

Keywords: airway ion transport, gel-on-brush model, mucins, muco-obstructive diseases, mucus
Abstract
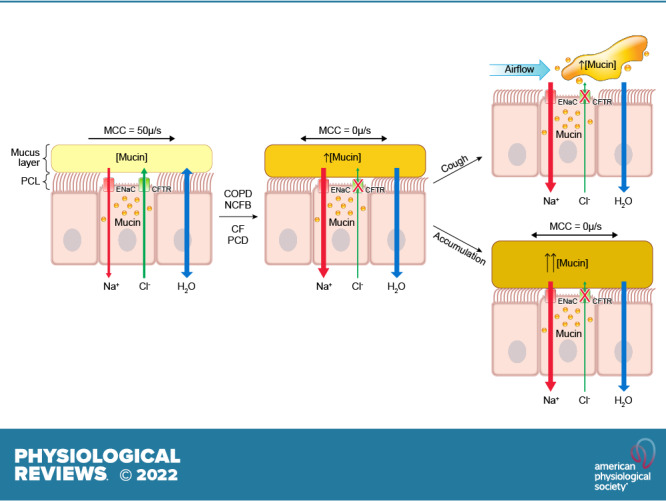
The mucus clearance system is the dominant mechanical host defense system of the human lung. Mucus is cleared from the lung by cilia and airflow, including both two-phase gas-liquid pumping and cough-dependent mechanisms, and mucus transport rates are heavily dependent on mucus concentration. Importantly, mucus transport rates are accurately predicted by the gel-on-brush model of the mucociliary apparatus from the relative osmotic moduli of the mucus and periciliary-glycocalyceal (PCL-G) layers. The fluid available to hydrate mucus is generated by transepithelial fluid transport. Feedback interactions between mucus concentrations and cilia beating, via purinergic signaling, coordinate Na+ absorptive vs Cl− secretory rates to maintain mucus hydration in health. In disease, mucus becomes hyperconcentrated (dehydrated). Multiple mechanisms derange the ion transport pathways that normally hydrate mucus in muco-obstructive lung diseases, e.g., cystic fibrosis (CF), chronic obstructive pulmonary disease (COPD), non-CF bronchiectasis (NCFB), and primary ciliary dyskinesia (PCD). A key step in muco-obstructive disease pathogenesis is the osmotic compression of the mucus layer onto the airway surface with the formation of adherent mucus plaques and plugs, particularly in distal airways. Mucus plaques create locally hypoxic conditions and produce airflow obstruction, inflammation, infection, and, ultimately, airway wall damage. Therapies to clear adherent mucus with hydrating and mucolytic agents are rational, and strategies to develop these agents are reviewed.
CLINICAL HIGHLIGHTS
Mucus accumulation represents a classic clinical problem in the chronic and exacerbating phases of many airway diseases. Recent translational data suggest that muco-obstructive diseases are typically associated with hyperconcentrated, i.e., dehydrated, mucus. Basic science studies have contributed two major insights into the mechanisms that mediate intrapulmonary mucus accumulation. First, electrophysiological studies suggest that imbalances in Na+/fluid absorption vs. Cl−/fluid secretion cause increased mucus concentrations. Second, polymer physics studies suggest that mucus hyperconcentration generates mucin polymer-dependent osmotic forces that compress the mucus layer onto airway surfaces, ultimately producing the mucus accumulation, particularly in small airways, that characterizes muco-obstructive diseases. Therapies for muco-obstructive diseases can be directed toward the upstream ion transport defects that produce hyperconcentrated mucus, as demonstrated by the highly effective modulators that restore CFTR function, and/or therapies designed to broadly rehydrate airway surfaces, including inhaled osmolytes. Therapeutic needs for the future include more durable/effective hydrating agents and agents that break up accumulated intrapulmonary mucus, i.e., effective mucolytics.
1. INTRODUCTION: SHORT HISTORY OF TRADITIONAL CONCEPTS OF NORMAL MUCUS, MUCUS CLEARANCE, AND MUCUS IN DISEASE
Mucus clearance constitutes the dominant mechanical host defense system of mammalian pulmonary airways (1–3). This system is required to protect the large proximal (bronchial) and smaller distal (bronchiolar) airways against the estimated 106–1010 bacteria and other particulates inhaled per day (4) (FIGURE 1A). It has been classically held since the 1930s that mucociliary clearance (MCC) reflects the movement of a mucus layer over a periciliary “sol” (liquid) layer in which cilia beat (5–7) (FIGURE 1Bi). This formulation, however, never adequately described why 1) a discrete mucus layer forms at all, e.g., why secreted mucins (100- to 300-nm radius of gyration) do not penetrate the interciliary spaces (250 nm) (8) and 2) how the mucus layer remains interactive with cilia under conditions of varying hydration, e.g., why the mucus layer does not “float” off cilia in periods of high airway hydration.
FIGURE 1.
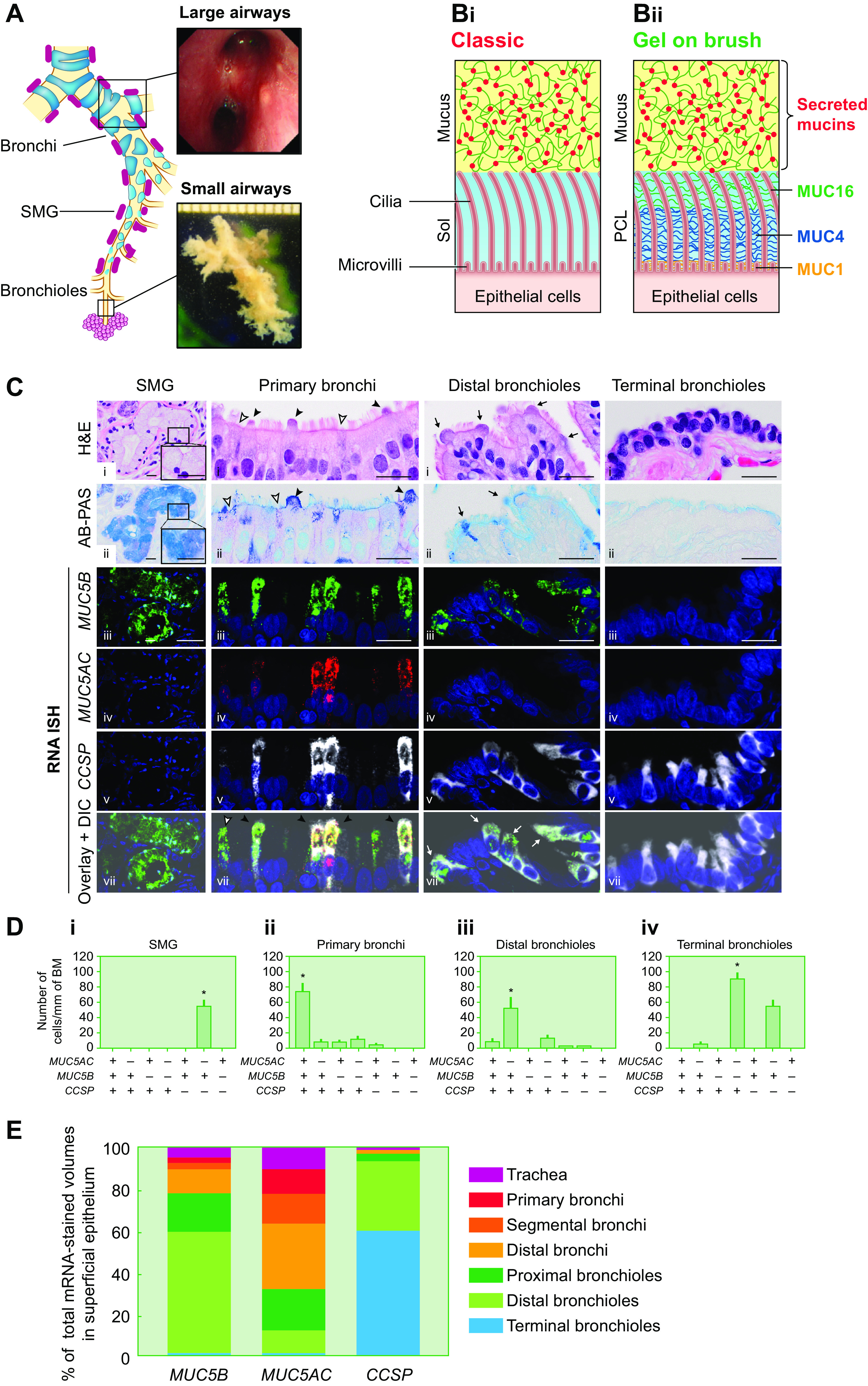
Mucin organization macroscopically and microscopically in the normal lung. A: regional airway and submucosal gland (SMG) distributions in the lung. Large airways, also defined as bronchial airways, are characterized by airway cartilage and SMGs. Small airways, also defined as bronchioles, typically are <2 mm in diameter and without cartilage and SMGs. B: microscopic organization of the mucociliary apparatus. Bi: schematic representation of the classic gel-on-liquid model showing a mucus layer (comprised of gel-forming mucins MUC5AC and MUC5B) and the periciliary layer (PCL) as a liquid-filled “sol” domain (4, 5). Bii: gel-on-brush model depicting a MUC5B- and MUC5AC-populated mucus layer juxtaposed to PCL comprised of “brushlike” epithelial-tethered mucins MUC1, 4, 16, and 20 (not shown), and likely other large glycopolymers. C: MUC5B, MUC5AC, and club cell secretory protein (CCSP) mRNA coexpression is region specific. Top: hematoxylin and eosin (H&E)- and Alcian blue and periodic acid-Schiff (AB-PAS)-stained sections of 4 airway regions. Bottom: localization of MUC5B and MUC5AC with CCSP mRNAs by RNA in situ hybridization (ISH) in the 4 different regions of normal/healthy human airways. For cellular localization, MUC5B (green), MUC5AC (red), and CCSP (white) mRNAs were visualized by fluorescent RNA ISH. Single-color images were merged (“overlay”) and the overlaid images superimposed on differential interference contrast (DIC) images. In SMGs, mucus cells exhibited large mucin granules definable by AB-PAS staining (insets in H&E- and AB-PAS-stained sections). In primary bronchial superficial epithelium, 2 nonciliated CCSP+ epithelial cell types were identified: 1) a nonciliated epithelial cell with an AB-PAS-stained apical bulge (black arrowheads) and 2) a nonciliated epithelial cell without the apical bulge (white arrowheads). In distal bronchioles, nonciliated epithelial cells with dome-shaped apical bulges (black arrows) dominated. Nuclei were stained with DAPI (blue). Scale bars, 20 µm. D: quantification of cell types expressing MUC5B, MUC5AC, and/or CCSP mRNAs in different regions of normal/healthy human airways. Data are expressed as the number of each cell type per millimeter of basement membrane (BM). Solid bars and error bars represent mean ± 2 SD. n = 5. *P < 0.05 compared with every other cell type. E: distinct regional distributions of MUC5AC, MUC5B, and CCSP mRNA localization in superficial epithelium of the normal/healthy lung. Data represent the calculated % of total MUC5AC, MUC5B, or CCSP mRNA-stained volumes for each airway region. Total MUC5AC, MUC5B, or CCSP mRNA-stained volumes for each airway region were calculated by multiplying the mean values of the RNA ISH volume densities for each airway region obtained from 5 normal/healthy lungs by the predicted total surface area of corresponding airway regions. Images in B from Refs. 8 and 25 and used with permission from Science and New England Journal of Medicine, respectively. Images in C–E from Ref. 35 and used with permission from American Journal of Respiratory Critical Care Medicine.
With respect to disease, there never has been an accurate nomenclature, or predictive pathophysiological schema, to describe diseases of failed mucus clearance of the lung. Neither the term “chronic bronchitis” (CB), which describes the cough and sputum production clinically associated with these diseases, nor “hypersecretory diseases,” which describes airway goblet cell metaplasia pathologically, adequately captures the small airway-dominated mucus obstruction, inflammation, airway remodeling, and symptoms associated with these diseases (9–22). “Muco-obstructive lung diseases” perhaps is a preferred term (23).
This review describes advances in the biochemical and especially biophysical understanding of the mucus clearance system in health, covering both cilia-dependent (mucociliary clearance, i.e., MCC) and airflow-dependent (gas-liquid pumping, cough) mechanisms. Because of the importance of mucus concentration in mucus biophysics/function, interactions between secreted mucus and airway epithelial ion/fluid transport are covered. The review also considers the mechanisms by which mucus clearance fails and produces muco-obstructive diseases. Because of its unique pathophysiology, and a recent Physiological Reviews article (24), asthma is not the focus of this review. Rather, the review focuses on more “suppurative” muco-obstructive lung diseases, e.g., chronic obstructive pulmonary disease (COPD), primary ciliary dyskinesia (PCD), cystic fibrosis (CF), and non-CF bronchiectasis (NCFB) (25). Finally, based on these novel concepts, general treatment paradigms for muco-obstructive diseases are reviewed.
2. MUCUS CLEARANCE
As reviewed below, mucus is a hydrogel with the biophysical properties required for transportability provided by the high-molecular-weight (MW) MUC5B and MUC5AC secreted mucins (26). The mucus clearance process in the lung can be viewed at multiple length scales. In this review, we start from the largest and proceed to more microscopic scales. In this context, we integrate mucus clearance across 1) lung-wide scales, describing the movement of mucus from the large surface area of small (bronchiolar) airway regions (∼1–2 m2) to the surface area choke point in third-generation proximal airways (∼ 50 cm2) (27); 2) cellular scales, i.e., microscopic (µm2); and 3) mucus mesh scales (10–100 nm) (28, 29).
2.1. Regions and Cell Types Mediating Mucin Secretion
Two high-MW polymeric mucins, i.e., MUC5AC and MUC5B, are the dominant secreted mucins that cover and are cleared from airway surfaces (30–33) (FIGURE 1B). A classic view of the mucociliary transport system assigned MUC5B secretion to the submucosal glands (SMGs) of the proximal airways, whereas the superficial epithelia of the proximal and distal airways were deemed the sites of MUC5AC secretion (20, 26) (FIGURE 1A). Recent data from mouse models, and, importantly, human studies, have led to a revision of this formulation (34, 35). In conditions of health, CCSP+ (SCGB1A1) club cells within the superficial epithelium of both human proximal and distal airways constitutively secrete MUC5B (FIGURE 1, C and D). Importantly, the club cell appears to constitutively secrete MUC5B via a small granule pathway (100 nm) (36, 37). The superficial epithelial MUC5B secretion is supplemented by MUC5B secretion from mucus cells within SMGs that may be in part tonic but likely can be greatly increased as part of the cough reflex (20, 38). A regional balance sheet of MUC5B secretion suggests that the distal small airways, because of their large surface area, dominate superficial airway epithelial MUC5B secretion in the healthy lung (FIGURE 1E). Notably, the capacity to secrete MUC5B in health disappears in the CCSP+ club cells in the most distal respiratory bronchioles (35). These club cells instead secrete a noncleaved form of surfactant protein B (SPB) and may buffer the alveolus from aspiration of secreted MUC5B (39, 40).
In contrast, the normal lung of humans or mice appears to secrete little MUC5AC in proximal airways and almost none distally (35, 41) (FIGURE 1, C and D). However, stresses to the lung, involving a wide spectrum of pathophysiologies, induce MUC5AC in the superficial epithelium (2, 32, 33, 42–49). Recent studies suggest that MUC5AC secretion is superimposed on CCSP+ club cells competent for basal MUC5B secretion, with intense upregulation of MUC5AC expression associated with metaplasia to a goblet cell phenotype and accumulation of intracellular mucin-containing granules ∼1 µm in diameter (35). The regulation of large granule/mucin secretion from goblet cells has been widely studied, and a number of G protein-coupled receptors trigger secretion via Ca2+- and phospholipase C-dependent mechanisms (32, 41). As implied by their definition, the regulation of secretion via the large granule pathway in goblet cells differs from the constitutive pathway in club cells (50, 51).
Finally, a balance sheet of mucin secretion and mucin clearance from the lung has not been established (52). Unlike surface active proteins in the alveolus, it has been assumed that there is no entero-airway cycle for mucins, i.e., secreted mucins are not recycled or proteolyzed by airway macrophages or epithelial cells as they traverse airway surfaces (53–55). Such an assumption needs to be tested. If true, it requires that all secreted mucins be cleared, and thus surface mucin fluxes have to increase from distal to proximal airway surfaces.
2.2. Macroscopic Anatomy of Mucus Clearance: Continuous vs. Discontinuous Layers
It is awkward that in 2022 a comprehensive description of the organization and topography of mucus transport macroscopically under basal or stressed conditions is not available. For example, it is not clear whether mucus lines airway surfaces as a continuous layer (“blanket”) or a discontinuous layer (“flakes,” “aggregates”) in health. Indeed, recent in vitro studies have questioned the need for a mucus layer at all for MCC (56, 57). Optical studies of the heavily glandular upper (nasal) airways support the notion that the mucus layer moves under basal conditions as a coordinated blanket in this region (58). However, data from the trachea, and especially more distal lower airways, are more mixed. Sturgess et al. (59) utilized scanning electron microscopy (SEM) to report that rabbit airways were covered by a continuous blanket/sheet. In the more distal airway of rodents, Iravani and colleagues (60, 61) reported from differential interference contrast (DIC) optical studies that mucus moves as flakes, i.e., insoluble masses of mucus 10–100 µm in diameter, that coalesced into larger aggregates as flakes moved proximally. Studies of clearance of inhaled radioactive particles from normal human lungs have reported data that are consistent with this notion (62).
More recently, studies have been performed in excised pig and human airways in which submucosal glands have been stimulated by cholinergic agonists, mimicking in part vagal-mediated cough reflex reactions to inhaled stressors. These studies have reported the differential transport of both a surface blanket (fast) and discontinuous “strands” or “bundles” (slow) emanating from glands (20, 63–66). The notion is that strands may function as “sweepers” to clear large particles deposited on upper airway surfaces. The use of inhaled fluorescent beads or deposited Alcian blue to visualize the strands raises the question of whether the strands were present de novo or reflect bundling of mucins in response to deposition of multivalent beads/Alcian blue. This experimental problem is difficult to resolve without internally labeled mucins because mucins are adapted to be highly interactive with, i.e., bind to, virtually any substance deposited on airway surfaces (see below).
A comprehensive description of the organization of mucus clearance in large and small airways will require more precise definitions of blankets, flakes, aggregates, and strands/bundles and new measurement techniques. Ultimately, the presence of blankets versus flakelike structures will be defined by a combination of experimental data and new theoretical concepts of mucus organization (23, 655). For example, recent experimental data suggest that entrainment and direction of cilia beat require continuous mucus sheets (67). In parallel, theoretical data suggest that newly secreted mucins may first localize via hydrophobic interactions at the mucus-air interface. It is as yet not clear whether mucin localization at the air-mucus interface is continuous (blanketlike) or discontinuous (flakelike). Thus, novel techniques to image the structure and concentration-dependent topological organization of mucins at the air-mucus interface will be a first step to ultimately describe the organization and topography of mucus on normal airway surfaces.
In human muco-obstructive disease, characterization of accumulated mucus in the lung should be easier to define, but data are limited because of the propensity to utilize intraluminal delivery of solutions to fix excised human diseased lungs. However, available human data and data from mouse models suggest that muco-obstruction occurs first in distal airways (bronchioles) as mucus plaques and intraluminal plugs (68–74).
2.3. Microscopic Anatomy of the Mucus Clearance Apparatus
The difficulties with the classic gel-on-liquid model led to a novel description of the mucociliary apparatus as comprised of a gel and a brush, i.e., a mucus layer and a periciliary/brush layer (PCL) (8) (FIGURE 1Bii). As with the classic model, a mucus layer is described that contains the secreted MUC5B and MUC5AC mucins and other molecules (see below). The important addition contributed by this model was the identification of a dense brush comprised of tethered cell surface mucins, including MUC1, MUC4, MUC16, and MUC20, and likely other large tethered biomolecules, on epithelial cell surfaces, microvilli, and cilia (8). The layers and how they interact are described below.
2.3.1. Mucus layer biochemistry and biophysics relative to airway function.
Normal human airway mucus is a hydrogel comprised of ∼97.5% water, 0.9% salt, ∼1.1% globular proteins, and ∼0.5% high-molecular-weight mucin polymers (30, 75). Note, we use 1.0% salt for simplicity in our calculations. We have historically defined the hydration status of mucus by the simple dry to wet weight content, i.e., dry weight/wet weight × 100 (% solids content) (14, 76). However, to describe the osmotic properties of the mucins and globular mucin proteins more accurately, the hydration status of mucus is better described as the wet-to-dry organic mucus content, i.e., the organic percent solids [(dry weight/wet weight) × 100% − 1% salt], and/or the absolute mucin concentration estimated by physical refractometry techniques (76, 77) (TABLE 1). The correlation between the gravimetric and physical measurements over a range of normal and disease values appears tight, so both measures are accurate (76). More recently, mass spectroscopy-based measures of the mucin sialic acid content, ratioed to urea, have also been reported and appear to correlate well with absolute mucin values and percent organic solids contents (78).
Table 1.
Metrics and values to describe mucus hydration/concentrations
| %TS | %OS | cos, mg/ml | cm, mg/mL (20%OS) | cm, mg/mL (50%OS) |
|---|---|---|---|---|
| 1.25 | 0.25 | 2.5 | 0.5 | 1.25 |
| 1.5 | 0.5 | 5 | 1 | 2.5 |
| 2.0 | 1 | 10 | 2 | 5 |
| 2.5 | 1.5 | 15 | 3 | 7.5 |
| 3.0 | 2 | 20 | 4 | 10 |
| 3.5 | 2.5 | 25 | 5 | 12.5 |
| 4 | 3 | 30 | 6 | 15 |
| 4.5 | 3.5 | 35 | 7 | 17.5 |
| 5 | 4 | 40 | 8 | 20 |
| 5.5 | 4.5 | 45 | 9 | 22.5 |
| 6 | 5 | 50 | 10 | 25 |
| 6.5 | 5.5 | 55 | 11 | 27.5 |
| 7 | 6 | 60 | 12 | 30 |
| 7.5 | 6.5 | 65 | 13 | 32.5 |
| 8 | 7 | 70 | 14 | 35 |
| 8.5 | 7.5 | 75 | 15 | 37.5 |
| 9 | 8 | 80 | 16 | 40 |
| 9.5 | 8.5 | 85 | 17 | 42.5 |
| 10 | 9 | 90 | 18 | 45 |
The hydration of mucus was initially reported as the percentage of a mucus sample that was not water, i.e., its percentage of total solids (%TS), calculated as dry weight/wet weight × 100 (8, 14, 76). Since mucus samples are typically isotonic, it is more informative to characterize the composition of mucus in terms of organic molecules that contribute to the osmotic pressures/moduli of mucus, i.e., the percentage of organic solids (%OS). To calculate %OS, the salt contribution of an isotonic liquid is subtracted from the total % solids, i.e., dry weight − 1% salt/wet weight × 100. For simplicity, we have approximated the salt contribution to total solids in Table 1 as 1% rather than 0.9%. The %OS can be converted to the overall concentration of organic solids (cos) as 1%OS = 10 mg/mL. The concentration of mucins (cm), the main gel-forming molecules in mucus, accounts for between 20% and 50% of the total cos in differing normal vs. disease states (76, 77, 79, 80).
2.3.1.1. fluid/solvent content of mucus: role of high airway epithelial water permeability.
The salt and water component of mucus is the product of the active ion transport and water permeability properties of the underlying airway epithelia. The airway surface liquid (ASL) under resting conditions is isotonic with respect to plasma (81–84). This property reflects the high water permeability (∼5 × 10−3 cm/s) of airway epithelia (83, 85–88) that is mediated in part by airway epithelial expression of aquaporins 3, 4, and 5 (89, 90). The ionic composition of the ASL/fluid component of mucus differs modestly from plasma (in mM): Na+ = ∼110 mM (plasma ≈ 140 mM); K+ = ∼30 mM (plasma ≈ 5 mM); Cl− = ∼110 mM (plasma ≈ 100 mM); = ∼30 mM (plasma ≈ 25 mM); and Ca2+ = ∼4 mM (2 mM bound; 2 mM free) (plasma Ca2+ ≈ 8–10 mM) (81). The raised K concentration reflects secretion by apical K+ channels that are coupled to epithelial Na+ channel (ENaC) activity via apical membrane electrochemical driving forces (83). The biologic role for the increased K+ concentrations is unknown, but extracellular K+ concentrations can regulate the rates of ATP release from pannexin I channels (91).
In addition to maintaining an isotonic ASL under resting conditions, the high water permeability of the airway epithelium is physiologically well adapted to replace water lost from proximal airway surfaces due to humidification of inspired air (83, 86, 92–94). For example, the evaporative loss of water from proximal airway surfaces during respiration can raise ASL osmolality/tonicity. The net result of the evaporation-induced increases in ASL osmolality/tonicity is to osmotically draw water into the lumen from the interstitium to replace evaporative water loss. The pathways that mediate airway surface water replenishment, however, are complex. Notably, the raised luminal osmolality is presented to an airway epithelium with asymmetric apical versus basolateral cell membrane water permeabilities (95, 96). Specifically, the high apical membrane versus low basolateral membrane water permeability produces cell shrinkage in response to luminal hyperosmolality, i.e., water flows faster from the cell across the apical membrane to the hyperosmolal lumen than water flows across a less water-permeable basolateral membrane into the hypertonic/hyperosmolal cytoplasm (94). Hence, the epithelium acts as an osmotic cell volume sensor to sense luminal ASL osmalality (96). The effector elements responding to luminal hyperosmolality-induced airway epithelial cell shrinkage include the synthesis and release of nitric oxide (NO) to the basolateral compartment. NO triggers an increase in submucosal blood flow that ensures adequate water availability to replenish airway surface evaporative water losses (97).
2.3.1.2. mucin polymer content of mucus.
The mucin polymers are extraordinary in size and impressive in their oligosaccharide content, i.e., ∼75% of total mucin mass (26, 98) (FIGURE 2A). Inserted between the NH2 and COOH termini are serine-threonine-rich domains that are heavily glycosylated with sugar side chains ranging from 2 to 12 sugars in length that are often terminally capped with negatively charged sialic acid or sulfate molecules (4, 99). The glycosylation domains of the secreted mucins provide 1) sites (hydroxyls) for hydration; 2) a combinatorial library of binding sites capable of binding virtually any inhaled material with a low but sufficient binding affinity to mediate clearance (100); and 3) the negative charges that make mucins negatively charged polymers. Cysteine-rich (CYS) domains are interspersed within the serine-threonine-rich domains and may provide hydrophobic regions for noncovalent intermucin and intramucin associations (101).
FIGURE 2.
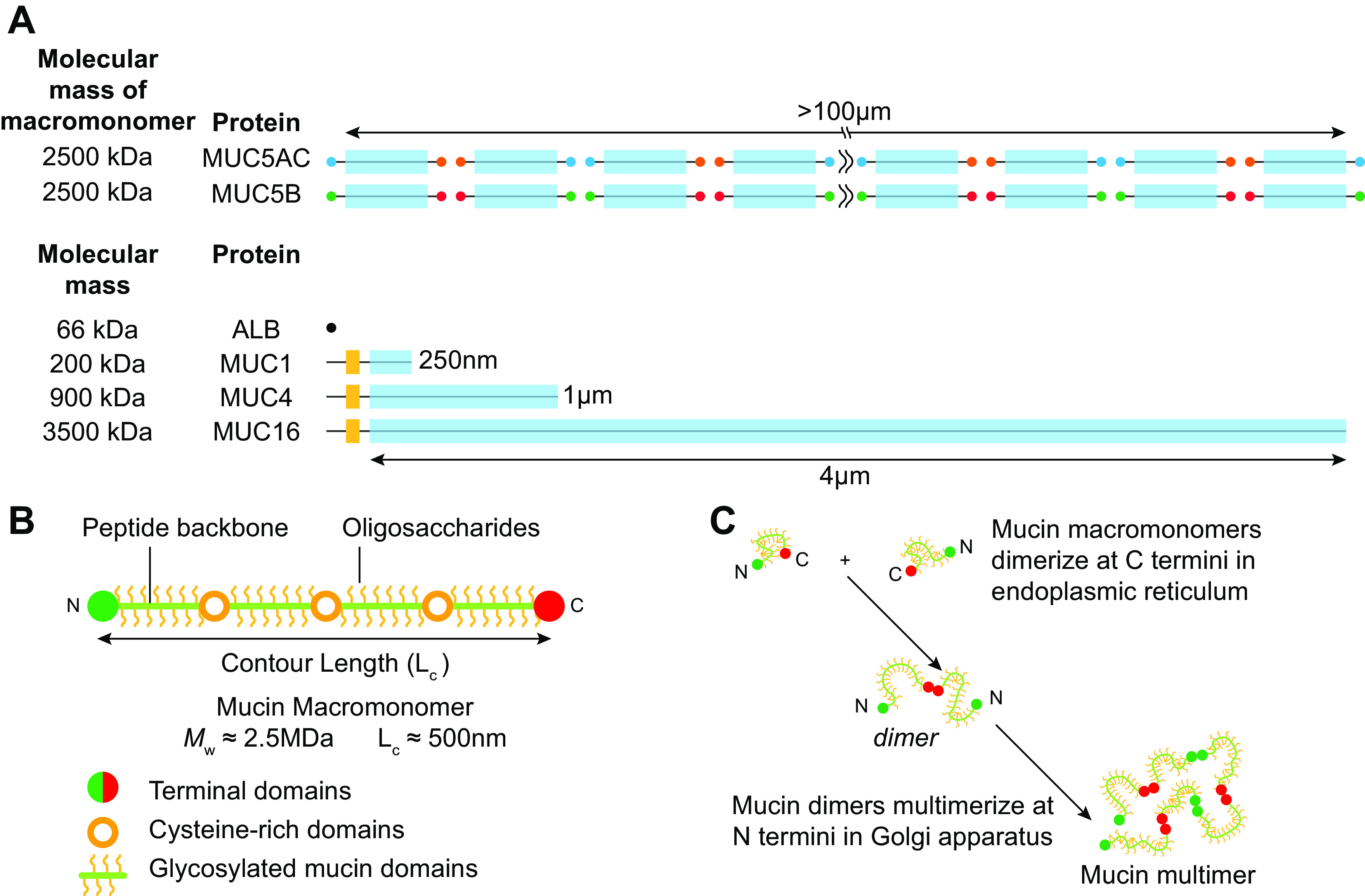
Characteristics of airway mucins. A: domain structures and relative sizes of the secreted mucins (MUC5AC and MUC5B) and tethered mucins (MUC1, MUC4, and MUC16). For reference, the globular protein albumin (ALB) is shown. The secreted mucins are composed of macromonomers with NH2 (N) terminals (MUC5AC, green; MUC5B, light blue), glycosylated domains, and COOH (C) terminals (MUC5AC, yellow; MUC5B, purple). Both the terminal C-C dimers and terminal N-N multimers are linked by cysteine-mediated S-S bonds. Note the molecular mass of the MUC5AC and MUC5B multimeric mucins typically exceeds 250 MDa. The tethered mucins have cytoplasmic NH2-terminal (dark gray), transmembrane (yellow), and heavily glycosylated extracellular domains (blue). For simplicity, unique domains and proteolytic cleavage sites are not shown. Estimated molecular weights are for the glycosylated forms of the secreted mucin macromonomers and tethered mucins. B: schematic of mucin glycoprotein macromonomer domains and sizes. Relationships of NH2- and COOH-terminal domains, cysteine-rich domains, and glycosylated domains not depicted to scale. C: biosynthesis of dimers in endoplasmic reticulum and multimers in the Golgi apparatus, respectively. Classic nonassociating mucin polymer shape depicted. A contains content from Ref. 25 and is used with permission from New England Journal of Medicine.
The individual mucin subunits, often called “macromonomers,” are composed of major domains required for specialized functions (FIGURE 2B). The NH2 and COOH termini are large domains, likely heavily folded, that contain von Willebrand-like domains (D domains) important for multimerization (FIGURE 2B). Mucin macromonomers are disulfide-linked via their COOH-terminal domains to form dimers before translocation to the Golgi, where they are disulfide-linked via their NH2-terminal domains, producing very large multimers (30, 32, 102–105).
Gene-targeted mouse studies have described important aspects of basal and regulated functions of Muc5b and Muc5ac. For example, Muc5b-knockout (KO) mice exhibit an absence of respiratory tract MCC and exhibit routine upper airway phenotypes (otitis media, nasal mucus plugs). The pulmonary phenotype of Muc5b-KO mice under naive conditions reflects the inability to clear intermittent aspiration (gastric or upper airway) and presents with a spectrum of disease ranging from health to sporadic fatalities. In contrast, no defects in MCC or spontaneous clinical phenotypes were detected in Muc5ac-KO mice (106). Subsequent studies of Muc5ac KO mice exposed to inhaled allergens, however, revealed the highly important role of Muc5ac in response to inhaled stresses (107–109).
There are likely differences between MUC5B and MUC5AC structure and function that explain the need for “two mucins.” For example, there are subtle differences in the NH2- and COOH-terminal domain structures of MUC5AC and MUC5B, numbers of Cys domains, VNTR lengths, and possibly domain-structure multimerization (33, 110, 111). MUC5B is a linear multimer of mucin macromonomers in size (∼2.5 MDa) that varies from 2 to ≥100 macromonomers [up to 50 µm in length and 250 MDa in mass (102, 112, 113)]. The covalent linear versus branching status of MUC5AC is not yet resolved but may exhibit linear and trimeric structures (114). Future studies are required to elucidate the functional aspects, e.g., adhesive interactions, of each mucin that may account for their distinct functions (115).
2.3.1.3. mucus biophysical properties.
The mucin polymers dominate the mucus biophysical properties required for efficient mucus clearance from the lung. The functional properties of mucus can be described by concepts from polymer physics that predict not only the viscoelastic properties of mucus but also other key biophysical properties, including the mucus osmotic modulus and adhesive, cohesive, and frictional properties (8, 29, 116). The biophysical properties of mucus related to transport, as with other polymers, scale to higher-order powers of mucin concentration (8). This feature of mucus predicts that relatively small changes in mucin/mucus concentration will produce profound effects on its biophysical and transport properties. In addition, the properties of mucus gels can be modified by conditions that generate interactions between adjacent mucin polymers and generate intermucin bonds of varying strength and durations (117–119). These interactions between associating polymers can produce “sticky” gels whose viscosity can scale as high powers of polymer concentration, e.g., with exponents as high as 6.8–8.5 (120).
2.3.1.4. mucin functions independent of biophysical properties.
Robust data suggest that secreted mucin polymers also have roles in host defense that are not directly related to their biophysical properties (98, 100, 121, 122). For example, the complex glycans associated with mucins may directly modify/limit bacterial pathogen virulence (121). Low-affinity, but high-abundance, interactions between bacteria and mucins, or between bacteria, immunoglobulins (IgA, IgG), and mucins, may increase the efficiency of clearance of bacteria from the lung via the mucus clearance pathway (56, 69, 123–125). Recent studies indicate that bacteriophage IgG-like folds may anchor phages to mucus, providing for an expanded role in mucus antimicrobial activities (126, 127).
In addition, mucin molecules organize a robust globular protein interactome. A substantial percentage (∼33%) of globular proteins in mucus may be bound to mucins under physiological conditions (113). Most of these proteins are involved in airway host defense (113) and include 1) antimicrobial proteins, e.g., LPLUNC, defensins, DMBT1, lysozyme, and C3; 2) antioxidant proteins, e.g., glutathione S-transferase, peroxiredoxin, and superoxide dismutase; and 3) cell signaling proteins, e.g., S100 and transgelin (113). The importance of the globular protein-mucin interactome was observed in studies in which a mouse model of chronic muco-obstructive disease (βENaC mice; see below) was crossed with Muc5b-KO mice (128). A predicted decrement in mucus accumulation was observed. However, this beneficial effect was offset by a loss of Muc5b-organized host defense functions as reflected in increased inflammation that produced a more severe phenotype in βENaC/Muc5b-KO than βENaC mice (128).
2.3.2. Periciliary layer-glycocalyx biochemical and biophysical properties.
The apical cell surfaces of respiratory epithelia are lined by another class of mucins, i.e., the tethered mucins (FIGURES 1Bii and 3Ai). The presence and functional importance of these mucins were first studied in detail in the ciliated cell (8, 113). These studies revealed that ciliated cells exhibit robust expression of MUC1 on microvilli, MUC4 on all aspects of the cilia shaft, and MUC16 on distal portions of the ciliary shaft. Because these tethered mucins are highly expressed in ciliated cells, they are part of the “periciliary layer” (PCL), and their biophysical (osmotic modulus) and molecular sieving (barrier) properties have been intensively studied (8). For example, the PCL brush exhibits osmotic forces that prevent dehydration by the osmotically active mucus layer (see below). The dense PCL brush also restricts entry of infectious/toxic particles with molecular radii > 40 nm into the layer, and its barrier properties become even more impressive near the cell surface, where only particles <5 nm (i.e., 5-nm mesh size) can reach the cell surface (FIGURE 3A). In addition to molecular barrier features, cell surface mucins have substantial roles in modulating inflammatory/immune responses to pathogens and likely signal via their EGF-ligand domains to regulate epithelial repair (14, 37, 129–132).
FIGURE 3.
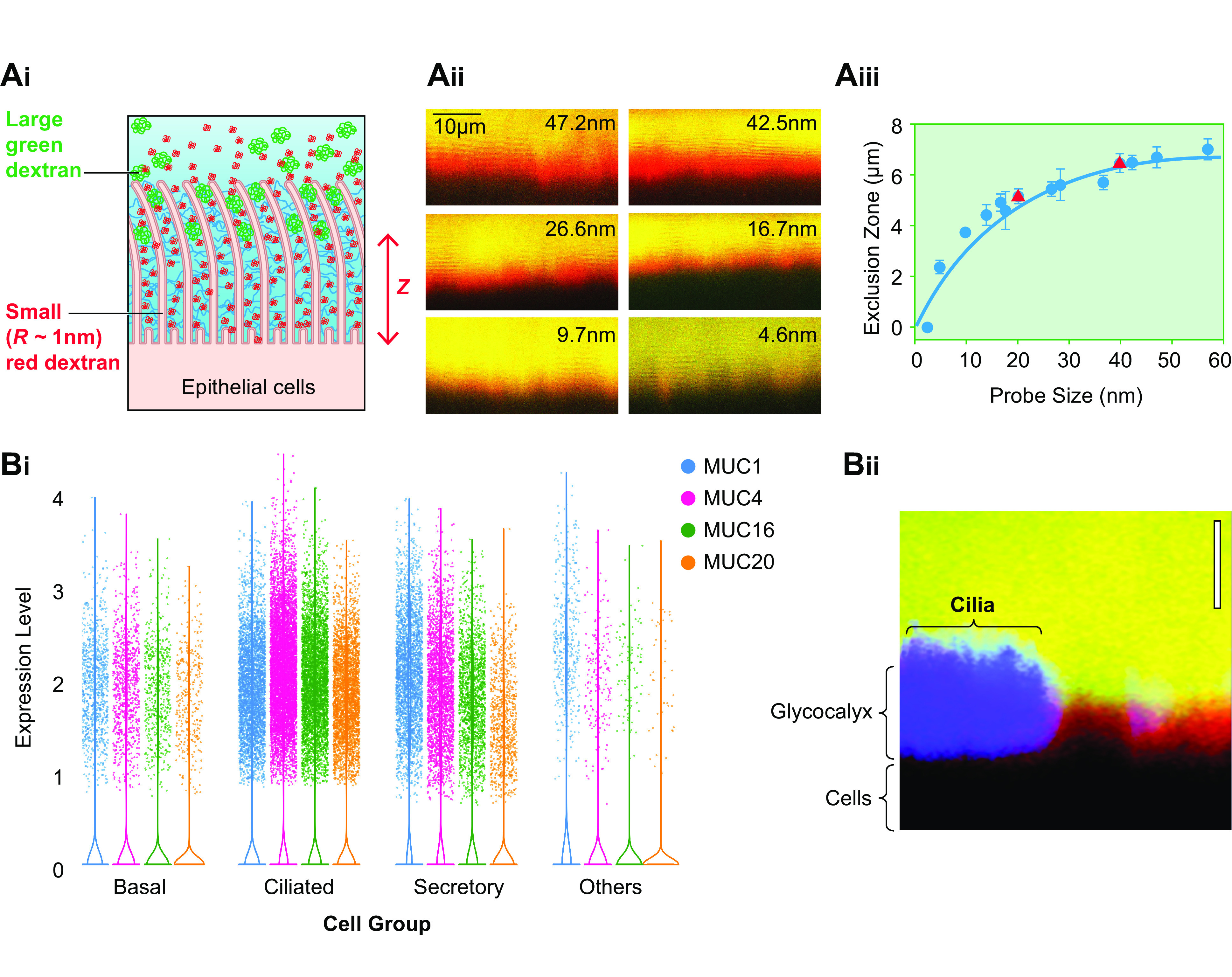
Properties of the periciliary layer-glycocalyx (PCL-G). A: mesh properties of the PCL-G. Ai: diagram depicting strategy to measure by confocal microscopy the molecular size sieving/permeability properties of the PCL-G. Fluorescent dextrans of varying sizes (radius of gyration, Rg) are utilized to 1) penetrate the PCL-G and define the epithelial surface (red dextran, Rg = 1 nm) and 2) characterize the graded mesh size/permeation characteristics of the PCL-G (larger green dextrans, Rg = 5–100 nm). Aii: X-Z confocal image of human airway cultures exposed to a small (1-nm Rg) red dextran with a graded series of larger (in nanometers) green dextrans. Aiii: graph depicting PCL height (z-axis) as defined by restriction of dextran probe (blue circles) access [exclusion zone (z)] to the cell surface. A similar relationship between size and PCL exclusion distance was observed with 20- and 40-nm nanoparticles (red triangles). The PCL-excluded molecules with Rg ∼40 nm are at the top of the PCL (the boundary) and define PCL height, and molecules with Rg ∼ 5 nm are near the cell surface. Note closer to the cell surface (lower z-axis number), the mesh becomes “tighter,” i.e., more restrictive. The PCL mesh size, i.e., correlation length (ξ), varies with the distance from the cell surface (z) as ξ = 17.5 nm·ln[7 µm/(7 µm–z)] where 17.5 nm was used as the average ξ for the PCL (blue line). B: properties of cell surface glycocalyx on nonciliated cells. Bi: forest plot depicting levels of RNA expression in major airway epithelial cell types based on single-cell RNA sequencing (RNAseq) experiments (35). Bii: comparison of height of PCL on ciliated cell (left, blue) and PCL-G on adjacent secretory cell (right) utilizing X-Z confocal imaging of dextran permeation. Green probe = 40 nm; red probe = 5 nm. Content in Ai to Aiii from Ref. 8 and used with permission from Science.
More recently, it has been recognized that tethered mucins are expressed on the apical surface of all lumen-facing airway epithelial cells. For example, single-cell RNA sequencing (scRNAseq) studies have revealed expression of MUC1, 4, 16, and 20 in secretory as well as ciliated cells (133) (FIGURE 3Bi). Confocal imaging studies have also identified the predicted molecular sieving properties of tethered mucins on the luminal surface of secretory cells (FIGURE 3Bii). Because tethered mucins dominate the PCL on ciliated cells and the “glycocalyx” (G) lining surfaces of secretory airway epithelial cells, the term “PCL-G” may best capture the widespread expression of tethered mucins on different luminal airway epithelial cell types (113). A focus on the PCL of the ciliated cell, however, is useful in analyses of muco-ciliary transport.
2.4. Mucus/PCL Osmotic Properties and Relationships to Health and Disease
Important concepts emerged from the gel-on-brush hypothesis that focused on the osmotic properties of polymer gels, solutions, and brushes, including mucins/mucus and the PCL. The osmotic pressure of the mucus layer was the first parameter to be directly measured and is discussed in this section. The subsequent measurements of PCL osmotic properties, and the relationships between the mucus layer and PCL osmotic properties, are also topics of this section.
2.4.1. Measurement of mucus layer osmotic pressure.
The osmotic pressure (Π) of a medium containing many components, e.g., the mucus layer, is defined by the rate of change of the free energy (F) of the medium upon changing its volume (V), when the number (Ni) of its components, e.g., mucin molecules and other globular proteins, is kept constant. These relationships can be described analytically as follows:
| (1) |
The osmotic pressure of the mucus layer (ΠML) is measured experimentally utilizing an osmometer that consists of hemichambers separated by a semipermeable membrane that allows solvent (water) and small solutes (salt, small proteins), but not larger solutes, e.g., mucins and large proteins, to pass through it (FIGURE 4A). The force per unit area exerted by mucus in one hemichamber, separated by a semipermeable membrane from the second hemichamber containing the solvent and salt, is measured by a pressure sensor as the osmotic pressure. Accordingly, the measured osmotic pressure difference reflects the contributions of all the solutes unable to permeate the semipermeable membrane. A typical measurement of osmotic pressure of cultured human bronchial epithelial (HBE) mucus with a 10-kDa semipermeable membrane as a function of mucus concentration is shown in FIGURE 4Di.
FIGURE 4.
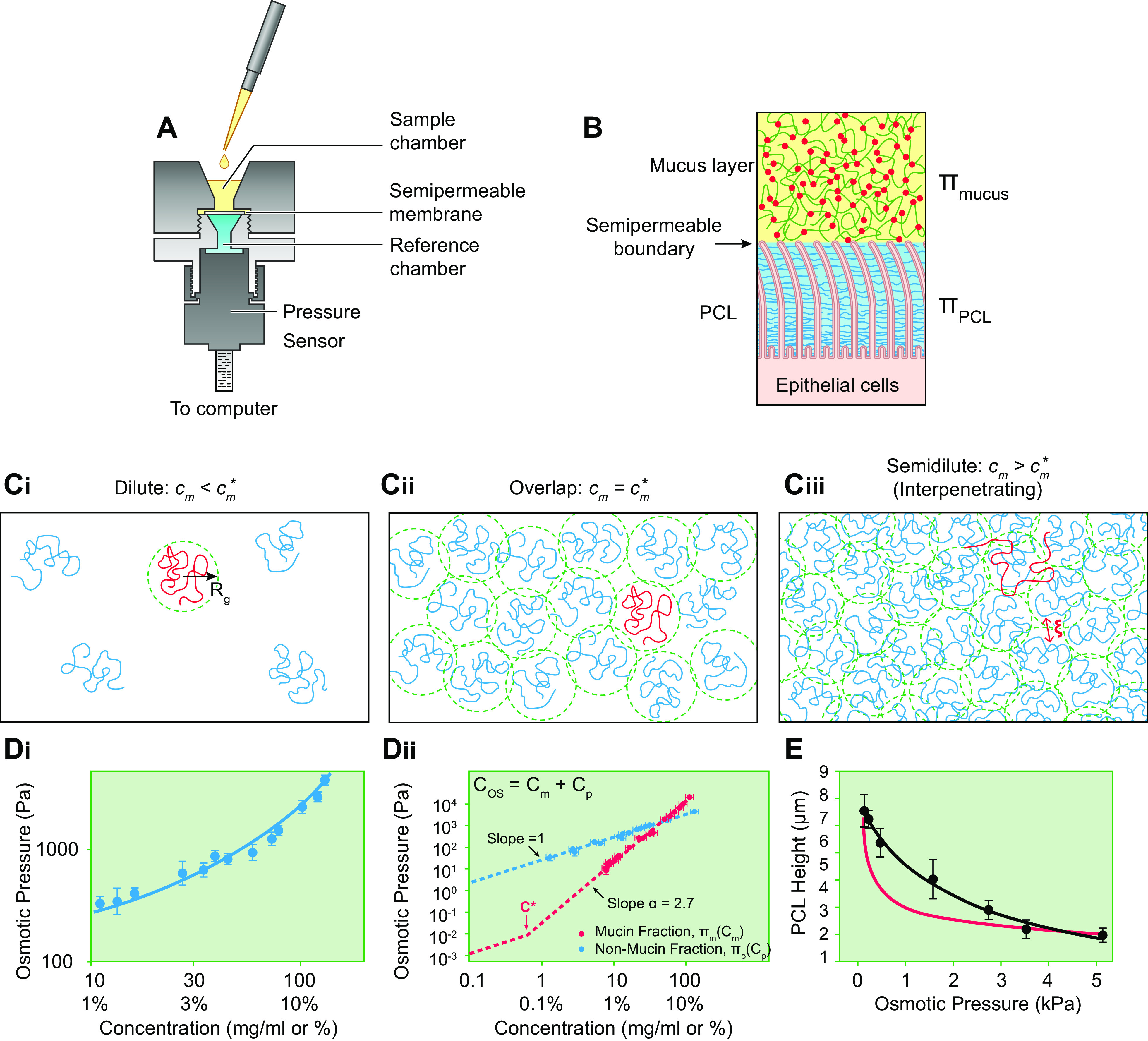
Mucus and PCL osmotic pressures. A: diagram of device modified to measure mucus osmotic pressure. Upper and lower hemichambers and pressure sensor depicted. A semipermeable membrane separates the hemichambers. B: depiction of the mucus layer-PCL interface as the semipermeable boundary separating the mucus layer and PCL compartments. C: concentration-dependent mucin polymeric interaction regimes. Ci: in the dilute regime, the mucin polymers are separated by distances greater than their size, Rg. Cii: at the overlap concentration (c*), the mucins polymers are separated by a distance on the order of their size, i.e., they just “touch.” Ciii: in the semidilute/interpenetrating regime, the centers of mass of mucin polymers are separated by distances smaller than their size, and thus mucins overlap with one another, i.e., interpenetrate (134). The average distance between nearest sections of mucin polymers is the correlation length (ξ), i.e., a measure of concentration. D: human bronchial epithelial (HBE) mucus osmotic pressure without and with fractionation by preparative chromatography into large molecular (“mucin”) and small molecular (“nonmucins” such as globular proteins) fractions. Di: measurement of total mucus osmotic pressure utilizing a 10 kDa semipermeable membrane of intact HBE mucus as a function of mucus concentration (total organic solids). Dii: measurements of the contributions of the mucin and globular protein fractions to HBE mucus osmotic pressure as a function of concentration. Cos, total organic solids concentration; Cm, mucin concentration; Cp, globular protein concentration. Osmotic pressure measurements of each component were performed utilizing 10 kDa MW cutoff semipermeable membranes. Blue dashed line is fitted to globular protein (Cp) measurements using the Van’t Hoff equation (Eq. 2) that predict a globular protein MW (i.e., number average protein mass, Mp) of 66 kDa. The red dashed line to the left of c* (0.6 mg/mL) depicts the osmotic pressure of mucins in dilute conditions (cm < c*) calculated from Eq. 2 with predicted mucin MW (i.e., number average mucin mass, Mm) = 100 MDa. The osmotic pressure values for mucin concentrations (cm) above c* are fitted by the dashed red line generated by the power law term of Eq. 3 describing the osmotic pressure of semidilute mucin solutions. (Courtesy Dr. Phillipe Lorchat.) E: PCL osmotic pressure. PCL height (distance of PCL surface/boundary to epithelial cell surface) dependence on PCL osmotic pressure (ΠPCL) as measured by PCL osmotic compression in response to luminal high-molar-mass dextran polymer administration (black symbols/line). Red line depicts PCL height dependence on osmotic pressure (Eq. 6) estimated from the height-dependent tethered mucin mesh size (ξ) variation within the PCL layer (see equation in legend for FIGURE 3Aiii). See glossary for other abbreviations. E uses content from Ref. 8, with permission from Science.
There are important technical and interpretative aspects of the measurement of the mucus layer osmotic pressure that must be addressed. On the airway surface, the mucus layer is apposed to the PCL (FIGURE 4B). The boundary between the two layers is analogous to the “semipermeable” membrane. Accordingly, the osmotic balance between the mucus and PCL layers is determined by macromolecules larger than the mesh size of the semipermeable PCL brush layer boundary (20–40 nm). Virtually all globular proteins in the mucus layer are smaller than the PCL boundary mesh size and freely exchange between the mucus and PCL layers (8). Therefore, the globular proteins that constitute the majority (∼2/3) of the organic component of mucus by weight do not contribute to the osmotic balance between the mucus layer and PCL. Unfortunately, semipermeable membranes for use in osmometers are not available in the very large molecule discrimination size ranges relevant to the PCL-mucus layer boundary. Therefore, the mucus layer osmotic pressure is typically measured with an osmometer equipped with a standard (10 kDa) semipermeable membrane, i.e., a membrane with a much smaller mesh size than the mucus layer-PCL boundary. The use of a 10-kDa semipermeable membrane renders some mucus layer globular proteins unable to penetrate the membrane, and hence they contribute to the experimentally measured mucus layer osmotic pressure (ΠML). Even with commercially available 100-kDa semipermeable membranes, mucus osmotic pressures higher than generated at the mucus layer-PCL interface are measured because 100-kDa semipermeable membranes also retain more globular proteins than are retained at the mucus layer-PCL boundary. Accordingly, let us consider the quantitative contributions of globular proteins to the experimentally measured mucus layer osmotic pressure.
The contribution of the globular proteins larger than the mesh size of the semipermeable membrane to the measured osmotic pressure of the mucus layer follows the Van’t Hoff law as
| (2) |
where cp is the concentration of the larger globular proteins, R is the molar gas constant (R = 8.314 J/K mol), T the absolute temperature, and Mp the number average protein mass (in grams/mole) (FIGURE 4Dii, blue dashed line). Thus, given knowledge of size (Rg) and concentration of globular proteins, coupled to the characteristics of the experimentally selected semipermeable membrane, the contributions of globular proteins to the measured mucus layer osmotic pressure can be calculated (see Eq. 2, FIGURE 4, Di and Dii).
2.4.2. Contributions of mucins to mucus layer osmotic pressure.
Because virtually all mucins are larger than any device semipermeable membrane, mucin osmotic properties can be measured by conventional device semipermeable membranes. However, the osmotic behavior of mucins in solutions is more complex than for globular proteins, as mucin osmotic properties strongly depend on the mucin concentration (cm). A parameter that describes the dramatic change in mucus osmotic behavior with concentration is termed the mucin “overlap concentration” (), the concentration at which mucins are forced to “touch” one another in solution (FIGURE 4Cii).
Below overlap concentrations, i.e., in dilute mucin conditions (cm < ), in which mucins do not physically “touch”/overlap (FIGURE 4Ci), the osmotic pressure of mucins (Πm) obeys the Van’t Hoff law as written for mucins from Eq. 2, i.e., , where Mm is the number average mucin mass (in grams/mole). Thus, the combined mucin and protein osmotic pressure measured by an osmometer with a relatively small mesh size (e.g., 10 kDa) at dilute mucin concentrations (cm < ) is
| (3) |
where ΠML, cp, Mp, cm, and Mm are as defined above. As noted below, the contribution of Πm to ΠML is so small in dilute conditions that Πm can be typically ignored.
The contributions of mucins to mucus osmotic pressures at mucin overlap concentrations can be estimated based on the physical characteristics of mucins (FIGURE 4Cii). The overlap concentration for mucins () in the mucus layer is estimated as , where the radius of gyration (Rg) quantitates mucin size (FIGURE 4Ci). The c* of HBE mucins, with Mm = 1 × 108 g/mol, i.e., 100 MDa (corresponding to 40-mer) with an Rg = 400 nm, is calculated to be ∼0.6 mg/mL, equivalent to ∼0.18% mucus organic solids (cos). The calculated osmotic pressure of mucins, with mass Mm = 1 × 108 g/mol at concentration cm = 0.6 mg/mL, in the mucus layer at c* is only 0.015 Pa (see Eq. 2), i.e., too small to be experimentally measured or have an effect on the PCL.
The concentration dependence of mucin osmotic pressure changes dramatically when mucins exceed overlap concentrations in the mucus layer, i.e., cm > (FIGURE 4Ciii). In this regime, new “semidilute” osmotic relationships emerge that reflect interactions between interpenetrating mucins (depicted in the steep component of the dashed red line to the right of c* in FIGURE 4Dii). The measured concentration (cm) dependence of mucin osmotic pressures (Πm) above the overlap concentration can be fitted in terms of cm, , and Mm. Combining dilute (Eq. 3) and semidilute/interpenetrating expressions for the osmotic pressure of mucins into a single approximate expression, we write
| (4) |
The first term (linear in concentration dependence) corresponds to a dilute (Van’t Hoff) mucin regime (Eq. 3). The second term (power law) reflects the contribution of interactions between semidilute/interpenetrating mucins. The power law exponent α describes the concentration-dependent contributions of semidilute mucins to osmotic pressure, i.e., (i.e., mucin concentration raised to the power of α). From data in FIGURE 4Dii, the exponent α for mucins in semidilute concentration ranges is calculated as 2.7. This large exponent reflects the strong concentration dependence of mucin osmotic pressure. For example, increasing mucin concentration by a factor of 5 leads to an osmotic pressure increase by a factor of 100 ! Note that Πm can also be estimated from the correlation length ξ (FIGURE 4Ciii), another measure of mucin concentration, as
| (5) |
2.4.3. Combined contributions of globular proteins and mucins to mucus layer osmotic pressure.
The experimentally measured osmotic pressure of the mucus layer (ΠML) increases linearly as a function of concentration in dilute regimes up to mucin overlap conditions ( ≈ 0.6 mg/mL or ≈ 1.8 mg/mL) (FIGURE 4, Di and Dii). As noted above, the value of Πm of mucins in the mucus layer remains relatively small compared with the osmotic pressure required to collapse the PCL even in the normal healthy mucus layer concentration ranges: Πm = 1.5 Pa at cm = 3.3 mg/mL or cos = 10 mg/mL (FIGURE 4Dii). Thus, the experimentally measured (with 10-kDa membrane) values of mucus layer osmotic pressure of a normal healthy mucus layer are dominated by globular proteins and therefore provide relatively little information about the osmotic pressure of the mucus layer relevant to PCL osmotic compression. However, Πm of mucins in the mucus layer strongly increases as a function of mucin concentrations (cm) above , reaching levels that may osmotically compress the PCL in disease states (FIGURE 4, Di and Dii). For example, if mucin concentrations increase by a factor of 6 to cm ≈ 20 mg/mL corresponding to diseased mucus with cos ≈ 60 mg/mL (i.e., 6% organic solids), the mucin osmotic pressure becomes Πm = ∼200 Pa. This value is close to the pressure at which mucins in the mucus layer begin to compress the PCL brush (see below). Accordingly, the experimentally measured ΠML is more informative with respect to PCL compression/mucus transport rates at total organic solids values in the range of disease, e.g., > 6% organic solids.
It is important to note that when mucus layer concentrations are raised by a factor of 6, the small globular protein concentration (cp) will be ≈ 40 mg/mL and the small protein osmotic pressure (Πp) will be ≈ 1,500 Pa. As noted above, the osmotic pressure generated by the small globular proteins does not compress the PCL because small proteins freely permeate the PCL. The small proteins, however, do not permeate cell membranes and hence compress the cell membrane, producing shrinkage of cell volume (see discussion in sect. 2.3).
2.4.4. Measurement of the PCL osmotic pressure.
In contrast to the ability to extract the mucus layer from culture surfaces or collect expectorated mucus for measurements of osmotic pressures with in vitro devices (FIGURE 4A), PCL osmotic properties had to be measured in situ in cell cultures, and novel techniques were developed to make these measurements (8).
Direct measurements of PCL osmotic pressure (ΠPCL) were performed by exposure of washed airway epithelial culture surfaces to solutions of polymeric dextrans with defined osmotic pressures and molecular weights much larger than the PCL mesh size. PCL heights were measured when equality between the applied polymeric solution and PCL osmotic pressures was achieved (8) (FIGURE 4E, black symbols/lines). This technique identified the onset of PCL compression (reduced PCL height) at polymeric dextran osmotic pressures of ∼350 Pa. A nonlinear relationship was observed between the osmotic pressure applied by the test polymeric dextran solution and PCL height/compression over the range of applied osmotic pressures (FIGURE 4E). This behavior in part represents the occupancy of the PCL by two components, an osmotically compressible grafted mucin brush and elastically deformable/bendable cilia.
Measurements of dextran size permeation properties were also utilized to estimate the contribution of tethered mucins to PCL osmotic pressure (ΠPCL). This approach utilized measurements of the correlation lengths (ξ), i.e., the mesh size, of the tethered mucins in the PCL; see FIGURE 3Aiii (8). A simple estimate of the average osmotic pressure of the PCL can be calculated from Eq. 5 utilizing an average PCL correlation length of 17.5 nm (see FIGURE 3Aiii). This estimate yields a PCL osmotic pressure of ∼180 Pa. However, the osmotic pressure of the PCL varies as a function of the PCL depth, reflecting the higher tethered mucin concentration toward the cell surface and hence smaller correlation length (FIGURE 3Aiii). Measurement of the correlation length profile ξ(z) within the PCL layer (FIGURE 3Aiii) allows simple estimates of 1) the relation between PCL correlation length and osmotic pressure ξ ≈ [3kT/(4πΠPCL)]1/3 and 2) the osmotic pressure profile ΠPCL(z).
