

Abstract
Purpose:
To report efficacy and safety of samotolisib (LY3023414; PI3K/mTOR dual kinase and DNA-dependent protein kinase inhibitor) plus enzalutamide in patients with metastatic castration-resistant prostate cancer (mCRPC) following cancer progression on abiraterone.
Patients and Methods:
In this double-blind, placebo-controlled phase Ib/II study (NCT02407054), following a lead-in segment for evaluating safety and pharmacokinetics of samotolisib and enzalutamide combination, patients with advanced castration-resistant prostate cancer with progression on prior abiraterone were randomized to receive enzalutamide (160 mg daily)/samotolisib (200 mg twice daily) or placebo. Primary endpoint was progression-free survival (PFS) assessed by Prostate Cancer Clinical Trials Working Group criteria (PCWG2). Secondary and exploratory endpoints included radiographic PFS (rPFS) and biomarkers, respectively. Log-rank tests assessed treatment group differences.
Results:
Overall, 13 and 129 patients were enrolled in phase Ib and II, respectively. Dose-limiting toxicity was not reported in patients during phase Ib and mean samotolisib exposures remained in the targeted range despite a 35% decrease when administered with enzalutamide. In phase II, median PCWG2-PFS and rPFS was significantly longer in the samotolisib/enzalutamide versus placebo/enzalutamide arm (3.8 vs. 2.8 months; P = 0.003 and 10.2 vs. 5.5 months; P = 0.03), respectively. Patients without androgen receptor splice variant 7 showed a significant and clinically meaningful rPFS benefit in the samotolisib/enzalutamide versus placebo/enzalutamide arm (13.2 months vs. 5.3 months; P = 0.03).
Conclusions:
Samotolisib/enzalutamide has tolerable side effects and significantly improved PFS in patients with mCRPC with cancer progression on abiraterone, and this may be enriched in patients with PTEN intact and no androgen receptor splice variant 7.
Translational Relevance.
The PI3K/mTOR pathway and the DNA-dependent protein kinase (DNA-PK) pathway have been implicated to play an important role in numerous cancers, including prostate cancer. This study investigated treatment with samotolisib (LY3023414), a potent, dual PI3K/mTOR and DNA-PK inhibitor, plus enzalutamide versus placebo plus enzalutamide in patients with metastatic castration-resistant prostate cancer (mCRPC) who had previously progressed on abiraterone. Results indicated that the addition of samotolisib to enzalutamide had acceptable tolerability and clinical benefit in some patients with mCRPC, particularly those who were androgen receptor splice variant 7–negative. The primary and exploratory biomarker results of samotolisib are promising and add notably to the existing literature to guide future research in mCRPC.
Introduction
Prostate cancer is the most prevalent cancer among men in the world (1). Although the majority of patients are diagnosed with localized prostate cancer, about 6% of patients present with metastatic disease with a 5-year survival rate of 29% (2). Androgen deprivation therapy (ADT) via medical or surgical castration has been the mainstay treatment for metastatic prostate cancer. However, prostate cancer cells develop resistance to ADT and progress to castration resistance, leading to poor prognosis and a median overall survival of about 3 to 5 years (3–5). Standard-of-care for patients with metastatic castration-resistant prostate cancer (mCRPC) has been abiraterone or enzalutamide (6, 7). Although abiraterone and enzalutamide do provide survival benefit in some patients with mCRPC in the first-line setting, the majority of patients with prior exposure to abiraterone or enzalutamide develop resistance to further androgen receptor targeting and have a poor prognosis with a hormonal therapy switch (8, 9). Detection of the androgen receptor splice variant 7 (AR-v7) is independently and strongly associated with resistance to enzalutamide and abiraterone in the second-line setting, with more aggressive clinical features (6, 10, 11) thereby indicating an unmet need for alternate therapy options. However, there is a minority who had a durable benefit with abiraterone who go on to have durable cancer control with switching to enzalutamide after abiraterone (12).
The PI3K/protein kinase B/mTOR (PI3K/AKT/ mTOR) signaling pathway is one of the most frequently activated pathways in solid and hematologic malignancies (13–15). Preclinical studies have demonstrated a potential association between the PI3K/AKT/mTOR pathway and androgen receptor (AR) signaling axes in prostate cancer cells developing resistance to ADT (13, 16). A similar relationship was demonstrated in breast cancer with the estrogen receptor pathway and PI3K/AKT/mTOR pathway. In a phase III study of women with breast cancer progression on an aromatase inhibitor, adding everolimus, an oral mTOR inhibitor to exemestane improved progression-free survival (PFS; ref. 17).
Several studies have shown promising results by inhibiting the PI3K/AKT/mTOR pathway in combination with enzalutamide. Inhibition of PI3K signaling with the dual PI3K/mTOR dual inhibitor NVP-BEZ235 (dactolisib) plus enzalutamide caused regression of tumors in mice with prostate-specific phosphatase and tensin homolog (PTEN) loss and PTEN-deficient human prostate cancer xenografts (13). The AKT inhibitor AZD5363 in combination with enzalutamide demonstrated similar synergistic activity in another preclinical study of prostate cancer cell lines, providing further support for combined targeting of AR and PI3K/mTOR pathways (18); combined inhibition of AR and PI3K/mTOR may result in improved benefit with mCRPC (13). Alternatively, an AKT inhibitor ipatasertib demonstrated antitumor activity in a model with glucocorticoid receptor upregulation associated with enzalutamide resistance (19); consequently, providing evidence of positive benefits of inhibiting the PI3K/AKT/mTOR pathway. Furthermore, the phase II (20) and phase III (21) trials showed improvement in radiographic PFS (rPFS) in patients with mCRPC and tumors with PTEN loss when ipatasertib was combined with abiraterone versus abiraterone alone. In addition, DNA-dependent protein kinase (DNA-PK), a key DNA repair protein, is deregulated in many cancers and promotes tumorigenesis. DNA-PK upregulation is associated with aggressive disease, resistance to therapy, and poor outcomes (22). In prostate cancer, evidence suggests the DNA-PK pathway contributes to prostate cancer progression and metastases (23). Notably, the DNA-PK pathway interacts with AR and works as a coregulator of AR in promoting double-strand break repair. This evidence supports the use of DNA-PK inhibitor simultaneously with an AR-targeted therapy for treatment of mCRPC (23).
Samotolisib (LY3023414) is a potent dual inhibitor of PI3K/mTOR and DNA-PK inhibitor with a potential of targeting mCRPC via 2 distinct pathways. Preclinically, samotolisib exhibits potent in vivo efficacy via intermittent target inhibition (24). The phase I study of this novel inhibitor demonstrated a favorable safety profile and single-agent activity in patients with advanced cancers (14). Here, we report the results of a randomized, placebo-controlled phase Ib/II study of samotolisib plus enzalutamide in patients with mCRPC with cancer progression on prior abiraterone. In addition, exploratory biomarker analyses were performed to evaluate the association of activity with AR-v7 emergence or PTEN loss status.
Patients and Methods
Study design
This multipart, phase Ib/II (NCT02407054) study investigated the treatment of samotolisib plus enzalutamide versus placebo plus enzalutamide in patients with mCRPC with cancer progression on prior abiraterone treatment (Fig. 1).
