

Summary
A detailed understanding of the developmental substates of human pluripotent stem cells (hPSCs) is needed to optimize their use in cell therapy and for modeling early development. Genetic instability and risk of tumorigenicity of primed hPSCs are well documented, but a systematic isogenic comparison between substates has not been performed. We derived four hESC lines in naive human stem cell medium (NHSM) and generated isogenic pairs of NHSM and primed cultures. Through phenotypic, transcriptomic, and methylation profiling, we identified changes that arose during the transition to a primed substate. Although early NHSM cultures displayed naive characteristics, including greater proliferation and clonogenic potential compared with primed cultures, they drifted toward a more primed-like substate over time, including accumulation of genetic abnormalities. Overall, we show that transcriptomic and epigenomic profiling can be used to place human pluripotent cultures along a developmental continuum and may inform their utility for clinical and research applications.
Subject areas: Stem cell plasticity, Genetics, Epigenetics, Developmental biology, Transcriptomics
Graphical abstract
Highlights
-
•
Molecular and phenotypic profiling of isogenic NHSM and primed hESC lines
-
•
Genetic aberrations accumulate over time in both NHSM and primed substates
-
•
DNA methylation levels of NHSM hESC lines drift toward a more primed-like substate
-
•
Molecular profiles place hPSC cultures along a developmental continuum
Stem cell plasticity; Genetics; Epigenetics; Developmental biology; Transcriptomics
Introduction
Cell transplantation therapy holds tremendous promise for regenerative purposes in patients suffering from severe injuries or chronic diseases.1 In some cases, the transplanted cells replace endogenous cells that have been irreversibly damaged or killed, whereas in others, they support and “nurse” endogenous damaged cells to restore their normal function.2 In either case, a starting cell type with the capacity to expand and differentiate into a variety of lineages, but also does not display a tendency toward malignant transformation, is of great value.3 The appeal of human pluripotent stem cells (hPSCs) arises from their capacity for essentially limitless proliferation and self-renewal, as well as their receptivity for directed differentiation to a broad range of cell types in response to the appropriate lineage-specific cues.4
Pluripotency, the potential to differentiate into all three embryonic germ layers, is a transient state in the natural course of embryonic development displayed by inner cell mass (ICM) cells in preimplantation blastocysts. However, it can be captured in vitro in the form of embryonic stem cells (ESCs)5 cultured from epiblast cells or by reprogramming somatic cells into induced pluripotent stem cells (iPSCs).6 In both cases, the resulting hPSCs must be maintained in culture conditions that stabilize their pluripotency.
It was first recognized in the mouse system that in vitro pluripotency can be divided into two major substates, termed naive and primed pluripotency. Naive mouse embryonic stem cells (mESCs) are derived from the ICM of a preimplantation blastocyst and are characterized by domed colonies, dependence on JAK/STAT signaling, high expression of early pluripotency genes, and more efficient contribution to chimeras.7,8 In contrast, mouse epiblast stem cells (mEpiSCs) are derived from the early post-implantation epiblast, representing a restricted pluripotent state, and are characterized by flattened colony morphology, dependence on TGFβ/ActivinA and FGF2 signaling, expression of lineage-specification genes and less efficient contribution to chimeras.9,10 One hallmark of naive mESCs is increased proliferation and higher clonogenicity (the ability of single cells to establish a colony) compared with primed mEpiSCs, properties that are useful for applications such as drug screening or cell therapies, which require large numbers of cells, or gene editing, which involves single-cell cloning.11,12
After the first report of in vitro conditions for the establishment and maintenance of hESCs,13 it was quickly recognized that hESCs differed markedly from mESCs, and that hESCs are more similar to mEpiSCs. Interestingly, hESCs are derived from the ICM of preimplantation embryos, and so it was immediately apparent that the primed state observed for hESCs is dictated by the culture conditions applied to them, and the more primed phenotype of the human ICM cells that are already polarized. Several groups, including ours, have since applied knowledge regarding differences in signaling pathways modulated by mESCs and mEpiSCs culture conditions to promote the naive or primed phenotypes of hESC cultures, respectively.14,15,16,17,18,19,20,21,22,23,24,25 Like mESCs, naive human ESCs have been shown to possess characteristics such as increased proliferation and higher clonogenicity.20,24,26,27,28 Differences in differentiation potential between naive and primed hPSCs may also drive the selection of a particular substate as the starting point for the production of cells for therapeutic applications.14,17,29 Recently, Lee reported that lineage-specific differentiation is influenced by the hPSCs substate.30
The majority of existing hESC lines were derived in primed conditions, and it has been shown that these primed hESCs can be transitioned to a naive state by transferring them to naive conditions (termed “converted naive”). A small number of hESC lines were directly derived in naive conditions. Notably, we were the first to derive hESC lines displaying naive characteristics directly from human blastocytes and termed them “virgin naive.”14 Recently, there is evidence to suggest that early development exists along a pluripotency continuum ranging between naive and primed, and that the various hPSC media stabilize cells at different points along this continuum.29,31,32
The enthusiasm for hPSCs as a source of cell therapy has been tempered by many studies reporting on their genetic instability, which may indicate an increased risk of tumorigenicity. Although most hPSC lines possess normal karyotypes when tested in early culture, they tend to acquire chromosomal duplications when cultured over many passages.33,34,35 Using high-resolution genotyping and DNA sequencing methods it was shown that over time in culture hESCs acquire not only large chromosomal duplications, but also small genetic aberrations, predominantly duplications.36,37 Moreover, comparisons with parental genomes and isogenic clones enabled us to show that even in early passage primed hESCs contain small genetic aberrations, mainly deletions and loss of heterozygosity,36 Some of the aberrations commonly found in primed hPSCs are also found in cancer cells and have been implicated as driver mutations for malignant processes.38,39,40,41,42,43,44 The genetic stability of hPSCs in developmentally earlier substates, however, has not been clearly established, as some studies have reported that naive hPSCs are genetically stable even after extended culture.14,21,24,45,46 It is important to note that none of these previous studies analyzed isogenic hESC lines that were cultured in parallel in conditions representing different substates. Likewise, numerous studies have compared the transcriptomes of hPSCs cultured in naive and primed conditions but have not studied the transcriptional stability of isogenic hESC lines derived in conditions representing an earlier substate and transitioned to primed conditions over time.15,23,47
In this study, we compared for the first time the cellular phenotype and transcriptomic, genomic, and epigenomic characteristics over time in a culture of four matched isogenic pairs of hESC cultures in different pluripotent substates originating from the same hESC lines derived in our laboratory. As expected, early passage NHSM hESCs displayed shorter doubling times, greater percentages of cells in S-phase, and higher clonogenicity compared with their primed counterparts. The transition from an earlier substate to the primed state was characterized by increased DNA methylation, decreased expression of previously reported naive marker genes, and decreased expression of encoded miRNAs on the X-chromosome of the two female lines and at certain imprinted loci. With increasing time in culture, both substates accumulated genetic aberrations, with trends toward copy number losses in late NHSM cultures and copy number gains in late primed cultures.
Overall, our results suggest that we have established hESCs that, compared with previously reported naive lines, have a moderately naive phenotype that can be transitioned to the primed state in a facile manner, and that this transition is characterized by expected shifts in mRNA and miRNA expression and DNA methylation. These findings lead us to suggest that these hESC lines may be a useful model to study the transition from an earlier substate to the primed substate, the differentiation potential of these states of pluripotency, and accordingly to determine what are the preferred conditions for the generation and culture of PSCs for clinical use in cell therapy.
Results
Confirmation of naive and primed markers in matched isogenic hESC cultures
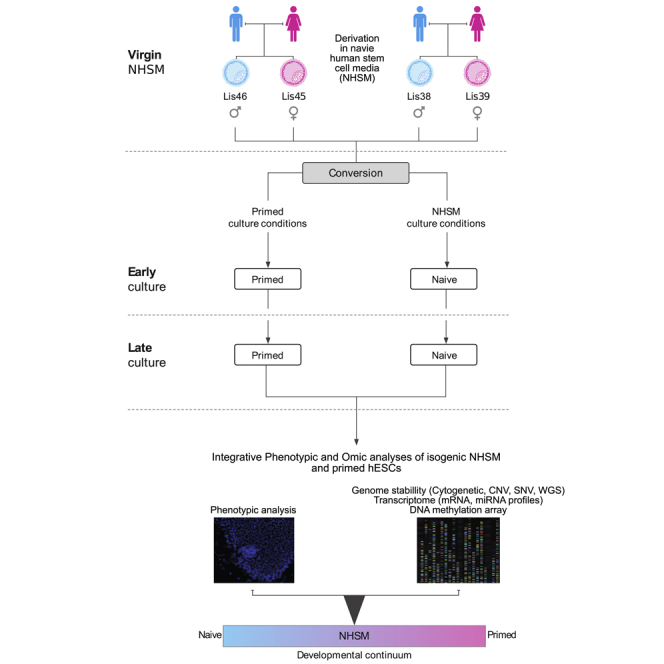
Four hESC lines were derived in our laboratory in NHSM conditions: Lis38_N, Lis39_N, Lis45_N, and Lis46_N. The Lis38 and Lis39 lines were derived from blastocysts donated from one set of parents and the Lis45 and Lis46 lines were derived from another set of parents; the Lis38 and Lis46 lines are male and the Lis39 and Lis45 lines are female. After these lines were established, they were split into three replicates, expanded, and collected for genomic analysis at passage 20 (p20 derivation) (Figure 1A). As the genetic background of hPSC lines may result in line-to-line variability, independently of their pluripotent state, we used these original hESC cultures to generate matched isogenic pairs of hESC cultures, by transitioning a portion of each NHSM culture to primed conditions, whereas another portion was kept in NHSM conditions. These converted primed and NHSM cultures were grown in parallel for another 10 passages and at p30 (Early culture), phenotyping and collection of samples for genomic analysis was performed (Figure 1A). Finally, these cultures were grown for an additional 20 passages, and at p50 (Late culture), phenotyping and collection of samples for genomic analysis was performed (Figure 1A).
Figure 1.

Validation of naive and primed states at derivation, and at early and late passages
(A) Study design – four hESC lines, namely Lis38, Lis39, Lis45, and Lis46, were derived and cultured in NHSM conditions. At passage 20 (p20, derivation), half of the cells from each line were transferred to primed conditions (conversion), and half continued to be cultured in NHSM conditions. After 10 passages cells in the NHSM (p30, Early NHSM) and primed (p30, Early primed) cultures were analyzed. Cells were analyzed again at after an additional 20 passages (p50, Late NHSM and Late primed).
(B) Colony morphology of NHSM-derived cells and their isogenic primed counterparts (scale bar – 200 mm) and immunofluorescence staining for the pluripotent markers OCT4, TRA-1-60, and SSEA4 and for the naive marker TFE3 (scale bar – 100 mm).
(C) Relative mRNA expression levels of the naive markers KLF17, TFCP2L1, and STELLA as determined by qRT-PCR in NHSM (red column) and their primed counterparts (blue column) lines, relative to primed control (black column). All data are expressed as mean ± SEM, ∗∗∗p < 0.005 by a paired t-test.
(D) In vivo differentiation was demonstrated by teratoma formation at p40. Tissues representing the three germ layers were identified using hematoxylin and eosin staining and marked by white arrows (scale bar – 50 μm).
The pluripotency of NHSM derived, Early NHSM, Early primed, Late NHSM, and Late primed cultures was confirmed by demonstrating similar expression of the pluripotency-associated markers OCT4, TRA-1-60, SSEA4, and NANOG (Figures 1B, S1A, S2B, and S3B). The naive status of all NHSM cultures was confirmed by demonstrating domed colony morphology and nuclear staining of TEF3. This was in contrast to flat colony morphology and cytoplasmic TEF3 staining in the primed cultures; (Figures 1B, S1B, S2A, S2B, S3A, and S3B), and higher expression of the naive markers KLF17, TFCPL1, STELLA, KLF5, and KLF4 compared with primed cultures (Figures 1C, S1C, S2C, and S3C). The differentiation capacity of the NHSM and primed hESC lines was demonstrated by their ability to generate mature teratomas comprised of cells representing all three germ layers following xenotransplantation into NSG mice (Figure 1D and S4).
Long-term culture has different effects on the phenotypes of hESCs in different substates
We compared the phenotypes of the early NHSM cultures and their isogenic early primed counterparts using two measures of cell proliferation (doubling time and EdU labeling) and an assessment of cell survival and proliferation (clonogenicity, or the ability to expand from single cells to form colonies). The early NHSM cultures showed evidence of greater proliferation and survival with significantly shorter doubling times (Figure 2A, left), higher fractions of cells in S phase (Figure 2B, left), and greater clonogenic potential (Figure 2C, left) when compared with the corresponding early primed cultures.
Figure 2.

Cytogenetic and phenotypic comparisons of proliferation rate and clonogenicity potential of NHSM derived and isogenic primed hESCs at early and late passages
(A) Proliferation rate was determined by the doubling time assay.
(B) Fraction of cells in the S-phase was analyzed using EdU incorporation and quantitative DNA staining followed by FACS analysis.
(C) Clonogenicity potential determined by alkaline phosphatase staining of colonies 7 days after seeding at low density. All data are expressed as mean ± SEM, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005 by a paired t-test.
(D) Karyotype analysis of derivation (p20) hESC lines.
(E) Validation of CMA results by WGS.
We then investigated the effect of extended culture on these phenotypic properties. The colony formation assay showed that late NHSM cells were again more clonogenic than their isogenic primed counterparts (Figure 2C, right). However, analysis of the proliferation rate, as indicated by either doubling time or fraction of cells in the S phase, demonstrated that only one hESC line (Lis38) maintained a higher proliferation rate in the late NHSM culture compared with the primed culture; for the other three lines (Lis39, Lis45, and Lis46), the late NHSM cells proliferated more slowly than their isogenic primed counterparts (Figures 2A and 2B, right). When the early NHSM cultures were compared with the late NHSM cultures, the proliferation rates were similar (Figures 2A and 2B), whereas the late primed cultures showed evidence of higher proliferation compared with the early primed cultures (Figures 2A and 2B). These results suggested that extended culture did not significantly alter the survival of hPSCs in either substate (with NHSM hPSCs having a higher clonogenic potential), but did have differential effects on the growth phenotypes, with stable proliferation of NHSM hPSCs, but increased proliferation of primed hPSCs in late cultures.
Transcriptome and methylome analysis places hESC cultures along a developmental continuum
mRNA transcriptomic profiling of all cultures was performed using RNA-seq. Triplicate cultures were collected and analyzed at the same three timepoints shown in Figure 1A, for a total of 60 samples. Principal component analysis (PCA) on normalized data revealed that the cultures separated along the first principal component by pluripotent state (Figure 3A). Differential expression analysis resulted in 278 genes upregulated and 668 genes downregulated (adj. p-value < 0.05, basemean >50, paired analysis by line and timepoint) in the NHSM cultures compared with their isogenic primed cultures at similar time points (Table S1 and Figure S5A). The genes upregulated in the NHSM cultures were enriched for the ectoderm differentiation pathway (adj. p-value < 0.005) whereas the top enriched pathways for genes downregulated in the NHSM cultures were Focal Adhesion and the VEGFA-VEGFR2 Signaling Pathway (Table S2).
Figure 3.

Transcriptomic profiling of early and late NHSM and primed cultures in the context of previously published reports
(A) Principal component analysis of all 60 (triplicates of each condition and timepoint) samples using all genes passing filters (see STAR methods)
(B) RNA-seq principal component analysis of all 60 (triplicates of each condition and timepoint) samples in this study along with previously published naive, intermediate, and primed lines (Table S3) using all genes passing filters (see STAR methods). Naive culture conditions used by study are listed in “( ).”
(C) Heatmap displaying the top 100 genes, ranked by their loadings on the first principal component (see Figure 3B). Samples and genes are hierarchically clustered using the Euclidean distance. A “∗” has been placed next to the five genes previously reported to be naive markers shared by three different naive conversion methods.
(D) Small RNA-seq principal component analysis of all samples in this study along with previously published naive and primed lines (Table S5) using all miRNAs passing filter (see STAR methods).
