

Summary
Immune cell subgroups in peripheral blood mononuclear cells (PBMCs) in primary Sjogren’s syndrome (pSS) are thought to regulate immune responses, but the nature and functions of these subgroups remain unclear. Here we performed single-cell RNA sequencing (scRNA-seq) of about 68,500 PBMCs from three patients with pSS and three healthy controls (HCs). We found that CD14+ monocytes from pSS patients expressed high levels of the transcription factor CEBPD, and the direct regulation of target genes expression by CEBPD tends to participate in the TNF-α signaling via NF-κB in monocytes. FOLR3 and IL1B were upregulated separately in CD14+ monocyte subsets from different pSS patients. We proposed a system for classifying CD56−CD16+ NK cells based on FCER1G expression. Compared with HCs, pSS patients showed a significantly higher ratio of CD56−CD16+FCER1G+ NK cells to CD56−CD16+FCER1G- NK cells. Our analysis provides a reference dataset and reveals its immune heterogeneity among PBMCs in pSS.
Subject areas: Immunology, Components of the immune system, Transcriptomics
Graphical abstract
Highlights
-
•
Generation of reference single cell atlas for primary Sjögren’s syndrome (pSS)
-
•
CD14+ monocytes from pSS patients expressed high levels of the transcription factor CEBPD
-
•
FOLR3 and IL1B were upregulated separately in CD14+ monocytes from different pSS patients
-
•
Proportion of CD56−CD16+FCER1G+ NK cells/CD56-CD16+FCER1G- NK cells increased in pSS
Immunology; Components of the immune system; Transcriptomics
Introduction
Sjögren’s syndrome (SS) is a systemic autoimmune disease, and it can be classified as primary Sjögren’s syndrome (pSS) or secondary Sjögren’s syndrome (sSS) according to the cause of the disease. The incidence of this disease varies greatly in different regions of the world, and the current incidence is approximately 0.01–3.0% (Narváez et al., 2020). pSS is a complex heterogeneous disease. There is evidence that both innate immune cells and adaptive immune cells are involved in pSS. Among these cells, innate immune cells have been shown to express type I interferons (IFNs). Proinflammatory environments further activate T cells and B cells, which produce auto-antibodies; these activated cells form ectopic germinal centers and participate in gland destruction, thereby promoting the occurrence and development of disease (Davies et al., 2019; Mavragani, 2017).
Peripheral blood mononuclear cells (PBMCs) are a subset of white blood cells that play an indispensable role in the human immune system, which defends against diseases. PBMCs are composed of a mixture of lymphocytes (T cells, B cells and natural killer (NK) cells) and myeloid cells. The abnormal immune responses in pSS are characterized by the infiltration of secretory glands by large numbers of T and B cells from the blood and the production of auto-antibodies, such as anti-SSA and anti-SSB antibodies (Mariette and Criswell, 2018). CD4+T cells are activated by antigen presentation by major histocompatibility complex (MHC) class II molecules, which leads to the differentiation of CD4+T cells into T helper 1 (Th1), T helper 17 (Th17) and T follicular helper (Tfh) cells (Verstappen et al., 2021). CD8+T cells contribute to acinar damage in the exocrine glands (Zhou et al., 2021). The number of B cells in the peripheral blood and exocrine glands significantly increases, and ectopic germinal centers are often observed (Nocturne and Mariette, 2013). Examination of the peripheral blood of patients with pSS can reveal leukopenia, thrombocytopenia, or occasionally hemolytic anemia (Meena and Bohra, 2019).
Single-cell RNA-sequencing (scRNA-seq) is a new technology for the high-throughput sequencing and analysis of RNA at the single-cell level. Unlike conventional tissue or bulk cell population sequencing, scRNA-seq facilitates the analysis of the complexity of changes between cells and within cell subsets and provides deeper insights into specific information. There have been a small number of studies that used scRNA-seq to analyze immune cells in the blood of pSS patients. They found that specific expansion of CD4+ cytotoxic T lymphocytes may be involved in the pathogenesis of pSS (Hong et al., 2021). Here, we performed a detailed cell subtype analysis of single-cell transcriptomes of PBMCs from pSS patients and compared the profiles of pSS patients and healthy controls (HCs), revealing the partially cell type-specific and common changes in gene expression.
Results
Single-cell transcriptional landscape of PBMCs from patients with pSS and HCs
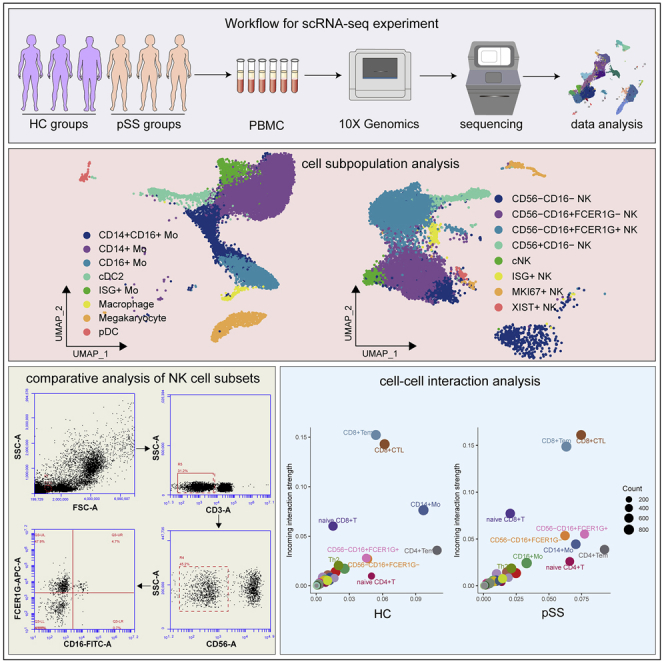
We first isolated and sequenced a total of 68,509 cells from PBMC suspensions from three pSS patients and three healthy controls (Figure 1A). After removing approximately 3.5% of the cells, which might have represented empty droplets and low-quality droplets, we obtained 66,141 total cells from the HCs (27,518 cells) and pSS patients (38,623 cells) that were used for further analysis Figure 1B). Unsupervised clustering using Seurat 4.0 identified four different cell clusters in both groups (Figures 1C–1E). Cluster 1 (∼5.78% of all cells) consisted of B cells expressing CD79A, CD79B and MS4A1. Cluster 2 (∼28.12% of all cells) contained myeloid cells expressing S100A8, S100A9, and CST3. Cluster 3 (∼21.57% of all cells) was composed of NK cells expressing NKG7, GNLY and CST7. Cluster 4 (∼44.53% of all cells) was composed of T cells expressing CD3D, CD3E and IL7R. The proportion of cells in each fraction is shown in Figure 1F.
Figure 1.

