

Abstract
Background:
Obesity predisposes individuals to metabolic syndrome, which increases the risk of cardiovascular diseases, non-alcoholic fatty liver disease (NAFLD) and type II diabetes. A pathological manifestation of obesity is the activation of the coagulation system. In turn, extravascular fibrin(ogen) deposits accumulate in adipose tissues and liver. These deposits promote adiposity and downstream sequelae by driving pro-inflammatory macrophage function through binding the leukocyte integrin receptor αMβ2.
Objectives:
An unresolved question is whether conversion of soluble fibrinogen to a crosslinked fibrin matrix is required to exacerbate obesity-driven diseases.
Methods:
Here, fibrinogen-deficient/depleted mice (Fib- or treated with siRNA against fibrinogen [siFga]), mice expressing fibrinogen that cannot polymerize to fibrin (FibAEK), and mice deficient in the fibrin crosslinking transglutaminase factor XIII (FXIII-) were challenged with a high-fat diet (HFD) and compared to mice expressing a mutant form of fibrinogen lacking the αMβ2-binding domain (Fib𝛾390-396A).
Results and Conclusions:
Consistent with prior studies, Fib𝛾390-396A mice were significantly protected from increased adiposity, NAFLD, hypercholesterolemia and diabetes while Fib- and siFga-treated mice gained as much weight and developed obesity-associated pathologies identical to wildtype mice. FibAEK and FXIII- mice displayed an intermediate phenotype with partial protection from some obesity-associated pathologies. Results here indicate that fibrin(ogen) lacking αMβ2 binding function offers substantial protection from obesity and associated disease that is partially recapitulated by preventing fibrin polymer formation or crosslinking of the wildtype molecule, but not by reduction or complete elimination of fibrinogen. Finally, these findings support the concept that fibrin polymerization and crosslinking are required for the full implementation of fibrin-driven inflammation in obesity.
Keywords: fibrinogen, factor XIII, obesity, fatty liver disease, diabetes
Introduction
Obesity, defined as body mass index of >30 kg/m2, is a global health issue and affects over 40% of the US population.1 Obesity is characterized by chronic low-grade inflammation and pathological accumulation of fatty acids in tissues that alters healthy metabolic function (e.g., glucose homeostasis). As such, obesity is a risk factor for metabolic syndrome, a constellation of pathological conditions that increases the risk for non-alcoholic fatty liver disease (NAFLD), type 2 diabetes, insulin resistance and cardiovascular diseases.2–4 Enlarged visceral adipose tissues, such as omental and mesenteric white adipose tissue (WAT) in humans and epididymal WAT (eWAT) in rodents, is associated with increased macrophage-driven inflammation that exacerbates insulin resistance and subsequently metabolism of glucose, cholesterol and lipids.5
A mechanistic underpinning for many of the obesity-associated sequelae is that obesity promotes a procoagulant and anti-fibrinolytic state that ultimately increases the risk of cardiovascular events. Circulating levels of procoagulant proteins such as von Willebrand factor, factor VII, and fibrinogen are commonly elevated in obese individuals.6–9 Further, elevated levels of biomarkers of coagulation activity, including thrombin-antithrombin complexes, thrombin f1+2, and D-dimer are routinely observed in obese individuals.10,11 Importantly, both initial risk and severe morbidity and mortality outcomes for vaso-occlusive diseases, including coronary artery disease and stroke, are positively correlated with the degree of obesity in patients.12,13 Collectively, these observations support a causal link between obesity and an increased risk for thromboembolic disease.
Elevated coagulation system activity is not just consequence of obesity but is also a contributing factor to the severity of obesity, metabolic syndrome and their associated sequelae. Extravascular fibrin(ogen) deposits accumulate in the adipose tissue and the liver of obese individuals.14 Fibrin(ogen) co-localizes with pro-inflammatory macrophages within crown-like structures of obese white adipose tissue and areas surrounding steatosis in the liver.14 Genetically modified Fibγ390-396A mice, which express fibrinogen with a mutated integrin αMβ2 binding motif, were previously shown to be protected from developing high fat diet (HFD)-induced obesity, insulin resistance and NAFLD.14 These studies supported a mechanism of action whereby fibrin-driven macrophage-mediated metabolic inflammation promotes HFD-induced obesity and associated pathologies, such as NAFLD and insulin resistance.
Fibrinogen contains a cryptic binding site for the integrin receptor αMβ2, expressed by macrophages and neutrophils, such that only immobilized fibrinogen or fibrin matrix, and not soluble fibrinogen, engages the receptor.15,16 An unresolved question is whether the αMβ2−binding motif requires fibrin polymerization and crosslinking for the full implementation of fibrin(ogen)-mediated HFD-induced obesity and associated pathologies. In this study, the contributions of specific molecular forms of fibrinogen in promoting HFD-driven obesity were explored using Fibγ390-396A mice, mice that are genetically deficient or pharmacologically depleted in fibrinogen (Fib- and siRNA targeting fibrinogen [siFga]-treated, respectively).17–19 In addition, to differentiate between distinct fibrin(ogen) molecular forms, we utilized mice expressing fibrinogen locked in the monomeric form (FibAEK) and mice deficient in the fibrin crosslinking transglutaminase FXIII (FXIII-).20,21
Materials and Methods
Mice.
Animal experiments and protocols were approved by the Animal Care and Use Committee of the University of North Carolina at Chapel Hill. Fibγ390-396A, Fib-, FibAEK and FXIII- mice were all previously described.18–21 Age-matched littermates of male mice between the ages of 6 and 10 weeks were used. For siFga experiments, male C57BL/6J mice were injected with 2 mg/kg of lipid nanoparticles containing siRNA against luciferase (siLuc) or fibrinogen Aα-chain (siFga) 1 week before the start of the HFD-challenge and subsequently every 10–11 days to minimize fluctuations in circulating fibrinogen levels.17 Mice were fed ad libitum low fat diet (LFD, 10–13% fat; Laboratory Autoclavable Rodent Diet 5010, LabDiet or D12450, Research Diets) or HFD (60% fat; catalog D12492, Research Diets). The total body weights of mice and the weight of food consumed were determined weekly. At the end of the observation period, mice were anesthetized before plasma and organs were harvested, weighed, and processed for further analysis.
RNA isolation, cDNA synthesis, and real-time qPCR.
Total RNA was isolated from tissues using Trizol (Ambion) according to the manufacturer’s protocol. RNA (1 μg) was used for cDNA synthesis with a High-Capacity cDNA Reverse Transcription kit (Applied Biosystems). Levels of Tnfa, Ccl2, Adgre1, Pparg, Cidea, Cd36, Hmgcr, Cyp7a1, Cyp27a1, Ucp1, and B2m were determined using TaqMan gene expression assays (Applied Biosystems) on an ABI StepOne Plus sequence detection system (Applied Biosystems). The expression of each gene was normalized relative to B2m expression levels using the Pfaffl method.22
Analysis of diabetes and insulin resistance.
Mice were fasted for at least 6 hours prior to analysis of circulating glucose levels, as previously described.14 For glucose tolerance tests (GTT) performed on week 16 of diet, dextrose was administered by i.p. injection (1 g/kg). For insulin tolerance tests (ITT) performed on mice on week 18, human recombinant insulin (Humulin; Eli Lilly) was administered by i.p. injection (1.5 U/kg). Blood glucose readings were taken at 15-minute intervals.
Analysis of NAFLD and hypercholesterolemia.
Hepatic triglyceride content was determined as described previously.14 Briefly, frozen liver tissue was homogenized and analyzed spectrophotometrically for triglycerides using commercially available reagents (Pointe Scientific). Plasma alanine transferase (ALT) activity was determined using a commercially available reagent (Thermo Fisher Scientific). ELISA was used to determine the circulating fibrinogen levels (Immunology Consultants Laboratory). Circulating levels of total, high-density lipoprotein (HDL) and free cholesterol were measured using commercially available kits (Wako). The left lateral lobe of the liver was used for histological analysis. Formalin-fixed tissue sections (5 μm) were stained with hematoxylin and eosin (H&E) stain as well as fibrin(ogen) antibody (DAKO). Extravascular deposition of fibrin was analyzed using automated capillary Western blotting as previously described.14
Statistics.
All analyses were performed using Prism 9. Comparisons of multiple groups were performed using 2-way ANOVA and Fisher’s LSD test. Results were considered significant when p<0.05.
Results
Obesity, adiposity, and white adipose tissue inflammation
To determine whether fibrinogen, fibrin or crosslinked fibrin matrix each contribute to the development of HFD-induced obesity, Fib-, FibAEK and FXIII- mice were fed LFD or HFD diet for 20 weeks and compared to Fibγ390-396A mice. Consistent with previous reports, Fibγ390-396A mice were protected from an HFD-driven increase in total body mass (Figure 1A–1B) and had smaller eWAT and subcutaneous inguinal WAT (iWAT) mass after 20 weeks on diet (Figure 1C–1D) relative to HFD-fed FibγWT mice.14 To examine the impact of fibrinogen deficiency on HFD-induced obesity, Fib- mice were challenged with HFD. Surprisingly, Fib- mice gained as much weight as Fib+ mice on HFD and had larger eWAT and iWAT mass than Fib+ mice (Figure 1E–1H). Complementary studies with pharmacological knockdown of fibrinogen were also performed. Here, by administering a lipid nanoparticle containing siRNA directed against the fibrinogen Aα-chain (siFga), circulating fibrinogen was reduced to ~10% compared to control mice treated with siRNA against luciferase (siLuc). Over the course of 20 weeks, siFga-treated mice gained as much weight as siLuc-treated mice on HFD and displayed an equivalent increase in eWAT and iWAT mass (Figure S1A–S1E). To examine the impact of fibrin polymerization, FibAEK mice were challenged with HFD; FibAEK mice displayed modest but significant protection from HFD-induced obesity, with a significant reduction in total body weight and iWAT mass at 20 weeks, but comparable eWAT mass relative to HFD-challenged FibWT mice (Figure 1I–1L). Finally, to examine the impact of fibrin crosslinking, FXIII- mice were challenged with HFD; FXIII- mice gained as much weight as FXIII+ mice on HFD (Figure 1M–N). HFD-fed FXIII mice had an eWAT mass comparable to FXIII+ mice, but the mass of the iWAT was significantly lower in FXIII- mice relative to FXIII+ mice after 20 weeks on a HFD (Figure 1O–1P), similar to previous reports.
