

Abstract
Purpose:
In a large cohort of 373 pediatric patients with Marfan syndrome (MFS) with a severe cardiovascular phenotype, we explored the proportion of patients with MFS with a pathogenic FBN1 variant and analyzed whether the type/location of FBN1 variants was associated with specific clinical characteristics and response to treatment. Patients were recruited on the basis of the following criteria: aortic root z-score > 3, age 6 months to 25 years, no prior or planned surgery, and aortic root diameter < 5 cm.
Methods:
Targeted resequencing and deletion/duplication testing of FBN1 and related genes were performed.
Results:
We identified (likely) pathogenic FBN1 variants in 91% of patients. Ectopia lentis was more frequent in patients with dominant-negative (DN) variants (61%) than in those with haploinsufficient variants (27%). For DN FBN1 variants, the prevalence of ectopia lentis was highest in the N-terminal region (84%) and lowest in the C-terminal region (17%). The association with a more severe cardiovascular phenotype was not restricted to DN variants in the neonatal FBN1 region (exon 25–33) but was also seen in the variants in exons 26 to 49. No difference in the therapeutic response was detected between genotypes.
Conclusion:
Important novel genotype–phenotype associations involving both cardiovascular and extra-cardiovascular manifestations were identified, and existing ones were confirmed. These findings have implications for prognostic counseling of families with MFS.
Keywords: Clinical genetics, Connective tissue disease, FBN1, Genotype–phenotype associations, Marfan syndrome
Introduction
Marfan syndrome (MFS, OMIM 154700) is an autosomal dominant connective tissue disorder with multisystemic manifestations, and the disorder mainly affects the skeletal (eg, long bone overgrowth, joint hypermobility, scoliosis, pectus deformity), ocular (eg, myopia, ectopia lentis [EL]), and cardiovascular systems. This disorder has an estimated prevalence between 1 in 3000 and 1 in 5000.1 The vast majority of patients with MFS ultimately develop an aortic aneurysm, typically at the level of the sinuses of Valsalva, with a propensity to aortic dissection.
Although MFS was initially described in 1898, it took approximately 100 years to discover its genetic cause. Pathogenic variants in the FBN1 gene, which codes for the extracellular matrix protein FBN1, are the cause of MFS.2 Over the past 25 years, >3000 pathogenic FBN1 variants have been described (FBN1-Universal Mutation Database). Today, variants curated as pathogenic or likely pathogenic (P/LP) are identified in over 90% to 95% of patients with MFS, and molecular testing has become an integral part of the diagnostic decision-making process (revised Ghent nosology).3
Although genetic analysis of FBN1 now contributes significantly to the diagnostic process, its predictive value to phenotypic outcomes is still limited. Nevertheless, uncovering genotype–phenotype associations facilitates genetic counseling and potentially allows the development of more personalized surgical and/or pharmacological patient-management strategies. FBN1 variants causing MFS are distributed across the entire gene, encompass all variant types, and can be categorized largely into 2 groups, namely haploinsufficiency (HI) and dominant-negative (DN) variants. HI variants lead to a decreased amount of FBN1 protein in the extracellular matrix. DN variants result in the production of a mutant protein that can interfere with the function of wild-type FBN1 derived from the normal allele of the gene. The effects of both HI and DN variants can contribute to the pathogenesis of MFS.4,5
In this study, we performed a molecular screening and sought genotype–phenotype associations in a cohort of well-characterized patients with MFS participating in the Pediatric Heart Network (PHN) Marfan trial (atenolol vs losartan).6–8 This is the largest pediatric and adolescent MFS genotype–phenotype study reported to date.
Materials and Methods
Patient cohort
The PHN Marfan Trial (atenolol vs losartan; ClinicalTrials.gov Identifier: NCT00429364) randomized 608 patients; of these patients, only a subset had previously undergone FBN1 genetic testing.6 Patients were recruited on the basis of the following criteria: aortic root z-score > 3, age 6 months to 25 years, no prior or planned surgery, and aortic root diameter < 5 cm. All patients were offered the opportunity to participate in this genetics ancillary cohort study by donating an additional blood sample after signing informed consent. In total, 304 patients (50%) donated a blood sample for genetic analysis, which resulted in bad quality DNA of 2 patients (patient identifier (ID) 372 and 373). For the genotype–phenotype analyses, we also incorporated the FBN1 analysis results from 69 patients who did not provide a blood sample but agreed to share their genetic test results through their treating physician. Overall, detailed genetic information and clinical phenotyping for 373 patients with MFS,6 as well as aortic measurements over 3 years of treatment with losartan or atenolol, were available at the start of this genetics ancillary study.7 At least 27 patients had 1 or 2 family members included in the genetics ancillary study (as indicated in Supplemental Tables 1A and 1B).
Molecular analysis
After standard DNA extraction methods and HaloPlex enrichment of target regions (Agilent Technologies) of 14 core thoracic aortic aneurysm and dissection (TAAD) genes (Supplemental Table 2, genes in bold), sequencing was performed on MiSeq System (Illumina).9 Data analysis was performed using an in-house developed pipeline (for details see Proost et al9). When no pathogenic variant in FBN1 could be identified, multiplex ligation-dependent probe amplification for the detection of deletions/duplications of FBN1 and TGFBR2 was performed (MRC Holland).9 If both sequencing and multiplex ligation-dependent probe amplification were negative, an analysis using an extended TAAD panel comprising 34 genes was carried out (Supplemental Table 2, underlined genes). Variants were classified as pathogenic, likely pathogenic, or variant of uncertain significance (VUS) according to the adjusted American College of Medical Genetics and Genomics /Association for Molecular Pathology criteria (Supplemental Tables 1A, 1B, and 3).10 P/LP FBN1 variants were grouped into 2 classes on the basis of molecular criteria only: DN and HI (Supplemental Table 4). All variants were submitted to ClinVar.
Statistical analysis
Statistical analyses were performed using SPSS Statistics (IBM SPSS Statistics for Windows version 27.0, IBM Corporation), and all hypothesis tests were 2-sided. Comparisons of the categorical clinical characteristics between different patient groups were performed using the χ2 or Fisher exact tests. Continuous clinical variables were compared using the unpaired t test. When there were >2 groups, differences in aortic root z-score were tested using 1-way analysis of variance. The effect of variant type on response to treatment (change in aortic root z-score/year) was modeled using analysis of covariance, including age (the only factor found significant in the main PHN clinical trial) as a covariate. Assumptions of the used tests were verified, including the use of Levene’s test to assess the equality of variances. Patients who were lost for follow-up were excluded from the response to treatment analysis. Missing data were left as missing, without imputation. We carried out a false discovery analysis to account for the multiple hypotheses testing in R (R version 4.0.3, R Foundation for Statistical Computing) and R-package Q-value (package version 2.18.0). Q ≤ 0.05 was considered significant.
