

Abstract
Epigenetic reader domains regulate chromatin structure and modulate gene expression through the recognition of post-translational modifications on histones. Recently, reader domains have also been found to harbor double-stranded (ds) DNA-binding activity, which is as functionally critical as histone association. Here, we explore the dsDNA recognition of the N-terminal bromodomain of the bromodomain and extra-terminal (BET) protein, BRD4. Using protein-observed 19F NMR, 1H-15N HSQC NMR, electrophoretic mobility shift assays (EMSA), and competitive-inhibition assays, we establish the binding surface of dsDNA and find it to be largely overlapping with the acetylated histone (KAc)-binding site. Rather than engaging in electrostatic contacts, we find dsDNA to interact competitively within the KAc-binding pocket. These interactions are distinct from the highly homologous BET bromodomain, BRDT. Nine additional bromodomains have also been characterized for interacting with dsDNA, including tandem BET bromodomains. Together, these studies help establish a binding model for dsDNA interactions with BRD4 bromodomains and elucidate the chromatin-recognition mechanisms of the BRD4 protein for regulating gene expression.
Graphical Abstract
INTRODUCTION
The eukaryotic genome is packaged in the cell nucleus in the form of a protein–DNA complex, chromatin.1 Its basic building block is the nucleosome, composed of a core of two copies of four histone proteins (H4, H3, H2A, and H2B) wrapped around by 147 base pairs of double-stranded (ds) DNA in a left-handed orientation.2 Histones have N-terminal unstructured and conformationally flexible tails projecting out from the nucleosome surface, which get post-translationally modified.3 Dynamic post-translational modifications (PTMs) of histones, such as acylation, methylation, and ubiquitination of lysine, influence the accessibility of the nucleosomal DNA, thereby exerting epigenetic control on gene transcription.4 For example, lysine acetylation (KAc) is associated with an open chromatin state and active gene expression.5 Apart from regulating electrostatic contacts between positively charged histone tails and phosphate groups on DNA, histone PTMs are recognized by reader domains, which work closely with the transcriptional machinery to regulate gene transcription.6
Reader domains are generally a part of multidomain chromatin-associating proteins, which can further modify or remodel chromatin.7 Though a considerable effort has been spent on elucidating the protein–protein interactions between modified histones and their cognate readers, a subset of the epigenetic readers have also been found to associate with DNA.8,9 Currently, members of the bromodomains (BDs),10–14 Tudor,15,16 chromodomain,17,18 PWWP (pro-line–tryptophan–tryptophan–proline),19,20 CW (four cysteine residues and two tryptophan residues) domain,21 PHD finger,22 PZP (PHD–zinc–knuckle–PHD),23 and SANT (Swi3, Ada2, NcoR, and TFIIIB)/Myb families24 exhibit nucleic acid-binding activity. There are different mechanisms through which reader domain–DNA interactions contribute to chromatin association. In one mechanism, the reader domain is part of a multidomain protein complex and forms weak, nonspecific contacts with the chromosomal DNA. These nonspecific interactions help the parent protein complex scan the chromosomal DNA and localize it at target sites.25 In another mechanism, the reader domain can associate with a given histone PTM and DNA multivalently to enhance selectivity and affinity for the respective histone PTM.12,15
Out of the 61 human bromodomains, members of the bromodomain and extra-terminal (BET) family have been studied in the most detail and are important epigenetic drug targets. BET proteins, BRD2, BRD3, BRD4, and BRDT, have two N-terminal tandem bromodomains (BD1 and BD2) followed by an extra-terminal (ET) domain.26 Through their BDs, BET proteins interact with acetylated histone proteins and transcription factors.27 Contrary to the general displacement of the transcriptional machinery and other BD-containing proteins from mitotic chromatin, BET proteins are retained on chromatin during mitosis.28 Thus, due to their significant role in regulating different cellular processes, it is essential to elucidate their chromatin-recognition mechanisms.
In 2014, Larue et al. reported high-affinity interactions of the short isoform of BRD4 (BRD4S) and its N-terminal BDs with dsDNA.13 Miller et al. subsequently reported an allosteric interaction between dsDNA and the first bromodomain of BRDT (BRDT–BD1) mediated via a basic patch of three lysines on the αZ-helix (Figure 1A).12 The authors proposed a bivalent mode of chromatin binding for BRDT–BD1 between acetylated histones and DNA. However, via electrophoretic mobility shift assay (EMSA), no interaction was seen between 167 bp dsDNA and BD1 of BRD4 tested at a single concentration (100 μM) of the protein, despite the high sequence similarity (BRD4–BD1: BRDT–BD1 = 76% similarity) between these two BDs (Figure 1C). While BRDT–BD1 interacted with acetylated nucleosomes with higher affinity as compared to acetylated peptides, BRD4–BD1 had comparable affinities for both of the binding partners further supporting a different binding mechanism. The tandem BDs of BRD4 were also found to interact with enhancer-directed transcripts (eRNAs) increasing the affinity of BRD4 for acetylated histones.29 Interestingly, a weaker interaction was seen for the tandem BDs of BRDT. More recently, Han et al. found BRD4S to interact with dsDNA in a sequence-independent manner.14 They attributed the high-affinity interactions with DNA to intrinsically disordered regions in the protein and a basic residue-enriched interaction domain (BID) present downstream to BD2.30 Using 1H-15N heteronuclear singular quantum coherence (HSQC) NMR spectroscopy, interactions between the tandem bromodomains of BRD4 and dsDNA showed that several residues on the structured domains were perturbed. However, interactions between isolated BDs and DNA were not structurally characterized. The interplay with acetylated histone recognition has also not been fully elaborated.
Figure 1.
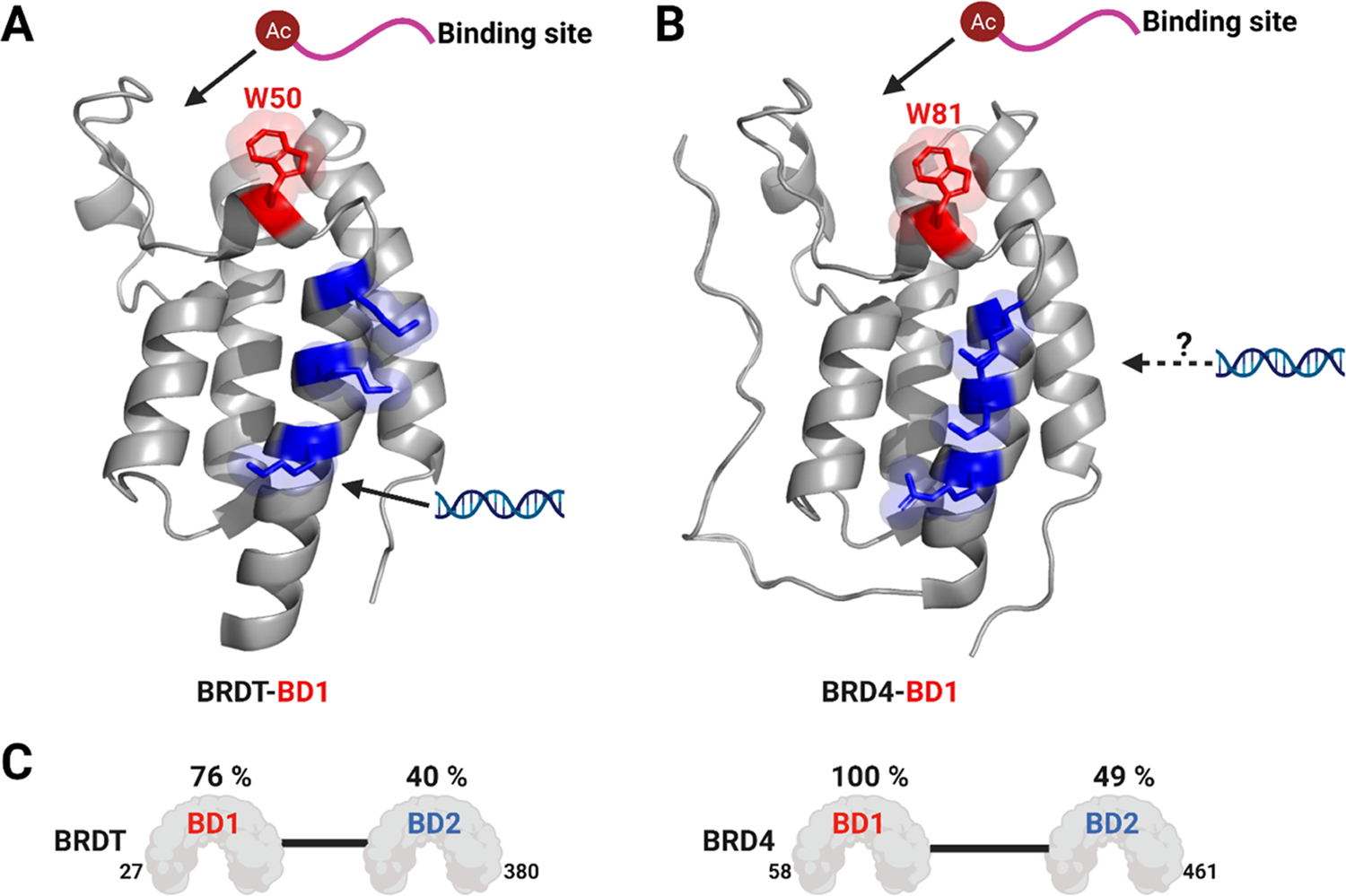
Acetylated histone-binding site, “WPF” shelf tryptophan (red), and a basic interaction site (blue) for (A) BRDT–BD1 (W50, K37, K41, and K45) and (B) BRD4–BD1 (W81, R68, K72, and K76). (C) Sequence similarity in each bromodomain of BRDT (left) and BRD4 (right) relative to BRD4–BD1. Percent similarities were relative to BRD4–BD1 calculated by the PDB sequence and structure alignment (PDB ID: 7L73 for BRDT–BD1 and 3UVW for BRD4–BD1).
Here, we performed a detailed structural analysis and used a variety of biophysical techniques to investigate interactions of BRD4–BD1 with dsDNA. Unexpectedly, given the high similarity between BRD4–BD1 and BRDT–BD1 (Figure 1C), we demonstrate dsDNA interactions via two distinct binding mechanisms. We used protein-observed 19F (PrOF) NMR and 1H-15N HSQC NMR spectroscopy to structurally characterize the binding interactions between BRD4–BD1 and DNA. Interestingly, rather than binding the protein via positively charged lysines, as in BRDT–BD1, we find dsDNA to interact with hydrophobic residues in the KAc-binding pocket. To further validate the site of interactions and to propose a new binding model, we conducted a mutational analysis to confirm our findings from different NMR studies. To establish the interplay between acetylated histone and DNA recognition, we further used a competitive-inhibition technique, AlphaScreen, and found dsDNA to compete with acetylated histone peptide binding with BRD4–BD1. Finally, we used PrOF NMR to assay the DNA-binding activity of nine additional BDs from three different classes: Class I (FALZ or BPTF BD), Class II (BET BDs), and Class III (CREBBP BD). We cross-validated our results from the PrOF NMR screen using EMSA. Thus, by performing detailed structural studies on BRD4–BD1, we now propose an alternative model to engage dsDNA by a BRD4 BD beyond the electrostatic patch model proposed by Miller et al. for BRDT–BD1.12 Such a model has yet to be confirmed with other BET BDs.
