

INTRODUCTION
RASopathies are a group of developmental multisystemic disorders caused by germline mutations in genes encoding signal transducers and regulatory proteins functionally linked to the RAS/mitogen-activated protein kinase (MAPK) pathway. Collectively, these disorders have an estimated prevalence of 1 in 1000 to 1 in 25001 among live births.
According to a large clinical registry,2 RASopathies may represent the underlying diagnosis in ~18% of childhood hypertrophic cardiomyopathy (HCM), particularly among infants younger than 1 year, where they account for ~42% of the cases.
The disorders constituting the RASopathies include Noonan syndrome (NS), NS with multiple lentigines (NSML, previously known as LEOPARD syndrome), cardiofaciocutaneous syndrome (CFCS), Mazzanti syndrome (also known as NS-like disorder with loose anagen hair), Costello syndrome (CS), type 1 neurofibromatosis, and Legius syndrome which are well recognized and clinically characterized3; however, other clinically related conditions are emerging.4
Although each RASopathy exhibits a unique clinical phenotype, these syndromes share many overlapping characteristics, including growth retardation, craniofacial features, cryptorchidism, cognitive deficits, renal malformations, bleeding disorders, variable predisposition to certain cancers, and congenital heart disease (CHD).5–8 Diagnosis of a RASopathy could be suggested by clinical clues (“red flags”),9 which could raise the suspicion of an underlying malformation syndrome and direct the clinician toward a specific genetic test (Table 1).
Table 1.
Clinical red flags for RASopathies
| Facial features | NS: broad forehead, down slanting palpebral fissures, hypertelorism, low set ears, pterygium colli, epicanthal folds, short and depressed nasal root CS: hypertelorism, down slanting palpebral fissures. Facial features are coarser. Feeding and suction difficulties are more prevalent. CFCS: macrocephaly, bitemporal narrowing, coarse facial features, small curly friable hair with sparse eyebrows with hyperkeratosis (ulerythema ophryogenes), broad nasal base, wide labial philtrum. Coarser than NS |
| Cardiovascular | Pulmonary valve stenosis, hypertrophic cardiomyopathy, mitral valve dysplasia, atrial septal defect, ventricular septal defect, atrioventricular septal defect, aortic coarctation, coronary arteries abnormalities |
| Electrocardiography | Extreme right-axis deviation (north west axis), left or right bundle branch block (NSML), prolonged QT (NSML), repolarization abnormalities, multifocal atrial tachyarrhythmia (CS) |
| Musculoskeletal | Short stature, hypotonia (CFCS), Pes planus (CFCS), scoliosis, pectus deformity, hip dysplasia, osteopenia/osteoporosis |
| Dermatologic | Cafè-au-lait spots, papillomas (CS), palmar keratoderma, acanthosis nigricans, keratosis pilaris (CFC), ulerythema ophryogenes (CFC), lentigines (NSML), multiple pigmented nevi |
| Genitourinary | Cryptorchidism, transitional cell carcinoma of the urinary bladder (CS) |
| Hematopoietic | Easy bruising, von Willebrand disease, coagulation defects, thrombocytopenia, NS/myeloproliferative disease (leukocytosis with monocytosis, thrombocytopenia, and hepatosplenomegaly) |
| Gastrointestinal | Failure to thrive, gastroesophageal reflux, intestinal malrotation, pyloric stenosis |
| Sensorineural | Hearing loss, laryngomalacia, cataract, strabismus |
| Neurologic | Seizures, infantile spasms, hydrocephalus, Chiari I malformation, intellectual disability |
| Endocrine | Growth hormone deficiency, hypoglycemia, delayed puberal spurt, short stature |
| Oncology | Juvenile myelomonocytic leukemia, lymphedema |
Abbreviations: CFCS, cardiofaciocutaneous syndrome; CS, Costello syndrome; HCM, hypertrophic cardiomyopathy; NS, Noonan syndrome.
It should be noted that the extent of clinical variability characterizing each RASopathy is strictly related to the extent of molecular variability and heterogeneity of these disorders. For example, NS, which is the most common disorder among the RASopathies. This disease is caused by mutations in more than 10 genes (ie, PTPN11, SOS1, SOS2, NRAS, KRAS, MRAS, RRAS2, RIT1, LZTR1, RAF1, MAP2K1), which are preferentially associated with certain features, including proper growth and cognition (SOS1, SOS2), high prevalence of pulmonary stenosis (PS) (PTPN11), or HCM (eg, RAF1, MRAS, and RIT1).4,10–15 On the contrary, other RASopathies are relatively homogeneous, being caused by a narrow spectrum of mutations in single genes, as in the case of CS and Mazzanti syndrome, which are caused by a bunch of mutations in HRAS and SHOC2, respectively.3
HYPERTROPHIC CARDIOMYOPATHY IN RASopathies
After CHD, HCM is the second most common cardiovascular abnormality observed in RASopathies,2,16–22 with worse clinical outcomes when associated with early onset presentation.
Compared with sarcomeric forms (S-HCM), HCM in RASopathies (R-HCM) shows increased prevalence and severity of left ventricular outflow tract obstruction (LVOTO)17,19,20,23 and higher rates of hospitalizations for heart failure or need for septal myectomy during childhood.23 R-HCM presents earlier in infancy, with a mean age at diagnosis of 6 months, whereas S-HCM mostly presents during adolescence.24 Congestive heart failure (CHF) is significantly more common in R-HCM (24% vs 9%)25 and accounts for a substantial early mortality. Patients affected by RASopathies are more likely to require heart failure hospitalizations or cardiovascular interventions,23 mainly septal myectomies and pulmonary valvuloplasties.26–28
In 5% to 10% of the cases, R-HCM is associated to a severe clinical presentation, particularly for infants with signs of heart failure, with a 70% one-year mortality. With the exception of these cases, clinical status tends to improve over time, and progression of left ventricular hypertrophy (LVH), described in S-HCM, seems uncommon in R-HCM.23,29 On the contrary, left ventricular reverse remodeling with regression of myocardial wall thickness z-scores over time on serial echocardiography has been reported in many clinical studies.23,26,28
LVOTO is common among patients with RASopathies, and its prevalence may be due to the contribution of other cardiovascular abnormalities such as displacement of papillary muscles, anomalous insertion of mitral chordae, or fibrous tissue causing midventricular obstruction.7,30,31 Mitral valve disease has been considered a marker of complexity in patients with HCM and may carry a negative prognosis, being associated with reintervention and mortality.32 In particular, compared with age- and sex-matched healthy controls, the length of the anterior leaflet of mitral valve is significantly increased in patients with R-HCM30; moreover, anterior displacement of the papillary muscles may cause distortion of the subvalvular apparatus.
