

SUMMARY
Background:
Malignant rhabdoid tumors (MRT) and Wilms tumors (WT) are rare and aggressive renal tumors of infants and young children comprising ~ 5% of all pediatric cancers. MRT are among the most genomically stable cancers and although WT are genomically heterogeneous, both generally lack therapeutically targetable genetic mutations.
Methods:
Comparative protein activity analysis of MRT (n=68) and WT (n=132) across TCGA and TARGET cohorts, using metaVIPER, revealed elevated Exportin 1 (XPO1) inferred activity. In vitro studies were performed on a panel of MRT and WT cell lines evaluating effects on proliferation and cell cycle progression following treatment with the selective XPO1 inhibitor selinexor. In vivo anti-tumor activity was assessed in patient-derived xenograft (PDX) models of MRT and WT.
Findings:
metaVIPER analysis identified markedly aberrant activation of XPO1 in MRT and WT compared to other tumor types. All MRT and most WT cell lines demonstrated baseline, aberrant XPO1 activity with in vitro sensitivity to selinexor via cell cycle arrest and induction of apoptosis. In vivo, XPO1 inhibitors significantly abrogated tumor growth in PDX models, inducing effective disease control with sustained treatment. Corroborating human relevance, we present a case report of a child with multiply relapsed WT with prolonged disease control on selinexor.
Conclusions:
We report on a novel systems biology-based, a comparative framework to identify non-genetically encoded vulnerabilities in genomically quiescent pediatric cancers. These results have provided preclinical rationale for investigation of XPO1 inhibitors in an upcoming investigator-initiated clinical trial of selinexor in children with MRT and WT and offer opportunities for exploration of inferred XPO1 activity as a potential predictive biomarker for response.
Funding:
CureSearch for Children’s Cancer, Alan B. Slifka Foundation, NIH (U01 CA217858, S10 OD012351, and S10 OD021764), Michael’s Miracle Cure, Hyundai Hope on Wheels, Cannonball Kids Cancer, Conquer Cancer the ASCO Foundation, Cycle for Survival, Paulie Strong Foundation, and the Grayson Fund.
Keywords: selinexor, eltanexor, malignant rhabdoid tumors, Wilms tumors, precision medicine, patient-derived xenograft (PDX) model, exportin 1
In Brief (eTOC blurb)
Coutinho et. al. describe a novel systems biology approach using tumor gene expression data that can effectively identify non-genetically encoded vulnerabilities in pediatric renal tumors. Results from a case report and preclinical models demonstrate significant anti-tumor effects of XPO1 inhibition, supporting its investigation in an upcoming clinical trial.
Graphical Abstract
INTRODUCTION
Genomic landscape studies across the spectrum of pediatric cancers have demonstrated relatively low frequencies of somatic mutations compared to adult cancers 1–6. With the increasing implementation and use of next-generation sequencing of patient tumors to inform diagnostics, prognosis, and therapeutic management, the identification of “druggable” targets for pediatric cancers has been variable (31 – 61% with actionable alterations) leaving a significant proportion of pediatric patients in need of therapeutic options, particularly in patients with rarer subtypes or relapsed/refractory disease 7–11.
Pediatric renal tumors comprise 5% of all childhood cancers with significant heterogeneity across tumor types 12. Wilms tumor (WT) is the most common childhood kidney tumor, with favorable and unfavorable (anaplastic) histologic variants. The genomic landscape of WT demonstrates significant heterogeneity and a lack of recurrent alterations in established targets 5. Additionally, single nucleotide polymorphisms (SNPs) involved in DNA repair may also play a role in Wilms tumor susceptibility and risk13. TP53 mutations occur in approximately half of high-risk diffusely anaplastic WT, and in a small subset of non-anaplastic WT with associated poor clinical outcomes 5,14–17. In addition, most WT exhibit broad chromosomal gains and losses with loss of heterozygosity (LOH) in 1p and 16q, or gain of 1q representing high-risk groups that benefit from augmented chemotherapy 18–21. Although the majority of children with this tumor will ultimately be cured, more than a quarter of patients will be left with severe long-term side effects, and outcomes are substantially worse for patients with high-risk disease, including anaplastic TP53 mutant tumors as well as those with high-risk cytogenetic findings 22. Indeed, approximately 1/3 of diffusely anaplastic stage IV WT patients remain uncured despite intensive multimodality therapy 15,16,23.
Malignant rhabdoid tumors (MRT) are rare and highly aggressive tumors most commonly occurring in the kidney (termed MRT of the kidney) and brain (termed atypical teratoid rhabdoid tumor, ATRT), but may also occur in soft tissues throughout the body. The peak incidence of MRT is in infancy, with a median age of 11 months at diagnosis 24. Despite multimodal therapy, outcomes remain poor with overall 4-year survival of 23.2%, and only 8.8% in patients younger than six months 25. MRT are characterized by bi-allelic loss-of-function of SMARCB1 with consequent loss of SMARCB1/INI1 protein expression in tumor cells 26. SMARCB1 is a highly conserved tumor suppressor which forms a core element of the SWI/SNF (SWItch/Sucrose Non-Fermentable) chromatin remodeling complex. When defective, SWI/SNF leads to deregulation and aberrant activation of several oncogenic programs including increased cell cycling via cyclin D/CDK4/6, sonic hedgehog and Wnt/β-catenin signaling pathway activation, as well as aberrant epigenetic silencing mediated by the PRC2 catalytic subunit EZH2 26,27. Furthermore, MRT are one of the most genomically stable of all pediatric cancers with a mutation rate of only 0.084 per megabase, and is 10 to 50-fold lower than most adult tumors 28.
Both genetic and epigenetic alterations, in combination with complex networks that regulate gene expression and post-translational modifications, contribute to dysregulated protein activity 29. The Virtual Inference of Protein-activity by Enriched Regulon analysis (VIPER) algorithm provides an accurate and highly reproducible measurement of protein activity in cancer, at the individual tumor level 30,31, by leveraging the tissue-specific transcriptional targets of a protein as a highly multiplexed gene reporter assay. VIPER has been extensively validated in several contexts and has been shown to compare favorably with antibody-based measurements 32. Specifically, it has been applied to identify: 1) candidate ‘Master Regulators’ (MRs) whose aberrant activity is critical to tumor viability and growth in vivo 33,34,35, 2) regulatory proteins whose differential activity associates with tumor subtype and outcome 36, metastatic state 37, treatment sensitivity 35, and mechanisms of resistance 38, and 3) drugs/combinations that disrupt the regulatory proteins responsible for a tumor’s homeostatic state providing predictions of drug sensitivity 35,39,40.
Given the relatively low frequency of identifiable actionable somatic mutations in many childhood cancers, we employed a mutation-agnostic, network-based methodology to identify candidate non-oncogene dependencies. Specifically, we used metaVIPER—an extension of the VIPER algorithm—to analyze whole transcriptome profiles from the Therapeutically Applicable Research To Generate Effective Treatments (TARGET) database to identify druggable proteins that were aberrantly activated in MRT and WT pediatric malignancies compared to all other childhood and adult cancers 41. The analysis identified the nuclear export protein Exportin 1 (XPO1) as among the most aberrantly activated proteins in virtually all MRT and in a significant proportion of WT, compared to other tumor types. XPO1’s core function involves mediating nuclear export of proteins and RNA 42. Increased efflux of tumor suppressor proteins, as facilitated by XPO1, is believed to contribute to its oncogenic role43. Selinexor, a specific XPO1 inhibitor and member of the emerging class of orally bioavailable selective inhibitors of nuclear export (SINE), has shown preclinical efficacy in several hematologic and solid malignancies and is FDA-approved for the treatment of multiple myeloma and relapsed/refractory diffuse large B-cell lymphoma (DLBCL) 44. Given the consistent aberrant XPO1 activity observed following metaVIPER analysis of MRT and WT cohorts, we hypothesize that XPO1 may represent a dependency and novel therapeutic target in these malignancies. An ongoing early phase pediatric clinical trial of the selective XPO1 inhibitor selinexor (NCT02323880) prompted our evaluation of its anti-tumor activity using a panel of MRT and WT cell line and patient-derived xenograft (PDX) models 45,46. As WT rarely metastasize to the central nervous system (only 0.5–0.6% of cases), the activity of a next-generation XPO1 inhibitor, eltanexor, with reduced CNS penetration to improve tolerability for this class of agents, was also evaluated 47–49.
RESULTS
Increased XPO1 Activity in MRT and WT
The VIPER algorithm is a computational systems biology approach to infer protein activity from whole transcriptomic data. VIPER leverages cancer type-specific gene regulatory networks, such as those constructed by ARACNe (Algorithm for the Reconstruction of Accurate Cellular Networks), with sample-specific gene expression signatures. The differential transcriptional activity of a protein is measured based on the differential expression of its transcriptional targets, akin to a highly multiplexed gene reporter assay (Fig. 1A) 30,31,50. To address the issue of rare/orphan malignancies with an insufficient number of samples to generate an accurate ARACNe network, we developed metaVIPER 41, which extends VIPER by effectively integrating the analysis across all available cancer networks.
Figure 1:
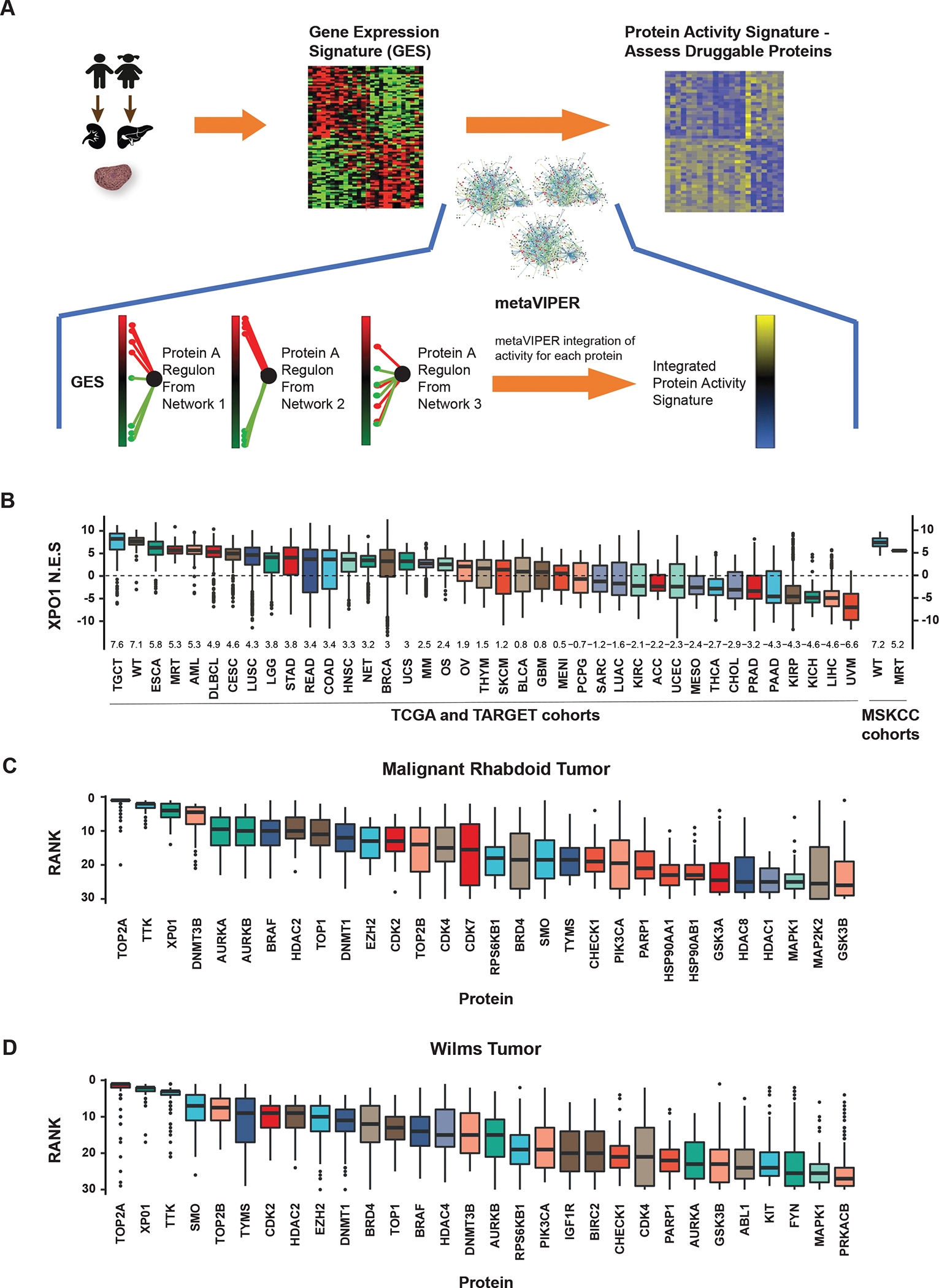
MetaVIPER inference of XPO1 activity in rhabdoid tumors and Wilms tumor cell lines. A) MetaVIPER analysis schema. Biopsy specimens undergo histopathologic review and cores with ≥ 70% tumor cellularity are reserved for RNAseq. A differential gene expression signature (GES) is computed by comparing the relative expression of each gene with a pan-cancer reference. Gene regulatory networks, e.g. interactomes generated by ARACNe from TCGA cancer cohorts, independently interrogate the GES by enrichment analysis, generating an NES score for each protein representing its transcriptional regulatory activity. The final activity score for each protein is computed by Fisher’s integration of the NES values derived using each network independently. B) Boxplots representing the distribution of metaVIPER inferred XPO1 activity for 33 TCGA tumor cohorts, 3 TARGET pediatric tumor cohorts (MRT, WT, and osteosarcoma), one neuroendocrine tumor cohort, and two MSK cohorts (WT and MRT). The median and interquartile range for NES values is represented by each box for the respective tumor cohort. NES values from enrichment analysis are comparable to Z-scores, with higher scores representing increased activity. C) Boxplots representing metaVIPER prediction rank for each of the top 30 ‘druggable’ proteins for MRT in TARGET (n=68). D) Boxplot of inferred activity for each of the top 30 proteins predicted for Wilms tumor in TARGET (n=132). XPO1 is a significantly activated protein in both MRT and WT. Boxplots representing the top 30 ‘druggable’ proteins in WT demonstrate the distribution of the rank of the protein in the 132 samples. XPO1 ranks as the third most frequently activated druggable protein in MRT and the second most frequently activated druggable protein in WT.
