

Abstract
The human gut microbiome is a complex system composed of hundreds of species, and metaproteomics can be used to explore their expressed functions. However, many lower abundance species are not detected by current metaproteomic techniques and represent the dark field of metaproteomics. We do not know the minimal abundance of a bacterium in a microbiome(depth) that can be detected by shotgun metaproteomics. In this study, we spiked 15N-labeled E. coli peptides at different percentages into peptides mixture derived from the human gut microbiome to evaluate the depth that can be achieved by shotgun metaproteomics. We observed that the number of identified peptides and peptide intensity from 15N-labeled E. coli were linearly correlated with the spike-in levels even when 15N-labeled E. coli was down to 0.5% of the biomass. Below that level, it was not detected. Interestingly, the match-between-run strategy significantly increased the number of quantified peptides even when 15N-labeled E. coli peptides were at low abundance. This is indicative that in metaproteomics of complex gut microbiomes many peptides from low abundant species are likely observable in MS1 but are not selected for MS2 by standard shotgun strategies.
Introduction
The human gut microbiome is an important factor for human health, and its dysbiosis is related to many chronic disease.1 The gut microbiome is a complex system having a relatively equal number of cells to that of its host.2 Metagenomics has been commonly used in human microbiome studies.4,5 Whereas it only reveals the functional potential of the microbiome, metaproteomics can reveal direct functional information and is more suitable to study functional alterations. However, the analysis of lower abundant proteins by metaproteomics remains an issue.
Genome sequencing has revealed that a healthy individual harbors around 200 bacterial species in their guts.6,7 However, bacterial abundances in the microbiome are not uniform with a few species forming the bulk of the microbiome.6 Therefore, most bacteria species in a microbiome are of low abundance and their proteins would also be in low abundance. Although gene copy number does not exactly correspond to protein biomass, it highlights the high diversity of the human gut microbiome.
The protein analysis depth that can be achieved by metaproteomics for the human gut microbiome is not well understood. Data-dependent acquisition (DDA) is extensively used in metaproteomics. In DDA, the most intense peaks in the mass spectrometer (MS)1 are selected for fragmentation and thus a bias based upon abundance is introduced. The selection of low abundant peptides is usually stochastic in DDA. As well, background proteins can decrease the sensitivity in DDA mode.8 Strategies like dynamic exclusion and match-between-run (MBR) can be used to increase the identification rate of DDA. Nevertheless, when DDA is used to analyze complex samples like microbiome communities, a great deal of information from low abundance peptides may be lost. To the best of our knowledge, the largest number of peptides identified in a single metaproteomic study is approximately 45,000 using an ultradeep analysis method with DDA mode.9 This method requires a prefractionation before loading samples onto the MS and much longer MS run times. However, in this study, we identified over 20,000 peptides from a single bacterial strain which account for ∼45% of the peptides previously identified by the ultradeep analysis. Considering the high diversity of the human gut microbiome, many peptides remain to be characterized. Therefore, the depth and resolution of MS-based metaproteomics on the human microbiome need to be evaluated.
In this study, we labeled an E. coli strain with 15N and then mixed the heavy-labeled E. coli peptides with unlabeled human gut microbiome peptides at different percentages. We then evaluated the performance of the DDA mode for the detection of 15N-labeled E. coli peptides at the different spike-in percentages. We also evaluated the MBR strategy used in DDA.
Experimental Section
Bacteria Culture and 15N Heavy Labeled E. coli
The Escherichia coli (DSM 101114; Leibniz Institute DSMZ- German collection of microorganisms and cell cultures) powder was rehydrated in LB broth (Millipore Sigma, ON, CAN) and then streaked onto sheep blood agar (tryptic soy agar (Fisher Scientific, ON, CAN) with 5% (v/v) sheep blood (Cedarlane, ON, CAN)) and was anaerobically cultured at 37 °C overnight. One isolated colony was transferred into 15N media (Cambridge Isotope Laboratories, Inc. CGM-1000-NS, QC, CAN) and cultured at 37 °C for 30 h. The samples were centrifuged at 14,000g at 4 °C for 5 min to pellet the cell. After the supernatant was removed, the pelleted cells were washed with ice-cold 1X phosphate-buffered saline (PBS; pH 7.4) three times and pellets were stored at −80 °C prior to protein extraction.