Biventricular hypertrophy may represent a useful clinical clue (“red flag”) for the suspicion of R-HCM,33 and its presence may indicate the co-occurrence of right ventricular outflow tract obstruction. The prevalence of PS ranges between ~25% and 70% in this subgroup of patients: The pulmonary valve is often dysplastic and shows signs of commissural fusion.27,34 In particular, PS is severe in ~30% of the cases and moderate in ~10%. Patients with mild PS are unlikely to require intervention, and their natural history is similar to that of patients without PS.28,35 In contrast, patients with moderate-to-severe PS often require percutaneous balloon valvuloplasty, but due to valvular dysplasia, the outcomes may be unfavorable, with high rates of reintervention.26,36
Beyond PS, which is observed in up to 65% of the patients, other CHDs commonly occur among patients with RASopathies: “Secundum atrial septal defect” (ASD) has been observed in ~8% to 30% of the cases,11,26–28,31,37–39 as well as ventricular septal defect (VSD) (~5%–10%)19,26,27,31,40 and atrioventricular canal defect (AVCD) (up to 15% of the cases). The association with AVCD is of pivotal importance: Mitral valve abnormalities (such as double orifice and parachute mitral valves) have a significant association with AVCD. Moreover, cranial displacement of the aortic valve anulus relative to the ventricular apex, which determines the so-called “gooseneck” deformity, may contribute to LVOTO.39,41,42 Complete AVCD is uncommon in RASopathies, and morphologic data are insufficient to define whether a specific AVC subtype, according to the Rastelli classification, is more prevalent.
Left-sided obstructive lesions are rare in RASopathies, although aortic stenosis, coarctation of the aorta, and isolated MV stenosis have been reported in the setting of NS.43–45
Coronary artery abnormalities are a relevant finding in RASopathies and may contribute to cause myocardial ischemia and worsen the imbalance between myocardial oxygen supply and demand. According to a registry,31 aneurysms in coronary arteries have been identified in ~15% of the cases, invariably involving left coronary artery but independently from a specific pathogenic variant.46 Long-term outcomes of coronary dilatation are not completely understood: As coronary artery aneurysms have been associated to athero-thrombosis, the role of antiplatelet drugs in primary prevention of myocardial infarction is debated, and no specific recommendation can be made.47
Although HCM, AVCD, and PS have been considered classic cardiovascular defects in the setting of RASopathies, recent data coming from a multicenter retrospective study seem to report a high prevalence of primary mitral regurgitation (24%, 4%) and aortic insufficiency (25%) or structural abnormalities of the ascending aorta, including kinking and aortic root dilatation.31
The summary of cardiovascular manifestations, involved genes, and their relative prevalence among RASopathies is shown in Table 2. The proposed algorithm for the diagnosis and management of R-HCM is shown in Fig. 1.
Table 2.
Clinical manifestations, mutated genes, and classical heart defects with their relative prevalence among RASopathies and relative prevalence of hypertrophic cardiomyopathy
| Gene | Clinical Features | HCM |
|---|---|---|
| Noonan syndrome | ||
| PTPN11 | PVS (65%), atrioventricular septal defects (18%), ASD (10%), VSD (9%), PDA (6%), mitral valve abnormalities (2.5%), Tetralogy of Fallot (<1%), aortic coarctation (<1%) | HCM (20%) |
| SOS1 | PVS (70%), ASD (14.5%), mitral valve abnormalities (4%) | HCM (18%) |
| RAF1 | PVS (21%), ASD (20%), mitral valve abnormalities (13%), Tetralogy of Fallot (2.8%), aortic coarctation (1.8%) | HCM (65%) |
| RIT1 | PVS (74%), ASD (30%), PDA (7%), mitral valve abnormalities (13%), biventricular obstruction (<1%) | HCM (36%) |
| SHOC2 | PVS (32%), ASD (32%), mitral valve abnormalities (27%), VSD (13%) | HCM (30%) |
| NRAS | PVS (14%) | HCM (39%) |
| CBL | PVS (50%), ASD (10%), mitral valve abnormalities (10%) | HCM (10%) |
| Noonan syndrome with multiple lentigines | ||
| PTPN11 | Biventricular hypertrophy (46%), LVOTO (40%), ventricular tachycardia, conduction abnormalities, mitral valve abnormalities (42%), PVS (21%), ASD (6%), atrioventricular septal defects, coronary artery abnormalities | HCM (85%) |
| RAF1 | Mitral valve abnormalities (100%), PVS (40%), high risk of SCD | HCM (100%) |
| BRAF | - | HCM (60%–85%) |
| Costello syndrome | ||
| HRAS | Atrial tachycardia (56%), PVS | HCM (65%) |
| Cardiofaciocutaneous syndrome | ||
| BRAF1 | PVS (45%), ASD (30%), VSD (8%). Tetralogy of Fallot (7%) | HCM (40%) |
| MAP2K1-MAP2K2 | PVS (65%–100%), septal defects (15%) | HCM (40%) |
| KRAS | PVS (40%), mitral valve abnormalities (26%), ASD (20%), VSD (13%) | HCM (20%) |
Abbreviations: ASD, atrial septal defect; HCM, hypertrophic cardiomyopathy; PDA, patent ductus arteriosus; PVS, pulmonary valve stenosis; VSD, ventricular septal defect.
Fig. 1.