MATERIAL AND METHODS
Unlabeled and Fluorinated Protein Expression.
The pNIC28-Bsa4 plasmid containing the first bromodomain of BRD4 (Addgene #38943), first bromodomain of BRDT (Addgene #38898), second bromodomain of BRD2 (Addgene #39074), bromodomain of CREB-binding protein (Addgene #38977), and bromodomain of BPTF (Addgene #39111) were a kind gift from Nicola Burgess-Brown. The pET-28a (+) plasmids containing the second bromodomain of BRD4 (residues 333–460), first and second bromodomains of BRD3 (residues 22–144 and 304–416, respectively), R68S K72S K76S BRD4–BD1, and K37S K41S K45S BRDT–BD1 were purchased from GenScript. The pET-28b(+) plasmid (Kanr) containing the tandem bromodomains of BRD4 (38–460) and the pET-15b plasmid (Ampr) containing the tandem domains of BRDT (2–416) were a kind gift from Prof. Ernst Schönbrunn (Moffitt Cancer Center). The procedure for fluorinated protein expression by Gee et al. was followed.31 The Escherichia coli strain BL21 Star (DE3) was transformed with the plasmid containing the desired protein gene and plated onto an agar plate containing the appropriate antibiotics. The plate was incubated overnight at 37 °C. A 5 mL LB culture containing antibiotics was inoculated using a single colony from this plate and grown overnight at 37 °C with shaking at 215 rpm. The primary culture was used to inoculate 1 L of LB media containing chloramphenicol (35 mg/L), kanamycin (100 mg/L), or ampicillin (100 mg/L). This secondary culture was grown at 37 °C at 215 rpm until the optical density at 600 nm had reached 0.6–0.8. At this point, for unlabeled protein expression, an equilibration time of 30 min at 20 °C and 215 rpm was followed by the addition of 1 mM IPTG to induce protein expression. For 5-fluorotryptophan (5FW) labeling, the cells were pelleted by centrifugation and resuspended in 1 L of defined media, and 5-fluoroindole (80 mg) dissolved in dimethyl sulfoxide (DMSO, 200 μL) was added. After a recovery time of 90 min at 37 °C and 215 rpm, followed by a 30 min cooling to the induction temperature of 20 °C, the culture was induced with 1 mM IPTG and allowed to shake for 16–20 h. Cells were pelleted by centrifugation at 10,000g and stored at −80°C until purification.
Expression of 15N-Labeled Proteins.
Following the procedure for unlabeled proteins, when the secondary culture reached an optical density at 600 nm of 0.6–0.8, cells were harvested via centrifugation and resuspended in minimal media (33.7 mM disodium phosphate, 22 mM potassium phosphate, 8.55 mM sodium chloride, and 1 g/L 15N ammonium chloride) with trace elements [134 μM ethylenediaminetetraacetic acid (EDTA), 31 μM iron(III) chloride (FeCl3), 6.2 μM zinc(III) chloride (ZnCl2), 0.72 μM copper(II) chloride (CuCl2), 0.42 μM cobalt(II) chloride (CoCl2), 1.62 μM boric acid (H3BO3), and 0.081 μM manganese(II) chloride (MnCl2)], 1 mM magnesium chloride (MgCl2), 0.3 mM calcium chloride (CaCl2), 1 μg/L biotin, 1 μg/L thiamine, 0.4% w/v glucose, and correct antibiotic. Cells were allowed to equilibrate at 37 °C, 250 RPM for 90 min. The temperature was reduced to 20 °C for 30 min before protein induction with 1 mM IPTG. Cells were harvested after 16–20 h by centrifugation at 10,000g and stored at −80°C until purification.
Protein-Observed Fluorine NMR (PrOF NMR).
Experiments were run on a Bruker 600 MHz Avance NEO (6002), equipped with a 5 mm triple resonance cryoprobe. 5FW-labeled bromodomains were diluted in 50 mM HEPES, 100 mM NaCl, and pH 7.4 buffer by the addition of D2O and 0.1% TFA/H2O to final concentrations of 4 and 0.4%, respectively. Two one-dimensional 19F NMR spectra were taken of the control protein sample at an O1P of −75 ppm, NS = 16, D1 =1.0 s, and AQ = 0.5 s (TFA reference set to −75.25 ppm) and an O1P of −125 ppm, NS = 1000–3000, D1 = 0.6 s, and AQ =0.05 s (protein resonances). Peptide stock solutions of 15–35 mM prepared in Milli-Q water or DNA stock solutions of 1000 μM prepared in Milli-Q water were titrated into bromodomain protein solutions (25–35 μM). The change in chemical shift of the protein resonance (Δδobs) was plotted as a function of ligand concentration to generate binding isotherms using eq 1 where (Δδmax) is the maximum change in fluorine chemical shift, [L] is the concentration of the ligand, [P] is the concentration of protein, and [PL] is the concentration of the bound complex. All titrations were performed in a single replicate unless otherwise specified.
| (1) |
1H-15N SOFAST HSQC.
1H-15N SOFAST HSQC spectra were obtained on a Bruker 600 MHz Avance NEO with a CryoProbe 5 mm TCI (1H, 13C, 15N, and 2H) w/ Z-gradient using Bruker’s IBS_SOFAST.x method with 32–64 scans, 0.05 s acquisition time, and 0.2 s delay. Data were acquired at 300 K and 45–55 μM protein in 50 mM phosphate, 100 mM NaCl, pH 7.4, and 5% (v/v) D2O. Data were processed with TopSpin. Δδ 1H–15N was calculated using eq 2.
| (2) |
Peptide Synthesis.
H4 K5Ac,K8Ac,K12Ac,K16Ac (H2N-YSGRGKacGGKacGLGKacGGAKacRHRK-C(O)NH2) were synthesized using standard N-9-fluorenylmethoxycarbonyl (Fmoc) solid-phase synthesis methods on NovaSyn TGR resin (Novabiochem, 0.25 mmol/g) using a Liberty Blue automated microwave synthesizer (CEM) and N,N′-diisopropylcarbodiimide (DIC) and Oxyma for amino acid activation. The peptide was cleaved from the solid support in a mixture of 95:2.5:2.5 trifluoroacetic acid (TFA)/triisopropylsilane-(TIPS)/water for 2–5 h followed by evaporation of the solvent under a nitrogen stream. The crude peptide was precipitated into cold diethyl ether ((C2H5)2O) and purified by reverse-phase (RP) high-performance liquid chromatography (HPLC) on a C-18 column using 0.1% TFA, water, and acetonitrile (CH3CN) as solvents (0–50% CH3CN gradient over 60 min). Peptide molecular weight was confirmed using an Ab-Sciex 5800 matrix-assisted laser desorption ionization (MALDI) time-of-flight mass spectrometer. Peptide theoretical and observed masses are listed in Table S2.
Nucleosome Preparation.
For nucleosome preparation, individual Xenopus laevis histones H2A, H2B, H3, and H4 were expressed and purified using a previously described one-pot protocol.32 In short, full-length X. laevis histones were transformed into BL21 (DE3) cells, except for H4, in which case, C41 (DE3) cells were used. H2A had the TEV-cleavage site present in the protein sequence. Bacterial cultures were allowed to grow at 37 °C until an OD600 of 0.6–0.8 was reached at which point protein expression was induced with 0.4 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) for 3 h (H2A, H2B, and H3) or 2 h (H4). Equimolar amounts of histones were combined in a single volume and isolated from inclusion bodies using denaturing conditions (8 M guanidinium hydrochloride, 20 mM sodium acetate, pH 5.2, 10 mM DTT). This mixture was subjected to sonication, centrifuged down, and the supernatant was dialyzed into a high salt buffer (20 mM Tris-HCl, pH 8.0, 2 M NaCl, 2 mM β-mercaptoethanol) overnight to allow for octamer refolding. Refolded solution was spun down to isolate the supernatant, and octamer concentration was determined with UV/vis at 280 nm. The nucleosome was reconstituted by combining the TEV-cleaved refolded octamer and 147 bp 601 Widom sequence33 in a 1:1 molar ratio using a linear salt gradient overnight at 4 °C, as previously described.34 Low salt buffer (20 mM Tris-HCl, pH 7.5, 1 mM EDTA, 1 mM DTT) was mixed in via a peristaltic pump running at 1 mL/min with a volume of at least 4 L. For long-term storage, reconstituted nucleosomes were further dialyzed against a cacodylic acid solution (20 mM cacodylic acid, pH 6.0, 1 mM EDTA) overnight at 4 °C.
Electrophoretic Mobility Shift Assay.
The binding reactions were set up in the reaction buffer (50 mM HEPES, 100 mM NaCl, pH 7.4) with either DNA (227 nM final concentration) or unmodified nucleosome (205 nM final concentration). The protein was added to the final concentrations indicated. Binding was performed for 90 min at 4°C. Protein–DNA complexes were resolved by PAGE (6% polyacrylamide and 1× TBE) at 110 V for ~30–45 min for DNA and ~2 h for nucleosomes at (4 °C). Gels were visualized via staining with ethidium bromide for 15 min followed by imaging using a GE Typhoon FLA 9500 gel scanner.
AlphaScreen assay.
Unlabeled His9-tagged BRD4–BD1 was expressed and purified, as described previously.35 The AlphaScreen assay procedure for the BRD4–BD1 bromodomain was adapted from the manufacturer’s protocol (PerkinElmer). Nickel-nitriloacetic acid (Ni-NTA) acceptor beads and streptavidin donor beads were purchased from PerkinElmer (Cat. #: 6760619M). The biotinylated histone H4 KAc5,8,12,16 peptide was purchased from EpiCypher, with the sequence: Ac-SGRGK(Ac)GGK(Ac)GLGK(Ac)GGAK(Ac)RHRKVLR-Peg(Biotin).