Blautia hydrogenotrophica (DSM10507; Leibniz Institute DSMZ) and Bacteroides uniformis (ATCC8492; Cedarlane, ON, CAN) were processed in the same method but cultured in 14N medium.
Human Stool Microbiome
The Human stool was collected from a healthy adult volunteer at the University of Ottawa, Ottawa, ON, CAN. The protocol (# 20160585-01H) was approved by Ottawa Health Science Network Research Ethics. Briefly, the fresh stool was resuspended to 20% (w/v) in cold PBS containing protease inhibitors and then mixed with 2.5 mm glass beads. The fecal slurry was centrifuged at 700g, 4 °C for 5 min to remove debris. The supernatant was collected and centrifuged at 14,000g for 30 min. The pellet was then resuspended with cold PBS and centrifuged for another 30 min at 14,000g, 4 °C. The pellets were stored at 80 °C for protein extraction.
Cultured Microbiome
The culture was based on our previous published method.10 Briefly, 100 μL of fecal inoculum were mixed with 900 μL of optimized medium in a 96-well plate. All the operations were carried out in an anaerobic chamber. Bacteria were harvested after 24 h culture at 37 °C. Following culture, the plate was centrifuged at 2272g at 4 °C for 45 min. The supernatant was removed. With the plate sitting on ice, 1 mL of cold PBS was added to each well and mixed thoroughly to wash the cells. Two additional washes were carried out. The plate was centrifuged at 300g at 4 °C for 5 min to spin down the debris following each wash. Following the 300g spins, the cell suspension was centrifuged at 2272g at 4 °C for 45 min. The supernatant was removed. Cell pellets were stored at −80 °C for protein extraction.
Protein Extraction and Tryptic Digestion
Bacteria cells were lysed in lysis buffer (6 M urea (Millipore Sigma) in 50 mM Tris-HCl buffer (pH = 8.0; Millipore Sigma), 4% (w/v) sodium dodecyl sulfate (SDS; Millipore Sigma), and Roche PhosSTOP and Roche cOmplete Mini tablets). The bacterial lysate was ultrasonicated for 10 min at 8 °C (Q125 Qsonica, USA) using a round of 10 s ultrasonication and 10 s cooling down at 50% amplitude. The lysate was centrifuged at 16,000g to remove the debris. Total protein was precipitated by adding ice-cold acetone/ethanol/acetic acid (50:50:0.1; Fisher Scientific) at a 1:5 (v/v) overnight at −20 °C. Proteins were pelleted at 16,000g and 4 °C. Protein pellets were washed with 100% acetone three times, and pellets were dissolved in 6 M urea, 50 mM ammonium bicarbonate (pH 8.0; Millipore Sigma). Protein concentration was measured by the detergent compatible (DC) assay (Bio-Rad, USA). A 50 μg amount of protein was reduced and alkylated with 10 mM dithiothreitol (37 °C for 1 h) and 20 mM iodoacetamide (room temperature in the dark for 45 min). After 10× dilution with 50 mM ammonium bicarbonate (pH 8.0), the protein was digested by 1 μg of trypsin (Worthington biochemicals, USA) per 50 μg of proteins at 37 °C for 24 h. Peptides were desalted by a 10 μm C18 column (Dr.Maisch HPLC GmbH, Ammerbuch, Germany). After freeze-drying, each sample was redissolved in 0.1% (v/v) formic acid (Millipore Sigma).
Generation of Serial Dilution Mixtures
Peptide concentrations were measured using Thermo Scientific Pierce Quantitative Colorimetric Peptide Assays according to the manufacturer’s directions. The serial dilution mixtures were prepared from 0.0005% to 100% of 15N labeled peptides with triplicates. Dried peptide mixtures were dissolved into an equal volume of 0.1% (v/v) formic acid before MS analysis.
