

Abstract
Pompe disease is a rare genetic neuromuscular disorder caused by acid α-glucosidase (GAA) deficiency resulting in lysosomal glycogen accumulation and progressive myopathy. Enzyme replacement therapy, the current standard of care, penetrates poorly into the skeletal muscles and the peripheral and central nervous system (CNS), risks recombinant enzyme immunogenicity, and requires high doses and frequent infusions. Lentiviral vector-mediated hematopoietic stem and progenitor cell (HSPC) gene therapy was investigated in a Pompe mouse model using a clinically relevant promoter driving nine engineered GAA coding sequences incorporating distinct peptide tags and codon optimizations. Vectors solely including glycosylation-independent lysosomal targeting tags enhanced secretion and improved reduction of glycogen, myofiber, and CNS vacuolation in key tissues, although GAA enzyme activity and protein was consistently lower compared with native GAA. Genetically modified microglial cells in brains were detected at low levels but provided robust phenotypic correction. Furthermore, an amino acid substitution introduced in the tag reduced insulin receptor-mediated signaling with no evidence of an effect on blood glucose levels in Pompe mice. This study demonstrated the therapeutic potential of lentiviral HSPC gene therapy exploiting optimized GAA tagged coding sequences to reverse Pompe disease pathology in a preclinical mouse model, providing promising vector candidates for further investigation.
Keywords: hematopoietic stem and progenitor cells, lentiviral vector, Pompe disease, tag technology, glycosylation-independent lysosomal targeting
Graphical abstract
Introduction
Pompe disease, or glycogenosis type II (OMIM: #232300), is an autosomal recessive metabolic myopathy caused by acid α-glucosidase (GAA) enzyme (EC 3.2.1.20) deficiency. It is characterized by lysosomal glycogen accumulation predominantly in the heart, skeletal muscle, and central nervous system (CNS).1,2 Infantile-onset Pompe disease (IOPD) patients have low-level GAA enzyme activity (<2% of normal) and display severe muscle weakness. Late-onset Pompe disease (LOPD) patients have higher residual GAA activity and show a more protracted disease progression, often becoming wheelchair and ventilation dependent, with an overall shortened life expectancy.1 The standard of care (SOC) is enzyme replacement therapy (ERT), which requires lifelong weekly to biweekly recombinant human GAA (rhGAA) administrations that can prolong the life of Pompe patients but cannot guarantee long-term symptom-free survival. ERT acts through cation-independent mannose 6-phosphate receptor (CI-M6PR) also known as insulin-like growth factor 2 (IGF2) receptor (IGF2R) mediated endocytosis to the lysosome,3,4 which is complicated by very low rhGAA concentrations reaching the interstitial space, hampering efficient uptake into affected muscle cells.5 In addition, the currently approved rhGAA has low abundance of mannose-6-phosphate (M6P), in particular biphosphorylated N-glycans, which are required for high affinity to the CI-MPR, leading to inefficient cellular uptake.5 Hence, other neo rhGAA with increased bis-mannose 6-phosphate levels have been investigated in clinical trials (avalglucosidase alfa; ClinicalTrials.gov ID: NCT02782741, NCT03019406, NCT02032524)6,7 with recent US Food and Drug Administration approval for LOPD patients. Furthermore, abnormal M6P trafficking may also hamper uptake.8,9 To address limitations of ERT and to enhance delivery to the skeletal muscle, a glycosylation-independent lysosomal targeting (GILT) tag based on IGF2 peptide that binds with high affinity to the IGF2R was fused to a truncated catalytic domain of GAA (reveglucosidase alfa) and demonstrated to be more effective in clearing glycogen in skeletal muscle in a Gaa−/− mouse model.10 Reveglucosidase alfa infusions in LOPD patients were reasonably well tolerated, and initial improvements of respiratory strength and ventilatory function were observed, but with limited effect on walking endurance (ClinicalTrials.gov ID: NCT01230801).11
Alternatively, there have been efforts to treat Pompe disease patients with adeno-associated virus (AAV) gene therapy,12 as well as preclinical attempts to use the hematopoietic system as a factory to produce recombinant human rhGAA enzyme.13,14,15 For treatment of patients with lysosomal storage disorders (LSDs), such as mucopolysaccharidosis I, allogeneic hematopoietic stem and progenitor cell (HSPC) transplantation is a therapeutic option for patients with a matched donor, which is not always available. The limited attempts of bone marrow transplantation for Pompe disease patients have been unsuccessful as a treatment,16 and the low-level expression of endogenous GAA enzyme in hematopoietic cells is insufficient for cross-correction. GAA activity in the hematopoietic system in mice is also low.13 Hence, allogeneic HSPC transplantation is unlikely to be beneficial, and high-level vector-driven ectopic enzyme expression in hematopoietic cells would be required to reach clinical efficacy. In clinical trials, lentiviral-mediated HSPC gene therapy for (neuro)metabolic diseases, such as X-linked adrenoleukodystrophy, metachromatic leukodystrophy (MLD), Fabry disease, and Hurler disease provided therapeutic benefit and proved to be safe.17,18,19,20,21,22 Moreover, preclinical studies using ex vivo lentiviral vector-mediated overexpression of GAA using the spleen focus-forming virus U3 promoter and partial chimerism of genetically modified cells demonstrated alleviation of clinical symptoms in Gaa−/− mice.13 Using the lineage-restricted locus control region of the β-globin chain fused to the ubiquitous elongation factor 1α short promoter (LCR-EFS) that boosts expression in the erythrocyte lineage, colleagues showed that it is safe in murine and human HSPCs, albeit providing partial therapeutic response in skeletal muscle and CNS in Gaa−/− mice.23 Furthermore, in contrast to ERT and AAV therapies, in which immune responses against either rhGAA, viral vector, or transgene product may reduce efficacy,24,25,26,27,28 allogeneic HSPC transplantation has been shown to promote immune tolerance induction to infusion of recombinant α-L-iduronidase in Hurler disease patients.29 In preclinical studies using HSPC gene therapy in Gaa−/− mice, robust immune tolerance induction against rhGAA was also observed.13,14,30 Importantly, liver-directed AAV gene therapy has been reported to promote immune tolerance induction to rhGAA in Gaa−/− mice by either restricting expression to hepatocytes,31 low-dose AAV vector administration,32 or a secretable GAA.33
Nevertheless, AAV serotypes targeting the muscle or liver33 typically do not effectively deliver transgene product to the CNS, and in turn AAV serotypes targeting the CNS may not completely address the systemic pathology, such as cardiac correction.34 The prominent CNS involvement, particularly in aging IOPD patients, could potentially be addressed through cross-correction of HSPC-derived genetically modified microglia that produce and secrete the transgene product locally.
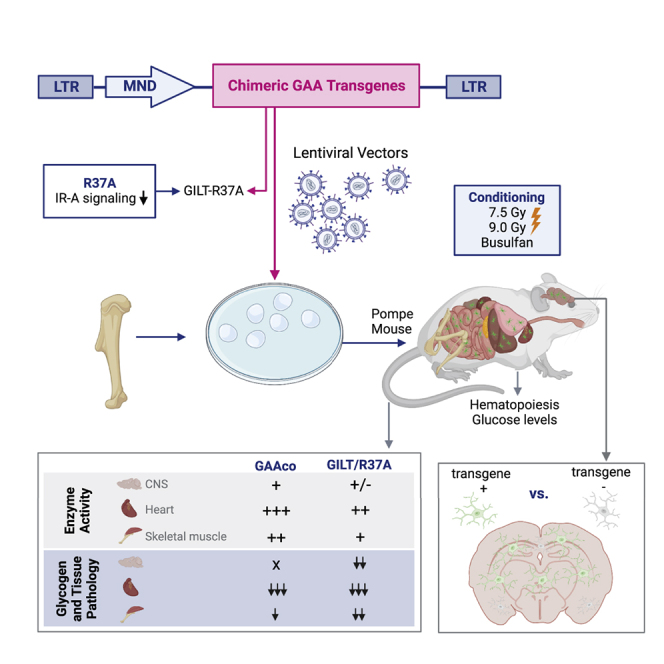
To enhance the efficacy of treatment by HSPC gene therapy, we tested ten lentiviral vectors expressing engineered human GILT-tagged GAA sequences in vitro. Nine of these lentiviral vectors were tested in ex vivo HSPC gene therapy in Gaa−/− mice. Interestingly, we demonstrated that two GILT-tagged vector sequences with an R37A mutein were particularly effective in providing biochemical correction in the heart, skeletal muscles, and CNS, revealing improved efficacy compared with non-tagged GAA sequence in HSPC gene therapy in Gaa−/− mice. Additionally, we show that the inclusion of an apolipoprotein E (ApoE) tag diminished efficacy particularly when positioned upstream of the GILT tag, and that a Gly-Ala-Pro (GAP) linker in the catalytic domain of GAA protein35 failed to enhance the GAA enzyme activity in Pompe mice, dismissing further applications.
Results
Secretion and uptake of genetically engineered GAA through IGF2R-mediated pathways
Eleven different lentiviral constructs encoding unique human GAA sequences were generated (Figures S1A and S1B). These included sequences with codon-optimized GAA (GAAco) and ten constructs containing an IGF2 tag fused to the N terminus of the catalytic GAA sequence (GILTco) as previously described.10 Derivatives of GILTco were generated to eliminate a consensus furin-cleavage site (R37A) in the IGF2 sequence.36 Native sequence (GILTm) and three coding sequences generated through two different codon-optimization algorithms (GILTco1-m or GILT-co2-) using GenBank : Y00839 and a third containing the consensus amino acid GAA sequence (CCDS database: CCDS32760.1 = GILT-co3-m). Another two lentiviral vectors contained a tandem repeat sequence of the ApoE either upstream of the GILT tag (GILTco1-m-ApoE1) or downstream of the GILT tag (GILTco1-m-ApoE2) to potentially facilitate enhanced crossing of the blood-brain barrier (BBB).37,38,39 Furthermore, we introduced a GAP peptide linker within the GAA amino acid sequence to enhance enzyme activity35,40 (GILTco1-m-L) and with the ApoE tag (GILTco1-m-ApoE1-L and GILTco2-m-ApoE2-L).
GAA is poorly secreted by cells.41 Using the signal peptide algorithm SignalP 5.042 to predict the likelihood of signal peptidase Sec/SPI cleavage, the likelihood of cleavage of the GILT-tagged GAA (0.98) was higher compared with the untagged GAA (0.26). Hence, we expected an increased secretion of the GILT-tagged protein. As anticipated, GAA enzyme activity in the cell lysates (Figure 1A) and in conditioned medium (Figure 1B) was detected in lentiviral vector transduced HAP1-GAA−/− cells, showing that all nine GILT-tagged constructs provided improved relative secretion of 3.3- to 16.3-fold compared with GAAco (Figure S2A), confirming that GILT-tagged GAA protein is secreted more efficiently than native GAA protein in vitro. Importantly, the transduced HAP1-GAA−/− cells showed both the precursor (110 kDa) and mature protein (75–70 kDa) in the cells (Figure S2B). We further investigated cellular uptake of the chimeric proteins by IGF2R (Figures 1C and S3). To that end, GAA-deficient K562 cells (K562-GAA−/−) were derived through targeted disruption of the GAA gene using CRISPR-Cas9 (Figures S3A–S3C). Subsequently, purified rhGAA, GILT-GAA, and GILT-R37A-GAA protein, which showed similar GAA enzyme activities relative to protein concentration (Figures S3D and S3E), were efficiently taken up by these K562-GAA−/− cells (Figure 1C). After an additional disruption of IGF2R expression by CRISPR-Cas9 gene editing in K562-GAA−/− cells, IGF2R protein and surface expression were diminished as shown by flow cytometry and western blot analysis (Figures S3F–S3I) and, consequently, uptake of rhGAA, rhGILT-GAA, and rhGILT-R37A-GAA proteins were also reduced (∼3-fold) at the highest concentrations (Figure 1C). Reintroduction of IGF2R by lentiviral transduction (Figures S3G and S3J) confirmed uptake through IGF2R by partially restoring cellular uptake of the three proteins (Figure 1C). Finally, IGF2 binds to a domain on the IGF2R other than M6P,10 and both GILT-GAA and GILT-R37A-GAA proteins showed specific competitive inhibition in a cellular uptake assay via IGF2 administration but not by M6P (Figure 1D).
Figure 1.

Effective secretion and uptake through IGF2R-mediated pathways
(A) GAA activity in transduced HAP1 GAA−/− cell lysates (n = 3) normalized to VCN, and (B) in conditioned medium from the cells (n = 2–3). (C) GAA activity after incubation with rhGAA proteins into (1) K562-GAA−/−, (2) GAA−/−IGF2R−/−, or (3) GAA−/−IGF2R−/− + IGF2R vector. Duplicate means connected by a non-linear fit line. (D) GAA activity after uptake of rhGAA proteins into human Pompe patient fibroblasts. Duplicate means ± SD are shown.
Genetically modified murine HSPCs provide robust expression of engineered GAA in Gaa−/− mice
Male donor bone marrow lineage-negative (Lin−) cells derived from Gaa−/− mice43 (Figures S4A and S4B) transduced with the chimeric GAA vectors (nine groups) or GAAco (Table S1) were intravenously infused into 7.5-Gy-irradiated female Gaa−/− mice. The GILTco1-m variant was also transplanted in 9-Gy-irradiated or Busulfex-conditioned Gaa−/− mice. Controls included treatment-naive Gaa−/− or Gaa+/+ mice, and transduction controls containing MND.GFP vector transduced Lin− cells. Lin− cells efficiently transduced at a vector copy number (VCN) range of 0.9–4.6 assessed on day 7 of liquid culture (Figure S5A). Total frequency of colony-forming units (CFU) of transduced Gaa−/− Lin− cells showed no significant differences between non-transduced, GFP vector or chimeric GAA vector variants (Figure S5B; p = 0.277, one-way ANOVA).