Study design and single-cell transcriptomic profiling of PBMCs from HCs and patients with pSS
(A) Schematic diagram of the experimental workflow for defining and comparing PBMCs between the two groups.
(B) The number of genes, number of transcripts and mitochondrial distribution profile of the cells in a sample after quality control filtration.
(C) Uniform manifold approximation and projection (UMAP) plot of a single-cell profile. Each cell uses different colors to distinguish the sample type and related cell types.
(D) The expression of marker genes for the cell types are defined above each panel.
(E) Distribution of the expression patterns of selected classic cell markers in four cell clusters.
(F) The ratios of the four cell types in HCs and pSS patients.
scRNA-seq analysis of changes in the distribution of eight myeloid cell subsets
Myeloid cells can play a key regulatory or immunosuppressive role. Among these cells, monocytes (Mos), macrophages and dendritic cells (DCs) are the first-line immune effectors. These cells are rapidly produced under conditions of infection or inflammation (Stegelmeier et al., 2019). Our data detected 14,674 myeloid cells in all 6 donors, and these myeloid cells were subclustered into eight subsets (Figures 2A and 2B). The identified clusters were designated as CD14+ Mos (CD14, LYZ), CD16+ Mos (FCGR3A), CD14+CD16+ Mos (APOBEC3A, ATF5 and HLA-DQA1, presumed to be a transitional feature between CD14+ Mos and CD16+ Mos), conventional type 2 DC cells (cDC2 cells, cD1C, FCER1A), macrophages cells (C1QA, C1QB and C1QC), pDC cells (DERL3, LRRC26, and TLR9), and megakaryocyte cells (PPBP, TUBB1 and PF4). Cluster 5 highly expressed MX1, IFIT1, IFI44L and IFIT3, indicating an important connection with the IFN signaling pathway. We named this cluster Interferon-stimulated genes (ISG) + Mos (Figure 2C). Compared with those in the HC group, the genes that were highly expressed by CD14+ Mos in the pSS group were mainly involved in activation during viral infection (Figure 2D). CD16+ Mos have exhibit enhanced Fc gamma R-mediated phagocytosis and regulate the polarization of Th1, Th2 and Th17 cells (Figure 2D). It was previously reported that CD16+ Mos can activate the Fc gamma R-mediated phagocytosis pathway and stimulate CD4+T cells to produce IL-4 (Wong et al., 2011). We found that in patients with pSS, the number of CD14+ Mos was decreased, whereas the numbers of CD14+CD16+ Mos and CD16+ Mos were increased (Figure 2E), which is consistent with previous reports (Ciccia et al., 2013; Lopes et al., 2021; Soret et al., 2021). The CD14+CD16+ Mo subpopulation is responsible for the proliferation and stimulation of T cells. These cells expressed higher levels of surface markers involved in antigen-presenting cell-T-cell interactions (Zawada et al., 2011).
Figure 2.

Analysis of myeloid cell subset
(A) The left image represents the distribution of myeloid cells in different samples, and the right image shows the clustering of myeloid cells in all samples.
(B) The distribution ratios of myeloid cell subsets.
(C) The highest differential gene expression in myeloid cells between HCs and pSS patients.
(D) Analysis of differential pathways in both bulk myeloid cells and myeloid cell subsets between pSS patients and HCs.
(E) Boxplots showing the proportions of each myeloid cell subtype in the pSS and HC groups. Cell types showing enrichment in the pSS or HC subgroups are marked with p values that were calculated by the two-sided Wilcoxon test.
Transcription factors (TFs) play critical roles in the regulation of target gene expression. To explore myeloid cell regulation, we performed SCENIC analysis, which revealed distinct gene regulatory networks in CD14+ Mos, CD14+CD16+ Mos and CD16+ Mos. TFs and their regulatory networks exhibited marked differences in CD14+ Mos and CD16+ Mos (Figures 3A and 3B). CEBPD directly regulates target genes that were upregulated in CD14+CD16+ Mos and CD16+ Mos compared with CD14+ Mos, and CEBPD was upregulated in the pSS group compared with the HC group in CD14+ Mos (Figure 3C). In addition, SPI1, IRF1, IRF7 and their target genes were upregulated in CD14+CD16+ Mos and CD16+ Mos. SPI1, IRF7 and ISG15 were upregulated in the pSS group compared with the HC group Figure 3D). GSEA results suggested that CEBPD, JUN, FOS and IRF1 might participate in the TNF-α signaling via NF-κB were significantly enriched in Mos (Figure 3E). ISGs including IRF1, IFI44L, IFIT3 and antiviral genes (OAS3, ADAR) with significant enrichment of IFN pathway gene sets including IFN-α and IFN-γ responses (Figure 3E, Tables S2 and S3). These results suggested that IFN signaling pathways may be activated in the pSS group. We further performed pseudotime trajectory analysis using Monocle3, which revealed that CD14+ Mos can further differentiate into CD14+CD16+ Mos and CD16+ Mos (Figures S1A and S1B). With the results of pseudo-temporal plots, trajectory analysis indicated that CD14, LYZ, FCGR3A, HLA-DRB1, HLA-DRA participates in monocytes differentiation (Figures S1C and S1D). In addition, IFNs such as IFITM3, IFITM2, IFITM1 and LY6E may contribute to the differentiation of CD16+Mos.
Figure 3.

Analysis of CD14+ Mos, CD14+CD16+ Mos and CD16+ Mos transcription factor activity
(A) Heatmap of cell viability in regulon subsets.
(B) Network of the regulation of expression by TFs and target genes. The top 100 genes were selected for the screen.
(C) Expression of CD14+ Mos TFs and their target genes in Mos.
(D) Expression of CD16+ Mos TFs and their target genes in Mos.
(E) GSEA plots from differentially regulated genes in scRNA-Seq data from Mos.
Next, by differential gene comparative analysis, we showed that FOLR3 was expressed at different levels in the different groups (Figure S2A, Table S2). Further analysis revealed that FOLR3 was upregulated and IL1B was downregulated in pSS_3 patients compared with pSS_1 and pSS_2 patients (Figures S2B and S2C). We performed data integration clustering of the GSE157278 dataset (Figure S2D), marked significantly differentially expressed genes (Figures S2E and S2F). The identified clusters were designated as T cells (CD3D, CD3E), B cells (CD79A, CD79B), NK cells (NKG7, GNLY and GZMB), CD14+ Mos (CD14), CD16+ Mos (FCGR3A), megakaryocyte cells (PPBP, TUBB1 and PF4). After subdivided the myeloid cells, and we found that FOLR3 was also highly expressed in pSS_1 patient (Figure S2G). In addition, the IL1B and LYZ genes were significantly upregulated in the CD14+ Mos of pSS_4 patients, indicating that even among the CD14+ Mos from pSS patients, there are different subtypes (Figure S2G). FOLR3 and IL1B exhibited opposite expression trends. In addition, CEBPD was significantly upregulated in CD14+ Mos from patients with pSS, further demonstrating its role in regulation of CD14+ Mos.