LC–MS/MS Analysis
Samples were analyzed by an UltiMate 3000 RSLCnano system (Thermo Fisher Scientific, USA) coupled to an Orbitrap Exploris 480 mass spectrometer (Thermo Fisher Scientific, USA). Peptides were loaded onto a tip column (75 μm inner diameter ×15 cm) packed with reverse phase beads (3 μm/120 Å ReproSil-Pur C18 resin, Dr. Maisch HPLC GmbH). A 60 min gradient of 5 to 35% (v/v) from buffer A (0.1% (v/v) formic acid) to B (0.1% (v/v) formic acid with 80% (v/v) acetonitrile) at a flow rate of 300 μL/min was used. The mass spectrometer was in data-dependent mode with top15 method. The dynamic exclusion repeat count was set to one, and the repeat exclusion duration was set to 20 s. The full mass scan was from 350 to 1200 (m/z). The samples were analyzed in a randomized order.
Database Searching and Peptides Quantification
The raw files were first searched against the human gut microbial IGC (integrated gene catalogue) database4 using Metalab 2.011 to generate a refined fasta database. The E. coli database was downloaded from the Uniprot by downloading all protein fasta sequences of Escherichia coli (strain K12). A combined database of the two databases above was generated which was used for the open search by pFind 3.012 for both 15N and background peptide identification. The quantification was performed by FlashLFQ13 with and without MBR. Isotope PPM tolerance was set at 5. At least 2 isotopes were required. The maximum window for MBR was 2.5 min. The peptides intensities used for analysis were raw intensity without any normalization or scaling.
Functional Analysis
All identified proteins were blasted against the latest Clusters of Orthologous Genes (COG) database.14 We carried out compositional analysis using an R Shiny app (https://shiny.imetalab.ca/playtable/) and calculated the Bray–Curtis distance using the R package “vegan”. Briefly, the identified proteins were annotated to their COG category. The intensity of the COG category in each sample was scaled by percentage by the total intensity. The averages of three replicates were used to represent the COG category percentage. The distance was calculated using the percentage between the 100% E. coli sample and the other samples.
Results and Discussion
Experiment Overview
Human gut microbiomes are complex systems composed of hundreds of microbial species.7 Metaproteomics has been used to better understand the biology of the gut microbiome. Nevertheless, metaproteomics principally provides information on the more abundant species representing the bulk of the biomass. Here we systematically evaluated the depth and coverage of the gut microbiome that can be achieved by DDA-based metaproteomics with 1-h nano-LC analysis. Briefly, a 15N-labeled E. coli proteome was spiked at different percentages (w/w) into a background peptide mixture either directly from human stool or a microbiome isolate following in vitro culture for 24 h10 (Figure 1). The mixtures were analyzed by HPLC-ESI-MS/MS. The peptides were then identified for 15N E. coli or the background human microbiome using pFind 3.0 in 15N or 14N mode. Unlike studies based on sequencing technologies which provide an indirect measure of cell numbers,15 MS-based proteomics directly provides protein biomass information.10 So, the intensity of 15N peptides is expected to be correlated with its quantity in the peptide mixture.
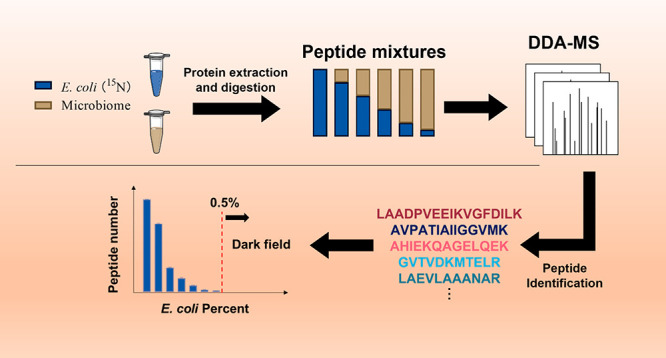
Figure 1.

