

ABSTRACT
Resistance to chemo-immunotherapy is a major issue for the treatment of non-small cell lung cancer. In a recent paper we unravel the role of MAPK in the capacity of restraining the therapeutic efficacy of chemo-immunotherapy. Inhibition of the MAPK pathway using a MAP2K/MEK inhibitor in combination with chemotherapy could promote OPTN (optineurin)-dependent mitophagy of cancer cells. Mitochondria then degrade via autophagosomes and amphisomes and release mitochondrial DNA, which interacts with TLR9 located in these compartments. TLR9 activation promotes the production of the chemokine CXCL10 by cancer cells, which could further improve T cell recruitment and improve the efficacy of immunotherapy.
KEYWORDS: Chemotherapy, CXCL10, immunotherapy, MAP2K/MEK inhibitor, mitophagy, optineurin, TLR9, ULK1
Chemotherapies affect mitochondrial biology, and drugs like cisplatin could alter mitochondrial function via the generation of mitochondrial reactive oxygen species (mtROS), leading to mitochondrial dysfunction, permeabilization and subsequent cell death. During such mitochondrial stress, mitochondrial fusion and fission play key roles in mitochondrial quality control. The selective autophagy process called mitophagy deals with damaged mitochondria, leading to their elimination via lysosome degradation. This catabolic pathway protects tumor cells against chemotherapy-induced cell death. In contrast, we unraveled a positive immune role of this catabolic pathway in cancer cells [1] (Figure 1).
Figure 1.
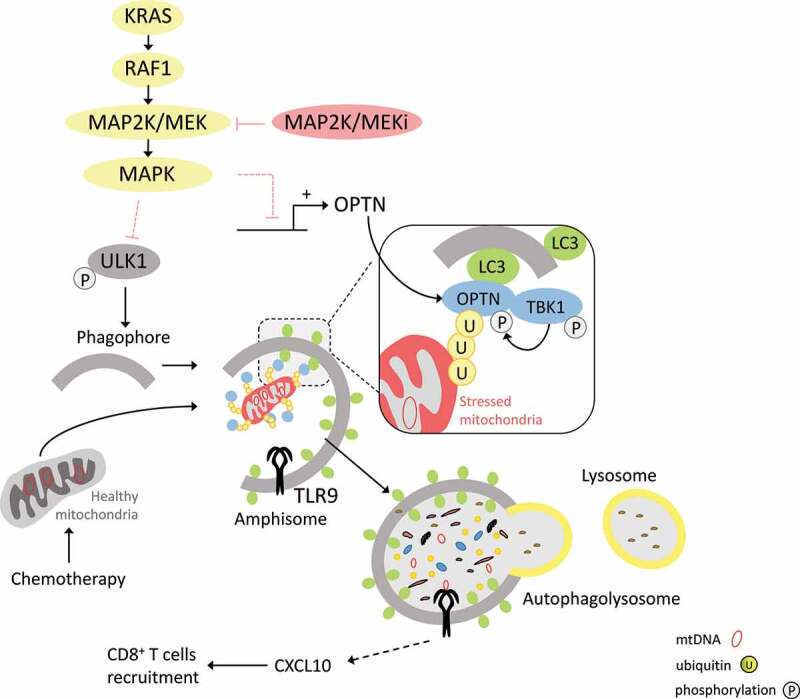
Interconnection between the MAPK pathway, mitophagy and CXCL10 expression. Blocking the KRAS oncogenic pathway with MAP2K/MEK inhibitors allows for the removal of chemo-induced mitophagy inhibition at different stages. First, phagophore formation is facilitated by loss of ULK1 inhibition and thus its increased phosphorylation on serine 555. Second, by restoring gene and protein expression of OPTN (optineurin), chemotherapy-damaged mitochondria can be more easily engulfed in phagophores. Phosphorylation of TBK1 on serine 172 also promotes activation of OPTN at ubiquitin (U) chains on the surface of mitochondria. Through the degradation of these in amphisomes or autolysosome-like structures, mitochondrial DNA activates TLR9, which in turn allows the expression of CXCL10 and the recruitment of CD8+ T cells.