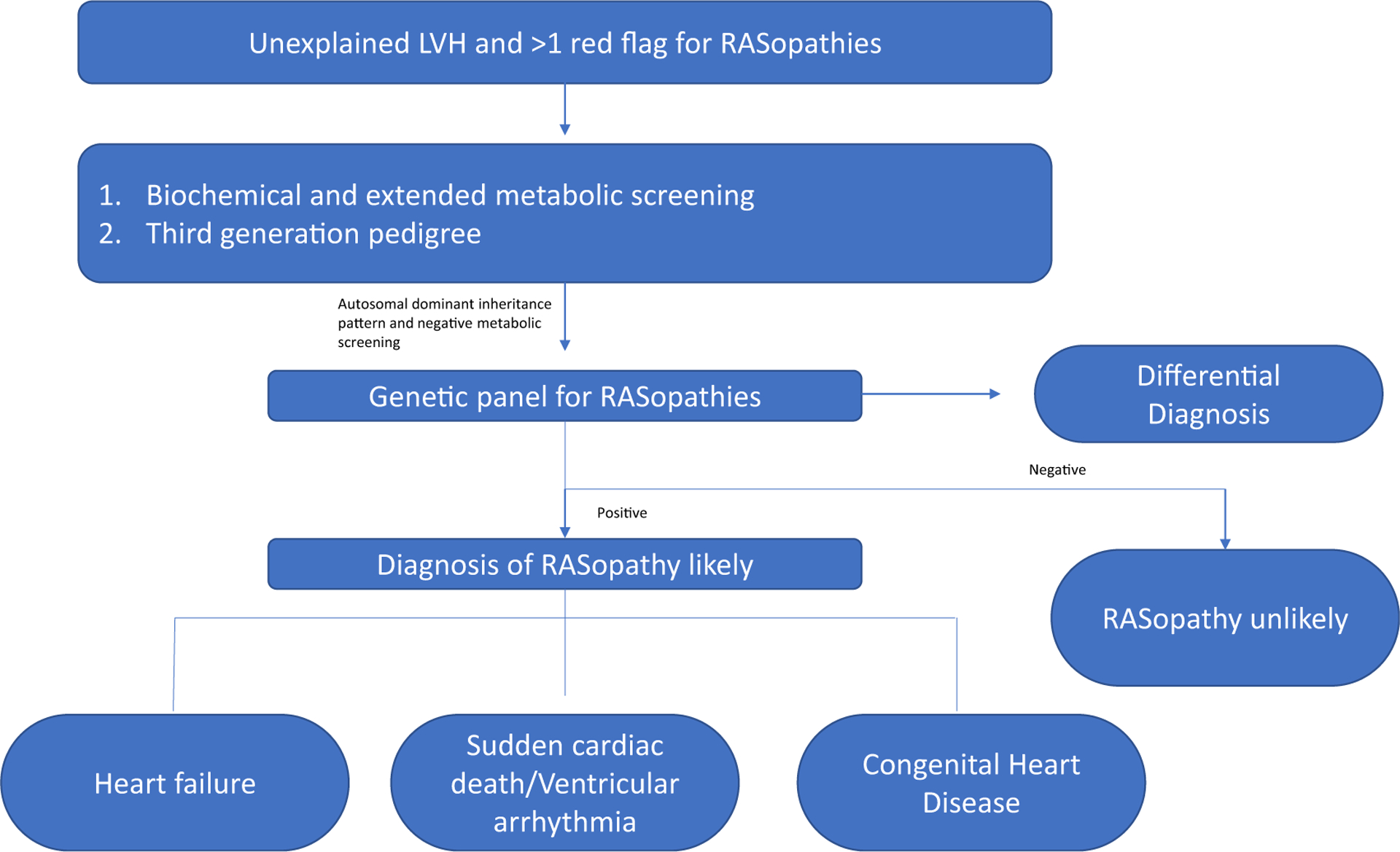
Proposed algorithm for the diagnosis and management of RASopathy-associated HCM (R-HCM).
Hypertrophic Cardiomyopathy in Noonan Syndrome with Multiple Lentigines
HCM is present in ~85% of the patients affected by NSML, among the highest rate among RASopathies. LVOTO may be associated to NSML in up to 40% of the cases, and biventricular hypertrophy is reported in nearly half of the patients. HCM is often diagnosed in early infancy, and the clinical phenotype almost invariably manifests before the appearance of the lentigines.19 Interestingly, patients with severe biventricular involvement often show high mortality rates, compared with mild asymptomatic cases, who have relatively benign clinical outcomes and frequent regression of LVH.27,48 Electrocardiographic abnormalities and progressive conduction abnormalities often coexist with HCM: In particular, a superiorly oriented QRS axis even in the absence of biventricular involvement, pseudo-infarction q waves, and prolonged corrected QT interval have been observed among NSML patients.19 A possible limitation in the existing study outcomes among patients with NSML is that, in some studies, diagnosis is based on clinical criteria without genotype corroboration. Of note, differentiating NSML from NS can be challenging, particularly in infants, for whom the prevalence of cutaneous manifestations is incomplete, and lentigines are not present.24 NSML is caused by a narrow spectrum of dominantly acting mutations in PTPN11. Many clinically relevant genotype-phenotype correlations should be considered: Patients harboring missense RASopathy variants in BRAF, LZTR1, RAF1, and RIT1 show an increased prevalence of LVH compared with carriers of PTPN11 pathogenic variants. In addition, some authors have reported worse HF and arrhythmic outcomes in a subgroup of patients harboring the missense Gln510Glu allele (PTPN11, exon 13).16 Interestingly, the molecular spectrum of NSML-causing PTPN11 mutations does not overlap the pattern of mutations observed in NS, and the biochemical behavior of these two classes of mutations is completely different, with the former impairing the catalytic activity of SHP2, the protein encoded by PTPN11, and the latter promoting enhanced activation of the phosphatase49–51
Hypertrophic Cardiomyopathy in Costello Syndrome
Among patients diagnosed with CS, approximately 65% fulfills diagnostic criteria for HCM: In this subgroup, the coexistence with CHD is reported in ~40% of the cases, and 60% may have LVOTO. Most patients seem to show subaortic septal thickening, although other patterns, such as biventricular or concentric hypertrophy, have been reported.52 The natural history of HCM in CS is variable, with a documented rate of progression in ~40% of the cases. Of note, a significant number of patients with severe HCM underwent septal myectomy (25%). Regression of LVH has been reported in 10% of the patients, although data reported do not allow the establishment of genotype-phenotype correlations.18,24,52,53 The comparison between the two commonest alleles for the HRAS gene (pG12S and pG13 C) with a pathogenic role in CS has shown no significant difference in the prevalence of HCM.54 Severe cardiomyopathy, associated with pleural and pericardial effusion as well as lung abnormalities has been observed in carriers of G12 C and G12D alleles.55 Atrial tachyarrhythmias are seen in more than 50% of the patients affected by CS, particularly non-reentrant atrial tachycardia and multifocal or ectopic atrial tachycardia.56 The natural history of atrial arrhythmias is usually benign in CS, with spontaneous regression within the first year of life and responsiveness to medical therapy, whereas ventricular arrhythmias are rare. Later-onset atrial fibrillation and atrial flutter have been reported, but their prognostic significance is unknown.57
Hypertrophic Cardiomyopathy in Cardiofaciocutaneous Syndrome
CFCS is caused by dominantly acting mutations in BRAF, MAP2K1, and MAP2K2. Nearly 75% of children with CFC have cardiovascular involvement, mainly PS, which can be diagnosed in 45% of the cases. HCM can be identified in ~40% of patients during infancy,6 with a variable phenotype, sometimes rapidly progressive and resulting in heart transplantation or death and, in other cases, with a mild phenotypic expression.6 Of note, the prevalence of HCM is not significantly different among patients who carry BRAF pathogenic variant mutation compared with carriers of MEK1 or MEK2 alleles.58,59 The most common coexisting forms of CHD are ASD (8%–18%) and VSD (11%–22%).60,61 Although uncommon in CFCS, arrhythmias can include ventricular preexcitation, atrioventricular block, or supraventricular tachycardia.59
Hypertrophic Cardiomyopathy in Noonan Syndrome and Clinically Related Disorders
Among RASopathies, HCM seems less common in NS, with a reported prevalence of ~20% among affected individuals. Interestingly patients with Mazzanti syndrome and harboring a recurrent missense substitution in SHOC2 seem to have a slightly higher prevalence of HCM (25%).62 Three independent studies reported that germline variants in CBL underlie a clinical syndrome with many overlapping features with NS, although HCM seems not to be associated to this condition.63–65 Among children with NS and HCM, asymmetrical septal hypertrophy is the most common presentation (75.6%), whereas apical involvement is rare.66 A 12-lead electrocardiogram can show extreme right heart deviation (“north west axis”), reflecting biventricular involvement.33 Genotype characterization can offer useful diagnostic clues in the management of NS-associated HCM. In particular, among patients harboring PTPN11 variants, the prevalence of HCM is low, whereas individuals affected by NS with causal variants in RAF1 and RIT1 are more likely to develop HCM.24
PATHOPHYSIOLOGY OF HYPERTROPHIC CARDIOMYOPATHY IN RASopathies