scRNA-seq analysis of changes in the distributions of NK-cell subsets
NK cells can directly exert cytotoxic effects, secrete immunomodulatory cytokines and chemokines, and play an important role in connecting innate immunity and adaptive immunity. A total of 11,057 NK cells were obtained, and these cells were mainly CD56−CD16+ NK cells (Figures 4A and 4B). These NK cells were subclustered into eight subsets. NK0 and NK1 expressed FCGR3A (CD16) at high levels, whereas NCAM1 (CD56) was shown to be expressed at low levels. FCER1G was highly expressed in NK0 cells, and the opposite trend was observed for NK1 cells. We defined NK0 and NK1 cells as CD56−CD16+FCER1G+ NK cells and CD56−CD16+FCER1G- NK cells, respectively (Figure 4C). CD56−CD16+FCER1G- NK cells showed enrichment of KLRC2 (NKG2) and KLRC3 (NKG3), indicating a memory-like NK-cell identity (Yang et al., 2019). Compared with those in the HC group, the genes that were highly expressed by CD56−CD16+FCER1G+ NK cells in the pSS patient group were involved in the activation of signaling pathways related to a variety of viral infections, including Epstein-Barr virus (EBV) infection, Kaposi sarcoma-associated herpes virus infection and hepatitis B infection (Figure 4D). Patients with pSS showed trends in which the proportion of CD56−CD16+FCER1G+ NK cells/CD56-CD16+FCER1G- NK cells was increased (Figure 4E). The results of the flow cytometric analysis agreed with those of the single-cell analysis (Figure 4F). The expression of IFI44L was slightly increased in CD56−CD16+FCER1G+ NK cells in the pSS group (Figure 4G), indicating enhanced IFN signaling pathway activity (Soret et al., 2021).
Figure 4.

Subgroup analysis of NK cells
(A) The left image represents the distribution of NK cells in different samples, and the right image shows the classification of NK cells in all samples.
(B) The distribution of NK-cell subset frequencies in each sample.
(C) Heatmap showing the marker genes with the highest expression in NK cells from the HC and pSS groups.
(D) Analysis of some differential pathways in both bulk NK cells and NK-cell subsets between pSS patients and HCs.
(E) Boxplots showing the proportions of CD56−CD16+ NK-cell subtypes in the two groups. Cell types showing enrichment in FCER1G+ or FCER1G-subgroups are marked. p values were calculated by the two-sided Wilcoxon test.
(F) Flow cytometric analysis of CD56−CD16+ NK cells. The cell markers used were CD3− (T cells) and CD56−CD16+FCER1G (NK cells).
(G) Different genes induced NK cells in HCs and pSS patients.
In addition, we showed that XCL1 and CXCR3, which are members of the chemokine family, were highly expressed in CD56+CD16−cell clusters. The production of XCL1 and CXCR3 by these NK cells may recruit other immune cells, such as DCs (Shimasaki et al., 2020). The NK4 cluster highly expressed genes related to cytotoxic NK cells, such as ZNF683, PTGDS, and CCL5Figure 4C), which were identified as markers of cytotoxic NK (cNK) cells (Zhao et al., 2020). Compared with those in the HC group, the genes that were highly expressed by these cells in the pSS patient group were involved in thermogenesis and ribosome pathways (Figure 4D). We found “inflamed NK” cell subsets with upregulated expression of IFIT1, IFIT3, RSAD2, MX1 and IFI44L. Recently published single-cell analysis of NK cells reported strong IFN characteristics (Shimasaki et al., 2020; Soret et al., 2021), and our data were consistent with this finding.
Analysis of changes in the distribution of B-cell subsets
A large number of studies have shown that B cells are one of the main groups of cells that produce proinflammatory cytokines and secrete auto-antibodies, and B cells play a critical role in pSS pathology (Du et al., 2021). Our study detected 3,208 B cells in all samples, and these B cells were subclustered into six subsets (Figures 5A and 5B). In our research, we found that B cells were mainly divided into two major groups: one is the initial B-cell subgroup comprising cells mainly expressing TCL1A, and the other is the active B-cell subgroup mainly comprising cells expressing TNFRSF13B. B0 cells expressed high levels of the activated naive B-cell markers TCL1A, SELL, IL4R, and CLECL1 (Figure 5C). B1 and B2 cells expressed high levels of TNFRSF13B. The high expression of CLECL1, CTSH and TCF4 in B1 cells indicated that these cells were memory B cells. We observed marked upregulation of IFI44L, RAC1, and ZFP36L2 in the patients compared with HCs (Figure 5D). Naive B cells and memory B cells participated in viral myocarditis, phagosome and leukocyte transendothelial migration and were highly enriched in pSS patients compared with HCs. Compared with those of HCs, the genes that were highly expressed by memory B cells and immature B cells of patients with pSS were involved in oxidative phosphorylation, and the antigen processing and presentation signaling pathway was highly enriched in pSS samples (Figure 5D). In addition, RPS4Y1, RPS10, and RPL36A were downregulated in the B-cell subpopulations of pSS patients, and the signaling pathways related to ribosomes were highly enriched in memory B cells and activated B cells (Figure 5E).
Figure 5.

B-cell clusters in PBMCs from HCs and pSS patients
(A) Two-dimensional UMAP visualization of two B-cell clusters. The left image represents the distribution of B cells in different samples, and the right image shows the subsets of B cells in all samples.
(B) The proportion of each cluster in the B-cell subpopulation.
(C) Heatmap showing the highest expression of genes in each B-cell subgroup from the HC and pSS groups.
(D) Analysis of some differential pathways in both bulk B cells and B-cell subsets between pSS patients and HCs.
(E) Some differentially expressed genes that were induced in B cells in HCs and pSS patients.
scRNA-seq analysis of changes in the distribution of eight T cell subsets
We conducted further analysis of T cells. After removing some of the duplicates, 28,957 T cells were detected, and T cells were the most prevalent cell type (Figures 6A and 6B). Reclustering revealed seven clusters, which were designated as effector memory CD4+T cells (CD4+ Tem cells, LTB, AQP3, CD40LG and KLRB1, cluster 0), naive CD4+T cells (CD4, CCR7, LEF1 and TCF7, cluster 1), CD8+ cytotoxic T cells (CD8+ CTLs, CD8A, GNLY and GZMB, cluster 2), effector memory CD8+T cells (CD8+ Tem cells, CD8A, GZMK, cluster 3), naive CD8+T cells (CD8B, CCR7, LEF1 and TCF7, cluster 4), T helper 2 (Th2) cells (GATA3, CAPG and CCR4, cluster 5), and regulatory T cell (Treg) cells (FOXP3, cluster 6) (Figure 6C). Both CD8+ CTLs and CD8+ Tem cells exhibited high expression of CCL5. CCL5 has a chemotactic effect on T cells, eosinophils and basophils and plays an active role in recruiting leukocytes to sites of inflammation. Human T lymphocyte virus-1 (HTLV-1)-infected T cells specifically express the CCL5 gene and produce CCL5 (Aqrawi et al., 2018). Th2 cells expressed high levels of GPR183 and CD40LG. The protein encoded by CD40LG is expressed on the surface of T cells. It regulates B cell function by engaging CD40 on the B cell surface (Sanosyan et al., 2019). Tregs works by keeping immune responses in check and avoiding uncontrolled responses that can harm the body (Keindl et al., 2022). The selective inhibition of the body’s auto-reactive T cells and effector T cells is an important mechanism for the formation and maintenance of immune homeostasis and for the prevention of autoimmune diseases.