Abbreviations: TCGA, The Cancer Genome Atlas; TARGET, Therapeutically Applicable Research to Generate Effective Treatments; NES, normalized enrichment score; MRT, malignant rhabdoid tumors; WT, Wilms tumors.
Given the paucity of samples in MRT and WT cohorts, we used metaVIPER analysis to measure the differential transcriptional activity of 180 proteins identified as ‘druggable’ in the DrugBank database 51, using aggregated gene expression profile data available in the TARGET and TCGA databases. Notably, analysis of 39 cancer cohorts identified MRT and WT among the top 4 cancer types in which XPO1 displays the most significant aberrant activity across the analyzed cohorts (Fig. 1B). Indeed, 89% of MRT (61 of 68 samples with FDR p-value < 0.01) and 97% of WT (128 of 132 samples with FDR p-value < 0.01) presented aberrant XPO1 activation. The median rank of XPO1 activity in MRT samples is 6, making it the third most aberrantly activated druggable protein, behind TOP2A and TTK (Fig. 1C). We validated these findings in an independent cohort of patients (n=14) whose tumors have been molecularly profiled at our institution (13 WT; 1 MRT). MetaVIPER analysis on this MSKCC cohort confirms that XPO1 is highly activated, recapitulating the results from our analysis of TARGET cases (Fig. 1B).
Unlike most other tumor cohorts, where XPO1 activity was distributed over a wide range of values, aberrant XPO1 activity in MRT was relatively uniform (interquartile range (IQR) for normalized enrichment score (NES): 4.59 to 6.08). Similarly, and despite its genomic heterogeneity, WT also exhibited even higher XPO1 activity compared to most other tumors (IQR for NES: 6.43 to 7.85), with a median rank of 3, making it the second most significantly activated druggable protein in WT, behind TOP2A (Fig. 1D). Ultimately, while TOP2A and TTK were identified as being top-ranked proteins, in addition to XPO1, we note that drugs targeting TOP2A include the cytotoxic chemotherapeutic agents etoposide and doxorubicin which are frequently incorporated into front-line regimens for WT and MRT. Hence, these agents are unlikely to be used in relapsed/salvage settings. While small molecule inhibitors exist for TTK, none have entered into the adult or pediatric clinical trial space. In contrast, XPO1 inhibitors are currently being explored in pediatric clinical trials of relapsed/refractory solid tumors, motivating our investigation into this molecular target.
To gain insight into whether genetic disruption of the SWI/SNF complex is associated with aberrant XPO1 activity, we performed co-segregation analysis between genomic events (i.e., somatic single nucleotide variants (SNVs) and copy number variants (CNV)) and XPO1 activity using 33 cancer cohorts in TCGA (N=8,348 tumors). Across this pan-cancer atlas, XPO1 activity was found to co-segregate with SNVs (p=0.00016) and deletions (p=0.00052) in genes encoding the SWI/SNF complex subunits (Fig. S1A). Notably, XPO1 activation occurs in association with mutations in SWI/SNF complex “core” subunits, including SMARCB1, and the AT-rich DNA binding subunits ARID1A and B, but not with accessory subunits 52, suggesting that a broader correlation exists between loss of SWI/SNF function and increased XPO1 activity.
MRT and WT cell lines demonstrate XPO1 activation and sensitivity to selinexor
To experimentally validate our in silico findings, we initially performed metaVIPER analysis using whole transcriptomic data obtained from a panel of MRT cell lines to determine if existing models recapitulate the signature of high inferred XPO1 activity in comparison to reference cell lines profiled in the Cancer Cell Line Encyclopedia (CCLE). Analysis of aggregated RNAseq data from 9 MRT cell lines sequenced by our group confirms high XPO1 activity signature (Fig. S1B), mirroring results from our analysis of patient tumors (Fig. 1B and 1C, Fig. S1C). Analysis of WT cell lines could not be performed given the lack of RNA-seq profiles available in the CCLE. Protein expression of XPO1 was evaluated in MRT and WT cell lines and found to be variably expressed (Fig. S2A, S2B).
To evaluate the cellular consequences of XPO1 inhibition, a panel of MRT (n=4) and WT cell lines (n=4) were treated with the selective XPO1 inhibitor selinexor. A reduction in cellular viability was uniformly observed across the panel of MRT and WT cell lines tested demonstrating differential sensitivity of MRT and WT cells following selinexor treatment when compared to non-tumor lines (Fig. 2A, Suppl. Table 1). The median IC50 for cell lines following 72 hour treatment with selinexor was 102 nM (IQR 75 – 137 nM) for WT (n = 4), 142 nM (IQR 128 – 181 nM) for MRT (n = 4), and 766 nM (IQR 406 nM – 1.17 μM) for ATRT (n = 3). Non-tumor cell lines BJ and RPE had comparatively higher IC50 values of 48 μM and 16.5 μM respectively. For comparison, multiple myeloma for which selinexor has been granted FDA approval had a median IC50 of 165 nM across 12 cell lines 53. Additionally, pharmacokinetic studies of selinexor have shown that the FDA-approved dose of 80 mg twice a week achieves a peak plasma concentration of 1–1.5 μM suggesting that the observed in vitro sensitivity to selinexor in MRT and WT cell lines are within a clinically achievable range 45,46.
Figure 2:
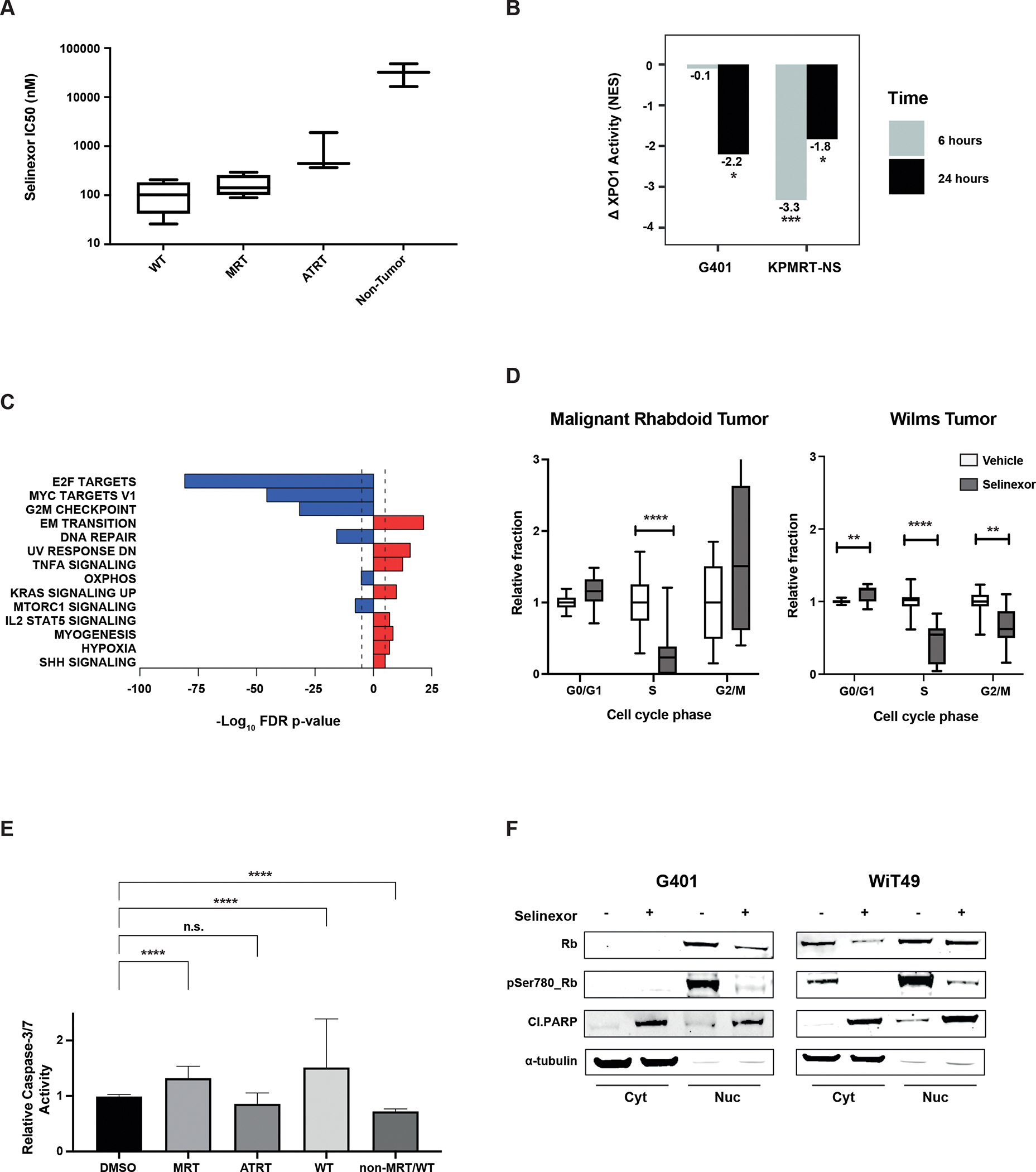
Pharmacodynamic analysis of selinexor treatment in WT and MRT cell lines A) Comparison of sensitivity to selinexor in Wilms, MRT, ATRT, and non-tumor cell lines (BJ and RPE) following 72 hours of treatment. B) Change in XPO1 activity (NES), as computed by metaVIPER, in two rhabdoid cell lines at baseline and after treatment with selinexor (30 nM) for 6 and 24 hours versus baseline. Significant decreases are observed in G401 at 24 hours (p=0.013, estimated from enrichment analysis) and in KPMRT-NS at 6 hours (p=0.0004) and 24 hours (p=0.033). C) “Cancer hallmark” pathway enrichment analysis on the differential protein activity signature following treatment of MRT cell lines. D) Cell cycle analysis across MRT and WT cell lines treated with 100 nM (WT) or 400 nM (MRT) selinexor for 48 hours demonstrates G1/S phase arrest. E) Relative caspase activity induction following treatment of MRT, WT and non-MRT/WT cell lines following 48 hours of treatment with selinexor. (F) Decrease in phosphorylated Rb and induction of cleaved PARP following selinexor treatment coincide with cell cycle arrest and apoptosis in MRT and WT cell lines. *=p<0.05, **=p<0.01, ***=p<0.001, ****=p<0.0001.
Selinexor inhibits XPO1 activity and induces cell cycle arrest and apoptosis
To examine the effects of selinexor treatment on XPO1 inferred activity, a subset of MRT cells were treated with either vehicle (DMSO) or a sublethal concentration of selinexor (30 nM) followed by post-perturbation RNAseq profiling and differential protein activity analysis by metaVIPER to evaluate the pharmacodynamic efficacy of XPO1 inhibition. Our results demonstrate that selinexor treatment was effective in reversing XPO1 activity when evaluated at 6 and 24 hours. Based on NES, there was a decrease in inferred XPO1 activity at 24 hours in both G401 (p=0.013) and KPMRT-NS (p=0.033) cell lines, as well as a decrease in inferred activity in KPMRT-NS at 6 hours (p=0.0004) (Fig. 2B). Additionally, the reduction in XPO1 inferred activity was associated with a compensatory increase in XPO1 mRNA expression (Fig. S3A). In order to survey “cancer hallmark” pathways affected by selinexor treatment, pathway enrichment analysis was performed on the differential protein activity signature which shows marked downregulation in pathways involving cell cycle progression and proliferation (E2F targets, MYC targets, G2M checkpoint) (Fig. 2C).
