

Abstract
The wild-type p53 induced phosphatase 1 (Wip1), a member of the serine/threonine-specific PP2C family, is overexpressed in numerous human cancers. Wip1 dephosphorylates p53 as well as several kinases (such as p38 MAPK, ATM, Chk1, and Chk2) in the DNA damage response pathway that are responsible for maintaining genomic stability and preventing oncogenic transformation. As a result, Wip1 is an attractive target for synthetic inhibitors that could be further developed into therapeutics to treat some cancers. In this study, we report a series of alkyl-substituted N-methylaryl-N’-aryl-4-aminobenzamides and their inhibitory activity of the Wip1 phosphatase. A straightforward synthetic route was developed to synthesize the target compounds from commercially available starting materials. Three different portions (R1, R2, R3) of the core scaffold were extensively modified to examine structure-activity relationships. This study revealed interesting trends about a new molecular scaffold to inhibit Wip1.
Keywords: Wip1 phosphatase, small molecule inhibitors, aminobenzamides, cancer treatment, cross coupling, phosphate
Graphical Abstract
1. Introduction
Phosphorylation is a reversible post-translational modification of proteins that regulates most cellular functions. The phosphorylation state of proteins is controlled by the opposing action of two types of enzymes: kinases and phosphatases. Kinases phosphorylate protein targets by transferring a phosphate (PO43−) group from ATP to specific amino acids on proteins, while phosphatases remove the modification through dephosphorylation. Although eukaryotic protein kinases share a common protein fold and catalytic mechanism, protein phosphatases are diverse and comprise ten distinct protein folds. [1] Protein phosphatases can be classified based on their substrate preferences: ones that target phosphorylated serine or threonine residues (PSPs/PSTPs), and others that target phosphorylated tyrosines (PTPs). [2] The PSPs/PSTPs can be further divided into two structurally unrelated classes: phosphoprotein phosphatases (PPPs) and metallo-dependent (Mg2+ or Mn2+ dependent) protein phosphatases (PPMs). The PPM phosphatases are present in both prokaryotes and eukaryotes and often regulate cellular stress response pathways. [3] Human cells express 19 PPM phosphatases. The PPM phosphatase Wip1 (wild type p53-induced phosphatase 1, also known as PPM1D or PP2Cδ) is of particular interest because of its association with human cancers, in addition to non-oncologic diseases. [4]
Wip1 is a multifunctional phosphatase that is generally expressed at low levels. Notably, its expression is significantly increased after cells are exposed to genotoxic stress. To counteract the effects of DNA damage, several kinases (such as ATM, ATR, and p38 MAPK) phosphorylate p53 directly or via intermediary proteins (such as Chk1 and Chk2). Once phosphorylated, p53 localizes in the cell nucleus where it activates context-dependent transcriptional induction of genes involved in apoptosis, cell cycle arrest and senescence. At the same time, p53 also induces expression of its own regulatory genes, namely Mdm2 and Wip1. The Mdm2 protein promotes degradation of p53 via ubiquitination. Wip1 suppresses p53 activity by dephosphorylating p53 at Ser 15 as well as dephosphorylating several upstream kinases (such as ATM, p38, Chk1, Chk2) that activate p53. [5 – 7] Furthermore, Wip1 dephosphorylates Mdm2 at Ser 395, which stabilizes the protein and enhances its ability to degrade p53. In general, Wip1 plays an important role in returning cells to a pre-stressed state after recovery from genotoxic stress. Overexpression of Wip1 through gene amplification, dysregulation, or protein truncation results in chronic suppression of p53 activity and promotes tumorigenesis in cancers such as breast, gastric, neuroblastoma, ovarian clear cell carcinoma, pancreatic adenocarcinoma and medulloblastoma. [8] Developing small molecules to inhibit Wip1 is a novel approach to restore the normal tumor suppressor function of p53 in cancers where Wip1 is overexpressed.
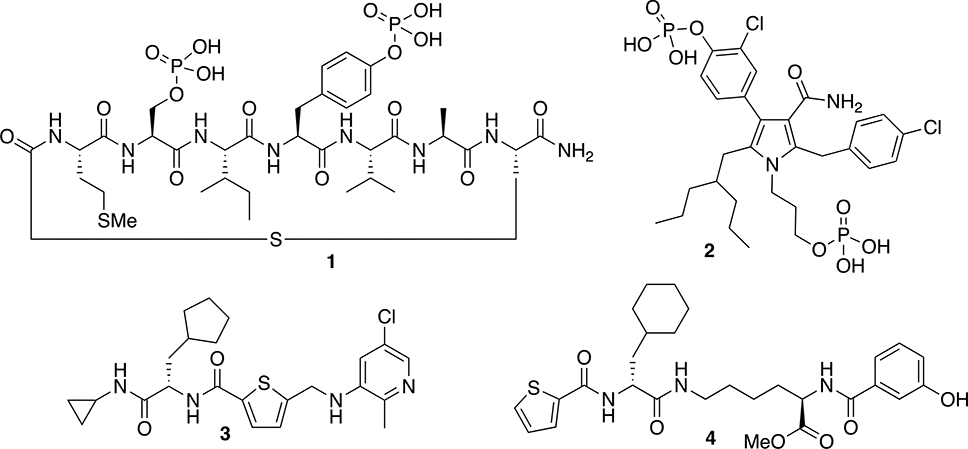
Until recently, investigations of Wip1 as a pharmacological target for cancer treatment have been limited by the lack of small-molecule inhibitors for Wip1. Studies focused on the screening of chemical libraries have identified a few good Wip1 inhibitors that show activity in both cell-based assays and in vivo mouse models of tumors. [9 – 11] The molecules in this study were inspired by three main classes of Wip1 inhibitors: phosphopeptides, phosphopyrroles, and allosteric peptides (Figure 1). A cyclic phosphopeptide (compound 1) with submicromolar IC50 inhibitory activity was initially described by Yamaguchi et al. as a competitive inhibitor, but it was not highly selective for Wip1 over the closely related phosphatase PP2Cα (Figure 1). [12] We reported a series of penta-substituted pyrroles as competitive and selective Wip1 inhibitors, with the best inhibitor represented by compound 2. [13] Gilmartin et al. reported an allosteric inhibitor of Wip1 (compound 3) that was identified using a DNA-encoded high throughput screening platform, [14] and a recent publication has elucidated more details about the unique mechanism of action. [15] Inspired by the work of Gilmartin et al., we explored structure-activity relationships among additional analogs, including compound 4, to better understand the molecular properties of the allosteric inhibitors. [16] In the current study, we attempted to combine the molecular features of compounds 1 to 4 into a single scaffold that could take advantage of the individual features of the previous Wip1 inhibitors. The basic scaffold we developed in this regard is alkyl-substituted N-methylaryl-N’-aryl-4-aminobenzamide.
Figure 1.
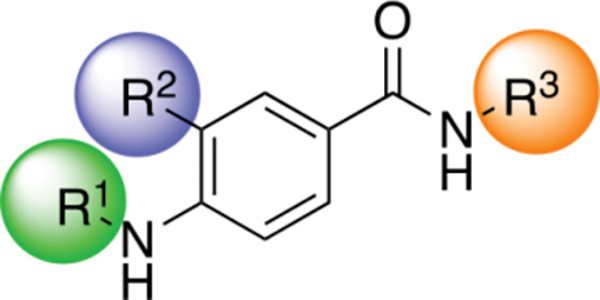
From the work of Gilmartin et al. with compound 3 and our own work with compound 4, it was clear that a cyclohexyl or cyclopentyl alanine with the L- stereochemistry was critical for the Wip1 inhibitory activity of these molecules via an allosteric mechanism. However, these amino acids were also detrimental to further development of 3 and 4 as drugs due to poor pharmacokinetics. [14, 16] We decided to explore a new series of simple molecular scaffolds that would incorporate the hydrophobic cycloalkyl group that was crucial for the activity of compounds 3 and 4 but use an achiral scaffold that was not based on an α-amino acid. We also wanted the flexibility to incorporate phosphate functionalities as these were critical for inhibitory activity of compounds 1 and 2 via a competitive mechanism (Figure 1). [12 – 13] Our general strategy was to combine the best molecular features from allosteric and competitive inhibitors into one scaffold. Initial tests were designed to determine the importance of a minimally functionalized cyclohexyl group (6 – 7), followed by successive incorporation of more functional groups until there was measurable inhibitory activity (11 – 12). After collecting the preliminary Wip1 inhibition data, we set out to synthesize a series of alkyl-substituted N-methylaryl-N’-aryl-4-aminobenzamides with the generalized structure shown in Figure 2, with the intention of incorporating some of the molecular features of earlier inhibitors 1 – 4. For instance, the chloroaniline group and the primary amide group in compound 3 were proposed to interact with residues of Wip1. Therefore, we intended to incorporate these features into our inhibitors. As mentioned previously, a cycloalkyl group appeared to be necessary for good inhibitory activity of compounds 3 and 4, even though its role and mode of interaction with Wip1 is not understood as the structure of Wip1 has not been reported to date. All the new inhibitors therefore contained a cycloakyl group. Inhibitors 1 and 2 needed phosphate groups to achieve good activity, so we also intended to include phosphates in the new inhibitors. The series of target compounds which embody these parameters is generally represented by three distinct structural domains shown in Figure 2: an aromatic portion 1 (R1), an alkyl portion which usually contains a cyclohexyl ring (R2), and aromatic portion 2 (R3).
Figure 2.
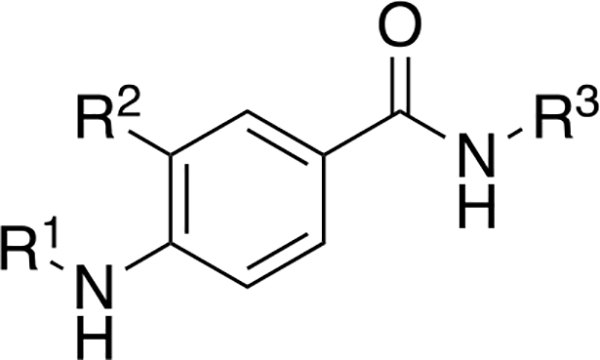
General structure of alkyl-substituted N-methylaryl-N’-aryl-4-aminobenzamide inhibitors.
2. Results and discussion
2.1. Design and synthesis
The simplest structures represented by molecules 6 and 7a – 7c were easily synthesized from 3-aminopropan-1-ol (5) as shown in Scheme 1a and consisted of one cyclohexyl group and a sidechain containing one amide and a hydroxy group in a linear arrangement. Subsequently, we expanded the scaffold by attaching the cyclohexyl group to aniline derivatives 8 – 9 using a Negishi coupling reaction to form methyl-cyclohexyl derivatives 10 – 11 and a Suzuki coupling reaction to obtain cyclohexylethyl derivatives 12 – 13 (Scheme 1b). After cross-coupling, other functional groups could subsequently be attached to the nitrogen on the aniline. In this regard, compounds 10 – 13 underwent reductive amination with 3-formylbenzoic acid to yield compounds 14 – 17.
Scheme 1a.
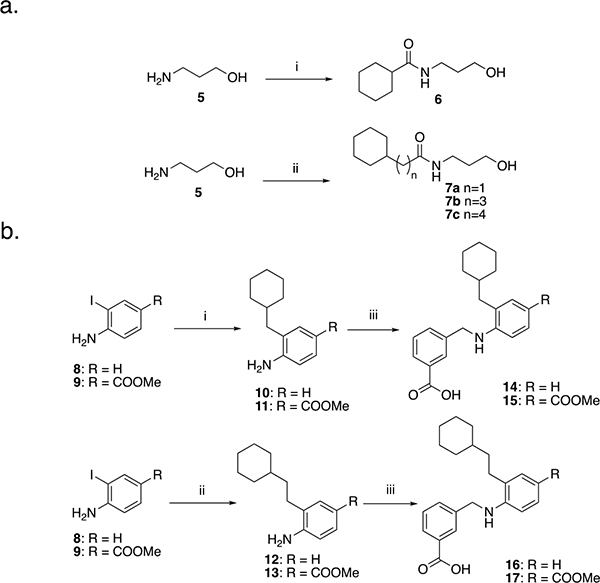
i) cyclohexanoyl chloride, CH2Cl2, RT, 30 min. ii) corresponding carboxylic acid, HOBt, i-Pr2Net, EDC, CH2Cl2, RT, 20 h. 1b. i) (cyclohexylmethyl)zinc bromide 0.5M in THF, Pd2dba3, SPhos, RT 15 min then reflux overnight. ii) (2-cyclohexylethyl)boronic acid, Pd2dba3, PPh3, K2CO3, toluene/H2O (20:1), reflux, 5 h. iii) 3-formylbenzoic acid, MeOH, reflux, 3 h. Cooled to RT, CH3COOH, 15 min then NaCNBH3, RT, overnight.
We then developed a straightforward and relatively short synthesis of the target compounds 22a – 22n (Scheme 2). As described in Scheme 1b, the first step is a Negishi coupling reaction between commercially available iodide 9 and cyclohexylmethyl zinc bromide to furnish the intermediate 11. Subsequent reductive amination with a variety of different aldehydes yields derivatives 18b – 18n. Esters 18b – 18n were treated with KOSiMe3 at 40 °C to afford the corresponding carboxylic acids 19b – 19n. Alternatively, 19a was directly obtained by deprotection of the ester 11. The derivatives 19a – 19n were then coupled to 3-aminophenol to yield 20a – 20n. To phosphorylate the phenolic hydroxy group, 20a – 20n were first treated with diethylchlorophosphate to provide the corresponding diethylphosphates 21a – 21n, which were purified before deprotection with TMSBr to afford the desired target compounds 22a – 22n. The same synthetic procedure was applied in Scheme 3a to obtain derivatives 29a – 29b and Scheme 3b to obtain compounds 32a – 32b. In Schemes 3a and 3b, the R1 group from compound 22g was maintained while groups at positions R2 (29a – 29b) and R3 (32a – 32b) were varied to determine the importance of the cyclohexyl group and to investigate how the position of the phosphate group impacts activity.
Scheme 2.
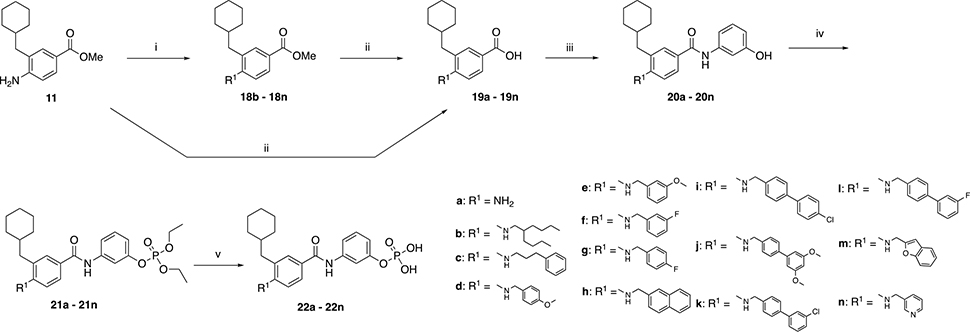
i) corresponding benzaldehyde, MeOH, reflux, 3 h. Cooled to RT, CH3COOH, 15 min then NaCNBH3, RT, overnight. ii) KOSiMe3, THF, 50 °C, overnight. iii) HATU, DIPEA, 3-aminophenol, DMF, RT, overnight. iv) diethyl chlorophosphate, DIPEA, CH2Cl2, 40 °C, overnight. v) TMSBr, CH2Cl2, RT, overnight. After evaporation, residue stirred in MeOH/H2O 9:1 for 1h.
Scheme 3a.
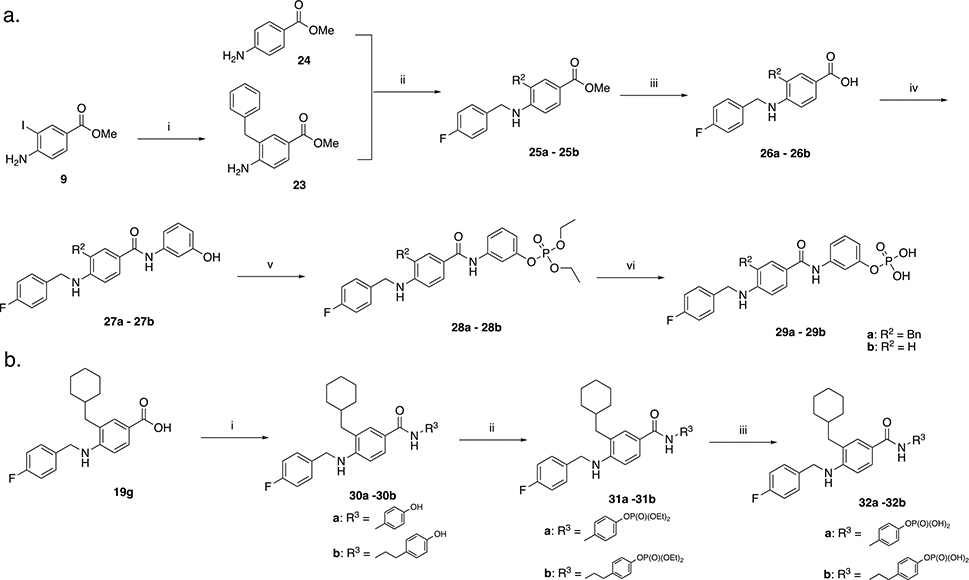
i) benzylzinc bromide 0.5M in THF, Pd2dba3, SPhos, RT 15 min then reflux overnight. ii) 4-fluorobenzaldehyde, MeOH, reflux, 3 h. Cooled to RT, CH3COOH, 15 min then NaCNBH3, RT, overnight. iii) KOSiMe3, THF, 50 °C, overnight. iv) HATU, DIPEA, 3-aminophenol, DMF, RT, overnight. v) diethyl chlorophosphate, DIPEA, CH2Cl2, 40 °C, overnight. vi) TMSBr, CH2Cl2, RT, overnight. After evaporation, residue stirred in MeOH/H2O 9:1 for 1 h. 3b. i) HATU, DIPEA, phenol, DMF, RT, overnight. v) diethyl chlorophosphate, DIPEA, CH2Cl2, 40 °C, overnight. vi) TMSBr, CH2Cl2, RT, overnight. After evaporation, residue stirred in MeOH/H2O 9:1 for 1 h.
Next, we focused on examining a range of groups at R1 with a few variations at R2. Starting with molecules 15 and 17 (Scheme 1b), we intended to use the carboxylic acid at position R1 to make additional analogs, also exploring para-substituted analogs represented by 33 which were made by using 4-formylbenzoic acid in the reductive amination step instead of 3-formylbenzoic acid which is used to make 15 and 17. We also introduced a norbornyl moiety at position R2 to explore a different type of hydrophobic group at this position. The synthetic route described in Scheme 4. Starting from the commercially available methyl 4-amino-3-iodobenzoate 9, we performed a Negishi coupling to obtain derivative 34. The second step involved a reductive amination to yield 35. The new step is the amide formation between the carboxylic acid groups of 15, 17, 33, and 35 with a variety of different anilines to yield derivatives 36a – 36f. Ester hydrolysis then provided derivatives 37a – 37h, which underwent coupling with 3-aminophenol to obtain 38a – 38f. The phenols were treated with diethylchlorophosphate to provide the corresponding diethylphosphates 39a – 39f, which were each isolated before subjecting to TMSBr to afford the desired target compounds 40a – 40f.
Scheme 4.
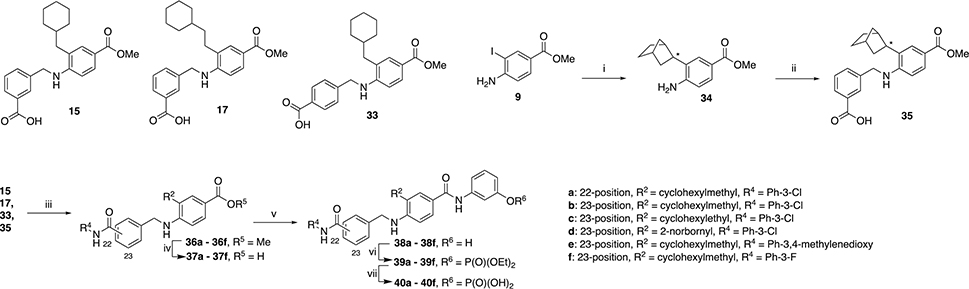
i) exo-2-norbornylzinc bromide 0.5M in THF, Pd2dba3, SPhos, RT 15 min then reflux overnight. ii) 3-formylbenzoic acid, MeOH, reflux, 3 h. Cooled to RT, CH3COOH, 15 min then NaCNBH3, RT, overnight. iii) HATU, DIPEA, aniline, DMF, RT, overnight. iv) KOSiMe3, THF, 50 °C, overnight. v) HATU, DIPEA, 3-aminophenol, DMF, RT, overnight. vi) diethyl chlorophosphate, DIPEA, CH2Cl2, 40 °C, overnight. vii) TMSBr, CH2Cl2, RT, overnight. After evaporation, residue stirred in MeOH/H2O 9:1 for 1h.
We then decided to evaluate the importance of the amine linker in aromatic portion 1 by introduction of a second amide bond (48a) and inversion of amide/amine bonds (48b). In order to do that, we needed to slightly modify the synthetic scheme as exemplified in Scheme 5. Starting from 3-formylbenzoic acid, acyl chloride 41 was obtained by reflux in toluene in presence of thionyl chloride and reacted with previously obtained 11 to achieve 42. Oxidation to carboxylic acid 43 and subsequent coupling with 3-chloroaniline yielded compound 44a, while reductive amination in presence of the same aniline gave derivative 44b. Ester hydrolysis performed with KOSiMe3 provided 45a – 45b, then coupled with 3-aminophenol to obtain 46a – 46b. Treatment with diethylchlorophosphate afforded the diethylphosphates 47a – 47b, which were purified and eventually reacted with TMSBr to afford the desired products 48a – 48b.
Scheme 5.
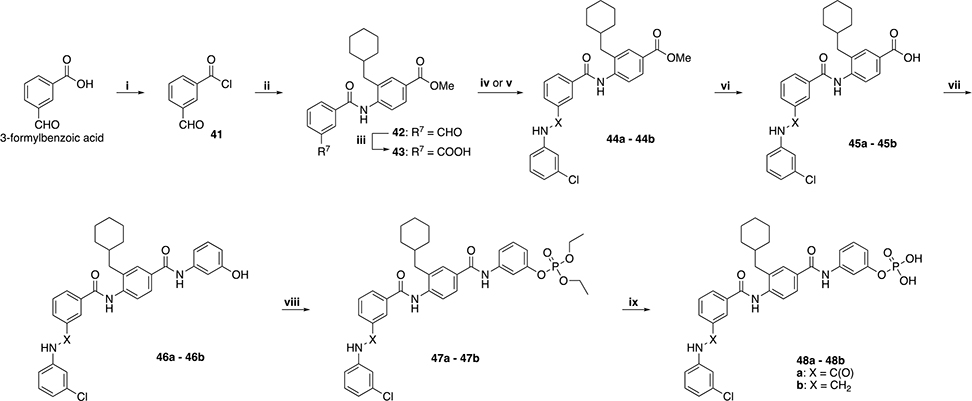
i) SOCl2, toluene, reflux, 1 h. ii) 11, Et3N, DCM, RT, overnight. iii) Oxone, DMF, RT, 3 h. iv) HBTU, DIPEA, 3-chloroaniline, DMF, RT, overnight. v) 3-chloroaniline, MeOH, reflux, 3h. Cooled to RT, CH3COOH, 15 min then NaCNBH3, RT, overnight. vi) KOSiMe3, THF, 50 °C, overnight. vii) HBTU, DIPEA, 3-aminophenol, DMF, RT, overnight. viii) diethyl chlorophosphate, DIPEA, CH2Cl2, 40 °C, overnight. ix) TMSBr, CH2Cl2, RT, overnight. After evaporation, residue stirred in MeOH/H2O 9:1 for 1h.
2.2. Wip1 inhibitory activity
One of the challenges in evaluating small molecule inhibition of Wip1 is the large range in exploratory compounds potency. For initial tests with inhibitors expected to have weak or almost no activity, a single concentration of 100 μM was used to determine the percent inhibition at this high concentration. If the inhibition at 100 μM was weak, no further tests were performed, and the percent inhibition at the high concentration was reported. For inhibitors of further interest, a nine- or ten-point dose-response experiment was performed. For weaker inhibitors, for which only a partial inhibition curve was obtained, nonlinear curve fitting methods were used to fit normalized percent inhibition data to a standard logistic inhibition curve with the IC50 value as the variable parameter. For stronger inhibitors, normalized percent inhibition data were fit to a variable slope logistic inhibition curve, with the IC50 and the Hill coefficient, n, as the parameters. With ideal behavior, the binding of an inhibitor to a single site on an enzyme results in a Hill coefficient with a value of 1.[17] Hill coefficients greater than 1 can result from multiple binding sites for the molecule on the enzyme or when the mechanism of inhibition involves concentration-dependent aggregation of the enzyme.[18 – 19] When available, Hill coefficients for inhibitors in this work are reported next to IC50 values.