Figure 1. Fibγ390-396A and FibAEK mice, but not Fib- and FXIII- mice, display significant protection from HFD-induced body weight gain.
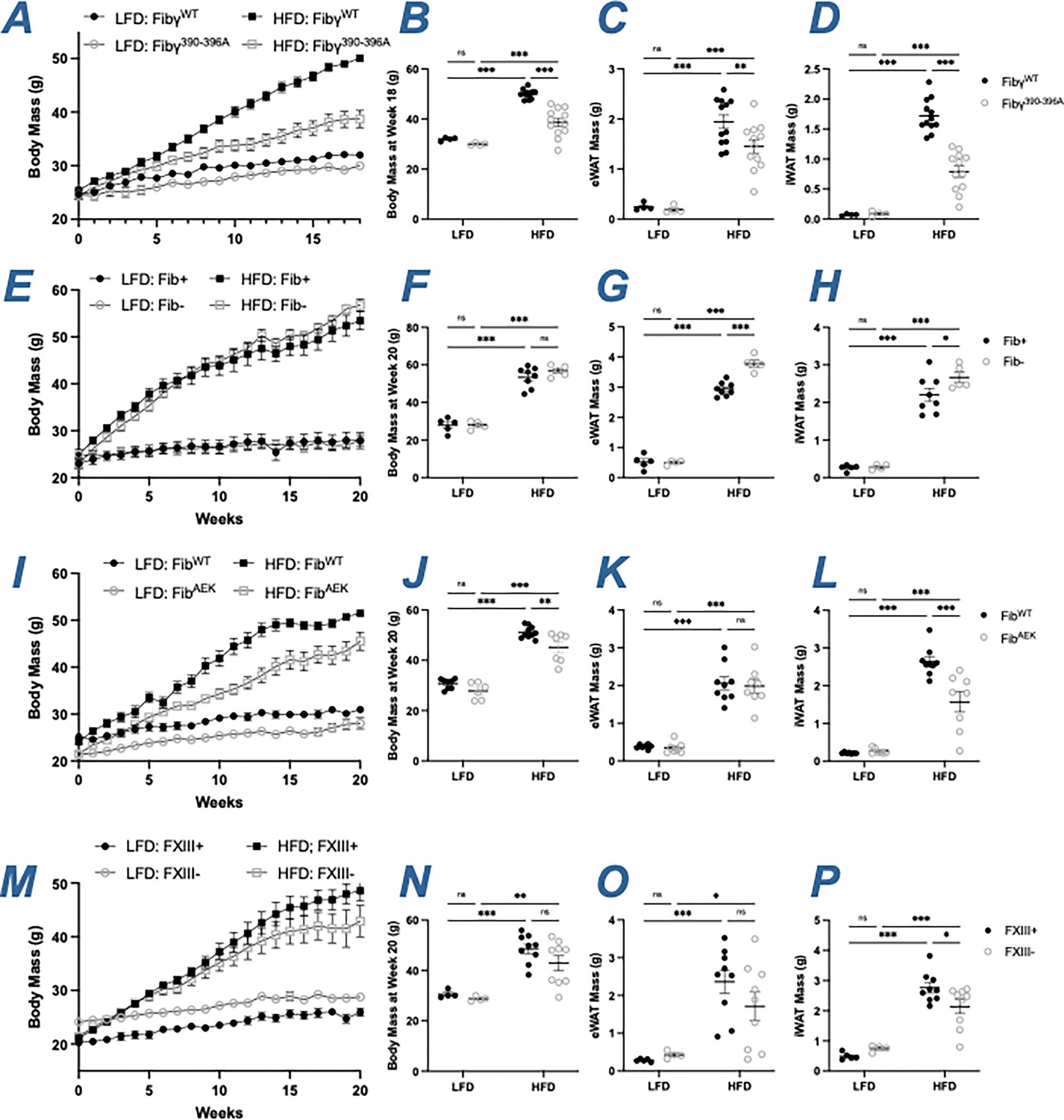
(A) Mean body weight over time of FibγWT and Fibγ390-396A mice. (B) Distribution of body weight for FibγWT and Fibγ390-396A mice after 18 weeks on diet. (C-D) Total fat pad mass of (C) eWAT and (D) iWAT for FibγWT and Fibγ390-396A mice (E) Mean body weight over time of Fib+ and Fib- mice. (F) Distribution of body weight for Fib+ and Fib- mice after 20 weeks on diet. (G-H) Total fat pad mass of (G) eWAT and (H) iWAT for Fib+ and Fib- mice. (I) Mean body weight over time of FibWT and FibAEK mice. (J) Distribution of body weight for FibWT and FibAEK mice after 20 weeks on diet. (K-L) Total fat pad mass of (K) eWAT and (L) iWAT for FibWT and FibAEK mice. (M) Mean body weight over time of FXIII+ and FXIII- mice. (N) Distribution of body weight for FXIII+ and FXIII- mice after 20 weeks on diet. (O-P) Total fat pad mass of (O) eWAT and (P) iWAT for FXIII+ and FXIII- mice. Data are expressed as mean±SEM and analyzed by 2-way ANOVA with Fisher’s LSD test. *P<0.05, **P<0.01, ***P<0.001. LFD: n=4–7; HFD: n=6–12
The development of metabolic syndrome is strongly associated with increased local inflammation in the eWAT.5 To examine local inflammatory changes, mRNA levels of proinflammatory genes Adgre1 (F4/80 receptor), Ccl2 (monocyte chemoattractant protein [MCP]-1) and Tnfa (tumor necrosis factor α) were measured. Comparative studies were performed in Fibγ390-396A, Fib-, FibAEK and FXIII- mice challenged with HFD to determine whether distinct molecular forms of fibrin(ogen) alter the local expression of macrophage-associated inflammatory mediators within the eWAT. As expected, levels of Adgre1, Ccl2 and Tnfa were low, with no genotype-dependent differences in LFD-fed mice across each of the genotypes analyzed (Figure 2A–2L). Adgre1, Ccl2 and Tnfa levels were significantly decreased in eWAT of HFD- fed Fibγ390-396A mice compared to HFD-fed FibγWT mice (Figure 2A–2C). However, there were no significant genotype-dependent differences in the expression of Adgre1, Ccl2 or Tnfa in HFD-fed Fib- and FibAEK mice relative to their respective controls. Ccl2 and Tnfa, but not Adgre1 levels were significantly decreased in eWAT of HFD-fed FXIII- relative to HFD-fed FXIII+ mice (Figure 2D–2L). Overall, protection from HFD-induced eWAT adiposity were associated with reduced local markers of macrophage-driven inflammation in eWAT.
Figure 2. Gene expression of inflammatory markers are reduced in eWAT of HFD-fed Fibγ390-396A and FXIII- mice, but not of Fib- and FibAEK mice.