All reagents were diluted in the assay buffer (50 mM Na-HEPES (ChemImpex), 100 mM NaCl (Sigma-Aldrich), 0.05% CHAPS (RPI), 0.1% BSA (Sigma-Aldrich), pH 7.4). The final assay concentrations (after the addition of all assay components) of 15 nM for the His9-tagged BRD4–BD1 bromodomain and 50 nM for the biotinylated peptide were used. Three-fold serial dilutions were prepared with varying concentrations of the test peptide and DNA and a fixed protein concentration. Five microliters of these solutions was added to a 384-well plate (ProxiPlate-384, PerkinElmer). This was followed by the addition of 5 μL of the biotinylated peptide. Five microliters of nickel chelate acceptor beads and 5 μL of streptavidin donor beads were added to each well under low light conditions (<100 lux) to a final concentration of 20 μg/mL. The plate was sealed and incubated at room temperature in the dark for 1 h. It was then read in AlphaScreen mode using a Tecan Spark plate reader. Test peptides and DNA were run in two technical replicates, N = 3. The data were normalized against the 0 μM inhibitor signal to obtain the % normalized AlphaScreen signal, and IC50 values were calculated in GraphPad Prism 5 using the sigmoidal four-parameter logistic (4PL) curve fit.
Site-Directed Mutagenesis.
Site-directed mutagenesis of N140A on BRD4–BD1 was conducted using previously reported transfer PCR procedures in 50 μL reaction mixtures.36 Reaction mixtures contained Phusion High-Fidelity Master Mix (NEB), template DNA, and 50 nM concentrations of each of the primers: the T7-forward primer (5′-TAATACGACTCACTATAGGG-3′) and the reverse-complement primer (N140A: 5′-CATCTCCAGGCTTCGCGTAGATGTAACAATT-3′). Successful mutagenesis was confirmed by DNA sequencing.
RESULTS
BRD4–BD1 Forms a Nonspecific Interaction with dsDNA in the Acetylated Histone-Binding Pocket.
NMR structural analyses of BD–DNA interactions were first evaluated to complement previously reported gel-shift assay data by Miller et al.12 In that study, BRDT–BD1 interacted with dsDNA with moderate affinity (Kd = 52 μM for a 25 bp dsDNA) via a basic patch of lysines (K37, K41, and K45) situated on the αZ-helix. However, the authors observed no interaction for BRD4–BD1 tested at 100 μM with a 167 bp Widom sequence. As an alternative method, protein-observed 19F (PrOF) NMR spectroscopy is a powerful technique to quantify weak protein–protein interactions with protein side chains and can provide additional structural information relative to gel-shift assays.37 Before moving on to investigate BRD4–DNA interactions, we first established PrOF NMR as a tool to detect protein–DNA interactions by studying the DNA-binding events of BRDT–BD1. To obtain fluorinated BRDT–BD1 for PrOF NMR, we incorporated 5-fluorotryptophan (5FW) via metabolic labeling. In this case, 5-fluoroindole is added during recombinant protein expression resulting in sequence-selective labeling of both tryptophan W44 and the “WPF shelf” tryptophan, which is located near the histone-binding site, W50.38 Figure S1 shows the PrOF NMR titration experiment of the previously tested 25 bp dsDNA by Miller et al.12 with 5FW BRDT–BD1. Stacked 19F NMR spectra show that the binding of the 25 bp dsDNA leads to chemical shift perturbations (CSPs) in the fast chemical exchange regime on the fluorine chemical shift time scale with a dose-dependent change in the chemical shift (Δδ) of W50. Plotting the Δδ, using a one-site binding model, yielded a dissociation constant (Kd) of 62 μM. Interestingly, W44 present on the αZ-helix proximal to the basic patch of lysines exhibited neither broadening nor a significant dose-dependent perturbation. As 5FW BRDT–BD1 has two fluorinated tryptophans to serve as 19F NMR probes, this technique is limited in resolution to structurally elucidate the binding of DNA to BRDT–BD1. Nevertheless, we could establish the use of the PrOF NMR assay to detect protein–DNA-binding interactions.
Having established that PrOF NMR can be used to determine comparable BD affinities with DNA to that established in the literature, we turned toward BRD4–BD1. In this case, BRD4–BD1 tryptophans, W75, W120, and the “WPF shelf” tryptophan, W81, were labeled with fluorine by the same protocol described above. We first investigated a 40 bp dsDNA sequence from the SMYD1 gene reported by Larue et al. to bind to BRD4S and the tandem bromodomains of BRD4 (BRD4-T).13 Figure 2 shows the PrOF NMR titration experiments of this 40 bp dsDNA with 5FW BRD4–BD1. Stacked 19F NMR spectra show that the binding of the 40 bp dsDNA leads to a reproducible and dose-dependent Δδ of W81 resulting in a Kd of 51 ± 12 μM (Figure 2B). Using PrOF NMR, we further analyzed additional dsDNA sequences reported in the literature (25 bp and 66 bp dsDNA from Miller et al.12 and 41 bp dsDNA from Han et al.14) and found BRD4–BD1 to interact with them in a sequence-independent manner (Figures S2–S4) with moderate affinities (18–118 μM) that are inversely related to the length of the dsDNA (66–25 bp).
Figure 2.
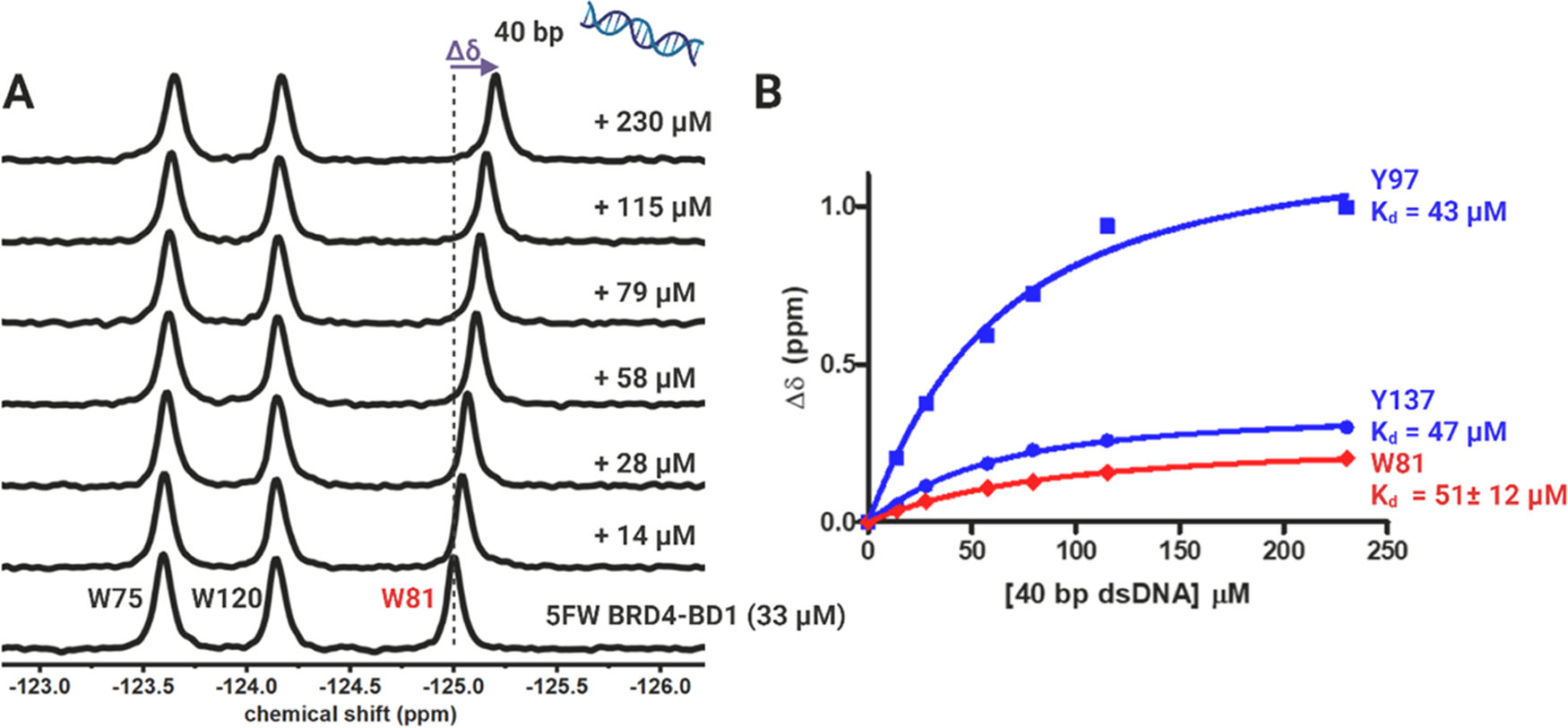
PrOF NMR experiment with 40 bp dsDNA binding to 5FW BRD4–BD1. (A) 19F NMR spectra and (B) binding isotherm of 40 bp dsDNA (14–230 μM) with 33 μM 5FW BRD4–BD1 and 25 μM 3FY BRD4–BD1. W81 is the “WPF shelf” tryptophan, and Y97 and Y137 are around the KAc-binding site, colored red and blue, respectively. The change in the chemical shift (Δδ) is shown in purple and is indicated by the dashed line. Kd calculated from the CSP of W81 is reported as the mean ± SD of three experimental replicates.
For better structural coverage, we expressed 3-fluorotyrosine (3FY)-labeled BRD4–BD1. There are seven tyrosines, Y65, Y97, Y98, Y118, Y119, Y137, and Y139, in BRD4–BD1 to serve as sensitive 19F NMR probes. Figure S5 shows the PrOF NMR titration experiment of 40 bp dsDNA with 3FY BRD4–BD1. 19F NMR spectra show that the binding of the 40 bp dsDNA leads to CSPs in fast chemical exchange with a dose-dependent Δδ of Y97 (Kd = 43 μM) and Y137 (Kd = 47 μM) (Figure 2B). Y97 forms a water-mediated hydrogen bond to acetyl-lysine groups on acetylated histones. Mishra et al. studied 19F NMR-binding signatures of various small molecules to 3FY BRD4–BD1 and found Y97 and Y137, near the histone-binding pocket, to show significant CSPs.38 In a parallel experiment here, to study the binding footprint of a native ligand, we also tested a tetra-acetylated peptide representing the N-terminal 21 amino acids of histone 4 (H4) against 3FY BRD4–BD1 and found Y137 to move downfield and Y97 broadened to the baseline (Figure S6). Thus, both 5FW- and 3FY-labeling strategies supported interactions between BRD4–BD1 and dsDNA around the KAc-binding site as an alternative site to the basic patch identified previously for BRDT–BD1.