The standard treatment for non-small cell lung cancer is evolving rapidly, and currently, an association of chemo-immunotherapy is the classical first line treatment. This approach improves response rates and survival in comparison to chemotherapy alone. The rationale for this synergy is linked to the capacity of chemotherapies to promote immune cell death and improve antitumor immune response. However, most patients fail to yield benefit from this combination. Multiple mechanisms could explain this resistance, but the main mechanism is the lack of tumor-infiltrating cytotoxic T lymphocytes (CTLs). Tumors with few if any CTLs, known as “cold” tumors, are intrinsically resistant to immunotherapy. Using a model of cold tumor in mice, we observed that despite the ability of cisplatin-based chemotherapy to trigger immunogenic cell death, chemo-immunotherapy does not induce immune recruitment at the tumor site, because of the inability of cancer cells to produce the major chemoattracting molecule CXCL10. Using drug screening, we observed that all MAP2K/MEK inhibitors, when given in combination with chemotherapy, can induce strong production of CXCL10. From a therapeutic point of view, CXCL10 production by cancer cells leads to accumulation of CD8+ T-cells at the tumor site. The treatment reverses resistance to chemo-immunotherapy in this lung cancer model. Importantly, the association of a MAP2K/MEK inhibitor plus chemo-immunotherapy demonstrates efficacy in various cancer models, such as carcinogen-induced lung cancer, colon cancer, and triple-negative breast cancer, thus generalizing the observation.
From a mechanistic point of view, we observed that chemotherapy induces mtROS, membrane depolarization, and MAP2K/MEK-MAPK/ERK pathway activation. Chemotherapies induce TBK1 phosphorylation, and this is further enhanced by MAP2K/MEK inhibitor treatment. TBK1 kinase activation leads to the phosphorylation of OPTN and other mitophagy receptors. This phosphorylation in turn enhances its binding affinity to ubiquitin chains on damaged mitochondria. Selective mitophagy can be mediated by various receptors such as SQSTM1/p62, NBR1, CALCOCO2/NDP52, TAX1BP1, and OPTN. RNAseq of cancer cells treated with chemotherapy plus MAP2K/MEK inhibitors underlined that only Calcoco2 and Optn gene expression are induced by this combination. Taken together, these findings underline the capacity of a combination of MAP2K/MEK inhibitor plus chemotherapy to promote mitophagy processes through transcriptional and post-translational effects on the TBK1-OPTN pathway.
Electron microscopy analysis showed that a combination of MAP2K/MEK inhibitor plus chemotherapy decreases the abundance of cytosolic mitochondria, and increases the number of those enclosed in a double membrane, suggesting induction of mitophagy. This treatment also leads to LC3 lipidation and ULK1 phosphorylation, suggesting an increase in autophagic flux. Importantly, inhibiting autophagy or mitophagy by inactivation of OPTN or ATG5 impedes the capacity of the treatment to trigger CXCL10 production by cancer cells and to recruit CD8+ T cells in the tumor. We hypothesize that mitochondrial DNA (mtDNA) is the mediator that triggers the generation of CXCL10 during mitophagy. Selective depletion of mtDNA using ethidum bromide impedes CXCL10 production. Fluorescence microscopy showed that chemotherapy plus a MAP2K/MEK inhibitor triggers colocalization of the mtDNA with TLR9 in LC3-positive autophagic structures. TLR9 genetic inactivation impairs the therapeutic efficacy of chemo-immunotherapy plus MAP2K/MEK inhibitors and impedes CXCL10 production by cancer cells.
Mitophagy is an essential catabolic pathway aimed at preserving cells from death induced by mitochondrial injury. We reveal the Janus face of this mechanism. While it can prevent cell death and death-dependent inflammation, mitochondrial DNA is processed in autophagosomes or amphisomes and detected by TLR9, which can induce production of inflammatory cytokines, such as CXCL10. These data suggest the biological complexity of mitophagy when viewed through the prism of inflammation. In the cancer setting, increasing the level of mitophagy might be deleterious in the context of a pure cytotoxic treatment. In contrast, our data underline a positive role in the context of chemo-immunotherapy. These data open avenues to developing mitophagy inducers to improve the efficacy of chemo-immunotherapy protocols.
Funding Statement
The author(s) reported there is no funding associated with the work featured in this article.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Reference
- [1].Limagne E, Nuttin L, Thibaudin M, et al. MEK inhibition overcomes chemoimmunotherapy resistance by inducing CXCL10 in cancer cells. Cancer Cell. 2022 Feb 14;40(2):136–152. [DOI] [PubMed] [Google Scholar]