We began by assessing the inhibitory activity of the simple derivatives 6 and 7a – 7c, consisting of one cyclohexyl group and a sidechain containing one amide and hydroxy group in a linear arrangement. These molecules, however, exhibited no or very weak inhibitory activity (Table 1) and suggested that a simple cyclohexyl group with a minimal number of nearby polar functional groups cannot function as Wip1 inhibitors. The unsubstituted anilines 10 and 12 also did not have significant inhibitory activity, while the derivatives 14 and 16, which contained the cyclohexyl ring as well as the aromatic carboxylic acid attached to the aniline nitrogen, displayed modest inhibition. Incorporation of an ester group on the central aromatic ring (15 and 17, Table 1) showed another slight improvement in the activity. The ester group also provided an additional position to functionalize, prompting us to focus on more complex derivatives where both the aniline nitrogen and the ester group were derivatized.
Table 1.
Initial screen for inhibitors with cyclohexyl fragment.
| Compound | Structure | IC50 [μM] | Hill coefficient [n] |
|---|---|---|---|
|
| |||
| 6 |
|
not active | -- |
| 7a |
|
not active | -- |
| 7b |
|
1% inhibition at 100 μM | -- |
| 7c |
|
4% inhibition at 100 μM | -- |
| 10 |
|
not active | --- |
| 12 |
|
3% inhibition at 100 μM | -- |
| 14 |
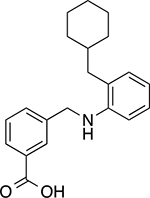
|
74.8 ± 8.3 | 1.9 ± 0.4 |
| 15 |
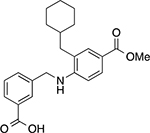
|
54.1 ± 3.8 | 2.8 ± 0.5 |
| 16 |
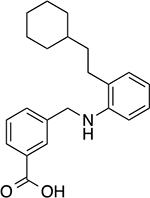
|
88% inhibition at 100 uM | -- |
| 17 |
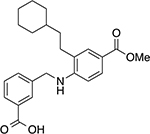
|
48.4 ± 0.8 | 4.4 ± 0.4 |
The series of molecules 22a – 22n was designed to explore how changes at position R1 impacted the inhibitory activity against the Wip1 phosphatase and the results are reported in Table 2. When R1 is a simple free amine (22a) the molecule has very low activity (>200 μM), while an alkyl-substituted derivative (22b) was inactive. Interestingly, elongation of the alkyl chain between aniline nitrogen atom and a phenyl group (22c) led to a slight improvement in the activity (165 μM). Introduction of a methoxy group on the phenyl ring at position R1 led to an improvement in inhibitory activity and placing this substituent at the para position on the aromatic ring was important for improving activity (compare 22d, IC50 = 37 ± 2 μM vs 22e, IC50 = 139 ± 28 μM). Fluorine substitution on the same aromatic ring demonstrated slightly better activity compared to the methoxy group, yet there was no substantial difference in activity when the fluorine was at the meta (22f) versus the para (22g) position (28.5 ± 0.9 μM vs 28.1 ± 0.98 μM, respectively). The naphthylmethyl derivative 22h displayed weak activity, while substituted biphenyls 22i – 22l were better. Interestingly, the position of the halogen within the biphenyl group seemed important (compare 22i (332 ± 100 μM) with 22k (48 ± 6 μM). Heteroaromatic groups provided varying levels of inhibitory activity with the benzofuranyl derivative 22m among the most active of this series (IC50 = 21.4 ± 1.6 μM) while the pyridinyl derivative 22n was 5-fold less active (IC50 = 105 ± 7 μM).
Table 2.
Structure-activity relationship of target compounds 22a – 22n with modifications in aromatic portion 1.
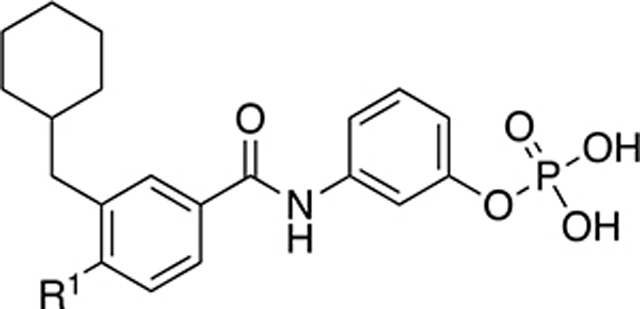
| |||
|---|---|---|---|
| Compound | R1 | IC50 [μM] | Hill coefficient [n] |
|
| |||
| 22a | H2N- | 330 ± 33 | 1.2 ± 0.10 |
| 22b |
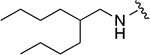
|
512 ± 230 | 1a |
| 22c |
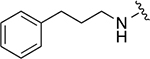
|
105 ± 23 | 1a |
| 22d |
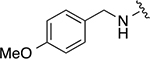
|
37 ± 2 | 2.0 ± 0.2 |
| 22e |
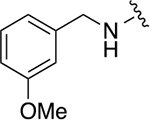
|
139 ± 28 | 1.3 ± 0.3 |
| 22f |
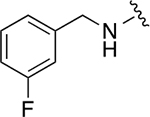
|
28.5 ± 0.9 | 3.4 ± 0.4 |
| 22g |
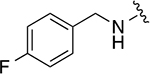
|
28.1 ± 1 | 3.0 ± 0.4 |
| 22h |
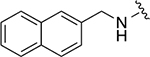
|
274 ± 42 | 1a |
| 22i |
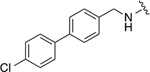
|
332 ± 100 | 1a -- |
| 22j |
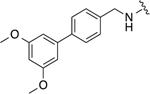
|
18.0 ± 0.4 | 1.8 ± 0.1 |
| 22k |
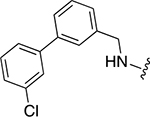
|
48 ± 6 | 1.3 ± 0.2 |
| 22l |
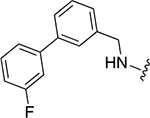
|
30 ± 4 | 1.7 ± 0.4 |
| 22m |
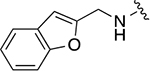
|
21.4 ± 1.6 | 1.5 ± 0.1 |
| 22n |
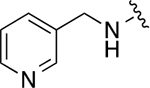
|
105 ± 7 | 1.6 ± 0.2 |
Normalized inhibition data were fit to a fixed-slope logistic curve, which assumes the Hill coefficient = 1.
After the last series of data, we decided to select one R1 group to hold constant while altering groups at some of the other positions to see how the activity would change. We selected compound 22g (IC50 = 28.1 ± 0.98, Hill coefficient, n = 3) as a starting point as it afforded a modest IC50 value and was relatively simple to synthesize. First, we altered position R2 to determine whether the cyclohexyl group was really necessary. Replacing the methyl-cyclohexyl moiety with a benzyl group (29a, Table 3) resulted in a 3-fold loss in activity while the removal of the alkyl group resulted in almost complete loss of inhibitory activity (29b, Table 3). Interestingly, these modifications lowered the Hill coefficient values, which suggests that the hydrophobic cyclohexyl group may be interacting in a non-specific manner with the protein. Regardless of this observation, it is clear that the cyclohexyl group is important for the inhibitors to achieve at least modest IC50 values and therefore this moiety was retained in most of the subsequent molecules.
Table 3.
Structure and biological activity of derivatives 22g and 29a – 29b.
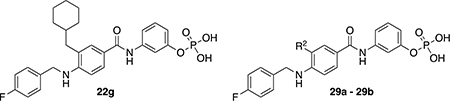
| |||
|---|---|---|---|
| Compound | R2 | IC50 [μM] | Hill coefficient [n] |
|
| |||
| 22g | - | 28.1 ± 1 | 3.0 ± 0.4 |
| 29a |
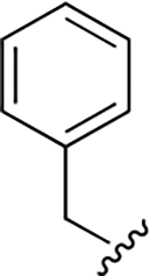
|
63 ± 3 | 1.7 ± 0.1 |
| 29b | H | 683 ± 72 | 1a |
Compound 22g was used as the starting point to further explore modifications at position R3. By using different aminophenols, we synthesized derivative 32a, where the phosphate group was moved from the meta to the para position on the phenyl ring, and compound 32b, in which two additional methylene groups were introduced to increase the distance between the phosphate group and the main scaffold. These two derivatives displayed comparable inhibitory activity with the parent compound 22g (Table 4). Noticeably, 32b had a better Hill coefficient value (n = 1 for 32b vs n = 3 for 22g). These results suggest that the phosphate-group may also mostly engage in non-specific interactions with Wip1.
Table 4.
Structures and biological activity of derivatives 22g and 32a – 32b.
| |||
|---|---|---|---|
| Compound | R3 | IC50 [μM] | Hill coefficient [n] |
|
| |||
| 22g | - | 28.1 ± 1 | 3.0 ± 0.4 |
| 32a |
|
29 ± 1 | 3.4 ± 0.4 |
| 32b |

|
36 ± 3.2 | 1.2 ± 0.1 |
From these results, it appeared that good inhibitory activity required a large hydrophobic group at R2 but was somewhat insensitive to small changes in the group at R3 as long as a phosphate was present. However, the higher Hill coefficients observed for several analogs suggested that the hydrophobic group at R2 and the phosphate at R3 could be contributing to non-specific binding with Wip1.
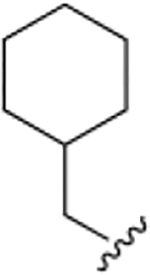
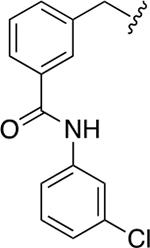
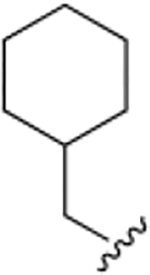
The chemical structures and inhibitory activities of the next series of synthesized molecules are reported in Table 5. The first compound we obtained was 40a, from the intermediate 33, which showed an improved activity with an IC50 value slightly above 10 μM. Remarkably, the meta-substituted derivative 40b had an IC50 value of 3.8 ± 0.3 μM, which represents a 7-fold improvement in Wip1 inhibition compared to 22g (28.2 ± 0.98 μM). This result prompted us to explore additional variations with the meta substitution. Furthermore, the Hill coefficient value ~1.5 suggested a reasonably specific interaction with Wip1. Next, we tested different alkyl groups, but both the ethyl-cyclohexyl (40c, IC50 = 32.7 ± 4 μM) and the norbornyl (40d, IC50 = 24.7 ± 1.9 μM) derivatives showed diminished activity compared to the cyclohexyl version. The importance of the phosphate group in R3 was also verified by the comparison of derivative 40c with its precursor 38c, demonstrating a considerable drop in activity when the phosphate is not present. Modifications focused on replacing the chlorine atom with different electron donating/withdrawing groups were also explored, but resulted in less active molecules (40e – 40f) compared with 40b.
Table 5.
Structures and biological activity of derivatives 38c, 40a – 40f, and 48a – 48b.
| ||||
|---|---|---|---|---|
| Compound | R1 | R2 | IC50 [μM] | Hill coefficient [n] |
|
| ||||
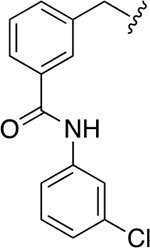
| 40a |
|
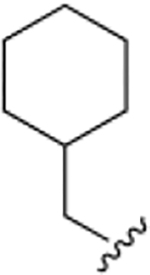
|
11.1 ± 0.7 | 1.9 ± 0.2 |
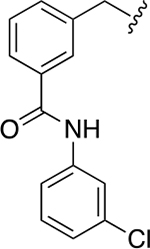
| 40b |
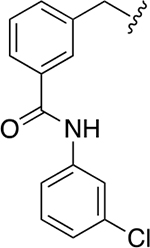
|
|
3.8 ± 0.3 | 1.4 ± 0.1 |
| 38c |
|
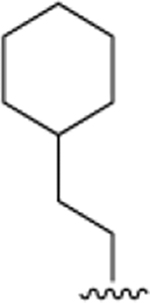
|
1600 ± 500 | 1a |
| 40c |
|
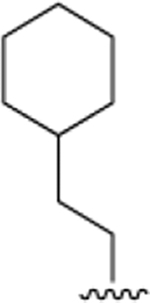
|
32.7 ± 4 | 1.0 ± 0.1 |
| 40d |
|
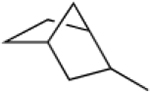
|
24.7 ± 1.9 | 2.3 ± 0.4 |
| 40e |
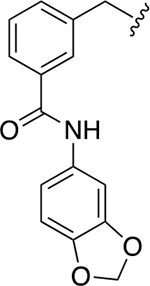
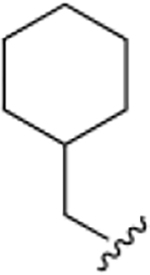
|
|
35.2 ± 0.4 | 2.9 ± 0.1 |
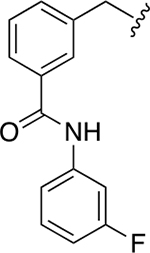
| 40f |
|
|
20.8 ± 0.4 | 3.6 ± 0.2 |
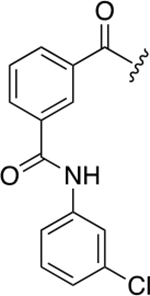
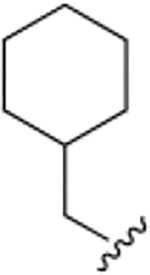
| 48a |
|
|
7.3 ± 0.2 | 5.1 ± 0.3 |
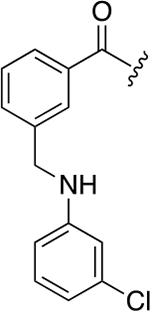
| 48b |
|
|
7.2 ± 0.3 | 2.3 ± 0.2 |
We then decided to evaluate the importance of the amine linker in aromatic portion 1. Introduction of a second amide bond (48a) and inversion of amide/amine bonds (48b) resulted in activity that was about the same as 40b (~7.3 μM vs 3.8 μM), but the Hill coefficient values for these two derivatives were higher, especially for 48a (n = 5).
2.3. Selectivity
The most potent inhibitors from this work were then evaluated for their ability to selectively inhibit Wip1 over a closely related serine/threonine phosphatase that belongs to the same family as Wip1. For these selectivity tests, we used PPM1A (also called PP2Cα), which has a catalytic domain that is 50% similar to that of Wip1.[12] Using the molecules 40b, 48a, and 48b, each one showed no inhibitory activity against PPM1A up to 100 μM concentrations. At the same time, these three molecules were able to inhibit Wip1 with IC50 values between 3.8 and 7.2 μM. (see Supporting Information).
3. Conclusion
In conclusion, we have successfully developed and synthesized a series of alkyl-substituted N-methylaryl-N’-aryl-4-aminobenzamides and discovered a novel molecule with satisfactory Wip1 inhibitory activity and specificity. The synthetic routes to make these inhibitors are highly modular, allowing for a large number of different functional groups to be tested. Starting from very simple molecules with no inhibitory activity, we systematically added different molecular portions to the scaffold to achieve an inhibitor with decent activity. As a result, compound 40b was identified as the lead compound having an IC50 value of 3.8 ± 0.3 μM and an acceptable Hill coefficient value of 1.5 (Figure 3). This is the first Wip1 inhibitor to incorporate both the cyclohexyl group and a phosphate group, which are two different groups that were important components of previous Wip1 inhibitors (1-4). There is currently no crystal structure of Wip1, which prevents accurate modeling of how 40b may bind to the protein. A recent publication by Miller et al. demonstrated that hydrogen-deuterium exchange mass spectrometry can be used to define the mechanism by which 3 binds to and inhibits Wip1.[15] This approach may be useful to identify how 40b interacts with Wip1. The synthetic routes to make the molecular scaffold presented in our work facilitated exploration of a range of different derivatives by modifying three different portions of the core structure (Figure 2), and this resulted in identification of 40b. Additional work to increase the molecular size and complexity of the R1 group of the scaffold may lead to more potent inhibitors that interact with a larger portion of the Wip1 binding site.
Figure 3.
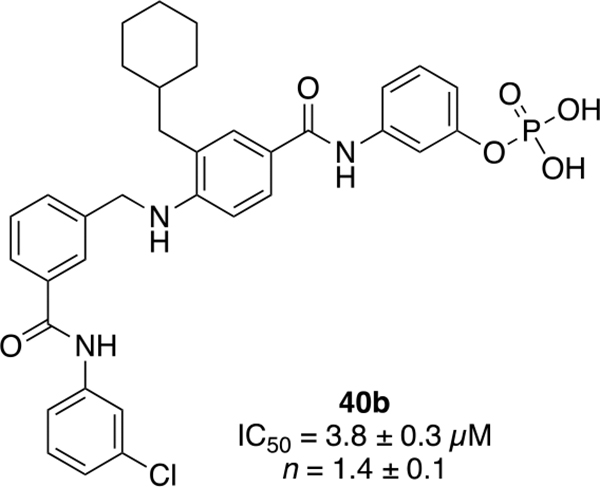
Structure of lead compound 40b.
Experimental
Chemicals and instrumentation
All reactions were carried out using oven-dried glassware under N2 atmosphere. All reagents were purchased from Sigma-Aldrich, AK Scientific or Creosalus depending on their availability and used without further purification. All solvents were purchased from Aldrich. Analytical thin-layered chromatography (TLC) was performed using plates pre-coated with silica gel (Sigma Aldrich; 60 Å, 17 mm particle size), impregnated with a fluorescent indicator (254 nm). TLC plates were visualized by exposure to ultraviolet (UV) light. Flash-column chromatography was performed using a Biotage Isolera One automated flash purification system with Biotage SNAP KP-Sil cartridges. 1H and 13C NMR spectra were recorded on Bruker (400 MHz, 500 MHz and 600 MHz) spectrometers. Chemical shifts (δ) are quoted in ppm and are referenced to residual protium in the NMR solvent (CDCl3, δ 7.26; DMSO-d6, δ 2.50; CD3OD, δ 3.31). Coupling constants (J) are quoted in Hz. Multiplicity recorded for 1H NMR as follows s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, m = multiplet, dd = doublet of doublets, dt = doublet of triplets, td = triplet of doublets, br = broad signal.
Phosphatase activity inhibition assay
Wip1 phosphatase activity was measured using purified Wip1 with a phosphopeptide substrate in a similar manner as described previously.[12, 14, 16, 20] The preparation and isolation of Wip1 has been extensively described.[21,22] Purified recombinant human Wip1 (1–420), in the absence or presence of the test inhibitor compound, was combined with 100 μM human ATM phosphopeptide (AFEEG-pS-QSTTIGY) in a buffer consisting of 50 mM Tris·HCl, pH 7.5, 0.1 mm EGTA, 1 mm DTT, 1mm CHAPS, 0.1 mg/mL BSA, and 30 mM MgCl2 for 7 min at 30 °C. Concentrations of the phosphate product were detected using the Biomol Green assay (Enzo Biosciences) in combination with a phosphate standard curve. Phosphatase activity was normalized by phosphate concentrations in the absence of inhibitor (100%) and in the absence of enzyme (0%). GSK2830371 (Sigma–Aldrich) was used as a positive control. Compounds were initially tested for inhibition of Wip1 phosphatase activity at the single high concentration of 100 μM. Compounds that exhibited no or weak inhibition were not further tested. Compounds with inhibitory activity of interest were tested in a 9- or 10-point dose-response experiment where each concentration was tested in triplicate. Normalized inhibition of phosphatase activity data were fit to fixed slope (weaker inhibiton) or variable slope (stronger inhibiton) concentration vs. normalized response logistic curves by non-linear least squares curve fitting using Prism 8.4 (GraphPad). Selected dose-response graphs for active inhibitors are presented in the Supporting Information, where the dotted lines show the 95% confidence limits. To test for phosphatase specificity, the most potent Wip1 inhibitors were tested for activity against PPM1A. PPM1A phosphatase activity was assayed as described previously.[23] In this assay, 100 μM human p38α (175–185, 180pT) phosphopeptide substrate was added with compounds (40b, 48a, and 48b) or DMSO prior to addition of 8 nM PPM1A.[24] All assays were performed in 50 mM Tris·HCl, pH 7.5, 0.1 mM EGTA, 1 mM DTT, 1mM CHAPS, 0.1 mg/mL BSA, and 30 mM MgCl2 by incubating with PPM1A for 7 min at 30 °C. The reaction was terminated using Biomol green (Enzo Life Sciences). The plate was incubated at room temperature for 20 min to allow the development of green color. The amount of phosphate released was quantified by measuring the absorbance at 620 nm using a SpectraMax iD3 microplate reader (Molecular Devices) in combination with a phosphate standard curve.
Synthesis of compounds
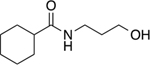
N-(3-hydroxypropyl)cyclohexanecarboxamide (6)
3-Amino-propan-1-ol (0.39 mL, 5.12 mmol) was added to a solution of cyclohexanecarbonyl chloride (0.46 mL, 3.41 mmol) in CH2Cl2 (13.6 mL) at 23°C. After stirring for 30 mins, the reaction mixture was diluted with CH2Cl2 (30.0 mL) and transferred in a separation funnel. Mixture was washed with an aq. sol. 1N HCl (4 × 15.0 mL), then the organic phase was collected and dried over MgSO4. After filtration and concentration, crude was purified by flash chromatography using a linear gradient 0 to 10% MeOH in CH2Cl2 to obtain the desired product as a colorless oil (280 mg, 1.51 mmol, 44%). 1H NMR (400 MHz, CDCl3) δ 6.48 (t, J = 5.8 Hz, 1H), 4.12 (s, 1H), 3.56 (t, 2H, J = 5.7 Hz), 3.34 (q, 2H, J = 6.1 Hz), 2.15 – 1.99 (m, 1H), 1.87 – 1.68 (m, 4H), 1.63 (quint, 3H, J = 5.8 Hz), 1.40 (m, 2H), 1.30 – 1.11 (m, 3H). 13C NMR (100 MHz, CDCl3) δ 177.82, 59.23, 45.54, 36.15, 32.20, 29.82, 29.14, 25.77, 25.55. HRMS–ESI (m/z): [M + H]+ calcd for C10H20NO2 186.1494; found, 186.1491.
Synthetic procedure for derivatives 7a-c
To a solution of appropriate carboxylic acid (1.00 eq) in CH2Cl2 (0.13 M) were added 3-amino-propan-1-ol (1.20 eq), DIPEA (3.50 eq), and HOBt (1.30 eq) at 23°C. Mixture was cooled to 0°C by ice-bath and EDC (1.30 eq) was added, then bath was removed to allow mixture reach 23°C. After stirring for 20h, mixture was cooled to 0°C and an aq. sol. 1N HCl (25.0 mL) was added. Organic layer was collected, and aqueous layer extracted with EtOAc (3x). The combined organic phase was dried over MgSO4 and concentrated. Crude was purified by flash chromatography using a linear gradient 0 to 10% MeOH in CH2Cl2.
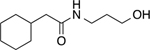
2-cyclohexyl-N-(3-hydroxypropyl)acetamide (7a)
Colorless oil (349 mg, 1.75 mmol, 83%). 1H NMR (400 MHz, CDCl3) δ 5.80 (s, 1H), 3.62 (t, 2H, J = 4.9 Hz), 3.41 (q, 2H, J = 6.1 Hz), 3.34 (s, 1H), 2.06 (d, 2H, J = 7.0 Hz), 1.84 – 1.59 (m, 8H), 1.35 – 1.19 (m, 2H), 1.18 – 1.06 (m, 1H), 1.01 – 0.85 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 173.97, 59.20, 44.97, 36.14, 35.42, 33.29, 32.60, 26.31, 26.18. HRMS–ESI (m/z): [M + H]+ calcd for C11H22NO2 200.1651; found, 200.1652.
4-cyclohexyl-N-(3-hydroxypropyl)butanamide (7b)
Colorless oil (41.2 mg, 0.18 mmol, 62%). 1H NMR (400 MHz, CDCl3) δ 5.91 (s, 1H), 3.61 (s, 1H), 3.43 – 3.37 (m, 3H), 2.21 – 2.12 (m, 2H), 1.74 – 1.56 (m, 9H), 1.27 – 1.09 (m, 6H), 0.91 – 0.81 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 174.73, 59.19, 37.53, 37.13, 36.15, 33.36, 32.51, 26.72, 26.42, 23.31. HRMS–ESI (m/z): [M + H]+ calcd for C13H26NO2 228.1964; found, 228.1962.
5-cyclohexyl-N-(3-hydroxypropyl)pentanamide (7c)
Colorless oil (52.6 mg, 0.22 mmol, 40%). 1H NMR (400 MHz, CDCl3) δ 6.80 (t, 1H, J = 5.6 Hz), 4.17 (t, 1H, J = 5.5 Hz), 3.54 (q, 2H, J = 5.1 Hz), 3.30 (q, 2H, J = 6.1 Hz), 2.15 – 2.12 (m, 2H), 1.67 – 1.46 (m, 9H), 1.27 – 1.20 (m, 2H), 1.18 – 1.03 (m, 6H), 0.78 (q, 2H, J = 10.5 Hz). 13C NMR (100 MHz, CDCl3) δ 174.86, 59.23, 37.52, 37.22, 36.71, 36.28, 33.41, 32.15, 26.72, 26.60, 26.41, 26.26. HRMS–ESI (m/z): [M + H]+ calcd for C14H28NO2 242.2120; found, 242.2121.