Experiment setup and identification profile. Experiment workflow. E. coli was cultured in 15N medium to label its proteins. Then the 15N-labeled proteome was extracted and digested into peptides. 15N peptides were mixed at different percentages with peptides from a human gut microbiome. The mixture was subjected to mass spectrometer analysis. Peptide identification was performed using Pfind3.0 with open search in 14N mode or 15N mode. FlashLFQ was used to do quantification with or without MBR.
From the pure 15N-labeled E. coli, on average, 22,148 15N labeled peptides (identification rate: 67%, Figure 2a) were found across 3 replicates, while only around 1,000 14N peptides were identified (identification rate: 2.80%). This suggested a high labeling efficiency of the E. coli culture. In contrast, 16,000 peptides were identified from the background microbiomes in the absence of 15N-labeled E. coli which is comparable to previous studies (Figures 2a and S2a).10
Figure 2.

Performance of DDA method for peptide identification and quantification with the stool protein as background. (a) The number of identified peptides in each sample. (b) Linear regression between log2 quantified peptide number and log2 of 15N peptide spike-in percentage (blue). Linear regression between log2 total peptide intensity and log2 of 15N peptide spike-in percentage(green). (c) The number of quantified peptides in each sample. Peptides identified directly by database searching (Pfind 3.0) are shown in red. Peptides transferred by MBR (FlashLFQ) are shown in blue. (d) Density plot of the intensity of all quantified 15N peptides (left). Distribution of LODs for peptide intensity (right). The figures above each LOD group represent the number of peptides in each group, which is also displayed by the color scale.
The number of identified peptides was correlated with concentrations of spiked-in labeled peptides
As expected, as the concentration of spike-in 15N E. coli peptide decreased, fewer 15N-labeled peptides were detected. On average 117 and 173 peptides were identified for 15N-labeled E. coli spiked at 0.5% in the two background microbiomes (Figures 2a and S2a). However, when 15N-labeled E. coli was spiked at 0.1% or lower, the identification of 15N-labeled peptides was nearly impossible. The number of quantified 15N-labeled E. coli peptides was linear-correlated with the spike-in percentage from 0.5% to 100% (Figures 2b and S2b). Low-abundant bacterial species are poorly characterized by metaproteomics. Due to the diversity of bacteria size and mass, it is difficult to identify the biomass composition of the human microbiome. However, composition derived from gene expression analysis can be used as a reference. One sequencing study including 120 healthy adults found that each individual harbored, on average, 186 species-level phylotypes (SLPs)7 (Figure S3a). Only around 30 SLPs had an abundance over 0.5% (Figure S3b). Moreover, those high abundant species (>0.5%) account for more than 80% of the total abundance (Figure S3c). This means the gut microbiome is dominated by several very high abundant species (Figure S4), and most species may represent less than 0.5% of the biomass in the microbiome and are undetectable in DDA mode using the parameters of this experiment. Although the gene expression composition of one species is not equal to its biomass composition, the extremely uneven composition indicated that a large amount of information has been neglected by the DDA mode. Moreover, a previous metaproteomic study indicated that ∼95% of the protein biomass in the gut microbiome is dominated by the top ∼100 species.16 This means that ∼50% of the bacterial species in the gut microbiome together contribute to about 5% of the protein biomass of the microbiome and are difficult to analyze by metaproteomics.
In DDA mode, the mass spectrometer is unlikely to consistently select the same ions for fragmentation across multiple experiments. Fortunately, it is possible to infer the identification of peptides across multiple runs using retention time and parent m/z information using the function match between runs (MBR).13,17 This function is clearly beneficial for lower concentrations of spike-in 15N E. coli and, in particular, from 5% to 0.5% (Figures 2c and S2c). At an E. coli spike-in of 0.5%, over 98% of peptides were from transferred peptide-spectrum matches (PSMs). We observed a sudden decrease in the number of transferred peptides when the 15N peptides percentage was below 0.5%. That is likely due to a threshold of the MBR algorithm, and the 0.5% spike-in approaches that threshold. Even so, the MBR strategy showed a remarkable effect on microbiome analysis.