Figure 6.

T cell subsets in PBMCs from HCs and pSS patients
(A) UMAP plot of T cells across all six donors. The left image represents the distribution of T cells in different donors, and the right image shows the subsets of T cells.
(B) The fraction of cells from eight cell types in the HC and pSS groups.
(C) The most highly expressed differentially expressed genes in T cell subtypes from HCs and pSS patients.
(D) Differentially expressed genes in T cell subsets between HCs and pSS patients. The expression level of each gene is indicated on a red (high)-blue (low) color scale.
(E) Analysis of some differential pathways in both bulk T cells and T cell subsets between pSS patients and HCs.
IFI44L, DUSP2, RAC1, and LAMP1 were upregulated, whereas RPS4Y1, RPS10 and RPL36A were downregulated in the pSS group compared with the HC group (Figure 6D). KEGG analysis showed that the genes that were highly expressed by naive CD4+T cells and CD4+ Tem cells in the pSS group were involved in pathways related to viral and bacterial infections. Both CD8+ CTL cells and CD8+ Tem cells play an important role in the T-cell receptor signaling pathway and the differentiation of Th1 cells, Th2 cells, and Th17 cells. In addition, in the pSS group, antigen processing and presentation and the intestinal immune network for IgA production were enriched in CD8+ CTL cells (Figure 6F, Table S3) compared with those in the HC group.
Analysis of the interactions between cells
Cell communication analysis mainly describes the relationship between cells. Next, we analyzed the interactions between cells in the different groups. The patterns of interactions among different cell types are shown in Figure 7A. CD14+ Mos, CD14+CD16+ Mos and CD16+ Mos had enhanced effects in the pSS group. CD16+ Mos communication with CD8+ CTLs and CD56−CD16+FCER1G+ NK cells was enhanced in pSS patients compared with HCs. Further analysis of the roles of ligands and receptors revealed that compared with those in the HC group, Mos and CD56−CD16+FCER1G+ NK cells in the pSS group exhibited significantly upregulated expression of the ligand-receptor pair CD52-SIGLEC10, which may be a stimulatory factor for CD56−CD16+FCER1G+ NK cells activation in inflamed sites Figure 7B). Compared with the HC group, the pSS patient group exhibited enhanced effects of the ligand-receptor pair CD74-COPA in interactions between Mos and naive B cells (Figure 7B). The same phenomenon was also observed in the myeloid cell cluster, where the CD74-APP pair mediated interactions between Mos and memory B cells and naive T cells. CellChat results showed that the effects of CD56−CD16+FCER1G+ NK cells, CD16+ Mos and CD8+ CTLs were enhanced, whereas the effect of CD14+ Mos was weakened (Figure 7C). These findings suggest that the pSS patients' ligand-receptor activity may encourage the autoimmune response. Further data and analysis are needed to reveal the mechanism involved in this.
Figure 7.

Cell-cell communication network in PBMCs
(A) Heatmap displaying the key cell-cell interaction pairs among PBMCs. The number of significantly enriched ligand-receptor pairs of each pair of cells is indicated by different colors.
(B) Bubble plots present ligand-receptor pairs of growth factors associated with HCs and pSS patients. The interaction intensity is presented by the bubble color.
(C) The strength of the interaction of these pairs between different groups of sender or recipient cells.
Discussion
In this paper, we used scRNA-Seq to reveal the complexity of the immune cell populations in the PBMCs of patients with pSS and HCs. The relevant subtypes of myeloid cells, NK cells, B cells and T cells were analyzed. Here, we discovered cell subsets and their related DE-Gs and signaling pathways, including viral infection, IFN system activation, antigen presentation, and activation of B cells and T cells.
We found that monocyte accounted for the major population of myeloid cells in patients with pSS. Similar analysis of PBMCs of the pSS with scRNA-seq was conducted by He et al. (2022), which mainly focused on transcriptomic changes in monocyte subsets showed that almost all monocyte subsets had increased expression of TNFSF10 (TRAIL). They also found that CEBPD was upregulated in Mos and our study further confirms that CEBPD was predominantly upregulated in CD14+ Mos from patients with pSS. Activation of CEBPD has been observed in many autoimmune diseases, and in the inflammatory environment, CEBPD is thought to be activated by inflammatory factors such as IL-6, IFN-α, IFN-γ, and IL1B (Ko et al., 2015). In addition, FOLR3 and IL1B were upregulated in CD14+ Mos of different patients. FOLR3 is involved in rheumatoid arthritis (Tseng et al., 2019) and SARS-CoV-2 (Sfikakis et al., 2021), but its relationship with pSS has not been reported. According to data from this study, it was found that FOLR3 showed the opposite trend of IL1B expression. Upregulated IL1B is observed in the lacrimal and salivary glands of pSS patients, and inhibition of the pathogenic function of IL1B may be beneficial for the treatment of pSS (Chen et al., 2012). However, the sample size of that clinical study was small, which needs to be further explored.
Currently, there are fewer reports of NK cells in pSS, including previous reports of pSS scRNA-seq that do not involve NK cells (Hong et al., 2021; He et al., 2022; Hou et al., 2022). Previous studies had reported that NK cells activity was decreased and that it may be partially restored by IFN-α in SS. Our investigation revealed an increased frequency of CD56−CD16+FCER1G+ NK cells and increased expression of IFI44L in pSS patients. Increased expression of IFI44L is part of the IFNs response and is closely related to the pathogenesis of pSS. Recent research findings show that FCER1G upregulation is dependent on cell proliferation progression mediated by IL-2, IL-15, or IL-12, is sensitive to mTOR suppression, and is inhibited by TGFβ or IFNα in NK cell subsets (Shemesh et al., 2022). How NK cells migrate to different sites and how NK cells participate in the pathogenesis of pSS remain to be fully elucidated.