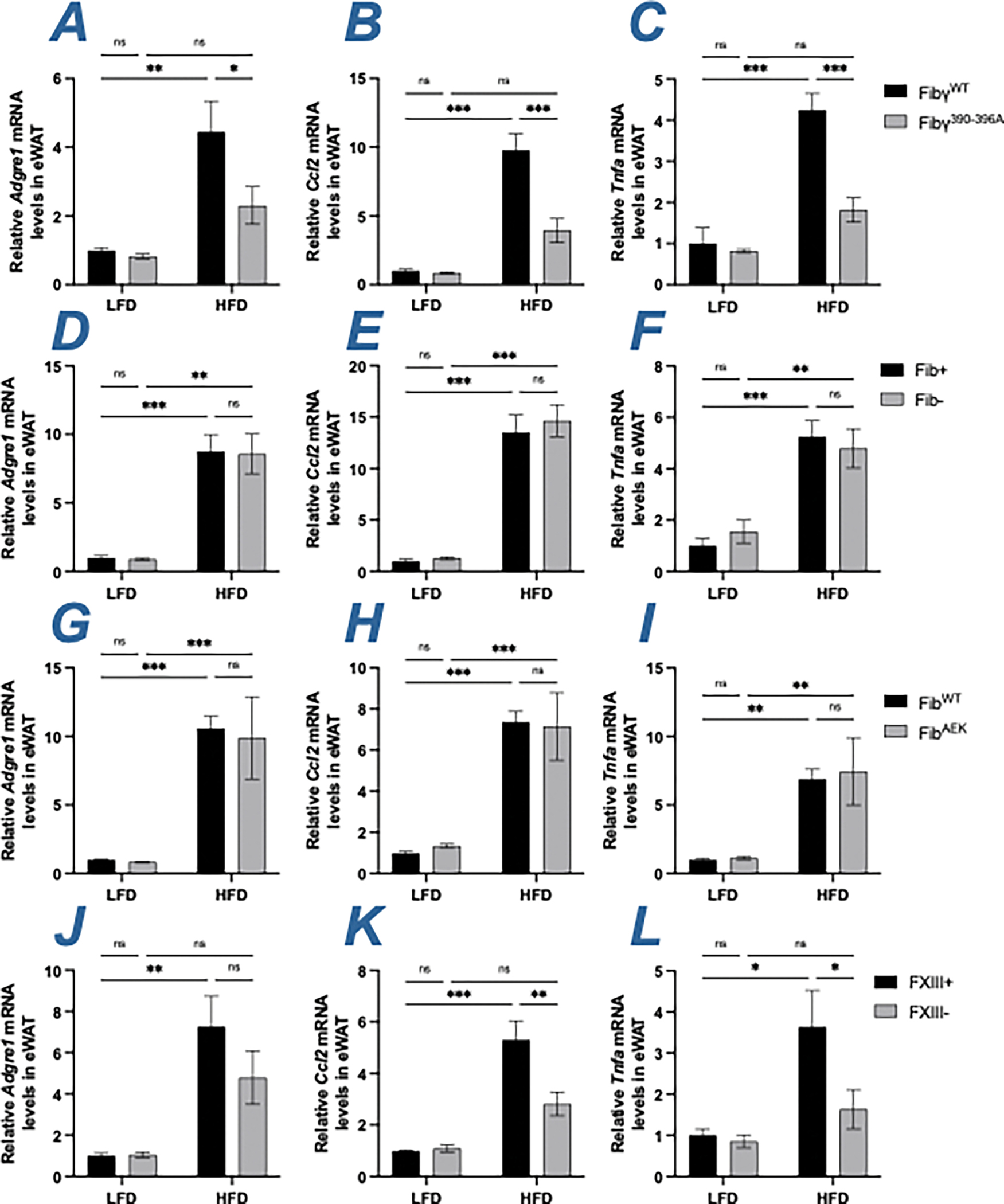
(A-C) Levels of mRNA encoding (A) F4/80 (Adgre1), (B) MCP-1 (Ccl2) and (C) Tnfα (Tnfa) in eWAT of FibγWT and Fibγ390-396A mice. (D-F) Levels of mRNA encoding (D) Adgre1, (E) Ccl2 and (F) Tnfa in eWAT of Fib+ and Fib- mice. (G-I) Levels of mRNA encoding (G) Adgre1, (H) Ccl2 and (I) Tnfa in eWAT of FibWT and FibAEK mice. (J-L) Levels of mRNA encoding (J) Adgre1, (K) Ccl2 and (L) Tnfa in eWAT of FXIII+ and FXIII- mice. Data are expressed as mean±SEM and analyzed by 2-way ANOVA with Fisher’s LSD test. *P<0.05, **P<0.01, ***P<0.001. LFD: n=3–7; HFD: n=6–10
NAFLD, hypercholesterolemia, and diabetes
NAFLD is a spectrum disorder that begins as elevated lipid accumulation within hepatocytes (steatosis) that can progress to hepatic inflammation and non-alcoholic steatohepatitis (i.e., NASH).4 Consistent with previous findings, HFD-fed Fibγ390-396A mice had smaller liver mass as well as reduced hepatic triglyceride accumulation and circulating ALT activity relative to HFD-challenged FibγWT mice, indicative of reduced hepatic steatosis and hepatocellular damage, respectively (Figure 3A–3C). Histological analysis of liver tissues also revealed evidence of fatty liver disease. Liver sections of HFD-challenged FibγWT mice displayed the accumulation of lipid within the hepatocytes as evidenced by the appearance of clear macrovesicular (large) and microvesicular (small) droplets within the cells. These droplets were either were reduced or absent in sections from HFD-challenged Fibγ390-396A mice and LFD-fed mice of each genotype (Figure 3D). In contrast, elimination of fibrinogen, either genetically or pharmacologically, did not reduce HFD-induced NAFLD development. No differences in liver mass, hepatic triglyceride accumulation, circulating ALT activity, or liver tissue histopathology were observed when comparing HFD-fed Fib+ and Fib- mice or HFD-fed siLuc-treated and siFga-treated mice (Figure 3E–3H, S1F–S1I). The severity of NAFLD in HFD-fed FibAEK mice appeared intermediate to wildtype (WT) and Fibγ390-396A mice. Liver mass and hepatic triglyceride accumulation were significantly reduced in HFD-fed FibAEK mice compared to FibWT mice (Figure 3I–3J). However, ALT activity induced by HFD only trended lower in FibAEK mice relative to FibWT animals (Figure 3K). The liver histology was also consistent with reduced hepatosteatosis in HFD-fed FibAEK mice. Intriguingly, FXIII- mice were protected from HFD-driven increase in hepatic triglyceride accumulation and circulating ALT activity (Figure 3M–3O). Moreover, liver histology revealed less apparent hepatosteatosis in HFD-fed FXIII- mice compared to FXIII+ mice (Figure 3P).
Figure 3. Fibγ390-396A mice are protected, FibAEK and FXIII- mice partially protected, and Fib- mice are not protected from developing NAFLD when challenged with HFD.
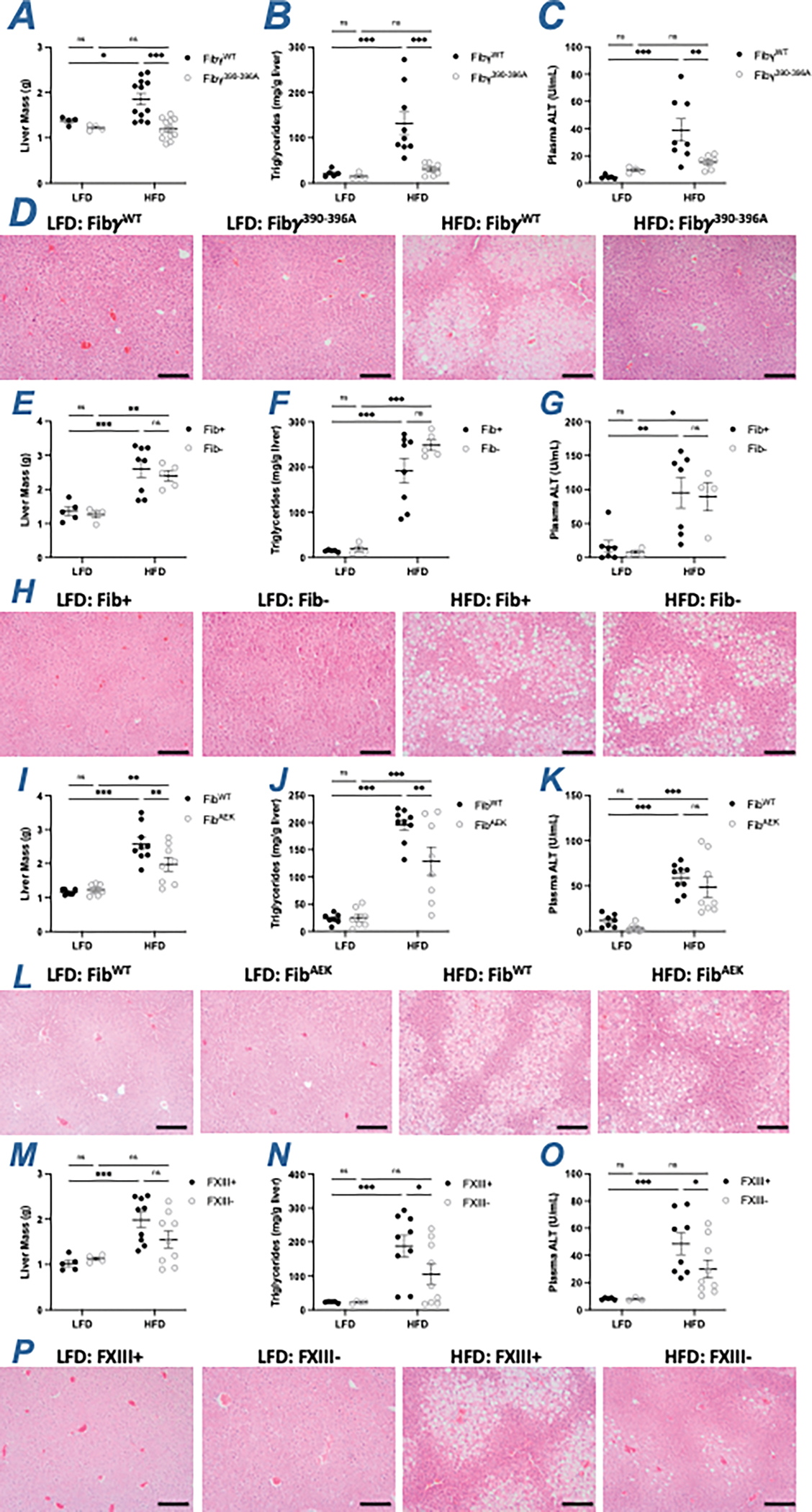
(A) Total liver mass, (B) level of hepatic triglyceride and (C) alanine transferase (ALT) activity in plasma of FibγWT and Fibγ390-396A mice. (D) Representative H&E-stained sections of livers isolated from FibγWT and Fibγ390-396A mice. (E) Total liver mass, (F) level of hepatic triglyceride and (G) ALT activity in plasma of Fib+ and Fib- mice. (H) Representative H&E-stained sections of livers isolated from Fib+ and Fib- mice. (I) Total liver mass, (J) level of hepatic triglyceride and (K) ALT activity in plasma of FibWT and FibAEK mice. (L) Representative H&E-stained sections of livers isolated from FibWT and FibAEK mice. (M) Total liver mass, (N) level of hepatic triglyceride and (O) ALT activity in plasma of FXIII+ and FXIII- mice. (P) Representative H&E-stained sections of livers isolated from FXIII+ and FXIII- mice. Magnification: 20X. Scale bar: 40 μm. Data are expressed as mean±SEM and analyzed by 2-way ANOVA with Fisher’s LSD test. *P<0.05, **P<0.01, ***P<0.001. LFD: n=4–7; HFD: n=6–12
Hepatic fibrin content was measured to determine whether NAFLD severity was associated with changes in the level of extravascular fibrin deposits in the liver. However, there were no genotype-dependent changes in hepatic fibrin(ogen) deposition in the liver measured immunohistochemically or biochemically using capillary Western blotting in HFD-fed Fibγ390-396A, FibAEK and FXIII- mice compared to their respective control cohorts (Figure S2). Local hepatic mRNA levels were measured for proinflammatory mediators, such as Adgre1, Ccl2, and Tnfa, as well as markers of lipid metabolism including the nuclear receptor Pparg, the fatty acid sensor Cidea and fatty acid transporter Cd36. As expected, the HFD-driven increases in mRNA levels of Pparg, Cidea and Cd36 observed in FibγWT mice were reduced in livers from HFD-fed Fibγ390-396A mice (Figure 4A–4F). In contrast, hepatic mRNA levels of Adgre1, Tnfa, Cidea and Cd36 were not significantly different between Fib+ and Fib- mice (Figure 4G–4L). In HFD-fed FibAEK mice, inflammatory mRNA levels were similar to FibWT mice. However, there was protection from HFD-driven increases in Pparg and Cidea levels HFD-fed FibAEK mice in comparison to HFD-fed FibWT mice. Additionally, Cd36 levels trended lower in HFD-fed FibAEK mice relative to HFD-fed FibWT mice (Figure 4M–4R). For FXIII- mice, HFD-induced hepatic mRNA levels of Adgre1, Ccl2, Tnfa and Cd36, but not Pparg and Cidea, were lower relative to HFD-fed FXIII+ mice (Figure 4S–4X). Collectively, studies of HFD-driven changes in liver weight, hepatic triglyceride accumulation, circulating ALT activity, liver histology, and hepatic gene expression suggest that prevention of fibrin polymerization and crosslinking help to suppress the development of NAFLD in a manner that mimics, but does not fully recapitulate, elimination of the fibrin(ogen)-αMβ2 binding motif.