The contribution of tethered mucins to the PCL osmotic pressure as a function of depth within the PCL can also be calculated by substituting the relationship between PCL correlation length and osmotic pressure into the PCL correlation length profile z = 7 µm [1 − exp(−ξ/17.5 nm)], presented in FIGURE 3Aiii, as
| (6) |
The estimates of the tethered mucin contribution to the PCL osmotic pressure as a function of PCL heights (z) calculated from Eq. 6 are depicted in FIGURE 4E (red line). Note that the osmotic pressure-PCL height values predicted from Eq. 6 are lower than those measured by dextran polymer PCL compression (black line in FIGURE 4E). This discrepancy likely reflects the elastic resistance of bending cilia that in addition to the grafted mucin gel contributes to PCL osmotic pressure (see above).
The PCL osmotic pressure values yielded by direct PCL compression measurements (black points and line in FIGURE 4E) and through ΠPCL estimates based on dextran permeation experiments (red line in FIGURE 4E) were initially thought to be surprisingly high compared with the measured osmotic pressure of a healthy mucus layer (ΠML ≈ 50–100 Pa). However, the higher PCL osmotic pressure ensures that the PCL remains well hydrated, which may be important for both mucus-PCL lubrication and liquid propulsion activities (see below). Another feature of tethered mucin brushes on each cilium important for mucus transport is that brushes exhibit excellent lubricant activities and reduce intercilial friction during ciliary beating (135).
Note that the osmotic pressure measurements of the airway surface have been analyzed in terms of the PCL, i.e., the tethered mucins covering ciliated cell surfaces/shafts. Although secretory cells facing the airway surface also exhibit tethered mucin layers (see sect. 2.3.2 and FIGURE 3B), neither the mean osmotic pressure nor osmotic pressure profiles of the secretory cell glycocalyx have been measured. Given the different geometries, i.e., cylindrical in ciliated cells and planar in secretory cells, and possible differences in densities of tethered mucin expression, there are likely to be differences in the osmotic pressures/profiles of the cell surface mucins/glycocalyx between these two cell types. Accordingly, the overall osmotic properties of airway surfaces may vary as densities of cell types change, e.g., in areas with reduced ciliated cell numbers as may be seen in COPD (see sect. 10). Knowledge of the relative cell surface osmotic pressures of different cell types may be useful in the future to predict changes in mucus transport rates and the likelihood of initial mucus accumulation in airway regions characterized by different cell populations.
2.4.5. Osmotic modulus of a mucus gel.
The osmotic modulus (K) of mucus is a useful parameter to define the water-drawing power of a gel. The osmotic modulus of the mucus layer or PCL gels is defined as the rate of change of osmotic pressure with the logarithm of concentration . As evident from this mathematical relationship, the units of osmotic pressure and modulus are the same, i.e., pascals (Pa). In conditions above c*, where mucus layer osmotic pressures increase as a power law of mucus concentration, i.e., , the mucus layer osmotic modulus (KML) can be estimated as
| (7) |
Thus, the osmotic modulus of the mucus layer in semidilute (>c*) mucin concentration conditions is ∼2.7-fold higher than the osmotic pressure. The osmotic moduli of the mucus layer and PCL are particularly useful parameters to describe relationships between airway epithelial ion transport, the relative hydration of the mucus versus PCL layers, and mucus transport rates (see below).
3. EPITHELIAL ION TRANSPORTAND AIRWAY SURFACE HYDRATION
The hydration status of airway mucus is controlled by the active ion transport and water permeability properties of airway epithelia. Airway epithelia regulate the mass of salt on airway surfaces and, because of their high epithelial water permeability, the volume of isotonic liquid/unit area, i.e., liquid height, on airway surfaces. As with the mucus clearance system, pulmonary epithelial ion/fluid transport can be viewed at multiple scales. Again, we start with the larger and proceed to smaller length scales.
3.1. Integrated Ion/Fluid Transport across Pulmonary Surfaces of the Lung
Like the macroscopic organization of the mucus layer, there are surprising gaps in our understanding of the macroscopic organization within the lung of ion and fluid transport. Indeed, little is known qualitatively or quantitatively about sites or magnitudes of integrated fluid transport throughout the respiratory tract. For example, it is not clear whether fluid is secreted by the alveolus, whether fluid if secreted is absorbed at other alveolar sites, i.e., there is an entero-alveolar cycle (136–141), or whether alveoli secrete fluid that moves onto small airway surfaces as suggested by Lindert et al. (142) and Bove et al. (143). Furthermore, little is known about the physical forces that may mediate fluid flow from the alveolus to the small airways if fluid flow indeed occurs. A clinical phenotype of primary cilia dyskinesia (PCD) subjects, i.e., failure to normally clear alveoli of fluid at birth, suggests a role for cilia in alveolar-small airway fluid coupling (144). In addition, Marangoni forces generated by surfactant gradients from regions of high surfactant concentration, i.e., alveolar surfaces, to regions of lower concentrations, i.e., distal airways, may also mediate bulk liquid flows (145, 146). The great disparity between alveolar (100 m2) and distal small airway (1–2 m2) surfaces suggests that tight regulation of coupled regional liquid flow by whatever mechanism(s) is required.
Similarly, Kilburn pointed out 50 years ago the implications for the great disparity between distal airway (1–2 m2) and proximal airway (50 cm2 at 3rd-generation airways) surfaces for mucus transport (27, 147). He suggested that progressive absorption of the liquid moving up airway surfaces was required to avoid fluid occlusion of proximal airway lumens, i.e., proximal airway “drowning” (147). Recent ion/fluid transport studies of cultured human distal small (bronchiolar) and proximal large (bronchial) airway epithelia suggest that the small and large airways exhibit similar transepithelial ion/fluid transport rates per unit surface area, reflecting similar levels of ENaC and cystic fibrosis transmembrane conductance regulator (CFTR) expression (133, 148–150). Based on surface area considerations, these data suggest that small airways have greater aggregate ion/fluid transport activities than large airways. To avoid proximal “drowning,” either 1) mucins, not water, must selectively be transported proximally or 2) airway epithelia must exhibit mechanisms to regulate intraregional fluid balance while adequately hydrating mucus for optimal transport. Part of the answer to this problem will require, as discussed above, resolution of the nature of the mucin layer versus flake controversy. However, airway epithelia appear to exhibit multiple mechanisms to continually establish proper local ASL volume/mucus hydration as mucus sweeps cephalad along converging surfaces, as postulated by Kilburn (147).
3.2. Regulation of Airway Transepithelial Ion/Fluid Transport
More is known about the microscopic regulation of airway epithelial surface hydration by active ion transport in health and disease. In health, airway epithelia can secrete or absorb ions and indeed likely do both simultaneously to finely tune airway surface volume (hydration) (151) (FIGURE 5A). Note that it is the ASL volume, as microliters per unit surface area (e.g., cm2), that is controlled by active ion transport. The quotient of this relationship, i.e., height, is typically measured by confocal microscopy to assess the volume status of airway surfaces. The typical 7-µm ASL height measured under basal, static conditions (see below) may reflect the biologic adaptation of airway epithelia to produce sufficient volume on airway surfaces to allow cilia to fully extend and efficiently propel mucus.
FIGURE 5.
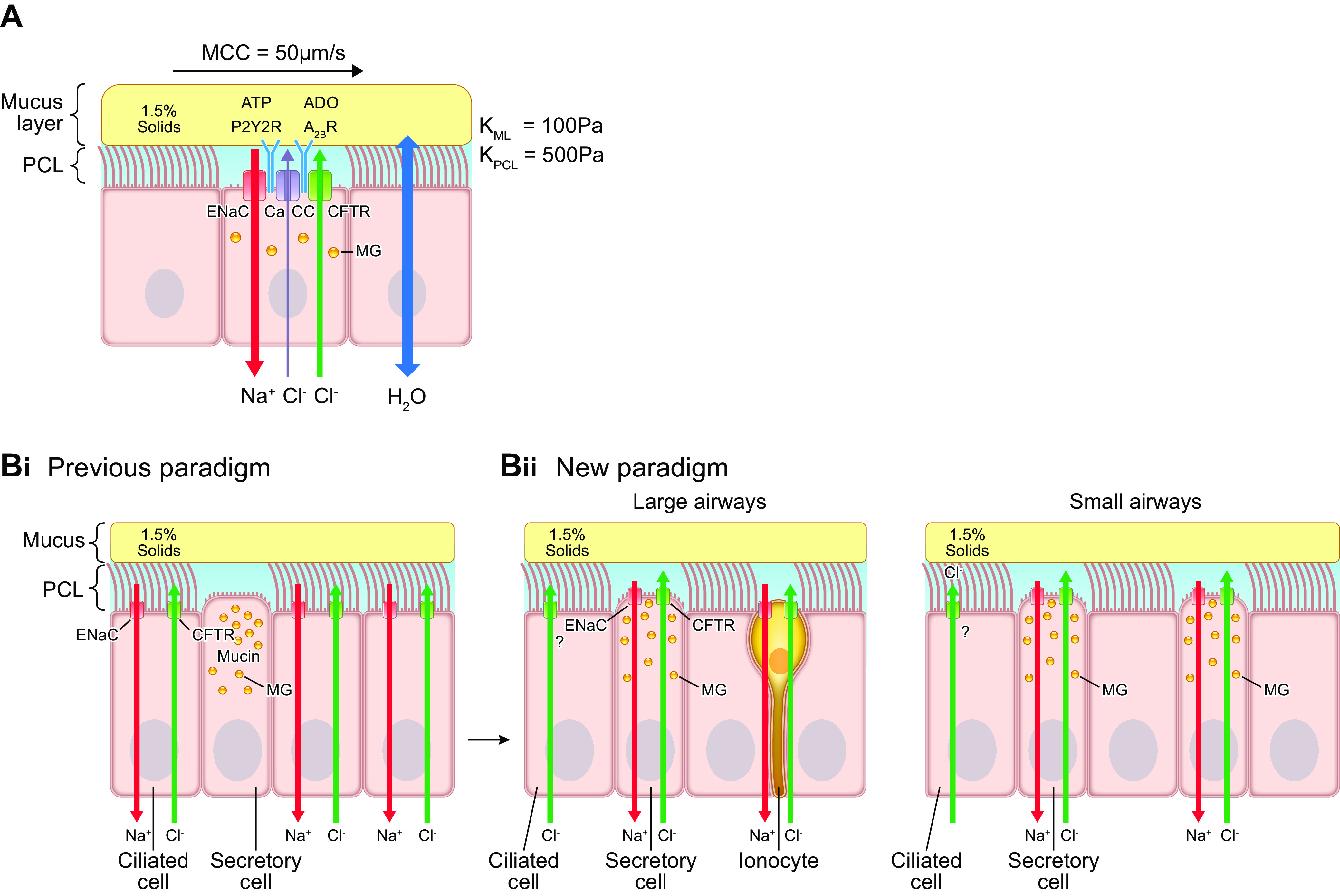
Airway epithelial ion transport regulation of airway surface liquid volume/surface area (height). A: diagram of the mucus layer and periciliary-glycocalyx layer in context of epithelial active ion transport pathways. The mucus layer is depicted with 1.5% organic solids concentration. Osmotic moduli (K) of the mucus layer (KML) and PCL (KPCL) in pascals (Pa) are noted. An epithelial Na+ channel (ENaC) on the apical airway epithelial membrane mediates Na+/liquid absorption. In parallel, the epithelium can secrete Cl−/anions (and liquid) to the lumen via the CFTR channel and a calcium-activated channel (CaCC) (93). The balance between active Na+ absorption and Cl− secretion is regulated in part by the concentrations of extracellular ATP, interacting with P2Y2 receptors (P2Y2Rs), and adenosine (ADO), interacting with A2B receptors. P2Y2R activation inhibits Na+ absorption and stimulates CFTR/CaCC-mediated Cl− secretion, producing/accelerating liquid secretion (152, 153). A2B activation accelerates CFTR-mediated Cl− secretion, also producing liquid secretion. B: expression of ion channels in human airway epithelial cells. Bi: the “classic”/previous paradigm depicted CFTR, ENaC, and related basolateral ion channels (not shown) as expressed in ciliated cells. Bii: a new paradigm has CFTR, ENaC, and related basolateral ion channels/transporters (not shown) mediating transepithelial ion flows expressed in CCSP+, mucin-expressing secretory cells in both large and small airways (133). The possibility that still some CFTR is expressed in ciliated cells is shown (133). A rare cell type, the ionocyte (yellow), is depicted in large airways that expresses high levels of ENaC and CFTR. See glossary for other abbreviations.
Previous models had assigned the major ion transport channels, i.e., the epithelial Na+ channel (ENaC) and CFTR, to ciliated cells (154) (FIGURE 5Bi). However, initial scRNAseq data of proximal mouse and human airways led to revision of this notion (155). These scRNAseq data from mice and humans identified a previously uncharacterized, unique cell type that expressed high levels of CFTR and V-type ATPases (as well as ENaC) (FIGURE 5Bii). This cell type bears functional and molecular similarities to mitochondrion-rich cells in amphibian skins and was termed an “ionocyte” (156, 157). The ionocyte appears more prevalent in proximal human airways and especially in the ductal regions of submucosal glands (155, 158–160). The precise functions of ionocytes in superficial airway epithelial function are not yet known, but they may play a role in producing a mildly hypotonic SMG secretion (see sect. 10).
The most recent single-cell RNAseq data describing the entire human respiratory tract, coupled to functional data, suggest that the CC10+ club cell expresses in addition to CC10 and MUC5B the major airway ion channels required for transepithelial fluid transport (133, 161–163) (FIGURE 5Bii). The club cell-mediated active Na+ absorption pathway includes the apical membrane epithelial Na+ channel (ENaC), which is typically rate limiting for Na+/fluid absorption, and the basolateral membrane Na+-K+-ATPase (93, 164). In parallel, the club cell has the capacity to secrete Cl−/anions to the lumen via apical membrane CFTR, a calcium-activated channel (CaCC), likely TMEM16a, and possibly SLC26A9 channels supported by a basolateral Na+-K+-2Cl− cotransporter (SLC12A2) (165–169). Apical and basolateral club cell K+ channels modulate the driving forces that determine the absolute rates of Na+ absorption and Cl− secretion as well as mediating transepithelial K+ secretion (170–172). ASL K+ may be recycled by an apical membrane H+-K+-ATPase that, in parallel with secretion by apical Cl− channels, including CFTR, and the paracellular permeability to and H+, regulates ASL pH (173–175). Quantitative biophysically based mathematical models of airway epithelial ion transport are available (164, 176–181).
The magnitudes of the active Na+ absorption and Cl− secretion rates are regulated by the number (N) of ion channels per epithelial cell apical membrane surface area and their activation state (open probability, Po). Channel activation states are regulated by intracellular processing, cell surface processing, and/or the concentrations of regulatory molecules in the ASL. More is known about the acute regulation of Po than chronic regulation of channel number (N), and, consequently, these acute regulatory activities are emphasized here.
Apical membrane ENaC Na+ conductance (NPo) is rate limiting for transepithelial Na+ absorption across pulmonary epithelia. ENaC is activated by proteolytic cleavage events, in part mediated intracellularly (furin) and in part at the apical membrane, e.g., PRSS8 (prostasin) (90, 153, 182–184). PRSS8 activity, and hence ENaC activity, is regulated by the concentration of poorly defined protease inhibitors in ASL (185). Proteolytic regulation of ENaC activity governs both the rate of Na+ absorption and the electrochemical driving force for Cl− secretion (93, 186–189). Superimposed on proteolytic regulation of ENaC are acute regulatory events mediated by G protein-coupled receptors. Perhaps most widely studied is the inhibition of ENaC activity produced by P2Y2R purinoceptor-mediated cleavage of apical membrane phosphatidylinositol bisphosphates (PIP2) (190–195).
Counterbalancing Na+ absorption are the number (N) and activity (Po) of apical membrane Cl− (anion) channels that, with favorable electrochemical driving forces, can convert the epithelium from net fluid absorptive to secretory. Regulation of CFTR channel number in airways is poorly understood, whereas there is abundant evidence that TMEM16a is strongly regulated by type 2 immune signaling (169, 196–200). The activation state of apical membrane Cl− channels is regulated by apical and basolateral membrane G protein-coupled receptors. The Gq class of receptors, e.g., nucleotide (e.g., ATP), histamine, bradykinin, and PAR receptors, activate CFTR via regulation of Ca2+-dependent adenylate cyclases and cAMP formation and TMEM16a by increases in intracellular Ca2+ and protein kinase C (201, 202). Stimulation of Gs-coupled receptors, e.g., β2 receptors, adenosine receptors, and VIP receptors, activates adenylate cyclase directly and raises cell cAMP (203–205). Cell cAMP levels regulate CFTR activity via PKA-dependent phosphorylation of the R domain (206, 207).
3.3. Coordinate Regulation of Ion Transport/Fluid Transport Mechanisms for Maintenance of Optimal Mucus Layer Concentration/Hydration: Superficial Epithelium
Coordinate regulation of Na+ absorptive and Cl− secretory pathways to maintain the proper hydration state of mucus, i.e., 97.5% water, 1% ions, and 1.5% organic molecules, is required to respond to the spectrum of conditions that confront the lung. In the distal airways, these functions are exclusive properties of the superficial epithelia. In the proximal airways, submucosal glands (SMGs) also contribute to these activities (see below). It must be stressed that net fluid transport across airway epithelia reflects the balance between simultaneous Na+-driven absorption versus Cl−-driven secretion (93). Insufficient hydration of airway surfaces thus can reflect an absolute increase of Na+ absorption superimposed on normal Cl− secretion rates (208), a normal rate of Na+ absorption superimposed on defective/absent Cl− secretion (209), or scenarios in between. Similarly, excessive fluid secretion can reflect a decrease in Na+ absorption, as observed in pseudohypoaldosteronism (210), or accelerated Cl− secretion, in response to cytokines and/or ATP released during inflammation (201, 211, 212). The pathways that regulate the balance between airway surface dehydration and flooding are therefore important to identify and quantitate for a comprehensive description of mucus hydration in health and disease.
It is important to note that there are multiple overlapping functional requirements of ASL volume homeostasis. First, there is a basal secretion of the liquid required to hydrate airway surfaces to provide water for conditioning inspired air. Second, there are the regulated components of ASL volume transport required to adjust ASL volumes intraregionally as mucus is transported cephalad up converging surface areas. Third, there is a need to adjust the liquid content of mucus locally at the cell level to maintain efficient local mucus transport. Because the extracellular purine nucleotide (NT, e.g., ATP) and purine nucleoside (NS, e.g., adenosine) pathways are the most extensively studied with reference to responses to different physiological conditions, their role in maintenance of ASL volume is emphasized in this section (FIGURE 6A). Note that this topic has also been quantitatively addressed in mathematical models describing 1) extracellular nucleotide/nucleoside release and metabolism and 2) relationships between extracellular nucleotide/nucleoside concentration, ion transport patterns/rates, and the net fluid transport required for ASL homeostasis in both health and disease (213–215).
FIGURE 6.
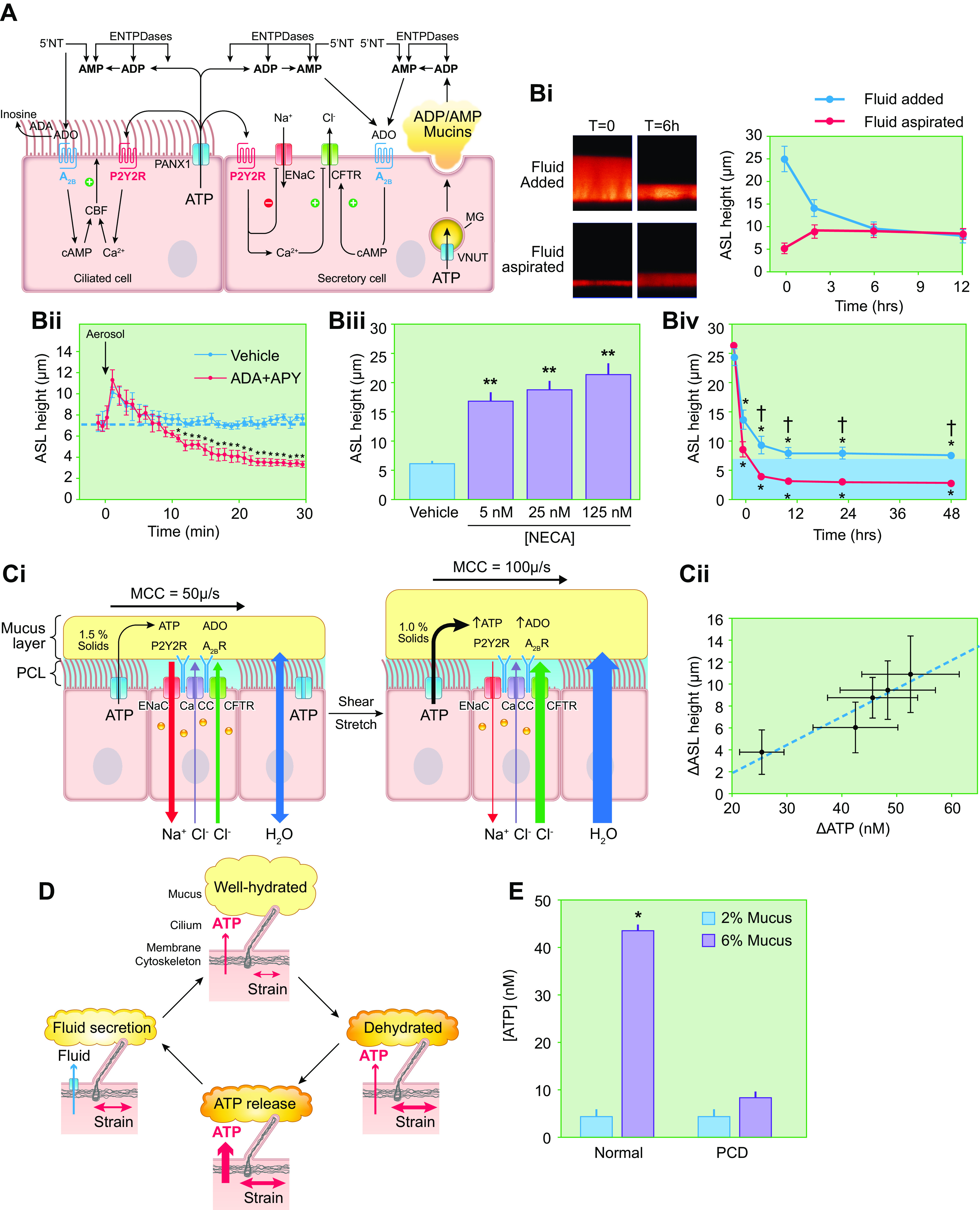
Airway surface liquid volume regulation. A: diagram of purinergic signaling pathways that coordinately control directions/rates of transepithelial ion and fluid transport and ciliary beat frequency (CBF) by the two major lumen-facing epithelial cell types, i.e., ciliated and secretory cells. The pannexin 1 (PANX1) ATP-releasing apical membrane hemichannel, cell surface/shed extracellular enzymes that metabolize ATP, ADP, AMP, and adenosine (ADO) (ENTPDases, 5′-NT), apical purinoceptors (nucleotide, P2Y2R; nucleoside, A2B), and regulated CFTR, ENaC, and Ca2+-activated (CaCC) ion channels shown. Purinoceptors and ecto-enzyme nucleotide/nucleoside metabolic enzymes are depicted as commonly expressed in both ciliated and secretory cells. Pannexin-1 (PANX-1)-mediated ATP release from ciliated cells consequent to cilia-mucus sensing (see FIGURE 6D) regulates cilial beat frequency in an autocrine fashion and regulates ion transport in secretory cells in a paracrine fashion. The extracellular metabolite of ATP, i.e., adenosine, also regulates CBF and ion transport in autocrine and paracrine fashions, respectively. In secretory cells, ATP is imported into mucus granules (MG) via the vesicular nucleotide transporter (VNUT) and metabolized to ADP and AMP. These nucleotides are co-released with mucins and, via metabolism to adenosine, trigger autocrine ion/fluid secretion to hydrate the newly secreted mucins. Note, although not depicted for simplicity, P2Y2R is expressed on secretory cells and when activated triggers mucin secretion. The absence of co-release of ATP with secreted mucins protects from excessive, autocrine-mediated ATP mucin secretion. Also not depicted for simplicity, ADP/AMP release from secretory cells likely also provides adenosine for regulation of ciliary beat frequency in adjacent ciliated cells. Thus nucleotide release exhibits critical autocrine and paracrine signaling properties for coordination of efficient mucus transport. B: control of airway surface liquid volume/surface area (height) by purinergic signaling as imaged by X-Z confocal microscopy. Bi: addition of fluorescent dextran-containing solution to cultured human airway surface without (top) or with (bottom) subsequent aspiration at time (t) 0 is followed by autoregulation over 4–6 h to a common height that is governed by rates of ATP release and conversion to ADO (courtesy of Dr. Robert Tarran, University of North Carolina-Chapel Hill). N = 8. Bii: basal ATP release and conversion to ADO governs Na+ vs. Cl− transport rates to generate basal ASL heights of ∼7 µm. Addition of enzymes to metabolize extracellular ATP (apyrase, APY) and adenosine (adenosine deaminase, ADA) abolishes the ability of the human bronchial epithelial (HBE) culture to maintain a 7-µm ASL height (blue dashed line). The default Na+ absorptive pathway removes all available ASL (216). Biii: dependence of ASL on concentration of added nonmetabolizable adenosine analog 5′-N-ethylcarboxamidoadenosine (NECA). Higher NECA concentrations stimulate higher fluid secretion rates and higher steady-state ASL height (216). Biv: CF vs. normal ASL homeostasis under static conditions. In contrast to normal cultures, CF cultures cannot maintain adequate airway surface volumes (height) under static conditions. Levels of ASL adenosine and A2B expression are similar in CF and normal HBE cultures (152). The failure of the mutant CFTR to secrete Cl−/fluid and moderate ENaC-mediated Na+ absorption drives ASL depletion in CF. C: control of airway surface hydration during tidal breathing. Ci: under static conditions, ATP release rates are fixed at ∼400 fmol/cm2/min and generate an ∼7-µm-deep ASL layer height. The mechanical shear and transmural stretch imparted onto airway epithelia during normal breathing increase ATP release rates via PANX-1 hemichannels. As a result, ASL ATP levels increase, which inhibits Na+ absorption, increases (via metabolism-generated ADO) Cl−/fluid secretion, and increases ASL volume/height. Fluid secretion dilutes mucus concentrations (depicted as % organic solids). Cii: relationship between the magnitude of changes in luminal ATP concentrations and ASL height. Human airway cultures were subjected to varying degrees of oscillatory stress during measurements of luminal [ATP] and steady-state ASL heights. These data revealed a direct relationship between [ATP] and changes in ASL height (correlation coefficient = 0.95 and slope = 0.3 mm height per steady-state change in ATP concentration) (216). D: airway epithelial autoregulation of proper surface hydration states, i.e., 97.5% water, 1.5% organic solids, 1% salt mucus, is mediated by mechanotransduction sensing by motile cilia. Motile cilia interact with mucus layer and transfer momentum to the layer. The yield stress of the mucus layer in response to ciliary beat is a function of mucus concentration. If mucus becomes hyperconcentrated/dehydrated, mucus layer yield stress increases (right). The decreased deformability (higher yield stress) of the mucus layer increases the strain on the ciliary shaft during ciliary beating, which promotes PANX-1-mediated increases in ATP release (bottom). Increased ATP release increases local ATP concentrations, increases fluid secretion (left), and rehydrates mucus to favorable homeostatic states in an autoregulatory feedback loop (top) (216). E: requirement for motile cilia in autoregulation of mucus hydration. Cilia from normal subjects respond to hyperconcentrated (6%) mucus with an increase in ATP release and a higher steady-state concentration of ATP within ASL. Primary ciliary dyskinesia (PCD) cultures, with absent cilial motility, fail to sense mucus hyperconcentration and accelerate ATP release/increase ATP concentrations within ASL. See glossary for other abbreviations. Content from Ref. 216 used with permission from Science Signaling.
3.3.1. Static in vitro conditions.
The control of ASL volume under static in vitro cell culture conditions has received much attention because of the simplicity of this condition for experimental manipulation (FIGURE 6, A and B). Functionally, this state may relate in vivo to an absence of ventilation of airways due to physical obstruction. Studies of static cultures revealed that airway epithelia, in the absence of ASL regulatory signals, exhibit Na+/fluid absorption as the default mode (152, 153). Experiments in which ion transport regulatory molecules in ASL were diluted by PBS boluses, or blocked by pharmacological agents, provided evidence for Na+ absorption as the default pathway (90, 152, 153) (FIGURE 6, Bi and Bii). In these experiments, ASL depletion by unregulated Na+ absorption occurred within 10–15 min of initiation of pharmacological block maneuvers, reflecting a basal ASL volume of ∼1 µL/cm2 (corresponding to 10 µm height) juxtaposed to active absorption rates in the unrestrained/default mode of ∼5 µL/cm2/h (or 0.8 µm/min) (93) (FIGURE 6Bii). It is possible that this default absorptive property is designed to keep airway/lumens patent for airflow when airway lumens are flooded in vivo and regulatory molecules diluted, e.g., during drowning episodes. However, unrestricted volume absorption under normal conditions would inappropriately dehydrate airway surfaces. Thus, a regulatory system is required to provide a brake on Na+/fluid absorption and stimulate sufficient Cl− (fluid) secretion to generate ASL fluid volumes suitable for steady-state mucus transport (see below; Ref. 152).
It is likely that apical membrane purinoceptors, activated by ATP [P2Y2 receptors (P2Y2Rs)] and adenosine (A2B), dominate this regulatory role (217) (FIGURE 6A). As required for ion transport synchronization, nucleotide/nucleoside receptors not only exhibit the capacity to inhibit ENaC but also activate Cl− channels/secretion (194, 210, 212). The concentrations of purinoceptor ligands, i.e., ATP and adenosine (ADO), are tightly controlled by complex NT release, extracellular metabolism, and nucleoside reuptake systems (214, 215, 217, 218). ATP is released from airway epithelia onto airway surfaces via pannexin-I hemichannels, vesicular pathways, e.g., the vesicular nucleotide uptake transporter (SLC17A9), and perhaps other pathways at a basal rate of ∼400 fmol/cm2/min under static conditions (17, 219–226) (FIGURE 6A). ATP released at this rate is rapidly metabolized by a complex of cell surface and secreted ecto-ATPases to produce basal ATP levels of ∼2 nM, too low to activate P2Y2Rs (152, 212, 215). However, ecto-metabolism of ATP generates ADO concentrations of ∼200 nM, sufficient to activate A2B receptors. Note that little if any ADO is directly released from airway epithelia, almost all being formed extracellularly via metabolism of ATP. Finally, nucleoside transport systems return ADO and its adenosine deaminase (ADA) metabolites, inosine and hypoxanthine, back into epithelial cells (227).
Importantly, there are also brakes on the nucleotide-regulated Cl− secretory system to prevent excessive Cl−/fluid secretion that may flood airway lumens. One brake is desensitization of the P2Y2R in response to the high ATP levels on pulmonary surfaces that may occur during periods of high mechanical forces (e.g., high pressure ventilation) or cell death (228–232).
Confocal microscopy measurements have established the ASL volume homeostatic roles of nucleotide signaling. Normal human airway culture surfaces are not volume depleted, i.e., are not dry, under basal static conditions (182, 183, 233, 234) (FIGURE 6B). They typically exhibit ∼7- to 10-µm-deep ASL layers, i.e., a volume per unit area of ∼1 µL/cm2. Remarkably, ASL height/volume will return to a physiological 7- to 10-µm height (1 µL/cm2 volume per unit area) after either bolus PBS addition or physical removal of ASL fluid, a steady-state volume governed by the adenosine concentrations produced by epithelial ATP release and ecto-enzyme conversion of ATP to adenosine (FIGURE 6Bi). The role of adenosine in static ASL volume homeostatic responses was revealed by the 1) reduction of steady-state ASL volumes in response to accelerated biochemical metabolism of ADO by exogenously added apyrase/adenosine deaminase (FIGURE 6Bii) or administration of pharmacological A2B receptor blockers (152) and 2) dose-effect relationships between ASL concentrations of an adenosine agonist [the metabolically inert adenosine analog 5′-N-ethylcarboxamidoadenosine (NECA)] and ASL height (152) (FIGURE 6Biii).
In contrast, study of CF airway cultures under static conditions revealed volume depletion (<3-µm layer thickness corresponding to 0.3 µL/cm2) of airway surfaces (FIGURE 6Biv). Extracellular adenosine concentrations or A2B receptor coupling to adenylate cycle were not different in CF versus normal cultures (152). Accordingly, these observations defined the role of functional CFTR as a downstream effector for ADO-regulated ASL volume homeostasis. The static CF culture system provided an important screening platform for the search for CFTR modulators (153, 233, 235, 236).
3.3.2. Phasic motion.
In vivo, the lung phasically exerts 1) stretch on airway walls/epithelia as airways dilate and constrict during breathing and 2) shear on airway surfaces due to phasic airflow (FIGURE 6Ci). Superimposed on basal ATP release rates, a variable ATP release component, governed in part by breathing-associated mechanical forces on airway surfaces, increases ATP release rates to generate the increased extracellular ATP and ADO concentration levels required for optimal mucus hydration during respiration. For example, under conditions that mimic tidal breathing, airway epithelial ATP release rates rise to levels that produce ATP concentrations of ∼30 nM and ∼300 nM ADO in ASL (234, 237). The increased levels of ATP/ADO promote an approximate doubling of ASL volume (and height), providing airway surfaces with a reserve of fluid to react to the diverse environmental conditions encountered during tidal ventilation (FIGURE 6Cii). A further increase in airflow-induced ATP release during exercise likely maintains airway surface hydration during periods of high pulmonary ventilation with increased micro-aerosol and increased evaporative water loss (238) (FIGURE 6Cii).
3.3.3. Local control of ASL hydration at the single-cell level.
Control of airway surface hydration (volume) is required to maintain a well-hydrated mucus layer and PCL-G atop an individual cell. Importantly, “local” control requires regulation of the volume/height of ASL by individual cells in the context of the mucus/volume load imposed on a cell by mucus funneling up from distal to proximal airway surfaces. Local airway epithelial sensing and transport properties are configured to deal with this load.
A key mechanism that mediates local control of ASL volume/hydration involves cilia-mucus concentration-dependent interactions (FIGURE 6D). As part of this process, motile cilia sense the concentration of mucus and adjust fluid secretion to maintain the overlying mucus layer at optimal concentrations for clearance via mechanisms similar in concept to glomerulotubular balance (239). Specifically, the shear-stress responses of the mucus layer to deformation by cilial beating vary monotonically with increasing mucus concentration (see below). The concentration-dependent shear stress characteristics of the mucus layer impose graded shear stress-dependent strain on beating cilial shafts, and hence ciliated cells, that regulates rates of ciliated cell ATP release via PANX-1 ATP channels (216, 240). Thus, beating cilia exhibit the capacity to sense mucus concentration and respond if needed with changes in ATP release rates, which adjust rates of local fluid absorption versus secretion to maintain a locally well-hydrated mucus (FIGURE 6D). The requirement for active cilia beating in this regulatory feedback mechanism was highlighted by the failure of dysmotile cilia on cultures from primary ciliary dyskinesia (PCD) subjects to sense increased mucus concentrations and accelerate ATP release to stimulate appropriate fluid secretion (FIGURE 6E and see sect. 11).
3.4. Synchronization of Superficial Epithelial Mucin and Fluid Secretory Rates
Airways have also evolved mechanisms to synchronize the rates of mucin and active ion/fluid transport. Although it now appears likely that both the ion transport/hydration and mucus secretory functions reside in the same cell type, i.e., CC10+ club cells (“secretory/mucus” cells), much of the known coupling of mucin and fluid secretion rates resides in the extracellular compartment (FIGURE 6A). Mucins are released from airway epithelial mucus granules via an extracellular Na+/intravesicular Ca2+ exchange mechanism that osmotically expels/“explodes” mucins from granules into the airway lumen (241–243). No evidence has been generated in airway epithelia to suggest that a proteolytic process is required to release mucins onto airway lumens as suggested for the intestine (244, 245). Ca2+, but likely little monovalent ions or water, is cosecreted with the mucin granule content. However, mucin granules corelease ADP and AMP as autocrine and paracrine regulators of liquid secretion (246) (FIGURE 6A). As described above, the release of ADP/AMP, via ecto-metabolism to ADO and activation of A2B receptors, inhibits Na+ absorption and accelerates anion/fluid secretion (214). Note that relatively little ATP is coreleased to avoid autocrine/paracrine mucin granule release. The net effect is to coordinate the rates of mucin and active ion transport to maintain the proper hydration balance of mucus in health.
Quantitative aspects of the physiology of mucin-fluid secretion synchronization are worthy of analysis. For example, how much fluid is required to hydrate newly secreted mucins on airway surfaces, and is paracrine/autocrine nucleotide/nucleoside-stimulated active ion/fluid secretion sufficient? For heuristic purposes, consider a small airway with a diameter of 2 mm and length of 5 mm (surface area ∼ 0.3 cm2) populated by goblet cells (8-µm2 apical surface area) containing 100 spherical granules each with an ∼1-µm diameter. As reported experimentally, each 1-µm-diameter granule (5 × 10−10 µL volume) requires sufficient available airway surface volume (water) to permit mucus granule content expansion by a factor of ∼300× upon secretion onto an airway surface (241–243). In health, ∼1% of the surface cells lining this small airway may be goblet cells, i.e., ∼3 × 104 goblet cells (2). If each goblet cell is stimulated to release all 100 granules within an hour, ∼0.58 µL of ambient ASL is required to hydrate newly secreted mucins. Active ion transport stimulated by adenosine can generate liquid secretion rates of ∼2.5 µL/cm2/h, so this process could within 40–60 min provide ∼0.8 µL of fluid (2.5 µL/cm2 × ∼0.3 cm2) (93). Thus, under conditions of health, the coupling of hydration to mucin secretory rates by ADO-mediated ion transport regulation likely is adequate for normal mucus hydration/homeostasis. However, in diseased airways, with for example 100% goblet cells (3 × 106 cells/airway), then ∼40–60 µL of volume is required on the surface to hydrate all newly secreted mucins. This volume exceeds that provided by active ion transport, rendering newly secreted mucus either hyperconcentrated, partially hydrated by mucus-driven osmotic transport to the lumen (see below), or both. Note, if mucus becomes hyperconcentrated and local airway mucus clearance fails, the 58 µL/h mucus secreted into the airway lumen (vol ∼15.7 µL) would produce rapid luminal filling and airways muco-obstruction as observed in disease.
3.5. Submucosal Gland Contributions to Proximal Airway Fluids
The role of the SMGs in controlling the volume and composition of airway surface liquids has been reviewed recently in Physiological Reviews (20) and is only briefly summarized here. SMGs appear to segregate mucin and fluid secretion into different cell types, i.e., mucus and serous cells, respectively. MUC5B is the exclusive mucin secreted by SMG in health and dominates in disease (26). The SMG MUC5B molecule may have significant differences in glycosylation and sulfation compared with superficial epithelial MUC5B. Approximately 50% of the SMG MUC5B after cholinergic stimulation is organized into bundles with diameters that are approximately the diameter of distal SMG ducts (∼15 µm), and these bundles do not swell or dissolve into PBS. Whether the bundle/strand feature of SMG mucus reflects differences in the SMG MUC5B mucin per se (e.g., more hydrophobic), or addition of novel proteins in SMGs to promote mucin bundle formation (e.g., proline-rich protein 4, PRR4), is not yet known (247, 248).
SMG mucus may have several features that adapt it well to augment protection of proximal airways. First, as discussed below, effective cough clearance requires accumulation of relatively large masses of mucus on proximal airway surfaces (∼5–10 mm high) (116). Secretion of bundles/strands is predicted to not only “sweep” airway surfaces clear of large particles but also promote the rapid accumulation of the large mucus masses in proximal airways required for effective cough clearance. Second, it appears that the SMG ducts produce a hypotonic SMG secretion (249). A hypotonic secretion may increase of the activity of salt-inhibitable antimicrobial peptides that would protect duct lumens from infection (250–253). Secretion of a hypotonic SMG mucus also allows for rapid concentration of secreted SMG mucus on airway surfaces, via transepithelial absorption of water down SMG mucus-interstitial osmotic gradients, to rapidly augment barrier function. Finally, it is possible that secretion of a MUC5B-only SMG mucin may buffer the putative adhesive qualities of superficial epithelial MUC5AC (115) and improve clearance.
Note that secretion of hypotonic mucus requires extraction of salt but not water from proximal SMG ductal regions. An absence of aquaporins, coupled to an ionocyte-like Cl− (and Na+)-selective absorption function, could accomplish this physiology in gland ducts (156). As noted above, ionocytes are reported to be most highly expressed in SMG ducts in mammalian airways (155, 159, 254).
4. INTEGRATED ION TRANSPORT/MUCUS BIOPHYSICAL MECHANISMS THAT REGULATE MUCUS CLEARANCE RATES IN HEALTH AND DISEASE
An understanding of mucus clearance requires integration of the epithelial ion/fluid transport mechanisms that control mucus concentration with the mucus concentration-dependent biophysical properties that govern mucus clearance rates. These analyses in particular are required to describe the relationships between 1) normal ion transport, mucus concentrations, and mucus transport in health and 2) relationships between abnormal ion transport, mucin secretion, mucus hyperconcentration, and mucus accumulation/plugging in disease.
4.1. Integrated Control of Mucus Transport by Active Ion Transport and Mucin/Globular Protein Osmotic Forces in Health and Disease
It is necessary to consider the sum of the forces generated by active ion transport and the mucin/globular protein-derived osmotic forces that govern airway surface hydration/mucus concentration to understand mucus transport in health and disease. In this section, we consider the osmotic forces exerted across the apical epithelial barrier (semipermeable barrier 1). In sect. 4.2, we consider the forces exerted across the juxtaposed mucus layer and PCL-G interface (FIGURE 7A, semipermeable barrier 2).
FIGURE 7.
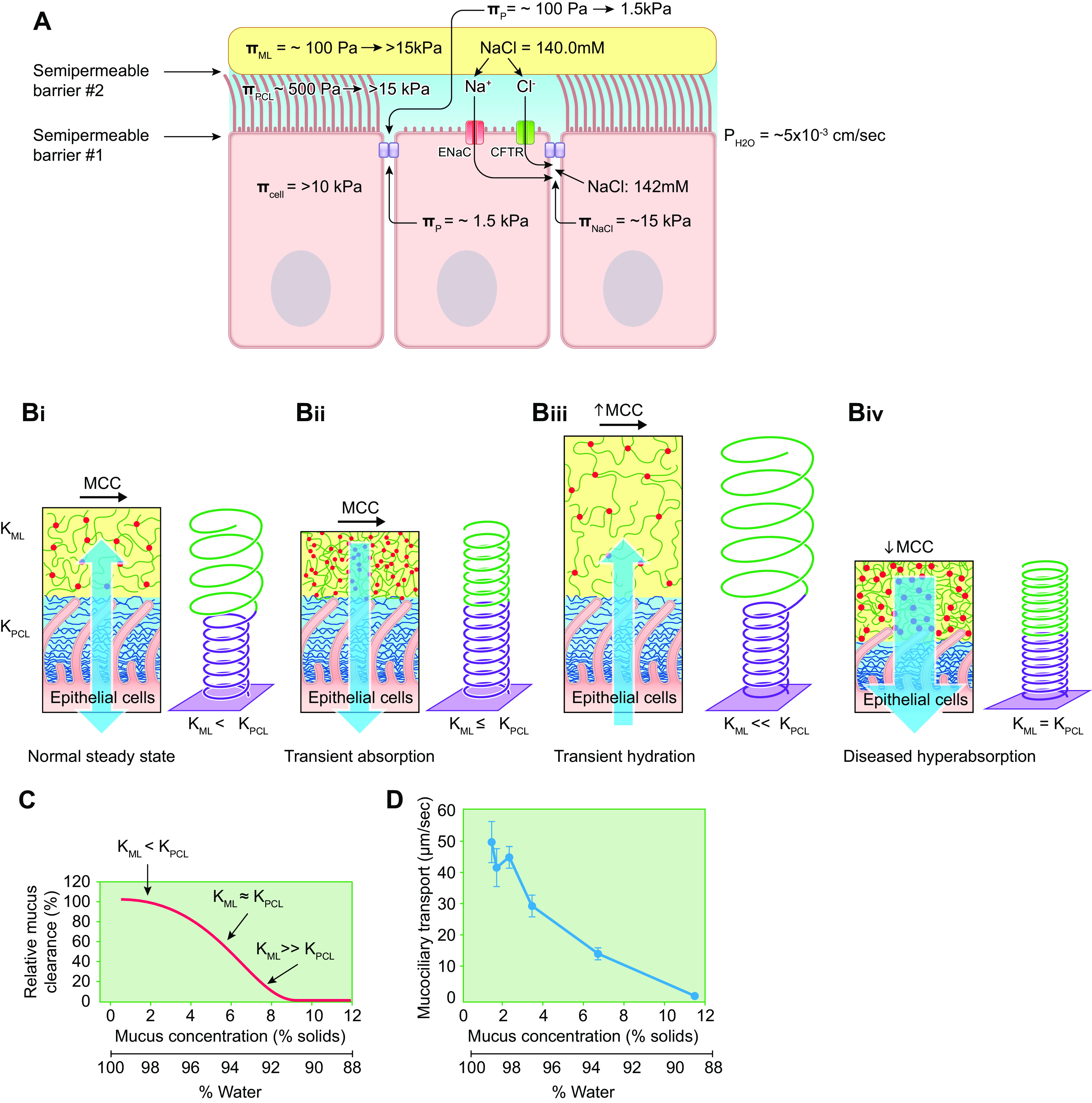
Integration of osmotic driving forces at the PCL-apical airway epithelial barrier, within airway surface liquid compartments, and relationships to mucus transport rates. A: schema depicting osmotic pressures/moduli related to mucus hydration. Note that because the osmotic forces of the cell (255), lateral space globular proteins (256, 257), and lateral space NaCl gradients (258) have been measured as osmotic pressures, for simplicity, all forces in this figure are depicted as pressures rather than moduli. Semipermeable barrier 1 depicts the airway apical epithelial barrier consisting of the apical cell membranes and tight junctions. Semipermeable barrier 2 describes the mucus layer-PCL interface. Three transapical epithelial osmotic forces govern water flow across semipermeable barrier 1, which has a high water permeability (). First, there are the osmotic pressures of the PCL and the cell. The cell osmotic pressure (πcell ∼10 kPa) is dominated by the cell cytoskeleton and not cytoplasm (30 Pa) (255). The PCL osmotic pressure (πPCL) is ∼250–500 Pa in basal, i.e., healthy, conditions. Second, there are the osmotic pressures generated by the small globular proteins (πp) secreted into airway surface liquid (e.g., CCSP) and proteins (e.g., albumin) passively permeating into ASL from the interstitial fluid bathing the basolateral aspects of the cell. The osmotic pressure of the globular protein component of the mucus layer has been measured at ∼40–100 Pa in health (FIGURES 3D and 12C). Because the PCL does not restrict permeation of globular proteins, the osmotic pressure of the globular proteins in the PCL is equivalent to the osmotic pressure of globular proteins measured in the mucus layer (see FIGURE 4). The globular protein osmotic pressure in the lateral space between airway epithelial cells is estimated at ∼ 60% of that of plasma proteins, i.e., ∼1.5 kPa, based on studies of lung lymph protein concentrations (256, 257). Third, active ion absorption generates small, i.e., ∼2 mM, NaCl gradients between the apical solution and the epithelial lateral space (258). The osmotic pressures, however, generated by the small ion gradients produced by active ion transport are large, i.e., ∼15 kPa. Note, ranges of πmucus, πPCL, and πp are depicted for basal conditions vs. severe airway surface liquid depletion, i.e., dehydration (values to right of arrows). Because the ion concentrations are isotonic under both basal and liquid (water)-depleted conditions, no range for extracellular NaCl concentration is depicted. B: relationships between net direction/magnitude of epithelial ion transport, the relative osmotic moduli of the mucus layer and PCL-G, and mucus transport rates. The relative water-drawing powers (osmotic moduli) of the mucus layer (KML) and PCL (KPCL) interfaced to epithelial cell-mediated fluid absorption or secretion (blue arrows) are depicted. The length of the Hookean springs (to right of the cell models) denotes the height of the PCL (purple) or mucus layer (green), and the spring diameter is inversely proportional to osmotic modulus. Bi: in the normal state, KML is lower than KPCL, Na+ absorption and Cl− secretion balanced, and MCC proceeds at physiologically adaptive rates. The Hookean spring model depicts this state with a green spring (KML) with a diameter larger than the purple spring (KPCL). Bii: transient period of fluid absorption (blue arrow) from apical surface by transepithelial Na+ transport. Modest amounts of fluid are removed from the compartment with the lower-osmotic modulus, i.e., the mucus layer. The KML remains lower than KPCL, so MCC is maintained at effective rates. Biii: transient period of fluid secretion mediated by transepithelial Cl− secretion (blue arrow). The PCL has the higher osmotic modulus, but its volume is fixed by the tethered mucins. Accordingly, the newly secreted fluid enters the lower-modulus, but expandable, mucus layer. The newly added hydration of the mucus layer is predicted to have small effects to increase MCC. In the context of both transient fluid absorption and fluid secretion, the mucus layer acts as a buffer/reservoir to store fluid on airway surfaces in a manner that preserves MCC. Biv: in a dehydrated state, persistent/abnormal absorption initially removes fluid from the lower-osmotic modulus mucus layer, but as KML = KPCL, fluid is removed coordinately from both the mucus layer and the PCL. The osmotic moduli of both layers are increased and equalized (smaller spring diameters) and volume depleted (springs shortened). This state osmotically compresses the cilia and produces mucus stasis. C: schema depicting relationships predicted by the gel-on-brush model between mucus percent solids, osmotic moduli of the mucus layer (KML), the periciliary layer (PCL) in the basal state (KPCL), and mucus clearance rates. Note, because the published data depicted in D were presented as total % solids, the values on the horizontal axis of FIGURE 7, C AND D are shown as total % solids. At normal mucus hydration (∼1.5% total solids), the osmotic modulus of the mucus layer is below basal PCL values (KPCL), the PCL is fully hydrated, and mucus transport is efficient. At modest levels of mucus dehydration (i.e., % total solids ∼3–4%) the osmotic modulus of the mucus layer slightly exceeds the basal PCL value, modest compression of the PCL results, and mucus transport slows. When mucus dehydration is severe (i.e., % total solids ∼7–8%), the mucus layer osmotically compresses and/or traps the cilia, producing mucus stasis and adhesion. D: relationships between mucus concentration and mucociliary transport measured in vitro in cultured human airway epithelia. Data are presented as means ± SE; n = 4–6 per group (14). See glossary for other abbreviations. A was created with BioRender.com, with permission. Bi, Biv, and C use content from Ref. 25, with permission from New England Journal of Medicine. C and D use content from Ref. 14, with permission from American Journal of Respiratory and Critical Care Medicine.
Three osmotic pressures/moduli are exerted across the apical surface of the epithelia lining airway surfaces (FIGURE 7A, semipermeable barrier 1). As noted in FIGURE 7A, the reports of the osmotic properties of the cell and plasma as pressures have directed the use of this parameter for this analysis. The first force is the transepithelial osmotic pressure generated by the high-MW mucin polymers at the barrier. Thus, the osmotic pressure of the PCL-G (ΠPCL-G) is opposed by the osmotic pressure of the cell (Πcell, which is a function of the cytoskeleton and free polymers in the cytoplasm) and the negligible soluble polymers in the interstitial space. As discussed in detail in sect. 4.2, the osmotic pressure of the PCL-G varies with surface hydration status. The osmotic pressure of the cell, largely a function of the actin cytoskeletal “shell,” has been estimated to be ∼5–10 kPa (255). The osmotic pressure of the polymers/large proteins in the cell cytoplasm in the absence of cytoskeletal components may be as low as 30 Pa (255). Like the PCL-G, the volume of the cell is constrained, in this case via the cytoskeleton, so there is likely asymmetry in its osmotic water-drawing properties, i.e., the cell may be able to donate water/shrink but exhibit limited capacity to absorb water/swell.
The second osmotic force reflects the concentration gradient for “small” (relative to mucins) globular proteins (cp). The osmotic pressure of these molecules has been measured in normal mucus with 10-kDa MW cutoff semipermeable membranes and is ∼40–100 Pa (8) (FIGURE 4D). Note that we estimate the osmotic pressure of the small globular proteins in the PCL-G based on measures of mucus layer protein/ion concentrations, reflecting the notion that small proteins are freely diffusible across the mucus-PCL-G interface (semipermeable barrier 2). The protein concentration of interstitial/lateral cell spaces in the lung has been estimated to be ∼60% of plasma proteins, suggesting that the osmotic pressure of plasma proteins in the lateral cell space is ∼1.5 kPa (256, 257).
The third osmotic gradient under basal conditions is generated by active Na+ transport. Given a fluid absorption rate of ∼5–7 µL/cm2 epithelial surface/h, a NaCl gradient of ∼2 mM NaCl from lumen to lateral epithelial space is predicted (258). Importantly, this gradient is predicted to generate a transepithelial ΠNaCl of ∼15 kPa. Note that all of these pressures are exerted across a semipermeable apical epithelial surface with a very high water permeability.
In health, the balance of osmotic pressures between the PCL-G mucin component (0.5 kPa) and the cell (>10 kPa) and the gradient of globular proteins freely soluble in the PCL (∼100 Pa) versus those in the lateral space (∼1.5 kPa) predict passive osmotic driving forces favoring fluid absorption. The NaCl gradient from the PCL-G to the intraepithelial lateral spaces generated by active ion transport is small in terms of ion concentration, e.g., 140 mM mucus layer versus 142 mM lateral space, but produces ∼15 kPa osmotic pressure favoring fluid absorption (258). Thus, all the passive transepithelial osmotic forces under basal conditions are aligned for fluid absorption, a state optimal perhaps for handling the volume loads imposed by distal to proximal MCC/fluid movement and/or maximizing the likelihood that airway lumens are fluid free to facilitate airflow. This analysis also emphasizes the critical role for transepithelial Cl− secretion to maintain airway surface hydration, i.e., shifting the sign of the 15-kPa ion transport osmotic force upon conversion from Na+ absorption to Cl− secretion will generate the osmotic forces required for fluid secretion.
4.2. Ion Transport/Mucus Concentration-Dependent Control of MCC Rates
A major advance afforded by the gel-on-brush hypothesis is that it has led to quantitative predictions between airway surface liquid status (hydration), mucus concentration, and mucus transport rates (FIGURE 7, B and C) (8). These predictions rest on analyses of the osmotic properties of the surface gel-on-brush layers (separated by semipermeable barrier 2) in series with the underlying ion/fluid transport function of airway epithelia. As noted above, the osmotic properties of the mucus layer and PCL are best described for these analyses in terms of the osmotic modulus of the two layers. Also note that the original publications of gravimetric mucus concentration reported dry weight values as the sum of the organic content plus salt content, i.e., total percent solids content (14, 76). The percent organic solids content nomenclature better describes the solids material containing the osmotically active molecules and eliminates a constant value of isotonic salt (0.9%) that obscures relationships particularly at lower mucus concentrations (TABLE 1).
In the normal state (1.5% organic solids, 1% salt, 97.5% water), the osmotic forces generated by the absorptive versus secretory ion transport rates are balanced, the mucus layer is well hydrated and contains sufficient fluid to act as a volume reservoir/buffer, and efficient mucus transport results (FIGURE 7Bi). If the epithelium transiently tips toward absorption of Na+/fluid from the surface, water is preferentially absorbed from the gel layer with the lower-osmotic modulus, i.e., the mucus layer, with little functional consequence on MCC (FIGURE 7Bii). Similarly, if the epithelium transiently tips toward Cl−/liquid secretion onto the airway surface, liquid preferentially moves into the mucus layer, again with little effect on MCC (FIGURE 7Biii). Note that the PCL is tethered to the cell surface and hence physically restrained and fully hydrated in the normal state, so it cannot accept newly secreted liquid, which is accommodated by the lower-osmotic modulus but expandable mucus layer. However, with disease-induced, persistent net epithelial Na+/liquid hyperabsorption, the osmotic modulus of the mucus layer rises rapidly as the modulus becomes dominated by the mucin component of mucus (see FIGURE 4D). Ultimately in this state, the modulus of the two layers becomes equal and water is absorbed coordinately from both layers with compression of cilia and slowing of MCC (FIGURE 7Biv). This state occurs when the mucus layer osmotic modulus rises to the value of the PCL, i.e., ∼500 Pa, 4–6% organic solids. With severe and unrelenting fluid absorption, mucus concentrations rise to 8–10% organic solids (>1,000 Pa osmotic modulus), osmotically flattening cilia and abolishing MCC. Thus, the gel-on-brush formulation 1) accurately predicts mucus transport rates in vitro based on mucus concentrations (FIGURE 7, C and D) and 2) explains why relatively small changes in hydration status (98% vs. 92% water, i.e., 1% vs. 8–10% organic solids) produce disease (FIGURE 7, C and D).
4.3. Mucus Concentrations in Disease
Three consequences of mucus hyperconcentration, failed mucus transport, and intraluminal mucus accumulation with respect to the interactions between the high osmotic forces generated by adherent hyperconcentrated mucus and active ion transport are worthy of note.
4.3.1. Failure of granular mucin secretion in disease.
As noted above, mucins are extruded from goblet cell granules onto airway surfaces by a Na+ for Ca2+ exchange-mediated osmotic expulsion mechanism (241–243). However, the magnitudes of the osmotic forces generated by mucin extrusion are unknown. This is an interesting question not only for basal physiology but also with reference to disease. For example, it is a common histological finding in muco-obstructive diseases that mucins are only partially extruded from goblet cells into airways filled with thickened, adherent mucus (259). It therefore is possible that the osmotic forces generated by initial granule expansion are lower than the forces generated by adherent, hyperconcentrated diseased mucus (K ≥ 1–5 kPa).
4.3.2. Maximal mucus concentrations in disease.
In disease, absolute mucus concentrations approach 15–20% solids in CF lungs and approach similar plateau levels of concentration (but not volume/height) over time in airway cultures in vitro (76) (FIGURE 8, Ai and Aii). These findings suggest that mucus layers, reflecting power law-dependent increases in osmotic moduli in regimes of >15% (150 mg/mL) organic solids (see Eq. 4 and FIGURE 4D), exhibit osmotic moduli (>15 kPa) sufficient to offset the osmotic forces generated by the active Na+ absorption (FIGURE 7A) and, consequently, limit the magnitude of mucus concentration on airway surfaces. Experimentally, this prediction was tested by exposing airway cultures to fluid challenges containing either blockers of active Na+ transport or impermeable dextrans at concentrations with osmotic moduli of 4.5 kPa or 17.8 kPa. As expected, Na+ transport blockers inhibited fluid absorption (FIGURE 8B). Strikingly, as predicted (see FIGURE 7A), dextrans at concentrations generating ∼17.8-kPa osmotic moduli also inhibited fluid absorption (FIGURE 8C). Thus, the balance between osmotic forces favoring fluid absorption (Na+ transport) versus fluid secretion (mucus osmotic modulus) produced the plateau of mucus concentration (FIGURE 8A).
FIGURE 8.

Relationships between osmotic forces generated by secreted polymeric mucins, active ion transport, and ASL height. A: measures of airway epithelial mucus concentration and height with time. Total % organic solids mucus concentration (Ai) and ASL height (Aii) measured over time in airway epithelial cultures allowed to secrete mucus without luminal washing. Note that mucus concentration reaches a quasi-steady-state plateau, whereas mucus height (mass) continues to increase with time. n = 7 (Ai); n = 4 (Aii). B: X-Z confocal ASL height measurements of human bronchial epithelial (HBE) culture exposed to hypertonic saline (HS) alone or HS in combination with an ENaC inhibitor (P1037). As predicted, pharmacological inhibition of ENaC slows/abolishes fluid absorption and, hence, reductions in ASL height following HS administration. C: X-Z confocal measurements of ASL height in HBE cultures exposed to PBS alone (control) or solutions containing 3 kDa MW dextrans at concentrations designated to produce ∼4.5 kPa or ∼17.8 kPa osmotic pressures. Strikingly, the osmotic pressure generated by active ion transport favoring fluid absorption (∼15 kPa; see FIGURE 7A) can be offset by administration to the apical surface of an apical membrane impermeable dextran solution with an osmotic pressure of ∼17.8 kPa. These data support the notion that hyperconcentrated mucus can generate osmotic forces sufficient to modulate the effects of active ion transport. n = 3 (8% and 16% dextran); n = 6 (control). See glossary for abbreviations. B uses data from Ref. 263, with permission from European Respiratory Journal.