Synthetic procedure for derivatives 10–11
To a commercially available 0.5M (cyclohexylmethyl)zinc bromide solution in THF (2.00 eq) were added appropriate iodide derivative (1.00 eq), Pd2dba3 (0.05 eq), and SPhos (0.10 eq) at 23°C. After 15 min, the reaction mixture was refluxed overnight. Reaction was quenched with aqueous saturated NH4Cl solution and extracted with EtOAc (3x). The combined organic layers were washed with brine and dried over Na2SO4. After filtration, solvent was removed under vacuum. The crude was then purified by flash chromatography using a gradient 0 to 20% EtOAc in Hex.
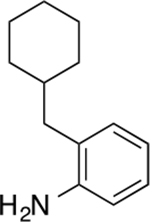
2-(cyclohexylmethyl)aniline (10)
Yellow oil (21.2 mg, 0.11 mmol, 49%). 1H NMR (400 MHz, CDCl3) δ 7.04 (td, 1H, J = 7.6, 1.6 Hz), 7.00 (dd, 1H, J = 7.4, 1.3 Hz), 6.73 (td, 1H, J = 7.4, 1.2 Hz), 6.68 (dd, 1H, J = 7.8, 1.1 Hz), 3.60 (s, 2H), 2.40 (d, 2H, J = 7.1 Hz), 1.82 – 1.63 (m, 5H), 1.63 – 1.52 (m, 1H), 1.28 – 1.15 (m, 3H), 1.10 – 0.93 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 144.44, 131.08, 127.00, 125.69, 118.58, 115.79, 39.73, 37.53, 33.73, 26.68, 26.45. HRMS–ESI (m/z): [M + H]+ calcd for C13H20N 190.1596; found, 190.1599.
Methyl 4-amino-3-(cyclohexylmethyl)benzoate (11)
Orange solid (2.45 g, 9.91 mmol, 55%). 1H NMR (400 MHz, CDCl3) δ 7.72 (dd, 1H, J = 8.3, 2.1 Hz), 7.68 (d, 1H, J = 1.8 Hz), 6.69 (d, 1H, J = 8.3 Hz), 3.85 (s, 3H), 2.40 (d, 1H, J = 7.3 Hz), 1.78 – 1.52 (m, 6H), 1.23 – 1.10 (m, 3H), 1.05 – 0.87 (m, 2H).
Synthetic procedure for derivatives 12–13
To solution of appropriate iodide derivative (1.00 eq) in toluene:H2O (20:1, 0.3M) were added 2-(cyclohexylethyl)boronic acid (5.00 eq), Pd(PPh3)4 (0.05 eq), and K2CO3 (3.00 eq). Reaction mixture was heated at reflux for 24h. After cooling at 23°C, reaction was quenched with aqueous saturated NH4Cl solution and extracted with EtOAc (3x). Organic phase was dried over MgSO4 and concentrated. Crude was purified by flash chromatography using a gradient 0 to 25% EtOAc in Hex.
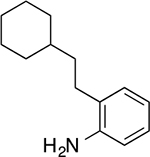
2-(2-cyclohexylethyl)aniline (12)
Yellow oil (26.5 mg, 0.13 mmol, 41%). 1H NMR (500 MHz, CDCl3) δ 7.09 – 7.02 (m, 2H), 6.75 (td, 1H, J = 7.5, 1.0 Hz), 6.69 (dd, 1H, J = 7.8, 0.9 Hz), 3.61 (br, 2H), 2.56 – 2.47 (m, 2H), 1.88 – 1.79 (m, 2H), 1.78 – 1.72 (m, 2H), 1.71 – 1.65 (m, 1H), 1.56 – 1.49 (m, 2H), 1.40 – 1.32 (m, 1H), 1.32 – 1.14 (m, 3H), 1.04 – 0.93 (m, 2H). 13C NMR (125 MHz, CDCl3) δ 144.10, 129.37, 127.34, 126.88, 118.88, 115.61, 37.82, 36.60, 33.45, 28.70, 26.80, 26.46. HRMS–ESI (m/z): [M + H]+ calcd for C14H22N 204.1752; found, 204.1752.
Methyl 4-amino-3-(2-cyclohexylethyl)benzoate (13)
Yellow oil (35.1 mg, 0.13 mmol, 52%). 1H NMR (400 MHz, CDCl3) δ 7.74 (s, 1H), 7.72 (d, 1H, J = 8.9 Hz), 6.62 (d, J = 8.2 Hz, 1H), 4.01 (br, 2H), 3.85 (s, 3H), 3.68 (t, J = 6.7 Hz, 2H), 2.54 – 2.42 (m, 2H), 1.79 (d, 2H, J = 12.5 Hz), 1.76 – 1.60 (m, 4H), 1.57 – 1.43 (m, 2H), 1.40 – 1.08 (m, 5H), 1.05 – 0.90 (m, 2H).
Synthetic procedure for derivatives 14–17
To a suspension of amine 10–13 (1.00 eq) in MeOH (0.15 M) under N2 atmosphere was added the appropriate aldehyde (1.05 eq) at 23°C and then refluxed for 3 hours. After cooling to room temperature, acetic acid (9.00 eq) was added and the mixture was stirred for 15 mins, then NaCNBH3 (1.10 eq) was added. The resulting mixture was stirred at 23°C overnight. Reaction mixture was filtered and the solid collected and purified by flash chromatography using a gradient 0 to 40% EtOAc in Hex to obtain the desired product.
3-(((2-(cyclohexylmethyl)phenyl)amino)methyl)benzoic acid (14)
Synthesized from 10. White solid (5.9 mg, 0.02 mmol, 17%). 1H NMR (500 MHz, CD3OD) δ 8.04 (s, 1H), 7.87 (dt, 1H, J = 7.6, 1.5 Hz), 7.59 – 7.55 (m, 1H), 7.38 (t, 1H, J = 7.7 Hz), 6.94 – 6.87 (m, 2H), 6.54 (td, 1H, J = 7.4, 1.1 Hz), 6.45 (dd, 1H, J = 8.6, 0.9 Hz), 4.45 (s, 2H), 2.45 (d, 2H, J = 7.2 Hz), 1.76 – 1.62 (m, 6H), 1.31 – 1.17 (m, 3H), 1.05 – 0.95 (m, 2H). 13C NMR (125 MHz, CD3OD) δ 170.01, 146.88, 142.60, 132.73, 132.19, 131.63, 129.48, 129.36, 129.14, 127.78, 126.43, 117.62, 111.97, 40.41, 38.46, 34.62, 27.77, 27.51. HRMS–ESI (m/z): [M + H]+ C21H26NO2 324.1964; found, 324.1962.
3-(((2-(cyclohexylmethyl)-4-(methoxycarbonyl)phenyl)amino)methyl)benzoic acid (15)
Synthesized from 11. White solid (376 mg, 0.99 mmol, 91%). 1H NMR (400 MHz, DMSO-d6) δ 12.90 (s, 1H), 7.92 (s, 1H), 7.77 (d, 1H, J = 7.7 Hz), 7.56 (d, 1H, J = 7.1 Hz), 7.52 – 7.47 (m, 2H), 7.43 (t, 1H, J = 7.6 Hz), 6.60 (t, 1H, J = 5.9 Hz), 6.38 (d, 1H, J = 8.8 Hz), 4.53 – 4.46 (m, 2H), 3.71 (s, 3H), 1.72 – 1.55 (m, 6H), 1.23 – 1.09 (m, 4H), 1.04 – 0.91 (m, 2H). 13C NMR (400 MHz, DMSO-d6) δ 167.79, 166.90, 150.23, 140.90, 132.01, 131.69, 131.55, 129.38, 128.98, 128.11, 127.99, 124.24, 116.21, 109.49, 51.66, 45.79, 38.60, 36.87, 33.15, 26.63, 26.24. HRMS–ESI (m/z): [M + H]+ calcd for C23H28NO4 382.2018; found, 382.2012.
3-(((2-(2-cyclohexylethyl)phenyl)amino)methyl)benzoic acid (16)
Synthesized from 12. Colorless oil (10.6 mg, 0.03 mmol, 24%). 1H NMR (500 MHz, CDCl3) δ 8.14 (s, 1H), 8.03 (d, 1H, J = 7.5 Hz), 7.64 (d, 1H, J = 7.5 Hz), 7.46 (t, 1H, J = 7.5 Hz), 7.10 – 7.03 (m, 2H), 6.71 (t, 1H, J = 7.2 Hz), 6.57 (d, 1H, J = 8.1 Hz), 4.46 (s, 2H), 2.56 – 2.48 (m, 2H), 1.80 (d, 2H, J = 12.2 Hz), 1.75 – 1.60 (m, 3H), 1.58 – 1.51 (m, 2H), 1.38 – 1.30 (m, 1H), 1.29 – 1.11 (m, 3H), 1.05 – 0.90 (m, 2H). 13C NMR (125 MHz, CDCl3) δ 171.84, 145.09, 140.38, 132.73, 129.68, 129.09, 129.06, 128.90, 126.98, 117.65, 110.57, 47.94, 37.67, 36.35, 33.35, 28.48, 26.66, 26.32. HRMS–ESI (m/z): [M + H]+ calcd for C22H28NO2 338.2120; found, 338.2114.
3-(((2-(2-cyclohexylethyl)-4-(methoxycarbonyl)phenyl)amino)methyl)benzoic acid (17)
Synthesized from 13. Colorless oil (12.7 mg, 0.03 mmol, 24%). 1H NMR (500 MHz, CDCl3) δ 8.11 (s, 1H), 8.04 (d, 1H, J = 7.6 Hz), 7.80 – 7.74 (m, 2H), 7.59 (d, 1H, J = 7.4 Hz), 7.48 (t, 1H, J = 7.4 Hz), 6.53 (d, 1H, J = 8.7 Hz), 4.53 (s, 2H), 3.85 (s, 3H), 2.56 – 2.47 (m, 2H), 1.79 (d, 2H, J = 12.0 Hz), 1.74 – 1.61 (m, 3H), 1.60 – 1.51 (m, 2H), 1.37 – 1.12 (m, 3H),1.02 – 0.89 (m, 2H). 13C NMR (125 MHz, CDCl3) δ 171.40, 167.56, 149.92, 139.33, 132.54, 130.43, 129.85, 129.53, 129.36, 129.07, 128.93, 125.93, 118.67, 109.29, 51.59, 47.39, 37.73, 36.02, 33.32, 28.27, 26.60, 26.28. HRMS–ESI (m/z): [M + H]+ calcd for C24H30NO4 396.2175; found, 396.2170.
Synthetic procedure for derivatives 18b-n
To a suspension of 11 (1.00 eq) in MeOH (0.15 M) under N2 atmosphere was added the appropriate aldehyde (1.05 eq) at 23°C and then the mixture was refluxed for 3 hours. After cooling to room temperature, acetic acid (9.00 eq) was added and the mixture was stirred for 15 mins, then NaCNBH3 (1.10 eq) was added. The resulting mixture was stirred at 23°C overnight. Reaction mixture was filtered and the solid collected, providing the desired product.
Methyl 4-((2-butylhexyl)amino)-3-(cyclohexylmethyl)benzoate (18b)
Crude mixture obtained was used in the next step without further purification.
Methyl 3-(cyclohexylmethyl)-4-((3-phenylpropyl)amino)benzoate (18c)
Crude mixture obtained was used in the next step without further purification.
Methyl 3-(cyclohexylmethyl)-4-((4-methoxybenzyl)amino)benzoate (18d)
White solid (119 mg, 0.32 mmol, 40%). 1H NMR (CDCl3, 400 MHz) δ 7.78 (dd, 1H, J = 8.6, 2.1 Hz), 7.69 (d, 1H, J = 2.1 Hz), 7.25 (dt, 2H, J = 8.8, 2.6 Hz), 6.89 (dt, 2H, J = 8.8, 2.6 Hz), 6.59 (d, 1H, J = 8.4 Hz), 4.35 (s, 2H), 3.84 (s, 3H), 3.80 (s, 3H), 2.38 (d, 2H, J = 7.1 Hz), 1.74 – 1.51 (m, 6H), 1.22 – 1.12 (m, 3H), 1.04 – 0.92 (m, 2H).
Methyl 3-(cyclohexylmethyl)-4-((3-methoxybenzyl)amino)benzoate (18e)
Yellow oil (116 mg, 0.32 mmol, 39%). 1H NMR (CDCl3, 400 MHz) δ 7.77 (dd, 1H, J = 8.4, 2.0 Hz), 7.70 (d, 1H, J = 1.7 Hz), 7.27 (t, 1H, J = 7.9 Hz), 6.92 (d, 1H, J = 7.5 Hz), 6.89 (s, 1H), 6.83 (dd, 1H, J = 8.2, 2.2 Hz), 6.60 (d, 1H, J = 7.8 Hz), 4.42 (s, 2H), 3.85 (s, 3H), 3.79 (s, 3H), 2.41 (d, 2H, J = 7.1 Hz), 1.75 – 1.63 (m, 6H), 1.22 – 1.14 (m, 3H), 1.05 – 0.95 (m, 2H).
Methyl 3-(cyclohexylmethyl)-4-((3-fluorobenzyl)amino)benzoate (18f)
Colorless residue (80.3 mg, 0.23 mmol, 28%). 1H NMR (CDCl3, 400 MHz) δ 7.76 (dd, 1H, J = 8.6, 1.8 Hz), 7.71 (s, 1H), 7.36 – 7.27 (m, 1H), 7.11 (d, 1H, J = 7.6 Hz), 7.03 (d, 1H, J = 9.7 Hz), 6.98 (t, 1H, J = 8.3 Hz), 6.54 (d, 1H, J = 6.5 Hz), 4.46 (s, 2H), 3.85 (s, 3H), 2.43 (d, 2H, J = 7.1 Hz), 1.76 – 1.52 (m, 6H), 1.24 – 1.14 (m, 3H), 1.06 – 0.96 (m, 2H).
Methyl 3-(cyclohexylmethyl)-4-((4-fluorobenzyl)amino)benzoate (18g)
White solid (117 mg, 0.33 mmol, 41%). 1H NMR (CDCl3, 400 MHz) δ 7.77 (dd, 1H, J = 8.5, 1.6 Hz), 7.71 (bs, 1H), 7.35 – 7.27 (m, 2H), 7.04 (t, 2H, J = 8.6 Hz), 6.55 (d, 1H, J = 8.6 Hz), 4.41 (s, 2H), 3.84 (s, 3H), 2.41 (d, 2H, J = 7.1 Hz), 1.75 – 1.53 (m, 6H), 1.23 – 1.13 (m, 3H), 1.06 – 0.94 (m, 2H).
Methyl 3-(cyclohexylmethyl)-4-((naphthalen-2-ylmethyl)amino)benzoate (18h)
White solid (122 mg, 0.32 mmol, 39%). 1H NMR (CDCl3, 400 MHz) δ 7.89 – 7.83 (m, 2H), 7.83 – 7.77 (m, 3H), 7.74 (d, 1H, J = 2.2 Hz), 7.55 – 7.45 (m, 3H), 6.66 (d, 1H, J = 8.4 Hz), 4.61 (s, 2H), 3.86 (s, 3H), 2.45 (d, 2H, J = 6.8 Hz), 1.78 – 1.59 (m, 6H), 1.25 – 1.14 (m, 3H), 1.09 – 0.96 (m, 2H).
Methyl 4-(((4’-chloro-[1,1’-biphenyl]-4-yl)methyl)amino)-3-(cyclohexylmethyl)benzoate (18i)
White solid (121 mg, 0.27 mmol, 33%). 1H NMR (CDCl3, 400 MHz) δ 7.78 (dd, 1H, J = 8.5, 2.1 Hz), 7.72 (d, 1H, J = 2.0 Hz), 7.56 – 7.48 (m, 5H), 7.43 – 7.38 (m, 4H), 4.50 (s, 2H), 3.85 (s, 3H), 2.44 (d, 2H, J = 7.1 Hz), 1.75 – 1.61 (m, 6H), 1.23 – 1.13 (m, 3H), 1.07 – 0.94 (m, 2H).
Methyl 3-(cyclohexylmethyl)-4-(((3’,5’-dimethoxy-[1,1’-biphenyl]-4-yl)methyl)amino)benzoate (18j)
White solid (121 mg, 0.27 mmol, 33%). 1H NMR (CDCl3, 400 MHz) δ 7.79 (dd, 1H, J = 8.7, 1.3 Hz), 7.75 – 7.70 (m, 2H), 7.55 (d, 2H, J = 7.9 Hz), 7.39 (d, 2H, J = 7.9 Hz), 7.14 (dd, 1H, J = 8.5, 1.4 Hz), 7.10 (bs, 1H), 6.95 (d, 1H, J = 8.3 Hz), 6.64 (bs, 1H), 4.48 (s, 2H), 3.95 (s, 3H), 3.92 (s, 3H), 3.85 (s, 3H), 2.43 (d, 2H, J = 7.0 Hz), 1.77 – 1.57 (m, 6H), 1.24 – 1.12 (m, 3H), 1.09 – 0.93 (m, 2H).
Methyl 4-(((3’-chloro-[1,1’-biphenyl]-4-yl)methyl)amino)-3-(cyclohexylmethyl)benzoate (18k)
White solid (134 mg, 0.30 mmol, 68%). 1H NMR (DMSO-d6, 400 MHz) δ 7.64 (bs, 2H), 7.55 (t, 2H, J = 7.9 Hz), 7.53 – 7.46 (m, 3H), 7.46 – 7.39 (m, 2H), 7.37 (d, 1H, J = 7.5 Hz), 7.60 (t, 1H, J = 5.2 Hz), 6.46 (d, 1H, J = 8.8 Hz), 4.50 (d, 2H J = 5.9 Hz), 3.70 (s, 3H), 1.68 – 1.55 (m, 6H), 1.15 – 1.07 (m, 3H), 1.04 −0.91 (m, 2H).
Methyl 3-(cyclohexylmethyl)-4-(((3’-fluoro-[1,1’-biphenyl]-4-yl)methyl)amino)benzoate (18l)
White solid (143 mg, 0.33 mmol, 69%). 1H NMR (DMSO-d6, 400 MHz) δ 7.64 (s, 1H), 7.55 (d, 1H, J = 7.4 Hz), 7.53 – 7.46 (m, 3H), 7.46 – 7.39 (m, 3H), 7.36 (d, 1H, J = 7.2 Hz), 7.18 (t, 1H, J = 8.3 Hz), 6.60 (t, 1H, J = 5.7 Hz), 6.46 (d, 1H, J = 7.9 Hz), 4.50 (d, 2H, J = 5.7 Hz), 3.70 (s, 3H), 1.69 – 1.57 (m, 6H), 1.15 – 1.06 (m, 3H), 1.04 – 0.91 (m, 2H).
Methyl 4-((benzofuran-2-ylmethyl)amino)-3-(cyclohexylmethyl)benzoate (18m)
White solid (117 mg, 0.31 mmol, 38%). 1H NMR (CDCl3, 400 MHz) δ 7.80 (dd, 1H, J = 8.6, 2.1 Hz), 7.71 (d, 1H, J = 2.0 Hz), 7.53 – 7.49 (m, 1H), 7.47 – 7.42 (m, 1H), 7.27 (td, 1H, J = 7.2, 1.4 Hz), 7.21 (td, 1H, J = 7.5, 1.2 Hz), 6.73 (d, 1H, J = 8.5 Hz), 6.61 (bs, 1H), 4.60 (s, 2H), 3.85 (s, 3H), 2.44 (d, 2H, J = 7.2 Hz), 1.75 – 1.62 (m, 6H), 1.24 – 1.12 (m, 3H), 1.06 – 0.95 (m, 2H).
Methyl 3-(cyclohexylmethyl)-4-((pyridin-3-ylmethyl)amino)benzoate (18n)
Colorless solid (167mg, 0.49 mmol, 61%). 1H NMR (CDCl3, 400 MHz) δ 8.63 (s, 1H), 8.56 (d, 1H, J = 4.9 Hz), 7.76 (dd, 1H, J = 8.5, 2.2 Hz), 7.70 (d, 1H, J = 2.2 Hz), 7.65 (dt, 1H, J = 7.6, 1.7 Hz), 7.32 – 7.27 (m, 1H), 6.52 (d, 1H, J = 8.6 Hz), 4.47 (s, 2H), 3.84 (s, 3H), 2.41 (d, 2H, J = 7.1 Hz), 1.76 – 1.61 (m, 6H), 1.22 – 1.12 (m, 3H), 1.05 – 0.92 (m, 2H).
Synthetic procedure for derivatives 19a-n
Methyl ester derivative 18a-n (1.00 eq) was dissolved in THF (0.10 M) at 23°C, then KOSiMe3 (5.00 eq) was added. Reaction mixture was heated at 50°C overnight. After cooling, solvent was removed under vacuum. The crude was dissolved in H2O and pH adjusted to ~2 using a 1M HCl aq. sol. and stirred for 30 mins. Precipitate formed was collected and dried to provide the desired product, which was used in the next step without further purification.
Synthetic procedure for derivatives 20a-n
Carboxylic acid 19a-n was dissolved in anh. DMF (0.10 M), then HATU (1.00 eq) and DIPEA (2.00 eq) were added. Reaction was stirred at 23°C for 10 mins, then 3-amino-phenol (1.00 eq) was added. Stirring continued overnight. Reaction was quenched with aqueous saturated NaHCO3 solution and extracted with EtOAc (3x). Organic phase was washed with H2O (1x) and brine (1x), then dried over MgSO4 and concentrated. Crude was purified by flash chromatography using a gradient 0 to 50% EtOAc in Hex.
4-amino-3-(cyclohexylmethyl)-N-(3-hydroxyphenyl)benzamide (20a)
Dark orange oil (71.7 mg, 0.22 mmol, 37%). 1H NMR (CD3OD, 400 MHz) δ 7.61 – 7.57 (m, 2H), 7.28 (t, 1H, J = 2.0 Hz), 7.12 (t, 1H, J = 8.0 Hz), 7.08 – 7.04 (m, 1H), 6.71 (dd, 1H, J = 7.3, 1.6 Hz), 6.58 – 6.53 (m, 1H), 2.42 (d, 2H, J = 7.2 Hz), 1.77 – 1.58 (m, 6H), 1.22 – 1.17 (m, 3H), 1.06 – 0.94 (m, 2H).
4-((2-butylhexyl)amino)-3-(cyclohexylmethyl)-N-(3-hydroxyphenyl)benzamide (20b)
Pale brown oil (58.2 mg, 0.12 mmol, 51% over 3 steps). 1H NMR (CD3OD, 400 MHz) δ 9.56 (s, 1H), 7.70 (dd, 1H, J = 8.5, 2.2 Hz), 7.57 (d, 1H, J = 2.2 Hz), 7.28 – 7.25 (m, 1H), 7.12 (t, 1H, J = 7.9 Hz), 7.08 – 7.03 (m, 1H), 6.60 (d, 1H, J = 8.7 Hz), 6.57 – 6.52 (m, 1H), 3.12 (d, 2H, J = 6.4 Hz), 2.44 (d, 2H, J = 7.1 Hz), 1.79 – 1.65 (m, 6H), 1.42 – 1.30 (m, 12H), 1.23 – 1.16 (m, 3H), 1.07 – 0.97 (m, 2H), 0.92 (t, 6H, J = 7.0 Hz).
3-(cyclohexylmethyl)-N-(3-hydroxyphenyl)-4-((3-phenylpropyl)amino)benzamide (20c)
Colorless oil (40.7 mg, 0.09 mmol, 27% over 3 steps). 1H NMR (CD3OD, 400 MHz) δ 7.66 (dd, 1H, J = 8.6, 2.2 Hz), 7.57 (d, 1H, J = 2.2 Hz), 7.29 – 7.02 (m, 8H), 6.57 – 6.52 (m, 2H), 3.22 (t, 2H, J = 7.5 Hz), 2.70 (t, 2H, J = 7.5 Hz), 2.40 (d, 2H, J = 7.1 Hz), 1.94 (quint, 2H, J = 6.9 Hz), 1.74 – 1.56 (m, 6H), 1.20 – 1.15 (m, 3H), 1.05 – 0.93 (m, 2H).
3-(cyclohexylmethyl)-N-(3-hydroxyphenyl)-4-((4-methoxybenzyl)amino)benzamide (20d)
Yellow solid (61.8 mg, 0.14 mmol, 65% over 2 steps). 1H NMR (CD3OD, 400 MHz) δ 7.61 – 7.57 (m, 2H), 7.28 – 7.20 (m, 3H), 7.11 (t, 1H, J = 8.0 Hz), 7.06 – 7.00 (m, 1H), 6.94 – 6.82 (m, 3H), 6.52 (d, 1H, J = 9.1 Hz), 4.39 (s, 2H), 3.75 (s, 3H), 2.49 (d, 2H, J = 7.0 Hz), 1.79 – 1.58 (m, 6H), 1.23 – 1.15 (m, 3H), 1.08 – 0.95 (m, 2H).