Figure 4. Gene expression of inflammatory markers and lipid metabolism in livers of HFD-fed Fibγ390-396A Fib-, FibAEK and FXIII- mice.
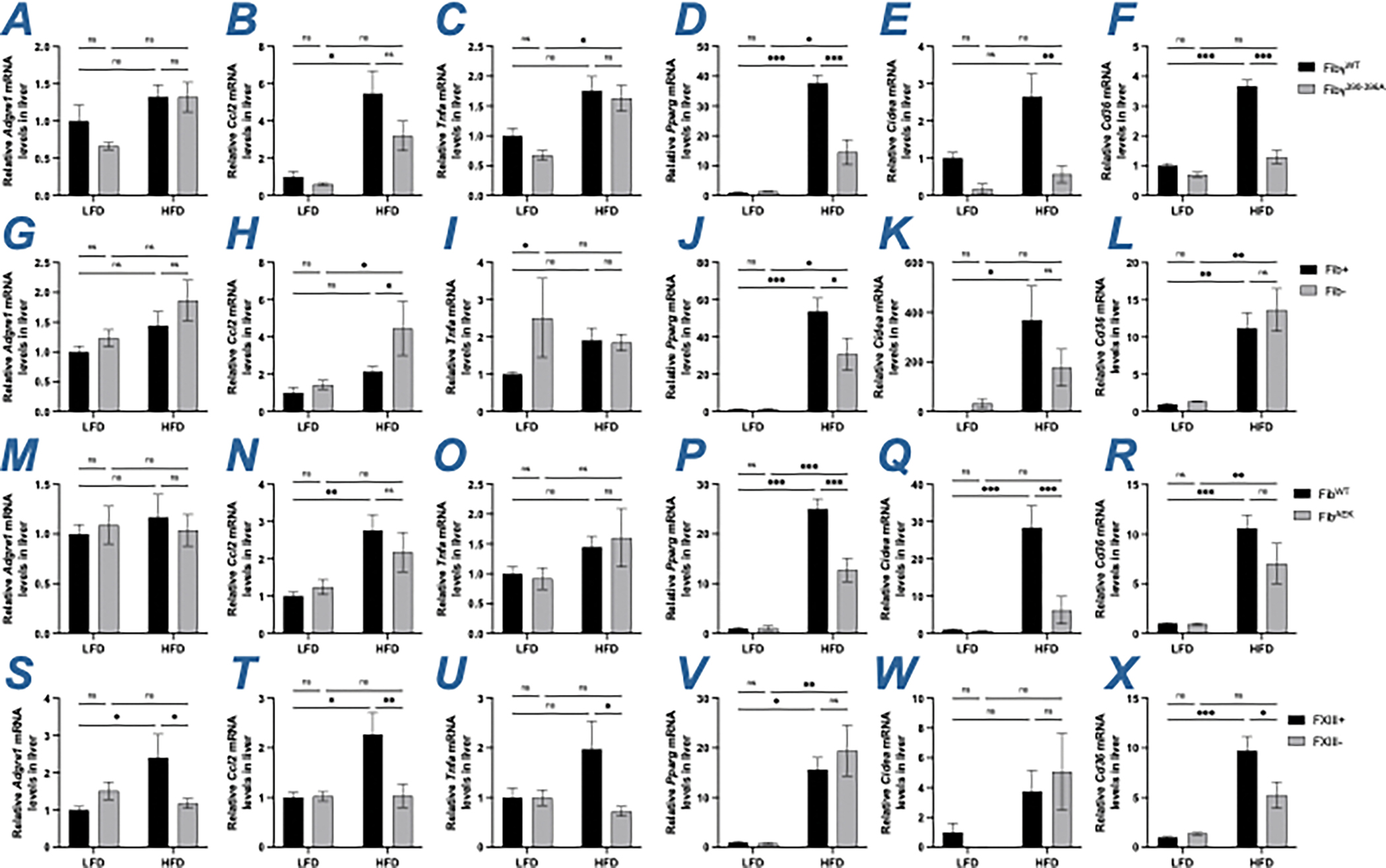
(A-F) Levels of mRNA encoding the genes (A) F4/80 (Adgre1), (B) MCP-1 (Ccl2), (C) Tnfα (Tnfa), (D) Pparγ (Pparg), (E) Cidea (Cidea) and (F) CD36 (Cd36) in livers of FibγWT and Fibγ390-396A mice. (G-L) Levels of mRNA encoding the genes (G) Adgre1, (H) Ccl2, (I) Tnfa, (J) Pparg, (K) Cidea and (L) Cd36 in livers of Fib+ and Fib- mice. (M-R) Levels of mRNA encoding the genes (M) Adgre1, (N) Ccl2, (O) Tnfa, (P) Pparg, (Q) Cidea and (R) Cd36 in livers of FibWT and FibAEK mice. (S-X) Levels of mRNA encoding the genes (S) Adgre1, (T) Ccl2), (U) Tnfa, (V) Pparg, (W) Cidea and (X) Cd36 in livers of FXIII+ and FXIII- mice. Data are expressed as mean±SEM and analyzed by 2-way ANOVA with Fisher’s LSD test. *P<0.05, **P<0.01, ***P<0.001. LFD: n=4–7; HFD: n=6–12
To evaluate the contribution of fibrin(ogen) in HFD-induced hypercholesterolemia, total cholesterol, HDL cholesterol and free cholesterol levels in plasma isolated from LFD- and HFD-challenged mice were measured. As expected, cholesterol was significantly increased in all mice following the HFD challenge (Fig 5A, 5D, 5G, 5J). HFD-fed Fibγ390-396A mice had significantly reduced levels of total, HDL and free cholesterol compared to HFD-challenged FibγWT mice (Figure 5A–5C). Fib- mice were modestly protected from HFD-driven increase in total circulating cholesterol, but not in HDL- and free cholesterol compared to Fib+ mice (Figure 5D–5F). Both FibAEK and FXIII- mice displayed modest protection against HFD-induced hypercholesterolemia, with significantly reduced levels of total, HDL, and free cholesterol relative to HFD-challenged control mice (Figure 5G–5L). The changes in cholesterol observed in Fibγ390-396A, FibAEK and FXIII- cohorts were not linked to changes in hepatic gene expression of key cholesterol regulatory molecules. Specifically, no genotype-dependent differences were observed in the hepatic mRNA levels of the cholesterol biosynthesis rate-limiting enzyme HMG-CoA reductase (Hmgcr) (Figure S3). In addition, hepatic gene expression of CYP7A1 (Cyp7a1) and CYP27A1 (Cyp27a1), key enzymes regulating the classical and alternative pathways of bile acid synthesis, respectively, were analyzed. For Cyp7a1, no differences were observed in HFD-fed Fibγ390-396A, Fib-, and FibAEK mice compared to their respective controls, whereas HFD-fed FXIII- mice had modestly higher hepatic levels than FXIII+ mice (Figure S3). For Cyp27a1, no differences in HFD-fed control and Fibγ390-396A, FibAEK, and FXIII- were observed whereas HFD-fed Fib- mice had modestly lower hepatic levels than Fib+ mice (Figure S3). These findings suggest that although fibrin(ogen) is a determinant of circulating cholesterol, the impact of fibrinogen occurs through mechanisms independent of altered hepatic cholesterol metabolism.
Figure 5. Fibγ390-396A, FibAEK and FXIII- mice, but not Fib- mice, mice are protected from HFD-associated hypercholesterolemia.