Results
Molecular screening of 302 patients with MFS and FBN1 variant spectrum
Figure 1 illustrates the workflow and results of the molecular screening. In total, we identified 270 variants in FBN1 (Supplemental Table 1A) and 10 patients with variants in other TAAD genes (Supplemental Table 1B). Furthermore, 17 deletions/duplications of (part of) the FBN1 gene were identified. Overall, 275 (91%) patients had a P/LP FBN1 variant, 12 (4%) patients had a FBN1-VUS, 10 (3%) patients had a P/LP variant or VUS in another gene associated with syndromic aortic aneurysms, and 5 (2%) patients had no causal variant identified. The clinical features of these TAAD panel–negative patients (n = 5) are listed in Supplemental Table 5. In 4 patients, 2 FBN1 variants were identified (always 1 P/LP FBN1 variant and 1 VUS).
Figure 1. Patient cohort and workflow of molecular characterization.
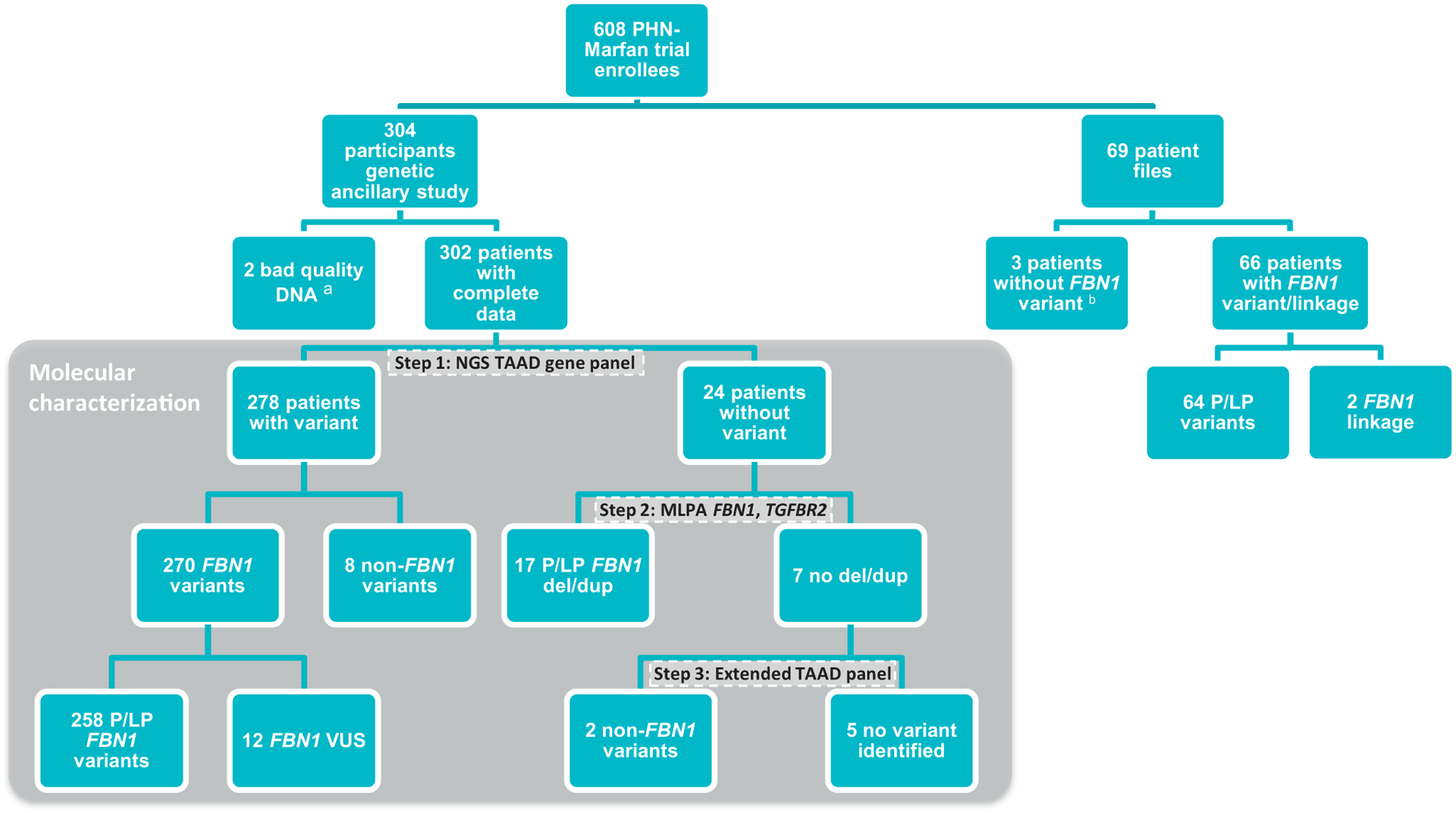
Overview of the participants included in this study and the results of their molecular screening. aIdentifiers (IDs) of patients with bad quality DNA: 372 and 373. bIDs of patients who did not have an FBN1 variant described in their patient report and were not sequenced in our genetic ancillary study: 369, 370, and 371. del, deletion; dup, duplication; MLPA, multiplex ligation-dependent probe amplification; NGS, next-generation sequencing; P/LP, pathogenic or likely pathogenic; PHN, Pediatric Heart Network; TAAD, Thoracic Aortic Aneurysm and Dissection; VUS, variant of uncertain significance
To study the FBN1 variant spectrum, we combined the results from our molecular screening with 64 patient reports that contained a FBN1 P/LP variant. In total, 339 P/LP FBN1 variants were identified in this extended cohort. These variants included 46% missense (of which 68% were cysteine-involving), 19% frameshift, 16% nonsense, and 13% splice site variants. In addition, 5% of subjects harbored a large deletion/duplication and 1% a small in-frame deletion/duplication (Figure 1). The overall distribution of the variant subtypes is comparable between the largest MFS cohort reported in the literature and our cohort (Supplemental eFigure 1).11
Genotype–phenotype associations
Overall cohort
No significant differences regarding major clinical characteristics, except positive family history (56% vs 71%, P = 2.4 × 10−4, Q = 0.003), were found between the genetics ancillary cohort (n = 373) and the trial patients not enrolled in this ancillary study (n = 235) (Supplemental Table 6).