Due to an alternative binding site on BRD4–BD1 distinct from the basic patch previously identified on the highly similar BRDT–BD1, we further mapped the binding interactions between dsDNA and non-fluorine-labeled BRD4–BD1 by 1H–15N HSQC NMR spectroscopy. 1H−15N HSQC NMR spectra were recorded on 15N BRD4–BD1 apo and upon titration of 40 bp dsDNA. The spectrum of 15N BRD4–BD1 was assigned based on literature assignments.39 The addition of dsDNA leads to CSPs of BD resonances in the fast chemical exchange regime on the proton/nitrogen chemical shift time scale similar to PrOF NMR, indicating binding in a similar affinity range (Figures 3 and S7). Mapping the CSPs onto the BD structure reveals that the CSPs are centered on the ZA loop (Q84, Q85, A89, and Y97) and αB (F133, N135, C136, and Y137) and αC helices (D144, I146, V147, and L153) (Figure 3A,B). These residues constitute the well-defined and largely hydrophobic KAc-binding pocket (Figure 3C) and support our findings by PrOF NMR. Kd values were calculated from normalized CSPs, and a residue-averaged Kd of 57 ± 6 μM was obtained (Figure S8). These results are similar to our affinities with fluorine-labeled proteins and are consistent with our prior findings that fluorination of the tyrosine and tryptophan residues is minimally perturbing to structure and function.38 Contrary to the basic patch model for BRDT–BD1, positively charged residues on the αZ-helix of BRD4–BD1, R68, K72, and K76 did not show statistically significant CSPs. Similar residues showed CSPs either in fast chemical exchange or exhibited slower exchange rates leading to broadening when a tetra-acetylated H4 peptide (H4 K5Ac,K8Ac,K12Ac,K16Ac) was titrated into 15N BRD4–BD1 (Figures 3D and S9–10). We also tested another reported dsDNA sequence, the slightly longer 66 bp dsDNA, to see if any additional contacts are made with a longer DNA. For 66 bp dsDNA, significant CSPs were found to be centered on the ZA loop and αB and αC helices as well (Figures S11–13). We concluded from these experiments that dsDNA engages with the N-terminal domain of BRD4 via residues in the conserved KAc-binding pocket.
Figure 3.
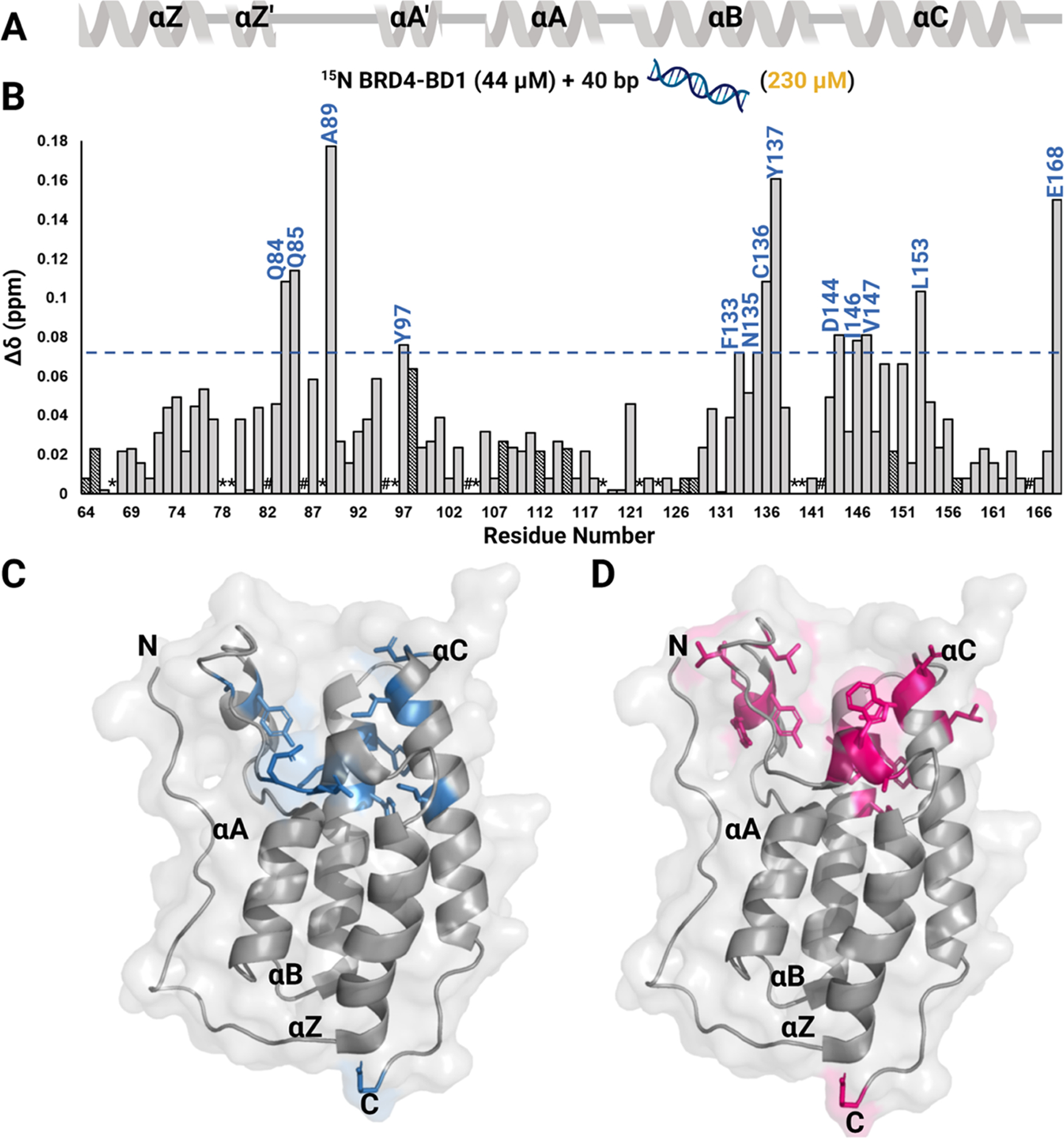
1H-15N HSQC NMR experiment with 40 bp dsDNA binding to 15N BRD4–BD1. (A) Secondary structure of the BD is denoted above the CSP plot. (B) Normalized CSPs are plotted as a function of residues upon titration of 40 bp dsDNA for the DNA concentration up to 230 μM and residues that are perturbed greater than the average plus one standard deviation are labeled in blue. A blue line marks this level of significance. Dashed bars indicate a CSP at a lower concentration of DNA (28 μM for D128, F157, 79 μM for Q64, G108, K112, and 115 μM for Y65, Y98, E115, Q127, and A150) as the chemical shift at 230 μM could not be assigned. These CSPs also accounted for average and standard deviation calculations. *indicates a missing resonance and # indicates a proline residue. A surface representation of the BRD4–BD1 structure (PDB ID: 3UVW) with residues that are significantly perturbed upon binding to (C) 40 bp dsDNA are colored in blue and (D) to H4 K5Ac,K8Ac,K12Ac,K16Ac are colored in pink.
In parallel to NMR assays, we explored a direct-binding gel-shift assay, electrophoretic mobility shift assay (EMSA), to monitor protein–DNA interactions with unmodified nucleosomes and dsDNA. These studies were conducted with bromodomains without poly-histidine tags (His6), as we found these led to artifacts in our experiments consistent with reports of others (Figure S17).40 To test our conditions for assay setup, we first analyzed the interaction between BRDT–BD1 and the unmodified nucleosome (Figure S14A) and 40 bp dsDNA (Figure S14B). As expected, we saw a shift for both ligands, indicating binding. As a negative control, we made the K37S K41S K45S (3KS) mutant of BRDT–BD1 and found a considerable reduction in affinity for both the unmodified nucleosome and dsDNA (Figure S15) consistent with prior reports.12 In the case of BRD4–BD1, we tested a wide range of protein concentrations in the assay and found a slight decrease in the intensity of the free nucleosome or dsDNA band at higher protein concentrations, indicating weaker-affinity protein–DNA interactions (Figure S16). From these experiments, we infer that EMSA, which is a non-equilibrium binding assay, can tend to miss weak protein–DNA interactions, as was the case for the analysis from Miller et al..22 Thus, for further experiments, we focused on our analyses using NMR.
Mutational Analysis on BRD4–BD1 to Validate the DNA-Binding Surface.
So far, our NMR studies utilizing 19F (5FW, 3FY) and 15N-labeling strategies have indicated dsDNA interactions with BRD4–BD1 in the KAc-binding site. To further validate these findings and investigate residue-specific contributions, we decided to test an N140A mutant for DNA binding. N140 forms a hydrogen bond to acetylated lysine and helps anchor the acetylated binding partner in the KAc-binding pocket. Mutation of N to A reduces the affinity for acetylated histones.41,42 We wanted to investigate whether N140 is a critical residue for DNA interactions as well.
First, we expressed 5FW N140A BRD4–BD1 and performed PrOF NMR titration of 40 bp dsDNA with the mutant protein. In the 19F NMR spectra, W81 did not exhibit a dose-dependent CSP leading to poor fitting of the binding isotherm (Figure S18). For further investigation, we also expressed 15N N140A BRD4–BD1 and assigned the spectrum of the mutant protein by overlaying it with the native protein’s spectrum. The mutant proteins are well folded as supported by the dispersed resonances in 19F NMR and 1H–15N HSQC spectra. 1H–15N HSQC spectra were collected for the titration of 40 bp dsDNA with 15N N140A BRD4–BD1 (Figures S19–21). Plotting the CSPs as a function of the BD residue revealed that CSPs were centered on the ZA loop (Q85, V87, N93, and A89) and αB (Y137) and αC helices (D144, I146, L148, and A152) (Figure 4B). Interestingly, we observed significant CSPs from K99, I100, and K102, situated on αA′, which were not observed for the titration with the native protein (Figures S19–20). However, similar residues were also perturbed when H4 K5Ac,K8Ac,K12Ac,K16Ac were titrated with the mutant protein indicating comparable binding profiles for dsDNA and the acetylated histone for the N140A mutant (Figures S22 and 23). Kd values were calculated from normalized CSPs, and a residue-averaged Kd of 780 ± 94 μM was obtained, indicating a >10-fold reduction in affinity for the N140A mutant (Figures 4A and S21). We conclude similar to histone binding, N140 may serve as a key hydrogen-bond anchoring residue for DNA.
Figure 4.