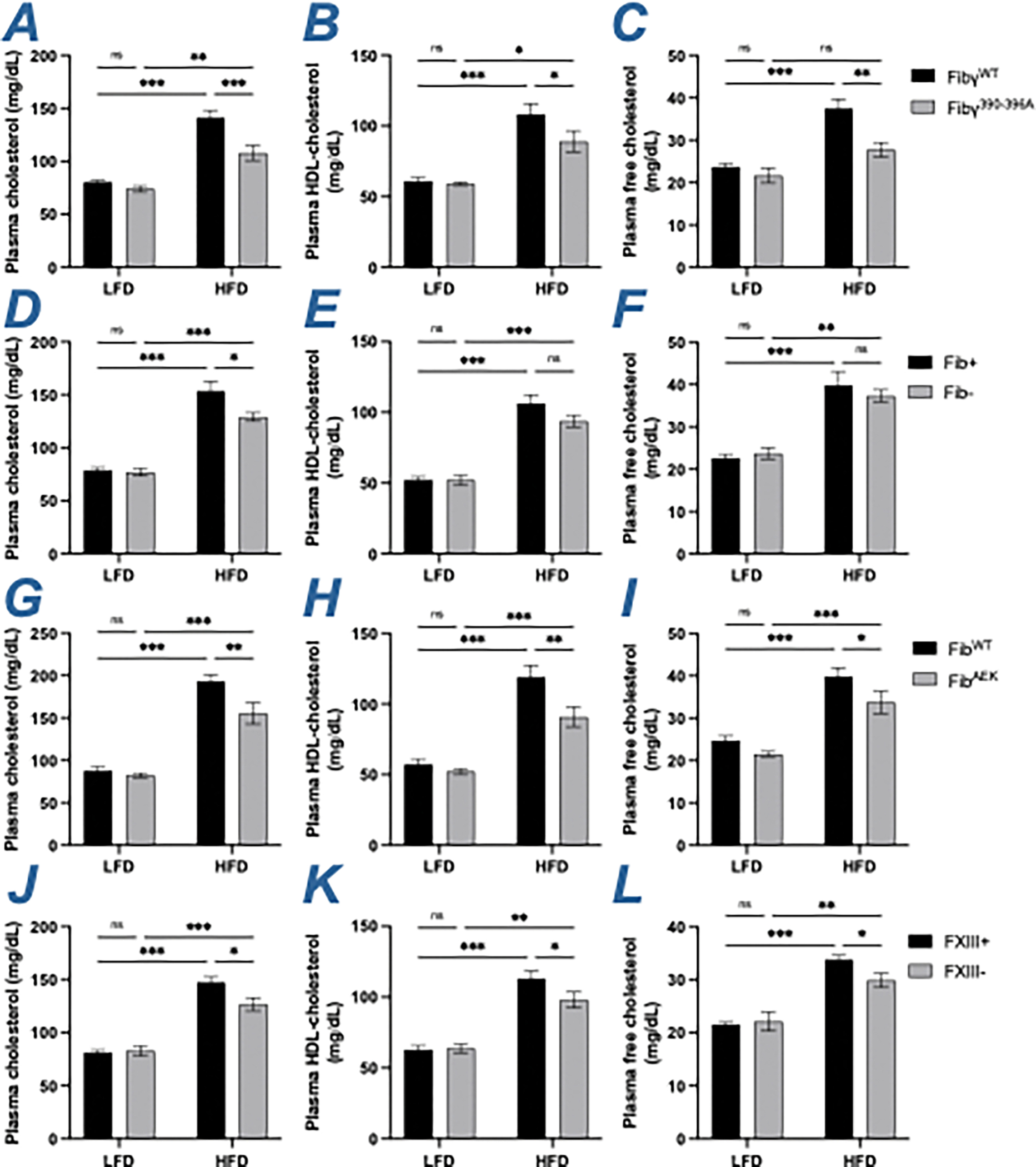
(A-C) Circulating levels of (A) total cholesterol, (B) high-density lipoprotein (HDL)-cholesterol and (C) free cholesterol in FibγWT and Fibγ390-396A mice. (D-F) Circulating levels of (D) total cholesterol, (E) HDL-cholesterol and (F) free cholesterol in Fib+ and Fib- mice. (G-I) Circulating levels of (G) total cholesterol, (H) HDL-cholesterol and (I) free cholesterol in FibWT and FibAEK mice. (J-L) Circulating levels of (J) total cholesterol, (K) HDL-cholesterol and (L) free cholesterol in FXIII+ and FXIII- mice. (V-X) Data are expressed as mean±SEM and analyzed by 2-way ANOVA with Fisher’s LSD test. *P<0.05, **P<0.01, ***P<0.001. LFD: n=4–7; HFD: n=6–12
The contribution of fibrin(ogen) to the development of HFD-driven diabetes and insulin resistance was also examined. As expected, control mice in each HFD group displayed a significant prolongation in blood glucose clearance relative to LFD control animals in glucose tolerance tests (Figure 6, S1J). Notably, only HFD-challenged Fibγ390-396A mice, but not Fib-, siFga-treated and FibAEK mice, exhibited a significantly improved glucose clearance relative to the HFD-challenged control mice (Figure 6A–6C, S1J). A previous study demonstrated that FXIII- also respond similar to WT mice following HFD-challenge in a glucose tolerance test.23 Similarly, the HFD-challenged Fibγ390-396A mice, but not HFD-challenged Fib-, siFga-treated, FibAEK and FXIII- mice, exhibited a significant improvement in response in an insulin tolerance test relative to their respective control groups (Figure S1K, S4), suggesting that changes in glucose homeostasis are particularly sensitive to elimination of the fibrin(ogen)-αMβ2 binding motif, but not perturbation of fibrin polymerization and crosslinking.
Figure 6. Fibγ390-396A mice, but not Fib- and FibAEK mice, are protected from HFD-induced glucose dysmetabolism.
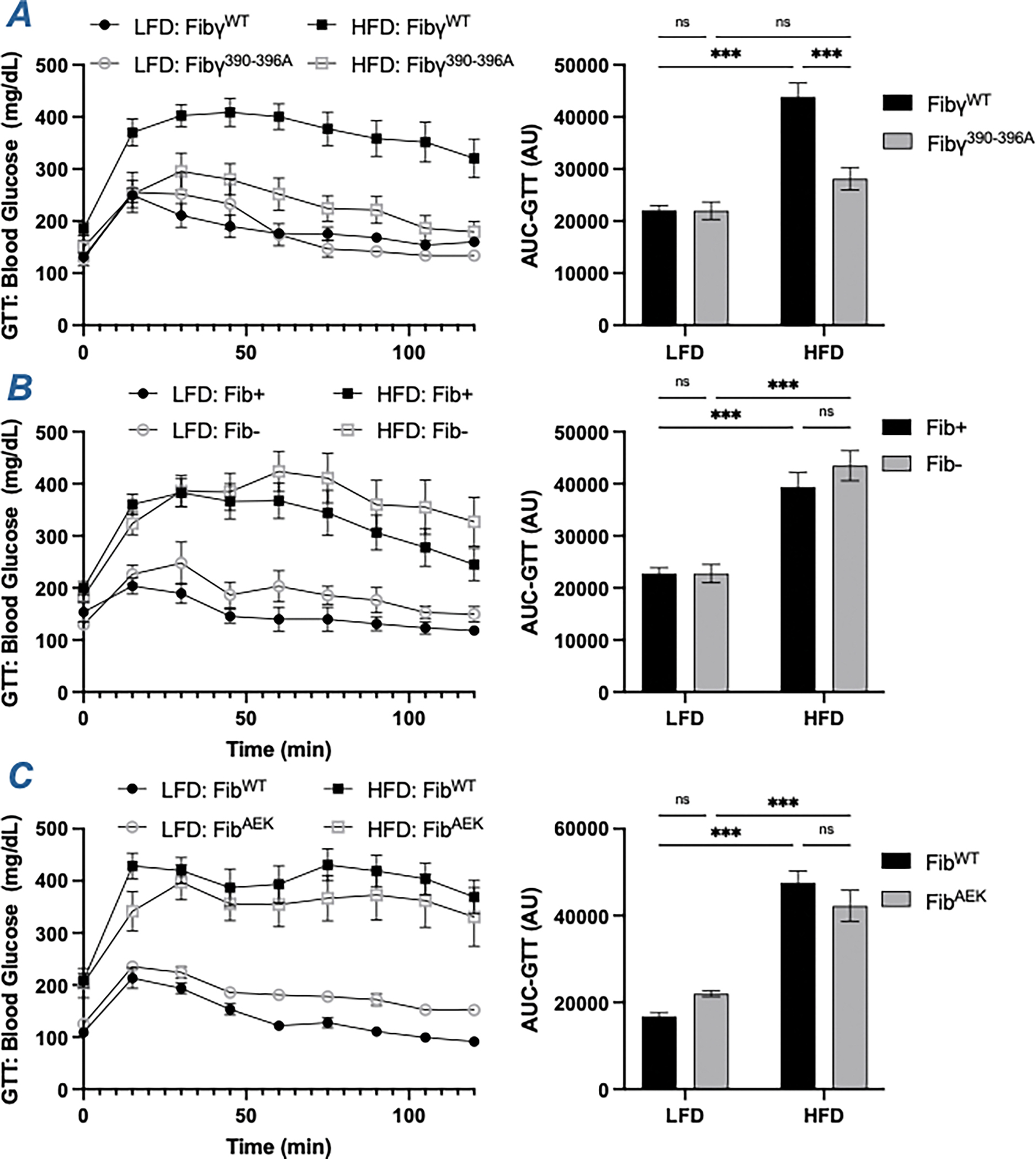
(A-C) Blood glucose levels following a glucose tolerance test at week 16 of (A) FibγWT and Fibγ390-396A mice, (B) Fib+ and Fib- mice and (C) FibWT and FibAEK mice. AUC was determined for each individual animal. Data are expressed as mean±SEM and analyzed by 2-way ANOVA with Fisher’s LSD test. *P<0.05, **P<0.01, ***P<0.001. LFD: n=4–7; HFD: n=6–9
Brown adipose tissue and thermogenesis
The contribution of fibrin(ogen) to HFD-driven reactive changes in brown adipose tissue (BAT) were also examined. In addition, mRNA levels of the regulator gene of facultative thermogenesis, uncoupling protein 1 (Ucp1), was determined. The HFD-driven increase in the BAT mass of HFD-challenged FibγWT mice was reduced in HFD-challenged Fibγ390-396A mice (Figure 7A). The mRNA level of Ucp1 was elevated in HFD-fed Fibγ390-396A mice relative to FibγWT mice (Figure 7B). In HFD-challenged Fib- and siFga-treated mice, there were no differences in BAT mass (Figure 7C, S1L) nor Ucp1 mRNA levels (Figure 7D). As with Fibγ390-396A mice, the BAT mass of HFD-challenged FibAEK mice was reduced relative to HFD-challenged FibWT mice (Figure 7E). However, Ucp1 mRNA levels were equivalent between the two genotypes (Figure 7F). HFD-fed FXIII- mice had smaller BAT mass as well as modest increase in Ucp1 expression relative to FXIII+ mice (Figure 7G–7H).