Note that, as mucus hyperconcentration occurs, globular proteins not bound to mucins in mucus (∼70%) are also concentrated, diminishing the transepithelial Πp gradient between the mucus layer/PCL and the lateral epithelial space that favors fluid absorption (FIGURE 7A). Functionally, both the high mucin and globular protein osmotic pressures in disease could generate sufficient passive osmotic forces to prevent airway surfaces from irretrievable dehydration/desiccation. An unresolved question is whether mucus layer/PCL osmotic pressures can exceed cytoplasm/cell osmotic pressures and produce cell shrinkage.
The increase in mucus height over time in culture (FIGURE 8Aii) suggests that persistent mucin secretion over the experimental period increased the mass of maximally concentrated mucus on airway surfaces. If replicated in vivo, continued mucin secretion is predicted to worsen occlusion of airway lumens with time.
CF lungs exhibit the highest percent solids mucus reported (76). In parallel, CF airway epithelia exhibit the highest active Na+ absorptive rates (260, 261). Experimentally, plateau values for mucus layer percent solids on normal cultures in vitro can be increased by raising Na+ transport rates. For example, treatment of normal airway epithelia with an “artificial” Na+ channel, i.e., nystatin, tripled Na+ transport rates and produced mucus percent solids ≈ 45% total solids content (262). Thus, maximal luminal mucus concentrations are sensitive to Na+ transport-dependent transepithelial osmotic forces, providing a likely explanation for the raised mucus concentrations in CF.
4.3.3. Response of dehydrated vs. hydrated mucus to topical osmotically active agents.
The studies of Goralski et al. measured with confocal microscopy the expansion of ASL height/volume to aerosolized hypertonic saline (HS) in human airway cultures. The authors noted that the rate of volume absorption after cessation for HS delivery was ∼50% slower from airway surfaces covered by hyperconcentrated mucus (12% organic solids) compared with normally hydrated (1% organic solids) mucus. They speculated that the osmotic forces generated by hyperconcentrated mucus layer were sufficient to offset the osmotic forces favoring volume absorption mediated by active transepithelial Na+ transport (263). This speculation is consistent with calculations presented above (FIGURE 7A).
5. INTEGRATION OF CILIAL FORCES AND MUCUS PROPERTIES TO GENERATE MUCUS TRANSPORT
Complex interactions are manifest between the forces generated by motile cilia and the mucus transport apparatus. Interactions between motile cilia, their tethered mucin brush, and secretory mucin-dominated mucus layers/aggregates are reviewed here.
5.1. Cilia Forces
In the lung, cilia beat in a coordinated fashion to propel mucus from the distal airways to the oropharynx (13, 264–267). The coordination of cilia beat occurs both within the ∼100 cilia on a ciliated cell (268) and between cilia on neighboring cells (265), with a metachronal wave propagating retrograde to the direction of mucus flow (FIGURE 9A, adapted from Ref. 269). Human airway cilia beat at frequencies between 10 and 20 Hz (270). The mechanisms that govern basal ciliary beat frequencies (CBFs) are poorly understood but likely include rates of basal ATP release (240, 271). Multiple pathways, including inflammatory mediators and cytokines, can accelerate CBF above basal frequencies, as reviewed in Refs. 272–275. Unlike cilia in other organisms, human airway cilia do not appear to cease beating physiologically, i.e., they maintain some degree of motility even under pathological conditions (276).
FIGURE 9.

Ciliary structure and function. A: components of ciliary beat cycle. Metachronal wave produced by 100 s of cilia beating in a coordinated fashion. The effect stroke (ES, blue arrow) is in the same direction as mucus flow (top black arrow), and the recovery stroke (RS, red arrow) is in the same direction as the propagation of the metachronal wave (bottom black arrow). Image from Ref. 269. B: molecular architecture of cilium showing the 9 outer microtubules (MTs, yellow) arranged around a central pair of MTs (green). Dynein motor proteins connect the outer MTs and generate the force that bends the cilium. C: cross section of cilium showing the spacing of MT (yellow circles) architecture of cilia. Dynein pairs between adjacent outer MTs on one side of the cilium drive the effective stroke (blue arrows), whereas the recovery stroke (red arrows) is driven by dynein pairs on the opposite side of the organelle. The angle column that motor proteins make with the bending direction dictates their contribution to the bending of the cilium. D: cartoon of the side view of the orange rectangle in B showing dynein motor proteins pulling MTs as they “walk” and bend the cilia. E: profile view of the cilia waveform during the effective and recovery strokes. Blue lines are the position of the cilia during the effective stroke, and red lines show the position of the cilia during the recovery stroke. Purple lines are the position of the cilia at the transitions between each stroke. F: displacement of a bead bound to the tip of a cilium during the effective (blue arrow) and recovery (red arrow) strokes along the direction of cilia beat (279). The maximum cilia tip velocity (Vtip) observed during the effective stroke is ∼200 µm/s, whereas the maximum Vtip during the recovery stroke is 150 µm/s. Images adapted from Refs. 279 and 280, with permission from Journal of Theoretical Biology, Biophysical Journal, and Biomedical Optics Express, respectively.
Structurally, cilia are organelles 7 μm in length with an internal architecture consisting of a 9 + 2 microtubular pattern, where 9 outer microtubule (MT) doublets surround a central pair of MTs (277, 278) (FIGURE 9, B and C). Thousands of dynein motor proteins, in pairs, apply force to the outer MTs, bending the MTs (FIGURE 9D) and generating the cilial waveform (FIGURE 9E).
The ciliary waveform, or beat pattern of an individual cilium, is divided into the effective stroke, which drives mucus from distal to proximal airways, and a recovery stroke, which returns the cilium to its starting position. The “effective” cilial stroke is produced when dyneins asymmetrically (i.e., motors on one side of the cilium) exert forces on the outer microtubules, bending the cilium (FIGURE 9, C and E). The effective stroke is characterized by the rapid acceleration in the direction of mucus flow and straightening of the cilium, with the tip reaching a maximum velocity of 200 µm/s within ∼10 ms (279) (FIGURE 9F). At the end of the effective stroke, the tip velocity decreases to 0 as the axoneme reaches its maximal displacement along the direction of mucus flow. This reduction in cilial tip velocity is mediated by disengagement of the dyneins from the MTs (282–284). The “recovery” stroke is initiated by activation of motors on the contralateral side of the cilium that return the cilium to its original position (FIGURE 9, C and E). The maximum velocity of the cilial tip is slower during the recovery stroke (i.e., ∼150 μm/s) (FIGURE 9F).
The force generation by cilia and flagella (which in eukaryotes are longer than cilia but share a similar 9 + 2 MT structure and whiplike beat pattern) has been characterized by several techniques, including optical (285) and magnetic (279) tweezers, atomic force microscopy (286, 287), and deflection of glass needles (288–290). These measurements have also been performed on flagella from multiple organisms (286, 289, 290), Mytilus edulis cilium (288), pulmonary epithelia excised from frogs (287), and human bronchial epithelium cell cultures (279). These studies suggest a wide range of forces generated by cilia, i.e., between 40 and 3,000 pN, with measurements for human airway cilia being reported between 60 and 210 pN (279, 287).
The high degree of variability between previous cilia and flagella force measurements, as well as the myriad of models used to interpret experimental data, points to the need for new measurements of the force generation of cilia that account for the influence of the PCL (discussed below) as well as consider the force distribution along the entire cilia and not just the cilial tip that come into contact with the mucus layer (279).
5.1.1. Cilia-PCL interactions during mucus transport.
Although the shape of the ciliary beat (264, 265, 291), tip motion, and force responses (279, 285–290) have been extensively studied, there are many unanswered questions regarding the transmission of the forces from cilia to structures in the airway surface environment required to generate mucus flow. For example, a question that emerged from the gel-on-brush description of MCC relates to the forces required to compress the PCL during the beat cycle and the possible fates of the water extruded/“pumped out” of a compressed PCL (FIGURE 10A). When cilia are vertically extended, the PCL has a height of 7 μm and the water in the environment is contained within the PCL. At the end of the effective stroke (FIGURE 9E), the cilia bend and the PCL is compressed by ∼3 µm (FIGURE 10A). We can calculate the stress that cilia apply to the PCL by dividing the ciliary force by the area over which the force is applied to the PCL, i.e., stress = force/area. The range of measured forces generated by cilia and flagella are up to 3,000 pN (279, 285–290). Given that there are 5 cilia per µm2, and that a cilial shaft occupies ∼0.2 µm2 of total surface area (FIGURE 10B), a simple estimate of cilial stress generation is 15,000 pN/µm2, or 15 kPa. Our previous work indicated that the osmotic modulus of the PCL compressed from 7 µm to 4 µm increases to 5,000 Pa (8). Thus, analyses of cilia-generated stress indicate that cilia can overcome the osmotic modulus of the PCL.
FIGURE 10.

PCL compression by cilia. A: as cilia beat, they must generate sufficient stress to compress the PCL (black arrow). Since water is incompressible, water is pumped out of the PCL by cilial compression (blue arrow). At the midpoint of the effective stroke (blue), the cilia are extended to their maximum height, and the cilial shaft-tethered mucins barely “touch” each other. In contrast, at the end of the effective stroke (red), the cilia compress the PCL by 3 µm of PCL. B: when cilia are extended from the cell surface (orange rectangle in A), the pore spacing of the PCL is large and all water within 7 μm of the surface is associated with the PCL. C: cilia at the end of the effective stoke (purple rectangle in A) compress one another, resulting in expulsion of water from the PCL. See glossary for abbreviations.
The potential importance of PCL compression by cilia has led to more detailed calculations of the forces required to extrude water from the PCL via ciliary forces. Like estimates of the forces required to compress PCL (based on osmotic moduli), the pressure required to extrude water from the PCL can be calculated and compared to the forces generated by dynein motors during the ciliary beat. The Darcy–Weisbach equation is used to calculate the flow of a Newtonian fluid through a pipe, which approximates the flow of water through the pores formed in a brush or gel mesh, including the PCL (292):
| (8) |
where ΔP is the pressure difference required for water to flow through a pipe, L is the length of the pipe, η is the viscosity of the solution (0.001 Pa·s for water), Q is the volumetric flow rate, and DC is the diameter of the pipe, which is on the order of the pore (mesh) size of the PCL brush, i.e., the correlation length, ξ. We assume that the PCL will be compressed by L = 3 µm. The average mesh pore size of the PCL at compressions ranging between 0 and 3 µm (FIGURE 10, B and C) is DC (30 nm), as determined from dextran size exclusion experiments measuring osmotic compression of the PCL (8) (FIGURE 3). If we assume cilia beat at a frequency (f) of 10 Hz and that PCL compression occurs during the effective stroke of each beat, i.e., on the timescale of a ½ cycle, the volumetric flow rate through a single “pipe” is given by . Thus, the pressure required to pump water out of the PCL is ≈ 10 kPa, indicating that cilia must generate a pressure (>10 kPa) to osmotically compress the PCL and pump water out of the layer. This pressure is consistent with simple estimates of ciliary stress generation described above (15 kPa). Interestingly, the pumping of fluid out of the PCL, and the stress requirements to do so within the context of the Darcy–Weisbach flow calculations described above, may be the physical constant that sets CBF rates at 10 Hz, as well as the magnitude of PCL compression during cilial beating.
So why does the cilium allocate a significant fraction of its generated stress to pumping water out of the PCL? This question has led us to hypothesize that the flow of water out of the PCL during the effective stroke (FIGURE 10A) may be a mechanism to produce mucus flow (56). Two previous reports are consistent with this hypothesis. First, measurements of the flow of both the mucus layer and the aqueous component of the PCL indicated that both components move in tandem (293), supporting the notion of water expulsion from the PCL as a mechanism for coordinately driving mucus transport. Second, if the mucus layer is unable to swell into the space created by ciliary beat/PCL compression (3 µm) on the timescale of ciliary beating (10 Hz), a watery “lake”/gap would form between the compressed cilia and the mucus layer. Such gaps between the cilia and mucus layer were observed by Sanderson and Sleigh with rapid freezing techniques (265). We therefore conclude that water pumping remains a viable mechanism to drive MCC.
5.1.2. Cilia-mucus interactions and mucus transport.
The speculated cilia, PCL compression, and hydraulic mucus layer transport mechanism requires consideration of previous paradigms describing cilia-mucus interactions and mucus transport. Classic notions posited that spikes/claws on cilial tips, based on observations generated in the female reproductive system, interact with the inner surface of mucus to propel mucus flow (294, 295). However, little evidence has been reported to support this notion for airway cilia. In part, the difficulty in describing cilia-mucus interactions reflects the absence of knowledge of the precise structure of the mucus covering airway surfaces, i.e., dilute or semidilute continuous or discontinuous gels (sect. 2.2), and the biochemical/structural characteristics of the ciliary tip (see below).
Recently, the motion of the cilia tip (279) and the flow of mucus near (i.e., parfocal with) the mucus-cilia interface have been studied in cell culture models covered by a continuous mucus layer (281) (FIGURE 11). With the use of particle imaging velocimetry to measure mucus flow, the motion beating cilia imparted to well-hydrated (1% organic solids) mucus was measured as a function of time (FIGURE 11A). From the end of the recovery stroke to the end of the effective stroke, the tip of cilia moved ∼5 µm (FIGURE 11B). We noted that the amplitude of the ciliary beat, i.e., 5 µm, measured by tracking a 2.8-µm bead (279) attached to the cilia tip, was roughly half the tip-to-tip displacement observed by Sanderson and Sleigh, i.e., ∼10 µm (265). We speculate that the difference in the amplitudes may be attributed to artifacts in the rapid freezing process. Recent studies of cilia dynamics have also reported ciliary beat amplitudes of 4–5 µm (291). Importantly, mucus at the mucociliary interface only moved forward ∼3 µm per effective stroke (FIGURE 11B). These data indicate that 60% of the motion of the cilia tip is imparted to the flowing mucus layer during the effective stroke. During the recovery stroke, ∼30% of the motion of the cilia tip was imparted to the mucus layer, as evidenced by mucus recoiling ∼1.5 µm. The recoil of mucus during the recovery stroke resulted in a net transport of mucus during the entire beat cycle of 1.5 µm, which limited the overall effectiveness of MCC to ∼30% of the motion of the cilia tip during the effective stroke. Whether the recoil of mucus during the recovery stroke is due to elastic recovery of mucus or coupling of mucus to cilia during the recovery stroke is unknown.
FIGURE 11.
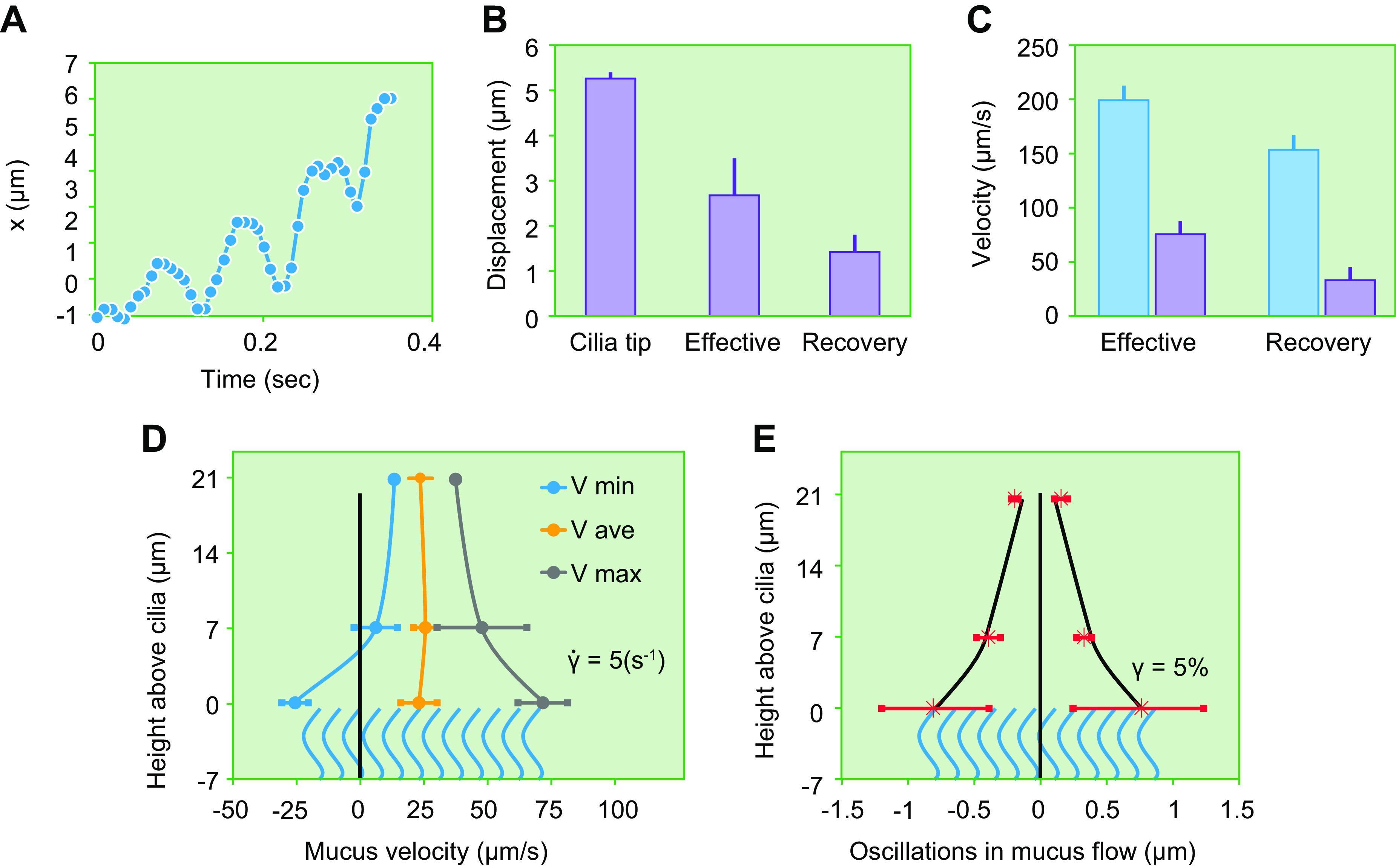
Mucociliary interactions. A: motion of beads embedded in mucus at the mucociliary interface (281). x in µm is linear displacement. B: the average absolute displacement of the beating cilium tip and the motion of the tracer particles embedded in mucus at the mucociliary interface during the effective and recovery strokes. C: maximum velocity of cilium tip during the effective and recovery strokes (blue) (279) and the average absolute velocity of beads in mucus (purple) during the effective and recovery strokes (281). D: strain rates imposed on flowing mucus. Oscillations in mucus transport (bead) velocity as a function of height above the mucus layer-ciliary interface. Blue is the average minimum (retrograde) velocity (Vmin), orange is the overall average velocity (Vave), and gray is the average maximum antegrade velocity (Vmax). These data indicate that near the mucus-cilia interface, the strain rate () is ∼5 s−1 and less than at the mucus-air interface. E: strains imposed on flowing mucus. Cilia induced strains (or the ratio of the displacement over height) in mucus during flow. Strains are calculated as oscillations about the mean bead transport rate and indicate that the strain (γ) at the mucociliary interface is ∼0.05, or 5%. D uses data from Ref. 281, with permission from Journal of Non-Newtonian Fluid Mechanics.
In addition to analyzing mucus displacement associated with MCC, the velocity of the cilia tip during the effective and recovery strokes was also compared to the velocity of transport of a 1% organic solids mucus layer. During the effective stroke, the cilia tip reached a maximum velocity of ∼200 µm/s (279) (FIGURE 11C) but the maximum velocity of mucus-embedded tracer particles at the mucociliary interface was ∼70 µm/s (281) (FIGURE 11C). This observation indicates that the mucus layer at the interface flows at a speed that is ∼35% of the speed of the cilia tip. During the recovery stroke, the cilia tip moved at ∼ −150 µm/s (FIGURE 11C), and mucus recoiled at a maximum speed of 30 µm/s, or at 20% of the speed of the cilia tip, during the recovery stroke. In summary, these data indicate that a relatively small fraction of the velocity of beating cilia during the effective stroke is communicated to flowing mucus.
These experiments also permitted study of the nature of cilium-driven mucus flow, i.e., does a mucus layer move as a uniform slab/plug or is it sheared? Measurements of the velocity of transport of beads embedded in mucus as a function of bead height above cilia revealed that oscillations in mucus velocities (i.e., the differences between the maximum and minimum observed bead velocities) decreased as a function of height above the mucociliary interface (FIGURE 11D). However, the mean transport rate was constant as a function of height (281). These findings indicate that mucus transport is grossly characterized by plug flow, i.e., the net transport rate of mucus at a concentration of 1% organic solids is the same at the mucus-air interface as at the mucus-cilia interface.
From these cell culture data, the strain rate, i.e., change in velocity of tracer particles flowing in mucus relative to the overall mean of mucus transport divided by height, that cilia impose on flowing mucus can also be calculated and was ∼5 s−1 (FIGURE 11D). By measuring the oscillations of tracer particle motion around its mean transport rate, the strain, or net displacement divided by height, imposed on mucus by cilia at the mucociliary interface can be estimated to be 5% (FIGURE 11E).
These observations characterize interesting features of cilia-mucus interactions but do not identify the physical nature of the cilia-mucus interactions that drive mucus flow, e.g., cilial tips “clawing” inner mucus layer surfaces, PCL compression and pumping water out of the layer, or other mechanisms. Understanding the nature of the ciliary-mucus interface and its coupling to mucus flow will require high-resolution imaging of the motion of beating cilia and flowing mucus. This approach will distinguish whether the mucus layer rises and falls with beating cilia or if a PCL-derived “pool” of water forms between the mucus and cilia (FIGURE 10A). Furthermore, such studies would measure the contact time and degree of slip between cilia and mucus, how mucus merges onto converging airway surfaces as it traverses the lung, and how cilia-layer/blanket versus cilia-flake/aggregates interact physically.
5.2. Rheology
The interactions of cilia with mucus to produce mucus flow have traditionally been related to the rheologic (viscoelastic) properties of mucus (296–299) (see appendix). As noted above, the high-molecular-weight (MW) secreted mucin glycoproteins MUC5B and MUC5AC are the main polymeric components of mucus that generate its characteristic viscoelastic properties (30). The rheologic properties of airway mucus have been characterized in terms of mucus storage (G′) and loss (G′′) moduli over a wide range of frequencies, as well as viscosities (η) (see TABLE 2 and appendix). Although the physical properties of mucus have been studied for decades and the overall conclusion that mucus is a viscoelastic fluid is widely accepted, the reported values for mucus viscoelastic properties vary by orders of magnitude (TABLE 2). Published zero shear rate viscosities (η0), measured by macroscopic cone and plate rheology (FIGURE 12A) of human respiratory mucus, vary from 0.01 Pa·s (10 times the viscosity of water at 0.5% organic solids) for mucus harvested from cell culture model systems (29), to 60 Pa·s (greater than the viscosity of honey) for sputum (10% organic solids) collected from CF patients (300) and up to 10,000 Pa·s for HBE mucus (301). These widely varying results arise from differences in measurement techniques and mucus sources and, most importantly, the absence of measuring, reporting, and controlling the concentration of measured mucus.
Table 2.
Published zero shear rate viscosities of human mucus and sputum
| Reference | Value as η0, Pa·s |
|---|---|
| Puchelle et al. 1981, human (recurrent bronchitis) (297) | 24.8 |
| Puchelle et al. 1981, human (mild chronic bronchitis) (297) | 11.4 |
| Puchelle et al. 1981, human (severe chronic bronchitis) (297) | 12.5 |
| Baconnais et al. 1999, human (302) | 0.71 |
| Baconnais et al. 1999, human (CF) (302) | 0.16 |
| Puchelle et al. 1983, human (298) | 24.7 |
| Dawson et al. 2003, human (CF) (300) | 60 |
| Tang 2021, 10% organic solids, human cell culture mucus (301) | 10,000 |
| Hill et al. 2014, 0.5% organic solids, human cell culture mucus (29) | 0.01 |
| Matsui et al. 2006, 7% organic solids, human cell culture mucus (80) | 3.3 |
CF, cystic fibrosis; η0, zero shear rate viscosity.
FIGURE 12.
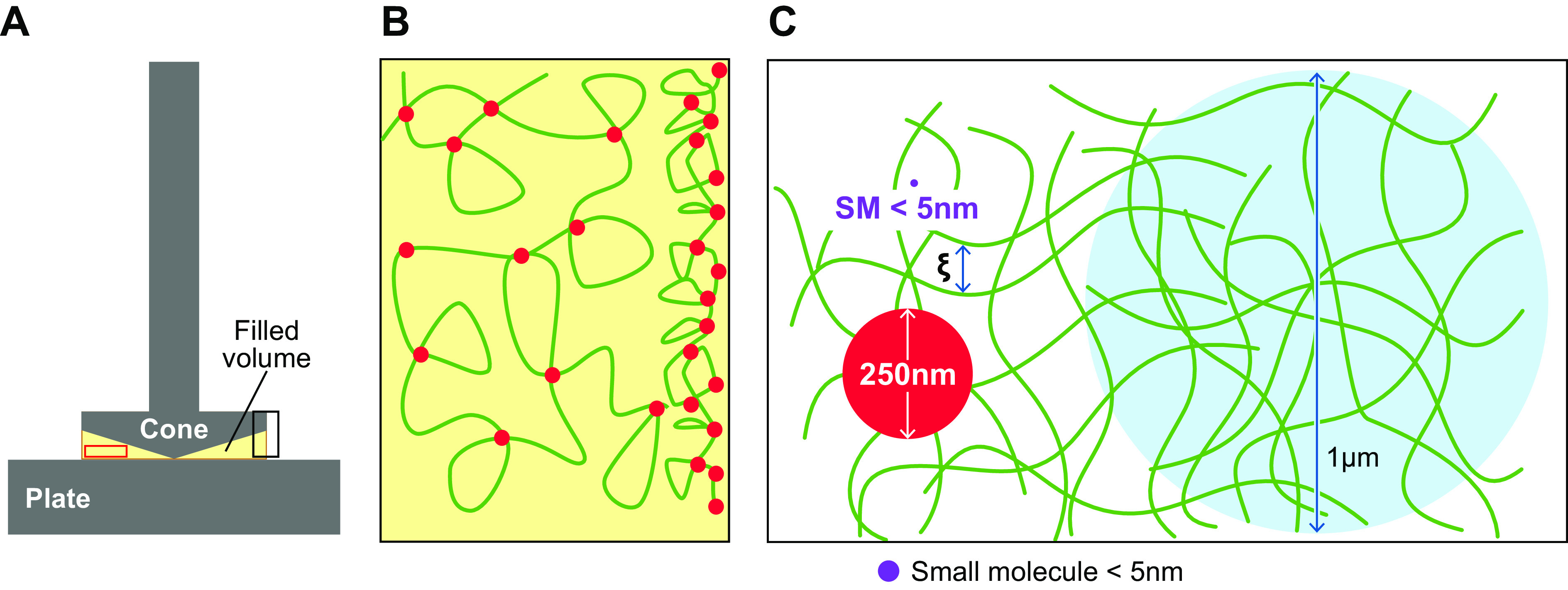
Rheological length scales referenced to measurement technologies. A: macroscopic data are collected on a cone and plate rheometer. Cone diameters range from 20 to 60 mm with fill volumes of 50 µL to 2 mL. B: microscopic and nanoscopic rheology are measures obtained by adding tracer particles or molecules to mucus (depicted in enlargement of red box from A). If the probe particle is larger than the mesh size of mucus (red, 250-nm probe; blue, 1-µm probe), the microscopic results theoretically will be similar in magnitude to macroscopic results. If the probes are much smaller than the mesh size, like the small molecules (SM) used fluorescence recovery after photobleaching experiments (purple, <5 nm), the probes will measure the rheological properties of the fluid component of mucus, not the polymeric properties. This illustration represents 1% organic solids mucus with a 50-nm correlation length (ξ, mesh size). C: effect of air interface on mucus. In a cone and plate rheometer, a fraction of the volume of the sample being assayed is in contact with air (depicted in enlargement of black box in A). At the air interface, associative mucin polymers (green) with hydrophobic domains (red) adsorb to the air interface, locally increasing the polymer concentration and, thereby, the viscosity of mucus. Cartoon courtesy of Scott Danielsen, PhD, Duke University. C uses content from Ref. 79, with permission from European Respiratory Journal.
The polymeric nature of overlapping mucins produces a mucus mesh with the spacing (ξm, correlation length; FIGURES 4C and 12C), i.e., porosity of the mucus mesh, depending on the concentration of the mucin glycoproteins (303). As reviewed in the context of osmotic pressure/modulus, the concentration at which the polymeric mucin molecules tessellate, i.e., just “touch” each other (i.e., c*, the overlap concentration), is dictated by the MW (250 ± 150 MDa) and radius of gyration (Rg ∼ 400 nm) of the mucins present in mucus (see FIGURE 4C). Note that this mucin MW includes the mass of bound globular proteins. Importantly, the overlap concentration (c*) of a polymeric solution marks the concentration at which the viscosity of mucus begins to rapidly increase as a function of concentration (303, 304). Based on rheological data from mucin solutions purified from HBE cultures, the overlap concentration (c*) of mucus is 1.8 mg/mL organic solids, i.e., a mucin concentration of ∼0.6 mg/mL (FIGURE 4, C and D, and TABLE 1).
5.2.1. Macrorheology.
The polymeric nature of mucus necessitates a matching of the length scales of a given measurement to the phenomena under investigation, e.g., bulk mucus transport, pathogen motility, and/or small-molecule diffusion. Macroscopic rheology assays performed in traditional cone and plate rheometers measure the gellike properties of bulk mucus (FIGURE 12A) as well as interactions of the mucin glycoproteins with the air interface (301) that is a feature of certain instruments (FIGURE 12B). Mucus gellike properties have often been correlated with independent MCC and cough clearance measurements (305). The macroscopic viscoelastic properties of mucus are characterized by the complex modulus G*(ω) (see appendix), comprised of the storage modulus G′(ω), which describes the solidlike properties of mucus, and the loss modulus G′′(ω), which describes the liquidlike properties of mucus. Both G′ and G′′ are dependent on the frequency ω (FIGURE 13A) at which the measurement is performed as well as the mucus concentration (FIGURE 13B). By combining frequency sweep data with creep recovery data, employing the method of Evans et al. (306), we were able to determine G′ and G′′ over 5 decades of frequency space (FIGURE 13A) (301). Additionally, data collected for mucus concentrations between 0.3% and 8% organic solids indicate that G′ and G′′ increase to a power of 1.2 as mucus concentration increases (301).
FIGURE 13.
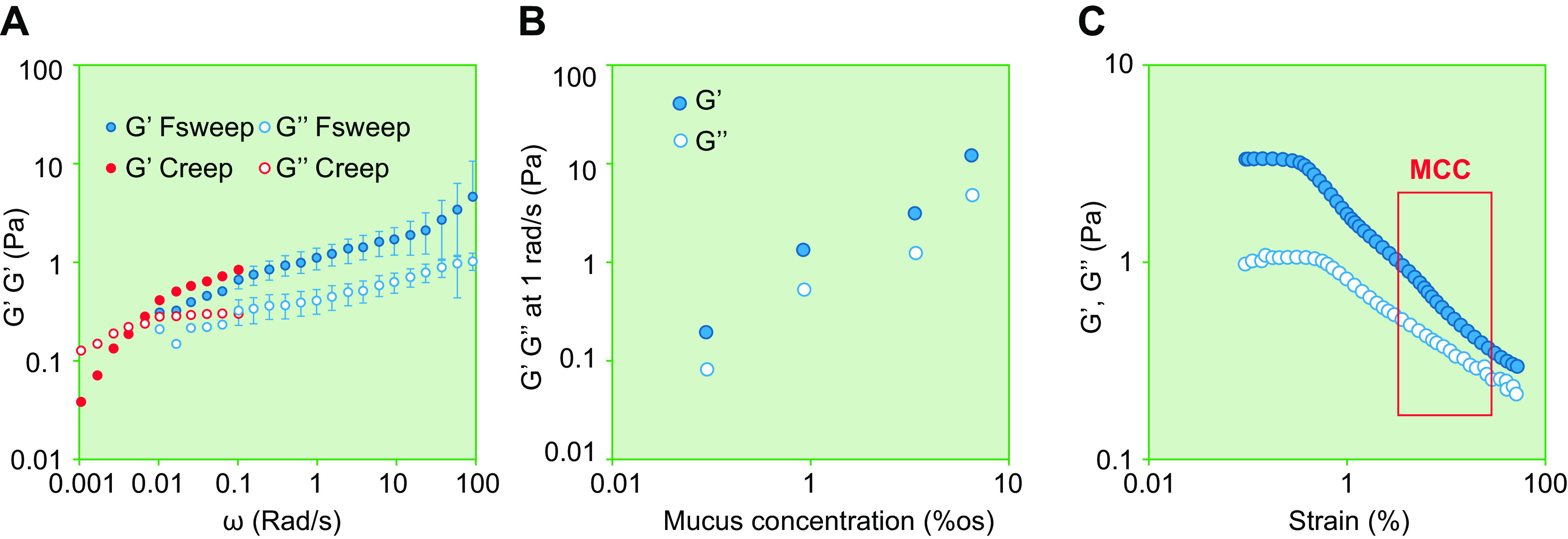
Macroscopic rheology of human airway mucus. A: frequency (ω) dependence of G′ elastic modulus (closed symbols) and G′′ viscous modulus (open symbols) of human bronchial epithelial (HBE) mucus at 1% organic solids. Five decades of frequency data are achieved by combining long-timescale, low-frequency creep recovery data (Creep, red) with data obtained from frequency sweep (Fsweep, blue) experiments (301). Frequency sweep data (Fsweep) are an average of n = 3, whereas creep data are from a single run. B: concentration-dependent mucus rheology (G′ and G′′ at 1 Hz) for 1%, 3%, and 5% organic solids (%os) HBE mucus (301). Graph represents the average of 3 frequency sweep experiments at each mucus concentration. C: strain dependence of rheologic parameters. Nonlinear reduction of G′ (blue, elastic modulus) and G′′ (white, viscous modulus) of CF sputum at high strains. At low strains (0.1−1%) the storage and loss moduli of sputum are greater than at strains associated with mucociliary clearance (MCC, red box) (307). n = 1 in 5% of CF sputum. A and B use data from Ref. 301, courtesy of Dr. Qishun Tang, and used with permission.
In addition to the frequency dependence of G′ and G′′ of mucus, the rheological properties of mucus are nonlinear, i.e., the measured values of these properties are both stress and strain dependent. In an experiment conducted at a single frequency of 1 Hz (FIGURE 13C), the storage and loss moduli of sputum decreases dramatically when the imposed strain exceeds those associated with the linear regime.
Typically, the rheological properties of mucus are reported in the linear viscoelastic regime (LVR) (79, 308–310), utilizing small-amplitude oscillatory shear protocols to avoid the complications of nonlinearity (FIGURE 13A). However, as discussed above, the strains imposed on mucus near the mucociliary interface by beating cilia approximate 5% (FIGURE 11D), which is greater than the strain rates in the LVR regime (<0.5%) (FIGURE 13C). We note that these results are dependent on the height of the mucus layer and CBF. These data highlight the need to match the selected experimental conditions to the biological questions studied. In the case of MCC, it is important to tailor strains to match those that cilia impose on mucus. Therefore, rheological measurements designed to explore mucociliary clearance conditions should impart stresses relevant to MCC at a frequency mimicking normal CBF, e.g., ∼10 Hz. In contrast, measurements designed to characterize how mucus responds to cough should be performed at higher frequencies (previous studies have used a frequency of 100 rad/s) (311–314) and stresses (100 Pa in larger airways and down to 1 Pa in small airways) (315, 316) to mimic cough. Finally, studies of vertical mucus flows are better assayed with constant-stress measurements that mimic the constant force of gravity (317–319).
An additional key consideration in measuring the macroscopic viscosity of mucus is to understand and measure the effect of air-mucus interfacial viscosities manifest in most macrorheologic devices (FIGURE 12, A and B). The viscosity of polymeric systems, including mucus (301), measured in a cone and plate rheometer is the sum of the bulk mucus viscosity, ηB, and the interfacial viscosity, ηi, times the ratio of the surface area of the air interface to the volume, SA/V, or η = ηB + ηi · SA/V (320). The most probable etiology of air-mucus interfacial viscosities with reference to mucus is hydrophobic adsorption of amphipathic mucins to the air interface (320), creating a locally higher mucin concentration (FIGURE 12B). These considerations indicate that previous measurements of the bulk viscosity of mucus may overestimate the true bulk viscosity of mucus and not accurately model bulk mucus/mucin viscosity profiles. New studies of the rheologic properties of mucus that account for interfacial surface properties are needed.
5.2.2. Microrheology.
Particle tracking microrheology (PTMR) measures the thermally driven diffusion of particles ranging from 100 nm to 1 µm in mucus (FIGURE 12C). PTMR utilizes beads to measure mucus microrheologic properties and diffusion of small particles of particular interest in mucus, e.g., viruses (321), bacteria (80), or gene transfer vectors (322–324) (FIGURE 12C). Particle tracking studies have reported that the magnitude of the ensemble average mean squared displacements of tracer microparticles embedded in mucus correlate with mucus concentration (29, 79, 99), the severity of pulmonary infections (325), FEV1 (326), and treatment with mucolytic compounds (23, 308, 327). PTMR assays are highly accessible, requiring only a video camera and a fluorescence microscope, and require much less sample volume (∼1–5 µL) than macroscopic assays (>40 µL).
Although PTMR assays are widely used in the study of mucus (23, 29, 79, 80, 99, 125, 322, 323, 327–332), they have significant limitations with respect to measurement of mucus rheologic parameters. For example, the surface chemistry of the particles will often promote interactions with the mucus network, i.e., “stickiness,” which will have pronounced effects on apparent rheologic results (323). Therefore, it is important to verify that any probe used in a PTMR assay does not interact with mucus at all or interacts with mucus in a manner that is similar to the biological object of interest. Notably, the viscosity and storage and loss moduli reported by microbead rheology vary with the particle selected for study and are typically smaller than those measured by macroscopic cone and plate rheology (333). Although one would expect that interactions between beads and the mucus network would increase the measured viscosity and storage and loss moduli, most studies show that microrheology reports rheological properties that are lower than those determined by macroscopic rheology (323, 334). The measured decrease in these properties likely reflects, as just discussed, differences between the surface and bulk rheology of mucus (301, 320). The problem of particle mucus interactions may be mitigated by performing two-point microrheology experiments, where the modulus of the fluid is determined by correlations of the fluctuations of the tracer particles (335). Although microrheology is limited by the factors described above, it can measure the inherent heterogeneity of mucus structure (23, 99, 336, 337).
In addition to thermally driven particle motion tracking techniques, several groups have used microbead-magnetic systems to measure the viscoelastic properties of biologically relevant fluids such as cytoplasm (338, 339), DNA solutions (340), mucus simulants, and mucus (341–346). These systems are advantageous because they control input stress and frequencies of oscillation and have no air-mucus interface. However, the addition of a large foreign object, e.g., a magnetic microsphere (1–100 µm), oscillated magnetically, may disrupt the microstructure of mucus on the length scale of the bead. Additionally, just as with passive, thermally driven techniques, the interaction of the bead with the mucus layer may affect results. One solution to these problems is to employ beads that are much larger than the correlation length, ξ, and have neutral interactions with the mucus network. Finally, to determine the accuracy of magnetic bead assays for the study of mucus rheology, it will be necessary to compare micro-magnetic data with macroscopic cone and plate assays that control for air-mucus interfaces utilizing both mucus and well-controlled polymeric reagents.
5.2.3. Fluorescence recovery after photobleaching.
Mucus can be further characterized on nanoscale levels with fluorescence recovery after photobleaching (FRAP) techniques, which employ thermally driven fluorescent probes (e.g., 5 K dextran, hydrodynamic radius of 2 nm) (8) and one-dimensional diffusion assays (80) (FIGURE 12C). The use of nanoscale probes, smaller than the correlation length of mucus even in pathological concentrations, allows for investigation of the diffusive properties of the interstitial space (i.e., solvent) between mucin polymers. Although these assays do not study mucus properties relevant to the length scales associated with MCC, they are appropriate for studies focused on the motility of small molecules in mucus, e.g., inhaled therapeutic agents and/or environmental toxins. Designing assays that employ probes of biological interest will ensure that the assays capture the steric (size)-dependent interactions of the molecules with mucus. These assays are critical to understand whether small molecules, including antibiotics (see below), interact with/are bound by the mucus layer or whether they freely diffuse and penetrate the mucus layer (322, 347, 348).
In summary, no single rheologic technology is capable of describing the broad-based behaviors of mucus in the myriad of mucus-centered biologies and physiologies in the lung. However, to provide useful descriptions, there are two key needs. First, there is a critical need to standardize rheological assays for the study of mucus to ensure reproducibility across laboratories. Second, it is imperative that all studies of the physical and physiological properties of mucus include characterizations of mucus concentration and would ideally include measures of the mucin concentration as well as mucin molecular weight.
5.3. Ciliary and Mucus Parameters That Govern Mucociliary Clearance Rates in Health and Disease
5.3.1. Cilia and MCC rates.
The biological requirement for MCC in lung health is well established, as is the role of impaired (or absent) MCC in pulmonary disease (25). Cilia are the driving force for MCC, and, within limits, faster rates of MCC are associated with more cilia present on an airway surface and/or faster ciliary beating. For example, MCC is positively correlated with the number of ciliated cells present on airway surfaces, i.e., percent ciliation. In mouse pups, an increase in the percent ciliation of the trachea between 10% and 50% was associated with an increase in rates of MCC from 4 to 10 µm/s (349). In the human nasal cavity, mean MCC rates increased from 5 to 10 mm/min, or from 80 to 160 µm/s, as the percent ciliation increased from 50% to nearly 100% as measured by intranasal optical coherence tomography (350). In the same studies, cilia beat frequency (CBF) was positively correlated with MCC, with clearance rates increasing from 80 to 150 µm/s as CBF increased from 6 to 15 Hz (331). It is important to note that there are fewer cilia in the small airways than in the large airways. Percent ciliation has been reported to decrease in more distal airway generations, decreasing from 56% ciliation in large airways to 43% in small airways (133). Cilia length also decreases as a function of airway generation, decreasing from 7 to 4 µm between generations 0 and 7 (351). Together, these changes in ciliated cell coverage area and ciliary length may contribute to the reported lower rates of MCC in the small compared to large airways (352).
With respect to disease, cilial density (353, 354), cilia beat frequency (353, 355), and ciliary length are all decreased with long-term cigarette smoke exposure (355), predominantly in the proximal airways. These findings are consistent with studies of slowed MCC in COPD subjects, particularly in large airways, and support a major role for cilial loss/dysfunction in the MCC dysfunction associated with COPD (356). However, it is well accepted that muco-obstructive lung disease dysfunction in general begins in the distal, small airways (357). In the case of cystic fibrosis, it has recently been demonstrated that mucus hyperconcentration occurs before the onset of structural lung damage and decreased pulmonary function (23). We therefore speculate that abnormal, hyperconcentrated mucus, not losses of cilia, dominates the early small airway pathology in muco-obstructive diseases (see below).
5.3.2. Concentration-independent mucus variables and MCC rates.
Variables other than mucus concentration-dependent biophysical properties can modify MCC rates. For example, alterations to the polymer composition of mucus likely derange mucus biophysical parameters relevant to transportability. One area of particularly active interest relates to the absolute and relative concentrations of the two secreted mucins, MUC5AC and MUC5B, in health and disease. As reviewed in sect. 2.1, MUC5B is the dominant mucin in health and MUC5AC concentrations increase with inhaled stresses/irritants (25, 77). However, the properties of MUC5AC versus MUC5B as related to mucus transportability are not yet known. Reports have suggested that cell cultures exposed to type 2 cytokines (e.g., IL-13) produce a MUC5AC mucin that is more adhesive and less transportable than MUC5B (115). However, the biophysical properties of purified MUC5AC and MUC5B, in the absence of potential interacting proteins related to mucus transport, have not yet been reported. Increasing concentrations of MUC5AC relative to MUC5B have been consistently reported in muco-obstructive diseases, where the MUC5AC-to-MUC5B ratio rises from ∼0.1 in health to ∼0.5 in disease (49, 76, 77, 358). In asthma, the MUC5AC-to-MUC5B ratio may exceed 1.0 (359, 656). Therefore, an understanding of the relative roles of MUC5AC versus MUC5B in mucus transport will be important for a comprehensive understanding of the role of mucins in muco-obstructive disease pathogenesis.
In pulmonary muco-obstructive diseases such as CF (360) and non-CF bronchiectasis (358), the concentration of extracellular DNA in mucus is also elevated (361). Although double-stranded DNA, as measured by the PicoGreen assay, is present in healthy mucus at a concentration of ∼2 µg/mL, or a 1:200 ratio of DNA to mucin (358), the ratio of DNA to mucins is increased in early CF lung disease to 1:100 (23). In adults with CF, the ratio of DNA to mucins is further increased to 1:20 (362). In patients with non-CF bronchiectasis, this ratio may approximate 1:13 (358). The addition of a small amount of DNA to mucus would not be predicted to alter the osmotic modulus of mucus. However, extracellular DNA has been reported to increase mucus viscosity (363) and decrease mucus transportability (362). We have recently shown that adding salmon sperm DNA to mucus at 1:100 DNA-to-mucin concentration ratios doubled mucus viscosity, as measured by microbead rheology (362). The addition of 1:100 DNA-to-mucin ratios also decreased MCC rates to 50% that of mucus without DNA (362). Understanding how the addition of small quantities of “stiff” polymers like DNA, as well as histones (364), to mucus alters mucus biophysical properties and its transportability independent of mucus concentration is an important future research area.
In addition to changes to the polymeric composition of mucus, interactions between mucins and other components of mucus have been reported to alter the rheological properties of mucus. Trefoil Factor 3 has been shown to act as a divalent lectin, interacting with the glycosylated domains in mucins, specifically with the terminal sugars N-acetylglucosamine (GlcNAc) and galactose (Gal) (118). These interactions have been shown to increase the viscosity of mucin solutions (365) as well as the storage and loss moduli of cervical mucus plugs (366). Another lectin, intelectin-1, has been shown to be a key component in fatal asthma (119) and to increase the elasticity of mucus (367).
Finally, oxidative stress (117) has been shown to increase the storage and loss moduli of mucus, likely because of intermucin cross linking. Oxidant stress has specifically been reported to cross link mucins covalently, increase mucin molecular weight, increase viscosity, and ultimately decrease MCC (117).
5.3.3. Multifaceted disease-induced mucus contributions to failed MCC.
Based on the reviewed data and our extrapolations, a framework describing the variables that produce the failure of MCC in muco-obstructive lung diseases can be constructed. We posit that mucus concentration is the most important determinant of MCC rates, with changes in the MCU5AC/MUC5B composition of mucus and “state” of the mucus (e.g., mucin molecular weight, oxidative stress, pH) exerting more modest effects on MCC rates. We hypothesize that as the concentration of mucus increases from 1% organic solids, a value typical for healthy individuals, to 4–6% organic solids in moderate disease, MCC is slowed because of the combination of increased resistance of mucus to flow (increased mucus viscosity) and modest compression of the PCL as the mucus osmotic modulus begins to exceed that of the healthy PCL. As disease-driven hyperconcentration of mucus exceeds 6–8% organic solids, the increased mucus layer osmotic modulus dominates and abolishes MCC via compression/collapse of the PCL, producing not only complete arrest of MCC but also adhesion of the mucus layer to ciliated airway surfaces.
6. AIRFLOW-DEPENDENT MUCUS CLEARANCE
Although cough is often considered the only airflow-dependent mucus clearance mechanism, gas-liquid pumping occurs at lower airflow rates. Both mechanisms are reviewed here.
6.1. Cough Clearance
In health, the clearance of mucus by the action of cilia beating is normally adequate to maintain airway sterility (FIGURE 14). However, cilium-dependent mucus clearance transiently fails during respiratory tract infections, and clearance of mucus by the high-speed airflow associated with cough is an important backup mechanism during periods of excessive mucus accumulation (130, 368). Interestingly, cough is the most common presenting symptom in the United States for pediatric medical office visits (369). Acute cough that is caused by a known insult, whether infectious or irritant, is treated by managing the precipitating cause, ideally removing the stimulus for cough. However, a persistent “productive” (of mucus) cough is a common symptom of muco-obstructive lung diseases, e.g., CF, NCFB, and COPD. Cough in these diseases functions as a critical backup mechanism to clear excessively concentrated airway mucus that persistently accumulates in the diseased lung (28, 370–372). Furthermore, in persons with primary cilia dyskinesia (PCD), a rare lung disease affecting cilium motility (see below), airflow-dependent clearance, including cough, represents the primary mechanism for mucus clearance (373).
FIGURE 14.
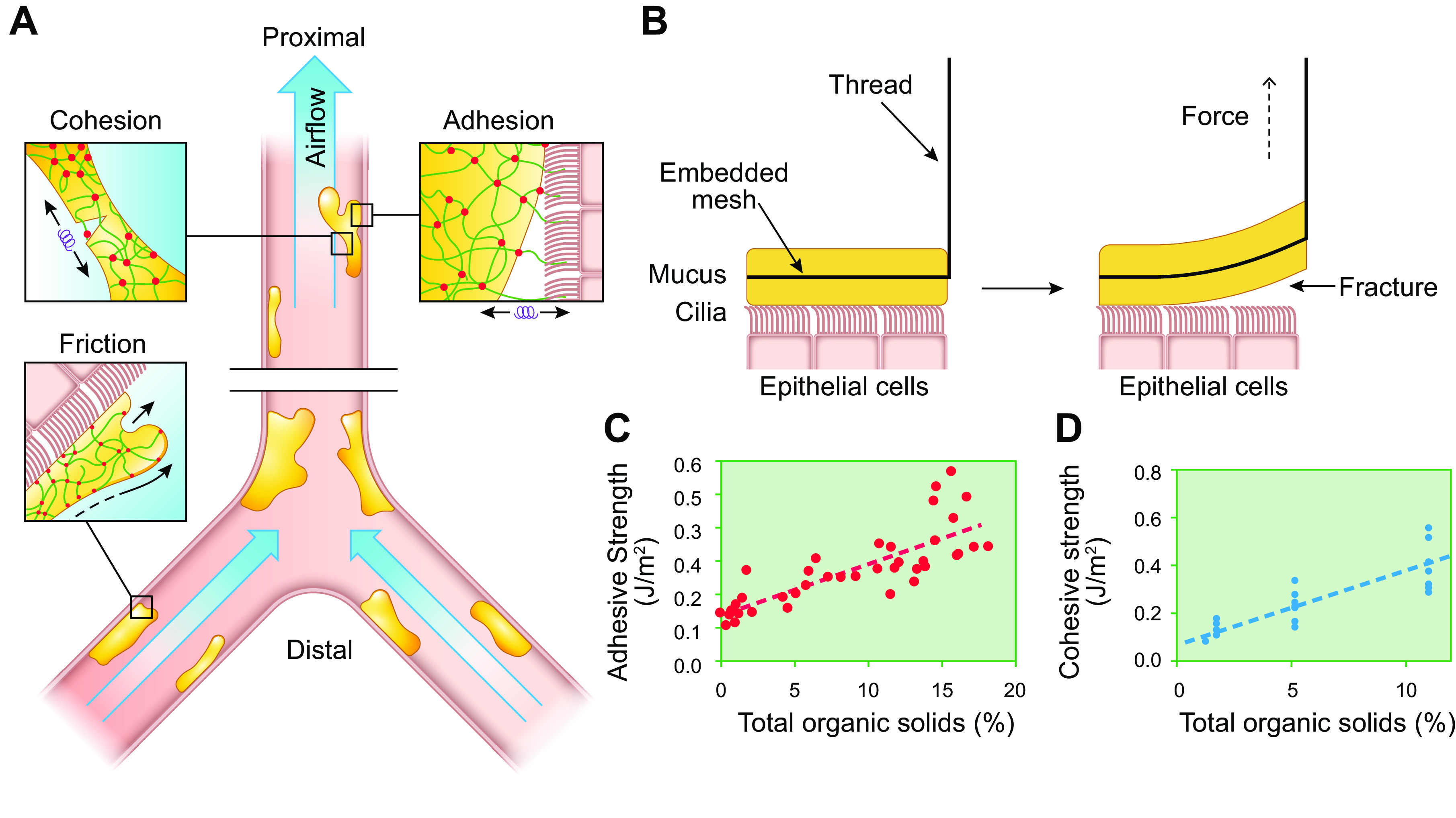
Cough clearance. A: biophysical properties of mucus-cell surface interactions relevant to cough clearance. In the small (distal) airways, the lower-velocity airflow can slide mucus along airway surfaces, a movement limited by mucus-cell surface friction. In large (proximal) airways, the higher airflow fractures intramucus cohesive forces and/or mucus-cell surface adhesive forces to expel mucus. B: peel testing device. Schematic diagram of the peel tester that was used to measure the force required to “peel” the mucus layer off the surface of the epithelium of well-differentiated human bronchial epithelial (HBE) cultures with an endogenous mucus layer. The embedded mesh in the mucus is connected by a thread (tail) to a motor that generates the force to fracture mucus-epithelial adhesive interactions. A force sensor (not shown) connected between mesh and motor measures the forces applied. C: effect of mucus concentration on mucus-PCL adhesive strength. Summary of the adhesion strength vs. mucus concentration over a range that spans normal (1% organic solids) to CF (5–15% organic solids) concentrations when peeled at 100 µm/s. D: effect of mucus concentration on intramucin cohesive strength. Summary of the cohesive strength vs. mucus concentration over a range that spans normal (1% organic solids) to CF (5–15% organic solids) concentrations when peeled at 100 µm/s. See glossary for abbreviations. Data from Ref. 116 used with permission from The Proceedings of the National Academy of Sciences of the United States of America.
The process of coughing is typically initiated as a reflex event reflecting stimulation of pulmonary irritant receptors and activation of vagal afferent nerves (15). Vagal irritant receptors in the airways respond to a wide array of noxious of stimuli, including mechanical and chemical irritants, inflammatory mediators, and large changes in lung volume (374). Coughing can also be voluntary, but voluntary cough is less effective than reflexive cough because of reduced airway dynamic changes and airflow velocities (375).
6.1.1. Airway shear stresses during cough.
The cough response is initiated with an inhalation of air above functional residual capacity (FRC), followed by closure of the glottis and rapid contraction of the abdominal muscles, generating intrapulmonary pressures of 100–250 cmH2O (9.8–24.5 kPa) (315). Upon subsequent opening of the glottis, subglottic pressures fall and a rapid expiratory flow of air from the lung is initiated. This high expiratory airflow develops within ∼100 ms and lasts for ∼200 ms (368). During the peak expiratory portion of cough, airflow velocities in the proximal airways can reach high subsonic velocities of ∼300 m/s (∼1,000 km/h) (376). In the more distal airways, the velocity of airflow during the expiratory phase of cough depends on the airway generation and the dynamic compliance of the airway during expiration. Moving from the central to the peripheral airways, the total airway diameter, the number of airway branches, and the overall effective cross-sectional area increase dramatically (316). Hence, airflow velocity is strikingly lower (∼2 orders of magnitude) in the smaller, higher-generation peripheral airways than in the larger, central airways (377).
Expiratory airflow during cough produces a spectrum of airway wall shear forces (376). The stress (σ) imparted by air on the mucus layer can be estimated from the dynamic airway pressure (pd) during cough (pd = ρ·/2) as
| (9) |
where f is the Darcy friction factor (378), ρ is the density of air, and vair is the airflow velocity during cough. Dynamic pressures vary from ∼1 Pa for vair ≈ 1 m/s during tidal breathing to ∼104 Pa for vair ≈ 100 m/s during cough (315). The Darcy friction factor f, a dimensionless value to describe losses due to friction along a surface, decreases with increasing Reynolds numbers as Re = vairD/μ, in an airway with diameter D and a kinematic viscosity of air (μ) of ≈ 10−5 m2/s. For laminar flow f = 64/Re, whereas for turbulent flow f decreases more slowly and saturates at a value of f ≈ 0.02–0.04, depending on the roughness of the mucus surface (379). As a result, the shear stress at mucus surfaces reaches σ ≈ 100 Pa at high velocities, e.g., vair ≈ 100 m/s, in airway generations 0–3. In contrast, shear stress (σ) in the smaller airways, i.e., generations > 7, is ∼1 Pa, or even lower, as a result of significantly lower cough-induced airflow velocities (vair ≈ 3 m/s) in small airways (315, 316).
6.1.2. Modes of mucus clearance by cough.
So how does airflow clear mucus from the lung? Whereas the high shear forces generated by the expiratory airflow of cough are predicted to propel mucus (and trapped pathogens and particles) toward the trachea and out of the lung, the exact mechanism(s) by which this transport occurs depends on a number of factors, including 1) the airway region, and hence the magnitude of airflow velocity and wall shear stresses, and 2) the physical properties of the non-Newtonian mucus layer and its interaction with the underlying epithelial surface.
6.1.3. Mucus-airway surface friction.
In the distal airways where airflow velocity and wall shear stresses are low, mucus likely is transported proximally predominantly as a result sliding of over airway surfaces. Consequently, the transport of mucus must overcome the “frictional” resistance to mucus sliding over airway surfaces and the resistance of mucus to shear flow (FIGURE 14A). The frictional resistance to airflow-induced mucus transport represents an integrative factor describing the strength of mucus-cilia interactions, including mucus interfacial properties and PCL roughness (380). Although the friction of a well-hydrated (1% organic solids) mucus sliding over a well-hydrated PCL is predicted to be low, increases in mucus concentration associated with muco-obstructive lung diseases significantly increase friction (8). Although currently unknown, it is likely that the collapsed PCL generates increased friction because of changes in its surface interfacial properties (i.e., roughness). When mucus is hyperconcentrated, the prediction is that an increase in frictional resistance to mucus flowing over the airway surface will result and produce reductions in the rates of both cough- and cilium-mediated mucus clearance.
6.1.4. Mucus-airway surface adhesion.
In the larger airways with higher cough-induced airflow velocities and wall shear stresses, mucus may be cleared by “disadhesion” when shear stresses are sufficiently large to overcome the “adhesive” interactions/bonds between mucus and the airway cell surface (FIGURE 14A). In the trachea, for example, airflow physically strips adherent mucus off airway surfaces. The adhesion strength (or fracture toughness), defined as the energy per unit area required to separate mucus from the cell surface, has been measured with a modified peel testing device (116, 381) (FIGURE 14B). An important observation from these studies is that at normal mucus concentrations (∼1% organic solids), the work of adhesion is only about three times the surface tension of water (382), i.e., ∼0.15 J/m2 (116). Therefore, despite mucus being characterized as “sticky,” the low mucin concentrations in the bulk-phase “normal” mucus only raise mucus-PCL adhesion to values modestly above water-water surface tensions. This property is consistent with a proposed high concentration of mucins at an air-mucus interface and a lower concentration in the bulk phase that is exposed to the cell surface (see sect. 5.2).
Cough, however, becomes a required mucus clearance mechanism in muco-obstructive lung diseases. Measurements of adhesion strength in disease have demonstrated that increased mucus concentrations produce significant increases in the strength of mucus adhesion to airway surfaces (FIGURE 14C). This observation suggests that when mucus concentration is increased with disease there is an increase in the density of mucins per unit area at the interface between the mucus and PCL layers, resulting in an increase in adhesive strength. Additionally, it is possible that when the PCL collapses (i.e., KML > KPCL), interpenetration of mucus layer mucins with those of the PCL increases the adhesive interactions between the two layers (8). The spectrum of the biochemical interactions between the mucus and PCL is unknown but could include the formation of chemical/physical bonds (i.e., carbohydrate-carbohydrate, protein-protein) and/or hydrophobic interactions.
6.1.5. Intramucin cohesive failure.
In addition to disadhesion of mucus, a second potential mechanism for mucus clearance mediated by high airflow velocity cough is the physical “tearing apart” of mucus, where intramucin “cohesive” bonds are broken (116). This event may occur when high-speed airflow produces shear stresses at the interface with large mucus accumulations/masses. The predicted result of this tearing is that fragments of mucus break off as discrete packets from adherent mucus and are propelled proximally (FIGURE 14A). Unlike disadhesion, this mode of failure during cough results in the airway surface still being covered by a residual layer of adherent mucus.
With a peel device similar to the adhesion peel tester described in sect. 6.1.4, the cohesive strength of mucus was characterized by measuring the magnitude of force required to tear a layer of mucus apart (116). As with adhesion, the cohesive strength of healthy mucus was found to be very low, i.e., slightly above twice the surface tension of mucus (382) (FIGURE 14D). However, when mucus was concentrated as typical of disease, significantly higher forces per unit area were required to tear mucus apart compared with mucus at normal (nondiseased) concentrations. The conjecture is that cohesion reflects the number density and strength of associative bonds between adjacent mucin stands that stabilize mucus gel structure. The number density of these associations, related to concentration-dependent correlation lengths, and their strength govern the generation of cohesive forces. These intermolecular associations can be formed by either direct mucin-mucin attractions or interactions between mucins mediated by globular proteins in the airway surface liquid milieu (383). When mucus becomes concentrated, the number density of these interactions increases, making it harder to pull mucus apart (induce cohesive fracture) by airflow-mediated shear stress (116).
