

Abstract
Histidyl-tRNA synthetase (HARS) is a highly conserved translation factor that plays an essential role in protein synthesis. HARS has been implicated in the human syndromes Charcot-Marie-Tooth (CMT) Type 2W and Type IIIB Usher (USH3B). The USH3B mutation, which encodes an Y454S substitution in HARS, is inherited in an autosomal recessive fashion and associated with childhood deafness, blindness and episodic hallucinations during acute illness. The biochemical basis of the pathophysiologies linked to USH3B is currently unknown. Here, we present a detailed functional comparison of wild type and Y454S HARS enzymes. Kinetic parameters for enzymes and canonical substrates were determined using both steady state and rapid kinetics. Enzyme stability was examined using differential scanning fluorimetry. Finally, enzyme functionality in primary cell culture was assessed. Our results demonstrate that the Y454S substitution leaves HARS amino acid activation, aminoacylation and tRNAHis binding functions largely intact compared with WT HARS, and the mutant enzyme dimerizes similarly to wild type. Interestingly, during our investigation it was revealed that the kinetics of amino acid activation differs from the previously characterized bacterial HisRS. Despite the similar kinetics, differential scanning fluorimetry revealed that Y454S is less thermally stable than WT HARS, and cells from Y454S patients grown at elevated temperature demonstrate diminished protein synthesis compared to wild type cells. The thermal sensitivity associated with the Y454S mutation represents offers a biochemical basis for understanding USH3B.
Graphical Abstract
All aminoacyl-tRNA synthetases (ARS) catalyze the attachment of amino acids to their cognate tRNAs in a two-step reaction that initiates protein synthesis1. The first amino acid activation step features the binding of amino acid and ATP to the active site, leading to the formation of the aminoacyl adenylate intermediate. Next, the amino acid is transferred from the enzyme bound aminoacyl adenylate intermediate to the 3′ end of the tRNA, producing an aminoacyl-tRNA and AMP. The ARSs are divided into two broad classes, based on structural topology and catalytic mechanism2. Aminoacyl-tRNA synthetases are universally required for protein synthesis and are therefore ubiquitously expressed in all tissue types. Accordingly, the tissue-specific nature and pathological mechanism of the numerous and complex human diseases linked to inherited mutations in ARS genes remains a critical gap in knowledge.
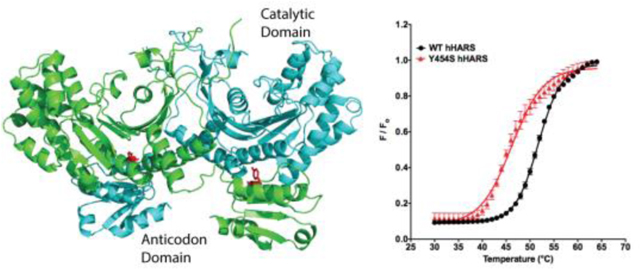
The human histidyl-tRNA synthetase (HARS, UniProtKB - P12081) is a class II aminoacyl tRNA synthetase featuring a homodimeric structure consisting of a class conserved catalytic domain organized around a seven-stranded antiparallel β-sheet and a C-terminal anticodon-binding domain based on an α/β fold that is positioned below the catalytic domain of the other monomer (Figure 1A,3, 4). HARS structures have been determined for a variety of species in various liganded states4–8. Six specific autosomal mutations linked to the HARS gene have been reported, and are associated with USH3B9 and peripheral neuropathies10, 11. Additionally, an inherited mutation in mitochondrial HARS2 has been reported to be associated with Perrault syndrome12. Interestingly, autoantibodies against HARS are the most prevalent autoantibodies in idiopathic inflammatory myopathies13.
Figure 1.

Location of the Y454S substitution in the structure of human HARS. A) The Y454S substitution is located in the anticodon-binding domain juxtaposed to the catalytic domain of the opposing monomer. B) Y454 hydrogen bonds with the conserved E439. C) Modeling the serine at Y454 position in the anticodon- binding domain creates a 6 Å gap between E439 and S454, potentially eliminating a structure-stabilizing hydrogen bonding interaction. D) A portion of the complete sequence alignment (Clustal Omega) of HARS protein sequences from Homo sapiens, Bos taurus, Mus musculus, Danio rerio, Drosophila melanogaster, Caenorhabditis elegans, Trypanosoma bruceii, Saccharomyces cerevisiae, Thermus thermophilus, Staphylococcus aureus and Escherichia coli.
The Y454S HARS substitution (c.1361A>C, Figure 1B, 1C) is inherited in an autosomal recessive manner in USH3B (OMIM #614504) and represents a striking example of a simple Mendelian trait encoding a single missense substitution that presents as a complex human disorder9. Patients with USH3B exhibit normal growth during infancy, but experience progressive loss of hearing and sight in the first and second decades of life, respectively. Rapid clinical deteriorations often occur in response to febrile illnesses. For most patients, acute viral infections induce vivid hallucinations. USH3B patients also exhibit mild truncal ataxia, wide based gait, and delayed motor development. No difference in either intracellular levels of expression or cytoplasmic distribution was observed between the wild type (WT) and the Y454S HARS enzyme9. Interestingly, another mammalian mutant also localized in the HARS anticodon binding domain of the enzyme, R362H, was obtained in an unbiased screen for temperature sensitive cell cycle arrest mutants14.
While the aminoacylation and tRNA recognition functions of the prokaryotic histidyl tRNA-synthetase (HisRS) have been studied by steady state and rapid kinetics15–19, relatively few studies have focused on the function of the human enzyme20. Multiple turnover kinetics have been reported for a number of other human ARS enzymes, including those associated with disease causing mutations21–25, but few have been characterized by rapid kinetics. In a preliminary aminoacylation comparison of murine versions of WT and Y454S HARS using crude human tRNA mix, only a minor decrease in aminoacylation by Y454S HARS was observed9. Thus, the preliminary conclusion was that the phenotype associated with Y454S HARS is a not straightforward loss of aminoacylation function. Given the limitations of the substrate used, the results were not decisive.
Here, we describe kinetic studies of the human WT and Y454S HARS enzymes using defined tRNA substrates that tested whether or not the USH3B phenotype could be ascribed to a loss in the canonical aminoacylation function. Additionally, we explored the hypothesis that WT and Y454S HARS differ with respect to thermal stability. The impact of the Y454S USH3B substitution may not be a simple loss of aminoacylation function, but a pathophysiological consequence of thermal instability.
EXPERIMENTAL PROCEDURES
Plasmid constructs, cell culture conditions and transfection.
HEK293 cells were grown in DMEM (Thermo) supplemented with 10% FBS (Gibco), 1% penicillin/streptomycin (Gibco), and 1% L-glutamine (Gibco) and maintained at 37 °C in a humidified incubator containing 5% CO2. The expression plasmids containing the genes for human WT and Y454S mutant N-terminal FLAG-tagged HARS were derived from plasmid pCAG/FLAG/RFC/A, which places the foreign open reading frame under control of the CAG promotor and inserts a FLAG-tag (derived from GateWay vector Reading Frame Cassette A) at the N-terminus. The plasmids bicistronically express green fluorescent protein, to monitor transfection efficiency. These were generous gifts from Dr. Robert Jinks. Cells were transfected when 50% confluent using polyethyleneimine (PEI) at a 3:1 (μg) ratio of PEI:DNA diluted in serum-free media, and incubated for 48 h post transfection.
Primary fibroblast cultures.
Primary fibroblast cultures were established from skin biopsies and cryogenically stored at −180 °C in liquid nitrogen vapor phase. Parents gave informed consent prior to skin biopsy. Cells were thawed and grown as monolayer cultures in T-25 flasks at 37 °C in a 5% CO2 incubator using Ham’s F-10 medium supplemented with 10% fetal bovine serum (Sigma-Aldrich Corp., St. Louis, Missouri). Three fibroblast cultures were established from patients with the Y454S HARS mutation and six control fibroblast lines were selected on the basis of similar ages to the patients and similar lengths of time spent in cryogenic storage. Cells were cultured in flasks at 37 °C in a 5% CO2 incubator.
Purification of human HARS.