Figure 7. Fibγ390-396A, FibAEK and FXIII- mice, but not Fib- mice, have smaller BAT than control mice following HFD-challenge.
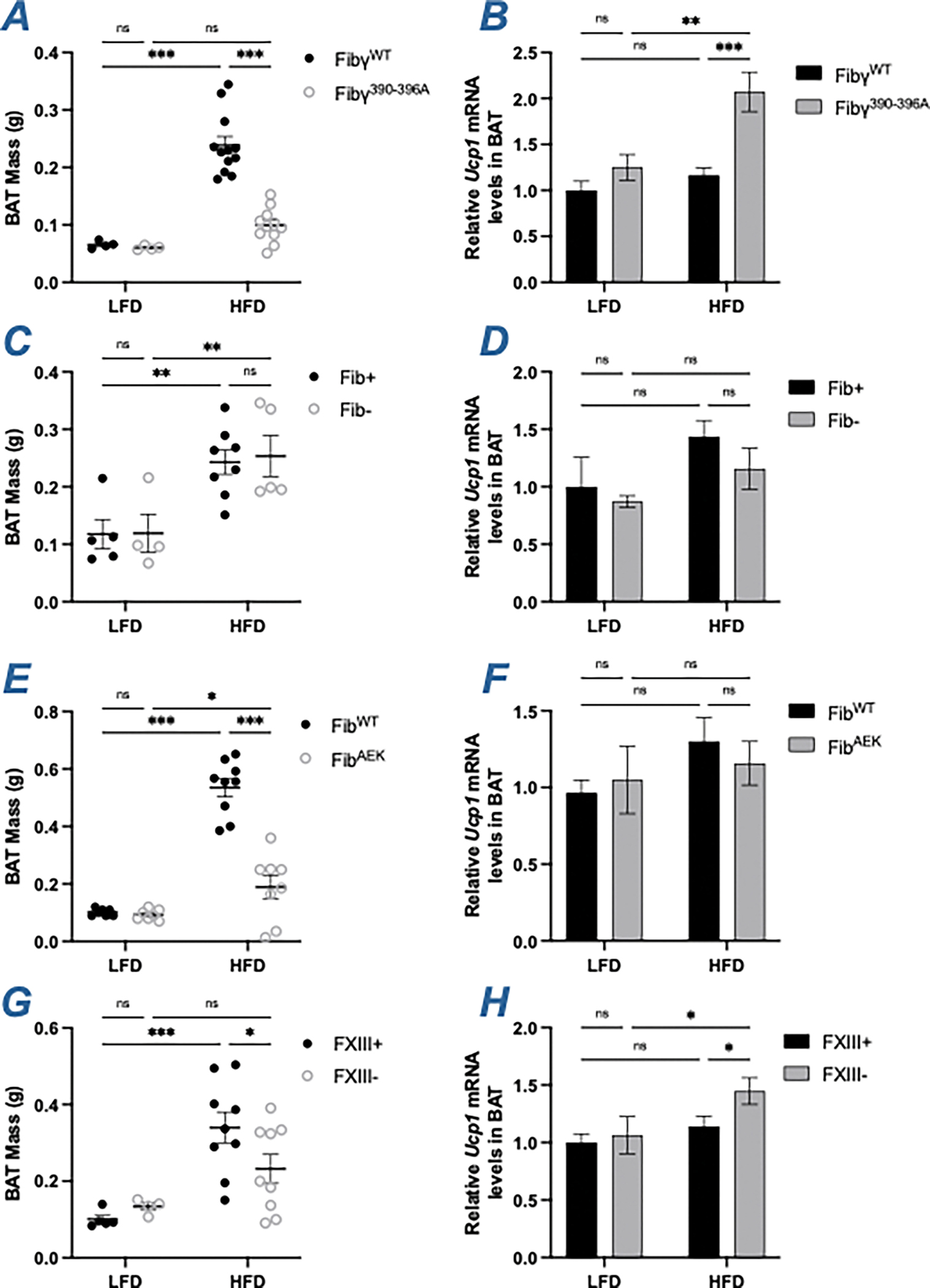
(A) Brown adipose tissue (BAT) mass of FibγWT and Fibγ390-396A mice. (B) Levels of mRNA in the BAT for uncoupling protein (UCP)-1 (Ucp1) of FibγWT and Fibγ390-396A mice. (C) BAT mass of Fib+ and Fib- mice. (D) Levels of mRNA in the BAT for Ucp1 of Fib+ and Fib- mice. (E) BAT mass of FibWT and FibAEK mice. (F) Levels of mRNA in the BAT for Ucp1 of FibWT and FibAEK mice. (G) BAT mass of FXIII+ and FXIII- mice. (H) Levels of mRNA in the BAT for Ucp1 of FXIII+ and FXIII- mice. Data are expressed as mean±SEM and analyzed by 2-way ANOVA with Fisher’s LSD test. *P<0.05, **P<0.01, ***P<0.001. LFD: n=4–7; HFD: n=5–12
Discussion
The key objective of this study was to determine the molecular forms of fibrin(ogen) that contribute to the development of HFD-induced obesity and associated metabolic sequelae in mice. The present analysis was motivated by previous observations that the fibrinogen integrin αMβ2 binding motif is a potent driver of metabolic inflammation, leading to increased adiposity, fatty liver disease, and diabetes.14 An unresolved question has been whether the different molecular forms of fibrinogen (i.e., immobilized monomer, fibrin matrix, crosslinked fibrin matrix) confer distinct or graded effects on disease processes that have been linked to the αMβ2 binding motif. In this report, we show that fibrin polymerization significantly promotes the development of HFD-induced adiposity, NAFLD and hypercholesterolemia while FXIII significantly promotes NAFLD and hypercholesterolemia. Moreover, contrary to our initial expectations, fibrinogen deficiency did not confer protection against HFD-driven obesity or any of the associated metabolic pathologies.
The binding site for the αMβ2 integrin on fibrinogen is cryptic such that it is inaccessible in soluble fibrinogen, but becomes exposed by immobilization on a surface or by conversion to an insoluble fibrin matrix.15,16 FibAEK mice express fibrinogen that cannot be converted to fibrin (i.e., locked in the monomeric form) due to a mutation in the α-chain thrombin cleavage site.20 Notably, the αMβ2 binding motif in fibrinogenAEK remains otherwise unperturbed. Here, we found that FibAEK mice were partially protected from an increase in HFD-driven adiposity and NAFLD, although Fibγ390-3906A mice had a greater degree of protection. Interestingly, FibAEK mice were partially protected from development of hepatic steatosis and iWAT adiposity, but not eWAT adiposity. Enlarged visceral adipose, such as the eWAT depot, is most associated with an increased chronic inflammation and subsequently exacerbated metabolic dysregulation.5 Consistently, gene expression of inflammatory markers in eWAT of HFD-fed FibAEK mice were similar to that of FibWT mice. The observed intermediate phenotype of HFD-fed FibAEK mice relative to that of HFD-fed WT mice and Fibγ390-396A mice is consistent with the concept that whereas fibrinogen can support αMβ2-driven activity on obesity and associated sequalae, fibrin polymerization is required for the maximum function of the γ-chain αMβ2 binding motif.
The observation that FibAEK mice display some protection from HFD-induced obesity and metabolic sequelae is also consistent with previous studies examining mice administered dabigatran, a direct thrombin inhibitor.14 In both prophylactic and therapeutic approaches, dabigatran-treated mice displayed protection from HFD-induced obesity and metabolic syndrome. Although it is difficult to make direct comparisons across studies, the protection conferred by the FibAEK mutation appears to be less pronounced than that provided by dabigatran. One possible explanation for this difference in phenotype could be that thrombin levels are lower in FibAEK mice compared to FibWT mice. However, analysis of plasma thrombin-antithrombin complexes (TATs), a marker of thrombin generation, revealed no genotype-dependent differences between control mice and Fib-, FibAEK and FXIII- mice on HFD (data not shown). Another explanation is that dabigatran would inhibit the activation of all downstream thrombin targets, including protease-activated receptor (PAR)-1 and FXIII.23,24 Notably, PAR-1−/− mice on Western diet have been shown to gain as much weight as WT mice but are completely protected from NAFLD.24 Similarly, as documented here and elsewhere, HFD-challenged FXIII- mice are protected from several aspects of metabolic syndrome.23 Studies examining the obesity phenotype of double or triple mutant mice could be designed (e.g., FibAEK/PAR-1−/− or FibAEK/FXIII- mice) to both better approximate the impact of thrombin inhibition and detail the impact of loss of multiple thrombin targets. Such studies will be the focus of future reports delineating the contribution of the coagulation system to obesity and metabolic syndrome.