Selinexor treatment of MRT and WT cell lines in vitro resulted in apoptosis and cell cycle arrest at the G1/S transition (Fig. 2D and E, Fig. S3C and E). Indeed G1/S phase arrest and apoptosis were associated with decreases in RB1 phosphorylation and induction of cleaved PARP (Fig. 2F). Evaluation of protein expression following selinexor treatment in WT and MRT cell lines showed variable changes in XPO1 and in tumor suppressors/cell cycle mediators such as p53, p27, and p21. (Fig. S3B). In the MRT cell line G401, while there do not appear to be significant changes in nuclear XPO1 and p21 levels with selinexor treatment, we observe loss of XPO1 and accumulation of p21 in the cytoplasmic fraction (Fig. S3B). In addition, there is an increase in nuclear p53 associated with selinexor treatment, but no significant changes in levels of c-Myc. In contrast, selinexor treatment of the Wilms tumor cell line WiT49 results in a decrease in nuclear XPO1 levels and a marginal increase in p21, but no appreciable changes in p27, p53 or c-Myc (Fig. S3B). No significant changes in nuclear γ-H2AX in either MRT or WT cell lines are seen (data not shown) suggesting that cell cycle arrest and apoptosis induction following selinexor monotherapy is unlikely associated with DNA damage response.
Effects of selinexor treatment in xenograft models of MRT and WT
To assess the in vivo activity of selinexor in MRT and WT, established cell line-derived xenograft (CDX) and patient-derived xenograft (PDX) models were generated for therapeutic studies. Initial studies using an MRT CDX model (G401) showed significant tumor growth inhibition (TGI= 71%) (p=4e−4) (Fig. 3A). Mutational analysis performed using MSK-IMPACT54 revealed that most cell lines, including G401, harbor mutations in TP53, which are uncommonly observed in rhabdoid patient tumors (Supplemental Table 2). Therefore, additional studies were conducted using two MRT PDX models. MSKMRT-14531 was generated from biopsy tissue of an 8 month-old girl with MRT and a hepatic primary. MSKMRT-31222 was generated from primary tumor tissue obtained from the resection of a pelvic soft tissue mass in a 16 year-old female with treatment-refractory MRT. Genomic characterization of each model confirmed homozygous deletion of 22q11.23 involving SMARCB1 with consequent loss of INI1 expression, but no mutations in TP53 (Supplemental Table 2).
Figure 3:
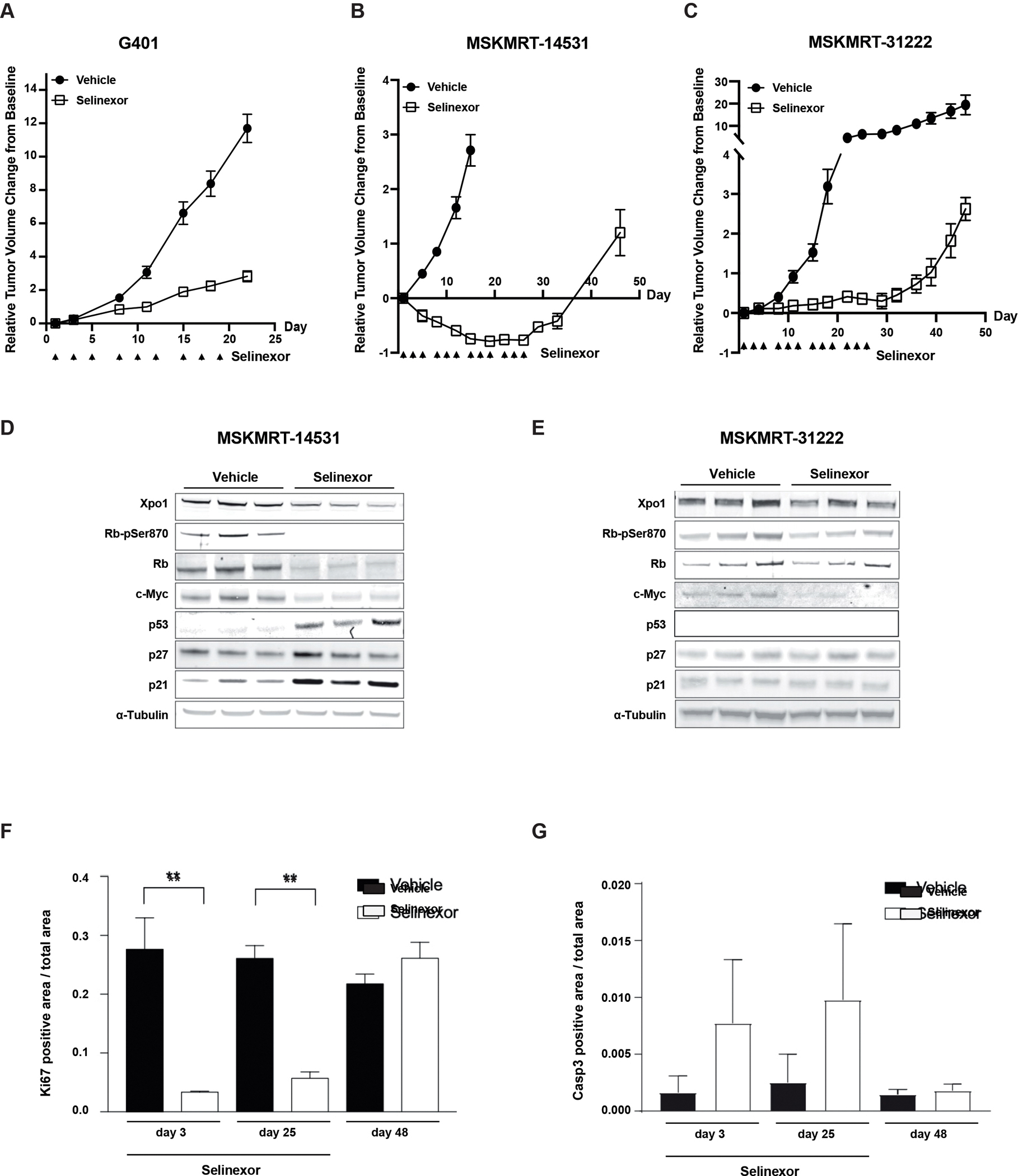
In vivo activity of selinexor in MRT xenografts. A) Tumor response in G401 xenograft (n= 8 per arm) treated with vehicle or selinexor for 21 days. Error bars: standard error of mean (SEM). B) and C) Tumor responses in MRT patient-derived xenograft (PDX) models (n=8/arm) treated with selinexor for 28 days. Error bars: SEM. D-E) Pharmacodynamic assessment of XPO1 and proliferative markers in MRT PDX models treated with selinexor. F) Immunohistochemical analysis of proliferation (Ki67) and G) apoptosis induction (caspase 3) in an MRT PDX model treated with selinexor (through Day 25) and following withdrawal of treatment.
Selinexor treatment of the two MRT PDX models demonstrated significant anti-tumor activity over 4 weeks of treatment with selinexor (Fig. 3B,C). However, abrogation of tumor growth was dependent upon sustained treatment. Compared to conventional chemotherapy (ifosfamide/etoposide) response, selinexor treatment is able to exert similar responses as observed in an MRT PDX model (Fig. S4A).
Similar to in vitro treatment of MRT cell lines, analysis of MSKMRT-14531 PDX tumors collected after 3 days of selinexor treatment showed a decrease in XPO1 and loss of phosphorylated RB1 (Fig. 3D) consistent with the anti-proliferative responses observed in vitro. Additionally, there are increases in p53, p27 and p21 levels which are more pronounced in MSKMRT-14531 which exhibited tumor regression with selinexor treatment. In contrast, only marginal changes in levels of XPO1, phosphorylated RB1 (RB1-pSer780), p27 and p21 are observed in MSKMRT-31222 which, in comparison to MSKMRT-14531, showed tumor stabilization on selinexor treatment (Fig 3E). Expression of p53 could not be detected in MSKMRT-31222 despite the lack of identifiable mutations in TP53 with genomic profiling of the PDX model. Immunohistochemical analysis of MSKMRT-31222 treated tumors showed significantly decreased Ki67 staining and induction of caspase 3 (Fig. 3F,G) consistent with the anti-proliferative effect of selinexor. However, the observed effects were reliant upon sustained treatment.
Effects of XPO1 inhibition in Wilms Tumor Models
The in vivo activity of selinexor, and the next-generation XPO1 inhibitor eltanexor, was evaluated across a panel of WT PDXs representing a diverse cohort of high-risk cases, including both favorable and anaplastic histologies (Fig. 4A,B). We found that eltanexor was equally tolerated in treated mice in spite of a more sustained dose schedule (5 daily doses weekly with eltanexor compared to 3 doses weekly with selinexor) (Fig. S5A,B). Treatment with either selinexor or eltanexor significantly decreased tumor growth compared to vehicle controls (p=0.003, p=0.004 respectively, Mann-Whitney). There is no significant difference between selinexor or eltanexor treated models (p=0.31, Mann-Whitney). There is also a significant effect on disease control in treated models (p<1e−4, log-rank) with no significant difference observed between either XPO1 inhibitor (p=0.13). Notably, WT models harboring high-risk biological features, including anaplasia and TP53 mutation, demonstrate significant tumor regression with either XPO1 inhibitor in contrast to disease stabilization observed in three favorable histology WT models (Fig. S4B).
Figure 4:
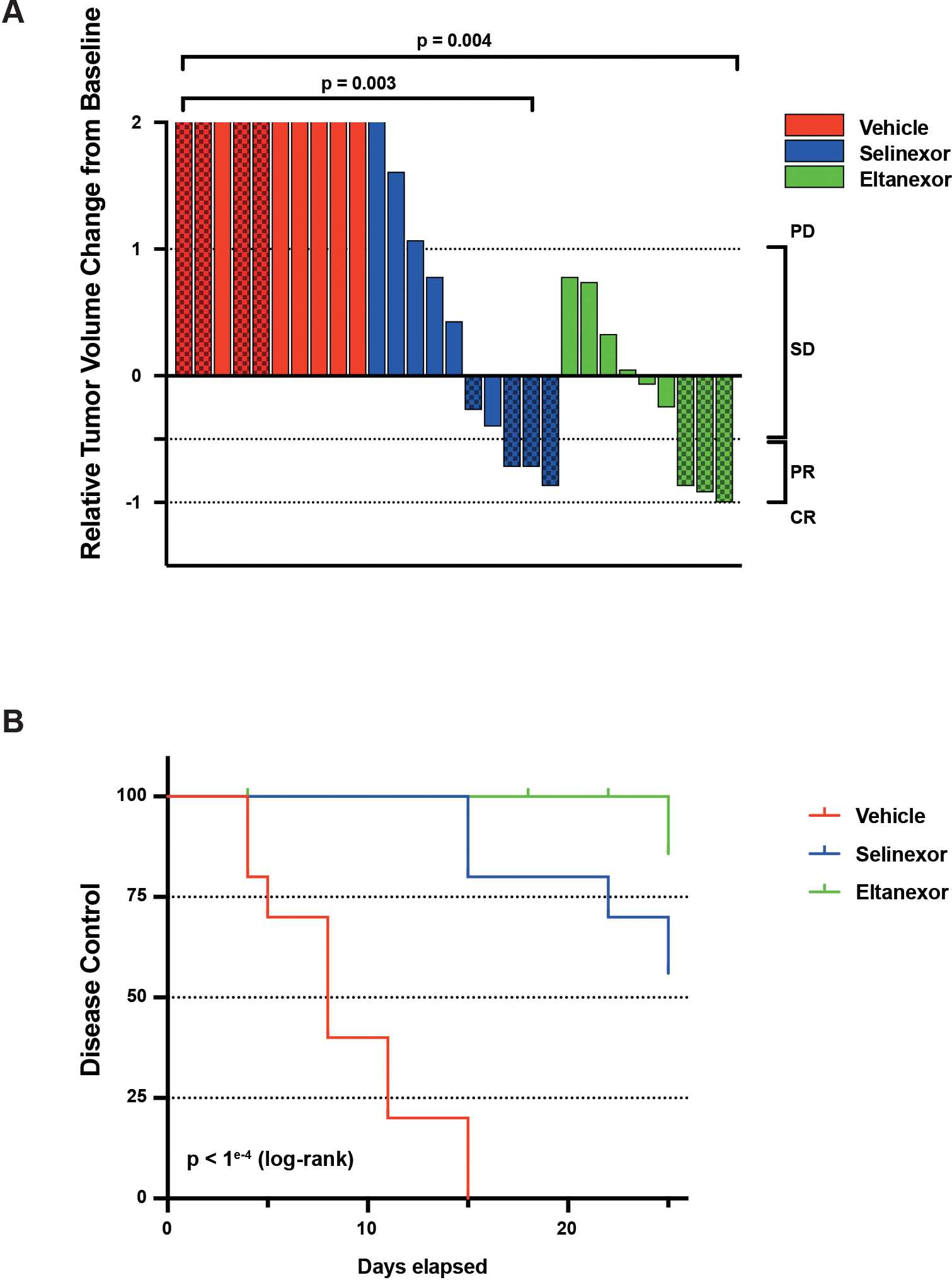
Wilms tumor PDX models show in vivo sensitivity to selinexor and eltanexor. A) Waterfall plot summarizing anti-tumor activity across a panel of Wilms tumor PDX models. Solid color bars denote models with favorable histology and textured bars denote anaplastic histology. Significant differences observed in either selinexor (p=0.003, Mann-Whitney) or eltanexor treated animals (p=0.004) compared to vehicle treatment, but no difference between selinexor or eltanexor treatment (p=0.31). B) Treatment with either selinexor or eltanexor results in significant disease control across a panel of WT PDX models (p<1e−4, log-rank). No significant differences were observed between selinexor- or eltanexor-treated cohorts (p=0.13).