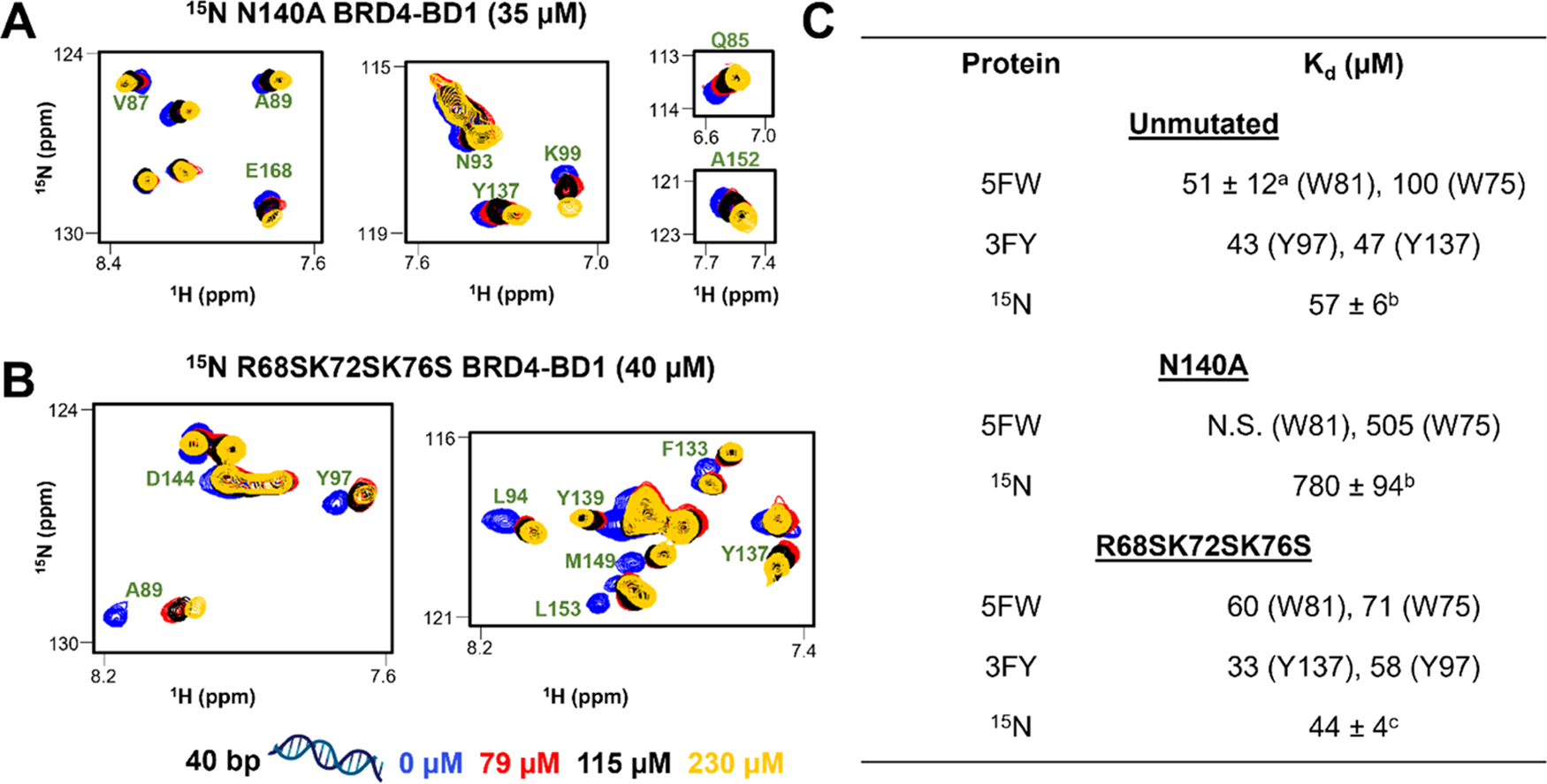
N140A mutation in BRD4–BD1 decreases affinity for dsDNA, whereas R68S K72S K76S mutations have a minimal effect. Overlay of 1H–15N HSQC spectra of (A) 15N N140A BRD4–BD1 (35 μM) and (B) 15N R68S K72S K76S BRD4–BD1 (40 μM) upon titration of 40 bp dsDNA. The selected region displays resonances of significant residues labeled in green. Titrations were performed at DNA concentrations of 0, 28, 58, 79, 115, and 230 μM. For clarity, only four points are displayed, and spectra are color-coded accordingly as shown in the legend. (C) Table summarizing dissociation constants of unmutated and mutant proteins binding to 40 bp dsDNA obtained using various labeled proteins (5FW, 3FY, and 15N). a: Kd is reported as the mean ± SD of three experimental replicates. b,c: Kd averaged over 12 and 9 resonances, respectively.
Following up on the findings from Miller et al.,12 to rule out a significant contribution of the basic, positively charged residues on the αZ-helix (Figure 1), we also made the triple mutant, R68S K72S K76S (called RKKS from hereafter). We expressed 5FW-, 3FY-, and 15N RKKS mutants and performed NMR titrations with 40 bp dsDNA. Plotting the dose-dependent CSPs of W81, Y97, and Y137 yielded Kd values of 60, 58, and 33 μM, respectively (Figures S24–25). Plotting the CSPs of 1H–15N cross-peaks as a function of the BD residue reveals that CSPs were centered on the ZA loop (Q84, Q85, A89, Y97, and Y98) and αB (F133, C136, and Y137) and αC helices (D144, I146, V147, and L153) (Figures S26 and 27). Kd values were calculated from normalized CSPs, and a residue-averaged Kd of 44 ± 6 μM was obtained, indicating a similar affinity as for the native protein (Figures 4A and S28). In conclusion, mutating R68, K72, and K76 on BRD4–BD1 to serine did not perturb the interactions with dsDNA and this supports the conclusion that, rather than electrostatic contacts from a basic patch, DNA binding is being primarily mediated by residues in the KAc-binding site. Along similar lines, various nucleobases and nucleosides such as thymine, thymidine, and 5-methyl uridine have been shown to bind to other bromodomains as acetylated lysine mimics.43 To analyze how dsDNA can serve as a KAc-mimetic, we also tested various nucleobases and nucleotides in PrOF NMR titrations with 5FW BRD4–BD1 but were unable to identify significant interactions (Figures S28–29).
In contrast, in the PrOF NMR titration of 5FW 3KS BRDT–BD1 with 40 bp dsDNA, W50 exhibited a non-saturating Δδ, indicating weaker affinity under the dsDNA concentrations tested (Figure S32) as compared to a Kd of 87 μM obtained after plotting the Δδ of W50 from the PrOF NMR titration of unmutated 5FW BRDT–BD1 (Figure S31). This result is consistent with the findings from Miller et al.12 Together these mutation studies indicate that the N-terminal homologous BDs of BRDT and BRD4 have different modes of engagement with dsDNA.
Interplay with Acetylated Histone Recognition.
Based on our new binding site hypothesis of BRD4–BD1 with dsDNA, we evaluated the interplay between DNA and acetylated histone recognition. From the NMR studies, it was evident that the acetylated histone and dsDNA-binding surfaces largely overlap. Miller et al. found that nonspecific DNA binding enhanced the affinity of BRDT–BD1 for acetylated nucleosomes over acetylated histone via bivalent binding.22 A similar trend was not observed for BRD4–BD1, which was attributed to the absence of interactions with dsDNA. This result could also be explained if the weaker dsDNA interaction is orthosteric to histone binding. Here, we investigated whether BRD4–BD1 bound the two ligands simultaneously or if the higher-affinity acetylated histone recognition affected the dsDNA binding by the BD.
To investigate a competitive binding model, we went back to our PrOF NMR experiments with fluorinated protein, 3FY BRD4 BD1. The addition of excess equivalents of H4 K5Ac,K8Ac,K12Ac,K16Ac broadened Y97, Y98, and Y139 into the baseline and moved Y137 downfield by 0.24 ppm (Figure S33). Upon the titration with 40 bp dsDNA, Y137 showed a minimal CSP of 0.03 ppm at 230 μM of dsDNA, as compared to 0.13 ppm when no histone was present. Similar results were obtained from 5FW BRD4–BD1 (Figure S34). For further analysis, we moved to 1H–15N HSQC NMR spectroscopy. We titrated 40 bp dsDNA into 15N-labeled BRD4–BD1 pre-bound to H4 K5Ac,K8Ac,K12Ac,K16Ac. We then compared 1H–15N HSQC NMR spectra of (1) apo protein, (2) DNA alone, (3) acetylated histone alone, and (4) both DNA and acetylated histone to get insights into competitive binding between both the ligands (Figures 5A and S35). One set of residues, Q85, V147, and N135, showed greater CSPs for DNA binding. Upon the addition of H4 K5Ac,K8Ac,K12Ac,K16Ac to DNA-bound protein, these resonances followed linear trajectories to the histone-bound state, indicating that the residues in the ternary binding experiment were in the same bound state as with the histone alone. Another subset of residues, G143, A80, and F83, was more sensitive to histone binding. Upon addition of DNA to histone-bound protein, these resonances showed CSPs along linear trajectories to the DNA-bound state. Notably, in the ternary system when the acetylated histone and DNA were present together, we did not observe a unique chemical shift for any residue, which was distinct from both the histone-bound and DNA-bound states. These analyses indicated that BRD4–BD1 could only interact with either an acetylated histone or dsDNA at one time and did not exhibit bivalent binding.
Figure 5.