Previous reports have suggested that instead of two pluripotent states, there exists a developmental continuum between the naive and primed states,15,31 including a formative intermediate state.29 To assess where our cultures lie in relation to this developmental continuum, we combined our transcriptomic data with that of nine previous studies (Table S3). On a PCA plot, the previously published naive and primed lines are separated largely on the first principal component, with our NHSM lines in the middle, whereas the published naive cultures are to their left and the published primed cultures are located on their right (Figure 3B). Our NHSM lines were positioned slightly to the left of hESCs cultured in similar NHSM culture conditions19 or conditions that result in an intermediate state;29 (Figure 3B and Table S3), whereas our primed cultures were positioned slightly to the right of those two reference cultures. Our NHSM and primed lines were also separated from each other on the second principal component (PCA2; Figure 3B). Performing hierarchical clustering using the top 100 genes ranked by their loadings on PCA1, our lines clustered together with all of the previously published primed lines, the intermediate lines (FTW) and, the four NHSM-derived lines (Figure 3C and Table S3). These 100 genes include five genes previously reported to be markers shared among three disparate naive conversion methods (DNMT3L, KHDC3L, TRIM60, KLF17, OLAH).47
We compared the DEGs that resulted from contrasting our late primed cultures to the derivation cultures to those also differentially expressed during preimplantation development in previously published data48 (adj. p-value < 0.05, log2 fold change > 2), and found that our NHSM lines most closely resembled the late blastocyst stage (Table S4).
We next analyzed how similar the miRNA profiles of our matched isogenic cultures were to those in the previously published miRNA datasets from naive (six HNES hESC lines) and primed hESC cultures (six HNES lines and two Encode WA01 (H1) lines) (Roadmap Epigenomics https://www.ncbi.nlm.nih.gov/bioproject/25958449) (Figure 3D and Table S5). The results show that our NHSM and primed cultures are separated from the HNES lines predominantly on PC1 and to a lesser extent on PC2. Our NHSM is located left of the naive in other publications, whereas our primed are right to the other primed lines. The primed WA01 cultures were positioned near the middle of both principal components. Differential expression analysis (adj. p-value < 0.05) revealed that the top five differentially expressed miRNA’s upregulated in the HNES naive cultures compared with HNES primed cultures were miR-143-3p, miR-92b-3p, miR-512-3p, miR-199, and miR-363-3p. Of these, miR-92b-3p and miR363-3p were also significantly upregulated in our NHSM cultures compared with our primed cultures (Figure 3D).
It is well established from DNA methylation studies that the preimplantation methylome is hypomethylated.50 Likewise, human naive hESCs converted from primed cells under a variety of conditions have reported global CpG methylation levels of about 30% compared with approximately 80% in primed cells.32 To interrogate our cultures’ global DNA methylation profiles, we performed whole genome EPIC DNA methylation array analysis (Illumina, Inc.) and found that the average methylation level of our NHSM cultures was close to 60%, approximately 2–4% lower than that of our primed cultures (Figure S5B), reinforcing the fact that our NHSM cells are at an intermediate substate.
Genetic stability of hESCs in different substates
High cellular proliferation rates may increase the susceptibility of cells to genetic instability, which in turn may increase the risk of tumorigenicity. Because prior studies have shown that derivation, extended culture, and culture conditions can impact the accumulation of genetic abnormalities in hPSCs, we compared the genomes of early and late passage hESC cultures in both substates, as well as the donors of the embryos from which the hESC lines were derived.
Cytogenetic analysis
Cytogenetic analysis showed that all four hESC lines had normal karyotypes at derivation as shown by their G-banding (Figure 2D). Consistent with the karyotype results, chromosomal microarray analysis (CMA) also showed no genetic aberrations in derivation or early NHSM hESCs or in isogenic early primed cultures (Figure S6).
At late passage, CMA analysis revealed no genetic aberrations for any of the cultures for two of the hESC lines (Lis38 and Lis39), but large copy number aberrations in both NHSM and primed cultures for the two other lines (Lis45 and Lis46) (Figure S6 and 2E). Specifically, CMA identified monosomy X in Lis45 NHSM cells (Lis45_N); partial duplications of chromosome 17 and 19 in Lis45 primed cells (Lis45_P); a partial duplication of chromosome 1, trisomy 6 and loss of chromosome X in Lis46_N; and duplication of chromosome X in Lis46_P (Figures S6 and 2E). Whereas CMA is a widely used method for detecting large chromosomal aberrations in clinical samples, WGS provides higher resolution and the ability to detect mosaic populations of cells, some of which contain a given genetic aberration, and others that do not. WGS confirmed the majority of the CMA results (monosomy X in late Lis45_N (female line), trisomy X in late Lis46_P (male line), normal single X in early Lis46_N (male), normal diploid chr19 in early and late Lis45_N, and normal diploid chr17 in early and late Lis45_N; Figure 2E). Moreover, WGS showed that some of the aberrations identified by CMA were, in fact, mosaic (monosomy X in late Lis46_N, partial chr19 duplication in late Lis46_P, chr17 duplication in late Lis46_P and trisomy 6 in late Lis46_N). In other cases, loci that were called normal by CMA were shown to have copy number alterations by WGS (mosaic monosomy X in early Lis45_N (female), mosaic trisomy X in late Lis45_P (female), a small duplication in chr6 in early Lis46_N, a mosaic duplication in chr6 in late Lis46_P, and a small duplication in chr1 in early Lis46_N). In one case, WGS detected a smaller aberration than was found by CMA (1 MB duplication in chr1 in late Lis46_N) (Figure 2E). Overall, a variety of chromosomal and large subchromosomal aberrations arose during extended culture in both NHSM and primed conditions, with the only pattern being a trend toward loss of the X chromosome in NHSM cultures and gain of the X chromosome in primed cultures.
Whole genome sequencing
Genetic stability was then analyzed in a higher resolution by performing WGS in order to compare de-novo CNVs and SNVs that could not be detected by CMA and arise in NHSM and isogenic primed hESCs over time in culture. As we had access to DNA samples of the donor parents, inherited genetic variants could be filtered out and further analysis could focus only on genetic variants that arose de-novo during hESC derivation and culture. CNV analysis revealed the presence of de-novo sub-chromosomal CNVs in both substates, with the primed cells having twice as many CNVs compared with the NHSM. Mapping the total amount of aberrations, the NHSM cultures tended to accumulate deletions over time, whereas the primed cultures tended to acquire duplications (Figures 4A and 4B). However, the total number of de novo CNVs varied markedly among hESC lines, with the Lis45 and Lis46 lines showing higher numbers of CNVs than Lis38 and Lis39. Most CNVs were found to be located in non-coding regions (315, 95.5%). Of the 15 CNVs that were found to be located in coding regions, analysis of RNA-seq data revealed that two of them were associated with differentially expressed genes (DEGs). Both CNVs were duplications on chromosome 16 in the same primed line (Lis46_P), and the corresponding upregulated gene was the autocrine motility factor receptor (AMFR). Notably, AMFR was significantly upregulated only in Lis 46_P compared to its NHSM counterparts, and not compared to the other primed cultures.
Figure 4.

CNVs and SNVs arising over time in culture and in regulatory regions
(A) Stacked bar chart shows the number of deletions and duplications arising during time in culture by whole genome sequencing.
(B) CNVs arising over time in culture. Genomic locations of duplications (green) and deletions (red) in late passage cultures. CNVs located within enhancer (C) or super-enhancer (D) regions in each line, including duplications (green) and deletions (red) with the respective DEG detected by RNA-seq. See also Table S6.
(E) SNVs located within enhancer (dark gray) or super-enhancer (light gray) regions had no transcriptional effect.
Next, we studied de-novo single nucleotide variations (SNVs). Our results showed that less than 50 SNVs arose during derivation or early culture, and most of these were not seen in the corresponding late passage cultures (Figure S7A). During long-term culture, Lis38 and Lis39 acquired approximately 60–90 additional SNVs, with no significant differences between the number of SNVs in late NHSM and isogenic primed cultures, whereas in Lis45 and Lis46, substantially more SNVs were observed in primed cells compared with late NHSM hESCs (>100 vs. <50 respectively; Figure S7A). Mapping the SNVs, we found that all were located in non-coding regions, mostly in introns and intergenic regions (Figure S7B).
Apart from mutations in the coding sequences that may affect the encoded protein structurally and functionally, genetic variants located in regulatory regions can also alter gene expression levels. Gene expression levels are modulated by cis-regulatory elements, such as enhancers, which regulate spatiotemporal expression of target genes by transcription factors binding, and super-enhancers that consist of clusters of enhancers. As the majority of CNVs and all SNVs observed at late passage were located in non-coding regions, we investigated whether they were associated with regulatory regions, particularly in light of a recent publication that reported that naive and primed hESCs can be distinguished by unique sets of enhancers and super-enhancers.51 We enumerated the number of CNVs in naive and primed substate-associated enhancers/super-enhancers in our late NHSM and primed cultures, respectively. We found that the number of CNVs in these regulatory regions was larger in the primed cells compared with their NHSM counterparts in all four lines, both within enhancers (Figure 4C) and super-enhancer regions (Figure 4D). Moreover, more SNVs were found in these enhancers and super-enhancer regions in all primed hESC lines compared with their NHSM counterparts (Figure 4E). We then analyzed the effects of these CNVs on the expression of genes that are close to the enhancer using RNA-seq and found that in three out of four lines (Lis 38, Lis 45, and Lis 46) the primed cells had more DEGs resulting from duplications or deletions within enhancer or super-enhancer regions containing CNVs (Table S6). A similar analysis for SNVs revealed that none were associated with DEGs.
We further investigated aberrations detected in enhancers and super-enhancers of cancer-promoting genes (classified as Tier1 in The Cancer Gene census of COSMIC v86 database). We found no CNVs in regulatory regions of Tier1 genes in the late NHSM cultures but found one in the enhancer region of the tumor suppressor FBXW7 in Lis39_P (Table S6). Not only was FBXW7 lower in Lis39_P compared with its isogenic NHSM counterpart, but it was also lower compared with the other late-primed cultures, in which expression was higher in the late primed compared with the late NHSM cultures (Figure S8).
We also explored whether our RNA-seq data could be used to identify SNVs present in mosaic populations of cells. In this analysis, we identified only two SNVs in transcripts from COSMIC Tier1 genes, which were found at low allelic fractions in two of the late-passage cultures: one SNV in POU5F1 in late Lis39_N and the other SNV in COL1A1 in late Lis39_P. Given that our culture conditions included MEFs, we examined these SNVs and determined that they corresponded to the murine sequences for these transcripts and thus can be attributed to a low level of contamination by MEFs.
Long-term culture has differential effects on the transcriptomes of hESCs in different substates
We next sought to explore the transcriptomic alterations that are associated with long-term NHSM culture conditions, by performing differential mRNA expression analysis comparing cultures from all time points. We identified 177 mRNAs that were specifically upregulated in derivation cultures compared with both early and late NHSM cells (adj. p-value < 0.05, log2 FoldChange > 1). These mRNAs were significantly enriched for gene ontology (GO) biological processes relating to the regulation of DNA transcription and pathway-restricted SMAD protein phosphorylation, including the previously reported naive markers NODAL and LEFTY224 (Table S7). The classical pluripotency markers OCT4/POU5F1 and NANOG, the pre-implantation embryo-associated gene TEAD4, and the naive marker KLF4 were also upregulated in derivation cultures. Using the same strategy, we identified 157 mRNAs that were downregulated in the early NHSM cultures but not in early primed cells. These mRNAs were significantly enriched for GO terms relating to development, differentiation, and negative regulation of both the Wnt and insulin-like growth factor receptor signaling pathways, consistent with their more naive state (Figure S9A and Table S8). These results indicate the loss of several previously reported transcriptomic hallmarks of naive pluripotency over time in culture in our NHSM cultures.
Next, because miRNAs are known to play important roles in the regulation of gene expression, and have recently been reported to influence human naive pluripotency,19,49,52 we performed an integrative analysis of miRNAs and mRNAs differentially expressed in NHSM cultures over time in culture. We first used STRING to create separate networks of mRNAs that were differentially expressed during prolonged culture and performed clustering to highlight closely related genes (networks in Figures S9B and S9C). The largest subcluster of genes that showed mRNA downregulation in the derivation cultures compared with early/late NHSM cultures (Figure S9B network) was highly enriched for the Hippo, TGF-beta, Wnt, and Hedgehog signaling pathways (Figure S9B table), all of which have been shown to be important in regulating the naive state, although TGF-beta is reported to be critical for both the naive and primed state25,26,52,53). Using hierarchical clustering to identify the miRNAs with the highest composite importance scores across the genes in the subcluster (Figure S9B heatmap), we applied miRPathDB 2.054 and found that the majority of these miRNAs significantly (p-value < 0.05) inhibited pathways governing naive pluripotency (Figure S9B table). The largest subcluster for the genes that showed mRNA upregulation in the derivation cultures compared with early/late NHSM cultures (Figure S9C network) was highly enriched for pathways related to pluripotency, namely the TGF-beta and PI3K-Akt signaling pathways,55,56,57 as well as pathways associated with cancer (Figure S9C network). Interestingly, 5 of the 18 miRNAs that had the highest importance scores for these two subclusters (4 out of 13 downregulated and 1 out of 5 upregulated miRNAs in the derivation cultures) are encoded by the same miRNA cluster on chromosome 7 (Figure S9D). The DNA methylation levels of the CpG islands in this region of chromosome 7 were lower for the derivation samples compared with the early and late NHSM and primed samples, although statistically significantly lower for only two of the four hESC lines (p-value < 0.05, Mann–Whitney U test) (Figure S9D). Moreover, 7 of these highest importance 18 miRNAs are included in the miRPathDB database, and 6 of these 7 were found to have highly similar sets of target genes54 (Table S9). Consistent with prior studies, we found that several let-7 family miRNAs were downregulated in derivation cultures compared with later passage cultures .49,52,58 However, the miR-302/miR-371-373 clusters, previously reported to be associated with the transition from naive to primed substates,49,52,58 were not differentially expressed between our early and late NHSM cultures. Collectively, this miRNA/mRNA integrated analyses suggest that miRNAs clustered on chromosome 7 and in the let-7 family, help regulate gene expression programs in our more naive cultures. Interrogation with DNA methylation data suggests that at least one of these miRNA clusters may be epigenetically regulated.
We next focused on studying differences in miRNA expression between NHSM and primed cultures and compared the results with recently reported human naive and primed small RNA-seq data.19,49,52 For this, differential expression analysis was performed between derivation cultures and late primed cultures and the results show that of the 72 miRNAs that were more highly expressed in derivation cultures, 20 (28%) were previously reported to be consistently upregulated in naive lines, and of the 74 miRNAs that were upregulated in the late passage primed cultures, 10 (14%) were previously reported to be consistently upregulated in primed lines (Table S10 and Figure 5A; in blue miRNAs previously reported). Notably, similar to our comparison between different time points of NHSM cultures, the previously reported naïve-associated miR-371-373 cluster was not upregulated in derivation cultures compared with late-primed cultures. To link changes between the differentially expressed miRNAs and mRNAs that might regulate the transition between substates, we combined the list of differentially expressed miRNAs with that of differentially expressed mRNAs using mirTarBase, a database of experimentally validated miRNA-target gene relations.59 GO enrichment analysis was performed on the 1,235 downregulated genes that were also targeted by a miRNA that was upregulated in the derivation cultures. The resulting enriched GO terms were then grouped into larger categories using Revigo and CirGO.60,61 The largest group of enriched GO terms was signaling pathways, and included numerous pluripotency-associated signaling pathways, including the MAPK cascade, and the Wnt, ERK, and TGF beta pathways62 (Figure 5B and Table S11). Notably, the TGF beta pathway was recently reported to be required to maintain the naive pluripotent state.63 Consistent with Wang (2019), we found that the Hedgehog Signaling pathway was significantly enriched (adj. p-value = 0.02, Wikipathway 2021) in our p20 derivation cultures, with the ciliary G-protein coupled receptor GPR161 targeted by hsa-miR-301a-3p, reported repressing the Hedgehog pathway in the primed substate, being upregulated in our primed cultures.52 Interestingly, the second and fourth largest GO term groups were the related cell migration and extracellular matrix organization term groups, respectively (Figure 5B). Recent reports have shown that membrane mechanics and cell migration may have important regulatory roles in the exit from naive pluripotency and help explain morphological differences between the naive and primed substates.64 The third largest group of GO terms related to cell proliferation, suggesting that miRNAs might be involved in regulating the doubling time and cell cycle differences that were noted between our substates (Figures 2A, 2B, and 5B).
Figure 5.

miRNA differences between our NHSM and primed cultures
(A) Differential expression analysis between our NHSM and primed cultures. Volcano plot shows miRNAs significantly different (adj. p-value < 0.05, log2 fold change > |1|, basemean > 10) between our derivation timepoint samples (p20) and our late (p50) primed cultures. The dotted lines represent a log2 fold change of |1| and an adjusted p-value of 0.05. Grey dots are not significant miRNAs, blue dots are miRNAs that have an adj. p-value < 0.05 but not a log2 fold change > |1|, green dots have a log2 fold change > |1| but not an adj. p-value < 0.05, and red dots are those miRNAs considered significantly different by both metrics. miRNAs in blue font are those that were previously reported to be differentially expressed between NHSM and primed lines.