The lentiviral vector transduced male Gaa−/− Lin− cells reconstituted Gaa−/− female mice and produced steady supraphysiological GAA enzyme activity levels in peripheral blood (PB) leukocytes ranging from ∼30-fold to over 100-fold increase when compared with Gaa+/+ mice (median 0.64 nmol/mg/h; weeks 5, 9, 13, and 15; Figure S5C). Likewise, in the GFP control groups, the percentage GFP-positive PB leukocytes remained steady (mean percentage of GFP between 62.7% ± 20.7% and 80.3% ± 4.5%) in recipient GFP-treated mice that were engrafted with donor cells until end of study at week 16 (Figure S5D) as well as in the bone marrow and spleen (Figure S5E), except for four out of nine mice that received Busulfex conditioning which presented with engraftment failure defined by <5% GFP+ cells in hematopoietic tissues. Remarkably, at week 16 GAA enzyme activity in the bone marrow was ∼16- to 275-fold higher in gene-therapy-treated mice compared with non-treated Gaa+/+ mice (median 7.42 nmol/mg/h, Figure 2A) and GFP-treated Gaa+/+ control mice. Supraphysiological GAA enzyme activity was also measured in splenocytes (Figure S5F). The GAAco exhibited consistently higher GAA enzyme activity than the GILT-tagged variants across PB, bone marrow (∼2- to 17-fold increase compared with all other GILT-tagged groups, Figures 2A and S5C), and spleen (∼1.6- to 10-fold increase compared with all other GILT-tagged groups, Figure S5F). No major difference in GAA enzyme activity between the GILTco and GILTco1-m (containing the R37A mutein) was detected in cell pellets. While the efficacy of the GILTco1-m was similar to that of the non-codon-optimized GILTm, the other codon-optimized GILTco2-m elicited slightly lower (∼2-fold) GAA enzyme activity in bone marrow and spleen (Figures 2A and S5F). Of note, the performance of the GILTco3-m, carrying the consensus GAA sequence, was significantly higher in spleen than that of the GILTco1-m, albeit representing a 2.4-fold increase in enzyme activity (Figure S5F). Surprisingly, the inclusion of the GAP linker within the GAA sequence in the GILTco1-m-L showed no enhanced GAA enzyme activity compared with the GILTco1-m, and the incorporation of ApoE upstream of the GILT tag resulted in much lower cellular GAA enzyme activity, consistent with our previous in vitro observations (Figures 2A, S5C, and S5F). However, when changing the position of the ApoE tag in the coding sequence, the GILTco1-m-ApoE2-L provided higher GAA enzyme activity, which was significant in spleen of mice (Figure S5F). Overall, GILTco3-m generally produced the highest activities of all the GILT-tagged constructs (Figures 2A, S5C, and S5F). The therapeutic transgenes contain the woodchuck hepatitis virus post-transcriptional regulatory element (WPRE) sequence, which was detected by WPRE fluorescence in situ hybridization (FISH) staining in the spleen (Figure S5G). In addition, immunohistochemical staining also showed clear localization of GAA and the GILT-tagged protein in a spleen of a GILTco1-m-treated mouse. Signal was not detected in a GFP-treated mouse (Figure S5G). We further investigated proper maturation of GILT-tagged GAA protein by western blot analysis, which showed variable ratios of the 110-kDa GAA precursor protein, as well as 95-kDa intermediate and lysosomal matured 70–75 kDa GAA forms in bone marrow (Figure 2B). GAA protein levels were generally lower in the GILT-tagged groups compared with the GAAco group (10 out of 11 groups with 90%–32% relative to GAAco group; Figure S5H). However, in the GILT-tagged groups, the relative abundance of the GAA precursor protein was ∼2.5-fold higher than in the GAAco group, and the GAAco group contained 44% more mature protein (Figure S5I).
Figure 2.

High levels of GAA in hematopoietic compartment
(A) GAA enzyme activity in bone marrow cells. Individual values, group medians, and interquartile range are presented; Gaa+/+ versus treated (dotted line); GAAco versus GILT-tagged variants; GILTco1-m versus treatment groups (n = 10).n-treated. (B) Western blot analysis of bone marrow lysates showing both precursor (110 kDa) and mature GAA species (75–70 kDa). Loading control: anti-β-actin (n = 3). (C) GAA enzyme activity in plasma at week 16 post transplant, comparing non-treated Gaa+/+with treated Gaa−/−--/-, and Gaa+/+ with treated groups (n = 8-13). (D) Western blot analysis of representative plasma samples (n = 3) at week 16 post transplant using an anti-GAA monoclonal antibody. (E) Correlation analysis between plasma GAA enzyme activity and plasma GAA protein concentration by simple linear regression. GILTco3-m is depicted by red triangles. (p < 0001). (F) VCN measured in bone marrow cells comparing GAAco-treated group with GILT-tagged variants, and GILTco1-m versus GILTco3-m (n = 9–10). (G) Urinary Hex4 at week 16 post transplant; individual values, group medians, and interquartile ranges are presented (n = 4–10). Untreated Gaa−/− mice versus GAAco Kruskal-Wallis test and Dunn’s test for multiple comparison between the treatment arms. (A, C, and F) Exact Wilcoxon rank-sum test comparing arms. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001. ns = not significant. Bu = Busulfex.
Furthermore, the GAA enzyme activity levels in plasma showed a 2.7- to 4.2-fold increase in the gene-therapy-treated groups compared with the baseline signal detected in untreated Gaa−/−, Gaa+/+, and treatment control MND.GFP mice (Figure 2C), and the secreted GAA precursor protein was confirmed in plasma as well (Figures 2D and S5J). Strikingly, the performance of the GILTco1-m group was indistinguishable from the GAAco group. Seemingly connected to the low GAA enzyme activity in the bone marrow cell pellets, the GAA enzyme activity originating from GILTco2-m and GILTco1-m-ApoE1 was consistently lower when compared with the GILTco1-m, while the enzyme activity obtained from the GILTco1-m-ApoE2-L construct was found to be significantly higher in mouse plasma.
Interestingly, the GILTco1-m groups had similar GAA enzyme activity levels in PB cellular pellets and plasma across the 7.5 Gy, 9 Gy, or Busulfex conditioning regimens, but bone marrow and spleen were lower in the 9 Gy or Busulfex groups (Figures 2A–2D, S5C, and S5F). Only one out of ten mice in the Busulfex group presented engraftment failure, suspected by undetectable GAA enzyme activity and very low VCN.
Our enzyme activity results in plasma were supported by a strong positive correlation with the GAA protein concentration in the plasma (Figure 2E; p < 0.0001). Further quantification of the GAA protein in the plasma confirmed the highest values in groups GILTco3-m (1.51 ± 0.4 μg/mL, n = 10) and GILTco1-m-ApoE2-L (1.81 ± 0.95 μg/mL, n = 10), while the lowest values were seen in the GILTco1-m-ApoE1 group (0.15 ± 0.05, n = 10). The GAA protein plasma concentration ranged from 0.03 to 4.4 μg/mL (Figure S5J) in all the groups and was predominantly higher in the GILT-tagged GAA groups. Furthermore, we estimated the in vivo GAA relative secretion in plasma by calculating the GAA enzyme activity in plasma/GAA enzyme activity in leukocytes, and found it to be significantly increased in seven out of nine vector groups ranging from 2.04- to 3.36-fold compared with the GAAco vector group (Figure S5K; GILTco3-m and GILTco1-m-ApoE2-L versus GAAco; not significant). Since the plasma only contains the precursor GAA protein, this was used to determine the GAA activity per protein content. In the GAAco group, the native GAA protein showed higher GAA activity per protein than all other GILT-tagged groups, except the GILT1co-m-ApoE construct with the ApoE feature downstream to the GILT sequence (Figure S5L).
Alongside the biochemical assays, we interrogated the lentiviral VCN per cell (or diploid genome) in bone marrow, which supported the restoration of the GAA enzyme activity in the hematopoietic tissues (Figure 2F). We observed in the bone marrow of GAAco and GILTco3-m groups the highest median VCN of 3.7 and 3.5, respectively. Besides, the animals in the GILTco3-m group presented significantly higher VCN than in the GILTco1-m group (1.3-fold) (Figure 2F). Higher VCNs were observed in the lentiviral GFP control groups (median and range in the Gaa−/− GFP group is 10.5 ± 5.8, n = 10; GFP [Bu] 10.9 ± 11.8, n = 5; and Gaa+/+ GFP 9.1 ± 13.8, n = 10; Figure S5M). We observed overall lower GAA enzymatic activity per VCN in the animals treated with the GILT-tagged vectors compared with the animals in the GAAco group, and expectedly the animals treated with GILTco1-m-ApoE1 exhibited the lowest VCN compared with the animals in other GILT-tagged groups (Figure S5N).
We also interrogated engraftment in our gene therapy sex-mismatched transplant model. We observed that male donor cell chimerism by Y chromosome qPCR showed similar chimerism across groups (Figure S5O), indicating that the GAA variants utilized in the study did not influence engraftment outcome. Besides, the animals which received 9 Gy conditioning presented, as anticipated, the highest donor chimerism (Figure S5O).
Consistent with robust GAA expression, Hex4, a urinary biomarker for monitoring ERT rhGAA efficacy, was significantly lower in the therapeutic groups encompassing GAAco, GILTco, GILTco1-m, and GILTco3-m compared with the Gaa−/− mice at 16 weeks (Figure 2G). Altogether, the data showed a supraphysiological activity that was maintained until 16 weeks after transplantation and an expected shift toward the secretory pathway using the GILT-tagged constructs.
Genetically modified murine HSPCs with engineered GAA clear glycogen in heart and skeletal muscles
At 16 weeks post infusion of genetically modified HSPCs, GAA activity in the cardiac tissue in the majority of gene-therapy-treated Gaa−/− mice was restored to close to normal levels as in the wild-type Gaa+/+ group and Gaa+/+ GFP mice (6.3–6.4 nmol/h/mg; Figure 3A), with the exception of the ApoE-tag-containing GILTco1-m-ApoE1 variant and the GILTco1-m-ApoE2 variant, which were lower performers (∼2.7-fold lower than the GILTco1-m group; p < 0.0001, and p < 0.0164, respectively). The highest restoration of GAA enzyme activity was observed in the mice transplanted with GAAco vector (21.5 nmol/mg/h), followed by mice in the GILTco-3-m group (14.7 nmol/mg/h), with 2.3-fold supranormal activity of wild-type Gaa+/+ mice (Figure 3A). Of note, GAA enzyme activity in the 7.5 Gy GILTco1-m mice was similar to the 9 Gy and Busulfex treatment groups, albeit significantly lower than the GILTco3-m mice. Furthermore, the heart GAA enzyme activity aligned with GAA protein content (Figures 3B and S6A), and the processed mature form of the lysosomal GAA protein of 70–75 kDa was the most prominently detected (Figure 3B).
Figure 3.

Robust correction in heart of Gaa−/− mice
(A) GAA enzyme activity in the heart of treated Gaa−/− mice; Gaa+/+ versus treatment groups. (B) Western blot analysis of heart tissue lysates with presence of mature GAA species (75–70 kDa; n = 3). (C) Reduction of lysosomal glycogen in the heart is shown with treated compared with non-treated Gaa−/− mice, and GILTco1-m to other treatment groups. (D) Representative PAS staining images of heart for glycogen (purple stain) of non-treated versus treated groups. Scale bars, 50 μm. (E) Reduction of heart mass with group comparison for Gaa−/− versus Gaa+/+ and versus GAA variants (n = 8–10). Individual values, group medians, and interquartile ranges are shown. Wilcoxon rank-sum test p value comparing arm versus Gaa+/+ or Gaa−/−. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001. NT = non-treated. Bu = Busulfex.
Previous studies showed that lentiviral HSPC gene therapy using a GAA transgene effectively reduced heart glycogen accumulation.13,15,23 In our study more than 99% reduction was observed in the animals in all the therapeutic groups, with the exception of mice in the GILTco1-m-ApoE1 group in which glycogen remained at 21% of untreated Gaa−/− mice (441.44 μg glycogen/mg protein; Figure 3C) at week 16. Remarkably, the GILTco1-m variant performed better than the GAAco treatment group. Similarly, robust glycogen reduction was observed in the heart of mice treated with the GILTm and GILTco3-m. In contrast, the animals treated with GILTco1-m-ApoE1 variant and the GILTco1-m-ApoE2 variant showed poor glycogen reduction (Figure 3C). Expectedly, quantification of periodic acid-Schiff (PAS) and hematoxylin and eosin (H&E) staining performed in hearts of selected groups confirmed significant reduction of glycogen and typical myofiber/vascular vacuolation in the GAAco group, albeit at greater extent in the GILTco, GILTco1-m, and GILTco3-m treatment groups (Figure 3D; Tables S2 and S3).
Cardiac hypertrophy is a hallmark of IOPD patients and Gaa−/− mice, and we investigated the efficacy of our constructs to reduce the heart pathology.1,43 Notably, significant heart muscle mass reduction was observed in the non-GILT-tagged and GILT-tagged treatment groups acquiring similar heart mass as the Gaa+/+ group, with the exception of animals in the GILTco1-m-ApoE1 and GILTco1-m-L groups, in which heart muscle mass remained at Gaa−/− mice levels (Figure 3E).
Next, we investigated the efficacy of our variants in reducing the accumulation of glycogen in several skeletal muscles. In the diaphragm and the hindleg muscle quadriceps, tibialis anterior, and gastrocnemius, the highest GAA enzyme activity was detected in the GAAco group followed by the GILTco3-m group (average all skeletal muscles: 5.7-fold and 3.5-fold increase to Gaa+/+, respectively; Figures 4A–4D, left). In the majority of other therapeutic groups, GAA enzyme activity was restored to normal levels in Gaa+/+ mice. Western blot analysis of gastrocnemius tissue (Figures 4E and S6B) showed more mature 70–75 kDa GAA protein in the GAAco and GILTco3-m group than in all other groups.
Figure 4.

Effective correction in skeletal muscles of Gaa−/− mice by GILT tag
(A–D) Left panel shows GAA activity and right panel shows glycogen content. (A) Diaphragm, (B) gastrocnemius, (C) quadriceps femoris, (D) tibialis anterior. Individual values, group medians, and interquartile ranges. Exact Wilcoxon rank-sum p value compared with Gaa+/+ or Gaa−/− group, and GILTco1-m compared with other groups. NT = non-treated. Bu = Busulfex.∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001. ns = not significant (n = 8–10). (E) Western blot analysis of representative gastrocnemius tissue lysates with presence of mature GAA species (75–70 kDa). Loading control: GAPDH (n = 3). (F) PAS staining of heart, diaphragm, and quadriceps for glycogen (purple stain) of treated versus non-treated groups. Scale bars, 50 μm.
Lentiviral targeted HSPC gene therapy carrying the GAA transgene historically presented lower efficacy in glycogen reduction in skeletal muscle such as the diaphragm, and more prominently hindleg muscles (quadriceps, tibialis anterior, and gastrocnemius).13 Using lentiviral-driven GAA variants we demonstrated that robust glycogen clearance was feasible in skeletal muscles of mice of all treatment groups (Figures 3A–3D, right), with the exception of the GILTco1-m-ApoE1. Of note, in skeletal muscles glycogen was significantly reduced in the GILT-tagged treatment groups. Particularly the GILTco1-m exhibited ∼3% in diaphragm and 24%–35% in hindleg muscles of Gaa−/− glycogen levels, while mice in the GILTco3-m treatment group presented ∼1% in diaphragm and 10%–17% in hindleg muscles of Gaa−/− glycogen levels. These values in GILTco1-m and GILTco3-m were ∼2.3-fold and ∼5-fold lower, respectively, compared with the GAAco group (p < 0.001; glycogen in GAAco group in diaphragm = 12%, gastrocnemius = 77%, quadriceps = 57%, and tibialis = 60% of Gaa−/− mice glycogen levels). In gastrocnemius, quadriceps, and tibialis, mice in the GILTco3-m treatment group achieved significantly better efficacy in glycogen reduction than mice in the GILTco1-m treatment group (∼40%–65% more glycogen reduction). PAS staining and quantification (Figures 4F and S7; Table S2) in selected samples confirmed the greater reduction in glycogen and myofiber vacuolation pathology in the GILTco1-m, and particularly in the GILTco3-m treatment group (Figure S8 and Table S3). The data in heart and skeletal muscle showed that heart pathology can be addressed effectively with GAAco and GILT-tagged GAA constructs, but that GILT-tagged GAA is more effective in resolving skeletal muscle pathology.