3-(cyclohexylmethyl)-N-(3-hydroxyphenyl)-4-((3-methoxybenzyl)amino)benzamide (20e)
Yellow solid (50.5 mg, 0.11 mmol, 46% over 2 steps). 1H NMR (CD3OD, 400 MHz) δ 7.60 – 7.54 (m, 2H), 7.21 (t, 1H, J = 7.8 Hz), 7.11 (t, 1H, J = 8.0 Hz), 7.02 (d, 1H, J = 8.0 Hz), 6.94 – 6.87 (m, 2H), 6.77 (dd, 1H, J = 8.3, 2.1 Hz), 6.54 (dd, 1H, J = 8.3, 2.1 Hz), 6.48 (d, 1H, J = 8.2 Hz), 6.26 – 6.21 (m, 1H), 4.46 (s, 2H), 3.74 (s, 3H), 2.52 (d, 2H, J = 6.8 Hz), 1.77 – 1.67 (m, 6H), 1.31 – 1.18 (m, 3H), 1.10 – 0.98 (m, 2H).
3-(cyclohexylmethyl)-4-((3-fluorobenzyl)amino)-N-(3-hydroxyphenyl)benzamide (20f)
Colorless residue (22.6 mg, 0.05 mmol, 31% over 2 steps). 1H NMR (CD3OD, 400 MHz) δ 7.61 – 7.54 (m, 2H), 7.34 – 7.27 (m, 1H), 7.27 – 7.22 (m, 1H), 7.18 – 7.12 (m, 2H), 7.10 (d, 1H, J = 8.0 Hz), 7.07 – 6.99 (m, 2H), 6.92 (dt, 1H, J = 8.4, 2.1 Hz), 4.49 (s, 2H), 2.53 (d, 2H, J = 6.8 Hz), 1.80 – 1.61 (m, 6H), 1.31 – 1.16 (m, 3H), 1.04 (q, 2H, J = 10.8 Hz).
3-(cyclohexylmethyl)-4-((4-fluorobenzyl)amino)-N-(3-hydroxyphenyl)benzamide (20g)
White solid (39.9 mg, 0.09 mmol, 41% over 2 steps). 1H NMR (CD3OD, 400 MHz) δ 7.62 – 7.54 (m, 2H), 7.37 – 7.30 (m, 2H), 7.25 (t, 1H, J = 2.0 Hz), 7.11 (t, 1H, J = 7.9 Hz), 7.06 – 6.98 (m, 2H), 6.54 (dd, 1H, J = 2.3, 0.9 Hz), 6.47 (d, 1H, J = 8.5 Hz), 6.25 – 6.20 (m, 1H), 4.45 (s, 2H), 2.51 (d, 2H, J = 6.9 Hz), 1.77 – 1.61 (m, 6H), 1.28 – 1.17 (m, 3H), 1.09 – 0.96 (m, 2H).
3-(cyclohexylmethyl)-N-(3-hydroxyphenyl)-4-((naphthalen-2-ylmethyl)amino)benzamide (20h)
Colorless residue (42.5 mg, 0.09 mmol, 37% over 2 steps). 1H NMR (CD3OD, 400 MHz) δ 7.79 (d, 2H, J = 8.5 Hz), 7.77 – 7.70 (m, 2H), 7.60 (d, 1H, J = 1.8 Hz), 7.53 (dd, 1H, J = 8.7, 2.1 Hz), 7.46 (d, 1H, J = 8.3 Hz), 7.43 – 7.38 (m, 2H), 7.25 (t, 1H, J = 2.0 Hz), 7.09 (t, 1H, J = 7.9 Hz), 7.02 (d, 1H, J = 8.2 Hz), 6.53 (d, 2H, J = 8.4 Hz), 4.63 (s, 2H), 2.54 (d, 2H, J = 6.8 Hz), 1.79 – 1.63 (m, 6H), 1.33 – 1.17 (m, 3H), 1.11 – 0.99 (m, 2H).
4-(((4’-chloro-[1,1’-biphenyl]-4-yl)methyl)amino)-3-(cyclohexylmethyl)-N-(3-hydroxyphenyl)benzamide (20i)
Yellow solid (42.4 mg, 0.08 mmol, 44% over 2 steps). 1H NMR (CDCl3, 400 MHz) δ 8.10 (t, 1H, J = 2.1 Hz), 7.81 (s, 1H), 7.56 – 7.48 (m, 5H), 7.42 – 7.36 (m, 4H), 7.14 (t, 1H, J = 7.1 Hz), 6.67 – 6.61 (m, 2H), 6.58 (d, 1H, J = 8.1 Hz), 4.46 (s, 2H), 2.43 (d, 2H, J = 7.1 Hz), 1.79 – 1.57 (m, 6H), 1.23 – 1.12 (m, 3H), 1.06 – 0.95 (m, 2H).
3-(cyclohexylmethyl)-4-(((3’,5’-dimethoxy-[1,1’-biphenyl]-4-yl)methyl)amino)-N-(3-hydroxyphenyl)benzamide (20j)
Colorless residue (37.0 mg, 0.07 mmol, 33% over 2 steps). 1H NMR (CD3OD, 400 MHz) δ 7.59 (d, 1H, J = 2.0 Hz), 7.56 (dd, 1H, J = 8.6, 2.1 Hz), 7.48 (d, 2H, J = 8.0 Hz), 7.33 (d, 2H, J = 7.8 Hz), 7.27 (t, 1H, J = 2.0 Hz), 7.13 – 7.01 (m, 4H), 6.93 (d, 1H, J = 8.1 Hz), 6.57 – 6.51 (m, 1H), 6.48 (d, 1H, J = 8.4 Hz), 4.45 (s, 2H), 3.82 (s, 3H), 3.79 (s, 3H), 2.48 (d, 2H, J = 6.7 Hz), 1.76 – 1.59 (m, 6H), 1.24 – 1.14 (m, 3H), 1.06 – 0.93 (m, 2H).
4-(((3’-chloro-[1,1’-biphenyl]-4-yl)methyl)amino)-3-(cyclohexylmethyl)-N-(3-hydroxyphenyl)benzamide (20k)
Colorless residue (38.0 mg, 0.07 mmol, 38% over 2 steps). 1H NMR (DMSO-d6, 400 MHz) δ 9.57 (s, 1H), 9.30 (s, 1H), 7.67 (d, 2H, J = 10.8 Hz), 7.61 – 7.51 (m, 4H), 7.48 (t, 1H, J = 7.8 Hz), 7.45 – 7.36 (m, 2H), 7.28 (s, 1H), 7.10 – 7.00 (m, 2H), 6.47 (d, 1H, J = 8.4 Hz), 6.44 – 6.39 (m, 2H), 4.51 (d, 2H, J = 5.5 Hz), 1.72 – 1.57 (m, 6H), 1.16 – 1.08 (m, 3H), 1.06 – 0.92 (m, 2H).
3-(cyclohexylmethyl)-4-(((3’-fluoro-[1,1’-biphenyl]-4-yl)methyl)amino)-N-(3-hydroxyphenyl)benzamide (20l)
Colorless residue (38.0 mg, 0.06 mmol, 30% over 2 steps). 1H NMR (DMSO-d6, 400 MHz) δ 9.57 (s, 1H), 9.30 (s, 1H), 7.68 (s, 1H), 7.58 – 7.51 (m, 3H), 7.51 – 7.37 (m, 4H), 7.28 (s, 1H), 7.19 (t, 1H, J = 8.1 Hz), 7.11 – 7.00 (m, 2H), 6.48 – 6.40 (m, 3H), 4.51 (d, 2H, J = 5.6 Hz), 1.73 – 1.57 (m, 6H), 1.17 – 1.08 (m, 3H), 1.07 – 0.93 (m, 2H).
4-((benzofuran-2-ylmethyl)amino)-3-(cyclohexylmethyl)-N-(3-hydroxyphenyl)benzamide (20m)
Yellow solid (53.5 mg, 0.12 mmol, 48% over 2 steps). 1H NMR (CD3OD, 400 MHz) δ 7.63 (dd, 1H, J = 8.5, 2.3 Hz), 7.59 (d, 1H, J = 2.2 Hz), 7.47 – 7.42 (m, 1H), 7.42 – 7.35 (m, 1H), 7.26 (t, 1H, J = 2.1 Hz), 7.20 (td, 1H, J = 7.3, 1.2 Hz), 7.14 (td, 1H, J = 7.5, 1.1 Hz), 7.10 (t, 1H, J = 7.8 Hz), 7.07 – 7.01 (m, 1H), 6.71 (d, 1H, J = 8.6 Hz), 6.58 – 6.51 (m, 2H), 4.60 (s, 2H), 2.49 (d, 2H, J = 6.9 Hz), 1.76 – 1.60 (m, 6H), 1.22 – 1.16 (m, 3H), 1.07 – 0.94 (m, 2H).
3-(cyclohexylmethyl)-N-(3-hydroxyphenyl)-4-((pyridin-3-ylmethyl)amino)benzamide (20n)
Yellow solid (118 mg, 0.29 mmol, 73% over 2 steps). 1H NMR (CD3OD, 400 MHz) δ 8.58 – 8.52 (m, 1H), 8.40 (dd, 1H, J = 4.9, 1.6 Hz), 7.87 – 7.79 (m, 1H), 7.64 – 7.56 (m, 2H), 7.39 (dd, 1H, J = 7.8, 5.5 Hz), 7.11 (t, 1H, J = 8.1 Hz), 7.06 – 7.00 (m, 1H), 6.64 (d, 1H, J = 8.8 Hz), 6.57 – 6.52 (m, 1H), 6.49 (d, 1H, J = 8.4 Hz), 4.57 (s, 2H), 2.55 (d, 2H, J = 6.7 Hz), 1.81 −1.65 (m, 6H), 1.33 – 1.25 (m, 3H), 1.12 – 1.00 (m, 2H).
Synthetic procedure for derivatives 21a-n
To a solution of phenol 20a-n (1.00 eq) in CH2Cl2 (0.1 M) was added diethyl chlorophosphate (1.00 eq) and DIPEA (3.00 eq). The resulting solution was stirred at room temperature overnight. Upon evaporation of the solvent, the residue was purified by silica gel chromatography (EtOAc:Hex = 7:3) to give the desired compound.
3-(4-amino-3-(cyclohexylmethyl)benzamido)phenyl diethyl phosphate (21a)
Colorless residue (33.4 mg, 0.07 mmol, 33%). 1H NMR (CD3OD, 400 MHz) δ 7.77 – 7.74 (m, 1H), 7.63 – 7.59 (m, 2H), 7.51 – 7.47 (m, 1H), 7.33 (t, 1H, J = 8.2 Hz), 6.97 – 6.93 (m, 1H), 6.72 (dd, 1H, J = 8.0, 0.6 Hz), 4.25 (q, 2H, J = 7.0 Hz), 4.23 (q, 2H, J = 7.1 Hz), 2.44 (d, 2H, J = 7.1 Hz), 1.78 – 1.60 (m, 6H), 1.37 (t, 3H, J = 7.1 Hz), 1.36 (t, 3H, J = 7.1 Hz), 1.30 – 1.15 (m, 3H), 1.08 – 0.95 (m, 2H).
3-(4-((2-butylhexyl)amino)-3-(cyclohexylmethyl)benzamido)phenyl diethyl phosphate (21b)
Colorless oil (37.6 mg, 0.06 mmol, 50%). 1H NMR (CD3OD, 400 MHz) δ 7.76 – 7.74 (m, 1H), 7.72 (dd, 1H, J = 8.6, 2.3 Hz), 7.59 (d, 1H, J = 2.1 Hz), 7.52 – 7.47 (m, 1H), 7.33 (t, 1H, J = 8.2 Hz), 6.97 – 6.93 (m, 1H), 6.62 (d, 1H, J = 8.8 Hz), 4.26 (q, 2H, J = 7.0 Hz), 4.24 (q, 2H, J = 7.1 Hz), 3.13 (d, 2H, J = 6.2 Hz), 2.45 (d, 2H, J = 7.0 Hz), 1.79 – 1.56 (m, 6H), 1.45 – 1.27 (m, 12H), 1.27 – 1.14 (m, 3H), 1.11 – 0.98 (m, 2H), 0.93 (t, 6H, J = 7.0 Hz).
3-(3-(cyclohexylmethyl)-4-((3-phenylpropyl)amino)benzamido)phenyl diethyl phosphate (21c)
Colorless oil (31.2 mg, 0.05 mmol, 59%). 1H NMR (CD3OD, 400 MHz) δ 7.69 (dd, 1H, J = 8.6, 2.0 Hz), 7.59 (d, 1H, J = 2.0 Hz), 7.55 – 7.45 (m, 2H), 7.37 – 7.14 (m, 6H), 6.95 (d, 1H, J = 8.1 Hz), 6.56 (d, 1H, J = 8.6 Hz), 4.24 (app quint, 4H, J = 7.4 Hz), 3.25 (t, 2H, J = 7.0 Hz), 2.72 (t, 2H, J = 7.5 Hz), 2.42 (d, 2H, J = 7.3 Hz), 1.96 (quint, 2H, J = 7.4 Hz), 1.78 – 1.58 (m, 6H), 1.36 (t, 6H, J = 7.0 Hz), 1.23 – 1.16 (m, 3H), 1.07 – 0.97 (m, 2H).
3-(3-(cyclohexylmethyl)-4-((4-methoxybenzyl)amino)benzamido)phenyl diethyl phosphate (21d)
White solid (31.6 mg, 0.05 mmol, 39%). 1H NMR (CD3OD, 400 MHz) δ 7.74 – 7.70 (m, 1H), 7.62 – 7.57 (m, 2H), 7.50 – 7.44 (m, 1H), 7.31 (t, 1H, J = 8.1 Hz), 7.25 (dt, 2H, J = 8.5, 2.4 Hz), 6.96 – 6.91 (m, 1H), 6.85 (dt, 2H, J = 8.5, 2.4 Hz), 6.55 – 6.50 (m, 1H), 4.40 (s, 2H), 4.24 (q, 2H, J = 7.1 Hz), 4.22 (q, 2H, J = 7.2 Hz), 3.75 (s, 3H), 2.50 (d, 2H, J = 7.4 Hz), 1.75 – 1.64 (m, 6H), 1.35 (t, 3H, J = 7.2 Hz), 1.35 (t, 3H, J = 7.2 Hz), 1.24 – 1.18 (m, 3H), 1.09 – 0.97 (m, 2H).
3-(3-(cyclohexylmethyl)-4-((3-methoxybenzyl)amino)benzamido)phenyl diethyl phosphate (21e)
White solid (36.4 mg, 0.06 mmol, 55%). 1H NMR (CD3OD, 400 MHz) δ 7.73 (s, 1H), 7.63 – 7.56 (m, 2H), 7.47 (d, 1H, J = 8.2 Hz), 7.30 (t, 1H, J = 8.2 Hz), 7.20 (t, 1H, J = 7.8 Hz), 6.97 – 6.87 (m, 3H), 6.76 (dd, 1H, J = 8.5, 2.3 Hz), 6.47 (d, 1H, J = 8.6 Hz), 4.45 (s, 2H), 4.22 (app quint, 4H, J = 7.5 Hz), 3.73 (s, 3H), 2.51 (d, 2H, J = 6.7 Hz), 1.79 – 1.63 (m, 6H), 1.35 (t, 6H, J = 7.1 Hz), 1.27 – 1.16 (m, 3H), 1.08 – 1.00 (m, 2H).
3-(3-(cyclohexylmethyl)-4-((3-fluorobenzyl)amino)benzamido)phenyl diethyl phosphate (21f)
Colorless residue (24.2 mg, 0.04 mmol, 82%). 1H NMR (CD3OD, 400 MHz) δ 7.74 – 7.71 (m, 1H), 7.61 (d, 1H, J = 2.1 Hz), 7.59 (dd, 1H, J = 8.4, 2.2 Hz), 7.50 – 7.44 (m, 1H), 7.34 – 7.29 (m, 2H), 7.16 (d, 1H, J = 7.6 Hz), 7.04 (dt, 1H, J = 10.0, 1.8 Hz), 6.97 – 6.90 (m, 2H), 6.46 (d, 1H, J = 8.5 Hz), 4.51 (s, 2H), 4.24 (q, 2H, J = 7.1 Hz), 4.22 (q, 2H, J = 7.1 Hz), 2.54 (d, 2H, J = 6.8 Hz), 1.78 – 1.65 (m 6H), 1.36 (t, 3H J = 7.1 Hz), 1.36 (t, 3H, J = 7.1 Hz), 1.27 – 1.20 (m, 3H), 1.12 – 0.99 (m, 2H).
3-(3-(cyclohexylmethyl)-4-((4-fluorobenzyl)amino)benzamido)phenyl diethyl phosphate (21g)
Colorless residue (29.2 mg, 0.05 mmol, 56%). 1H NMR (CD3OD, 400 MHz) δ 7.75 – 7.71 (m, 1H), 7.62 – 7.57 (m, 2H), 7.49 – 7.45 (m, 1H), 7.37 – 7.28 (m, 3H), 7.05 – 6.99 (m, 2H), 6.96 – 6.92 (m, 1H), 6.47 (d, 1H, J = 8.4 Hz), 4.24 (q, 2H, J = 7.1 Hz), 4.22 (q, 2H, J = 7.1 Hz), 2.51 (d, 2H, J = 6.8 Hz), 1.77 – 1.64 (m, 6H), 1.35 (t, 3H, J = 7.1 Hz), 1.35 (t, 3H, J = 7.1 Hz), 1.28 – 1.18 (m, 3H), 1.09 – 0.97 (m, 2H).
3-(3-(cyclohexylmethyl)-4-((naphthalen-2-ylmethyl)amino)benzamido)phenyl diethyl phosphate (21h)
Colorless residue (25.7 mg, 0.43 mmol, 47%). 1H NMR (CD3OD, 400 MHz) δ 7.80 (d, 2H, J = 8.0 Hz), 7.77 – 7.70 (m, 3H), 7.61 (d, 1H, J = 2.2 Hz), 7.55 (dd, 1H, J = 8.6, 2.3 Hz), 7.50 – 7.40 (m, 4H), 7.29 (t, 1H, J = 8.1 Hz), 6.93 (d, 1H, J = 7.8 Hz), 6.54 (d, 1H, J = 8.5 Hz), 4.65 (s, 2H), 4.22 (app quint, 4H, J = 7.4 Hz), 2.56 (d, 2H, J = 6.9 Hz), 1.83 – 1.64 (m, 6H), 1.34 (t, 6H, J = 7.2 Hz), 1.28 – 1.20 (m, 3H), 1.13 – 0.99 (m, 2H).
3-(4-(((4’-chloro-[1,1’-biphenyl]-4-yl)methyl)amino)-3-(cyclohexylmethyl)benzamido)phenyl diethyl phosphate (21i)
Colorless residue (31.7 mg, 0.05 mmol, 59%). 1H NMR (CDCl3, 400 MHz) δ 7.92 (s, 1H), 7.61 – 7.58 (m, 2H), 7.57 – 7.53 (m, 2H), 7.53 – 7.47 (m, 3H), 7.43 – 7.38 (m, 4H), 7.27 (t, 1H, J = 8.1 Hz), 6.97 – 6.93 (m, 1H), 6.59 (d, 1H, J = 9.2 Hz), 4.49 (s, 2H), 4.26 – 4.16 (m, 4H), 2.45 (d, 2H, J = 7.2 Hz), 1.77 – 1.57 (m, 6H), 1.35 (t, 3H, J = 7.2 Hz), 1.35 (t, 3H, J = 7.2 Hz), 1.22 – 1.14 (m, 3H), 1.07 – 0.96 (m, 2H).
3-(3-(cyclohexylmethyl)-4-(((3’,5’-dimethoxy-[1,1’-biphenyl]-4-yl)methyl)amino)benzamido)phenyl diethyl phosphate (21j)
White residue (10.8 mg, 0.02 mmol, 23%). 1H NMR (CD3OD, 400 MHz) δ 7.74 – 7.71 (m, 1H), 7.62 – 7.58 (m, 2H), 7.53 (dt, 2H, J = 8.2, 1.9 Hz), 7.49 – 7.45 (m, 1H), 7.41 – 7.36 (m, 2H), 7.31 (t, 1H, J = 8.2 Hz), 7.17 – 7.13 (m, 2H), 6.99 (d, 1H, J = 8.0 Hz), 6.96 – 6.92 (m, 1H), 6.53 (d, 1H, J = 8.2 Hz), 4.52 (s, 2H), 4.24 (q, 2H, J = 7.1 Hz), 4.22 (q, 2H, J = 7.1 Hz), 3.88 (s, 3H), 3.85 (s, 3H), 2.54 (d, 2H, J = 6.9 Hz), 1.79 – 1.63 (m, 6H), 1.36 (t, 3H, J = 7.1 Hz), 1.35 (t, 3H, J = 7.1 Hz), 1.27 – 1.19 (m, 3H), 1.11 – 0.99 (m, 2H).
3-(4-(((3’-chloro-[1,1’-biphenyl]-4-yl)methyl)amino)-3-(cyclohexylmethyl)benzamido)phenyl diethyl phosphate (21k)
White residue (30.0 mg, 0.05 mmol, 63%). 1H NMR (DMSO-d6, 400 MHz) δ 9.85 (s, 1H), 8.53 (s, 1H), 7.73 (s, 1H), 7.67 (d, 2H, J = 12.0 Hz), 7.61 – 7.51 (m, 5H), 7.47 (t, 1H, J = 7.8 Hz), 7.45 – 7.37 (m, 2H), 7.29 (t, 1H, J = 8.2 Hz), 6.85 (d, 1H, J = 7.5 Hz), 6.51 – 6.45 (m, 2H), 4.51 (d, 2H, J = 5.4 Hz), 4.14 (app quint, 4H, J = 7.7 Hz), 1.72 – 1.55 (m, 6H), 1.27 (t, 6H, J = 7.2 Hz), 1.16 – 1.08 (m, 3H), 1.07 – 0.93 (m, 2H).
3-(3-(cyclohexylmethyl)-4-(((3’-fluoro-[1,1’-biphenyl]-4-yl)methyl)amino)benzamido)phenyl diethyl phosphate (21l)
White solid (38.0 mg, 0.06 mmol, 84%). 1H NMR (DMSO-d6, 400 MHz) δ 9.86 (s, 1H), 8.54 (s, 2H), 7.73 (s, 1H), 7.68 (s, 1H), 7.62 – 7.37 (m, 7H), 7.29 (t, 1H, J = 8.2 Hz), 7.22 – 7.15 (m, 1H), 6.85 (d, 1H, J = 8.8 Hz), 6.51 – 6.45 (m, 2H), 4.51 (d, 2H, J = 5.7 Hz), 4.14 (app quint, 4H, J = 8.0 Hz), 1.72 – 1.54 (m, 6H), 1.27 (t, 6H, J = 8.1 Hz), 1.15 – 1.07 (m, 3H), 1.06 – 0.95 (m, 2H).
3-(4-((benzofuran-2-ylmethyl)amino)-3-(cyclohexylmethyl)benzamido)phenyl diethyl phosphate (21m)
Yellow residue (44.7 mg, 0.08 mmol, 64%). 1H NMR (CDCl3, 400 MHz) δ 8.07 (s, 1H), 7.65 – 7.58 (m, 3H), 7.53 – 7.46 (m, 2H), 7.46 – 7.42 (m, 1H), 7.30 – 7.18 (m, 2H), 6.98 – 6.92 (m, 1H), 6.69 (d, 1H, J = 8.7 Hz), 6.58 (bs, 1H), 4.57 (s, 2H), 4.25 – 4.15 (m, 4H), 2.44 (d, 2H, J = 7.1 Hz), 1.76 – 1.61 (m, 6H), 1.33 (t, 3H, J = 7.1 Hz), 1.33 (t, 3H, J = 7.1 Hz), 1.21 – 1.12 (m, 3H), 1.06 – 0.93 (m, 2H).
3-(3-(cyclohexylmethyl)-4-((pyridin-3-ylmethyl)amino)benzamido)phenyl diethyl phosphate (21n)
Yellow solid (38.4 mg, 0.07 mmol, 31%). 1H NMR (CD3OD, 400 MHz) δ 8.53 (d, 1H, J = 1.4 Hz), 8.40 (dd, 1H, J = 5.1, 1.4 Hz), 7.81 (td, 1H, J = 8.0, 1.9 Hz), 7.75 – 7.72 (m, 1H), 7.65 – 7.58 (m, 2H), 7.50 – 7.44 (m, 1H), 7.41 – 7.35 (m, 1H), 7.31 (t, 1H, J = 8.2 Hz), 6.97 – 6.91 (m, 1H), 6.49 (d, 1H, J = 8.5 Hz), 4.56 (s, 2H), 4.24 (q, 2H, J = 7.1 Hz), 4.22 (q, 2H, J = 7.1 Hz), 2.54 (d, 2H, J = 7.0 Hz), 1.80 – 1.62 (m, 6H), 1.36 (t, 3H, J = 7.1 Hz), 1.35 (t, 3H, J = 7.1Hz), 1.25 – 1.18 (m, 3H), 1.11 – 0.98 (m, 2H).
Synthetic procedure for derivatives 22a-n
To the phosphate derivative 21a-n was added a commercially available solution of TMSBr (6.50 eq) in CH2Cl2 (0.25 M) and stirred at room temperature overnight. Upon evaporation of the solvent, the residue was saturated with MeOH/H2O 9:1 and the resulting mixture was stirred at room temperature for 1 hour. Precipitate formed was filtered and collected to give the desired product.