In the earliest paper reported by Hong, CD4+ CTLs cytotoxic T lymphocyte was revealed to may be involved in the pathogenesis of pSS (Hong et al., 2021). They found Th17 cell subsets and no Th2 cell subsets in patients. In contrast, Th2 cells were detected in pSS group whereas Th17 cells were not clearly detected in our study. In the pre-pSS stage, Th1 and Th17 cells trigger pSS, and as pSS progress, Th2 and Tfh cells dominate (Psianou et al., 2018). which also showed that cell subpopulations might differ among patients with different stages of pSS. Th2 cells in turn promote the activation of B cells, and patients with advanced pSS mainly exhibit symptoms because of B cell activation (Ibrahem, 2019). In addition, Th2 cell differentiation is affected by the presence of Tregs, and defects in Tregs lead to syndromes characterized by hypersensitivity and autoimmunity (Das et al., 2011).
Our study found that pathways related to a variety of virus infections were enriched in the pSS group. This study suggested that virus infection-associated pathways were key up-regulated pathways in the B cells of pSS patients, consistent with previous report (Hong et al., 2021). B cells are the main target of EBV infection, and EBV is one of the factors that induces pSS (Maślińska, 2019). EBV infection contributes to the differentiation of B and T cells into the typical effector phenotypes observed in pSS patients (Barcelos et al., 2021). There are several infectious agents that can cause disease manifestations similar to Sjogren’s syndrome, including hepatitis C virus, EBV, cytomegalovirus and HTLV-1, and these four pathogens have been found to cause persistent infection of the salivary glands after the first infection; this persistent infection subsequently leads to organ destruction and xerostomia syndrome (Björk et al., 2020; Utomo and Putri, 2020). However, virus affects the intrinsic immune response of patients with pSS remains to be explored.
In this study, we collected PBMCs from HCs and patients with pSS and analyzed the pSS-driven changes in the composition and function of PBMCs through scRNA-seq. CD14+ Mos, CD14+CD16+ Mos, CD16+ Mos, CD8+ CTLs, and CD56−CD16+FCER1G+ NK cells were found to play major roles in pSS. These results not only illustrate the molecular and cellular immune characteristics in the clinical progression of pSS but also provide a way to identify urgently needed biomarkers and therapeutic targets in pSS.
Limitations of the study
The interpretation of this study had important limitations. First, because only a limited number of patients were examined in our study, we need to perform larger clinical trials and further studies to verify the differences between pSS patients and HCs. Second, our research focused on single-cell transcriptomic data from PBMCs in the blood. If we can combine this information with data from lesions (such as those in the salivary glands), our analysis will be more systematic and comprehensive.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| PBMC | This paper | N/A |
| Critical commercial assays | ||
| Chromium Next GEM Single-Cell 5′ Reagent Kits v1.1 | 10X Genomics | # PN-1000165 |
| Chromium Single Cell 5′ Library Construction Kit | 10X Genomics | #PN-1000020 |
| Deposited data | ||
| pSS scRNAseq (GSE157278) | GEO | GSE157278 |
| Software and algorithms | ||
| R (v4.1.2) | R CRAN | https://cran.r-project.org/ |
| Seurat R package (v4.0.5) | R CRAN | https://cran.r-project.org/web/packages/Seurat/index.html |
| pheatmap R package (v1.0.12) | R CRAN | https://cran.r-project.org/web/packages/ |
| CellChat R package (v1.1.3) | Github | https://github.com/sqjin/CellChat |
| SCENIC R package (v1.2.4) | Github | https://github.com/aertslab/SCENIC |
| ClusterProfiler R package (v4.2.0) | Bioconductor | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| GseaVis (v0.0.1) | Github | https://github.com/junjunlab/GseaVis |
| Scillus (v0.5.0) | Github | https://github.com/xmc811/Scillus |
| CellPhoneDB (v3.0) | Github | https://github.com/Teichlab/cellphonedb |
| Monocle3 (v 1.2.9) | Github | https://github.com/cole-trapnell-lab/monocle3 |
| NicheNet (v1.1.0) | Github | https://github.com/saeyslab/nichenetr |
| Antibodies | ||
| PE anti-human CD56 (NCAM) Antibody | Biolegend | Cat#362524; RRID:AB_2564161 |
| FITC anti-human CD16 Antibody | Biolegend | Cat#302006; RRID:AB_314206 |
| Anti-Human CD3, violetFluor 450 (SK7) | Multi-Sciences | Cat#70-AH00307-100 |
| Anti-FCER1G/APC Conjugated antibody | Bioss | Cat#bs-13167R-APC |
Resource availability
Lead contact
Further information and requests for resources and data should be directed to and will be fulfilled by the lead contact, Bin Wu (wuubinn@126.com).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Study design and patients
The study was approved by the Ethics Committee of Chongqing Hospital of Traditional Chinese Medicine (Chongqing Traditional Chinese Medicine Hospital Ethics Committee Approval Number, 2021-KY-KS-WB). After obtaining written informed consent from all the patients, we collected blood from three pSS patients and three HCs who were recruited at the Rheumatology Department of Chongqing Traditional Chinese Medicine Hospital between October 2020 and March 2021. Patient’s clinical and laboratory features were analyzed. All patients fulfilled 2016 American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) criteria for pSS. The EULAR SS disease activity index (ESSDAI) score of pSS patients were calculated. All clinical information, including demographic data, medical history, symptoms, signs and laboratory data, was derived from the patient’s medical history. Laboratory data included data about routine blood parameters, lymphocyte subsets, and inflammatory cytokines (Table S1).
The collection, processing and laboratory testing of the samples followed the World Health Organization (WHO) guidelines. For both the pSS patients and HCs, 5 mL of peripheral blood was collected from each person into a heparin K test tube. All the samples were placed on ice after collection and were processed within 2–3 h of collection.
Data collection
The Human scRNA-seq data of GSE157278 were obtained from the NIH Gene Expression Omnibus (GEO) database.
Method details
PBMC isolation
Peripheral blood was diluted with Hanks' balanced salt solution (HBSS) without Ca2+ and Mg2+ (containing 0.04% BSA, 400 μg/mL). The diluted blood was centrifuged at 500 g and 20°C for 20 min. The interface layer containing PBMCs was collected along the periphery of the centrifuge tube and transferred into another centrifuge tube. Then, 5 mL of HBSS was added to wash the samples, the PBMCs were centrifuged at 300 × g for 10 min, and the supernatants were discarded; these steps were repeated 6 times. Then, 1 mL of HBSS was added to resuspend the cells, and the cell suspensions were mixed gently. Cell viability was examined by 0.4% trypan blue staining, and it was confirmed to be higher than 90% per sample. The concentration of live cells was adjusted to the ideal concentration (1,000–2,000 cells/μL).