GILT-R37A tag rescues CNS pathology
We then investigated whether HSPC gene therapy could penetrate the CNS and provide biochemical correction through local presence of genetically modified microglia. GAA enzyme activity in the cerebellum in the GAAco, GILTco, GILTco-1-m (9 Gy), GILTco1-m-ApoE2, and GILTco1-m-ApoE2-L groups was significantly higher compared with untreated Gaa−/− mice (p < 0.0001, p = 0.0279, p = 0.0029, p = 0.0029, and p = 0.0288; respectively) (Figure 5A, left). However, the GAA enzyme activity levels never came close to that of non-treated Gaa+/+ mice, e.g., wild-type levels of ∼30% with GAAco, 18% with GILTco1-m (9 Gy), and less than ∼6% in the other therapeutic groups. In contrast, in the cerebrum only the GAAco group showed detectable GAA activity that was above non-specific signal in Gaa−/− controls (1.16 nmol/mg/h versus 0.36 nmol/mg/h; p = 0.0001, GAAco versus Gaa−/−, respectively) (Figure 5B, left). Mature GAA protein was predominantly observed in cerebrum, which was ∼6.7-fold higher in the GAAco group when compared with the GILT-tagged groups (Figures 5C and S6C).
Figure 5.

GILT-tag GAA required for resolving pathology in CNS: GAA enzyme activity in the cerebrum and cerebellum of Gaa−/− mice
(A and B) Left: GAA activity; right: glycogen. Values are for individual animals, represented as group medians and interquartile range. Exact Wilcoxon rank-sum p value comparing treatment groups versus Gaa+/+ or Gaa−/− or GAAco (n = 8–10). ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001. NT = non-treated. Bu = Busulfex. (C) Western blot analysis of cerebrum detecting mature GAA species (75–70 kDa). Loading control: GAPDH (n = 3). (D) PAS staining images of brain and spinal cord sections for glycogen (purple stain) depicted in non-treated versus treated groups. Top three panels: cerebral cortex, cerebellum, and spinal motor neurons. Cerebellum sections show the molecular and granular layers; arrow indicates Purkinje cells. Scale bars, 50 μm. Bottom panel: PAS staining of the cervical spinal cord regions, indicating the dorsal horn region (top right panel) and the ventral horn region (bottom right panel). Scale bars, 250 μm for high magnification and 50 μm for low magnification. (E) Neuronal/axonal vacuolation severity scores (n = 5 per group).
Despite the low GAA activity and protein content in cerebellum and cerebrum, glycogen was significantly reduced (e.g., <99% in GILTco-1-m and GILTco-3-m), except in the GAAco (p not significant, both 74% versus Gaa−/− (NT) groups) and GILTco1-m-ApoE1 (p not significant) groups, which were indistinguishable from Gaa−/− mice (Figures 5A and 5B, right), with the GILT-tagged variants showing superior performance, which was maintained with the R37A substitution. Furthermore, reduction in PAS staining intensity of the cerebral cortex, cerebellum, and cervical spinal cord was observed, including in spinal cord motor neurons (Figure 5D, for quantification see Figure S7 and Table S2). As a consequence of glycogen reduction, neuronal/axonal vacuolation was reduced in the GILT-tagged groups (Figure 5E). In addition, vascular vacuolation ranged from minimal to moderate in the Gaa−/− controls; however, in GILT-tagged treated mice vascular vacuolation was minimal to mild (Table S4).
To measure the efficacy of conditioning to promote engraftment of donor genetically modified cells in the brain of Gaa−/− mice, we utilized the GFP vector groups. Of all the GFP-positive mononuclear cells in the whole brain across all the groups that received GFP+ HSPCs (Busulfex group and both 7.5 Gy in Gaa−/− and Gaa+/+ mice) 53.3% ± 17.2% (n = 19) co-expressed the microglial marker Iba1, and these cells that showed colocalization exhibited a microglial morphology (Figure 6A). The extent of genetically modified microglia in the microglial population in the whole brain was on average similarly low across the groups (GFP [Bu] 0.91 ± 0.76, n = 5, GFP 1.87 ± 0.95, n = 7, Gaa+/+ GFP 2.5 ± 1.0, n = 7), although the Busulfex-treated group resulted in the lowest GFP+ percentages, as assessed by Iba1+/GFP+ cells within the Iba+ population, comparable with percentages in cortex and hippocampus (Figure 6B).
Figure 6.

Lentiviral vector expression in brain structures
(A) GFP-positive microglia in brain tissues. Representative images of regions of interest (ROIs) showing Iba1-positive microglia (yellow) with amoeboid to ramified morphology (higher magnification). ROIs included piriform cortex dentate gyrus, hypothalamic nucleus, adjacent to lateral ventricle, hypothalamus, reticular nucleus, and cortex. Gaa+/+ animals served as controls. Bu = Busulfex. (B) Percentage of GFP+ cells in the microglia (Iba1+) cells in the cortex, hippocampus, and total brain (n = 5–7). Non-engrafted mice with no GFP+ cells in peripheral blood leukocytes, bone marrow, and spleen were excluded from the analysis. Exact Wilcoxon rank-sum p value comparing treatment groups. NT = non-treated. ∗p < 0.05 (C) FISH colocalization of WPRE RNA (yellow) signal within the microglial cells (Iba1+, green immunofluorescence) in one gene-therapy-treated group. Top: Gaa−/− animals. Bottom: GILTco1-m. Representative ROIs include cortex (all groups), striatum (Gaa−/−), and dentate gyrus (GILTco1-m). (D) Percentage of WPRE+ cells in the Iba1+ microglial cell population for the cortex and hippocampus. Means ± SD are shown (n = 4–7).
Furthermore, colocalization of Iba1+ and WPRE-containing transcripts by FISH in multiple brain regions of GILTco1-m-treated mice showed rare positive cells in multiple brain regions CA2, CA3/CA4, corpus callosum, and dentate gyrus, although the presence of Iba1+/WPRE+ cells was more prominent in cortex and hippocampus. However, the signal was highly variable between animals (Figure 6C), e.g., percentages of Iba1+/WPRE+ cells were highest in the GILTco1-m (9 Gy) group (0.87% ± 1.07%; n = 7) and in hippocampus (2.96% ± 3.49%, n = 7) (Figure 6D), and lowest in the GILTco1-m (Bu) group (<0.3% on average). Total levels of Iba1+ cells in the cerebral cortex and hippocampus showed no significant difference between control Gaa−/−, Gaa+/+, or the 7.5 Gy, 9 Gy, or Busulfex GILTco1-m groups (Figure S9). These data showed that GILT-tagged GAA was required for robust restoration of CNS pathology.
GILT-R37A does not perturb insulin signaling, blood glucose, or hematopoiesis
The use of reveglucosidase alfa, an IGF2-tagged GAA analog, induced transient hypoglycemia at high-dose infusions in a proportion of treated LOPD patients.11 Hence, we developed a reporter assay to investigate the ability of the GILT-R37A-tag modification to induce insulin signaling (Figure S10). High rhGILT-R37A-GAA protein concentrations did not produce a reporter signal, as compared with strong responses detected by low concentrations of insulin, and 10- to 1,000-fold lower activation by IGF2 and GILT-GAAco protein incubation (Figure 7A). Additionally, if off-target effects such as insulin signaling occurred by application of the GILT-R37A tag, we would expect a reduction in blood glucose levels in mice treated with GILT-R37A-tag variant, and therefore increased group mortality. Nevertheless, blood glucose levels were similar across treatment groups and controls until the end of the study (Figure 7B, glycemia prior to termination; and weeks 5, 9, and 13 in Table S5). In addition, the overall survival in the GILT-tagged treatment groups (N = 110 mice) until week 16 was high. Only three mice were preterminally fated (one in the GILT.GAAco group on day 3, one in the GILT.R37A.GAAco on day 58, and one in the GILT.R37A.GAAco on day 103), and no mouse died in the GILTco3-m group which had the highest GAA enzyme activity and VCN/diploid genome. In the GFP transplanted control groups (N = 33), one animal was found dead on day 9. Taken together, these data show that there is no unusual sign or trend indicating that hypoglycemia has occurred in the treatment groups.
Figure 7.

GILT-R37A does not evoke an insulin signaling response
(A) RAT2 fibroblast insulin reporter cells were treated with 0.01 nM to 100 μM concentrations of insulin, IGF2, rhGILT-GAA, or rhGILT-R37A-GAA protein. Each data point is presented as mean (triplicate) ± SD. Results were normalized to untreated cells. (B) Glycemia and (C) and HbA1c blood levels in experimental mice prior to termination; group medians with interquartile ranges (dotted lines show normal ranges) (n = 9–13). NT = non-treated. Bu = Busulfex.
Other potential long-term blood glucose perturbations assessed by glycated hemoglobin levels (%HbA1c) were within normal range in therapeutic and controls groups, suggesting no obvious evidence of blood glucose dysregulation in the GILT-tagged GAA-treated mice (Figure 7C).
Finally, hematopoiesis was assessed in PB at monthly intervals and at termination. The erythroid and platelet, as well as myeloid and lymphoid populations, i.e., CD11b+Gr1+ myeloid cells, B220+ B lymphocytes, and CD3e+CD4+ and CD3e+CD8+ T lymphocyte populations were comparable between the different study groups and conditioning regimens over time (Figure S11 and Table S6, week 16). At termination, bone marrow and spleen populations were similar across study groups (Tables S7 and S8). Overall, this demonstrated that lentiviral HSPC gene therapy using GILT-R37A or other tag combinations did not perturb engraftment and differentiation of mouse HSPCs in Gaa−/− recipient mice. The supraphysiological GAA levels were well tolerated with no increased mortality. Only one animal in each of the GILTm, GILTco1-m, GFP, and GILTco treatment groups were found dead or preterminally euthanized, but these were isolated events unrelated to the vector constructs tested. The safety assessment of the R37A substitution confirmed the enhanced safety profile of the GILT-R37A tag with reduced insulin signaling in an in vitro assay, with no measurable deleterious effect on glucose levels or hematopoiesis.
Discussion
The SOC for Pompe disease patients is ERT with biweekly rhGAA infusions over the course of the patient’s lifetime, requiring high doses of 20 mg/kg body weight, which represents ∼10- to 20-fold higher than ERT requirements of Fabry and Gaucher disease.44,45 Furthermore, weekly ERT at 40 mg/kg is administered to selected IOPD Pompe disease patients to reach therapeutic efficacy.24,46 The clinical efficacy of the approved SOC is limited by suboptimal GAA enzyme uptake in target cells, poor receptor availability on target tissue, and inability of rhGAA to cross the BBB to address the neurological impairments. New therapies, including gene therapy, are currently being developed to address these limitations and to enhance delivery to the CNS and the skeletal muscles. Alternatively, in neurometabolic LSDs, such as MLD and Hurler syndrome, allogeneic stem cell transplantation may be applied, but this risks transplant-related complications and is dependent on endogenous expression of the lysosomal protein in hematopoietic cells, which may restrict effective cross-correction, emphasizing the need to improve clinical outcomes by boosting therapeutic protein levels.47,48
To this end, nine lentiviral vectors with a clinically relevant promoter driving the expression of engineered GILT-tagged GAA variants designed to improve efficacy in mouse HSPC-mediated gene therapy for Pompe disease were investigated in this study. Most of the variants investigated conferred supraphysiological expression of GAA enzyme activity and increased GAA secretion from hematopoietic cells in Gaa−/− mice, which resulted in glycogen reduction and resolved Pompe disease pathology in key target tissues including heart, skeletal muscle, and CNS of animals in the treatment groups. Using GILT-tag technology, high-abundance biphosphorylated glycans are not required for uptake in target tissues, and we showed that the GILT-R37A-GAA protein, devoid of the furin-cleavage site in the IGF2 moiety, was effectively taken up by cultured cells through its IGF2-binding site on IGF2R. We further interrogated the efficacy of tag technology incorporating additional tags, such as a tandem repeat ApoE peptide, which was fused to the N terminus upstream or downstream of the IGF2 moiety of the GAA sequence separated by a short GAP linker, aiming to enhance BBB crossing and CNS delivery in vivo in Gaa−/− mice. However, these sequence incorporations did not enhance CNS pharmacodynamics in vivo in our study, in contrast to results reported by Gleitz et al. showing that ApoE-tagged iduronate-2-sulfatase enzyme resulted in improved reduction of substrate and inflammation in CNS of Ids−/− mice.39 We suspect that positioning of IGF2, ApoE, and GAA peptide sequences affected effective production, at least for the ApoE1 construct, in our study or hampered the capacity of the ApoE-tagged proteins to dock to their cognate receptors,38 although this would require further investigation. Likewise, including a GAP linker in the protease 3 site of our therapeutic transgene,35,40 designed to boost the GAA enzymatic activity in GILTco1-m-L and GILTco1-m-ApoE2-L treatment groups, did not enhance biochemical correction. Further attempts were directed to recoding the transgene to enhance expression and protein translation, which can augment up to 10-fold higher transgene expression and production,49,50,51,52 and this could improve the therapeutic enzyme biodistribution. However, similarly to another study using codon-optimized GAA for AAV therapy,33 the codon-optimization algorithms investigated in our study did not dramatically enhance GILT-R37A-GAA protein production, which suggests that the GAA sequence may already be optimal for mammalian transcription and translation.