Figure 1.

Study design: Figure describes the study design followed in the phase Ib and phase II part of the trial. BID, twice a day; ENZ, enzalutamide; mCRPC, metastatic castration-resistant prostate cancer; n, number of patients; QD, every day; RR, response rate.
This study consisted of a nonrandomized, open-label lead-in phase; phase Ib, where patients were given samotolisib monotherapy twice daily during the initial week. Thereafter, patients received 200 mg of samotolisib twice daily in combination with 160 mg of enzalutamide once daily for a 28-day cycle. A safety internal monitoring committee evaluated the safety and pharmacokinetics (PK) interactions of the samotolisib and enzalutamide combination. The phase Ib part of the study was designed to evaluate a minimum of 6 patients, determine the recommended phase II dose of samotolisib in combination with enzalutamide, and assess any dose limiting toxicity [DLT; an adverse event (AE) related to the study drug that occurred with the first cycle of a 28-day treatment] before the double-blind, randomized, phase II portion of the study.
Patients were randomized (1:1) to receive enzalutamide 160 mg once daily plus 200 mg samotolisib or placebo twice daily on a 28-day treatment cycle. Randomization was stratified by visceral disease status and prior chemotherapy in the hormone-sensitive setting. Patients were treated until disease progression, death, AEs, or if any other withdrawal criteria were met.
The primary objective in the phase Ib part of the study was to assess the safety, tolerability, and PK of samotolisib with or without enzalutamide. In the phase II, the primary objective was to compare PFS measured by prostate-specific antigen (PSA) or radiographic or symptomatic progression per Prostate Cancer Clinical Trials Working Group (PCWG2) criteria (25) between the two treatment arms. As a coprimary, PFS defined by PSA progression and rPFS were evaluated. The secondary objectives included comparing overall response rates (ORR) among patients who had measurable disease at baseline using Response Evaluation criteria in Solid Tumors (RECIST) version 1.1 (26), time to clinical or PSA progression, and maximum decline in PSA between the treatment arms. Exploratory analyses examined blood AR-v7 and PTEN tumor status as potential biomarkers to predict clinical efficacy of samotolisib and disease progression.
Patients
Men ≥18 years of age with history of histologically and cytologically confirmed mCRPC with progression on prior abiraterone treatment, underwent surgical or medical castration, had testosterone levels <50 ng/dL, and an Eastern Cooperative Oncology Group performance status (ECOG PS) of 0 or 1 were considered eligible for this study. Patients required a minimum 6-week washout period for long-acting agents including nilutamide and bicalutamide and a minimum 4-week washout period for the short-acting agent flutamide. Patients on prior treatment for CRPC, known brain metastases, serious preexisting conditions, and comorbidities such as seizure, hypertension, insulin-dependent diabetes mellitus, or acute or chronic leukemia were excluded.
This study was conducted in accordance with the International Conference on Harmonization requirements for Good Clinical Practice and with the consensus ethics principles derived from the International Ethics Guidelines outlined in the Declaration of Helsinki and Council for International Organizations of Medical Sciences (27). All patients provided written informed consent prior to study enrollment.
Study assessments
PK evaluation
PK blood samples for patients participating in phase Ib were taken on the last day of the single-agent samotolisib week treatment (that is 1 day prior to first combination treatment), on day 1 of the combination treatment, on day X (any day between day 15 and day 28), and day X+1 (any day between days 16 and day 29) in cycle 1. Moreover, PK samples were also collected on day 1 of cycle 2 and 3. Standard noncompartmental PK analysis was performed using Phoenix 8.1 WinNonlin (Certara) to derive area under curve (AUC), maximum observed concentration (Cmax), and other standard PK parameters including apparent clearance (CL/F), volume of distribution (Vz/F), and half-life (t1/2).
Efficacy
All patients randomized in phase II comprised the full analysis set (FAS). Progression-free survival by PCWG2 criteria (25), response to treatment using the RECIST version 1.1 (26), and monthly PSA were evaluated every 2 cycles for patients with bone disease. PSA levels were evaluated monthly for the course of the study and were the primary efficacy endpoints for patients with no measurable disease at baseline. PSA was collected on day 1 of each cycle and, if PSA progression occurred, it was confirmed at the next study visit, 4 weeks later. Tumor response was assessed by CT scans or MRI according to RECIST v1.1 (26) at screening, and thereafter every two cycles through follow-up visits. Lymph node and bone lesions were evaluated using PCWG2 criteria (25). For patients with progression based on PCWG2 and RECIST v1.1 criterion, the earliest determined date of progression was used to derive progression-based efficacy outcomes.
Safety and tolerability
Safety and tolerability were assessed through clinical and laboratory evaluations on days 1 and 15 of cycle 1 and on the first day of each subsequent cycle for the duration of the study. Adverse events were graded according to the Common Terminology Criteria for Adverse Events (CTCAE v.4.03). During the phase Ib part, related AEs occurring during the first cycle were defined as DLTs, if they met any of the following criteria — grade 4 thrombocytopenia, neutropenia (≥7 days) and grade 3 febrile neutropenia, grade ≥3 thrombocytopenia with grade >2 hemorrhage, or grade ≥3 nonhematologic toxicity despite maximal medical management were considered a DLT. A treatment delay of ≥14 days due to unresolved AE or any other clinically significant drug-related AE not responding to supportive care were considered DLTs.
Exploratory biomarkers
AR-v7 splice variant status (positive or negative) was assessed in baseline plasma exosome mRNA, by quantitative PCR (qPCR; ref. 14), validated in the Clinical Diagnostics Laboratory (Eli Lilly and Company) prior to use.
To define the patient population with PTEN loss tumors in the exploratory analyses, PTEN status was assessed by an IHC assay performed on archival soft tumor tissues at Neogenomics, using a validated assay. PTEN homolog protein expression was determined on formalin-fixed and paraffin-embedded sections using PTEN, Clone 6H2.1 (Dako). A qualified pathologist evaluated results according to prespecified interpretation guideline: a negative PTEN specimen was defined as having <5% of cells that exhibited staining in nuclear and/or cytoplasmic pattern at any intensity and a positive PTEN specimen as having ≥5% of cells that exhibited staining in nuclear and/or cytoplasmic pattern at any intensity. The assay was not validated for bone metastases due to the need for a decalcification step and only soft tissue samples were used. The analysis was conducted prior to unblinding. rPFS by PTEN status was examined.
Statistical analyses
A total of 92 PFS events were needed to have ≥80% power to test the primary hypothesis using a one-sided log-rank test at the 0.2 significance level. All efficacy analyses were conducted using the FAS, whereas the sensitivity analyses were performed on the per protocol analysis set (PPAS; patients on treatment without major protocol deviations). Treatment group differences for the primary endpoint were tested using unstratified log-rank tests. The stratified analysis was performed as sensitivity analysis. The HR of the treatment effect was calculated using Cox proportional hazards models, and median PFS was estimated using the Kaplan–Meier method. Alternatively, treatment group differences for ORR, defined as the best objective response per RECIST v1.1 (26), were evaluated using χ2 and Cochran–Mantel–Haenszel test adjusted by stratification factors. All statistical analyses were conducted using SAS version 9.2 or higher in Unix.