HEK293 cells transiently transfected with HARS-expressing plasmids were harvested and lysed in CelLytic M buffer (Sigma-Aldrich) containing mammalian protease inhibitor cocktail (Sigma-Aldrich) for 20 min at 4 °C. FLAG-tagged HARS was purified by binding to an anti-DYDDDDK resin per manufacturer protocol (GenScript) and eluted by competition with 3X-DYKDDDDK peptide in buffer containing 50 mM Tris-HCl pH 7.4, and 150 mM NaCl. Isolated FLAG-HARS was further purified by HiTrapQ HP column (GE Healthcare) and eluted by a NaCl gradient to 150–500 mM. Individual fractions containing HARS were identified by SDS-PAGE, pooled, and dialyzed at 4 °C into Standard Buffer, consisting of 50 mM HEPES pH 7.5, 150 mM KCl, 10 mM MgCl2, and 5 mM β-mercaptoethanol (β-ME). After dialysis, samples were concentrated using Amicon Ultra-4 centrifugal filters (Millipore), then diluted by the addition of 80% glycerol for a final glycerol concentration of 40%. Protein concentrations were determined by A280 and stored at −20 °C. Preparations were greater than 99% HARS as analyzed on SDS-PAGE (data not shown). The activity of each enzyme preparation was determined by active site titration26, 27 monitoring the appearance of α-labeled 32P-AMP in the presence of histidine under pre-steady state conditions with rapid chemical quench. Enzyme at 5 μM was incubated in one syringe with saturating histidine, 5 mM MgCl2 and 8 U/mL of pyrophosphatase (PPiase) in Standard Buffer, while the second syringe contained ATP in Standard Buffer. The reactions were quenched with 400 mM NaOAc, pH 4.5, 0.1% SDS, and then the products were analyzed by thin-layer chromatography. A plot of AMP produced versus time yields a burst phase and linear phase. The amplitude of the burst phase measures the first turnover, and the concentration of AMP produced is compared to the concentration of enzyme in the assay to yield the fraction of active enzyme molecules.
Western blot analysis of purified HARS proteins.
To quantify endogenous WT HARS dimerization with purified FLAG-tagged HARS proteins, purified FLAG-tagged HARS proteins and lysates from HEK293 cells transfected with FLAG-tagged WT or Y454S HARS were separated by 12% SDS-PAGE and subjected to Western blot analysis. FLAG-tagged transfected HARS (WT and Y454S) was visualized with murine anti-FLAG antibody (1:3000, Sigma F1804, Lot SLBF6631V), and both endogenously produced HARS and FLAG-tagged transfected HARS were visualized with rabbit anti-HARS antibody (1:3000 dilution, Abcam ab137591 LotGR204396–1). Signal was developed by secondary antibody conjugated to HRP (1:10000, Santa Cruz) and Super Signal West Femto (Thermo). Blots were visualized by Versadoc and quantified by Quantity One (Bio-Rad) software as a ratio of endogenous HARS to total HARS (endogenous plus FLAG-tagged). Endogenous HARS runs faster than FLAG-tagged HARS on SDS-PAGE, but little was detected associated with FLAG-tagged HARS after purification (Supporting Information Figure S1).
In vitro transcription, purification and radiolabeling of human tRNAHis.
Human tRNAHis was constructed by overlap PCR incorporating EcoRI and BamHI sites and cloned into a pUC19 derivative, pJA1519, 27. Purified plasmid was restricted with FokI (New England Biolabs) to produce a linearized tRNAHis sequence with a T7 polymerase binding site. In vitro transcription reactions were performed as previously described16, 27, 28 and the tRNA transcripts were precipitated with ethanol and NaOAc. The tRNA was re-suspended in 50% 10 mM HEPES (pH 6.0), 50% formamide, and then purified on 12% acrylamide (1:19 acrylamide:bis-acrylamide), 6M urea, Tris-borate-EDTA gels. tRNAs were electroeluted from gel slices with an Elutrap apparatus (Whatman/Schleicher & Schuell). Purified tRNAHis was then stored at −80 °C. For enzyme kinetic studies purified tRNAHis was labeled at the A76 nucleotide with α-ATP [32P] as previously described29. tRNA was not annealed for kinetic experiments, as experiments demonstrated it did not improve tRNA aminoacylation (data not shown, annealing protocol below). Plateau charging (aminoacylation under single-turnover conditions) indicates that approximately 50–60% of tRNAHis prepared by this method can be aminoacylated.
tRNA Electrophoretic Mobility Shift Assay (EMSA).
These experiments employed radiolabeled tRNA, either 32P-tRNAHis transcripts prepared as above, or total human placental 32P-tRNA9. The tRNAs, at concentrations (30 nM) much below enzyme concentration, were incubated with various concentrations (0.1–30 μM) of WT or Y454S HARS at room temperature. The binding reactions were carried out in Standard Buffer containing 0.0001% Triton-X. Binding reactions included 5 mM histidine and 5 mM AMP to mimic adenylate structural features while precluding aminoacylation of tRNAHis. Prior to be being added to binding reactions, tRNAHis was annealed by heating to 95 °C for two minutes in a water bath, adding MgCl2 to 5 mM, and cooling to room temperature. Following a ten-minute incubation period, samples were brought to 25% glycerol with loading dye, and then loaded onto a native polyacrylamide gel (10% polyacrylamide, 50 mM Tris borate, 5 mM MgCl2, 0.0001% Triton X-100). The gel was electrophoresed at 10 mA per gel for 3–4 hours, dried, exposed to a phosphorimaging screen, and then scanned using a Pharos Bio-Rad Molecular Imager FX™. The data was analyzed using Prism from GraphPad Software, employing a one-site heterologous model with depletion for the competition assay, as described in30. Modeling of the competition assay determined that less than 1.25% of the binding sites were non-specific, so we used a one site-specific binding equation (Equation 1) to measure binding of the transcribed tRNAHis to enzyme, where Y = specific binding, X = radiolabel concentration, and Bmax is maximum binding.
| Equation 1 |
Multiple turnover aminoacylation kinetics.
Multiple turnover aminoacylation assays were performed using a modified version of the Uhlenbeck-Wolfson assay29. The reactions were carried out at 37 °C in a 20 μL reaction composed of Standard Buffer with 5 mM β-ME, 2 U/ml PPiase, and 0.5 pM 32P-labeled tRNAHis 31. Steady-state kinetic constants were determined in a range of substrate concentrations from 1 μM to 5 mM in histidine and 100 nM to 15 μM in tRNAHis. Reactions were initiated with the addition of 2 nM HARS enzyme, and were terminated by introducing 2 μL of the reaction mixture into 8 μL of a quenching buffer (400 mM NaOAc pH 4.5, 0.1% SDS). The tRNA was subsequently digested to single nucleotides in a 40–60 minute incubation with 0.1 μg P1 nuclease (Sigma) at ambient temperature, and then radiolabeled aminoacylated A76 was separated from non-aminoacylated A76 by thin layer chromatography on PEI-cellulose plates (Scientific Adsorbents) using a mobile phase of 0.1M ammonium acetate/5% acetic acid. Radioactive products were detected by radioisotopic imaging on a phosphor screen (Bio-Rad Molecular Imager FX™). The concentration of aminoacylated tRNAHis was quantified by comparing the ratio of the relative amount of aminoacylated A76 (aa) (determined as counts * mm2) to total radiolabeled product (aa + A76) according to equation (2):
| Equation 2 |
Pre-steady state and single turnover rapid chemical quench kinetics.
Pre-steady state amino acid activation was characterized using a KinTek RQF-3 chemical quench flow apparatus (KinTek, Austin, Texas). The reaction design and the data analysis were carried out essentially as described previously17, 19. In a typical experiment, one syringe contained WT or Y454S HARS enzyme at a final concentration of 5 μM in HARS monomer, 8 U/mL PPiase, a saturating concentration of histidine, and 25 μM (final) tRNAHis (if present) in Reaction Buffer, while the second syringe contained 100 μM [α-32P] ATP in Reaction Buffer. The fractional conversion of ATP to AMP on the TLC plates was analyzed by radioisotopic imaging using a Bio-Rad phosphor K-screen and Bio-Rad Molecular Imager FX™. Single turnover aminoacylation kinetics of WT and Y454S were also determined using the KinTek RQF-3 chemical quench flow apparatus. In all single turnover reactions, the enzyme:histidyl-adenylate complex was generated in situ prior to the reaction by the incubation of HARS (2 μM), ATP (2.5 mM), histidine (10 mM), and PPiase (2 U/mL) at 37 °C for 10 min. The enzyme adenylate reaction mix was loaded into one syringe, and then mixed with 200 nM of tRNAHis in the second syringe, for a final concentration of 100 nM final. The reactions were quenched as described above. After P1 nuclease digestion, aminoacylated tRNAHis product was detected and quantified as described above. The single turnover aminoacyl transfer rate data were fit to a single exponential equation from which rate and amplitude parameters could be extracted, as described27. The amount of aminoacylated tRNAHis product was plotted as a function of time and the single turnover aminoacyl transfer rate data were determined by use of equation (3):
| Equation 3 |
where A represents the amplitude, C is the y intercept offset, and ktrans is the rate of histidine transfer.
Analytical centrifugation and sedimentation velocity analysis.