Variant type: HI vs DN
We compared the phenotypes of individuals in the predicted DN (n = 174) and HI (n = 125) FBN1 groups (Figure 2, Table 1). In total, 40 P/LP FBN1 variants could not be classified in either of these groups because of uncertainty about their effect and were excluded from this comparative analysis. We observed no difference in aortic root diameters (absolute or z-score) or aortic root growth progression between the 2 groups. DN FBN1 variants in the entire gene were significantly associated with EL (61% [99/161]) when compared with HI variants (27% [29/109]) (P = 1.8 × 10−8, Q = 5.4 × 10−7), especially when only the cysteine-involving variants (71% [72/102]) were compared with HI variants (27% [29/109]) (P = 1.6 × 10−10, Q = 1.0 × 10−8). Skeletal features were more pronounced in patients harboring HI variants; pectus excavatum was more frequent in patients with HI variants (36% [45/125]) than in patients harboring DN variants (21% [36/174]) (P = .003, Q = 0.03), and the patients harboring HI variants had a taller stature (height z-score = 2.4 ± 1.1) than those heterozygous for DN variants (1.9 ± 1.2) (P = .001, Q = 0.01). A similar trend was observed for joint hypermobility (HI: 89% [109/122] vs DN: 80% [134/168]) (P = .03, Q = 0.18), but this P-value did not survive multiple hypotheses analysis. The P-values for these skeletal features did not change significantly when only the subset of patients with cysteine-involving or other missense variants were compared with patients with HI variants.
Figure 2. Workflow of genotype–phenotype analysis.
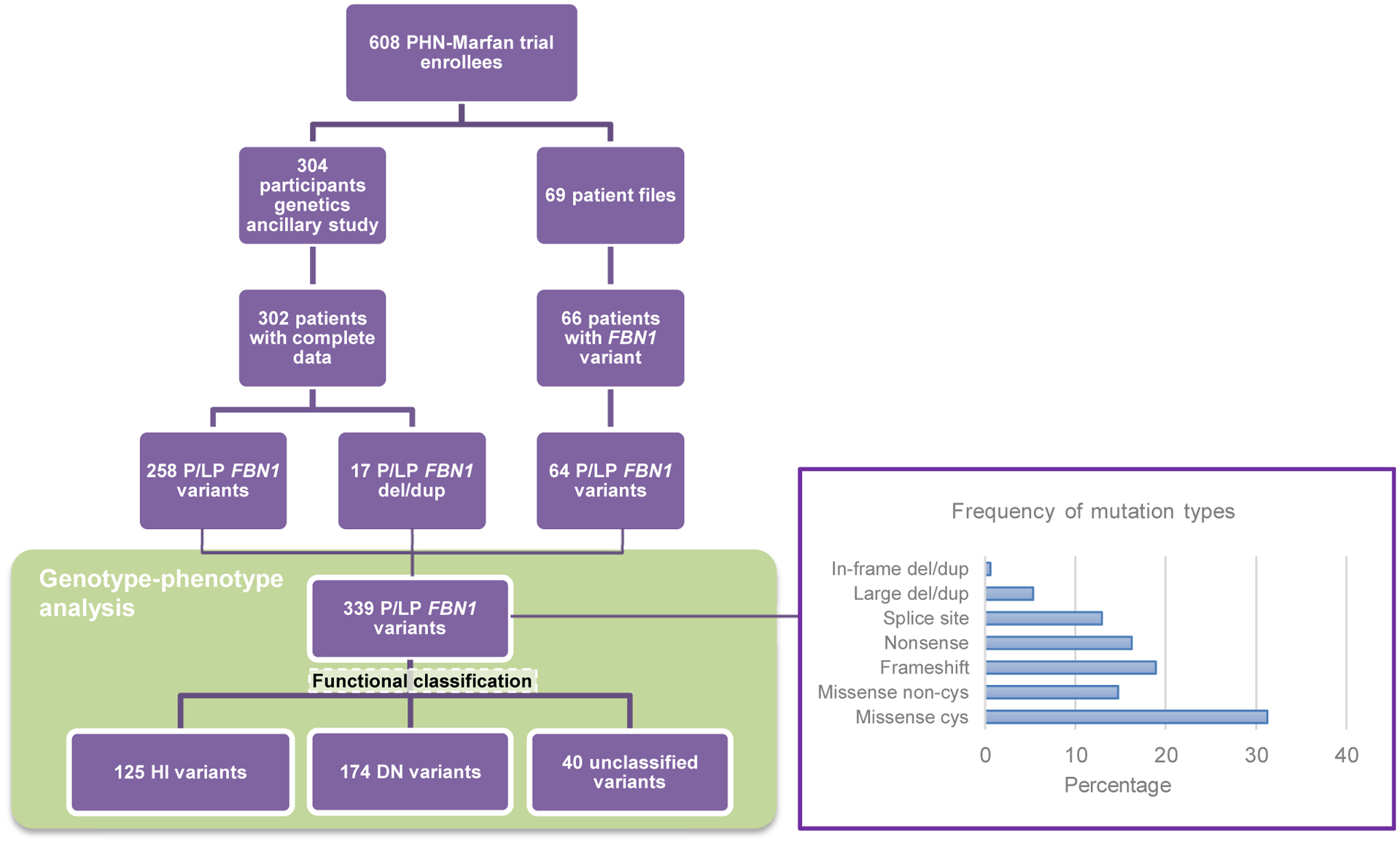
Overview ofthe variant workflow, functional classification, and variant types. cys, cysteine; del, deletion; DN, dominant negative; dup, duplication; HI, haploinsufficient; P/LP, pathogenic or likely pathogenic; PHN, Pediatric Heart Network.
Table 1.