Data sharing statement
Lilly provides access to all individual participant data collected during the trial, after anonymization, with the exception of pharmacokinetic or genetic data. Data are available to request 6 months after the indication studied has been approved in the United States and European Union and after primary publication acceptance, whichever is later. No expiration date of data requests is currently set once data are made available. Access is provided after a proposal has been approved by an independent review committee identified for this purpose and after receipt of a signed data sharing agreement. Data and documents, including the study protocol, statistical analysis plan, clinical study report, blank or annotated case report forms, will be provided in a secure data sharing environment. For details on submitting a request, see the instructions provided at www.vivli.org.
Results
A total of 142 patients were enrolled in the study and treated between April 2015 and April 2020. Phase Ib consisted of 13 patients and phase II enrolled 129 patients.
Patients and disease characteristics
Phase Ib
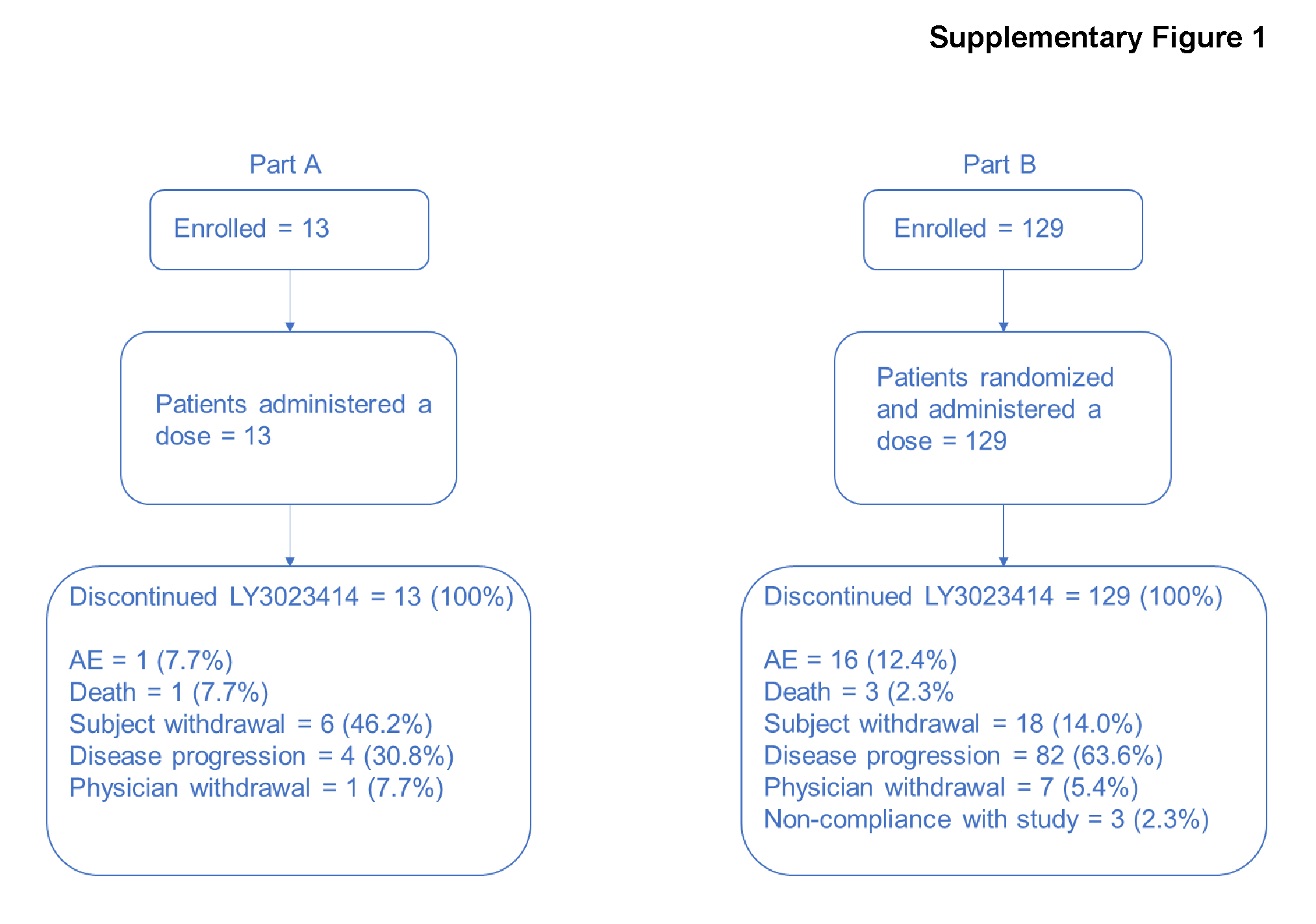
The median patient age was 77.0 years (range, 61–88 years) and majority of the patients were white (84.6%). All 13 patients in this phase had an ECOG PS of either 0 (53.8%) or 1 (46.2%). Of the 13 patients enrolled, 11 patients received samotolisib (200 mg twice daily) monotherapy followed by samotolisib (200 mg twice daily) plus enzalutamide (160 mg once daily) in phase Ib. The remaining 2 patients discontinued after samotolisib monotherapy due to physician decision and withdrawal of patient consent, respectively. Withdrawal of consent (53.8%), progressive disease (38.5%), and nonstudy treatment-related death (7.7%) were primary reasons for study discontinuation. The primary reason for samotolisib discontinuation was withdrawal of consent (46.2%; Supplementary Fig. S1).
Phase II
For the overall population in phase II, the median age was 70.0 (45–91) years, and the majority of patients were white (83.7%) with an ECOG score of either 0 (53.5%) or 1 (45.0%). Most patients had adenocarcinoma (85.3%) with bone metastasis (93.0%). The median time from initial diagnosis was 59.0 months (range, 4–287). Prior docetaxel use for metastatic hormone-sensitive prostate cancer was reported in 26.4% of patients and 20.2% had visceral disease (Table 1). A total of 129 patients were randomized to samotolisib (200 mg twice daily) plus enzalutamide (160 mg once daily) arm (n = 65) and the placebo plus enzalutamide (160 mg once daily) arm (n = 64). Patients in phase 2 discontinued the study (72.1%) and samotolisib (63.6%) predominantly due to progressive disease (Supplementary Fig. S1).
Table 1.