PD – progressive disease; SD – stable disease; PR – partial response; CR – complete response.
XPO1 inhibitor therapy in Wilms tumor – a case report
Based upon the promising preclinical data from this study, a child with relapsed and subsequently progressive Wilms tumor was placed on selinexor monotherapy (Fig. 5). The patient originally presented with an unresectable large unilateral renal mass and underwent six weeks of standard chemotherapy (DD4A; vincristine, dactinomycin, and doxorubicin). The patient subsequently underwent a unilateral complete nephrectomy followed by flank radiation therapy. While the tumor demonstrated favorable histology, it exhibited the high-risk feature of blastemal predominance prompting augmentation of systemic therapy (Regimen M; vincristine, dactinomycin, doxorubicin, cyclophosphamide, and etoposide) as per AREN0533 (NCT00379340). Complete remission was achieved after initial therapy, but the tumor relapsed just over 1 year off therapy, presenting with a massive intra-abdominal tumor with diffuse peritoneal implants and pulmonary nodules. Although there was an initial response to salvage chemotherapy (topotecan, cyclophosphamide, doxorubicin, vincristine, ifosfamide, carboplatin, etoposide), the patient was found to have multiple progressive abdominal lesions after 6 months of therapy. He underwent an R1 resection of numerous peritoneal implants followed by whole lung and abdominal radiation. The patient’s tumor was submitted for FoundationOne® genomic profiling, which demonstrated a BRCA1 rearrangement in intron 18. At that point, the patient’s medical team and family felt that additional systemic conventional chemotherapy would provide little benefit. Based upon the promising preclinical data presented in this report, the patient was transitioned to selinexor monotherapy via the Karyopharm Expanded Access Program 55. The patient remains disease-free now 14 months after initiation of selinexor with excellent performance status.
Figure 5:
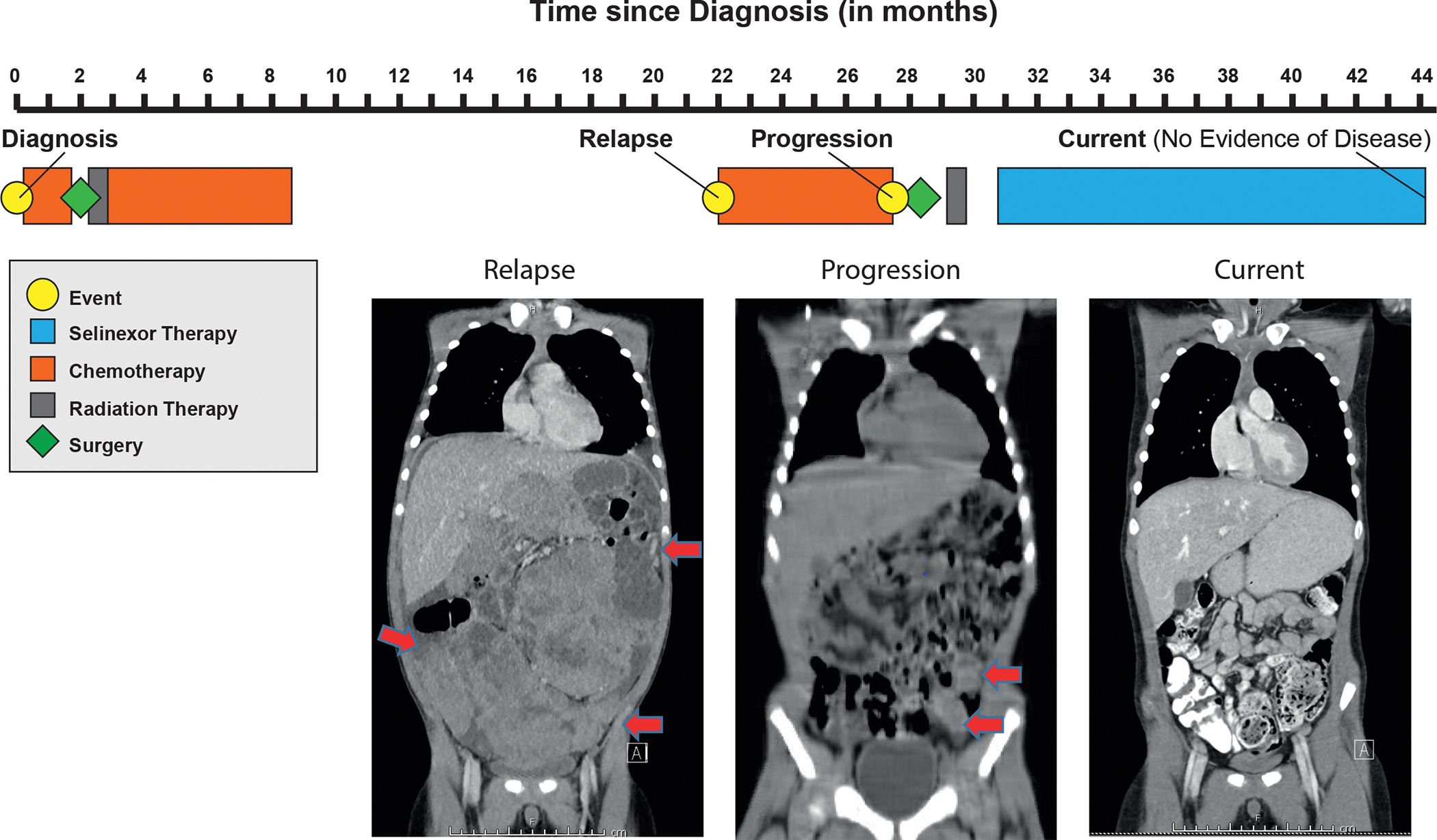
Treatment timeline of a pediatric patient with relapsed Wilms tumor treated with selinexor. Red arrows indicate peritoneal implants on MRI performed at relapse and CT performed at time of disease progression. At present, patient has no evidence of disease on selinexor monotherapy.
DISCUSSION
There is no current standard approach for the treatment of recurrent or progressive disease in MRT, WT, and other pediatric solid tumors. In pediatrics, this challenge is compounded by the general lack of targetable somatic alterations. The current approach of identifying and matching “actionable” mutations with molecularly-targeted drugs (e.g., NCI-MATCH) has resulted in modest preliminary results 56. Furthermore, while the FDA has approved over 120 targeted antineoplastic drugs over the past two decades, there are only 15 unique genomic alterations linked as companion diagnostics to these drugs 44. Hence, application of novel analytical approaches that can interrogate the molecular landscape of pediatric malignancies for non-genetically encoded “druggable” targets may offer a novel and unique opportunity to identify candidate therapies for high-risk pediatric cancers. We report on the application of a novel in silico approach that identified the nuclear export protein XPO1 as a non-oncogene vulnerability in MRT and WT.
The application of VIPER analysis, a novel systems biology approach for inferring protein activity from whole transcriptomic sequencing data, has been successfully validated across a spectrum of adult solid tumors 32,34,36,37,41,57–60. To address the challenge of identifying druggable targets in pediatric tumors, we describe how a related approach, metaVIPER, can be employed for identifying molecular vulnerabilities in pediatric cancers despite the absence of “actionable” mutations. metaVIPER infers the relative activity of over 6,000 proteins, including ~400 currently druggable proteins. This allows for the identification not only of proteins that are directly activated by a genomic alteration, but also of proteins that become highly activated as the result of a complex interplay of expression, protein-protein interactions, and post-translational modifications. The primary output of VIPER/metaVIPER analysis is a rank order list of “Master Regulator” (MR) proteins that represent critical nodes that are essential for maintaining the homeostatic state of any given tumor. Hence, targeted inhibition of any MR, or collection of MRs, can disrupt and ultimately collapse the MR architecture of the tumor cell resulting in cell death. Critically, the approach presented in this manuscript has been implemented in a NY and CA Departments of Health approved, CLIA compliant test (OncoTarget) 61, thus supporting its use in the design of clinical trials targeting proteins identified as aberrantly activated in patients. As such, this approach constitutes a direct mutation-agnostic extension of the oncogene addiction paradigm 62.
The studies presented detail the identification of XPO1 as a consistent and highly activated MR protein in MRT and WT, leading to the hypothesis that direct inhibition of XPO1 activity may represent a molecular vulnerability in these tumors. Interestingly, metaVIPER is able to identify proteins with markedly aberrant activation even when differences are not readily apparent in protein abundance, and indeed, we observe that XPO1 activity does not necessarily correlate with increased protein expression levels across MRT and WT. Evaluation of XPO1 as a viable therapeutic target in MRT and WT was achieved by the treatment of MRT and WT cell line and PDX models with selective inhibitors of XPO1, selinexor and eltanexor. We demonstrate that MRT and WT cells are sensitive to XPO1 inhibition, which induces cell cycle arrest at G1/S phase and consequently apoptosis. These results are similar to responses described in other cancers following selinexor treatment 63–66. The observed G1/S phase arrest appears to be mediated through RB1, with loss of phospho-RB1 consistently observed in selinexor-treated MRT and WT models.
It has been proposed that the dysregulation of p53 and c-MYC may mediate aberrant XPO1 activity67, and that XPO1 inhibitors exert their anti-tumor effect through nuclear sequestration of cargo proteins, including several critical tumor suppressors 68–71. However, we do not consistently observe nuclear accumulation of specific tumor suppressor proteins in either MRT or WT models after selinexor treatment. Further, while we identify a marked decrease in XPO1 activity via the application of metaVIPER to short-term selinexor treated samples, we have not discovered a clear signal of down or upregulation in the activity of these critical cargo proteins. Akin to the challenge of precisely understanding how specific genomic alterations lead to the development of MRT (SMARCB1) and WT (aneuploidy), we suspect that there are multiple overlapping, context-specific mechanisms by which XPO1 inhibitors have activity in these tumors.
Both selinexor and eltanexor induced impressive anti-tumor effects in MRT and WT PDX models, but the effects observed require sustained treatment and can range from tumor regression to stable disease, suggesting that combinatorial therapies may be needed to enhance and sustain treatment responses in some patients. Additional studies will be needed to determine the extent to which epigenetic subtypes of MRT72 and histological and mutation-based subtypes of WT, modulate treatment response to XPO1 inhibition and may guide the development of combination strategies. Pre-clinical studies of acute myelogenous leukemia (AML) suggest selinexor may help overcome resistance to topoisomerase II inhibitors, as increased nuclear retention of topoisomerase II is observed following XPO1 inhibition 73. As TOP2A is identified by metaVIPER analysis to be the most activated protein across the TARGET MRT and WT cohorts and as the topoisomerase II inhibitors etoposide and doxorubicin are standard agents used for MRT and WT, combining these with XPO1 inhibition would be a sensible strategy. Furthermore, sequential treatment with selinexor following administration of DNA damaging agents shows enhanced cytotoxicity, as opposed to the reverse order or concurrent treatment, in preclinical models of AML and multiple myeloma, presenting an additional therapeutic strategy to explore 74. Combination strategies incorporating chemotherapy75,76, CDK4/6 inhibitors which may further enhance XPO1 inhibition induced G1/S phase arrest in RB1-wildtype tumors77, and proteasome inhibitors78,79 have biological relevance in MRT and WT, but will require further preclinical validation. As a future direction, we would like to study tumor samples at the time of acquired resistance to help elucidate 1) whether XPO1 activity continues to be suppressed by the drug in spite of resistance (i.e. on vs. off-target resistance); and 2) other acquired protein activity changes associated with resistance that potentially could be targeted synergistically. Ultimately, combination strategies will need to balance synergistic activity with manageable toxicity.
Selinexor has demonstrated efficacy in patients with refractory hematologic malignancies, as well as those with advanced or metastatic solid tumors 45,46,80. It is currently being evaluated in adult trials for the treatment of a range of cancers including AML, sarcoma, and other solid tumors, and received FDA accelerated approval for the treatment of multiple myeloma and diffuse large B-cell lymphoma (DLBCL) 44. Notably, our analysis showed that AML, DLBCL, and multiple myeloma also have high XPO1 activity, though not as uniformly high as in MRT or WT, supporting the idea that XPO1 activation may predict response to selinexor. In a multiple myeloma trial of selinexor and dexamethasone, metaVIPER was used with a linear discriminant analysis classifier that identified a four-protein classifier with high predictive performance of patient response to selinexor 81. In contrast, the use of differential gene expression data did not produce an effective classifier suggesting the potential application of metaVIPER analysis for biomarker identification in other cancers. Currently, three pediatric clinical trials have evaluated the safety and efficacy of selinexor in children (NCT02323880, NCT04898894, NCT02091245) 82–84. Data from these studies have informed the development of future clinical trials exploring the activity of XPO1 inhibition in patients with MRT and WT.
Eltanexor (KPT-8602) is a derivative of selinexor, but with 30-fold less CNS penetration resulting in an improved tolerability profile and allows for more sustained dosing 49. Indeed, preclinical studies demonstrate that this decreased CNS penetration may lead to an improved toxicity profile and improved anti-tumor activity 49. We evaluated eltanexor across a panel of WT PDX models demonstrating anti-tumor activity similar to that observed with selinexor. Additionally, eltanexor was equally tolerated in treated mice in spite of a more sustained dose schedule. The first-in-human study of eltanexor, “Study of the safety, tolerability, and efficacy of KPT-8602 in patients with relapsed/refractory cancer indications” (NCT02649790) is now completed, although no studies in children have opened to date.