Spike-in composition of 15N E. coli peptides can be quantified accurately
In this experiment, the decrease of total peptide intensity is due to the decrease of the number of detected peptides and the decrease of their concentrations. Interestingly, a positive linear relationship was observed between the total 15N peptide intensity versus the spike-in percentage of E. coli in the background microbiome (Figure 2b). This means that the MS can be used to accurately characterize the biomass contributed by individual bacterial species in a microbiome. The biomass of the same source measured by MS is totally comparable across samples/conditions. Nevertheless, in the regular experiment without the 15N labeling, it would be very hard to assign all the peptides to E. coli as many of the peptides would be also present in other species.
We then evaluate the quantification of each peptide. We defined the limit of repeatability (LOR) for each 15N-labeled peptide as the lowest mixture percentage at which it is quantified in at least 2 out of 3 replicates. Peptides observed in only one of the three 100% E. coli replicates were considered as false identification and excluded in this analysis (20,224 peptides remained). Interestingly, there was a strong correlation between the peptide intensity observed in the 100% 15N E. coli sample and their LOR (Figures 2d and S2d). Those results showed that high abundant peptides have lower LORs which reinforced the character of DDA. Most peptides had their LORs at 50%. Only a fraction of peptides (121 and 81 for each group) can reach LORs of 0.5%. The distribution of peptides among LOR groups can be partially explained by the distribution of the peptide intensity. The average log2 intensity (Orbitrap Exploris 480 mass spectrometer) of E. coli peptides is around 25 which is close to the 50% group (Figure 2d). We also found that low-intensity peptides always had high LORs, but not all the high-intensity peptides had low LORs (Figure 2d). Some high-intensity peptides have their LORs at 100%. That means one peptide selected at 100% is not necessarily found at 50% even if its intensity is higher than most of the peptides in the 50% group. Even so, the low LOR peptides showed good reproducibility. Most low LOR sequences were shared in both protein backgrounds (Figure S5).
We selected all the peptides whose LORs could reach 0.5% and analyzed their correlation with E. coli percentage. Because the LOR could accept one missing value, we employed a robust regression model which can assign the missing value a lower weight.18 The result showed that most of the correlations for these peptides had R2 values >0.99 (Figure S6) with the lowest R2 value = 0.93. Although only a small number of peptides can be detected in the 0.5% spike-in sample, all the identified peptides can be quantified accurately.
The functional composition based on detected peptides is not consistent across all the percentages
As fewer 15N peptides were found in the low spike-in percentage samples, we questioned whether the microbiome functional profile could be maintained. We annotated the detected protein group to each COG category. We found that as the percentage of spike-in decreased, some COG categories disappeared. And there was significant alteration across samples (Figure S7). We used Bray–Curtis distance to describe the dissimilarity of COG composition between each sample and 100% E. coli sample. The distance was significantly increased when the spike-in percentage was below 50% (Figure S8).
This result showed that the current DDA-based metaproteomics approach may not achieve sufficient depth to study the functional composition of low abundance species. Of note, when studying taxon-specific functional profiles, the approach that we applied enabled accurate between-sample comparisons of functions of higher abundance taxa.
Evaluation of Match-between-Run
MBR is a strategy using retention time and mass to charge ratios to transfer PSMs across samples. It is based on the assumption that the samples in the same batch are all similar and their proteins belong to the same proteome. The power of MBR depends on data quality and sample number which vary in different experiments. In this study, the three 100% 15N E. coli replicates were an ideal reference for MBR. Using this approach, we noticed that the number of quantified peptides was increased significantly and the bias favoring high-intensity peptides reduced (Figure 3a). The number of peptides with LOR of 0.5% increased 50-fold (stool: from 121 to 5511 and cultured: from 81 to 4146). These results suggested that MBR can greatly improve the performance of DDA by mining the information in MS1. It also revealed that peptide features are present in MS1 even for a bacterium that only represents 0.5% of the biomass. So, in the metaproteomics experiment from gut microbiomes, there are a large number of peptides observed in MS1 only within an MS experiment. Therefore, alternative strategies for the selection of peptides for MS2 analysis might be invaluable for deeper metaproteomics.