Demographic and disease characteristics.
| Phase Ib | Samotolisib + enzalutamide | Placebo + enzalutamide | Total, phase II | |
|---|---|---|---|---|
| Parameters | n = 13 | n = 65 | n = 64 | N = 129 |
| Age in years, median (range) | 77 (61–88) | 69 (49–86) | 71 (45–91) | 70 (45–91) |
| Race, white | 11 (84.6) | 53 (81.5) | 55 (85.9) | 108 (83.7) |
| Time since initial diagnosis, median (range), months | 57.5 (13–256) | 67.1 (4–287) | 59.0 (4–287) | |
| ECOG PS, 0 / 1, n | 7/6 | 34/30 | 39/25 | 69/58 |
| Prior docetaxel for mHSPC | 17 (26.2) | 17 (26.6) | 34 (26.4) | |
| Visceral disease | 12 (18.5) | 14 (21.9) | 26 (20.2) | |
| Disease sites | ||||
| Bone | 59 (90.8) | 61 (95.3) | 120 (93.0) | |
| Distant lymph nodes | 19 (29.2) | 14 (21.9) | 33 (25.6) | |
| Local/regional lymph nodes | 18 (27.7) | 17 (26.6) | 35 (27.1) | |
| Liver | 5 (7.7) | 4 (6.3) | 9 (7.0) | |
| Lung | 7 (10.8) | 8 (12.5) | 15 (11.6) | |
Note: All values are represented in n (%), unless otherwise stated.
Abbreviations: mHSPC, metastatic hormone-sensitive prostate cancer; N, number of patients; n, number of patients per category.
PK
PK data showed an increase in samotolisib apparent clearance and decrease in samotolisib AUC by 35% following concomitant administration of samotolisib and enzalutamide compared with samotolisib monotherapy administration. The reported AUC, Cmax, and t1/2 of single-agent samotolisib were 3,230 ng/h/mL (59%), 1,020 ng/mL (55%), and 1.72 hours (27%), respectively. The PK parameters AUC, Cmax, and t1/2 for samotolisib in the combination of samotolisib and enzalutamide were 1,820 ng/h/mL (56%), 541 ng/mL (68%), and 2.07 hours (35%), respectively. These exposures are in the efficacious range, corresponding to average PK profile with samotolisib concentration above the EC50 (determined by the pharmacokinetic/pharmacodynamic model) for approximately 8 to 9 hours per day (under twice-daily dosing).
Efficacy
PFS
As of the data cutoff, a total of 46 (70.8%) patients in the samotolisib plus enzalutamide arm compared with 56 (87.5%) patients in the placebo plus enzalutamide arm had disease progression (radiographic, symptomatic, or PSA) or death. Median PCWG-PFS in the samotolisib plus enzalutamide arm was statistically significantly longer compared with the placebo plus enzalutamide arm [3.78 months vs. 2.83 months; HR, 0.59; 95% confidence interval (CI), 0.40–0.87; P = 0.003; Fig. 2A]. This effect was consistent across subgroups evaluated. In the poststratified analysis based on presence of visceral disease and administration of prior chemotherapy in the hormone-sensitive setting, a significantly longer PFS was observed in the samotolisib plus enzalutamide arm compared with placebo plus enzalutamide (HR, 0.60; 95% CI, 0.41–0.89; P = 0.005).
Figure 2.

PFS and rPFS of patients from phase II. A, The PFS of samotolisib + enzalutamide arm versus placebo + enzalutamide arm. B, The rPFS of samotolisib + enzalutamide arm versus placebo + enzalutamide arm.
Overall, 32 (49.2%) patients in the samotolisib plus enzalutamide arm compared with 44 (68.8%) patients in the placebo plus enzalutamide arm had radiologic progression. Median rPFS was 10.2 months in the samotolisib plus enzalutamide arm and 5.5 months in the placebo plus enzalutamide arm (HR, 0.64; 95% CI, 0.41–1.01; P = 0.03; Fig. 2B).
Symptomatic progression was observed in 27 (41.5%) patients in the samotolisib plus enzalutamide arm and 29 (45.3%) patients in the placebo plus enzalutamide arm. The median PFS was 8.74 months in samotolisib plus enzalutamide arm and 9.82 months in the placebo plus enzalutamide arm. PSA progression was observed in 33 (50.8%) patients with median PFS of 5.06 months in the samotolisib plus enzalutamide arm and in 42 (65.6%) patients with median PFS of 3.61 months in the placebo plus enzalutamide. No significant difference in symptomatic and PSA progression was observed between the arms.
Overall response
Among patients with measurable disease, 3 (4.6%) patients each in both groups demonstrated partial response per RECIST v1.1. The disease control rate per RECIST v1.1 was 80% in the samotolisib plus enzalutamide arm and 84.3% in the placebo plus enzalutamide arm. Among patients taking samotolisib, 13 (20.0%) patients had a 50% decline in PSA compared with 16 (25.0%) patients in the placebo arm (Fig. 3).
Figure 3.

Waterfall plot of best prostate-specific antigen (PSA) response.
Safety
Phase Ib
All 13 patients had at least 1 drug-related AE. Ten (76.9%) patients had CTCAE grade ≥3, of which 5 (38.5%) patients had drug related CTCAE grade ≥3 AEs. Nausea (76.9%), fatigue (53.8%), and diarrhea (53.8%) were commonly observed among patients. Severe AEs occurred in 5 (38.5%) patients, 1 (7.7%) of which was related to the study treatment. No DLTs were reported in the phase Ib portion of the study.
A total of 6 patients (3 each for samotolisib and enzalutamide) had dose reductions due to AEs. Dose interruptions were observed in 9 (69.2%) patients treated with samotolisib and 5 (38.5%) patients treated with enzalutamide. The mean dose intensity for samotolisib and enzalutamide was 348.8 mg/days and 138.4 mg/days, respectively. Two patients discontinued study treatment due to an AE (Table 2).
Table 2.
Safety profile of patients in phase Ib and phase II.
| Phase II | |||
|---|---|---|---|
| Samotolisib+enzalutamide | Placebo+enzalutamide | ||
| Phase Ib | n = 65 | n = 64 | |
| Total AEsa | 13 (100.0) | 64 (98.5) | 63 (98.4) |
| Grade ≥3 AEs | 10 (76.9) | 35 (53.8) | 33 (51.6) |
| Discontinuation due to AE | 2 (15.4) | 13 (20.0) | 4 (6.3) |
| Serious AEs | 5 (38.5) | 13 (20.0) | 12 (18.8) |
| Death | 0 (0.0) | 0 (0.0) | 3 (4.7) |
| TEAEs, >15% of all patients, any grade | |||
| Fatigue | 7 (53.8) | 41 (63.1) | 36 (56.3) |
| Nausea | 10 (76.9) | 38 (58.5) | 25 (39.1) |
| Diarrhea | 7 (53.8) | 40 (61.5) | 10 (15.6) |
| Decreased appetite | 5 (38.5) | 20 (30.8) | 14 (21.9) |
| Anemia | 2 (15.4) | 12 (18.5) | 19 (29.7) |
| Constipation | 2 (15.4) | 11 (16.9) | 12 (18.8) |
Note: All values are represented in n (%), unless otherwise specified.
Abbreviation: n, number of patients in each category.
aTotal AEs could be drug-related or non–drug-related.
On the basis of safety, tolerability, and PK of samotolisib monotherapy in the phase I trial (14) and the samotolisib plus enzalutamide combination in this study, the samotolisib dose of 200 mg twice daily was considered for the phase II part of the study.