In conclusion, we identify XPO1 as an aberrantly activated protein in MRT and WT and provide preliminary evidence that it is a viable non-genetically encoded therapeutic target. These findings demonstrate the value of metaVIPER analysis in tumors lacking actionable oncogenic alterations to identify potential therapeutic targets and biomarkers of response and XPO1 inhibition as a promising therapeutic strategy for the treatment of MRT, WT, and other pediatric tumors. An investigator-initiated trial of selinexor for children and young adults with advanced MRT and WT is in final stages of trial initiation to evaluate the clinical benefit of this strategy.
LIMITATIONS OF STUDY
While VIPER and metaVIPER overcome the high technical and biological variation of RNA-based analyses by providing accurate and highly reproducible inference of protein activity, there remain certain limitations in the clinical setting. Stromal infiltration may dilute or even introduce false signals when evaluating for dependencies in the tumor cell compartment, a concern that is currently only partly mitigated through histopathologic selection of biopsy specimens for high tumor cellularity. Further, temporal and spatial tumor heterogeneity may require sequential biopsies and sampling of multiple metastatic sites to determine if inferred vulnerabilities are conserved before choosing optimal therapies.
While FoundationOne® CDx genomic testing was performed on the tumor tissue from the WT case report, additional germline testing and sequential cell-free DNA (cfDNA) analyses, which may have further informed our understanding of the benefit of selinexor, were not performed. Current WT clinical guidelines recommend germline testing only for children with physical findings consistent with a WT predisposition condition85, which our patient did not exhibit. At this time, we do not know if certain germline variants correspond to XPO1 activation. Likewise, careful analysis of sequential cfDNA samples could potentially inform of genomic alterations and clonal selection corresponding to selinexor response or predating frank resistance.
Despite the promising anti-tumor effects observed in MRT and WT models, the precise mechanism of action of selinexor in these tumor types requires further definition. Proteins implicated as contributing to selinexor’s mechanism of action in other tumor types - especially tumor suppressors and cell cycle mediators, such as p53, p21, p27, and c-Myc – have not been shown to be consistently affected following treatment with selinexor in our studies. Furthermore, in vivo activity of selinexor in CDX and PDX models require sustained treatment to induce disease control suggesting that identification of selinexor combination therapies may be key to inducing durable treatment responses.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Filemon S. Dela Cruz (delacrf1@mskcc.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Oncotarget predictions and RNA-seq data have been deposited at Figshare and Gene Expression Omnibus (GEO), respectively, and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CRM1 antibody (XPO1); 1:1000 | Abcam | Cat# ab24189, RRID: AB 2257217 |
| SNF5 antibody [EPR6966]; 1:1000 | Abcam | Cat# ab126734, RRID: AB 11127897 |
| gamma H2A.X (pS139) antibody [9F3]; 1:1000 | Abcam | Cat# ab26350, RRID: AB 470861 |
| p53 (7F5) Rabbit mAb antibody; 1:1000 | CST | Cat# 2527, RRID: AB 10695803 |
| Rb (4H1) Mouse mAb antibody; 1:2000 | CST | Cat# 9309, RRID: AB 823629 |
| Phospho-Rb (Ser780) antibody; 1:1000 | CST | Cat# 9307, RRID: AB 330015 |
| p21 Waf1/Cip1 (12D1) antibody; 1:1000 | CST | Cat# 2947, RRID: AB 823586 |
| p27 Kip1 (D37H1) mAb antibody; 1:1000 | CST | Cat# 3688, RRID: AB 2077836 |
| Cleaved PARP (Asp214) (D64E10) antibody; 1:1000 | CST | Cat# 5625, RRID: AB 10699459 |
| c-Myc (D84C12) mAb antibody; 1:1000 | CST | Cat# 5605, RRID: AB 1903938 |
| Anti-β-Actin antibody; 1:5000 | Sigma-Aldrich | Cat# A5441, RRID: AB 476744 |
| Anti-α-Tubulin antibody; 1:1000 | Sigma-Aldrich | Cat# T9026, RRID: AB 477593 |
| IRDye 680RD Donkey anti-Mouse IgG antibody; 1:5000 | LI-COR Biosciences | Cat# 926-68072, RRID: AB 10953628 |
| IRDye 800CW Donkey anti-Rabbit IgG antibody; 1:15000 | LI-COR Biosciences | Cat# 926-32213, RRID: AB 621848 |
| Monoclonal Mouse Anti-Human Ki-67 Antigen; 1:150 | Agilent | Cat# M7240, RRID: AB 2142367 |
| Cleaved Caspase-3 (Asp175) antibody; 1:1000 | CST | Cat# 9661, RRID: AB 2341188 |
| Biotinylated horse anti-mouse IgG; 1:50 | Vector Labs | Cat# MKB-22258 |
| Biotinylated goat anti-rabbit; 1:50 | Vector Labs | Cat# PK6101 |
| Tyramide Alexa Fluor 568 | Invitrogen | Cat# T20949 |
| DAPI | Sigma Aldrich | Cat# D9542 |
| Mowiol | Sigma Aldrich | Cat# 81381 |
| Chemicals, peptides, and recombinant proteins | ||
| Dulbecco’s Modified Eagle Medium (DMEM) | ThermoFisher Scientific | Cat# 12491015 |
| DMEM/F-12 | ThermoFisher Scientific | Cat# 11320033 |
| Iscove’s Modification of DMEM (IMDM) | Corning | Cat# 10-016 |
| Fetal Bovine Serum (FBS) | Corning | Cat# 35-010 |
| Antibiotic-Antimycotic (100X) | ThermoFisher Scientific | Cat# 15240062 |
| L-Glutamine (200 mM) | ThermoFisher Scientific | Cat# 25030081 |
| Selinexor | Selleckchem | Cat# S7252 |
| Eltanexor | Selleckchem | Cat# S8397 |
| Ethanol | Merck | Cat# 100983 |
| Phosphate Buffered Saline | Gibco | Cat# 10010023 |
| Ribonuclease H (RNase H) | Sigma-Aldrich | Cat# 10786357001 |
| Propidium iodide | Sigma-Aldrich | Cat# P4170 |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | Cat# D2650 |
| RIPA Buffer | Sigma-Aldrich | Cat# R0278 |
| Pierc BCA Protein Assay Kit | Thermofisher Scientific | Cat# 23225 |
| Formalin solution, neutral buffered, 10% | Sigma-Aldrich | Cat# HT501128 |
| EZ Prep Concentrate | Ventana | Cat# 950-102 |
| ULTRA Cell Conditioning (CC1) | Ventana | Cat# 950-224 |
| Background Buster | Innovex | Cat# NB306 |
| Critical commercial assays | ||
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | Cat# G7572 |
| Caspase-Glo® 3/7 Assay | Promega | Cat# G8091 |
| Nuclear Extraction Kit | Abcam | Cat# ab113474 |
| NuPAGE™ 4 to 12%, Bis-Tris, 1.0-1.5 mm, Mini Protein Gels | Invitrogen | Cat# NP0321BOX |
| QIAGEN QIAamp Tissue Kit | Qiagen | Cat# 51404 |
| RNeasy Micro Kit | Qiagen | Cat# 74004 |
| Agilent SureSelectXT All Exon V5 + UTRs | Agilent | Cat# 5191-7408 |
| TruSeq Stranded Total RNA LT Sample Prep Kit | Illumina | Cat# 20020596 |
| Deposited data | ||
| RNA-seq data from cell lines and used for perturbation screen | Gene Expression Omnibus (GEO) | GEO Accession: GSE198000 |
| Oncotarget predictions | Figshare | https://figshare.com/sZ3213cfaf6b2a2aeb7783 |
| Experimental models: Cell lines | ||
| 786-O | ATCC | Cat# CRL-1932 |
| BJ | Dr. Alex Kentsis; MSKCC | N/A |
| BT12 | Dr. Alex Kentsis; MSKCC | N/A |
| BT16 | Dr. Alex Kentsis; MSKCC | N/A |
| CHLA-266 | COG cell line repository | RRID: CVCL_M149 |
| COGW408 | Dr. C. Patrick Reynolds; Texas Tech University | N/A |
| G401 | ATCC | Cat# 87042204, RRID: CVCL 0270 |
| KP-MRT-AN | Dr. Hajime Hosoi’s; Kyoto Prefectural University of Medicine | N/A |
| KP-MRT-NS | Dr. Hajime Hosoi’s; Kyoto Prefectural University of Medicine | N/A |
| KP-MRT-YM | Dr. Hajime Hosoi’s; Kyoto Prefectural University of Medicine | N/A |
| RH-30 | ATCC | CVCL 0041 |
| RPE | Dr. Alex Kentsis; MSKCC | N/A |
| TC-71 | ATCC | CVCL 2213 |
| Wilms 1 | Dr. Brigitte Royer-Pokora; Heinrich-Heine-University | N/A |
| Wilms 10 | Dr. Brigitte Royer-Pokora; Heinrich-Heine-University | N/A |
| WiT49 | Dr. Herman Yeger; University of Toronto | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ | The Jackson Laboratory | Cat# 005557; RRID: IMSR_JAX: 005557 |
| Software and algorithms | ||
| Prism Software Version 9 | GraphPad | N/A |
This study did not generate any new codes.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Human samples
Informed and signed consent to publish the data was obtained and archived for the research performed and publication of the results, including the case report. Memorial Sloan Kettering Cancer Center (MSKCC) patients were enrolled onto the MSKCC targeted gene sequencing research study (Genomic profiling in cancer patients; NCT01775072) with approval from the MSKCC Institutional Review Board under protocol IRB# 12–245, #06–107, and #17–387. For xenografts generated at Columbia University Medical Center, patient consent was obtained for clinical sequencing at Columbia University Medical Center (CUMC) through the Precision in Pediatric Sequencing Program (PIPseq) and PDX tumor mouse model generation under the CUMC Institutional Review Board (IRB)-approved protocol AAAN8404.
Xenograft models
All animal studies were performed in accordance with institutional guidelines and under an approved protocol from the CUMC Institutional Animal Care and Use Committee (Protocol AAAF5850) or the MSKCC Institutional Animal Care and Use Committee (#16–08-011).
The patient-derived xenograft (PDX) models were developed by transplanting a 2 mm fragment from a patient’s biopsy specimen into the flank of 8 – 10 week old NOD (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) SCID gamma (NSG) mice to generate the passage 0 (P0) generation. Tumors were serially propagated and expanded from the PDX P0 generation for therapeutic studies. The G401 cell line xenograft models were developed by injecting 2 million G401 cells into the flank of NSG mice.
Cell line models
Rhabdoid cell line G401 (ECACC Cat# 87042204, RRID:CVCL_0270) was obtained from the American Type Culture Collection (ATCC). Rhabdoid cell lines KP-MRT-AN (RRID:CVCL_7049), KP-MRT-NS (RRID:CVCL_7050), and KP-MRT-YM (RRID:CVCL_7052) were obtained courtesy of Dr. H. Hosoi 86–89. The ATRT cell line CHLA-266 (RRID:CVCL_M149) was obtained from the COG cell line repository. WiT49 and COGW408 were provided courtesy of Dr. Herman Yeger (Toronto, Canada) and Dr. C. Patrick Reynolds (Lubbock, TX, United States) respectively. Dr. Brigitte Royer-Pokora (Dusseldorf, Germany) provided the Wilms1 and Wilms10 cell lines. BT12, BT16, BJ, and RPE cell lines were provided by Dr. Alex Kentsis (New York City, NY, United States). RH-30, TC-71, and 786–0 were obtained from ATCC. Cell lines were authenticated using STR profiling. Rhabdoid tumor cell lines were cultured in DMEM with 10% FBS and 1% Antibiotic-Antimycotic (Thermo Fisher Scientific). COGW408 was cultured in IMDM with 10% FBS, 1% L-glutamine, and 1% penicillin/streptomycin. WiT49, Wilms1, and Wilms10 were grown in DMEM-F12 with 10% FBS, 1% L-glutamine, and 1% penicillin/streptomycin. All cells were tested for Mycoplasma 90.
METHOD DETAILS
Cell viability assay
Cells (250 −2,000 cells/well) were seeded in 384-well plates and after 24 hours were incubated with selinexor (Selleckchem) for 72 hours. The viability of the cells was determined using CellTiter-Glo Luminescent Cell Viability Assay (Promega) according to the manufacturer’s recommendations. IC50 concentrations were determined using GraphPad Prism Software Version 7.0.
Cell cycle analysis
Cells (300,000 cells/well) were seeded in 6-well plates. MRT and WT cells were treated with 400 nM and 100 nM Selinexor, respectively, for 48 hours. Cells were then fixed in 70% ethanol, washed, and stained in PBS 1X containing 50 μg/ml RNase H (Sigma Aldrich) and 50 μg/ml propidium iodide (PI) (Sigma Aldrich), followed by flow cytometric analysis.
Apoptosis assay
Cells were seeded in 384-well plated with with either DMSO or 200 nM selinexor for 48 hours. Apoptosis of cells was determined using Caspase-Glo 3/7 Assay (Promega) according to the manufacturer’s recommendations.