Figure 3.

Performance of DDA method on peptide quantification with match-between-run. (a) Distribution of LOD on peptide intensity stool microbiome group. The figures above each LOD group represent the number of peptides in that group, which is also displayed by the color scale. (b) R value from robust regression between log2 peptide intensity and log2 sample percentage of peptides whose LOD were at 0.5% when performing MBR. The figures above represent the number of peptides in each group. The figures on both sides represent the number (percent) of peptides whose R value larger than 0.93.
To validate this point, we further evaluated how many transferred peptides were reliable. When not using MBR, all the identified peptides had good linear relationships with the E. coli spike-in percentage (Figure S5). If the transferred peptides were selected correctly, they should also have a good linear relationship with the E. coli spike-in percentage. We did linear regression using transferred peptides whose LOR was at 0.5% with the E. coli spike-in percentage. The R2 values distribution was not as good as those without MBR (Figure 3b). But to our delight, if we use 0.93 (the lowest R2 value when not using MBR) as the threshold, 2294 (stool) and 1512 (cultured) transferred peptides have good linear relationships (Figures S9 and S10) between their intensity and the spike-in percentage indicating that they were likely proper matches. For the 0.5% spike-in sample, the features added by MBR represents a 20-fold increase in quantified peptides from 121 to 2294 (stool) and 81 to 1512 (culture), respectively. This means that the MBR with proper quality control could be an invaluable approach for deeper metaproteomics.
We further tested whether a MBR based strategy could be developed to go deeper into the human gut microbiome by focusing on specific reference bacterial strains. To test this strategy, we performed a proof-of-concept study by performing three consecutive DDA analysis of two bacterial strains (Blautia hydrogenotrophica, DSM10507 and Bacteroides uniformis, ATCC84892) and a human gut microbiome. First, the DDA MS/MS analysis of the human gut microbiome identified 16488 peptides of which 10 and 15 were from Blautia hydrogenotrophica, DSM10507; Bacteroides uniformis, ATCC84892. Interestingly, the MBR transferred another 9597 peptides from the individual analysis of the two bacterial strains to the human gut microbiome analysis (Figure S11). Although not all the transferred peptides were reliable, we showed the potential of this strategy using the reference samples that are of interest to researchers to increase the quantified peptides.
Conclusion
Species representing less than 0.5% of the biomass of a microbiome are not likely to be detected using top N based DDA approaches in a 1-h nano-LC metaproteomic experiment. Although by tweaking parameters, such as gradient time, target value, resolution and Boxcar, the DDA method could increase the number of identified peptides, marginal increases in the number of identified species as the top N DDA experiments are biased toward higher abundance species. We found that although fewer peptides were found from low abundant bacteria, all the identified peptides could be quantified accurately.
Gut microbiome samples are very complex with a huge dynamic range on each level, and it is likely that peptides from low abundant species are present in MS1 but are not prioritized for MS2 analysis in DDA experiments. Therefore, alternative approaches to top N DDA are needed for deeper metaproteomic analysis. The data-independent acquisition (DIA) is an alternative approach. The DIA method may identify more peptides from the microbiome sample and allow deeper metaproteomics. Although some software has been developed for the DIA data analysis,19,20−22 we still lack a method specific to microbiome data generated in DIA mode.22 Here we demonstrated a strategy using MBR to match MS1 features between metaproteomics of human gut microbiomes as well as between specific bacterial strains and human gut microbiomes. We found that MBR can significantly increase the number of quantified peptides with half of them appearing to be correct matches. This proof-of-concept experiment establishes that MBR is a promising strategy to increase quantification for low abundant bacteria. MBR can be a strategy in metaproteomics with careful experimental planning. It also establishes that low abundant bacteria are predominantly represented in MS1 spectra and not MS2 spectra during DDA analysis of human gut microbiome. This means that lower abundance bacteria can be detected in metaproteomic, but that typical DDA experiments are not sufficient and further strategies to characterize these low abundant species are needed.
