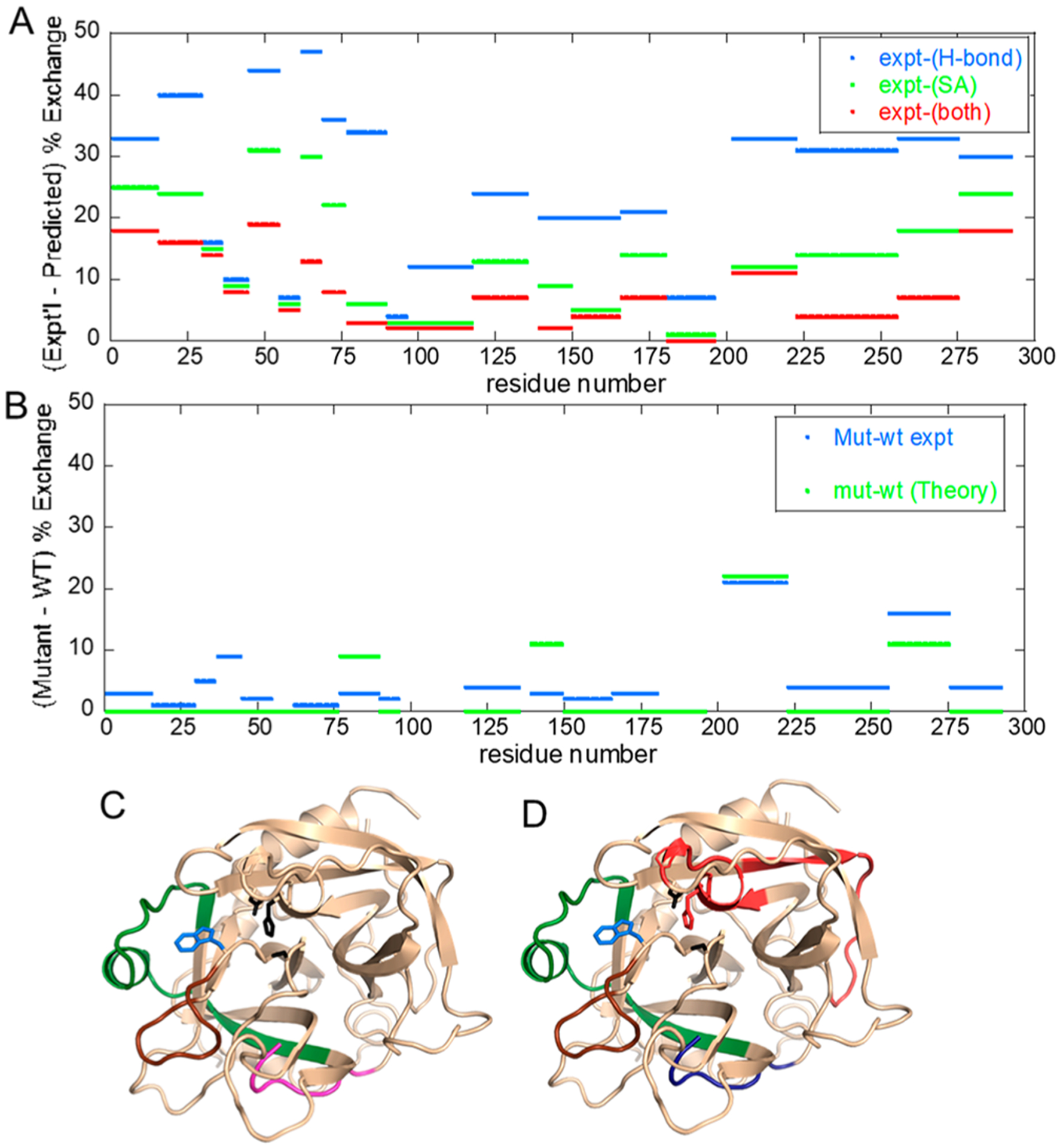
Data from ref 41 in which aMD simulations were performed and then solvent accessibility and H-bonding were evaluated across the simulation. (A) Plot of the difference between experimental and predicted amide exchange for wild-type thrombin. The aMD simulations recapitulate the amide exchange best when both H-bonding (blue) and solvent accessibility (green) are combined in the computation of amide exchange (red). (B) Plot of the difference between experimental and predicted amide exchange for the W215A mutant thrombin. The amide exchange for the mutant differs primarily from the wild type at the 170s (sequential residues 200–225) and 220s (sequential residues 255–275) loops. The theory and experiment agree well for these two loops. (C) Structure of thrombin showing the regions of thrombin (180s loop, green; 220s loop, brown; N-terminus of the heavy chain, magenta) that are allosterically affected by the mutation of W215 (blue sticks). (D) The aMD simulations overestimated the dynamics of the region of thrombin (red) as compared to experiment, and they underestimated the dynamics of the N-terminus of the heavy chain (blue).