(B) Gene ontology (GO) enrichment analysis of downregulated genes (adj. p-value < 0.05, baseMean > 50) in the derivation timepoint that were also targeted by an miRNA upregulated in the derivation cultures. Revigo and CirGO were used to provide a non-redundant and biologically succinct visualization of GO terms with an adjusted p-value < 0.05.
(C) Genomic locations of miRNA clusters containing multiple miRNAs upregulated in derivation cultures (p20).
(D) Boxplots displaying methylation levels of the regions containing the chromosome 14 and chromosome X miRNA clusters in (C). Boxes span the top 75th percentile to bottom 25th percentile with the line in the middle of the boxes representing the median. Whiskers contain 95% of the data. “∗” represents a significant difference between cultures (p-value < 0.05 Mann–Whitney U).
Finally, we noted that there were three genomic locations that contained multiple NHSM upregulated miRNAs: one miRNA cluster on chromosome 14 q32 in the pluripotency-associated DLK1-DIO3 locus, which contained 17 of the miRNAs that were upregulated in the p20 derivation cultures, and whose 167 target genes were enriched for the miRNA-regulated DNA damage repair pathway (adj. p-value = 0.002); another miRNA cluster on the X chromosome, which contained six p20 upregulated miRNAs, and whose 71 target genes were enriched for regulation of cell migration (adj. p-value = 0.01); and a third smaller cluster on Chr.7 (analysis performed using miRTarBase) (Figure 5C). Both the chr14 and chrX loci are known to be epigenetically regulated (imprinted and X-linked, respectively). We therefore analyzed the DNA methylation in the CpG island regions associated with the miRNAs in these two clusters. Consistent with a pattern of downregulation of gene expression by DNA methylation, we found that the methylation levels of the early NHSM cultures for Lis39 and Lis45 (the two female lines) were markedly lower than their late corresponding primed cultures, whereas the p20 derivation cultures for Lis38 and Lis46 (the two male lines) had higher methylation levels (Figure 5D). These results suggest that mRNA regulation by miRNAs, which are themselves regulated by DNA methylation, plays a role in the establishment and maintenance of substates of pluripotency and may contribute to the phenotypic differences seen in our cultures.
Genome-wide DNA methylation analysis reveals alterations that differ by pluripotency state and time in culture
Recent studies have reported that, similar to human preimplantation embryos,45,50,65,66 the human naive stem cell state is characterized by genome-wide DNA hypomethylation.20,65,67 As a result, previous studies have reported a loss of DNA methylation and biallelic expression at imprinting sites in the naive substate and a failure to re-establish DNA methylation at these sites upon culturing in primed conditions.45 Furthermore, loss of DNA methylation and imprinting have been linked to genomic instability.45,68 We therefore analyzed DNA methylation levels at regions associated with single-isoform imprinted genes that were differentially methylated among naive and primed cultures according to a previous report.69 We found that our NHSM cultures were modestly hypomethylated compared with the corresponding primed cultures, but that there was high variability between hESC lines, culture conditions, and passage numbers (Figure S10A). Moreover, unlike previously reported 5i naive lines,45,65 several imprinted loci that were unmethylated in our derivation (p20) cultures converted to a hemi-methylated status after the transition to a primed state, and in some cases also after extended time in culture in our NHSM conditions (e.g., MAGEL2, SNRPN, FAM50B). With the exception of a handful of loci (such as GNAS 1A and ZDBF2, which were hypomethylated and hypermethylated in nearly all cultures, respectively), our primed cultures were hemimethylated at nearly all of the evaluated imprinted loci. In contrast, the later passage NHSM cultures displayed sporadic losses of DNA methylation at imprinted loci, suggesting greater epigenetic stability at imprinted loci in our primed cultures.
X chromosome inactivation (XCI) is characterized by methylation of XCI-linked CpG islands (CGI) on one of the two X chromosomes in female somatic cells.70 Thus, female somatic cells are hemimethylated at XCI-linked CGIs, whereas male somatic cells are unmethylated at these loci. Previous reports have shown that both female preimplantation embryos and female human naive hPSCs contain two actively transcribed X chromosomes65,71,72 and are unmethylated at XCI-linked CGIs. On the other hand, female primed lines typically contain one active X chromosome and have hemimethylated XCI-linked CGIs. We assessed the methylation at the CGIs of XCI-linked, non-XCI-linked X chromosome, and autosomal genes in our cultures (Figure S10B). As expected, the male lines were largely unmethylated on the X chromosome. For the two female lines (Lis39 and Lis45), we saw similar DNA methylation levels between the XCI-linked and non-XCI-linked CGIs for both NHSM and primed cultures. Also in the female lines, the derivation cultures showed slightly lower levels of DNA methylation on the X chromosome compared with the early and late NHSM and primed cultures, which all had similar X chromosome methylation levels with the exception of the late Lis45 cultures, which were essentially unmethylated on the X chromosome, consistent with the X chromosome deletion seen in the CMA and WGS data (Figure 4B). The median beta values in all of the NHSM female cultures (except for late passage Lis45) were close to 0.3–0.6, consistent with a largely hemimethylated state. Despite this somewhat unexpected hemimethylated pattern, the inspection of the RNA-seq data showed that NHSM cultures of both female hESC lines showed biallelic expression of XCI-linked genes (Table S12), suggesting that they contain two active X chromosomes. Interestingly, only Lis45 primed cultures showed unexpected biallelic expression of XCI-linked genes.
As our NHSM cultures were only slightly hypomethylated compared with their corresponding isogenic primed cultures (Figure S5B), we examined which loci were consistently differentially methylated between our NHSM and primed cultures. We found there were 674 loci, associated with 307 genes, which were consistently hypomethylated in our early NHSM cultures compared with their matched primed cultures and only 81 loci, associated with 47 genes, which were hypomethylated in the early primed cultures compared with their matched NHSM cultures (Figure 6A). A similar analysis in our late cultures revealed 1,124 (462 genes) hypomethylated loci in our NHSM cultures compared with just 307 (157 genes) in our primed cultures (Figure 6A). PCA using either the early or late passage differentially methylated sites separated the NHSM and primed samples along PC1 (Figure 6B). We also noted that for three of the four hESC lines, the derivation cultures (indicated by “∗”) separated from their early and late passage NHSM counterparts along PC2 (Figure 6B). This suggested to us that, in contrast to mRNA expression changes (Figure 3A), DNA methylation changes resulting from time in culture occurred predominantly during early culture and stabilized by passage 30.
Figure 6.

Methylation differences between NHSM and primed cultures
(A) Venn diagrams showing differential methylation, defined as a beta difference (methylated/(methylated + unmethylated)) of at least |0.3| in all four of the paired NHSM/primed comparisons, between early and late passage NHSM and primed pairs. Venn diagrams show that NHSM cultures contain more differentially hypomethylated probes than primed cultures and that there are more differentially methylated probes in the late passage comparisons compared with the early passage comparisons.
(B) Principal component analysis using either the early or late passage differentially methylated probes from Figure 6A. The “∗” shows the location of the derivation (p20) NHSM culture samples.
To identify functional DNA methylation changes occurring during the early period after derivation, we focused on the three hESC lines that showed separation along PC2 between derivation cultures and early/late cultures (Lis38, Lis39, and Lis45) in Figure 6B. We identified loci with a >|0.3β| difference between both the matched derivation (p20) vs early NHSM (p30) and derivation (p20) vs late NHSM (p50) cultures for these three lines. We found that there were markedly more sites that were hypomethylated in the derivation cultures than in the later cultures, with 4,682 sites showing lower DNA methylation levels in derivation cultures and 1,956 sites showing lower DNA methylation levels in early/late cultures (Figure 7A).
Figure 7.

Methylation differences between NHSM cultures due to time in culture
(A) Venn diagrams showing methylation differences between derivation (p20) cultures and both early (p30) and late (p50) NHSM cultures. Lis46 was removed from the analysis (Figure 6B).
(B) Heatmap showing the gene expression of genes that were both differentially hypomethylated (see A) and were differentially upregulated (log2 fold change > 1) at the derivation (p20) timepoint when compared with both the early (p30) and late (p50) NHSM timepoints.
(C) Scatterplot showing the correlation between methylation levels and expression levels of all genes associated with probes that were hypomethylated (beta difference (methylated/(methylated + unmethylated)) of at least |0.3|) in derivation cultures (p20) compared with late cultures (p50). The red dots are genes that are both differentially hypomethylated (see A) and were differentially upregulated (log2 fold change > 1) at the derivation (p20) timepoint when compared with both the early (p30) and late (p50) NHSM timepoints.
To find functional differences related to the differentially methylated sites, we identified 1,581 mRNAs that were differentially expressed (log2 fold Change > |1|) between both the matched derivation (p20) vs early NHSM (p30) and derivation (p20) vs late NHSM (p50) cultures for these three lines, of which 57 were associated with differentially methylated loci: 38 genes showed low DNA methylation and high gene expression at the derivation timepoint compared with early/late; 8 showed high DNA methylation and low gene expression at derivation; 10 showed low DNA methylation and low gene expression at derivation; and 1 gene that showed high DNA methylation and high gene expression at derivation. Thus, the largest group of genes that showed both differential DNA methylation and gene expression was hypomethylated and upregulated in the derivation cultures, and this group contained several previously identified naive marker genes, including LEFTY2 and DPPA3, as well as TRIM61, a gene closely related to the previously reported naive marker gene TRIM6027,47 (Figures 7B and 7C).
Gene enrichment analysis of the genes that were hypomethylated and showed higher expression in the derivation cultures compared with the early/late cultures revealed significant enrichments for pathways relating to NODAL and TGF-beta signaling, which have been associated with naive pluripotency,24,27 SMAD protein phosphorylation and BMP signaling pathway, which have been reported to be important in the acquisition of multi-lineage competence in cells departing naive pluripotency,73 and maintenance of pluripotency (SMAD)74 (Figure S11). Taken together, our results showed that the early NHSM cultures were modestly hypomethylated compared with their primed counterparts, and an increase in DNA methylation in the NHSM cultures over time, with the largest changes between derivation and early passage and largely stable DNA methylation thereafter. Integrative DNA methylation/RNA-seq analysis showed that increased DNA methylation over time in culture was correlated with decreased expression of naïve-associated genes.
Discussion
The potential therapeutic utility of hPSCs and their ability to recapitulate and model early development depends on a thorough understanding of both their pluripotent substate and stability. This is the first study to quantitatively and systematically compare the cellular phenotype and genetic, epigenetic, and transcriptional changes in isogenic hESCs of different substates across time from derivation through a long-term passage. In contrast to the large majority of previous studies on naive hPSCs that were established in primed conditions and only later converted to naive, in this study all four hESC lines were derived in NHSM conditions, and then each line was split and subjected to long-term culture in NHSM and primed conditions in parallel, with longitudinal samples collected for analysis. The analysis included phenotypic and genomic comparisons between matched cultures, at different substates of pluripotency and at different time points: derivation, early and late passages. Our unique access to blastocyst-stage preimplantation embryos, as well as to their parental DNA enabled definitive identification of both de novo genetic aberrations and informative loci for evaluation of allelic expression.
Consistent with prior studies, our early passage NHSM cultures displayed expression of naïve-associated marker genes and typical characteristics of naive hPSCs, such as higher proliferation and clonogenicity compared with isogenic matched primed cultures. The proliferation of the NHSM cells remained stable from the early to late passage, and the clonogenicity of both NHSM and primed cultures was stable as well, but for the primed cultures cell proliferation increased across time. These results suggest that cell proliferation and survival are at least partially separable processes for hPSCs. This is consistent with previous studies that have shown that although certain signaling pathways, such as PI3K, TGF-β, and MAPK, regulate both proliferation and survival in hESCs,75,76,77 other pathways such as Rho-ROCK and Bcl-2 signaling, regulate cell survival by blocking apoptosis without influencing cell proliferation.78,79,80
Given the strong associations between genetic instability and cancer, it is crucial to ensure the genetic integrity of cells designated for clinical use. It has been shown that primed hESCs accumulate genomic aberrations throughout derivation and culture,33,36,81,82 and that culture conditions such as passaging method, presence of a feeder layer, media type, and time in culture can influence the rate that mutations appear.35,37,39,40,41,83,84,85,86,87,88,89,90 Previous studies that compared the genetic stability of naive and primed hESCs have reported conflicting results, with some reporting a higher frequency of genetic changes in naive cells,21,27,45,46 whereas others showed the opposite.14,24,91,92,93 To our knowledge, a well-controlled molecular examination of genetic stability in different pluripotent substates has not been previously reported. To address this gap in the literature, we performed a detailed genomic comparison between isogenic hESCs in different substates. Karyotype and CMA analyses of early NHSM and their primed counterparts were normal, whereas WGS revealed several small aberrations in predominantly non-coding regions. At late passage, we found no significant differences between the numbers of large or small genetic aberrations between the genetic stability of NHSM and primed cultures. Overall, the only patterns observed were a trend toward small and large copy number losses (including small deletions and loss of the X chromosome) in late NHSM cultures and copy number gains (including small duplications and gain of the X chromosome) in late primed cultures. These tendencies are interesting given prior studies showing that X chromosome duplications are common in hPSCs cultured in a variety of primed conditions,35,37,81,94 and that deletions are more common during derivation, whereas duplications are more common during extended culture in primed conditions.95
Many of the aberrations found in the cultures of either substate were located in loci that have been previously reported to be frequently mutated in primed hESCs, such as chromosomes 1,35,37,81 17,33,35,37,81,88 and X.35,37,81,96 However, only 15 of the many identified CNVs were found to be located in coding regions, and of these, the only two associated with differential expression of an associated transcript were duplications linked to upregulation of the same gene, AMFR. Overexpression of AMFR has been observed in several types of human cancer and its expression has been found to be correlated with more advanced tumor stages and decreased survival rates.97,98,99,100,101
The fact that the vast majority of the observed aberrations in both substates were located in non-coding regions suggested that cell proliferation or survival advantages conferred by genetic aberrations might be due to disruption of gene regulation, rather than altering protein-coding sequences. We found that our primed hESC cultures had more aberrations in previously reported primed substate-associated enhancer and super-enhancer regions, compared with the number of aberrations in naive substate-associated regions in our late NHSM cultures.51 Several of the CNVs, but none of the SNVs, in these regulatory regions were associated with alterations in gene expression. Moreover, primed cultures possessed more DEG-associated CNVs than their more naive counterparts. Looking specifically at COSMIC Tier1 cancer-related genes, a deletion in the enhancer of the tumor suppressor gene FBXW7102,103 was found in one of the late passage primed cultures and was correlated with decreased expression compared with both the corresponding NHSM culture and the other primed cultures; interestingly, the other three hESC lines showed the opposite pattern of FBXW7 expression (i.e., higher in the late primed cultures compared with the late NHSM cultures).
In addition to variant calling using WGS, we performed variant calling on RNA-seq data to detect SNVs of low allelic fraction in transcribed regions. A similar approach was used in a previous study, which reported that hESCs that were derived in primed conditions and converted to naive at later passages acquired more SNV mutations in COSMIC Tier1 genes, compared with cells that were kept in primed conditions;104 these results were subsequently found to be likely an artifact of contamination of hPSC lines by mouse embryonic fibroblast (MEF) feeder cells.105 In our cultures, this strategy revealed only two SNVs in COSMIC Tier1 genes, both of which were attributed to low levels of MEF contamination.