Different signaling pathways seem to be involved in the molecular pathogenesis of HCM in RASopathies. Almost 50% of the cases of NS are caused by missense gain-of-function mutations in PTPN11, which typically cluster around the phosphotyrosine phosphatase domains of SHP2, causing constitutive activation of the protein.67 Interestingly, HCM is underrepresented in patients harboring missense changes in PTPN11 or SOS1, whereas it is overrepresented is RAF1-mutated NS, where it seems to be allele-specific.14,68 A distinct class of variants in PTPN11 was associated to NSML, which carries a higher lifetime risk of HCM.49 While NS-causing variants may promote upregulation of RAS/MAPK cascade, alleles involved in NSML should be considered as dominant negative mutants promoting enhanced signal flow through the PI3K-AKT-mTOR pathway. In a murine model of NS bearing the p.L613 V mutation in the Raf1 gene, increased RAS/MAPK signaling was observed both in fibroblasts and in neonatal cardiomyocytes. Consistent with that observation, treatment with an MEK inhibitor seemed to induce positive reverse remodeling in Raf1L613V mice.69 Treatment with trametinib, an MEK inhibitor, has been associated to reversal of LVH and LVOTO in two cases of RIT1-mutated NS within 4 months after initiation of the therapy. Remodeling was associated with a favorable clinical response and a catch-up in somatic growth, which may be attributable to recovery from severe HF.70 In contrast, a mouse model of NSML bearing the p.Y279 C mutation in the Ptpn11 gene showed impaired ligand-evoked ERK phosphorylation and increased signal flow through the PI3K-AKT-mTOR pathway. Treatment with rapamycin, an mTOR inhibitor, rescued the cardiac phenotype in Ptpn11Y279C mice.71 In the last years, everolimus was used to prevent CHF in patients with severe HCM, but regression of LVH was not documented.48
PROGNOSTIC IMPLICATIONS OF HYPERTROPHIC CARDIOMYOPATHY IN RASopathies
The diagnosis of HCM significantly affects the outcome in patients affected by NS: Long-term studies have demonstrated that severity of cardiovascular involvement is associated with a lower survival rate, with higher risk of death in patients younger than 2 years and adolescents. Specific risk factors for early mortality include evidence of HF during the first 6 months of life, low cardiac output, significant diastolic disfunction, and several cardiac interventions.7,25
Taken together, the outcomes of children affected by R-HCM seem to reflect a distinct disease course compared with carriers of sarcomere pathogenic variants: Patients with R-HCM can be severely affected during the perinatal and infancy periods, are more likely to require septal myectomy or pulmonary balloon valvuloplasty, and lack the typical progression of LVH during adolescence and young age.23,28
Histopathologic studies have demonstrated that patients with R-HCM had a similar amount of quantified fibrosis compared with nonsyndromic familial HCM caused by sarcomere protein mutations. Of note, fibrotic changes are already present in an early phase of disease, in contrast to S-HCM, in which myocardial disarray and fibrosis develop over time.23
Apical aneurysms have been described in R-HCM, nonetheless their overall prevalence and clinical significance have not been assessed yet.72
HCM is the main cause of sudden cardiac death (SCD) among adolescents and young adults.73 Among patients with R-HCM, sustained ventricular tachycardia (VT) is frequent and related to the risk of SCD.74–76 Although the overall risk of SCD appears lower than that in HCM caused by sarcomere mutations, disease-specific risk factors are currently an area of debate.23,28 A previous history of VT or cardiac arrest, unexplained syncope, non-sustained VT, or massive LVH has been associated with higher risk of SCD and may require Implantable cardioverter defibrillator implantation.73 However, the weight of isolated risk factors, irrespective of their magnitude (ie, massive LVH), is not clear, and the risk could be higher when many risk factors coexist in the same patient. Interestingly, other conventional risk factors such as age, family history of SCD, and left atrial z score should be evaluated carefully in children, whereas LVOT gradient seems not associated to SCD.77 Recently, two scores have been validated for risk stratification in pediatric patients.77,78
MANAGEMENT AND FUTURE PERSPECTIVES
Medical therapy remains the first-line option in patients with R-HCM. Nonvasodilating beta blockers should be titrated to the maximum tolerated dose in patients, and diuretic dose should be accurately set to reduce congestive status and HF symptoms, while avoiding hypovolemia, which could increase LVOTO. When beta blockers are ineffective or not tolerated, nonvasodilating calcium channel blockers could be administered in children older than 6 months. Of note, there are several reports of severe bradycardia and HF worsening in infants with LVOT gradients greater than 100 mm Hg, suggesting that verapamil could be harmful in this clinical setting.79,80
In patients who remain symptomatic despite beta-blockers, disopyramide could be a therapeutic option to reduce obstruction. Recently, mavacamten has shown to improve functional status in S-HCM, nevertheless further studies are needed to assess its role in nonsarcomeric variants. Surgical myectomy may be considered in patients who remain symptomatic for significant LVOTO despite maximal medical therapy and should be performed in specialized centers with high-volume experience.81,82 Orthotopic heart transplantation (OHT) is rarely performed in patients with NS,83 although it should be considered in patients with severe HF symptoms with evidence of refractoriness to medical therapy, intractable ventricular arrhythmias, cardiogenic shock requiring inotropes, and in case of severe diastolic dysfunction.84,85 Early multiorgan damage should not be considered a contraindication for heart transplantation, but careful evaluation of absolute contraindication is required, to exclude the risk of futility in case of moderate or severe end organ dysfunction.86 According to the Pediatric Cardiomyopathy Registry,87,88 cumulative waitlist mortality was significantly high in infants affected by HCM, suggesting a malignant course of disease and the presence of comorbidities in critically ill infants. Poorer outcomes were observed in patients listed for priority in status 1 or younger than 1 year than in older children and patients listed in status 2.89 Therefore, OHT should be considered as early as possible and before clinical deterioration in patients with R-HCM.