Among the variants tested, the GAAco construct consistently resulted in significantly higher GAA enzyme activity and protein in hematopoietic cells and tissues, but not in plasma, suggesting that the GAA protein seems less amenable to being secreted. This was confirmed by the relatively lower levels of precursor protein and the predominant presence of the mature GAA protein in the bone marrow of the GAAco group. This in part may explain the higher enzyme activities measured in the hematopoietic tissues of the GAAco group, especially because GAA maturation increases affinity for glycogen by 7- to 10-fold.35 In the transduced cells in the GAAco group, the GAA protein is expected to be preferentially targeted to the lysosome by the GAA signal peptide and retained within the cells, as opposed to targeted to the secretory pathway through the IGF2 signal peptide in the GILT-GAA groups. In addition, this may contribute to the higher enzyme activities and GAA protein measured in the tissues, since hematopoietic stem cell (HSC)-derived residual transduced hematopoietic cells and resident tissue macrophages may produce part of the GAA enzyme activity measured in the tissue homogenates. Furthermore, in the study by Maga et al.,10 a correlation was also not found between glycogen clearance and GAA enzyme activity when rhGAA and BMN 701 were compared. It was hypothesized that this could be due to preferential accumulation of GAA enzyme in capillary endothelial vasculature in muscle tissue, glycosylation deficits, lower affinity to the CI-M6PR, and autophagic buildup that affects lysosomal targeting to muscle fibers in Pompe disease, as discussed in the literature,53,54,55 which could hamper GAA protein being effectively taken up in muscle fibers.56 Owing to the high affinity IGF2 tag, GILT-tagged GAA may overcome these barriers more efficiently. As suggested previously for BMN 701,10 it may be that M6P on the GILT-tag protein is considerably lower than the levels on native GAA, which may have affected protein activity, as observed in plasma in our study, but not uptake and lysosomal targeting of GILT-GAA. Although we showed a change in relative distribution of GAA species in bone marrow cells, improved secretion, and lower GAA activity per protein in the plasma in the GILT-tagged groups, we did not find experimental evidence that factors, such as glycosylation deficits, autophagy or unproductive targeting to endothelial vasculature, played a role in the lower enzyme activities measured in the target tissues, although higher efficacy achieved in CNS and skeletal muscle using the GILT-tagged constructs, and this would require further investigation.
In our study, and consistent with prior publications which reported that heart and diaphragm typically respond better than skeletal muscle to treatment in both Gaa−/− mice and IOPD patients,31,57 almost all the chimeric constructs reduced the glycogen deposits to undetectable levels in the heart (>99%). Similar efficacy was observed in the diaphragm and gastrocnemius of the GILT-tagged GAA vector-treated Gaa−/− mice with near complete myofiber glycogen clearance. The quadriceps and tibialis anterior contained more residual glycogen at 16 weeks after transplantation of genetically modified cells, likely related to the different myofiber type and/or short duration of the follow-up period. Therefore, we postulate that improved efficacy may be obtained with the GILT-tagged vectors in long-term follow-up studies, similar to observations in AAV vector studies.33,34 Among the GILT-tagged GAA treatment groups, greater glycogen reduction was obtained in mice treated with GILTco3-m variant, likely explained by the modestly higher in vivo VCN/cell, because the GAA activity per protein was not higher in the GILTco3-m group and similar to the other GILT-tagged-containing groups. In addition to the reduction of glycogen, a reversal of myofiber and vascular pathology in the skeletal muscle was observed, with decreased vacuolation incidence and severity in all animals treated with GILT-tagged GAA compared with animals treated with the non-tagged GAAco construct. Hex4, an efficacy biomarker of ERT rhGAA,41,58 was significantly lower compared with pretreatment levels in urine in the treatment groups analyzed, confirming improvement of biochemical parameters monitored during our study.
In a clinical trial infusing 20 mg/kg reveglucosidase alfa (BMN 701) in LOPD patients, hypoglycemia was observed because IGF2 binds to the insulin receptor (IR), and particularly with higher affinity to the IR absence (IR-A) variant that lacks exon 11.11,59 Using an in vitro IR reporter assay, insulin signaling by GILT-R37A-GAA protein was remarkably undetectable. Our results align with prior works of others in which reduced binding affinity of the R37A mutein to the IR was reported in a competitive receptor binding assay.40
Moreover, hypoglycemia was not observed in vivo during multiple interim laboratory assessments in our study. Blood glucose levels were in the range of untreated Gaa−/− mice and treatment controls, as well as average blood glucose levels assessed by glycated hemoglobin A1c. In addition, treatment-related mortality in mice was not observed, which strongly accounts for the safety of GILT-R37A-GAA.
It is becoming more apparent that ERT cannot address CNS involvement of this neuromuscular disorder, which is an emerging key component in the Pompe disease phenotype, also contributing to respiratory dysfunction.11,60,61,62 The BBB limits the effectiveness of large-molecule therapeutics.63 Consequently, rhGAA does not cross the BBB effectively and results in significant motor neuron glycogen depositions in the ventral horn of the spinal cord, as observed in a long-term ERT-treated LOPD patient after autopsy.64 Prior non-clinical lentiviral HSPC gene therapy studies to deliver non-tagged GAA into the brain achieved suboptimal efficacy at clinically high VCN/cell using a known genotoxic promoter13,15 or with an erythroid-lineage-restricted physiological promoter.23 In our study, GAA enzyme activity in the GAAco group was at 11% and 30% of wild-type levels in cerebrum and cerebellum, respectively, but glycogen deposits were not reduced. Effective delivery of GAA protein necessitates M6P receptors for uptake and lysosomal trafficking, which decrease physiologically during adulthood.65 In addition, non-clinical studies using long-term transgene product exposure have shown beneficial effects on the neuronal compartment using AAV8,15,33 and increased effectivity of an IGF2-tag approach for CNS delivery via intralingual AAV9 injection.66 In our study, GILT-tagged GAA did show reduced glycogen accumulation in both neurons and glial cells in the cerebral cortex, corpus callosum, hippocampus, and cerebellum, and in brainstem and spinal cord. PAS staining revealed extensive glycogen reduction in the large neurons, especially in the motoneurons, similarly to wild-type mice. In contrast, histopathological analysis of the non-tagged GAAco-treated group failed to show CNS improvement. Surprisingly, the fluorometric 4-methylumbelliferyl-α-D-glucosidase (4-MU) GAA enzyme activity measured in brains of the GILT-tagged groups was below detectable levels. Subsequently, western blot protein quantification on cerebral homogenates was performed to overcome the low assay sensitivity, which revealed clear presence of GILT-tagged GAA protein averaging 5%–30% of the levels in the GAAco group, sufficient to reduce glycogen deposits and vacuolization in the brain.
We propose that the mechanism of action in the brain is local production of GAA by genetically modified microglia, derived from HSPCs that migrate and differentiate in the CNS. Microglia constitute 5%–12% of rodent brain and can be identified by Iba1+ staining.67 In our study, we examined colocalization of GFP with the microglial Iba1+ marker to estimate the frequency of donor-derived transduced microglia in mice, and found that 0.1%–3.7% of the total Iba1+ cells were GFP+ regardless the conditioning regimen applied to the Gaa−/− mice. More importantly, WPRE FISH confirmed the presence of genetically modified Iba1+ cells in the brain in the GILTco1-m group, and consequently local secretion and uptake of GILT-tagged GAA could explain the lysosomal glycogen reduction observed in the CNS. Based on these results, we conclude that low brain donor chimerism, even at ∼1% of genetically modified cells, can produce sufficient enzyme to reduce glycogen deposits and mitigate the Pompe pathology in Gaa−/− mice. This may also explain that with the conventional 4-MU assay, GAA enzyme activity levels were below the limit of detection. Literature review shows that in non-clinical MLD and Hunter syndrome studies, a broad range of enzyme activity results in substrate reduction in the CNS but that enzyme activity is typically lower than endogenous expression in other tissues.39,68,69 In our study, CNS GAA enzyme activity and transgene product levels did not directly correlate with glycogen reduction because the native GAA was insufficient to alleviate tissue pathology of Pompe disease. Under healthy conditions the CNS contains a low amount of glycogen, mainly located in the astrocytes that support several key physiological processes.70 In Pompe disease patients glycogen accumulates in neurons and glial cells, such as astrocytes.60 In studies of mucopolysaccharidosis type IIIB (Sanfilippo type B syndrome) using mouse primary astrocytes and cortical neurons, recombinant human α-N-acetylglucosaminidase (rhNAGLU) was less effective than IGF2-tagged NAGLU (BMN 250) in breaking down heparan sulfate.71,72 The improved uptake by these specific cell types at similar concentrations of rhNAGLU and BMN 250 were IGF2 receptor dependent, in contrast to microglia, in which IGF2 receptor-independent mechanisms also provided uptake of rhNAGLU. This mechanism of action may explain the observed efficient glycogen reduction using GILT-tag technology in treated Pompe mice in our study.
There have also been investigational efforts to test AAV gene therapy in preclinical models in Gaa−/− mice using AAV1, AAV8, and AAV9 serotypes targeting liver, muscle, or CNS, reviewed by Salabarria et al. and Unnisa et al.73,74 In an AAV-mediated gene therapy study, replacing the endogenous GAA signal peptide with a signal peptide of a secreted protein enhanced excretion from hepatocytes, thereby improving cross-correction to target tissues, particularly skeletal muscles.33,41 In another study, an antibody domain fused to GAA targeting CD63 was effective in delivery through ERT and liver-directed AAV gene therapy to skeletal muscles as well.75 Besides using the liver as a depot for effective correction of skeletal muscle,76 muscle-directed AAV gene therapy has also shown efficacious effects.77 Importantly, in a clinical trial directing AAV1 serotype to the diaphragm, gene therapy led to an increase in subject tolerance for duration of unassisted breathing,12 showing potential for AAV gene therapy to treat Pompe disease. However, the long-term efficacy can be hampered by the complexity of immune responses to the transgene products or CD8+ T cell immune responses to the AAV vector capsid.78 More recently, the use of AAV vectors with restricted expression to hepatocytes induced immune tolerance against the transgene product and the rhGAA.32,41,79,80 Immunogenicity to rhGAA has previously been investigated in Gaa−/− mice in HSPC gene therapy settings.13,14,30 In our 16-week follow-up study with stabilized hematopoietic reconstitution of genetically modified cells in Gaa−/− mice, the GAA enzyme activity in PB leukocytes and plasma was supraphysiological and sustained without evidence of immune responses to the recombinant proteins in the treatment groups, suggesting the occurrence of advantageous immune tolerance to the transgene product.
The genotoxic risk in HSPC gene therapy is likely multifactorial and carefully monitored in current human studies.81,82 Extensive literature discusses contributions of the vector type/design, promoter and splice signals, disease background, transgene function, bone marrow inflammation, or proliferative stress.81,83,84,85 To provide robust expression of our chimeric GAA coding sequences, the MND promoter was incorporated in our third-generation self-inactivating lentiviral vector backbone aiming to achieve high-level transgene product expression in CNS tissues of Gaa−/− mice. There are clinical trials using the MND promoter to drive the expression of therapeutic transgenes with an acceptable safety profile.17,20,86 However, for future HSC gene therapy application in Pompe disease patients this will require further risk-benefit evaluation.
Finally, in our study we used sublethal conditioning, leading to a partial chimerism thereby leaving the residual host compartment intact to ensure survival. A substantial number of mice (four out of nine) in the Busulfex conditioning treatment group had failure to engraft, likely due to the sex-mismatched model of transplantation utilized in our study.87 Also, the 9 Gy treatment group had higher donor cell chimerism levels which, however, did not lead to an obvious improved therapeutic effect. Overall, donor cell engraftment was consistent in the therapeutic and control groups, hence it did not influence efficacy. We anticipate that the translation to clinical protocols will require careful evaluation of the conditioning regimens for Pompe disease patients to balance optimal efficacy and subject safety. Careful therapeutic drug monitoring to provide adequate space in the bone marrow for successful engraftment of genetically modified cells while minimizing risks of conditioning-related morbidities is required in clinical studies.88,89 Recently, non-toxic conditioning drugs such as humanized antibodies against HSPC receptors or mobilization-based chemotherapy-free engraftment have been developed to vacate niches and create space and engraftment in bone marrow, and are being intensively investigated in non-clinical and clinical trials (ClinicalTrial.Gov ID: NCT04429191).90,91,92 Furthermore, in vivo HSC transduction infusing vector directly in the bone marrow niche are rapidly progressing93 as well as the use of G-CSF1R inhibitor drugs to make space for genetically modified HSC-derived microglia in the brain.94,95
In summary, our results showed robust expression and secretion using GILT-tagged GAA transgenes, achieving supraphysiological and sustainable GAA enzyme activity levels in vivo in Gaa−/− mice. We presented in vitro and in vivo evidence that the GILT-tag R37A-GAA, i.e., GILTco1-m and GILTco-3-m, clearly emerged as more suitable chimeric sequence variants that displayed robust reduction of skeletal muscle and CNS glycogen accumulation and tissue pathology in Gaa−/− mice, more so than the native GAA (GAAco), which completely lacked CNS correction. The mechanism of action of native GAA versus GILT-tag GAA requires further investigation as to whether secretion, uptake, or other mechanisms improved biodistribution in key tissues and subsequently the performance of the GILT-tag GAA in skeletal muscle and CNS. The GILTco1-m and GILTco-3-m exhibited performance similar to that of non-mutated GILT tag (GILTco) but with the advantage of the R37A safety feature which minimizes risks of hypoglycemia via IR signaling. The codon-optimization approach could still be beneficial to reduce sequence similarity with native human GAA sequence and removing cryptic splice sites to improve vector safety, despite it failing to provide overly translational advantages in our study. Investigation of ApoE-tag and GAP-linker additions were demonstrated to be ineffective in vivo, since the former consistently hampered GAA enzyme activity particularly when positioned upstream of the GILT tag, and the linker failed to confer enhanced substrate reduction. Consequently, simplified transgene configurations are preferred in the absence of additional benefit. No evidence of immunogenicity of the transgene product was detected in Gaa−/− mice during the course of the study. From a safety perspective, keeping VCN low is preferred; however, GILTco3-m performed apparently better with a higher VCN/cell. Altogether, our data indicate that lentiviral vectors containing an IGF2 tag with R37A substitution (GILTco1-m and GILTco3-m) are considered lead candidate vectors and warrant further long-term efficacy and safety preclinical investigation for application in first-in-human lentiviral HSPC gene therapy for Pompe disease.
Materials and methods
Plasmid construction and lentiviral vector production
Human codon-optimized GAA (GAAco), and GILT-tagged chimeric GAA coding sequence variants GILTco, GILTco1-m, GILTco-m-ApoE1, GILTco-m-ApoE2, GILTco1-m-L, GILTco1-m-ApoE1-L, and GILTco1-m-ApoE2-L were designed using GenSmart Codon Optimization Tool (GenScript). Human GAA coding sequence variant GILTco2-m was designed using the GeneArt codon-optimization algorithm (Thermo Fisher Scientific). The human GAA coding sequence variant GILTco3-m was designed using a consensus sequence (CCDS32760.1). GILTm contained the native nucleotide sequences. All GAA coding sequence variants were synthesized and cloned into a lentiviral vector backbone described previously.96 The EFS promoter from this construct was replaced by the myeloproliferative sarcoma virus enhancer, negative control region deleted, dl587rev primer-binding site substituted (MND) promoter.97 Plasmids were fully verified by Sanger sequencing and produced at Aldevron.