Phase II
Overall, 127 (98.4%) patients experienced at least 1 AE across both the samotolisib plus enzalutamide arm and placebo plus enzalutamide arm, 88.4% of patients experienced at least 1 drug-related event and 29.5% experienced AEs with CTCAE grade ≥3. The most common treatment-emergent AEs (TEAE) per system organ class were gastrointestinal disorders (81.7%), general disorders and administrative site conditions (74.6%), and musculoskeletal and connective tissue disorders (57.0%). Fatigue (63.1%), diarrhea (61.5%), and nausea (58.5%) were the most common AEs in the samotolisib plus enzalutamide arm, whereas fatigue (56.3%), nausea (39.1%), and anemia (29.7%) were commonly observed in the placebo plus enzalutamide arm. Serious adverse events were observed in 19.4% of patients with 3.1% of patients suffering from drug-related SAEs. Seventeen (13.2%) patients in both treatment groups discontinued treatment due to AEs (Table 2). Seven (5.4%) patients died within 30 days of treatment discontinuation. No deaths in the samotolisib plus enzalutamide and 3 (2.3%) deaths in the placebo plus enzalutamide arm due to AEs including 1 event each of sepsis, anemia with atrial fibrillation and elevated troponin I, and intracranial hemorrhage were observed. The other 4 patients died due to underlying disease. Notably, the prevalence of glucose events was low. Grade 1 and grade 2 TEAEs of hyperglycemia occurred in 4 (6.2%) patients and 2 (3.1%) patients in the samotolisib plus enzalutamide arm and 5 (7.8%) patients and 1 (1.6%) patient in the placebo plus enzalutamide arm, respectively. The average HbA1c level did not increase over time in the experimental treatment arm (data not shown).
Exploratory biomarkers
Assessments of the AR-v7 variant were performed in 122 baseline plasma samples using qualitative reporting. The prevalence of AR-v7 per plasma exosome mRNA qPCR assay at baseline was 14%. A total of 9 (13.8%) and 8 (12.5%) patients in the samotolisib plus enzalutamide and placebo plus enzalutamide arms, respectively, had AR-v7–positive results. Conversely, 51 (78.5%) patients in the samotolisib plus enzalutamide arm and 54 (84.4%) patients in the placebo plus enzalutamide arm had AR-v7–negative results. Among patients with AR-v7 negative status, the median rPFS was significantly longer in the samotolisib plus enzalutamide arm compared with the placebo arm (13.2 months vs. 5.3 months; HR, 0.52; 95% CI, 0.28–0.95; P = 0.03; Fig. 4A). Among patients with AR-v7–positive status, no significant difference in the median rPFS was observed between the samotolisib plus enzalutamide and placebo plus enzalutamide arms (5.5 months vs. 3.6 months; HR, 0.99; 95% CI, 0.27–3.63; P = 0.99; Fig. 4B).
Figure 4.

rPFS by AR-v7 status. A, The PFS of samotolisib + enzalutamide arm versus placebo + enzalutamide arm in patients without AR-v7. B, The PFS of samotolisib + enzalutamide arm versus placebo + enzalutamide arm in patients with AR-v7.
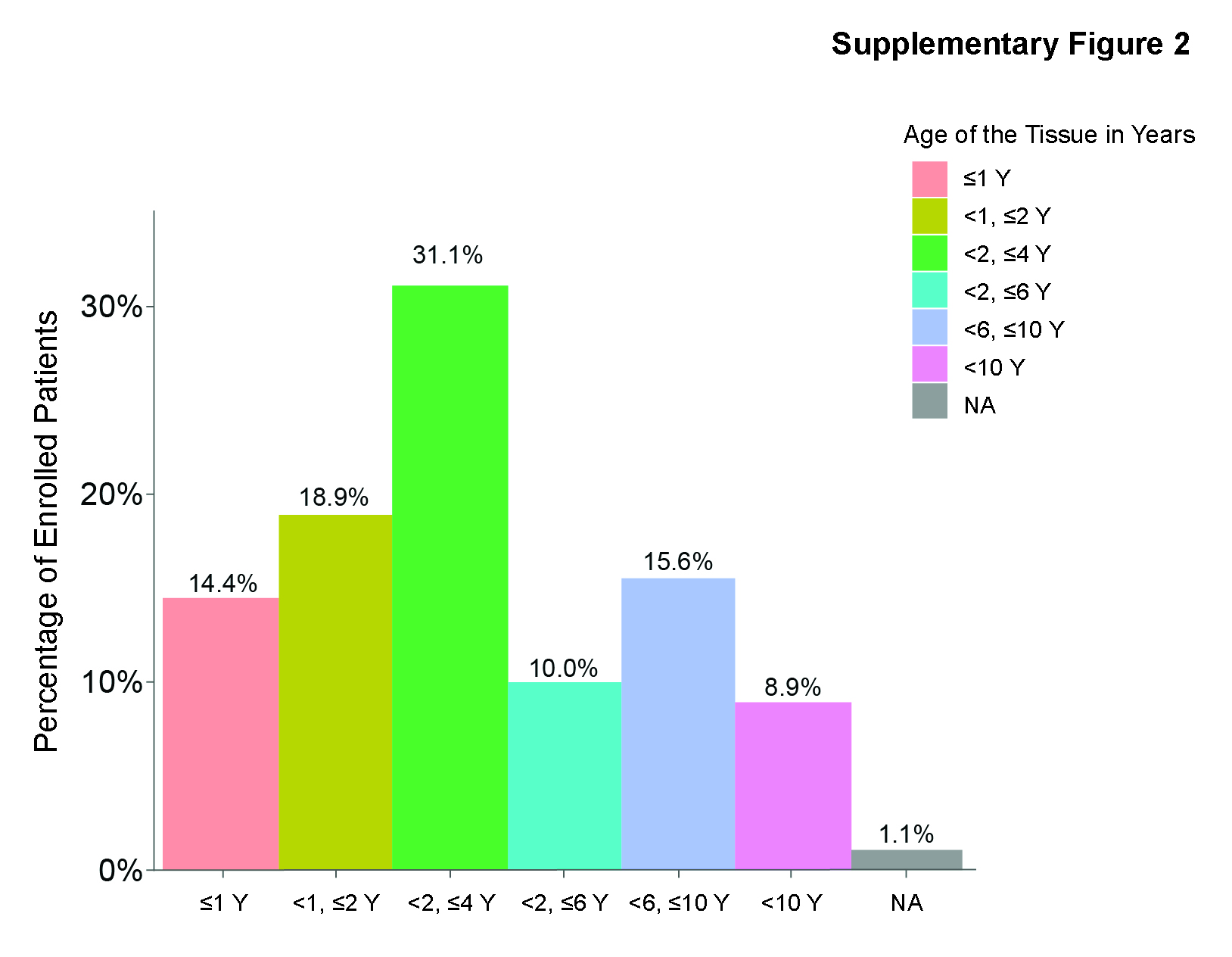
Evaluation of PTEN status by IHC was carried out on 90 archival soft tumor tissues collected from phase II patients. The average time between collection of archival tissue samples and initiation of treatment was 4.6 years (≤1 year – >10 years; Supplementary Fig. S2) with 71 samples obtained from pre-ADT localized disease and 19 samples from metastatic disease (Supplementary Table S1). Of the 90 patients whose tissue samples were stained by IHC, 30 (33.3%) patients had tumors with PTEN loss (n = 23 primary and n = 7 metastatic lesions), of which 18 (27.7%) patients were in the samotolisib plus enzalutamide and 12 (18.8%) were in the placebo plus enzalutamide arm. Among patients with tumors with no PTEN loss, median rPFS was 13.2 months in the samotolisib plus enzalutamide arm compared with 3.6 months in the placebo plus enzalutamide arm (HR, 0.49; 95% CI, 0.22–1.08; P = 0.07; Fig. 5A). Among patients with PTEN loss, median rPFS was 7.2 months in the samotolisib plus enzalutamide arm compared with 4.5 months in the placebo plus enzalutamide arm (HR, 0.66; 95% CI, 0.25–1.72; P = 0.40; Fig. 5B).