A solution of all three HARS samples was measured at 0.3 OD 230 nm (0.7 μM), 20 ºC, 35 K rpm, and measured by UV intensity detection in a Beckman Optima XLI analytical ultracentrifuge at the Center for Analytical Ultracentrifugation of Macromolecular Assemblies at the University of Texas Health Science Center at San Antonio, using an An60Ti rotor and standard 2-channel epon centerpieces (Beckman-Coulter). All data were analyzed with UltraScan-III ver. 3.5, release 217032, 33. All samples were measured in a 10 mM potassium phosphate buffer containing 50 mM KCl. Hydrodynamic corrections for buffer density and viscosity were estimated by UltraScan to be 1.0019 g/ml and 0.998 cP. The partial specific volume of HARS (0.745 ml/g) was estimated by UltraScan from protein sequence analogous to methods outlined in Laue et al.34. Experimental SV data were pre-processed by 2-dimensional spectrum analysis (2DSA)35 with simultaneously removal of time- and radially-invariant noise contributions36, and to fit the meniscus position as described37. The resulting data were fitted by the parametrically constrained spectrum analysis (PCSA), using a straight-line parameterization coupled with a Monte Carlo approach38, 39. The calculations are computationally intensive and are carried out on high-performance computing platforms40. All calculations were performed on the Lonestar cluster at the Texas Advanced Computing Center at the University of Texas at Austin and on Comet and Gordon at San Diego Supercomputing Center.
Differential scanning fluorimetry.
The DSF experiments were carried out as described41, 42 with some minor modifications. An Applied Biosystems 7500 fast real-time PCR device was used to monitor protein unfolding by the increase in fluorescence of SYPRO Orange (Invitrogen). HARS protein samples (10 μM) were incubated in a 96-well microplate (Costar) with 6X SYPRO orange dye with DSF reaction buffer (25 mM HEPES pH 7.5, 50 mM KCl) in a final volume of 20 μL. Enzyme was incubated with 5 mM histidine, 5 mM ATP, or 10 μM tRNAHis (20 μM with R362H). Plates were sealed with optical-quality sealing tape (Bio-Rad) while in the RT-PCR instrument. Samples were heated at 1 °C per minute, from 26 °C to 95 °C and fluorescence intensity was measured every 1 °C. Wavelengths for excitation and emission were 490 and 575 nm, respectively. The melting temperature (Tm), defined as the midpoint of the protein unfolding transition curve, was determined using the Boltzmann model as previously described43. GraphPad Prism software was used to fit the fluorescence data (excluding data after maximal fluorescence intensity) to the equation (4):
| Equation 4 |
where I is the fluorescence intensity at temperature T, A is pre-transitional fluorescence, B is post-transitional fluorescence, and C is the slope factor. In the absence of substrate Tm = T0 and substrate dependent changes in Tm were calculated as equation (5):
| Equation 5 |
Tritiated histidine incorporation and [3H]-histidine incorporation analysis.
L-[2,5-3H] histidine ([3H]-histidine, specific activity 50 Ci/mmol) was obtained from Perkin Elmer (Boston, MA). The fibroblast cell types (three patient, six wild-type) were passaged into three (patient) or six (wild-type) 24-well plates (five wells per type, three types per plate) at an approximate concentration of 0.05 × 106 cells/mL, equally split to ensure that all wells had the same cell concentration. The plates were grown at 37 °C degrees. Once all the plates reached 50–70% confluence, plates were assigned treatment; one plate (patient) or two plates (wild type) at each temperature 37 °C and 43 °C. The cells in the remaining plates were lysed and the initial DNA content was measured.
The 37 °C plate and the 43 °C plate underwent the same treatment. The cells were washed three times with Hank’s Balanced Salt Solution (HBSS) and then starved of histidine for 24 hours using histidine-deficient tissue culture media prepared by supplementing Basal Medium Eagle with vitamins and an amino acid mixture to approximate Ham’s F10 media minus histidine. After this period, the media was changed to a complete media with 5% fetal bovine serum and [3H]-histidine (5.0 μCi/well). Wells used for DNA determination were grown in complete media with 5% fetal bovine serum without the added trace [3H]-histidine. Fibroblasts were cultured for an additional 24 hours.
Medium was removed and each well washed with HBSS. Cells were then lysed by addition of 200 μL of distilled H2O and a single freeze/thaw was performed at −80 oC. After thawing, the bottoms of the wells were scraped to collect cell lysates, which were sonicated. The total DNA content for each non-tritiated cell lysate was measured using Quant-IT DNA assay (Thermo Fisher Scientific). For each cell type, the DNA was measured in duplicate.
Bovine serum albumin was added to the [3H]-histidine labelled cell lysates to add mass for the protein precipitation. Protein was precipitated with ice cold 5% trichloroacetic acid and collected on circular 24 mm glass-fiber filters (Whatman GF/C, VWR International) using vacuum flask filtration. Filters were dried overnight and radioactivity was counted on a Beckman LS-1801 scintillation counter.
RESULTS
Purified wild type and Y454S enzymes prepared from human cell culture are active.
In order to compare the kinetics of WT and Y454S HARS, we expressed and purified human FLAG-tagged WT and Y454S HARS from human embryonic kidney (HEK293) cells. The successful purification sequence included affinity and ion exchange chromatography. On the basis of active site titration (described in Experimental Procedures), purified enzymes contain at least 75% active protein (data not shown). A Western blot analysis showed that FLAG-HARS constituted 93% of the purified protein, and less than 7% represented the endogenous untagged WT HARS (Supporting Information Figure S1).
Wild type and Y454S HARS bind tRNA with comparable affinities.
Based on its location in helix 15, the Y454S substitution might indirectly alter tRNA binding, leading to an effect on tRNA discrimination. We tested this hypothesis by the use of an EMSA to measure the tRNAHis binding of the Y454S mutant relative to WT. Binding reactions were conducted under conditions of excess protein with the tRNA at a limiting concentration. Under the conditions reported, WT and Y454S HARS exhibited identical apparent Kds of 7.9 ± 1.1 μM (Figure 2A)These apparent Kds were dependent on assay conditions, most notably the percent of acrylamide gel used in the electrophoresis (data not shown). Therefore, we can use this method only to determine an upper limit of the Kd, and to compare the Kd of enzymes analyzed similarly.
Figure 2.

tRNAHis and total tRNA binding are unaffected by Y454S. A) Y454S and HARS tRNAHis binding forward curve. HARS (circles) and Y454S (triangles) varied from 0.1 to 30 μM. 32P-tRNAHis concentration was 30 nM. B) Competition Plot where transcribed tRNAHis bound to either HARS or Y454S was competed by increasing concentrations of total tRNA purified from human placenta. 32P-tRNAHis concentration was 30 nM, enzyme (HARS (circles) and Y454S (triangles)) was 5 μM and placental tRNA varied from 0 to 100 μM. Values reported are the mean ± standard error of two independent experiments.
In the cell, each ARS must select its correct tRNA from a pool including high concentrations of non-cognate tRNAs. In addition, tRNAHis produced in vivo is modified by conversion of the first guanosine in the anticodon to queuosine, relative to transcribed tRNAHis 44. In order to test whether WT and Y454S differ with respect to non-cognate tRNAs, we carried out a binding competition experiment using 32P labeled tRNAHis transcript as the primary ligand and total human placental tRNA as competitor. (A caveat in the design of this experiment is that total tRNA contains ~1.4% native tRNAHis 9.) As shown in Figure 2B) WT and Y454S responded in an identical fashion to the presence of increasing concentrations of unlabeled competitor tRNA. Thus, under the conditions explored, differences in tRNA binding or tRNA binding specificity that could account for the phenotype of Y454S were not observed.
Multiple turnover kinetics indicates Y454S HARS mutant has comparable kinetic parameters with wild type HARS.
The binding experiments suggested that the Y454S substitution does not significantly affect tRNA recognition at the binding step. To more fully investigate the effect of the substitution on aminoacylation function directly, steady state kinetic parameters were determined for the tRNAHis transcript and histidine substrates. Under steady state conditions where tRNA was the variable substrate, we observed that WT and Y454S HARS had essentially equivalent Km values (WT, 1.2 ± 0.5 μM vs. Y454S, 0.9 ± 0.2 μM) and kcat values (WT, 3.9 ± 0.5 s−1 vs. Y454S, 4.4 ± 0.3 s−1) (Table 1). When histidine was titrated as the variable substrate, the value of Km for Y454S was 1.7-fold decreased relative to WT, and the value of kcat for Y454S was 1.4-fold greater than WT (Table 1). In the previous comparison of mouse WT and Y454S enzymes with bulk human tRNA, the ratio of relative kcat/Km for WT and Y454S HARS was approximately 1.5. Here, with purified human enzymes and substrates, the ratios of kcat/Km for tRNA and for histidine were larger for Y454S than WT, by factors of 1.6 and 2.4 respectively.
Table 1.