All third-generation self-inactivating lentiviral vectors described above were produced by transient transfection of 293T cells using packaging and transfer plasmids at the Viral Vector Core of Cincinnati Children’s Hospital. HOS cells used to determine vector titers were acquired from ATCC (cat. #CRL-1543). Titers determined on HOS cells by qPCR were in the range of 2.25 × 107 to 6.67 × 107 transducing units per mL and were subsequently titrated on lineage-negative bone marrow cells to determine the multiplicity of infection (MOI) needed to achieve a VCN of ∼4.
In vitro experiments for secretion and uptake of chimeric proteins
The vector configurations were tested in HAP1-GAA−/− cells for protein production and secretion and K562 cell lines were generated to assess uptake by the GILT tag. HAP1 GAA−/− cells (Horizon Discovery, ID: HZGHC004204c004, parental ID: C631) were transduced with 11 lentiviral vectors encoding GAA variants (Figure S1B) and GFP at an MOI of 3. Conditioned media of vector transduced cells and cell pellets were collected at day 11 for VCN normalized GAA enzyme activity measurement.
Recombinant human GILT-R37A-GAA protein was produced by transfection of HD Chinese hamster ovary cells (GenScript). Cell-culture supernatant was centrifuged at 5,000 × g for 5 min, followed by filtration through a 0.2-μm Supor-Mach filter (Nalgene), and loaded onto a Phenyl-Sepharose 6 Low-Sub Fast-Flow (GE Healthcare) column prepared with HIC Load Buffer (50 mM citrate [pH 6.0], 1M AMSO4). Samples from the elution peaks were pooled, and buffer was exchanged into PBS using centricon spin concentrators (Amicon) and Bio-Spin-6 desalting columns (Bio-Rad). Purified protein was analyzed by SDS-PAGE, western blot analysis, and Bradford assay to determine concentration, molecular weight, and purity.
K562 cells (ATCC), K562 cells GAA−/− clone 20, GAA−/−IGF2R−/− clone 25, GAA−/−IGF2R−/− clone 25, and lentiviral vector IGF2R (clone 51) were used for uptake assays. All knockout cell lines were generated by CRISPR-Cas9 gene editing using ribonucleoprotein complexes (Figure S3). Knockout efficiency was assessed with surveyor assay, and single cells were sorted, expanded, and screened for IGF2R protein by anti-IGF2R antibody (BioLegend) by flow cytometry.
K562 cell lines were incubated with concentrations of 125, 31.3, 7.8, and 2.0 nM purified rhGAA, rhGILT-GAA, and rhGILT-R37A-GAA proteins at 37°C and 5% CO2 for 18 h. Subsequently, cells were pelleted, washed three times with Dulbecco’s PBS (DPBS), and lysed with 100 μL of PC-T lysis buffer. After removal of debris by centrifugation at 10,000 × g for 10 min, duplicate lysate samples were used to measure GAA activity with 4-methylumbelliferyl (4MU) normalized to protein concentration with bicinchoninic acid protein (BCA) assay (Pierce).
Healthy and Pompe-affected human fibroblast cells were obtained through the Coriell Institute (cat. #GM07525 and #GM00244, respectively). Fibroblast cells were seeded and grown for 24 h to approximately 90% confluency. Cell medium was replaced with medium containing 50 nM purified rhGAA, rhGILT-GAA, and rhGILT-R37A-GAA proteins and incubated at 37°C and 5% CO2 for 18 h. Select wells also contained inhibitors M6P (1 nM to 10 mM; Calbiochem) or IGF2 (5e−2 pM to 5e2 nM; Cell Sciences). Subsequently, cells were trypsinized, pelleted, washed three times with DPBS, and run in a GAA activity assay.
Insulin reporter assay
A cellular reporter assay to monitor IR kinase activity based on signal transducer and activator of transcription 5B (STAT5b)-dependent luciferase gene expression, similar to previously reported,98 was generated to assess insulin signaling by the IGF2 and IGF2-R37A moieties. RAT2 fibroblast cells (ATCC) were genetically modified with a lentiviral vector to express human insulin receptor A (IR-A) isoform, and STAT5b and mCherry (VectorBuilder). Cells were sorted for IR-A and mCherry, and subsequently transduced with a puromycin-selectable STAT5 response element luciferase reporter lentiviral vector (G&P Bioscience). Following puromycin selection, the RAT2 reporter cell line was plated at a concentration of 100,000 cells/well in a 96-well plate in Dulbecco’s modified Eagle’s medium, 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. After 12 h the medium was replaced, supplemented with 0.01-nM to 100-μM concentrations of insulin, IGF2, or GILT-GAA or GILT-R37A-GAA protein. Following 8 h of activation with the supplemented medium, One-Glo Luciferase Assay reagent was added to the wells. Following 10 min of lysis, lysates were transferred to a white-walled 96-well assay plate for measurement on the Softmax i3x plate reader. Results were normalized by dividing each treated well value by the mean of the untreated values.
Mice
Gaatm1Rabn/J mice (Gaa−/− mice, Pompe mice) were used for the study.43 Mice were obtained from an on-site breeding colony (kindly provided by Dr. Nina Raben, NIH, Bethesda, MD). Control B6129SF1/J mice (Gaa+/+ mice, wild-type mice) were obtained from The Jackson Laboratories (stock no. 101043). All mice were maintained in clean rooms and fed with irradiated certified commercial chow and sterile acidified water ad libitum. Assessment of animal health status, body weight, and examinations during the in-life term of the study were conducted by veterinarian personnel and documented. All protocols were approved by the Institutional Animal Use and Care Committee at the Canadian Council on Animal Care.
Assessment of dosing formulations and infusion in conditioned Gaa−/− mice
After in vitro assessment, nine GILT-tagged GAA containing lentiviral vectors were selected for in vivo testing in female B6; 129-Gaatm1Rabn/J [6neo] (Gaa knockout; Gaa−/−) mice. After sublethal conditioning the mice with 7.5 Gy or 9 Gy γ-irradiation (1.6 Gy/min) or by 4 × 25 mg/kg Busulfex (Otsuka Pharmaceutical) injections days −4 to −1 before cell dosing, 6- to 9-week-old mice were injected intravenously with enriched male donor Lin− HSPCs transduced with engineered GAA vectors encoding for native or chimeric GAA proteins. Female Gaa−/− and Gaa+/+ mice GFP vector and non-transduced control groups were included. Mice were monitored for 16 weeks with interim blood collections. Bone marrow cells were harvested from femurs and tibias of 6- to 12-week-old male Gaa−/− donor mice, and Lin− enriched for HSPCs using RoboSep (STEMCELL Technologies). After enrichment, cells were transduced overnight with ten lentiviral vectors (Figure S1) at a density of 106 cells/mL (MOI of 4) in serum-free StemMACS medium containing 100 ng/mL recombinant murine stem cell factor (SCF); 50 ng/mL recombinant human FMS-like tyrosine kinase 3 ligand (Flt-3), and 10 ng/mL recombinant human thrombopoietin (STEMCELL Technologies). On the following day the cells were washed, counted, and assessed for viability. A 3 × 103 cell fraction of non-transduced and transduced Lin− cells were transferred into 3 mL of methylcellulose-based medium with mouse cytokines optimized for hematopoietic progenitor cell differentiation (STEMCELL Technologies) and plated at 1 mL/well for a CFU assay. CFUs were scored using STEMvision (STEMCELL Technologies) at day 7.
Preconditioned female Gaa−/− recipients were injected intravenously with 0.5 × 106 cells per mouse. At four interim time points after cell infusion, leukocyte and plasma GAA enzyme activity, blood glucose, and complete blood counts using a Hemavet (Drew Scientific) analyzer were measured.
Analysis of glucotetrasaccharide Hex4 in urine samples via liquid chromatography-tandem mass spectrometry
Mice were fasted before urine collection for glucotetrasaccharide Hex4. The quantification of 6-α-D-glucopyranosyl maltotriose (Hex4) was conducted in a Shimadzu (Columbia, MD) instrument via protein precipitation using acetonitrile and 4 mM uric acid with 0.2% NH4OH as surrogate matrix. Calibration standards and quality controls were run in surrogate matrix and mouse urine matrix in duplicate. Results were normalized by measurement of creatinine in the urine sample using a commercial kit.
Blood glucose and glycated hemoglobin monitoring
Blood glucose was measured at different time points during the in-life study and prior to scheduled termination with glucometer Accu-chek AVIVA. Animals were fasted overnight prior to collections, with results reported as nmol/L (standard Canadian units) and converted to mg/dL by multiplying by a factor of 8. Similarly, glycated hemoglobin (HbA1c) was measured using A1cNow (pts Diagnostics, Indianapolis, IN), with results reported in %HbA1c. Quality controls of the A1cNow kit were performed with NOVA-ONE kit levels 1 and 2 (NOVA-ONE Diagnostics, Calabazas, CA).
Flow cytometry
PB, bone marrow, thymus, and spleen cells underwent red blood cell lysis and were stained with monoclonal antibodies against surface markers CD45.1, CD45.2, CD3e, CD4, CD8a, B220, CD11b, Gr-1 (Ly6G and Ly6C), TER119, and CD41, or Sca-1c-Kit, and a hematopoietic lineage cocktail (BD Biosciences, BioLegend) and analyzed on a FACSLyric flow cytometer using Fixable Viability Dye eFluor 506 (FVD506) (Invitrogen) for dead cell exclusion.
Scheduled termination procedure
Scheduled termination was week 16 post transplant. Animals were fasted 8–12 h prior to euthanasia and anesthetized with isoflurane. After cardiac puncture blood collection, the animals were perfused with 1× PBS (pH 7.4) until internal organs were pale in appearance. The heart mass was weighed, and the dissected tissues were snap frozen and stored at −80°C for GAA enzyme activity and glycogen measurement or processed for histopathology. Therapeutic endpoints included biochemical GAA enzyme activity, tissue glycogen content, western blotting analysis, VCN analysis, histological evaluation, blood glucose, glycated HbA1c, clinical pathology, immunophenotyping via flow cytometry, and immunofluorescence and FISH analysis of gene-modified cells in brain sections.
Measurement of GAA enzyme activity and glycogen by biochemical analysis
For GAA enzyme activity and glycogen measurement, tissue samples of heart, diaphragm, gastrocnemius, quadriceps femoris, tibialis anterior, cerebellum, and cerebrum collected at necropsy were processed by chilling to homogenization in sterile dH2O, centrifugation, and removal of clear supernatants, then stored at −80°C until the assay day. Similarly, terminal plasma and cell pellets from PB, spleen, and bone marrow samples were collected and kept at −80°C for GAA enzyme activity.
The GAA enzyme activity was measured similarly as described previously.99,100 Study samples were assayed in a 96-well plate using fluorescent synthetic substrate 4-MU at 6 mM concentration, in the presence of 9 μM acarbose and 90 min of incubation. A 0.5 M carbonate buffer (pH 10.7) was used to stop the reaction, and assay plates were read at 365 nm excitation and 450 nm emission in a SpectraMax i3X (Molecular Devices, San Jose, CA). This method was qualified over the range of 0.1–81 nmol/mL. Results were normalized for protein concentration in the sample using a BCA kit (Pierce, Thermo Fisher Scientific).
The glycogen in tissues was estimated by treating samples with and without Aspergillus niger amyloglucosidase that generates β-D-glucose, which is oxidized to release gluconic acid and hydrogen peroxide.13 In the presence of horseradish peroxidase (HRP), hydrogen peroxide reacts with o-dianisidine hydrochloride to generate a colored product detectable at 540 nm in a spectrophotometer and is proportional to the glucose concentration in the sample. The qualified assay had a dynamic range of 5.6–160 μg/mL. Results were normalized for protein concentration in the sample using a BCA kit (Pierce, Thermo Fisher Scientific).
VCN analysis and donor cell chimerism
Lin− cell dosing formulations cultured until day 7 and bone marrow samples collected at termination were processed for gDNA and quantified with Quant-iT assay kit or Nanodrop One. This qPCR assay consisted of oligonucleotide primers and probe mix containing either a TaqMan 6-carboxyfluorescenin (FAM) or VIC fluorescent probe designed to amplify HIV Psi vector and Gtdc1 housekeeping gene sequences, which has been previously reported (Table S9). A plasmid containing both sequences was used as a reference standard in a range of 50 to 5 × 107 copies. Data were reported as VCN/diploid genome.
Comparative male donor cell chimerism was determined by PCR on bone marrow gDNA using Y chromosome gene Zfy1 and Bcl2 gene to normalize the total gDNA input per reaction following An and Kang101 (Table S9). Donor-derived male cells engraftment in female recipient mice was calculated using a gDNA standard curve with known percentage of male versus female gDNA.
Western blot analysis
Monoclonal antibodies were generated by immunizing rabbits or mice with GAA or GILT-tag-containing peptides (GenScript and Thermo Fisher Scientific). Plasma was collected to assess antibody development by ELISA. Spleens of selected rabbits and mice were further processed to create hybridomas for monoclonal production of antibodies. Mouse plasma samples were screened for the presence of GAA protein using Jess ProteinSimple and following the manufacturer’s protocol (ProteinSimple, Bio-Techne Brand). The sensitivity of the method is on the order of picograms. Samples were denatured at 95°C before analysis. For tissues, roughly 200 ng of cell lysate was loaded per well, calculated based on the results of a BCA kit (Pierce, Thermo Fisher Scientific). Mouse antibody clone 1C12C11.F9 (Thermo Fisher Scientific) was used as detection antibody for GAA, and rabbit antibody clone 7C10 was used as detection antibody for the IGF2 amino acid sequence, i.e., GILT-tag-containing precursor protein. Dilutions of rhGILT-GAA protein were included either as a positive control (tissues) or standard curve (plasma) for all assay runs. A monoclonal anti-β-actin antibody (ab8224; Abcam) or rabbit anti-GAPDH antibody (clone 14C10; Cell Signaling Technologies) were used as controls. The products were visualized via anti-mouse or anti-rabbit secondary antibodies conjugated to HRP (ProteinSimple 042-205 and 042-206, respectively). All Jess detection was performed as single-plex reactions in the chemiluminescent channel. Relative GAA quantification of tissues was calculated using the loading control and reported as a percentage in reference to GAAco. GAA absolute quantification in the plasma was determined by interpolating values using a rhGILT-GAA protein standard curve, multiplying by the dilution factor and adjusted per milliliter of plasma.