Immunoblotting
Cells treated with Selinexor for 48h and PDX tumor samples were resuspended in either RIPA Buffer with SDS 1% (Sigma Aldrich) to extract whole protein or Nuclear Extraction Kit (Abcam) to obtain proteins from cytosolic and nuclear fractions. Protein concentrations were determined using the Pierce BCA Protein Assay Kit (Thermofisher) according to manufacturer’s recommendations. The isolated protein samples were separated on 4–12% SDS-PAGE gels and were subsequently transferred to a nitrocellulose membrane using the iBlot Gel Transfer Device and iBlot Gel Transfer Stacks (Thermo Scientific). Membranes were stained with the antibodies listed in Key Resources Table.
In vivo study
Tumor growth was measured biweekly using calipers. Treatments were started when flank tumors achieved a tumor volume (TV) of ~ 100 mm3 (TV = width2 * length/2). PDX models were dosed with selinexor by oral gavage three times a week (Monday, Wednesday, Friday) at a dose of 15 mg/kg. Eltanexor treatment was 15 mg/kg by oral gavage five times per week. Tumors were collected and fragments were either fixed in 10% formalin for histologic analysis or snap frozen in liquid nitrogen for subsequent DNA, RNA, and protein isolation and analyses.
Immunofluorescence
Immunofluorescence staining was performed in the Molecular Cytology Core Facility of MSKCC using the Discovery XT processor (Ventana Medical Systems). The tissue sections were deparaffinized with EZPrep buffer (Ventana Medical Systems), and antigen retrieval was performed with CC1 buffer (Ventana Medical Systems). Sections were blocked for 30 minutes with Background Buster solution (Innovex), followed by avidin-biotin blocking for 8 minutes (Ventana Medical Systems). Sections were incubated with human-specific anti-Ki67 (DAKO, cat# M7240) or human specific anti-ClCaspase3 (CST, cat# 9661), followed by a 60 minutes incubation with biotinylated horse anti-mouse IgG (Vector labs, cat# MKB-22258) or biotinylated goat anti-rabbit (Vector Labs, cat# PK6101) respectively. The detection was performed with Streptavidin-HRP D (part of DABMap kit, Ventana Medical Systems), followed by incubation with Tyramide Alexa Fluor 568 (Invitrogen) prepared according to manufacturer’s instructions with predetermined dilutions. After staining, all slides were counterstained with DAPI (Sigma Aldrich) for 10 min and cover-slipped with Mowiol (Sigma Aldrich).
Nucleic acid extraction, clinical sequencing, and analysis
DNA from frozen tissue or paraffin embedded tissue was extracted using the QIAGEN QIAamp Tissue Kit (for tissue samples) on the QIAcube system. RNA was extracted using the QIAGEN RNeasy Kit (fresh frozen tissue). All slides were evaluated by a pathologist to ensure that a minimum of 50 % viable tumor was present for subsequent extraction and analyses. Whole exome sequencing (WES) was performed using the Agilent SureSelectXT All Exon V5 + UTRs capture kit for library generation, and sequenced on the HiSeq 2500 System (Illumina), using paired-end 100 cycle × 2 sequencing. RNA was sequenced using the TruSeq Stranded Total RNA LT Sample Prep Kit (Illumina), with 100 cycles × 2 paired-end sequencing on the HiSeq 2500. DNA sequencing reads were de-multiplexed and converted to FASTQ files using CASAVA from Illumina. Following mapping and variant calling of both tumor and normal samples by NextGENe, resulting variants were subject to filtering. Variants in normal DNA were passed through a “reference range filter” of cancer predisposition genes, genes relevant to pharmacogenomics, and variants relevant to patient care; a “reportable range filter” which includes COSMIC variants in the patient’s mutation report file and variants in genes on the list of ACMG (American College of Medical Genetics and Genomics) recommendations for reporting of secondary findings; as well as a frequency filter. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT) testing was performed as described previously 54.
MetaVIPER Analysis
All regulatory networks used for metaVIPER analysis were reverse engineered by ARACNe 50. Twenty four core TCGA RNA-Seq derived networks are publicly available in the R Bioconductor package aracne.networks. In the case of signaling proteins, ARACNe detects maximum information path targets to define its set of “indirectly regulated” target genes.
After standard read alignment of RNA-Seq data by STAR 91 to the GRCh38 reference genome build and summarization of expression quantities at the gene count level, gene expression was normalized by variance stabilization. The stabilization parameters are stored from a single run of the Variance Stabilizing Transformation (VST), as implemented in the DESeq2 package 92, across all TCGA and TARGET samples and these parameters are then applied to individual samples. In the case of novel samples, post-normalization gene expression profiles are compared between the novel samples and tissue-relevant samples from TCGA/TARGET through methods such as principal component analysis and t-SNE to confirm the absence of any significant batch effects that may be attributed to instrument/library-specific biases. In the case of cell line samples, an analogous procedure is performed through the initial application of VST to all CCLE samples.
For the purposes of computing differential protein activity of TARGET MRT, a gene expression signature (GES) was computed between each MRT versus a pan-cancer reference group that includes gene expression profiles of 11,289 tumors belonging to 33 cancer types, followed by application of the analytic Rank-based enrichment analysis (aREA) using each of the available networks 30,31,41. As VIPER is a rank based enrichment method, GES’s are generated through a double-rank transformation process. First, the normalized expression of each gene in the sample is ranked relative to the distribution of the expression of this gene across all TCGA and TARGET samples. Expression distributions for all genes have been fitted to a spline-model to ensure comparable ranks. Next, genes within a sample are ranked based on their relative expressions. Following generation of a GES, aREA tests for a global shift in the positions of a regulatory protein’s target genes in the rank-sorted GES and is an extension of other efforts at computationally efficient method to approximate GSEA 30,93. aREA uses the mean of the rank-transformed positions as the test statistic (enrichment score). The enrichment score is computed first by a 1-tail approach, rank-transforming the absolute values of the GES and secondly by a 2-tail approach, where positions of the repressed target genes (as inferred in the network) are inverted in the gene expression signature before computing the enrichment score. The contribution of each target gene to the enrichment score is also weighted based on the regulator-target gene mutual information computed by ARACNe. In the final step of aREA, in the case of single sample analysis, statistical significance for the enrichment score is computed by an analytic approach that approximates shuffling the genes in the signature at random, outputting a normalized enrichment score (NES). Gene shuffling is approximated as follows: according to the central limit theorem, the mean of a sufficiently large number of independent random variables is normally distributed. We ensure a mean of zero and variance equal to one for the enrichment score under the null hypothesis by applying a quantile transformation based on the normal distribution to the rank-transformed GES before computing the enrichment score. Then, under the null hypothesis, the enrichment score will be normally distributed with mean of zero and variance n−1, where n is the regulon size, which is further scaled to the standard variable. In the single sample analysis, aREA cannot directly account for biological correlation between the expression of various genes in the given biological context, and so conservative NES/p-value thresholds are empirically used with the assumption that this approach underestimates the p-value. In the final step of metaVIPER analysis, NES’s generated by the application of VIPER using each of the available networks are integrated at the individual regulatory protein level by a weighted average, with greater absolute NES’s contributing a greater weight to the final integrated score to maximize the contribution of signal over noise, as benchmarked in 41. As noted, alternative methods including a simple average of NES scores appear to provide an almost equivalent result.
Similarly, metaVIPER was used to infer differential protein activity between samples from each of four selinexor treated MRT/ATRT cell lines and corresponding DMSO treated controls. As these experiments were done in replicate, the GES is computed through gene-wise modified student t-test between condition and control.
Pathway analysis on the Selinexor induced integrated differential protein activity signature in MRT/ATRT cell lines was performed with ‘Cancer Hallmark’, ‘Gene Ontology’, and ‘Oncogenic Signature’ gene sets provided in the Broad MSigDB collections 94. Pathway enrichment analysis was performed by using the single-tail aREA method described above as a rapid approximation to the KS-test used in GSEA, inputting the pathway genes and the sorted differential protein activity signature. Unlike other statistical tests used for pathway enrichment analysis, such as the Fisher’s exact test on a thresholded list of differentially expressed genes or the repeated application of the Kolmogorov-Smirnov test in classical GSEA, aREA does not require the arbitrary binarization into significant/non-significant hits for target genes in the signature but takes into account their relative position in the signature.
Co-Segregation Analysis of XPO1 Activity
In order to identify potential genomic events that causally or incidentally associate with increased XPO1 activity, a co-segregation analysis was performed to determine somatic events that are enriched in tumors that are inferred to have high XPO1 activity by metaVIPER across 33 TCGA cancer type cohorts. Publicly available somatic SNV and CNV (GISTIC) calls from the Broad TCGA Genome Data Analysis Center (GDAC) Firehose were downloaded in order to perform this analysis. Enrichment analysis by aREA was performed by arranging tumors in order of lowest to highest XPO1 activity; a null model derived from enrichment analysis of each genomic event with all proteins that metaVIPER infers activity on was developed to estimate the statistical significance of the co-segregation. Analogously, the co-segregation analysis was performed between increased XPO1 activity and the activity of about 411 other ‘druggable’ proteins in both the TARGET malignant rhabdoid tumors dataset and the TCGA datasets to develop hypotheses on biologically relevant protein interactions and potential synergistic drug combinations to treat tumors with elevated XPO1 activity.
Quantification and Statistical Analysis
For MetaVIPER analysis, enrichment scores are first computed for the targets of each protein in the GES for each network independently, using aREA. Each enrichment score is computed using a two-tailed approach. The absolute value of the GES for the positively regulated target genes are rank transformed, and the signatures are then inverted to compute enrichment of the repressed target genes. The enrichment score is a weighted average based on targets’ ranks in the signature, and the protein-target gene mutual information assignments in the network, with stronger interactions contributing more heavily to the score. Subsequently, statistical significance for the enrichment score is computed by an analytic approach. Under the null hypothesis, the enrichment score is normally distributed with mean of zero and variance proportional to 1/(n−1), where n is the number of targets, scaled to the standard variable. This approximates shuffling the genes in the signature at random, outputting a normalized enrichment score (NES). While aREA cannot directly account for biological correlation between the expression of various genes (i.e. covariance) in the given biological context, a very conservative NES/p-value threshold is empirically used with the assumption that this approach underestimates the p-value. In the final step of metaVIPER analysis, NES’s generated using each of the available networks are integrated at the individual protein level by a weighted Stouffer’s Z-score method, with greater absolute NES’s contributing a greater weight to the final integrated NES score for the protein, as benchmarked in 41.
Statistical analyses and visualizations of in vitro and in vivo experiments were performed using GraphPad Prism 9. IC50 values obtained from dose-response assays were compared across cell lines using the Kruskal-Wallis test. Pairwise comparisons were made using Dunn’s test, and multiplicity adjusted p-values were reported. XPO1 activity was analyzed using the same methodology. Cell cycle analyses comparing average relative fractions of cells in different stages treated with vehicle against Selinexor were run using several unpaired t-tests. A multiple testing correction to maintain a false discovery rate of 0.01 was implemented using a two-stage step up procedure. Differences in relative tumor volume, measured as a change from baseline, were evaluated in various PDX models using a Mann-Whitney U test. The Kruskal-Wallis test was used to compare Ki67 and caspase measurements through IHC. Overall disease control probabilities were estimated using Kaplan-Meier analysis. The global test for differences in disease control across treatment arms, as well as individual comparisons of survival curves, were carried out using a log-rank test. Waterfall plots were produced to visualize differences in relative tumor volume across treatment arms, and differences were evaluated using unpaired t-tests. Statistical significance was defined as p values < 0.05.
Supplementary Material
HIGHLIGHTS.
A network-based algorithm identifies molecular vulnerabilities in pediatric tumors
Pediatric renal tumors exhibit aberrant activation of Exportin 1 (XPO1)
Inhibiting XPO1 in pediatric renal tumor models induces cell cycle arrest and apoptosis
Selinexor has antitumor effect in PDX models and leads to disease control in a patient
CONTEXT AND SIGNIFICANCE.
We need new tools to identify “druggable” targets in pediatric cancers, since they have a lower frequency of somatic mutations compared to adult cancers. Here, the authors apply a mutation-independent methodology to identify abnormally activated proteins in pediatric renal cancers with low tumor mutational frequency. In silico analysis identified XPO1 as consistently activated, and its inhibition with the drug Selinexor reduced tumor proliferation in vitro and in vivo in relevant preclinical models. Corroborating human relevance, the authors present the case report of a child with multiply relapsed Wilms tumor with prolonged disease control following treatment with selinexor. These findings support further clinical investigation of XPO1 inhibition for pediatric renal tumors and provide a novel approach to identifying therapeutic targets for other pediatric cancers.
ACKNOWLEDGMENTS
This research was supported by CureSearch for Children’s Cancer, Alan B. Slifka Foundation, the Gold Ribbon Riders, Cancer Center Support grant P30 CA008748, NIH grant U01 CA217858 to Andrea Califano and Andrew Kung, and NIH grants S10 OD012351 and S10 OD021764 to Andrea Califano. Michael Ortiz acknowledges support on this project from Michael’s Miracle Cure, Hyundai Hope on Wheels, Cannonball Kids Cancer, and Conquer Cancer the ASCO Foundation. Filemon Dela Cruz acknowledges support from Cycle for Survival, the Paulie Strong Foundation, and the Grayson Fund.