Comparison of clinical phenotype in DN and HI variants patient groups
| Clinical Features | DN (n = 174) | HI (n = 125) | P | Q |
|---|---|---|---|---|
| Age ± SD (y) | 11.1 ± 6.1 | 10.9 ± 6.1 | .81 | 0.75 |
| Height ± SD (z-score) | 1.9 ± 1.2 | 2.4 ± 1.1 | .001 | 0.01 |
| BMI ± SD (kg/m2) | 16.7 ± 3.5 | 16.5 ± 4.1 | .80 | 0.75 |
| Ectopia lentis, n (%) | 99 (62) | 29 (27) | 1.78 × 10−8 | 5.43 × 10−7 |
| Highly arched palate, n (%) | 147 (87) | 109 (88) | .81 | 0.75 |
| Pulmonary artery diameter z-score > 2.0, n (%) | 20 (24) | 19 (33) | .22 | 0.44 |
| Dural ectasia, n (%) | 13 (42) | 5 (26) | .26 | 0.46 |
| Striae, n (%) | 64 (37) | 48 (39) | .69 | 0.75 |
| Arachnodactyly, n (%) | 114 (68) | 85 (69) | .77 | 0.75 |
| Pes plani, n (%) | 115 (68) | 95 (78) | .07 | 0.23 |
| Pectus excavatum, n (%) | 36 (21) | 45 (36) | .003 | 0.03 |
| Pectus carinatum, n (%) | 78 (45) | 49 (39) | .33 | 0.53 |
| Scoliosis, n (%) | 49 (29) | 34 (28) | .85 | 0.75 |
| Joint hypermobility, n (%) | 134 (80) | 109 (89) | .03 | 0.18 |
| Aortic root diameter ± SD (z-score ± SD) | 3.4 ± 0.7 cm (4.4 ± 1.3) | 3.4 ± 0.7 cm (4.2 ± 1.1) | .89 (.27) | 0.75 (0.46) |
| Change in aortic root diameter (z-score/y) ± SD | −0.15 ± 0.25 | −0.15 ± 0.24 | .95 | 0.77 |
BMI, body mass index; DN, dominant negative; HI, haploinsufficient.
Variant location
Exon 25 to 33 vs other
This neonatal FBN1 region is formerly described to include exons 24 to 32. However, in the most recent reference transcript of FBN1 (NM_000138.4), the noncoding exon 1 is added, which causes a shift in the numbering. Therefore, the neonatal region in this manuscript is now referred to as exon 25 to 33. Although we observed a higher aortic root z-score for patients with P/LP DN FBN1 variants in the so-called neonatal or middle region of the FBN1 gene (exon 25–33, 4.9 ± 1.5, n = 33) than for patients with HI and other P/LP DN FBN1 variants (4.2 ± 1.1, n = 265, P = .02, Q = 0.11, Supplemental Table 7), this did not survive multiple hypotheses analysis. We also observed a lower prevalence of striae (21% [7/33] vs 40% [105/264]) (P = .04, Q = 0.21) and higher prevalence of arachnodactyly (84% [26/31] vs 66% [173/261]) (P = .05, Q = 0.22) in the P/LP DN FBN1 neonatal group than in the HI and other P/LP DN FBN1 variant group. However, none of these 3 P-values survived multiple hypotheses analysis.
Exon 26 to 49 vs other
When visualizing the average aortic root z-score per exon for DN variants, the region with a more severe aortic phenotype appeared to be bigger than just the exon 25 to 33 region (Supplemental Table 8, Supplemental eFigure 2). We observed a significantly higher aortic root z-score for patients with P/LP DN variants in exons 26 to 49 (4.8 ± 1.4, n = 76) than for patients with other P/LP DN or HI FBN1 variants (4.1 ± 1.0, n = 223, P = 5.0 × 10−5, Q = 7.6 × 10−4).
5′-End vs middle vs 3′-end
On visualizing the proportion of EL in patients with DN P/LP variants on the basis of their location, a novel genotype–phenotype association emerged. A clear gradient was observed when comparing the proportion of EL between individuals with DN P/LP pathogenic variants across the FBN1 gene. On the basis of this gradient, 3 statistically different regions were defined with the following frequencies of EL: exon 2 to 24: 84% (31/37); exon 25 to 57: 64% (63/99); and exon 58 to 66: 17% (4/24) (Figure 3) (P = 7.3 × 10−7, Q = 1.5 × 10−5). Within this gradient, 2 specific subregions with 100% penetrance for EL were identified, which included all patients with cysteine-involving variants between residues 611 to 921 (n = 23) and 1348 to 1429 (n = 13).
Figure 3. Effect of location of DN variants on the proportion of ectopia lentis.
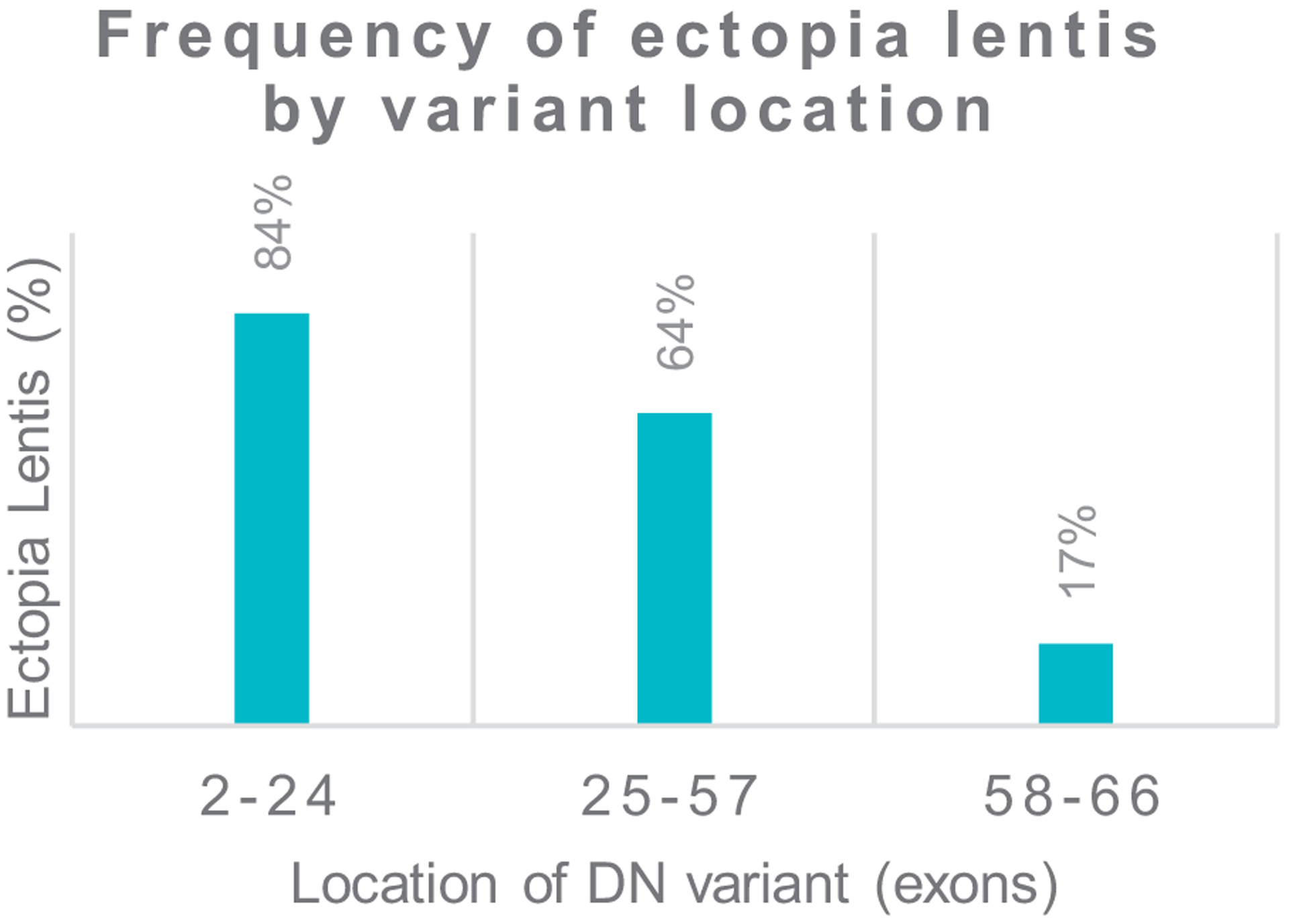
Groups were made on the basis of the proportion of ectopia lentis. DN, dominant negative.