Steady state aminoacylation kinetics of tRNAHis by human and bacterial histidyl-tRNA synthetases
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Variable Substrate | |||||||
|
|
|||||||
| tRNA | histidine | ||||||
|
|
|||||||
| Enzyme | Km (μM) | kcat (s−1) | kcat/Km (μM−1 s−1) | Km (μM) | kcat (s−1) | kcat/Km (μM−1 s−1) | References |
| WT HARS | 1.2 ± 0.5 | 3.9 ± 0.6 | 3.2 ± 1.0 | 8.0 ± 4.0 | 4.1 ± 0.4a | 0.5 ± 0.4 | This work |
| Y454S HARS | 0.9 ± 0.2 | 4.4 ± 0.3 | 5.1 ± 1.0 | 4.7 ± 2.5 | 5.9 ± 0.5a | 1.2 ± 0.4 | This work |
| E. coli HisRS | 0.34 ± 0.05 | 1.71 ± 0.06 | 5.0 | 35 ± 4b | 133 ± 2b | 3.8b | 16 |
Values reported are the mean ± standard error of three independent experiments.
Significantly different (extra sum-of-squares F test) p<0.05.
Data are for histidine in the pyrophosphate exchange reaction.
Y454S variant has similar rates of amino acid activation and aminoacyl transfer relative to wild type.
While not large, these differences prompted us to examine the adenylation and aminoacylation half reactions separately. First, we examined the production of AMP under pre-steady state conditions where ATP was the limiting substrate, but still in excess over enzyme. As seen in Figure 3A and 3B, WT and Y454S HARS produced 3.4 μM ± 0.2 and 3.6 μM ± 0.1 of AMP respectively in the first five seconds of the reaction. Based on a concentration of 2.5 μM in HARS dimers or 5 μM monomers in these reactions, this suggests a product stoichiometry of 0.75 molecules of AMP formed per active site. At the end of five seconds, the slopes of the progress curves approached zero suggesting that further production of AMP had ceased. Prior to this plateau period, the rates of product formation were derived by fitting the progress curves in the first 500–800 milliseconds to a single exponential followed by a linear phase. (Other equations, such as an isolated single exponential or a double exponential, produced substantially poorer fits.) These fits provided rates of 18.9 ± 15 s−1 and 18.2 ± 6 s−1, in the exponential phases for WT and Y454S respectively, followed by rates of 2.8 ± 1.2 s−1 and 2.9 ± 0.7 s−1 for the respective linear phases (Table 2). The values of the exponential phases for WT and Y454S were essentially equivalent to the kobs1 for the first exponential for the rate of adenylate formed by the E.coli enzyme under pre-steady state conditions18. This value is thought to represent the rate of activation in the first subunit of the dimer, with the second considerably slower. Consistent with the conclusions from the steady state analysis, these measurements confirm that WT and Y454S catalyze the activation of histidine in the absence of tRNA at the same rate. In the presence of tRNA, the rates of the exponential and linear phases are increased by 2.5 −3 fold for WT and 1.2 −2.6 fold for Y454.
Figure 3.


Amino acid activation and the rate of histidine transfer is unaffected by the Y454S mutation even in the presence of tRNAHis. Amino acid activation (WT: panel A; Y454S: panel B) was monitored by mixing 5 μM monomer HARS enzyme (WT: filled circles; Y454S: filled triangles) with 100 μM ATP and saturating histidine. tRNAHis (25 μM) addition (empty circles and empty triangles, insets: note 0.5 s time scale) increased the rate of amino acid activation, which was determined by linear fit within the first turnover. The rate constant for histidine transfer ktrans (WT: panel C; Y454S: panel D) was determined by single turnover with 2 μM enzyme, preformed with histidyl-adenylate, and mixed with 100 nM tRNAHis. ktrans was determined by fit to a single exponential. The number of experiments is reported in Tables 2 and 3.
Table 2.
Pre-steady state rate for amino acid activation.
|
|
||
|---|---|---|
| Rate of amino acid activation | ||
|
| ||
| Reactants | Rate of exponential phase (s−1) | Rate of linear phase k1 (s−1) |
| WT HARS (n=3) | 19 ± 15 | 3 ± 1a |
| WT HARS + tRNAHis (n=2) | 47 ± 21 | 9.1 ± 0.7a |
| Y454S (n=2) | 18 ± 6 | 2.8 ± 0.7b |
| Y454S HARS + tRNAHis (n=2) | 21 ± 5 | 6.4 ± 0.6b |
Values reported are the mean ± standard error of n independent experiments.
Significantly different at p<0.05.
Significantly different at p<0.01 (extra sum-of-squares F test).
Owing to the fact that the experiments were performed under conditions where the ATP concentration (100 μM) was below Km, the rates reported are unlikely to represent the maximal rate of amino acid activation. Values for the Km for ATP in aminoacylation reactions previously reported in the literature include a value of 380 μM for rat HARS45 and 80 μM for rabbit HARS46. As the HARS sequence has high identity across mammalian species, we expect these values to be a good estimate for human HARS. Accordingly, the maximal possible rates of amino acid activation catalyzed by both the WT and the mutant are likely to be higher than reported here18.
In order to address whether the WT and mutant enzymes differ with respect to the rate of aminoacyl transfer, a single turnover experiment was performed. As described in Methods, WT and Y454S HARS were each pre-incubated with ATP and histidine to pre-form the enzyme-adenylate complex in situ. Using rapid chemical quench techniques, rates of 30 ± 6 s−1 and 30 ± 4 s−1 were determined (Table 3, Figure 3C and 3D). Notably, the single turnover rates of both human enzymes were essentially equivalent, further supporting the kinetic equivalence of WT and Y454S HARS. It is also potentially of interest that these rates were both considerably faster than the 18.1 s−1 previously reported for E. coli HisRS17, 19. In summary, the small increase in Y454S steady state kcat is not accounted for by differences in either isolated pre-steady state amino acid activation or aminoacyl transfer partial reactions.
Table 3.
ktrans for Y454S and WT HARS
| Single turnover aminoacyl transfer | ||
|---|---|---|
|
| ||
| Enzyme | ktrans (s−1) | References |
| WT HARS (n=4) | 30 ± 6 | This work |
| Y454S (n=6) | 30 ± 4 | This work |
| E.coli HARS | 18.8 ± 2.5 | 17 |
Values reported are the mean ± standard error of n independent experiments. No difference by extra sum-of-squares F test.
The Y454S substitution reduces the thermal stability of HARS but not dimerization.
The observations reported above suggested that the Y454S substitution has minimal consequences for HARS function as determined in the standard assays for aminoacylation. Inspection of the human HARS structure leaves open the possibility that the substitution nevertheless influences protein stability by weakening inter-domain dimeric interactions. To further assess the dimer stability of Y454S HARS, we performed sedimentation velocity experiments (SV) and compared the results to those obtained from WT. As an additional comparison, we also evaluated the SV of a temperature sensitive (ts) version of HARS originally isolated in BHK21 cells (tsBN250) that encodes an R362H substitution14. We hypothesized that if a mutant dimer were less stable than the WT, it would exhibit an increased Kd concentration and display a shift in the sedimentation coefficient distribution when it more readily dissociates into monomer. This effect would be enhanced at low concentration. Therefore, we measured the SV experiments at 230 nm where the extinction coefficient is significantly higher compared to the standard 280 nm absorbance, and allows us to monitor at lower concentration. Data quality at 230 nm is also higher due to the high light emission intensity of the Xenon lamp used in the ultracentrifuge.
Molar mass distributions from HARS mutants and WT are shown in Figure 4A, demonstrating remarkable similarity between WT and HARS mutants. The molar mass of the major species observed for each sample is in excellent agreement with the dimer form of HARS. Detailed integration results for the Monte Carlo analysis are reported in Table 4. Pseudo-3D overlays of the anisotropy vs. sedimentation coefficient for each species are shown in Figure 4B. These results conclusively show that the mutant forms of HARS are equally stable under the conditions used for the SV experiments as HARS WT.
Figure 4.

A. Molar mass distributions determined by sedimentation velocity, showing the PCSA-Monte Carlo distributions for HARS WT (black), HARS Y454S (red), and HARS R362H (blue). Molar masses are in excellent agreement with a dimer of each species. B. Frictional ratio anisotropies for HARS WT (black), HARS Y454S (red), and HARS R362H (blue) as a function of the sedimentation coefficient. Pseudo-3D plots derived from PCSA-Monte Carlo analyses of sedimentation velocity experiments. The right vertical axis indicates the partial concentration in terms of color density. All three samples show remarkable similarity.
Table 4.
PCSA – Monte Carlo results (straight-line parameterization) from the SV experiment.
| Species | Molar mass (kDa) | D (× 10−7 cm/sec2) | s (× 10−13 sec) | f/f0 | RMSD |
|---|---|---|---|---|---|
| WT | 118.5 (111.6, 125.5) | 4.54 (4.43, 4.66) | 5.67 (5.48, 5.85) | 1.44 (1.43, 1.45) | 0.002484 |
| Y454S | 116.5 (108.6, 124.3) | 4.62 (4.46, 4.77) | 5.66 (5.47, 5.84) | 1.43 (1.41, 1.44) | 0.002468 |
| R362H | 117.7 (109.8, 125.5) | 4.55 (4.40, 4.71) | 5.63 (5.45, 5.81) | 1.44 (1.43, 1.46) | 0.002825 |
Values in parenthesis are 95% confidence intervals from the Monte Carlo analysis. The theoretical molar mass of HARS WT dimer is 114.9 kDa, indicating the observed species is a dimer. The residual mean square deviation (RMSD) of the fit is shown in units of absorbance at 230 nm.