Consistent with a previous study, FXIII- mice were not protected from HFD-driven total body weight gain, but had a modest, albeit statistically significant, reduction in iWAT mass and a trend in reduced eWAT mass.23 FXIII- mice were previously shown to be protected from insulin resistance and hyperinsulinemia. In contrast, we observe that HFD-fed FXIII- mice were not significantly protected from insulin resistance. The difference in ITT results may be explained by the different dosage and source of insulin used (0.5 U/kg of Novolin vs 1.5 U/kg of Humulin); the higher insulin concentration used in our experiments may have masked the subtle differences in insulin sensitivity. It is possible that local inflammation within other organs may perturb metabolic function and contribute to the development of obesity and obesity-associated pathologies. For example, in a preliminary analysis we found that gene expression of inflammatory markers trended towards reduction in the pancreas of HFD-fed Fibγ390-396A compared to FibγWT mice (data not shown). The complex network of contributions from various organ systems to fibrin(ogen)-mediated regulation of glucose homeostasis will require detailed analyses that may be addressed in future studies.
Moreover, we showed for the first time that the development of NAFLD and hypercholesterolemia is attenuated in HFD-challenged FXIII- mice. A key question is the mechanism by which FXIII exacerbates obesity and metabolic sequelae. One possibility is FXIII contributing to fibronectin assembly and collagen accumulation in WAT that alters adipocyte proliferation and macrophage activation/infiltration.23 However, studies specifically analyzing fibronectin as an experimental variable (e.g., imposing fibronectin deficiency or a functional mutation) in the context of FXIII deficiency remain to be performed. We postulate that the differences in the obesity phenotype (e.g., adiposity, NAFLD, hypercholesterolemia) observed in HFD-fed FXIII- mice are primarily due to a loss of extravascular fibrin crosslinking in adipose and/or liver tissue and that changes in fibronectin are secondary. Such a model is supported by recent studies suggesting that (i) extravascular fibrin deposition and FXIII-mediated crosslinking both influence the severity of liver injury following other challenges and (ii) transglutaminase-crosslinked fibrin versus non-crosslinked fibrin exerts unique effects on macrophage inflammatory function.25–28 Definitive analyses on whether FXIII alters the obesity phenotype through mechanisms linked to fibrin, fibronectin, or both will be a focal point of future studies.
An unexpected finding was the observation that neither genetic fibrinogen deficiency nor pharmacological knockdown of fibrinogen to ~10% of normal was protective against HFD-induced obesity and associated metabolic sequelae. In contrast, elimination of the fibrinogen-αMβ2 integrin binding motif was highly effective in preventing HFD-induced adiposity, NAFLD, diabetes, and hypercholesterolemia. Distinct responses, and thus unique pathogenic mechanisms, between Fib- mice and Fibγ390-396A have been reported in other disease models, including acetaminophen-induced liver injury.29 Collectively, these findings suggest that the impact of fibrin matrices on HFD-induced obesity can be multifactorial. In the native form, the capacity of fibrin to drive inflammatory activity through the integrin αMβ2-binding motif appears to be a dominant mechanism to exacerbate obesity and metabolic syndrome. However, when fibrin is stripped of its αMβ2-proinflammatory function, as with fibrinogen γ390-396A, the presence of ‘non-inflammatory’ fibrin appears to serve a protective role in suppressing HFD-driven adiposity and associated metabolic sequelae. It could be that fibrin deposits, in the absence of its αMβ2-proinflammatory functions, act as a physical barrier that restrains adipocyte hypertrophy and hyperplasia, such that fibrin deficiency promotes unregulated eWAT and iWAT expansion, as observed in Figure 1G and 1H. Consistently, adipocytes and adipocyte precursors express ICAM-1 and integrin αVβ3, known fibrinogen receptors, that could regulate preadipocyte proliferation, maturation, and hypertrophy.30,31 Alternatively, fibrinogen deficiency or depletion may lead to compensatory increases in other extracellular matrix (ECM)-associated proteins such as fibronectin. As noted, fibronectin is a FXIII substrate that can support adipocyte differentiation and hypertrophy.32 Compensatory increases in fibronectin protein and function have been identified in mice with reduced fibrinogen. For example, α-granules of platelets isolated from Fib- mice have significantly higher fibronectin than those of Fib+ mice, resulting in similar in vitro platelet aggregation function following PAR activation.33,34 Moreover, in models of thrombosis, plasma fibronectin appears to compensate for the loss of fibrinogen to promote the formation of platelet-rich thrombi.35 Whether similar compensatory changes occur as a consequence of fibrinogen deficiency remains an open question.
Here, we show that fibrin polymerization and potentially crosslinking are required for full fibrin(ogen)-αMβ2 integrin function in driving the development of HFD-induced adiposity, NAFLD, and hypercholesterolemia in mice. However, the beneficial effects of fibrin(ogen) mutations on HFD-driven obesity and metabolic sequelae are not conferred by partial or full elimination of fibrinogen itself. The findings presented here suggest that selective reduction of fibrin-driven inflammation, through modulation of fibrin polymerization and β2 integrin binding, could be a novel therapeutic strategy for the treatment of obesity and associated pathologies, particularly for NAFLD and hypercholesterolemia.
Supplementary Material
(A-C) Levels of mRNA in the liver for (A) HMG-CoA reductase (Hmgcr), (B) Cyp7a1 (Cyp7a1) and (C) Cyp27a1 (Cyp27a1) in FibγWT and Fibγ390-396A mice. (D-F) Levels of mRNA in the liver for (D) Hmgcr, (E) Cyp7a1 and (F) Cyp27a1 in Fib+ and Fib- mice. (G-I) Levels of mRNA in the liver for G) Hmgcr, (H) Cyp7a1 and (I) Cyp27a1 in FibWT and FibAEK mice. (J-L) Levels of mRNA in the liver for (J) Hmgcr, (K) Cyp7a1 and (L) Cyp27a1 in FXIII+ and FXIII- mice. Data are expressed as mean±SEM and analyzed by 2-way ANOVA with Fisher’s LSD test. *P<0.05, **P<0.01, ***P<0.001. LFD: n=4–7; HFD: n=6–12
(A) Representative fibrin(ogen)-stained sections of liver of HFD- fed Fibγ390-396A, FibAEK and FXIII- mice. Magnification: 20X. Scale bar: 40 μm. (B) Quantification of fibrin accumulation in the insoluble fraction of the liver homogenate normalized to total soluble protein in livers of HFD- fed Fibγ390-396A, FibAEK and FXIII- mice. Data are expressed as mean±SEM and analyzed by 1-way ANOVA.
(A) Circulating levels of fibrinogen after 20 weeks on LFD or HFD with continuous dosing of siLuc or siFga as described in the Materials and Methods. (B) Mean body weight over time of LFD- or HFD-fed siLuc- and siFga-treated mice. (C) Distribution of body weight after 20 weeks on diet. (D-F) Total mass of (D) eWAT, (E) iWAT and (F) liver. (G) Hepatic levels of triglycerides. (H) Activity of alanine transferase (ALT) in plasma. (I) Representative H&E-stained sections of livers. Magnification: 20X. Scale bar: 40 μm. (J-K) Blood glucose levels following (J) a glucose tolerance test (GTT) at week 16 and (K) an insulin tolerance (ITT) at week 18. (L) Total BAT mass. Data are expressed as mean±SEM and analyzed by 2-way ANOVA with Fisher’s LSD test. *P<0.05, **P<0.01, ***P<0.001. LFD: n=5; HFD: n=10
(A-D) Blood glucose levels following an insulin tolerance test (ITT) at week 18 of (A) FibγWT and Fibγ390-396A mice, (B) Fib+ and Fib- mice, (C) FibWT and FibAEK mice and (D) FXIII+ and FXIII- mice. AUC was determined for each individual animal. Data are expressed as mean±SEM and analyzed by 2-way ANOVA with Fisher’s LSD test. *P<0.05, **P<0.01, ***P<0.001. LFD: n=4–7; HFD: n=6–10
Essentials.
Fibrin(ogen) deposits in tissues promote obesity and associated metabolic sequalae.
Fibrinogen deficiency or depletion does not protect against high-fat diet induced obesity.
Selectively eliminating fibrin polymerization and crosslinking partially protects against obesity-related pathologies.
Acknowledgements
The authors thank Sara Abrahams, Alyssa Dandridge and Colton Strong for their technical assistance. This work was supported by the Canadian Institutes of Health Research (MFE181897 to W.S.H.), National Institutes of Health grants R01DK112778 to M.J.F., R01CA211098 to M.J.F., and R01HL160046 to M.J.K., A.S.W. and C.J.K. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflict of Interest
C.J.K. is a director and shareholder of NanoVation Therapeutics, Inc., which is developing RNA-based therapies. C.J.K., L.J.J. and J.L. have filed intellectual property on RNA-based therapies, with the intention of commercializing these inventions. The remaining authors declare no competing financial interests.