6.1.6. Peel rate dependence of adhesive and cohesive strength.
Studies by Button et al. (116) demonstrated that, in addition to concentration dependence, the magnitudes of both adhesive and cohesive strength (fracture toughness) were peel rate velocity dependent. The faster the rate of peeling, i.e., increased peeling velocity, the greater the force required to separate the mucus from the airway surface (adhesion) or tear it apart (cohesion). Interestingly, adhesive and cohesive strengths exhibited similar velocity and concentration dependencies, suggesting that the viscous dissipation of energy within mucus during cough determines the efficiency of cough clearance of diseased, hyperconcentrated mucus. The magnitude of both adhesive and cohesive fracture toughness (Γ) and its dependence on mucus concentration (c) and crack propagation velocity (v) can be written as
| (10) |
where γ is the surface tension of mucus (34 mN/m) (382), c0 is the characteristic mucus concentration at which the work of adhesion (Wa)/cohesion (Wc) doubles (at v = 0), v0 is the characteristic peeling velocity at which the dissipative component of fracture toughness is comparable to the work of adhesion/cohesion (at c = c0), and β is the dynamic exponent. For adhesion, β = 0.43 ± 0.04, c0 = 0.078 ± 0.020 g/mL, and c0v0β = 7.1 ± 1.1 g/mL(m/s)0.43. For cohesion, β = 0.38 ± 0.11, c0 = 0.49 ± 2.03 g/mL, and c0v0β = 10.6 ± 1.6 g/mL(m/s)0.38. Importantly, the dynamic exponents (β) of the dissipative component of fracture toughness obtained from the fits of adhesion and cohesion data are similar, i.e., within the errors of measurement. This result suggests that viscous dissipation represents the common major contributor to both adhesive and cohesive fracture toughness.
6.1.7. Cough clearance by adhesive and cohesive failure.
Based on the studies quantitating the adhesive and cohesive strengths of mucus, the probability of mucus being cleared by forces that overcome the adhesive and/or cohesive strengths of mucus can be calculated. The force (f) per unit length applied to mucus accumulated on an airway surface can be calculated as
| (11) |
where σ is the surface shear stress (as calculated in Eq. 9) and t the thickness of the mucus layer. In the upper airways (i.e., generations 1–3), where shear stresses are on the order of σ ≈ 100 Pa, the force applied to a 1-cm-thick (tall) discrete mucus mass is ∼1.0 J/m2, which is predicted to clear diseased/concentrated mucus by overcoming both adhesive and cohesive forces (see FIGURE 14, B and C). This prediction is supported by the observation that the mass of sputum expectorated by cough in subjects with muco-obstructive lung disease averages ∼1 g (corresponding to the volume ∼1 cm3) (384). Notably, for smaller (i.e., less tall) masses of accumulated mucus, e.g., 1 mm, the force per unit length generated by cough in large airways is only ∼0.1 J/m2, a value below the adhesive/cohesive fracture toughness of mucus at any concentration. For this reason, repetitive cough events occurring in humans in response to difficulty in clearing intrapulmonary mucus may serve to accumulate mucus in large airways to a volume, and hence height, that can be cleared by cough (311). Notably, in smaller airways (<2 mm, i.e., generations 7 and greater), mucus cannot reach a volume/height that would permit cough-induced shear stress forces to exceed mucus adhesive and cohesive fracture toughness at any mucin concentration. The predicted failure to clear hyperconcentrated mucus from small airways via cilial or cough-dependent mechanisms in disease is consistent with the characteristic early small airway mucus plugging in muco-obstructive diseases (18, 25, 68, 71, 72). A recent radiographic study of subjects with severe asthma also demonstrated the failure to clear adherent distal airway mucus plugs by mucociliary or cough-dependent mechanisms (385).
6.2. Gas-Liquid Mucus Transport
Although clearance of mucus by high-speed airflow associated with cough has been well established, another mode of mucus clearance, referred to as cilium-independent two-phase concurrent flow, or “gas-liquid transport” (GLT), has been proposed (386, 387). Like cough clearance, GLT is produced by a two-phase gas-liquid flow whereby gas (exhaled air) moves a liquid (mucus) layer out of the airways (386, 388). However, unlike the high-speed airflow generated by cough, GLT is proposed to occur at velocities associated with tidal breathing. Using various “test tube” model systems with mucus simulants, C. S. Kim and colleagues concluded that an average linear airflow velocity of ∼3 m/s was required for GLT (386). Indeed, such airflow velocities are achieved during normal tidal breathing throughout the airways, including bronchioles (386). Mucus clearance by GLT is potentially important in airway regions devoid of functional cilia as the result of damage and in persons with primary ciliary dyskinesia (PCD) who exhibit defects in cilia motility (389).
The physical basis for mucus clearance mediated by tidal breathing is that airflow velocities during expiration exceed those during inspiration. Although volumetric flow rates are similar during the inhalation and exhalation periods of tidal breathing, the diameters of airways are smaller during exhalation, as a result of thoracic cavity contraction, than during inhalation (390, 391). Thus, under conditions of relatively low Reynolds numbers (i.e., Re ≈ 850 to 2,100), the asymmetric airflow velocities generated during the respiratory cycle, with higher velocities characterizing exhalation, produce directional fluid transport. Notably, even greater asymmetries in transport were observed when utilizing viscoelastic solutions of mucus and mucus simulants (130, 387, 388, 392). Given the low shear stresses generated by tidal breathing [0.5 dyn/cm2, 0.05 Pa (153)], GLT mucus clearance likely reflects the ability of low shear stress-associated airflow to overcome the low frictional resistance to flow of normal mucus and generate a directional flow (FIGURE 14A). Increases in breathing rate, lung volume, and airflow velocity associated with deep breathing and exercise are predicted to increase GLT-mediated mucus clearance. Other potential factors determining the rate of GLT have been suggested, including the thickness and rheological properties of the mucus layer, surface tension, and airway wall compliance (387, 391, 392).
7. BASIC SCIENCE GAPS IN KNOWLEDGE OF MUCUS PROPERTIES/FUNCTION
Despite what may hopefully seem like a comprehensive review, there are surprising gaps in our knowledge of basic mechanisms that mediate mucus clearance in health. For example, we need to know basic information describing the structure of mucins in free solution, i.e., are they linear, as often depicted, or are they associative polymers that “loop” because of formation of intramucin reversible bonds? Neutron-scattering techniques may be required to resolve this issue. Similarly, there are surprising gaps in our knowledge of the organization of mucins within putative mucus layers or flakes (23, 655). Is it possible that amphipathic mucins are preferentially concentrated at the hydrophobic air-mucus interface and the mucus layer and/or flakes incorporate a “rubber skin” surrounding a fluidlike internal milieu? Or are mucins homogeneously intertwined throughout the mucus layer or flakes as commonly depicted? Such organizational attributes have major implications for the barrier properties of mucus, the transfer of momentum from cilia to mucus, and the functional interactions between the mucus layer and the PCL. As a related question, do cilia transfer momentum to mucus by “clawing” via rigid cilial tip structures entering a mucus layer, as often depicted (294, 295)? Or is mucus is “pushed” by water squeezed out of the PCL during the active cilial stroke (see above)? Does each mechanism have a variable contribution with respect to horizontal versus vertical MCC? Finally, do these mechanisms have implications for why cilia beat at 10–20 Hz?
At the more macroscopic basic level, we do need to understand the organization of the mucus covering airways, particularly in the distal airways. Specifically, is mucus organized in continuous layers/“skins” or in discontinuous flakes and aggregates? In a correlated question, how does mucus remodel at bifurcations, i.e., how does mucus accommodate to smaller surface areas as it moves from the very large distal to smaller-surface area proximal airways? Does viscous remodeling dominate this convergence process? Could there actually be reabsorption of mucins by airway epithelia/macrophages in more proximal airway surfaces? Finally, what do SMGs uniquely add to proximal airway defense? Does their unique geometry and vagal innervation allow them to “fire,” i.e., rapidly secrete, in a unique fashion that allows them to produce mucus bundles/strands that may be critical for effective cough clearance?
With respect to control of mucus clearance in disease, what are the relative roles of viscoelasticity and osmotic modulus/compression of the PCL in controlling MCC rates in disease ranging from mild to severe? Are these contributions disease specific? In that context, why are there two secreted mucins, i.e., MUC5B and MUC5AC, and do they have different properties that govern mucus transport rates, propensity to adhesion, and/or other functions? Finally, what is the role of inflammation in mucus clearance in disease? What does the addition of stiff polymers, e.g., DNA as free double-stranded polymers or in neutrophils NETs, do to mucus transport, and what role do histones play in altering mucus rheology and clearance? Similarly, what are the roles of inflammatory cells and/or microvesicles in mucus, e.g., are they trapped bystanders, do they serve as a scaffold for flake formation, and/or do they provide the enzymes/oxidants that “cook” (cross link) mucus flakes (117)?
8. PATHOGENESIS OF MUCO-OBSTRUCTIVE LUNG DISEASES
It has not been clear heretofore how the “chronic bronchitic”/bronchiectatic diseases could be grouped from a pathophysiological perspective (23). This section proposes mucus hyperconcentration as a unifying theme, and sects. 9–12 analyze four muco-obstructive diseases in terms of the mucus hyperconcentration pathophysiological perspective.
8.1. Concept of Mucus Hyperconcentration (Dehydration) as Final Common Pathway for Muco-obstructive Lung Diseases
A spectrum of pulmonary diseases that affect the airways of the lung, including COPD, cystic fibrosis (CF), primary ciliary dyskinesia (PCD), and non-CF bronchiectasis (NCFB), can be broadly characterized as part of a “muco-obstructive disease syndrome” (144, 393–395). Several features unify these muco-obstructive diseases. The symptoms associated with this syndrome typically are cough, sputum production, dyspnea/shortness of breath, and episodic clinical pulmonary “exacerbations” marked by worsening of disease symptoms (10). The pathology of this syndrome includes the accumulation of intraluminal mucus, particularly in the small airways, airway inflammation, and airway epithelial remodeling (18, 68, 69, 357, 396–399). The functional correlates of this syndrome reflect airflow obstruction and are manifest earliest and most severely as reductions in small-airway flows, e.g., FEF25–75% followed later by reductions in FEV1 and FEV1-to-FVC ratios (18).
A general description of muco-obstructive disease pathogenesis is depicted in FIGURE 15A. In health, a protective mucus layer is depicted as being transported rapidly (50 µm/s) from the lung, a process that requires coordination of epithelial mucin secretion, ion/water transport rates, and ciliary activity (FIGURE 15A, left). In muco-obstructive diseases, epithelial defects in ion transport/water transport and/or mucin secretion produce hyperconcentrated mucus that slows mucus transport and eventually produces mucus stasis (FIGURE 15A, center). Mucus/mucin concentrations have been measured and are abnormally raised in CB/COPD, non-CF bronchiectasis (NCFB), primary ciliary dyskinesia (PCD), and cystic fibrosis (CF) (23, 76, 77, 99, 373) (FIGURE 15B). Raised mucin concentrations produce tracheal mucus stasis/accumulation that can be expectorated by cough as “phlegm” or “sputum” (FIGURE 15A, top right). The mucus in the small airways that cannot be cleared by cough continues to accumulate, becoming the nidus for the airflow obstruction, infection, and inflammation that are the hallmarks of this disease syndrome (FIGURE 15A, bottom right).
FIGURE 15.
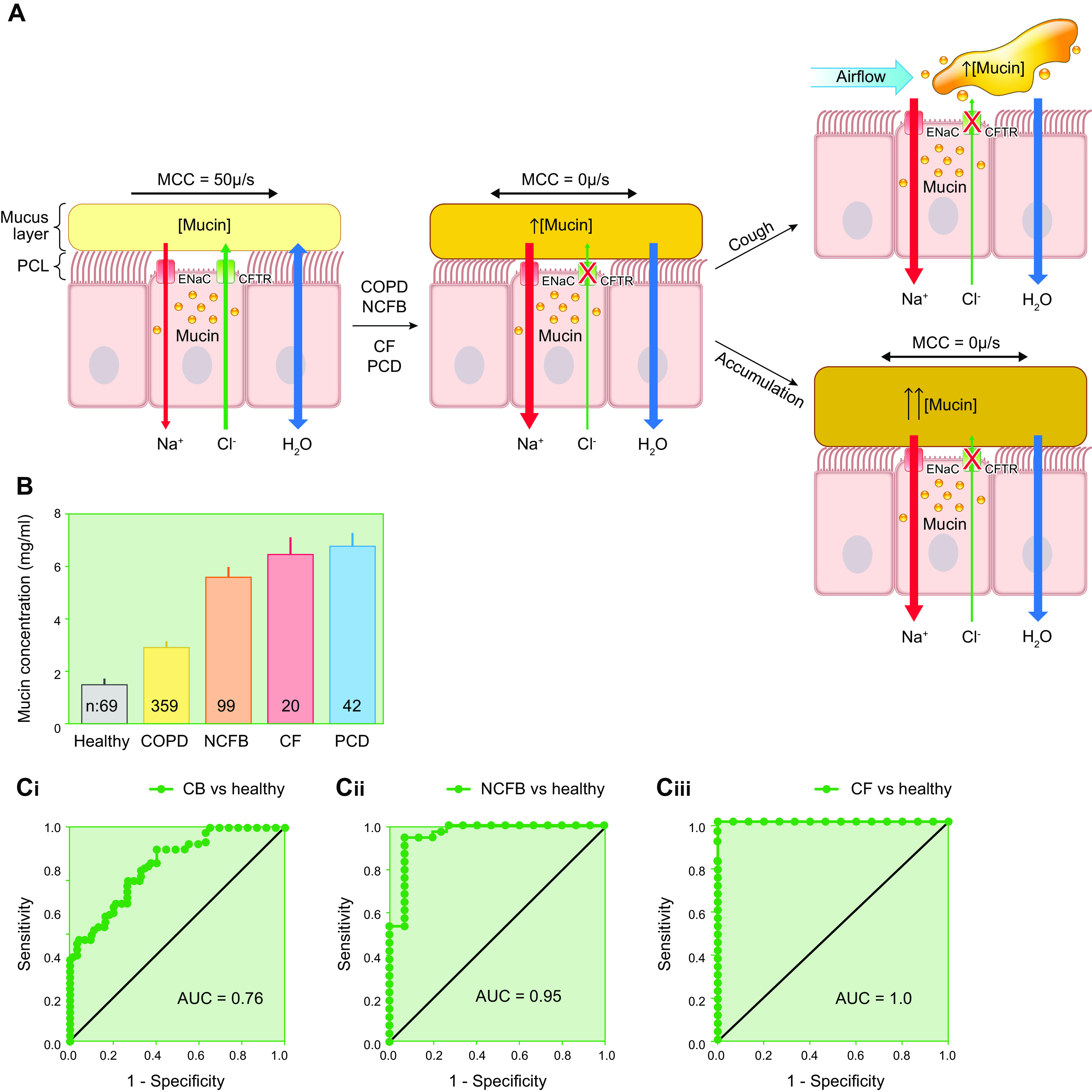
Muco-obstructive disease pathogenesis and total mucin concentrations in health and disease. A: progression from normal to muco-obstructed airway states. In healthy persons (left), well-balanced epithelial sodium (Na+) absorption and chloride (Cl−) secretion hydrates airway surfaces and promotes efficient mucociliary clearance (MCC). In persons with muco-obstructive lung disease (center), an imbalance of ion/fluid transport coupled with mucin hypersecretion increases mucin concentrations in the mucus layer, osmotically compresses the periciliary layer (PCL), and slows/eventually abolishes MCC. Note, the red X over the cystic fibrosis transmembrane regulator (CFTR) depicts an acquired structural or regulatory defect in CFTR Cl− secretory function, and the increased epithelial Na+ channel (ENaC) activity depicts acceleration of Na+ transport due to failures of regulatory inhibition (25). The adherent mucus may be expelled as sputum by cough (top right). Mucus that cannot be expelled by cough accumulates, concentrates, obstructs airflow, and becomes the nidus for infection (bottom right). CF, cystic fibrosis; CFTR, cystic fibrosis transmembrane conductance regulator; COPD, chronic obstructive pulmonary disease; ENaC, epithelial sodium channel; NCFB, non-cystic fibrosis bronchiectasis; PCD, primary ciliary dyskinesia (25). B: total sputum mucin concentrations in healthy persons and subjects with COPD, NCFB, CF, or PCD. Sputum mucin concentrations were measured by high-performance liquid chromatography and refractometry (400, 401). Bars indicate SE, and n = number of subjects studied. C: receiver operating characteristic (ROC) curves for sputum mucus concentration in diseased vs. healthy subjects. Ci: cigarette smoke-induced chronic bronchitis (CB) vs. healthy subjects. Area under the curve (AUC) = 0.76. Cii: NCFB vs. healthy subjects. AUC = 0.95. Ciii: CF vs. healthy subjects. AUC = 1.0. Content from Refs. 25 and 77 used with permission from New England Journal of Medicine. Cii uses data from Ref. 358, with permission from American Journal of Respiratory and Critical Care Medicine.
8.2. Pathogenesis of Muco-obstruction: Plaque Formation
An important feature of muco-obstructive lung diseases is the heterogeneity of expression of disease within an individual lung (18, 23). It is unlikely that this heterogeneity reflects heterogeneous defects in intrinsic innate host defense mechanisms within the lung. It is more likely that the heterogeneity results from the “oral-pulmonary axis,” i.e., the heterogeneous (“patchy”) aspiration of oral/gastric contents and viruses into the lung (402–408).
A common feature that unifies muco-obstructive diseases is the onset of mucus-obstructive disease in the small airways (18, 68, 69, 357, 396, 409, 410). This phenomenon likely reflects at least in part “physics,” i.e., the inability to generate the airflow rates required to clear hyperconcentrated, adhesive mucus from the small airways of COPD, CF, PCD, and non-CF bronchiectasis patients (see sect. 6; Refs. 116, 411).
The intrapulmonary airways are defended against infection by both mechanical mucus clearance and the activities of secreted antimicrobial proteins/peptides and immunoglobulins (15, 314, 412). The ability of antimicrobial activities to suppress bacterial infection replication is likely finite, e.g., hours (412). Consequently, a kinetic “horse race” is manifest in the lung, pitting the kinetics of acquisition of bacterial resistance to antimicrobial suppression against the kinetics of mechanical (mucus) clearance of bacteria. Slowing of clearance alone, however, is probably not sufficient to produce infection/disease. The pathogenesis of muco-obstructive diseases likely requires the formation of stationary mucus plaques and plugs within airway lumens (128, 208). Animal models have been generated that produce 1) mucus concentration-dependent, heterogeneous airway mucus plaque/plug formation with moderate defects in average lung MCC (βENaC mice) and 2) a complete cessation of MCC without mucin plaques/plugs (Muc5b−/− mice). The full spectrum of muco-obstructive diseases, including airflow obstruction, inflammation, and intermittent/persistent infection, was observed in mucus-plugged βENaC mice but not in Muc5b−/− mice, demonstrating the relative importance of mucus stasis/plaque formation versus defective MCC alone (128, 208, 413–415).
Mucus plaque/plug formation often is triggered by mucin secretory and inflammatory responses associated with viral infections and/or aspiration (23, 402, 403, 416–420) (FIGURE 16, A and B). A key component of plaque/plug formation is the secretion of mucins without sufficient fluid to hydrate newly secreted mucins and “flush” them with trapped toxicants from airway surfaces. In CF (see below), the failure to secrete fluid reflects genetic defects in CFTR ion transport function. In non-CF diseases, the mechanisms that mediate failure to secrete sufficient fluid to adequately hydrate newly secreted mucins are more diverse and complex. Disease-specific mechanisms are described below, but a common mechanism may involve local airway obstruction, due to either aspiration or virus-induced mucus/cellular plugging, with airway epithelial hypoxia and hypoxia-induced abnormalities in the regulation of ion transport that limit the ability to hydrate airway surfaces appropriately (421–423).
FIGURE 16.

Pathogenetic sequence linking chronic injury, mucin secretion but defective hydration, and hypoxia-induced and IL-1α/β-dominated positive feedback cycles. A: chronic injury produces a muco-obstructed epithelium with limited fluid secretory activity, modest mucin hypersecretion, and moderately hyperconcentrated mucin/mucus. B: heterogeneous acute insults to the lung stimulate mucin secretion without adequate fluid secretion. The effect is to increase mucus concentration, produce mucus accumulation and adhesion to airway surfaces, and generate distances for O2 diffusion that limit O2 availability to airway epithelia, resulting in luminal/epithelial hypoxia (blue). C: luminal hypoxia stimulates epithelial release of IL-1α and macrophage release of IL-1β. Both IL-1 cytokines stimulate mucin secretion via interactions with epithelial IL1 receptors (IL1R1) without adequate fluid secretion, producing a further accumulation of hyperconcentrated mucus on airway surfaces and worsened hypoxia (increased epithelial cell blue color). The positive feedback cycles generated are depicted as red plus signs. Note that IL-1α/β also stimulate polymorphoneutrophilic (PMN) inflammation, which may trigger parallel/additional positive-feedback cycles (424, 425). CXCL1, chemokine (CXC motif) ligand 1; ERN2, endoribonuclease 2; SPDEF, SAM-pointed domain-containing Ets transcription factor.
With the mismatch of mucin versus airway hydration/fluid secretion, mucus becomes hyperconcentrated, static mucus plaques form, and positive feedback (“vicious”) muco-inflammatory cycles are initiated (426) (FIGURE 16C). As one limb of this cycle, mucus plaques/plugs further limit the availability of oxygen to underlying airway epithelial cells, producing worsened epithelial hypoxia, release of preformed IL-1α, and stimulation of additional mucin secretion (427, 428). As a second component of the cycle, macrophages trapped in thickened mucus release IL-1β, which also accelerates mucin secretion (211). In part, this response may be a response to hypoxia and hypoxia-inducible factor (HIF)-1/2α activation (429, 430). In part, the macrophage activation may reflect a more general epigenetic reprogramming of macrophages in response to hyperconcentrated mucus (431).
Persistent airway luminal plaque/plug formation also stimulates neutrophilic inflammation, in part via IL-1/IL-8 signaling, which also increases mucin secretion, e.g., via released neutrophil elastase (371, 425, 432, 433), and produces airway epithelial remodeling, including goblet cell metaplasia (12, 434). Undoubtedly, other macrophage products (TNF-α, IL-6) and other inflammatory cells in both the airway lumen and submucosa participate in this cycle, especially with time (435). Ultimately, severe/persistent muco-obstructed plaques become infected with bacteria, which is typical of these diseases (see below).
8.3. Bacterial Infection of Mucus Plaques
Bacterial infection is a common feature of muco-obstructive lung diseases and likely accentuates the production of a human neutrophil elastase (HNE)-rich mucus milieu that promotes airway wall damage and, ultimately, bronchiectasis (see below). In contrast to bacterial infection of other mucosal surfaces, e.g., ocular or bladder surfaces, most airway bacterial infections do not involve direct attachment to airway epithelial surfaces (436). Furthermore, it is rare for bacteria to penetrate airway walls and produce sepsis, even in diseases with very high intensity (>108 cfu/mL mucus) infections as noted in CF (437). The most prominent exception to this rule may be Burkholderia cepacia, which appears to secrete sufficient mucinases to penetrate the glycocalyx, disrupt tight junctions, and seed the vascular compartment in some people with CF (438).
Data from CF and other muco-obstructive diseases, including COPD, indicate that the major sites of infection in muco-obstructive lung diseases are intraluminal mucus plaques and plugs (259, 436). Mucus plaques promote bacterial infection by multiple mechanisms in addition to being associated with a failure of clearance. For example, hyperconcentrated mucus plaques exhibit “tightened”/smaller mucin mesh sizes (smaller correlation lengths) that limit the ability of neutrophils to penetrate mucus plaques to capture and kill bacteria (439). The tight mucin mesh also promotes the formation of biofilms by restricting both bacterial mobility and the diffusion of autoinducers from bacterial sites of synthesis to promote high local autoinducer concentrations (80, 440, 441). Importantly, mucus plaques tend to be anaerobic because of the limitation of oxygen diffusion through plaques to actively O2-consuming airway epithelial cells. Notably, the presence of O2-consuming bacteria within a mucus plaque can also greatly reduce intramucus O2 concentrations (442–444).
A wealth of microbiome data from CF, NCFB, PCD, and COPD subjects suggests that an oropharynx-lung aspiration axis exists in which anaerobes are the first bacteria to infect hypoxic mucus plaques in the muco-obstructed lung (445–449). An important recent concept is that anaerobes may modify the static/hypoxic mucus environment by releasing sugars from mucins via glycosidase activities and ferment them, with the fermentation products acting as substrates for invasion by fermentation-defective classical pathogens, e.g., Staphylococcus and Pseudomonas (122, 450–452). These findings have clinical implications with respect to the importance of a report of “oral pharyngeal flora” in sputum samples from muco-obstructive subjects and their importance in disease pathogenesis.
8.4. Airway Wall Ectasia
Airway ectasia is defined as dilations of airway lumens/diameters due to airway damage and likely is a consequence of long-standing, intensely inflamed/infected intraluminal mucus plaques/plugs (296, 453, 454). Traditionally, airway wall ectasia has been assigned anatomically by computed tomography (CT) to bronchial regions, although pathological studies suggest that bronchioles may be a major site of ectasia in muco-obstructive diseases (68, 455). Typically, ectatic airways are associated pathologically with a loss of airway wall elastin and collagen and, if bronchial in location, diminution/loss of cartilage (296, 397). A second major feature of ectasia is an increase in the size/caliber of the bronchial circulation (456, 457). A striking feature of ectatic airways is the preservation of the lining airway epithelium, i.e., ectasia does not typically represent an ulcerlike lesion with a loss of the epithelium. This observation suggests that the proteases that damage airway walls do not permeate from infected mucus plaques/plugs through the epithelium to the submucosa (458). Rather, epithelial preservation suggests that the epithelium itself may orchestrate destruction of the airway wall directly via secretion of proteases and/or by secreting chemokines that direct protease-laden inflammatory cells into the submucosal compartment (424, 425). Notably, airway epithelia that line ectatic airway regions may be chronically hypoxic and, by analogy to other hypoxic tissues, exhibit the proteolytic, proangiogenesis, and preinflammatory features that orchestrate airway wall ectasia (459–467). Future studies of chronic airway epithelial hypoxia appear warranted.
8.5. Diagnostic Utility of Mucus Hyperconcentration in Muco-obstructive Diseases
Dating from the original description of chronic bronchitis by Dr. William Stokes in 1837, the principal tools to diagnose “chronic bronchitis” have been patient-related outcomes (468). Typical questions relating to cough and sputum production are contained in the St. George’s Respiratory Questionnaire (SGRQ) (10, 469, 470). These tools are useful but exhibit the inevitable problems of recall time, recall reproducibility, and repeatability. Consequently, an objective biomarker of the cough and sputum features of the muco-obstructive syndrome is needed.
The observation that mucus hyperconcentration is a requisite feature of sputum production suggests that a measure of mucus concentration may provide such a biomarker (see sect. 6). Both sputum percent total solids and total mucin concentration data have been reported as potential biomarkers of muco-obstructive disease (14, 77). Receiver operating characteristic (ROC) curve analyses for the mucus hyperconcentrative component of COPD, NCFB, and CF are shown in FIGURE 15C. Sputum concentration in general appears to be a good biomarker for the muco-obstructive clinical syndrome. Notably, as muco-obstructive disease severity worsens, e.g., comparing CF with COPD, the performance of sputum concentration as a biomarker improves. Current studies suggest that MUC5AC, because of its lower basal levels in health and high reactivity to stresses, may provide a particularly sensitive mucin-based biomarker for muco-obstructive diseases (49). Measurement of muco-obstructive lung disease-specific globular protein biomarkers, as well as novel MRI-based sputum measures, may also improve the sensitivity and specificity of mucus concentration-dependent biomarkers for muco-obstructive lung diseases in general and for specific muco-obstructive lung diseases (49, 471).
9. CYSTIC FIBROSIS: AN OVERVIEW OF A PROTOTYPIC MUCUS HYPERCONCENTRATION DISEASE
CF is a monogenetic disease that is caused by mutations in the CFTR gene (395, 472–474). CF is a multisystem disease that affects diverse body epithelial surfaces, but chronic lung disease, reflecting muco-obstruction and bacterial infection, is the most common cause of death. As reflected in its alternative name, i.e., mucoviscidosis (11, 475), abnormal mucus is a common abnormality in most affected organs. Based on this observation, an intensive investigation of mucus abnormalities in CF lung disease has been pursued, including studies of the relationship of the gel-on-brush hypothesis to the pathogenesis of CF lung disease.
9.1. CF-CFTR-Mediated Failure of Airway Surface Hydration
The presence of abnormally concentrated mucus in CF lungs almost undoubtedly reflects a primary abnormality of CF airway epithelial ion transport. A defect in CFTR-mediated anion secretion (Cl−/bicarbonate) has been universally described (395, 437, 476, 477). The measured rates of Na+ absorption in CF airway epithelia have varied from “normal” to elevated (260, 478–482). As noted above, even a “normal” rate of Na+ absorption juxtaposed to the absence of CFTR-mediated anion/fluid secretion would render CF airway epithelia vulnerable to volume (fluid) hyperabsorption.
In periods of clinical stability, it is probable that the singular activity of extracellular ATP to inhibit ENaC and activate CACC (TMEM16a)-mediated Cl− secretion via P2Y2R activation is sufficient to maintain airway surface liquid hydration adequate for mucociliary transport (214) (FIGURE 17Ai, left). This notion is consistent with evidence of normal mucociliary transport rates in the central regions of the CF lung from radionuclide scanning studies (401). As discussed below, the sole dependence of CF airways on extracellular ATP to regulate ASL volume renders the CF lung vulnerable to exogenous insults that adversely perturb the extracellular ATP signaling pathway (FIGURES 16Biv and 17Ai, right). When such insults occur, CF mucus is predicted to become hyperconcentrated. Failure to resolve mucus hyperconcentration and consequent plaque/plug formation is predicted to be a key factor driving CF pathogenesis (see FIGURES 15A, 16, and 17Ai).
FIGURE 17.
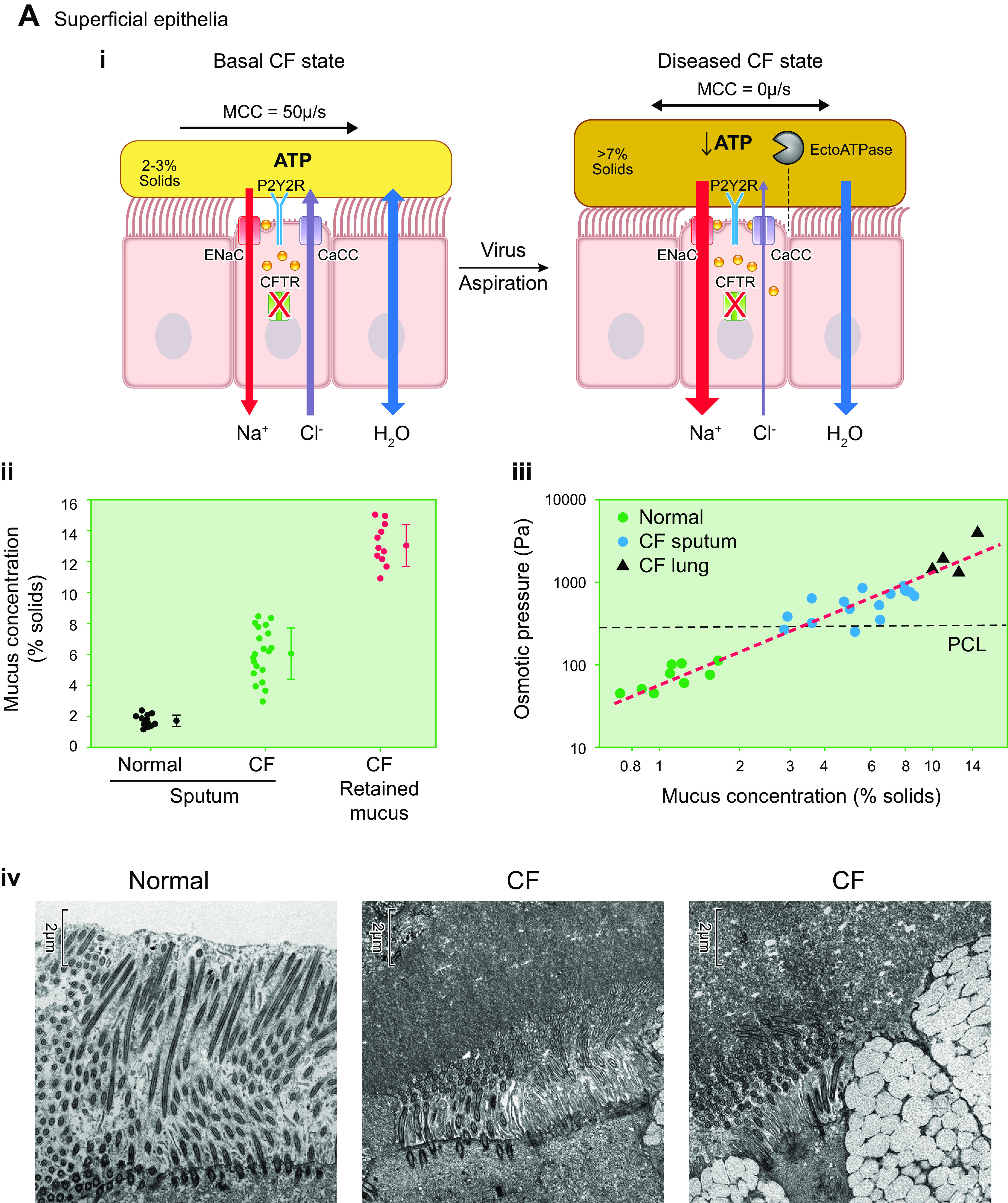
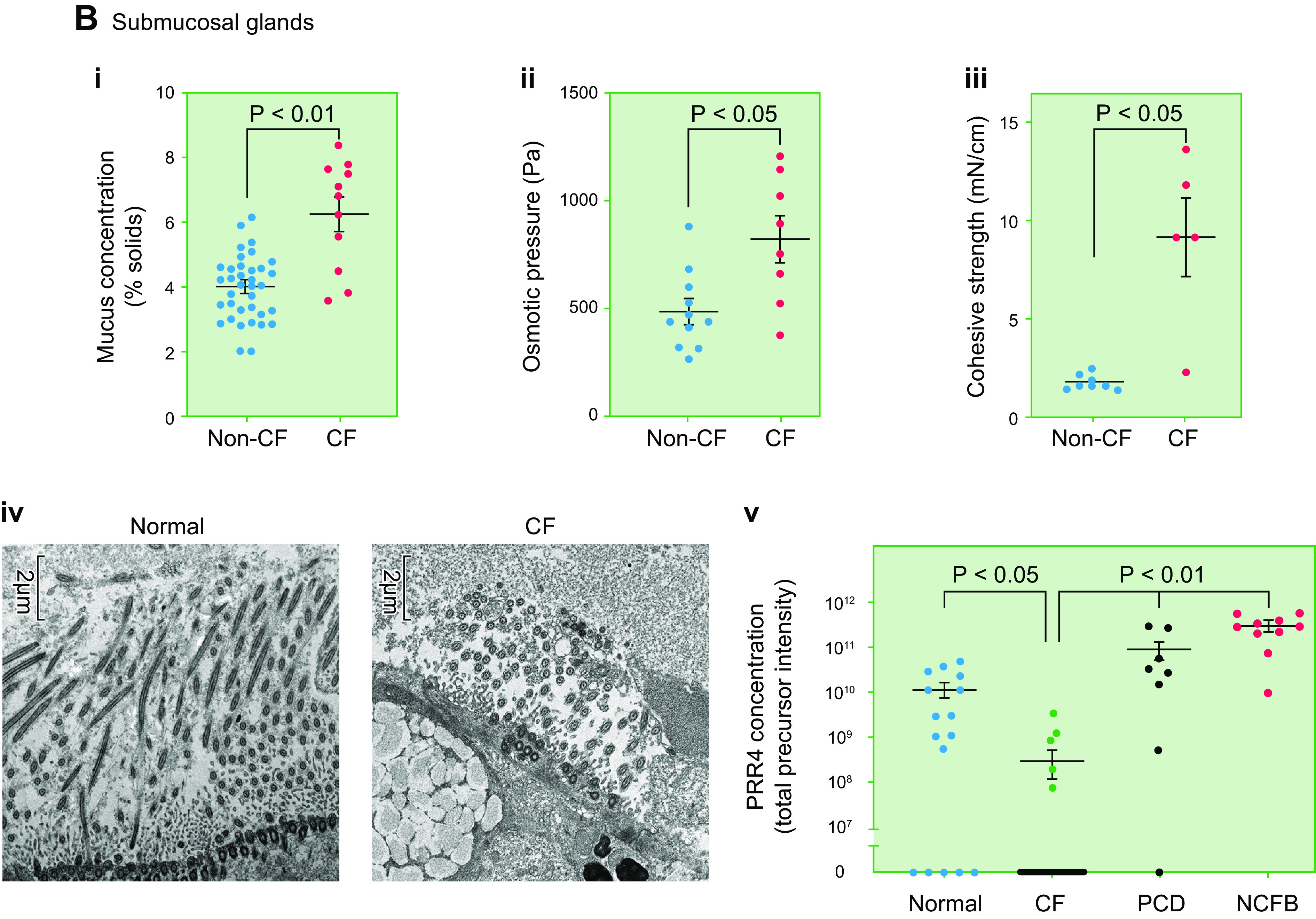
Unified pathogenesis of CF airways disease based on mucus hyperconcentration. A: superficial airway epithelial mucus hyperconcentration contribution to CF airways disease. Ai: one scenario that depicts the generation of hyperconcentrated mucus on CF airway surfaces. Ai, left: in the basal CF state, CFTR fails (green bars with red X) to mature and localize to the apical cell membrane consequent to defective folding/maturation. In basal periods of health (clinical stability), adenosine (ADO)-regulated CFTR-mediated Cl− secretion is missing, but it is probable that extracellular ATP-dependent regulation alone of ENaC-mediated fluid absorption (inhibition) and CaCC-mediated Cl−/fluid secretion (accelerated) maintains mucus hydration adequate for mucociliary transport. This notion is consistent with normal ASL heights (volumes) in CF airway cultures with cyclic compression stress (mimicking breathing) and preserved central airway MCC rates in vivo (153, 401). Ai, right: with a viral infection and likely aspiration, extracellular ATPase expression (e.g., ecto-ATPases) increases, extracellular ATP levels fall, and, in the absence of downstream adenosine-CFTR compensatory mechanisms, the balance of ion and fluid transport is tipped toward excessive fluid absorption with consequent mucus hyperconcentration and stasis (153). Aii: total % solids content of respiratory secretions for adult normal and CF subjects. Sputum was obtained for normal (non-CF subjects) and CF subjects, whereas “retained” CF mucus was removed from excised CF lungs at time of transplant (76). n = 11–20. Aiii: relationship between the total % solids content of human-derived mucus samples and osmotic pressure. Samples depicted were from normal (green) and CF (blue) sputum, and retained airway mucus samples were obtained from excised CF lungs at the time of transplantation (black). Red dashed line represents the best fit of Eq. 3. Aiv: electron micrographs of airways resected during transplant procedures. Left: normal proximal airway with fully extended cilia. Middle: thickened, hyperconcentrated mucus layer compressing cilia in CF airway. Right: thickened, hyperconcentrated mucus compressing cilia over ciliated cell and PCL-G over secretory (goblet) cell in CF airway. Scale bars, 2 µm. See glossary for abbreviations. Content from Ref. 25 used with permission from New England Journal of Medicine. Aiii uses content from Ref. 76, with permission from Journal of Clinical Investigation. B: submucosal gland (SMG) mucus hyperconcentration contribution to proximal airway CF airways disease. Bi: SMG mucus concentration expressed as total percent solids for SMG mucus obtained from normal vs. CF excised lungs. Bii: osmotic pressure measured in SMG mucus samples obtained from normal vs. CF excised lungs. Biii: cohesive strength of SMG mucus obtained from healthy vs. CF excised lungs. Biv: transmission electron micrographs of SMG ducts obtained from normal (left) vs. CF (right) excised lungs. Scale bars = 2 µm. Mean cilial height in normals = 6.25 ± 0.22 μm SE; mean ciliary height in CF = 3.78 ± 0.26 µm SE; P < 0.001 (531). Note, all data displayed in Bi-Biv obtained from mucus/ducts of normal vs. excised CF lungs exposed to acetylcholine (10−5 M, 2 h) to facilitate mucus collection (531). Bv: proline-rich peptide 4 (PRR4) as a biomarker for submucosal gland secretion in human subjects in vivo. PRR4 levels were measured in sputum collected from normal subjects, people with CF, and non-CF bronchiectasis (NCFB) and primary ciliary dyskinesia (PCD) disease control subjects. PRR4 levels were measured by mass spectroscopy. B uses content from Ref. 531, with permission from Science Advances.
9.2. Evidence for Mucus Hyperconcentration as a Disease-Causing Pathway
Mucus hyperconcentration as a feature of CF lung disease is perhaps best documented in CF adults. Direct measures of sputum mucin concentrations, intrapulmonary mucus percent solids, and mucus osmotic pressure measurements have been made in adult CF versus normal subjects (76, 373) (FIGURE 17, Aii and Aiii). These data show that in health the measured mucus/sputum osmotic pressure (100 Pa, dominated by globular protein at 1% organic solids) was, as predicted, below that of the osmotic modulus of the PCL (500 Pa). In contrast, CF sputa exhibited osmotic pressures (>500 Pa, reflecting a significant mucin osmotic component at > 6% organic solids) on average above those of basal PCL modulus values. The spontaneous production of sputum by CF subjects suggests that the high airflows in the CF trachea associated with cough can overcome mucus hyperconcentration-dependent adhesive interactions with tracheal surfaces and expectorate mucus (116). Importantly, mucus was also obtained from CF lungs excised at transplant, allowing evaluation of mucus that could not be coughed out of the lung (76) (FIGURE 17, Aii and Aiii). Notably, the noncoughable CF lung mucus had higher organic solids concentrations and very high osmotic pressures, e.g., >1,000 Pa, dominated by mucin osmotic pressures at 10–14% organic solids. These parameters are consistent with intrapulmonary mucus that was retained because it could not be cleared by MCC or cough.
9.3. Consequences of Mucus Hyperconcentration vs. pH on Mucus Biophysical Properties
It is well established that the mucus layer of CF patients has abnormal rheological properties, e.g., raised viscosity and elasticity, compared with normal subjects (79, 373). There are currently competing hypotheses describing the mechanisms producing these abnormalities. One hypothesis, as described above, states that impaired Cl− secretion by defective CFTR, coupled to intact and/or raised Na+ transport, produces ion/fluid hyperabsorption and a highly concentrated mucus with increased viscoelastic (and osmotic) properties (29, 80, 153, 233, 373, 483, 484). A competing hypothesis posits that the defective bicarbonate transport by CFTR produces an acidification of the ASL, deranging the rheological properties of the mucus layer and impeding clearance (307, 485–487).
There is ample evidence that the rheological properties of mucus are highly correlated with mucus concentration in CF patients (79, 322, 360, 373) as well as patients with other muco-obstructive pulmonary disease, e.g., asthma (488, 489), COPD (490–492), and non-CF bronchiectasis (358). Over the relatively neutral pH ranges relevant to CF, the macroscopic rheologic properties of airway mucus more closely correlate with mucus concentration than pH. When a modest increase was made in the concentration of mucus, increasing from 1% to 3% organic solids, the complex viscosity of the fluid increased sixfold (79), consistent with data shown in FIGURE 13B. By contrast, when the pH of mucus was acidified 100-fold, from 8 to 6, the complex viscosity of mucus increased only 1.5-fold (79). Given that mucus concentrations associated with CF are typically 5–8% solids (29) and pH changes associated with CF are typically ∼0.5 pH unit (174, 493), these data confirm that mucus rheology is more tightly correlated with concentration than pH. Similar patterns were observed in microrheology experiments utilizing particle bead tracking (79). Expanding these findings over a wider range of frequencies and mucus concentrations is a future need.
The results favoring the dominance of mucus concentration versus pH on airway mucus rheologic properties are consistent with previous studies of mucus rheology in other organs. For example, reductions of pH by ∼4 pH units (relevant to the stomach) were required to produce order of magnitude increases in G′ and G′′ in gastric mucus (304, 334, 487, 494). In contrast, Georgiades et al. (304) reported that a 1-log increase in isolated gastric mucin concentration increased the viscosity of the solution by two orders of magnitude. In sum, these studies indicate that mucus concentration, not pH, is the most important factor in determining the viscoelastic rheologic properties of mucus relevant to airway clearance.
Fluorescence recovery after photobleaching (FRAP) techniques, which as discussed above probe nanoscopic length scales well below mucin mesh size, reported pH-dependent increases in the viscosity of CF pig airway mucus (487). These measurements likely probed pH-dependent interactions between probe molecules and the solvent, not the mucin mesh component of mucus (79). This conclusion was confirmed in FRAP studies comparing viscosity of whole sputum versus sputum cleared of mucins by centrifugation and filtering (79).
Finally, macroscopic mucus transport measurements directly measuring the effects of changes in concentration and pH on mucus transportability have been made. In contrast to large effects of mucus hyperconcentration, reductions in pH in ranges relevant to airway pH had no detectable effects on mucociliary transport rates or osmotic pressure (79). Furthermore, studies of mucus adhesion and cohesion, relevant to cough clearance, suggested that adhesive and cohesive strengths were not altered by acidification of or reduction in mucus bicarbonate concentration in ranges relevant to airway physiology (1). In contrast, mucus concentration had demonstrable, large effects on both adhesion and cohesion (79).
9.4. Mucus in Early CF Pathogenesis
Studies from the Australian Respiratory Early Surveillance Team for Cystic Fibrosis (AREST CF) study in Australia have identified important aspects of the role of abnormal mucus in early disease acquisition in the neonatal/early childhood CF lung (23). It appears that viral infection and/or gastric aspiration in infants/neonates stimulate inflammation and accelerate mucin secretion (23, 402, 403, 416–418). The inability of the dysregulated CF epithelium to properly hydrate newly secreted mucins (see FIGURE 16B and below) triggers the formation of hyperconcentrated mucus plaques on CF airway surfaces that become the nidus for persistent airflow obstruction and inflammation. Importantly, there is a failure in CF to resolve the muco-inflammatory state compared with resolution in normal infants. Although there has been robust discussion on whether the CF epithelium is intrinsically hyperinflammatory (495–498), studies of well-differentiated CF airway cells and animal models do not support such a concept (499–502). Rather, the apparent hyperinflammation likely reflects failure to limit the continued secretion of mucin, in the absence of adequate fluid, generating positive feedback cycles that include airway mucus accumulation/plugging, epithelial hypoxia, and inflammation (503, 504) (FIGURE 16C).
The AREST studies also addressed other areas key to early CF pathogenesis. For example, failure to secrete bicarbonate might suggest a role of airway surface pH in early CF lung disease. However, direct lower airway pH measurements in normal versus CF infants in the AREST cohort failed to detect a difference in airway pH (79, 173). Well documented in this cohort was that the persistent muco-inflammatory environment can precede bacterial infection, an observation consistent with animal models of CF (23, 505). As evidenced by the fact that the first bacteria that invade and proliferate in young CF subject lungs are strict anaerobes, e.g., Prevotella and Veillonella, this persistent anaerobic muco-inflammatory environment constitutes a major risk factor for the onset of polymicrobial bacterial infection in the CF lung (23, 445, 449, 506).
9.5. Evolution to Established (Later) CF Lung Disease
It should be noted that early CF lung disease is heterogeneous, with only 5–6% of the lung exhibiting areas of bronchiectasis by age 6 yr (507, 508). CF lung diseases likely progress in part via intermittent disease “exacerbations” that reflect at least two mechanisms. First, there is heterogeneous spread of disease to new areas (509). This spread likely reflects aspiration of respiratory viruses via a nasal-oral axis into previously “normal” areas of the CF lung that produce “exacerbations” of disease (510–513). Mechanistically, viruses stimulate an upregulation of extracellular ecto-ATPases that reduce ATP levels, abolish fluid secretion, and produce new areas of mucus hyperconcentration that often fail to resolve (FIGURE 17Ai, right). Gastric aspiration may produce similar consequences (514). Second, CF lung disease can intensify locally within the lung. It is well documented that the acquisition of the classical CF bacterial pathogens, i.e., Staphylococcus and Pseudomonas aeruginosa, produces a downhill clinical course in CF lung health and pulmonary function (395). Recent data have suggested that new positive-feedback mucin secretion without fluid secretion cycles triggered by bacterial products, including LPS and flagellin (515), may interact with hypoxia/IL-1α/β feedback cycles to mediate this downhill trend in CF subjects (211, 516, 517). The final results are mucus-plugged airways that perpetuate airway obstruction, hypoxia, inflammation, and infection (FIGURE 16).
9.6. Unification of Superficial Epithelial and Submucosal Gland Contributions to CF Lung Pathogenesis
Although it is likely that the aglandular small airways are the earliest and most severely affected region of the CF lung, there is also a large airway component to CF lung disease. As reviewed above, small airway disease pathogenesis reflects the failure of the superficial epithelium to properly hydrate secreted mucins, with consequent mucus hyperconcentration and mucus plaque/plug formation. The role of the SMG in CF large airway disease pathogenesis is more controversial. Proposed CF pathophysiologies include 1) failure to secrete sufficient SMG mucus to provide adequate hydration and host antimicrobial activities to airway surfaces (20, 518–521) and 2) secretion of bundles/strands onto airway surfaces that fail to detach from SMG orifices to “cleanse” airway surfaces and/or adhere to airway surfaces and become the nidus for persistent infection (63, 64, 522–525).
Studies of freshly excised normal and CF SMGs, coupled to studies of the anion secretion-inhibited excised pig model of CF, provided data that link SMG disease pathogenesis to that of the superficial epithelium. Notably, Ballard and colleagues (526–529) reported that block of anion secretion in pig SMGs inhibited fluid but not mucin/protein secretion, producing a hyperconcentrated SMG mucus. Ballard, and subsequently others, also reported that human CF SMG mucus was hyperconcentrated compared with normal human SMG mucus (524, 526, 527, 530). More recently, Kato et al. (531) reported that both the anion secretion-inhibited pig SMG model and human CF SMGs exhibited SMG mucus hyperconcentration in ranges associated with osmotic pressures and cohesive forces predicted to slow/abolish SMG ductal mucus flow (FIGURE 17, Bi-Biii). Transmission electron microscopic studies of SMGs from both species revealed evidence of intraductal mucus retention and PCL flattening, consistent with SMG mucus osmotic compression of ductal surfaces and cessation of flow (FIGURE 17 Biv).
Parallel clinical studies measured the concentrations of a SMG-specific protein biomarker, PRR4, by mass spectroscopy in sputum obtained from normal subjects, people with CF, and two disease control bronchiectatic populations (NCFB, PCD) (FIGURE 17 Bv). Consistent with reported SMG hypertrophy and perhaps slowed mucus clearance, sputum PRR4 levels were increased ∼10-fold in disease control bronchiectatic versus normal subjects. In striking contrast, PRR4 levels in CF sputum were ∼30-fold lower than in normal subjects and 300-fold lower than in bronchiectatic disease control subjects. These data strongly support the notion that the SMG defect in CF does not reflect production of strands that accumulate on airway surfaces but reflects the failure to secrete normal volumes of SMG mucus onto CF airway surfaces.
Collectively, the data from superficial epithelia and now SMGs support the notion that the unifying defect with respect to CF lung pathophysiology is the failure to adequately hydrate secreted mucins. The biophysical correlates of mucus hyperconcentration, i.e., increased osmotic pressure, cohesion, and adhesion, satisfactorily describe the failure of hyperconcentrated mucus to transport atop superficial airway surfaces or within SMG ducts. Interestingly, whereas mucus hyperconcentration is a common defect, the functional consequences of this abnormality in superficial epithelia and SMGs are different, but complementary, with respect to defective host defense. Superficial epithelial dysfunction produces airflow obstruction, inflammation, and infection throughout the airways. The failure of proximal airway SMGs to secrete adequate volumes of mucus limits the ability to clear proximally deposited foreign materials (see sect. 6.1) and add key antimicrobial activities to proximal airway surfaces.
9.7. Data Consistent with the Gel-on-Brush Model for CF
Data from a number of studies employing different technologies in addition to measures of mucus concentration are consistent with predictions of the gel-on-brush hypothesis that CF lung disease is characterized by heterogeneous hyperconcentrated mucus plugging, initially in small airways. First, radionuclide-based MCC measurements have demonstrated the early and dominant reduction of both cilial and cough-dependent mucus clearance in the peripheral zones versus central zones of CF subjects (401, 532). Second, micro-CT studies of excised CF lungs have identified profound mucus plugging, most predominant in the peripheral (<2 mm) airways (69). Third, the lung clearance index (LCI), which captures with high sensitivity peripheral airway disease, is perhaps the most sensitive functional measurement of early CF airflow defects (533, 534). Fourth, novel 19F-MRI technologies have detected evidence of peripheral ventilation defects, that precede decrements in FEV1, and perhaps LCI, in CF subjects with mild disease (535). Fifth, micro-optical coherence tomography (micro-OCT) measurements in vivo in animal models of CF and the human CF nasal cavity have yielded evidence of hyperconcentrated mucus, a reduction in PCL height (osmotic compression), and reductions in mucociliary transport (331, 536, 537). Finally, dual-isotope imaging studies in vivo have demonstrated fluid hyperabsorption in CF compared with normal airways, mirroring isotope flux measurements of human CF airway epithelia, that was coupled to decreased MCC (260, 261, 538).
10. COPD: AN ACQUIRED DISEASE OF MODEST MUCUS HYPERCONCENTRATION
COPD is a prevalent muco-obstructive disease, characterized by significant genetic risk, that in response to environmental agents, typically inhaled cigarette smoke, exhibits persistent airflow obstruction (19, 393, 539–541). COPD is associated with hyperconcentrated mucus (77) (FIGURE 15B). Recent studies suggest that a major trigger of COPD, e.g., cigarette smoke, is associated with an increase in mucus concentration in COPD (49, 77, 542). Importantly, mucus hyperconcentration is associated with COPD disease severity as indexed by associations between increased sputum total mucin concentrations and higher Global Initiative for Chronic Obstructive Lung Disease (GOLD) status and prospective exacerbation rates (14, 49). Increased COPD sputum total mucin concentrations were also associated with 1) CB symptoms and 2) a subgroup of early COPD subjects, i.e., GOLD 0 subjects with symptoms but normal airflow and an increased risk for accelerated CB/COPD disease (77). A major recent data set describing the importance of mucus plugging in airflow obstruction in COPD emerged from CT studies sensitive to bronchial airway mucus obstruction in COPD subjects (543–545). These studies reported striking associations between the magnitude of mucus plugging and airflow obstruction, including in COPD subjects with few/modest CB symptoms.
The epithelial defects that produce mucus hyperconcentration and plugging in COPD are complex, reflecting in part the complexity of the >4,000 materials contained in cigarette smoke (18). Exposure to cigarette smoke has been reported to produce abnormalities in CFTR-mediated Cl−/anion-mediated fluid secretion via oxidant-induced reduction of CFTR transcriptional rates and direct damage of CFTR protein in the apical membrane, causing CFTR endocytosis and degradation (546, 547). Furthermore, reductions in extracellular ATP (reflecting increased extracellular metabolism) and adenosine levels (also reflecting increased metabolism) suggest that there is also defective purinoceptor regulation of airway surface hydration in COPD (14). These defects in epithelial/ion water transport (hydration) may be amplified by cigarette smoke induction of mucin hypersecretion, as reported for both MUC5AC and MUC5B (32, 49, 542, 548, 549). Mucin hypersecretion may be particularly important in COPD because the accelerated metabolism of adenosine, coupled with defective CFTR function, may limit the ADO-CFTR hydration response to mucin secretion in COPD (246) (FIGURE 6A). Thus, multiple mechanisms focused on ion channels, regulation of ion channels, and mucin secretion rates may produce hyperconcentrated mucus in COPD.
As discussed with respect to cilia-mucus concentration interactions (see sect. 5.3), in addition to mucus hyperconcentration, there are likely important ciliary contributions to reduced MCC in COPD. Studies of radioparticle clearance in COPD subjects have reported striking defects in central airway clearance that likely reflect reduced numbers and dysfunction of cilia due to cigarette smoke exposure in this region (32–39, 356). This central clearance defect may contribute mostly to problems with expectoration (a central airways function; see sect. 6) more than airflow obstruction (a peripheral airways problem).
Note that the mucus hyperconcentration that characterizes COPD mucus is perhaps the mildest example quantitatively of the muco-obstructive disease spectrum (FIGURE 15B). Interestingly, COPD subjects also tend to have the least severe muco-obstructive lung disease, particularly with respect to the lower incidence of frank bronchiectasis in this disease population (31, 550).
11. PRIMARY CILIARY DYSKINESIA: A DISEASE OF CILIA-MUCUS INTERACTION DYSFUNCTION(S)
Primary ciliary dyskinesia (PCD) has traditionally been considered to be solely a “motor problem” with respect to defective MCC (144). Genetic defects in >50 genes that code for proteins in cilial shafts and basal bodies, with consequent abnormalities in cilial formation/beat, have been reported (551–555). However, recent data indicate that PCD sputum is abnormally concentrated, suggesting that a “second hit” may contribute to the severity of PCD lung disease (373) (FIGURES 6D and 15B). The in vitro studies that revealed the mechanotransduction pathways linking mucus concentration to cilial beat/shaft strain-induced ATP release in normal subjects also reported that this pathway was defective in airway specimens from PCD subjects with abnormal cilial motility (FIGURE 6E) (216). Thus, these data raise the possibility that the absence of the cilial motility-dependent sensing of mucus concentration limits the release of local autocrine/paracrine hydrating mediators, e.g., ATP, producing a dehydrated PCD airway surface and sputum/mucus hyperconcentration.
Interestingly, the phenotype of lung disease in PCD subjects, aside from the more basilar distribution of bronchiectasis, is similar to CF but is shifted to older ages with respect to age versus disease severity relationships (144, 556). Notable similarities to CF include a predominance of early small airway disease and the presence of anaerobes in early lung disease. Later, there is acquisition of classic muco-obstructive/bronchiectatic pathogens, e.g., Staphylococcus, H. influenza, Pseudomonas aeruginosa, and severe airway neutrophilic inflammation with raised extracellular DNA concentrations (373, 557). It is not clear whether the milder phenotype of PCD patients reflects an admixture of mild versus severe PCD genotypes, the lesser systemic disease burden than CF, the persistence of CFTR functions other than hydration, and/or other factors (558).
12. NCFB: A HOST-ENVIRONMENT DISEASE WITH A FINAL COMMON PATHWAY
NCFB is a syndrome defined by ectatic airways on CT scans (31, 296, 453, 559–564). NCFB likely reflects environmental/host interactions, reflecting a complex of host defense gene risk alleles, that produces muco-obstruction and airway ectasia as a final common pathway (397). Notably, NCFB can genetically reflect the influences of impaired intraluminal host defense, immune deficiency, and connective tissue risk alleles (565).
The recent reports that NCFB sputum is hyperconcentrated suggest that epithelial defects in the regulation of mucin hydration may also contribute to NCFB pathogenesis (99) (FIGURE 15B). However, genome-wide association studies (GWASs) have not linked ion transport genes to NCFB, and direct measurements of CFTR function have suggested that CFTR, indeed, can function normally in the regions of NCFB airway epithelia accessible for study (124). Gastric aspiration is associated with NCFB as with other muco-obstructive lung diseases (566, 567). Indeed, children with neurological disease but apparently normal lungs develop NCFB from repetitive aspiration (568, 569), features that have been phenocopied in neurological mouse models associated with aspiration (570). In parallel, “severe” viral infections have traditionally been associated with idiopathic NCFB. It is plausible that aspirated small “foreign bodies” and/or virus-associated plugs of mucus and shed cells may produce airway obstruction and local hypoxia, which may perturb airway surface hydration by an unexpected HIF-1/2α-dependent increase in Na+ absorption and a failure to produce sufficient extracellular ATP or adenosine under hypoxic conditions to activate CFTR-mediated Cl− secretion (416, 421–423). Thus, gastric aspiration and/or viral infection in people with NCFB may produce focal areas of obstruction, hypoxia-induced defective fluid transport, and hyperconcentrated mucus that produce airway damage via the hypoxic airway epithelial mechanisms described above.
Interestingly, the classic diagnostic feature of NCFB is bronchial dilation on CT scans, defined without rigorous pathological correlations that identified structures are indeed ectatic bronchi. Like other muco-obstructive diseases, NCFB is characterized by a major component of peripheral/small airway disease (397, 455, 571). As noted above, it will be important to determine unambiguously the pathological site of early NCFB, e.g., small airways, because more advanced dynamic ventilation (19F based) MRI technologies and CT algorithms are now available for early detection of small airway disease to identify and follow NCFB subjects (535, 572–574).