Footnotes
DECLARATION OF INTERESTS
A.C. is co-founder, equity holder, and consultant of DarwinHealth Inc., which has licensed IP related to the VIPER algorithms from Columbia University. Columbia University is an equity holder in DarwinHealth Inc. A.L.K. is on the Scientific Advisory Board of Emendo Biotherapeutics, Karyopharm Therapeutics, Imago BioSciences, and DarwinHealth; is co-Founder and on the Board Directors of Isabl Technologies; and has equity interest in Imago BioSciences, Emendo Biotherapeutics, and Isabl Technologies.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Lawrence MS, Stojanov P, Polak P, et al. : Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499:214–218, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grobner SN, Worst BC, Weischenfeldt J, et al. : The landscape of genomic alterations across childhood cancers. Nature 555:321–327, 2018 [DOI] [PubMed] [Google Scholar]
- 3.Vogelstein B, Papadopoulos N, Velculescu VE, et al. : Cancer genome landscapes. Science 339:1546–58, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shern JF, Chen L, Chmielecki J, et al. : Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov 4:216–31, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gadd S, Huff V, Walz AL, et al. : A Children’s Oncology Group and TARGET initiative exploring the genetic landscape of Wilms tumor. Nat Genet 49:1487–1494, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chun HE, Lim EL, Heravi-Moussavi A, et al. : Genome-Wide Profiles of Extracranial Malignant Rhabdoid Tumors Reveal Heterogeneity and Dysregulated Developmental Pathways. Cancer Cell 29:394–406, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Forrest SJ, Geoerger B, Janeway KA: Precision medicine in pediatric oncology. Curr Opin Pediatr 30:17–24, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mody RJ, Wu YM, Lonigro RJ, et al. : Integrative Clinical Sequencing in the Management of Refractory or Relapsed Cancer in Youth. JAMA 314:913–25, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oberg JA, Glade Bender JL, Sulis ML, et al. : Implementation of next generation sequencing into pediatric hematology-oncology practice: moving beyond actionable alterations. Genome Med 8:133, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harris MH, DuBois SG, Glade Bender JL, et al. : Multicenter Feasibility Study of Tumor Molecular Profiling to Inform Therapeutic Decisions in Advanced Pediatric Solid Tumors: The Individualized Cancer Therapy (iCat) Study. JAMA Oncol 2:608–615, 2016 [DOI] [PubMed] [Google Scholar]
- 11.Sweet-Cordero EA, Biegel JA: The genomic landscape of pediatric cancers: Implications for diagnosis and treatment. Science 363:1170–1175, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crist WM, Kun LE: Common solid tumors of childhood. N Engl J Med 324:461–71, 1991 [DOI] [PubMed] [Google Scholar]
- 13.Zhu J, Fu W, Jia W, et al. : Association between NER Pathway Gene Polymorphisms and Wilms Tumor Risk. Molecular therapy. Nucleic acids 12:854–860, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maschietto M, Williams RD, Chagtai T, et al. : TP53 mutational status is a potential marker for risk stratification in Wilms tumour with diffuse anaplasia. PLoS One 9:e109924, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dome JS, Cotton CA, Perlman EJ, et al. : Treatment of anaplastic histology Wilms’ tumor: results from the fifth National Wilms’ Tumor Study. J Clin Oncol 24:2352–8, 2006 [DOI] [PubMed] [Google Scholar]
- 16.Ooms AH, Gadd S, Gerhard DS, et al. : Significance of TP53 Mutation in Wilms Tumors with Diffuse Anaplasia: A Report from the Children’s Oncology Group. Clin Cancer Res 22:5582–5591, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wegert J, Vokuhl C, Ziegler B, et al. : TP53 alterations in Wilms tumour represent progression events with strong intratumour heterogeneity that are closely linked but not limited to anaplasia. J Pathol Clin Res 3:234–248, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grundy PE, Breslow NE, Li S, et al. : Loss of heterozygosity for chromosomes 1p and 16q is an adverse prognostic factor in favorable-histology Wilms tumor: a report from the National Wilms Tumor Study Group. J Clin Oncol 23:7312–21, 2005 [DOI] [PubMed] [Google Scholar]
- 19.Dix DB, Fernandez CV, Chi YY, et al. : Augmentation of Therapy for Combined Loss of Heterozygosity 1p and 16q in Favorable Histology Wilms Tumor: A Children’s Oncology Group AREN0532 and AREN0533 Study Report. J Clin Oncol 37:2769–2777, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gratias EJ, Dome JS, Jennings LJ, et al. : Association of Chromosome 1q Gain With Inferior Survival in Favorable-Histology Wilms Tumor: A Report From the Children’s Oncology Group. J Clin Oncol 34:3189–94, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chagtai T, Zill C, Dainese L, et al. : Gain of 1q As a Prognostic Biomarker in Wilms Tumors (WTs) Treated With Preoperative Chemotherapy in the International Society of Paediatric Oncology (SIOP) WT 2001 Trial: A SIOP Renal Tumours Biology Consortium Study. J Clin Oncol 34:3195–203, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dome JS, Fernandez CV, Mullen EA, et al. : Children’s Oncology Group’s 2013 blueprint for research: renal tumors. Pediatr Blood Cancer 60:994–1000, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daw NC, Chi YY, Kalapurakal JA, et al. : Activity of Vincristine and Irinotecan in Diffuse Anaplastic Wilms Tumor and Therapy Outcomes of Stage II to IV Disease: Results of the Children’s Oncology Group AREN0321 Study. J Clin Oncol 38:1558–1568, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weeks DA, Beckwith JB, Mierau GW, et al. : Rhabdoid tumor of kidney. A report of 111 cases from the National Wilms’ Tumor Study Pathology Center. Am J Surg Pathol 13:439–58, 1989 [PubMed] [Google Scholar]
- 25.Tomlinson GE, Breslow NE, Dome J, et al. : Rhabdoid tumor of the kidney in the National Wilms’ Tumor Study: age at diagnosis as a prognostic factor. J Clin Oncol 23:7641–5, 2005 [DOI] [PubMed] [Google Scholar]
- 26.Roberts CW, Biegel JA: The role of SMARCB1/INI1 in development of rhabdoid tumor. Cancer Biol Ther 8:412–6, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim KH, Roberts CW: Mechanisms by which SMARCB1 loss drives rhabdoid tumor growth. Cancer Genet 207:365–72, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Milholland B, Auton A, Suh Y, et al. : Age-related somatic mutations in the cancer genome. Oncotarget 6:24627–35, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Califano A, Alvarez MJ: The recurrent architecture of tumour initiation, progression and drug sensitivity. Nat Rev Cancer 17:116–130, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alvarez MJ, Shen Y, Giorgi FM, et al. : Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nature genetics 48:838–47, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alvarez MJ: Virtual Inference of Protein-activity by Enriched Regulon analysis, 2016
- 32.Obradovic A, Chowdhury N, Haake SM, et al. : Single-cell protein activity analysis identifies recurrence-associated renal tumor macrophages. Cell 184:2988–3005.e16, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carro MS, Lim WK, Alvarez MJ, et al. : The transcriptional network for mesenchymal transformation of brain tumours. Nature 463:318–25, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rajbhandari P, Lopez G, Capdevila C, et al. : Cross-Cohort Analysis Identifies a TEAD4-MYCN Positive Feedback Loop as the Core Regulatory Element of High-Risk Neuroblastoma. Cancer Discov 8:582–599, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alvarez MJ, Subramaniam PS, Tang LH, et al. : A precision oncology approach to the pharmacological targeting of mechanistic dependencies in neuroendocrine tumors. Nat Genet 50:979–989, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paull EO, Aytes A, Jones SJ, et al. : A modular master regulator landscape controls cancer transcriptional identity. Cell 184:334–351.e20, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walsh LA, Alvarez MJ, Sabio EY, et al. : An Integrated Systems Biology Approach Identifies TRIM25 as a Key Determinant of Breast Cancer Metastasis. Cell Rep 20:1623–1640, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Piovan E, Yu J, Tosello V, et al. : Direct reversal of glucocorticoid resistance by AKT inhibition in acute lymphoblastic leukemia. Cancer Cell 24:766–76, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mitrofanova A, Aytes A, Zou M, et al. : Predicting Drug Response in Human Prostate Cancer from Preclinical Analysis of In Vivo Mouse Models. Cell Rep 12:2060–71, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bansal M, Yang J, Karan C, et al. : A community computational challenge to predict the activity of pairs of compounds. Nat Biotechnol 32:1213–22, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ding H, Douglass EF Jr., Sonabend AM, et al. : Quantitative assessment of protein activity in orphan tissues and single cells using the metaVIPER algorithm. Nat Commun 9:1471, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Azizian NG, Li Y: XPO1-dependent nuclear export as a target for cancer therapy. J Hematol Oncol 13:61, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Turner JG, Dawson J, Sullivan DM: Nuclear export of proteins and drug resistance in cancer. Biochem Pharmacol 83:1021–32, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.FDA: Hematology/Oncology (Cancer) Approvals & Safety Notifications, 2019
- 45.Garzon R, Savona M, Baz R, et al. : A phase I clinical trial of single-agent selinexor in acute myeloid leukemia. Blood, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuruvilla J, Savona M, Baz R, et al. : Selective inhibition of nuclear export with selinexor in patients with non-Hodgkin’s lymphoma. Blood, 2017 [DOI] [PubMed] [Google Scholar]
- 47.Lowis SP, Foot A, Gerrard MP, et al. : Central nervous system metastasis in Wilms’ tumor: a review of three consecutive United Kingdom trials. Cancer 83:2023–9, 1998 [DOI] [PubMed] [Google Scholar]
- 48.Venkatramani R, Chi YY, Coppes MJ, et al. : Outcome of patients with intracranial relapse enrolled on national Wilms Tumor Study Group clinical trials. Pediatr Blood Cancer 64, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Etchin J, Berezovskaya A, Conway AS, et al. : KPT-8602, a second-generation inhibitor of XPO1-mediated nuclear export, is well tolerated and highly active against AML blasts and leukemia-initiating cells. Leukemia 31:143–150, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Margolin AA, Nemenman I, Basso K, et al. : ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC bioinformatics 7 Suppl 1:S7, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wishart DS, Knox C, Guo AC, et al. : DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res 36:D901–6, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roberts CW, Orkin SH: The SWI/SNF complex--chromatin and cancer. Nat Rev Cancer 4:133–42, 2004 [DOI] [PubMed] [Google Scholar]
- 53.Hall PA, Lane DP: p53 in tumour pathology: can we trust immunohistochemistry?--Revisited! J Pathol 172:1–4, 1994 [DOI] [PubMed] [Google Scholar]
- 54.Cheng DT, Mitchell TN, Zehir A, et al. : Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn 17:251–64, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ortiz MV, Siddiquee A, You D, et al. : Preclinical evaluation of XPO1 inhibition in Wilms tumors. Journal of Clinical Oncology 38:3580–3580, 2020 [Google Scholar]
- 56.Jhaveri K MV, Wang XV, Chen AP, Flaherty K, Conley BA: Results from the National Cancer Institute (NCI) Molecular Analysis for Therapy Choice (MATCH) trial. Journal of Clinical Oncology 36, 2018 [Google Scholar]
- 57.Landsburg DJ, Barta SK, Ramchandren R, et al. : Fimepinostat (CUDC-907) in patients with relapsed/refractory diffuse large B cell and high-grade B-cell lymphoma: report of a phase 2 trial and exploratory biomarker analyses. Br J Haematol 195:201–209, 2021 [DOI] [PubMed] [Google Scholar]