It is important to reiterate that the higher-energy accessible state that corresponds to the allosteric state will appear very different from the HDX-MS versus NMR perspective. NMR chemical shift changes reported in relaxation dispersion experiments typically report on the pathway of allostery from one site to another. However, NMR can miss the end points of a pathway, particularly if they are dynamic surface loops that adopt a range of conformations and have an ensemble of resonances that are too broad to be observed in the NMR spectrum. In contrast, it is unlikely that the H-bonding state of the pathway residues will change (recall that these are minimally frustrated and well-folded regions of the protein), so HDX-MS typically reveals the end points of the allosteric pathway because it captures the changes in H-bonding and solvent accessibility at the ends of the pathway. A clear example of this is our recent study of the thrombin–TM complex by NMR and HDX-MS.27 TM binds to the N-terminal β-barrel and allosterically reduces the dynamics of the C-terminal β-barrel (Figure 5). The 170s loop (residues 160–189CT) did not show a reduction in H/D exchange upon TM binding, whereas the 220s loop (residues 212–227CT) did show a reduction (Figure 5B). Both loops had NH groups that showed reduced microsecond to millisecond time scale dynamics as indicated by relaxation dispersion experiments (Figure 5C).
Figure 5.
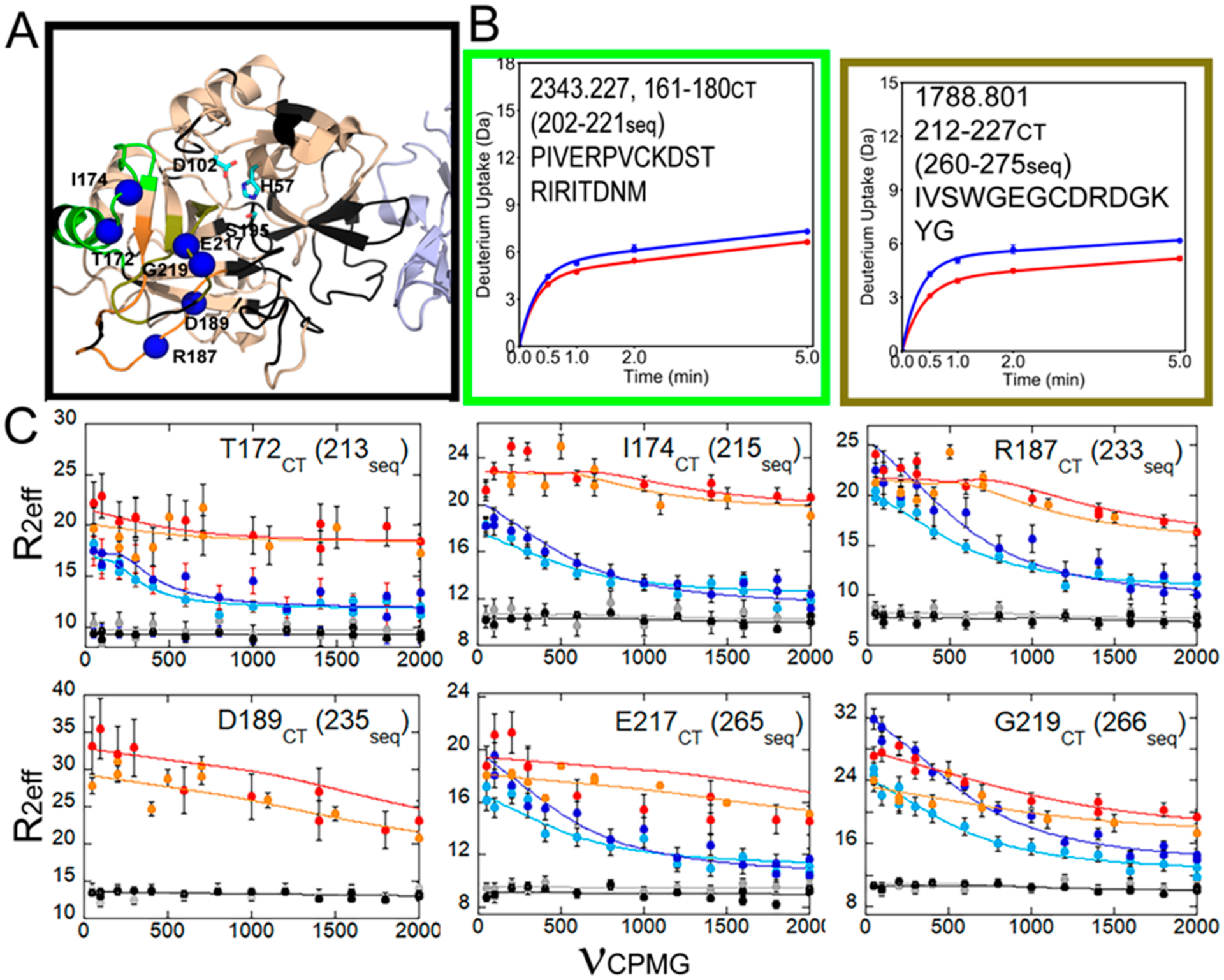
Data from ref 27 for the C-terminal region of thrombin, which is allosterically altered by TM binding. (A) Structure of the thrombin (wheat)–TM (light blue) complex showing the catalytic triad (cyan sticks) and the loops as in Figure 2. Missing resonances resulted in a lack of assignments for some loops (black). (B) Deuterium uptake curves for the 180s loop were not different between thrombin (blue) and thrombin bound to TM (red), whereas the level of deuterium uptake was lower into the 220s loop when TM was bound. (C) Relaxation dispersion curves for the NH groups of residues marked by blue spheres in panel A. All of these groups showed microsecond to millisecond motions in apo-thrombin (light blue, 600 MHz data; dark blue, 800 MHz data) that were quenched upon binding of the active site inhibitor, PPACK (gray, 600 MHz data; black, 800 MHz data) or TM (orange, 600 MHz data; red, 800 MHz data). We note that R0 is non-uniform in the thrombin–TM complex likely due to the fact that the TM was made in Pichia pastoris and is glycosylated at two sites and probably tumbles non-uniformly.