3-(4-amino-3-(cyclohexylmethyl)benzamido)phenyl dihydrogen phosphate (22a)
White solid (22.5 mg, 0.05 mmol, 77%). 1H NMR (CD3OD, 500 MHz) δ 7.66 – 7.62 (m, 2H), 7.61 (bs, 1H), 7.46 (d, 1H, J = 8.3 Hz), 7.30 (t, 1H, J = 8.2 Hz), 7.00 (d, 1H, J = 8.1 Hz), 6.82 (d, 1H, J = 8.4 Hz), 2.47 (d, 2H, J = 7.2 Hz), 1.78 – 1.57 (m, 6H), 1.32 –1.18 (m, 3H), 1.09 – 0.98 (m, 2H). 13C NMR (CD3OD, 125 MHz) δ 169.95, 153.32 (JP = 6.8 Hz), 149.08, 141.38, 131.86, 130.39, 127.98, 126.72, 124.94, 118.09, 116.95, 116.92, 116.57, 114.49, 114.45, 40.04, 38.38, 34.45, 27.67, 27.44. HRMS–ESI (m/z): [M + H]+ calcd for C20H26N2O5P 405.1579; found, 405.1584.
3-(4-((2-butylhexyl)amino)-3-(cyclohexylmethyl)benzamido)phenyl dihydrogen phosphate (22b)
White solid (17.7 mg, 0.03 mmol, 52%). 1H NMR (CD3OD, 400 MHz) δ 7.78 (dd, 1H, J = 8.8, 2.4 Hz), 7.69 (d, 1H, J = 2.4 Hz), 7.68 – 7.65 (m, 1H), 7.48 – 7.44 (m, 1H), 7.30 (t, 1H, J = 8.4 Hz), 7.02 – 6.97 (m, 1H), 6.84 (d, 1H, J = 8.6 Hz), 3.17 (d, 2H, J = 6.8 Hz), 2.50 (d, 2H, J = 6.8 Hz), 1.80 – 1.64 (m, 6H), 1.49 – 1.38 (m, 3H), 1.38 – 1.31 (m, 9H), 1.28 – 1.17 (m, 3H), 1.10 – 1.00 (m, 2H), 0.94 (t, 6H, J = 3.2 Hz). 13C NMR (CD3OD, 100 MHz) δ 168.72, 152.97 (JP = 6.5 Hz), 148.20, 141.45, 131.67, 130.51, 128.49, 127.70, 125.13, 118.29, 116.91, 114.45, 113.19, 50.34, 39.92, 38.49, 37.93, 34.56, 32.81, 30.01, 27.60, 27.48, 24.18, 14.47. HRMS–ES (m/z): [M + H]+ calcd for C30H44N2O5P 543.2988; found, 543.2994.
3-(3-(cyclohexylmethyl)-4-((3-phenylpropyl)amino)benzamido)phenyl dihydrogen phosphate (22c)
White solid (16.8 mg, 0.03 mmol, quant.). 1H NMR (CD3OD, 500 MHz) δ 7.69 (dd, 1H, J = 8.4, 2.2 Hz), 7.61 – 7.56 (m, 2H), 7.46 (d, 1H, J = 8.1 Hz), 7.31 – 7.25 (m, 3H), 7.23 – 7.20 (m, 2H), 7.19 – 7.15 (m, 1H), 7.01 – 6.97 (m, 1H), 6.58 (d, 1H, J = 8.8 Hz), 3.27 (t, 2H, J = 7.0 Hz), 2.74 (t, 2H, J = 7.6 Hz), 2.43 (d, 2H, J = 6.9 Hz), 1.98 (quint, 2H, J = 7.2 Hz), 1.76 – 1.58 (m, 6H), 1.27 – 1.18 (m, 3H), 1.07 – 0.97 (m, 2H). 13C NMR (CD3OD, 125 MHz) δ 169.23, 153.45 (JP = 6.5 Hz), 151.00, 143.21, 141.46, 131.39, 130.33, 129.46, 128.43, 126.95, 125.52, 121.86, 117.99, 116.83 (JP = 3.9 Hz), 114.45 (JP = 5.0 Hz), 109.85, 43.62, 40.01, 38.27, 34.55, 34.40, 31.85, 27.69, 27.52. HRMS–ES (m/z): [M + H]+ calcd for C29H36N2O5P 523.2362; found 523.2366.
3-(3-(cyclohexylmethyl)-4-((4-methoxybenzyl)amino)benzamido)phenyl dihydrogen phosphate (22d)
Pale yellow solid (28.5 mg, 0.05 mmol, quant.). 1H NMR (CD3OD, 500 MHz) δ 7.86 – 7.81 (m, 2H), 7.70 – 7.68 (m, 1H), 7.50 – 7.46 (m, 1H), 7.33 (t, 1H, J = 8.2 Hz), 7.25 – 7.18 (m, 3H), 7.05 – 7.00 (m, 1H), 6.88 (dt, 2H, J = 8.8, 2.4 Hz), 4.56 (s, 2H), 3.78 (s, 3H), 2.46 (d, 2H, J = 7.0 Hz), 1.76 – 1.57 (m, 6H), 1.30 – 1.16 (m, 3H), 1.07 – 0.95 (m, 2H). 13C NMR (CD3OD, 100 MHz) δ167.47, 162.15, 152.99 (JP = 6.2 Hz), 140.99, 139.31, 134.75, 134.41, 132.66, 131.87, 130.64, 127.99, 124.60, 122.85, 118.32, 117.42, 115.41, 114.47 (JP = 4.0 Hz), 55.82, 54.92, 39.05, 38.70, 34.08, 27.45, 27.28. HRMS–ES (m/z): [M + H]+ calcd for C28H32N2O6P 523.1998; found, 523.2000.
3-(3-(cyclohexylmethyl)-4-((3-methoxybenzyl)amino)benzamido)phenyl dihydrogen phosphate (22e)
White solid (16.8 mg, 0.03 mmol, 51%) 1H NMR (CD3OD, 500 MHz) δ 7.63 – 7.61 (m, 1H), 7.61 – 7.58 (m, 2H), 7.45 – 7.42 (m, 1H), 7.29 (t, 1H, J = 8.1 Hz), 7.22 (t, 1H, J = 7.9 Hz), 7.00 – 6.95 (m, 1H), 6.93 – 6.89 (m, 2H), 6.78 (dd, 1H, J = 8.1, 2.3 Hz), 6.51 (d, 1H, J = 8.4 Hz), 4.47 (s, 2H), 3.74 (s, 3H), 2.53 (d, 2H, J = 6.9 Hz), 1.82 – 1.64 (m, 6H), 1.30 – 1.18 (m, 3H), 1.09– 0.99 (m, 2H). 13C NMR (CD3OD, 125 MHz) δ 169.23, 161.49, 152.96 (JP = 6.6 Hz), 150.60, 142.63, 141.58, 131.44, 130.55, 130.43, 128.15, 125.83, 122.44, 120.18, 118.24, 116.71 (JP = 4.1 Hz), 114.36 (JP = 5.2 Hz), 113.41, 113.32, 111.04, 55.55, 47.96, 40.12, 38.33, 34.53, 27.70, 27.53. HRMS–ESI (m/z): [M + H]+ calcd for C28H34N2O6P 525.2154; found, 525.2161.
3-(3-(cyclohexylmethyl)-4-((3-fluorobenzyl)amino)benzamido)phenyl dihydrogen phosphate (22f)
White solid (17.2 mg, 0.03 mmol, 79%). 1H NMR (500 MHz, MeOD) δ 7.61 (d, 1H, J = 2.3 Hz), 7.60 – 7.57 (m, 2H), 7.44 (d, 1H, J = 8.1 Hz), 7.34 – 7.30 (m, 1H), 7.28 (t, 1H, J = 8.1 Hz), 7.17 (dt, 1H, J = 8.0, 1.0 Hz), 7.05 (dt, 1H, J = 10.2, 2.0 Hz), 7.00 – 6.96 (m, 1H), 6.94 (td, 1H, J = 8.2, 2.3 Hz), 6.46 (d, 1H, J = 8.5 Hz), 4.51 (s, 2H), 2.56 (d, 2H, J = 6.9 Hz), 1.80 – 1.61 (m, 6H), 1.27 – 1.19 (m, 3H), 1.06 (q, 2H, J = 11.5 Hz). 13C NMR (CD3OD, 125 MHz) δ 169.23, 164.53 (JF = 243 Hz), 153.23 (JP = 6.4 Hz), 150.58, 144.49 (JP = 6.4 Hz), 141.49, 131.50, 131.23 (JF = 8.0 Hz), 130.37, 128.16, 125.74, 123.58, 122.43, 118.07, 116.77, 115.37 (JF = 22 Hz), 115.16 (JF = 21 Hz), 114.44 (JP = 4.6 Hz), 110.68, 47.26, 40.14, 38.26, 34.52, 27.72, 27.53. HRMS–ES (m/z): [M + H]+ calcd for C27H29FN2O5P 511.1798; found, 511.1801.
3-(3-(cyclohexylmethyl)-4-((4-fluorobenzyl)amino)benzamido)phenyl dihydrogen phosphate (22g)
White solid (25.5 mg, 0.05 mmol, quant.). 1H NMR (CD3OD, 500 MHz) δ 7.62 – 7.61 (m, 1H), 7.61 – 7.58 (m, 2H), 7.45 – 7.41 (m, 1H), 7.37 – 7.33 (m, 2H), 7.29 (t, 2H, J = 8.4 Hz), 7.03 (tt, 2H, J = 8.6, 2.4 Hz), 7.00 – 6.96 (m, 1H), 6.49 (d, 1H, J = 8.4 Hz), 4.48 (s, 2H), 2.53 (d, 2H, J = 7.0 Hz), 1.81 – 1.62 (m, 6H), 1.30 – 1.18 (m, 3H), 11.05 (q, 2H, J = 10.8 Hz). 13C NMR (CD3OD, 125 MHz) δ 169.20, 163.30 (JF = 244 Hz), 152.99 (JP = 6.2 Hz), 150.66, 141.58, 137.01, 131.44, 130.42, 129.65 (JP = 7.9 Hz), 128.16, 125.77, 122.35, 118.22, 116.71, 116.19, 116.02, 114.38 (JP = 4.0 Hz), 110.77, 47.18, 40.11, 38.26, 34.52, 27.71, 27.52. HRMS–ES (m/z): [M + H]+ calcd for C27H29FN2O5P 511.1798; found, 511.1805.
3-(3-(cyclohexylmethyl)-4-((naphthalen-2-ylmethyl)amino)benzamido)phenyl dihydrogen phosphate (22h)
Pale yellow solid (24.9 mg, 0.04 mmol, quant.). 1H NMR (CD3OD, 500 MHz) δ 7.81 (d, 2H, J = 8.0 Hz), 7.78 – 7.71 (m, 2H), 7.62 (bs, 2H), 7.56 (d, 1H, J = 8.5 Hz), 7.48 (d, 1H, J = 8.5 Hz), 7.45 – 7.38 (m, 2H), 7.27 (t, 1H, J = 8.0 Hz), 6.97 (d, 1H, J = 7.5 Hz), 6.57 (d, 1H, J = 8.5 Hz), 4.67 (s, 2H), 2.57 (d, 2H, J = 6.4 Hz), 1.87 – 1.64 (m, 6H), 1.37 – 1.18 (m, 3H), 1.07 (q, 2H, J = 11.2 Hz). 13C NMR (CD3OD, 125 MHz) δ 169.26, 152.95 (JP = 6.0 Hz), 150.70, 141.58, 138.48, 134.98, 134.20, 131.48, 130.41, 129.26, 128.64 (JP = 4.9 Hz), 128.15, 127.12, 126.59, 126.34, 125.86, 122.44, 118.21, 116.71, 114.34 (JP = 4.3 Hz), 111.00, 48.11, 40.16, 38.30, 34.55, 27.72, 27.56. HRMS–ESI (m/z): [M + H]+ calcd for C31H34N2O5P 545.2200; found, 545.2203.
3-(4-(((4’-chloro-[1,1’-biphenyl]-4-yl)methyl)amino)-3-(cyclohexylmethyl)benzamido)phenyl dihydrogen phosphate (22i)
Pale yellow solid (27.4 mg, 0.04 mmol, 94%). 1H NMR (CD3OD, 500 MHz) δ 7.63 – 7.60 (m, 2H), 7.60 – 7.53 (m, 4H), 7.46 – 7.38 (m, 5H), 7.28 (t, 1H, J = 8.2 Hz), 7.00 – 6.96 (m, 1H), 6.53 (d, 1H, J = 8.4 Hz), 4.54 (s, 1H), 2.54 (d, 2H, J = 6.8 Hz), 1.82 – 1.64 (m, 6H), 1.35 – 1.18 (m, 3H), 1.12 – 1.00 (m, 1H). 13C NMR (CD3OD, 125 MHz) δ 169.20, 152.96 (JP = 6.8 Hz), 150.70, 141.57, 140.86, 140.70, 139.78, 134.26, 131.44, 130.41, 129.90, 129.37, 128.54, 128.15, 128.01, 125.78, 122.32, 118.21, 116.69 (JP = 3.7 Hz), 114.36 (JP = 5.1 Hz), 110.92, 47.61, 40.14, 38.25, 34.53, 27.70, 27.52. HRMS–ESI (m/z): [M + H]+ calcd for C33H35ClN2O5P 605.1972; found, 605.1971.
3-(3-(cyclohexylmethyl)-4-(((3’,5’-dimethoxy-[1,1’-biphenyl]-4-yl)methyl)amino)benzamido)phenyl dihydrogen phosphate (22j)
Yellow solid (7.20 mg, 0.01 mmol, 73%). 1H NMR (CD3OD, 400 MHz) δ 7.80 – 7.71 (m, 2H), 7.68 – 7.63 (m, 1H), 7.57 (d, 2H, J = 8.0 Hz), 7.50 – 7.43 (m, 1H), 7.36 (d, 2H, J = 8.0 Hz), 7.31 (t, 1H, J = 8.1 Hz), 7.22 – 7.14 (m, 2H), 7.05 – 6.98 (m, 2H), 6.97 – 6.92 (m, 1H), 4.60 (s, 2H), 3.88 (s, 3H), 3.86 (s, 3H), 2.52 (d, 2H, J = 6.8 Hz), 1.79 – 1.62 (m, 6H), 1.31 – 1.18 (m, 3H), 1.10 – 0.98 (m, 2H). 13C NMR (CD3OD, 100 MHz) δ 168.32, 152.97 (JP = 7.0 Hz), 150.79, 150.42, 142.28, 141.28, 134.84, 131.66, 130.54, 130.10, 130.01, 128.11, 128.01, 120.62, 118.30, 117.08, 114.45, 113.41, 113.33, 112.00, 111.91, 56.59, 39.43, 38.62, 34.32, 27.57, 27.41. HRMS–ES (m/z): [M + H]+ calcd for C35H38N2O7P 629.2417; found, 629.2405.
3-(4-(((3’-chloro-[1,1’-biphenyl]-4-yl)methyl)amino)-3-(cyclohexylmethyl)benzamido)phenyl dihydrogen phosphate (22k)
White solid (2.13 mg, 0.01 mmol, 8%). 1H NMR (500 MHz, DMSO-d6) δ 9.79 (s, 1H), 7.68 (s, 1H), 7.65 (t, 1H, J = 1.8 Hz), 7.63 (s, 1H), 7.98 – 7.51 (m, 4H), 7.49 – 7.46 (m, 2H), 7.44 – 7.38 (m, 3H), 7.23 (t, 1H, J = 8.1 Hz), 6.84 – 6.82 (m, 1H), 6.47 (m, 2H), 4.51 (d, 2H, J = 5.5 Hz), 1.68 – 1.60 (m, 6H), 1.14 – 1.11 (m, 3H), 1.04 – 0.97 (m, 2H).13C NMR (125 MHz, DMSO-d6) δ 166.02, 151.99 (JP = 6.2 Hz), 149.16, 142.89, 141.58, 141.31, 139.09, 134.27, 131.50, 131.31, 130.79, 129.58, 127.83, 127.57, 127.30, 126.12, 125.80, 125.69, 124.27, 121.29, 116.11, 115.10, 112.59, 109.50, 46.15, 38.83, 37.17, 33.25, 26.70, 26.36. HRMS-ESI+ (m/z): [M + H]+ calcd for C33H35ClN2O5P 605.1972; found, 605.1962.
3-(3-(cyclohexylmethyl)-4-(((3’-fluoro-[1,1’-biphenyl]-4-yl)methyl)amino)benzamido)phenyl dihydrogen phosphate (22l)
White solid (3.20 mg, 0.01 mmol, 9%). 1H NMR (500 MHz, DMSO-d6) δ 9.79 (s, 1H), 7.68 (s, 1H), 7.63 (bs, 1H), 7.57 – 7.54 (m, 3H), 7.51 – 7.38 (m, 6H), 7.23 (t, 1H, J = 8.2 Hz), 7.21 – 7.15 (m, 1H), 6.84 – 6.82 (m, 1H), 6.47 (d, 2H, J = 8.4 Hz), 4.51 (d, 2H, J = 5.0 Hz), 1.69 – 1.61 (m, 6H), 1.15 – 1.12 (m, 3H), 1.04 – 1.00 (m, 2H). 13C NMR (125 MHz, DMSO-d6) δ 166.00, 149.16, 141.52, 141.33, 139.23, 131.43, 130.79, 129.54, 127.57, 127.22, 125.72, 125.64, 124.25, 123.12, 121.32, 116.09, 115.08, 114.78, 114.61, 113.82, 113.64, 112.55, 109.48, 46.18, 37.19, 33.26, 26.69, 26.35. HRMS-ESI+ (m/z): [M + H]+ calcd for C33H35FN2O5P 589.2270; found, 589.2268.
3-(4-((benzofuran-2-ylmethyl)amino)-3-(cyclohexylmethyl)benzamido)phenyl dihydrogen phosphate (22m)
Yellow solid (10.0 mg, 0.02 mmol, 33%). 1H NMR (CD3OD, 500 MHz) δ 7.66 (dd, 1H, J = 8.4, 2.4 Hz), 7.63 – 7.62 (m, 1H), 7.61 (d, 1H, J = 2.3 Hz), 7.48 – 7.46 (m, 1H), 7.45 – 7.42 (m, 1H), 7.42 – 7.39 (m, 1H), 7.29 (t, 1H, J = 8.4 Hz), 7.21 (td, 1H, J = 7.2, 1.2 Hz), 7.16 (td, 1H, J = 7.6, 1.2 Hz), 7.00 – 6.96 (m, 1H), 6.74 (d, 1H, J = 8.7 Hz), 6.57 (d, 1H, J = 0.8 Hz), 4.63 (s, 2H), 2.53 (d, 2H, J = 6.8 Hz), 1.77 – 1.62 (m, 6H), 1.26 – 1.16 (m, 3H), 1.10 – 0.92 (m, 2H). 13C NMR (CD3OD, 125 MHz) δ 169.20, 157.52, 156.32, 152.97 (JP = 6.3 Hz), 150.45, 141.56, 131.54, 130.43, 129.85, 128.15, 126.12, 124.82, 123.76, 122.88, 121.69, 118.22, 116.73 (JP = 4.1 Hz), 114.36 (JP = 5.0 Hz), 111.76, 110.85, 104.45, 41.69, 40.06, 38.32, 34.47, 27.69, 27.50. HRMS-ESI+ (m/z): [M + H]+ calcd for C29H32N2O6P 535.1998; found, 535.1994
3-(3-(cyclohexylmethyl)-4-((pyridin-3-ylmethyl)amino)benzamido)phenyl dihydrogen phosphate (22n)
White solid (7.44 mg, 0.01 mmol, 22%). 1H NMR (CD3OD, 400 MHz) δ 8.78 (br, 2H), 8.66 – 8.60 (m, 1H), 8.09 (dd, 1H, J = 8.0, 6.0 Hz), 7.70 – 7.63 (m, 3H), 7.46 – 7.42 (m, 1H), 7.30 (t, 1H, J = 8.2 Hz), 7.01 – 6.97 (m, 1H), 6.49 (d, 1H, J = 8.4 Hz), 4.79 (s, 2H), 2.60 (d, 2H, J = 6.8 Hz), 1.80 – 1.67 (m, 6H), 1.33 – 1.23 (m, 3H), 1.14 – 1.05 (m, 2H). 13C NMR (CD3OD, 100 MHz) δ 168.82, 152.95 (JP = 6.7 Hz), 149.47, 146.75, 143.15, 141.44, 141.30, 141.18, 131.72, 130.49, 128.51, 126.48, 123.71, 118.32, 116.86, 114.46, 114.42, 110.61, 44.74, 39.99, 38.41, 34.47, 27.68, 27.48. HRMS–ESI (m/z): [M + H]+ calcd for C26H31N3O5P 496.2001; found, 496.1999.
Methyl 4-amino-3-benzylbenzoate (23)
To a commercially available 0.5M benzylzinc bromide solution in THF (4.34 mL, 2.17 mmol) were added 9 (300 mg, 1.08 mmol), Pd2dba3 (49.4 mg, 0.05 mmol), and SPhos (44.3 mg, 0.11 mmol) at 23°C. After 15 min, the reaction mixture was refluxed for 7.5h. Reaction was quenched with aqueous saturated NH4Cl solution and extracted with EtOAc (3 × 20.0 mL). The combined organic layers were washed with brine and dried over Na2SO4. After filtration, solvent was removed under vacuum. The crude was then purified by flash chromatography using a gradient 0 to 10% EtOAc in Hex to obtain a mixture of the desired product 23 and benzyl alcohol which was submitted to next step without further purification.
Methyl 3-benzyl-4-(4-fluorobenzamido)benzoate (25a)
To a suspension of 23 (261 mg, 1.08 mmol) in MeOH (0.15 M) under N2 atmosphere was added 4-fluorobenzaldehyde (0.12 mL, 1.05 eq) at 23°C and then the mixture was refluxed for 2 hours. After cooling to room temperature, acetic acid (0.54 mL, 9.40 mmol) was added and the mixture was stirred for 15 mins, then NaCNBH3 (71.0 mg, 1.13 mmol) was added. The resulting mixture was stirred at 23°C for 1h. Reaction volume was reduced to 1/3 and H2O was added resulting in a slurry, which was extracted with EtOAc (x3 × 15.0 mL). The combined organic layers were washed with brine and dried over Na2SO4. After filtration, solvent was removed under vacuum. The crude was then purified by flash chromatography using a gradient 0 to 10% EtOAc in Hex to provide the desired product as a white solid (176 mg, 0.51 mmol, 47% over 2 steps). 1H NMR (CDCl3, 400 MHz) δ 7.89 – 7.85 (m, 2H), 7.32 – 7.24 (m, 3H), 7.19 – 7.14 (m, 2H), 6.97 – 6.92 (m, 3H), 6.95 (d, 1H, J = 9.1 Hz), 4.34 (t, 1H, J = 5.2 Hz), 4.25 (d, 2H, J = 5.6 Hz), 3.95 (s, 2H), 3.86 (s, 3H).
Methyl 4-(4-fluorobenzamido)benzoate (25b)
To a suspension of commercially available methyl 4-aminobenzoate 24 (300 mg, 1.98 mmol) in MeOH (0.15 M) under N2 atmosphere was added 4-fluorobenzaldehyde (0.22 mL, 1.05 eq) at 23°C and then the mixture was refluxed for 2 hours. After cooling to room temperature, acetic acid (0.88 mL, 17.2 mmol) was added and the mixture was stirred for 15 mins, then NaCNBH3 (130 mg, 2.08 mmol) was added. The resulting mixture was stirred at 23°C for 2.5h. Reaction volume was reduced to 1/3 and H2O was added resulting in a slurry, which was stirred for 1h. Precipitate was filtered yielding the desired product as a white solid (489 mg, 1.89 mmol, 95%). 1H NMR (CDCl3, 400 MHz) δ 7.87 (dt, 2H, J = 8.8, 2.3 Hz), 7.35 – 7.29 (m, 2H), 7.08 – 7.00 (m, 2H), 6.58 (dt, 2H, J = 8.7, 2.3 Hz), 4.47 (t, 1H, J = 5.1 Hz), 4.36 (d, 2H, J = 5.1 Hz), 3.86 (s, 3H).
Synthetic procedure for derivatives 26a-b
Methyl ester 25a-b (1.00 eq) was dissolved in THF (0.10 M) at 23°C, then KOSiMe3 (5.00 eq) was added. Reaction mixture was heated at 50°C overnight. After cooling, solvent was removed under vacuum. The crude was dissolved in H2O and pH adjusted to ~2 using a 1M HCl aq. sol. and stirred for 30 mins. Precipitate formed was collected and dried to provide the desired product, which was used in the next step without further purification.
Synthetic procedure for derivatives 27a-b
Carboxylic acid 26a-b was dissolved in anh. DMF (0.10 M), then HATU (1.00 eq) and DIPEA (2.00 eq) were added. Reaction was stirred at 23°C for 10 mins, then 3-amino-phenol (1.00 eq) was added. Stirring continued overnight. Reaction was quenched with aqueous saturated NaHCO3 solution and extracted with EtOAc (3x). Organic phase was dried over MgSO4 and concentrated. Crude was purified by flash chromatography using a gradient 0 to 10% EtOAc in Hex.