Association of genotype with response to treatment
The patients included in the genetics ancillary study cohort showed comparable phenotypic characteristics in both treatment groups (losartan: n = 189, atenolol: n = 184), including aortic z-scores at the start of the treatment (Supplemental Table 9). Within the treatment groups, no difference in baseline aortic z-scores between DN and HI genotypes was observed. When analyzing the effect of variant type (ie, DN and HI) and treatment (ie, losartan and atenolol) on treatment outcome, we found no significant difference after accounting for age (change in aortic root z-score/year; HI + atenolol: −0.16 ± 0.04; HI + losartan: −0.15 ± 0.03; DN + atenolol: −0.17 ± 0.03; DN + losartan −0.12 ± 0.03, P = .18, Q = 0.39, Figure 4).
Figure 4. Effect of variant type (DN vs HI) and treatment (atenolol vs losartan) on response to treatment.
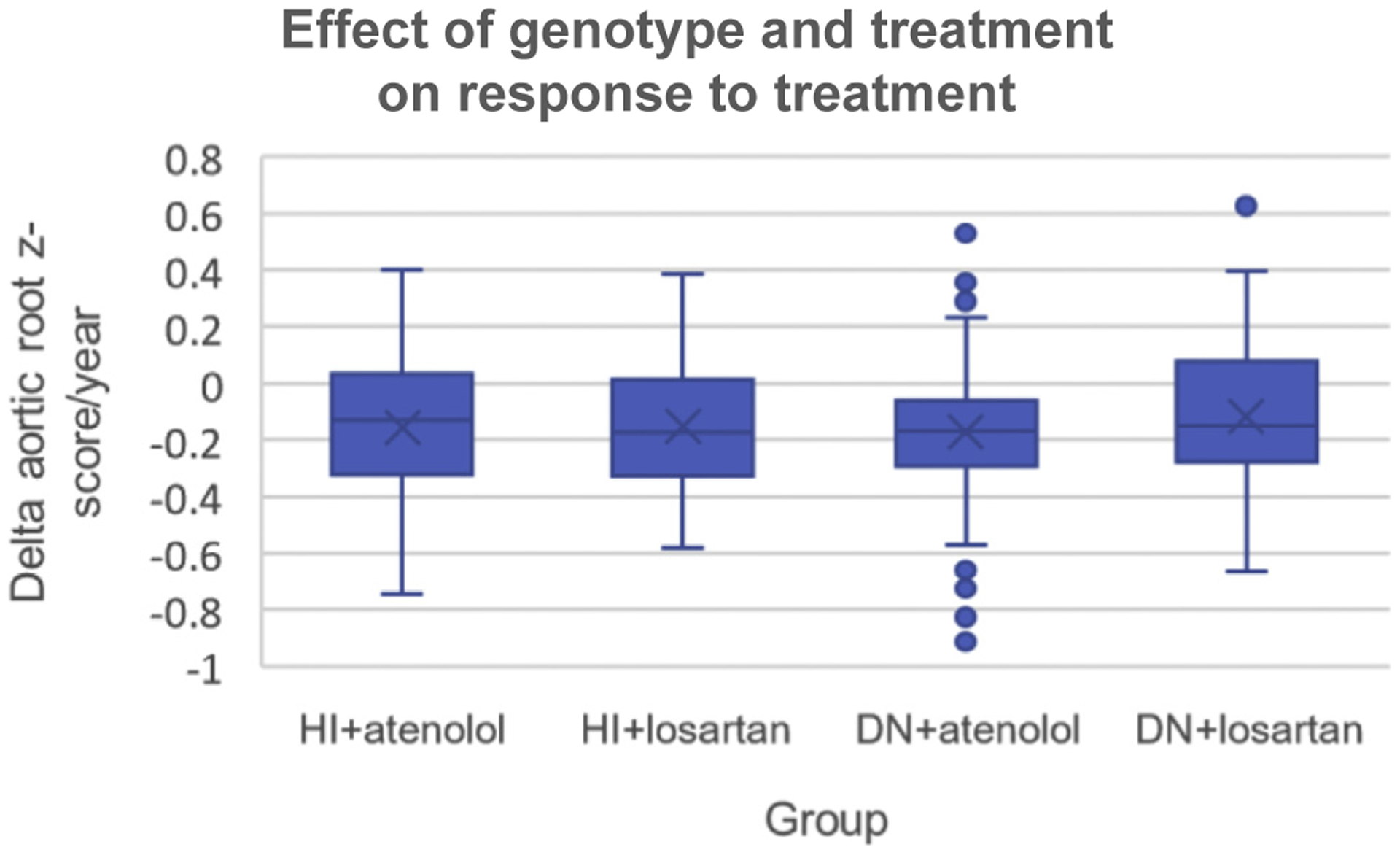
Boxplot of the change of aortic root z-score per year for the different groups.The thick line in the middle is the median. The green box shows the first and third quartiles. The whiskers show the maximum and minimum values, with the exceptions of outliers (circles). DN, dominant negative; HI, haploinsufficient.
No association between the nature or location (cysteine missense variants vs other missense variants, DN variants in the middle region [exon 26–49] vs other variants [including HI and DN in other exons], and DN variants in neonatal vs non-neonatal region) of the P/LP FBN1 variants and the treatment outcome was detected when accounting for age, but subgroups were smaller in size (data not shown). Throughout analysis of covariance analyses, we never observed any significant interaction between the independent variables.