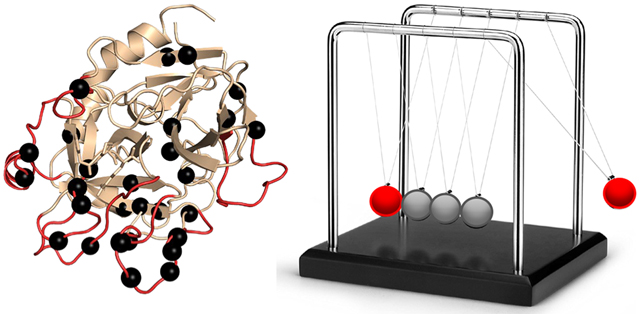
These data show that HDX-MS and NMR relaxation dispersion provide complementary and sometimes overlapping data due to the difference in the dynamically sensitive phenomena they measure. Both reveal allostery, but as one can see in the Newton’s cradle analogy, NMR reveals the pathway of the allostery (the middle spheres in Newton’s cradle) whereas HDX-MS is much more likely to reveal allostery at the ends of the pathway in the less-structured parts of the protein. Newton’s cradle provides an apt analogy for protein allostery because momentum and energy are largely conserved, and the ramifications of this conservation are that the middle spheres do not appear to move while the force is transmitted through them from one end to the other (Figure 6). Allostery would not occur through proteins if the thermodynamic force induced by ligand binding to the allosteric site was dissipated through the interior of the protein structure before impacting the active site. The frustration patterns in allosteric proteins bear this out. The core of a folded protein is minimally frustrated, whereas highly frustrated patches of residues that correlate with allosteric sites occur on the surface42 (Figure 2D). Thus, a minimally frustrated protein core is optimized to transduce an allosteric signal between highly frustrated sites as analogously occurs in soft matter.43
Figure 6.
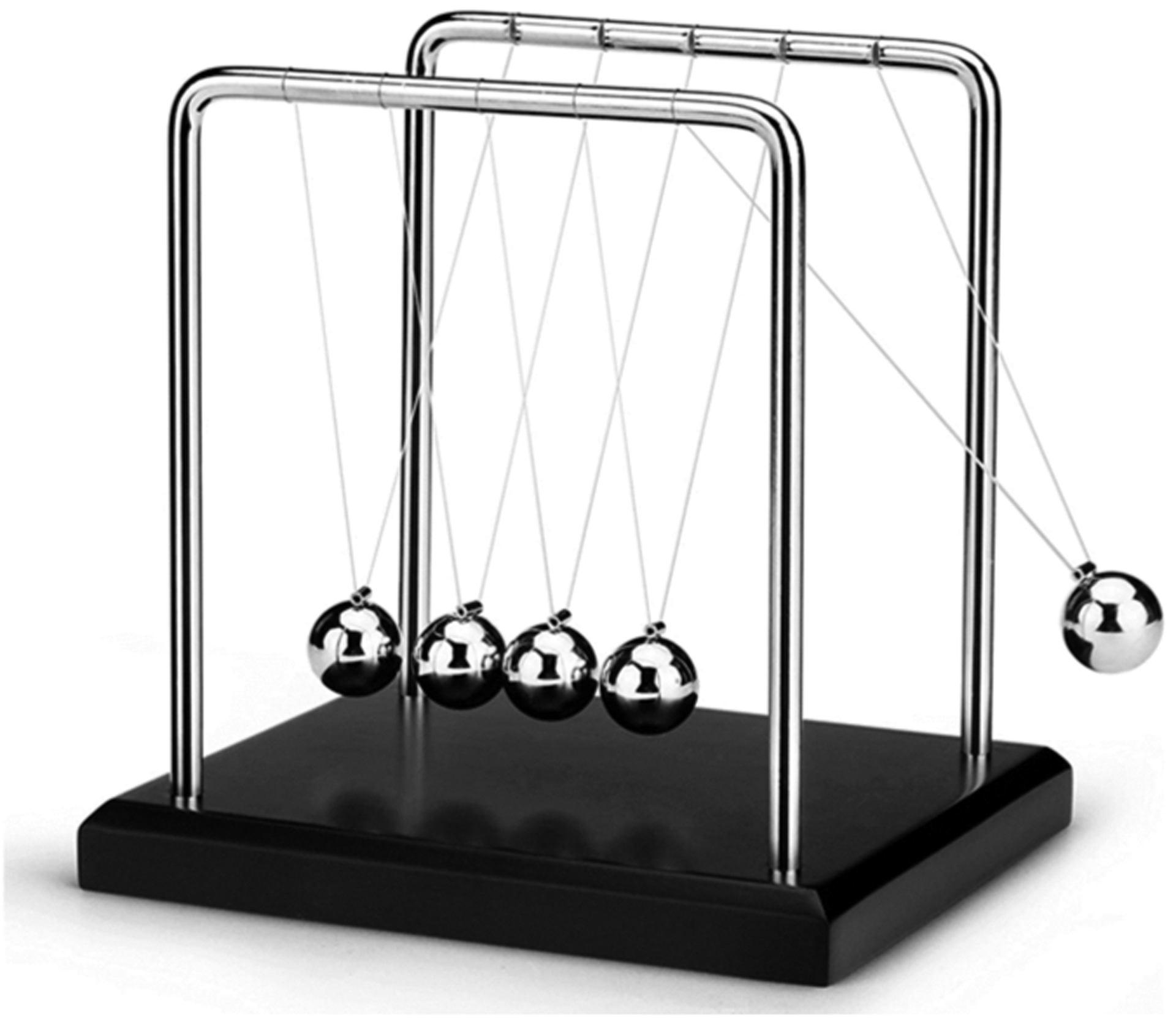
Newton’s cradle helps show the conservation of momentum and energy with swinging spheres. When one of the end spheres strikes the stationary spheres, it transmits a force through the stationary spheres that pushes the last sphere out. This phenomenon is similar to what happens in allosteric proteins.