The microbiome of NCFB subjects is typically dominated (50%) by anaerobic bacteria, with ∼25% of patients exhibiting H. flu/Staphylococcus aureus and another 25% of patients exhibiting Pseudomonas aeruginosa as well (31, 575–577). In this context, NCFB resembles “early” CF bronchiectatic lung disease. Notably, NCFB patients may have a higher incidence of nontuberculous mycobacteria infection than other muco-obstructive diseases, perhaps approaching 40–60% of all subjects (563, 578, 579). Finally, NCFB subjects exhibit the typical behavior of recurrent acute exacerbations that are characterized, like CF, by rather small changes in the microbiome (576, 580, 581).
13. THERAPIES OF MUCO-OBSTRUCTIVE DISEASES
The general care for subjects with muco-obstructive diseases, e.g., oral and inhaled antibiotics, bronchodilators, physiotherapy, and nutrition, has been well reviewed elsewhere (582–585). With respect to the unique aspects of intraluminal airway mucus to disease management, intraluminal mucus can both 1) block aerosol delivery of therapeutic agents to affected regions of the lung and 2) bind drugs, reducing active free fractions of therapeutic agents. Both features of mucus obstruction are relevant to all therapeutic classes of agents, but these principles have been particularly well studied in the context of antibiotics (586–588). The block to delivery of inhaled antibiotics to muco-infected airway regions presumably can be bypassed by parenteral delivery. However, antibiotic binding to intraluminal mucus may reduce the free fraction of antibiotics available for antimicrobial suppression/killing and limit the effectiveness of both parenteral and inhaled antibiotics. This consideration has led to use of very high concentrations of inhaled antibiotics in nebulized solutions (589–592). Ironically, based on the observations that virtually all bacteria infecting the airways of muco-obstructive lung patients are resident in the intraluminal mucus (see sect. 7.3), the best antibiotic regimen for treatment of muco-obstructive lung disease may not include antibiotics. Rather, agents that clear bacterially infected mucus plaques and plugs may be the more effective regimen.
Recently, extraordinary disease-specific therapies for muco-obstruction, e.g., CFTR potentiators and correctors, have become available for people with CF (pwCF). These therapeutic agents exhibit striking associations between in vitro measures of airway surface hydration, increases in central and especially peripheral mucociliary clearance in vivo, and improvements in clinically relevant outcomes, e.g., FEV1, validating a mucus clearance/disease pathogenesis paradigm in CF (593, 594). Importantly, the availability of the new triple-combination corrector/potentiator compounds (Trikafta) has made highly effective modulator therapy available for ∼90% of pwCF (595–602). The gratifyingly large increases in FEV1, body weight, and symptom scores observed in phase III clinical trials with Trikafta have been mirrored in clinical practice and forecast a remarkable transformation in the health of treated pwCF (603, 604).
It is possible that the use of CFTR modulators/correctors may be extended to treat other muco-obstructive diseases. The probable best candidate is COPD because of the reported adverse effects of cigarette smoke on CFTR cell biology/function (see above). Phase II studies are exploring this concept in COPD subjects (605–607).
The focus of the remainder of this review is on alternative strategies to restore mucus hydration and clearance in muco-obstructed patients in need.
13.1. Hydrators
From a broad disease category perspective, perhaps the most direct approach to treat muco-obstructive lung diseases is to reduce the concentration of pathological mucus, i.e., rehydrate it. The simplest currently available approach to achieve this goal is to inhale osmotically active solutes, e.g., hypertonic saline or mannitol (36, 129, 401, 608–610). In clinical development are modulators of ion transport that may redirect ion transport from net absorptive to secretory directions, providing a mechanism for endogenous restoration of airway surface hydration (428, 611–618).
Hypertonic saline (HS) is the most widely investigated inhaled osmotic therapy for muco-obstructive lung diseases (FIGURE 18). The use of HS is best described in pwCF (401, 619, 620). In vitro studies have shown that both the initial increase in ASL height (volume) in response to apically delivered HS and the duration of the ASL expansion response are greater in CF than normal cultures (FIGURE 18A). The increased initial efficacy and prolonged durability of action reflect the fact that absorption of the NaCl deposited on CF airway surfaces is electrochemically slowed because the absence of an “open” Cl− channel (CFTR) for Cl− absorption (234, 401) (FIGURE 18, B and C). In accord with these in vitro findings, HS inhalation in pwCF produces more robust immediate and sustained increases (>4 h) in mucociliary clearance compared with normal/healthy subjects (FIGURE 18D), which correlates with improvements in FEV1 and FEF25–75% in pwCF (401, 621). Long-term administration of inhaled HS improves FEV1 and reduces exacerbation frequency in pwCF by ∼50% (620, 621). The Cochrane Review rates the evidence for HS therapy in CF as a “B” (622).
FIGURE 18.
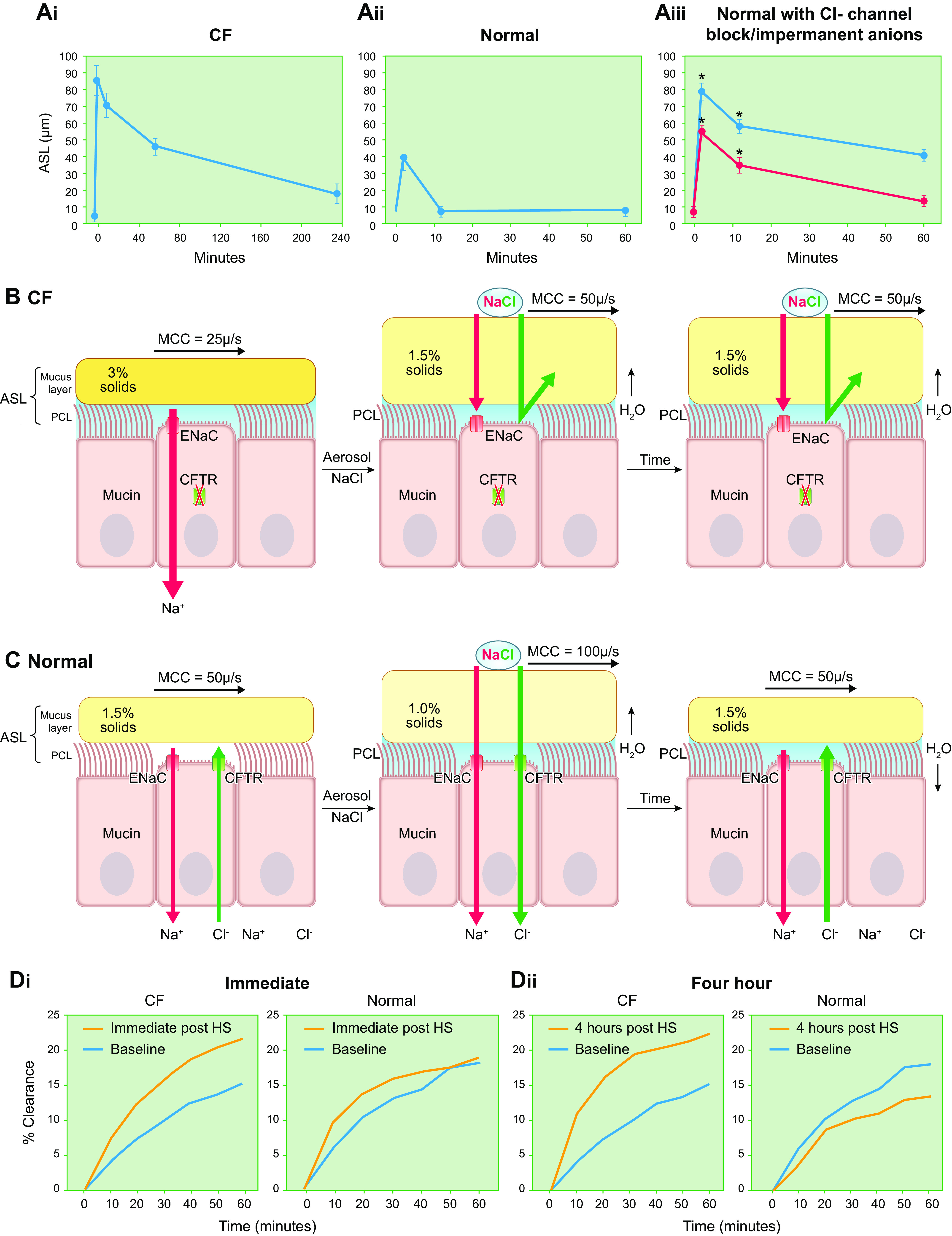
Effectiveness of inhaled hypertonic saline (HS) for airway hydration in CF vs. normal subjects. A: comparison of CF vs. normal subject human bronchial epithelial (HBE) culture airway surface liquid (ASL) height/volume responses to NaCl deposited on apical surfaces. The height of ASL covering HBE culture surfaces was measured by labeling ASL with Texas Red dextran and X-Z confocal microscopy (401). The ASL height/volume response to a common mass of apically deposited NaCl is shown for CF (Ai) vs. normal (Aii) airway epithelia. Note the larger and more long-lasting, i.e., durable, ASL height (volume) responses in CF compared with normal HBEs. The greater and more durable responses to apical osmolyte deposition in normal HBE cultures pretreated with a CFTR channel blocker (CF172, red) or exposed to a dosing solution with Cl− replaced by a CFTR impermanent anion (Na+-gluconate, blue) are consistent with the notion that Cl− absorption via CFTR is limiting for NaCl absorption in normal epithelia (Aiii). B: schema depicting deposition of hypertonic NaCl aerosols on CF airway surfaces. The deposited NaCl increases ASL osmolality and osmotically draws water to airway surfaces. In contrast to normals (C), the deposited NaCl remains for longer periods on CF airway surfaces because the transcellular route for Cl− absorption, i.e., CFTR, is absent/dysfunctional. (Ca2+-activated channels are not shown for simplicity.) Na+ is retained on airway surfaces for electrostatic reasons. The net result is that inhaled HS (NaCl) is more effective in CF in restoring MCC and clearing obstructing mucus because 1) the mass of deposited NaCl aerosols on airway surfaces at the end of nebulization is greater than in normals, reflecting the absence of NaCl absorption during nebulization, and 2) the deposited NaCl remains aerosols on airway surfaces for longer intervals, providing a more durable hydrating/therapeutic effect. C: fate of deposited HS (NaCl) on normal airway surfaces. The NaCl deposited during aerosol administration acutely increases ASL osmolality and osmotically draws water to the airway surface, diluting mucin concentrations and accelerating mucus clearance rates (MCC). However, the deposited NaCl is rapidly absorbed across the normal epithelium via “open” ENaC and CFTR channels coordinately down the NaCl gradient (NaCl lumen > cell/interstitium) generated by NaCl deposition. The net result is rapid absorption of NaCl, limited duration of airway surface hydration and short-lived acceleration of MCC. D: human whole lung in vivo mucociliary measurements utilizing inhaled radiotracer (621). Di: rates of central lung in vivo mucociliary clearance of inhaled radiotracer particles in people with CF (pwCF) and normal subjects immediately (0–60 min) after administration of aerosolized 7% HS. Dii: rates of mucociliary clearance of inhaled radiotracer particles in pwCF vs. normal subjects 4–5 h after inhalation of 7% HS. Note the greater immediate response and much more durable (4 h) response in CF compared with normal subjects. % Clearance, rate of clearance of deposited radiotracer particles. See glossary for other abbreviations. A contains data from Ref. 401 and is used with permission from New England Journal of Medicine.
Inhaled HS exhibits a shorter duration of action in non-CF airways (234, 263, 401, 623). This shortened durability of hydrating action in non-CF airway epithelia reflects the rapid absorption of deposited NaCl via “open” CFTR and ENaC channels (FIGURE 18, A and C). HS has been shown to be safe in COPD subjects for sputum induction, both during baseline periods and during exacerbations (624–626). Aerosolized HS also increased the probability of obtaining a COPD subject sputum sample, i.e., increased expectoration, and was associated with a 25% reduction in sputum concentration, i.e., sputum was diluted (627). Recent data, however, suggest that the effects of inhaled HS to restore the slowed MCC in COPD were very short in duration (∼10 min) (356). Repetitive administration of aerosolized HS to COPD subjects over a 2-wk interval was associated with minor improvements in symptoms but not in pulmonary function (356). Longer-term trials of inhaled HS in COPD have not been reported. HS appears to be relatively widely used in PCD, but a vehicle control study measuring the effects of HS in PCD reported no clinical benefit over a 12-wk interval (628, 629). Finally, data describing HS inhalation in NCFB are fragmentary (630, 631). Larger chronic duration studies with perhaps more sensitive disease indexes, e.g., LCI, are in progress.
13.2. Mucolytics
Mucolytic agents have been advocated for years for the therapy of muco-obstructive lung diseases (237, 632–637). These agents broadly target the mucins and associated proteins that contribute to mucus concentration-independent biophysical abnormalities. The most investigated class of inhaled agents to modify mucus properties is the thiol-reducing class, e.g., N-acetylcysteine (NAC) (23, 108, 237, 638, 639). Disulfide bond reducing agents have as a target reduction of S-S bonds mediating N-N and C-C terminus mucin multimerization, i.e., they reduce mucin chain lengths, i.e., molecular weight (FIGURE 2). However, the rationale for and the execution of this strategy are complex, and this strategy has not heretofore been successful for several reasons.
First, a targeted/“therapeutic” reduction of chain length may have differential effects on mucus function in health versus disease or, indeed, healthy versus diseased regions of lungs of subjects with heterogeneous muco-obstructive disease (FIGURE 19). For example, therapeutic reduction of mucin chain length from multimers to dimers increases mucin overlap concentrations (c*), reflecting the smaller mucin size (Rg) (see sect. 2 and FIGURE 19, B and C). Thus, biochemically reduced mucins in healthy subjects, or airway regions with normal mucus concentrations, may be in <c* conditions, with an attendant loss of elastic properties and reduction in viscosity. The loss of elastic modulus in <c* conditions is predicted to slow MCC and perhaps produce mucus accumulation in healthy subjects/regions with normal mucus concentrations (657). In muco-obstructed disease subjects with hyperconcentrated mucus, reductions in polymer size may be achieved while maintaining mucin concentrations above the c* values for reduced dimers (FIGURE 19C). Thus in disease, mucolytics are predicted to reduce both the elastic and viscous properties of mucus that may promote cilium- and cough-dependent clearance of accumulated mucus.
FIGURE 19.
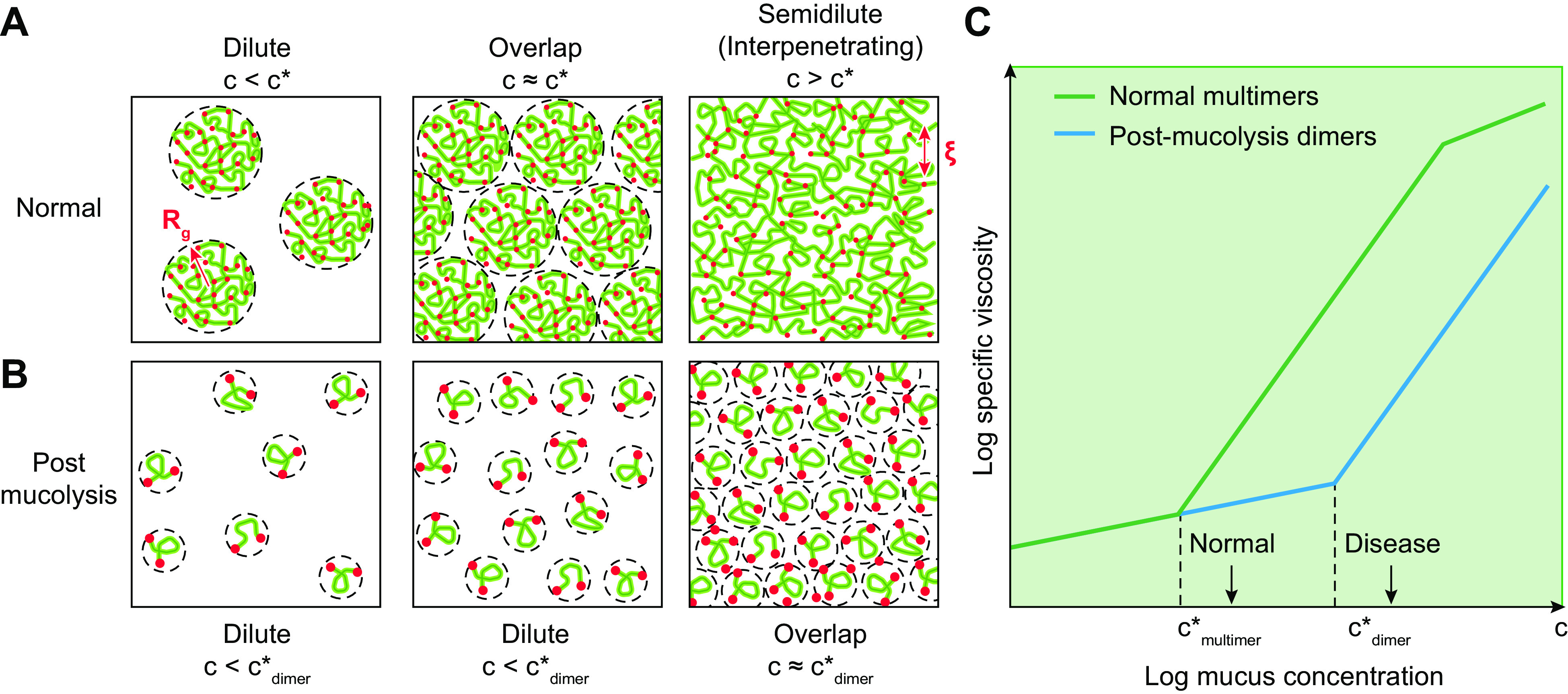
Biophysical consequences of mucolytic therapies. A: normal conditions. Normal mucins are >250 MDa in mass (molecular weight, MW) and >250 nm in size (radius of gyration, Rg, depicted by red arrow). Under normal mucus concentrations (c), i.e., 10–20 mg/mL total organic solids (∼3–6 mg/mL mucin concentrations), mucins exceed dilute conditions and are in overlap (c*) or semidilute/interpenetrating conditions and exhibit gel properties, e.g., elasticity and viscosity (η). B: mucins reduced to dimers by prototypic mucolytic agents. Dimers exhibit a MW of ∼ 5 MDa and an Rg of ∼50 nm. The mucin concentrations required to achieve overlap (c*) conditions are described as c* ≈ MW/(). Thus, at normal mucin concentrations, reduced dimeric mucins with Rg ∼5× smaller than multimeric mucins will be below c*, i.e., remain in dilute conditions. Reduced mucins (dimers) below c* will exhibit 1) a greatly reduced viscosity, predicting improved mucus coughability, and 2) an absence of an elastic modulus (not shown), predicting reduced cilium-dependent transport. C: relationships between concentrations of normal mucin multimers vs. post-mucolysis dimers and mucus-specific viscosity. The ∼3× rightward shift in the mucus concentration (c) required to achieve c* conditions for reduced (dimeric) mucins compared with multimeric mucins is shown on the x-axis (note log scale mucus concentration). A reduction in mucus viscosity is predicted for reduced (dimeric) mucins under normal and disease conditions (hyperconcentrated mucus) relative to multimeric mucins. Gellike properties, including an elastic modulus, are only predicted for reduced (dimeric) mucins in hyperconcentrated disease conditions where mucus concentrations exceed by >3× normal mucus concentrations. Figure courtesy of Dr. Scott P. O. Danielsen, Duke University, and used with permission.
Second, there is the issue of the specificity of thiol reduction by mucolytic agents. For example, there are >190 S-S bonds within a mucin macromonomer, whereas only 5 (2 N-N; 3 C-C) mediate multimerization. Accordingly, there is a large number of “off-target,” i.e., non-chain length-reducing S-S bonds, which may minimize the selectivity of chemical reducing agents. Conceptually, off-target reductions of mucins could have adverse effects. For example, reduction of S-S bonds in the CK domains could unwind this hydrophobic domain and create “sticky” sites, potentially producing novel intermucin interactions that would paradoxically increase the average MW of “reduced” mucins.
As a target for muco-obstructive diseases, reducing agents may be most useful in improving cough clearance. Recent studies of cough have demonstrated that mucus viscosity is a key variable governing mucus cohesive and adhesive properties relevant to cough efficacy (see sect. 6). In these studies, reducing agents decreased both mucus viscosity and adhesive strength independently of concentration and improved cough clearance (116). It should be noted, however, that reducing agents have also been shown to decrease the storage (elastic) modulus of mucus (measured at 1 Hz) and accelerate transport of hyperconcentrated mucus in vitro, consistent with a role for mucolysis in restoring cilia-dependent mucus clearance in disease (638). Expanding these studies over a wider frequency and concentration range is a goal for future research.
A novel target for mucolytics emerged from studies of CF infants. These studies showed that the mucus flakes and plaques recovered from CF subjects during bronchoalveolar lavage (BAL) were unable to dissolve in excess solvent, i.e., these flakes were “permanent” gels (23). Permanency can be conferred to a gel by the generation of strong intermucin S-S bonds, e.g., mediated by oxidants (117, 334, 487, 494). Treatment of CF mucus flakes with reducing agents caused them to dissolve, a response predicted to facilitate flake clearance from the lung. Unfortunately, inhaled N-acetylcysteine (NAC) does not exhibit the intrinsic activity or durability of action in human subjects in vivo required for efficacy (237, 635).
Strategies to alter the state of abnormal mucus by disrupting interactions between mucins and the other components of mucus have also been reported (16). For example, inhaled surfactants have been tested, but a favorable therapeutic ratio has not yet been reported for this class of agents (299). Because of the need for effective mucolytics in clinical practice, multiple therapies with a wide range of therapeutic targets in mucus are under development (108, 308, 309, 327, 331, 428, 637, 640–645).
Although not a mucin target, the rheological parameters of CF sputum, e.g., viscosity, are improved by cleaving extracellular DNA (363). Inhaled recombinant DNase was the first approved CF-specific inhaled therapy and has been shown to improve FEV1, reduce exacerbation frequencies, and likely modify the course of CF lung disease (646, 647). Somewhat surprisingly, DNase was not effective in NCFB and was, perhaps, even associated with an increased number of adverse events (648).
14. GAPS IN CLINICAL TRANSLATION OF MUCUS BIOCHEMICAL/BIOPHYSICAL CONCEPTS
The translation of the basic biochemical and biophysical properties of mucus to the clinic is fraught with gaps in knowledge and practice. Perhaps first and foremost, how does one measure mucus concentration in the clinic? Devices to measure the water content of small quantities of food products have been developed, so could such technologies become widely available to the pulmonary community? It is odd that we cannot measure clinically in 2022 one of the most important concentrations related to a major body organ, i.e., mucus concentration as it relates to lung function/disease.
Similarly, there are gaps in our ability to measure the quantity/distribution of mucus accumulation in the lung. CT studies/new CT algorithms hold promise of becoming automated and available to measure mucus accumulation in mid-sized airways (385, 544). However, new, more sensitive technologies are needed to measure mucus accumulation in the more vulnerable small airways. Dynamic 19F-MRI imaging holds promise to measure small-airway ventilation defects due to airway obstruction but is not specific for mucus-mediated obstruction (535).
Finally, we need better agents to clear mucus from pulmonary subjects in need. The activity of inhaled HS is likely too short to be broadly effective outside of the CF field as a global mucus hydrator, and hence newer hydrating agents must be developed. In addition, there is a total absence of agents that can break up accumulated in mucus in patients, i.e., mucolytics. The field needs such agents to debulk muco-obstructed patients, to treat acute exacerbations of mucus-associated disease, and for general ICU applications. Only when complementary hydrating and mucolytic agents are available will the field be able to test the hypothesis that clearing mucus accumulated in the lung will broadly improve airway obstruction, inflammation, and, indeed, infection.
Perhaps the most optimistic assessment of where we are at the time of this review with respect to the clinic is that we have a good start in understanding relevant aspects of mucus from genetic, cell biologic, and biophysical perspectives. Importantly, our current knowledge has allowed us to ask the key questions required to fill in the gaps in the field so that pulmonary subjects with muco-obstructive and other mucus-related disease can be treated more effectively in the future.
15. CONCLUSIONS
The innate defense of airway surfaces from inhaled infectious agents and toxicants is dominated by the mechanical clearance activity of the mucus transport system. The normal mucus transport system relies heavily on a well-orchestrated homeostasis of epithelial ion/fluid transport, airway mucin secretion, mucus concentration, and ciliary beating. Failed mucus clearance in muco-obstructive diseases follows a general final common pathophysiological pathway that reflects abnormal hydration of secreted mucins. The resultant mucus hyperconcentration produces osmotic compression of cilia, mucus adhesion, and the formation of persistent mucus plaques and plugs, which are particularly vulnerable to formation in small airways because of the low airflow-induced shear in this region. Adherent mucus plaques ultimately become the nidus for airflow obstruction, inflammation, and persistent infection.
The four diseases grouped into the muco-obstructive syndrome, i.e., COPD, NCFB, CF, and PCD, differ with respect to the epithelial abnormalities that produce mucus hyperconcentration, reflecting a range of abnormalities in ion channel function, ion channel regulation, and/or mucin secretion rates. However, these diseases exhibit common mucus hyperconcentration and inflammation-mediated modifications of mucus that render mucus more viscous, more elastic, and/or permanent/insoluble, all features that impede mucus clearance.
Therapies designed to rehydrate and restore mucous osmotic and viscous/elastic properties are rational. The challenge is to deliver these therapies to the small airways where airflow obstruction may be complete and the physics of aerosol deposition daunting. However, the concept that these diseases may be highly treatable in the future, i.e., much of the dysfunction is reversible, emanates from studies in CF in which highly effective modulators have stunningly reversed mucus obstruction in both large and small airways and produced astonishing improvements in lung health and quality of life (218, 649).
GLOSSARY
- A2B
Adenosine 2b receptor. Linked via Gs to activation of adenylate cyclase
- Adhesion strength
Energy per unit area required to separate mucus from epithelial surface
- Mucus aggregate
Collection of mucus flakes
- ASL
Airway surface liquid, i.e., the liquid in the mucus layer and PCL compartments
- Bundle
A nonswelling mucus structure with a large aspect ratio, indicative of a cylindrical geometry
- c*
Overlap concentration
- C3
Third component of complement
- CaCC
Calcium-activated chloride channel
- CBF
Ciliary beat frequency
- CF
Cystic fibrosis
- CFTR
Cystic fibrosis transmembrane conductance regulator
- Ciliated cell
Multiciliated (100–200 cilia/cell) cell responsible for providing motive force for mucus transport
- c m
Concentration of mucins
- Cohesion strength (fracture toughness)
Energy per unit area to fracture mucus
- Complex modulus
Storage [G′(ω)] and loss [G′′(ω)] moduli are real and imaginary components of complex modulus G*(ω), which is a function of oscillation frequency ω. G′(ω) and G′′(ω) are the in-phase and out-of-phase components of stress divided by strain amplitudes of shear oscillations at frequency ω.
- Cone and plate rheometer
Cone and plate is a traditional geometry employed in macroscopic rheometers. The lower interface is a flat “plate,” and the upper, rotating interface is conical. This configuration is employed to ensure constant strain (or deformation divided by height) in the entire volume.
- COPD
Chronic Obstructive Pulmonary Disease
- c os
Concentration of organic solids
- c p
Concentration of small proteins
- DMBT1
Deleted in Malignant Brain Tumors 1
- Darcy friction factor
Quantifies frictional losses due to flow through the pipe relating pressure gradient and flux
- D c
Pipe diameter
- ENaC
Epithelial sodium channel
- F
Free energy
- FC
Force generated by cilia
- FEF25–75%
Forced expiratory flow rates between 25% and 75% of total lung capacity
- FEV1
Forced expiratory volume in 1 s
- Flake
An amorphous, nonswelling mucus structure 5–100 µm in diameter
- FRC
Functional residual capacity
- FS
Stall force of dynein
- FVC
Forced vital capacity
- Gal
Galactose
- Gas-liquid transport (GLT)
Two-phase gas-liquid flow-mediated mucus clearance at airflow velocities associated with tidal breathing
- GlcNAc
N-acetylglucosamine
- Interfacial viscosity
The viscosity of a fluid at an air interface due to solute, such as mucin, adsorption to the interface
- Ionocyte
Minor cell type in airway epithelia characterized by expression of the FOXil transcription factor and high expression levels of CFTR, V-type ATPases, and ENaC
- K
Osmotic modulus
- k
Boltzmann constant
- KO
Mouse models exhibiting genetic ablation of targeted genes, i.e., knockouts
- L
Length
- Loss modulus
G′′(ω), the viscous component of the overall resistance of a material to deformation; see Complex modulus for detail
- LVR
Linear viscoelastic regime
- MCC
Mucociliary clearance
- M eff
Number average mass of osmotically active molecules in mucus
- M m
Mucin mass
- MOLD
Muco-obstructive lung disease
- M P
Number average protein mass
- MUC5AC
Human MUC5AC mucin
- Muc5ac
Murine Muc5ac mucin
- MUC5B
Human MUC5B mucin
- Muc5b
Murine Muc5b mucin
- NAC
N-acetylcysteine
- N Av
Avogadro number
- NCFB
Non-cystic fibrosis bronchiectasis
- Ni
Number of components
- NO
Nitric oxide
- N motor
Number of dynein motors per 96-nm repeat
- Osmotic modulus
The rate change of osmotic pressure with the logarithm of concentration [∂Π/∂ln(c)]
- Osmotic pressure (Π)
Rate of change of solution free energy with volume at constant number of solute molecules
- P2Y2R
Seven transmembrane purinoceptor linked via Gq to activation of phospholipase C
- Pannexin 1
A plasma membrane hemichannel that conducts ATP
- PCL
Periciliary layer
- PCL-G
Periciliary layer-glycocalyx
- Peel tester
Device to measure adhesive or cohesive strength
- Polymers
Very large molecules composed of many repeating subunits
- PRR4
Proline-Rich Protein 4
- PRSS8
Prostasin
- PTMR
Particle tracking microrheology
- Purinoceptors
As relevant to airway epithelia, seven transmembrane receptors that are activated by triphosphate (ATP, UTP) nucleotide (P2Y2 receptors) or nucleoside (adenosine) molecules (A2B)
- Q
Volumetric flow rate
- Reynolds number
A ratio of inertial to viscous forces within a fluid that helps predict whether a flow is laminar (sheetlike) or turbulent
- R g
Radius of gyration
- Secreted mucin
In airways, MUC5B and MUC5AC are dominant secreted mucins; little MUC2 protein observed in the respiratory system in health or disease
- Secretory cell
An epithelial cell that is classically defined as expressing the globular club cell secretory protein (CCSP), known as SCGB1A1 at the gene level; This cell type also expresses MUC5B, variable MUC5AC, major ion transport proteins, e.g., CFTR and ENaC, and their regulators, e.g., purinoceptors
- Shear
Deformation of a material in a direction that is parallel to the face of the material
- Shear rate
The velocity of deforming of a material in shear divided by the thickness of the material
- Shear strain
The amount of deformation of a material in a direction parallel to its surface divided by the thickness of the material
- Shear stress
The force per unit area applied in a direction parallel to the surface of the material
- SMG
Submucosal gland
- Storage modulus
The elastic component of a material resistance to deformation, denoted as G′(ω); see Complex modulus for detail
- Stress
Force per unit area
- T
Absolute temperature
- Tethered mucin
Mucin anchored to apical cell membrane via transmembrane domains; MUC1, 4, 16, and 20 are dominant tethered mucins in airway epithelia
- Viscosity
Quantity describing the resistance of material to deformation at a given rate; the ratio of shear stress to shear rate
- ΔP
Pressure difference
- η
Viscosity of a fluid
- ηbulk
Bulk viscosity of a fluid
- ηinterfacial
Viscosity of a fluid at an air interface
- ξ
Correlation length of mucins
- Π
Osmotic pressure
- ΠML
Osmotic pressure of mucus layer
- ΠPCL
Osmotic pressure of periciliary layer
GRANTS
This work was supported by National Institutes of Health Grants UH3 HL123645 and R01 HL136961 (R.C.B.); P30 DK065988 (R.C.B., B.B., D.B.H.); P01 HL108808 (R.C.B., B.B., M.R, D.B.H.); and R01 HL125280 (B.B.); by National Science Foundation Grant EFMA-1830957 (M.R.); and by Cystic Fibrosis Foundation Grants BOUCHE19R0 (R.C.B.); BOUCHE19XX0 (R.C.B., B.B., and D.B.H.); BUTTON19G0 (B.B.); and HILL19G0 and HILL20Y2-OUT (D.B.H.).
DISCLOSURES
R. C. Boucher is Chairman of the Board of Parion Sciences; is on the Scientific Advisory Board, Enterprise Therapeutics; is on the Scientific Advisory Board, Concentrix Pharma; and consults for Novartis. None of the other authors has any conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
D.B.H., B.B., M.R., and R.C.B. prepared figures; drafted manuscript; edited and revised manuscript; and approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Eric C. Roe for editorial assistance. We also thank the UNC Virtual Lung Group for two decades of discussions, Drs. William Davis and Erik Hviid Larsen for insightful comments, and all the research coworkers that generated data pertinent to the review. We further acknowledge the contributions of QiShun Tang, Dr. Scott P. O. Danielsen, and Dr. Phillipe Lorchat to this work.
APPENDIX: RHEOLOGY PRIMER
Rheology is the study of the flow of matter. Practically, rheology applies known stresses (force per unit area) or deformations to a solid or fluid and measures its response. In a traditional rheometer, an upper surface is moved relative to a fixed lower surface, or plate. The sample in immediate contact with the plate is assumed to have the same motion as the plate, a condition termed “no-slip,” which must be verified. The deformation of the sample, or strain, γ, is defined as the displacement of the upper interface between the sample and the upper plate relative to the lower sample/plate interface, δx, divided by the distance between the two interfaces, H, i.e., γ = δx/H. For the strain on a material to be constant throughout the sample, the upper surface of the rheometer is often conical, so that the height, H, between the upper and bottom interfaces increases linearly with radius, compensating for the increased displacement of the upper interface of the rheometer as a function of radius during its rational motion. The rate of deformation, or strain rate, is defined as the velocity, v, at which the upper plate moves relative to the lower plate divided by the height, H. By convention, the velocity, which is the time derivative of the displacement δx, is written as , defining the strain rate as (FIGURE A1). For liquids, the viscosity, or resistance to flow, η, of the fluid is defined as the ratio of the stress to the strain rate, or . For a solid, the shear modulus, G, or measure of the resistance of the material to deformation, is defined by the ratio of the shear stress to the strain, or G = σ/γ. However, each of these properties is dependent upon the frequency at which it is measured. For viscoelastic materials like mucus, the dynamic modulus, G*(ω), incorporates both the storage modulus, G′(ω), which is a measure of a fluid’s ability to store energy upon small oscillatory deformation with frequency ω, and the loss modulus, G′′( ω), which is a measure of a fluid’s ability to dissipate energy.
FIGURE A1.

Definition of rheological terms. During a rheological assay, a fluid is deformed by applying stress, σ, or force per unit area to deform a sample. This deformation is quantified as strain γ, or the amount of deformation (δx) divided by the gap height H.
In a cone and plate rheometer, there are several types of experiments that can be employed to characterize the physical properties of mucus. In viscometry assays, a unidirectional stress or strain rate (rotation of a plate in a particular direction—clockwise or counterclockwise) is applied to a fluid for a given period of time to measure its viscosity. These assays have been previously used to characterize the effect of surfactants and chaotropic agents such as guanidinium hydrochloride on the viscosity of mucus as a function of applied stress (650). Creep recovery assays apply a stress to a fluid for a fixed period of time, after which the applied stress is removed and the recovery of the fluid is measured. Data in a creep recovery assay are typically reported by plotting the compliance of the fluid, J, as a function of time, where J(t) = γ(t)/σ, with the initial strain at the beginning of the application of stress defined to be 0. Unlike a viscometry assay, which is only able to measure the viscosity of a liquid, a creep recovery assay can measure the physical properties of liquids, solids (like rubber), and viscoelastic materials (FIGURE A2). For a liquid, viscosity is defined by the inverse of the slope of the compliance, i.e., its time derivative , assuming that the slope of J is constant. Furthermore, for a liquid, there is no recovery. For an elastic solid, the material deforms to a constant compliance upon the application of stress where the modulus, G, is proportional to the inverse of the compliance, or G ∼ 1/J. The material then fully recovers after the removal of the applied stress. For viscoelastic materials like polymeric liquids, both a terminal, long-timescale viscosity as well as recovery are observed (651).
FIGURE A2.

Example of creep and recovery experiments. The compliance, J, is plotted vs. time during a period of active stress or creep (blue rectangle) and recovery (yellow rectangle). The viscosity of the fluid (η) is defined by the inverse of the time derivative of the compliance, and the modulus is defined as the inverse of the recoverable compliance (JR).
In addition to these unidirectional assays, the viscoelastic properties of fluids can be measured with oscillatory assays. In these assays, shear stress or stain is applied to the sample by the rheometer cone in the form of a sine wave. Measurements are taken as the magnitude (amplitude) of the response of the sample at a particular frequency of the sine wave. The rheological properties of most materials, including mucus, are nonlinear at larger strain amplitudes for oscillatory measurements or for higher strain rates for unidirectional studies, as evidenced by the shear-thinning (311, 323, 652) of mucus at high shear rates. Note that the storage and loss moduli of complex fluids, such as mucus, must be measured in the linear viscoelastic regime (LVR). The LVR is defined by the strain or stress range over which G′(ω) and G′′(ω) are independent of the amplitude of oscillation (FIGURE A3). Once the LVR is determined, the frequency dependence of G′(ω) and G′′(ω) is determined by inputting sine waves over a prescribed range of frequencies at a strain or stress that has been determined to be within the LVR (see FIGURE 7B). Although the LVR formally has no lower bound, instrument noise at low strains makes data unreliable below a given threshold.
FIGURE A3.

Determination of the linear viscoelastic regime (LVR). Top: 2 strain-sweep experiments were run at frequencies of 0.1 and 10 Hz. The LVR (black box) is determined to be the region of the strain sweep at which the least variation is observed across G′ and G′′ at both frequencies. A strain that falls above the instrumental noise threshold and prior to the onset of nonlinear effects (between 0.2% and 1%) is then selected for the frequency sweep experiment (bottom) ranging from 0.1 Hz to 10 Hz.
Particle tracking microrheology (PTMR) uses the thermally driven motion of embedded tracer particles to measure the viscoelastic properties of a fluid. Operationally, the mean squared displacement (MSD) of a particle in two dimensions is calculated over a series of time lags (τ) and then converted into the modulus through the generalized Stokes–Einstein equation (653): , where f = 1/τ. The modulus is then converted into the storage and loss moduli based on the power law dependence of the MSD, , where α = 1 for a liquid and 0 for a solid.
Typically, the viscoelastic properties of a fluid are calculated from either the ensemble average MSD or the median MSD. However, to analyze the heterogeneity of a given sample, the per-particle MSD or G′ and G′′ can be further analyzed with Bayesian methods (654) or Gaussian mixture modeling (23) to identify statistically distinct populations with a fluid.
REFERENCES
- 1. Knowles MR, Boucher RC. Mucus clearance as a primary innate defense mechanism for mammalian airways. J Clin Invest 109: 571–577, 2002. doi: 10.1172/JCI0215217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fahy JV, Dickey BF. Airway mucus function and dysfunction. N Engl J Med 363: 2233–2247, 2010. doi: 10.1056/NEJMra0910061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Albert RE, Lippmann M, Peterson HT Jr, Berger J, Sanborn K, Bohning D. Bronchial deposition and clearance of aerosols. Arch Intern Med 131: 115–127, 1973. doi: 10.1001/archinte.1973.00320070111013. [DOI] [PubMed] [Google Scholar]
- 4. Chatterjee M, van Putten JP, Strijbis K. Defensive properties of mucin glycoproteins during respiratory infections-relevance for SARS-CoV-2. mBio 11: e02374-20, 2020. doi: 10.1128/mBio.02374-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lucas AM, Douglas LC. Principles underlying ciliary activity in the respiratory tract: II. A comparison of nasal clearance in man, monkey and other mammals. Arch Otolaryngol 20: 518–541, 1934. doi: 10.1001/archotol.1934.03600040074006. [DOI] [Google Scholar]
- 6. Proctor DF, Wagner HN Jr.. Clearance of particles from the human nose. Preliminary report. Arch Environ Health 11: 366–371, 1965. doi: 10.1080/00039896.1965.10664231. [DOI] [PubMed] [Google Scholar]
- 7. Wood PB, Nagy E, Pearson FG, Rae S. Measurement of mucociliary clearance from the lower respiratory tract of normal dogs. Can Anaesth Soc J 20: 192–206, 1973. doi: 10.1007/BF03027208. [DOI] [PubMed] [Google Scholar]
- 8. Button B, Cai LH, Ehre C, Kesimer M, Hill DB, Sheehan JK, Boucher RC, Rubinstein M. A periciliary brush promotes the lung health by separating the mucus layer from airway epithelia. Science 337: 937–941, 2012. doi: 10.1126/science.1223012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rogers DF. Physiology of airway mucus secretion and pathophysiology of hypersecretion. Respir Care 52: 1134–1146, 2007. [PubMed] [Google Scholar]
- 10. Jones PW. St George’s Respiratory Questionnaire: MCID. COPD 2: 75–79, 2005. doi: 10.1081/copd-200050513. [DOI] [PubMed]
- 11.Anonymous. Respiratory tract involvement in cystic fibrosis of pancreas (mucoviscidosis). JAMA 150: 1407, 1952. doi: 10.1001/jama.1952.03680140045014. [DOI] [PubMed] [Google Scholar]
- 12. Abdullah LH, Coakley R, Webster MJ, Zhu Y, Tarran R, Radicioni G, Kesimer M, Boucher RC, Davis CW, Ribeiro CM. Mucin production and hydration responses to mucopurulent materials in normal versus cystic fibrosis airway epithelia. Am J Respir Crit Care Med 197: 481–491, 2018. doi: 10.1164/rccm.201706-1139OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Aiello E, Sleigh M. Ciliary function of the frog oro-pharyngeal epithelium. Cell Tissue Res 178: 267–278, 1977. doi: 10.1007/BF00219053. [DOI] [PubMed] [Google Scholar]
- 14. Anderson WH, Coakley RD, Button B, Henderson AG, Zeman KL, Alexis NE, Peden DB, Lazarowski ER, Davis CW, Bailey S, Fuller F, Almond M, Qaqish B, Bordonali E, Rubinstein M, Bennett WD, Kesimer M, Boucher RC. The relationship of mucus concentration (hydration) to mucus osmotic pressure and transport in chronic bronchitis. Am J Respir Crit Care Med 192: 182–190, 2015. doi: 10.1164/rccm.201412-2230OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ando A, Farrell MJ, Mazzone SB. Cough-related neural processing in the brain: a roadmap for cough dysfunction? Neurosci Biobehav Rev 47: 457–468, 2014. doi: 10.1016/j.neubiorev.2014.09.018. [DOI] [PubMed] [Google Scholar]
- 16. Anzueto A, Jubran A, Ohar JA, Piquette CA, Rennard SI, Colice G, Pattishall EN, Barrett J, Engle M, Perret KA, Rubin BK. Effects of aerosolized surfactant in patients with stable chronic bronchitis: a prospective randomized controlled trial. JAMA 278: 1426–1431, 1997. doi: 10.1001/jama.1997.03550170056032. [DOI] [PubMed] [Google Scholar]
- 17. Bao L, Locovei S, Dahl G. Pannexin membrane channels are mechanosensitive conduits for ATP. FEBS Lett 572: 65–68, 2004. doi: 10.1016/j.febslet.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 18. Hogg JC, Paré PD, Hackett TL. The contribution of small airway obstruction to the pathogenesis of chronic obstructive pulmonary disease. Physiol Rev 97: 529–552, 2017. doi: 10.1152/physrev.00025.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Widdicombe JH. A brief history of bronchitis in England and Wales. Chronic Obstr Pulm Dis 7: 303–314, 2020. doi: 10.15326/jcopdf.7.4.2020.0135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Widdicombe JH, Wine JJ. Airway gland structure and function. Physiol Rev 95: 1241–1319, 2015. doi: 10.1152/physrev.00039.2014. [DOI] [PubMed] [Google Scholar]
- 21. Hiemstra PS, McCray PB Jr, Bals R. The innate immune function of airway epithelial cells in inflammatory lung disease. Eur Respir J 45: 1150–1162, 2015. doi: 10.1183/09031936.00141514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Albert RE, Lippmann M, Briscoe W. The characteristics of bronchial clearance in humans and the effects of cigarette smoking. Arch Environ Health 18: 738–755, 1969. doi: 10.1080/00039896.1969.10665482. [DOI] [PubMed] [Google Scholar]
- 23. Esther CR Jr, Muhlebach MS, Ehre C, Hill DB, Wolfgang MC, Kesimer M, Ramsey KA, Markovetz MR, Garbarine IC, Forest MG, Seim I, Zorn B, Morrison CB, Delion MF, Thelin WR, Villalon D, Sabater JR, Turkovic L, Ranganathan S, Stick SM, Boucher RC. Mucus accumulation in the lungs precedes structural changes and infection in children with cystic fibrosis. Sci Transl Med 11: eaav3488, 2019. doi: 10.1126/scitranslmed.aav3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ray A, Camiolo M, Fitzpatrick A, Gauthier M, Wenzel SE. Are we meeting the promise of endotypes and precision medicine in asthma? Physiol Rev 100: 983–1017, 2020. doi: 10.1152/physrev.00023.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Boucher RC. Muco-obstructive lung diseases. N Engl J Med 380: 1941–1953, 2019. doi: 10.1056/NEJMra1813799. [DOI] [PubMed] [Google Scholar]
- 26. McShane A, Bath J, Jaramillo AM, Ridley C, Walsh AA, Evans CM, Thornton DJ, Ribbeck K. Mucus. Curr Biol 31: R938–R945, 2021. doi: 10.1016/j.cub.2021.06.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Weibel ER. Morphometry of the Human Lung. New York: Academic Press, 1963. [Google Scholar]
- 28. Nadel JA. Mucous hypersecretion and relationship to cough. Pulm Pharmacol Ther 26: 510–513, 2013. doi: 10.1016/j.pupt.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 29. Hill DB, Vasquez PA, Mellnik J, McKinley SA, Vose A, Mu F, Henderson AG, Donaldson SH, Alexis NE, Boucher RC, Forest G. A biophysical basis for mucus solids concentration as a candidate biomarker for airways disease. PLoS One 9: e87681, 2014. doi: 10.1371/journal.pone.0087681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Thornton DJ, Rousseau K, McGuckin MA. Structure and function of the polymeric mucins in airways mucus. Annu Rev Physiol 70: 459–486, 2008. doi: 10.1146/annurev.physiol.70.113006.100702. [DOI] [PubMed] [Google Scholar]
- 31. Aksamit TR, O’Donnell AE, Barker A, Olivier KN, Winthrop KL, Daniels MLA, Johnson M, Eden E, Griffith D, Knowles M, Metersky M, Salathe M, Thomashow B, Tino G, Turino G, Carretta B, Daley CL; Bronchiectasis Research Registry Consortium. Adult patients with bronchiectasis: a first look at the US Bronchiectasis Research Registry. Chest 151: 982–992, 2017. doi: 10.1016/j.chest.2016.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rose MC, Voynow JA. Respiratory tract mucin genes and mucin glycoproteins in health and disease. Physiol Rev 86: 245–278, 2006. doi: 10.1152/physrev.00010.2005. [DOI] [PubMed] [Google Scholar]
- 33. Bonser LR, Erle DJ. Airway mucus and asthma: the role of MUC5AC and MUC5B. J Clin Med 6: 112, 2017. doi: 10.3390/jcm6120112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhu Y, Ehre C, Abdullah LH, Sheehan JK, Roy M, Evans CM, Dickey BF, Davis CW. Munc13-2-/- baseline secretion defect reveals source of oligomeric mucins in mouse airways. J Physiol 586: 1977–1992, 2008. doi: 10.1113/jphysiol.2007.149310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Okuda K, Chen G, Subramani DB, Wolf M, Gilmore RC, Kato T, Radicioni G, Kesimer M, Chua M, Dang H, Livraghi-Butrico A, Ehre C, Doerschuk CM, Randell SH, Matsui H, Nagase T, O'Neal WK, Boucher RC. Localization of secretory mucins MUC5AC and MUC5B in normal/healthy human airways. Am J Respir Crit Care Med 199: 715–727, 2019. doi: 10.1164/rccm.201804-0734OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nevitt SJ, Thornton J, Murray CS, Dwyer T. Inhaled mannitol for cystic fibrosis. Cochrane Database Syst Rev 2: CD008649, 2018. doi: 10.1002/14651858.CD008649.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhou X, Kinlough CL, Hughey RP, Jin M, Inoue H, Etling E, Modena BD, Kaminski N, Bleecker ER, Meyers DA, Jarjour NN, Trudeau JB, Holguin F, Ray A, Wenzel SE. Sialylation of MUC4beta N-glycans by ST6GAL1 orchestrates human airway epithelial cell differentiation associated with type-2 inflammation. JCI Insight 4: e122475, 2019. doi: 10.1172/jci.insight.122475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sturgess J, Reid L. Secretory activity of the human bronchial mucous glands in vitro. Exp Mol Pathol 16: 362–381, 1972. doi: 10.1016/0014-4800(72)90011-1. [DOI] [PubMed] [Google Scholar]
- 39. Brasch F, Johnen G, Winn-Brasch A, Guttentag SH, Schmiedl A, Kapp N, Suzuki Y, Müller KM, Richter J, Hawgood S, Ochs M. Surfactant protein B in type II pneumocytes and intra-alveolar surfactant forms of human lungs. Am J Respir Cell Mol Biol 30: 449–458, 2004. doi: 10.1165/rcmb.2003-0262OC. [DOI] [PubMed] [Google Scholar]
- 40. Guttentag S. Posttranslational regulation of surfactant protein B expression. Semin Perinatol 32: 367–370, 2008. doi: 10.1053/j.semperi.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Davis CW, Dickey BF. Regulated airway goblet cell mucin secretion. Annu Rev Physiol 70: 487–512, 2008. doi: 10.1146/annurev.physiol.70.113006.100638. [DOI] [PubMed] [Google Scholar]
- 42. Chen Y, Garvin LM, Nickola TJ, Watson AM, Colberg-Poley AM, Rose MC. IL-1β induction of MUC5AC gene expression is mediated by CREB and NF-κB and repressed by dexamethasone. Am J Physiol Lung Cell Mol Physiol 306: L797–L807, 2014. doi: 10.1152/ajplung.00347.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Oguma T, Asano K, Tomomatsu K, Kodama M, Fukunaga K, Shiomi T, Ohmori N, Ueda S, Takihara T, Shiraishi Y, Sayama K, Kagawa S, Natori Y, Lilly CM, Satoh K, Makimura K, Ishizaka A. Induction of mucin and MUC5AC expression by the protease activity of Aspergillus fumigatus in airway epithelial cells. J Immunol 187: 999–1005, 2011. doi: 10.4049/jimmunol.1002257. [DOI] [PubMed] [Google Scholar]
- 44. Young HW, Williams OW, Chandra D, Bellinghausen LK, Pérez G, Suárez A, Tuvim MJ, Roy MG, Alexander SN, Moghaddam SJ, Adachi R, Blackburn MR, Dickey BF, Evans CM. Central role of Muc5ac expression in mucous metaplasia and its regulation by conserved 5' elements. Am J Respir Cell Mol Biol 37: 273–290, 2007. doi: 10.1165/rcmb.2005-0460OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Thai P, Chen Y, Dolganov G, Wu R. Differential regulation of MUC5AC/Muc5ac and hCLCA-1/mGob-5 expression in airway epithelium. Am J Respir Cell Mol Biol 33: 523–530, 2005. doi: 10.1165/rcmb.2004-0220RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhao J, Maskrey B, Balzar S, Chibana K, Mustovich A, Hu H, Trudeau JB, O’Donnell V, Wenzel SE. Interleukin-13-induced MUC5AC is regulated by 15-lipoxygenase 1 pathway in human bronchial epithelial cells. Am J Respir Crit Care Med 179: 782–790, 2009. doi: 10.1164/rccm.200811-1744OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Koeppen M, McNamee EN, Brodsky KS, Aherne CM, Faigle M, Downey GP, Colgan SP, Evans CM, Schwartz DA, Eltzschig HK. Detrimental role of the airway mucin Muc5ac during ventilator-induced lung injury. Mucosal Immunol 6: 762–775, 2013. doi: 10.1038/mi.2012.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kanoh S, Tanabe T, Rubin BK. IL-13-induced MUC5AC production and goblet cell differentiation is steroid resistant in human airway cells. Clin Exp Allergy 41: 1747–1756, 2011. doi: 10.1111/j.1365-2222.2011.03852.x. [DOI] [PubMed] [Google Scholar]
- 49. Radicioni G, Ceppe A, Ford AA, Alexis NE, Barr RG, Bleecker ER, Christenson SA, Cooper CB, Han MK, Hansel NN, Hastie AT, Hoffman EA, Kanner RE, Martinez FJ, Ozkan E, Paine R 3rd, Woodruff PG, O’Neal WK, Boucher RC, Kesimer M. Airway mucin MUC5AC and MUC5B concentrations and the initiation and progression of chronic obstructive pulmonary disease: an analysis of the SPIROMICS cohort. Lancet Respir Med 9: 1241–1254, 2021. doi: 10.1016/S2213-2600(21)00079-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jaramillo AM, Azzegagh Z, Tuvim MJ, Dickey BF. Airway mucin secretion. Ann Am Thorac Soc 15: S164–S170, 2018. doi: 10.1513/AnnalsATS.201806-371AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jaramillo AM, Piccotti L, Velasco WV, Delgado ASH, Azzegagh Z, Chung F, Nazeer U, Farooq J, Brenner J, Parker-Thornburg J, Scott BL, Evans CM, Adachi R, Burns AR, Kreda SM, Tuvim MJ, Dickey BF. Different Munc18 proteins mediate baseline and stimulated airway mucin secretion. JCI Insight 4: e124815, 2019. doi: 10.1172/jci.insight.124815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Karamaoun C, Sobac B, Mauroy B, Van Muylem A, Haut B. New insights into the mechanisms controlling the bronchial mucus balance. PLoS One 13: e0199319, 2018. doi: 10.1371/journal.pone.0199319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fessler MB, Summer RS. Surfactant lipids at the host-environment interface. Metabolic sensors, suppressors, and effectors of inflammatory lung disease. Am J Respir Cell Mol Biol 54: 624–635, 2016. doi: 10.1165/rcmb.2016-0011PS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Weaver TE, Conkright JJ. Function of surfactant proteins B and C. Annu Rev Physiol 63: 555–578, 2001. doi: 10.1146/annurev.physiol.63.1.555. [DOI] [PubMed] [Google Scholar]
- 55. Wright JR. Host defense functions of pulmonary surfactant. Biol Neonate 85: 326–332, 2004. doi: 10.1159/000078172. [DOI] [PubMed] [Google Scholar]
- 56. Bermbach S, Weinhold K, Roeder T, Petersen F, Kugler C, Goldmann T, Rupp J, König P. Mechanisms of cilia-driven transport in the airways in the absence of mucus. Am J Respir Cell Mol Biol 51: 56–67, 2014. doi: 10.1165/rcmb.2012-0530OC. [DOI] [PubMed] [Google Scholar]
- 57. Hussong J, Lindken R, Faulhammer P, Noreikat K, Sharp KV, Kummer W, Westerweel J. Cilia-driven particle and fluid transport over mucus-free mice tracheae. J Biomech 46: 593–598, 2013. doi: 10.1016/j.jbiomech.2012.08.020. [DOI] [PubMed] [Google Scholar]
- 58. Morgan KT, Jiang XZ, Patterson DL, Gross EA. The nasal mucociliary apparatus. Correlation of structure and function in the rat. Am Rev Respir Dis 130: 275–281, 1984. doi: 10.1164/arrd.1984.130.2.275. [DOI] [PubMed] [Google Scholar]
- 59. Sturgess JM. The mucous lining of major bronchi in the rabbit lung. Am Rev Respir Dis 115: 819–827, 1977. doi: 10.1164/arrd.1977.115.5.819. [DOI] [PubMed] [Google Scholar]
- 60. Iravani J, Van As A. Mucus transport in the tracheobronchial tree of normal and bronchitic rats. J Pathol 106: 81–93, 1972. doi: 10.1002/path.1711060204. [DOI] [PubMed] [Google Scholar]
- 61. Iravani J, Melville GN. Mucociliary activity in the respiratory tract as influenced by prostaglandin E. Respiration 32: 305–315, 1975. doi: 10.1159/000193659. [DOI] [PubMed] [Google Scholar]
- 62. Bennett WD, Wu J, Fuller F, Balcazar JR, Zeman KL, Duckworth H, Donn KH, O’Riordan TG, Boucher RC, Donaldson SH. Duration of action of hypertonic saline on mucociliary clearance in the normal lung. J Appl Physiol (1985) 118: 1483–1490, 2015. doi: 10.1152/japplphysiol.00404.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ermund A, Meiss LN, Dolan B, Bähr A, Klymiuk N, Hansson GC. The mucus bundles responsible for airway cleaning are retained in cystic fibrosis and by cholinergic stimulation. Eur Respir J 52: 1800457, 2018. doi: 10.1183/13993003.00457-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ermund A, Meiss LN, Rodriguez-Pineiro AM, Bähr A, Nilsson HE, Trillo-Muyo S, Ridley C, Thornton DJ, Wine JJ, Hebert H, Klymiuk N, Hansson GC. The normal trachea is cleaned by MUC5B mucin bundles from the submucosal glands coated with the MUC5AC mucin. Biochem Biophys Res Commun 492: 331–337, 2017. doi: 10.1016/j.bbrc.2017.08.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ermund A, Trillo-Muyo S, Hansson GC. Assembly, release, and transport of airway mucins in pigs and humans. Ann Am Thorac Soc 15: S159–S163, 2018. doi: 10.1513/AnnalsATS.201804-238AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Meyerholz DK, Stoltz DA, Namati E, Ramachandran S, Pezzulo AA, Smith AR, Rector MV, Suter MJ, Kao S, McLennan G, Tearney GJ, Zabner J, McCray PB Jr, Welsh MJ. Loss of cystic fibrosis transmembrane conductance regulator function produces abnormalities in tracheal development in neonatal pigs and young children. Am J Respir Crit Care Med 182: 1251–1261, 2010. doi: 10.1164/rccm.201004-0643OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gsell S, Loiseau E, D’Ortona U, Viallat A, Favier J. Hydrodynamic model of directional ciliary-beat organization in human airways. Sci Rep 10: 8405, 2020. doi: 10.1038/s41598-020-64695-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, Cherniack RM, Rogers RM, Sciurba FC, Coxson HO, Paré PD. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med 350: 2645–2653, 2004. doi: 10.1056/NEJMoa032158. [DOI] [PubMed] [Google Scholar]
- 69. Boon M, Verleden SE, Bosch B, Lammertyn EJ, McDonough JE, Mai C, Verschakelen J, Kemner-van de Corput M, Tiddens HA, Proesmans M, Vermeulen FL, Verbeken EK, Cooper J, Van Raemdonck DE, Decramer M, Verleden GM, Hogg JC, Dupont LJ, Vanaudenaerde BM, De Boeck K. Morphometric analysis of explant lungs in cystic fibrosis. Am J Respir Crit Care Med 193: 516–526, 2016. doi: 10.1164/rccm.201507-1281OC. [DOI] [PubMed] [Google Scholar]
- 70. Simel DL, Mastin JP, Pratt PC, Wisseman CL, Shelburne JD, Spock A, Ingram P. Scanning electron microscopic study of the airways in normal children and in patients with cystic fibrosis and other lung diseases. Pediatr Pathol 2: 47–64, 1984. doi: 10.3109/15513818409041187. [DOI] [PubMed] [Google Scholar]