Gel bead-in-emulsions generation and barcoding
Cellular suspensions were loaded on a 10x Genomics GemCode single-cell instrument to generate single-cell gel beads-in-emulsions (GEMs). Libraries were generated from the cDNAs and sequenced with Chromium Next GEM Single-Cell 5′ Reagent Kits v1.1. Upon dissolution of the gel bead in a GEM, primers containing (i) an Illumina R1 sequence (read1 sequencing primer), (ii) a 16 nt 10x barcode, (iii) a 10 nt unique molecular identifier (UMI), and (iv) a poly-dT primer sequence were mixed with the cell lysate and Master Mix. Barcoded, full-length cDNAs were then reverse-transcribed from polyadenylated mRNA.
Library construction and sequencing
The prepared cell suspensions were processed with a 10x Chromium Single-Cell 5′ library kit. The Gel Bead and Multiplex Kit and Chip Kit (10x Genomics) were used to convert the single-cell suspensions of scRNA-seq samples into a barcoded scRNA-seq library. The DNA libraries were sequenced on an Illumina sequencing platform by Genedenovo Biotechnology Co., Ltd. (Guangzhou, China). Each library was sequenced using an Illumina NovaSeq6000 sequencer with a paired-end 150 bp read strategy. The Single-Cell 5′ protocol was followed to produce Illumina-ready sequencing libraries according to the manufacturer’s instructions.
scRNA-seq data processing
10x Genomics Cell Ranger software (version 3.0.2) was used to convert raw base call (BCL) files to FASTQ files and perform alignment and count quantification. Reads with low-quality barcodes and UMIs were filtered out, and then, the remaining reads were mapped to the reference genome GRCh38.p5. Reads that were uniquely mapped to the transcriptome and had at least 50% overlap with an exon were considered for UMI counting. Before quantification, the UMI sequences were corrected for sequencing errors, and valid barcodes were identified based on the EmptyDrops method. Cells were produced by gene matrices via UMI counting and cell barcode calling.
Cells with an unusually high number of UMIs (≥10,000) or percentage of mitochondrial genes (≥10%) were filtered out. We also excluded cells with fewer than 200 or more than 3,000 genes detected. To minimize the effects of batch effects and behavioral conditions on clustering, we used Seurat 4.0 (Stuart et al., 2019), which utilizes canonical correlation analysis and mutual nearest neighbor analysis, to aggregate all samples. A total of 2,000 highly variable genes were selected in each sample based on a variance-stabilizing transformation. Anchors between individual datapoints were identified, and correction vectors were calculated to generate an integrated expression matrix, which was used for subsequent clustering.
To validate our results, we also analyzed another scRNA-seq dataset. scRNA-seq data from 5 pSS patients and 5 controls in GSE157278 from GEO were used in our study. This study was treated and filtered using the same approach as described.
Differentially expressed gene analysis
The expression value of each gene in a given cluster was compared against that in the rest of the cells using the Wilcoxon rank-sum test. Significantly upregulated genes were identified using a number of criteria. An adjusted p< 0.05 and |log2FC| > 0.36 were used to define significant differentially expressed genes (DE-Gs). For DE-Gs, Gene Ontology (GO) analysis, Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis and Gene Set Enrichment Analysis (GSEA) were performed with clusterProfiler4.0 (Wu et al., 2021).
Transcription factor analysis
Single-cell regulatory network inference and clustering (SCENIC) was used to predict transcription factors (TFs) and corresponding target genes, build a transcription factor regulatory network module (regulon), quantify the activity of each regulon in cells based on the expression of TFs and target genes in the regulon, and finally determine the transcriptional regulatory activity of TFs in different cells (Aibar et al., 2017). We performed SCENIC by starting from the raw counts and following the proposed workflow using the default parameters and retaining those with a cis-regulatory binding motif for upstream regulators. Custom scripts for analysing data are available at https://github.com/aertslab/SCENIC.
Ligand-receptor expression abundance analysis
CellPhoneDB (Efremova et al., 2020) and CellChat (Jin et al., 2021) software were used to construct the cell communication network based on the single-cell gene expression matrix. CellPhoneDB contains ligand-receptor information that is used to analyze the expression of ligand-receptor pairs in two cell types on the basis of the expression of the receptor by one cell type and that of the ligand by another cell type. Only receptors and ligands that were expressed in a percentage of cells in a specific cluster that was higher than a user-specified threshold were considered for the analysis (default was 10%).
Prediction of ligand-target gene regulation
Ligand activity analysis was performed with NicheNet (Browaeys et al., 2020) to prioritize which sender cell ligands were most likely to affect gene expression in interacting recipient cells. This procedure, called ligand activity prediction, was used to rank ligands according to how well their prior target gene predictions corresponded to the observed changes in gene expression resulting from communication with sender cells.
Antibodies and flow cytometric analysis
Six pSS patients and seven healthy controls were recruited. A total volume of 100 μL whole blood from these participants was incubated with 2.5 μL antibody and treated with 2 mL erythrocyte lysis buffer for 15 min, and the cells were collected by centrifugation (300 g/min, 5 min). The monoclonal antibodies used were antibodies specific for human CD56 (Biolegend, 362524), CD16 (Biolegend, 302006), CD3 (Multi-Sciences, 70-AH00307-100) and FCER1G (Bioss, bs-13167R-APC). The cells were analyzed using a BD Accuri C6 Cytometer (BD Biosciences). All the samples were processed within 2–3 h of collection. The percentages of CD56−CD16+FCER1G + NK cells and CD56−CD16+FCER1G- NK cells were calculated by a two-sided Wilcoxon test, and the difference was considered significant if the p value was less than 0.05.
Quantification and Statistical analysis
Statistical analysis was carried out in R (v4.1.2). We used either two-sided non-parametric Wilcoxon rank sum tests or Wilcoxon signed rank tests to compare two groups of values.
Acknowledgments
The study was supported by Natural Science Foundation of China (81904042), Chongqing Science and Technology Committee (cstc2018jxjl130084, cstc2021jcyj-msxmX0852, CSTB2022BSXM-JCX0076), Chongqing Technology Innovation and Application Development Special Key Project (cstc2019jscx-dxwtBX0023) and Chongqing Hospital of Traditional Chinese Medicine of young top-notch talents (CQSZYY1146). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We gratefully acknowledge the patients and control subjects for their donation of peripheral blood samples. We are grateful to thank Genedenovo Biotechnology Co., Ltd for assisting in sequencing and bioinformatics analysis. We thank Dr. Jianming Zeng (University of Macau), and all the members of his bioinformatics team, biotrainee, for generously sharing their experience and codes.
Author contributions
B.W., J.L., and H.G. designed and supervised the study. B.W., C.L., F.Z., M.W., and Y.X. collected the clinical samples. J.L. and H.G. performed the sequencing experiments, carried out experiments. J.L. wrote the original manuscript and performed bioinformatics. All authors contributed to the article and approved the submitted version.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list. We avoided “helicopter science” practices by including the participating local contributors from the region where we conducted the research as authors on the paper.