The work described thus far suggests that the Y454S substitution has minimal effect on substrate recognition (when assessed kinetically) and overall dimer formation. However, these prior analyses might not be able to detect subtler structural effects, such as those associated with localized protein unfolding. To test this hypothesis, we used differential scanning fluorimetry (DSF) to measure the transition temperature for molten globular formation, which will be referred to herein as Tm. Melting profiles were initially performed in the absence of substrates, yielding Tm’s of 50.7 ± 0.4 °C and 45.5 ± 0.7 °C for WT and Y454S HARS, respectively (Figure 5B, summarized in Table 5). The 5 °C temperature decrease observed with the mutant relative to WT provided the first evidence in temperature instability for Y454S. The unliganded melting profile for Y454S was only 2°C higher than the value determined for ts R362H (44.1 ± 0.2 °C), a bonafide temperature sensitive mutant in HARS described earlier14. The close Tm values for Y454S and R362H support the conclusion that the Y454S substitution destabilizes the HARS enzyme structure in a manner similar to R362H and in fact confers temperature sensitivity.
Figure 5.

Y454S mutation is thermally less stable relative to WT HARS. 10 μM enzyme (WT black circles, Y454S red triangles and R362H blue squares) was incubated with Sypro Orange dye in the absence (filled symbols) of substrate, or presence of 5 mM histidine (empty symbols). A) Wild type ± histidine. B) Wild type vs. Y454S. C) Y454S ± histidine. D) R362H ± histidine. Thermal shift results are from two preparations of WT and Y454S enzymes each measured in triplicate.
Table 5.
Thermal stability of HARS enzymes with substrates
| Enzyme | Tm °C | Δ Tm °Ca | Δ Tm °Cb |
|---|---|---|---|
| WT | 51.7 ± 0.4c | - | |
| WT + histidine | 58.8 ± 0.4 | 7.0 | - |
| WT + ATP | 53.8 ± 0.2 | 2.0 | - |
| WT + tRNAHis | 53.4 ± 0.4 | 1.7 | - |
| Y454S | 46.3 ± 0.7c | - | −5.4 |
| Y454S + histidine | 54.4 ± 0.5 | 8.1 | −4.4 |
| Y454 + ATP | 47.0 ± 0.5 | 0.7 | −6.8 |
| Y454S + tRNAHis | 48.5 ± 0.4 | 2.2 | −4.9 |
| R362H | 44.1 ± 0.2c | - | −7.7 |
| R362H + histidine | 50.4 ± 0.4 | 6.3 | −8.4 |
| R362H + ATP | 48.2 ± 0.5 | 4.1 | −5.6 |
| R362H + tRNAHis | 51.3 ± 0.3 | 7.2 | −2.1 |
Relative to the apo form of the same enzyme.
Relative to the same form of WT HARS enzyme. Values reported are the mean ± standard error of 2 independent experiments in triplicate (1 experiment in triplicate for R362H).
Values that are significantly different WT to Y454S or R362H, p<0.0001 (extra sum-of-squares F test).
Previously, it has been shown that histidine is a stabilizing substrate for HARS4. We therefore evaluated the effect of all three HARS substrates on the apparent melting transition of the enzymes. Consistent with the conclusions that histidine stabilizes the HARS active site, the addition of 5 mM histidine increased the Tm for all three enzymes by 6.3 – 8 °C. By contrast, ATP and tRNAHis showed less pronounced stabilizing effects on Tm, providing increases in the Tm in the range of 1 – 2 °C for both WT and Y454S mutant enzymes. Interestingly, the R362H substitution exhibited larger stabilizing effects for these substrates, corresponding to 4.1 and 7.2 °C for ATP and tRNA, respectively. In summary, both Y454S and R362H HARS displayed significant decreases in thermal stability relative to the WT protein, in the presence and absence of substrates. While both substitutions could be described as conferring temperature sensitivity, the two mutations were distinguished by different responses to individual substrates, with R362H exhibiting substantially increased stability with binding all three substrates, and Y454S responding only to histidine (Table 5 and Figure 5C).
Elevated temperature results in lowered de novo protein synthesis in patient fibroblasts.
The observed reduced thermal stability of the Y454S substituted HARS and the clinical association of acute illnesses including hallucinations and interstitial lung disease with febrile events in USH3B patients prompted us to assess the incorporation of 3H–histidine into total protein, a measure of cellular protein translation, in cultured WT and Y454S human primary fibroblasts. WT and Y454S fibroblasts incorporated 3H–histidine into cultured fibroblasts at the same rate at 37 °C. However, when the fibroblasts were cultured at 43 °C, the incorporation of 3H –histidine into total cellular protein in the WT fibroblasts was about 7 –fold greater than in the Y454S fibroblasts. (Figure 6). Thus, the temperature sensitivity of Y454S detected in vitro has consequences for protein synthesis in vivo.
Figure 6.

Human fibroblasts from patients with the Y454S mutation demonstrate less de novo protein synthesis at elevated temperatures than wild type cells. Scatter plot showing 3H-histidine incorporation into total cellular protein (values normalized to cellular DNA), Y454S cells (circles) and wild type cells (squares). Values shown include three replicates each for three Y454S cell lines and three replicates each for six wild type cell lines (culture temperature 37 °C solid symbols, 43 °C open symbols). Bars indicate the mean +/− SEM. Statistical significances were determined by Welch’s t-test.
DISCUSSION
In this study, we critically evaluated the hypotheses that USH3B results from a decrease in canonical HARS catalyzed aminoacylation function. Rates were determined for both partial reactions, including the amino acid activation and aminoacyl transfer steps, and the overall rate of aminoacylation. The main conclusion from these studies is that, under the conditions examined, no significant differences in aminoacylation could be observed in a comparison of WT and Y454S. At the gross structural level, sedimentation velocity experiments revealed no difference in the strength of dimer formation. By contrast, the DSF assay indicated that the two proteins exhibited significant differences in the Tm. In addition, translation was significantly impaired in USH3B patient fibroblasts at elevated temperatures. These results provide a new context to rationalize the linkage of the Y454S HARS mutant to USH3B.
Structural context of the Y454S substitution, and its apparent minimal effect on aminoacylation.
The USH3B Y454S substitution is located on helix 15 on the surface of the anticodon-binding domain, which is located proximal to the dimer surface of the catalytic domain of the opposing monomer3. The substitution of serine for Tyr454 (Figure 1C) changes the hydrophobic character of this position, while also removing a likely hydrogen bond with Glu 439. Tyr454 is conserved among eukaryotic species extending from the human to yeast (Saccharomyces) (Figure 1D)4. In bacterial species (such as Escherichia coli and Staphylococcus aureus) however, the residue corresponding to Y454 positions is a charged residue (Figure 1D), and in the case of Thermus thermophilus, hydrophobic. The helix harboring the Y454S substitution in the anticodon-binding domain is predicted to make direct or indirect contacts to the phosphodiester backbone of the tRNA anticodon arm5. While initial expectations were that the mutation might affect binding affinity to tRNA, our results indicate that tRNA binding specificity of Y454S was not disrupted (Figure 2). Previously, an Escherichia coli HisRS mutant with the double substitution of G96D and R104H was uncovered in a genetic selection for altered tRNA binding specificity47. Kinetic characterization of the mutant protein showed that it exhibits reduced discrimination for the specific sequence of the tRNAHis anticodon47. While the similar location of R104 and Y454 in the catalytic:anticodon-binding domain interface encourages one to speculate about similar functional effects, the E. coli mutant enzyme with the single R104H mutation remains to be characterized. Modeling studies of the human HARS:tRNAHis complex (not shown) suggest that Y454 is likely to have at most only an indirect effect on tRNA binding through perturbations of contacts to the phosphodiester backbone, which is in agreement with our results.
A detailed analysis of human HARS kinetics showed that with respect to the individual half reactions of aminoacylation and with respect to all parameters examined, WT and Y454S were virtually equivalent. Key parameters that were essentially equivalent for the two enzymes included the Km for tRNA and the overall kcat for aminoacylation when tRNA was the variable substrate. When histidine was the variable substrate in the presence of saturating tRNA Y454S HARS was slightly more active, as reflected in a higher kcat and slightly lower Km.