A deeper understanding of how HDX-MS reveals allostery is important because HDX-MS can reveal allostery in systems that are not amenable to NMR spectroscopy. In the past few years, complexes such as E3 ligases that contain upward of 10 different proteins have been analyzed by HDX-MS, revealing that allostery can be transmitted through several proteins into others far away in the complex (Figure 5).44,45 Allostery was also observed in AAA+ complexes46 and even in whole viruses.47 Finally, HDX-MS can also reveal allostery in membrane proteins.48 Allostery has even been observed from the intracellular nucleotide binding domains through the transmembrane region to the extracellular domain of P-glycoprotein.49
CONCLUSIONS
As new methodologies developed, our conception of the thermodynamic mechanisms that drive allostery has also evolved, making it necessary to expand beyond the models originally constructed that reflected structural changes. NMR and HDX-MS experiments are unique in their abilities to experimentally report on the conformational fluctuations relevant to protein allostery. Despite this similarity, the chemical phenomena measured by these two techniques are fundamentally distinct, and therefore, their information content overlaps but is also complementary. Thus, the advancements being made in both NMR and HDX-MS enable allostery to be described in numerous protein systems that would not otherwise fit the traditional models, revealing multiple ways to modulate energy landscapes of proteins and protein complexes to elicit diverse functions and regulatory mechanisms.
Funding
This work was supported by National Institutes of Health Grants R01 R01HL127041 and S10 OD016234. R.B.P. acknowledges support from National Institutes of General Medical Sciences Molecular Biophysics Training Grant T32 GM008326.
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.biochem.1c00277
The authors declare no competing financial interest.
Contributor Information
Riley B. Peacock, Department of Chemistry and Biochemistry, University of California, San Diego, La Jolla, California 92093-0378, United States
Elizabeth A. Komives, Department of Chemistry and Biochemistry, University of California, San Diego, La Jolla, California 92093-0378, United States.