Proof of concept that inhibition of the RAS/MAPK cascade could induce regression of LVH and cardiac remodeling in patients affected by RASopathies has been achieved by clinical evaluation of trametinib, an MEK inhibitor, in two patients with RIT1-induced NS, with significant regression of LVH within 4 months after initiation of treatment.70 On the contrary, among children with NSML with germline loss-of-function variation in PTPN11, the overactivation in PI3K-AKT-mTOR pathway was partially reversed with the use of rapamycin.48,71 The design of therapeutic trials is challenging in patients with RASopathies because of the rarity of disease, patient variability in disease progression and regression, and the heterogeneity of molecular mechanisms.90 Possible future treatment candidates may include farnesil transferase inhibitors, pan-RAF inhibitors, SHP2 inhibitors, and, because copper is required for catalytic activity of MEK kinases, chelation with ammonium tetrathiomolybdate.
KEY POINTS.
RASopathies are developmental multisystemic disorders caused by germline mutation in genes linked to the RAS/mitogen-activated protein kinase pathway.
Hypertrophic cardiomyopathy is the second most common cardiovascular manifestation in RASopathies and exhibits unique features such as the coexistence of congenital heart disease and early-onset congestive heart failure and accounts for significant mortality rates.
Diagnosis of a RASopathy could be suggested by clinical clues (“red flags”), which could raise the suspicion of an underlying malformation syndrome and direct the clinician toward a specific genetic test.
CLINICS CARE POINTS.
RASopathy should be suspected in infants (<1 year) with new-onset HCM and clinical red flags.
Patients affected by RASopathies are more likely to require HF hospitalizations or cardiovascular interventions.
In 5% to 10% of the cases, R-HCM is associated to a severe clinical presentation, particularly for infants with signs of heart failure.
The prevalence of pulmonary stenosis (PS) ranges between ~25% and 70% in this subgroup of patients.
Other CHDs commonly occur among patients with RASopathies: “secundum atrial septal defect”, as well as ventricular septal defect, atrioventricular canal defect (AVCD), mitral valve abnormalities, coronary arteries abnormalities, and left-sided obstructive lesions.
In selected cases, rapamycin or MEK1 inhibitors may promote regression of left ventricular hypertrophy and should be considered as a therapeutic option.
ACKNOWLEDGMENTS
The authors would like to acknowledge Michela Piscopo, Daniela Lafera, and Ciro De Prisco who made possible our research and clinical activity.
Footnotes
DISCLOSURE
The authors declare that they have no conflict of interest.
REFERENCES
- 1.Mendez HMM, Opitz JM. Noonan syndrome: A review. Am J Med Genet 1985;21(3):493–506. [DOI] [PubMed] [Google Scholar]
- 2.Norrish G, Field E, Mcleod K, et al. Clinical presentation and survival of childhood hypertrophic cardiomyopathy: a retrospective study in United Kingdom. Eur Heart J 2019;40(12):986–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tartaglia M, Gelb BD, Zenker M. Noonan syndrome and clinically related disorders. Best Pract Res Clin Endocrinol Metab 2011;25(1):161–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Motta M, Pannone L, Pantaleoni F, et al. Enhanced MAPK1 Function Causes a Neurodevelopmental Disorder within the RASopathy Clinical Spectrum. Am J Hum Genet 2020;107(3):499–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hennekam RCM. Costello syndrome: An overview. Am J Med Genet C Semin Med Genet 2003;117 C(1):42–8. [DOI] [PubMed] [Google Scholar]
- 6.Pierpont MEM, Magoulas PL, Adi S, et al. Cardio-facio-cutaneous syndrome: clinical features, diagnosis, and management guidelines. Pediatrics 2014;134(4):e1149–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calcagni G, Limongelli G, D’Ambrosio A, et al. Cardiac defects, morbidity and mortality in patients affected by RASopathies. CARNET study results. Int J Cardiol 2017;245:92–8. [DOI] [PubMed] [Google Scholar]
- 8.Roberts AE, Allanson JE, Tartaglia M, et al. Noonan syndrome. Lancet 2013;381(9863):333–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Limongelli G, Monda E, Tramonte S, et al. Prevalence and clinical significance of red flags in patients with hypertrophic cardiomyopathy. Int J Cardiol 2020;299:186–91. [DOI] [PubMed] [Google Scholar]
- 10.Tartaglia M, Pennacchio LA, Zhao C, et al. Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat Genet 2007;39(1):75–9. [DOI] [PubMed] [Google Scholar]
- 11.Tartaglia M, Kalidas K, Shaw A, et al. PTPN11 mutations in noonan syndrome: Molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity. Am J Hum Genet 2002;70(6): 1555–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lepri F, De Luca A, Stella L, et al. SOS1 mutations in Noonan syndrome: Molecular spectrum, structural insights on pathogenic effects, and genotype-phenotype correlations. Hum Mutat 2011;32(7): 760–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cordeddu V, Yin JC, Gunnarsson C, et al. Activating Mutations Affecting the Dbl Homology Domain of SOS2 Cause Noonan Syndrome. Hum Mutat 2015; 36(11):1080–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pandit B, Sarkozy A, Pennacchio LA, et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet 2007;39(8):1007–12. [DOI] [PubMed] [Google Scholar]