The discrepancy in values of kcat between conditions where tRNA was saturating and conditions where histidine was saturating could arise from several sources. One possibility is that there is a discrepancy in the calculation of active sites for different preparations, which would affect the values of kcat. Alternatively, the Y454S mutant could have a bona fide differential interaction with tRNA, leading to either an increased percentage of correctly folded protein in the presence of saturating tRNA, or a faster dissociation of aminoacylated product. Either scenario highlights the fact that kcat measures the limiting rate in the overall catalytic cycle, and incorporates all elementary steps including product release. Clearly, not all of these features nor the full catalytic contribution of the dimeric structure (e.g. potential alternating catalysis in the two active sites of the dimer19) are captured in the pre-steady state experiments. In any event, the increase in kcat seen with Y454S HARS is relatively slight, and supports the conclusion that the effect of the mutant in Usher syndrome is unlikely to be due to a loss in catalytic efficiency, as measured in a purified system. These findings support the conclusions of an earlier study that reported less than a two-fold difference in aminoacylation functions of the WT and Y454S enzymes9. Notably, the values of Km for tRNAHis determined in the two studies were virtually identical, despite the fact that the earlier study employed murine enzyme preparations and total human tRNA as substrate.
Adenylation kinetics determined in the presence of tRNA highlighted a potential difference from the E. coli enzyme. In the E.coli enzyme, the progress curve for the formation of AMP in the absence of tRNA is best described as a double exponential followed by a linear rate18. In the presence of tRNA, these complex kinetics reduce to a linear progress curve over multiple turnovers17. This was interpreted to suggest that the presence of tRNA inhibits the rate of amino acid activation in the first subunit of the dimer, while increasing the same rate in the second subunit. The equalization of rates is thought to reflect the coupling of activities of the two active sites in the dimer and the linkage of dissociation of product from subunit one to formation of product in subunit two18. The rate of adenylation in the human HARS enzyme did not show the same attenuation by the presence of tRNA, suggesting a potential mechanistic difference between these structurally similar enzymes. This difference remains to be explored further in additional experiments.
The Y454S substitution is less thermally stable compared to wild type HARS.
While the results of standard aminoacylation assays argue strongly against the model that the USH3B phenotype arises from an aminoacylation defect, the potential structural changes (Figure 1) suggest a possible decrease in protein stability by virtue of the weakening intra- or inter-domain interactions. Here, we used sedimentation velocity analysis to assess the dimer status of the mutant, and saw no apparent weakening of dimeric structure (Figure 4). SV experiments characterize the solution behavior of macromolecules and report on the sedimentation and diffusion behavior of all species in a mixture. Information is also provided about the partial concentrations of various species, buoyant molar masses, and anisotropies. Even at low concentrations, HARS and the temperature sensitive mutants maintained their dimeric structure, indicating the mutation alone does not alter enzyme dimerization, consistent with the binding and kinetic analyses.
Originally developed as a high throughput method for quantitatively assessing the strength of small molecule protein interactions, the DSF assay has proven useful in other contexts, such as profiling protein constructs for their suitability in crystallographic studies. Here, DSF assays were used to indirectly compare the Tm of WT and Y454S HARS, and the effects of substrates on protein stability. The clear result was that the Y454S substitution decreases the Tm for HARS in the range of 5–6 °C. Moreover, as evidenced by the gain in Tm, each of the three canonical substrates (His, ATP and tRNAHis) provided the same enhancement of protein stability as seen with WT HARS. This supports the earlier conclusion that Y45S has minimal effect on the recognition of any of individual substrates, versus overall stability. By contrast, the R362H mutant showed increased Tm in the presence of ATP and tRNA relative to the other proteins, suggesting a greater propensity to stabilization by these substrates. It will be of interest to determine whether this signature holds true for other bona fide temperature sensitive mutants.
The inability of fibroblast cultures from patients with the HARS Y454S mutation to incorporate [3H]-histidine into total cellular protein at elevated temperature, when compared to control fibroblasts, is a key finding. Notably, there was no difference in incorporation at 37 °C, but only when the cell was stressed at 43 °C. Although the temperature used in the cell culture experiments cannot be directly compared with febrile conditions in patients or with the DSF results, this finding comports with the clinical observation that febrile illnesses evoke the USH3B phenotype9,
These observations and the qualitatively similar results obtained with R362H HARS strongly suggest that decreased temperature stability underlies the molecular basis of the USH3B syndrome. This interpretation is supported by the direct experimental comparison of Y454S HARS to the R362H substitution (Figure 5D). The latter mutant was derived from the tsBN250 BHK cell line, which was isolated in an unbiased screen for cell cycle arrest mutants14, 48. Consistent with a temperature sensitive phenotype, tsBN250 cells exhibited decreased cell growth and increased apoptosis at elevated temperatures. In both the tsBN250 and related tsBN269 BHK cells (which carry a ts LysRS allele), cell cycle arrest was attributed to decreased cyclin D1 production, which resulted from decreased 3H incorporation into the total protein pool. Growth defects could be overcome by supplementation of high concentrations of histidine or inducing expression of cyclin D114. Given that Y454S and R362H HARS are accompanied by similar decreases in protein stability, we can hypothesize that elevated temperatures (or other physiological stresses) might cause reduced protein synthesis in the mutant cells, leading to cell cycle arrest and apoptosis. In a specific G296D ValRS mutant isolated in C. elegans, altered editing domain function is predicted to reduce global translation by inappropriately deacylating Val-tRNAVal at high temperatures. The reduction in aminoacylated Val-tRNAVal subsequently results in mitotic cell arrest of stem cells within the organism49.
This hypothesis might at first glance appear to contradict some observed features of Y454S HARS, including the lack of an apparent effect on intracellular protein levels9 and a lack of an effect on primary aminoacylation function (this work). Of note, the intracellular protein levels were determined in unstressed cells cultured at normal temperature 9. Again, the comparison with R362H HARS is instructive. Notably, no apparent degradation of the R362H enzyme was observed in tsBN250 BHK at increased temperatures14, 48. Further, thermal sensitive aminoacyl-tRNA synthetase mutations were originally described as mutations that disrupt normal cellular growth and division, and sensitivity to increased temperature in CHO cell lines with ARS defects could be alleviated by high concentrations of cognate amino acid50. These CHO cell lines are His-1, with a temperature sensitive mutation in HisRS, and Arg-1, with a temperature and arginine-sensitive mutation in arginyl-tRNA synthetase50. Curiously, while His-1 and Arg-1 CHO cell lines showed decreased 3H incorporation into the total protein pool, there was not a clear impairment of aminoacylation activity in vivo50. Accordingly, our inability to detect decreased aminoacylation function with Y454S HARS under specified in vitro conditions may not rule out the possibility of decreased function in vivo, particularly at elevated temperatures. Along these lines, it should prove instructive to conduct a kinetic characterization of the R362H enzyme, and determine whether or not it too exhibits relatively robust aminoacylation function in vitro.
Comparison to other ARS linked diseases.
Our study adds to a growing body of literature in which mutations encoding missense substitutions in aminoacyl-tRNA synthetases have been linked to a variety of neurological disorders, most notably CMT syndrome51, 52. Among the tRNA synthetases implicated in this way are GlyRS24, 52, 53, AlaRS22, 25, GlnRS54, LysRS23, and TyrRS55. While for many of these examples a significant loss in aminoacylation function has been documented24, 52, 53, there are also a few notable reports where retention of aminoacylation activity has been claimed56, 57. These latter examples raise the possibility that, at least for some ARS-linked diseases, the molecular basis of pathophysiology may arise from features that are distinct from a loss of primary function under normal physiological conditions.
CONCLUSIONS
Here, we have shown that perturbation of the primary aminoacylation function is unlikely to fully account for the basis of disease for the Y454S mutation, as compared to other ARS mutants linked to CMT that exhibit a clear loss of aminoacylation function when measured in vitro24. Further work will be necessary to determine how the thermal instability of the Y454S HARS enzyme plays a role in USH3B disease progression in the context of the cellular environment. A multi-factorial approach will be required, with investigations into the role of Y454S in the cellular stress response and Y454S HARS interactions with other proteins, which may entail HARS secondary functions, yet to be identified.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Karen Lounsbury for use of her cell culture facilities; Dr. Jeff Bond for helpful discussions on kinetics and statistics; Dr. D. Holmes Morton, Dr. Robert Jinks for the kind gifts of plasmids and Wendy McCaul for culturing the fibroblasts at the London Health Sciences Centre.
Funding Sources
This research is funded by the Hearing Health Foundation to S.R.B.; and the National Institutes of Health [GM54899 to C.S.F. and T32 HL007594–30 to J.A.A]. The automated DNA sequencing, molecular imaging, digital western visualizations and differential scanning fluorimetry were performed in the University of Vermont Cancer Center Advanced Genome Technologies Core. BD acknowledges the support of the San Antonio Cancer Institute grant P30 CA054174 for support of the Center for Analytical Ultracentrifugation of Macromolecular Assemblies at the University of Texas Health Science Center at San Antonio. A Rare Disease Foundation grant to CAR and VMS supported the primary fibroblast work.