Figure 5.

rPFS by PTEN (IHC) assay. A, The PFS of samotolisib + enzalutamide arm versus placebo + enzalutamide arm in patients with PTEN loss. B, The PFS of samotolisib + enzalutamide arm versus placebo + enzalutamide arm in patients without PTEN loss.
Discussion
This randomized placebo-controlled trial demonstrated that samotolisib could be combined with enzalutamide with an acceptable toxicity profile and a modest improvement in PFS (PSA, radiographic, and death) and rPFS (radiographic and death) in patients with mCRPC with cancer progression on prior abiraterone treatment.
Samotolisib is a novel PI3k/mTOR inhibitor with intermittent target inhibition leading to potent antitumor activity in vivo. A potential advantage of the intermittent “quick-on/quick-off” inhibition is enhanced clinical tolerability and reduced resistance (14). An assessment of the AE profile and outcome by biomarker have further provided insights into its clinical activity.
As enzalutamide is a CYP3A4 inducer and samotolisib a CYP3A4 substrate, the combination of samotolisib plus enzalutamide showed an increase in apparent clearance and decrease in AUC compared with monotherapy administration. Despite that decrease in samotolisib exposure when administered in combination with enzalutamide, the observed AUC, Cmax, and t1/2 of 20 mg twice daily were considered within efficacious range. As single-agent monotherapy in the phase I part of the study, samotolisib PK parameters including AUC, Cmax, and t1/2 were similar in this prostate cancer population compared with the data reported in the phase I trial (14). Similar results were observed in a phase II trial of buparlisib (28). The authors suggested that insufficient drug levels were observed when buparlisib was concurrently used with enzalutamide, limiting the activity and consequently the efficacy (28). Another phase I study of capivasertib demonstrated a mean 40% decrease in the concentration of capivasertib when administered with enzalutamide (29). The mean state AUC of enzalutamide was consistent with reported data, thereby indicating that enzalutamide PK was unaffected by the administration by samotolisib.
This study met its primary endpoint of PCGW2-PFS by demonstrating a longer PFS for samotolisib plus enzalutamide versus placebo plus enzalutamide. Despite the small absolute differences in median and the small sample size, the overall HR was statistically significant. Although this study was not powered a priori to assess the rPFS, the primary endpoint was supported by a longer median rPFS in the samotolisib plus enzalutamide arm. The initial decision to use the composite endpoint with PSA as part of the analysis was made to expedite the readout of efficacy phase II screening study, we would not recommend this for future studies.
A clinically meaningful delay in rPFS in patients with AR-v7–negative status was observed in this study. Interestingly, treatment effects with improved rPFS were observed without changes to the PSA levels. This would suggest PSA-based metrics to assess efficacy of potent AR inhibition in mCRPC post-abiraterone setting should not be used as a sole metric. The exploratory biomarker suggests that samotolisib plus enzalutamide combination was more effective than placebo plus enzalutamide in patients who were negative for AR-v7, a disease setting when second-line hormonal therapy is more likely to have activity (6). Moreover, it appeared patients with no PTEN loss treated with samotolisib plus enzalutamide compared with the patients treated with placebo plus enzalutamide group also had a delay in progression as measured by rPFS. In contrast, a phase II study of abiraterone plus ipatasertib and a follow-up randomized phase III study demonstrated an improved rPFS versus abiraterone plus placebo in patients with tumors with PTEN loss as assessed by IHC (20, 21). The discordance in these results could be due to the age of available archival tissue samples (which might underrepresent the actual biology of the patient at the time of collection), tumor heterogeneity (which is exacerbated by obtaining a biopsy from only one site) and small sample size. The latter was due to the assay only being validated for soft tissue (primary or metastatic disease) and does not represent bone metastases. Unlike the ipatasertib data where the benefit is enriched in patients with PTEN loss (20, 30), it is notable that samotolisib also has activity via DNA-PK inhibition and this may account for the benefit seen in patients with PTEN intact. Moreover, the data strongly suggests that the samotolisib does not overcome the resistance associated with AR-v7.
The findings of the samotolisib plus enzalutamide combination is of particular interest as the median rPFS of 10.2 months in the samotolisib plus enzalutamide arm compares favorably with the observations from the CARD trial. Specifically, the hormone switch arm of abiraterone or enzalutamide after the other hormonal agent plus docetaxel had a median rPFS of 3.7 months, which approximates the 5.5 months in the placebo plus enzalutamide arm of this study (8).
The safety profile of samotolisib plus enzalutamide is consistent with previous reports for samotolisib and enzalutamide as single agents (14, 31). As demonstrated by a similar dose intensity of enzalutamide in both the treatment and placebo arms, dosing of enzalutamide was not impacted by administration of samotolisib. Hyperglycemia is a known class effect of PI3K inhibitors (32); however, similar to the phase I study of samotolisib (14), infrequent and mild cases of hyperglycemia were observed in this trial. Treatment emergent adverse events of hyperglycemia did not differ by study arm, hypothetically due to the short half-life of samotolisib and the ability to intermittently target PI3K, unlike other PI3K inhibitors (14). It is also possible that we are not seeing a pronounced glucose effect while seeing some clinical activity because samotolisib is also working through the DNA-PK pathway and not solely PI3K pathway (24). Similarly, another commonly observed AE, mucositis (14, 33), was not observed frequently in patients treated with samotolisib. The absence of DLTs supported the further development of samotolisib twice daily as a potential inhibitor of multiple cancer promoting pathways. Enzalutamide has strong clinical drug-drug interactions potential as a CY3A4 inducer, which may diminish the efficacy of medications given concurrently (34). However, samotolisib was able to maintain clinically relevant exposure in combination with enzalutamide. More research is needed to determine the potential mechanisms underlying this finding.
Some limitations to this study should be considered when interpreting these results. This study did not preselect patients based on pathway-relevant biomarkers. Survival data post protocol therapy was not collected, and thus there is no overall survival data at this time. It should also be noted the biomarker work is exploratory and needs to be confirmed in future studies.