REFERENCES
- (1).Sekhar A, and Kay LE (2019) An NMR View of Protein Dynamics in Health and Disease. Annu. Rev. Biophys 48, 297–319. [DOI] [PubMed] [Google Scholar]
- (2).Hammes GG, Chang Y-C, and Oas TG (2009) Conformational selection or induced fit: A flux description of reaction mechanism. Proc. Natl. Acad. Sci. U. S. A 106, 13737–13741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Silva DA, Bowman GR, Sosa-Peinado A, and Huang X (2011) A role for both conformational selection and induced fit in ligand binding by the LAO protein. PLoS Comput. Biol 7, e1002054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Fuglestad B, Gasper PM, Tonelli M, McCammon JA, Markwick PR, and Komives EA (2012) The dynamic structure of thrombin in solution. Biophys. J 103, 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ferreiro DU, Hegler JA, Komives EA, and Wolynes PG (2007) Localizing frustration in native proteins and protein assemblies. Proc. Natl. Acad. Sci. U. S. A 104, 19819–19824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Fuglestad B, Gasper PM, McCammon JA, Markwick PR, and Komives EA (2013) Correlated Motions and Residual Frustration in Thrombin. J. Phys. Chem. B 117, 12857–12863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Bode W, Mayr I, Baumann U, Huber R, Stone SR, and Hofsteenge J (1989) The refined 1.9 A crystal structure of human alpha-thrombin: interaction with D-Phe-Pro-Arg chloromethylketone and significance of the Tyr-Pro-Pro-Trp insertion segment. EMBO J. 8, 3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Stivers JT, Abeygunawardana C, Mildvan AS, and Whitman CP (1996) 15N NMR relaxation studies of free and inhibitor-bound 4-oxalocrotonate tautomerase: backbone dynamics and entropy changes of an enzyme upon inhibitor binding. Biochemistry 35, 16036–16047. [DOI] [PubMed] [Google Scholar]
- (9).Palmer A. G. r. (2014) Chemical exchange in biomacromolecules: Past, present, and future. J. Magn. Reson 241, 3–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Farber PJ, and Mittermaier A (2015) Relaxation dispersion NMR spectroscopy for the study of protein allostery. Biophys. Rev 7, 191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Palmer A. G. r., and Koss H (2019) Chemical Exchange. Methods Enzymol. 615, 177–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).East KW, Skeens E, Cui JY, Belato HB, Mitchell B, Hsu R, Batista VS, Palermo G, and Lisi GP (2020) NMR and computational methods for molecular resolution of allosteric pathways in enzyme complexes. Biophys. Rev 12, 155–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Handley LD, Fuglestad B, Stearns K, Tonelli M, Fenwick RB, Markwick PRL, and Komives EA (2017) NMR reveals a dynamic allosteric pathway in thrombin. Sci. Rep 7, 39575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Carver JP, and Richards RE (1972) A general two-site solution for the chemical exchange produced dependence of T2 upon the Carr-Purcell pulse separation. J. Magn. Reson 6, 89–105. [Google Scholar]
- (15).Gasper PM, Fuglestad B, Komives EA, Markwick PR, and McCammon JA (2012) Allosteric networks in thrombin distinguish procoagulant vs. anticoagulant activities. Proc. Natl. Acad. Sci. U. S. A 109, 21216–21222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Luz Z, and Meiboom S (1963) Nuclear Magnetic Resonance Study of the Protolysis of Trimethylammonium Ion in Aqueous Solution–Order of the Reaction with Respect to Solvent. J. Chem. Phys 39, 366–370. [Google Scholar]