ABBREVIATIONS
- ARS
aminoacyl-tRNA synthetases
- HARS
histidyl-tRNA synthetase
- USH3B
Type IIIB Usher Syndrome
- CMT
Charcot-Marie-Tooth
- HisRS
prokaryotic histidyl tRNA-synthetase
- pCAG/FLAG/RFC/A
plasmid with CAG promotor and FLAG-tag from GateWay vector Reading Frame Cassette A
- DSF
differential scanning fluorimetry
- Tm
thermal melting temperature
- ts
temperature sensitive
- PEI
polyethyleneimine
- β-ME
β-mercaptoethanol
- PPiase
inorganic pyrophosphatase
- NaOAc
sodium acetate, sedimentation velocity experiments (SV)
- EMSA
electromobility shift assay
Footnotes
The authors declare no competing financial interest.
SUPPORTING INFORMATION
Western blot analysis of purified HARS proteins (Figure S1)
REFERENCES
- [1].Ibba M, and Soll D. (2004) Aminoacyl-tRNAs: setting the limits of the genetic code, Genes Dev. 18, 731–738. [DOI] [PubMed] [Google Scholar]
- [2].Carter CW Jr. (1993) Cognition, mechanism, and evolutionary relationships in aminoacyl-tRNA synthetases., Annu. Rev. Biochem 62, 715–748. [DOI] [PubMed] [Google Scholar]
- [3].Xu Z, Wei Z, Zhou JJ, Ye F, Lo WS, Wang F, Lau CF, Wu J, Nangle LA, Chiang KP, Yang XL, Zhang M, and Schimmel P. (2012) Internally deleted human tRNA synthetase suggests evolutionary pressure for repurposing, Structure 20, 1470–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Koh CY, Wetzel AB, de van der Schueren WJ, and Hol WG (2014) Comparison of histidine recognition in human and trypanosomatid histidyl-tRNA synthetases, Biochimie 106C, 111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Tian Q, Wang C, Liu Y, and Xie W. (2015) Structural basis for recognition of G-1-containing tRNA by histidyl-tRNA synthetase, Nucleic Acids Res. [DOI] [PMC free article] [PubMed]
- [6].Merritt EA, Arakaki TL, Gillespie JR, Larson ET, Kelley A, Mueller N, Napuli AJ, Kim J, Zhang L, Verlinde CL, Fan E, Zucker F, Buckner FS, van Voorhis WC, and Hol WG (2010) Crystal structures of trypanosomal histidyl-tRNA synthetase illuminate differences between eukaryotic and prokaryotic homologs, J. Mol. Biol 397, 481–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Arnez JG, Harris DC, Mitschler A, Rees B, Francklyn CS, and Moras D. (1995) Crystal structure of histidyl-tRNA synthetase from Escherichia coli complexed with histidyl-adenylate, The EMBO journal 14, 4143–4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Aberg A, Yaremchuk A, Tukalo M, Rasmussen B, and Cusack S. (1997) Crystal structure analysis of the activation of histidine by Thermus thermophilus histidyl-tRNA synthetase, Biochemistry 36, 3084–3094. [DOI] [PubMed] [Google Scholar]
- [9].Puffenberger EG, Jinks RN, Sougnez C, Cibulskis K, Willert RA, Achilly NP, Cassidy RP, Fiorentini CJ, Heiken KF, Lawrence JJ, Mahoney MH, Miller CJ, Nair DT, Politi KA, Worcester KN, Setton RA, Dipiazza R, Sherman EA, Eastman JT, Francklyn C, Robey-Bond S, Rider NL, Gabriel S, Morton DH, and Strauss KA (2012) Genetic mapping and exome sequencing identify variants associated with five novel diseases, PLoS One 7, e28936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Vester A, Velez-Ruiz G, McLaughlin HM, Lupski JR, Talbot K, Vance JM, Zuchner S, Roda RH, Fischbeck KH, Biesecker LG, Nicholson G, Beg AA, and Antonellis A. (2013) A loss-of-function variant in the human histidyl-tRNA synthetase (HARS) gene is neurotoxic in vivo, Hum. Mutat 34, 191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Safka Brozkova D, Deconinck T, Beth Griffin L, Ferbert A, Haberlova J, Mazanec R, Lassuthova P, Roth C, Pilunthanakul T, Rautenstrauss B, Janecke AR, Zavadakova P, Chrast R, Rivolta C, Zuchner S, Antonellis A, Beg AA, De Jonghe P, Senderek J, Seeman P, and Baets J. (2015) Loss of function mutations in HARS cause a spectrum of inherited peripheral neuropathies, Brain. [DOI] [PMC free article] [PubMed]
- [12].Pierce SB, Chisholm KM, Lynch ED, Lee MK, Walsh T, Opitz JM, Li W, Klevit RE, and King MC (2011) Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome, Proc. Natl. Acad. Sci. U. S. A 108, 6543–6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Mahler M, Miller FW, and Fritzler MJ (2014) Idiopathic inflammatory myopathies and the anti-synthetase syndrome: a comprehensive review, Autoimmun Rev 13, 367–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Motomura S, Fukushima K, Nishitani H, Nawata H, and Nishimoto T. (1996) A hamster temperature-sensitive G1 mutant, tsBN250 has a single point mutation in histidyl-tRNA synthetase that inhibits an accumulation of cyclin D1, Genes Cells 1, 1101–1112. [DOI] [PubMed] [Google Scholar]
- [15].Himeno H, Hasegawa T, Ueda T, Watanabe K, Miura K, and Shimizu M. (1989) Role of the extra G-C pair at the end of the acceptor stem of tRNA(His) in aminoacylation, Nucleic Acids Res. 17, 7855–7863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hawko SA, and Francklyn CS (2001) Covariation of a specificity-determining structural motif in an aminoacyl-tRNA synthetase and a tRNA identity element, Biochemistry 40, 1930–1936. [DOI] [PubMed] [Google Scholar]
- [17].Guth E, Connolly SH, Bovee M, and Francklyn CS (2005) A substrate-assisted concerted mechanism for aminoacylation by a class II aminoacyl-tRNA synthetase, Biochemistry 44, 3785–3794. [DOI] [PubMed] [Google Scholar]
- [18].Guth E, Farris M, Bovee M, and Francklyn CS (2009) Asymmetric amino acid activation by class II histidyl-tRNA synthetase from Escherichia coli, J. Biol. Chem 284, 20753–20762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Guth EC, and Francklyn CS (2007) Kinetic discrimination of tRNA identity by the conserved motif 2 loop of a class II aminoacyl-tRNA synthetase, Mol. Cell 25, 531–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Sang Lee J, Gyu Park S, Park H, Seol W, Lee S, and Kim S. (2002) Interaction network of human aminoacyl-tRNA synthetases and subunits of elongation factor 1 complex, Biochem. Biophys. Res. Commun 291, 158–164. [DOI] [PubMed] [Google Scholar]
- [21].Kaminska M, Deniziak M, Kerjan P, Barciszewski J, and Mirande M. (2000) A recurrent general RNA binding domain appended to plant methionyl-tRNA synthetase acts as a cis-acting cofactor for aminoacylation, EMBO J. 19, 6908–6917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].McLaughlin HM, Sakaguchi R, Giblin W, Program NCS, Wilson TE, Biesecker L, Lupski JR, Talbot K, Vance JM, Zuchner S, Lee YC, Kennerson M, Hou YM, Nicholson G, and Antonellis A. (2012) A recurrent loss-of-function alanyl-tRNA synthetase (AARS) mutation in patients with Charcot-Marie-Tooth disease type 2N (CMT2N), Hum. Mutat 33, 244–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].McLaughlin HM, Sakaguchi R, Liu C, Igarashi T, Pehlivan D, Chu K, Iyer R, Cruz P, Cherukuri PF, Hansen NF, Mullikin JC, Biesecker LG, Wilson TE, Ionasescu V, Nicholson G, Searby C, Talbot K, Vance JM, Zuchner S, Szigeti K, Lupski JR, Hou YM, Green ED, and Antonellis A. (2010) Compound heterozygosity for loss-of-function lysyl-tRNA synthetase mutations in a patient with peripheral neuropathy, Am. J. Hum. Genet 87, 560–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Griffin LB, Sakaguchi R, McGuigan D, Gonzalez MA, Searby C, Zuchner S, Hou YM, and Antonellis A. (2014) Impaired function is a common feature of neuropathy-associated glycyl-tRNA synthetase mutations, Hum. Mutat 35, 1363–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Simons C, Griffin LB, Helman G, Golas G, Pizzino A, Bloom M, Murphy JL, Crawford J, Evans SH, Topper S, Whitehead MT, Schreiber JM, Chapman KA, Tifft C, Lu KB, Gamper H, Shigematsu M, Taft RJ, Antonellis A, Hou YM, and Vanderver A. (2015) Loss-of-function alanyl-tRNA synthetase mutations cause an autosomal-recessive early-onset epileptic encephalopathy with persistent myelination defect, Am. J. Hum. Genet 96, 675–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Fersht AR, Ashford JS, Bruton CJ, Jakes R, Koch GLE, and Hartley BS (1975) Active site titration and aminoacyl adenylate binding stoichiometry of aminoacyl-tRNA synthetases, Biochemistry 14, 1–4. [DOI] [PubMed] [Google Scholar]
- [27].Francklyn CS, First EA, Perona JJ, and Hou YM (2008) Methods for kinetic and thermodynamic analysis of aminoacyl-tRNA synthetases, Methods 44, 100–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Milligan JR, Groebe DR, Witherell GW, and Uhlenbeck OC (1987) Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates, Nucleic Acids Res. 15, 8783–8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wolfson AD, Pleiss JA, and Uhlenbeck OC (1998) A new assay for tRNA aminoacylation kinetics, RNA 4, 1019–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Swillens S. (1995) Interpretation of binding curves obtained with high receptor concentrations: practical aid for computer analysis, Mol. Pharmacol 47, 1197–1203. [PubMed] [Google Scholar]
- [31].Ledoux S, and Uhlenbeck OC (2008) [3’−32P]-labeling tRNA with nucleotidyltransferase for assaying aminoacylation and peptide bond formation, Methods 44, 74–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Demeler B, Gorbet G, Zollars D, Dubbs B, Brookes E, and Cao W. (2016) UltraScan-III version 3.5: A comprehensive data analysis software package for analytical ultracentrifugation experiments, http://www.ultrascan3.uthscsa.edu/.