As the phenotypic changes observed in our late passage cultures could not be explained by genetic changes alone, we further explored the possibility that some differences might be the result of culture adaptations driven by epigenetic and/or transcriptomic changes, which has previously been reported in hESCs,106 mESCs,107 primary mammalian cell lines,108,109 and human cancer cells.110 Our NHSM and primed cultures exhibited differential expression of mRNAs and miRNAs, as well as differences in DNA methylation. Both the transcriptional and DNA methylation profiles of our NHSM lines showed that they are more similar to recently reported intermediate pluripotent cells29 than to the majority of naive hPSCs. Interestingly, the DNA methylation profiles of our NHSM cultures were markedly different from hPSCs converted to naive pluripotency using 5i/t2iL + PKCi-like protocols,20,29,65,67 but were quite similar to those derived in NHSM media,14,45 which is more similar to the media used in our study. Most of the p20 NHSM hESC lines cultured in primed conditions displayed a rapid transition to a primed-like DNA methylation signature, including modestly increased global methylation and strong increases in DNA methylation in known naive pluripotency-associated genes and imprinted regions. Over time in culture, even the cultures maintained in NHSM conditions experienced epigenetic drift toward a more primed-like substate. The only line (Lis46) that did not undergo the same rapid increase in DNA methylation after transitioning to primed conditions already displayed a higher degree of DNA methylation at early passage compared with the other lines, suggesting that it might have either a genetic predisposition toward higher DNA methylation or undergone a transient environmental exposure that resulted in a premature increase in DNA methylation. Similarly, our mRNA and miRNA profiling results, particularly when examined in the context of previously published naive and primed hPSC datasets (Figures 2B and S6A), suggest that our derivation (p20) hESC lines drift toward a more primed-like substate over time in culture. By passage 30, our NHSM cultures appear to stabilize in a slightly more naive state than the recently described intermediate substate.29
Based on these results, we suggest that molecular profiling can be used to distinguish between distinct substates that exist along the pluripotency continuum. Rather than any one of these substates being universally “better” than the others, the ability to stabilize hPSCs in a range of substates might be advantageous by providing researchers with multiple developmental options depending on their desired applications. In our previous study, using cultures generated using a similar NHSM media to the one used here, the more naive cells were able to form interspecies chimeras.14 Recent reports suggest that the hPSCs in the naive substate show little to no chimeric contribution and may differentiate to trophoblast more readily than primed hPSCs,111 but may not efficiently differentiate into mesoderm and endoderm.30 However, considering the epigenetic and transcriptional drift of our cultures over time, we would be interested to know if both early and late passage NHSM lines show a high differentiation potential and are therefore functionally closer to a formative-like substate of pluripotency. We speculate that at very early passage, NHSM-derived hPSCs may display molecular profiles that are more similar to hPSCs cultured in 5i and t2iL + PKCi media, and also possess differentiation capacities that are more similar to the ICM of the blastocyst,20,48,65,67 whereas late passage NHSM hPSCs and hPSCs that have been transitioned to primed culture conditions may represent embryonic cells at a later stage of development. Clearly, additional research using larger numbers of cell lines, cultured in a variety of media and analyzed longitudinally over extended time in culture to determine their differentiation capacities and perform molecular profiling, will be required to test these hypotheses and determine whether it is possible to stabilize hPSCs in different in vitro pluripotent substates.
The findings from this study expand our knowledge of the human pluripotency continuum and have important implications for regenerative medicine. The higher proliferation rates, clonogenic potential, and lack of culture- and conversion-induced genetic defects of early passage hESCs derived in NHSM make them attractive for therapeutic purposes. However, we note that the efficacy and safety of hPSCs for clinical use largely depends on their potential to differentiate efficiently and uniformly into different types of functional cells, to optimize functionality and minimize the potential for continued proliferation and/or malignancy. Therefore, the ideal substate of pluripotency of source cells may not be the same for different target lineages.
Limitations of the study
Given that this work included a relatively small sample size of four cell lines from two sets of gamete donors that were all derived and cultured in the NHSM conditions, it is unknown whether our conclusions would be applicable to a larger number of cell lines, cell lines that are more genetically diverse, or to cell lines cultured in conditions that favor a more naive state. As shown in this study, the cell lines cultured in NHSM in the present work are consistent with characteristics of a pluripotency state in-between a naïve and primed substate. Cells lines derived and cultured in an analogous manner (NHSM) to the present lines can contribute to interspecies chimaeras. To allow for this study to be mapped to previously published studies, future experiments that determined when in culture this property is established should be performed. Additionally, molecular profiling experiments alongside functional experiments such as differentiation potential experiments of early and late passage NHSM cultures would help assess how similar the present lines are to bona fide naïve human stem cells and the intermediate pluripotent substate. Likewise, a more thorough investigation into the present cells X-chromosome status using RNA FISH for XIST and XACT along with deeper WGS/RNA-seq for biallelic expression determination, would better determine where the present cells sit on the pluripotency continuum. Lastly, our sample size was not large enough to draw definitive conclusions regarding differences in genetic stability between different substates. Inconsistencies between our observations and those from previous reports may result from differences in the origin of the cells, the conversion technique, the specific culture media compositions, or the methods used for genetic analyses. Our study used single base-pair resolution sequencing of both the whole genome and the whole mRNA transcriptome of hESCs derived directly in NHSM conditions that were subjected to long-term culture in NHSM or primed conditions. Our culture conditions include a version of the NHSM that contains PD0325901, an MEK inhibitor not found in primed medium and recently reported to be associated with genetic instability of naïve cells.46 It is important to note prior studies that suggest that other components of naive culture media can also promote genetic instability.16,21,24,92,112 To distinguish between the effects of the directionality of the conversion process (primed toward naive vs naive toward primed), specific culture conditions, and genomic methods, future studies using larger numbers of isogenic lines, with some derived in parallel in naive conditions and others derived in primed conditions, each of which is then split into parallel naive and primed cultures, with parental genomes available, would need to be done.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| mouse anti-OCT4 | Santa-Cruz | sc-5279 |
| mouse anti-SSEA4 | cell signaling technology | CST-4755S |
| mouse anti-TRA-1-60 | Abcam | ab16288 |
| rabbit anti-TFE3 | Sigma-Aldrich | HPA023881 |
| Biological samples | ||
| Parental DNA/RNA for Lis38/39 and Lis45/4 | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Neurobasal medium | Gibco | 21103-049 |
| DMEM/F-12 | Gibco | 21331-020 |
| Pen/Strep | Biological industries | 03-033-1B |
| L-Glutamine | Biological industries | 03-020-1B |
| 1% non-essential amino acids | Biological industries | 01-340-1B |
| sodium pyruvate | Biological industries | 03-042-1B |
| B27 supplement | This paper | N/A |
| 0.2% ml defined lipid concentrate | Gibco | 11905-031 |
| β-ME | Gibco | 31350-010 |
| Insulin | Sigma-Aldrich | I-1882 |
| apo-transferrin | Sigma-Aldrich | T-1147 |
| progesterone | Sigma-Aldrich | P8783 |
| Putrescine | Sigma-Aldrich | P5780 |
| sodium selenite | Sigma-Aldrich | S5261 |
| L-ascorbic acid 2-phosphate | Sigma-Aldrich | A8960 |
| BSA | Gibco | 1526-037 |
| Human LIF | Peprotech | 300-05 |
| IWR1 | Biotest | 3532 |
| human activin A | Peprotech | 120-14E |
| FGF2 | Peprotech | 100-18B |
| Chir99021 | Axon Medchem | 1386 |
| PD0325901 | Axon Medchem | 1408 |
| BIRB796 | Axon Medchem | 1358 |
| SP600125 | Tocris | 1496 |
| PKCi | Tocris | 2285 |
| Thiazovivin | Peprotech | 1535 |
| CGP77675 | Axon Medchem | 2097 |
| 20% Knockout Serum Replacement | Gibco | 10828028 |
| trypsin + EDTA | Biological industries | 03-053-1B |
| Y27632 ROCK inhibitor | Axon Medchem | 1683 |
| Triton X-100 | Sigma-Aldrich | X100 |
| FxCycle Violet | Invitrogen | F10347 |
| Colcemid | Biological industries | 12-004-1D |
| Ampure XP beads | Beckman Coulter | A63881 |
| Critical commercial assays | ||
| RNeasy mini kit | Qiagen | 74104 |
| Superscript IV RT-PCR kit | Invitrogen | 18091050 |
| FAST SYBR Green Master Mix | Quanta bio | 95072-012 |
| Click-iT EdU Alexa Fluor 647 Flow Cytometry Kit | Life Technologies | C10420 |
| AP staining kit | Stemgent | 00-0055 |
| DNeasy Blood & Tissue Kit | Qiagen | 20-69504 |
| Kapa HyperPlus Kit | Roche | KK8510 |
| Qubit dsDNA BR | ThermoFisher Scientific | Q32853 |
| Agilent DNA 7500 Kit | Agilent | 5067-1506 |
| mirVana miRNA Isolation Kit | ThermoFisher Scientific | AM1560 |
| KAPA mRNA HyperPrep Kit | Roche | KK8581 |
| RNA 6000 Nano Kit | Agilent | 5067-1511 |
| NEBNext Multiplex Small RNA Library Prep Set for Illumina | New England Biolabs, Inc. | E7300L |
| Deposited data | ||
| Whole genome sequencing (PRJNA859118) | This paper | https://www.ncbi.nlm.nih.gov/ |
| RNA-seq (GSE208300) | This paper | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE208300 |
| Small RNA-seq (GSE208301) | This paper | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE208301 |
| Methylation array data (GSE208299) | This paper | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE208299 |
| Experimental models: Cell lines | ||
| hESC lines Lis38 and Lis39 | Tel-Aviv Sourasky Medical Center | N/A |
| hESC lines Lis45 and Lis46 | Tel-Aviv Sourasky Medical Center | N/A |
| hESC line WA09 (H9) | University of Wisconsin | WiCell |
| hESC line WIBR3 | Whitehead Institute for Biomedical Research | N/A |
| MEF cells – DR4 on ICR background | This paper | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: NSG | Harlan Ltd. | N/A |
| Oligonucleotides | ||
| Primer: ACTIN Forward: CCACGAAACTAC CTTCAACTCC Reverse: GTGATCTCCTTC TGCATCCTGT |
This paper | N/A |
| Primer: STELLA Forward: CGCATGA AAGAAGACCAACAAACAA Reverse: TTAGACACGCAGAAACTGCAGGGA |
This paper | N/A |
| Primer: TFCP2L1 Forward: TCCTTCTTTA GAGGAGAAGC Reverse: ACCAACGTTG ACTGTAATTC |
This paper | N/A |
| Primer: KLF17 Forward: AGCAAGAGATG ACGATTTTC Reverse: GTGGGACATTATT GGGATTC |
This paper | N/A |
| Primer: KLF4 Forward: CGCTCCATTA CCAAGAGCTCAT Reverse: CACGATC GTCTTCCCCTCTTT |
This paper | N/A |
| Primer: KLF5 Forward: CACACTGGTGAAAAGCCATACAA Reverse: GCCTGTGTGCTTCCGGTAGT | This paper | N/A |
| Primer: NANOG Forward: GCAGAAGGCCTCAGCACCTA Reverse: AGGTTCCCAGTCGGGTTCA | This paper | N/A |
| Software and algorithms | ||
| BD FACSDiva Software | BD Biosciences | https://www.bdbiosciences.com/en-us/products/software/instrument-software/bd-facsdiva-softwa |
| BlueFuse Multi software | Illumina | https://www.illumina.com/clinical/clinical_informatics/bluefuse.html |
| FASTQC v.0.11.8 | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Trim Galore v. 0.4.1 | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
| Bowtie2 v.2.3.4.3 | Langmead and Salzberg,113 | https://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Picard Tools v.2.18.15 | Broad Institute | https://broadinstitute.github.io/picard/ |
| GATK4 v. 4.0.11.0 | Van der Auwera et al.,114 | https://gatk.broadinstitute.org/hc/en-us/sections/360007226651-Best-Practices-Workflows |
| ANNOVAR v. 2019Oct24 | Wang et al,115 | https://annovar.openbioinformatics.org/en/latest/ |
| vcftools v. 0.1.16 | 1000 Genomes Project | https://vcftools.github.io/index.html |
| Bedtools v. 2.25.0 | Quinlan Lab University of Utah | https://bedtools.readthedocs.io/en/latest/ |
| CNVkit v. 0.9.6 | Talevich et al.,116 | https://cnvkit.readthedocs.io/en/stable/ |
| ERDS v. 1.1 | Zhu et al.,117 | https://github.com/igm-team/ERDS |
| PURPLE v. 2.34 | Priestley et al.,118 | https://github.com/hartwigmedical/hmftools/blob/master/purple/README.md |
| Homer v. 4.10.4 | Heinz et al.,119 | http://homer.ucsd.edu/homer/ |
| STAR v. 2.7.3a | Dobin et al.,120 | https://github.com/alexdobin/STAR |
| featureCounts (subread v. 2.6.3) | Liao et al,121 | https://subread.sourceforge.net/ |
| BiomaRt v. 2.42.1 | Ensembl | http://uswest.ensembl.org/info/data/biomart/index.html |
| DESeq2 v. 1.26.0 | Love et al.,122 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| fgsea v. 1.12.0 | Korotkevich et al.,123 | https://github.com/ctlab/fgsea |
| Genboree Workbench | Rozowsky et al.,124 | https://www.genboree.org/site/ |
| Enrichr | Chen et al.,125; Kuleshov et al.,126; Xie et al.,127 | https://maayanlab.cloud/Enrichr/ |
| Revigo | Supek et al.,60 | http://revigo.irb.hr/ |
| CirGO | Kuznetsova et al.,61 | https://github.com/IrinaVKuznetsova/CirGO |
| Cytoscape v. 3.8.0 | Cytoscape Organization | https://cytoscape.org/ |
| stringApp v. 1.5.1 | Cytoscape App store | https://apps.cytoscape.org/apps/stringapp |
| Arboreto v. 0.1.5 | Moerman et al.,128 | https://github.com/aertslab/arboreto |
| miRPathDB 2.0 | Kehl et al.,54 | https://mpd.bioinf.uni-sb.de/ |
| mirTarBase (release 8.0) | Hsu et al.,129; Huang et al.,130 | https://miRTarBase.cuhk.edu.cn:443/ |
| Scanpy v. 1.3.2 | Wolf et al.,131 | https://scanpy.readthedocs.io/en/stable/ |
| Minfi v. 1.32.0 | Aryee et al.,132 | https://bioconductor.org/packages/release/bioc/html/minfi.html |
| CpGtools | Wei et al.,133 | https://github.com/liguowang/cpgtools |
| Other | ||
| IX51 inverted light microscope | Olympus | https://www.olympus-lifescience.com/en/microscopes/inverted/ |
| Rotor Gene 6000 Series | Corbett | https://www.qiagen.com/us/corbett/welcome/productinfo/ |
| BD FACSCanto II Flow Cytometer | BD Biosciences | https://www.bdbiosciences.com/en-us/products/instruments/flow-cytometers/clinical-cell-analyzers/facscanto |
| 24sure V3 microarray | Illumina | https://www.illumina.com/content/dam/illumina-marketing/documents/products/technotes/24sure-validation-tech-note-1570-2014-026.pdf |
| Agilent G2565CA scanner | Agilent | https://www.agilent.com/cs/library/usermanuals/Public/G2505-90019_ScannerC_User.pdf |
| Agilent BioAnalyzer | Agilent | https://www.agilent.com/en/product/automated-electrophoresis/bioanalyzer-systems/bioanalyzer-instrument/2100-bioanalyzer-instrument-228250 |
| NovaSeq S4 | Illumina | https://www.illumina.com/systems/sequencing-platforms/novaseq/specifications.html |
| HiSeq 4000 | Illumina | https://www.illumina.com/systems/sequencing-platforms/hiseq-3000-4000/specifications.html |
| Pippin Prep | Sage Science | https://sagescience.com/products/pippin-prep/#description2d90-f633 |
| Illumina Infinium MethylationEPIC array | Illumina | https://www.illumina.com/products/by-type/microarray-kits/infinium-methylation-epic.html |
Resource availability
Lead contact
Requests for further information, resources, and reagents should be directed to and will be fulfilled by the lead contacts, Louise Laurent (llaurent@ucsd.edu).
Materials availability
This study did not generate new unique reagents. Cell lines Lis45 and Lis46 are available upon request.
Experimental model and subject details
Cell lines
Establishment of hESCs
The use of excess IVF-derived embryos following PGD for the generation of hESCs was approved by the Israeli National Ethics Committee (7/04–043) and is in accordance with the guidelines released by the Bioethics Advisory Committee of the Israel Academy of Sciences and Humanities. Signed permissions for the use of parental genomic DNA were given by the parents, according to the protocol approved by the Israeli National Ethics Committee (0399/09). Human ESC lines were derived in our laboratory at Tel-Aviv Sourasky Medical Center as described in14 (Lis38_N & Lis39_N published in Gafni et al., 2013), and Lis 45_N and Lis 46_N. Lis38 and Lis46 are both male lines and Lis39 and Lis45 are female lines. These cell lines have not been authenticated by an outside service. Primed WA09 (H9 - female) and WIBR3 (female) hESC lines were kindly provided by WiCell, University of Wisconsin and Whitehead Institute for Biomedical Research, respectively).