Published: December 22, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105509.
Supplemental information
Data and code availability
The authors declare that all datasupporting the findings of this study are available within the article, the supplementary data, and the data repository or from the corresponding author upon reasonable request. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request. The raw sequence data reported in this paper had deposited in the Genome Sequence Archive of the BIG Data Center at the Beijing Institute of Genomics, under accession number HRA001355 (accessible at http://bigd.big.ac.cn/gsa-human). The codes used to perform including processing, clustering, batch effect correction and subclustering are available on GitHub: https://github.com/tudou666/pSS.
References
- Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.C., Geurts P., Aerts J., et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017;14:1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aqrawi L.A., Jensen J.L., Øijordsbakken G., Ruus A.K., Nygård S., Holden M., Jonsson R., Galtung H.K., Skarstein K. Signalling pathways identified in salivary glands from primary Sjögren’s syndrome patients reveal enhanced adipose tissue development. Autoimmunity. 2018;51:135–146. doi: 10.1080/08916934.2018.1446525. [DOI] [PubMed] [Google Scholar]
- Barcelos F., Martins C., Monteiro R., Cardigos J., Prussiani T., Sítima M., Alves N., Vaz-Patto J., Cunha-Branco J., Borrego L.M. Association between EBV serological patterns and lymphocytic profile of SjS patients support a virally triggered autoimmune epithelitis. Sci. Rep. 2021;11:4082. doi: 10.1038/s41598-021-83550-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björk A., Mofors J., Wahren-Herlenius M. Environmental factors in the pathogenesis of primary Sjögren’s syndrome. J. Intern. Med. 2020;287:475–492. doi: 10.1111/joim.13032. [DOI] [PubMed] [Google Scholar]
- Browaeys R., Saelens W., Saeys Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods. 2020;17:159–162. doi: 10.1038/s41592-019-0667-5. [DOI] [PubMed] [Google Scholar]
- Chen Y.T., Lazarev S., Bahrami A.F., Noble L.B., Chen F.Y.T., Zhou D., Gallup M., Yadav M., McNamara N.A. Interleukin-1 receptor mediates the interplay between CD4+ T cells and ocular resident cells to promote keratinizing squamous metaplasia in Sjögren’s syndrome. Lab. Invest. 2012;92:556–570. doi: 10.1038/labinvest.2011.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia F., Alessandro R., Rodolico V., Guggino G., Raimondo S., Guarnotta C., Giardina A., Sireci G., Campisi G., De Leo G., Triolo G. IL-34 is overexpressed in the inflamed salivary glands of patients with Sjögren’s syndrome and is associated with the local expansion of pro-inflammatory CD14brightCD16+ monocytes. Rheumatology. 2013;52:1009–1017. doi: 10.1093/rheumatology/kes435. [DOI] [PubMed] [Google Scholar]
- Das R., Moss J.E., Robinson E., Roberts S., Levy R., Mizue Y., Leng L., McDonald C., Tigelaar R.E., Herrick C.A., Bucala R. Role of macrophage migration inhibitory factor in the Th2 immune response to epicutaneous sensitization. J. Clin. Immunol. 2011;31:666–680. doi: 10.1007/s10875-011-9541-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies R., Sarkar I., Hammenfors D., Bergum B., Vogelsang P., Solberg S.M., Gavasso S., Brun J.G., Jonsson R., Appel S. Single cell based phosphorylation profiling identifies alterations in toll-like receptor 7 and 9 signaling in patients with primary sjogren’s syndrome. Front. Immunol. 2019;10:281–318. doi: 10.3389/fimmu.2019.00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du W., Han M., Zhu X., Xiao F., Huang E., Che N., Tang X., Zou H., Jiang Q., Lu L. The multiple roles of B cells in the pathogenesis of Sjögren’s syndrome. Front. Immunol. 2021;12:684999. doi: 10.3389/fimmu.2021.684999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efremova M., Vento-Tormo M., Teichmann S.A., Vento-Tormo R. CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 2020;15:1484–1506. doi: 10.1038/s41596-020-0292-x. [DOI] [PubMed] [Google Scholar]
- He Y., Chen R., Zhang M., Wang B., Liao Z., Shi G., Li Y. Abnormal changes of monocyte subsets in patients with Sjögren’s syndrome. Front. Immunol. 2022;13:864920. doi: 10.3389/fimmu.2022.864920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong X., Meng S., Tang D., Wang T., Ding L., Yu H., Li H., Liu D., Dai Y., Yang M. Single-cell RNA sequencing reveals the expansion of cytotoxic CD4+ T lymphocytes and a landscape of immune cells in primary Sjögren’s syndrome. Front. Immunol. 2020;11:594658. doi: 10.3389/fimmu.2020.594658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou X., Hong X., Ou M., Meng S., Wang T., Liao S., He J., Yu H., Liu L., Yin L., et al. Analysis of gene expression and TCR/B cell receptor profiling of immune cells in primary Sjögren’s syndrome by single-cell sequencing. J. Immunol. 2022;209:238–249. doi: 10.4049/jimmunol.2100803. [DOI] [PubMed] [Google Scholar]
- Ibrahem H.M. B cell dysregulation in primary Sjögren’s syndrome: a review. Jpn. Dent. Sci. Rev. 2019;55:139–144. doi: 10.1016/j.jdsr.2019.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S., Guerrero-Juarez C.F., Zhang L., Chang I., Ramos R., Kuan C.H., Myung P., Plikus M.V., Nie Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021;12:1088. doi: 10.1038/s41467-021-21246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keindl M., Davies R., Bergum B., Brun J.G., Hammenfors D., Jonsson R., Lyssenko V., Appel S. Impaired activation of STAT5 upon IL-2 stimulation in Tregs and elevated sIL-2R in Sjögren’s syndrome. Arthritis Res. Ther. 2022;24:101. doi: 10.1186/s13075-022-02769-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko C.Y., Chang W.C., Wang J.M. Biological roles of CCAAT/enhancer-binding protein delta during inflammation. J. Biomed. Sci. 2015;22:6. doi: 10.1186/s12929-014-0110-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes A.P., Bekker C.P.J., Hillen M.R., Blokland S.L.M., Hinrichs A.C., Pandit A., Kruize A.A., Radstake T.R.D.J., van Roon J.A.G. The transcriptomic profile of monocytes from patients with Sjögren’s syndrome is associated with inflammatory parameters and is mimicked by circulating mediators. Front. Immunol. 2021;12:701656. doi: 10.3389/fimmu.2021.701656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariette X., Criswell L.A. Primary Sjögren’s syndrome. N. Engl. J. Med. 2018;378:931–939. doi: 10.1056/NEJMcp1702514. [DOI] [PubMed] [Google Scholar]