- 71. Aikawa T, Shimura S, Sasaki H, Ebina M, Takishima T. Marked goblet cell hyperplasia with mucus accumulation in the airways of patients who died of severe acute asthma attack. Chest 101: 916–921, 1992. doi: 10.1378/chest.101.4.916. [DOI] [PubMed] [Google Scholar]
- 72. Aikawa T, Shimura S, Sasaki H, Takishima T, Yaegashi H, Takahashi T. Morphometric analysis of intraluminal mucus in airways in chronic obstructive pulmonary disease. Am Rev Respir Dis 140: 477–482, 1989. doi: 10.1164/ajrccm/140.2.477. [DOI] [PubMed] [Google Scholar]
- 73. Kuyper LM, Paré PD, Hogg JC, Lambert RK, Ionescu D, Woods R, Bai TR. Characterization of airway plugging in fatal asthma. Am J Med 115: 6–11, 2003. doi: 10.1016/S0002-9343(03)00241-9. [DOI] [PubMed] [Google Scholar]
- 74. Reid L. Pathology of chronic bronchitis. Proc R Soc Med 49: 771–773, 1956. [PubMed] [Google Scholar]
- 75. Matthews LW, Spector S, Lemm J, Potter JL. Studies on pulmonary secretions. I. The over-all chemical composition of pulmonary secretions from patients with cystic fibrosis, bronchiectasis, and laryngectomy. Am Rev Respir Dis 88: 199–204, 1963. doi: 10.1164/arrd.1963.88.2.199. [DOI] [PubMed] [Google Scholar]
- 76. Henderson AG, Ehre C, Button B, Abdullah LH, Cai LH, Leigh MW, DeMaria GC, Matsui H, Donaldson SH, Davis CW, Sheehan JK, Boucher RC, Kesimer M. Cystic fibrosis airway secretions exhibit mucin hyperconcentration and increased osmotic pressure. J Clin Invest 124: 3047–3060, 2014. doi: 10.1172/JCI73469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kesimer M, Ford AA, Ceppe A, Radicioni G, Cao R, Davis CW, Doerschuk CM, Alexis NE, Anderson WH, Henderson AG, Barr RG, Bleecker ER, Christenson SA, Cooper CB, Han MK, Hansel NN, Hastie AT, Hoffman EA, Kanner RE, Martinez F, Paine R 3rd, Woodruff PG, O’Neal WK, Boucher RC. Airway mucin concentration as a marker of chronic bronchitis. N Engl J Med 377: 911–922, 2017. doi: 10.1056/NEJMoa1701632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Esther CR Jr, Hill DB, Button B, Shi S, Jania C, Duncan EA, Doerschuk CM, Chen G, Ranganathan S, Stick SM, Boucher RC. Sialic acid-to-urea ratio as a measure of airway surface hydration. Am J Physiol Lung Cell Mol Physiol 312: L398–L404, 2017. doi: 10.1152/ajplung.00398.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hill DB, Long RF, Kissner WJ, Atieh E, Garbarine IC, Markovetz MR, Fontana NC, Christy M, Habibpour M, Tarran R, Forest MG, Boucher RC, Button B. Pathological mucus and impaired mucus clearance in cystic fibrosis patients result from increased concentration, not altered pH. Eur Respir J 52: 1801297, 2018. doi: 10.1183/13993003.01297-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Matsui H, Wagner VE, Hill DB, Schwab UE, Rogers TD, Button B, Taylor RM, Superfine R, Rubinstein M, Iglewski BH, Boucher RC. A physical linkage between cystic fibrosis airway surface dehydration and Pseudomonas aeruginosa biofilms. Proc Natl Acad Sci USA 103: 18131–18136, 2006. doi: 10.1073/pnas.0606428103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Knowles MR, Robinson JM, Wood RE, Pue CA, Mentz WM, Wager GC, Gatzy JT, Boucher RC. Ion composition of airway surface liquid of patients with cystic fibrosis as compared with normal and disease-control subjects. J Clin Invest 100: 2588–2595, 1997. doi: 10.1172/JCI119802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Jayaraman S, Song Y, Verkman AS. Airway surface liquid osmolality measured using fluorophore-encapsulated liposomes. J Gen Physiol 117: 423–430, 2001. doi: 10.1085/jgp.117.5.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Jayaraman S, Song Y, Vetrivel L, Shankar L, Verkman AS. Noninvasive in vivo fluorescence measurement of airway-surface liquid depth, salt concentration, and pH. J Clin Invest 107: 317–324, 2001. doi: 10.1172/JCI11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Caldwell RA, Grubb BR, Tarran R, Boucher RC, Knowles MR, Barker PM. In vivo airway surface liquid Cl- analysis with solid-state electrodes. J Gen Physiol 119: 3–14, 2002. doi: 10.1085/jgp.119.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Claas GJ, Jülg B, Goebel FD, Bogner J. Metabolic and anthropometric changes one year after switching from didanosine/stavudine to tenofovir in HIV-infected patients. Eur J Med Res 12: 54–60, 2007. [PubMed] [Google Scholar]
- 86. Song Y, Jayaraman S, Yang B, Matthay MA, Verkman AS. Role of aquaporin water channels in airway fluid transport, humidification, and surface liquid hydration. J Gen Physiol 117: 573–582, 2001. doi: 10.1085/jgp.117.6.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Folkesson HG, Matthay MA, Frigeri A, Verkman AS. Transepithelial water permeability in microperfused distal airways. Evidence for channel-mediated water transport. J Clin Invest 97: 664–671, 1996. doi: 10.1172/JCI118463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Pedersen PS, Procida K, Larsen PL, Holstein-Rathlou NH, Frederiksen O. Water permeability in human airway epithelium. Pflugers Arch 451: 464–473, 2005. doi: 10.1007/s00424-005-1484-5. [DOI] [PubMed] [Google Scholar]
- 89. Kreda SM, Gynn MC, Fenstermacher DA, Boucher RC, Gabriel SE. Expression and localization of epithelial aquaporins in the adult human lung. Am J Respir Cell Mol Biol 24: 224–234, 2001. doi: 10.1165/ajrcmb.24.3.4367. [DOI] [PubMed] [Google Scholar]
- 90. Schmidt H, Michel C, Braubach P, Fauler M, Neubauer D, Thompson KE, Frick M, Mizaikoff B, Dietl P, Wittekindt OH. Water permeability adjusts resorption in lung epithelia to increased apical surface liquid volumes. Am J Respir Cell Mol Biol 56: 372–382, 2017. doi: 10.1165/rcmb.2016-0161OC. [DOI] [PubMed] [Google Scholar]
- 91. Chiu YH, Schappe MS, Desai BN, Bayliss DA. Revisiting multimodal activation and channel properties of Pannexin 1. J Gen Physiol 150: 19–39, 2018. doi: 10.1085/jgp.201711888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Boucher RC. Human airway ion transport. Part two. Am J Respir Crit Care Med 150: 581–593, 1994. doi: 10.1164/ajrccm.150.2.8049852. [DOI] [PubMed] [Google Scholar]
- 93. Boucher RC. Human airway ion transport. Part one. Am J Respir Crit Care Med 150: 271–281, 1994. doi: 10.1164/ajrccm.150.1.8025763. [DOI] [PubMed] [Google Scholar]
- 94. Boucher RC, Stutts MJ, Bromberg PA, Gatzy JT. Regional differences in airway surface liquid composition. J Appl Physiol Respir Environ Exerc Physiol 50: 613–620, 1981. doi: 10.1152/jappl.1981.50.3.613. [DOI] [PubMed] [Google Scholar]
- 95. Matsui H, Davis CW, Tarran R, Boucher RC. Osmotic water permeabilities of cultured, well-differentiated normal and cystic fibrosis airway epithelia. J Clin Invest 105: 1419–1427, 2000. doi: 10.1172/JCI4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Willumsen NJ, Davis CW, Boucher RC. Selective response of human airway epithelia to luminal but not serosal solution hypertonicity. Possible role for proximal airway epithelia as an osmolality transducer. J Clin Invest 94: 779–787, 1994. doi: 10.1172/JCI117397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Prazma J, Coleman CC, Shockley WW, Boucher RC. Tracheal vascular response to hypertonic and hypotonic solutions. J Appl Physiol (1985) 76: 2275–2280, 1994. doi: 10.1152/jappl.1994.76.6.2275. [DOI] [PubMed] [Google Scholar]
- 98. Wagner CE, Wheeler KM, Ribbeck K. Mucins and their role in shaping the functions of mucus barriers. Annu Rev Cell Dev Biol 34: 189–215, 2018. doi: 10.1146/annurev-cellbio-100617-062818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Markovetz MR, Subramani DB, Kissner WJ, Morrison CB, Garbarine IC, Ghio A, Ramsey KA, Arora H, Kumar P, Nix DB, Kumagai T, Krunkosky TM, Krause DC, Radicioni G, Alexis NE, Kesimer M, Tiemeyer M, Boucher RC, Ehre C, Hill DB. Endotracheal tube mucus as a source of airway mucus for rheological study. Am J Physiol Lung Cell Mol Physiol 317: L498–L509, 2019. doi: 10.1152/ajplung.00238.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Bakshani CR, Morales-Garcia AL, Althaus M, Wilcox MD, Pearson JP, Bythell JC, Burgess JG. Evolutionary conservation of the antimicrobial function of mucus: a first defence against infection. NPJ Biofilms Microbiomes 4: 14, 2018. doi: 10.1038/s41522-018-0057-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Demouveaux B, Gouyer V, Robbe-Masselot C, Gottrand F, Narita T, Desseyn JL. Mucin CYS domain stiffens the mucus gel hindering bacteria and spermatozoa. Sci Rep 9: 16993, 2019. doi: 10.1038/s41598-019-53547-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Javitt G, Khmelnitsky L, Albert L, Bigman LS, Elad N, Morgenstern D, Ilani T, Levy Y, Diskin R, Fass D. Assembly mechanism of mucin and von Willebrand factor polymers. Cell 183: 717–729.e6, 2020. doi: 10.1016/j.cell.2020.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Perez-Vilar J, Eckhardt AE, DeLuca A, Hill RL. Porcine submaxillary mucin forms disulfide-linked multimers through its amino-terminal D-domains. J Biol Chem 273: 14442–14449, 1998. doi: 10.1074/jbc.273.23.14442. [DOI] [PubMed] [Google Scholar]
- 104. Perez-Vilar J, Eckhardt AE, Hill RL. Porcine submaxillary mucin forms disulfide-bonded dimers between its carboxyl-terminal domains. J Biol Chem 271: 9845–9850, 1996. doi: 10.1074/jbc.271.16.9845. [DOI] [PubMed] [Google Scholar]
- 105. Bansil R, Turner BS. The biology of mucus: Composition, synthesis and organization. Adv Drug Deliv Rev 124: 3–15, 2018. doi: 10.1016/j.addr.2017.09.023. [DOI] [PubMed] [Google Scholar]
- 106. Evans CM, Raclawska DS, Ttofali F, Liptzin DR, Fletcher AA, Harper DN, McGing MA, McElwee MM, Williams OW, Sanchez E, Roy MG, Kindrachuk KN, Wynn TA, Eltzschig HK, Blackburn MR, Tuvim MJ, Janssen WJ, Schwartz DA, Dickey BF. The polymeric mucin Muc5ac is required for allergic airway hyperreactivity. Nat Commun 6: 6281, 2015. doi: 10.1038/ncomms7281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Roy MG, Livraghi-Butrico A, Fletcher AA, McElwee MM, Evans SE, Boerner RM, , et al. Muc5b is required for airway defence. Nature 505: 412–416, 2014. doi: 10.1038/nature12807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Morgan LE, Jaramillo AM, Shenoy SK, Raclawska D, Emezienna NA, Richardson VL, Hara N, Harder AQ, NeeDell JC, Hennessy CE, El-Batal HM, Magin CM, Grove Villalon DE, Duncan G, Hanes JS, Suk JS, Thornton DJ, Holguin F, Janssen WJ, Thelin WR, Evans CM. Disulfide disruption reverses mucus dysfunction in allergic airway disease. Nat Commun 12: 249, 2021. doi: 10.1038/s41467-020-20499-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Xue Y, Bao W, Zhou Y, Fu Q, Hao H, Han L, Yin D, Zhang Y, Zhang X, Zhang M. Small-Airway dysfunction is involved in the pathogenesis of asthma: evidence from two mouse models. J Asthma Allergy 14: 883–896, 2021. doi: 10.2147/JAA.S312361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Desseyn JL, Aubert JP, Porchet N, Laine A. Evolution of the large secreted gel-forming mucins. Mol Biol Evol 17: 1175–1184, 2000. doi: 10.1093/oxfordjournals.molbev.a026400. [DOI] [PubMed] [Google Scholar]
- 111. Thornton DJ, Sharpe C, Ridley C. Intracellular processing of human secreted polymeric airway mucins. Ann Am Thorac Soc 15: S154–S158, 2018. doi: 10.1513/AnnalsATS.201802-143AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Ridley C, Lockhart-Cairns MP, Collins RF, Jowitt TA, Subramani DB, Kesimer M, Baldock C, Thornton DJ. The C-terminal dimerization domain of the respiratory mucin MUC5B functions in mucin stability and intracellular packaging before secretion. J Biol Chem 294: 17105–17116, 2019. doi: 10.1074/jbc.RA119.010771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Kesimer M, Ehre C, Burns KA, Davis CW, Sheehan JK, Pickles RJ. Molecular organization of the mucins and glycocalyx underlying mucus transport over mucosal surfaces of the airways. Mucosal Immunol 6: 379–392, 2013. doi: 10.1038/mi.2012.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Carpenter J, Wang Y, Gupta R, Li Y, Haridass P, Subramani DB, Reidel B, Morton L, Ridley C, O’Neal WK, Buisine MP, Ehre C, Thornton DJ, Kesimer M. Assembly and organization of the N-terminal region of mucin MUC5AC: indications for structural and functional distinction from MUC5B. Proc Natl Acad Sci USA 118: e2104490118, 2021. doi: 10.1073/pnas.2104490118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Bonser LR, Zlock L, Finkbeiner W, Erle DJ. Epithelial tethering of MUC5AC-rich mucus impairs mucociliary transport in asthma. J Clin Invest 126: 2367–2371, 2016. doi: 10.1172/JCI84910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Button B, Goodell HP, Atieh E, Chen YC, Williams R, Shenoy S, Lackey E, Shenkute NT, Cai LH, Dennis RG, Boucher RC, Rubinstein M. Roles of mucus adhesion and cohesion in cough clearance. Proc Natl Acad Sci USA 115: 12501–12506, 2018. doi: 10.1073/pnas.1811787115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Yuan S, Hollinger M, Lachowicz-Scroggins ME, Kerr SC, Dunican EM, Daniel BM, Ghosh S, Erzurum SC, Willard B, Hazen SL, Huang X, Carrington SD, Oscarson S, Fahy JV. Oxidation increases mucin polymer cross-links to stiffen airway mucus gels. Sci Transl Med 7: 276ra27, 2015. doi: 10.1126/scitranslmed.3010525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Järvå MA, Lingford JP, John A, Soler NM, Scott NE, Goddard-Borger ED. Trefoil factors share a lectin activity that defines their role in mucus. Nat Commun 11: 2265, 2020. doi: 10.1038/s41467-020-16223-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Kerr SC, Carrington SD, Oscarson S, Gallagher ME, Solon M, Yuan S, Ahn JN, Dougherty RH, Finkbeiner WE, Peters MC, Fahy JV. Intelectin-1 is a prominent protein constituent of pathologic mucus associated with eosinophilic airway inflammation in asthma. Am J Respir Crit Care Med 189: 1005–1007, 2014. doi: 10.1164/rccm.201312-2220LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Rubinstein M, Semenov AN. Dynamics of entangled solutions of associating polymers. Macromolecules 34: 1058–1068, 2001. doi: 10.1021/ma0013049. [DOI] [Google Scholar]
- 121. Wheeler KM, Cárcamo-Oyarce G, Turner BS, Dellos-Nolan S, Co JY, Lehoux S, Cummings RD, Wozniak DJ, Ribbeck K. Mucin glycans attenuate the virulence of Pseudomonas aeruginosa in infection. Nat Microbiol 4: 2146–2154, 2019. doi: 10.1038/s41564-019-0581-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Meldrum OW, Chotirmall SH. Mucus, microbiomes and pulmonary disease. Biomedicines 9: 675, 2021. doi: 10.3390/biomedicines9060675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Chen A, McKinley SA, Wang S, Shi F, Mucha PJ, Forest MG, Lai SK. Transient antibody-mucin interactions produce a dynamic molecular shield against viral invasion. Biophys J 106: 2028–2036, 2014. doi: 10.1016/j.bpj.2014.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Bienvenu T, Sermet-Gaudelus I, Burgel PR, Hubert D, Crestani B, Bassinet L, Dusser D, Fajac I. Cystic fibrosis transmembrane conductance regulator channel dysfunction in non-cystic fibrosis bronchiectasis. Am J Respir Crit Care Med 181: 1078–1084, 2010. doi: 10.1164/rccm.200909-1434OC. [DOI] [PubMed] [Google Scholar]
- 125. Birket SE, Chu KK, Liu L, Houser GH, Diephuis BJ, Wilsterman EJ, Dierksen G, Mazur M, Shastry S, Li Y, Watson JD, Smith AT, Schuster BS, Hanes J, Grizzle WE, Sorscher EJ, Tearney GJ, Rowe SM. A functional anatomic defect of the cystic fibrosis airway. Am J Respir Crit Care Med 190: 421–432, 2014. doi: 10.1164/rccm.201404-0670OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Almeida GM, Laanto E, Ashrafi R, Sundberg LR. Bacteriophage adherence to mucus mediates preventive protection against pathogenic bacteria. mBio 10: e01984-19, 2019. doi: 10.1128/mBio.01984-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Bond MC, Vidakovic L, Singh PK, Drescher K, Nadell CD. Matrix-trapped viruses can prevent invasion of bacterial biofilms by colonizing cells. Elife 10: e65355, 2021. doi: 10.7554/eLife.65355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Livraghi-Butrico A, Grubb BR, Wilkinson KJ, Volmer AS, Burns KA, Evans CM, O’Neal WK, Boucher RC. Contribution of mucus concentration and secreted mucins Muc5ac and Muc5b to the pathogenesis of muco-obstructive lung disease. Mucosal Immunol 10: 395–407, 2017. doi: 10.1038/mi.2016.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Anderson SD, Daviskas E, Brannan JD, Chan HK. Repurposing excipients as active inhalation agents: the mannitol story. Adv Drug Deliv Rev 133: 45–56, 2018. doi: 10.1016/j.addr.2018.04.003. [DOI] [PubMed] [Google Scholar]
- 130. Bennett WD, Foster WM, Chapman WF. Cough-enhanced mucus clearance in the normal lung. J Appl Physiol (1985) 69: 1670–1675, 1990. doi: 10.1152/jappl.1990.69.5.1670. [DOI] [PubMed] [Google Scholar]
- 131. Bose M, Mukherjee P. Microbe-MUC1 crosstalk in cancer-associated infections. Trends Mol Med 26: 324–336, 2020. doi: 10.1016/j.molmed.2019.10.003. [DOI] [PubMed] [Google Scholar]
- 132. Zhou LB, Zheng YM, Liao WJ, Song LJ, Meng X, Gong X, Chen G, Liu WX, Wang YQ, Han DM, Zhong NS, Lu WJ, Yang PC, Zhang XW. MUC1 deficiency promotes nasal epithelial barrier dysfunction in subjects with allergic rhinitis. J Allergy Clin Immunol 144: 1716–1719.e5, 2019. doi: 10.1016/j.jaci.2019.07.042. [DOI] [PubMed] [Google Scholar]
- 133. Okuda K, Dang H, Kobayashi Y, Carraro G, Nakano S, Chen G, , et al. Secretory cells dominate airway CFTR expression and function in human airway superficial epithelia. Am J Respir Crit Care Med 203: 1275–1289, 2021. doi: 10.1164/rccm.202008-3198OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Davidson JD. Multiscale Modeling and Simulation of Crosslinked Polymers (Dissertation). Ann Arbor, MI: University of Michigan, 2014. [Google Scholar]
- 135. de Beer S, Kutnyanszky E, Schön PM, Vancso GJ, Müser MH. Solvent-induced immiscibility of polymer brushes eliminates dissipation channels. Nat Commun 5: 3781, 2014. doi: 10.1038/ncomms4781. [DOI] [PubMed] [Google Scholar]
- 136. Matthay MA, Robriquet L, Fang X. Alveolar epithelium: role in lung fluid balance and acute lung injury. Proc Am Thorac Soc 2: 206–213, 2005. doi: 10.1513/pats.200501-009AC. [DOI] [PubMed] [Google Scholar]
- 137. Folkesson HG, Matthay MA. Alveolar epithelial ion and fluid transport: recent progress. Am J Respir Cell Mol Biol 35: 10–19, 2006. doi: 10.1165/rcmb.2006-0080SF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Johnson MD, Widdicombe JH, Allen L, Barbry P, Dobbs LG. Alveolar epithelial type I cells contain transport proteins and transport sodium, supporting an active role for type I cells in regulation of lung liquid homeostasis. Proc Natl Acad Sci USA 99: 1966–1971, 2002. doi: 10.1073/pnas.042689399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Matalon S. Mechanisms and regulation of ion transport in adult mammalian alveolar type II pneumocytes. Am J Physiol Cell Physiol 261: C727–C738, 1991. doi: 10.1152/ajpcell.1991.261.5.C727. [DOI] [PubMed] [Google Scholar]
- 140. Matalon S, Bridges RJ, Benos DJ. Amiloride-inhibitable Na+ conductive pathways in alveolar type II pneumocytes. Am J Physiol Lung Cell Mol Physiol 260: L90–L96, 1991. doi: 10.1152/ajplung.1991.260.2.L90. [DOI] [PubMed] [Google Scholar]
- 141. Matalon S, Bartoszewski R, Collawn JF. Role of epithelial sodium channels in the regulation of lung fluid homeostasis. Am J Physiol Lung Cell Mol Physiol 309: L1229–L1238, 2015. doi: 10.1152/ajplung.00319.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Lindert J, Perlman CE, Parthasarathi K, Bhattacharya J. Chloride-dependent secretion of alveolar wall liquid determined by optical-sectioning microscopy. Am J Respir Cell Mol Biol 36: 688–696, 2007. doi: 10.1165/rcmb.2006-0347OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Bove PF, Grubb BR, Okada SF, Ribeiro CM, Rogers TD, Randell SH, O’Neal WK, Boucher RC. Human alveolar type II cells secrete and absorb liquid in response to local nucleotide signaling. J Biol Chem 285: 34939–34949, 2010. doi: 10.1074/jbc.M110.162933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Knowles MR, Zariwala M, Leigh M. Primary ciliary dyskinesia. Clin Chest Med 37: 449–461, 2016. doi: 10.1016/j.ccm.2016.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Stetten AZ, Iasella SV, Corcoran TE, Garoff S, Przybycien TM, Tilton RD. Surfactant-induced Marangoni transport of lipids and therapeutics within the lung. Curr Opin Colloid Interface Sci 36: 58–69, 2018. doi: 10.1016/j.cocis.2018.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Stetten AZ, Moraca G, Corcoran TE, Tristram-Nagle S, Garoff S, Przybycien TM, Tilton RD. Enabling Marangoni flow at air-liquid interfaces through deposition of aerosolized lipid dispersions. J Colloid Interface Sci 484: 270–278, 2016. doi: 10.1016/j.jcis.2016.08.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Kilburn KH. A hypothesis for pulmonary clearance and its implications. Am Rev Respir Dis 98: 449–463, 1968. doi: 10.1164/arrd.1968.98.3.449. [DOI] [PubMed] [Google Scholar]
- 148. Blouquit S, Regnier A, Dannhoffer L, Fermanian C, Naline E, Boucher R, Chinet T. Ion and fluid transport properties of small airways in cystic fibrosis. Am J Respir Crit Care Med 174: 299–305, 2006. doi: 10.1164/rccm.200506-987OC. [DOI] [PubMed] [Google Scholar]
- 149. Flores-Delgado G, Lytle C, Quinton PM. Site of fluid secretion in small airways. Am J Respir Cell Mol Biol 54: 312–318, 2016. doi: 10.1165/rcmb.2015-0238RC. [DOI] [PubMed] [Google Scholar]
- 150. Quinton PM. Both Ways at once: keeping small airways clean. Physiology (Bethesda) 32: 380–390, 2017. doi: 10.1152/physiol.00013.2017. [DOI] [PubMed] [Google Scholar]
- 151. Shamsuddin AK, Quinton PM. Concurrent absorption and secretion of airway surface liquids and bicarbonate secretion in human bronchioles. Am J Physiol Lung Cell Mol Physiol 316: L953–L960, 2019. doi: 10.1152/ajplung.00545.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Lazarowski ER, Tarran R, Grubb BR, van Heusden CA, Okada S, Boucher RC. Nucleotide release provides a mechanism for airway surface liquid homeostasis. J Biol Chem 279: 36855–36864, 2004. doi: 10.1074/jbc.M405367200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Tarran R, Button B, Picher M, Paradiso AM, Ribeiro CM, Lazarowski ER, Zhang L, Collins PL, Pickles RJ, Fredberg JJ, Boucher RC. Normal and cystic fibrosis airway surface liquid homeostasis. The effects of phasic shear stress and viral infections. J Biol Chem 280: 35751–35759, 2005. doi: 10.1074/jbc.M505832200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Kreda SM, Mall M, Mengos A, Rochelle L, Yankaskas J, Riordan JR, Boucher RC. Characterization of wild-type and deltaF508 cystic fibrosis transmembrane regulator in human respiratory epithelia. Mol Biol Cell 16: 2154–2167, 2005. doi: 10.1091/mbc.e04-11-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Montoro DT, Haber AL, Biton M, Vinarsky V, Lin B, Birket SE, Yuan F, Chen S, Leung HM, Villoria J, Rogel N, Burgin G, Tsankov AM, Waghray A, Slyper M, Waldman J, Nguyen L, Dionne D, Rozenblatt-Rosen O, Tata PR, Mou H, Shivaraju M, Bihler H, Mense M, Tearney GJ, Rowe SM, Engelhardt JF, Regev A, Rajagopal J. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 560: 319–324, 2018. doi: 10.1038/s41586-018-0393-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Larsen EH. Dual skin functions in amphibian osmoregulation. Comp Biochem Physiol A Mol Integr Physiol 253: 110869, 2021. doi: 10.1016/j.cbpa.2020.110869. [DOI] [PubMed] [Google Scholar]
- 157. Purdom T, Wayland A, Nicoll JX, Ludwar B, Fry AC, Shanle E, Giles J. Intramuscular hyaluronic acid expression following repetitive power training induced nonfunctional overreaching. Appl Physiol Nutr Metab 46: 1563–1566, 2021. doi: 10.1139/apnm-2020-0708. [DOI] [PubMed] [Google Scholar]
- 158. Engelhardt JF, Yang Y, Stratford-Perricaudet LD, Allen ED, Kozarsky K, Perricaudet M, Yankaskas JR, Wilson JM. Direct gene transfer of human CFTR into human bronchial epithelia of xenografts with E1-deleted adenoviruses. Nat Genet 4: 27–34, 1993. doi: 10.1038/ng0593-27. [DOI] [PubMed] [Google Scholar]
- 159. Scudieri P, Musante I, Venturini A, Guidone D, Genovese M, Cresta F, Caci E, Palleschi A, Poeta M, Santamaria F, Ciciriello F, Lucidi V, Galietta LJ. Ionocytes and CFTR chloride channel expression in normal and cystic fibrosis nasal and bronchial epithelial cells. Cells 9: 2090, 2020. doi: 10.3390/cells9092090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Zaragosi LE, Deprez M, Barbry P. Using single-cell RNA sequencing to unravel cell lineage relationships in the respiratory tract. Biochem Soc Trans 48: 327–336, 2020. doi: 10.1042/BST20191010. [DOI] [PubMed] [Google Scholar]
- 161. Okuda K, Kobayashi Y, Dang H, Nakano S, Cardenas BS, O’Neal VK, Kato T, Chen G, Gilmore RC, Chua M, Livraghi-Butrico A, Doerschuk C, Grubb B, Randell SH, Tata P, O’Neal W, Boucher R. Regional regulation of CFTR and ionocyte expression in normal human airways (Abstract). Pediatr Pulmonol 54: S173, 2019. [Google Scholar]
- 162. Goldfarbmuren KC, Jackson ND, Sajuthi SP, Dyjack N, Li KS, Rios CL, Plender EG, Montgomery MT, Everman JL, Bratcher PE, Vladar EK, Seibold MA. Dissecting the cellular specificity of smoking effects and reconstructing lineages in the human airway epithelium. Nat Commun 11: 2485, 2020. doi: 10.1038/s41467-020-16239-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Deprez M, Zaragosi LE, Truchi M, Becavin C, Ruiz García S, Arguel MJ, Plaisant M, Magnone V, Lebrigand K, Abelanet S, Brau F, Paquet A, Pe'er D, Marquette CH, Leroy S, Barbry P. A single-cell atlas of the human healthy airways. Am J Respir Crit Care Med 202: 1636–1645, 2020. doi: 10.1164/rccm.201911-2199OC. [DOI] [PubMed] [Google Scholar]
- 164. Garcia GJ, Boucher RC, Elston TC. Biophysical model of ion transport across human respiratory epithelia allows quantification of ion permeabilities. Biophys J 104: 716–726, 2013. doi: 10.1016/j.bpj.2012.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Lam AN, Aksit MA, Vecchio-Pagan B, Shelton CA, Osorio DL, Anzmann AF, Goff LA, Whitcomb DC, Blackman SM, Cutting GR. Increased expression of anion transporter SLC26A9 delays diabetes onset in cystic fibrosis. J Clin Invest 130: 272–286, 2020. doi: 10.1172/JCI129833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Frizzell RA, Hanrahan JW. Physiology of epithelial chloride and fluid secretion. Cold Spring Harb Perspect Med 2: a009563, 2012. doi: 10.1101/cshperspect.a009563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Bertrand CA, Zhang R, Pilewski JM, Frizzell RA. SLC26A9 is a constitutively active, CFTR-regulated anion conductance in human bronchial epithelia. J Gen Physiol 133: 421–438, 2009. doi: 10.1085/jgp.200810097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Pedemonte N, Galietta LJ. Structure and function of TMEM16 proteins (anoctamins). Physiol Rev 94: 419–459, 2014. doi: 10.1152/physrev.00039.2011. [DOI] [PubMed] [Google Scholar]
- 169. Caputo A, Caci E, Ferrera L, Pedemonte N, Barsanti C, Sondo E, Pfeffer U, Ravazzolo R, Zegarra-Moran O, Galietta LJ. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science 322: 590–594, 2008. doi: 10.1126/science.1163518. [DOI] [PubMed] [Google Scholar]
- 170. Vega G, Guequén A, Philp AR, Gianotti A, Arzola L, Villalón M, Zegarra-Moran O, Galietta LJ, Mall MA, Flores CA. Lack of Kcnn4 improves mucociliary clearance in muco-obstructive lung disease. JCI Insight 5: e140076, 2020. doi: 10.1172/jci.insight.140076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Kis A, Krick S, Baumlin N, Salathe M. Airway hydration, apical K+ secretion, and the large-conductance, Ca2+-activated and voltage-dependent potassium (BK) channel. Ann Am Thorac Soc 13, Suppl 2: S163–S168, 2016. doi: 10.1513/AnnalsATS.201507-405KV. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Zhao KQ, Xiong G, Wilber M, Cohen NA, Kreindler JL. A role for two-pore K+ channels in modulating Na+ absorption and Cl- secretion in normal human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol 302: L4–L12, 2012. doi: 10.1152/ajplung.00102.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Schultz A, Puvvadi R, Borisov SM, Shaw NC, Klimant I, Berry LJ, Montgomery ST, Nguyen T, Kreda SM, Kicic A, Noble PB, Button B, Stick SM. Airway surface liquid pH is not acidic in children with cystic fibrosis. Nat Commun 8: 1409, 2017. doi: 10.1038/s41467-017-00532-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Coakley RD, Grubb BR, Paradiso AM, Gatzy JT, Johnson LG, Kreda SM, O’Neal WK, Boucher RC. Abnormal surface liquid pH regulation by cultured cystic fibrosis bronchial epithelium. Proc Natl Acad Sci USA 100: 16083–16088, 2003. doi: 10.1073/pnas.2634339100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Rehman T, Thornell IM, Pezzulo AA, Thurman AL, Romano Ibarra GS, Karp PH, Tan P, Duffey ME, Welsh MJ. TNFα and IL-17 alkalinize airway surface liquid through CFTR and pendrin. Am J Physiol Cell Physiol 319: C331–C344, 2020. doi: 10.1152/ajpcell.00112.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Hartmann T, Verkman AS. Model of ion transport regulation in chloride-secreting airway epithelial cells. Integrated description of electrical, chemical, and fluorescence measurements. Biophys J 58: 391–401, 1990. doi: 10.1016/S0006-3495(90)82385-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Duszyk M, French AS. An analytical model of ionic movements in airway epithelial cells. J Theor Biol 151: 231–247, 1991. doi: 10.1016/s0022-5193(05)80362-5. [DOI] [PubMed] [Google Scholar]
- 178. Novotny JA, Jakobsson E. Computational studies of ion-water flux coupling in the airway epithelium. I. Construction of model. Am J Physiol Cell Physiol 270: C1751–C1763, 1996. doi: 10.1152/ajpcell.1996.270.6.C1751. [DOI] [PubMed] [Google Scholar]
- 179. Novotny JA, Jakobsson E. Computational studies of ion-water flux coupling in the airway epithelium. II. Role of specific transport mechanisms. Am J Physiol Cell Physiol 270: C1764–C1772, 1996. doi: 10.1152/ajpcell.1996.270.6.C1764. [DOI] [PubMed] [Google Scholar]
- 180. Warren NJ, Tawhai MH, Crampin EJ. A mathematical model of calcium-induced fluid secretion in airway epithelium. J Theor Biol 259: 837–849, 2009. doi: 10.1016/j.jtbi.2009.04.026. [DOI] [PubMed] [Google Scholar]
- 181. Falkenberg CV, Jakobsson E. A biophysical model for integration of electrical, osmotic, and pH regulation in the human bronchial epithelium. Biophys J 98: 1476–1485, 2010. doi: 10.1016/j.bpj.2009.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Myerburg MM, Harvey PR, Heidrich EM, Pilewski JM, Butterworth MB. Acute regulation of the epithelial sodium channel in airway epithelia by proteases and trafficking. Am J Respir Cell Mol Biol 43: 712–719, 2010. doi: 10.1165/rcmb.2009-0348OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Myerburg MM, McKenna EE, Luke CJ, Frizzell RA, Kleyman TR, Pilewski JM. Prostasin expression is regulated by airway surface liquid volume and is increased in cystic fibrosis. Am J Physiol Lung Cell Mol Physiol 294: L932–L941, 2008. doi: 10.1152/ajplung.00437.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Nimishakavi S, Besprozvannaya M, Raymond WW, Craik CS, Gruenert DC, Caughey GH. Activity and inhibition of prostasin and matriptase on apical and basolateral surfaces of human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol 303: L97–L106, 2012. doi: 10.1152/ajplung.00303.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Garcia-Caballero A, Rasmussen JE, Gaillard E, Watson MJ, Olsen JC, Donaldson SH, Stutts MJ, Tarran R. SPLUNC1 regulates airway surface liquid volume by protecting ENaC from proteolytic cleavage. Proc Natl Acad Sci USA 106: 11412–11417, 2009. doi: 10.1073/pnas.0903609106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Willumsen NJ, Boucher RC. Shunt resistance and ion permeabilities in normal and cystic fibrosis airway epithelia. Am J Physiol Cell Physiol 256: C1054–C1063, 1989. doi: 10.1152/ajpcell.1989.256.5.C1054. [DOI] [PubMed] [Google Scholar]
- 187. Willumsen NJ, Boucher RC. Activation of an apical Cl- conductance by Ca2+ ionophores in cystic fibrosis airway epithelia. Am J Physiol Cell Physiol 256: C226–C233, 1989. doi: 10.1152/ajpcell.1989.256.2.C226. [DOI] [PubMed] [Google Scholar]
- 188. Willumsen NJ, Davis CW, Boucher RC. Cellular Cl- transport in cultured cystic fibrosis airway epithelium. Am J Physiol Cell Physiol 256: C1045–C1053, 1989. doi: 10.1152/ajpcell.1989.256.5.C1045. [DOI] [PubMed] [Google Scholar]
- 189. Willumsen NJ, Davis CW, Boucher RC. Intracellular Cl- activity and cellular Cl- pathways in cultured human airway epithelium. Am J Physioll Cell Physiol 256: C1033–C1044, 1989. doi: 10.1152/ajpcell.1989.256.5.C1033. [DOI] [PubMed] [Google Scholar]
- 190. Kunzelmann K, Bachhuber T, Regeer R, Markovich D, Sun J, Schreiber R. Purinergic inhibition of the epithelial Na+ transport via hydrolysis of PIP2. FASEB J 19: 142–143, 2005. doi: 10.1096/fj.04-2314fje. [DOI] [PubMed] [Google Scholar]
- 191. Ma HP, Saxena S, Warnock DG. Anionic phospholipids regulate native and expressed epithelial sodium channel (ENaC). J Biol Chem 277: 7641–7644, 2002. doi: 10.1074/jbc.C100737200. [DOI] [PubMed] [Google Scholar]
- 192. Yue G, Edinger RS, Bao HF, Johnson JP, Eaton DC. The effect of rapamycin on single ENaC channel activity and phosphorylation in A6 cells. Am J Physiol Cell Physiol 279: C81–C88, 2000. doi: 10.1152/ajpcell.2000.279.1.C81. [DOI] [PubMed] [Google Scholar]
- 193. Devor DC, Singh AK, Bridges RJ, Frizzell RA. Psoralens: novel modulators of Cl- secretion. Am J Physiol Cell Physiol 272: C976–C988, 1997. doi: 10.1152/ajpcell.1997.272.3.C976. [DOI] [PubMed] [Google Scholar]
- 194. Leipziger J. Control of epithelial transport via luminal P2 receptors. Am J Physiol Renal Physiol 284: F419–F432, 2003. doi: 10.1152/ajprenal.00075.2002. [DOI] [PubMed] [Google Scholar]
- 195. Kota P, Gentzsch M, Dang YL, Boucher RC, Stutts MJ. The N terminus of alpha-ENaC mediates ENaC cleavage and activation by furin. J Gen Physiol 150: 1179–1187, 2018. doi: 10.1085/jgp.201711860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Scudieri P, Caci E, Bruno S, Ferrera L, Schiavon M, Sondo E, Tomati V, Gianotti A, Zegarra-Moran O, Pedemonte N, Rea F, Ravazzolo R, Galietta LJ. Association of TMEM16A chloride channel overexpression with airway goblet cell metaplasia. J Physiol 590: 6141–6155, 2012. doi: 10.1113/jphysiol.2012.240838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Galietta LJ. The TMEM16 protein family: a new class of chloride channels? Biophys J 97: 3047–3053, 2009. doi: 10.1016/j.bpj.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Zhang Y, Wang X, Wang H, Jiao J, Li Y, Fan E, Zhang L, Bachert C. TMEM16A-mediated mucin secretion in IL-13-induced nasal epithelial cells from chronic rhinosinusitis patients. Allergy Asthma Immunol Res 7: 367–375, 2015. doi: 10.4168/aair.2015.7.4.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Dulin NO. Calcium-activated chloride channel ANO1/TMEM16A: regulation of expression and signaling. Front Physiol 11: 590262, 2020. doi: 10.3389/fphys.2020.590262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Danahay H, Atherton H, Jones G, Bridges RJ, Poll CT. Interleukin-13 induces a hypersecretory ion transport phenotype in human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol 282: L226–L236, 2002. doi: 10.1152/ajplung.00311.2001. [DOI] [PubMed] [Google Scholar]
- 201. Clarke LL, Paradiso AM, Mason SJ, Boucher RC. Effects of bradykinin on Na+ and Cl- transport in human nasal epithelium. Am J Physiol Cell Physiol 262: C644–C655, 1992. doi: 10.1152/ajpcell.1992.262.3.C644. [DOI] [PubMed] [Google Scholar]
- 202. Namkung W, Thiagarajah JR, Phuan PW, Verkman AS. Inhibition of Ca2+-activated Cl- channels by gallotannins as a possible molecular basis for health benefits of red wine and green tea. FASEB J 24: 4178–4186, 2010. doi: 10.1096/fj.10-160648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Salathe M. Effects of beta-agonists on airway epithelial cells. J Allergy Clin Immunol 110: S275–S281, 2002. doi: 10.1067/mai.2002.129412. [DOI] [PubMed] [Google Scholar]
- 204. Nguyen LP, Al-Sawalha NA, Parra S, Pokkunuri I, Omoluabi O, Okulate AA, Windham Li E, Hazen M, Gonzalez-Granado JM, Daly CJ, McGrath JC, Tuvim MJ, Knoll BJ, Dickey BF, Bond RA. β2-Adrenoceptor signaling in airway epithelial cells promotes eosinophilic inflammation, mucous metaplasia, and airway contractility. Proc Natl Acad Sci USA 114: E9163–E9171, 2017. doi: 10.1073/pnas.1710196114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Boucher RC, Cheng EH, Paradiso AM, Stutts MJ, Knowles MR, Earp HS. Chloride secretory response of cystic fibrosis human airway epithelia. Preservation of calcium but not protein kinase C- and A-dependent mechanisms. J Clin Invest 84: 1424–1431, 1989. doi: 10.1172/JCI114316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Chappe V, Irvine T, Liao J, Evagelidis A, Hanrahan JW. Phosphorylation of CFTR by PKA promotes binding of the regulatory domain. EMBO J 24: 2730–2740, 2005. doi: 10.1038/sj.emboj.7600747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Hegedus T, Aleksandrov A, Mengos A, Cui L, Jensen TJ, Riordan JR. Role of individual R domain phosphorylation sites in CFTR regulation by protein kinase A. Biochim Biophys Acta 1788: 1341–1349, 2009. doi: 10.1016/j.bbamem.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 208. Mall M, Grubb BR, Harkema JR, O’Neal WK, Boucher RC. Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat Med 10: 487–493, 2004. doi: 10.1038/nm1028. [DOI] [PubMed] [Google Scholar]
- 209. Blouquit S, Morel H, Hinnrasky J, Naline E, Puchelle E, Chinet T. Characterization of ion and fluid transport in human bronchioles. Am J Respir Cell Mol Biol 27: 503–510, 2002. doi: 10.1165/rcmb.4869. [DOI] [PubMed] [Google Scholar]
- 210. Knowles MR, Clarke LL, Boucher RC. Activation by extracellular nucleotides of chloride secretion in the airway epithelia of patients with cystic fibrosis. N Engl J Med 325: 533–538, 1991. doi: 10.1056/NEJM199108223250802. [DOI] [PubMed] [Google Scholar]
- 211. Chen G, Sun L, Kato T, Okuda K, Martino MB, Abzhanova A, Lin JM, Gilmore RC, Batson BD, O’Neal YK, Volmer AS, Dang H, Deng Y, Randell SH, Button B, Livraghi-Butrico A, Kesimer M, Ribeiro CM, O'Neal WK, Boucher RC. IL-1beta dominates the promucin secretory cytokine profile in cystic fibrosis. J Clin Invest 129: 4433–4450, 2019. doi: 10.1172/JCI125669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Mason SJ, Paradiso AM, Boucher RC. Regulation of transepithelial ion transport and intracellular calcium by extracellular ATP in human normal and cystic fibrosis airway epithelium. Br J Pharmacol 103: 1649–1656, 1991. doi: 10.1111/j.1476-5381.1991.tb09842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Garcia GJ, Picher M, Zuo P, Okada SF, Lazarowski ER, Button B, Boucher RC, Elston TC. Computational model for the regulation of extracellular ATP and adenosine in airway epithelia. Subcell Biochem 55: 51–74, 2011. doi: 10.1007/978-94-007-1217-1_3. [DOI] [PubMed] [Google Scholar]
- 214. Sandefur CI, Boucher RC, Elston TC. Mathematical model reveals role of nucleotide signaling in airway surface liquid homeostasis and its dysregulation in cystic fibrosis. Proc Natl Acad Sci USA 114: E7272–E7281, 2017. doi: 10.1073/pnas.1617383114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Zuo P, Picher M, Okada SF, Lazarowski ER, Button B, Boucher RC, Elston TC. Mathematical model of nucleotide regulation on airway epithelia. Implications for airway homeostasis. J Biol Chem 283: 26805–26819, 2008. doi: 10.1074/jbc.M801516200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Button B, Okada SF, Frederick CB, Thelin WR, Boucher RC. Mechanosensitive ATP release maintains proper mucus hydration of airways. Sci Signal 6: ra46, 2013. doi: 10.1126/scisignal.2003755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Lazarowski ER, Boucher RC. Purinergic receptors in airway hydration. Biochem Pharmacol 187: 114387, 2021. doi: 10.1016/j.bcp.2020.114387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Rowe SM, Heltshe SL, Gonska T, Donaldson SH, Borowitz D, Gelfond D, Sagel SD, Khan U, Mayer-Hamblett N, Van Dalfsen JM, Joseloff E, Ramsey BW; GOAL Investigators of the Cystic Fibrosis Foundation Therapeutics Development Network. Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. Am J Respir Crit Care Med 190: 175–184, 2014. doi: 10.1164/rccm.201404-0703OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Okada SF, Nicholas RA, Kreda SM, Lazarowski ER, Boucher RC. Physiological regulation of ATP release at the apical surface of human airway epithelia. J Biol Chem 281: 22992–23002, 2006. doi: 10.1074/jbc.M603019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Ransford GA, Fregien N, Qiu F, Dahl G, Conner GE, Salathe M. Pannexin 1 contributes to ATP release in airway epithelia. Am J Respir Cell Mol Biol 41: 525–534, 2009. doi: 10.1165/rcmb.2008-0367OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Seminario-Vidal L, Okada SF, Sesma JI, Kreda SM, van Heusden CA, Zhu Y, Jones LC, O’Neal WK, Penuela S, Laird DW, Boucher RC, Lazarowski ER. Rho signaling regulates pannexin 1-mediated ATP release from airway epithelia. J Biol Chem 286: 26277–26286, 2011. doi: 10.1074/jbc.M111.260562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Dunn PJ, Salm EJ, Tomita S. ABC transporters control ATP release through cholesterol-dependent volume-regulated anion channel activity. J Biol Chem 295: 5192–5203, 2020. doi: 10.1074/jbc.RA119.010699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Ma Z, Tanis JE, Taruno A, Foskett JK. Calcium homeostasis modulator (CALHM) ion channels. Pflugers Arch 468: 395–403, 2016. doi: 10.1007/s00424-015-1757-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Workman AD, Carey RM, Chen B, Saunders CJ, Marambaud P, Mitchell CH, Tordoff MG, Lee RJ, Cohen NA. CALHM1-mediated ATP release and ciliary beat frequency modulation in nasal epithelial cells. Sci Rep 7: 6687, 2017. doi: 10.1038/s41598-017-07221-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Sawada K, Echigo N, Juge N, Miyaji T, Otsuka M, Omote H, Yamamoto A, Moriyama Y. Identification of a vesicular nucleotide transporter. Proc Natl Acad Sci USA 105: 5683–5686, 2008. doi: 10.1073/pnas.0800141105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Sesma JI, Kreda SM, Okada SF, van Heusden C, Moussa L, Jones LC, O’Neal WK, Togawa N, Hiasa M, Moriyama Y, Lazarowski ER. Vesicular nucleotide transporter regulates the nucleotide content in airway epithelial mucin granules. Am J Physiol Cell Physiol 304: C976–C984, 2013. doi: 10.1152/ajpcell.00371.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Hirsh AJ, Stonebraker JR, van Heusden CA, Lazarowski ER, Boucher RC, Picher M. Adenosine deaminase 1 and concentrative nucleoside transporters 2 and 3 regulate adenosine on the apical surface of human airway epithelia: implications for inflammatory lung diseases. Biochemistry 46: 10373–10383, 2007. doi: 10.1021/bi7009647. [DOI] [PubMed] [Google Scholar]
- 228. Rich PB, Douillet CD, Mahler SA, Husain SA, Boucher RC. Adenosine triphosphate is released during injurious mechanical ventilation and contributes to lung edema. J Trauma 55: 290–297, 2003. doi: 10.1097/01.TA.0000078882.11919.AF. [DOI] [PubMed] [Google Scholar]
- 229. Rich PB, Douillet CD, Hurd H, Boucher RC. Effect of ventilatory rate on airway cytokine levels and lung injury. J Surg Res 113: 139–145, 2003. doi: 10.1016/s0022-4804(03)00195-1. [DOI] [PubMed] [Google Scholar]
- 230. Flores RV, Hernández-Pérez MG, Aquino E, Garrad RC, Weisman GA, Gonzalez FA. Agonist-induced phosphorylation and desensitization of the P2Y2 nucleotide receptor. Mol Cell Biochem 280: 35–45, 2005. doi: 10.1007/s11010-005-8050-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Brown HA, Lazarowski ER, Boucher RC, Harden TK. Evidence that UTP and ATP regulate phospholipase C through a common extracellular 5'-nucleotide receptor in human airway epithelial cells. Mol Pharmacol 40: 648–655, 1991. [PubMed] [Google Scholar]
- 232. Herold CL, Qi AD, Harden TK, Nicholas RA. Agonist versus antagonist action of ATP at the P2Y4 receptor is determined by the second extracellular loop. J Biol Chem 279: 11456–11464, 2004. doi: 10.1074/jbc.M301734200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Matsui H, Grubb BR, Tarran R, Randell SH, Gatzy JT, Davis CW, Boucher RC. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell 95: 1005–1015, 1998. doi: 10.1016/S0092-8674(00)81724-9. [DOI] [PubMed] [Google Scholar]
- 234. Tarran R, Grubb BR, Parsons D, Picher M, Hirsh AJ, Davis CW, Boucher RC. The CF salt controversy: in vivo observations and therapeutic approaches. Mol Cell 8: 149–158, 2001. doi: 10.1016/s1097-2765(01)00286-6. [DOI] [PubMed] [Google Scholar]
- 235. Button B, Picher M, Boucher RC. Differential effects of cyclic and constant stress on ATP release and mucociliary transport by human airway epithelia. J Physiol 580: 577–592, 2007. doi: 10.1113/jphysiol.2006.126086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Tarran R, Button B, Boucher RC. Regulation of normal and cystic fibrosis airway surface liquid volume by phasic shear stress. Annu Rev Physiol 68: 543–561, 2006. doi: 10.1146/annurev.physiol.68.072304.112754. [DOI] [PubMed] [Google Scholar]
- 237. Tarrant BJ, Le Maitre C, Romero L, Steward R, Button BM, Thompson BR, Holland AE. Mucoactive agents for chronic, non-cystic fibrosis lung disease: A systematic review and meta-analysis. Respirology 22: 1084–1092, 2017. doi: 10.1111/resp.13047. [DOI] [PubMed] [Google Scholar]
- 238. Wu D, Boucher RC, Button B, Elston T, Lin CL. An integrated mathematical epithelial cell model for airway surface liquid regulation by mechanical forces. J Theor Biol 438: 34–45, 2018. doi: 10.1016/j.jtbi.2017.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Burnstock G, Evans LC, Bailey MA. Purinergic signalling in the kidney in health and disease. Purinergic Signal 10: 71–101, 2014. doi: 10.1007/s11302-013-9400-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. van Heusden C, Grubb BR, Button B, Lazarowski ER. Airway epithelial nucleotide release contributes to mucociliary clearance. Life (Basel) 11: 430, 2021. doi: 10.3390/life11050430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Verdugo P. Mucin exocytosis. Am Rev Respir Dis 144: S33–S37, 1991. doi: 10.1164/ajrccm/144.3_pt_2.S33. [DOI] [PubMed] [Google Scholar]
- 242. Verdugo P. Supramolecular dynamics of mucus. Cold Spring Harb Perspect Med 2: a009597, 2012. doi: 10.1101/cshperspect.a009597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Verdugo P, Aitken M, Langley L, Villalon MJ. Molecular mechanism of product storage and release in mucin secretion. II. The role of extracellular Ca++. Biorheology 24: 625–633, 1987. doi: 10.3233/bir-1987-24615. [DOI] [PubMed] [Google Scholar]
- 244. Hansson GC, Johansson ME. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Gut Microbes 1: 51–54, 2010. doi: 10.4161/gmic.1.1.10470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Schütte A, Ermund A, Becker-Pauly C, Johansson ME, Rodriguez-Pineiro AM, Bäckhed F, Müller S, Lottaz D, Bond JS, Hansson GC. Microbial-induced meprin β cleavage in MUC2 mucin and a functional CFTR channel are required to release anchored small intestinal mucus. Proc Natl Acad Sci USA 111: 12396–12401, 2014. doi: 10.1073/pnas.1407597111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Kreda SM, Seminario-Vidal L, van Heusden CA, O’Neal W, Jones L, Boucher RC, Lazarowski ER. Receptor-promoted exocytosis of airway epithelial mucin granules containing a spectrum of adenine nucleotides. J Physiol 588: 2255–2267, 2010. doi: 10.1113/jphysiol.2009.186643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Fischer AJ, Goss KL, Scheetz TE, Wohlford-Lenane CL, Snyder JM, McCray PB. Jr.. Differential gene expression in human conducting airway surface epithelia and submucosal glands. Am J Respir Cell Mol Biol 40: 189–199, 2009. doi: 10.1165/rcmb.2008-0240OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Joo NS, Evans IA, Cho HJ, Park IH, Engelhardt JF, Wine JJ. Proteomic analysis of pure human airway gland mucus reveals a large component of protective proteins. PLoS One 10: e0116756, 2015. doi: 10.1371/journal.pone.0116756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Luan X, Tam JS, Jagadeeshan S, Grishchenko N, Hassan N, Gioino P, Shipley AM, Machen TE, Ianowski JP. Airway submucosal glands from cystic fibrosis swine suffer from abnormal ion transport across the serous acini, collecting duct, and ciliated duct. Am J Physiol Lung Cell Mol Physiol 318: L931–L942, 2020. doi: 10.1152/ajplung.00219.2019. [DOI] [PubMed] [Google Scholar]
- 250. Bals R. Epithelial antimicrobial peptides in host defense against infection. Respir Res 1: 141–150, 2000. doi: 10.1186/rr25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Bals R, Wang X, Wu Z, Freeman T, Bafna V, Zasloff M, Wilson JM. Human beta-defensin 2 is a salt-sensitive peptide antibiotic expressed in human lung. J Clin Invest 102: 874–880, 1998. doi: 10.1172/JCI2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Goldman MJ, Anderson GM, Stolzenberg ED, Kari UP, Zasloff M, Wilson JM. Human beta-defensin-1 is a salt-sensitive antibiotic in lung that is inactivated in cystic fibrosis. Cell 88: 553–560, 1997. doi: 10.1016/s0092-8674(00)81895-4. [DOI] [PubMed] [Google Scholar]
- 253. Ostedgaard LS, Price MP, Whitworth KM, Abou Alaiwa MH, Fischer AJ, Warrier A, Samuel M, Spate LD, Allen PD, Hilkin BM, Romano Ibarra GS, Ortiz Bezara ME, Goodell BJ, Mather SE, Powers LS, Stroik MR, Gansemer ND, Hippee CE, Zarei K, Goeken JA, Businga TR, Hoffman EA, Meyerholz DK, Prather RS, Stoltz DA, Welsh MJ. Lack of airway submucosal glands impairs respiratory host defenses. Elife 9: e59653, 2020. doi: 10.7554/eLife.59653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Engelhardt JF, Yankaskas JR, Ernst SA, Yang Y, Marino CR, Boucher RC, Cohn JA, Wilson JM. Submucosal glands are the predominant site of CFTR expression in the human bronchus. Nat Genet 2: 240–248, 1992. doi: 10.1038/ng1192-240. [DOI] [PubMed] [Google Scholar]
- 255. Guo M, Pegoraro AF, Mao A, Zhou EH, Arany PR, Han Y, Burnette DT, Jensen MH, Kasza KE, Moore JR, Mackintosh FC, Fredberg JJ, Mooney DJ, Lippincott-Schwartz J, Weitz DA. Cell volume change through water efflux impacts cell stiffness and stem cell fate. Proc Natl Acad Sci USA 114: E8618–E8627, 2017. doi: 10.1073/pnas.1705179114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Ohkuda K, Nakahara K, Weidner WJ, Binder A, Staub NC. Lung fluid exchange after uneven pulmonary artery obstruction in sheep. Circ Res 43: 152–161, 1978. doi: 10.1161/01.res.43.2.152. [DOI] [PubMed] [Google Scholar]
- 257. Zarins CK, Rice CL, Peters RM, Virgilio RW. Lymph and pulmonary response to isobaric reduction in plasma oncotic pressure in baboons. Circ Res 43: 925–930, 1978. doi: 10.1161/01.res.43.6.925. [DOI] [PubMed] [Google Scholar]
- 258. Larsen EH, Sørensen JN. Stationary and nonstationary ion and water flux interactions in kidney proximal tubule: mathematical analysis of isosmotic transport by a minimalistic model. Rev Physiol Biochem Pharmacol 177: 101–147, 2020. doi: 10.1007/112_2019_16. [DOI] [PubMed] [Google Scholar]
- 259. Fernández-Blanco JA, Fakih D, Arike L, Rodríguez-Piñeiro AM, Martínez-Abad B, Skansebo E, Jackson S, Root J, Singh D, McCrae C, Evans CM, Astrand A, Ermund A, Hansson GC. Attached stratified mucus separates bacteria from the epithelial cells in COPD lungs. JCI Insight 3: e120994, 2018. doi: 10.1172/jci.insight.120994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Boucher RC, Stutts MJ, Knowles MR, Cantley L, Gatzy JT. Na+ transport in cystic fibrosis respiratory epithelia. Abnormal basal rate and response to adenylate cyclase activation. J Clin Invest 78: 1245–1252, 1986. doi: 10.1172/JCI112708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Knowles MR, Stutts MJ, Spock A, Fischer N, Gatzy JT, Boucher RC. Abnormal ion permeation through cystic fibrosis respiratory epithelium. Science 221: 1067–1070, 1983. doi: 10.1126/science.6308769. [DOI] [PubMed] [Google Scholar]
- 262. Livraghi A, Mall M, Paradiso AM, Boucher RC, Ribeiro CM. Modelling dysregulated Na+ absorption in airway epithelial cells with mucosal nystatin treatment. Am J Respir Cell Mol Biol 38: 423–434, 2008. doi: 10.1165/rcmb.2007-0177OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Goralski JL, Wu D, Thelin WR, Boucher RC, Button B. The in vitro effect of nebulised hypertonic saline on human bronchial epithelium. Eur Respir J 51: 1702652, 2018. doi: 10.1183/13993003.02652-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Satir P, Sleigh MA. The physiology of cilia and mucociliary interactions. Annu Rev Physiol 52: 137–155, 1990. doi: 10.1146/annurev.ph.52.030190.001033. [DOI] [PubMed] [Google Scholar]
- 265. Sanderson MJ, Sleigh MA. Ciliary activity of cultured rabbit tracheal epithelium: beat pattern and metachrony. J Cell Sci 47: 331–347, 1981. doi: 10.1242/jcs.47.1.331. [DOI] [PubMed] [Google Scholar]
- 266. Sleigh MA. The form of beat in cilia of Stentor and Opalina. J Exp Biol 37: 1–10, 1960. doi: 10.1242/jeb.37.1.1. [DOI] [Google Scholar]
- 267. Sleigh MA. Metachronism and frequency of beat in the peristomial cilia of stentor. J Exp Biol 33: 15–28, 1956. doi: 10.1242/jeb.33.1.15. [DOI] [Google Scholar]
- 268. Sorokin SP. Reconstructions of centriole formation and ciliogenesis in mammalian lungs. J Cell Sci 3: 207–230, 1968. doi: 10.1242/jcs.3.2.207. [DOI] [PubMed] [Google Scholar]
- 269. Fulford GR, Blake JR. Mucociliary transport in the lung. J Theor Biol 121: 381–402, 1986. doi: 10.1016/S0022-5193(86)80098-4. [DOI] [PubMed] [Google Scholar]