Discussion
In the current study, our molecular analyses identified a P/LP FBN1 variant and VUS in the overwhelming majority (91% and 4%, respectively) of 302 clinically diagnosed pediatric patients with MFS. Only 3% presented with non-FBN1 variants and, as such, confirmed the solid association between MFS and the FBN1 gene.12 In total, 4 patients presented with 2 FBN1 variants, of which always 1 was a P/LP variant and 1 was a VUS. Owing to the use of a short read next-generation sequencing technology, the fact that the identified variants were located in different exons, and the fact that no complementary DNA was available for testing, we were not able to determine whether the FBN1 variants were located in cis or in trans. Furthermore, owing to the low number of patients (ie, 4) with 2 variants in FBN1, we were not able to confirm the association with a more severe phenotype, as has been suggested in literature.13
Despite significant progress in the understanding of the molecular defects underlying MFS, only a limited number of convincing genotype–phenotype associations have emerged. Several studies have attempted to define possible associations, but these were often refuted by subsequent analyses.2–17
Previous studies have suggested an association between DN FBN1 variants and EL.14,18,19 Our study convincingly confirmed that patients with EL are significantly enriched for DN variants (Q = 5.4 × 10−7), a finding even more pronounced when only cysteine-involving DN variants were considered (Q = 1.0 × 10−8). Overall, a cysteine-involving missense variant gives a 2.7-fold higher risk for developing EL than HI variants. For the first time, when considering DN variants, we found that EL was more frequent in the 5′-end than in the 3′-end. In their study, Pees et al14 described that EL occurs in 80% of cases with a pathogenic variant in exon 1 to 21, but we now describe a clear gradient that implies that DN variants that are located more toward the 5′-end of the gene are more likely to lead to EL. Furthermore, the following 2 subregions showed 100% penetrance of EL: cysteine-involving variants between residues 611 to 921 and 1348 to 1429. To confirm our observations in an independent cohort, we used the clinically classified patients by Groth et al.15 Our observations of 100% EL penetrance in patients with cysteine-involving variants in these subregions were confirmed in 49 and 14 patients. These regions overlap with LTBP2- and ADAMTS10-binding sites in FBN1.16,17 Pathogenic variants in those genes are linked to nonsyndromic autosomal recessive EL18–21 and Weill-Marchesani syndrome, respectively, in which EL is frequently observed.22 These observations have important clinical implications and aid patient-tailored management.
Another historical observation is that the vast majority of pathogenic variants in the central region of the FBN1 gene (exons 25–33) is linked to more severe MFS presentations,23 although exceptions have been reported.24 While analyzing this neonatal region of the FBN1 gene in our cohort, we did not observe a significant result for severity of cardiovascular involvement (aortic root z-score) for DN neonatal variants. However, it should also be noted that children at the most severe end of the MFS spectrum are underrepresented in this cohort on the basis of the exclusion criteria including aortic root diameters >5 cm and the presence of prior or planned aortic surgery. When we extended this region to exons 26 to 49, on the basis of the average aortic z-score per exon, we did observe a highly significant result, which also survived multiple hypotheses analysis (Q = 7.6 × 10−4). This indicates that on average patients with MFS with P/LP DN variant in exons 26 to 49 have a more severe aortic phenotype than patients with either HI variants or DN variants in other exons. This finding is in contrast with previous literature findings that suggest that patients with HI variants may have higher aortic risk.25–28 A recent study (n = 207) suggested that truncating and splice site variants more frequently correlate with aortic events at young age.26 The study by Franken et al27 also looked into the effect of the FBN1 variant subtype (HI vs DN) on prospective cardiovascular outcome, including survival and time free of dissection in patients with MFS. Overall, 357 adult patients with MFS were included. The adjusted hazard ratios (for age, sex, and prior events) showed a 2.5-fold increased risk of cardiovascular death (P = .05) and a 2.4-fold increased risk of the combined end point, comprising cardiovascular death and dissection (P < .001), in patients with MFS with HI FBN1 variants compared with those with DN variants. In the same cohort, the effect of variant type on treatment outcome was analyzed. As indicated by aortic root dilatation rate, patients with MFS with HI FBN1 variants seemed to be more responsive to losartan therapy than patients with DN variants.28 In our study cohort we were not able to replicate these findings because all patients received treatment (either losartan or atenolol) and could not be compared with patients without treatment.
Possible explanations for the different observations between our study and the study by Franken et al27 are the different ages of the patients with MFS (6 months-25 years vs >18 years), the greater cardiovascular severity at baseline in our study (z-score ≥ 3 as inclusion criterion), and the larger contribution of multiple individuals from the same families in the study by Franken et al.27 Our study classified FBN1 variants as DN and HI on the basis of sequence information alone, when in fact experimental evidence has shown that an accurate prediction of the ultimate effect of a FBN1 variant at the protein level is difficult to achieve. For example, it was shown that upregulation of the wild-type FBN1 allele can (partially) compensate for the loss of protein derived from a mutant FBN1 nonsense allele.29 In addition, FBN1 premature termination codon alleles can escape decay of the mutant messenger RNA through alternative splicing or other mechanisms and hence produce a truncated protein with DN potential. Alternatively, predicted DN variants, including cysteine-replacing missense variants, can encode mutant proteins that stall in the secretion process, limiting their ability to interfere with normal FBN1 in the extracellular matrix.30 Therefore, without experimental protein work, the effect of a specific FBN1 variant remains speculative.
Taken together, the presented and published data suggest that the nature and location of the implicated FBN1 defect can only partially explain phenotypic variability. Validation of observed genotype–phenotype associations is further hampered by the observation that related individuals harboring an identical pathogenic FBN1 variant vary widely with respect to disease severity. It has been speculated that differences in expression levels between the variant FBN1 allele and the wild-type FBN1 allele contribute to intrafamilial clinical variability.31–33 However, residual FBN1 expression in cultured cells has not proven to be a reliable predictor of phenotypic severity in small studies.5,29,31 In the largest systematic investigation to date (n = 80), low levels of wild-type FBN1 messenger RNA have been shown to associate with increased risk of EL and pectus abnormalities and a trend toward an increased risk of aortic dilatation.32 An alternative explanation for the intrafamilial variability is the involvement of modifier genes.34
Limitations
The uniqueness of this cohort lies in the fact that all patients with MFS are younger than 25 years and have an aortic root z-score > 3, which makes them rather severely affected. However, patients with an aortic dissection, a prior or planned aortic surgery, or an aortic root diameter > 5 cm were excluded from the study.6 As such, the most severe end of the spectrum was excluded, and consequently cardiovascular events were rare. Overall, these selection criteria could have created a bias regarding genotype–phenotype associations. Owing to the fact that this MFS cohort is very young, some phenotypic features could still develop later in life as reported in literature that most aortic events occur above the age 20 years.35 Therefore, it is possible that some genotype–phenotype associations are missed in this cohort. Furthermore, classification into DN and HI variants is based on predictions. No functional analyses were performed to confirm these predictions, and therefore, some variants could have been misclassified. Finally, no complementary DNA was available for analysis, and therefore, deep intronic variants could have been missed.