- (17).Lumpkin RJ, Ahmad AS, Blake R, Condon CJ, and Komives EA (2021) The Mechanism of NEDD8 Activation of CUL5 Ubiquitin E3 Ligases. Mol. Cell. Proteomics 20, 100019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Engen JR, and Komives EA (2020) Complementarity of Hydrogen/Deuterium Exchange Mass Spectrometry and Cryo-Electron Microscopy. Trends Biochem. Sci 45, 906–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Mandell JG, Falick AM, and Komives EA (1998) Identification of protein-protein interfaces by decreased amide proton solvent accessibility. Proc. Natl. Acad. Sci. U. S. A 95, 14705–14710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Chalmers MJ, Busby SA, Pascal BD, He Y, Hendrickson CL, Marshall AG, and Griffin PR (2006) Probing protein ligand interactions by automated hydrogen/deuterium exchange mass spectrometry. Anal. Chem 78, 1005–1014. [DOI] [PubMed] [Google Scholar]
- (21).Bai Y, Milne JS, Mayne L, and Englander SW (1993) Primary structure effects on peptide group hydrogen exchange. Proteins: Struct., Funct., Genet 17, 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Zhang YZ, Paterson Y, and Roder H (1995) Rapid amide proton exchange rates in peptides and proteins measured by solvent quenching and two-dimensional NMR. Protein Sci. 4, 804–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Englander SW, Englander JJ, McKinnie RE, Ackers GK, Turner GJ, Westrick JA, and Gill SJ (1992) Hydrogen exchange measurement of the free energy of structural and allosteric change in hemoglobin. Science 256, 1684–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Mandell JG, Baerga-Ortiz A, Akashi S, Takio K, and Komives EA (2001) Solvent accessibility of the thrombin-thrombomodulin interface. J. Mol. Biol 306, 575–589. [DOI] [PubMed] [Google Scholar]
- (25).Baerga-Ortiz A, Bergqvist SP, Mandell JG, and Komives EA (2004) Two different proteins that compete for binding to thrombin have opposite kinetic and thermodynamic profiles. Protein Sci. 13, 166–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Fuentes-Prior P, Iwanaga Y, Huber R, Pagila R, Rumennik G, Seto M, Morser J, Light DR, and Bode W (2000) Structural basis for the anticoagulant activity of the thrombin-thrombomodulin complex. Nature 404, 518–525. [DOI] [PubMed] [Google Scholar]
- (27).Peacock RJ, McGrann T, Tonelli M, and Komives EA (2021) Serine protease dynamics revealed by NMR analysis of the thrombin-thrombomodulin complex. Sci. Rep, 9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Hoofnagle AN, Resing KA, Goldsmith EJ, and Ahn NG (2001) Changes in protein conformational mobility upon activation of extracellular regulated protein kinase-2 as detected by hydrogen exchange. Proc. Natl. Acad. Sci. U. S. A 98, 956–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Byun JA, VanSchouwen B, Akimoto M, and Melacini GJ (2020) Allosteric inhibition explained through conformational ensembles sampling distinct “mixed” states. Comput. Struct. Biotechnol. J 18, 3803–3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Hilser VJ, Wrabl JO, and Motlagh HN (2012) Structural and energetic basis of allostery. Annu. Rev. Biophys 41, 585–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kasinath V, Sharp KA, and Wand AJ (2013) Microscopic insights into the NMR relaxation-based protein conformational entropy meter. J. Am. Chem. Soc 135, 15092–15100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Tsai C-J, and Nussinov R (2014) A unified view of “how allostery works. PLoS Comput. Biol 10, e1003394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Handley LD, Treuheit NA, Venkatesh VJ, and Komives EA (2015) Thrombomodulin binding selects the catalytically active form of thrombin. Biochemistry 54, 6650–6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Deredge D, Li J, Johnson KA, and Wintrode PL (2016) Hydrogen/Deuterium Exchange Kinetics Demonstrate Long Range Allosteric Effects of Thumb Site 2 Inhibitors of Hepatitis C Viral RNA-dependent RNA Polymerase. J. Biol. Chem 291, 10078–10088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Deredge DJ, Huang W, Hui C, Matsumura H, Yue