- 270. Satir P, Christensen ST. Structure and function of mammalian cilia. Histochem Cell Biol 129: 687–693, 2008. doi: 10.1007/s00418-008-0416-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Rogers TD, Ostrowski LE, Livraghi-Butrico A, Button B, Grubb BR. Mucociliary clearance in mice measured by tracking trans-tracheal fluorescence of nasally aerosolized beads. Sci Rep 8: 14744, 2018. doi: 10.1038/s41598-018-33053-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Bustamante-Marin XM, Ostrowski LE. Cilia and mucociliary clearance. Cold Spring Harb Perspect Biol 9: a028241, 2017. doi: 10.1101/cshperspect.a028241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Delmotte P, Sanderson MJ. Ciliary beat frequency is maintained at a maximal rate in the small airways of mouse lung slices. Am J Respir Cell Mol Biol 35: 110–117, 2006. doi: 10.1165/rcmb.2005-0417OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Lorenzo IM, Liedtke W, Sanderson MJ, Valverde MA. TRPV4 channel participates in receptor-operated calcium entry and ciliary beat frequency regulation in mouse airway epithelial cells. Proc Natl Acad Sci USA 105: 12611–12616, 2008. doi: 10.1073/pnas.0803970105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Salathe M. Regulation of mammalian ciliary beating. Annu Rev Physiol 69: 401–422, 2007. doi: 10.1146/annurev.physiol.69.040705.141253. [DOI] [PubMed] [Google Scholar]
- 276. Yaghi A, Dolovich MB. Airway epithelial cell cilia and obstructive lung disease. Cells 5: 40, 2016. doi: 10.3390/cells5040040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Grigg G, Hodge A. Electron microscopic studies of spermatozoa I. The morphology of the spermatozoon of the common domestic fowl (Gallus domesticus). Aust J Biol Sci 2: 271–286, 1949. doi: 10.1071/BI9490271. [DOI] [Google Scholar]
- 278. Nicastro D, Schwartz C, Pierson J, Gaudette R, Porter ME, McIntosh JR. The molecular architecture of axonemes revealed by cryoelectron tomography. Science 313: 944–948, 2006. doi: 10.1126/science.1128618. [DOI] [PubMed] [Google Scholar]
- 279. Hill DB, Swaminathan V, Estes A, Cribb J, O’Brien ET, Davis CW, Superfine R. Force generation and dynamics of individual cilia under external loading. Biophys J 98: 57–66, 2010. doi: 10.1016/j.bpj.2009.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Oldenburg AL, Chhetri RK, Hill DB, Button B. Monitoring airway mucus flow and ciliary activity with optical coherence tomography. Biomed Opt Express 3: 1978–1992, 2012. doi: 10.1364/BOE.3.001978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Vasquez PA, Jin Y, Vuong K, Hill DB, Forest MG. A new twist on Stokes’ second problem: Partial penetration of nonlinearity in sheared viscoelastic layers. J Non-Newton Fluid 196: 36–50, 2013. doi: 10.1016/j.jnnfm.2012.12.016. [DOI] [Google Scholar]
- 282. Lindemann CB. Testing the geometric clutch hypothesis. Biol Cell 96: 681–690, 2004. doi: 10.1016/j.biolcel.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 283. Lindemann CB. Structural-functional relationships of the dynein, spokes, and central-pair projections predicted from an analysis of the forces acting within a flagellum. Biophys J 84: 4115–4126, 2003. doi: 10.1016/S0006-3495(03)75136-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Lindemann CB. Geometric Clutch model version 3: the role of the inner and outer arm dyneins in the ciliary beat. Cell Motil Cytoskeleton 52: 242–254, 2002. doi: 10.1002/cm.10049. [DOI] [PubMed] [Google Scholar]
- 285. König K, Svaasand L, Liu Y, Sonek G, Patrizio P, Tadir Y, Berns MW, Tromberg BJ. Determination of motility forces of human spermatozoa using an 800 nm optical trap. Cell Mol Biol (Noisy-le-grand) 42: 501–509, 1996. [PubMed] [Google Scholar]
- 286. Sakakibara HM, Kunioka Y, Yamada T, Kamimura S. Diameter oscillation of axonemes in sea-urchin sperm flagella. Biophys J 86: 346–352, 2004. doi: 10.1016/S0006-3495(04)74110-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Teff Z, Priel Z, Gheber LA. Forces applied by cilia measured on explants from mucociliary tissue. Biophys J 92: 1813–1823, 2007. doi: 10.1529/biophysj.106.094698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Yoneda M. Force exerted by a single cilium of Mytilus-edulis. J Exp Biol 37: 461–468, 1960. doi: 10.1242/jeb.37.3.461. [DOI] [Google Scholar]
- 289. Tani T, Kamimura S. Dynein-ADP as a force-generating intermediate revealed by a rapid reactivation of flagellar axoneme. Biophys J 77: 1518–1527, 1999. doi: 10.1016/S0006-3495(99)76999-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Schmitz KA, Holcomb-Wygle DL, Oberski DJ, Lindemann CB. Measurement of the force produced by an intact bull sperm flagellum in isometric arrest and estimation of the dynein stall force. Biophys J 79: 468–478, 2000. doi: 10.1016/S0006-3495(00)76308-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Sears PR, Thompson K, Knowles MR, Davis CW. Human airway ciliary dynamics. Am J Physiol Lung Cell Mol Physiol 304: L170–L183, 2013. doi: 10.1152/ajplung.00105.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. O’Neal WK, Gallins P, Pace RG, Dang H, Wolf WE, Jones LC, Guo X, Zhou YH, Madar V, Huang J, Liang L, Moffatt MF, Cutting GR, Drumm ML, Rommens JM, Strug LJ, Sun W, Stonebraker JR, Wright FA, Knowles MR. Gene expression in transformed lymphocytes reveals variation in endomembrane and HLA pathways modifying cystic fibrosis pulmonary phenotypes. Am J Hum Genet 96: 318–328, 2015. doi: 10.1016/j.ajhg.2014.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293. Matsui H, Randell SH, Peretti SW, Davis CW, Boucher RC. Coordinated clearance of periciliary liquid and mucus from airway surfaces. J Clin Invest 102: 1125–1131, 1998. doi: 10.1172/JCI2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294. Norwood JT, Anderson RG. Evidence that adhesive sites on the tips of oviduct cilia membranes are required for ovum pickup in situ. Biol Reprod 23: 788–791, 1980. doi: 10.1095/biolreprod23.4.788. [DOI] [PubMed] [Google Scholar]
- 295. Norwood JT, Hein CE, Halbert SA, Anderson RG. Polycationic macromolecules inhibit cilia-mediated ovum transport in the rabbit oviduct. Proc Natl Acad Sci USA 75: 4413–4416, 1978. doi: 10.1073/pnas.75.9.4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. King PT. The pathophysiology of bronchiectasis. Int J Chron Obstruct Pulmon Dis 4: 411–419, 2009. doi: 10.2147/copd.s6133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. Puchelle E, Zahm JM, Aug F. Viscoelasticity, protein content and ciliary transport rate of sputum in patients with recurrent and chronic bronchitis. Biorheology 18: 659–666, 1981. doi: 10.3233/bir-1981-183-628. [DOI] [PubMed] [Google Scholar]
- 298. Puchelle E, Zahm JM, Duvivier C. Spinability of bronchial mucus. Relationship with viscoelasticity and mucous transport properties. Biorheology 20: 239–249, 1983. doi: 10.3233/bir-1983-20214. [DOI] [PubMed] [Google Scholar]
- 299. Rubin BK. Aerosol medications for treatment of mucus clearance disorders. Respir Care 60: 825–829, 2015. doi: 10.4187/respcare.04087. [DOI] [PubMed] [Google Scholar]
- 300. Dawson M, Wirtz D, Hanes J. Enhanced viscoelasticity of human cystic fibrotic sputum correlates with increasing microheterogeneity in particle transport. J Biol Chem 278: 50393–50401, 2003. doi: 10.1074/jbc.M309026200. [DOI] [PubMed] [Google Scholar]
- 301. Tang Q. Structural and Rheological Properties of Airway Mucus & Counterion Condensation around Charged Nanoparticles. Chapel Hill, NC: University of North Carolina, 2021. [Google Scholar]
- 302. Baconnais S, Tirouvanziam R, Zahm JM, de Bentzmann S, Péault B, Balossier G, Puchelle E. Ion composition and rheology of airway liquid from cystic fibrosis fetal tracheal xenografts. Am J Respir Cell Mol Biol 20: 605–611, 1999. doi: 10.1165/ajrcmb.20.4.3264. [DOI] [PubMed] [Google Scholar]
- 303. Rubinstein M, Colby RH. Polymer Physics. Oxford: Oxford University Press, 2003. [Google Scholar]
- 304. Georgiades P, Pudney PD, Thornton DJ, Waigh TA. Particle tracking microrheology of purified gastrointestinal mucins. Biopolymers 101: 366–377, 2014. doi: 10.1002/bip.22372. [DOI] [PubMed] [Google Scholar]
- 305. King M, Zahm JM, Pierrot D, Vaquez-Girod S, Puchelle E. The role of mucus gel viscosity, spinnability, and adhesive properties in clearance by simulated cough. Biorheology 26: 737–745, 1989. doi: 10.3233/bir-1989-26406. [DOI] [PubMed] [Google Scholar]
- 306. Evans RM, Tassieri M, Auhl D, Waigh TA. Direct conversion of rheological compliance measurements into storage and loss moduli. Phys Rev E Stat Nonlin Soft Matter Phys 80: 012501, 2009. doi: 10.1103/PhysRevE.80.012501. [DOI] [PubMed] [Google Scholar]
- 307. Quinton PM. Viscosity versus composition in airway pathology. Am J Respir Crit Care Med 149: 6–7, 1994. doi: 10.1164/ajrccm.149.1.8111599. [DOI] [PubMed] [Google Scholar]
- 308. Rouillard KR, Hill DB, Schoenfisch MH. Antibiofilm and mucolytic action of nitric oxide delivered via gas or macromolecular donor using in vitro and ex vivo models. J Cyst Fibros 19: 1004–1010, 2020. doi: 10.1016/j.jcf.2020.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309. Ahonen MJ, Hill DB, Schoenfisch MH. Nitric oxide-releasing alginates as mucolytic agents. ACS Biomater Sci Eng 5: 3409–3418, 2019. doi: 10.1021/acsbiomaterials.9b00482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310. Ma JT, Tang C, Kang L, Voynow JA, Rubin BK. Cystic fibrosis sputum rheology correlates with both acute and longitudinal changes in lung function. Chest 154: 370–377, 2018. doi: 10.1016/j.chest.2018.03.005. [DOI] [PubMed] [Google Scholar]
- 311. Zahm JM, King M, Duvivier C, Pierrot D, Girod S, Puchelle E. Role of simulated repetitive coughing in mucus clearance. Eur Respir J 4: 311–315, 1991. [PubMed] [Google Scholar]
- 312. King M, Brock G, Lundell C. Clearance of mucus by simulated cough. J Appl Physiol (1985) 58: 1776–1782, 1985. doi: 10.1152/jappl.1985.58.6.1776. [DOI] [PubMed] [Google Scholar]
- 313. King M. The role of mucus viscoelasticity in cough clearance. Biorheology 24: 589–597, 1987. doi: 10.3233/bir-1987-24611. [DOI] [PubMed] [Google Scholar]
- 314. Agarwal M, King M, Shukla AB. Mucous gel transport in a simulated cough machine - effects of longitudinal grooves representing spacings between arrays of cilia. Biorheology 31: 11–19, 1994. doi: 10.3233/bir-1994-31102. [DOI] [PubMed] [Google Scholar]
- 315. Leith DE. Cough. Phys Ther 48: 439–447, 1968. doi: 10.1093/ptj/48.5.439. [DOI] [PubMed] [Google Scholar]
- 316. Leith DE. The development of cough. Am Rev Respir Dis 131: S39–S42, 1985. doi: 10.1164/arrd.1985.131.S5.S39. [DOI] [PubMed] [Google Scholar]
- 317. Wong JW, Keens TG, Wannamaker EM, Crozier DN, Levison H, Aspin N. Effects of gravity on tracheal mucus transport rates in normal subjects and in patients with cystic fibrosis. Pediatrics 60: 146–152, 1977. [PubMed] [Google Scholar]
- 318. Naylor JM, McLean A, Chow CM, Heard R, Ting I, Avolio A. A modified postural drainage position produces less cardiovascular stress than a head-down position in patients with severe heart disease: a quasi-experimental study. Aust J Physiother 52: 201–209, 2006. doi: 10.1016/S0004-9514(06)70029-0. [DOI] [PubMed] [Google Scholar]
- 319. Carpenter J, Lynch SE, Cribb JA, Kylstra S, Hill DB, Superfine R. Buffer drains and mucus is transported upward in a tilted mucus clearance assay. Am J Physiol Lung Cell Mol Physiol 315: L910–L918, 2018. doi: 10.1152/ajplung.00274.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320. Utomo NW, Nazari B, Parisi D, Colby RH. Determination of intrinsic viscosity of native cellulose solutions in ionic liquids. J Rheol 64: 1063–1073, 2020. doi: 10.1122/8.0000015. [DOI] [Google Scholar]
- 321. Hida K, Lai SK, Suk JS, Won SY, Boyle MP, Hanes J. Common gene therapy viral vectors do not efficiently penetrate sputum from cystic fibrosis patients. PLoS One 6: e19919, 2011. doi: 10.1371/journal.pone.0019919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322. Duncan GA, Jung J, Hanes J, Suk JS. The mucus barrier to inhaled gene therapy. Mol Ther 24: 2043–2053, 2016. doi: 10.1038/mt.2016.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323. Lai SK, Wang YY, Wirtz D, Hanes J. Micro- and macrorheology of mucus. Adv Drug Deliv Rev 61: 86–100, 2009. doi: 10.1016/j.addr.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324. Kim N, Kwak G, Rodriguez J, Livraghi-Butrico A, Zuo X, Simon V, Han E, Shenoy SK, Pandey N, Mazur M, Birket SE, Kim A, Rowe SM, Boucher R, Hanes J, Suk JS. Inhaled gene therapy of preclinical muco-obstructive lung diseases by nanoparticles capable of breaching the airway mucus barrier. Thorax 2021: thoraxjnl-2020-215185, 2021. doi: 10.1136/thoraxjnl-2020-215185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325. Murgia X, Kany AM, Herr C, Ho DK, De Rossi C, Bals R, Lehr CM, Hirsch AK, Hartmann RW, Empting M, Röhrig T. Micro-rheological properties of lung homogenates correlate with infection severity in a mouse model of Pseudomonas aeruginosa lung infection. Sci Rep 10: 16502, 2020. doi: 10.1038/s41598-020-73459-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326. Duncan GA, Jung J, Joseph A, Thaxton AL, West NE, Boyle MP, Hanes J, Suk JS. Microstructural alterations of sputum in cystic fibrosis lung disease. JCI Insight 1: e88198, 2016. doi: 10.1172/jci.insight.88198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327. Hancock LA, Hennessy CE, Solomon GM, Dobrinskikh E, Estrella A, Hara N, Hill DB, Kissner WJ, Markovetz MR, Grove Villalon DE, Voss ME, Tearney GJ, Carroll KS, Shi Y, Schwarz MI, Thelin WR, Rowe SM, Yang IV, Evans CM, Schwartz DA. Muc5b overexpression causes mucociliary dysfunction and enhances lung fibrosis in mice. Nat Commun 9: 5363, 2018. doi: 10.1038/s41467-018-07768-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328. Wang YY, Lai SK, Ensign LM, Zhong W, Cone R, Hanes J. The microstructure and bulk rheology of human cervicovaginal mucus are remarkably resistant to changes in pH. Biomacromolecules 14: 4429–4435, 2013. doi: 10.1021/bm401356q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329. Lai SK, Wang YY, Hida K, Cone R, Hanes J. Nanoparticles reveal that human cervicovaginal mucus is riddled with pores larger than viruses. Proc Natl Acad Sci USA 107: 598–603, 2010. doi: 10.1073/pnas.0911748107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330. Lai SK, O’Hanlon DE, Harrold S, Man ST, Wang YY, Cone R, Hanes J. Rapid transport of large polymeric nanoparticles in fresh undiluted human mucus. Proc Natl Acad Sci USA 104: 1482–1487, 2007. doi: 10.1073/pnas.0608611104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331. Fernandez-Petty CM, Hughes GW, Bowers HL, Watson JD, Rosen BH, Townsend SM, Santos C, Ridley CE, Chu KK, Birket SE, Li Y, Leung HM, Mazur M, Garcia BA, Evans TI, Libby EF, Hathorne H, Hanes J, Tearney GJ, Clancy JP, Engelhardt JF, Swords WE, Thornton DJ, Wiesmann WP, Baker SM, Rowe SM. A glycopolymer improves vascoelasticity and mucociliary transport of abnormal cystic fibrosis mucus. JCI Insight 4: e125954, 2019. doi: 10.1172/jci.insight.125954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332. Donaldson SH, Zeman K, Laube B, Corcoran T, Locke LW, Pilewski J, Hanes J, Schuster B, Kanzawa M, Rowe SM, Bennett WD. Effect of ivacaftor on mucociliary clearance and mucus rheology in patients with a G551D CFTR mutation. Pediatr Pulmonol 48: 279–280, 2013. [Google Scholar]
- 333. MacKintosh FC, Schmidt CF. Microrheology. Curr Opin Colloid Interface Sci 4: 300–307, 1999. doi: 10.1016/S1359-0294(99)90010-9. [DOI] [Google Scholar]
- 334. Wagner CE, Turner BS, Rubinstein M, McKinley GH, Ribbeck K. A rheological study of the association and dynamics of MUC5AC gels. Biomacromolecules 18: 3654–3664, 2017. doi: 10.1021/acs.biomac.7b00809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335. Crocker JC, Valentine MT, Weeks ER, Gisler T, Kaplan PD, Yodh AG, Weitz DA. Two-point microrheology of inhomogeneous soft materials. Phys Rev Lett 85: 888–891, 2000. doi: 10.1103/PhysRevLett.85.888. [DOI] [PubMed] [Google Scholar]
- 336. Mellnik JW, Lysy M, Vasquez PA, Pillai NS, Hill DB, Cribb J, McKinley SA, Forest MG. Maximum likelihood estimation for single particle, passive microrheology data with drift. J Rheol 60: 379–392, 2016. doi: 10.1122/1.4943988. [DOI] [Google Scholar]
- 337. Mellnik J, Vasquez PA, McKinley SA, Witten J, Hill DB, Forest MG. Micro-heterogeneity metrics for diffusion in soft matter. Soft Matter 10: 7781–7796, 2014. doi: 10.1039/c4sm00676c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338. Crick FH, Hughes AF. The physical properties of cytoplasm. A study by means of the magnetic particle method. Part I. experimental. Exp Cell Res 1: 37–80, 1950. doi: 10.1016/0014-4827(50)90048-6. [DOI] [Google Scholar]
- 339. Crick FH. The physical properties of cytoplasm. A study by means of the magnetic particle method. Part II. theoretical treatment. Exp Cell Res 1: 505–533, 1950. doi: 10.1016/0014-4827(50)90002-4. [DOI] [Google Scholar]
- 340. Cribb JA, Vasquez PA, Moore P, Norris S, Shah S, Forest MG, Superfine R. Nonlinear signatures of entangled polymer solutions in active microbead rheology. J Rheol (NY NY) 57: 1247, 2013. doi: 10.1122/1.4811477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341. King M, Macklem PT. Rheological properties of microliter quantities of normal mucus. J Appl Physiol Respir Environ Exerc Physiol 42: 797–802, 1977. doi: 10.1152/jappl.1977.42.6.797. [DOI] [PubMed] [Google Scholar]
- 342. Khan MA, Wolf DP, Litt M. Effect of mucolytic agents on the rheological properties of tracheal mucus. Biochim Biophys Acta 444: 369–373, 1976. doi: 10.1016/0304-4165(76)90380-9. [DOI] [PubMed] [Google Scholar]
- 343. Litt M, Khan MA, Wolf DP. Mucus rheology—relation to structure and function. Biorheology 12: 102–103, 1975. [DOI] [PubMed] [Google Scholar]
- 344. Litt M. Mucus rheology. Relevance to mucociliary clearance. Arch Intern Med 126: 417–423, 1970. doi: 10.1001/archinte.126.3.417. [DOI] [PubMed] [Google Scholar]
- 345. De Sanctis GT, Tomkiewicz RP, Rubin BK, Schürch S, King M. Exogenous surfactant enhances mucociliary clearance in the anesthetized dog. Eur Respir J 7: 1616–1621, 1994. doi: 10.1183/09031936.94.07091616. [DOI] [PubMed] [Google Scholar]
- 346. Rubin BK, Ramirez O, Zayas JG, Finegan B, King M. Collection and analysis of respiratory mucus from subjects without lung disease. Am Rev Respir Dis 141: 1040–1043, 1990. doi: 10.1164/ajrccm/141.4_Pt_1.1040. [DOI] [PubMed] [Google Scholar]
- 347. Mastorakos P, da Silva AL, Chisholm J, Song E, Choi WK, Boyle MP, Morales MM, Hanes J, Suk JS. Highly compacted biodegradable DNA nanoparticles capable of overcoming the mucus barrier for inhaled lung gene therapy. Proc Natl Acad Sci USA 112: 8720–8725, 2015. doi: 10.1073/pnas.1502281112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348. Suh J, Dawson M, Hanes J. Real-time multiple-particle tracking: applications to drug and gene delivery. Adv Drug Deliv Rev 57: 63–78, 2005. doi: 10.1016/j.addr.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 349. Francis RJ, Chatterjee B, Loges NT, Zentgraf H, Omran H, Lo CW. Initiation and maturation of cilia-generated flow in newborn and postnatal mouse airway. Am J Physiol Lung Cell Mol Physiol 296: L1067–L1075, 2009. doi: 10.1152/ajplung.00001.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350. Leung HM, Birket SE, Hyun C, Ford TN, Cui D, Solomon GM, Shei RJ, Adewale AT, Lenzie AR, Fernandez-Petty CM, Zheng H, Palermo JH, Cho DY, Woodworth BA, Yonker LM, Hurley BP, Rowe SM, Tearney GJ. Intranasal micro-optical coherence tomography imaging for cystic fibrosis studies. Sci Transl Med 11: eaav3505, 2019. doi: 10.1126/scitranslmed.aav3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351. Stannard W, O’Callaghan C. Ciliary function and the role of cilia in clearance. J Aerosol Med 19: 110–115, 2006. doi: 10.1089/jam.2006.19.110. [DOI] [PubMed] [Google Scholar]
- 352. Tomkiewicz RP, Albers GM, De Sanctis GT, Ramirez OE, King M, Rubin BK. Species differences in the physical and transport properties of airway secretions. Can J Physiol Pharmacol 73: 165–171, 1995. doi: 10.1139/y95-025. [DOI] [PubMed] [Google Scholar]
- 353. Simet SM, Sisson JH, Pavlik JA, Devasure JM, Boyer C, Liu X, Kawasaki S, Sharp JG, Rennard SI, Wyatt TA. Long-term cigarette smoke exposure in a mouse model of ciliated epithelial cell function. Am J Respir Cell Mol Biol 43: 635–640, 2010. doi: 10.1165/rcmb.2009-0297OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354. Milara J, Armengot M, Bañuls P, Tenor H, Beume R, Artigues E, Cortijo J. Roflumilast N-oxide, a PDE4 inhibitor, improves cilia motility and ciliated human bronchial epithelial cells compromised by cigarette smoke in vitro. Br J Pharmacol 166: 2243–2262, 2012. doi: 10.1111/j.1476-5381.2012.01929.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355. Elliott MK, Sisson JH, Wyatt TA. Effects of cigarette smoke and alcohol on ciliated tracheal epithelium and inflammatory cell recruitment. Am J Respir Cell Mol Biol 36: 452–459, 2007. doi: 10.1165/rcmb.2005-0440OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356. Bennett WD, Henderson AG, Ceppe A, Zeman KL, Wu J, Gladman C, Fuller F, Gazda S, Button B, Boucher RC, Donaldson SH. Effect of hypertonic saline on mucociliary clearance and clinical outcomes in chronic bronchitis. ERJ Open Res 6: 00269-2020, 2020. doi: 10.1183/23120541.00269-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357. Hogg JC, Macklem PT, Thurlbeck WM. Site and nature of airway obstruction in chronic obstructive lung disease. N Engl J Med 278: 1355–1360, 1968. doi: 10.1056/NEJM196806202782501. [DOI] [PubMed] [Google Scholar]
- 358. Ramsey KA, Chen AC, Radicioni G, Lourie R, Martin M, Broomfield A, Sheng YH, Hasnain SZ, Radford-Smith G, Simms LA, Burr L, Thornton DJ, Bowler SD, Livengood S, Ceppe A, Knowles MR, Noone PG, Donaldson SH, Hill DB, Ehre C, Button B, Alexis NE, Kesimer M, Boucher RC, McGuckin MA. Airway mucus hyperconcentration in non-cystic fibrosis bronchiectasis. Am J Respir Crit Care Med 201: 661–670, 2020. doi: 10.1164/rccm.201906-1219OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359. Cho JL, Ling MF, Adams DC, Faustino L, Islam SA, Afshar R, Griffith JW, Harris RS, Ng A, Radicioni G, Ford AA, Han AK, Xavier R, Kwok WW, Boucher R, Moon JJ, Hamilos DL, Kesimer M, Suter MJ, Medoff BD, Luster AD. Allergic asthma is distinguished by sensitivity of allergen-specific CD4+ T cells and airway structural cells to type 2 inflammation. Sci Transl Med 8: 359ra132, 2016. doi: 10.1126/scitranslmed.aag1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360. Rubin BK. Mucus structure and properties in cystic fibrosis. Paediatr Respir Rev 8: 4–7, 2007. doi: 10.1016/j.prrv.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 361. Keir HR, Shoemark A, Dicker AJ, Perea L, Pollock J, Giam YH, Suarez-Cuartin G, Crichton ML, Lonergan M, Oriano M, Cant E, Einarsson GG, Furrie E, Elborn JS, Fong CJ, Finch S, Rogers GB, Blasi F, Sibila O, Aliberti S, Simpson JL, Huang JT, Chalmers JD. Neutrophil extracellular traps, disease severity, and antibiotic response in bronchiectasis: an international, observational, multicohort study. Lancet Respir Med 9: 873–884, 2021. doi: 10.1016/S2213-2600(20)30504-X. [DOI] [PubMed] [Google Scholar]
- 362. Sears PR, Bustamante-Marin XM, Gong H, Markovetz MR, Superfine R, Hill DB, Ostrowski LE. Induction of ciliary orientation by matrix patterning and characterization of mucociliary transport. Biophys J 120: 1387–1395, 2021. doi: 10.1016/j.bpj.2021.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363. Zahm JM, Girod de Bentzmann S, Deneuville E, Perrot-Minnot C, Dabadie A, Pennaforte F, Roussey M, Shak S, Puchelle E. Dose-dependent in vitro effect of recombinant human DNase on rheological and transport properties of cystic fibrosis respiratory mucus. Eur Respir J 8: 381–386, 1995. doi: 10.1183/09031936.95.08030381. [DOI] [PubMed] [Google Scholar]
- 364. Marcos V, Zhou-Suckow Z, Önder Yildirim A, Bohla A, Hector A, Vitkov L, Krautgartner WD, Stoiber W, Griese M, Eickelberg O, Mall MA, Hartl D. Free DNA in cystic fibrosis airway fluids correlates with airflow obstruction. Mediators Inflamm 2015: 408935, 2015. doi: 10.1155/2015/408935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365. Thim L, Madsen F, Poulsen SS. Effect of trefoil factors on the viscoelastic properties of mucus gels. Eur J Clin Invest 32: 519–527, 2002. doi: 10.1046/j.1365-2362.2002.01014.x. [DOI] [PubMed] [Google Scholar]
- 366. Bastholm SK, Samson MH, Becher N, Hansen LK, Stubbe PR, Chronakis IS, Nexo E, Uldbjerg N. Trefoil factor peptide 3 is positively correlated with the viscoelastic properties of the cervical mucus plug. Acta Obstet Gynecol Scand 96: 47–52, 2017. doi: 10.1111/aogs.13038. [DOI] [PubMed] [Google Scholar]
- 367. Kerr SC, Carrington SD, Yuan S, Solon M, Fahy JV. Galactose binding lectins cross-link airway mucins and are a novel mediator of mucus plug formation in acute asthma (Abstract). Am J Respir Crit Care Med 181: A5510, 2010. doi: 10.1164/ajrccm-conference.2010.181.1_MeetingAbstracts.A5510. [DOI] [Google Scholar]
- 368. Fontana GA, Widdicombe J. What is cough and what should be measured? Pulm Pharmacol Ther 20: 307–312, 2007. doi: 10.1016/j.pupt.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 369. Chung KF, Pavord ID. Prevalence, pathogenesis, and causes of chronic cough. Lancet 371: 1364–1374, 2008. doi: 10.1016/S0140-6736(08)60595-4. [DOI] [PubMed] [Google Scholar]
- 370. Bennett W, Zeman K, Donaldson SH, Donohue JF, Knowles MR, Boucher RC. Cough clearance is less effective in cystic fibrosis than chronic bronchitis (Abstract). Am J Respir Crit Care Med 169: A386, 2004. [Google Scholar]
- 371. Bennett WD, Chapman WF, Gerrity TR. Ineffectiveness of cough for enhancing mucus clearance in asymptomatic smokers. Chest 102: 412–416, 1992. doi: 10.1378/chest.102.2.412. [DOI] [PubMed] [Google Scholar]
- 372. McCool FD, Leith DE. Pathophysiology of cough. Clin Chest Med 8: 189–195, 1987. [PubMed] [Google Scholar]
- 373. Bush A, Payne D, Pike S, Jenkins G, Henke MO, Rubin BK. Mucus properties in children with primary ciliary dyskinesia: comparison with cystic fibrosis. Chest 129: 118–123, 2006. doi: 10.1378/chest.129.1.118. [DOI] [PubMed] [Google Scholar]
- 374. Mazzone SB, Canning BJ, Widdicombe JG. Sensory pathways for the cough reflex. In: Cough: Causes, Mechanisms and Therapy, edited by Chung KF, Widdicombe JG, Boushey HA.. Malden, MA: Blackwell Publishing, 2008, p. 159–172. [Google Scholar]
- 375. Brandimore AE, Hegland KW, Okun MS, Davenport PW, Troche MS. Voluntary upregulation of reflex cough is possible in healthy older adults and Parkinson’s disease. J Appl Physiol (1985) 123: 19–26, 2017. doi: 10.1152/japplphysiol.00612.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376. Farzan S. Cough and sputum production. In: Clinical Methods: The History, Physical, and Laboratory Examinations, edited by Walker HK, Hall WD, Hurst JW.. Boston, MA: Butterworths, 1990. [PubMed] [Google Scholar]
- 377. Hasani A, Pavia D, Agnew JE, Clarke SW. Regional lung clearance during cough and forced expiration technique (FET): effects of flow and viscoelasticity. Thorax 49: 557–561, 1994. doi: 10.1136/thx.49.6.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378. Massey BS, Ward-Smith J. Mechanics of Fluids. London: Spon Press, 2012. [Google Scholar]
- 379. Haaland SE. Simple and explicit formulas for the friction factor in turbulent pipe flow. J Fluids Eng 105: 89–90, 1983. doi: 10.1115/1.3240948. [DOI] [Google Scholar]
- 380. Yashima S, Takase N, Kurokawa T, Gong JP. Friction of hydrogels with controlled surface roughness on solid flat substrates. Soft Matter 10: 3192–3199, 2014. doi: 10.1039/c3sm52883a. [DOI] [PubMed] [Google Scholar]
- 381. Goodell HP, Shenoy SK, Shenkute NT, Lackey E, Dennis RG, Button B. Adhesive and cohesive peel force measurement of human airway mucus. Bio Protoc 9: e3287, 2019. doi: 10.21769/BioProtoc.3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382. Tarran R, Grubb BR, Gatzy JT, Davis CW, Boucher RC. The relative roles of passive surface forces and active ion transport in the modulation of airway surface liquid volume and composition. J Gen Physiol 118: 223–236, 2001. doi: 10.1085/jgp.118.2.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383. Cao R, Wang TT, DeMaria G, Sheehan JK, Kesimer M. Mapping the protein domain structures of the respiratory mucins: a mucin proteome coverage study. J Proteome Res 11: 4013–4023, 2012. doi: 10.1021/pr300058z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384. Alexis NE, Wells H, Siperly E, Goldstein B, Henderson AG, Peden DB. Baseline sputum parameters in normals, asthmatics, COPD, atopics, smokers and ex-smokers (Abstract). J Allergy Clin Immunol 137: AB208, 2016. doi: 10.1016/j.jaci.2015.12.1121. [DOI] [Google Scholar]
- 385. Dunican EM, Elicker BM, Gierada DS, Nagle SK, Schiebler ML, Newell JD, Raymond WW, Lachowicz-Scroggins ME, Di Maio S, Hoffman EA, Castro M, Fain SB, Jarjour NN, Israel E, Levy BD, Erzurum SC, Wenzel SE, Meyers DA, Bleecker ER, Phillips BR, Mauger DT, Gordon ED, Woodruff PG, Peters MC, Fahy JV; National Heart Lung and Blood Institute (NHLBI) Severe Asthma Research Program (SARP). Mucus plugs in patients with asthma linked to eosinophilia and airflow obstruction. J Clin Invest 128: 997–1009, 2018. doi: 10.1172/JCI95693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386. Kim CS, Greene MA, Sankaran S, Sackner MA. Mucus transport in the airways by two-phase gas-liquid flow mechanism: continuous flow model. J Appl Physiol (1985) 60: 908–917, 1986. doi: 10.1152/jappl.1986.60.3.908. [DOI] [PubMed] [Google Scholar]
- 387. Kim CS, Rodriguez CR, Eldridge MA, Sackner MA. Criteria for mucus transport in the airways by two-phase gas-liquid flow mechanism. J Appl Physiol (1985) 60: 901–907, 1986. doi: 10.1152/jappl.1986.60.3.901. [DOI] [PubMed] [Google Scholar]
- 388. Scherer PW, Burtz L. Fluid mechanical experiments relevant to coughing. J Biomech 11: 183–187, 1978. doi: 10.1016/0021-9290(78)90011-8. [DOI] [PubMed] [Google Scholar]
- 389. Baum GL, Zwas ST, Katz I, Roth Y. Mucociliary clearance from central airways in patients with excessive sputum production with and without primary ciliary dyskinesia. Chest 98: 608–612, 1990. doi: 10.1378/chest.98.3.608. [DOI] [PubMed] [Google Scholar]
- 390. Fink JB. Forced expiratory technique, directed cough, and autogenic drainage. Respir Care 52: 1210–1221, 2007. [PubMed] [Google Scholar]
- 391. Wanner A. Does chest physical therapy move airway secretions? Am Rev Respir Dis 130: 701–702, 1984. doi: 10.1164/arrd.1984.130.5.701. [DOI] [PubMed] [Google Scholar]
- 392. Benjamin RG, Chapman GA, Kim CS, Sackner MA. Removal of bronchial secretions by two-phase gas-liquid transport. Chest 95: 658–663, 1989. doi: 10.1378/chest.95.3.658. [DOI] [PubMed] [Google Scholar]
- 393. Han MK, Agusti A, Calverley PM, Celli BR, Criner G, Curtis JL, Fabbri LM, Goldin JG, Jones PW, Macnee W, Make BJ, Rabe KF, Rennard SI, Sciurba FC, Silverman EK, Vestbo J, Washko GR, Wouters EF, Martinez FJ. Chronic obstructive pulmonary disease phenotypes: the future of COPD. Am J Respir Crit Care Med 182: 598–604, 2010. doi: 10.1164/rccm.200912-1843CC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394. King PT, Holdsworth SR, Freezer NJ, Villanueva E, Holmes PW. Characterisation of the onset and presenting clinical features of adult bronchiectasis. Respir Med 100: 2183–2189, 2006. doi: 10.1016/j.rmed.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 395. Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med 352: 1992–2001, 2005. doi: 10.1056/NEJMra043184. [DOI] [PubMed] [Google Scholar]
- 396. Tiddens HA, Donaldson SH, Rosenfeld M, Paré PD. Cystic fibrosis lung disease starts in the small airways: can we treat it more effectively? Pediatr Pulmonol 45: 107–117, 2010. doi: 10.1002/ppul.21154. [DOI] [PubMed] [Google Scholar]
- 397. Whitwell F. A study of the pathology and pathogenesis of bronchiectasis. Thorax 7: 213–239, 1952. doi: 10.1136/thx.7.3.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398. Burgel PR, Bourdin A, Chanez P, Chabot F, Chaouat A, Chinet T, de Blic J, Devillier P, Deschildre A, Didier A, Garcia G, Jebrak G, Laurent F, Morel H, Perez T, Pilette C, Roche N, Tillie-Leblond I, Verbanck S, Dusser D. Update on the roles of distal airways in COPD. Eur Respir Rev 20: 7–22, 2011. doi: 10.1183/09059180.10010610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399. Pisi R, Aiello M, Frizzelli A, Calzetta L, Marchi L, Bertorelli G, Pisi G, Chetta A. Detection of small airway dysfunction in asymptomatic smokers with preserved spirometry: the value of the impulse oscillometry system. COPD 16: 2585–2590, 2021. doi: 10.2147/COPD.S319972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400. Boucher RC. Airway surface dehydration in cystic fibrosis: pathogenesis and therapy. Annu Rev Med 58: 157–170, 2007. doi: 10.1146/annurev.med.58.071905.105316. [DOI] [PubMed] [Google Scholar]
- 401. Donaldson SH, Bennett WD, Zeman KL, Knowles MR, Tarran R, Boucher RC. Mucus clearance and lung function in cystic fibrosis with hypertonic saline. N Engl J Med 354: 241–250, 2006. doi: 10.1056/NEJMoa043891. [DOI] [PubMed] [Google Scholar]
- 402. Liesman RM, Buchholz UJ, Luongo CL, Yang L, Proia AD, DeVincenzo JP, Collins PL, Pickles RJ. RSV-encoded NS2 promotes epithelial cell shedding and distal airway obstruction. J Clin Invest 124: 2219–2233, 2014. doi: 10.1172/JCI72948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403. Aherne W, Bird T, Court SD, Gardner PS, McQuillin J. Pathological changes in virus infections of the lower respiratory tract in children. J Clin Pathol 23: 7–18, 1970. doi: 10.1136/jcp.23.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404. Piccione JC, McPhail GL, Fenchel MC, Brody AS, Boesch RP. Bronchiectasis in chronic pulmonary aspiration: risk factors and clinical implications. Pediatr Pulmonol 47: 447–452, 2012. doi: 10.1002/ppul.21587. [DOI] [PubMed] [Google Scholar]
- 405. Huxley EJ, Viroslav J, Gray WR, Pierce AK. Pharyngeal aspiration in normal adults and patients with depressed consciousness. Am J Med 64: 564–568, 1978. doi: 10.1016/0002-9343(78)90574-0. [DOI] [PubMed] [Google Scholar]
- 406. Cardasis JJ, MacMahon H, Husain AN. The spectrum of lung disease due to chronic occult aspiration. Ann Am Thorac Soc 11: 865–873, 2014. doi: 10.1513/AnnalsATS.201310-360OC. [DOI] [PubMed] [Google Scholar]
- 407. Franquet T, Giménez A, Rosón N, Torrubia S, Sabaté JM, Pérez C. Aspiration diseases: findings, pitfalls, and differential diagnosis. Radiographics 20: 673–685, 2000. doi: 10.1148/radiographics.20.3.g00ma01673. [DOI] [PubMed] [Google Scholar]
- 408. Hou YJ, Okuda K, Edwards CE, Martinez DR, Asakura T, Dinnon KH 3rd, , et al. SARS-CoV-2 reverse genetics reveals a variable infection gradient in the respiratory tract. Cell 182: 429–446.e14, 2020. doi: 10.1016/j.cell.2020.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409. Landau LI, Phelan PD, Williams HE. Ventilatory mechanics in patients with bronchiectasis starting in childhood. Thorax 29: 304–312, 1974. doi: 10.1136/thx.29.3.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410. Mellins RB. The site of airway obstruction in cystic fibrosis. Pediatrics 44: 315–318, 1969. [PubMed] [Google Scholar]
- 411. Romanò F, Muradoglu M, Fujioka H, Grotberg JB. The effect of viscoelasticity in an airway closure model. J Fluid Mech 913: A31, 2021. doi: 10.1017/jfm.2020.1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412. Cole AM, Liao HI, Stuchlik O, Tilan J, Pohl J, Ganz T. Cationic polypeptides are required for antibacterial activity of human airway fluid. J Immunol 169: 6985–6991, 2002. doi: 10.4049/jimmunol.169.12.6985. [DOI] [PubMed] [Google Scholar]
- 413. Zhou Z, Duerr J, Johannesson B, Schubert SC, Treis D, Harm M, Graeber SY, Dalpke A, Schultz C, Mall MA. The ENaC-overexpressing mouse as a model of cystic fibrosis lung disease. J Cyst Fibros 10: S172–S182, 2011. doi: 10.1016/S1569-1993(11)60021-0. [DOI] [PubMed] [Google Scholar]
- 414. Mall MA, Harkema JR, Trojanek JB, Treis D, Livraghi A, Schubert S, Zhou Z, Kreda SM, Tilley SL, Hudson EJ, O’Neal WK, Boucher RC. Development of chronic bronchitis and emphysema in beta-epithelial Na+ channel-overexpressing mice. Am J Respir Crit Care Med 177: 730–742, 2008. doi: 10.1164/rccm.200708-1233OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415. Lewis BW, Choudhary I, Paudel K, Mao Y, Sharma R, Wang Y, Deshane JS, Boucher RC, Patial S, Saini Y. The innate lymphoid system is a critical player in the manifestation of mucoinflammatory airway disease in mice. J Immunol 205: 1695–1708, 2020. doi: 10.4049/jimmunol.2000530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416. Johnson JE, Gonzales RA, Olson SJ, Wright PF, Graham BS. The histopathology of fatal untreated human respiratory syncytial virus infection. Mod Pathol 20: 108–119, 2007. doi: 10.1038/modpathol.3800725. [DOI] [PubMed] [Google Scholar]
- 417. Pickles RJ, DeVincenzo JP. Respiratory syncytial virus (RSV) and its propensity for causing bronchiolitis. J Pathol 235: 266–276, 2015. doi: 10.1002/path.4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418. Lee AS, Lee JS, He Z, Ryu JH. Reflux-aspiration in chronic lung disease. Ann Am Thorac Soc 17: 155–164, 2020. doi: 10.1513/AnnalsATS.201906-427CME. [DOI] [PubMed] [Google Scholar]
- 419. Hewson CA, Haas JJ, Bartlett NW, Message SD, Laza-Stanca V, Kebadze T, Caramori G, Zhu J, Edbrooke MR, Stanciu LA, Kon OM, Papi A, Jeffery PK, Edwards MR, Johnston SL. Rhinovirus induces MUC5AC in a human infection model and in vitro via NF-κB and EGFR pathways. Eur Respir J 36: 1425–1435, 2010. doi: 10.1183/09031936.00026910. [DOI] [PubMed] [Google Scholar]
- 420. Zhu L, Lee PK, Lee WM, Zhao Y, Yu D, Chen Y. Rhinovirus-induced major airway mucin production involves a novel TLR3-EGFR-dependent pathway. Am J Respir Cell Mol Biol 40: 610–619, 2009. doi: 10.1165/rcmb.2008-0223OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421. Guimbellot JS, Fortenberry JA, Siegal GP, Moore B, Wen H, Venglarik C, Chen YF, Oparil S, Sorscher EJ, Hong JS. Role of oxygen availability in CFTR expression and function. Am J Respir Cell Mol Biol 39: 514–521, 2008. doi: 10.1165/rcmb.2007-0452OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422. Zheng W, Kuhlicke J, Jäckel K, Eltzschig HK, Singh A, Sjöblom M, Riederer B, Weinhold C, Seidler U, Colgan SP, Karhausen J. Hypoxia inducible factor-1 (HIF-1)-mediated repression of cystic fibrosis transmembrane conductance regulator (CFTR) in the intestinal epithelium. FASEB J 23: 204–213, 2009. doi: 10.1096/fj.08-110221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423. Mikami Y, Grubb BR, Rogers TD, Dang H, Kota P, Gilmore RC, Okuda K, Asakura T, Kato T, Gentzsch M, Stutts JM, Randell SH, O’Neal WK, Boucher RC. Airway obstruction produces hypoxia-dependent sodium absorption in human airway epithelial cells. J Cyst Fibros 20: S173–S174, 2021. doi: 10.1016/S1569-1993(21)01790-2. [DOI] [Google Scholar]
- 424. Mincham KT, Bruno N, Singanayagam A, Snelgrove RJ. Our evolving view of neutrophils in defining the pathology of chronic lung disease. Immunology 164: 701–721, 2021. doi: 10.1111/imm.13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425. Voynow JA, Shinbashi M. Neutrophil elastase and chronic lung disease. Biomolecules 11: 1065, 2021. doi: 10.3390/biom11081065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426. Cole PJ. Inflammation: a two-edged sword–the model of bronchiectasis. Eur J Respir Dis Suppl 147: 6–15, 1986. [PubMed] [Google Scholar]
- 427. Fritzsching B, Zhou-Suckow Z, Trojanek JB, Schubert SC, Schatterny J, Hirtz S, Agrawal R, Muley T, Kahn N, Sticht C, Gunkel N, Welte T, Randell SH, Länger F, Schnabel P, Herth FJ, Mall MA. Hypoxic epithelial necrosis triggers neutrophilic inflammation via IL-1 receptor signaling in cystic fibrosis lung disease. Am J Respir Crit Care Med 191: 902–913, 2015. doi: 10.1164/rccm.201409-1610OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428. Mall MA, Danahay H, Boucher RC. Emerging concepts and therapies for mucoobstructive lung disease. Ann Am Thorac Soc 15: S216–S226, 2018. doi: 10.1513/AnnalsATS.201806-368AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429. Codo AC, Davanzo GG, Monteiro LB, de Souza GF, Muraro SP, Virgilio-da-Silva JV, , et al. Elevated glucose levels favor SARS-CoV-2 infection and monocyte response through a HIF-1α/glycolysis-dependent axis. Cell Metab 32: 437–446.e5, 2020. doi: 10.1016/j.cmet.2020.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430. Mills EL, Kelly B, Logan A, Costa AS, Varma M, Bryant CE, Tourlomousis P, Däbritz JH, Gottlieb E, Latorre I, Corr SC, McManus G, Ryan D, Jacobs HT, Szibor M, Xavier RJ, Braun T, Frezza C, Murphy MP, O’Neill LA. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 167: 457–470.e13, 2016. doi: 10.1016/j.cell.2016.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431. Hey J, Paulsen M, Toth R, Weichenhan D, Butz S, Schatterny J, Liebers R, Lutsik P, Plass C, Mall MA. Epigenetic reprogramming of airway macrophages promotes polarization and inflammation in muco-obstructive lung disease. Nat Commun 12: 6520, 2021. doi: 10.1038/s41467-021-26777-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432. Kohri K, Ueki IF, Nadel JA. Neutrophil elastase induces mucin production by ligand-dependent epidermal growth factor receptor activation. Am J Physiol Lung Cell Mol Physiol 283: L531–L540, 2002. doi: 10.1152/ajplung.00455.2001. [DOI] [PubMed] [Google Scholar]
- 433. Park JA, He F, Martin LD, Li Y, Chorley BN, Adler KB. Human neutrophil elastase induces hypersecretion of mucin from well-differentiated human bronchial epithelial cells in vitro via a protein kinase Cdelta-mediated mechanism. Am J Pathol 167: 651–661, 2005. doi: 10.1016/S0002-9440(10)62040-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434. Voynow JA, Fischer BM, Malarkey DE, Burch LH, Wong T, Longphre M, Ho SB, Foster WM. Neutrophil elastase induces mucus cell metaplasia in mouse lung. Am J Physiol Lung Cell Mol Physiol 287: L1293–L1302, 2004. doi: 10.1152/ajplung.00140.2004. [DOI] [PubMed] [Google Scholar]
- 435. Andelid K, Öst K, Andersson A, Mohamed E, Jevnikar Z, Vanfleteren L, Göransson M. Lung macrophages drive mucus production and steroid-resistant inflammation in chronic bronchitis. Respir Res 22: 172, 2021. doi: 10.1186/s12931-021-01762-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436. Worlitzsch D, Tarran R, Ulrich M, Schwab U, Cekici A, Meyer KC, Birrer P, Bellon G, Berger J, Weiss T, Botzenhart K, Yankaskas JR, Randell S, Boucher RC, Döring G. Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J Clin Invest 109: 317–325, 2002. doi: 10.1172/JCI13870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437. Shteinberg M, Haq IJ, Polineni D, Davies JC. Cystic fibrosis. Lancet 397: 2195–2211, 2021. doi: 10.1016/S0140-6736(20)32542-3. [DOI] [PubMed] [Google Scholar]
- 438. Schwab U, Abdullah LH, Perlmutt OS, Albert D, Davis CW, Arnold RR, Yankaskas JR, Gilligan P, Neubauer H, Randell SH, Boucher RC. Localization of Burkholderia cepacia complex bacteria in cystic fibrosis lungs and interactions with Pseudomonas aeruginosa in hypoxic mucus. Infect Immun 82: 4729–4745, 2014. doi: 10.1128/IAI.01876-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439. Matsui H, Verghese MW, Kesimer M, Schwab UE, Randell SH, Sheehan JK, Grubb BR, Boucher RC. Reduced three-dimensional motility in dehydrated airway mucus prevents neutrophil capture and killing bacteria on airway epithelial surfaces. J Immunol 175: 1090–1099, 2005. doi: 10.4049/jimmunol.175.2.1090. [DOI] [PubMed] [Google Scholar]
- 440. Moreau-Marquis S, Stanton BA, O’Toole GA. Pseudomonas aeruginosa biofilm formation in the cystic fibrosis airway. Pulm Pharmacol Ther 21: 595–599, 2008. doi: 10.1016/j.pupt.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441. Weeks JR, Staples KJ, Spalluto CM, Watson A, Wilkinson TM. The role of non-typeable Haemophilus influenzae biofilms in chronic obstructive pulmonary disease. Front Cell Infect Microbiol 11: 720742, 2021. doi: 10.3389/fcimb.2021.720742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442. Glasser NR, Oyala PH, Osborne TH, Santini JM, Newman DK. Structural and mechanistic analysis of the arsenate respiratory reductase provides insight into environmental arsenic transformations. Proc Natl Acad Sci USA 115: E8614–E8623, 2018. doi: 10.1073/pnas.1807984115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443. Jorth P, Spero MA, Livingston J, Newman DK. Quantitative visualization of gene expression in mucoid and nonmucoid Pseudomonas aeruginosa aggregates reveals localized peak expression of alginate in the hypoxic zone. mBio 10: e02622-19, 2019. doi: 10.1128/mBio.02622-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444. Kopf SH, Sessions AL, Cowley ES, Reyes C, Van Sambeek L, Hu Y, Orphan VJ, Kato R, Newman DK. Trace incorporation of heavy water reveals slow and heterogeneous pathogen growth rates in cystic fibrosis sputum. Proc Natl Acad Sci USA 113: E110–E116, 2016. doi: 10.1073/pnas.1512057112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445. Muhlebach MS, Zorn BT, Esther CR, Hatch JE, Murray CP, Turkovic L, Ranganathan SC, Boucher RC, Stick SM, Wolfgang MC. Initial acquisition and succession of the cystic fibrosis lung microbiome is associated with disease progression in infants and preschool children. PLoS Pathog 14: e1006798, 2018. doi: 10.1371/journal.ppat.1006798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446. Tunney MM, Field TR, Moriarty TF, Patrick S, Doering G, Muhlebach MS, Wolfgang MC, Boucher R, Gilpin DF, McDowell A, Elborn JS. Detection of anaerobic bacteria in high numbers in sputum from patients with cystic fibrosis. Am J Respir Crit Care Med 177: 995–1001, 2008. doi: 10.1164/rccm.200708-1151OC. [DOI] [PubMed] [Google Scholar]
- 447. Rogers JD, Ajami NJ, Fryszczyn BG, Estes MK, Atmar RL, Palzkill T. Identification and characterization of a peptide affinity reagent for detection of noroviruses in clinical samples. J Clin Microbiol 51: 1803–1808, 2013. doi: 10.1128/JCM.00295-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448. Huang YJ, Erb-Downward JR, Dickson RP, Curtis JL, Huffnagle GB, Han MK. Understanding the role of the microbiome in chronic obstructive pulmonary disease: principles, challenges, and future directions. Transl Res 179: 71–83, 2017. doi: 10.1016/j.trsl.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449. Bozzella MJ, Chaney H, Sami I, Koumbourlis A, Bost JE, Zemanick ET, Freishtat RJ, Crandall KA, Hahn A. Impact of anaerobic antibacterial spectrum on cystic fibrosis airway microbiome diversity and pulmonary function. Pediatr Infect Dis J 40: 962–968, 2021. doi: 10.1097/INF.0000000000003211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450. Flynn JM, Niccum D, Dunitz JM, Hunter RC. Evidence and role for bacterial mucin degradation in cystic fibrosis airway disease. PLoS Pathog 12: e1005846, 2016. doi: 10.1371/journal.ppat.1005846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451. Flynn JM, Cameron LC, Wiggen TD, Dunitz JM, Harcombe WR, Hunter RC. Disruption of cross-feeding inhibits pathogen growth in the sputa of patients with cystic fibrosis. mSphere 5: e00343-20, 2020. doi: 10.1128/mSphere.00343-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452. Hoffman CL, Lalsiamthara J, Aballay A. Host mucin is exploited by Pseudomonas aeruginosa to provide monosaccharides required for a successful infection. mBio 11: e00060-20, 2020. doi: 10.1128/mBio.00060-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453. Lonni S, Chalmers JD, Goeminne PC, McDonnell MJ, Dimakou K, De Soyza A, Polverino E, Van de Kerkhove C, Rutherford R, Davison J, Rosales E, Pesci A, Restrepo MI, Torres A, Aliberti S. Etiology of non-cystic fibrosis bronchiectasis in adults and its correlation to disease severity. Ann Am Thorac Soc 12: 1764–1770, 2015. doi: 10.1513/AnnalsATS.201507-472OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454. Moulton BC, Barker AF. Pathogenesis of bronchiectasis. Clin Chest Med 33: 211–217, 2012. doi: 10.1016/j.ccm.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 455. Reid LM. Reduction in bronchial subdivision in bronchiectasis. Thorax 5: 233–247, 1950. doi: 10.1136/thx.5.3.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456. Cockett FB, Vass CC. A comparison of the role of the bronchial arteries in bronchiectasis and in experimental ligation of the pulmonary artery. Thorax 6: 268–275, 1951. doi: 10.1136/thx.6.3.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457. Lee SH, Lee SH, Kim CH, Yang KS, Lee EJ, Min KH, Hur GY, Lee SH, Lee SY, Kim JH, Shin C, Shim JJ, In KH, Kang KH, Lee SY. Increased expression of vascular endothelial growth factor and hypoxia inducible factor-1α in lung tissue of patients with chronic bronchitis. Clin Biochem 47: 552–559, 2014. doi: 10.1016/j.clinbiochem.2014.01.012. [DOI] [PubMed] [Google Scholar]
- 458. Chalmers JD, Moffitt KL, Suarez-Cuartin G, Sibila O, Finch S, Furrie E, Dicker A, Wrobel K, Elborn JS, Walker B, Martin SL, Marshall SE, Huang JT, Fardon TC. Neutrophil elastase activity is associated with exacerbations and lung function decline in bronchiectasis. Am J Respir Crit Care Med 195: 1384–1393, 2017. doi: 10.1164/rccm.201605-1027OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459. Colgan SP, Furuta GT, Taylor CT. Hypoxia and innate immunity: keeping up with the HIFsters. Annu Rev Immunol 38: 341–363, 2020. doi: 10.1146/annurev-immunol-100819-121537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460. Watts ER, Walmsley SR. Inflammation and hypoxia: HIF and PHD isoform selectivity. Trends Mol Med 25: 33–46, 2019. doi: 10.1016/j.molmed.2018.10.006. [DOI] [PubMed] [Google Scholar]
- 461. Shivaraju M, Chitta UK, Grange RM, Jain IH, Capen D, Liao L, Xu J, Ichinose F, Zapol WM, Mootha VK, Rajagopal J. Airway stem cells sense hypoxia and differentiate into protective solitary neuroendocrine cells. Science 371: 52–57, 2021. doi: 10.1126/science.aba0629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462. Woodworth BA. Resveratrol ameliorates abnormalities of fluid and electrolyte secretion in a hypoxia-induced model of acquired CFTR deficiency. Laryngoscope 125, Suppl 7: S1–S13, 2015. doi: 10.1002/lary.25335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463. Blount A, Zhang S, Chestnut M, Hixon B, Skinner D, Sorscher EJ, Woodworth BA. Transepithelial ion transport is suppressed in hypoxic sinonasal epithelium. Laryngoscope 121: 1929–1934, 2011. doi: 10.1002/lary.21921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464. O’Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol 16: 553–565, 2016. doi: 10.1038/nri.2016.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465. Lee JW, Ko J, Ju C, Eltzschig HK. Hypoxia signaling in human diseases and therapeutic targets. Exp Mol Med 51: 1–13, 2019. doi: 10.1038/s12276-019-0235-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466. Colgan SP, Taylor CT. Hypoxia: an alarm signal during intestinal inflammation. Nat Rev Gastroenterol Hepatol 7: 281–287, 2010. doi: 10.1038/nrgastro.2010.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467. Page LK, Staples KJ, Spalluto CM, Watson A, Wilkinson TM. Influence of hypoxia on the epithelial-pathogen interactions in the lung: implications for respiratory disease. Front Immunol 12: 653969, 2021. doi: 10.3389/fimmu.2021.653969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468. Stokes W. A Treatise on the Diagnosis and Treatment of Diseases of the Chest. Dublin: Hodges and Smith, 1837. [Google Scholar]
- 469. Puhan MA, Guyatt GH, Goldstein R, Mador J, McKim D, Stahl E, Griffith L, Schünemann HJ. Relative responsiveness of the Chronic Respiratory Questionnaire, St. Georges Respiratory Questionnaire and four other health-related quality of life instruments for patients with chronic lung disease. Respir Med 101: 308–316, 2007. doi: 10.1016/j.rmed.2006.04.023. [DOI] [PubMed] [Google Scholar]
- 470. Welling JB, Hartman JE, Ten Hacken NH, Klooster K, Slebos DJ. The minimal important difference for the St George’s Respiratory Questionnaire in patients with severe COPD. Eur Respir J 46: 1598–1604, 2015. doi: 10.1183/13993003.00535-2015. [DOI] [PubMed] [Google Scholar]
- 471. Abrami M, Maschio M, Conese M, Confalonieri M, Gerin F, Dapas B, Farra R, Adrover A, Torelli L, Ruaro B, Grassi G, Grassi M. Combined use of rheology and portable low-field NMR in cystic fibrosis patients. Respir Med 189: 106623, 2021. doi: 10.1016/j.rmed.2021.106623. [DOI] [PubMed] [Google Scholar]
- 472. Ramsey BW, Downey GP, Goss CH. Update in cystic fibrosis 2018. Am J Respir Crit Care Med 199: 1188–1194, 2019. doi: 10.1164/rccm.201902-0310UP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473. Stoltz DA, Meyerholz DK, Welsh MJ. Origins of cystic fibrosis lung disease. N Engl J Med 372: 351–362, 2015. doi: 10.1056/NEJMra1300109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474. Boucher RC. New concepts of the pathogenesis of cystic fibrosis lung disease. Eur Respir J 23: 146–158, 2004. doi: 10.1183/09031936.03.00057003. [DOI] [PubMed] [Google Scholar]
- 475. Richmond RC, Shwachman H. Studies of fibrocystic disease of the pancreas (mucoviscidosis): chymotrypsin activity of duodenal fluid. Pediatrics 16: 207–214, 1955. [PubMed] [Google Scholar]
- 476. Bear CE, Li CH, Kartner N, Bridges RJ, Jensen TJ, Ramjeesingh M, Riordan JR. Purification and functional reconstitution of the cystic fibrosis transmembrane conductance regulator (CFTR). Cell 68: 809–818, 1992. doi: 10.1016/0092-8674(92)90155-6. [DOI] [PubMed] [Google Scholar]
- 477. Clarke LL, Grubb BR, Gabriel SE, Smithies O, Koller BH, Boucher RC. Defective epithelial chloride transport in a gene-targeted mouse model of cystic fibrosis. Science 257: 1125–1128, 1992. doi: 10.1126/science.257.5073.1125. [DOI] [PubMed] [Google Scholar]