Conclusion
In conclusion, we present novel genotype–phenotype data in the largest pediatric and adolescent MFS cohort reported to date. A more severe aortic phenotype was not limited to the neonatal region because patients with (likely) pathogenic DN variants in a more extended central region of the gene (exons 26–49) had a significantly worse aortic phenotype than other patients. Furthermore, we observed a clear difference in the proportion of EL on the basis of the location of the DN variant. Patients with MFS with cysteine-involving variants have 2.6-fold higher risk of EL than those carrying HI variant. Furthermore, patients with cysteine-involving variants between residues 611 to 921 and 1348 to 1429 probably have a near 100% chance of developing EL. These findings could guide frequency of ocular follow-up. According to our data, both the location and type of the FBN1 variant influence the phenotype of patients with MFS. No effect of variant type on treatment outcome was detected in this cohort.
Supplementary Material
Acknowledgments
We are very grateful to the families and the Pediatric Heart Network investigators who participated in this study. This research was supported by funding from the Marfan Foundation (US); the University of Antwerp (GOA, Methusalem-OEC grant “GENOMED” FFB190208); the Research Foundation - Flanders (FWO, Belgium, G.0356.17); The Dutch Heart Foundation (2013T093); and the Fondation Leducq (MIBAVA Leducq 12CVD03). Bart L. Loeys is a senior clinical investigator of the Research Foundation - Flanders and holds a consolidator grant from the European Research Council (Genomia-ERC-CoG-2017-771945). Bart L. Loeys and Aline Verstraeten are members of European Reference Network on rare vascular disorders (VASCERN). Josephina A.N. Meester, Lotte Van Den Heuvel, Silke Peeters, and Marjolijn Renard are funded by the Research Foundation - Flanders. Seema Mital received funding from the National Heart, Lung, and Blood Institute (NHLBI) 1UG1HL135680-01, Heart and Stroke Foundation of Canada Chair, the Canadian Institutes of Health Research (CIHR) European Research Area (ERA) PerMed Joint Transnational Call, and the Ted Rogers Centre for Heart Research.
Footnotes
Ethics Declaration
All patients from the Pediatric Heart Network Marfan Trial were offered the opportunity to participate in this genetics ancillary cohort study by donating an additional blood sample after signing informed consent. Institutional Review Board approval was received form Ethical Committee University Hospital Ghent (Belgian Registry number B67020084735).
Conflict of Interest
The authors declare no conflicts of interest.
Additional Information
The online version of this article (https://doi.org/10.1016/j.gim.2021.12.015) contains supplementary material, which is available to authorized users.
Data Availability
All data relevant to the study are included in the article or uploaded as Supplemental Tables 1–9 and Supplemental Figures 1 and 2. Additional data are available from the corresponding author upon request. All variants observed in the present cohort were submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/): accession numbers SCV002025277 - SCV002025581.
References
- 1.Robinson PN, Arteaga-Solis E, Baldock C, et al. The molecular genetics of Marfan syndrome and related disorders. J Med Genet. 2006;43(10):769–787. 10.1136/jmg.2005.039669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dietz HC, Cutting GR, Pyeritz RE, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352(6333):337–339. 10.1038/352337a0. [DOI] [PubMed] [Google Scholar]
- 3.Loeys BL, Dietz HC, Braverman AC, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47(7):476–485. 10.1136/jmg.2009.072785. [DOI] [PubMed] [Google Scholar]
- 4.Judge DP, Biery NJ, Keene DR, et al. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J Clin Invest. 2004;114(2):172–181. 10.1172/JCI20641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mátyás G, Alonso S, Patrignani A, et al. Large genomic fibrillin-1 (FBN1) gene deletions provide evidence for true haploinsufficiency in Marfan syndrome. Hum Genet. 2007;122(1):23–32. 10.1007/s00439-007-0371-x. [DOI] [PubMed] [Google Scholar]
- 6.Lacro RV, Guey LT, Dietz HC, et al. Characteristics of children and young adults with Marfan syndrome and aortic root dilation in a randomized trial comparing atenolol and losartan therapy. Am Heart J. 2013;165(5):828–835.e3. 10.1016/j.ahj.2013.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lacro RV, Dietz HC, Sleeper LA, et al. Atenolol versus losartan in children and young adults with Marfan’s syndrome. N Engl J Med. 2014;371(22):2061–2071. 10.1056/NEJMoa1404731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lacro RV, Dietz HC, Wruck LM, et al. Rationale and design of a randomized clinical trial of beta-blocker therapy (atenolol) versus angiotensin II receptor blocker therapy (losartan) in individuals with Marfan syndrome. Am Heart J. 2007;154(4):624–631. 10.1016/j.ahj.2007.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Proost D, Vandeweyer G, Meester JA, et al. Performant mutation identification using targeted next-generation sequencing of 14 thoracic aortic aneurysm genes. Hum Mutat. 2015;36(8):808–814. 10.1002/humu.22802. [DOI] [PubMed] [Google Scholar]
- 10.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Faivre L, Collod-Beroud G, Loeys BL, et al. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet. 2007;81(3):454–466. 10.1086/520125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loeys B, De Backer J, Van Acker P, et al. Comprehensive molecular screening of the FBN1 gene favors locus homogeneity of classical Marfan syndrome. Hum Mutat. 2004;24(2):140–146. 10.1002/humu.20070. [DOI] [PubMed] [Google Scholar]