3-benzyl-4-(4-fluorobenzamido)-N-(3-hydroxyphenyl)benzamide (27a)
White solid (86.8 mg, 0.21 mmol, 52% over 2 steps). 1H NMR (CD3OD, 400 MHz) δ 7.94 (s, 1H), 7.71 – 7.62 (m, 2H), 7.30 – 7.17 (m, 5H), 7.13 – 7.01 (m, 4H), 6.95 – 6.87 (m, 2H), 6.57 – 6.50 (m, 1H), 6.25 – 6.20 (m, 1H), 4.31 (s, 2H), 3.80 (s, 2H).
4-fluoro-N-(4-((3-hydroxyphenyl)carbamoyl)phenyl)benzamide (27b)
White solid (59.8 mg, 0.18 mmol, 50% over 2 steps). 1H NMR (CD3OD, 400 MHz) δ 7.71 (dt, 2H, J = 8.8, 2.3 Hz), 7.41 – 7.34 (m, 2H), 7.24 (t, 1H, J = 2.1 Hz), 7.11 (t, 1H, J = 8.1 Hz), 7.07 – 7.00 (m, 3H), 6.64 (dt, 2H, J = 8.8, 2.2 Hz), 6.57 – 6.52 (m, 1H), 4.37 (s, 2H).
Synthetic procedure for derivatives 28a-b
To a solution of phenol 27a-b (1.00 eq) in CH2Cl2 (0.1 M) was added diethyl chlorophosphate (1.00 eq) and DIPEA (3.00 eq). The resulting solution was stirred at room temperature overnight. Upon evaporation of the solvent, the residue was purified by silica gel chromatography using a gradient 0 to 75% EtOAc in Hex to give the desired compound.
3-(3-benzyl-4-(4-fluorobenzamido)benzamido)phenyl diethyl phosphate (28a)
White solid (50.4 mg, 0.09 mmol, 44%). 1H NMR (CD3OD, 400 MHz) δ 7.74 – 7.70 (m, 2H), 7.67 (dd, 1H, J = 8.4, 2.3 Hz), 7.48 – 7.44 (m, 1H), 7.31 (t, 1H, J = 8.2 Hz), 7.30 – 7.25 (m, 2H), 7.24 – 7.19 (m, 3H), 7.10 – 7.04 (m, 2H), 6.96 – 6.89 (m, 3H), 6.54 (d, 1H, J = 8.6 Hz), 4.35 (s, 2H), 4.24 (q, 2H, J = 7.1 Hz), 4.22 (q, 2H, J = 7.1 Hz), 4.01 (s, 2H), 1.35 (t, 3H, J = 7.1 Hz), 1.35 (t, 3H, J = 7.1 Hz).
Diethyl (3-(4-(4-fluorobenzamido)benzamido)phenyl) phosphate (28b)
White solid (32.1 mg, 0.07 mmol, 56%). 1H NMR (CD3OD, 400 MHz) δ 7.76 – 7.70 (m, 3H), 7.49 – 7.43 (m, 1H), 7.40 – 7.35 (m, 2H), 7.32 (t, 1H, J = 8.2 Hz), 7.07 – 7.00 (m, 2H), 7.98 – 7.92 (m, 1H), 6.64 (dt, 2H, J = 8.8, 2.3 Hz), 4.37 (s, 2H), 4.24 (q, 2H, J = 7.1 Hz), 4.22 (q, 2H, J = 7.1 Hz), 1.36 (t, 3H, J = 7.1 Hz), 1.36 (t, 3H, J = 7.1 Hz).
Synthetic procedure for derivatives 29a-b
To the phosphate intermediate 28a-b was added a commercially available solution of TMSBr (6.50 eq) in CH2Cl2 (0.25 M) and stirred at room temperature overnight. Upon evaporation of the solvent, the residue was saturated with MeOH/H2O (9:1) and the resulting mixture was stirred at room temperature for 1 hour. Precipitate formed was filtered and collected to give the desired product.
3-(3-benzyl-4-((4-fluorobenzyl)amino)benzamido)phenyl dihydrogen phosphate (29a)
White solid (40.5 mg, 0.08 mmol, quant.). 1H NMR (CD3OD, 400 MHz) δ 7.73 – 7.69 (m, 2H), 7.64 – 7.61 (m, 1H), 7.44 – 7.41 (m, 1H), 7.33 – 7.26 (m, 3H), 7.25 – 7.19 (m, 3H), 7.13 – 7.07 (m, 2H), 7.00 – 6.93 (m, 3H), 6.67 (d, 1H, J = 9.2 Hz), 4.37 (s, 2H), 4.03 (s, 2 H). 13C NMR (CD3OD, 100 MHz) δ 168.56, 163.73 (JF = 244.8 Hz), 152.95 (JP = 6.5 Hz), 147.06, 141.36, 140.19, 134.10, 131.91, 130.93, 130.85, 130.55, 129.88, 129.77, 128.86, 128.49, 127.70, 126.63, 118.27, 117.00, 116.42, 116.21, 114.80, 114.38 (JP = 3.9 Hz), 38.13. HRMS–ESI (m/z): [M + H]+ calcd for C27H25FN2O5P 507.1480; found, 507.1482.
3-(4-((4-fluorobenzyl)amino)benzamido)phenyl dihydrogen phosphate (29b)
Yellow solid (29.6 mg, 0.07 mmol, quant.). 1H NMR (CD3OD, 400 MHz) δ 7.99 (d, 2H, J = 8.4 Hz), 7.72 – 7.67 (m, 1H), 7.50 – 7.41 (m, 3H), 7.37 – 7.29 (m, 3H), 7.12 (t, 2H, J = 8.8 Hz), 7.04 – 7.00 (m, 1H), 4.60 (s, 2H). 13C NMR (CD3OD, 100 MHz) δ 167.33, 163.44, 152.98 (JP = 6.4 Hz), 141.00, 133.21, 130.72, 122.54, 118.23, 117.38, 117.00, 116.78, 114.35, 54.27. HRMS–ESI (m/z): [M + H]+ calcd for C20H19FN2O5P 417.1016; found, 417.1019.
Synthetic procedure for derivatives 30a-b
Carboxylic acid 19i was dissolved in anh. DMF (0.10 M), then HATU (1.00 eq) and DIPEA (2.00 eq) were added. Reaction was stirred at 23°C for 10 mins, then appropriate phenol (1.00 eq) was added. Stirring continued overnight. Reaction was quenched with aqueous saturated NaHCO3 solution and extracted with EtOAc (3x). Organic phase was dried over MgSO4 and concentrated. Crude was purified by flash chromatography using a gradient 0 to 50% EtOAc in Hex.
3-(cyclohexylmethyl)-4-((4-fluorobenzyl)amino)-N-(4-hydroxyphenyl)benzamide (30a)
Colorless residue (34.1 mg, 0.08 mmol, 60%). 1H NMR (CDCl3, 400 MHz) δ 7.77 (s, 1H), 7.60 – 7.54 (m, 2H), 7.34 – 7.25 (m, 3H), 7.14 (bs, 1H), 7.07 – 6.99 (m, 2H), 6.57 – 6.50 (m, 2H), 6.53 (d, 1H, J = 9.2 Hz), 4.38 (s, 2H), 2.39 (d, 1H, J = 7.1 Hz), 1.74 – 1.50 (m, 6H), 1.22 – 1.10 (m, 3H), 1.04 – 0.92 (m, 2H).
3-(cyclohexylmethyl)-4-(4-fluorobenzamido)-N-(4-hydroxyphenethyl)benzamide (30b)
Colorless residue (37.1 mg, 0.08 mmol, 61%). 1H NMR (CDCl3, 400 MHz) δ 7.44 (d, 1H, J = 2.1 Hz), 7.38 (dd, 1H, J = 8.5, 2.2 Hz), 7.31 – 7.24 (m, 2H), 7.06 – 6.99 (m, 4H), 6.85 – 6.78 (m, 2H), 6.48 (d, 1H, J = 8.4 Hz), 6.15 (t, 1H, J = 5.7 Hz), 4.36 (s, 2H), 3.62 (q, 2H, J = 6.4 Hz), 2.79 (t, 2H, J = 7.0 Hz), 2.36 (d, 2H, J = 7.0 Hz), 1.72 – 1.45 (m, 6H), 1.21 – 1.08 (m, 3H), 1.01 – 0.87 (m, 2H).
Synthetic procedure for derivatives 31a-b
To a solution of phenol 30a-b (1.00 eq) in CH2Cl2 (0.1 M) was added diethyl chlorophosphate (1.00 eq) and DIPEA (3.00 eq). The resulting solution was stirred at room temperature overnight. Upon evaporation of the solvent, the residue was purified by silica gel chromatography using a gradient 0 to 90% EtOAc in Hex to give the desired compound.
4-(3-(cyclohexylmethyl)-4-((4-fluorobenzyl)amino)benzamido)phenyl diethyl phosphate (31a)
Colorless residue (28.5 mg, 0.05 mmol, 64%). 1H NMR (CDCl3, 400 MHz) δ 7.99 (s, 1H), 7.62 – 7.56 (m, 4H), 7.34 – 7.27 (m, 2H), 7.19 – 7.13 (m, 2H), 7.09 – 7.01 (m, 2H), 6.53 (dd, 1H, J = 6.0, 2.9 Hz), 4.40 (s, 2H), 4.23 – 4.13 (m, 4H), 2.42 (d, 2H, J = 7.0 Hz), 1.75 – 1.51 (m, 6H), 1.33 (t, 3H, J = 7.1 Hz), 1.32 (t, 3H, J = 7.1 Hz), 1.22 – 1.13 (m, 3H), 1.04 – 0.93 (m, 2H).
4-(2-(3-(cyclohexylmethyl)-4-(4-fluorobenzamido)benzamido)ethyl)phenyl diethyl phosphate (31b)
Colorless residue (25.2 mg, 0.04 mmol, 52%). 1H NMR (CD3OD, 400 MHz) δ 7.41 (td, 2H, J = 8.1, 2.1 Hz), 7.35 – 7.29 (m, 2H), 7.27 – 7.22 (m, 2H), 7.04 – 6.98 (m, 2H), 6.41 (d, 1H, J = 8.6 Hz), 4.25 (s, 2H), 4.23 – 4.13 (m, 4H), 3.52 (t, 2H, J = 7.2 Hz), 2.86 (t, 2H, J = 7.2 Hz), 2.47 (d, 2H, J = 6.9 Hz), 1.78 – 1.62 (m, 6H), 1.31 (t, 3H, J = 7.1 Hz), 1.31 (t, 3H, J = 7.1 Hz), 1.25 – 1.19 (m, 3H), 1.06 – 0.94 (m, 2H).
Synthetic procedure for derivatives 32a-b
To the phosphate intermediate 31a-b was added a commercially available solution of TMSBr (6.50 eq) in CH2Cl2 (0.25 M) and stirred at room temperature overnight. Upon evaporation of the solvent, the residue was saturated with MeOH/H2O (9:1) and the resulting mixture was stirred at room temperature for 1 hour. Precipitate formed was filtered and collected to give the desired product.
4-(3-(cyclohexylmethyl)-4-((4-fluorobenzyl)amino)benzamido)phenyl dihydrogen phosphate (32a)
White solid (22.2 mg, 0.04 mmol, 86%). 1H NMR (CD3OD, 400 MHz) δ 7.65 – 7.57 (m, 4H), 7.36 – 7.30 (m, 2H), 7.17 (dd, 2H, J = 9.2, 1.2 Hz), 7.03 (t, 2H, J = 8.8 Hz), 6.55 (d, 1H, J = 8.4 Hz), 4.47 (s, 2H), 2.51 (d, 2H, J = 6.8 Hz), 1.79 – 1.62 (m, 6H), 1.30 – 1.18 (m, 3H), 1.10 – 0.86 (m, 2H). 13C NMR (CD3OD, 100 MHz) δ 168.95, 163.43 (JF = 243 Hz), 149.47, 149.00 (JP = 6.9 Hz), 136.86, 136.25, 131.43, 130.03 (JF = 7.6 Hz), 128.08, 126.64, 123.57, 121.50, 116.19 (JF = 22.1 Hz), 111.96, 47.90, 39.98, 38.34, 34.48, 27.68, 27.49. HRMS–ESI (m/z): [M + H]+ calcd for C27H31FN2O5P 513.1955; found, 513.1956.
4-(2-(3-(cyclohexylmethyl)-4-(4-fluorobenzamido)benzamido)ethyl)phenyl dihydrogen phosphate (32b)
White solid (21.1 mg, 0.04 mmol, 90%). 1H NMR (CD3OD, 400 MHz) δ 7.56 – 7.48 (m, 2H), 7.32 (d, 1H, J = 8.4 Hz), 7.31 (d, 1H, J = 8.4 Hz), 7.22 (d, 2H, J = 8.4 Hz), 7.12 (d, 2H, J = 8.4 Hz), 7.04 (t, 2 H, J = 8.4 Hz), 6.73 (d, 1H, J = 8.4 Hz), 4.49 (s, 2H), 3.55 (t, 2H, J = 7.2 Hz), 2.87 (t, 2H, J = 7.2 Hz), 2.46 (d, 2H, J = 6.8 Hz), 1.80 – 1.59 (m, 6H), 1.33 – 1.17 (m, 3H), 1.10 – 0.95 (m, 2H). 13C NMR (CD3OD, 100 MHz) δ170.13, 165.07, 162.63, 151.33 (JP = 6.3 Hz), 146.12, 137.07, 133.99, 131.15, 131.01, 129.07, 127.72, 126.33, 121.37, 116.41 (JF = 21.8 Hz), 115.34, 49.90, 42.71, 39.55, 38.54, 35.88, 34.34, 27.60, 27.43. HRMS–ESI (m/z): [M + H]+ calcd for C29H35FN2O5P 541.2262; found, 541.2268.
4-(((2-(cyclohexylmethyl)-4-(methoxycarbonyl)phenyl)amino)methyl)benzoic acid (33)
To a suspension of 11 (127 mg, 0.52 mmol) in MeOH (0.15 M) under N2 atmosphere was added 4-formylbenzoic acid (81.4 mg, 0.54 mmol) at 23°C and then the mixture was refluxed for 3 hours. After cooling to room temperature, acetic acid (0.25 mL, 4.49 mmol) was added and the mixture was stirred for 15 mins, then NaCNBH3 (34.1 mg, 0.54 mmol) was added. The resulting mixture was stirred at 23°C for 30 mins. Reaction volume was reduced to 1/3 and H2O was added resulting in a white suspension, which was stirred overnight. Precipitate was filtered yielding the desired product as a white solid, which was used in the subsequent step without purification.
Methyl 4-amino-3-(bicyclo[2.2.1]heptan-2-yl)benzoate (34)
To a commercially available 0.5M exo-2-norbornylzinc bromide in THF (25.0 mL, 12.5 mmol) were added methyl 4-amino-3-iodobenzoate (1.73 g, 6.25 mmol), Pd2dba3 (286 mg, 0.31 mmol), and SPhos (257 mg, 0.63 mmol) and stirred at 23°C for 4 days. Reaction was quenched with aqueous saturated NH4Cl solution and extracted with EtOAc (5 × 50.0 mL). Organic layer was dried over Na2SO4, filtered and concentrated. The crude was then purified by flash chromatography using a gradient 0 to 20% EtOAc in Hex to obtain the desired product as a brown solid (440 mg, 1.79 mmol, 29%). 1H NMR (CD3OD, 400 MHz, mixture of diastereomers) δ 7.81 (d, 0.4H, J = 1.4 Hz), 7.76 (s, 0.6H, J = 1.4 Hz), 7.62 (td, 1H, J = 8.5, 1.3 Hz), 3.84 (s, 1.4H), 3.83 (s, 1.6H)3.25 – 3.15 (m, 1H), 2.59 (dd, 0.5H, J = 8.3, 5.5 Hz), 2.50 (br, 0.5H), 2.44 (d, 0.5H, J = 2.8 Hz), 2.35 (d, 1H, J = 2.8 Hz), 1.95 – 1.93 (m, 1H), 1.75 – 1.09 (m, 7H).
3-(((2-(bicyclo[2.2.1]heptan-2-yl)-4-(methoxycarbonyl)phenyl)amino)methyl)benzoic acid (35)
To a suspension of 34 (440 mg, 1.79 mmol) in MeOH (0.5 M) under N2 atmosphere was added 3-formylbenzoic acid (282 mg, 1.88 mmol) at 23°C and then the mixture was refluxed for 3 hours. After cooling to room temperature, acetic acid (0.92 mL, 16.1 mmol) was added and the mixture was stirred for 15 mins, then NaCNBH3 (118 mg, 1.88 mmol) was added. The resulting mixture was stirred at 23°C for 30 mins. Reaction volume was reduced to 1/3 and H2O was added resulting in a white suspension, which was stirred overnight and then extracted with EtOAc (3 × 15.0 mL). Organic layer was dried over Na2SO4, filtered and concentrated. The crude was then purified by flash chromatography using a gradient 0 to 100% EtOAc in Hex to yield the desired product as a yellow solid (364 mg, 0.96 mmol, 54%). 1H NMR (DMSO-d6, 400 MHz, mixture of diastereomers) δ 12.88 (s, 1H), 7.90 (s, 1H), 7.77 (d, 1H, J = 7.7 Hz), 7.68 (s, 0.6H), 7.62 (s, 0.4H), 7.57 – 7.46 (m, 2H), 7.42 (t, 1H, J = 7.9 Hz), 6.69 (t, 0.5H, J = 5.9 Hz), 6.43 (t, 0.3H, J = 5.9 Hz), 6.41 – 6.34 (m, 1H), 4.52 – 4.45 (m, 2H), 3.71 (s, 2H), 3.70 (s, 1H), 2.75 – 2.68 (m, 0.3H), 2.32 (s, 1H), 2.09 (t, 0.5H, J = 10.0 Hz), 1.88 (t, 1H, J = 11.5 Hz), 1.69 – 1.40 (m, 4H), 1.35 – 1.10 (m, 3H), 1.07 – 0.95 (m, 1H).
Synthetic procedure for derivatives 36a-f
Carboxylic acid (15, 17, 33, 35) was dissolved in anh. DMF (0.10 M), then HATU (1.00 eq) and DIPEA (2.00 eq) were added. Reaction was stirred at 23°C for 10 mins, then appropriate aniline (1.00 eq) was added. Stirring continued overnight. Reaction was quenched with aqueous saturated NaHCO3 solution and extracted with EtOAc (3x). Organic phase was washed with H2O (1x) and brine (1x), then dried over MgSO4 and concentrated. Crude was purified by flash chromatography using a gradient 0 to 100% EtOAc in Hex.
Methyl 4-((4-((3-chlorophenyl)carbamoyl)benzyl)amino)-3-(cyclohexylmethyl)benzoate (36a)
Synthesized from 33. White foam (67.5 mg, 0.14 mmol, 52% over 2 steps). 1H NMR (400 MHz, CDCl3) δ 7.84 (dt, 2H, J = 8.4, 1.8 Hz), 7.80 – 7.74 (m, 2H), 7.72 (dd, 1H, J = 8.9, 2.1 Hz), 7.50 – 7.42 (m, 3H), 7.29 (t, 1H, J = 8.0 Hz), 7.15 – 7.11 (m, 1H), 7.47 (d, 1H, J = 8.5 Hz), 4.55 (s, 2H), 3.84 (s, 3H), 2.44 (d, 2H, J = 7.1 Hz), 1.79 – 1.56 (m, 6H), 1.25 – 1.14 (m, 3H), 1.10 – 0.96 (m, 2H).
Methyl 4-((3-((3-chlorophenyl)carbamoyl)benzyl)amino)-3-(cyclohexylmethyl)benzoate (36b)
Synthesized from 15. White solid (205 mg, 0.42 mmol, 42%). 1H NMR (400 MHz, CDCl3) δ 8.52 (s, 1H), 7.84 (br, 1H), 7.78 – 7.73 (m, 2H), 7.71 – 7.63 (m, 2H), 7.55 – 7.49 (m, 1H), 7.47 – 7.42 (m, 1H), 7.37 (t, 1H, J = 7.7 Hz), 7.21 (t, 1H, J = 7.7 Hz), 7.10 – 7.05 (m, 1H), 6.41 (d, 1H, J = 8.4 Hz), 4.58 (t, 1H, J = 5.2 Hz), 4.41 (d, 2H, J = 5.2 Hz), 3.79 (s, 3H), 2.38 (d, 2H, J = 7.1 Hz), 1.73 – 1.49 (m, 6H), 1.21 – 1.08 (m, 3H), 1.04 – 0.89 (m, 2H).
Methyl 4-((3-((3-chlorophenyl)carbamoyl)benzyl)amino)-3-(2-cyclohexylethyl)benzoate (36c)
Synthesized from 17. White solid (20.0 mg, 0.04 mmol, 79%). 1H NMR (400 MHz, CDCl3) δ 9.07 (s, 1H), 8.22 (t, 1H, J = 2.2 Hz), 7.94 (t, 1H, J = 2.2 Hz), 7.89 (br, 1H), 7.84 (s, 1H), 7.79 (dt, 1H, J = 7.6, 1.6 Hz), 7.66 – 7.60 (m, 1H), 7.43 (dt, 1H, J = 7.6, 1.5 Hz), 7.39 (d, 1H, J = 7.6 Hz), 7.36 (d, 1H, J = 1.7 Hz), 7.29 – 7.22 (m, 2H), 7.14 (t, 1H, J = 8.0 Hz), 7.12 – 7.09 (m, 1H), 6.67 – 6.60 (m, 1H), 6.60 – 6.56 (m, 1H), 6.31 – 6.21 (m, 2H), 4.17 – 4.08 (m, 2H), 4.03 (br, 1H), 2.24 – 2.14 (m, 2H), 1.71 – 1.54 (m, 6H), 1.44 – 1.32 (m, 2H), 1.22 – 1.06 (m, 3H), 0.93 – 0.78 (m, 2H).
Methyl 3-(bicyclo[2.2.1]heptan-2-yl)-4-((3-((3-chlorophenyl)carbamoyl)benzyl)amino)benzoate (36d)
Synthesized from 35. White solid (98.0 mg, 0.20 mmol, 26%). 1H NMR (DMSO-d6, 400 MHz, mixture of diastereomers) δ 10.38 (s, 1H), 7.94 (t, 1H, J = 2.0 Hz), 7.91 (bs, 1H), 7.80 (dt, 1H, J = 7.3, 1.5 Hz), 7.74 – 7.61 (m, 2H), 7.57 – 7.44 (m, 3H), 7.38 (t, 1H, J = 8.1 Hz), 7.15 (dd, 1H, J = 8.0, 1.2 Hz), 6.70 (t, 1H, J = 6.0 Hz), 6.48 – 6.36 (m, 2H), 4.57 – 4.50 (m, 2H), 3.73 (s, 2H), 3.72 (s, 1H), 2.78 – 2.70 (m, 0.3H), 2.34 (bs, 1H), 2.16 – 2.07 (m, 0.3H), 1.90 (t, 1H, J = 11.8 Hz), 1.71 – 1.41 (m, 4H), 1.37 – 1.18 (m, 3H), 1.10 – 0.98 (m, 1H).
Methyl 4-((3-(benzo[d][1,3]dioxol-5-ylcarbamoyl)benzyl)amino)-3-(cyclohexylmethyl)benzoate (36e)
Synthesized from 15. Yellow solid (160 mg, 0.32 mmol, 71%). 1H NMR (DMSO-d6, 400 MHz) δ 10.13 (s, 1H), 7.89 (s, 1H), 7.76 (d, 1H, J = 7.6 Hz), 7.53 – 7.39 (m, 5H), 7.15 (dd, 1H, J = 8.3, 1.7 Hz), 6.89 (d, 1H, J = 8.1 Hz), 6.60 (t, 1H, J = 6.2 Hz), 6.41 (d, 1H, J = 8.1 Hz), 6.00 (s, 3H), 4.51 (d, 2H, J = 5.9 Hz), 3.70 (s, 3H), 1.72 – 1.54 (m, 6H), 1.18 – 1.10 (m, 3H), 1.05 – 0.89 (m, 2H).
Methyl 3-(cyclohexylmethyl)-4-((3-((3-fluorophenyl)carbamoyl)benzyl)amino)benzoate (36f)
Synthesized from 15. White solid (142 mg, 0.30 mmol, 94%). 1H NMR (DMSO-d6, 400 MHz) δ 10.41 (s, 1H), 7.91 (s, 1H), 7.79 (d, 1H, J = 7.2 Hz), 7.72 (dt, 1H, J = 11.8, 2.0 Hz), 7.57 – 7.44 (m, 5H), 7.41 – 7.33 (m, 1H), 6.92 (td, 1H, J = 8.4, 2.0 Hz), 6.60 (t, 1H, J = 6.1 Hz), 6.41 (d, 1H, J = 8.4 Hz), 4.52 (d, 2H, J = 5.7 Hz), 3.71 (s, 3H), 1.70 – 1.54 (m, 6H), 1.20 – 1.08 (m, 3H), 1.04 – 0.91 (m, 2H).
Synthetic procedure for derivatives 37a-f
Methyl ester 36a-f (1.00 eq) was dissolved in THF (0.10 M) at 23°C, then KOSiMe3 (5.00 eq) was added. Reaction mixture was heated at 50°C overnight. After cooling, solvent was removed under vacuum. The crude was dissolved in H2O and pH adjusted to ~2 using a 1M HCl aq. sol. and stirred for 30 mins. Precipitate formed was collected and dried to provide the desired product, which was used in the next step without further purification.