PAS and H&E staining of mouse tissues
Following scheduled necropsy, tissues from Gaa−/− and Gaa+/+ mice were divided for different endpoint analyses, and a portion of each was collected, preserved, trimmed, and placed in separate cassettes. Tissues were fixed in 10% neutral buffered formalin (NBF) for up to 32 h and post-fixed in 1% periodic acid (PA)/10% NBF for 48 h at 4°C. One hemisphere from each brain sample along with heart, diaphragm, tibialis anterior, gastrocnemius, and quadriceps femoris tissues were fixed in 10% NBF for up to 32 h and further post-fixed in 1% PA/10% NBF for 48 h at 4°C and processed into formalin-fixed paraffin-embedded (FFPE) blocks. Blocks of cerebral cortex, cerebellum, hippocampus, and/or brainstem, thoracic and cervical spinal cord, heart, quadriceps femoris, diaphragm, gastrocnemius, and tibialis anterior were sectioned at 4 μm, mounted onto glass slides, and stained with PAS and H&E, and evaluated for glycogen accumulation and vacuolation by light microscopy. Tissues stained via PAS/H&E protocols were scanned at 20× magnification using a Hamamatsu NanoZoomer whole-slide scanner. Scan files were imported into Visiopharm for quantitative image analysis. A region of interest (ROI) surrounding individual tissue sections was automatically applied to all scan files using a Visiopharm Analysis Protocol Package (APP). Automated ROIs were manually refined to optimize anatomic homology across animals. Automated image analysis APPs were used to detect the dark purple PAS+ signal contrasted against a pink PAS− background. The following equations were performed in Visiopharm to generate quantitative endpoints: , , and . Quantitative immunofluorescent data were exported from Visiopharm as Excel spreadsheets. For severity scoring of vacuolation, score was assigned as minimally (score 1) affected tissues having <50% of cells within the section with small discrete centralized regions of cytoplasmic vacuolation involving <10% of the cytoplasmic volume; mildly (score 2) affected tissues having larger regions of vacuolation involving ≥10% of the cytoplasmic volume affecting <50% of cells within the section and none to rare myofibers that were diffusely enlarged with overall decreased cytoplasmic staining intensity; moderately (score 3) affected tissues having regions of cytoplasmic vacuolation involving >10% of cells with >50% of myofibers showing evidence of myofiber degeneration characterized by enlargement of myofibers and overall decreased staining intensity; and markedly (score 4) affected tissues having overall enlargement and decreased staining intensity of the majority of myofibers with both centralized regions of cytoplasmic vacuolation and evidence of myofiber degeneration.
Immunofluorescence and vector RNAscope imaging of brain
The brain tissue sections were processed into three levels (forebrain, midbrain, and hindbrain) following necropsy. Spleen and brain tissues were fixed fresh in 10% NBF for 16–32 h at room temperature and then processed into FFPE blocks to be used for immunofluorescence detection of GFP with rabbit polyclonal anti-GFP antibody (ab290, Abcam) or rabbit monoclonal anti-Iba1 antibody (ab178846, Abcam). For Iba1 antigen, an immunofluorescence assay was used with detection reagents and fluorescent kits DISCOVERY OmniMap anti-Rb HRP (RUO) (Ventana) and DISCOVERY FAM Kit (Ventana), and for GFP antigen DISCOVERY OmniMap anti-Rb HRP (RUO) (Ventana) and DISCOVERY Rhodamine Kit (RUO) (Ventana). For the detection of lentiviral vector sequences within the tissue, an untranslated mRNA sequence of WPRE was used as a probe in fluorescent RNAscope in situ hybridization assay. The detection was validated by using Fluorescent RNAscope VS Universal HRP Assay using RNAscope 2.5 VS Probe-WPRE-O3 (ACD, reference no. 518629) with number of probe pairs = 10, and with detection reagents and fluorescent kits RNAscope VS Universal HRP Reagent Kit (ACD) and DISCOVERY Red610 Kit (RUO) (Ventana) on a Ventana Discovery ULTRA machine (Roche). Fluorescein isothiocyanate (FITC) and tetramethylrhodamine isothiocyanate (TRTC) channels were used to measure fluorescent signals, and 4′,6-diamidino-2-phenylindole (DAPI) was used as a nuclear stain. The images were scanned on a Hamamatsu NanoZoomer whole-slide scanner. Brain tissues were scanned at 40× magnification, and scans were imported into the Visiopharm software system. ROIs for the GFP quantifications were drawn around cerebral cortex, the hippocampus, or including all regions of each sample. The first Visiopharm configuration was designed to detect individual cells by nuclear DAPI stain. The second configuration was designed to classify each cell as Iba−, Iba+ or GFP+, or dual positive. Iba1+GFP+, Iba+GFP−, Iba1−GFP+, Iba−GFP−, and surface area were quantified. Mice that did not have peripheral engraftment in PB, bone marrow, and spleen based on the lack of flow-cytometric presence of GFP+ cells were excluded from the analysis. A similar analysis was performed for the FISH for WPRE-positive cells and colocalization with Iba1+ in brain, with ROIs CA2, CA3/4, corpus callosum, cortex, dentate gyrus, and hippocampus. Standard inclusion/exclusion criteria were applied to all tissue sections during the manual refinement processes. Each ROI was manually refined to exclude meninges and histological artifacts. Only coherent and clearly defined ROIs were included whereas regions containing visible folds, cracks, and bubbles were excluded. Tissue sections where the ROI could not be identified within the sample profile were excluded from image analysis. The optimal threshold for each fluorescent channel (FITC/TRTC) was calculated for each region using a modified Otsu’s method for stable, unsupervised, mathematical approach to calculating the optimal threshold for segmenting background and foreground pixels with single-channel intensity values.102 A modified approach for Otsu’s method uses the median absolute deviation to calculate the optimal threshold from highly skewed, unimodal distributions.103 This mathematical inference is used to determine the threshold value where the foreground and background are most dissimilar without a priori knowledge of the object of interest. In brief, median values for foreground and background pixel intensities are used to calculate the threshold at which the two populations display the greatest degree of variation.
Statistical analysis
Experimental groups were sized to allow for statistical analysis; not all the animals were included in the analysis, and select outliers were excluded. Mice were assigned randomly to the experimental groups based on weights. GAA enzyme activity, glycogen, and Hex4 measurement were analyzed by VERISTAT applying median and interquartile range using Kruskal-Wallis and exact Wilcoxon rank-sum tests for groups comparison and SAS/STAT software v9.4. A result of <0.05 was indicative of significant difference in the groups. For correlation analysis, the Pearson R coefficient and p value were used.
Quantitative immunofluorescent data were collected in Visiopharm, and statistical analysis was performed using SAS software (v9.4). Continuous variables were analyzed via Levene’s test for equality of variance. Where Levene’s test was not significant, one-way ANOVA was used to detect differences among three more group means. Tukey’s test was used to explore pairwise group comparisons when one-way ANOVA was significant. Where Levene’s test was significant, the Kruskal-Wallis test was used to detect differences between three or more group means. The Dwass-Steel-Critchlow-Fligner method was used to explore pairwise group comparisons when the Kruskal-Wallis test was significant. Significance was set to p < 0.05 for all statistical tests. In the figures, asterisks indicate ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
Acknowledgments
We thank all the members of AVROBIO for their continued support of our work. In particular, we would like to thank Maurine Braun, Bianling Liu, Robert Plasschaert, Mark DeAndrade, Claudia Fiorini, Vicky Chen, Tara Peterson, Daniella Pizzurro, Nicole Del Signore, Haydy George Leha, Maria Grigorova, Steven Tyler, Becky Reese, and Carolina Romano for scientific support and assistance. We would like to also thank Jon Lebowitz (BioMarin Pharmaceutical) for providing recombinant human GILT-GAA protein (BMN 701) and valuable input for the project. We would like to thank Charles River Laboratories, including Maia Araujo Abrahim, Karen Wong, Romain Genard, Lauriane Padet, Anne Larrivée, Camille Laure-Pittet, Simon Authier, and Daphne Gordon for study execution and diligent care of study animals. Additionally, we thank Li Na, Kyle Takayama, and Lyn Wancket (Charles River Laboratories), Jennifer Dannehl, Leonard Jared, Rathna Veeramachaneni, and Dawn Dufield (KCAS Bioanalytical & Biomarker Services) for their contributions to sample analysis, and Douglas Arbetter (VERISTAT) for biostatistical analysis. Thanks go to Allison Cole for creating the graphical abstract, which was created by Biorender.com. Finally, we would like to thank Nina Raben (National Institutes of Health) for supplying the Gaa knockout mice.
Author contributions
Y.D., C.N.B., J.K.Y., and J.W.S. contributed to the experimental design and execution, biochemical and molecular assays development and qualification, data analysis and interpretation, and wrote the manuscript; Z.U., S.G., and M.E.J. conducted experiments and performed data analysis; D.L.C. organized production of lentiviral vectors; A.S. contributed to vector, cDNA, and experimental design; C.O., R.P., and C.H. contributed to assay development and study logistics; T.M. performed statistical analysis; C.M. contributed to the design and the reporting of all work presented; N.P.v.T. designed the study plan and vector configurations, performed data interpretation, and wrote the manuscript.
Declaration of interests
All authors are current or former employees of AVROBIO, Inc., Cambridge, MA, USA during the conception and writing of the manuscript, except A.S. AVROBIO, Inc. has a preclinical gene therapy program for Pompe disease (AVR-RD-03) based on a genetically modified HSPC platform using lentiviral vectors. Collection of data and analysis was performed as part of the program. This research received no external funding and was sponsored by AVROBIO, Inc.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2022.10.017.
Contributor Information
Chris Mason, Email: chris.mason@ucl.ac.uk.
Niek P. van Til, Email: n.p.vantil@amsterdamumc.nl.
Supplemental information
Data availability
Data supporting the studies presented in this paper can be found in the main text or the supplemental information. Additional information, as appropriate, may be made available by request directed to the corresponding author and, as appropriate, following execution of a suitable confidentiality agreement with AVROBIO, Inc.
References
- 1.van der Ploeg A.T., Reuser A.J. Pompe's disease. Lancet. 2008;372:1342–1353. doi: 10.1016/S0140-6736(08)61555-X. [DOI] [PubMed] [Google Scholar]
- 2.Elmallah M.K., Falk D.J., Nayak S., Federico R.A., Sandhu M.S., Poirier A., Byrne B.J., Fuller D.D. Sustained correction of motoneuron histopathology following intramuscular delivery of AAV in pompe mice. Mol. Ther. 2014;22:702–712. doi: 10.1038/mt.2013.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bijvoet A.G., Van Hirtum H., Vermey M., Van Leenen D., Van Der Ploeg A.T., Mooi W.J., Reuser A.J. Pathological features of glycogen storage disease type II highlighted in the knockout mouse model. J. Pathol. 1999;189:416–424. doi: 10.1002/(SICI)1096-9896(199911)189:3<416::AID-PATH445>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 4.Van den Hout H., Reuser A.J., Vulto A.G., Loonen M.C., Cromme-Dijkhuis A., Van der Ploeg A.T. Recombinant human alpha-glucosidase from rabbit milk in Pompe patients. Lancet. 2000;356:397–398. doi: 10.1016/s0140-6736(00)02533-2. [DOI] [PubMed] [Google Scholar]
- 5.Do H.V., Khanna R., Gotschall R. Challenges in treating Pompe disease: an industry perspective. Ann. Transl. Med. 2019;7:291. doi: 10.21037/atm.2019.04.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pena L.D.M., Barohn R.J., Byrne B.J., Desnuelle C., Goker-Alpan O., Ladha S., Laforêt P., Mengel K.E., Pestronk A., Pouget J., et al. Safety, tolerability, pharmacokinetics, pharmacodynamics, and exploratory efficacy of the novel enzyme replacement therapy avalglucosidase alfa (neoGAA) in treatment-naive and alglucosidase alfa-treated patients with late-onset Pompe disease: a phase 1, open-label, multicenter, multinational, ascending dose study. Neuromuscul. Disord. 2019;29:167–186. doi: 10.1016/j.nmd.2018.12.004. [DOI] [PubMed] [Google Scholar]
- 7.Diaz-Manera J., Kishnani P.S., Kushlaf H., Ladha S., Mozaffar T., Straub V., Toscano A., van der Ploeg A.T., Berger K.I., Clemens P.R., et al. Safety and efficacy of avalglucosidase alfa versus alglucosidase alfa in patients with late-onset Pompe disease (COMET): a phase 3, randomised, multicentre trial. Lancet Neurol. 2021;20:1012–1026. doi: 10.1016/S1474-4422(21)00241-6. [DOI] [PubMed] [Google Scholar]
- 8.Lim J.A., Li L., Raben N. Pompe disease: from pathophysiology to therapy and back again. Front. Aging Neurosci. 2014;6:177. doi: 10.3389/fnagi.2014.00177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cardone M., Porto C., Tarallo A., Vicinanza M., Rossi B., Polishchuk E., Donaudy F., Andria G., De Matteis M.A., Parenti G. Abnormal mannose-6-phosphate receptor trafficking impairs recombinant alpha-glucosidase uptake in Pompe disease fibroblasts. Pathogenetics. 2008;1:6. doi: 10.1186/1755-8417-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maga J.A., Zhou J., Kambampati R., Peng S., Wang X., Bohnsack R.N., Thomm A., Golata S., Tom P., Dahms N.M., et al. Glycosylation-independent lysosomal targeting of acid alpha-glucosidase enhances muscle glycogen clearance in pompe mice. J. Biol. Chem. 2013;288:1428–1438. doi: 10.1074/jbc.M112.438663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Byrne B.J., Geberhiwot T., Barshop B.A., Barohn R., Hughes D., Bratkovic D., Desnuelle C., Laforet P., Mengel E., Roberts M., et al. A study on the safety and efficacy of reveglucosidase alfa in patients with late-onset Pompe disease. Orphanet J. Rare Dis. 2017;12:144. doi: 10.1186/s13023-017-0693-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corti M., Liberati C., Smith B.K., Lawson L.A., Tuna I.S., Conlon T.J., Coleman K.E., Islam S., Herzog R.W., Fuller D.D., et al. Safety of intradiaphragmatic delivery of adeno-associated virus-mediated alpha-glucosidase (rAAV1-CMV-hGAA) gene therapy in children affected by pompe disease. Hum. Gene Ther. Clin. Dev. 2017;28:208–218. doi: 10.1089/humc.2017.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Til N.P., Stok M., Aerts Kaya F.S.F., de Waard M.C., Farahbakhshian E., Visser T.P., Kroos M.A., Jacobs E.H., Willart M.A., van der Wegen P., et al. Lentiviral gene therapy of murine hematopoietic stem cells ameliorates the Pompe disease phenotype. Blood. 2010;115:5329–5337. doi: 10.1182/blood-2009-11-252874. [DOI] [PubMed] [Google Scholar]