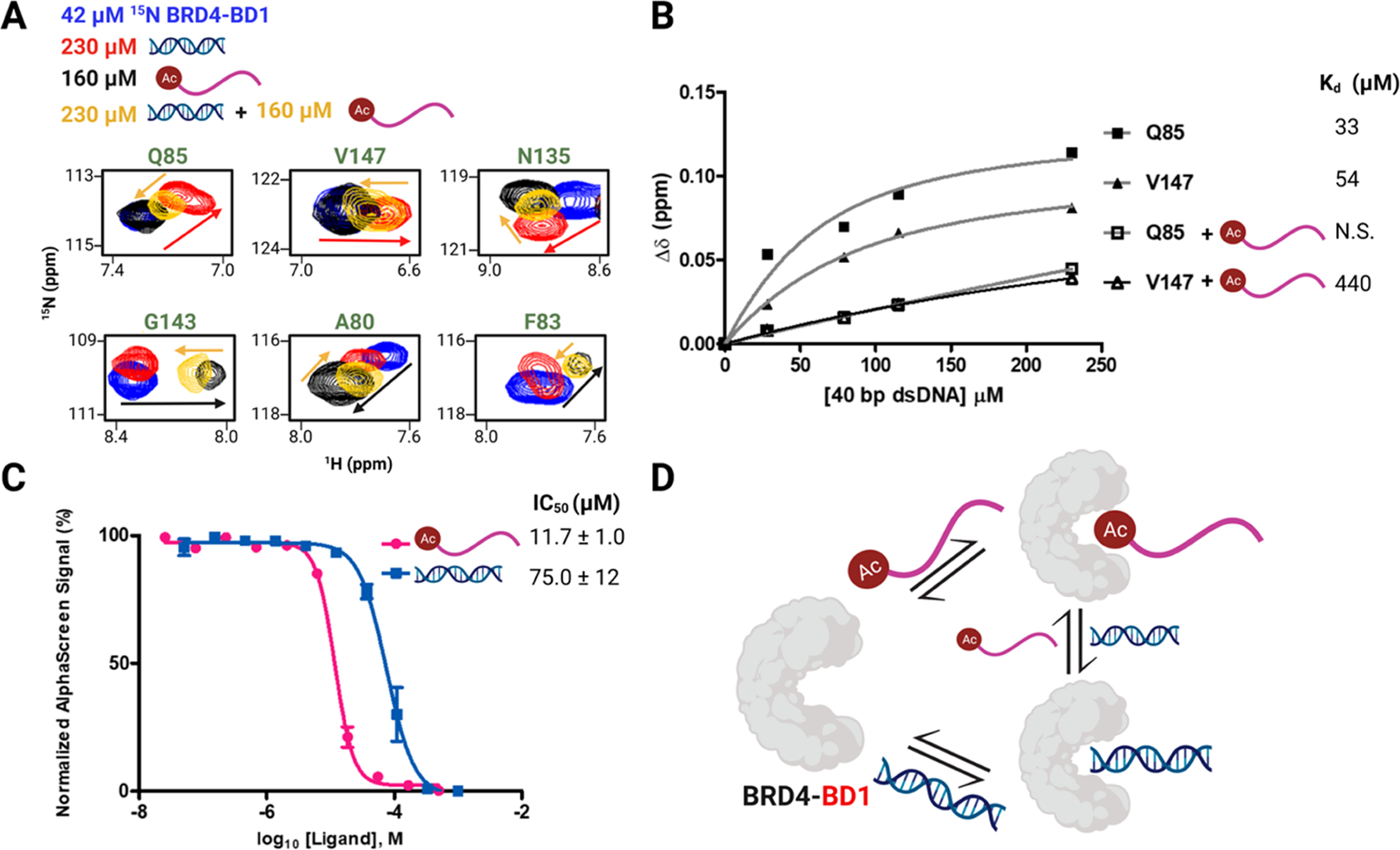
Acetylated histone and dsDNA exhibit competitive binding to BRD4–BD1. (A) Subset of residues showing the overlay of 1H–15N HSQC NMR spectra of 42 μM 15N BRD4–BD1 (blue) and in the presence of 230 μM 40 bp dsDNA (red), 160 μM H4 K5Ac,K8Ac,K12Ac,K16Ac (black), or both 230 μM 40 bp dsDNA and 160 μM H4 K5Ac,K8Ac,K12Ac,K16Ac (yellow). Arrows trace the linear trajectory between apo and histone-bound BRD4–BD1 (black) or apo and DNA-bound BRD4–BD1 (red) and upon titration of dsDNA into histone-bound BRD4–BD1 (yellow) and vice-versa. (B) Binding isotherms of residues Q85 and V147 in the absence and presence of 160 μM H4 K5Ac,K8Ac,K12Ac,K16Ac and corresponding Kd values (N.S. stands for nonsaturating). (C) AlphaScreen competition experiments with 9xHis–BRD4–BD1 using a biotinylated histone peptide reported as mean ± SD of three experimental replicates performed in duplicate. (D) Cartoon depicting the competitive binding between the acetylated histone and dsDNA to interact with BRD4–BD1.
When the dsDNA was titrated into 15N BRD4–BD1 pre-bound to acetylated histone, a subset of residues exhibited reduced CSPs as compared to when the histone was absent (Figures 5B and S36–38). Plotting the CSP of residues Q85 and V147, we observed a nine-fold reduction in affinity indicating weaker associations with DNA in the presence of the acetylated histone. However, since histones are rich in basic residues, such as lysine and arginine, they might also interact with DNA to lower its effective concentration. Thus, we performed related experiments with the pan-BET BD inhibitor, (+)-JQ1. In the presence of an excess of (+)-JQ1, minimal CSPs were observed for the titration of dsDNA to 5FW BRD4–BD1 (Figure S39). Similar results were obtained from 1H–15N HSQC NMR titration (Figure S40). Together, these experiments indicated that the high-affinity KAc binders inhibit the interactions between BRD4–BD1 and dsDNA.
Finally, we used an orthogonal competitive-inhibition assay, AlphaScreen, using a biotinylated histone peptide and BRD4–BD1 (Figures 5C and S41). Due to the assay format, the IC50 values tend to approximate the ligand affinity.37 In this case, we obtained an IC50 of 75 ± 12 μM in good agreement with our NMR-based Kd values. Together these data demonstrated competitive binding between the acetylated histone and dsDNA (Figure 5D).
Investigation of DNA Interactions with Other Bromodomains.
Bromodomains are generally a part of larger multidomain chromatin-binding proteins having different functions. In some cases, interactions with DNA mediated by the BD might facilitate the parent protein complex to diffuse along chromatin for localization on specific sites. Along similar lines, we wanted to investigate the generality of the DNA-recognition ability of BDs both within the Class I BET BDs and outside this family of BDs. These included the BD of bromodomain PHD finger transcription factor (BPTF) (Class I), BET BDs: BRD4–BD1/BD2, BRD2–BD1/BD2, BRD3–BD1/BD2, and BRDT–BD1/BD2 (Class II), and the BD of CREB-binding protein (CREBBP) (Class III). We expressed all of the BDs as 5FW-labeled proteins and performed PrOF NMR titrations with 40 bp dsDNA (Figures S42–S52). From the 19F NMR spectra, we plotted the dose-dependent CSP of the tryptophan closest to the KAc-binding pocket to obtain Kd values (Figure 6). The C-terminal BET BDs (BD2s) were found to have a moderately weaker affinity for dsDNA as compared to BET BD1s. For the non-BET BDs, BPTF BDs, and CREBBP BDs, we could not fit the binding isotherm data to obtain a Kd indicating weaker affinity or interactions happening in regions insensitive to reporting on DNA binding from PrOF NMR.
Figure 6.
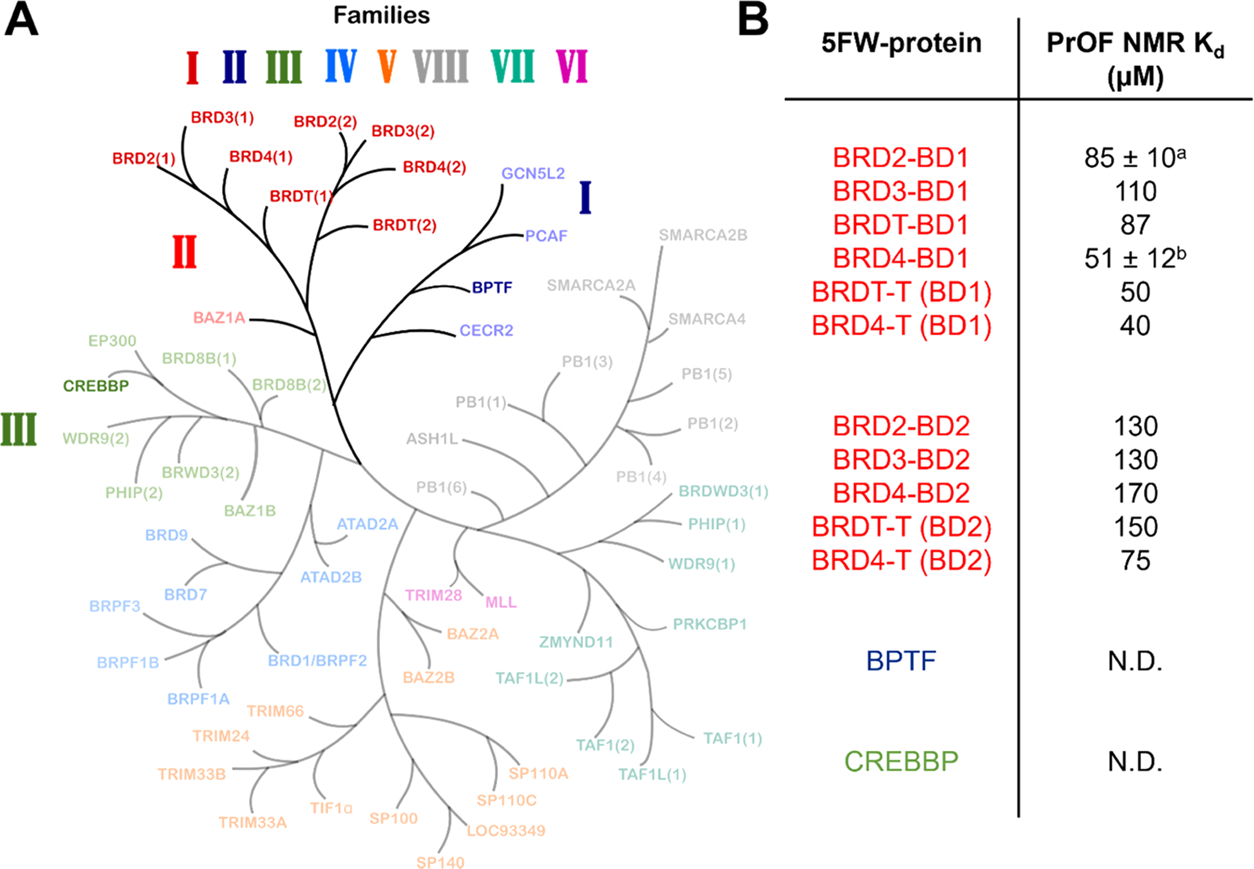
(A) Bromodomain phylogenetic tree adapted from Pomerantz et al..37 BDs tested in the PrOF NMR screen are highlighted. (B) Table showing PrOF NMR Kd values from the titration of 40 bp dsDNA with 5FW-labeled proteins. N.B. = binding affinity could not be determined. a: Kd is reported as the mean ± SD of two experimental replicates. b: Kd is reported as the mean ± SD of three experimental replicates.
PrOF NMR experiments with fluorinated tryptophans as reporter residues only report interactions around the labeled residues and can potentially exclude protein–DNA interactions happening at other sites on the protein. Thus, we employed EMSA as a secondary assay to cross-validate our findings from the PrOF NMR screen. EMSA of 5FW BPTF BDs with 40 bp dsDNA indicated minimal interaction as neither smearing nor gel shift was seen for the free DNA band (Figure S50A). However, in the EMSA of 5FW CREBBP BDs with dsDNA, a slight decrease in the intensity of the free DNA band could be seen at higher protein concentrations, indicating weak interactions (Figure S50B). It can be inferred that for CREBBP since the WPF shelf is not fully defined and the three tryptophans (W1151, W1158, and W1165) are situated on the αB-helix, DNA-binding events happening at other sites on the protein can be missed in the PrOF NMR-binding experiment. Thus, additional structural techniques would be necessary to fully investigate the DNA interactions with the BD of CREBBP.
Han et al.14 found the tandem domains of BRD4 (BRD4-T) to interact with a 41 bp dsDNA with high affinity (Kd = 480 nM from EMSA) but the interactions of individual BDs were not studied though their residues showed significant CSPs in a 1H–15N HSQC NMR titration. As a final experiment, we wanted to investigate if this is also reflected in our PrOF NMR assay. Thus, we tested 40 bp dsDNA with BRD4-T (Figure S51) and the tandem domains of BRDT, BRDT-T (Figures 6B and S52). For BDs in the tandem proteins, we found comparable Kd values to the individual domains. However, from EMSA, we found BRD4-T (Figure S53) to bind to unmodified nucleosomes and dsDNA very strongly. Similar results were observed with BRDT-T (Figure S54). This is an interesting result, as both the individual BDs of BRD4, BD1 (Figure S16), and BD2 (Figure S55) showed only weak interactions in EMSA, indicating that the disordered region connecting the structured BDs might be contributing to higher-affinity binding with dsDNA.