- 15.Yaoita M, Niihori T, Mizuno S, et al. Spectrum of mutations and genotype–phenotype analysis in Noonan syndrome patients with RIT1 mutations. Hum Genet 2016;135(2):209–22. [DOI] [PubMed] [Google Scholar]
- 16.Limongelli G, Sarkozy A, Pacileo G, et al. Genotype-phenotype analysis and natural history of left ventricular hypertrophy in LEOPARD syndrome. Am J Med Genet A 2008;146(5):620–8. [DOI] [PubMed] [Google Scholar]
- 17.Cerrato F, Pacileo G, Limongelli G, et al. A standard echocardiographic and tissue Doppler study of morphological and functional findings in children with hypertrophic cardiomyopathy compared to those with left ventricular hypertrophy in the setting of Noonan and LEOPARD syndromes. Cardiol Young 2008;18(6):575–80. [DOI] [PubMed] [Google Scholar]
- 18.Gripp KW, Lin AE. Costello syndrome: A Ras/mitogen activated protein kinase pathway syndrome (rasopathy) resulting from HRAS germline mutations. Genet Med 2012;14(3):285–92. [DOI] [PubMed] [Google Scholar]
- 19.Sarkozy A, Digilio MC, Dallapiccola B. Leopard syndrome. Orphanet J Rare Dis 2008;3(1). 10.1186/1750-1172-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Calcagni G, Adorisio R, Martinelli S, et al. Clinical Presentation and Natural History of Hypertrophic Cardiomyopathy in RASopathies. Heart Fail Clin 2018;14(2):225–35. [DOI] [PubMed] [Google Scholar]
- 21.Monda E, Rubino M, Lioncino M, et al. Hypertrophic Cardiomyopathy in Children: Pathophysiology, Diagnosis, and Treatment of Non-sarcomeric Causes. Front Pediatr 2021;9:632293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caiazza M, Rubino M, Monda E, et al. Combined ptpn11 and mybpc3 gene mutations in an adult patient with noonan syndrome and hypertrophic cardiomyopathy. Genes (Basel) 2020;11(8):1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaltenecker E, Schleihauf J, Meierhofer C, et al. Long-term outcomes of childhood onset Noonan compared to sarcomere hypertrophic cardiomyopathy. Cardiovasc Diagn Ther 2019;9(S2):S299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gelb BD, Roberts AE, Tartaglia M. Cardiomyopathies in Noonan syndrome and the other RASopathies. Prog Pediatr Cardiol 2015;39(1):13–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilkinson JD, Lowe AM, Salbert BA, et al. Outcomes in children with Noonan syndrome and hypertrophic cardiomyopathy: A study from the Pediatric Cardiomyopathy Registry. Am Heart J 2012;164(3):442–8. [DOI] [PubMed] [Google Scholar]
- 26.Prendiville TW, Gauvreau K, Tworog-Dube E, et al. Cardiovascular disease in Noonan syndrome. Arch Dis Child 2014;99(7):629–34. [DOI] [PubMed] [Google Scholar]
- 27.Linglart L, Gelb BD. Congenital heart defects in Noonan syndrome: Diagnosis, management, and treatment. Am J Med Genet C Semin Med Genet 2020;184(1):73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Colquitt JL, Noonan JA. Cardiac Findings in Noonan Syndrome on Long-term Follow-up. Congenit Heart Dis 2014;9(2):144–50. [DOI] [PubMed] [Google Scholar]
- 29.Maron BJ, Tajik AJ, Ruttenberg HD, et al. Hypertrophic cardiomyopathy in infants: Clinical features and natural history. Circulation 1982;65(1 I):7–17. [DOI] [PubMed] [Google Scholar]
- 30.Maron MS, Olivotto I, Harrigan C, et al. Mitral valve abnormalities identified by cardiovascular magnetic resonance represent a primary phenotypic expression of hypertrophic cardiomyopathy. Circulation 2011;124(1):40–7. [DOI] [PubMed] [Google Scholar]
- 31.Calcagni G, Gagliostro G, Limongelli G, et al. Atypical cardiac defects in patients with RASopathies: Updated data on CARNET study. Birth Defects Res 2020;112(10):725–31. [DOI] [PubMed] [Google Scholar]
- 32.Marino B, Gagliardi MG, Digilio MC, et al. Noonan syndrome: Structural abnormalities of the mitral valve causing subaortic obstruction. Eur J Pediatr 1995;154(12):949–52. [DOI] [PubMed] [Google Scholar]
- 33.Rapezzi C, Arbustini E, Caforio ALP, et al. Diagnostic work-up in cardiomyopathies: bridging the gap between clinical phenotypes and final diagnosis. A position statement from the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2013;34(19):1448–58. [DOI] [PubMed] [Google Scholar]
- 34.Pierpont ME, Digilio MC. Cardiovascular disease in Noonan syndrome. Curr Opin Pediatr 2018;30(5): 601–8. [DOI] [PubMed] [Google Scholar]
- 35.Shaw AC, Kalidas K, Crosby AH, et al. The natural history of Noonan syndrome: A long-term follow-up study. Arch Dis Child 2007;92(2):128–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holzmann J, Tibby SM, Rosenthal E, et al. Results of balloon pulmonary valvoplasty in children with Noonan’s syndrome. Cardiol Young 2018;28(5):647–52. [DOI] [PubMed] [Google Scholar]
- 37.Ishizawa A, Oho SI, Dodo H, et al. Cardiovascular abnormalities in Noonan syndrome: The clinical findings and treatments. Pediatr Int 1996;38(1):84–90. [DOI] [PubMed] [Google Scholar]
- 38.Bertola DR, Sugayama SM, Albano LM, et al. Noonan syndrome: a clinical and genetic study of 31 patients. Rev Hosp Clin Fac Med Sao Paulo 1999;54(5):147–50. [DOI] [PubMed] [Google Scholar]
- 39.Marino B, Digilio MC, Toscano A, et al. Congenital heart diseases in children with Noonan syndrome: An expanded cardiac spectrum with high prevalence of atrioventricular canal. J Pediatr 1999; 135(6):703–6. [DOI] [PubMed] [Google Scholar]