Acknowledgments
The art image used in the paper is from https://smart.servier.com/. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/). The line charts and bar graphs in Figure 2a, Figure 2c, Figure S1, Figure S2a, Figure S2c, Figure S3 and Figure S8 were performed using GraphPad Prism version 8.0.0 for Windows, GraphPad Software, San Diego, California USA, www.graphpad.com. This work was supported by funding from the Natural Sciences and Engineering Research Council of Canada (NSERC), and the Government of Canada through Genome Canada and the Ontario Genomics Institute (OGI-114 and OGI-149). D.F. acknowledges a Distinguished Research Chair from the University of Ottawa. H.D. was supported by the NSERC-CREATE TECHNOMISE program.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.2c02452.
The number of 15N protein groups found in each sample (Figure S1); Performance of DDA method on peptide identification and quantification with the cultured protein as background (Figure S2); Abundance distribution of gut microbiome (Figure S3); Representative microbiome composition (Figure S4); Intersected peptides with LOR at 0.5% between two groups (Figure S5); R2 value from robust regression between log2 intensity of peptides and E. coli percentage and representative linear regression plots (Figure S6); Evaluation of taxonomy and function analysis (Figure S7); Bray–Curtis distance between each sample and 100% E. coli on COG composition (Figure S8); Representative well-fitted linear regression plot from cultured microbiome group when using MBR (Figure S9); Representative well-fitted linear regression plot from stool microbiome group when using MBR (Figure S10); The number of quantified peptides by MBR (Figure S11). (PDF)
The authors declare the following competing financial interest(s): D.F. has a co-founded MedBiome, a clinical microbiome company. The remaining authors declare no competing interests.
Supplementary Material
References
- Clemente J. C.; Ursell L. K.; Parfrey L. W.; Knight R. The impact of the gut microbiota on human health: an integrative view. Cell 2012, 148 (6), 1258–1270. 10.1016/j.cell.2012.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sender R.; Fuchs S.; Milo R. Are We Really Vastly Outnumbered? Revisiting the Ratio of Bacterial to Host Cells in Humans. Cell 2016, 164 (3), 337–340. 10.1016/j.cell.2016.01.013. [DOI] [PubMed] [Google Scholar]
- Sender R.; Fuchs S.; Milo R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14 (8), e1002533 10.1371/journal.pbio.1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh P. J.; Ley R. E.; Hamady M.; Fraser-Liggett C. M.; Knight R.; Gordon J. I. The human microbiome project. Nature 2007, 449 (7164), 804–810. 10.1038/nature06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Jia H.; Cai X.; Zhong H.; Feng Q.; Sunagawa S.; Arumugam M.; Kultima J. R.; Prifti E.; Nielsen T.; et al. An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 2014, 32 (8), 834–841. 10.1038/nbt.2942. [DOI] [PubMed] [Google Scholar]
- Qin J.; Li R.; Raes J.; Arumugam M.; Burgdorf K. S.; Manichanh C.; Nielsen T.; Pons N.; Levenez F.; Yamada T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464 (7285), 59–65. 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Pu J.; Lu S.; Bai X.; Wu Y.; Jin D.; Cheng Y.; Zhang G.; Zhu W.; Luo X.; et al. Species-Level Analysis of Human Gut Microbiota With Metataxonomics. Front Microbiol 2020, 11, 2029. 10.3389/fmicb.2020.02029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer M.; Ahrne E.; Baron A. P.; Glatter T.; Fava L. L.; Santamaria A.; Nigg E. A.; Schmidt A. Evaluation of data-dependent and -independent mass spectrometric workflows for sensitive quantification of proteins and phosphorylation sites. J. Proteome Res. 2014, 13 (12), 5973–5988. 10.1021/pr500860c. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Chen W.; Ning Z.; Mayne J.; Mack D.; Stintzi A.; Tian R.; Figeys D. Deep Metaproteomics Approach for the Study of Human Microbiomes. Anal. Chem. 2017, 89 (17), 9407–9415. 10.1021/acs.analchem.7b02224. [DOI] [PubMed] [Google Scholar]