- 58.Risom T, Langer EM, Chapman MP, et al. : Differentiation-state plasticity is a targetable resistance mechanism in basal-like breast cancer. Nat Commun 9:3815, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Alvarez MJ, Shen Y, Giorgi FM, et al. : Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat Genet 48:838–47, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shen Y, Alvarez MJ, Bisikirska B, et al. : Systematic, network-based characterization of therapeutic target inhibitors. PLoS Comput Biol 13:e1005599, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Alvarez MJ, Califano A: Darwin OncoTarget/OncoTreat: NY CLIA certified tests to identify effective drugs on an individual cancer patient basis from RNASeq profiles Dpt of Pathology and Cell Biology Web Site, Columbia University, 2018 [Google Scholar]
- 62.Weinstein IB: Cancer. Addiction to oncogenes--the Achilles heal of cancer. Science 297:63–4, 2002 [DOI] [PubMed] [Google Scholar]
- 63.Abeykoon JP, Wu X, Nowakowski KE, et al. : Salicylates enhance CRM1 inhibitor antitumor activity by induction of S-phase arrest and impairment of DNA-damage repair. Blood 137:513–523, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Baek HB, Lombard AP, Libertini SJ, et al. : XPO1 inhibition by selinexor induces potent cytotoxicity against high grade bladder malignancies. Oncotarget 9:34567–34581, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Garg M, Kanojia D, Mayakonda A, et al. : Selinexor (KPT-330) has antitumor activity against anaplastic thyroid carcinoma in vitro and in vivo and enhances sensitivity to doxorubicin. Sci Rep 7:9749, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Garg M, Kanojia D, Mayakonda A, et al. : Molecular mechanism and therapeutic implications of selinexor (KPT-330) in liposarcoma. Oncotarget 8:7521–7532, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Golomb L, Bublik DR, Wilder S, et al. : Importin 7 and exportin 1 link c-Myc and p53 to regulation of ribosomal biogenesis. Molecular cell 45:222–232, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Galinski B, Alexander TB, Mitchell DA, et al. : Therapeutic Targeting of Exportin-1 in Childhood Cancer. Cancers (Basel) 13, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Subhash VV, Yeo MS, Wang L, et al. : Anti-tumor efficacy of Selinexor (KPT-330) in gastric cancer is dependent on nuclear accumulation of p53 tumor suppressor. Sci Rep 8:12248, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sun H, Hattori N, Chien W, et al. : KPT-330 has antitumour activity against non-small cell lung cancer. Br J Cancer 111:281–91, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tai YT, Landesman Y, Acharya C, et al. : CRM1 inhibition induces tumor cell cytotoxicity and impairs osteoclastogenesis in multiple myeloma: molecular mechanisms and therapeutic implications. Leukemia 28:155–65, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chun HE, Johann PD, Milne K, et al. : Identification and Analyses of Extra-Cranial and Cranial Rhabdoid Tumor Molecular Subgroups Reveal Tumors with Cytotoxic T Cell Infiltration. Cell Rep 29:2338–2354 e7, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ranganathan P, Kashyap T, Yu X, et al. : XPO1 Inhibition using Selinexor Synergizes with Chemotherapy in Acute Myeloid Leukemia by Targeting DNA Repair and Restoring Topoisomerase IIalpha to the Nucleus. Clin Cancer Res 22:6142–6152, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kashyap T, Argueta C, Unger T, et al. : Selinexor reduces the expression of DNA damage repair proteins and sensitizes cancer cells to DNA damaging agents. Oncotarget 9:30773–30786, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Turner JG, Cui Y, Bauer AA, et al. : Melphalan and Exportin 1 Inhibitors Exert Synergistic Antitumor Effects in Preclinical Models of Human Multiple Myeloma. Cancer Res 80:5344–5354, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Furtwangler R, Kager L, Melchior P, et al. : High-dose treatment for malignant rhabdoid tumor of the kidney: No evidence for improved survival-The Gesellschaft fur Padiatrische Onkologie und Hamatologie (GPOH) experience. Pediatr Blood Cancer 65, 2018. [DOI] [PubMed] [Google Scholar]
- 77.Hong AL, Tseng YY, Cowley GS, et al. : Integrated genetic and pharmacologic interrogation of rare cancers. Nat Commun 7:11987, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Carugo A, Minelli R, Sapio L, et al. : p53 Is a Master Regulator of Proteostasis in SMARCB1-Deficient Malignant Rhabdoid Tumors. Cancer Cell 35:204–220 e9, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Turner JG, Kashyap T, Dawson JL, et al. : XPO1 inhibitor combination therapy with bortezomib or carfilzomib induces nuclear localization of IkappaBalpha and overcomes acquired proteasome inhibitor resistance in human multiple myeloma. Oncotarget 7:78896–78909, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gounder MM, Zer A, Tap WD, et al. : Phase IB Study of Selinexor, a First-in-Class Inhibitor of Nuclear Export, in Patients With Advanced Refractory Bone or Soft Tissue Sarcoma. J Clin Oncol 34:3166–74, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chari A, Vogl DT, Gavriatopoulou M, et al. : Oral Selinexor-Dexamethasone for Triple-Class Refractory Multiple Myeloma. N Engl J Med 381:727–738, 2019 [DOI] [PubMed] [Google Scholar]
- 82.Alexander TB, Lacayo NJ, Choi JK, et al. : Phase I Study of Selinexor, a Selective Inhibitor of Nuclear Export, in Combination With Fludarabine and Cytarabine, in Pediatric Relapsed or Refractory Acute Leukemia. J Clin Oncol 34:4094–4101, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Place AE, Blonquist TM, Stieglitz E, et al. : hase I Study of the Selinexor in Relapsed/Refractory Childhood Acute Leukemia. Blood 132, 2018 [Google Scholar]
- 84.Green AL, Minard CG, Liu X, et al. : Abstract P162: Phase 1 trial of selinexor in children and adolescents with recurrent/refractory solid and CNS tumors (ADVL1414): A Children’s Oncology Group Phase 1 Consortium trial, AACR, 2021 [Google Scholar]
- 85.Balis F, Green DM, Anderson C, et al. : Wilms Tumor (Nephroblastoma), Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 19:945–977, 2021 [DOI] [PubMed] [Google Scholar]
- 86.Sugimoto T, Hosoi H, Horii Y, et al. : Malignant rhabdoid-tumor cell line showing neural and smooth-muscle-cell phenotypes. Int J Cancer 82:678–86, 1999 [DOI] [PubMed] [Google Scholar]
- 87.Misawa A, Hosoi H, Imoto I, et al. : Translocation (1;22)(p36;q11.2) with concurrent del(22)(q11.2) resulted in homozygous deletion of SNF5/INI1 in a newly established cell line derived from extrarenal rhabdoid tumor. J Hum Genet 49:586–9, 2004 [DOI] [PubMed] [Google Scholar]
- 88.Kuroda H, Moritake H, Sawada K, et al. : Establishment of a cell line from a malignant rhabdoid tumor of the liver lacking the function of two tumor suppressor genes, hSNF5/INI1 and p16. Cancer Genet Cytogenet 158:172–9, 2005 [DOI] [PubMed] [Google Scholar]
- 89.Katsumi Y, Kuwahara Y, Tamura S, et al. : Trastuzumab activates allogeneic or autologous antibody-dependent cellular cytotoxicity against malignant rhabdoid tumor cells and interleukin-2 augments the cytotoxicity. Clin Cancer Res 14:1192–9, 2008 [DOI] [PubMed] [Google Scholar]
- 90.Mariotti E, Mirabelli P, Di Noto R, et al. : Rapid detection of mycoplasma in continuous cell lines using a selective biochemical test. Leuk Res 32:323–6, 2008 [DOI] [PubMed] [Google Scholar]
- 91.Dobin A, Davis C, Schlesinger F, et al. : STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Love MI, Huber W, Anders S: Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kim SY, Volsky DJ: PAGE: parametric analysis of gene set enrichment. BMC Bioinformatics 6:144, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liberzon A: A description of the Molecular Signatures Database (MSigDB) Web site. Methods Mol Biol 1150:153–60, 2014 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Oncotarget predictions and RNA-seq data have been deposited at Figshare and Gene Expression Omnibus (GEO), respectively, and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CRM1 antibody (XPO1); 1:1000 | Abcam | Cat# ab24189, RRID: AB 2257217 |
| SNF5 antibody [EPR6966]; 1:1000 | Abcam | Cat# ab126734, RRID: AB 11127897 |
| gamma H2A.X (pS139) antibody [9F3]; 1:1000 | Abcam | Cat# ab26350, RRID: AB 470861 |
| p53 (7F5) Rabbit mAb antibody; 1:1000 | CST | Cat# 2527, RRID: AB 10695803 |
| Rb (4H1) Mouse mAb antibody; 1:2000 | CST | Cat# 9309, RRID: AB 823629 |
| Phospho-Rb (Ser780) antibody; 1:1000 | CST | Cat# 9307, RRID: AB 330015 |
| p21 Waf1/Cip1 (12D1) antibody; 1:1000 | CST | Cat# 2947, RRID: AB 823586 |
| p27 Kip1 (D37H1) mAb antibody; 1:1000 | CST | Cat# 3688, RRID: AB 2077836 |
| Cleaved PARP (Asp214) (D64E10) antibody; 1:1000 | CST | Cat# 5625, RRID: AB 10699459 |
| c-Myc (D84C12) mAb antibody; 1:1000 | CST | Cat# 5605, RRID: AB 1903938 |
| Anti-β-Actin antibody; 1:5000 | Sigma-Aldrich | Cat# A5441, RRID: AB 476744 |
| Anti-α-Tubulin antibody; 1:1000 | Sigma-Aldrich | Cat# T9026, RRID: AB 477593 |
| IRDye 680RD Donkey anti-Mouse IgG antibody; 1:5000 | LI-COR Biosciences | Cat# 926-68072, RRID: AB 10953628 |
| IRDye 800CW Donkey anti-Rabbit IgG antibody; 1:15000 | LI-COR Biosciences | Cat# 926-32213, RRID: AB 621848 |
| Monoclonal Mouse Anti-Human Ki-67 Antigen; 1:150 | Agilent | Cat# M7240, RRID: AB 2142367 |
| Cleaved Caspase-3 (Asp175) antibody; 1:1000 | CST | Cat# 9661, RRID: AB 2341188 |
| Biotinylated horse anti-mouse IgG; 1:50 | Vector Labs | Cat# MKB-22258 |
| Biotinylated goat anti-rabbit; 1:50 | Vector Labs | Cat# PK6101 |
| Tyramide Alexa Fluor 568 | Invitrogen | Cat# T20949 |
| DAPI | Sigma Aldrich | Cat# D9542 |
| Mowiol | Sigma Aldrich | Cat# 81381 |
| Chemicals, peptides, and recombinant proteins | ||
| Dulbecco’s Modified Eagle Medium (DMEM) | ThermoFisher Scientific | Cat# 12491015 |
| DMEM/F-12 | ThermoFisher Scientific | Cat# 11320033 |
| Iscove’s Modification of DMEM (IMDM) | Corning | Cat# 10-016 |
| Fetal Bovine Serum (FBS) | Corning | Cat# 35-010 |
| Antibiotic-Antimycotic (100X) | ThermoFisher Scientific | Cat# 15240062 |
| L-Glutamine (200 mM) | ThermoFisher Scientific | Cat# 25030081 |
| Selinexor | Selleckchem | Cat# S7252 |
| Eltanexor | Selleckchem | Cat# S8397 |
| Ethanol | Merck | Cat# 100983 |
| Phosphate Buffered Saline | Gibco | Cat# 10010023 |
| Ribonuclease H (RNase H) | Sigma-Aldrich | Cat# 10786357001 |
| Propidium iodide | Sigma-Aldrich | Cat# P4170 |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | Cat# D2650 |
| RIPA Buffer | Sigma-Aldrich | Cat# R0278 |
| Pierc BCA Protein Assay Kit | Thermofisher Scientific | Cat# 23225 |
| Formalin solution, neutral buffered, 10% | Sigma-Aldrich | Cat# HT501128 |
| EZ Prep Concentrate | Ventana | Cat# 950-102 |
| ULTRA Cell Conditioning (CC1) | Ventana | Cat# 950-224 |
| Background Buster | Innovex | Cat# NB306 |
| Critical commercial assays | ||
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | Cat# G7572 |
| Caspase-Glo® 3/7 Assay | Promega | Cat# G8091 |
| Nuclear Extraction Kit | Abcam | Cat# ab113474 |
| NuPAGE™ 4 to 12%, Bis-Tris, 1.0-1.5 mm, Mini Protein Gels | Invitrogen | Cat# NP0321BOX |
| QIAGEN QIAamp Tissue Kit | Qiagen | Cat# 51404 |
| RNeasy Micro Kit | Qiagen | Cat# 74004 |
| Agilent SureSelectXT All Exon V5 + UTRs | Agilent | Cat# 5191-7408 |
| TruSeq Stranded Total RNA LT Sample Prep Kit | Illumina | Cat# 20020596 |
| Deposited data | ||
| RNA-seq data from cell lines and used for perturbation screen | Gene Expression Omnibus (GEO) | GEO Accession: GSE198000 |
| Oncotarget predictions | Figshare | https://figshare.com/sZ3213cfaf6b2a2aeb7783 |
| Experimental models: Cell lines | ||
| 786-O | ATCC | Cat# CRL-1932 |
| BJ | Dr. Alex Kentsis; MSKCC | N/A |
| BT12 | Dr. Alex Kentsis; MSKCC | N/A |
| BT16 | Dr. Alex Kentsis; MSKCC | N/A |
| CHLA-266 | COG cell line repository | RRID: CVCL_M149 |
| COGW408 | Dr. C. Patrick Reynolds; Texas Tech University | N/A |
| G401 | ATCC | Cat# 87042204, RRID: CVCL 0270 |
| KP-MRT-AN | Dr. Hajime Hosoi’s; Kyoto Prefectural University of Medicine | N/A |
| KP-MRT-NS | Dr. Hajime Hosoi’s; Kyoto Prefectural University of Medicine | N/A |
| KP-MRT-YM | Dr. Hajime Hosoi’s; Kyoto Prefectural University of Medicine | N/A |
| RH-30 | ATCC | CVCL 0041 |
| RPE | Dr. Alex Kentsis; MSKCC | N/A |
| TC-71 | ATCC | CVCL 2213 |
| Wilms 1 | Dr. Brigitte Royer-Pokora; Heinrich-Heine-University | N/A |
| Wilms 10 | Dr. Brigitte Royer-Pokora; Heinrich-Heine-University | N/A |
| WiT49 | Dr. Herman Yeger; University of Toronto | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ | The Jackson Laboratory | Cat# 005557; RRID: IMSR_JAX: 005557 |
| Software and algorithms | ||
| Prism Software Version 9 | GraphPad | N/A |
This study did not generate any new codes.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.