Conclusions
The combination of samotolisib with enzalutamide significantly improved PFS in patients with mCRPC with cancer progression on abiraterone. In addition, the rPFS benefit with the combination appeared to be most pronounced in patients with negative AR-v7 status, indicating that samotolisib may enhance the activity of enzalutamide in the post-abiraterone setting especially when the disease is likely to still have some reliance on AR signaling and greater chance of benefit from the hormone switch strategy. Moreover, the findings of this study suggested that enhancing the activity of potent direct AR inhibition in mCRPC post-abiraterone along with inhibition of compensatory oncogenic pathways might be more effective in patients likely to respond to AR inhibition. However, other strategies to overcome resistance mechanisms associated with the presence AR-v7 may be required for patients who have a more cell-autonomous disease.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
We thank all the patients, their families, the study sites, and the study personnel who participated in this study. We would like to thank Shaleen Multani, an employee of Eli Lilly Services India Private Limited, for providing writing support. This work was supported by grants from Eli Lilly and Company.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Disclosures
C.J. Sweeney reports personal fees from Lilly and Genentech and grants and personal fees from Bayer, Sanofi, Pfizer, Astellas, and Janssen during the conduct of the study; in addition, C.J. Sweeney has a patent for KDM5D and ZFP36 in prostate cancer issued to Dana-Farber Cancer Institute, a patent for Cabozantinib and Abiraterone in prostate cancer issued to Exelixis, and a patent for Parthenolide in Cancer issued to Indiana University. S. Babu reports grants from Eli Lilly paid to Fort Wayne Medical Oncology & Hematology during the conduct of the study. B. Mehlhaff reports other support from Lilly during the conduct of the study; and personal fees from Pfizer, Astellas, AstraZeneca, Bayer, Dendreon, and Merck outside the submitted work. O.B. Goodman reports personal fees from Pfizer, AstraZeneca, Bayer, and Janssen outside the submitted work. D. Morris reports personal fees from Astellas, Pfizer, Bayer, AstraZeneca, Dendreon, and Janssen outside the submitted work. P. Sieber reports personal fees from Pfizer, Astellas, Dendreon, Bayer, and Myovant outside the submitted work. W. Zhang reports non-financial support from Sarah Cannon Research Institute during the conduct of the study; and is a shareholder of Eli Lilly and Company. V. Wacheck reports other support from Eli Lilly and Company during the conduct of the study. A.M. Szpurka reports other support from Eli Lilly and Company outside the submitted work (and Eli Lilly and Company employment and salary). S. Callies reports other support from Eli Lilly and Company outside the submitted work. B.K. Lin reports personal fees from Eli Lilly and Company during the conduct of the study; and owns stock in Eli Lilly and Company. J.C. Bendell reports grants from Gilead, Genentech/Roche, BMS, Five Prime, Lilly, Merck, MedImmune, Celgene, EMD Serono, Taiho, Macrogenics, GSK, Novartis, OncoMed, LEAP, TG Therapeutics, AstraZeneca, BI, Daiichi Sankyo, Bayer, Incyte, Apexigen, Koltan, SynDevRex, Forty Seven, AbbVie, Array, Onyx, Sanofi, Takeda, Celldex, Eisai, Agios, Cytomx, Nektar, ARMO, Boston Biomedical, Ipsen, Merrimack, Tarveda, Tyrogenex, Oncogenex, Marshall Edwards, Pieris, Mersana, Calithera, Blueprint, Evelo, FORMA, Merus, Jacobio, Effector, Novocare, Arrys, Tracon, Sierra, Innate, Arch Oncology, Prelude Oncology, Unum Therapeutics, Vyriad, Harpoon, ADC, Amgen, Pfizer, Millennium, Imclone, Acerta Pharma, Rgenix, Bellicum, Gossamer Bio, Arcus Bio, Seattle Genetics, TempestTx, Shattuck Labs, Synthorx Inc, Revolution Medicines, Inc., Bicycle Therapeutics, Zymeworks, Relay Therapeutics, Scholar Rock, NGM Biopharma, Stemcentrx, Beigene, CALGB, Cyteir Therapeutics, Foundation Bio, Innate Pharma, Morphotex, OncXerna, NuMab, AtlasMedx, Treadwell Therapeutics, IGM Biosciences, Mabspace, Hutchinson MediPharma, REPARE Therapeutics, NeoImmune Tech, Regeneron, PureTech Health, Phoenix Bio, Cyteir, Molecular Partners, Innate, Torque, Tizona, Janssen, Tolero, Amgen, Moderna Therapeutics, Agios, Continuum Clinical, Samsung Bioepios, Pfizer, and Fusion Therapeutics during the conduct of the study and outside the submitted work. No disclosures were reported by the other authors.
Authors' Contributions
C.J. Sweeney: Conceptualization, investigation, writing–original draft, writing–review and editing. I.J. Percent: Investigation, writing–review and editing. S. Babu: Investigation, writing–original draft, writing–review and editing. J.L. Cultrera: Conceptualization, investigation, methodology, writing–review and editing. B.A. Mehlhaff: Investigation, writing–review and editing. O.B. Goodman: Investigation, writing–review and editing. D.S. Morris: Investigation, writing–review and editing. I.D. Schnadig: Investigation, writing–review and editing. C. Albany: Investigation, writing–original draft, writing–review and editing. N.D. Shore: Investigation, writing–review and editing. P.R. Sieber: Investigation, writing–review and editing. S.C. Guba: Conceptualization, investigation, methodology, writing–original draft, writing–review and editing. W. Zhang: Formal analysis, writing–original draft, writing–review and editing. V. Wacheck: Conceptualization, investigation, methodology, writing–review and editing. G.P. Donoho: Conceptualization, formal analysis, writing–original draft, writing–review and editing. A.M. Szpurka: Formal analysis, investigation, writing–original draft, writing–review and editing. S. Callies: Formal analysis, methodology, writing–original draft, writing–review and editing. B.K. Lin: Formal analysis, investigation, writing–original draft, writing–review and editing. J.C. Bendell: Conceptualization, investigation, methodology, writing–review and editing.