- [33].Demeler B, and Gorbet G. (2016) In Analytical Ultracentrifugation Data Analysis with UltraScan-III. Analytical Ultracentrifugation: Instrumentation, Software, and Applications (Uchiyama S, Stafford WF, and Laue T), pp 119–143, Springer. [Google Scholar]
- [34].Laue TM, Shah BD, Ridgeway TM, and Pelletier SL (1992) Analytical Ultracentrifugation in Biochemistry and Polymer Science, (Harding S, and Rowe A, Eds.), pp 90–125, Royal Society of Chemistry.
- [35].Brookes E, Cao W, and Demeler B. (2010) A two-dimensional spectrum analysis for sedimentation velocity experiments of mixtures with heterogeneity in molecular weight and shape, Eur. Biophys. J 39, 405–414. [DOI] [PubMed] [Google Scholar]
- [36].Schuck P, and Demeler B. (1999) Direct sedimentation analysis of interference optical data in analytical ultracentrifugation, Biophys. J 76, 2288–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Demeler B. (2010) Methods for the design and analysis of sedimentation velocity and sedimentation equilibrium experiments with proteins, Curr Protoc Protein Sci Chapter 7, Unit 7 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Gorbet G, Devlin T, Hernandez Uribe BI, Demeler AK, Lindsey ZL, Ganji S, Breton S, Weise-Cross L, Lafer EM, Brookes EH, and Demeler B. (2014) A parametrically constrained optimization method for fitting sedimentation velocity experiments, Biophys. J 106, 1741–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Demeler B, and Brookes E. (2008) Monte Carlo analysis of sedimentation experiments, Colloid Polym Sci 286, 129–137. [Google Scholar]
- [40].Brookes EH, and Demeler B. (2008) Parallel computational techniques for the analysis of sedimentation velocity experiments in UltraScan, Colloid Polym. Sci 286, 138–148. [Google Scholar]
- [41].Williams TL, Yin YW, and Carter CW Jr. (2016) Selective Inhibition of Bacterial Tryptophanyl-tRNA Synthetases by Indolmycin Is Mechanism-based, J. Biol. Chem 291, 255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Abbott JA, Livingston NM, Egri SB, Guth E, and Francklyn CS (2017) Characterization of aminoacyl-tRNA synthetase stability and substrate interaction by differential scanning fluorimetry, Methods 113, 64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Niesen FH, Berglund H, and Vedadi M. (2007) The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability, Nat. Protoc 2, 2212–2221. [DOI] [PubMed] [Google Scholar]
- [44].Katze JR, and Farkas WR (1979) A factor in serum and amniotic fluid is a substrate for the tRNA-modifying enzyme tRNA-guanine transferase, Proc. Natl. Acad. Sci. U. S. A 76, 3271–3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Fahoum SK, and Yang DC (1987) Purification of mammalian histidyl-tRNA synthetase and its interaction with myositis-specific anti-Jo-1 antibodies, Biochemistry 26, 5871–5877. [DOI] [PubMed] [Google Scholar]
- [46].Kane SM, Vugrincic C, Finbloom DS, and Smith DW (1978) Purification and some properties of the histidyl-tRNA synthetase from the cytosol of rabbit reticulocytes, Biochemistry 17, 1509–1514. [DOI] [PubMed] [Google Scholar]
- [47].Yan W, Augustine J, and Francklyn C. (1996) A tRNA identity switch mediated by the binding interaction between a tRNA anticodon and the accessory domain of a class II aminoacyl-tRNA synthetase, Biochemistry 35, 6559–6568. [DOI] [PubMed] [Google Scholar]
- [48].Fukushima K, Motomura S, Kuraoka A, Nakano H, and Nishimoto T. (1996) A single point mutation of hamster aminoacyl-tRNA synthetase causes apoptosis by deprivation of cognate amino acid residue, Genes Cells 1, 1087–1099. [DOI] [PubMed] [Google Scholar]
- [49].Rastogi S, Borgo B, Pazdernik N, Fox P, Mardis ER, Kohara Y, Havranek J, and Schedl T. (2015) Caenorhabditis elegans glp-4 Encodes a Valyl Aminoacyl tRNA Synthetase, G3 (Bethesda) 5, 2719–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Thompson LH, Lofgren DJ, and Adair GM (1977) CHO cell mutants for arginyl-, asparagyl-, glutaminyl-, histidyl- and methionyl-transfer RNA synthetases: identification and initial characterization, Cell 11, 157–168. [DOI] [PubMed] [Google Scholar]
- [51].Sivakumar K, Kyriakides T, Puls I, Nicholson GA, Funalot B, Antonellis A, Sambuughin N, Christodoulou K, Beggs JL, Zamba-Papanicolaou E, Ionasescu V, Dalakas MC, Green ED, Fischbeck KH, and Goldfarb LG (2005) Phenotypic spectrum of disorders associated with glycyl-tRNA synthetase mutations, Brain 128, 2304–2314. [DOI] [PubMed] [Google Scholar]
- [52].Antonellis A, Lee-Lin SQ, Wasterlain A, Leo P, Quezado M, Goldfarb LG, Myung K, Burgess S, Fischbeck KH, and Green ED (2006) Functional analyses of glycyl-tRNA synthetase mutations suggest a key role for tRNA-charging enzymes in peripheral axons, J. Neurosci 26, 10397–10406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Deng X, Qin X, Chen L, Jia Q, Zhang Y, Zhang Z, Lei D, Ren G, Zhou Z, Wang Z, Li Q, and Xie W. (2016) Large Conformational Changes of Insertion 3 in Human Glycyl-tRNA Synthetase (hGlyRS) during Catalysis, J. Biol. Chem 291, 5740–5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Zhang X, Ling J, Barcia G, Jing L, Wu J, Barry BJ, Mochida GH, Hill RS, Weimer JM, Stein Q, Poduri A, Partlow JN, Ville D, Dulac O, Yu TW, Lam AT, Servattalab S, Rodriguez J, Boddaert N, Munnich A, Colleaux L, Zon LI, Soll D, Walsh CA, and Nabbout R. (2014) Mutations in QARS, encoding glutaminyl-tRNA synthetase, cause progressive microcephaly, cerebral-cerebellar atrophy, and intractable seizures, Am. J. Hum. Genet 94, 547–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Jordanova A, Irobi J, Thomas FP, Van Dijck P, Meerschaert K, Dewil M, Dierick I, Jacobs A, De Vriendt E, Guergueltcheva V, Rao CV, Tournev I, Gondim FA, D’Hooghe M, Van Gerwen V, Callaerts P, Van Den Bosch L, Timmermans JP, Robberecht W, Gettemans J, Thevelein JM, De Jonghe P, Kremensky I, and Timmerman V. (2006) Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy, Nat. Genet 38, 197–202. [DOI] [PubMed] [Google Scholar]
- [56].Froelich CA, and First EA (2011) Dominant Intermediate Charcot-Marie-Tooth disorder is not due to a catalytic defect in tyrosyl-tRNA synthetase, Biochemistry 50, 7132–7145. [DOI] [PubMed] [Google Scholar]
- [57].van Berge L, Kevenaar J, Polder E, Gaudry A, Florentz C, Sissler M, van der Knaap MS, and Scheper GC (2013) Pathogenic mutations causing LBSL affect mitochondrial aspartyl-tRNA synthetase in diverse ways, Biochem. J 450, 345–350. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.