Maintenance and culture of hESCs
A modification of the naive human stem cell medium (NHSM) developed by the Hanna laboratory14 included culture on MEFs, consisting of: 1:1 Neurobasal medium and DMEM/F-12 (Gibco); 1X Pen/Strep (Biological industries); 2 mM L-Glutamine (Biological industries); 1% non-essential amino acids (Biological industries); 0.7 mM sodium pyruvate (Biological industries); 2% ml B27 supplement (in house produced); 0.2% ml defined lipid concentrate (Gibco); 0.05 mM β-ME (Gibco); 12.5 μg/mL insulin (Sigma-Aldrich); 100 μg/mL apo-transferrin (Sigma-Aldrich); 0.02 μg/mL progesterone (Sigma-Aldrich); 16 μg/mL putrescine (Sigma-Aldrich); 30 μM sodium selenite (Sigma-Aldrich); 50 μg/mL L-ascorbic acid 2-phosphate (Sigma-Aldrich); 0.07% BSA (Gibco); 20 ng/mL human LIF (Peprotech); 5 μM IWR1 (Biotest); 20 ng/mL human activin A (Peprotech); 1.7 ng/mL FGF2 (Peprotech); 0.2 μM Chir99021 (Axon Medchem); 1 μM PD0325901 (Axon Medchem); 0.2 μM BIRB796 (Axon Medchem); 2 μM SP600125 (Tocris); 2 μM PKCi (Tocris); 0.4 μM Thiazovivin (Peprotech); and 1.5 μM CGP77675 (Axon Medchem). Primed conditions included culturing on mitotically inactivated mouse embryo fibroblast (MEF) feeder layers in primed hESC culture medium, consisting of: DMEM/F-12 with 20% Knockout Serum Replacement (Gibco); 2 mM L-glutamine (Biological industries); 0.1 mM β-ME (Gibco); 1% non-essential amino acids (Biological industries); 1X Pen/Strep (Biological industries) and 8 ng/mL FGF2 (Peprotech). For both NHSM and primed cultures, the medium was changed daily, passaging was performed every 3–5 days using 0.05% trypsin + EDTA (Biological industries), and 5 μM of Y27632 ROCK inhibitor (Axon Medchem) were used for 24 hr. before and after passaging. The cells were kept at 37°C. Both NHSM and primed cultures were grown without MEFs for two passages prior to collection of cells for molecular analysis.
Mouse model
NSG female mice (age 6–8 weeks; Harlan Ltd.) had free access to food and water. All procedures involving mice used in this study were performed according to institutional guidelines under approval by the Weizmann Institute IACUC (approval #00960212-3).
Method details
Quantitative RT-PCR
Total RNA was isolated using the RNeasy mini kit (Qiagen), followed by random hexamer-primed reverse transcription using the Superscript IV RT-PCR kit (Invitrogen). Quantitative Real Time PCR (qRT-PCR) was performed using the FAST SYBR Green Master Mix (Quanta bio). Cycling and analysis were performed using a Rotor Gene 6000 Series instrument (Corbett) and its dedicated data analysis software. Standard curves were performed for each gene in every run, and all PCR reactions were performed for two independent experiments with three technical replicates for each experiment. All qRT-PCR assays included no-template control (NTC) and ACTIN served as the control for normalization of target gene expression. Primer sequences are listed in Table S13. RNA from a primed WIBR3 culture was used as the reference.
Immunofluorescence
Cells were grown on Matrigel-coated glass cover slips (13 mm; Marienfeld) in 24-well plates, fixed with 4% paraformaldehyde, and incubated in blocking solution (2.5% BSA) with 0.1% Triton X-100 to enable staining of intracellular markers. Cells were incubated with primary antibody for 1 hr. at RT, washed and incubated with secondary antibodies for 1 hr. at RT, counterstained with DAPI for nuclear staining, and imaged using an Olympus IX51 inverted light microscope. The following antibodies were used at the indicated dilutions: mouse anti-OCT4 (sc-5279, Santa-Cruz, 1:60), mouse anti-SSEA4 (CST-4755S, cell signaling technology, 1:200), mouse anti-TRA-1-60 (ab16288, Abcam, 1:200) and rabbit anti-TFE3 (HPA023881, Sigma-Aldrich, 1:60).
Teratoma formation
hESCs were harvested, resuspended in their respective medium condition with 10% Matrigel and 20 uM ROCKi, and injected subcutaneously into 6–8-week-old NSG mice (Harlan Ltd.). Teratomas generally developed within 7–10 weeks and animals were sacrificed before tumor size exceeded 1.5 cm in diameter. Teratomas were dissected and prepared for conventional FFPE and H&E histology. All animal experiments were conducted according to institutional guidelines under approval by the Weizmann Institute IACUC (approval #00960212-3).
Proliferation and clonogenicity assays
Matched sets of NHSM and primed hESCs were seeded onto 6-well plates (200,000 cells/well on Matrigel coated wells). Two and four days following plating, cells were collected using trypsin, and the doubling time was calculated as duration (hours)×log(2)/(log2(final concentration)-log(initial concentration))27 using number of cells at day 4 normalized by number of cells at day 2.
Cell cycle distribution was determined using the Click-iT EdU Alexa Fluor 647 Flow Cytometry Kit (Life Technologies) according to the manufacturer's protocol. Cells were also stained with FxCycle Violet (Invitrogen) for total DNA content and analyzed using a BD FACSCanto II Flow Cytometer (BD Biosciences) with BD FACSDiva Software (BD Biosciences).
Clonogenicity was analyzed following plating of 100 cells/well on MEF coated 24-well plates. After 7 days, colonies were stained for alkaline phosphatase using the AP staining kit (Stemgent).
Karyotyping
Cells were incubated with 100 ng/mL colcemid (Biological Industries) for 30 min at 37°C, collected by trypsin, incubated with 0.075 mol/L potassium chloride for 10 min at 37°C, fixed with methanol and acetic acid (1:3) and dropped onto glass slides. Karyotype analysis of chromosome spreads was determined by Giemsa staining of at least 20 different metaphase-stage cells for each culture.
Chromosomal microarray analysis (CMA)
Genomic DNA was extracted from samples using the DNeasy Blood & Tissue Kit (Qiagen). The DNA was amplified, labeled, and hybridized to a 24sure V3 microarray (Illumina) according to the manufacturer’s protocol. Scanning was performed using Agilent G2565CA scanner and the arrays were analyzed using the BlueFuse Multi software. The detected CNVs were interpreted by referring to key public databases (ISCA, DGV, Ensembl, and Decipher).
Whole genome sequencing (WGS)
WGS libraries were constructed using the Kapa HyperPlus Kit (Roche Holding AG). Briefly, up to one ug of EDTA-free dsDNA was incubated with the fragmentation enzyme for ten minutes at 37 degrees. The fragmented samples were then end-repaired and A-Tailed and Illumina indexed adapters were ligated onto the ends of the dsDNA. The libraries were then quanted (Qubit, ThermoFisher Scientific) and run on the BioAnalyzer (Agilent Technologies Inc.) for quality control purposes. The libraries were then size selected using Ampure XP beads (Beckman Coulter) using a 0.65 Ampure XP to DNA ratio followed by a 0.9 ratio to achieve an average library size of about 400 bp. Due to small input amounts for several of the libraries, library amplification (4 cycles) was performed on a selection of samples. Libraries were sequenced using paired-end 100 bp reads on the NovaSeq S4, and six of the 16 samples were re-sequenced on the HiSeq 4000 to achieve greater depth. Samples were sequenced to an average depth of 27x and assessed for quality using FASTQC (v. 0.11.8). Reads were quality trimmed using Trim Galore (v. 0.4.1) with 30 as the quality cutoff. Bowtie 2 (v. 2.3.4.3)113 was used to map the reads to GRCh38 (v. GCA_000001405.15) and Picard Tools (v. 2.18.15) was used to fix mate information, merge Bam files for samples with two sequencing runs, and mark duplicates.
WGS single nucleotide variant & InDel calling
SNVs and InDels were identified using the best practice instructions of GATK4 (v. 4.0.11.0) HaplotypeCaller.114 Bam files were recalibrated using GATK BaseRecalibrator with the “known-sites” dbsnp138, 1000G phase1 high confidence SNPs, and Mills 1000G gold standard indels. The data was then run through the HaplotypeCaller and combined using CombineGVCFs for joint genotyping. Following joint genotyping the SNPs and InDels were recalibrated using the following options for the SNPs: --resource hapmap, known = false, training = true, truth = true, prior = 15.0:/hapmap_3.3.hg38.vcf.gz --resource omni, known = false, training = true, truth = true, prior = 12.0:/1000G_omni2.5.hg38.vcf.gz --resource 1000G, known = false, training = true, truth = false, prior = 10.0:/1000G_phase1.snps.high_confidence.hg38.vcf.gz --resource dbsnp, known = true, training = false, truth = false, prior = 2.0:/Homo_sapiens_assembly38.dbsnp138.vcf -an QD -an FS -an SOR -an MQ -an MQRankSum -an ReadPosRankSum -an DP --mode SNP -tranche 100.0 -tranche 99.9 -tranche 99.0 -tranche 90.0 and for the InDels: --resource mills, known = false, training = true, truth = true, prior = 12.0:/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz --resource dbsnp, known = true, training = false, truth = false, prior = 2.0:/Homo_sapiens_assembly38.dbsnp138.vcf -an QD -an FS -an SOR -an MQRankSum -an ReadPosRankSum -an DP --mode INDEL -tranche 100.0 -tranche 99.9 -tranche 99.0 -tranche 90.0 --max-gaussians 4. Genotype posteriors using the sample trios as priors was calculated for each trio and possible de novo mutations were annotated by VariantAnnotator. Finally, low-quality genotypes (q < 20) were removed and high confidence de novo variants were selected. Variants were then annotated using ANNOVAR (v. 2019Oct24).115 Filtered variant files were compared using vcftools (v. 0.1.16), vcf-isec. Bedtools (v. 2.25.0) was used to find variants overlapping specific genomic regions.
Copy number variation analysis
Regions of the genome with copy number variations (CNV’s) were called using three different algorithms, CNVkit (v. 0.9.6),116 ERDS (v. 1.1),117 and PURPLE (v. 2.34).118 CNVkit: A joint reference using the parental lines was created using the options: --method WGS, --access access-excludes.GRCh38.bed followed by an estimation of the integer copy number of each segment using cnvkit.py call. The input sample was first deduplicated using the samtools (v. 1.9) command rmdup. The integer copy number output was then filtered for only CNV calls and regions of the same copy number were combined. ERDS: erds_pipeline.pl was used with the filtered vcf files for each sample along with the merged and duplicate marked bam files as input. The output from CNVkit and ERDS were split into deletion and duplication bed files. Bedtools (v. 2.25.0) intersect was then used to look for copy number overlaps between the output of the two CNV calling tools, determine if a duplication or deletion was a de novo event, and find CNVs overlapping regions of interest. A CNV was called if it was larger than 2000 bp and found by both CNVkit and ERDS. CNV regions of the same copy number were merged if they were within 100 kb. The filtered CNV calls were then annotated using Homer (v. 4.10.4)119 annotatePeaks.pl with hg38 (v. 6.0). PURPLE was used to validate differences in calls between the CMA array and WGS CNV calls.
RNA sequencing
RNA was extracted using mirVana miRNA Isolation Kit (ThermoFisher Scientific), following the manufacturers' instructions. RNA quality was assessed using a BioAnalyzer 2100 (Agilent Technologies, Inc.). All samples had a RIN greater than 8.5. RNA-seq libraries were constructed in triplicate using the KAPA mRNA HyperPrep Kit (Roche) with 500 ng of input RNA. Libraries were sequenced on a HiSeq 4000 (Illumina, Inc.) with paired-end (2X100 bp) reads. Samples were sequenced to an average depth of 20 million uniquely mapped reads per sample and assessed for quality using FASTQC (v.0.11.8). The reads were mapped to GRCh38.p10 (GENCODE release 26) using STAR (v. 2.7.3a)120 and annotated using featureCounts (subread v.1.6.3, GENCODE release 26 primary assembly annotation).121 The STAR parameters used were: --runMode alignReads --outSAMmode Full --outSAMattributes Standard --genomeLoad LoadAndKeep --clip3pAdapterSeq AGATCGGAAGAGC --clip3pAdapterMMp 1. The featureCounts parameters were: -s 2 -p -t exon -T 13 -g gene_id. Ensembl genes without at least three samples with more than 10 reads were removed from the analysis leaving about 20k genes. BiomaRt (v. 2.42.1) was used to convert Ensembl gene IDs to HUGO gene names. The R (v. 3.6.3) package DESeq2 (v.1.26.0),122 using a multifactor design formula to account for experimental design variables, was used to perform differential expression analysis and normalize the count matrix. Genes with an adjusted p-value of less than 0.05 were considered significant unless otherwise noted. Data from all lines created for this manuscript were compared to previously published naïve and primed lines listed in (Table S3). Raw fastq files were downloaded and processed using a similar pipeline to that listed above. Samples sequenced using single end reads were mapped separately from paired-end samples and all samples were combined on Ensembl gene IDs. PCA plots were created using DESeq2, VSD transformed values with limma removeBatchEffect. Gene Set Enrichment Analysis134,135 was performed using the R package fgsea (v. 1.12.0)123 with the hallmark gene sets136 and the C5 GO gene sets downloaded from the gsea-msigdb (v. 7.1). Transcriptional Element (TEs) regions were taken from,65 and a liftover from hg19 to GRCh38 was performed using UCSCs Lift Genome Annotation tool (https://genome.ucsc.edu/cgi-bin/hgLiftOver). TEs were mapped and analyzed using the same tools as were used for the gene expression data.
Small RNA-seq
Small RNA-seq libraries were constructed using the NEBNext® Multiplex Small RNA Library Prep Set for Illumina® (New England Biolabs, Inc., Ipswich, MA). The libraries were pooled, size-selected for products that were 120–135 nucleotides in length using a Pippin Prep with a 3% agarose gel cassette (Sage Science, Beverly, Massachusetts), and run on a MiSeq instrument (Illumina, San Diego, California) at the UC San Diego UCSD Institute for Genomic Medicine (IGM) Genomics Core. Samples that produced adequate numbers of miRNA read counts were then rebalanced to produce similar numbers of miRNA reads and sequenced on a HiSeq 4000 instrument using 1 × 75 bp reads (Illumina, San Diego, California) at the UC San Diego IGM Genomics Core. The data were trimmed and mapped to GRCh38 using the exceRpt Small RNA-seq Pipeline Workflow implemented in the Genboree Workbench.124 Micro RNAs were filtered such that at least five reads in at least two samples were retained, resulting in 1035 pass-filter miRNAs. Differential expression was carried out in DESeq2 (v.1.26.0)122 requiring an adjusted p-value < 0.05 and log2 FoldChange > 1 considered differentially expressed unless otherwise noted. As with the long RNA-seq, data from all lines created for this manuscript were compared to previously published naive and primed lines listed in (Table S5).
Differential mRNA expression analysis
Transcriptional changes due to long term culturing, NHSM derivation (p20) samples vs early (p30) and late (p50) NHSM samples, were determined by performing differential expression analysis using DESeq2 (v.1.26.0)122 with an adjusted p-value < 0.05, a log2 fold change > 1, and a base mean > 50. Genes considered to be significantly upregulated/downregulated in p20 samples were required to be upregulated/downregulated in both the early (p20 vs p30) and late (p20 vs p50) differential expression analysis comparisons as well as not upregulated/downregulated in a similar early (p30) primed vs late (p50) primed comparison. The differentially upregulated/downregulated gene lists were then separately input into Enrichr125,126,127 to obtain the gene ontology significantly (adj. p-value < 0.05) enriched biological process terms. The terms were then input into Revigo to summarize and find representative subsets of terms using the semantic similarity algorithm SimRel and using the adjusted p-value with every term.60 The resulting tree map was then input into CirGO, which allows for the visualization of the most enriched terms in an informative 2D circular graph.61
Integrative analysis of miRNAs and mRNAs
mRNAs that were either upregulated or downregulated in the derivation p20 cultures compared to the later cultures (the same differentially expressed mRNAs identified in the section above, “differential mRNA expression analysis”) were used to create protein-protein interaction networks using the stringApp (v. 1.5.1, confidence (score) cutoff = 0.4, max additional interactors = 0, use smart delimiters) application in Cytoscape (v. 3.8.0). The networks were then clustered using MCL clustering with the clusterMaker2 application (v. 1.3.1, inflation value = 2.0, assumption that edges were undirected, and loops were adjusted before clustering). The largest clusters for the mRNAs that were downregulated and upregulated in the p20 cultures are shown in Figure S9B and S9C, respectively. Functional enrichment analysis was used to identify pathways enriched for the genes in these clusters.
To integrate the miRNA and mRNA data in relation to the transcriptional changes due to long term culturing, p20 samples vs p30 and p50 NHSM samples, we performed differential expression of the small RNA-seq data, requiring miRNAs to be differentially expressed between both the p20 and p30 timepoints and the p20 and p50 timepoints. Additionally, miRNAs that were differentially expressed between the primed p30 and primed p50 timepoints were filtered out of the resulting miRNA lists. This differentially expressed miRNA list and the differentially expressed mRNA list from the section above (“differential mRNA expression analysis”) were each converted to counts per million and combined by pairing the differentially upregulated mRNA genes with the differentially downregulated miRNAs and vice versa. These combined datasets were then used as input into the gene regulatory network inference algorithm GRNBoost2 using Arboreto (v. 0.1.5).128 For each target gene, the algorithm uses a tree-based regression model to predict its expression profile using the expression values of the set of miRNAs. The algorithm outputs importance scores that reflect the degree to which each potential mRNA target is regulated by each miRNAs in the dataset.