- Li L.; Ning Z.; Zhang X.; Mayne J.; Cheng K.; Stintzi A.; Figeys D. RapidAIM: a culture- and metaproteomics-based Rapid Assay of Individual Microbiome responses to drugs. Microbiome 2020, 8 (1), 33. 10.1186/s40168-020-00806-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng K.; Ning Z.; Zhang X.; Li L.; Liao B.; Mayne J.; Figeys D. MetaLab 2.0 Enables Accurate Post-Translational Modifications Profiling in Metaproteomics. J. Am. Soc. Mass Spectrom. 2020, 31 (7), 1473–1482. 10.1021/jasms.0c00083. [DOI] [PubMed] [Google Scholar]
- Chi H.; Liu C.; Yang H.; Zeng W.-F.; Wu L.; Zhou W.-J.; Wang R.-M.; Niu X.-N.; Ding Y.-H.; Zhang Y. Comprehensive identification of peptides in tandem mass spectra using an efficient open search engine. Nature biotechnology 2018, 36 (11), 1059–1061. 10.1038/nbt.4236. [DOI] [PubMed] [Google Scholar]
- Millikin R. J.; Solntsev S. K.; Shortreed M. R.; Smith L. M. Ultrafast Peptide Label-Free Quantification with FlashLFQ. J. Proteome Res. 2018, 17 (1), 386–391. 10.1021/acs.jproteome.7b00608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galperin M. Y.; Wolf Y. I.; Makarova K. S.; Vera Alvarez R.; Landsman D.; Koonin E. V. COG database update: focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Res. 2021, 49 (D1), D274–D281. 10.1093/nar/gkaa1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiner M.; Thorson E.; Sharp C. E.; Dong X.; Liu D.; Li C.; Strous M. Assessing species biomass contributions in microbial communities via metaproteomics. Nat. Commun. 2017, 8 (1), 1558. 10.1038/s41467-017-01544-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Figeys D. Perspective and Guidelines for Metaproteomics in Microbiome Studies. J. Proteome Res. 2019, 18 (6), 2370–2380. 10.1021/acs.jproteome.9b00054. [DOI] [PubMed] [Google Scholar]
- Cox J.; Hein M. Y.; Luber C. A.; Paron I.; Nagaraj N.; Mann M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell Proteomics 2014, 13 (9), 2513–2526. 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maronna R. A.; Martin R. D.; Yohai V. J.; Salibián-Barrera M.. Robust statistics: theory and methods (with R); John Wiley & Sons: 2019. [Google Scholar]
- Tsou C. C.; Avtonomov D.; Larsen B.; Tucholska M.; Choi H.; Gingras A. C.; Nesvizhskii A. I. DIA-Umpire: comprehensive computational framework for data-independent acquisition proteomics. Nat. Methods 2015, 12 (3), 258–264. (257 p following 264) 10.1038/nmeth.3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demichev V.; Messner C. B.; Vernardis S. I.; Lilley K. S.; Ralser M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 2020, 17 (1), 41–44. 10.1038/s41592-019-0638-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinitcyn P.; Hamzeiy H.; Salinas Soto F.; Itzhak D.; McCarthy F.; Wichmann C.; Steger M.; Ohmayer U.; Distler U.; Kaspar-Schoenefeld S.; et al. MaxDIA enables library-based and library-free data-independent acquisition proteomics. Nat. Biotechnol. 2021, 39 (12), 1563–1573. 10.1038/s41587-021-00968-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietilä S.; Suomi T.; Elo L. L. Introducing untargeted data-independent acquisition for metaproteomics of complex microbial samples. ISME Communications 2022, 2 (1), 1–8. 10.1038/s43705-022-00137-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.