DISCUSSION
BET proteins bind acetylated lysines on histones and nonhistone proteins regulating cellular processes, chromatin dynamics, and gene expression. Recently, similar to other epigenetic readers, BDs have also been found to have nucleic acid-binding activity in addition to the canonical KAc recognition. Morrison et al. reported BDs of hBRM and BRG1 to have dual histone/DNA-binding activity.11 Around the same time, Miller et al. investigated the N-terminal BD of BRDT, BRDT–BD1, as a model case to explore BD–DNA recognition mechanisms and implications on chromatin regulation.12 BRDT–BD1 was found to bind to linear and nucleosomal DNA through a basic site comprising three lysines. The histone-binding site and DNA-binding site are comprised of different surfaces, giving the protein an ~six-fold enhancement in affinity for acetylated nucleosomes over acetylated histones. However, the highly homologous BRD4–BD1 though retaining a similar positively charged surface did not interact with dsDNA in a gel-shift assay with an affinity measurable in their experiments. Though Han et al. found the tandem domains of BRD4 to interact with DNA, the role of individual BDs in binding to chromatin was not defined.14 To further understand the molecular mechanisms associated with gene expression and to identify potential new sites for functional inhibition, it is important to understand how epigenetic readers interact with chromatin via multivalent interactions.
In this study, we structurally elucidate the dsDNA binding to BRD4–BD1. Though EMSA detected weaker interactions, extensive NMR analyses using PrOF NMR and 1H–15N HSQC NMR were used to investigate the interactions of dsDNA and BRD4–BD1. Interestingly, rather than binding positively charged lysines and arginine on the αZ-helix, we found dsDNA to interact via residues around the conserved KAc-binding pocket and the histone-binding and DNA-binding pockets were found to be largely overlapping. Similar observations have also been reported by Musselman et al. when they found dsDNA to perturb mostly hydrophobic residues around the H3K36me3-binding pocket in the PHF1 Tudor domain, rather than making solely electrostatic contacts.11 In a related study, Charier et al. found the tandem Tudor domains of 53BP1 to bind to dsDNA and RG-rich peptides using the same set of hydrophobic residues.44 Thus, a surface electrostatic potential analysis may serve as the starting point for predicting BD–DNA interactions; however, as with our results, DNA binding might also be mediated via additional noncovalent contacts. Hence, extensive structural and functional studies would be needed to understand BD–DNA-binding mechanisms.
Since BDs are conserved KAc readers, it is imperative to understand the interplay between the newly discovered DNA-binding activity and canonical histone recognition. BRD4–BD1 has an affinity of 6.8 μM for H4 K5Ac,K8Ac and 2.8 μM for H4 K5Ac,K8Ac,K12Ac,K16Ac.45 In a few cases, histone binding and DNA binding have been found to function independently.12 In other studies, histone association enhances affinity for DNA and vice-versa.46 Contrary to these reports, we found dsDNA to show antagonistic binding to BRD4–BD1 with H4 K5Ac,K8Ac,K12Ac,K16Ac. We probe this using NMR analyses on ternary binding experiments taking protein, histone, and DNA. In no case did we find any residue in a unique environment and found the histone or small-molecule inhibitor to significantly reduce the affinity of the protein for DNA. We further investigated this using a competitive-inhibition assay, AlphaScreen, and found dsDNA to compete with the acetylated histone giving an IC50 comparable to Kd values obtained from NMR studies.
Finally, we also performed a PrOF NMR screen testing on BDs of three different classes (Class I, II, and III). Though BDs of BPTF and CREBBP showed no interaction from PrOF NMR, an EMSA experiment of CREBBP BDs with dsDNA indicated weaker-affinity interactions. Extensive NMR analyses would be necessary to fully investigate this.
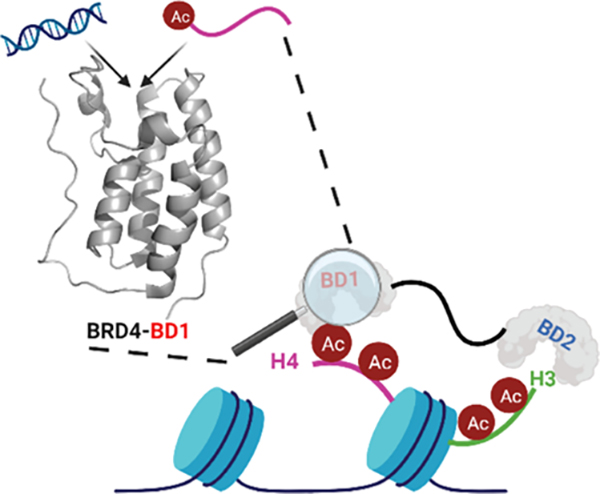
Through our PrOF NMR and 1H–15N HSQC NMR analyses on individual BRD4 BDs and the tandem domains of BRD4, we propose a model for the protein to interact with chromatin (Figure 7). Though the two BDs in BRD4-T bind to dsDNA with comparable affinity as to individual BDs, the protein exhibits submicromolar binding affinity as observed in EMSA. This is also similar to the reported results on BRD4-T14 and was observed for BRDT-T here as well. We believe that the intrinsically disordered region connecting the BDs largely contributes to a higher affinity for dsDNA.47,48 This has also been postulated before as the linker is rich in basic residues and has an A-motif (281–300),13 which is speculated to phosphorylate the Pol II C-terminal domain (CTD) via direct engagement, though nothing is known about its structure.49
Figure 7.
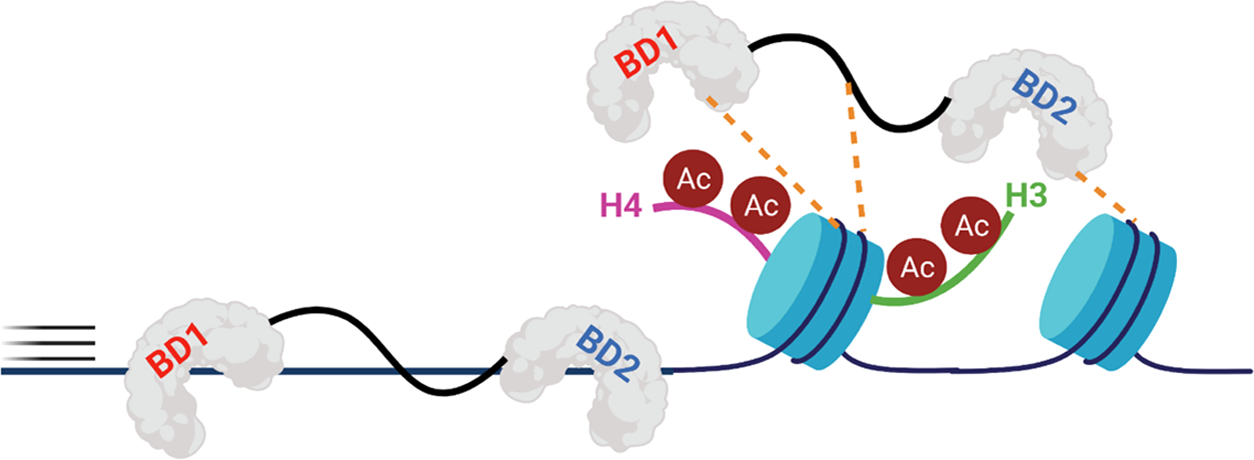
BRD4-T uses its BDs and disordered region to scan chromosomal dsDNA and associate with acetylated nucleosomes. Though KAc recognition is the dominant interaction, weak, nonspecific contacts with dsDNA may help the protein to efficiently sample chromatin.
Our observations and those of others raise questions about the importance of bromodomain nucleic acid interactions. In one context, dsDNA binding by BRD4 has been found to promote its liquid–liquid phase separation and subsequent chromatin condensation.14 These processes are typically governed by multiple weak interactions such as histone and/or the nucleic acid interactions shown here. However, given their similar modes of recognition with BRD4, it remains difficult to tease apart the exact contributions of both the dsDNA recognition and acetylated histone binding to the chromatin association of BRD4.
In a second context, despite the higher-affinity linker interactions observed here, bromodomain binding to chromatin is essential. Prior fluorescence recovery after photobleaching (FRAP) studies found that BRD4-binding site mutants, N140F and N433F, led to a faster recovery of the fluorescence signal from the full-length protein, implying that the BRD4 BDs play a part in anchoring the protein to chromatin.50,51 Most likely, BRD4’s chromatin binding is a multivalent effect of combined nucleosomal dsDNA and acetylated histone binding in the canonical binding pocket, as well as additional interactions with the unstructured linker between the two bromodomains. Though most of the structural studies were done with a 40 bp dsDNA sequence here, the highest affinity of 18 μM from PrOF NMR and 14 ± 2 μM from 1H–15N HSQC NMR was measured for a 66 bp dsDNA sequence, which is approaching the affinity of BRD4–BD1 for acetylated histones (2.8–6.8 μM for H4 K5Ac,K8Ac,K12Ac,K16Ac and H4 K5Ac,K8Ac) supporting an interaction strength of physiological importance. Thus, our studies have been able to further refine the model by elaborating on a new binding mode of interaction of dsDNA with BRD4 BDs. These studies also highlight the value of using PrOF NMR as a rapid structural method to assess BD–DNA-binding interactions when the crystallization conditions have not yet been established.
In summary, we have explored the chromatin-recognition mechanisms of the epigenetic protein, BRD4, by performing detailed biophysical analyses on the dsDNA-binding activity of the N-terminal BD of BRD4. Using a suite of structural techniques such as PrOF NMR and 1H–15N HSQC NMR and mutational analyses, we have established that dsDNA interacts with BRD4–BD1 with moderate affinity (Kd = 18–118 μM) via residues around the KAc-binding site. Moreover, using AlphaScreen, we establish that dsDNA and acetylated histones exhibit competitive binding to BRD4–BD1. Using PrOF NMR and EMSA, we have also surveyed the dsDNA-binding ability of BDs from different classes (Class I, II, and III). Based on our studies, we have refined a model of BRD4–nucleic acid interactions and find that the protein employs its BDs and unstructured region to localize on chromatin. We have found this model to be distinct from a highly homologous BET bromodomain BRDT–BD1 and thus warrants further investigation of additional BET proteins. Knowledge of the native interactions of epigenetic readers is essential to dissect their biological mechanisms and design better therapeutics.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank Dr Jorden Johnson for assistance with the 1H–15N HSQC NMR experiments. Figures were created using GraphPad Prism and BioRender.