- 14.Douillard-Guilloux G., Richard E., Batista L., Caillaud C. Partial phenotypic correction and immune tolerance induction to enzyme replacement therapy after hematopoietic stem cell gene transfer of alpha-glucosidase in Pompe disease. J. Gene Med. 2009;11:279–287. doi: 10.1002/jgm.1305. [DOI] [PubMed] [Google Scholar]
- 15.Stok M., de Boer H., Huston M.W., Jacobs E.H., Roovers O., Visser T.P., Jahr H., Duncker D.J., van Deel E.D., Reuser A.J.J., et al. Lentiviral hematopoietic stem cell gene therapy corrects murine pompe disease. Mol. Ther. Methods Clin. Dev. 2020;17:1014–1025. doi: 10.1016/j.omtm.2020.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Watson J.G., Gardner-Medwin D., Goldfinch M.E., Pearson A.D. Bone marrow transplantation for glycogen storage disease type II (Pompe's disease) N. Engl. J. Med. 1986;314:385. doi: 10.1056/nejm198602063140611. [DOI] [PubMed] [Google Scholar]
- 17.Cartier N., Hacein-Bey-Abina S., Bartholomae C.C., Veres G., Schmidt M., Kutschera I., Vidaud M., Abel U., Dal-Cortivo L., Caccavelli L., et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326:818–823. doi: 10.1126/science.1171242. [DOI] [PubMed] [Google Scholar]
- 18.Biffi A., Montini E., Lorioli L., Cesani M., Fumagalli F., Plati T., Baldoli C., Martino S., Calabria A., Canale S., et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341:1233158. doi: 10.1126/science.1233158. [DOI] [PubMed] [Google Scholar]
- 19.Gentner B., Tucci F., Galimberti S., Fumagalli F., De Pellegrin M., Silvani P., Camesasca C., Pontesilli S., Darin S., Ciotti F., et al. Hematopoietic stem- and progenitor-cell gene therapy for Hurler syndrome. N. Engl. J. Med. 2021;385:1929–1940. doi: 10.1056/NEJMoa2106596. [DOI] [PubMed] [Google Scholar]
- 20.Eichler F., Duncan C., Musolino P.L., Orchard P.J., De Oliveira S., Thrasher A.J., Armant M., Dansereau C., Lund T.C., Miller W.P., et al. Hematopoietic stem-cell gene therapy for cerebral adrenoleukodystrophy. N. Engl. J. Med. 2017;377:1630–1638. doi: 10.1056/NEJMoa1700554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khan A., Barber D.L., Huang J., Rupar C.A., Rip J.W., Auray-Blais C., Boutin M., O'Hoski P., Gargulak K., McKillop W.M., et al. Lentivirus-mediated gene therapy for Fabry disease. Nat. Commun. 2021;12:1178. doi: 10.1038/s41467-021-21371-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fumagalli F., Calbi V., Natali Sora M.G., Sessa M., Baldoli C., Rancoita P.M.V., Ciotti F., Sarzana M., Fraschini M., Zambon A.A., et al. Lentiviral haematopoietic stem-cell gene therapy for early-onset metachromatic leukodystrophy: long-term results from a non-randomised, open-label, phase 1/2 trial and expanded access. Lancet. 2022;399:372–383. doi: 10.1016/S0140-6736(21)02017-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Piras G., Montiel-Equihua C., Chan Y.K.A., Wantuch S., Stuckey D., Burke D., Prunty H., Phadke R., Chambers D., Partida-Gaytan A., et al. Lentiviral hematopoietic stem cell gene therapy rescues clinical phenotypes in a murine model of pompe disease. Mol. Ther. Methods Clin. Dev. 2020;18:558–570. doi: 10.1016/j.omtm.2020.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Desai A.K., Kazi Z.B., Bali D.S., Kishnani P.S. Characterization of immune response in Cross-Reactive Immunological Material (CRIM)-positive infantile Pompe disease patients treated with enzyme replacement therapy. Mol. Genet. Metab. Rep. 2019;20:100475. doi: 10.1016/j.ymgmr.2019.100475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Gelder C.M., Hoogeveen-Westerveld M., Kroos M.A., Plug I., van der Ploeg A.T., Reuser A.J.J. Enzyme therapy and immune response in relation to CRIM status: the Dutch experience in classic infantile Pompe disease. J. Inherit. Metab. Dis. 2015;38:305–314. doi: 10.1007/s10545-014-9707-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poelman E., Hoogeveen-Westerveld M., van den Hout J.M.P., Bredius R.G.M., Lankester A.C., Driessen G.J.A., Kamphuis S.S.M., Pijnappel W.W.M., van der Ploeg A.T. Effects of immunomodulation in classic infantile Pompe patients with high antibody titers. Orphanet J. Rare Dis. 2019;14:71. doi: 10.1186/s13023-019-1039-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herzog R.W. Complexity of immune responses to AAV transgene products - example of factor IX. Cell. Immunol. 2019;342:103658. doi: 10.1016/j.cellimm.2017.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ronzitti G., Gross D.A., Mingozzi F. Human immune responses to adeno-associated virus (AAV) vectors. Front. Immunol. 2020;11:670. doi: 10.3389/fimmu.2020.00670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saif M.A., Bigger B.W., Brookes K.E., Mercer J., Tylee K.L., Church H.J., Bonney D.K., Jones S., Wraith J.E., Wynn R.F. Hematopoietic stem cell transplantation improves the high incidence of neutralizing allo-antibodies observed in Hurler's syndrome after pharmacological enzyme replacement therapy. Haematologica. 2012;97:1320–1328. doi: 10.3324/haematol.2011.058644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liang Q., Vlaar E.C., Catalano F., Pijnenburg J.M., Stok M., van Helsdingen Y., Vulto A.G., Unger W.W.J., van der Ploeg A.T., Pijnappel W.W.M.P., et al. Lentiviral gene therapy prevents anti-human acid alpha-glucosidase antibody formation in murine Pompe disease. Mol. Ther. Methods Clin. Dev. 2022;25:520–532. doi: 10.1016/j.omtm.2022.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun B., Bird A., Young S.P., Kishnani P.S., Chen Y.T., Koeberl D.D. Enhanced response to enzyme replacement therapy in Pompe disease after the induction of immune tolerance. Am. J. Hum. Genet. 2007;81:1042–1049. doi: 10.1086/522236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Han S.O., Ronzitti G., Arnson B., Leborgne C., Li S., Mingozzi F., Koeberl D. Low-dose liver-targeted gene therapy for pompe disease enhances therapeutic efficacy of ERT via immune tolerance induction. Mol. Ther. Methods Clin. Dev. 2017;4:126–136. doi: 10.1016/j.omtm.2016.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Puzzo F., Colella P., Biferi M.G., Bali D., Paulk N.K., Vidal P., Collaud F., Simon-Sola M., Charles S., Hardet R., et al. Rescue of Pompe disease in mice by AAV-mediated liver delivery of secretable acid alpha-glucosidase. Sci. Transl. Med. 2017;9:eaam6375. doi: 10.1126/scitranslmed.aam6375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hordeaux J., Dubreil L., Robveille C., Deniaud J., Pascal Q., Dequéant B., Pailloux J., Lagalice L., Ledevin M., Babarit C., et al. Long-term neurologic and cardiac correction by intrathecal gene therapy in Pompe disease. Acta Neuropathol. Commun. 2017;5:66. doi: 10.1186/s40478-017-0464-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moreland R.J., Higgins S., Zhou A., VanStraten P., Cauthron R.D., Brem M., McLarty B.J., Kudo M., Canfield W.M. Species-specific differences in the processing of acid alpha-glucosidase are due to the amino acid identity at position 201. Gene. 2012;491:25–30. doi: 10.1016/j.gene.2011.09.011. [DOI] [PubMed] [Google Scholar]
- 36.Kan S.H., Aoyagi-Scharber M., Le S.Q., Vincelette J., Ohmi K., Bullens S., Wendt D.J., Christianson T.M., Tiger P.M.N., Brown J.R., et al. Delivery of an enzyme-IGFII fusion protein to the mouse brain is therapeutic for mucopolysaccharidosis type IIIB. Proc. Natl. Acad. Sci. USA. 2014;111:14870–14875. doi: 10.1073/pnas.1416660111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spencer B.J., Verma I.M. Targeted delivery of proteins across the blood-brain barrier. Proc. Natl. Acad. Sci. USA. 2007;104:7594–7599. doi: 10.1073/pnas.0702170104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang D., El-Amouri S.S., Dai M., Kuan C.Y., Hui D.Y., Brady R.O., Pan D. Engineering a lysosomal enzyme with a derivative of receptor-binding domain of apoE enables delivery across the blood-brain barrier. Proc. Natl. Acad. Sci. USA. 2013;110:2999–3004. doi: 10.1073/pnas.1222742110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gleitz H.F., Liao A.Y., Cook J.R., Rowlston S.F., Forte G.M., D'Souza Z., O'Leary C., Holley R.J., Bigger B.W. Brain-targeted stem cell gene therapy corrects mucopolysaccharidosis type II via multiple mechanisms. EMBO Mol. Med. 2018;10:e8730. doi: 10.15252/emmm.201708730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.LeBowitz J.H., Maga J.A. 2008. Lysosomal Targeting Peptides and Uses Thereof. US9469683B2. [Google Scholar]
- 41.Sun B., Zhang H., Benjamin D.K., Jr., Brown T., Bird A., Young S.P., McVie-Wylie A., Chen Y.T., Koeberl D.D. Enhanced efficacy of an AAV vector encoding chimeric, highly secreted acid alpha-glucosidase in glycogen storage disease type II. Mol. Ther. 2006;14:822–830. doi: 10.1016/j.ymthe.2006.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Almagro Armenteros J.J., Tsirigos K.D., Sønderby C.K., Petersen T.N., Winther O., Brunak S., von Heijne G., Nielsen H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019;37:420–423. doi: 10.1038/s41587-019-0036-z. [DOI] [PubMed] [Google Scholar]
- 43.Raben N., Nagaraju K., Lee E., Kessler P., Byrne B., Lee L., LaMarca M., King C., Ward J., Sauer B., et al. Targeted disruption of the acid alpha-glucosidase gene in mice causes an illness with critical features of both infantile and adult human glycogen storage disease type II. J. Biol. Chem. 1998;273:19086–19092. doi: 10.1074/jbc.273.30.19086. [DOI] [PubMed] [Google Scholar]
- 44.Weidemann F., Krämer J., Duning T., Lenders M., Canaan-Kühl S., Krebs A., Guerrero González H., Sommer C., Üçeyler N., Niemann M., et al. Patients with Fabry disease after enzyme replacement therapy dose reduction versus treatment switch. J. Am. Soc. Nephrol. 2014;25:837–849. doi: 10.1681/ASN.2013060585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Burrow T.A., Grabowski G.A. Velaglucerase alfa in the treatment of Gaucher disease type 1. Clin. Investig. 2011;1:285–293. doi: 10.4155/cli.10.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Landis J.L., Hyland H., Kindel S.J., Punnoose A., Geddes G.C. Pompe disease treatment with twice a week high dose alglucoside alfa in a patient with severe dilated cardiomyopathy. Mol. Genet. Metab. Rep. 2018;16:1–4. doi: 10.1016/j.ymgmr.2018.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boucher A.A., Miller W., Shanley R., Ziegler R., Lund T., Raymond G., Orchard P.J. Long-term outcomes after allogeneic hematopoietic stem cell transplantation for metachromatic leukodystrophy: the largest single-institution cohort report. Orphanet J. Rare Dis. 2015;10:94. doi: 10.1186/s13023-015-0313-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aldenhoven M., Jones S.A., Bonney D., Borrill R.E., Coussons M., Mercer J., Bierings M.B., Versluys B., van Hasselt P.M., Wijburg F.A., et al. Hematopoietic cell transplantation for mucopolysaccharidosis patients is safe and effective: results after implementation of international guidelines. Biol. Blood Marrow Transplant. 2015;21:1106–1109. doi: 10.1016/j.bbmt.2015.02.011. [DOI] [PubMed] [Google Scholar]
- 49.Pike-Overzet K., Rodijk M., Ng Y.Y., Baert M.R.M., Lagresle-Peyrou C., Schambach A., Zhang F., Hoeben R.C., Hacein-Bey-Abina S., Lankester A.C., et al. Correction of murine Rag1 deficiency by self-inactivating lentiviral vector-mediated gene transfer. Leukemia. 2011;25:1471–1483. doi: 10.1038/leu.2011.106. [DOI] [PubMed] [Google Scholar]
- 50.Huston M.W., van Til N.P., Visser T.P., Arshad S., Brugman M.H., Cattoglio C., Nowrouzi A., Li Y., Schambach A., Schmidt M., et al. Correction of murine SCID-X1 by lentiviral gene therapy using a codon-optimized IL2RG gene and minimal pretransplant conditioning. Mol. Ther. 2011;19:1867–1877. doi: 10.1038/mt.2011.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van Til N.P., de Boer H., Mashamba N., Wabik A., Huston M., Visser T.P., Fontana E., Poliani P.L., Cassani B., Zhang F., et al. Correction of murine Rag2 severe combined immunodeficiency by lentiviral gene therapy using a codon-optimized RAG2 therapeutic transgene. Mol. Ther. 2012;20:1968–1980. doi: 10.1038/mt.2012.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dahl M., Doyle A., Olsson K., Månsson J.E., Marques A.R.A., Mirzaian M., Aerts J.M., Ehinger M., Rothe M., Modlich U., et al. Lentiviral gene therapy using cellular promoters cures type 1 Gaucher disease in mice. Mol. Ther. 2015;23:835–844. doi: 10.1038/mt.2015.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Raval K.K., Tao R., White B.E., De Lange W.J., Koonce C.H., Yu J., Kishnani P.S., Thomson J.A., Mosher D.F., Ralphe J.C., et al. Pompe disease results in a Golgi-based glycosylation deficit in human induced pluripotent stem cell-derived cardiomyocytes. J. Biol. Chem. 2015;290:3121–3136. doi: 10.1074/jbc.M114.628628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nascimbeni A.C., Fanin M., Tasca E., Angelini C., Sandri M. Impaired autophagy affects acid alpha-glucosidase processing and enzyme replacement therapy efficacy in late-onset glycogen storage disease type II. Neuropathol. Appl. Neurobiol. 2015;41:672–675. doi: 10.1111/nan.12214. [DOI] [PubMed] [Google Scholar]