- 40.Zenker M, Buheitel G, Rauch R, et al. Genotype-phenotype correlations in Noonan syndrome. J Pediatr 2004;144(3):368–74. [DOI] [PubMed] [Google Scholar]
- 41.Digilio MC, Romana Lepri F, Lisa Dentici M, et al. Atrioventricular canal defect in patients with RASopathies. Eur J Hum Genet 2013;21(2):200–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pradhan AK, Pandey S, Usman K, et al. Noonan syndrome with complete atrioventricular canal defect with pulmonary stenosis. J Am Coll Cardiol 2013; 62(20):1905. [DOI] [PubMed] [Google Scholar]
- 43.Zmolikova M, Puchmajerova A, Hecht P, et al. Coarctation of the aorta in Noonan-like syndrome with loose anagen hair. Am J Med Genet Part A 2014; 164(5):1218–21. [DOI] [PubMed] [Google Scholar]
- 44.Lam J, Corno A, Oorthuys HWE, et al. Unusual combination of congenital heart lesions in a child with Noonan’s syndrome. Pediatr Cardiol 1982;3(1):23–6. [DOI] [PubMed] [Google Scholar]
- 45.Digilio MC, Marino B, Giannotti A, et al. Noonan syndrome with cardiac left-sided obstructive lesions. Hum Genet 1997;99(2):289. [DOI] [PubMed] [Google Scholar]
- 46.Calcagni G, Baban A, De Luca E, et al. Coronary artery ectasia in Noonan syndrome: Report of an individual with SOS1 mutation and literature review. Am J Med Genet Part A 2016;170(3):665–9. [DOI] [PubMed] [Google Scholar]
- 47.Kawsara A, Núñez Gil IJ, Alqahtani F, et al. Management of Coronary Artery Aneurysms. JACC Cardiovasc Interv 2018;11(13):1211–23. [DOI] [PubMed] [Google Scholar]
- 48.Hahn A, Lauriol J, Thul J, et al. Rapidly progressive hypertrophic cardiomyopathy in an infant with Noonan syndrome with multiple lentigines: Palliative treatment with a rapamycin analog. Am J Med Genet Part A 2015;167(4):744–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Calcagni G, Digilio MC, Marino B, et al. Pediatric patients with RASopathy-associated hypertrophic cardiomyopathy: The multifaceted consequences of PTPN11 mutations. Orphanet J Rare Dis 2019; 14(1):163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tartaglia M, Martinelli S, Stella L, et al. Diversity and functional consequences of germline and somatic PTPN11 mutations in human disease. Am J Hum Genet 2006;78(2):279–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hanna N, Montagner A, Lee WH, et al. Reduced phosphatase activity of SHP-2 in LEOPARD syndrome: Consequences for PI3K binding on Gab1. FEBS Lett 2006;580(10):2477–82. [DOI] [PubMed] [Google Scholar]
- 52.Lin AE, Alexander ME, Colan SD, et al. Clinical, pathological, and molecular analyses of cardiovascular abnormalities in Costello syndrome: A Ras/MAPK pathway syndrome. Am J Med Genet Part A 2011;155(3):486–507. [DOI] [PubMed] [Google Scholar]
- 53.Gripp KW, Rauen KA. Costello Syndrome. 1993. Available at: http://www.ncbi.nlm.nih.gov/pubmed/20301680. Accessed September 5, 2020.
- 54.Gripp KW, Hopkins E, Sol-Church K, et al. Phenotypic analysis of individuals with Costello syndrome due to HRAS p.G13C. Am J Med Genet Part A 2011;155(4):706–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kerr B, Delrue MA, Sigaudy S, et al. Genotype-phenotype correlation in Costello syndrome: HRAS mutation analysis in 43 cases. J Med Genet 2006; 43(5):401–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Levin MD, Saitta SC, Gripp KW, et al. Nonreentrant atrial tachycardia occurs independently of hypertrophic cardiomyopathy in RASopathy patients. Am J Med Genet Part A 2018;176(8):1711–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gripp KW, Morse LA, Axelrad M, et al. Costello syndrome: Clinical phenotype, genotype, and management guidelines. Am J Med Genet Part A 2019; 179(9):1725–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abe Y, Aoki Y, Kuriyama S, et al. Prevalence and clinical features of Costello syndrome and cardio-facio-cutaneous syndrome in Japan: Findings from a nationwide epidemiological survey. Am J Med Genet Part A 2012;158 A(5):1083–94. [DOI] [PubMed] [Google Scholar]
- 59.Allanson JE, Annerén G, Aoki Y, et al. Cardio-faciocutaneous syndrome: Does genotype predict phenotype? Am J Med Genet C Semin Med Genet 2011;157(2):129–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kavamura MI, Peres CA, Alchorne MMA, et al. CFC index for the diagnosis of cardiofaciocutaneous syndrome. Am J Med Genet 2002;112(1):12–6. [DOI] [PubMed] [Google Scholar]
- 61.Armour CM, Allanson JE. Further delineation of cardio-facio-cutaneous syndrome: Clinical features of 38 individuals with proven mutations. J Med Genet 2008;45(4):249–54. [DOI] [PubMed] [Google Scholar]
- 62.Mazzanti L, Cacciari E, Cicognani A, et al. Noonan-like syndrome with loose anagen hair: A new syndrome? Am J Med Genet 2003;118 A(3):279–86. [DOI] [PubMed] [Google Scholar]
- 63.Martinelli S, De Luca A, Stellacci E, et al. Heterozygous germline mutations in the CBL tumor-suppressor gene cause a noonan syndrome-like phenotype. Am J Hum Genet 2010;87(2):250–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Niemeyer CM, Kang MW, Shin DH, et al. Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic Leukemia. Nat Genet 2010;42(9):641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pérez B, Mechinaud F, Galambrun C, et al. Germline mutations of the CBL gene define a new genetic syndrome with predisposition to juvenile myelomonocytic leukaemia. J Med Genet 2010;47(10): 686–91. [DOI] [PubMed] [Google Scholar]