References
- 1.Hales CM, Carroll MD, Fryar CD & Ogden CL Prevalence of Obesity Among Adults and Youth: United States, 2015–2016. NCHS Data Brief, 1–8 (2017). [PubMed] [Google Scholar]
- 2.Wilson PW, D’Agostino RB, Sullivan L, Parise H & Kannel WB Overweight and obesity as determinants of cardiovascular risk: the Framingham experience. Arch Intern Med 162, 1867–1872, doi: 10.1001/archinte.162.16.1867 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Wu H & Ballantyne CM Metabolic Inflammation and Insulin Resistance in Obesity. Circ Res 126, 1549–1564, doi: 10.1161/CIRCRESAHA.119.315896 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Woo Baidal JA & Lavine JE The intersection of nonalcoholic fatty liver disease and obesity. Sci Transl Med 8, 323rv321, doi: 10.1126/scitranslmed.aad8390 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Kahn CR, Wang G & Lee KY Altered adipose tissue and adipocyte function in the pathogenesis of metabolic syndrome. J Clin Invest 129, 3990–4000, doi: 10.1172/JCI129187 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosito GA et al. Association between obesity and a prothrombotic state: the Framingham Offspring Study. Thromb Haemost 91, 683–689, doi: 10.1160/th03-01-0014 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Lijnen HR Role of fibrinolysis in obesity and thrombosis. Thromb Res 123 Suppl 4, S46–49, doi: 10.1016/S0049-3848(09)70143-4 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Abdollahi M, Cushman M & Rosendaal FR Obesity: risk of venous thrombosis and the interaction with coagulation factor levels and oral contraceptive use. Thromb Haemost 89, 493–498 (2003). [PubMed] [Google Scholar]
- 9.Klovaite J, Benn M & Nordestgaard BG Obesity as a causal risk factor for deep venous thrombosis: a Mendelian randomization study. J Intern Med 277, 573–584, doi: 10.1111/joim.12299 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Asakawa H, Tokunaga K & Kawakami F Elevation of fibrinogen and thrombin-antithrombin III complex levels of type 2 diabetes mellitus patients with retinopathy and nephropathy. J Diabetes Complications 14, 121–126, doi: 10.1016/s1056-8727(00)00075-1 (2000). [DOI] [PubMed] [Google Scholar]
- 11.Kaye SM et al. Obesity-related derangements of coagulation and fibrinolysis: a study of obesity-discordant monozygotic twin pairs. Obesity (Silver Spring) 20, 88–94, doi: 10.1038/oby.2011.287 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Powell-Wiley TM et al. Obesity and Cardiovascular Disease: A Scientific Statement From the American Heart Association. Circulation 143, e984–e1010, doi: 10.1161/CIR.0000000000000973 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morange PE & Alessi MC Thrombosis in central obesity and metabolic syndrome: mechanisms and epidemiology. Thromb Haemost 110, 669–680, doi: 10.1160/TH13-01-0075 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Kopec AK et al. Thrombin promotes diet-induced obesity through fibrin-driven inflammation. J Clin Invest 127, 3152–3166, doi: 10.1172/JCI92744 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lishko VK, Kudryk B, Yakubenko VP, Yee VC & Ugarova TP Regulated unmasking of the cryptic binding site for integrin alpha M beta 2 in the gamma C-domain of fibrinogen. Biochemistry 41, 12942–12951, doi: 10.1021/bi026324c (2002). [DOI] [PubMed] [Google Scholar]
- 16.Loike JD et al. The role of protected extracellular compartments in interactions between leukocytes, and platelets, and fibrin/fibrinogen matrices. Ann N Y Acad Sci 667, 163–172, doi: 10.1111/j.1749-6632.1992.tb51608.x (1992). [DOI] [PubMed] [Google Scholar]
- 17.Juang LJ et al. Suppression of fibrin(ogen)-driven pathologies in disease models through controlled knockdown by lipid nanoparticle delivery of siRNA. Blood 139, 1302–1311, doi: 10.1182/blood.2021014559 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suh TT et al. Resolution of spontaneous bleeding events but failure of pregnancy in fibrinogen-deficient mice. Genes Dev 9, 2020–2033, doi: 10.1101/gad.9.16.2020 (1995). [DOI] [PubMed] [Google Scholar]
- 19.Flick MJ et al. Leukocyte engagement of fibrin(ogen) via the integrin receptor alphaMbeta2/Mac-1 is critical for host inflammatory response in vivo. J Clin Invest 113, 1596–1606, doi: 10.1172/JCI20741 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prasad JM et al. Mice expressing a mutant form of fibrinogen that cannot support fibrin formation exhibit compromised antimicrobial host defense. Blood 126, 2047–2058, doi: 10.1182/blood-2015-04-639849 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lauer P et al. Targeted inactivation of the mouse locus encoding coagulation factor XIII-A: hemostatic abnormalities in mutant mice and characterization of the coagulation deficit. Thromb Haemost 88, 967–974 (2002). [PubMed] [Google Scholar]
- 22.Pfaffl MW A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29, e45, doi: 10.1093/nar/29.9.e45 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Myneni VD, Mousa A & Kaartinen MT Factor XIII-A transglutaminase deficient mice show signs of metabolically healthy obesity on high fat diet. Sci Rep 6, 35574, doi: 10.1038/srep35574 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kassel KM et al. Protease-activated receptor 1 and hematopoietic cell tissue factor are required for hepatic steatosis in mice fed a Western diet. Am J Pathol 179, 2278–2289, doi: 10.1016/j.ajpath.2011.07.015 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Groeneveld D et al. Intrahepatic fibrin(ogen) deposition drives liver regeneration after partial hepatectomy in mice and humans. Blood 133, 1245–1256, doi: 10.1182/blood-2018-08-869057 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poole LG, Kopec AK, Flick MJ & Luyendyk JP Cross-linking by tissue transglutaminase-2 alters fibrinogen-directed macrophage proinflammatory activity. J Thromb Haemost 20, 1182–1192, doi: 10.1111/jth.15670 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poole LG et al. Factor XIII cross-links fibrin(ogen) independent of fibrin polymerization in experimental acute liver injury. Blood 137, 2520–2531, doi: 10.1182/blood.2020007415 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poole LG et al. Chronic liver injury drives non-traditional intrahepatic fibrin(ogen) crosslinking via tissue transglutaminase. J Thromb Haemost 17, 113–125, doi: 10.1111/jth.14330 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kopec AK et al. Fibrin(ogen) drives repair after acetaminophen-induced liver injury via leukocyte alphaMbeta2 integrin-dependent upregulation of Mmp12. J Hepatol 66, 787–797, doi: 10.1016/j.jhep.2016.12.004 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brake DK, Smith EO, Mersmann H, Smith CW & Robker RL ICAM-1 expression in adipose tissue: effects of diet-induced obesity in mice. Am J Physiol Cell Physiol 291, C1232–1239, doi: 10.1152/ajpcell.00008.2006 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Morandi EM et al. ITGAV and ITGA5 diversely regulate proliferation and adipogenic differentiation of human adipose derived stem cells. Sci Rep 6, 28889, doi: 10.1038/srep28889 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Myneni VD, Hitomi K & Kaartinen MT Factor XIII-A transglutaminase acts as a switch between preadipocyte proliferation and differentiation. Blood 124, 1344–1353, doi: 10.1182/blood-2013-12-543223 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Ni H et al. Persistence of platelet thrombus formation in arterioles of mice lacking both von Willebrand factor and fibrinogen. J Clin Invest 106, 385–392, doi: 10.1172/JCI9896 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hur WS et al. Hypofibrinogenemia with preserved hemostasis and protection from thrombosis in mice with an Fga truncation mutation. Blood 139, 1374–1388, doi: 10.1182/blood.2021012537 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ni H, Papalia JM, Degen JL & Wagner DD Control of thrombus embolization and fibronectin internalization by integrin alpha IIb beta 3 engagement of the fibrinogen gamma chain. Blood 102, 3609–3614, doi: 10.1182/blood-2003-03-0850 (2003). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A-C) Levels of mRNA in the liver for (A) HMG-CoA reductase (Hmgcr), (B) Cyp7a1 (Cyp7a1) and (C) Cyp27a1 (Cyp27a1) in FibγWT and Fibγ390-396A mice. (D-F) Levels of mRNA in the liver for (D) Hmgcr, (E) Cyp7a1 and (F) Cyp27a1 in Fib+ and Fib- mice. (G-I) Levels of mRNA in the liver for G) Hmgcr, (H) Cyp7a1 and (I) Cyp27a1 in FibWT and FibAEK mice. (J-L) Levels of mRNA in the liver for (J) Hmgcr, (K) Cyp7a1 and (L) Cyp27a1 in FXIII+ and FXIII- mice. Data are expressed as mean±SEM and analyzed by 2-way ANOVA with Fisher’s LSD test. *P<0.05, **P<0.01, ***P<0.001. LFD: n=4–7; HFD: n=6–12
(A) Representative fibrin(ogen)-stained sections of liver of HFD- fed Fibγ390-396A, FibAEK and FXIII- mice. Magnification: 20X. Scale bar: 40 μm. (B) Quantification of fibrin accumulation in the insoluble fraction of the liver homogenate normalized to total soluble protein in livers of HFD- fed Fibγ390-396A, FibAEK and FXIII- mice. Data are expressed as mean±SEM and analyzed by 1-way ANOVA.
(A) Circulating levels of fibrinogen after 20 weeks on LFD or HFD with continuous dosing of siLuc or siFga as described in the Materials and Methods. (B) Mean body weight over time of LFD- or HFD-fed siLuc- and siFga-treated mice. (C) Distribution of body weight after 20 weeks on diet. (D-F) Total mass of (D) eWAT, (E) iWAT and (F) liver. (G) Hepatic levels of triglycerides. (H) Activity of alanine transferase (ALT) in plasma. (I) Representative H&E-stained sections of livers. Magnification: 20X. Scale bar: 40 μm. (J-K) Blood glucose levels following (J) a glucose tolerance test (GTT) at week 16 and (K) an insulin tolerance (ITT) at week 18. (L) Total BAT mass. Data are expressed as mean±SEM and analyzed by 2-way ANOVA with Fisher’s LSD test. *P<0.05, **P<0.01, ***P<0.001. LFD: n=5; HFD: n=10
(A-D) Blood glucose levels following an insulin tolerance test (ITT) at week 18 of (A) FibγWT and Fibγ390-396A mice, (B) Fib+ and Fib- mice, (C) FibWT and FibAEK mice and (D) FXIII+ and FXIII- mice. AUC was determined for each individual animal. Data are expressed as mean±SEM and analyzed by 2-way ANOVA with Fisher’s LSD test. *P<0.05, **P<0.01, ***P<0.001. LFD: n=4–7; HFD: n=6–10