- 55.Myerowitz R., Puertollano R., Raben N. Impaired autophagy: the collateral damage of lysosomal storage disorders. EBioMedicine. 2021;63:103166. doi: 10.1016/j.ebiom.2020.103166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fukuda T., Ahearn M., Roberts A., Mattaliano R.J., Zaal K., Ralston E., Plotz P.H., Raben N. Autophagy and mistargeting of therapeutic enzyme in skeletal muscle in Pompe disease. Mol. Ther. 2006;14:831–839. doi: 10.1016/j.ymthe.2006.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen L.R., Chen C.A., Chiu S.N., Chien Y.H., Lee N.C., Lin M.T., Hwu W.L., Wang J.K., Wu M.H. Reversal of cardiac dysfunction after enzyme replacement in patients with infantile-onset Pompe disease. J. Pediatr. 2009;155:271–275.e2. doi: 10.1016/j.jpeds.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 58.Young S.P., Zhang H., Corzo D., Thurberg B.L., Bali D., Kishnani P.S., Millington D.S. Long-term monitoring of patients with infantile-onset Pompe disease on enzyme replacement therapy using a urinary glucose tetrasaccharide biomarker. Genet. Med. 2009;11:536–541. doi: 10.1097/GIM.0b013e3181a87867. [DOI] [PubMed] [Google Scholar]
- 59.Andersen M., Nørgaard-Pedersen D., Brandt J., Pettersson I., Slaaby R. IGF1 and IGF2 specificities to the two insulin receptor isoforms are determined by insulin receptor amino acid 718. PLoS One. 2017;12:e0178885. doi: 10.1371/journal.pone.0178885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Korlimarla A., Lim J.A., Kishnani P.S., Sun B. An emerging phenotype of central nervous system involvement in Pompe disease: from bench to bedside and beyond. Ann. Transl. Med. 2019;7:289. doi: 10.21037/atm.2019.04.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.DeRuisseau L.R., Fuller D.D., Qiu K., DeRuisseau K.C., Donnelly W.H., Jr., Mah C., Reier P.J., Byrne B.J. Neural deficits contribute to respiratory insufficiency in Pompe disease. Proc. Natl. Acad. Sci. USA. 2009;106:9419–9424. doi: 10.1073/pnas.0902534106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Colella P., Sellier P., Gomez M.J., Biferi M.G., Tanniou G., Guerchet N., Cohen-Tannoudji M., Moya-Nilges M., van Wittenberghe L., Daniele N., et al. Gene therapy with secreted acid alpha-glucosidase rescues Pompe disease in a novel mouse model with early-onset spinal cord and respiratory defects. EBioMedicine. 2020;61:103052. doi: 10.1016/j.ebiom.2020.103052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pardridge W.M. Treatment of Alzheimer's disease and blood-brain barrier drug delivery. Pharmaceuticals. 2020;13:E394. doi: 10.3390/ph13110394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Byrne B.J., Fuller D.D., Smith B.K., Clement N., Coleman K., Cleaver B., Vaught L., Falk D.J., McCall A., Corti M. Pompe disease gene therapy: neural manifestations require consideration of CNS directed therapy. Ann. Transl. Med. 2019;7:290. doi: 10.21037/atm.2019.05.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Begley D.J., Pontikis C.C., Scarpa M. Lysosomal storage diseases and the blood-brain barrier. Curr. Pharm. Des. 2008;14:1566–1580. doi: 10.2174/138161208784705504. [DOI] [PubMed] [Google Scholar]
- 66.Doyle B.M., Turner S.M.F., Sunshine M.D., Doerfler P.A., Poirier A.E., Vaught L.A., Jorgensen M.L., Falk D.J., Byrne B.J., Fuller D.D. AAV gene therapy utilizing glycosylation-independent lysosomal targeting tagged GAA in the hypoglossal motor system of pompe mice. Mol. Ther. Methods Clin. Dev. 2019;15:194–203. doi: 10.1016/j.omtm.2019.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mittelbronn M., Dietz K., Schluesener H.J., Meyermann R. Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol. 2001;101:249–255. doi: 10.1007/s004010000284. [DOI] [PubMed] [Google Scholar]
- 68.Biffi A., Capotondo A., Fasano S., del Carro U., Marchesini S., Azuma H., Malaguti M.C., Amadio S., Brambilla R., Grompe M., et al. Gene therapy of metachromatic leukodystrophy reverses neurological damage and deficits in mice. J. Clin. Invest. 2006;116:3070–3082. doi: 10.1172/JCI28873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Visigalli I., Delai S., Politi L.S., Di Domenico C., Cerri F., Mrak E., D'Isa R., Ungaro D., Stok M., Sanvito F., et al. Gene therapy augments the efficacy of hematopoietic cell transplantation and fully corrects mucopolysaccharidosis type I phenotype in the mouse model. Blood. 2010;116:5130–5139. doi: 10.1182/blood-2010-04-278234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bak L.K., Walls A.B., Schousboe A., Waagepetersen H.S. Astrocytic glycogen metabolism in the healthy and diseased brain. J. Biol. Chem. 2018;293:7108–7116. doi: 10.1074/jbc.R117.803239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Prill H., Luu A., Yip B., Holtzinger J., Lo M.J., Christianson T.M., Yogalingam G., Aoyagi-Scharber M., LeBowitz J.H., Crawford B.E., Lawrence R. Differential uptake of NAGLU-IGF2 and unmodified NAGLU in cellular models of Sanfilippo syndrome type B. Mol. Ther. Methods Clin. Dev. 2019;14:56–63. doi: 10.1016/j.omtm.2019.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yogalingam G., Luu A.R., Prill H., Lo M.J., Yip B., Holtzinger J., Christianson T., Aoyagi-Scharber M., Lawrence R., Crawford B.E., et al. BMN 250, a fusion of lysosomal alpha-N-acetylglucosaminidase with IGF2, exhibits different patterns of cellular uptake into critical cell types of Sanfilippo syndrome B disease pathogenesis. PLoS One. 2019;14:e0207836. doi: 10.1371/journal.pone.0207836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Salabarria S.M., Nair J., Clement N., Smith B.K., Raben N., Fuller D.D., Byrne B.J., Corti M. Advancements in AAV-mediated gene therapy for pompe disease. J. Neuromuscul. Dis. 2020;7:15–31. doi: 10.3233/JND-190426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Unnisa Z., Yoon J.K., Schindler J.W., Mason C., van Til N.P. Gene therapy developments for pompe disease. Biomedicines. 2022;10:302. doi: 10.3390/biomedicines10020302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Baik A.D., Calafati P., Zhang X., Aaron N.A., Mehra A., Moller-Tank S., Miloscio L., Praggastis M., Giovannone N., Pan C., et al. Cell type-selective targeted delivery of a recombinant lysosomal enzyme for enzyme therapies. Mol. Ther. 2021;29:3512–3524. doi: 10.1016/j.ymthe.2021.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Costa-Verdera H., Collaud F., Riling C.R., Sellier P., Nordin J.M.L., Preston G.M., Cagin U., Fabregue J., Barral S., Moya-Nilges M., et al. Hepatic expression of GAA results in enhanced enzyme bioavailability in mice and non-human primates. Nat. Commun. 2021;12:6393. doi: 10.1038/s41467-021-26744-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Eggers M., Vannoy C.H., Huang J., Purushothaman P., Brassard J., Fonck C., Meng H., Prom M.J., Lawlor M.W., Cunningham J., et al. Muscle-directed gene therapy corrects Pompe disease and uncovers species-specific GAA immunogenicity. EMBO Mol. Med. 2022;14:e13968. doi: 10.15252/emmm.202113968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Manno C.S., Pierce G.F., Arruda V.R., Glader B., Ragni M., Rasko J.J., Ozelo M.C., Hoots K., Blatt P., Konkle B., et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 79.Kishnani P.S., Koeberl D.D. Liver depot gene therapy for Pompe disease. Ann. Transl. Med. 2019;7:288. doi: 10.21037/atm.2019.05.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Monahan P.E., Négrier C., Tarantino M., Valentino L.A., Mingozzi F. Emerging immunogenicity and genotoxicity considerations of adeno-associated virus vector gene therapy for hemophilia. J. Clin. Med. 2021;10:2471. doi: 10.3390/jcm10112471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bushman F.D. Retroviral insertional mutagenesis in humans: evidence for four genetic mechanisms promoting expansion of cell clones. Mol. Ther. 2020;28:352–356. doi: 10.1016/j.ymthe.2019.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Six E., Guilloux A., Denis A., Lecoules A., Magnani A., Vilette R., Male F., Cagnard N., Delville M., Magrin E., et al. Clonal tracking in gene therapy patients reveals a diversity of human hematopoietic differentiation programs. Blood. 2020;135:1219–1231. doi: 10.1182/blood.2019002350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Williams D.A., Bledsoe J.R., Duncan C.N., Eichler F.S., Grzywacz B., Gupta A.O., Lund T., Orchard P.J., Slauson S., Whitney D., et al. Myelodysplastic syndromes after eli-cel gene therapy for cerebral adrenoleukodystrophy (CALD) Mol. Ther. 2022;30:6–7. [Google Scholar]
- 84.Hsieh M.M., Bonner M., Pierciey F.J., Uchida N., Rottman J., Demopoulos L., Schmidt M., Kanter J., Walters M.C., Thompson A.A., et al. Myelodysplastic syndrome unrelated to lentiviral vector in a patient treated with gene therapy for sickle cell disease. Blood Adv. 2020;4:2058–2063. doi: 10.1182/bloodadvances.2019001330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nguyen G.N., Everett J.K., Kafle S., Roche A.M., Raymond H.E., Leiby J., Wood C., Assenmacher C.A., Merricks E.P., Long C.T., et al. A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nat. Biotechnol. 2021;39:47–55. doi: 10.1038/s41587-020-0741-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Reinhardt B., Habib O., Shaw K.L., Garabedian E., Carbonaro-Sarracino D.A., Terrazas D., Fernandez B.C., De Oliveira S., Moore T.B., Ikeda A.K., et al. Long-term outcomes after gene therapy for adenosine deaminase severe combined immune deficiency (ADA SCID) Blood. 2021;138:1304–1316. doi: 10.1182/blood.2020010260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Drysdale C.M., Tisdale J.F., Uchida N. Immunoresponse to gene-modified hematopoietic stem cells. Mol. Ther. Methods Clin. Dev. 2020;16:42–49. doi: 10.1016/j.omtm.2019.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bernardo M.E., Aiuti A. The role of conditioning in hematopoietic stem-cell gene therapy. Hum. Gene Ther. 2016;27:741–748. doi: 10.1089/hum.2016.103. [DOI] [PubMed] [Google Scholar]
- 89.Tucci F., Galimberti S., Naldini L., Valsecchi M.G., Aiuti A. A systematic review and meta-analysis of gene therapy with hematopoietic stem and progenitor cells for monogenic disorders. Nat. Commun. 2022;13:1315. doi: 10.1038/s41467-022-28762-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Logan A.C., Weissman I.L., Shizuru J.A. The road to purified hematopoietic stem cell transplants is paved with antibodies. Curr. Opin. Immunol. 2012;24:640–648. doi: 10.1016/j.coi.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.George B.M., Kao K.S., Kwon H.S., Velasco B.J., Poyser J., Chen A., Le A.C., Chhabra A., Burnett C.E., Cajuste D., et al. Antibody conditioning enables MHC-mismatched hematopoietic stem cell transplants and organ graft tolerance. Cell Stem Cell. 2019;25:185–192.e3. doi: 10.1016/j.stem.2019.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Omer-Javed A., Pedrazzani G., Albano L., Ghaus S., Latroche C., Manzi M., Ferrari S., Fiumara M., Jacob A., Vavassori V., et al. Mobilization-based chemotherapy-free engraftment of gene-edited human hematopoietic stem cells. Cell. 2022;185:2248–2264.e21. doi: 10.1016/j.cell.2022.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Richter M., Stone D., Miao C., Humbert O., Kiem H.P., Papayannopoulou T., Lieber A. In vivo hematopoietic stem cell transduction. Hematol. Oncol. Clin. North Am. 2017;31:771–785. doi: 10.1016/j.hoc.2017.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Xu Z., Rao Y., Huang Y., Zhou T., Feng R., Xiong S., Yuan T.F., Qin S., Lu Y., Zhou X., et al. Efficient strategies for microglia replacement in the central nervous system. Cell Rep. 2020;33:108443. doi: 10.1016/j.celrep.2020.108443. [DOI] [PubMed] [Google Scholar]
- 95.Shibuya Y., Kumar K.K., Mader M.M.D., Yoo Y., Ayala L.A., Zhou M., Mohr M.A., Neumayer G., Kumar I., Yamamoto R., et al. Treatment of a genetic brain disease by CNS-wide microglia replacement. Sci. Transl. Med. 2022;14:eabl9945. doi: 10.1126/scitranslmed.abl9945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dahl M., Smith E.M.K., Warsi S., Rothe M., Ferraz M.J., Aerts J.M.F.G., Golipour A., Harper C., Pfeifer R., Pizzurro D., et al. Correction of pathology in mice displaying Gaucher disease type 1 by a clinically-applicable lentiviral vector. Mol. Ther. Methods Clin. Dev. 2021;20:312–323. doi: 10.1016/j.omtm.2020.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Halene S., Wang L., Cooper R.M., Bockstoce D.C., Robbins P.B., Kohn D.B. Improved expression in hematopoietic and lymphoid cells in mice after transplantation of bone marrow transduced with a modified retroviral vector. Blood. 1999;94:3349–3357. [PMC free article] [PubMed] [Google Scholar]
- 98.Storz P., Döppler H., Horn-Müller J., Groner B., Pfizenmaier K., Müller G. A cellular reporter assay to monitor insulin receptor kinase activity based on STAT 5-dependent luciferase gene expression. Anal. Biochem. 1999;276:97–104. doi: 10.1006/abio.1999.4345. [DOI] [PubMed] [Google Scholar]
- 99.Jack R.M., Gordon C., Scott C.R., Kishnani P.S., Bali D. The use of acarbose inhibition in the measurement of acid alpha-glucosidase activity in blood lymphocytes for the diagnosis of Pompe disease. Genet. Med. 2006;8:307–312. doi: 10.1097/01.gim.0000217785.19262.9e. [DOI] [PubMed] [Google Scholar]
- 100.Okumiya T., Keulemans J.L.M., Kroos M.A., Van der Beek N.M.E., Boer M.A., Takeuchi H., Van Diggelen O.P., Reuser A.J.J. A new diagnostic assay for glycogen storage disease type II in mixed leukocytes. Mol. Genet. Metab. 2006;88:22–28. doi: 10.1016/j.ymgme.2005.10.016. [DOI] [PubMed] [Google Scholar]
- 101.An N., Kang Y. Using quantitative real-time PCR to determine donor cell engraftment in a competitive murine bone marrow transplantation model. J. Vis. Exp. 2013:e50193. doi: 10.3791/50193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Otsu N. A threshold selection method from gray-level histograms. IEEE Trans. Syst. Man Cybern. 1979;9:62–66. [Google Scholar]
- 103.Yang X., Shen X., Long J., Chen H. An improved median-based Otsu image thresholding algorithm. AASRI Procedia. 2012;3:468–473. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data supporting the studies presented in this paper can be found in the main text or the supplemental information. Additional information, as appropriate, may be made available by request directed to the corresponding author and, as appropriate, following execution of a suitable confidentiality agreement with AVROBIO, Inc.