Z, Moënne-Loccoz P, Shen J, Wintrode PL, and Wilks A (2017) Ligand-induced allostery in the interaction of the Pseudomonas aeruginosa heme binding protein with heme oxygenase. Proc. Natl. Acad. Sci. U. S. A 114, 3421–3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Ghosh M, Wang LC, Ramesh R, Morgan LK, Kenney LJ, and Anand GS (2017) Lipid-Mediated Regulation of Embedded Receptor Kinases via Parallel Allosteric Relays. Biophys. J 112, 643–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Ramsey KM, Dembinski HE, Chen W, Ricci CG, and Komives EA (2017) DNA and IκBα Both Induce Long-Range Conformational Changes in NFκB. J. Mol. Biol 429, 999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Kromann-Hansen T, Lange EL, Sørensen HP, Hassanzadeh-Ghassabeh G, Huang M, Jensen JK, Muyldermans S, Declerck PJ, Komives EA, and Andreasen PA (2017) Discovery of a novel conformational equilibrium in urokinase-type plasminogen activator. Sci. Rep 7, 3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Lučić I, Rathinaswamy MK, Truebestein L, Hamelin DJ, Burke JE, and Leonard TA (2018) Conformational sampling of membranes by Akt controls its activation and inactivation. Proc. Natl. Acad. Sci. U. S. A 115, E3940–E3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Amatya N, Wales TE, Kwon A, Yeung W, Joseph RE, Fulton DB, Kannan N, Engen JR, and Andreotti AH (2019) Lipid-targeting pleckstrin homology domain turns its autoinhibitory face toward the TEC kinases. Proc. Natl. Acad. Sci. U. S. A 116, 21539–21544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Markwick PRL, Peacock RB, and Komives EA (2019) Accurate Prediction of Amide Exchange in the Fast Limit Reveals Thrombin Allostery. Biophys. J 116, 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Ferreiro DU, Hegler JA, Komives EA, and Wolynes PG (2011) On the role of frustration in the energy landscapes of allosteric proteins. Proc. Natl. Acad. Sci. U. S. A 108, 3499–3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Ghode A, Gross LZF, Tee WV, Guarnera E, Berezovsky IN, Biondi RM, and Anand GS (2020) Synergistic Allostery in Multiligand-Protein Interactions. Biophys. J 119, 1833–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Faull SV, Lau AMC, Martens C, Ahdash Z, Hansen K, Yebenes H, Schmidt C, Beuron F, Cronin NB, Morris EP, and Politis A (2019) Structural basis of Cullin 2 RING E3 ligase regulation by the COP9 signalosome. Nat. Commun 10, 3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Lumpkin RJ, Baker RW, Leschziner AE, and Komives EA (2020) Structure and dynamics of the ASB9 CUL-RING E3 Ligase. Nat. Commun 11, 2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Bhat JY, Miličić G, Thieulin-Pardo G, Bracher A, Maxwell A, Ciniawsky S, Mueller-Cajar O, Engen JR, Hartl FU, Wendler P, and Hayer-Hartl M (2017) Mechanism of Enzyme Repair by the AAA+ Chaperone Rubisco Activase. Mol. Cell 67, 744–756.e6. [DOI] [PubMed] [Google Scholar]
- (47).Lim XX, Chandramohan A, Lim XE, Crowe JEJ, Lok SM, and Anand GS (2017) Epitope and Paratope Mapping Reveals Temperature-Dependent Alterations in the Dengue-Antibody Interface. Structure 25, 1391–1402.e3. [DOI] [PubMed] [Google Scholar]
- (48).Pirrone GF, Emert-Sedlak LA, Wales TE, Smithgall TE, Kent MS, and Engen JR (2015) Membrane-Associated Conformation of HIV-1 Nef Investigated with Hydrogen Exchange Mass Spectrometry at a Langmuir Monolayer. Anal. Chem 87, 7030–7035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Kopcho N, Chang G, and Komives EA (2019) Dynamics of ABC Transporter P-glycoprotein in Three Conformational States. Sci. Rep 9, 15092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Zweckstetter M (2008) NMR: prediction of molecular alignment from structure using the PALES software. Nat. Protoc 3, 679–690. [DOI] [PubMed] [Google Scholar]