For the largest clusters from both the downregulated (Figure S9B) and upregulated (Figure S9C) p20 mRNA networks, heatmaps comprised of the mRNAs in the clusters plotted against the anticorrelated differentially expressed miRNAs were generated, where each cell is color-coded according to the importance score for its respective miRNA/mRNA pair. Hierarchical clustering was then used to visually identify the miRNAs with the highest composite importance scores across their mRNA targets in each cluster. We then used miRPathDB 2.054 to identify pathways that are predicted to be regulated by these miRNAs, and mapped those to the pathways that are enriched for the mRNA targets in each cluster. The similarity between the miRNAs was assessed by Jaccard similarity coefficients, using “Similar miRNAs” function in miRPathDB. The 6 out of 7 miRNAs found to be similar were ranked in the top 20 in the “Jaccard coefficient (‘target genes strong)” column out of the over 4000 total miRNAs in the database.
Integration of miRNA and mRNA data in relation to the transcriptional changes between derivation p20 cultures vs late p50 primed cultures was carried out using mirTarBase (Release 8.0).129,130 Directionally opposite differentially expressed miRNAs (adj. p-value < 0.05, log2 FoldChange > 1; normalized basemean > 10) and corresponding differentially expressed mRNAs (target genes) (adj. p-value < 0.05, normalized basemean > 50) were used in this comparison. Pathway analysis on the genes within the largest clustered network and miRNA-targets were analyzed using Enrichr.125,126,127
Mapping RNA-seq data to scRNA-seq
RNA-seq samples were compared to single cell expression data from human embryonic stages in an equivalent manner to Theunissen 2016.65 Briefly, raw single cell mRNA-seq counts48 were downloaded from https://www.ebi.ac.uk/gxa/sc/experiments/E-GEOD-36552/downloads, processed in scanpy (v. 1.3.2)131 and differential expression was performed in R (v.3.6.3) using the package DESeq2 (v. 1.26.0) for each cell state compared against all other cell states. A gene was determined to be expressed in a specific cell state if it had an adjusted p-value less than 0.05 and a log2fold change greater than 3. Genes determined to be expressed in a specific cell state were divided between up and down regulated and compared to the genes that were determined to be up or down regulated in our early NHSM cell lines compared to our late primed cell lines using an adjusted p-value cutoff of less than 0.05 and a log2fold change greater than 2.
Variant analysis using RNA-seq data
Samples were aligned to the GRCh38 reference genome as described above. Variants were identified using the best practice instructions of GATK4 HaplotypeCaller.114 Briefly, Picard Tools (v. 2.18.15) was used to first merge bam files and mark duplicates. Next, GATK (v. 4.0.11.0) SplitNCigarReads was used to reformat reads that span introns and base quality recalibration was done to detect and correct for patterns of systematic errors in the base quality scores. The following “known-sites” were used in the BaseRecalibrator step: Homo_sapiens_assembly38.dbsnp138.vcf, 1000G_phase1.snps.high_confidence.hg38.vcf, and Mills_and_1000G_gold_standard.indels.hg38.vcf. HaplotypeCaller with a –stand-call-conf 20 was then used to call variants. To find high certainty de novo variants, the HaplotypeCaller results were filtered not dissimilar to.104 First, variants were required to be called in two of the three replicates for each sample and not in the low passage NHSM sample, unless the samples in question were the low passage NHSM samples. Next, only positions with over 20 reads, are uncommon in the general population (allele frequency lower than 0.0001 in the Exome Aggregation Consortium (ExAC Database) (v.0.3),137 and cause nonsynonymous single nucleotide variations or stop-gain mutations were assessed. InDels were not used in the analysis. Annotations were obtained using ANNOVAR (v. 2019Oct24)115 and only variants in genes listed as “Tier 1” in Cancer Gene Census of the Catalogue of Somatic Mutations in Cancer (COSMIC) v. 90 database (https://cancer.sanger.ac.uk/census)138 with FATHMM scores below −1.5 were considered pathogenic. Single nucleotide variants passing these filters were then checked against DNA-seq reads as well as assessed for possible murine contamination.
Methylation profiling
DNA from every line and time point was sent to UC San Diego UCSD Institute for Genomic Medicine (IGM) Genomics Core for processing and imaging. The samples were processed according to standard protocols and hybridized to the Illumina Infinium MethylationEPIC array, which interrogates over 850,000 methylation sites across the genome, and imaged on the Illumina iScan (Illumina Inc., San Diego, CA). The data was normalized in R (v. 3.6.3) using the minfi package (v. 1.32.0)132 with the “funnorm” function. Following normalization, probes that were found to cross-reactive on the EPIC array, probes overlapping genetic variants at targeted CpG sites, and probes overlapping genetic variants at single base extension sites for Infinium Type I probes were filtered out139 leaving a total of 811,063 probes. Annotation BED files were download and analysis was performed using python scripts from the CpGtools package.133 Imprinted regions (iDMRs) were taken from,69 and X-chromosome inactivation (XCI) and escape regions (non-XCI) were lifted over to GRCh38 from.140
Quantification and statistical analysis
For each experiment, data were obtained from 2-3 independent biological experiments (with each experiment including at least three replicates). P-values were calculated by Mann Whitney U test and paired or unpaired two-tailed Student’s t test using SPSS software, are ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. The statistical details of experiments can be found in the figure legends, figures, and results.
Acknowledgments
Whole Genome Sequencing, sequencing of RNA-seq and small RNA-seq libraries, and DNA methylation microarray analysis was conducted at the Institute for Genomic Medicine (IGM) Genomics Center, University of California, San Diego, La Jolla, CA. Small RNA-seq libraries were generated by Aishwarya Vuppala under the supervision of Peter De Hoff. This publication includes data generated at the UC San Diego IGM Genomics Center utilizing an Illumina NovaSeq 6000 that was purchased with funding from a National Institutes of Health SIG grant (#S10 OD026929). This work used the Extreme Science and Engineering Discovery Environment (XSEDE) Expanse at the San Diego Super Computer through allocation MCB140074. RM was supported by a grant from the National Institutes of Health, USA (NIH grant T32GM008806). Small RNA-seq data processing was performed using the exceRpt pipeline on the Genboree Workbench developed by the Data Integration and Analysis Component (DIAC) of the Extracellular RNA Communication Consortium. The authors would also like to thank the dedicated team of embryologists, geneticists, and medical professionals at the Institution of Reproduction and IVF, Lis Maternity Hospital, Tel Aviv Sourasky Medical Center. Additionally, we are grateful for the following funding agencies, which supported our work and enabled us to conduct this study: The Sagol fund for embryos and stem cells as part of the Sagol Network and the Israel Science Foundation Physician-Scientist Grant (Grant No. 2089/15).
Author contributions
C.D. and R.M. designed, conducted, analyzed, and interpreted the experiments (wet lab and dry lab, respectively) and drafted the paper. J.H. provided materials and guidance. D.B., L.L., and H.A. designed the project and experiments, interpreted the results, and drafted the paper.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: December 22, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105469.
Contributor Information
Louise C. Laurent, Email: llaurent@ucsd.edu.
Dalit Ben-Yosef, Email: dalitb@tlvmc.gov.il.
Hadar Amir, Email: hadarnmb@gmail.com.
Supplemental information
Differentially expressed genes (adj. p-value < 0.05, basemean >50, paired analysis by the line and timepoint) between NHSM and isogenic primed cultures at similar timepoints. The table contains normalized data from all 60 samples with a column indicating whether the gene was up or downregulated in our NHSM cultures compared with our primed.
Gene set enrichment analysis for upregulated and downregulated genes in the NHSM cultures compared with their isogenic primed cultures at similar time points using WikiPathways 2019 in Enrichr.
Gene ontology enrichment analysis (biological processes) for the 177 mRNAs uniquely upregulated at the earliest NHSM derived (p20) timepoint.
Gene ontology enrichment analysis (biological processes) for the 157 mRNAs uniquely downregulated at the earliest NHSM (p20, NHSM derived) timepoint.
The table shows six of the seven miRPathDB included miRNAs are in the top 20 most similar miRNAs by “Jaccard coefficient (target genes strong)”.
Gene ontology enrichment analysis (biological process 2021) of downregulated genes also targeted by a miRNA upregulated in NHSM derived (p20) cultures. Refers to data in Figure 5B.
Alleles with at least 10 reads were considered in analysis. An allele was considered biallelic if the allelic frequency of both the reference and alternative allele were over 30%.
Data and code availability
-
•
Whole genome sequencing data has been deposited at NCBI’s SRA (BioProject: PRJNA859118). RNA-seq (GSE208300), small RNA-seq (GSE208301), and methylation array data (GSE208299) have been deposited at GEO and are publicly available under the accession number GSE208302. All data can be found under the umbrella project PRJNA859148.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Mason C., Dunnill P. A brief definition of regenerative medicine. Regen. Med. 2008;3:1–5. doi: 10.2217/17460751.3.1.1. [DOI] [PubMed] [Google Scholar]
- 2.Yoshida Y., Yamanaka S. Induced pluripotent stem cells 10 years later: for cardiac applications. Circ. Res. 2017;120:1958–1968. doi: 10.1161/circresaha.117.311080. [DOI] [PubMed] [Google Scholar]
- 3.Neofytou E., O'Brien C.G., Couture L.A., Wu J.C. Hurdles to clinical translation of human induced pluripotent stem cells. J. Clin. Invest. 2015;125:2551–2557. doi: 10.1172/JCI80575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Desgres M., Menasché P. Clinical translation of pluripotent stem cell therapies: challenges and considerations. Cell Stem Cell. 2019;25:594–606. doi: 10.1016/j.stem.2019.10.001. [DOI] [PubMed] [Google Scholar]
- 5.Nichols J., Smith A. Pluripotency in the embryo and in culture. Cold Spring Harb. Perspect. Biol. 2012;4:a008128. doi: 10.1101/cshperspect.a008128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 7.Nichols J., Smith A. Naive and primed pluripotent states. Cell Stem Cell. 2009;4:487–492. doi: 10.1016/j.stem.2009.05.015. [DOI] [PubMed] [Google Scholar]
- 8.Hassani S.N., Totonchi M., Gourabi H., Schöler H.R., Baharvand H. Signaling roadmap modulating naive and primed pluripotency. Stem Cells Dev. 2014;23:193–208. doi: 10.1089/scd.2013.0368. [DOI] [PubMed] [Google Scholar]
- 9.Brons I.G.M., Smithers L.E., Trotter M.W.B., Rugg-Gunn P., Sun B., Chuva de Sousa Lopes S.M., Howlett S.K., Clarkson A., Ahrlund-Richter L., Pedersen R.A., Vallier L. Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature. 2007;448:191–195. doi: 10.1038/nature05950. [DOI] [PubMed] [Google Scholar]
- 10.Tesar P.J., Chenoweth J.G., Brook F.A., Davies T.J., Evans E.P., Mack D.L., Gardner R.L., McKay R.D.G. New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature. 2007;448:196–199. doi: 10.1038/nature05972. [DOI] [PubMed] [Google Scholar]
- 11.Kumari D. In: Pluripotent Stem Cells - From the Bench to the Clinic. Tomizawa, editor. IntechOpen; 2016. States of pluripotency: naïve and primed pluripotent stem cells. [Google Scholar]
- 12.Mahla R.S. Stem cells applications in regenerative medicine and disease therapeutics. Int. J. Cell Biol. 2016;2016:6940283. doi: 10.1155/2016/6940283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomson J.A., Itskovitz-Eldor J., Shapiro S.S., Waknitz M.A., Swiergiel J.J., Marshall V.S., Jones J.M. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 14.Gafni O., Weinberger L., Mansour A.A., Manor Y.S., Chomsky E., Ben-Yosef D., Kalma Y., Viukov S., Maza I., Zviran A., et al. Derivation of novel human ground state naive pluripotent stem cells. Nature. 2013;504:282–286. doi: 10.1038/nature12745. [DOI] [PubMed] [Google Scholar]
- 15.Weinberger L., Ayyash M., Novershtern N., Hanna J.H. Dynamic stem cell states: naive to primed pluripotency in rodents and humans. Nat. Rev. Mol. Cell Biol. 2016;17:155–169. doi: 10.1038/nrm.2015.28. [DOI] [PubMed] [Google Scholar]
- 16.Guo G., von Meyenn F., Santos F., Chen Y., Reik W., Bertone P., Smith A., Nichols J. Naive pluripotent stem cells derived directly from isolated cells of the human inner cell mass. Stem Cell Rep. 2016;6:437–446. doi: 10.1016/j.stemcr.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu Z., Li H., Jiang H., Ren Y., Yu X., Qiu J., Stablewski A.B., Zhang B., Buck M.J., Feng J. Transient inhibition of mTOR in human pluripotent stem cells enables robust formation of mouse-human chimeric embryos. Sci. Adv. 2020;6:eaaz0298. doi: 10.1126/sciadv.aaz0298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang W.W.C., Dietmann S., Irie N., Leitch H.G., Floros V.I., Bradshaw C.R., Hackett J.A., Chinnery P.F., Surani M.A. A unique gene regulatory network resets the human germline epigenome for development. Cell. 2015;161:1453–1467. doi: 10.1016/j.cell.2015.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sperber H., Mathieu J., Wang Y., Ferreccio A., Hesson J., Xu Z., Fischer K.A., Devi A., Detraux D., Gu H., et al. The metabolome regulates the epigenetic landscape during naive-to-primed human embryonic stem cell transition. Nat. Cell Biol. 2015;17:1523–1535. doi: 10.1038/ncb3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takashima Y., Guo G., Loos R., Nichols J., Ficz G., Krueger F., Oxley D., Santos F., Clarke J., Mansfield W., et al. Resetting transcription factor control circuitry toward ground-state pluripotency in human. Cell. 2014;158:1254–1269. doi: 10.1016/j.cell.2014.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Theunissen T.W., Powell B.E., Wang H., Mitalipova M., Faddah D.A., Reddy J., Fan Z.P., Maetzel D., Ganz K., Shi L., et al. Systematic identification of culture conditions for induction and maintenance of naive human pluripotency. Cell Stem Cell. 2014;15:471–487. doi: 10.1016/j.stem.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Y., Lai D. Pluripotent states of human embryonic stem cells. Cell. Reprogram. 2015;17:1–6. doi: 10.1089/cell.2014.0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ware C.B., Nelson A.M., Mecham B., Hesson J., Zhou W., Jonlin E.C., Jimenez-Caliani A.J., Deng X., Cavanaugh C., Cook S., et al. Derivation of naive human embryonic stem cells. Proc. Natl. Acad. Sci. USA. 2014;111:4484–4489. doi: 10.1073/pnas.1319738111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duggal G., Warrier S., Ghimire S., Broekaert D., Van der Jeught M., Lierman S., Deroo T., Peelman L., Van Soom A., Cornelissen R., et al. Alternative routes to induce naïve pluripotency in human embryonic stem cells. Stem Cell. 2015;33:2686–2698. doi: 10.1002/stem.2071. [DOI] [PubMed] [Google Scholar]