- 478. Benedetto R, Centeio R, Ousingsawat J, Schreiber R, Janda M, Kunzelmann K. Transport properties in CFTR-/- knockout piglets suggest normal airway surface liquid pH and enhanced amiloride-sensitive Na+ absorption. Pflugers Arch 472: 1507–1519, 2020. doi: 10.1007/s00424-020-02440-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479. Olivença DV, Fonseca LL, Voit EO, Pinto FR. Thickness of the airway surface liquid layer in the lung is affected in cystic fibrosis by compromised synergistic regulation of the ENaC ion channel. J R Soc Interface 16: 20190187, 2019. doi: 10.1098/rsif.2019.0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480. Strandvik B. Is the ENaC dysregulation in CF an effect of protein-lipid interaction in the membranes? Int J Mol Sci 22: 2739, 2021. doi: 10.3390/ijms22052739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481. Itani OA, Chen JH, Karp PH, Ernst S, Keshavjee S, Parekh K, Klesney-Tait J, Zabner J, Welsh MJ. Human cystic fibrosis airway epithelia have reduced Cl- conductance but not increased Na+ conductance. Proc Natl Acad Sci USA 108: 10260–10265, 2011. doi: 10.1073/pnas.1106695108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482. Luan X, Le Y, Jagadeeshan S, Murray B, Carmalt JL, Duke T, Beazley S, Fujiyama M, Swekla K, Gray B, Burmester M, Campanucci VA, Shipley A, Machen TE, Tam JS, Ianowski JP. cAMP triggers Na+ absorption by distal airway surface epithelium in cystic fibrosis swine. Cell Rep 37: 109795, 2021. doi: 10.1016/j.celrep.2021.109795. [DOI] [PubMed] [Google Scholar]
- 483. Boucher RC. Regulation of airway surface liquid volume by human airway epithelia. Pflugers Arch 445: 495–498, 2003. doi: 10.1007/s00424-002-0955-1. [DOI] [PubMed] [Google Scholar]
- 484. Boucher RC. An overview of the pathogenesis of cystic fibrosis lung disease. Adv Drug Deliv Rev 54: 1359–1371, 2002. doi: 10.1016/s0169-409x(02)00144-8. [DOI] [PubMed] [Google Scholar]
- 485. Reddy MM, Wang XF, Quinton PM. Effect of cytosolic pH on epithelial Na+ channel in normal and cystic fibrosis sweat ducts. J Membr Biol 225: 1–11, 2008. doi: 10.1007/s00232-008-9126-4. [DOI] [PubMed] [Google Scholar]
- 486. Quinton PM. Cystic fibrosis: impaired bicarbonate secretion and mucoviscidosis. Lancet 372: 415–417, 2008. doi: 10.1016/S0140-6736(08)61162-9. [DOI] [PubMed] [Google Scholar]
- 487. Tang XX, Ostedgaard LS, Hoegger MJ, Moninger TO, Karp PH, McMenimen JD, Choudhury B, Varki A, Stoltz DA, Welsh MJ. Acidic pH increases airway surface liquid viscosity in cystic fibrosis. J Clin Invest 126: 879–891, 2016. doi: 10.1172/JCI83922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488. Reid L. Sputum rheology in evaluation of anti-asthmatic drugs. Scand J Respir Dis Suppl 103: 90–95, 1979. [PubMed] [Google Scholar]
- 489. Keal EE. Biochemistry and rheology of sputum in asthma. Postgrad Med J 47: 171–177, 1971. doi: 10.1136/pgmj.47.545.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490. Rogers DF. Mucociliary dysfunction in COPD: effect of current pharmacotherapeutic options. Pulm Pharmacol Ther 18: 1–8, 2005. doi: 10.1016/j.pupt.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 491. Henke MO, Shah SA, Rubin BK. The role of airway secretions in COPD—clinical applications. COPD 2: 377–390, 2005. doi: 10.1080/15412550500218148. [DOI] [PubMed] [Google Scholar]
- 492. Chisholm JF, Shenoy SK, Shade JK, Kim V, Putcha N, Carson KA, Wise R, Hansel NN, Hanes JS, Suk JS, Neptune E. Nanoparticle diffusion in spontaneously expectorated sputum as a biophysical tool to probe disease severity in COPD. Eur Respir J 54: 1900088, 2019. doi: 10.1183/13993003.00088-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493. Garland AL, Walton WG, Coakley RD, Tan CD, Gilmore RC, Hobbs CA, Tripathy A, Clunes LA, Bencharit S, Stutts MJ, Betts L, Redinbo MR, Tarran R. Molecular basis for pH-dependent mucosal dehydration in cystic fibrosis airways. Proc Natl Acad Sci USA 110: 15973–15978, 2013. doi: 10.1073/pnas.1311999110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494. Celli JP, Turner BS, Afdhal NH, Ewoldt RH, McKinley GH, Bansil R, Erramilli S. Rheology of gastric mucin exhibits a pH-dependent sol-gel transition. Biomacromolecules 8: 1580–1586, 2007. doi: 10.1021/bm0609691. [DOI] [PubMed] [Google Scholar]
- 495. Chmiel JF, Berger M, Konstan MW. The role of inflammation in the pathophysiology of CF lung disease. Clin Rev Allergy Immunol 23: 5–27, 2002. doi: 10.1385/CRIAI:23:1:005. [DOI] [PubMed] [Google Scholar]
- 496. Cohen-Cymberknoh M, Kerem E, Ferkol T, Elizur A. Airway inflammation in cystic fibrosis: molecular mechanisms and clinical implications. Thorax 68: 1157–1162, 2013. doi: 10.1136/thoraxjnl-2013-203204. [DOI] [PubMed] [Google Scholar]
- 497. Machen TE. Innate immune response in CF airway epithelia: hyperinflammatory? Am J Physiol Cell Physiol 291: C218–C230, 2006. doi: 10.1152/ajpcell.00605.2005. [DOI] [PubMed] [Google Scholar]
- 498. Cantin AM, Hartl D, Konstan MW, Chmiel JF. Inflammation in cystic fibrosis lung disease: pathogenesis and therapy. J Cyst Fibros 14: 419–430, 2015. doi: 10.1016/j.jcf.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 499. Bartlett JA, Ramachandran S, Wohlford-Lenane CL, Barker CK, Pezzulo AA, Zabner J, Welsh MJ, Meyerholz DK, Stoltz DA, McCray PB Jr.. Newborn cystic fibrosis pigs have a blunted early response to an inflammatory stimulus. Am J Respir Crit Care Med 194: 845–854, 2016. doi: 10.1164/rccm.201510-2112OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500. Becker MN, Sauer MS, Muhlebach MS, Hirsh AJ, Wu Q, Verghese MW, Randell SH. Cytokine secretion by cystic fibrosis airway epithelial cells. Am J Respir Crit Care Med 169: 645–653, 2004. doi: 10.1164/rccm.200207-765OC. [DOI] [PubMed] [Google Scholar]
- 501. Ribeiro CM, Paradiso AM, Schwab U, Perez-Vilar J, Jones L, O’Neal W, Boucher RC. Chronic airway infection/inflammation induces a Ca2+i-dependent hyperinflammatory response in human cystic fibrosis airway epithelia. J Biol Chem 280: 17798–17806, 2005. doi: 10.1074/jbc.M410618200. [DOI] [PubMed] [Google Scholar]
- 502. Stoltz DA, Meyerholz DK, Pezzulo AA, Ramachandran S, Rogan MP, Davis GJ, Hanfland RA, Wohlford-Lenane C, Dohrn CL, Bartlett JA, Nelson GA 4th, Chang EH, Taft PJ, Ludwig PS, Estin M, Hornick EE, Launspach JL, Samuel M, Rokhlina T, Karp PH, Ostedgaard LS, Uc A, Starner TD, Horswill AR, Brogden KA, Prather RS, Richter SS, Shilyansky J, McCray PB Jr, Zabner J, Welsh MJ. Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci Transl Med 2: 29ra31, 2010. doi: 10.1126/scitranslmed.3000928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503. Mall MA, Hwang TC, Braakman I. Cystic fibrosis research topics featured at the 14th ECFS Basic Science Conference: Chairman’s summary. J Cyst Fibros 17: S1–S4, 2018. doi: 10.1016/j.jcf.2017.11.008. [DOI] [PubMed] [Google Scholar]
- 504. Rosen BH, Evans TIA, Moll SR, Gray JS, Liang B, Sun X, Zhang Y, Jensen-Cody CW, Swatek AM, Zhou W, He N, Rotti PG, Tyler SR, Keiser NW, Anderson PJ, Brooks L, Li Y, Pope RM, Rajput M, Hoffman EA, Wang K, Harris JK, Parekh KR, Gibson-Corley KN, Engelhardt JF. Infection is not required for mucoinflammatory lung disease in CFTR-knockout ferrets. Am J Respir Crit Care Med 197: 1308–1318, 2018. doi: 10.1164/rccm.201708-1616OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505. Sun X, Yi Y, Yan Z, Rosen BH, Liang B, Winter MC, Evans TIA, Rotti PG, Yang Y, Gray JS, Park SY, Zhou W, Zhang Y, Moll SR, Woody L, Tran DM, Jiang L, Vonk AM, Beekman JM, Negulescu P, Van Goor F, Fiorino DF, Gibson-Corley KN, Engelhardt JF. In utero and postnatal VX-770 administration rescues multiorgan disease in a ferret model of cystic fibrosis. Sci Transl Med 11: eaau7531, 2019. doi: 10.1126/scitranslmed.aau7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506. O’Toole GA, Crabbé A, Kümmerli R, LiPuma JJ, Bomberger JM, Davies JC, , et al. Model systems to study the chronic polymicrobial infections in cystic fibrosis: current approaches and exploring future directions. mBio 12: e0176321, 2021. doi: 10.1128/mBio.01763-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507. Oudraad MC, Kuo W, Rosenow T, Andrinopoulou ER, Stick SM, Tiddens H. Assessment of early lung disease in young children with CF: a comparison between pressure-controlled and free-breathing chest computed tomography. Pediatr Pulmonol 55: 1161–1168, 2020. doi: 10.1002/ppul.24702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508. Rosenow T, Oudraad MC, Murray CP, Turkovic L, Kuo W, de Bruijne M, Ranganathan SC, Tiddens HA, Stick SM; Australian Respiratory Early Surveillance Team for Cystic Fibrosis (AREST CF). PRAGMA-CF. A quantitative structural lung disease computed tomography outcome in young children with cystic fibrosis. Am J Respir Crit Care Med 191: 1158–1165, 2015. doi: 10.1164/rccm.201501-0061OC. [DOI] [PubMed] [Google Scholar]
- 509. Lubamba BA, Jones LC, O’Neal WK, Boucher RC, Ribeiro CM. X-Box-Binding Protein 1 and innate immune responses of human cystic fibrosis alveolar macrophages. Am J Respir Crit Care Med 192: 1449–1461, 2015. doi: 10.1164/rccm.201504-0657OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510. Boucher RC. On the pathogenesis of acute exacerbations of mucoobstructive lung diseases. Ann Am Thorac Soc 12, Suppl 2: S160–S163, 2015. doi: 10.1513/AnnalsATS.201507-460AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 511. Asner S, Waters V, Solomon M, Yau Y, Richardson SE, Grasemann H, Gharabaghi F, Tran D. Role of respiratory viruses in pulmonary exacerbations in children with cystic fibrosis. J Cyst Fibros 11: 433–439, 2012. doi: 10.1016/j.jcf.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 512. Somayaji R, Goss CH, Khan U, Neradilek M, Neuzil KM, Ortiz JR. Cystic fibrosis pulmonary exacerbations attributable to respiratory syncytial virus and influenza: a population-based study. Clin Infect Dis 64: 1760–1767, 2017. doi: 10.1093/cid/cix203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 513. Wat D, Gelder C, Hibbitts S, Cafferty F, Bowler I, Pierrepoint M, Evans R, Doull I. The role of respiratory viruses in cystic fibrosis. J Cyst Fibros 7: 320–328, 2008. doi: 10.1016/j.jcf.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 514. Caparrós-Martín JA, Flynn S, Reen FJ, Woods DF, Agudelo-Romero P, Ranganathan SC, Stick SM, O’Gara F. The detection of bile acids in the lungs of paediatric cystic fibrosis patients is associated with altered inflammatory patterns. Diagnostics (Basel) 10: 282, 2020. doi: 10.3390/diagnostics10050282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515. Pearson JP, Feldman M, Iglewski BH, Prince A. Pseudomonas aeruginosa cell-to-cell signaling is required for virulence in a model of acute pulmonary infection. Infect Immun 68: 4331–4334, 2000. doi: 10.1128/IAI.68.7.4331-4334.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516. Sanders DB, Emerson J, Ren CL, Schechter MS, Gibson RL, Morgan W, Rosenfeld M; EPIC Study Group. Early childhood risk factors for decreased FEV1 at age six to seven years in young children with cystic fibrosis. Ann Am Thorac Soc 12: 1170–1176, 2015. doi: 10.1513/AnnalsATS.201504-198OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517. Zemanick ET, Emerson J, Thompson V, McNamara S, Morgan W, Gibson RL, Rosenfeld M; EPIC Study Group. Clinical outcomes after initial pseudomonas acquisition in cystic fibrosis. Pediatr Pulmonol 50: 42–48, 2015. doi: 10.1002/ppul.23036. [DOI] [PubMed] [Google Scholar]
- 518. Joo NS, Cho HJ, Khansaheb M, Wine JJ. Hyposecretion of fluid from tracheal submucosal glands of CFTR-deficient pigs. J Clin Invest 120: 3161–3166, 2010. doi: 10.1172/JCI43466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519. Joo NS, Irokawa T, Robbins RC, Wine JJ. Hyposecretion, not hyperabsorption, is the basic defect of cystic fibrosis airway glands. J Biol Chem 281: 7392–7398, 2006. doi: 10.1074/jbc.M512766200. [DOI] [PubMed] [Google Scholar]
- 520. Joo NS, Irokawa T, Wu JV, Robbins RC, Whyte RI, Wine JJ. Absent secretion to vasoactive intestinal peptide in cystic fibrosis airway glands. J Biol Chem 277: 50710–50715, 2002. doi: 10.1074/jbc.M208826200. [DOI] [PubMed] [Google Scholar]
- 521. Joo NS, Lee DJ, Winges KM, Rustagi A, Wine JJ. Regulation of antiprotease and antimicrobial protein secretion by airway submucosal gland serous cells. J Biol Chem 279: 38854–38860, 2004. doi: 10.1074/jbc.M407077200. [DOI] [PubMed] [Google Scholar]
- 522. Fischer AJ, Pino-Argumedo MI, Hilkin BM, Shanrock CR, Gansemer ND, Chaly AL, Zarei K, Allen PD, Ostedgaard LS, Hoffman EA, Stoltz DA, Welsh MJ, Abou Alaiwa MH. Mucus strands from submucosal glands initiate mucociliary transport of large particles. JCI Insight 4: e124863, 2019. doi: 10.1172/jci.insight.124863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 523. Ostedgaard LS, Moninger TO, McMenimen JD, Sawin NM, Parker CP, Thornell IM, Powers LS, Gansemer ND, Bouzek DC, Cook DP, Meyerholz DK, Abou Alaiwa MH, Stoltz DA, Welsh MJ. Gel-forming mucins form distinct morphologic structures in airways. Proc Natl Acad Sci USA 114: 6842–6847, 2017. doi: 10.1073/pnas.1703228114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524. Xie Y, Lu L, Tang XX, Moninger TO, Huang TJ, Stoltz DA, Welsh MJ. Acidic submucosal gland pH and elevated protein concentration produce abnormal cystic fibrosis mucus. Dev Cell 54: 488–500.e5, 2020. doi: 10.1016/j.devcel.2020.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 525. Hoegger MJ, Fischer AJ, McMenimen JD, Ostedgaard LS, Tucker AJ, Awadalla MA, Moninger TO, Michalski AS, Hoffman EA, Zabner J, Stoltz DA, Welsh MJ. Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis. Science 345: 818–822, 2014. doi: 10.1126/science.1255825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526. Inglis SK, Corboz MR, Taylor AE, Ballard ST. Effect of anion transport inhibition on mucus secretion by airway submucosal glands. Am J Physiol Lung Cell Mol Physiol 272: L372–L377, 1997. doi: 10.1152/ajplung.1997.272.2.L372. [DOI] [PubMed] [Google Scholar]
- 527. Inglis SK, Corboz MR, Ballard ST. Effect of anion secretion inhibitors on mucin content of airway submucosal gland ducts. Am J Physiol Lung Cell Mol Physiol 274: L762–L766, 1998. doi: 10.1152/ajplung.1998.274.5.L762. [DOI] [PubMed] [Google Scholar]
- 528. Cooper JL, Quinton PM, Ballard ST. Mucociliary transport in porcine trachea: differential effects of inhibiting chloride and bicarbonate secretion. Am J Physiol Lung Cell Mol Physiol 304: L184–L190, 2013. doi: 10.1152/ajplung.00143.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 529. Ballard ST, Trout L, Bebök Z, Sorscher EJ, Crews A. CFTR involvement in chloride, bicarbonate, and liquid secretion by airway submucosal glands. Am J Physiol Lung Cell Mol Physiol 277: L694–L699, 1999. doi: 10.1152/ajplung.1999.277.4.L694. [DOI] [PubMed] [Google Scholar]
- 530. Martens CJ, Inglis SK, Valentine VG, Garrison J, Conner GE, Ballard ST. Mucous solids and liquid secretion by airways: studies with normal pig, cystic fibrosis human, and non-cystic fibrosis human bronchi. Am J Physiol Lung Cell Mol Physiol 301: L236–L246, 2011. doi: 10.1152/ajplung.00388.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531. Kato T, Radicioni G, Papanikolas MJ, Stoychev GV, Markovetz MR, Aoki K, Porterfield M, Okuda K, Barbosa Cardenas SM, Gilmore RC, Morrison CB, Ehre C, Burns KA, White KK, Brennan TA, Goodell HP, Thacker H, Loznev HT, Forsberg LJ, Nagase T, Rubinstein M, Randell SH, Tiemeyer M, Hill DB, Kesimer M, O’Neal WK, Ballard ST, Freeman R, Button B, Boucher RC. Mucus concentration-dependent biophysical abnormalities unify submucosal gland and superficial airway dysfunction in cystic fibrosis. Sci Adv 8: eabm9718, 2022. doi: 10.1126/sciadv.abm9718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532. Donaldson SH, Corcoran TE, Laube BL, Bennett WD. Mucociliary clearance as an outcome measure for cystic fibrosis clinical research. Proc Am Thorac Soc 4: 399–405, 2007. doi: 10.1513/pats.200703-042BR. [DOI] [PubMed] [Google Scholar]
- 533. Ratjen F. How best to capture early cystic fibrosis lung disease? Am J Respir Crit Care Med 195: 283–284, 2017. doi: 10.1164/rccm.201609-1836ED. [DOI] [PubMed] [Google Scholar]
- 534. Stanojevic S, Ratjen F. Physiologic endpoints for clinical studies for cystic fibrosis. J Cyst Fibros 15: 416–423, 2016. doi: 10.1016/j.jcf.2016.05.014. [DOI] [PubMed] [Google Scholar]
- 535. Goralski JL, Chung SH, Glass TM, Ceppe AS, Akinnagbe-Zusterzeel EO, Trimble AT, Boucher RC, Soher BJ, Charles HC, Donaldson SH, Lee YZ. Dynamic perfluorinated gas MRI reveals abnormal ventilation despite normal FEV1 in cystic fibrosis. JCI Insight 5: e133400, 2020. doi: 10.1172/jci.insight.133400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 536. Birket SE, Chu KK, Houser GH, Liu L, Fernandez CM, Solomon GM, Lin V, Shastry S, Mazur M, Sloane PA, Hanes J, Grizzle WE, Sorscher EJ, Tearney GJ, Rowe SM. Combination therapy with cystic fibrosis transmembrane conductance regulator modulators augment the airway functional microanatomy. Am J Physiol Lung Cell Mol Physiol 310: L928–L939, 2016. doi: 10.1152/ajplung.00395.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 537. Birket SE, Davis JM, Fernandez-Petty CM, Henderson AG, Oden AM, Tang L, Wen H, Hong J, Fu L, Chambers A, Fields A, Zhao G, Tearney GJ, Sorscher EJ, Rowe SM. Ivacaftor reverses airway mucus abnormalities in a rat model harboring a humanized G551D-CFTR. Am J Respir Crit Care Med 202: 1271–1282, 2020. doi: 10.1164/rccm.202002-0369OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 538. Markovetz MR, Corcoran TE, Locke LW, Myerburg MM, Pilewski JM, Parker RS. A physiologically-motivated compartment-based model of the effect of inhaled hypertonic saline on mucociliary clearance and liquid transport in cystic fibrosis. PLoS One 9: e111972, 2014. doi: 10.1371/journal.pone.0111972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 539. Agustí A, Hogg JC. Update on the pathogenesis of chronic obstructive pulmonary disease. N Engl J Med 381: 1248–1256, 2019. doi: 10.1056/NEJMra1900475. [DOI] [PubMed] [Google Scholar]
- 540. Hikichi M, Mizumura K, Maruoka S, Gon Y. Pathogenesis of chronic obstructive pulmonary disease (COPD) induced by cigarette smoke. J Thorac Dis 11: S2129–S2140, 2019. doi: 10.21037/jtd.2019.10.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541. MacNee W. Pathogenesis of chronic obstructive pulmonary disease. Proc Am Thorac Soc 2: 258–266, 2005. doi: 10.1513/pats.200504-045SR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 542. Groiss S, Somvilla I, Daxböck C, Fuchs J, Lang-Olip I, Stiegler P, Leber B, Liegl-Atzwanger B, Brislinger D. Quantification of increased MUC5AC expression in airway mucus of smoker using an automated image-based approach. Microsc Res Tech 85: 5–18, 2022. doi: 10.1002/jemt.23879. [DOI] [PubMed] [Google Scholar]
- 543. Park J, Hobbs BD, Crapo JD, Make BJ, Regan EA, Humphries S, Carey VJ, Lynch DA, Silverman EK; COPDGene Investigators. Subtyping COPD by using visual and quantitative CT imaging features. Chest 157: 47–60, 2020. doi: 10.1016/j.chest.2019.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544. Dunican EM, Elicker BM, Henry T, Gierada DS, Schiebler ML, Anderson W, Barjaktarevic I, , et al. Mucus plugs and emphysema in the pathophysiology of airflow obstruction and hypoxemia in smokers. Am J Respir Crit Care Med 203: 957–968, 2021. doi: 10.1164/rccm.202006-2248OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 545. Chassagnon G, Burgel PR. Mucus plugs in medium-sized airways: a novel imaging biomarker for phenotyping COPD. Am J Respir Crit Care Med 203: 932–934, 2021. doi: 10.1164/rccm.202011-4178ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 546. Clunes LA, Davies CM, Coakley RD, Aleksandrov AA, Henderson AG, Zeman KL, Worthington EN, Gentzsch M, Kreda SM, Cholon D, Bennett WD, Riordan JR, Boucher RC, Tarran R. Cigarette smoke exposure induces CFTR internalization and insolubility, leading to airway surface liquid dehydration. FASEB J 26: 533–545, 2012. doi: 10.1096/fj.11-192377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 547. Cantin AM, Bilodeau G, Ouellet C, Liao J, Hanrahan JW. Oxidant stress suppresses CFTR expression. Am J Physiol Cell Physiol 290: C262–C270, 2006. doi: 10.1152/ajpcell.00070.2005. [DOI] [PubMed] [Google Scholar]
- 548. Shao MX, Nakanaga T, Nadel JA. Cigarette smoke induces MUC5AC mucin overproduction via tumor necrosis factor-alpha-converting enzyme in human airway epithelial (NCI-H292) cells. Am J Physiol Lung Cell Mol Physiol 287: L420–L427, 2004. doi: 10.1152/ajplung.00019.2004. [DOI] [PubMed] [Google Scholar]
- 549. Kato K, Chang EH, Chen Y, Lu W, Kim MM, Niihori M, Hecker L, Kim KC. MUC1 contributes to goblet cell metaplasia and MUC5AC expression in response to cigarette smoke in vivo. Am J Physiol Lung Cell Mol Physiol 319: L82–L90, 2020. doi: 10.1152/ajplung.00049.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 550. Murphy TF, Brauer AL, Eschberger K, Lobbins P, Grove L, Cai X, Sethi S. Pseudomonas aeruginosa in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 177: 853–860, 2008. doi: 10.1164/rccm.200709-1413OC. [DOI] [PubMed] [Google Scholar]
- 551. Horani A, Ferkol TW, Dutcher SK, Brody SL. Genetics and biology of primary ciliary dyskinesia. Paediatr Respir Rev 18: 18–24, 2016. doi: 10.1016/j.prrv.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 552. Horani A, Ferkol TW. Advances in the genetics of primary ciliary dyskinesia: clinical implications. Chest 154: 645–652, 2018. doi: 10.1016/j.chest.2018.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 553. Lucas JS, Davis SD, Omran H, Shoemark A. Primary ciliary dyskinesia in the genomics age. Lancet Respir Med 8: 202–216, 2020. doi: 10.1016/S2213-2600(19)30374-1. [DOI] [PubMed] [Google Scholar]
- 554. Shapiro AJ, Davis SD, Polineni D, Manion M, Rosenfeld M, Dell SD, Chilvers MA, Ferkol TW, Zariwala MA, Sagel SD, Josephson M, Morgan L, Yilmaz O, Olivier KN, Milla C, Pittman JE, Daniels MLA, Jones MH, Janahi IA, Ware SM, Daniel SJ, Cooper ML, Nogee LM, Anton B, Eastvold T, Ehrne L, Guadagno E, Knowles MR, Leigh MW, Lavergne V; American Thoracic Society Assembly on Pediatrics. Diagnosis of primary ciliary dyskinesia. An official American Thoracic Society Clinical Practice Guideline. Am J Respir Crit Care Med 197: e24–e39, 2018. doi: 10.1164/rccm.201805-0819ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 555. Wheway G, Thomas NS, Carroll M, Coles J, Doherty R, Goggin P, Green B, Harris A, Hunt D, Jackson CL, Lord J, Mennella V, Thompson J, Walker WT, Lucas JS; Genomics England Research Consortium. Whole genome sequencing in the diagnosis of primary ciliary dyskinesia. BMC Med Genomics 14: 234, 2021. doi: 10.1186/s12920-021-01084-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 556. Maglione M, Bush A, Nielsen KG, Hogg C, Montella S, Marthin JK, Di Giorgio A, Santamaria F. Multicenter analysis of body mass index, lung function, and sputum microbiology in primary ciliary dyskinesia. Pediatr Pulmonol 49: 1243–1250, 2014. doi: 10.1002/ppul.22984. [DOI] [PubMed] [Google Scholar]
- 557. Rogers GB, Carroll MP, Zain NM, Bruce KD, Lock K, Walker W, Jones G, Daniels TW, Lucas JS. Complexity, temporal stability, and clinical correlates of airway bacterial community composition in primary ciliary dyskinesia. J Clin Microbiol 51: 4029–4035, 2013. doi: 10.1128/JCM.02164-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 558. Knowles MR, Daniels LA, Davis SD, Zariwala MA, Leigh MW. Primary ciliary dyskinesia. Recent advances in diagnostics, genetics, and characterization of clinical disease. Am J Respir Crit Care Med 188: 913–922, 2013. doi: 10.1164/rccm.201301-0059CI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 559. Flume PA, Chalmers JD, Olivier KN. Advances in bronchiectasis: endotyping, genetics, microbiome, and disease heterogeneity. Lancet 392: 880–890, 2018. doi: 10.1016/S0140-6736(18)31767-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560. Meerburg JJ, Veerman GD, Aliberti S, Tiddens H. Diagnosis and quantification of bronchiectasis using computed tomography or magnetic resonance imaging: a systematic review. Respir Med 170: 105954, 2020. doi: 10.1016/j.rmed.2020.105954. [DOI] [PubMed] [Google Scholar]
- 561. Kurashima A. [Clinical study on development of nontuberculous mycobacterial lung disease]. Kekkaku 79: 737–741, 2004. [PubMed] [Google Scholar]
- 562. Ledda RE, Balbi M, Milone F, Ciuni A, Silva M, Sverzellati N, Milanese G. Imaging in non-cystic fibrosis bronchiectasis and current limitations. BJR Open 3: 20210026, 2021. doi: 10.1259/bjro.20210026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 563. Szymanski EP, Leung JM, Fowler CJ, Haney C, Hsu AP, Chen F, Duggal P, Oler AJ, McCormack R, Podack E, Drummond RA, Lionakis MS, Browne SK, Prevots DR, Knowles M, Cutting G, Liu X, Devine SE, Fraser CM, Tettelin H, Olivier KN, Holland SM. Pulmonary nontuberculous mycobacterial infection. a multisystem, multigenic disease. Am J Respir Crit Care Med 192: 618–628, 2015. doi: 10.1164/rccm.201502-0387OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 564. Aliberti S, Goeminne PC, O’Donnell AE, Aksamit TR, Al-Jahdali H, Barker AF, , et al. Criteria and definitions for the radiological and clinical diagnosis of bronchiectasis in adults for use in clinical trials: international consensus recommendations. Lancet Respir Med 10: 298–306, 2022. doi: 10.1016/S2213-2600(21)00277-0. [DOI] [PubMed] [Google Scholar]
- 565. Marciano BE, Holland SM. Primary immunodeficiency diseases: current and emerging therapeutics. Front Immunol 8: 937, 2017. doi: 10.3389/fimmu.2017.00937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 566. Chalmers JD, Aliberti S, Filonenko A, Shteinberg M, Goeminne PC, Hill AT, Fardon TC, Obradovic D, Gerlinger C, Sotgiu G, Operschall E, Rutherford RM, Dimakou K, Polverino E, De Soyza A, McDonnell MJ. Characterization of the “frequent exacerbator phenotype” in bronchiectasis. Am J Respir Crit Care Med 197: 1410–1420, 2018. doi: 10.1164/rccm.201711-2202OC. [DOI] [PubMed] [Google Scholar]
- 567. Lee AL, Button BM, Denehy L, Roberts SJ, Bamford TL, Ellis SJ, Mu FT, Heine RG, Stirling RG, Wilson JW. Proximal and distal gastro-oesophageal reflux in chronic obstructive pulmonary disease and bronchiectasis. Respirology 19: 211–217, 2014. doi: 10.1111/resp.12182. [DOI] [PubMed] [Google Scholar]
- 568. Kapur N, Mackay IM, Sloots TP, Masters IB, Chang AB. Respiratory viruses in exacerbations of non-cystic fibrosis bronchiectasis in children. Arch Dis Child 99: 749–753, 2014. doi: 10.1136/archdischild-2013-305147. [DOI] [PubMed] [Google Scholar]
- 569. Piccione JC, Boesch RP. Bronchiectasis due to chronic pulmonary aspiration in children (Abstract). Chest 136: 8S, 2009. doi: 10.1378/chest.136.4_MeetingAbstracts.8S-f. [DOI] [Google Scholar]
- 570. Kida H, Takahashi T, Nakamura Y, Kinoshita T, Hara M, Okamoto M, Okayama S, Nakamura K, Kosai KI, Taniwaki T, Yamashita Y, Matsuishi T. Pathogenesis of lethal aspiration pneumonia in Mecp2-null mouse model for Rett syndrome. Sci Rep 7: 12032, 2017. doi: 10.1038/s41598-017-12293-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 571. Roberts HR, Wells AU, Milne DG, Rubens MB, Kolbe J, Cole PJ, Hansell DM. Airflow obstruction in bronchiectasis: correlation between computed tomography features and pulmonary function tests. Thorax 55: 198–204, 2000. doi: 10.1136/thorax.55.3.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 572. Labaki WW, Gu T, Murray S, Hatt CR, Galbán CJ, Ross BD, Martinez CH, Curtis JL, Hoffman EA, Pompe E, Lynch DA, Kazerooni EA, Martinez FJ, Han MK. Reprint of: Voxel-wise longitudinal parametric response mapping analysis of chest computed tomography in smokers. Acad Radiol 26: 306–312, 2019. doi: 10.1016/j.acra.2019.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 573. Pompe E, Galbán CJ, Ross BD, Koenderman L, Ten Hacken NH, Postma DS, van den Berge M, de Jong PA, Lammers JW, Mohamed Hoesein FA. Parametric response mapping on chest computed tomography associates with clinical and functional parameters in chronic obstructive pulmonary disease. Respir Med 123: 48–55, 2017. doi: 10.1016/j.rmed.2016.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 574. Vasilescu DM, Martinez FJ, Marchetti N, Galbán CJ, Hatt C, Meldrum CA, Dass C, Tanabe N, Reddy RM, Lagstein A, Ross BD, Labaki WW, Murray S, Meng X, Curtis JL, Hackett TL, Kazerooni EA, Criner GJ, Hogg JC, Han MK. Noninvasive imaging biomarker identifies small airway damage in severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med 200: 575–581, 2019. doi: 10.1164/rccm.201811-2083OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 575. Serisier DJ, Martin ML, McGuckin MA, Lourie R, Chen AC, Brain B, Biga S, Schlebusch S, Dash P, Bowler SD. Effect of long-term, low-dose erythromycin on pulmonary exacerbations among patients with non-cystic fibrosis bronchiectasis: the BLESS randomized controlled trial. JAMA 309: 1260–1267, 2013. doi: 10.1001/jama.2013.2290. [DOI] [PubMed] [Google Scholar]
- 576. Tunney MM, Einarsson GG, Wei L, Drain M, Klem ER, Cardwell C, Ennis M, Boucher RC, Wolfgang MC, Elborn JS. Lung microbiota and bacterial abundance in patients with bronchiectasis when clinically stable and during exacerbation. Am J Respir Crit Care Med 187: 1118–1126, 2013. doi: 10.1164/rccm.201210-1937OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 577. Choate R, Aksamit TR, Mannino D, Addrizzo-Harris D, Barker A, Basavaraj A, Daley CL, Daniels ML, Eden E, DiMango A, Fennelly K, Griffith DE, Johnson MM, Knowles MR, McShane PJ, Metersky ML, Noone PG, O’Donnell AE, Olivier KN, Salathe MA, Schmid A, Thomashow B, Tino G, Winthrop KL, Stone G. Pseudomonas aeruginosa associated with severity of non-cystic fibrosis bronchiectasis measured by the modified bronchiectasis severity score (BSI) and the FACED: The US bronchiectasis and NTM Research Registry (BRR) study. Respir Med 177: 106285, 2020. doi: 10.1016/j.rmed.2020.106285. [DOI] [PubMed] [Google Scholar]
- 578. Reich JM, Johnson RE. Mycobacterium avium complex pulmonary disease presenting as an isolated lingular or middle lobe pattern. the Lady Windermere syndrome. Chest 101: 1605–1609, 1992. doi: 10.1378/chest.101.6.1605. [DOI] [PubMed] [Google Scholar]
- 579. Prince DS, Peterson DD, Steiner RM, Gottlieb JE, Scott R, Israel HL, Figueroa WG, Fish JE. Infection with Mycobacterium avium complex in patients without predisposing conditions. N Engl J Med 321: 863–868, 1989. doi: 10.1056/NEJM198909283211304. [DOI] [PubMed] [Google Scholar]
- 580. Amati F, Simonetta E, Gramegna A, Tarsia P, Contarini M, Blasi F, Aliberti S. The biology of pulmonary exacerbations in bronchiectasis. Eur Respir Rev 28: 190055, 2019. doi: 10.1183/16000617.0055-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 581. Mac Aogáin M, Narayana JK, Tiew PY, Ali N, Yong VF, Jaggi TK, Lim AY, Keir HR, Dicker AJ, Thng KX, Tsang A, Ivan FX, Poh ME, Oriano M, Aliberti S, Blasi F, Low TB, Ong TH, Oliver B, Giam YH, Tee A, Koh MS, Abisheganaden JA, Tsaneva-Atanasova K, Chalmers JD, Chotirmall SH. Integrative microbiomics in bronchiectasis exacerbations. Nat Med 27: 688–699, 2021. doi: 10.1038/s41591-021-01289-7. [DOI] [PubMed] [Google Scholar]
- 582. Singh D, Agusti A, Anzueto A, Barnes PJ, Bourbeau J, Celli BR, Criner GJ, Frith P, Halpin DMG, Han M, López Varela MV, Martinez F, Montes de Oca M, Papi A, Pavord ID, Roche N, Sin DD, Stockley R, Vestbo J, Wedzicha JA, Vogelmeier C. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease: the GOLD science committee report 2019. Eur Respir J 53: 1900164, 2019. doi: 10.1183/13993003.00164-2019. [DOI] [PubMed] [Google Scholar]
- 583. Polverino E, Goeminne PC, McDonnell MJ, Aliberti S, Marshall SE, Loebinger MR, Murris M, Cantón R, Torres A, Dimakou K, De Soyza A, Hill AT, Haworth CS, Vendrell M, Ringshausen FC, Subotic D, Wilson R, Vilaró J, Stallberg B, Welte T, Rohde G, Blasi F, Elborn S, Almagro M, Timothy A, Ruddy T, Tonia T, Rigau D, Chalmers JD. European Respiratory Society guidelines for the management of adult bronchiectasis. Eur Respir J 50: 1700629, 2017. doi: 10.1183/13993003.00629-2017. [DOI] [PubMed] [Google Scholar]
- 584. McShane PJ, Naureckas ET, Tino G, Strek ME. Non-cystic fibrosis bronchiectasis. Am J Respir Crit Care Med 188: 647–656, 2013. doi: 10.1164/rccm.201303-0411CI. [DOI] [PubMed] [Google Scholar]
- 585. Lucas JS, Burgess A, Mitchison HM, Moya E, Williamson M, Hogg C; National PCD Service, UK. Diagnosis and management of primary ciliary dyskinesia. Arch Dis Child 99: 850–856, 2014. doi: 10.1136/archdischild-2013-304831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 586. Paredes Aller S, Quittner AL, Salathe MA, Schmid A. Assessing effects of inhaled antibiotics in adults with non-cystic fibrosis bronchiectasis–experiences from recent clinical trials. Expert Rev Respir Med 12: 769–782, 2018. doi: 10.1080/17476348.2018.1503540. [DOI] [PubMed] [Google Scholar]
- 587. Hubert D, Leroy S, Nove-Josserand R, Murris-Espin M, Mely L, Dominique S, Delaisi B, Kho P, Kovarik JM. Pharmacokinetics and safety of tobramycin administered by the PARI eFlow rapid nebulizer in cystic fibrosis. J Cyst Fibros 8: 332–337, 2009. doi: 10.1016/j.jcf.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 588. Sands D, Sapiejka E, Gąszczyk G, Mazurek H; T100 Study Group. Comparison of two tobramycin nebuliser solutions: pharmacokinetic, efficacy and safety profiles of T100 and TNS. J Cyst Fibros 13: 653–660, 2014. doi: 10.1016/j.jcf.2014.04.006. [DOI] [PubMed] [Google Scholar]
- 589. Magréault S, Mankikian J, Marchand S, Diot P, Couet W, Flament T, Grégoire N. Pharmacokinetics of colistin after nebulization or intravenous administration of colistin methanesulphonate (Colimycin®) to cystic fibrosis patients. J Cyst Fibros 19: 421–426, 2020. doi: 10.1016/j.jcf.2019.08.027. [DOI] [PubMed] [Google Scholar]
- 590. Huang JX, Blaskovich MA, Pelingon R, Ramu S, Kavanagh A, Elliott AG, Butler MS, Montgomery AB, Cooper MA. Mucin binding reduces colistin antimicrobial activity. Antimicrob Agents Chemother 59: 5925–5931, 2015. doi: 10.1128/AAC.00808-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 591. Bos AC, Passé KM, Mouton JW, Janssens HM, Tiddens HA. The fate of inhaled antibiotics after deposition in cystic fibrosis: how to get drug to the bug? J Cyst Fibros 16: 13–23, 2017. doi: 10.1016/j.jcf.2016.10.001. [DOI] [PubMed] [Google Scholar]
- 592. Dalhoff A. Pharmacokinetics and pharmacodynamics of aerosolized antibacterial agents in chronically infected cystic fibrosis patients. Clin Microbiol Rev 27: 753–782, 2014. doi: 10.1128/CMR.00022-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 593. Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Dřevínek P, Griese M, McKone EF, Wainwright CE, Konstan MW, Moss R, Ratjen F, Sermet-Gaudelus I, Rowe SM, Dong Q, Rodriguez S, Yen K, Ordoñez C, Elborn JS; VX08-770-102 Study Group. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 365: 1663–1672, 2011. doi: 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 594. Van Goor F, Hadida S, Grootenhuis PD, Burton B, Cao D, Neuberger T, Turnbull A, Singh A, Joubran J, Hazlewood A, Zhou J, McCartney J, Arumugam V, Decker C, Yang J, Young C, Olson ER, Wine JJ, Frizzell RA, Ashlock M, Negulescu P. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci USA 106: 18825–18830, 2009. doi: 10.1073/pnas.0904709106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 595. Keating D, Marigowda G, Burr L, Daines C, Mall MA, McKone EF, Ramsey BW, Rowe SM, Sass LA, Tullis E, McKee CM, Moskowitz SM, Robertson S, Savage J, Simard C, Van Goor F, Waltz D, Xuan F, Young T, Taylor-Cousar JL; VX16-659-101 Study Group. VX-445-tezacaftor-ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N Engl J Med 379: 1612–1620, 2018. doi: 10.1056/NEJMoa1807120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 596. Middleton PG, Mall MA, Dřevínek P, Lands LC, McKone EF, Polineni D, Ramsey BW, Taylor-Cousar JL, Tullis E, Vermeulen F, Marigowda G, McKee CM, Moskowitz SM, Nair N, Savage J, Simard C, Tian S, Waltz D, Xuan F, Rowe SM, Jain R; VX17-445-102 Study Group. Elexacaftor-Tezacaftor-Ivacaftor for cystic fibrosis with a single Phe508del allele. N Engl J Med 381: 1809–1819, 2019. doi: 10.1056/NEJMoa1908639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 597. Barry PJ, Mall MA, Álvarez A, Colombo C, de Winter-de Groot KM, Fajac I, McBennett KA, McKone EF, Ramsey BW, Sutharsan S, Taylor-Cousar JL, Tullis E, Ahluwalia N, Jun LS, Moskowitz SM, Prieto-Centurion V, Tian S, Waltz D, Xuan F, Zhang Y, Rowe SM, Polineni D; VX18-445-104 Study Group. Triple therapy for cystic fibrosis Phe508del-Gating and -residual function genotypes. N Engl J Med 385: 815–825, 2021. doi: 10.1056/NEJMoa2100665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 598. Davies JC, Sermet-Gaudelus I, Naehrlich L, Harris RS, Campbell D, Ahluwalia N, Short C, Haseltine E, Panorchan P, Saunders C, Owen CA, Wainwright CE; VX16-661-115 Investigator Group. A phase 3, double-blind, parallel-group study to evaluate the efficacy and safety of tezacaftor in combination with ivacaftor in participants 6 through 11 years of age with cystic fibrosis homozygous for F508del or heterozygous for the F508del-CFTR mutation and a residual function mutation. J Cyst Fibros 20: 68–77, 2021. doi: 10.1016/j.jcf.2020.07.023. [DOI] [PubMed] [Google Scholar]
- 599. Lopes-Pacheco M. CFTR modulators: the changing face of cystic fibrosis in the era of precision medicine. Front Pharmacol 10: 1662, 2019. doi: 10.3389/fphar.2019.01662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 600. Rowe SM, Daines C, Ringshausen FC, Kerem E, Wilson J, Tullis E, Nair N, Simard C, Han L, Ingenito EP, McKee C, Lekstrom-Himes J, Davies JC. Tezacaftor-ivacaftor in residual-function heterozygotes with cystic fibrosis. N Engl J Med 377: 2024–2035, 2017. doi: 10.1056/NEJMoa1709847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 601. Flume PA, Biner RF, Downey DG, Brown C, Jain M, Fischer R, De Boeck K, Sawicki GS, Chang P, Paz-Diaz H, Rubin JL, Yang Y, Hu X, Pasta DJ, Millar SJ, Campbell D, Wang X, Ahluwalia N, Owen CA, Wainwright CE; VX14-661-110 Study Group. Long-term safety and efficacy of tezacaftor-ivacaftor in individuals with cystic fibrosis aged 12 years or older who are homozygous or heterozygous for Phe508del CFTR (EXTEND): an open-label extension study. Lancet Respir Med 9: 733–746, 2021. doi: 10.1016/S2213-2600(20)30510-5. [DOI] [PubMed] [Google Scholar]
- 602. Heijerman HG, McKone EF, Downey DG, Van Braeckel E, Rowe SM, Tullis E, Mall MA, Welter JJ, Ramsey BW, McKee CM, Marigowda G, Moskowitz SM, Waltz D, Sosnay PR, Simard C, Ahluwalia N, Xuan F, Zhang Y, Taylor-Cousar JL, McCoy KS, McCoy K, Donaldson S, Walker S, Chmiel J, Rubenstein R, Froh DK, Neuringer I, Jain M, Moffett K, Taylor-Cousar JL, McCoy KS; VX17-445-103 Trial Group. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Lancet 394: 1940–1948, 2019. doi: 10.1016/S0140-6736(19)32597-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 603. Bell SC, Mall MA, Gutierrez H, Macek M, Madge S, Davies JC, , et al. The future of cystic fibrosis care: a global perspective. Lancet Respir Med 8: 65–124, 2020. doi: 10.1016/S2213-2600(19)30337-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 604. Mall MA, Mayer-Hamblett N, Rowe SM. Cystic fibrosis: emergence of highly effective targeted therapeutics and potential clinical implications. Am J Respir Crit Care Med 201: 1193–1208, 2020. doi: 10.1164/rccm.201910-1943SO. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 605. Solomon GM, Fu L, Rowe SM, Collawn JF. The therapeutic potential of CFTR modulators for COPD and other airway diseases. Curr Opin Pharmacol 34: 132–139, 2017. doi: 10.1016/j.coph.2017.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 606. Solomon GM, Raju SV, Dransfield MT, Rowe SM. Therapeutic approaches to acquired cystic fibrosis transmembrane conductance regulator dysfunction in chronic bronchitis. Ann Am Thorac Soc 13, Suppl 2: S169–S176, 2016. doi: 10.1513/AnnalsATS.201509-601KV. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 607. Rowe SM, Jones I, Dransfield MT, Haque N, Gleason S, Hayes KA, Kulmatycki K, Yates DP, Danahay H, Gosling M, Rowlands DJ, Grant SS. Efficacy and safety of the CFTR potentiator Icenticaftor (QBW251) in COPD: results from a phase 2 randomized trial. Int J Chron Obstruct Pulmon Dis 15: 2399–2409, 2020. doi: 10.2147/COPD.S257474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 608. Flume PA, Amelina E, Daines CL, Charlton B, Leadbetter J, Guasconi A, Aitken ML. Efficacy and safety of inhaled dry-powder mannitol in adults with cystic fibrosis: an international, randomized controlled study. J Cyst Fibros 20: 1003–1009, 2021. doi: 10.1016/j.jcf.2021.02.011. [DOI] [PubMed] [Google Scholar]
- 609. Zhang Y, Song A, Liu J, Dai J, Lin J. Therapeutic effect of nebulized hypertonic saline for muco-obstructive lung diseases: a systematic review and meta-analysis with trial sequential analysis. J Investig Med 69: 742–748, 2021. doi: 10.1136/jim-2020-001479. [DOI] [PubMed] [Google Scholar]
- 610. Ademhan Tural D, Yalçın E, Emiralioglu N, Ozsezen B, Sunman B, Nayir Buyuksahin H, Guzelkas I, Dogru D, Ozcelik U, Kiper N. Comparison of inhaled mannitol/dornase alfa combination and daily dornase alfa alone in children with cystic fibrosis. Pediatr Pulmonol 57: 142–151, 2022. doi: 10.1002/ppul.25740. [DOI] [PubMed] [Google Scholar]
- 611. Shei RJ, Peabody JE, Kaza N, Rowe SM. The epithelial sodium channel (ENaC) as a therapeutic target for cystic fibrosis. Curr Opin Pharmacol 43: 152–165, 2018. doi: 10.1016/j.coph.2018.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 612. Hagner M, Albrecht M, Guerra M, Braubach P, Halle O, Zhou-Suckow Z, Butz S, Jonigk D, Hansen G, Schultz C, Dittrich AM, Mall MA. IL-17A from innate and adaptive lymphocytes contributes to inflammation and damage in cystic fibrosis lung disease. Eur Respir J 57: 1900716, 2021. doi: 10.1183/13993003.00716-2019. [DOI] [PubMed] [Google Scholar]
- 613. Reihill JA, Douglas LE, Martin SL. Modulation of ion transport to restore airway hydration in cystic fibrosis. Genes 12: 453, 2021. doi: 10.3390/genes12030453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 614. Fujikawa H, Kawakami T, Nakashima R, Nasu A, Kamei S, Nohara H, Eto Y, Ueno-Shuto K, Takeo T, Nakagata N, Suico MA, Kai H, Shuto T. Azithromycin inhibits constitutive airway epithelial sodium channel activation in vitro and modulates downstream pathogenesis in vivo. Biol Pharm Bull 43: 725–730, 2020. doi: 10.1248/bpb.b19-01091. [DOI] [PubMed] [Google Scholar]
- 615. Mall MA. ENaC inhibition in cystic fibrosis: potential role in the new era of CFTR modulator therapies. Eur Respir J 56: 2000946, 2020. doi: 10.1183/13993003.00946-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 616. Goss CH, Fajac I, Jain R, Seibold W, Gupta A, Hsu MC, Sutharsan S, Davies JC, Mall MA. Efficacy and safety of inhaled ENaC inhibitor BI 1265162 in patients with cystic fibrosis: BALANCE-CF™ 1—a randomised, Phase II study. Eur Respir J 59: 2100746, 2022. doi: 10.1183/13993003.00746-2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 617. Mackie A, Rascher J, Schmid M, Endriss V, Brand T, Seibold W. First clinical trials of the inhaled epithelial sodium channel inhibitor BI 1265162 in healthy volunteers. ERJ Open Res 7: 00447-2020, 2021. doi: 10.1183/23120541.00447-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 618. Mall MA, Galietta LJ. Targeting ion channels in cystic fibrosis. J Cyst Fibros 14: 561–570, 2015. doi: 10.1016/j.jcf.2015.06.002. [DOI] [PubMed] [Google Scholar]
- 619. Donaldson SH, Danielle Samulski T, LaFave C, Zeman K, Wu J, Trimble A, Ceppe A, Bennett WD, Davis SD. A four week trial of hypertonic saline in children with mild cystic fibrosis lung disease: effect on mucociliary clearance and clinical outcomes. J Cyst Fibros 19: 942–948, 2020. doi: 10.1016/j.jcf.2020.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 620. Elkins MR, Robinson M, Rose BR, Harbour C, Moriarty CP, Marks GB, Belousova EG, Xuan W, Bye PT; National Hypertonic Saline in Cystic Fibrosis (NHSCF) Study Group. A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. N Engl J Med 354: 229–240, 2006. doi: 10.1056/NEJMoa043900. [DOI] [PubMed] [Google Scholar]
- 621. Trimble AT, Whitney Brown A, Laube BL, Lechtzin N, Zeman KL, Wu J, Ceppe A, Waltz D, Bennett WD, Donaldson SH. Hypertonic saline has a prolonged effect on mucociliary clearance in adults with cystic fibrosis. J Cyst Fibros 17: 650–656, 2018. doi: 10.1016/j.jcf.2018.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 622. Elkins M, Dentice R. Timing of hypertonic saline inhalation for cystic fibrosis. Cochrane Database Syst Rev 2: Cd008816, 2020. doi: 10.1002/14651858.CD008816.pub4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 623. Bennett WD, Burbank A, Almond M, Wu J, Ceppe A, Hernandez M, Boucher RC, Peden DB. Acute and durable effect of inhaled hypertonic saline on mucociliary clearance in adult asthma. ERJ Open Res 7: 00062-2021, 2021. doi: 10.1183/23120541.00062-2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 624. Bhowmik A, Seemungal TA, Sapsford RJ, Devalia JL, Wedzicha JA. Comparison of spontaneous and induced sputum for investigation of airway inflammation in chronic obstructive pulmonary disease. Thorax 53: 953–956, 1998. doi: 10.1136/thx.53.11.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 625. Bhowmik A, Seemungal TA, Sapsford RJ, Wedzicha JA. Relation of sputum inflammatory markers to symptoms and lung function changes in COPD exacerbations. Thorax 55: 114–120, 2000. doi: 10.1136/thorax.55.2.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 626. Tangedal S, Aanerud M, Persson LJ, Brokstad KA, Bakke PS, Eagan TM. Comparison of inflammatory markers in induced and spontaneous sputum in a cohort of COPD patients. Respir Res 15: 138, 2014. doi: 10.1186/s12931-014-0138-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 627. Henderson AG, Anderson WH, Ceppe A, Coakley RD, Button B, Alexis NE, Peden DB, Lazarowski ER, Davis CW, Fuller F, Almond M, Qaqish B, Kesimer M, Boucher RC. Mucus hydration in subjects with stable chronic bronchitis: a comparison of spontaneous and induced sputum. COPD 15: 572–580, 2018. doi: 10.1080/15412555.2019.1566892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 628. Paff T, Omran H, Nielsen KG, Haarman EG. Current and future treatments in primary ciliary dyskinesia. Int J Mol Sci 22: 9834, 2021. doi: 10.3390/ijms22189834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 629. Paff T, Daniels JM, Weersink EJ, Lutter R, Vonk Noordegraaf A, Haarman EG. A randomised controlled trial on the effect of inhaled hypertonic saline on quality of life in primary ciliary dyskinesia. Eur Respir J 49: 1601770, 2017. doi: 10.1183/13993003.01770-2016. [DOI] [PubMed] [Google Scholar]
- 630. Anuradha K, Gunathilaka PK, Wickramasinghe VP. Effectiveness of hypertonic saline nebulization in airway clearance in children with non-cystic fibrosis bronchiectasis: a randomized control trial. Pediatr Pulmonol 56: 509–515, 2021. doi: 10.1002/ppul.25206. [DOI] [PubMed] [Google Scholar]
- 631. Máiz Carro L, Martínez-García MA. Nebulized hypertonic saline in noncystic fibrosis bronchiectasis: a comprehensive review. Ther Adv Respir Dis 13: 1753466619866102, 2019. doi: 10.1177/1753466619866102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 632. Rogers DF. Mucoactive drugs for asthma and COPD: any place in therapy? Expert Opin Investig Drugs 11: 15–35, 2002. doi: 10.1517/13543784.11.1.15. [DOI] [PubMed] [Google Scholar]
- 633. Rogers DF, Barnes PJ. Treatment of airway mucus hypersecretion. Ann Med 38: 116–125, 2006. doi: 10.1080/07853890600585795. [DOI] [PubMed] [Google Scholar]
- 634. Ha EV, Rogers DF. Novel therapies to inhibit mucus synthesis and secretion in airway hypersecretory diseases. Pharmacology 97: 84–100, 2016. doi: 10.1159/000442794. [DOI] [PubMed] [Google Scholar]
- 635. Tam J, Nash EF, Ratjen F, Tullis E, Stephenson A. Nebulized and oral thiol derivatives for pulmonary disease in cystic fibrosis. Cochrane Database Syst Rev 1: CD007168, 2013. doi: 10.1002/14651858.CD007168.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 636. Lo Bello F, Ieni A, Hansbro PM, Ruggeri P, Di Stefano A, Nucera F, Coppolino I, Monaco F, Tuccari G, Adcock IM, Caramori G. Role of the mucins in pathogenesis of COPD: implications for therapy. Expert Rev Respir Med 14: 465–483, 2020. doi: 10.1080/17476348.2020.1739525. [DOI] [PubMed] [Google Scholar]
- 637. Muñoz Castro G, Balañá Corberó A. Airway clearance and mucoactive therapies. Semin Respir Crit Care Med 42: 616–622, 2021. doi: 10.1055/s-0041-1730922. [DOI] [PubMed] [Google Scholar]
- 638. Ehre C, Rushton ZL, Wang B, Hothem LN, Morrison CB, Fontana NC, Markovetz MR, Delion MF, Kato T, Villalon D, Thelin WR, Esther CR Jr, Hill DB, Grubb BR, Livraghi-Butrico A, Donaldson SH, Boucher RC. An improved inhaled mucolytic to treat airway muco-obstructive diseases. Am J Respir Crit Care Med 199: 171–180, 2019. doi: 10.1164/rccm.201802-0245OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 639. Schwalfenberg GK. N-acetylcysteine: a review of clinical usefulness (an old drug with new tricks). J Nutr Metab 2021: 9949453, 2021. doi: 10.1155/2021/9949453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 640. Ermund A, Recktenwald CV, Skjåk-Braek G, Meiss LN, Onsøyen E, Rye PD, Dessen A, Myrset AH, Hansson GC. OligoG CF-5/20 normalizes cystic fibrosis mucus by chelating calcium. Clin Exp Pharmacol Physiol 44: 639–647, 2017. doi: 10.1111/1440-1681.12744. [DOI] [PubMed] [Google Scholar]
- 641. Horsley A, Rousseau K, Ridley C, Flight W, Jones A, Waigh TA, Thornton DJ. Reassessment of the importance of mucins in determining sputum properties in cystic fibrosis. J Cyst Fibros 13: 260–266, 2014. doi: 10.1016/j.jcf.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 642. Pritchard MF, Oakley JL, Brilliant CD, Rye PD, Forton J, Doull IJ, Ketchell I, Hill KE, Thomas DW, Lewis PD. Mucin structural interactions with an alginate oligomer mucolytic in cystic fibrosis sputum. Vib Spectrosc 103: 102932, 2019. doi: 10.1016/j.vibspec.2019.102932. [DOI] [Google Scholar]
- 643. Pritchard MF, Powell LC, Menzies GE, Lewis PD, Hawkins K, Wright C, Doull I, Walsh TR, Onsøyen E, Dessen A, Myrvold R, Rye PD, Myrset AH, Stevens HN, Hodges LA, MacGregor G, Neilly JB, Hill KE, Thomas DW. A new class of safe oligosaccharide polymer therapy to modify the mucus barrier of chronic respiratory disease. Mol Pharm 13: 863–872, 2016. doi: 10.1021/acs.molpharmaceut.5b00794. [DOI] [PubMed] [Google Scholar]
- 644. Budai-Szűcs M, Berkó S, Kovács A, Jaikumpun P, Ambrus R, Halász A, Szabó-Révész P, Csányi E, Zsembery Á. Rheological effects of hypertonic saline and sodium bicarbonate solutions on cystic fibrosis sputum in vitro. BMC Pulm Med 21: 225, 2021. doi: 10.1186/s12890-021-01599-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 645. Ferrera L, Capurro V, Delpiano L, Gianotti A, Moran O. The application of bicarbonate recovers the chemical-physical properties of airway surface liquid in cystic fibrosis epithelia models. Biology (Basel) 10: 278, 2021. doi: 10.3390/biology10040278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 646. Fuchs HJ, Borowitz DS, Christiansen DH, Morris EM, Nash ML, Ramsey BW, Rosenstein BJ, Smith AL, Wohl ME. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group. N Engl J Med 331: 637–642, 1994. doi: 10.1056/NEJM199409083311003. [DOI] [PubMed] [Google Scholar]
- 647. Yang C, Chilvers M, Montgomery M, Nolan SJ. Dornase alfa for cystic fibrosis. Cochrane Database Syst Rev 4: CD001127, 2016. doi: 10.1002/14651858.CD001127.pub3. [DOI] [PubMed] [Google Scholar]
- 648. O’Donnell AE, Barker AF, Ilowite JS, Fick RB. Treatment of idiopathic bronchiectasis with aerosolized recombinant human DNase I. rhDNase Study Group. Chest 113: 1329–1334, 1998. doi: 10.1378/chest.113.5.1329. [DOI] [PubMed] [Google Scholar]
- 649. Donaldson SH, Laube BL, Corcoran TE, Bhambhvani P, Zeman K, Ceppe A, Zeitlin PL, Mogayzel PJ Jr, Boyle M, Locke LW, Myerburg MM, Pilewski JM, Flanagan B, Rowe SM, Bennett WD. Effect of ivacaftor on mucociliary clearance and clinical outcomes in cystic fibrosis patients with G551D-CFTR. JCI Insight 3: e122695, 2018. doi: 10.1172/jci.insight.122695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 650. Radicioni G, Cao R, Carpenter J, Ford AA, Wang T, Li L, Kesimer M. The innate immune properties of airway mucosal surfaces are regulated by dynamic interactions between mucins and interacting proteins: the mucin interactome. Mucosal Immunol 9: 1442–1454, 2016. doi: 10.1038/mi.2016.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 651. Macosko CW. Rheology Principles, Measurements, and Applications. New York: Weinheim Cambridge VCH, 1994. [Google Scholar]
- 652. Cone RA. Barrier properties of mucus. Adv Drug Deliv Rev 61: 75–85, 2009. doi: 10.1016/j.addr.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 653. Mason TG. Estimating the viscoelastic moduli of complex fluids using the generalized Stokes-Einstein equation. Rheologica Acta 39: 371–378, 2000. doi: 10.1007/s003970000094. [DOI] [Google Scholar]
- 654. Lysy M, Pillai NS, Hill DB, Forest MG, Mellnik JW, Vasquez PA, McKinley SA. Model comparison and assessment for single particle tracking in biological fluids. J Am Stat Assoc 111: 1413–1426, 2016. doi: 10.1080/01621459.2016.1158716. [DOI] [Google Scholar]
- 655. Markovetz MR, Garbarine IC, Morrison CB, Kissner WJ, Seim I, Forest MG, Papanikolas MJ, Freeman R, Ceppe A, Ghio A, Alexis NE, Stick SM, Ehre C, Boucher RC, Esther CR, Muhlebach MS, Hill DB. Mucus and mucus flake composition and abundance reflect inflammatory and infection status in cystic fibrosis. J Cyst Fibros. In press. doi: 10.1016/j.jcf.2022.04.008. [DOI] [PubMed] [Google Scholar]
- 656. Batson B, Zorn B, Radicioni G, Livengood S, Kumagai T, Dang H, Ceppe A, Clapp P, Tunney M, Elborn S, McElvaney G, Muhlebach M, Boucher RC, Tiemeyer M, Wolfgang M, Kesimer M. Cystic fibrosis airway mucus hyperconcentration produces a vicious cycle of mucin, pathogen, and inflammatory interactions that promote disease persistence. Am J Respir Cell Mol Biol. In press. doi: 10.1165/rcmb.2021-0359OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 657. Shih CK, Litt M, Khan MA, Wolf DP. Effect of nondialyzable solids concentration and viscoelasticity on ciliary transport of tracheal mucus. Am Rev Respir Dis 115: 989–95, 1997. doi: 10.1164/arrd.1977.115.6.989. [DOI] [PubMed] [Google Scholar]