- 66.Nishikawa T, Ishiyama S, Shimojo T, et al. Hypertrophic cardiomyopathy in Noonan syndrome. Pediatr Int 1996;38(1):91–8. [DOI] [PubMed] [Google Scholar]
- 67.Tartaglia M, Mehler EL, Goldberg R, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet 2001;29(4):465–8. [DOI] [PubMed] [Google Scholar]
- 68.Tartaglia M, Gelb BD. Disorders of dysregulated signal traffic through the RAS-MAPK pathway: Phenotypic spectrum and molecular mechanisms. Ann N Y Acad Sci 2010;1214(1):99–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wu X, Simpson J, Hong JH, et al. MEK-ERK pathway modulation ameliorates disease phenotypes in a mouse model of Noonan syndrome associated with the Raf1L613V mutation. J Clin Invest 2011;121(3): 1009–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Andelfinger G, Marquis C, Raboisson MJ, et al. Hypertrophic Cardiomyopathy in Noonan Syndrome Treated by MEK-Inhibition. J Am Coll Cardiol 2019; 73(17):2237–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Marin TM, Keith K, Davies B, et al. Rapamycin reverses hypertrophic cardiomyopathy in a mouse model of LEOPARD syndrome-associated PTPN11 mutation. J Clin Invest 2011;121(3):1026–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hudsmith LE, Petersen SE, Francis JM, et al. Hypertrophic cardiomyopathy in Noonan syndrome closely mimics familial hypertrophic cardiomyopathy due to sarcomeric mutations. Int J Cardiovasc Imaging 2006;22(3–4):493–5. [DOI] [PubMed] [Google Scholar]
- 73.Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy. Circulation 2020. 10.1161/cir.0000000000000937. [DOI] [Google Scholar]
- 74.Petrin Z, Soffer B, Daniels SJ. Sudden cardiac arrest in the field in an 18-year-old male athlete with Noonan syndrome: Case presentation and 5-year follow-up. Cardiol Young 2019;29(9):1214–6. [DOI] [PubMed] [Google Scholar]
- 75.Aydin A, Yilmazer MS, Gurol T. Sudden death in a patient with Noonan syndrome. Cardiol Young 2011;21(2):233–4. [DOI] [PubMed] [Google Scholar]
- 76.Eichhorn C, Voges I, Daubeney PEF. Out-of-hospital cardiac arrest and survival in a patient with Noonan syndrome and multiple lentigines: A case report. J Med Case Rep 2019;13(1). 10.1186/s13256-019-2096-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Miron A, Lafreniere-Roula M, Steve Fan CP, et al. A validated model for sudden cardiac death risk prediction in pediatric hypertrophic cardiomyopathy. Circulation 2020;142(3):217–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kaski JP, Norrish G, Ding T, et al. Development of a Novel Risk Prediction Model for Sudden Cardiac Death in Childhood Hypertrophic Cardiomyopathy (HCM Risk-Kids). JAMA Cardiol 2019;4(9):918–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Moran AM, Colan SD. Verapamil therapy in infants with hypertrophic cardiomyopathy. Cardiol Young 1998;8(3):310–9. [DOI] [PubMed] [Google Scholar]
- 80.Spicer RL, Rocchini AP, Crowley DC, et al. Hemodynamic effects of verapamil in children and adolescents with hypertrophic cardiomyopathy. Circulation 1983;67(2):413–20. [DOI] [PubMed] [Google Scholar]
- 81.Poterucha JT, Johnson JN, O’Leary PW, et al. Surgical Ventricular Septal Myectomy for Patients with Noonan Syndrome and Symptomatic Left Ventricular Outflow Tract Obstruction. Am J Cardiol 2015; 116(7):1116–21. [DOI] [PubMed] [Google Scholar]
- 82.Hemmati P, Dearani JA, Daly RC, et al. Early Outcomes of Cardiac Surgery in Patients with Noonan Syndrome. Semin Thorac Cardiovasc Surg 2019. 10.1053/j.semtcvs.2018.12.004. [DOI] [PubMed] [Google Scholar]
- 83.McCallen LM, Ameduri RK, Denfield SW, et al. Cardiac transplantation in children with Noonan syndrome. Pediatr Transplant 2019;23(6). 10.1111/petr.13535. [DOI] [PubMed] [Google Scholar]
- 84.Mehra MR, Canter CE, Hannan MM, et al. The 2016 International Society for Heart Lung Transplantation listing criteria for heart transplantation: A 10-year update. J Heart Lung Transplant 2016;35(1):1–23. [DOI] [PubMed] [Google Scholar]
- 85.Rossano JW, Cherikh WS, Chambers DC, et al. The Registry of the International Society for Heart and Lung Transplantation: Twentieth Pediatric Heart Transplantation Report-2017; Focus Theme: Allograft ischemic time. J Heart Lung Transplant 2017. 10.1016/j.healun.2017.07.018. [DOI] [PubMed] [Google Scholar]
- 86.Topilsky Y, Pereira NL, Shah DK, et al. Left ventricular assist device therapy in patients with restrictive and hypertrophic cardiomyopathy. Circ Hear Fail 2011;4(3):266–75. [DOI] [PubMed] [Google Scholar]
- 87.Lipshultz SE, Sleeper LA, Towbin JA, et al. The Incidence of Pediatric Cardiomyopathy in Two Regions of the United States. N Engl J Med 2003;348(17):1647–55. [DOI] [PubMed] [Google Scholar]
- 88.Colan SD, Lipshultz SE, Lowe AM, et al. Epidemiology and cause-specific outcome of hypertrophic cardiomyopathy in children: Findings from the Pediatric Cardiomyopathy Registry. Circulation 2007; 115(6):773–81. [DOI] [PubMed] [Google Scholar]
- 89.Gajarski R, Naftel DC, Pahl E, et al. Outcomes of Pediatric Patients With Hypertrophic Cardiomyopathy Listed for Transplant. J Heart Lung Transplant 2009;28(12):1329–34. [DOI] [PubMed] [Google Scholar]
- 90.Gross AM, Frone M, Gripp KW, et al. Advancing RAS/RASopathy therapies: An NCI-sponsored intramural and extramural collaboration for the study of RASopathies. Am J Med Genet Part A 2020; 182(4):866–76. [DOI] [PMC free article] [PubMed] [Google Scholar]