References
- 1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2021;71:209–49. [DOI] [PubMed] [Google Scholar]
- 2. Kelly SP, Anderson WF, Rosenberg PS, Cook MB. Past, current, and future incidence rates and burden of metastatic prostate cancer in the United States. Eur Urol Focus 2018;4:121–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Omlin A, Pezaro C, Mukherji D, Mulick Cassidy A, Sandhu S, Bianchini D, et al. Improved survival in a cohort of trial participants with metastatic castration-resistant prostate cancer demonstrates the need for updated prognostic nomograms. Eur Urol 2013;64:300–6. [DOI] [PubMed] [Google Scholar]
- 4. Patrikidou A, Loriot Y, Eymard JC, Albiges L, Massard C, Ileana E, et al. Who dies from prostate cancer? Prostate Cancer Prostatic Dis 2014;17:348–52. [DOI] [PubMed] [Google Scholar]
- 5. Ritch C, Cookson M. Recent trends in the management of advanced prostate cancer. F1000Res 2018;7:F1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med 2014;371:1028–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Eisenberger MA, Antonarakis ES. Hormonal therapy or chemotherapy for metastatic prostate cancer - Playing the right CARD. N Engl J Med 2019;381:2564–6. [DOI] [PubMed] [Google Scholar]
- 8. de Wit R, de Bono J, Sternberg CN, Fizazi K, Tombal B, Wulfing C, et al. Cabazitaxel versus abiraterone or enzalutamide in metastatic prostate cancer. N Engl J Med 2019;381:2506–18. [DOI] [PubMed] [Google Scholar]
- 9. de Bono J, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. Olaparib for metastatic castration-resistant prostate cancer. N Engl J Med 2020;382:2091–102. [DOI] [PubMed] [Google Scholar]
- 10. Armstrong AJ, Halabi S, Luo J, Nanus DM, Giannakakou P, Szmulewitz RZ, et al. Prosepctive multicenter validation of androgen receptor splice variant 7 and hormone therapy resistance in high-risk castration-resistant prostate cancer: The PROPHECY Study. J Clin Oncol 2019;37:1120–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bastos DA, Antonarakis ES. CTC-derived AR-V7 detection as a prognostic and predictive biomarker in advanced prostate cancer. Expert Rev Mol Diagn 2018;18:155–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. de Bono JS, Chowdhury S, Feyerabend S, Elliott T, Grande E, Melhem-Bertrandt A, et al. Antitumour activity and safety of enzalutamide in patients with metastatic castration-resistant prostate cancer previously treated with abiraterone acetate plus prednisone for ≥24 weeks in Europe. Eur Urol 2018;74:37–45. [DOI] [PubMed] [Google Scholar]
- 13. Carver BS, Chapinski C, Wongvipat J, Kieronymus H, Chen Y, Chandarlapaty S, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 2011;19:575–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bendell JC, Varghese AM, Hyman DM, Bauer TM, Pant S, Callies S, et al. A first-in-human phase 1 study of LY3023414, an oral PI3K/mTOR dual inhibitor, in patients with advanced cancer. Clin Cancer Res 2018;24:3253–62. [DOI] [PubMed] [Google Scholar]
- 15. Braglia L, Zavatti M, Vinceti M, Martelli AM, Marmiroli S. Deregulated PTEN/PI3K/AKT/mTOR signaling in prostate cancer: Still a potential druggable target? Biochim Biophys Acta Mol Cell Res 2020;1867:118731. [DOI] [PubMed] [Google Scholar]
- 16. Bitting RL, Armstrong AJ. Targeting the PI3K/Akt/mTOR pathway in castration-resistant prostate cancer. Endocr-Relat Cancer 2013;20:R83–99. [DOI] [PubMed] [Google Scholar]
- 17. Baselga J, Campone M, Piccart M, Burris HA, Rugo HS, Sahmoud T, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med 2012;366:520–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thomas C, Lamoureux F, Crafter C, Davies BR, Beraldi E, Fazli L, et al. Synergistic targeting of PI3K/Akt pathway and androgen receptor axis significantly delays castration-resistant prostate cancer progression in vivo. Mol Cancer Ther 2013;12:2342. [DOI] [PubMed] [Google Scholar]
- 19. Adelaiye-Ogala R, Gryder BE, Nguyen YTM, Alilin AN, Grayson AR, Bajwa W, et al. Targeting the PI3K/AKT pathway overcomes enzalutamide resistance by inhibiting induction of the glucocorticoid receptor. Mol Cancer Ther 2020;19:1436–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. de Bono JS, De Giorgi U, Rodrigues DN, Massard C, Bracarda S, Font A, et al. Randomized phase II study evaluating Akt blockad with ipatasertib, in combination with abiraterone, in patients with metastatic prostate cancer with and without PTEN loss. Clin Cancer Res 2019;25:928–36. [DOI] [PubMed] [Google Scholar]
- 21. de Bono JS, Bracarda S, Sternberg CN, Chi KN, Olmos D, Sandhu S, et al. IPATential150: Phase III study of ipatasertib (ipat) plus abiraterone (abi) vs placebo (pbo) plus abi in metastatic castration-resistant prostate cancer (mCRPC). Ann Oncol 2020;31:S1142–S215. [Google Scholar]
- 22. Dylgjeri E, McNair C, Goodwin JF, Raymon HK, McCue PA, Shafi AA, et al. Pleiotropic impact of DNA-PK in cancer and implications for therapeutic strategies. Clin Cancer Res 2019;25:5623–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Goodwin JF, Kothari V, Drake JM, Zhao S, Dylgjeri E, Dean JL, et al. DNA-PKcs-mediated transcriptional regulation drives prostate cancer progression and metastasis. Cancer Cell 2015;28:97–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Smith MC, Mader MM, Cook JA, Iversen P, Ajamie R, Perkins E, et al. Characterization of LY3023414, a novel PI3K/mTOR dual inhibitor eliciting transient target modulation to impede tumor growth. Mol Cancer Ther 2016;15:2344–56. [DOI] [PubMed] [Google Scholar]
- 25. Scher HI, Halabi S, Tannock I, Morris M, Sternberg CN, Carducci MA, et al. Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: Recommendations of the Prostate Cancer Clinical Trials Working Group. J Clin Oncol 2008;26:1148–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criterai in solid tumors: Revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228–47. [DOI] [PubMed] [Google Scholar]
- 27. World Medical Association. Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA 2013;310:2191–4. [DOI] [PubMed] [Google Scholar]
- 28. Armstrong AJ, Halabi S, Healy P, Alumkal JJ, Winters C, Kephart J, et al. Phase II trial of the PI3 kinase inhibitor buparlisib (BKM-120) with or without enzalutamide in men with metastatic castration resistant prostate cancer. Eur J Cancer 2017;81:228–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kolinsky MP, Rescigno P, Bianchini D, Zafeiriou Z, Mehra N, Mateo J, et al. A phase I dose-escalation study of enzalutamide in combination with the AKT inhibitor AZD5363 (capivasertib) in patients with metastatic castration-resistant prostate cancer. Ann Oncol 2020;31:619–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sweeney C, Bracarda S, Sternberg CN, Chi KN, Olmos D, Sandhu S, et al. Ipatasertib plus abiraterone and prednisolone in metastatic castration-resistant prostate cancer (IPATential150): a multicentre, randomised, double-blind, phase 3 trial. Lancet 2021;398:131–42. [DOI] [PubMed] [Google Scholar]
- 31. Xtandi (enzalutamide) package insert. Astellas, Pfizer; 2020. Available from: https://wwwxtandihcpcom/safety-profile.
- 32. Rodon J, Brana I, Shiu LL, De Jonge MJ, Homji N, Mills D, et al. Phase I dose-escalation and -expansion study of buparlisib (BKM120), an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. Invest New Drugs 2014;32:670–81. [DOI] [PubMed] [Google Scholar]
- 33. Wei XX, Hsieh AC, Kim W, Friedlander T, Lin AM, Louttit M, et al. A phase I study of abiraterone acetate combined with BEZ235, a dual PI3K/mTOR inhibitor, in metastatic castration resistant prostate cancer. Oncologist 2017;22:503–e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee EK, Jamani R, Berry SR, DeAngelis C, Giotis A, Emmenegger U. Characterizing the risk of drug-drug interactions in patients receiving enzalutamide for castration-resistant prostate cancer. J Clin Oncol 2015;33:261. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.