- 25.Qin H., Hejna M., Liu Y., Percharde M., Wossidlo M., Blouin L., Durruthy-Durruthy J., Wong P., Qi Z., Yu J., et al. YAP induces human naive pluripotency. Cell Rep. 2016;14:2301–2312. doi: 10.1016/j.celrep.2016.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu Z., Robitaille A.M., Berndt J.D., Davidson K.C., Fischer K.A., Mathieu J., Potter J.C., Ruohola-Baker H., Moon R.T. Wnt/β-catenin signaling promotes self-renewal and inhibits the primed state transition in naïve human embryonic stem cells. Proc. Natl. Acad. Sci. USA. 2016;113:E6382–E6390. doi: 10.1073/pnas.1613849113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Warrier S., Van der Jeught M., Duggal G., Tilleman L., Sutherland E., Taelman J., Popovic M., Lierman S., Chuva De Sousa Lopes S., Van Soom A., et al. Direct comparison of distinct naive pluripotent states in human embryonic stem cells. Nat. Commun. 2017;8:15055. doi: 10.1038/ncomms15055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J., Xie G., Singh M., Ghanbarian A.T., Raskó T., Szvetnik A., Cai H., Besser D., Prigione A., Fuchs N.V., et al. Primate-specific endogenous retrovirus-driven transcription defines naive-like stem cells. Nature. 2014;516:405–409. doi: 10.1038/nature13804. [DOI] [PubMed] [Google Scholar]
- 29.Yu L., Wei Y., Sun H.X., Mahdi A.K., Pinzon Arteaga C.A., Sakurai M., Schmitz D.A., Zheng C., Ballard E.D., Li J., et al. Derivation of intermediate pluripotent stem cells amenable to primordial germ cell specification. Cell Stem Cell. 2021;28:550–567.e12. doi: 10.1016/j.stem.2020.11.003. [DOI] [PubMed] [Google Scholar]
- 30.Lee J.H., Laronde S., Collins T.J., Shapovalova Z., Tanasijevic B., McNicol J.D., Fiebig-Comyn A., Benoit Y.D., Lee J.B., Mitchell R.R., Bhatia M. Lineage-specific differentiation is influenced by state of human pluripotency. Cell Rep. 2017;19:20–35. doi: 10.1016/j.celrep.2017.03.036. [DOI] [PubMed] [Google Scholar]
- 31.Smith A. Formative pluripotency: the executive phase in a developmental continuum. Development. 2017;144:365–373. doi: 10.1242/dev.142679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Collier A.J., Rugg-Gunn P.J. Identifying human naïve pluripotent stem cells - evaluating state-specific reporter lines and cell-surface markers. Bioessays. 2018;40:e1700239. doi: 10.1002/bies.201700239. [DOI] [PubMed] [Google Scholar]
- 33.Maitra A., Arking D.E., Shivapurkar N., Ikeda M., Stastny V., Kassauei K., Sui G., Cutler D.J., Liu Y., Brimble S.N., et al. Genomic alterations in cultured human embryonic stem cells. Nat. Genet. 2005;37:1099–1103. doi: 10.1038/ng1631. [DOI] [PubMed] [Google Scholar]
- 34.Imreh M.P., Gertow K., Cedervall J., Unger C., Holmberg K., Szöke K., Csöregh L., Fried G., Dilber S., Blennow E., Ahrlund-Richter L. In vitro culture conditions favoring selection of chromosomal abnormalities in human ES cells. J. Cell. Biochem. 2006;99:508–516. doi: 10.1002/jcb.20897. [DOI] [PubMed] [Google Scholar]
- 35.Baker D.E.C., Harrison N.J., Maltby E., Smith K., Moore H.D., Shaw P.J., Heath P.R., Holden H., Andrews P.W. Adaptation to culture of human embryonic stem cells and oncogenesis in vivo. Nat. Biotechnol. 2007;25:207–215. doi: 10.1038/nbt1285. [DOI] [PubMed] [Google Scholar]
- 36.Ben-Yosef D., Boscolo F.S., Amir H., Malcov M., Amit A., Laurent L.C. Genomic analysis of hESC pedigrees identifies de novo mutations and enables determination of the timing and origin of mutational events. Cell Rep. 2013;4:1288–1302. doi: 10.1016/j.celrep.2013.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garitaonandia I., Amir H., Boscolo F.S., Wambua G.K., Schultheisz H.L., Sabatini K., Morey R., Waltz S., Wang Y.C., Tran H., et al. Increased risk of genetic and epigenetic instability in human embryonic stem cells associated with specific culture conditions. PLoS One. 2015;10:e0118307. doi: 10.1371/journal.pone.0118307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nguyen H.T., Geens M., Mertzanidou A., Jacobs K., Heirman C., Breckpot K., Spits C. Gain of 20q11.21 in human embryonic stem cells improves cell survival by increased expression of Bcl-xL. Mol. Hum. Reprod. 2014;20:168–177. doi: 10.1093/molehr/gat077. [DOI] [PubMed] [Google Scholar]
- 39.Avery S., Hirst A.J., Baker D., Lim C.Y., Alagaratnam S., Skotheim R.I., Lothe R.A., Pera M.F., Colman A., Robson P., et al. BCL-XL mediates the strong selective advantage of a 20q11.21 amplification commonly found in human embryonic stem cell cultures. Stem Cell Rep. 2013;1:379–386. doi: 10.1016/j.stemcr.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.International Stem Cell Initiative. Amps K., Andrews P.W., Anyfantis G., Armstrong L., Avery S., Baharvand H., Baker J., Baker D., Munoz M.B., et al. Screening ethnically diverse human embryonic stem cells identifies a chromosome 20 minimal amplicon conferring growth advantage. Nat. Biotechnol. 2011;29:1132–1144. doi: 10.1038/nbt.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Amir H., Touboul T., Sabatini K., Chhabra D., Garitaonandia I., Loring J.F., Morey R., Laurent L.C. Spontaneous single-copy loss of TP53 in human embryonic stem cells markedly increases cell proliferation and survival. Stem Cell. 2017;35:872–885. doi: 10.1002/stem.2550. [DOI] [PubMed] [Google Scholar]
- 42.Lefort N., Feyeux M., Bas C., Féraud O., Bennaceur-Griscelli A., Tachdjian G., Peschanski M., Perrier A.L. Human embryonic stem cells reveal recurrent genomic instability at 20q11.21. Nat. Biotechnol. 2008;26:1364–1366. doi: 10.1038/nbt.1509. [DOI] [PubMed] [Google Scholar]
- 43.Brosh R., Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat. Rev. Cancer. 2009;9:701–713. doi: 10.1038/nrc2693. [DOI] [PubMed] [Google Scholar]
- 44.Andrews P.W. The selfish stem cell. Nat. Biotechnol. 2006;24:325–326. doi: 10.1038/nbt0306-325. [DOI] [PubMed] [Google Scholar]
- 45.Pastor W.A., Chen D., Liu W., Kim R., Sahakyan A., Lukianchikov A., Plath K., Jacobsen S.E., Clark A.T. Naive human pluripotent cells feature a methylation landscape devoid of blastocyst or germline memory. Cell Stem Cell. 2016;18:323–329. doi: 10.1016/j.stem.2016.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Di Stefano B., Ueda M., Sabri S., Brumbaugh J., Huebner A.J., Sahakyan A., Clement K., Clowers K.J., Erickson A.R., Shioda K., et al. Reduced MEK inhibition preserves genomic stability in naive human embryonic stem cells. Nat. Methods. 2018;15:732–740. doi: 10.1038/s41592-018-0104-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Messmer T., von Meyenn F., Savino A., Santos F., Mohammed H., Lun A.T.L., Marioni J.C., Reik W. Transcriptional heterogeneity in naive and primed human pluripotent stem cells at single-cell resolution. Cell Rep. 2019;26:815–824.e4. doi: 10.1016/j.celrep.2018.12.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yan L., Yang M., Guo H., Yang L., Wu J., Li R., Liu P., Lian Y., Zheng X., Yan J., et al. Single-cell RNA-seq profiling of human preimplantation embryos and embryonic stem cells. Nat. Struct. Mol. Biol. 2013;20:1131–1139. doi: 10.1038/nsmb.2660. [DOI] [PubMed] [Google Scholar]
- 49.Dodsworth B.T., Hatje K., Rostovskaya M., Flynn R., Meyer C.A., Cowley S.A. Profiling of naïve and primed human pluripotent stem cells reveals state-associated miRNAs. Sci. Rep. 2020;10:10542. doi: 10.1038/s41598-020-67376-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smith Z.D., Chan M.M., Humm K.C., Karnik R., Mekhoubad S., Regev A., Eggan K., Meissner A. DNA methylation dynamics of the human preimplantation embryo. Nature. 2014;511:611–615. doi: 10.1038/nature13581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barakat T.S., Halbritter F., Zhang M., Rendeiro A.F., Perenthaler E., Bock C., Chambers I. Functional dissection of the enhancer repertoire in human embryonic stem cells. Cell Stem Cell. 2018;23:276–288.e8. doi: 10.1016/j.stem.2018.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang Y., Hussein A.M., Somasundaram L., Sankar R., Detraux D., Mathieu J., Ruohola-Baker H. microRNAs regulating human and mouse naïve pluripotency. Int. J. Mol. Sci. 2019;20:E5864. doi: 10.3390/ijms20235864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hassani S.N., Totonchi M., Sharifi-Zarchi A., Mollamohammadi S., Pakzad M., Moradi S., Samadian A., Masoudi N., Mirshahvaladi S., Farrokhi A., et al. Inhibition of TGFβ signaling promotes ground state pluripotency. Stem Cell Rev. Rep. 2014;10:16–30. doi: 10.1007/s12015-013-9473-0. [DOI] [PubMed] [Google Scholar]
- 54.Kehl T., Kern F., Backes C., Fehlmann T., Stöckel D., Meese E., Lenhof H.P., Keller A. miRPathDB 2.0: a novel release of the mirna pathway dictionary database. Nucleic Acids Res. 2020;48:D142–D147. doi: 10.1093/nar/gkz1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Romorini L., Garate X., Neiman G., Luzzani C., Furmento V.A., Guberman A.S., Sevlever G.E., Scassa M.E., Miriuka S.G. AKT/GSK3β signaling pathway is critically involved in human pluripotent stem cell survival. Sci. Rep. 2016;6:35660. doi: 10.1038/srep35660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vallier L., Touboul T., Brown S., Cho C., Bilican B., Alexander M., Cedervall J., Chandran S., Ahrlund-Richter L., Weber A., Pedersen R.A. Signaling pathways controlling pluripotency and early cell fate decisions of human induced pluripotent stem cells. Stem Cell. 2009;27:2655–2666. doi: 10.1002/stem.199. [DOI] [PubMed] [Google Scholar]
- 57.Xu R.H., Sampsell-Barron T.L., Gu F., Root S., Peck R.M., Pan G., Yu J., Antosiewicz-Bourget J., Tian S., Stewart R., Thomson J.A. NANOG is a direct target of TGFbeta/activin-mediated SMAD signaling in human ESCs. Cell Stem Cell. 2008;3:196–206. doi: 10.1016/j.stem.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gu K.L., Zhang Q., Yan Y., Li T.T., Duan F.F., Hao J., Wang X.W., Shi M., Wu D.R., Guo W.T., Wang Y. Pluripotency-associated miR-290/302 family of microRNAs promote the dismantling of naive pluripotency. Cell Res. 2016;26:350–366. doi: 10.1038/cr.2016.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chou C.H., Shrestha S., Yang C.D., Chang N.W., Lin Y.L., Liao K.W., Huang W.C., Sun T.H., Tu S.J., Lee W.H., et al. miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018;46:D296–D302. doi: 10.1093/nar/gkx1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Supek F., Bošnjak M., Škunca N., Šmuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One. 2011;6:e21800. doi: 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kuznetsova I., Lugmayr A., Siira S.J., Rackham O., Filipovska A. CirGO: an alternative circular way of visualising gene ontology terms. BMC Bioinf. 2019;20:84. doi: 10.1186/s12859-019-2671-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gordeeva O. TGFβ family signaling pathways in pluripotent and teratocarcinoma stem cells' fate decisions: balancing between self-renewal, differentiation, and cancer. Cells. 2019;8:E1500. doi: 10.3390/cells8121500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Osnato A., Brown S., Krueger C., Andrews S., Collier A.J., Nakanoh S., Quiroga Londoño M., Wesley B.T., Muraro D., Brumm A.S., et al. TGFβ signalling is required to maintain pluripotency of human naïve pluripotent stem cells. Elife. 2021;10:e67259. doi: 10.7554/eLife.67259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bergert M., Lembo S., Sharma S., Russo L., Milovanović D., Gretarsson K.H., Börmel M., Neveu P.A., Hackett J.A., Petsalaki E., Diz-Muñoz A. Cell surface mechanics gate embryonic stem cell differentiation. Cell Stem Cell. 2021;28:209–216.e4. doi: 10.1016/j.stem.2020.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Theunissen T.W., Friedli M., He Y., Planet E., O'Neil R.C., Markoulaki S., Pontis J., Wang H., Iouranova A., Imbeault M., et al. Molecular criteria for defining the naive human pluripotent state. Cell Stem Cell. 2016;19:502–515. doi: 10.1016/j.stem.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sahakyan A., Kim R., Chronis C., Sabri S., Bonora G., Theunissen T.W., Kuoy E., Langerman J., Clark A.T., Jaenisch R., Plath K. Human naive pluripotent stem cells model X chromosome dampening and X inactivation. Cell Stem Cell. 2017;20:87–101. doi: 10.1016/j.stem.2016.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Guo G., von Meyenn F., Rostovskaya M., Clarke J., Dietmann S., Baker D., Sahakyan A., Myers S., Bertone P., Reik W., et al. Epigenetic resetting of human pluripotency. Development. 2017;144:2748–2763. doi: 10.1242/dev.146811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Haaf T. The effects of 5-azacytidine and 5-azadeoxycytidine on chromosome structure and function: implications for methylation-associated cellular processes. Pharmacol. Ther. 1995;65:19–46. doi: 10.1016/0163-7258(94)00053-6. [DOI] [PubMed] [Google Scholar]
- 69.Bar S., Schachter M., Eldar-Geva T., Benvenisty N. Large-scale Analysis of loss of imprinting in human pluripotent stem cells. Cell Rep. 2017;19:957–968. doi: 10.1016/j.celrep.2017.04.020. [DOI] [PubMed] [Google Scholar]
- 70.Sharp A.J., Stathaki E., Migliavacca E., Brahmachary M., Montgomery S.B., Dupre Y., Antonarakis S.E. DNA methylation profiles of human active and inactive X chromosomes. Genome Res. 2011;21:1592–1600. doi: 10.1101/gr.112680.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Okamoto I., Patrat C., Thépot D., Peynot N., Fauque P., Daniel N., Diabangouaya P., Wolf J.P., Renard J.P., Duranthon V., Heard E. Eutherian mammals use diverse strategies to initiate X-chromosome inactivation during development. Nature. 2011;472:370–374. doi: 10.1038/nature09872. [DOI] [PubMed] [Google Scholar]
- 72.Petropoulos S., Edsgärd D., Reinius B., Deng Q., Panula S.P., Codeluppi S., Plaza Reyes A., Linnarsson S., Sandberg R., Lanner F. Single-cell RNA-seq reveals lineage and X chromosome dynamics in human preimplantation embryos. Cell. 2016;165:1012–1026. doi: 10.1016/j.cell.2016.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mulas C., Kalkan T., Smith A. NODAL secures pluripotency upon embryonic stem cell progression from the ground state. Stem Cell Rep. 2017;9:77–91. doi: 10.1016/j.stemcr.2017.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sakaki-Yumoto M., Liu J., Ramalho-Santos M., Yoshida N., Derynck R. Smad2 is essential for maintenance of the human and mouse primed pluripotent stem cell state. J. Biol. Chem. 2013;288:18546–18560. doi: 10.1074/jbc.M112.446591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Armstrong L., Hughes O., Yung S., Hyslop L., Stewart R., Wappler I., Peters H., Walter T., Stojkovic P., Evans J., et al. The role of PI3K/AKT, MAPK/ERK and NFkappabeta signalling in the maintenance of human embryonic stem cell pluripotency and viability highlighted by transcriptional profiling and functional analysis. Hum. Mol. Genet. 2006;15:1894–1913. doi: 10.1093/hmg/ddl112. [DOI] [PubMed] [Google Scholar]
- 76.Dalton S. Signaling networks in human pluripotent stem cells. Curr. Opin. Cell Biol. 2013;25:241–246. doi: 10.1016/j.ceb.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yu J.S.L., Cui W. Proliferation, survival and metabolism: the role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development. 2016;143:3050–3060. doi: 10.1242/dev.137075. [DOI] [PubMed] [Google Scholar]
- 78.Bai H., Chen K., Gao Y.X., Arzigian M., Xie Y.L., Malcosky C., Yang Y.G., Wu W.S., Wang Z.Z. Bcl-xL enhances single-cell survival and expansion of human embryonic stem cells without affecting self-renewal. Stem Cell Res. 2012;8:26–37. doi: 10.1016/j.scr.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xu Y., Zhu X., Hahm H.S., Wei W., Hao E., Hayek A., Ding S. Revealing a core signaling regulatory mechanism for pluripotent stem cell survival and self-renewal by small molecules. Proc. Natl. Acad. Sci. USA. 2010;107:8129–8134. doi: 10.1073/pnas.1002024107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Watanabe K., Ueno M., Kamiya D., Nishiyama A., Matsumura M., Wataya T., Takahashi J.B., Nishikawa S., Nishikawa S.i., Muguruma K., Sasai Y. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat. Biotechnol. 2007;25:681–686. doi: 10.1038/nbt1310. [DOI] [PubMed] [Google Scholar]
- 81.Laurent L.C., Ulitsky I., Slavin I., Tran H., Schork A., Morey R., Lynch C., Harness J.V., Lee S., Barrero M.J., et al. Dynamic changes in the copy number of pluripotency and cell proliferation genes in human ESCs and iPSCs during reprogramming and time in culture. Cell Stem Cell. 2011;8:106–118. doi: 10.1016/j.stem.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lund R.J., Närvä E., Lahesmaa R. Genetic and epigenetic stability of human pluripotent stem cells. Nat. Rev. Genet. 2012;13:732–744. doi: 10.1038/nrg3271. [DOI] [PubMed] [Google Scholar]