- Maślińska M. The role of Epstein-Barr virus infection in primary Sjögren’s syndrome. Curr. Opin. Rheumatol. 2019;31:475–483. doi: 10.1097/BOR.0000000000000622. [DOI] [PubMed] [Google Scholar]
- Mavragani C.P. Mechanisms and new strategies for primary Sjögren’s syndrome. Annu. Rev. Med. 2017;68:331–343. doi: 10.1146/annurev-med-043015-123313. [DOI] [PubMed] [Google Scholar]
- Meena D.S., Bohra G.K. Primary sjogren’s syndrome presenting as autoimmune cytopenia. Clin. Pract. 2019;9:1190–2110. doi: 10.4081/cp.2019.1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narváez J., Sánchez-Fernández S.Á., Seoane-Mato D., Díaz-González F., Bustabad S. Prevalence of Sjögren’s syndrome in the general adult population in Spain: estimating the proportion of undiagnosed cases. Sci. Rep. 2020;10:10627. doi: 10.1038/s41598-020-67462-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nocturne G., Mariette X. Advances in understanding the pathogenesis of primary Sjögren’s syndrome. Nat. Rev. Rheumatol. 2013;9:544–556. doi: 10.1038/nrrheum.2013.110. [DOI] [PubMed] [Google Scholar]
- Psianou K., Panagoulias I., Papanastasiou A.D., de Lastic A.L., Rodi M., Spantidea P.I., Degn S.E., Georgiou P., Mouzaki A. Clinical and immunological parameters of Sjögren’s syndrome. Autoimmun. Rev. 2018;17:1053–1064. doi: 10.1016/j.autrev.2018.05.005. [DOI] [PubMed] [Google Scholar]
- Sanosyan A., Daien C., Nutz A., Bollore K., Bedin A.S., Morel J., Zimmermann V., Nocturne G., Peries M., Guigue N., et al. Discrepancy of serological and molecular patterns of circulating Epstein-Barr virus reactivation in primary Sjögren’s syndrome. Front. Immunol. 2019;10:1153. doi: 10.3389/fimmu.2019.01153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfikakis P.P., Verrou K.M., Ampatziadis-Michailidis G., Tsitsilonis O., Paraskevis D., Kastritis E., Lianidou E., Moutsatsou P., Terpos E., Trougakos I., et al. Blood transcriptomes of anti-SARS-CoV-2 antibody-positive healthy individuals who experienced asymptomatic versus clinical infection. Front. Immunol. 2021;12:746203. doi: 10.3389/fimmu.2021.746203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shemesh A., Su Y., Calabrese D.R., Chen D., Arakawa-Hoyt J., Roybal K.T., Heath J.R., Greenland J.R., Lanier L.L. Diminished cell proliferation promotes natural killer cell adaptive-like phenotype by limiting FcεRIγ expression. J. Exp. Med. 2022;219:e20220551. doi: 10.1084/jem.20220551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimasaki N., Jain A., Campana D. NK cells for cancer immunotherapy. Nat. Rev. Drug Discov. 2020;19:200–218. doi: 10.1038/s41573-019-0052-1. [DOI] [PubMed] [Google Scholar]
- Soret P., Le Dantec C., Desvaux E., Foulquier N., Chassagnol B., Hubert S., Jamin C., Barturen G., Desachy G., Devauchelle-Pensec V., et al. A new molecular classification to drive precision treatment strategies in primary Sjögren’s syndrome. Nat. Commun. 2021;12:3523. doi: 10.1038/s41467-021-23472-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegelmeier A.A., van Vloten J.P., Mould R.C., Klafuric E.M., Minott J.A., Wootton S.K., Bridle B.W., Karimi K. Myeloid cells during viral infections and inflammation. Viruses. 2019;11:168. doi: 10.3390/v11020168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng C.C., Lin Y.Z., Lin C.H., Li R.N., Yen C.Y., Chan H.C., Tsai W.C., Ou T.T., Wu C.C., Sung W.Y., Yen J.H. Next-generation sequencing profiles of the methylome and transcriptome in peripheral blood mononuclear cells of rheumatoid arthritis. J. Clin. Med. 2019;8:1284. doi: 10.3390/jcm8091284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utomo S.W., Putri J.F. Infections as risk factor of Sjögren’s syndrome. Open Access Rheumatol. 2020;12:257–266. doi: 10.2147/OARRR.S276727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstappen G.M., Kroese F.G.M., Bootsma H. T cells in primary Sjögren’s syndrome: targets for early intervention. Rheumatology. 2019;60:3088–3098. doi: 10.1093/rheumatology/kez004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong K.L., Tai J.J.Y., Wong W.C., Han H., Sem X., Yeap W.H., Kourilsky P., Wong S.C. Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood. 2011;118:e16–e31. doi: 10.1182/blood-2010-12-326355. [DOI] [PubMed] [Google Scholar]
- Wu T., Hu E., Xu S., Chen M., Guo P., Dai Z., Feng T., Zhou L., Tang W., Zhan L., et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation. 2021;2:100141. doi: 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C., Siebert J.R., Burns R., Gerbec Z.J., Bonacci B., Rymaszewski A., Rau M., Riese M.J., Rao S., Carlson K.S., et al. Heterogeneity of human bone marrow and blood natural killer cells defined by single-cell transcriptome. Nat. Commun. 2019;10:3931. doi: 10.1038/s41467-019-11947-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawada A.M., Rogacev K.S., Rotter B., Winter P., Marell R.R., Fliser D., Heine G.H. SuperSAGE evidence for CD14++CD16+ monocytes as a third monocyte subset. Blood. 2011;118:e50–e61. doi: 10.1182/blood-2011-01-326827. [DOI] [PubMed] [Google Scholar]
- Zhao J., Zhang S., Liu Y., He X., Qu M., Xu G., Wang H., Huang M., Pan J., Liu Z., et al. Single-cell RNA sequencing reveals the heterogeneity of liver-resident immune cells in human. Cell Discov. 2020;6:22. doi: 10.1038/s41421-020-0157-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H., Yang J., Tian J., Wang S. CD8+ T lymphocytes: crucial players in Sjögren’s syndrome. Front. Immunol. 2020;11:602823. doi: 10.3389/fimmu.2020.602823. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all datasupporting the findings of this study are available within the article, the supplementary data, and the data repository or from the corresponding author upon reasonable request. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request. The raw sequence data reported in this paper had deposited in the Genome Sequence Archive of the BIG Data Center at the Beijing Institute of Genomics, under accession number HRA001355 (accessible at http://bigd.big.ac.cn/gsa-human). The codes used to perform including processing, clustering, batch effect correction and subclustering are available on GitHub: https://github.com/tudou666/pSS.