- 13.McInerney-Leo AM, West J, Wheeler L, et al. Compound heterozygous mutations in FBN1 in a large family with Marfan syndrome. Mol Genet Genomic Med. 2020;8(3):e1116. 10.1002/mgg3.1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pees C, Michel-Behnke I, Hagl M, Laccone F. Detection of 15 novel mutations in 52 children from 40 families with the Marfan or Loeys-Dietz syndrome and phenotype-genotype correlations. Clin Genet. 2014;86(6):552–557. 10.1111/cge.12314. [DOI] [PubMed] [Google Scholar]
- 15.Groth KA, Von Kodolitsch Y, Kutsche K, et al. Evaluating the quality of Marfan genotype-phenotype correlations in existing FBN1 databases. Genet Med. 2017;19(7):772–777. 10.1038/gim.2016.181. [DOI] [PubMed] [Google Scholar]
- 16.Hirani R, Hanssen E, Gibson MA. LTBP-2 specifically interacts with the amino-terminal region of fibrillin-1 and competes with LTBP-1 for binding to this microfibrillar protein. Matrix Biol. 2007;26(4):213–223. 10.1016/j.matbio.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 17.Kutz WE, Wang LW, Bader HL, et al. ADAMTS10 protein interacts with fibrillin-1 and promotes its deposition in extracellular matrix of cultured fibroblasts. J Biol Chem. 2011;286(19):17156–17167. 10.1074/jbc.M111.231571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ali M, McKibbin M, Booth A, et al. Null mutations in LTBP2 cause primary congenital glaucoma. Am J Hum Genet. 2009;84(5):664–671. 10.1016/j.ajhg.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Narooie-Nejad M, Paylakhi SH, Shojaee S, et al. Loss of function mutations in the gene encoding latent transforming growth factor beta binding protein 2, LTBP2, cause primary congenital glaucoma. Hum Mol Genet. 2009;18(20):3969–3977. 10.1093/hmg/ddp338. [DOI] [PubMed] [Google Scholar]
- 20.Désir J, Sznajer Y, Depasse F, et al. LTBP2 null mutations in an autosomal recessive ocular syndrome with megalocornea, spherophakia, and secondary glaucoma. Eur J Hum Genet. 2010;18(7):761–767. 10.1038/ejhg.2010.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumar A, Duvvari MR, Prabhakaran VC, Shetty JS, Murthy GJ, Blanton SH. A homozygous mutation in LTBP2 causes isolated microspherophakia. Hum Genet. 2010;128(4):365–371. 10.1007/s00439-010-0858-8. [DOI] [PubMed] [Google Scholar]
- 22.Dagoneau N, Benoist-Lasselin C, Huber C, et al. ADAMTS10 mutations in autosomal recessive Weill-Marchesani syndrome. Am J Hum Genet. 2004;75(5):801–806. 10.1086/425231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tiecke F, Katzke S, Booms P, et al. Classic, atypically severe and neonatal Marfan syndrome: twelve mutations and genotype-phenotype correlations in FBN1 exons 24–40. Eur J Hum Genet. 2001;9(1):13–21. 10.1038/sj.ejhg.5200582. [DOI] [PubMed] [Google Scholar]
- 24.Loeys B, Nuytinck L, Delvaux I, De Bie S, De Paepe A. Genotype and phenotype analysis of 171 patients referred for molecular study of the fibrillin-1 gene FBN1 because of suspected Marfan syndrome. Arch Intern Med. 2001;161(20):2447–2454. 10.1001/archinte.161.20.2447. [DOI] [PubMed] [Google Scholar]
- 25.Arnaud P, Milleron O, Hanna N, et al. Clinical relevance of genotype-phenotype correlations beyond vascular events in a cohort study of 1500 Marfan syndrome patients with FBN1 pathogenic variants. Genet Med. 2021;23(7):1296–1304. 10.1038/s41436-021-01132-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baudhuin LM, Kotzer KE, Lagerstedt SA. Increased frequency of FBN1 truncating and splicing variants in Marfan syndrome patients with aortic events. Genet Med. 2015;17(3):177–187. 10.1038/gim.2014.91. [DOI] [PubMed] [Google Scholar]
- 27.Franken R, Groenink M, de Waard V, et al. Genotype impacts survival in Marfan syndrome. Eur Heart J. 2016;37(43):3285–3290. 10.1093/eurheartj/ehv739. [DOI] [PubMed] [Google Scholar]
- 28.Franken R, den Hartog AW, Radonic T, et al. Beneficial outcome of losartan therapy depends on type of FBN1 mutation in Marfan syndrome. Circ Cardiovasc Genet. 2015;8(2):383–388. 10.1161/CIRCGENETICS.114.000950. [DOI] [PubMed] [Google Scholar]
- 29.De Backer J, Loeys B, Leroy B, Coucke P, Dietz H, De Paepe A. Utility of molecular analyses in the exploration of extreme intrafamilial variability in the Marfan syndrome. Clin Genet. 2007;72(3):188–198. 10.1111/j.1399-0004.2007.00845.x. [DOI] [PubMed] [Google Scholar]
- 30.Whiteman P, Handford PA. Defective secretion of recombinant fragments of fibrillin-1: implications of protein misfolding for the pathogenesis of Marfan syndrome and related disorders. Hum Mol Genet. 2003;12(7):727–737. 10.1093/hmg/ddg081. [DOI] [PubMed] [Google Scholar]
- 31.Hutchinson S, Furger A, Halliday D, et al. Allelic variation in normal human FBN1 expression in a family with Marfan syndrome: a potential modifier of phenotype? Hum Mol Genet. 2003;12(18):2269–2276. 10.1093/hmg/ddg241. [DOI] [PubMed] [Google Scholar]
- 32.Aubart M, Gross MS, Hanna N, et al. The clinical presentation of Marfan syndrome is modulated by expression of wild-type FBN1 allele. Hum Mol Genet. 2015;24(10):2764–2770. 10.1093/hmg/ddv037. [DOI] [PubMed] [Google Scholar]
- 33.Aoyama T, Francke U, Gasner C, Furthmayr H. Fibrillin abnormalities and prognosis in Marfan syndrome and related disorders. Am J Med Genet. 1995;58(2):169–176. 10.1002/ajmg.1320580216. [DOI] [PubMed] [Google Scholar]
- 34.Aubart M, Gazal S, Arnaud P, et al. Association of modifiers and other genetic factors explain Marfan syndrome clinical variability. Eur J Hum Genet. 2018;26(12):1759–1772. 10.1038/s41431-018-0164-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Groth KA, Stochholm K, Hove H, et al. Aortic events in a nationwide Marfan syndrome cohort. Clin Res Cardiol. 2017;106(2):105–112. 10.1007/s00392-016-1028-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data relevant to the study are included in the article or uploaded as Supplemental Tables 1–9 and Supplemental Figures 1 and 2. Additional data are available from the corresponding author upon request. All variants observed in the present cohort were submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/): accession numbers SCV002025277 - SCV002025581.