Synthetic procedure for derivatives 38a-f
Carboxylic acid 37a-f was dissolved in anh. DMF (0.10 M), then HATU (1.00 eq) and DIPEA (2.00 eq) were added. Reaction was stirred at 23°C for 10 mins, then 3-amino-phenol (1.00 eq) was added. Stirring continued overnight. Reaction was quenched with aqueous saturated NaHCO3 solution and extracted with EtOAc (3x). Organic phase was dried over MgSO4 and concentrated. Crude was purified by flash chromatography using a gradient 0 to 100% EtOAc in Hex.
4-((4-((3-chlorophenyl)carbamoyl)benzyl)amino)-3-(cyclohexylmethyl)-N-(3-hydroxyphenyl)benzamide (38a)
White solid (26.5 mg, 0.05 mmol, 73% over 2 steps). 1H NMR (400 MHz, CD3OD) δ 7.90 – 7.85 (m, 2H), 7.60 (d, 1H, J = 2.1 Hz), 7.59 – 7.53 (m, 2H), 7.47 (d, 1H, J = 8.4 Hz), 7.31 (t, 1H, J = 8.2 Hz), 7.26 – 7.22 (m, 1H), 7.15 – 7.11 (m, 1H), 7.11 (t, 1H, J =8.1 Hz), 7.05 – 7.00 (m, 1H), 6.58 – 6.51 (m, 1H), 6.44 (d, 1H, J = 8.6 Hz), 4.57 (s, 2H), 2.55 (d, 2H, J = 6.7 Hz), 1.81 – 1.64 (m, 6H), 1.30 – 1.19 (m, 3H), 1.09 – 0.99 (m, 2H).
4-((3-((3-chlorophenyl)carbamoyl)benzyl)amino)-3-(cyclohexylmethyl)-N-(3-hydroxyphenyl)benzamide (38b)
Yellow residue (51.5 mg, 0.91 mmol, 58% over 2 steps). 1H NMR (400 MHz, CDCl3) δ 8.65 (s, 1H), 8.03 (t, 1H, J = 2.1 Hz), 7.88 – 7.79 (m, 3H), 7.74 (dt, 1H, J = 7.4, 1.6 Hz), 7.58 – 7.53 (m, 1H), 7.49 – 7.33 (m, 5H), 7.27 – 7.22 (m, 1H), 7.17 – 7.09 (m, 2H), 6.68 – 6.59 (m, 2H), 6.36 (d, 1H, J = 8.5 Hz), 4.28 (s, 2H), 2.26 (d, 2H, J = 7.0 Hz), 1.70 – 1.56 (m, 6H), 1.20 – 1.07 (m, 3H), 0.97 – 0.85 (m, 2H).
4-((3-((3-chlorophenyl)carbamoyl)benzyl)amino)-3-(2-cyclohexylethyl)-N-(3-hydroxyphenyl)benzamide (38c)
Yellow residue (20.0 mg, 0.03 mmol, 79% over 2 steps). 1H NMR (400 MHz, CDCl3) δ 9.06 (s, 1H), 8.22 (t, 1H, J = 2.2 Hz), 7.94 (t, 1H, J = 2.1 Hz), 7.89 (br, 1H), 7.84 (s, 1H), 7.79 (dt, 1H, J = 7.6, 1.6 Hz), 7.66 – 7.61 (m, 1H), 7.47 – 7.41 (m, 1H), 7.41 – 7.34 (m, 2H), 7.25 – 7.22 (m, 2H), 7.14 (t, 1H, J = 8.0 Hz), 7.12 – 7.09 (m, 1H), 6.63 (dd, 1H, J = 8.1, 1.8 Hz), 6.58 (dd, 1H, J = 8.0, 1.1 Hz), 6.31 – 6.22 (m, 2H), 4.12 (s, 2H), 4.04 (s, 1H), 2.24 – 2.13 (m, 2H), 1.72 – 1.54 (m, 5H), 1.43 – 1.32 (m, 2H), 1.22 – 1.08 (m, 4H), 0.92 – 0.78 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 168.23, 166.03, 157.59, 148.65, 148.00, 139.74, 139.64, 139.13, 134.91, 134.74, 131.27, 130.45, 130.11, 129.82, 129.34, 127.33, 126.83, 126.73, 126.32, 124.54, 122.66, 120.58, 118.52, 111.38, 109.72, 108.00, 107.34, 105.84, 102.37, 47.72, 37.81, 35.61, 33.38, 28.15, 26.67, 26.37.
3-(bicyclo[2.2.1]heptan-2-yl)-4-((3-((3-chlorophenyl)carbamoyl)benzyl)amino)-N-(3-hydroxyphenyl)benzamide (38d)
Pale yellow solid (51.0 mg, 0.09 mmol, 45% over 2 steps). 1H NMR (DMSO-d6, 400 MHz, mixture of diastereomers) δ 10.39 (s, 1H), 9.64 (s, 0.5H), 9.62 (s, 0.5H), 9.28 (s, 1H), 7.94 (br, 2H), 7.80 (d, 1H, J = 7.4 Hz), 7.73 – 7.66 (m, 1H), 7.63 – 7.45 (m,4H), 7.38 (t, 1H, J = 8.1 Hz), 7.26 (dt, 1H, J = 8.8, 2.2 Hz), 7.16 (d, 1H, J = 7.8 Hz), 7.12 – 7.01 (m, 2H), 6.52 (t, 0.4 H, J = 5.5 Hz), 6.46 – 6.37 (m, 2H), 6.25 (t, 0.3H, J = 5.5 Hz), 4.55 (br, 2H), 2.76 (t, 0.5H, J = 7.0 Hz), 2.35 (br, 1H), 1.89 (t, 0.5H, J = 11.0 Hz), 1.72 – 1.44 (m, 5H), 1.43 – 1.13 (m, 4H).
4-((3-(benzo[d][1,3]dioxol-5-ylcarbamoyl)benzyl)amino)-3-(cyclohexylmethyl)-N-(3-hydroxyphenyl)benzamide (38e)
White solid (37.0 mg, 0.06 mmol, 19% over 2 steps). 1H NMR (DMSO-d6, 400 MHz) δ 10.13 (s, 1H), 9.57 (s, 1H), 9.28 (s, 1H), 7.91 (s, 1H), 7.76 (d, 1H, J = 7.7 Hz), 7.55 – 7.49 (m, 3H), 7.45 (d, 1H, J = 7.7 Hz), 7.44 – 7.40 (m, 1H), 7.28 (s,1H), 7.19 – 7.14 (m, 1H), 7.10 – 7.01 (m, 2H), 6.88 (d, 1H, J = 8.2 Hz), 6.46 – 6.38 (m, 2H), 6.00 (s, 2H), 4.51 (d, 2H, J = 5.7 Hz), 1.73 – 1.55 (m, 6H), 1.20 – 1.10 (m, 3H), 1.06 – 0.93 (m, 2H).
3-(cyclohexylmethyl)-4-((3-((3-fluorophenyl)carbamoyl)benzyl)amino)-N-(3-hydroxyphenyl)benzamide (38f)
White solid (105 mg, 0.19 mmol, 64% over 2 steps). 1H NMR (DMSO-d6, 400 MHz) δ 10.42 (s, 1H), 9.57 (s, 1H), 9.28 (s, 1H), 7.93 (s, 1H), 7.7 (d, 1H, J = 8.1 Hz), 7.76 – 7.70 (m, 1H), 7.58 – 7.51 (m, 3H), 7.48 (t, 1H, J = 7.3 Hz), 7.42 – 7.34 (m, 1H), 7.28 (br, 1H), 7.11 – 7.00 (m, 2H), 6.93 (t, 1H, J = 8.5 Hz), 6.42 (d, 2H, J = 8.5 Hz), 4.53 (d, 2H, J = 5.8 Hz), 1.73 – 1.53 (m, 6H), 1.21 – 1.10 (m, 3H), 1.06 – 0.92 (m, 2H).
Synthetic procedure for derivatives 39a-f
To a solution of phenol 38a-f (1.00 eq) in CH2Cl2 (0.1 M) was added diethyl chlorophosphate (1.00 eq) and DIPEA (3.00 eq). The resulting solution was stirred at room temperature overnight. Upon evaporation of the solvent, the residue was purified by silica gel chromatography using a gradient 0 to 90% EtOAc in Hex to give the desired compound.
3-(4-((4-((3-chlorophenyl)carbamoyl)benzyl)amino)-3-(cyclohexylmethyl)benzamido)phenyl diethyl phosphate (39a)
White solid (9.40 mg, 0.01 mmol, 60%). 1H NMR (400 MHz, CD3OD) δ 7.39 (dt, 1H, J = 8.5, 1.7 Hz), 7.86 (t, 1H, J = 2.0 Hz), 7.74 – 7.71 (m, 1H), 7.62 (d, 1H, J = 2.2 Hz), 7.60 – 7.56 (m, 2H), 7.50 (d, 2H, J = 8.5 Hz), 7.49 – 7.44 (m, 1H), 7.31 (t, 2H, J = 8.1 Hz), 7.15 – 7.10 (m, 1H), 6.97 – 6.92 (m, 1H), 6.45 (d, 1H, J = 8.6 Hz), 4.59 (s, 2H), 4.24 (q, 2H, J = 6.9 Hz), 4.23 (q, 2H, J = 6.9 Hz), 2.56 (d, 2H, J = 6.9 Hz), 1.82 – 1.65 (m, 6H), 1.36 (t, 3H, J =6.9 Hz), 1.35 (t, 3H, J = 6.9 Hz), 1.31 – 1.23 (m, 3H), 1.12 – 1.01 (m, 2H).
3-(4-((3-((3-chlorophenyl)carbamoyl)benzyl)amino)-3-(cyclohexylmethyl)benzamido)phenyl diethyl phosphate (39b)
Yellow solid (31.1 mg, 0.04 mmol, 49%). 1H NMR (400 MHz, CD3OD) δ 7.92 (t, 1H, J = 1.7 Hz), 7.85 (t, 1H, J = 2.0 Hz), 7.77 (dt, 1H, J = 7.7, 1.5 Hz), 7.73 – 7.71 (m, 1H), 7.61 – 7.54 (m, 5H), 7.48 – 7.42 (m, 2H), 7.30 (td, 1H, J = 8.1, 2.8 Hz), 7.15 – 7.10 (m, 1H), 6.95 – 6.90 (m, 1H), 6.49 (d, 1H, J = 8.4 Hz), 4.57 (s, 2H), 4.23 (q, 2H, J = 7.1 Hz), 4.21 (q, 2H, J = 7.1 Hz), 2.53 (d, 2H, J = 6.9 Hz), 1.77 – 1.58 (m, 6H), 1.34 (t, 3H, J = 7.1 Hz), 1.34 (t, 3H, J = 7.1 Hz), 1.22 – 1.17 (m, 3H), 1.09 – 0.98 (m, 2H).
3-(4-((3-((3-chlorophenyl)carbamoyl)benzyl)amino)-3-(2-cyclohexylethyl)benzamido)phenyl diethyl phosphate (39c)
Colorless oil (15.5 mg, 0.02 mmol, 76%). 1H NMR (400 MHz, CDCl3) δ 8.44 (s, 1H), 7.95 (s, 1H), 7.88 (br, 1H), 7.79 – 7.73 (m, 2H), 7.59 – 7.55 (m, 2H), 7.55 – 7.51 (m, 1H), 7.50 – 7.43 (m, 3H), 7.40 (t, 1H, J = 7.6 Hz), 7.25 – 7.20 (m, 2H), 7.12 – 7.06 (m, 1H), 6.95 – 6.89 (m, 1H), 6.41 (d, 1H, J = 8.5 Hz), 4.51 (t, 1H, J = 5.5 Hz), 4.44 (d, 2H, J = 4.6 Hz), 4.24 – 4.13 (m, 4H), 2.50 – 2.42 (m, 2H), 1.80 – 1.59 (m, 6H), 1.55 – 1.45 (m, 2H), 1.33 (t, 3H, J =7.0 Hz), 1.32 (t, 3H, J = 7.0 Hz), 1.24 – 1.07 (m, 3H), 0.99 – 0.81 (m, 2H).
3-(3-(bicyclo[2.2.1]heptan-2-yl)-4-((3-((3-chlorophenyl)carbamoyl)benzyl)amino)benzamido)phenyl diethyl phosphate (39d)
Colorless oil (24.0 mg, 0.03 mmol, 38%). 1H NMR (400 MHz, CD3OD, mixture of diastereomers) δ 9.82 (s, 1H), 7.92 (s, 1H), 7.88 – 7.80 (m, 1H), 7.77 (d, 1H, J = 7.7 Hz), 7.73 (d, 1H, J = 10.9 Hz), 7.61 – 7.50 (m, 3H), 7.48 – 7.39 (m, 2H), 7.29 (td, 2H, J = 7.8, 2.3 Hz), 7.11 (dd, 1H, J = 8.0, 1.0 Hz), 6.93 (d, 1H, J = 8.0 Hz), 6.46 (dd, 1H, J = 8.6, 3.1 Hz), 4.56 (s, 2H), 7.21 (app quint, 4H, J = 7.2 Hz), 2.73 – 2.66 (m, 0.4H), 2.65 – 2.55 (m, 1H), 2.33 (s, 1H), 1.91 (t, 0.5H, J = 12.0 Hz), 1.77 – 1.13 (m, 6H), 1.34 (t, 6H, J = 7.1 Hz).
3-(4-((3-(benzo[d][1,3]dioxol-5-ylcarbamoyl)benzyl)amino)-3-(cyclohexylmethyl)benzamido)phenyl diethyl phosphate (39e)
Yellow oil (15.0 mg, 0.02 mmol, 35%). 1H NMR (600 MHz, DMSO-d6) δ 10.15 (s, 1H), 9.87 (s, 1H), 7.93 (s, 1H), 7.77 (d, 1H, J = 7.7 Hz), 7.74 (br, 1H), 7.59 – 7.51 (m, 4H), 7.46 (t, 1H, J = 7.3 Hz), 7.42 (d, 1H, J = 1.8 Hz), 7.31 (t, 1H, J = 8.2 Hz), 7.17 (dd, 1H, J = 8.6, 1.2 Hz), 6.90 (d, 1H, J = 8.0 Hz), 6.88 – 6.84 (m, 1H), 6.54 (s, 1H), 6.49 (t, 1H, J = 6.2 Hz), 6.44 (d, 1H, J = 8.4 Hz), 6.01 (s, 2H), 4.53 (d, 2H, J = 5.7 Hz), 4.15 (q, 2H, J = 7.1 Hz), 4.14 (q, 2H, J = 7.1 Hz), 1.72 – 1.57 (m, 6H), 1.28 (t, 6H, J = 7.1 Hz), 1.18 – 1.14 (m, 3H), 1.05 – 0.96 (m, 2H).
3-(3-(cyclohexylmethyl)-4-((3-((3-fluorophenyl)carbamoyl)benzyl)amino)benzamido)phenyl diethyl phosphate (39f)
White solid (103 mg, 0.15 mmol, 78%). 1H NMR (400 MHz, DMSO-d6) δ 10.42 (s, 1H), 9.86 (s, 1H), 7.94 (s, 1H), 7.79 (d, 1H, J = 8.0 Hz), 7.76 – 7.70 (m, 1H), 7.60 – 7.51 (m, 4H), 7.48 (t, 1H, J = 7.6 Hz), 7.42 – 7.34 (m, 1H), 7.30 (t, 1H, J = 8.5 Hz), 6.92 (td, 1H, J = 8.4, 2.0 Hz), 6.85 (dd, 1H, J = 8.2, 1.1 Hz), 6.48 (t, 1H, J = 5.6 Hz), 6.44 (d, 1H, J = 8.1 Hz), 4.54 (d, 2H, J = 5.4 Hz), 4.20 – 4.09 (m, 4H), 1.74 – 1.52 (m, 6H), 1.27 (t, 6H, J = 7.1 Hz), 1.18 – 1.12 (m, 3H), 1.06 – 0.94 (m, 2H).
Synthetic procedure for derivatives 40a-f
To the phosphate intermediate 39a-f (1.00 eq) was added a commercially available solution of TMSBr (6.50 eq) in CH2Cl2 (0.25 M) and stirred at room temperature overnight. Upon evaporation of the solvent, the residue was saturated with MeOH/H2O (9:1) and the resulting mixture was stirred at room temperature for 1 hour. Precipitate formed was filtered and collected to give the desired product.
3-(4-((4-((3-chlorophenyl)carbamoyl)benzyl)amino)-3-(cyclohexylmethyl)benzamido)phenyl dihydrogen phosphate (40a)
White solid (8.80 mg, 0.01 mmol, quant.).1H NMR (CD3OD, 400 MHz) δ 7.89 (dt, 2H, J = 8.0, 1.7 Hz), 7.87 (t, 1H, J = 2.0 Hz), 7.66 – 7.56 (m, 4H), 7.49 (d, 2H, J = 8.0 Hz), 7.43 (d, 1H, J = 8.0 Hz), 7.35 – 7.27 (m, 2H), 7.15 – 7.11 (m, 1H), 7.01 – 6.96 (m, 1H), 6.54 (d, 1H, J = 8.4 Hz), 4.60 (s, 1H), 2.57 (d, 2H, J = 6.8 Hz), 1.82 – 1.66 (m, 6H), 1.38 – 1.21 (m, 3H), 1.13 – 1.00 (m, 2H). 13C NMR (CD3OD, 100 MHz) δ 169.01, 168.65, 153.00, 152.93, 149.67, 141.52, 141.43, 135.37, 134.78, 131.56, 131.06, 130.47, 129.00, 128.34, 128.18, 126.57, 125.26, 123.42, 121.85, 120.10, 118.29, 116.83, 114.45, 111.77, 48.12, 40.03, 38.35, 34.50, 27.70, 27.53. HRMS–ESI (m/z): [M + H]+ calcd for C34H36ClN3O6P 648.2030; found, 648.2040.
3-(4-((3-((3-chlorophenyl)carbamoyl)benzyl)amino)-3-(cyclohexylmethyl)benzamido)phenyl dihydrogen phosphate (40b)
White solid (24.7 mg, 0.04, quant.). 1H NMR (CD3OD, 400 MHz) δ 7.93 (t, 1H, J = 1.8 Hz), 7.85 (t, 1H, J = 2.0 Hz), 7.77 (dt, 1H, J = 7.7, 1.3 Hz), 7.63 – 7.55 (m, 5H), 7.46 (t, 1H, J = 7.7 Hz), 7.44 – 7.40 (m, 1H), 7.32 (t, 1H, J = 8.1 Hz), 7.28 (t, 1H, J = 8.1 Hz), 7.16 – 7.10 (m, 1H), 7.00 – 6.94 (m, 1H), 6.51 (d, 1H, J = 8.4 Hz), 4.58 (s, 2H), 2.55 (d, 2H, J = 6.8 Hz), 1.80 – 1.60 (m, 6H), 1.31 – 1.16 (m, 3H), 1.12 – 0.99 (m, 2H). 13C NMR (CD3OD, 100 MHz) δ 168.43, 168.28, 149.16 (JP = 10 Hz), 145.21, 141.27, 138.10, 136.57, 136.48, 135.36, 133.03, 131.66, 131.08, 130.07, 129.06, 128.21, 128.11, 125.37, 123.66, 121.86, 121.54, 120.12, 116.44, 50.96, 39.47, 38.67, 34.29, 27.55, 27.40. HRMS–ESI (m/z): [M + H]+ calcd for C34H36ClN3O6P 648.2030; found, 648.2021.
3-(4-((3-((3-chlorophenyl)carbamoyl)benzyl)amino)-3-(2-cyclohexylethyl)benzamido)phenyl dihydrogen phosphate (40c)
White solid (14.0 mg, 0.02 mmol, quant.).1H NMR (400 MHz, CD3OD) δ 7.94 (s, 1H), 7.86 (t, 1H, J = 2.1 Hz), 7.79 (d, 1H, J = 7.8 Hz), 7.66 (d, 1H, J = 1.7 Hz), 7.64 – 7.55 (m, 4H), 7.47 (t, 1H, J = 7.4 Hz), 7.42 (d, 1H, J = 8.2 Hz), 7.32 (t, 1H, J = 8.2 Hz), 7.28 (t, 1H, J = 8.2 Hz), 7.13 (d, J = 8.2 Hz, 1H), 6.97 (d, 1H, J = 7.9 Hz), 6.53 (d, 1H, J = 8.6 Hz), 4.59 (s, 1H), 2.71 – 2.62 (m, 2H), 1.86 (d, 1H, J = 12.1 Hz), 1.75 – 1.55 (m, 5H), 1.44 – 1.15 (m, 4H), 1.00 (q, J = 11.6 Hz, 1H). 13C NMR (100 MHz, CD3OD) δ 169.16, 168.91, 152.94 (JP = 6.4 Hz), 149.91, 141.74, 141.53, 141.38, 136.40, 135.37, 131.69, 131.06, 130.43, 129.85, 129.80, 128.11, 128.06, 127.57, 127.19, 125.28, 123.23, 121.83, 120.07, 118.22, 116.75, 114.36 (JP = 4.6 Hz), 111.05, 47.90, 38.91, 37.44, 34.55, 29.38, 27.77, 27.48. HRMS–ESI (m/z): [M + H]+ calcd for C35H36ClN3O6P 660.2030; found 660.2035.
3-(3-(bicyclo[2.2.1]heptan-2-yl)-4-((3-((3-chlorophenyl)carbamoyl)benzyl)amino)benzamido)phenyl dihydrogen phosphate (40d)
White solid (17.2 mg, 0.03 mmol, 78%).1H NMR (500 MHz, CD3OD, mixture of diastereomers) δ 7.96 – 7.91 (m, 1H), 7.86 (s, 1H), 7.81 (d, 1H, J = 13.6 Hz), 7.77 (d, 1H, J = 11.0 Hz), 7.61 – 7.56 (m, 4H), 7.47 (td, 1H, J = 7.6 Hz, 3.3 Hz), 7.42 (d, 1H, J = 8.1 Hz), 7.33 (t, 1H J = 8.1 Hz), 7.29 (t, 1H, J = 8.3 Hz), 7.14 (d, 1H, J = 8.0 Hz), 6.97 (d, 1H, J = 7.9 Hz), 6.52 (dd, 1H, J = 8.5 Hz, 3.0 Hz), 4.61 (s, 2H), 3.39 – 3.35 (m, 0.6 H), 2.76 – 2.73 (m, 0.4H), 2.64 – 2.63 (m, 1H), 2.39 – 2.38 (m, 1H), 2.11 – 2.06 (m, 0.4H), 1.99 – 1.92 (m, 0.4H), 1.77 – 1.21 (m, 7H). 13C NMR (125 MHz, CD3OD, mixture of diastereomers) δ 169.48, 168.92, 152.93 (JP = 6.1 Hz), 149.87, 141.58, 141.39, 136.32, 132.21, 131.50, 131.08, 130.43, 129.85, 127.98, 127.64, 127.47, 127.40, 127.07, 126.11, 125.28, 122.54, 122.22, 121.80, 120.06, 118.27, 116.72, 114.36, 110.71, 110.67, 47.72, 42.66, 41.92, 41.62, 41.58, 40.74, 39.85, 39.68, 39.52, 39.44, 39.35, 39.18, 38.63, 38.28, 37.47, 34.06, 31.10, 30.65, 30.13, 24.22. HRMS–ESI (m/z): [M + H]+ calcd for C34H34ClN3O6P 646.1874; found, 646.1874.
3-(4-((3-(benzo[d][1,3]dioxol-5-ylcarbamoyl)benzyl)amino)-3-(cyclohexylmethyl)benzamido)phenyl dihydrogen phosphate (40e)
White solid (13.0 mg, 0.02 mmol, quant.).1H NMR (400 MHz, DMSO-d6) δ 10.13 (s, 1H), 9.79 (s, 1H), 7.92 (s, 1H), 7.76 (d, 1H, J = 7.0 Hz), 7.63 (s, 1H), 7.55 – 7.42 (m, 6H), 7.23 (t, 1H, J = 7.9 Hz), 7.16 (d, 1H, J = 7.5 Hz), 6.88 (d, 1H, J = 8.4 Hz), 6.83 (d, 1H, J = 7.8 Hz), 6.42 (d, 2H, J = 8.9 Hz), 6.00 (s, 2H), 4.52 (s, 2H), 1.70–1.60 (m, 6H), 1.44 – 1.20 (m, 3H), 1.04 – 0.98 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 165.96, 165.84, 151.97 (JP = 5.5 Hz), 149.07, 147.41, 143.68, 141.28, 141.04, 135.64, 133.94, 130.69, 130.24, 129.49, 128.72, 127.47, 126.87, 126.14, 124.30, 121.41, 116.07, 115.04, 113.76, 112.57, 109.44, 108.37, 102.91, 101.45, 46.19, 38.77, 36.96, 33.23, 26.66, 26.27. HRMS-ESI+ (m/z): [M + Na]+ calcd for C35H36N3NaO8P 680.2138; found, 680.2134.