- 83.Bai Q., Ramirez J.M., Becker F., Pantesco V., Lavabre-Bertrand T., Hovatta O., Lemaître J.M., Pellestor F., De Vos J. Temporal analysis of genome alterations induced by single-cell passaging in human embryonic stem cells. Stem Cells Dev. 2015;24:653–662. doi: 10.1089/scd.2014.0292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kim M., Rhee J.K., Choi H., Kwon A., Kim J., Lee G.D., Jekarl D.W., Lee S., Kim Y., Kim T.M. Passage-dependent accumulation of somatic mutations in mesenchymal stromal cells during in vitro culture revealed by whole genome sequencing. Sci. Rep. 2017;7:14508. doi: 10.1038/s41598-017-15155-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Guo F., Li J., Du W., Zhang S., O'Connor M., Thomas G., Kozma S., Zingarelli B., Pang Q., Zheng Y. mTOR regulates DNA damage response through NF-κB-mediated FANCD2 pathway in hematopoietic cells. Leukemia. 2013;27:2040–2046. doi: 10.1038/leu.2013.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Goldring C.E.P., Duffy P.A., Benvenisty N., Andrews P.W., Ben-David U., Eakins R., French N., Hanley N.A., Kelly L., Kitteringham N.R., et al. Assessing the safety of stem cell therapeutics. Cell Stem Cell. 2011;8:618–628. doi: 10.1016/j.stem.2011.05.012. [DOI] [PubMed] [Google Scholar]
- 87.Taapken S.M., Nisler B.S., Newton M.A., Sampsell-Barron T.L., Leonhard K.A., McIntire E.M., Montgomery K.D. Karotypic abnormalities in human induced pluripotent stem cells and embryonic stem cells. Nat. Biotechnol. 2011;29:313–314. doi: 10.1038/nbt.1835. [DOI] [PubMed] [Google Scholar]
- 88.Draper J.S., Moore H.D., Ruban L.N., Gokhale P.J., Andrews P.W. Culture and characterization of human embryonic stem cells. Stem Cells Dev. 2004;13:325–336. doi: 10.1089/scd.2004.13.325. [DOI] [PubMed] [Google Scholar]
- 89.Martins-Taylor K., Nisler B.S., Taapken S.M., Compton T., Crandall L., Montgomery K.D., Lalande M., Xu R.H. Recurrent copy number variations in human induced pluripotent stem cells. Nat. Biotechnol. 2011;29:488–491. doi: 10.1038/nbt.1890. [DOI] [PubMed] [Google Scholar]
- 90.Merkle F.T., Ghosh S., Kamitaki N., Mitchell J., Avior Y., Mello C., Kashin S., Mekhoubad S., Ilic D., Charlton M., et al. Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nature. 2017;545:229–233. doi: 10.1038/nature22312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Carter M.G., Smagghe B.J., Stewart A.K., Rapley J.A., Lynch E., Bernier K.J., Keating K.W., Hatziioannou V.M., Hartman E.J., Bamdad C.C. A primitive growth factor, NME7AB , is sufficient to induce stable naïve state human pluripotency; reprogramming in this novel growth factor confers superior differentiation. Stem Cell. 2016;34:847–859. doi: 10.1002/stem.2261. [DOI] [PubMed] [Google Scholar]
- 92.Zimmerlin L., Park T.S., Huo J.S., Verma K., Pather S.R., Talbot C.C., Jr., Agarwal J., Steppan D., Zhang Y.W., Considine M., et al. Tankyrase inhibition promotes a stable human naïve pluripotent state with improved functionality. Development. 2016;143:4368–4380. doi: 10.1242/dev.138982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yang Y., Liu B., Xu J., Wang J., Wu J., Shi C., Xu Y., Dong J., Wang C., Lai W., et al. Derivation of pluripotent stem cells with in vivo embryonic and extraembryonic potency. Cell. 2017;169:243–257.e25. doi: 10.1016/j.cell.2017.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bar S., Seaton L.R., Weissbein U., Eldar-Geva T., Benvenisty N. Global characterization of X chromosome inactivation in human pluripotent stem cells. Cell Rep. 2019;27:20–29.e3. doi: 10.1016/j.celrep.2019.03.019. [DOI] [PubMed] [Google Scholar]
- 95.Iuchi S., Cole S.T., Lin E.C. Multiple regulatory elements for the glpA operon encoding anaerobic glycerol-3-phosphate dehydrogenase and the glpD operon encoding aerobic glycerol-3-phosphate dehydrogenase in Escherichia coli: further characterization of respiratory control. J. Bacteriol. 1990;172:179–184. doi: 10.1128/jb.172.1.179-184.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Inzunza J., Sahlén S., Holmberg K., Strömberg A.M., Teerijoki H., Blennow E., Hovatta O., Malmgren H. Comparative genomic hybridization and karyotyping of human embryonic stem cells reveals the occurrence of an isodicentric X chromosome after long-term cultivation. Mol. Hum. Reprod. 2004;10:461–466. doi: 10.1093/molehr/gah051. [DOI] [PubMed] [Google Scholar]
- 97.Chiu C.G., St-Pierre P., Nabi I.R., Wiseman S.M. Autocrine motility factor receptor: a clinical review. Expert Rev. Anticancer Ther. 2008;8:207–217. doi: 10.1586/14737140.8.2.207. [DOI] [PubMed] [Google Scholar]
- 98.Nakajima K., Raz A. Autocrine motility factor and its receptor expression in musculoskeletal tumors. J. Bone Oncol. 2020;24:100318. doi: 10.1016/j.jbo.2020.100318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tsai Y.C., Mendoza A., Mariano J.M., Zhou M., Kostova Z., Chen B., Veenstra T., Hewitt S.M., Helman L.J., Khanna C., Weissman A.M. The ubiquitin ligase gp78 promotes sarcoma metastasis by targeting KAI1 for degradation. Nat. Med. 2007;13:1504–1509. doi: 10.1038/nm1686. [DOI] [PubMed] [Google Scholar]
- 100.Jiang W.G., Raz A., Douglas-Jones A., Mansel R.E. Expression of autocrine motility factor (AMF) and its receptor, AMFR, in human breast cancer. J. Histochem. Cytochem. 2006;54:231–241. doi: 10.1369/jhc.5A6785.2005. [DOI] [PubMed] [Google Scholar]
- 101.Huang Z., Zhang N., Zha L., Mao H.C., Chen X., Xiang J.F., Zhang H., Wang Z.W. Aberrant expression of the autocrine motility factor receptor correlates with poor prognosis and promotes metastasis in gastric carcinoma. Asian Pac. J. Cancer Prev. 2014;15:989–997. doi: 10.7314/apjcp.2014.15.2.989. [DOI] [PubMed] [Google Scholar]
- 102.Nakayama K.I., Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat. Rev. Cancer. 2006;6:369–381. doi: 10.1038/nrc1881. [DOI] [PubMed] [Google Scholar]
- 103.Deng P., Wu Y. Knockdown of miR-106a suppresses migration and invasion and enhances radiosensitivity of hepatocellular carcinoma cells by upregulating FBXW7. Int. J. Clin. Exp. Pathol. 2019;12:1184–1193. [PMC free article] [PubMed] [Google Scholar]
- 104.Avior Y., Eggan K., Benvenisty N. Cancer-related mutations identified in primed and naive human pluripotent stem cells. Cell Stem Cell. 2019;25:456–461. doi: 10.1016/j.stem.2019.09.001. [DOI] [PubMed] [Google Scholar]
- 105.Stirparo G.G., Smith A., Guo G. Cancer-related mutations are not enriched in naive human pluripotent stem cells. Cell Stem Cell. 2021;28:164–169.e2. doi: 10.1016/j.stem.2020.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Allegrucci C., Wu Y.Z., Thurston A., Denning C.N., Priddle H., Mummery C.L., Ward-van Oostwaard D., Andrews P.W., Stojkovic M., Smith N., et al. Restriction landmark genome scanning identifies culture-induced DNA methylation instability in the human embryonic stem cell epigenome. Hum. Mol. Genet. 2007;16:1253–1268. doi: 10.1093/hmg/ddm074. [DOI] [PubMed] [Google Scholar]
- 107.Choi J., Huebner A.J., Clement K., Walsh R.M., Savol A., Lin K., Gu H., Di Stefano B., Brumbaugh J., Kim S.Y., et al. Prolonged Mek1/2 suppression impairs the developmental potential of embryonic stem cells. Nature. 2017;548:219–223. doi: 10.1038/nature23274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nestor C.E., Ottaviano R., Reinhardt D., Cruickshanks H.A., Mjoseng H.K., McPherson R.C., Lentini A., Thomson J.P., Dunican D.S., Pennings S., et al. Rapid reprogramming of epigenetic and transcriptional profiles in mammalian culture systems. Genome Biol. 2015;16:11. doi: 10.1186/s13059-014-0576-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Casey T., Patel O.V., Plaut K. Transcriptomes reveal alterations in gravity impact circadian clocks and activate mechanotransduction pathways with adaptation through epigenetic change. Physiol. Genomics. 2015;47:113–128. doi: 10.1152/physiolgenomics.00117.2014. [DOI] [PubMed] [Google Scholar]
- 110.Arthur S.E., Sorgeloos F., Hosmillo M., Goodfellow I.G. Epigenetic suppression of interferon lambda receptor expression leads to enhanced human norovirus replication in vitro. mBio. 2019;10 doi: 10.1128/mBio.02155-19. 021555-e2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dong C., Beltcheva M., Gontarz P., Zhang B., Popli P., Fischer L.A., Khan S.A., Park K.M., Yoon E.J., Xing X., et al. Derivation of trophoblast stem cells from naïve human pluripotent stem cells. Elife. 2020;9:e52504. doi: 10.7554/eLife.52504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chan Y.S., Göke J., Ng J.H., Lu X., Gonzales K.A.U., Tan C.P., Tng W.Q., Hong Z.Z., Lim Y.S., Ng H.H. Induction of a human pluripotent state with distinct regulatory circuitry that resembles preimplantation epiblast. Cell Stem Cell. 2013;13:663–675. doi: 10.1016/j.stem.2013.11.015. [DOI] [PubMed] [Google Scholar]
- 113.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Van der Auwera G.A., Carneiro M.O., Hartl C., Poplin R., Del Angel G., Levy-Moonshine A., Jordan T., Shakir K., Roazen D., Thibault J., et al. From FastQ data to high confidence variant calls: the genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinformatics. 2013;43:11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Talevich E., Shain A.H., Botton T., Bastian B.C. CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput. Biol. 2016;12:e1004873. doi: 10.1371/journal.pcbi.1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhu M., Need A.C., Han Y., Ge D., Maia J.M., Zhu Q., Heinzen E.L., Cirulli E.T., Pelak K., He M., et al. Using ERDS to infer copy-number variants in high-coverage genomes. Am. J. Hum. Genet. 2012;91:408–421. doi: 10.1016/j.ajhg.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Priestley P., Baber J., Lolkema M.P., Steeghs N., de Bruijn E., Shale C., Duyvesteyn K., Haidari S., van Hoeck A., Onstenk W., et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature. 2019;575:210–216. doi: 10.1038/s41586-019-1689-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 122.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Korotkevich G., Sukhov V., Budin N., Shpak B., Artyomov M.N., Sergushichev A. Fast gene set enrichment analysis. bioRxiv. 2021 doi: 10.1101/060012. Preprint at. [DOI] [Google Scholar]
- 124.Rozowsky J., Kitchen R.R., Park J.J., Galeev T.R., Diao J., Warrell J., Thistlethwaite W., Subramanian S.L., Milosavljevic A., Gerstein M. exceRpt: a comprehensive analytic platform for extracellular RNA profiling. Cell Syst. 2019;8:352–357.e3. doi: 10.1016/j.cels.2019.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chen E.Y., Tan C.M., Kou Y., Duan Q., Wang Z., Meirelles G.V., Clark N.R., Ma'ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinf. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kuleshov M.V., Jones M.R., Rouillard A.D., Fernandez N.F., Duan Q., Wang Z., Koplev S., Jenkins S.L., Jagodnik K.M., Lachmann A., et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–W97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Xie Z., Bailey A., Kuleshov M.V., Clarke D.J.B., Evangelista J.E., Jenkins S.L., Lachmann A., Wojciechowicz M.L., Kropiwnicki E., Jagodnik K.M., et al. Gene set knowledge discovery with Enrichr. Curr. Protoc. 2021;1:e90. doi: 10.1002/cpz1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Moerman T., Aibar Santos S., Bravo González-Blas C., Simm J., Moreau Y., Aerts J., Aerts S. GRNBoost2 and Arboreto: efficient and scalable inference of gene regulatory networks. Bioinformatics. 2019;35:2159–2161. doi: 10.1093/bioinformatics/bty916. [DOI] [PubMed] [Google Scholar]
- 129.Hsu S.D., Lin F.M., Wu W.Y., Liang C., Huang W.C., Chan W.L., Tsai W.T., Chen G.Z., Lee C.J., Chiu C.M., et al. miRTarBase: a database curates experimentally validated microRNA-target interactions. Nucleic Acids Res. 2011;39:D163–D169. doi: 10.1093/nar/gkq1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Huang H.Y., Lin Y.C.D., Li J., Huang K.Y., Shrestha S., Hong H.C., Tang Y., Chen Y.G., Jin C.N., Yu Y., et al. miRTarBase 2020: updates to the experimentally validated microRNA-target interaction database. Nucleic Acids Res. 2020;48:D148–D154. doi: 10.1093/nar/gkz896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wolf F.A., Angerer P., Theis F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Aryee M.J., Jaffe A.E., Corrada-Bravo H., Ladd-Acosta C., Feinberg A.P., Hansen K.D., Irizarry R.A. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–1369. doi: 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wei T., Nie J., Larson N.B., Ye Z., Eckel-Passow J.E., Robertson K.D., Kocher J.P.A., Wang L. CpGtools: a python package for DNA methylation analysis. Bioinformatics. 2021;37:1598–1599. doi: 10.1093/bioinformatics/btz916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mootha V.K., Lindgren C.M., Eriksson K.F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstråle M., Laurila E., et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 136.Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J.P., Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O'Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B., et al. Analysis of protein-coding genetic variation in 60, 706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Forbes S.A., Beare D., Boutselakis H., Bamford S., Bindal N., Tate J., Cole C.G., Ward S., Dawson E., Ponting L., et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017;45:D777–D783. doi: 10.1093/nar/gkw1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Pidsley R., Zotenko E., Peters T.J., Lawrence M.G., Risbridger G.P., Molloy P., Van Djik S., Muhlhausler B., Stirzaker C., Clark S.J. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016;17:208. doi: 10.1186/s13059-016-1066-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Balaton B.P., Cotton A.M., Brown C.J. Derivation of consensus inactivation status for X-linked genes from genome-wide studies. Biol. Sex Differ. 2015;6:35. doi: 10.1186/s13293-015-0053-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Differentially expressed genes (adj. p-value < 0.05, basemean >50, paired analysis by the line and timepoint) between NHSM and isogenic primed cultures at similar timepoints. The table contains normalized data from all 60 samples with a column indicating whether the gene was up or downregulated in our NHSM cultures compared with our primed.
Gene set enrichment analysis for upregulated and downregulated genes in the NHSM cultures compared with their isogenic primed cultures at similar time points using WikiPathways 2019 in Enrichr.
Gene ontology enrichment analysis (biological processes) for the 177 mRNAs uniquely upregulated at the earliest NHSM derived (p20) timepoint.
Gene ontology enrichment analysis (biological processes) for the 157 mRNAs uniquely downregulated at the earliest NHSM (p20, NHSM derived) timepoint.
The table shows six of the seven miRPathDB included miRNAs are in the top 20 most similar miRNAs by “Jaccard coefficient (target genes strong)”.
Gene ontology enrichment analysis (biological process 2021) of downregulated genes also targeted by a miRNA upregulated in NHSM derived (p20) cultures. Refers to data in Figure 5B.
Alleles with at least 10 reads were considered in analysis. An allele was considered biallelic if the allelic frequency of both the reference and alternative allele were over 30%.
Data Availability Statement
-
•
Whole genome sequencing data has been deposited at NCBI’s SRA (BioProject: PRJNA859118). RNA-seq (GSE208300), small RNA-seq (GSE208301), and methylation array data (GSE208299) have been deposited at GEO and are publicly available under the accession number GSE208302. All data can be found under the umbrella project PRJNA859148.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.