3-(3-(cyclohexylmethyl)-4-((3-((3-fluorophenyl)carbamoyl)benzyl)amino)benzamido)phenyl dihydrogen phosphate (40f)
White solid (79.2 mg, 0.12 mmol, 84%).1H NMR (400 MHz, DMSO-d6) δ 10.42 (s, 1H), 9.79 (s, 1H), 7.94 (s, 1H), 7.79 (d, 1H, J = 7.5 Hz), 7.73 (dt, 1H, J = 11.7 Hz, 2.2 Hz), 7.63 (s, 1H), 7.57 – 7.53 (m, 4H), 7.50 – 7.46 (m, 2H), 7.38 (q, 1H, J = 7.5 Hz), 7.23 (t, 1H, J = 8.1 Hz), 6.92 (td, 1H, J = 8.1 Hz, 2.2 Hz), 6.83 (d, 1H, J = 8.2 Hz), 6.46 – 6.42 (m, 2H), 4.53 (s, 2H), 1.70 – 1.59 (m, 6H), 1.19 – 1.11 (m, 3H), 1.04 – 0.99 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 166.44, 165.94, 162.53 (JF = 241.04 Hz), 151.97 (JP = 6.0 Hz), 149.05, 141.47, 141.35, 141.27, 141.15, 135.34, 130.73, 130.69, 130.65, 130.58, 129.48, 128.81, 127.48, 126.95, 126.30, 124.31, 121.44, 116.38, 116.08, 115.04, 112.58, 110.63, 110.42, 109.44, 107.48, 107.22, 46.16, 38.78, 36.97, 33.23, 26.65, 26.27. 19F NMR (376 MHz, DMSO-d6) δ −112.26. HRMS-ESI+ (m/z): [M + H]+ calcd for C34H35FN3O6P 632.2324; found 632.2326.
3-formylbenzoyl chloride (41)
A solution of 3-formylbenzoic acid (343 mg, 2.29 mmol) and thionyl chloride (1.66 mL, 22.9 mmol) in toluene (40 mL) was heated under reflux for 1 hour. Upon evaporation of the solvent under the reduced pressure, the crude product 41 was used in the next step without any further purification.
Methyl 3-(cyclohexylmethyl)-4-(3-formylbenzamido)benzoate (42)
To a solution of the crude 41 in CH2Cl2 (40 mL) was added the aniline 11 (436 mg, 1.76 mmol) and triethylamine (0.25 mL, 1.76 mmol). The resulting solution was stirred at room temperature overnight. The resulting mixture was extracted between CH2Cl2 (3 × 40.0 mL) and saturated aqueous NaHCO3 solution (40.0 mL). The combined organic layers were washed with brine, dried over Na2SO4, and filtered. Upon evaporation of the solvent under the reduced pressure, the crude product 42 was used in the next step without any further purification.
3-((2-(cyclohexylmethyl)-4-(methoxycarbonyl)phenyl)carbamoyl)benzoic acid (43)
To a solution of the crude aldehyde 42 in DMF (8.00 mL) was added Oxone (541 mg, 1.76 mmol). The resulting mixture was stirred at room temperature for 3 hours. The resulting mixture was extracted between EtOAc (3 × 40.0 mL) and aq. 1N HCl sol. The combined organic layers were washed with brine, dried over Na2SO4, and filtered. Upon evaporation of the solvent under the reduced pressure, the crude product 43 was used in the next step without any further purification.
Methyl 4-(3-((3-chlorophenyl)carbamoyl)benzamido)-3-(cyclohexylmethyl)benzoate (44a)
To a solution of crude 43 in DMF (10.0 mL) was added 3-aminophenol (0.20 mL, 2.11 mmol), HBTU (803 mg, 2.11 mmol), and DIPEA (0.62 mL, 3.52 mmol). The resulting mixture was stirred at room temperature overnight. The resulting mixture was extracted between EtOAc (3 × 40.0 mL) and saturated aqueous NaHCO3 solution. The combined organic layers were washed with brine, dried over Na2SO4, filtered, and evaporated under reduced pressure. The residue was purified by silica gel chromatography (EtOAc:Hex = 3:7) to give the desired compound 44a as a white solid (551 mg, 1.09 mmol, 48% over 4 steps). 1H NMR (CD3OD with three drops of DMSO-d6, 500 MHz) δ 8.52 (s, 1H), 8.22 (d, 1H, J = 1.6 Hz), 8.19 (d, 1H, J = 1.6 Hz), 7.97 – 7.90 (m, 2H), 7.78 – 7.67 (m, 4H), 7.35 (t, 1H, J = 8.2 Hz), 7.20 – 7.15 (m, 1H), 3.89 (s, 3H), 2.75 (d, 2H, J = 6.7 Hz), 1.71 – 1.62 (m, 6H), 1.20 – 1.10 (m, 3H), 1.08 – 0.94 (m, 2H).
Methyl 4-(3-(((3-chlorophenyl)amino)methyl)benzamido)-3-(cyclohexylmethyl)benzoate (44b)
To a solution of the crude aldehyde 42 in MeOH (15.0 mL) was added 3-chloroaniline (0.18 mL, 1.76 mmol). The resulting mixture was heated under reflux for 3 hours. After the reaction mixture was cooled down to around 35°C, NaCNBH3 (111 mg, 1.76 mmol) and acetic acid (0.87 mL, 15.3 mmol) were added to the mixture. The resulting mixture was stirred at 35°C for 30 mins. Upon evaporation of the solvent, the residue was suspended in water (50.0 mL) overnight to provide a precipitation, which was filtered and washed with water (3 × 40.0 mL) to provide the title compound 44b as a gray solid (467 mg, 0.95 mmol, 41% over 3 steps). 1H NMR (DMSO-d6 with one drop of D2O, 500 MHz) δ 10.10 (br, 1H), 7.97 (s, 1H), 7.94 – 7.82 (m, 2H), 7.80 – 7.75 (m, 2H), 7.53 – 7.49 (m, 2H), 7.18 (t, 1H, J = 8.1 Hz), 6.89 (s, 1H), 6.75 – 6.70 (m, 2H), 4.36 (s, 2H), 3.89 (s, 3H), 2.63 (d, 2H, J = 6.1 Hz), 1.60 – 1.50 (m, 6H), 1.12 – 1.02 (m, 3H), 0.97 – 0.81 (m, 2H).
Synthetic procedure for compounds 45a-b
To a solution of 44a-b (1.00 eq) in THF was added KOSiMe3 (5.00 eq). The resulting mixture was heated at 50°C overnight. Upon the evaporation of the solvent, the residue was saturated with H2O (30.0 mL) and adjusted to pH = 2 using 1M aqueous HCl solution. A precipitation was formed and collected through a vacuum filtration to provide the crude 45a-b as an off-white solid, which was used in the next step without any further purification.
Synthetic procedure for compounds 46a-b
To a solution of crude 45a-b in anh. DMF (10.0 mL) was added 3-aminophenol (91.0 mg, 0.83 mmol), HBTU (315 mg, 0.83 mmol), and DIPEA (0.29 mL, 1.66 mmol). The resulting mixture was stirred at room temperature overnight. The resulting mixture was extracted between EtOAc (3x) and saturated aqueous NaHCO3 solution. The combined organic layers were washed with brine, dried over Na2SO4, filtered, and evaporated under reduced pressure. The residue was purified by silica gel chromatography (EtOAc:Hex = 1:1) to give the desired compound 46a-b.
N1-(3-chlorophenyl)-N3-(2-(cyclohexylmethyl)-4-((3-hydroxyphenyl)carbamoyl)phenyl)isophthalamide (46a)
White solid (157 mg, 0.27 mmol, 50% over 2 steps). 1H NMR (CD3OD with three drops of DMSO-d6, 500 MHz) δ 8.59 (s, 1H), 8.24 (d, 1H, J = 1.6 Hz), 8.20 (d, 1H, J = 1.6 Hz), 8.01 (t, 1H, J = 2.0 Hz), 7.91 – 7.85 (m, 2H), 7.75 (s, 1H), 7.72 (t, 1H, J = 7.8 Hz), 7.71 – 7.67 (m, 1H), 7.62 (d, 1H, J = 8.0 Hz), 7.59 – 7.55 (m, 1H), 7.44 (t, 1H, J = 8.1 Hz), 7.35 (t, 1H, J = 8.2 Hz), 7.20 – 7.15 (m, 1H), 7.05 – 7.00 (m, 1H), 2.73 (d, 2H, J = 6.7 Hz), 1.70 – 1.60 (m, 6H), 1.25 – 1.10 (m, 3H), 1.08 – 0.92 (m, 2H).
4-(3-(((3-chlorophenyl)amino)methyl)benzamido)-3-(cyclohexylmethyl)-N-(3-hydroxyphenyl)benzamide (46b)
White solid (429 mg, 0.76 mmol, 91% over 2 steps). 1H NMR (DMSO-d6 with one drop of D2O, 500 MHz) δ 10.37 (br, 1H), 10.10 (br, 1H), 7.97 (s, 1H), 7.85 – 7.78 (m, 3H), 7.72 (s, 1H), 7.60 – 7.54 (m, 2H), 7.53 – 7.49 (m, 2H), 7.34 (t, 1H, J = 8.2 Hz), 7.08 (t, 1H, J = 8.1 Hz), 6.96 (d, 1H, J = 8.0 Hz), 6.64 – 6.54 (m, 3H), 4.36 (s, 2H), 2.61 (d, 2H, J = 6.1 Hz), 1.63 – 1.50 (m, 6H), 1.13 – 1.03 (m, 3H), 0.96 – 0.80 (m, 2H).
Synthetic procedure for compounds 47a-b
To a solution of phenol 46a-b (1.00 eq) in CH2Cl2 (0.1 M) was added diethyl chlorophosphate (1.00 eq) and DIPEA (3.00 eq). The resulting solution was stirred at room temperature overnight. Upon evaporation of the solvent, the residue was purified by silica gel chromatography (EtOAc:Hex = 7:3) to give the desired compound.
3-(4-(3-((3-chlorophenyl)carbamoyl)benzamido)-3-(cyclohexylmethyl)benzamido)phenyl diethyl phosphate (47a)
White foam (115 mg, 0.16 mmol, 84%). 1H NMR (CD3OD with three drops of DMSO-d6, 500 MHz) δ 8.59 (s, 1H), 8.22 (d, 1H, J = 1.6 Hz), 8.18 (d, 1H, J = 1.6 Hz), 8.00 (t, 1H, J = 2.0 Hz), 7.90 – 7.82 (m, 2H), 7.77 (s, 1H), 7.74 (t, 1H, J = 7.8 Hz), 7.72 – 7.64 (m, 1H), 7.62 (d, 1H, J = 8.0 Hz), 7.58 – 7.54 (m, 1H), 7.41 (t, 1H, J = 8.1 Hz), 7.36 (t, 1H, J = 8.2 Hz), 7.20 – 7.17 (m, 1H), 7.07 – 7.02 (m, 1H), 4.25 (q, J = 7.2 Hz, 2H), 4.23 (q, J = 7.3 Hz, 2H), 2.73 (d, 2H, J = 6.7 Hz), 1.70 – 1.60 (m, 6H), 1.33 (t, J = 7.1 Hz, 3H), 1.31 (t, J = 7.2 Hz, 3H), 1.24 – 1.11 (m, 3H), 1.09 – 0.92 (m, 2H).
3-(4-(3-(((3-chlorophenyl)amino)methyl)benzamido)-3-(cyclohexylmethyl)benzamido)phenyl diethyl phosphate (47b)
White foam (132 mg, 0.19 mmol, 55%). 1H NMR (DMSO-d6 with one drop of D2O, 500 MHz) δ 10.37 (br, 1H), 10.08 (br, 1H), 7.97 (s, 1H), 7.89 – 7.80 (m, 3H), 7.71 (s, 1H), 7.61 – 7.56 (m, 2H), 7.56 – 7.50 (m, 2H), 7.31 (t, 1H, J = 8.2 Hz), 7.09 (t, 1H, J = 8.1 Hz), 6.94 (d, 1H, J = 8.0 Hz), 6.60 – 6.51 (m, 3H), 4.32 (s, 2H), 4.23 (q, J = 7.1 Hz, 2H), 4.21 (q, J = 7.1 Hz, 2H), 2.61 (d, 2H, J = 6.1 Hz), 1.60 – 1.50 (m, 6H), 1.37 (t, J = 7.2 Hz, 3H), 1.33 (t, J = 7.1 Hz, 3H), 1.10 – 1.03 (m, 3H), 0.97 – 0.82 (m, 2H).
Synthetic procedure for compounds 48a-b
To the phosphate intermediate 47a-b (1.00 eq) was added a commercially available solution of TMSBr (6.50 eq) in CH2Cl2 (0.25 M) and stirred at room temperature overnight. Upon evaporation of the solvent, the residue was saturated with MeOH/H2O (9:1) and the resulting mixture was stirred at room temperature for 1 hour. Upon evaporation of the solvent, the residue was saturated with CH2Cl2/Et2O (1:1) providing a precipitate, which was filtered and washed with Et2O (3x) to provide the title compound.
3-(4-(3-((3-chlorophenyl)carbamoyl)benzamido)-3-(cyclohexylmethyl)benzamido)phenyl dihydrogen phosphate (48a)
Pale yellow solid (65.0 mg, 0.10 mmol, 63%).1H NMR (CD3OD with three drops of DMSO-d6, 500 MHz) δ 8.58 (s, 1H), 8.21 (d, 1H, J = 1.6 Hz), 8.19 (d, 1H, J = 1.6 Hz), 7.99 (t, 1H, J = 2.0 Hz), 7.90 – 7.86 (m, 2H), 7.77 (s, 1H), 7.75 (t, 1H, J = 7.8 Hz), 7.72 – 7.68 (m, 1H), 7.63 (d, 1H, J = 8.0 Hz), 7.59 – 7.55 (m, 1H), 7.40 (t, 1H, J = 8.1 Hz), 7.37 (t, 1H, J = 8.2 Hz), 7.21 – 7.17 (m, 1H), 7.07 – 7.03 (m, 1H), 2.71 (d, 2H, J = 6.7 Hz), 1.72 – 1.60 (m, 6H), 1.25 – 1.11 (m, 3H), 1.09 – 0.96 (m, 2H). 13C NMR (CD3OD with three drops of DMSO-d6, 125 MHz) δ 166.44 (two carbon signals are overlapped here), 166.15, 151.89, 140.37, 140.18, 139.23, 137.24, 135.47, 135.01, 133.89, 132.70, 130.81, 130.65, 130.17, 130.12, 129.42, 128.95, 127.14, 126.99, 125.71, 123.97, 120.30, 118.77, 116.66, 115.84, 112.98, 39.43, 38.83, 33.13, 26.25, 26.08. HRMS–ESI (m/z): [M + H]+ calcd for C34H34ClN3O7P 662.1823; found, 662.1814.
3-(4-(3-(((3-chlorophenyl)amino)methyl)benzamido)-3-(cyclohexylmethyl)benzamido)phenyl dihydrogen phosphate (48b)
Pale yellow solid (110 mg, 0.17 mmol, 87%). 1H NMR (DMSO-d6 with one drop of D2O, 500 MHz) δ 10.35 (br, 1H), 10.06 (br, 1H), 7.96 (s, 1H), 7.88 – 7.80 (m, 3H), 7.71 (s, 1H), 7.62 – 7.56 (m, 2H), 7.55 – 7.50 (m, 2H), 7.32 (t, 1H, J = 8.2 Hz), 7.06 (t, 1H, J = 8.1 Hz), 6.95 (d, 1H, J = 8.0 Hz), 6.62 – 6.52 (m, 3H), 4.38 (s, 2H), 2.62 (d, 2H, J = 6.1 Hz), 1.63 – 1.50 (m, 6H), 1.13 – 1.03 (m, 3H), 0.96 – 0.85 (m, 2H).13C NMR (DMSO-d6 with one drop of D2O, 125 MHz) δ 166.18, 165.80, 152.34, 150.43, 140.86, 140.66, 139.71, 137.08, 135.07, 134.10, 132.49, 130.90, 130.44, 129.73, 129.07, 127.37, 127.04, 126.38, 126.13, 116.26, 115.84, 112.87, 111.92, 111.46, 46.41, 40.22, 38.78, 33.19, 26.46, 26.22. HRMS–ESI (m/z): [M + H]+ calcd for C34H34ClN3O7P 662.1823; found, 662.1814.
Supplementary Material
Novel molecular scaffold to inhibit Wip1 phosphatase
New chemical synthesis to make phosphatase inhibitors
Inhibition optimized to low micromolar activity
Acknowledgments
We gratefully acknowledge Dr. Robert O’Connor for his assistance in NMR analysis and Dr. John Lloyd together with the mass spectrometry core facility of NIDDK for analysis of all compounds.
Funding Sources
This research was supported by the Intramural Research Programs (IRPs) of NIDDK (National Institute of Diabetes and Digestive and Kidney Diseases) and NCI (National Cancer Institute) at NIH.
Footnotes
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Declaration of interests
☒ The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Supporting Information
The Supporting Information is available free of charge on the website.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Esser D; Hoffmann L; Pham TK; Bräsen C; Qiu W; Wright PC; Albers S-V; Siebers B Protein phosphorylation and its role in archaeal signal transduction. FEMS Microbiol. Rev. 2016, 40, 625–647. doi: 10.1093/femsre/fuw020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Fahs S; Lujan P; Köhn M Approaches to Study Phosphatases. ACS Chem. Biol. 2016, 11, 11, 2944–2961. doi: 10.1021/acschembio.6b00570. [DOI] [PubMed] [Google Scholar]
- [3].Lu X; Nguyen T-A; Moon S-H; Darlington Y; Sommer M; Donehower LA The type 2C phosphatase Wip1: An oncogenic regulator of tumor suppressor and DNA damage response pathways. Cancer Metastasis Rev. 2008, 27, 123–135. doi: 10.1007/s10555-008-9127-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chen MJ, Dixon JE; Manning G Genomics and evolution of protein phosphatases. Sci Signal 2017, 10, eaag1796. doi: 10.1126/scisignal.aag1796. [DOI] [PubMed] [Google Scholar]
- [5].Bulavin DV; Deminov ON; Saito S; Kauraniemi P; Phillips C; Amundson SA; Ambrosino C; Sauter G; Nebreda AR; Anderson CW; Kallioniemi A; Fornace AJ Jr.; Appella E Amplification of PPM1D in human tumors abrogates p53 tumor-suppressor activity. Nat. Genet, 2002, 31, 210–215. doi: 10.1038/ng894. [DOI] [PubMed] [Google Scholar]
- [6].Fiscella M; Zhang H; Fan S; Sakaguchi K; Shen S; Mercer WE; Vande Woude GF; O’Connor PM; Appella E Wip1, a novel human protein phosphatase that is induced inresponse to ionizing radiation in a p53-dependent manner. Proc. Natl. Acad. Sci. U.S.A, 1997, 94, 6048–6053. doi: 10.1073/pnas.94.12.6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Choi J; Appella E; Donehower LA The Structure and Expression of the Murine Wildtype p53-Induced Phosphatase 1 (Wip1). Gene. Genomics, 2000, 64, 298–306. doi: 10.1006/geno.2000.6134. [DOI] [PubMed] [Google Scholar]
- [8].Deng W; Li J; Dorrah K; Jimenez-Tapia D; Arriaga B; Hao Q; Cao W; Gao Z; Vadgama J; Wu Y The role of PPM1D in cancer and advances in studies of its inhibitors. Biomedicine & Pharmacotherapy 2020, 125, 109956. doi: 10.1016/j.biopha.2020.109956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Rayter S; Elliott R; Travers J; Rowlands MG; Richardson TB; Boxall K; Jones K; Linardopoulos S; Workman P; Aherne W; Lord CJ; Ashworth A A chemical inhibitor of PPM1D that selectively kills cells overexpressing PPM1D. Oncogene. 2008, 27, 1036–44. doi: 10.1038/sj.onc.1210729. [DOI] [PubMed] [Google Scholar]
- [10].Yagi H; Chuman Y; Kozakai Y; Imagawa T; Takahashi Y; Yoshimura F; Tanino K; Sakaguchi K A small molecule inhibitor of p53-inducible protein phosphatase PPM1D. Bioorg Med Chem Lett. 2012, 22, 729–32. doi: 10.1016/j.bmcl.2011.10.084. [DOI] [PubMed] [Google Scholar]
- [11].Ogasawara S; Kiyota Y; Chuman Y; Kowata A; Yoshimura F; Tanino K; Kamada R; Sakaguchi K Novel inhibitors targeting PPM1D phosphatase potently suppress cancer cell proliferation. Bioorg Med Chem. 2015, 23, 6246–9. doi: 10.1016/j.bmc.2015.08.042. [DOI] [PubMed] [Google Scholar]
- [12].Yamaguchi H; Durell SR; Feng H; Bai Y; Anderson CW; Appella E Development of a Substrate-Based Cyclic Phosphopeptide Inhibitor of Protein Phosphatase 2Cd, Wip1. Biochemistry, 2006, 45 (44), 13193–13202. doi: 10.1021/bi061356b [DOI] [PubMed] [Google Scholar]
- [13].Bang J; Yamaguchi H; Durell SR; Appella E; Appella DH A Small Molecular Scaffold for Selective Inhibition of Wip1 Phosphatase. ChemMedChem 2008, 3, 230–232. doi: 10.1002/cmdc.200700281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gilmartin AG; Faitg TH; Richter M; Groy A; Seefeld MA; Darcy MG, Peng X; Federowicz K; Yang J; Zhang S-Y; Minthorn E; Jaworski J-P; Schaber M; Martens S; McNulty DE; Sinnamon RH; Zhang H; Kirkpatrick RB; Neysa N; Cui G; Pietrak B; Diaz E; Jones A; Brandt M; Schwartz B; Heerding DA; Kumar R Allosteric Wip1 phosphatase inhibition through flap-subdomain interaction. Nat. Chem. Biol. 2014, 10, 181–187. doi: 10.1038/nchembio.1427 [DOI] [PubMed] [Google Scholar]
- [15].Miller PG, Sathappa M, Moroco JA, Jiang W; Qian Y; Iqbal S; Guo Q; Giacomelli AO; Shaw S; Vernier C; Bajrami B; Yang X; Raffier C; Sperling AS; Gibson CJ; Kahn J; Jin C; Ranaghan M; Caliman A; Brosseau M; Fischer ES; Lintner R; Piccioni F; Campbell AJ; Root DE; Garvie CW; Ebert BL Allosteric inhibition of PPM1D serine/threonine phosphatase via an altered conformational state. Nat. Commun. 2022, 13, 3778. doi: 10.1038/s41467-022-30463-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tagad HD; Debnath S; Clausse V; Saha M; Mazur SJ; Appella E; Appella DH Chemical Features Important for Activity in a Class of Inhibitors Targeting the Wip1 Subdomain. ChemMedChem 2018, 13, 894–901. doi: 10.1002/cmdc.201700779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Prinz HJ Chem. Biol. 2010, 3, 37–44. doi: 10.1007/s12154-009-0029-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Shoichet BK Interpreting Steep Dose-Response Curves in Early Inhibitory Discovery. J. Med. Chem. 2006, 49, 7274–7277. doi: 10.1021/jm061103g [DOI] [PubMed] [Google Scholar]
- [19].Allen SJ; Dower CM; Liu AX; Lumb KJ Detection of Small-Molecule Aggregation with High-Throughput Microplate Biophysical Methods. Curr. Protoc. Chem. Biol. 2020, 12, e78. doi: 10.1002/cpch.78 [DOI] [PubMed] [Google Scholar]
- [20].Hayashi R; Tanoue K; Durell SR; Chatterjee DK; Miller Jenkins LM; Appella DH; Appella E Optimization of a cyclic peptide inhibitor of Ser/Thr phosphatase PPM1D (Wip1). Biochemistry 2011, 50, 4537–4549. doi: 10.1021/bi101949t [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yamaguchi H, Minopoli G, Demidov ON, Chatterjee DK, Anderson CW, Durell SR, Appella E, Substrate Specificity of the Human Protein Phosphatase 2Cδ, Wip1, Biochemistry. 44 (2005) 5285–5294. doi: 10.1021/bi0476634. [DOI] [PubMed] [Google Scholar]
- [22].Clausse V, Tao D, Debnath S, Fang Y, Tagad HD, Wang Y, Sun H, LeClair CA, Mazur SJ, Lane K, Shi ZD, Vasalatiy O, Eells R, Baker LK, Henderson MJ, Webb MR, Shen M, Hall MD, Appella E, Appella DH, Coussens NP, Physiologically relevant orthogonal assays for the discovery of small-molecule modulators of WIP1 phosphatase in high-throughput screens, Journal of Biological Chemistry. 294 (2019) 17654–17668. doi: 10.1074/jbcRA119.010201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tanoue LM, Jenkins Miller, Durell SR, Debnath S, Sakai H, Tagad HD, Ishida K, Appella E, Mazur SJ, Binding of a third metal ion by the human phosphatases PP2Cα and Wip1 is required for phosphatase activity, Biochemistry. 52 (2013) 5830–5843. doi: 10.1021/bi4005649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Debnath D. Kosek, Tagad HD, Durell SR, Appella DH, Acevedo R, Grishaev A, Dyda F, Appella E, Mazur SJ, A trapped human PPM1A–phosphopeptide complex reveals structural features critical for regulation of PPM protein phosphatase activity, Journal of Biological Chemistry. 293 (2018) 7993–8008. doi: 10.1074/jbc.RA117.001213. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.