Funding
This work was funded by the NIH MIRA award, R35 GM140837–01 (W.C.K.P) and R01 CA250329 (Y.D.), and a University of Minnesota doctoral dissertation fellowship (H.Z.)
ABBREVIATIONS USED
- BPTF
bromodomain- and PHD finger-containing transcription factor
- PHD
plant homeodomain
- PrOF NMR
protein-observed fluorine nuclear magnetic resonance
- 5FW
5-fluorotryptophan
- PTM
post-translational modification
- EMSA
electrophoretic mobility shift assay
- HSQC NMR
heteronuclear single quantum coherence nuclear magnetic resonance
- AlphaScreen
amplified luminescent proximity homogeneous assay
Footnotes
The authors declare no competing financial interest.
■ ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biochem.2c00157.
Methods: Protein purification; Figures S1–S55; Tables S1–S3; HPLC Purity Traces of Peptides (PDF)
Accession Codes
Accession IDs BRD2: P25440 BRD3: Q15059 BRD4: O60885 BRDT: Q58F21 BPTF: Q12830 CBP: Q92793
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.biochem.2c00157
Contributor Information
Prakriti Kalra, Department of Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States.
Huda Zahid, Department of Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States.
Alex Ayoub, Department of Pathology, University of Michigan, Ann Arbor, Michigan 48109, United States.
Yali Dou, Norris Comprehensive Cancer Center, University of Southern California, Los Angeles, California 90089, United States.
William C. K. Pomerantz, Department of Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States; Department of Medicinal Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States.
REFERENCES
- (1).Allis CD; Muir TW Spreading Chromatin into Chemical Biology. ChemBioChem 2011, 12, 264–279. [DOI] [PubMed] [Google Scholar]
- (2).Luger K; Mäder AW; Richmond RK; Sargent DF; Richmond TJ Crystal Structure of the Nucleosome Core Particle at 2.8 Å Resolution. Nature 1997, 389, 251–260. [DOI] [PubMed] [Google Scholar]
- (3).Strahl BD; Allis CD The Language of Covalent Histone Modifications. Nature 2000, 403, 41–45. [DOI] [PubMed] [Google Scholar]
- (4).Allis CD; Jenuwein T The Molecular Hallmarks of Epigenetic Control. Nat. Rev. Genet. 2016, 17, 487–500. [DOI] [PubMed] [Google Scholar]
- (5).Loidl P Histone Acetylation: Facts and Questions. Chromosoma 1994, 103, 441–449. [DOI] [PubMed] [Google Scholar]
- (6).Arrowsmith CH; Bountra C; Fish PV; Lee K; Schapira M Epigenetic Protein Families: A New Frontier for Drug Discovery. Nat. Rev. Drug Discov. 2012, 11, 384–400. [DOI] [PubMed] [Google Scholar]
- (7).Filippakopoulos P; Picaud S; Mangos M; Keates T; Lambert JP; Barsyte-Lovejoy D; Felletar I; Volkmer R; Müller S; Pawson T; Gingras AC; Arrowsmith CH; Knapp S Histone Recognition and Large-Scale Structural Analysis of the Human Bromodomain Family. Cell 2012, 149, 214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Weaver TM; Morrison EA; Musselman CA Reading More than Histones: The Prevalence of Nucleic Acid Binding among Reader Domains. Molecules 2018, 23, No. 2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Vann KR; Klein BJ; Kutateladze TG ScienceDirect Structural Biology Mechanistic Similarities in Recognition of Histone Tails and DNA by Epigenetic Readers. Curr. Opin. Struct. Biol. 71, 1–6. DOI: 10.1016/j.sbi.2021.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Vögeli B; Dykhuizen EC; Musselman CA PBRM1 BD2 and BD4 Associate with RNA to Facilitate Chromatin Association. bioRxiv 2022, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Morrison EA; Sanchez JC; Ronan JL; Farrell DP; Varzavand K; Johnson JK; Gu BX; Crabtree GR; Musselman CA DNA Binding Drives the Association of BRG1/HBRM Bromodomains with Nucleosomes. Nat. Commun. 2017, 8, No. 16080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Miller TCR; Simon B; Rybin V; Grötsch H; Curtet S; Khochbin S; Carlomagno T; Müller CW A Bromodomain-DNA Interaction Facilitates Acetylation-Dependent Bivalent Nucleosome Recognition by the BET Protein BRDT. Nat. Commun. 2016, 7, No. 13855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Larue RC; Plumb MR; Crowe BL; Shkriabai N; Sharma A; DiFiore J; Malani N; Aiyer SS; Roth MJ; Bushman FD; Foster MP; Kvaratskhelia M Bimodal High-Affinity Association of Brd4 with Murine Leukemia Virus Integrase and Mononucleosomes. Nucleic Acids Res. 2014, 42, 4868–4881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Han X; Yu D; Gu R; Jia Y; Wang Q; Jaganathan A; Yang X; Yu M; Babault N; Zhao C; Yi H; Zhang Q; Zhou MM; Zeng L Roles of the BRD4 Short Isoform in Phase Separation and Active Gene Transcription. Nat. Struct. Mol. Biol. 2020, 27, 333–341. [DOI] [PubMed] [Google Scholar]
- (15).Musselman CA; Gibson MD; Hartwick EW; North JA; Gatchalian J; Poirier MG; Kutateladze TG Binding of PHF1 Tudor to H3K36me3 Enhances Nucleosome Accessibility. Nat. Commun. 2013, 4, No. 2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Musselman CA; Avvakumov N; Watanabe R; Abraham CG; Lalonde ME; Hong Z; Allen C; Roy S; Nuñez JK; Nickoloff J; Kulesza CA; Yasui A; Côté J; Kutateladze TG Molecular Basis for H3K36me3 Recognition by the Tudor Domain of PHF1. Nat. Struct. Mol. Biol. 2012, 19, 1266–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Ishida M; Shimojo H; Hayashi A; Kawaguchi R; Ohtani Y; Uegaki K; Nishimura Y; Nakayama JI Intrinsic Nucleic Acid-Binding Activity of Chp1 Chromodomain Is Required for Heterochromatic Gene Silencing. Mol. Cell 2012, 47, 228–241. [DOI] [PubMed] [Google Scholar]
- (18).Kim D; Blus BJ; Chandra V; Huang P; Rastinejad F; Khorasanizadeh S Corecognition of DNA and a Methylated Histone Tail by the MSL3 Chromodomain. Nat. Struct. Mol. Biol. 2010, 17, 1027–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Yang J; Everett AD Hepatoma Derived Growth Factor Binds DNA through the N-Terminal PWWP Domain. BMC Mol. Biol. 2007, 8, No. 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Van Nuland R; Van Schaik FMA; Simonis M; Van Heesch S; Cuppen E; Boelens R; Timmers HTM; Van Ingen H Nucleosomal DNA Binding Drives the Recognition of H3K36-Methylated Nucleosomes by the PSIP1-PWWP Domain. Epigenet. Chromatin 2013, 6, No. 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Tencer AH; Cox KL; Wright GM; Zhang Y; Petell CJ; Klein BJ; Strahl BD; Black JC; Poirier MG; Kutateladze TG Molecular Mechanism of the MORC4 ATPase Activation. Nat. Commun. 2020, 11, No. 5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Liu L; Qin S; Zhang J; Ji P; Shi Y; Wu J Solution Structure of an Atypical PHD Finger in BRPF2 and Its Interaction with DNA. J. Struct. Biol. 2012, 180, 165–173. [DOI] [PubMed] [Google Scholar]
- (23).Klein BJ; Vann KR; Andrews FH; Wang WW; Zhang J; Zhang Y; Beloglazkina AA; Mi W; Li Y; Li H; Shi X; Kutateladze AG; Strahl BD; Liu WR; Kutateladze TG Structural Insights into the π-π-π Stacking Mechanism and DNA-Binding Activity of the YEATS Domain. Nat. Commun. 2018, 9, No. 4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Pilotto S; Speranzini V; Tortorici M; Durand D; Fish A; Valente S; Forneris F; Mai A; Sixma TK; Vachette P; Mattevi A Interplay among Nucleosomal DNA, Histone Tails, and Corepressor CoREST Underlies LSD1-Mediated H3 Demethylation. Proc. Natl. Acad. Sci. U.S.A. 2015, 112, 2752–2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Van Royen ME; Zotter A; Ibrahim SM; Geverts B; Houtsmuller AB Nuclear Proteins: Finding and Binding Target Sites in Chromatin. Chromosome Res. 2011, 19, 83–98. [DOI] [PubMed] [Google Scholar]
- (26).Wang C-Y; Filippakopoulos P Beating the Odds: BETs in Disease. Trends Biochem. Sci. 2015, 40, 468–479. [DOI] [PubMed] [Google Scholar]
- (27).Cheung KL; Kim C; Zhou MM The Functions of BET Proteins in Gene Transcription of Biology and Diseases. Front. Mol. Biosci. 2021, 8, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Dey A; Ellenberg J; Farina A; Coleman AE; Maruyama T; Sciortino S; Lippincott-Schwartz J; Ozato K A Bromodomain Protein, MCAP, Associates with Mitotic Chromosomes and Affects G2-to-M Transition. Mol. Cell. Biol. 2000, 20, 6537–6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Rahnamoun H; Lee J; Sun Z; Lu H; Ramsey KM; Komives EA; Lauberth SM Chromatin Engagement and Transcription Activation. Nat. Struct. Mol. Biol. 2018, 25, 687–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Wu SY; Lee AY; Lai HT; Zhang H; Chiang CM Phospho Switch Triggers Brd4 Chromatin Binding and Activator Recruitment for Gene-Specific Targeting. Mol. Cell 2013, 49, 843–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Gee Clifford T. A. Protein-Observed 19F-NMR for Fragment Screening, Affinity Quantification and Druggability Assessment. Nat. Protoc. 2016, 11, 1414–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Lee YT; Gibbons G; Lee SY; Nikolovska-Coleska Z; Dou Y One-Pot Refolding of Core Histones from Bacterial Inclusion Bodies Allows Rapid Reconstitution of Histone Octamer. Protein Expression Purif. 2015, 110, 89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Lowary PT; Widom J New DNA Sequence Rules for High Affinity Binding to Histone Octamer and Sequence-Directed Nucleosome Positioning. J. Mol. Biol. 1998, 276, 19–42. [DOI] [PubMed] [Google Scholar]
- (34).Dyer PN; Edayathumangalam RS; White CL; Bao Y; Chakravarthy S; Muthurajan UM; Luger K Reconstitution of Nucleosome Core Particles from Recombinant Histones and DNA. Methods Enzymol. 2003, 375, 23–44. [DOI] [PubMed] [Google Scholar]
- (35).Ycas PD; Zahid H; Chan A; Olson NM; Johnson JA; Talluri SK; Schonbrunn E; Pomerantz WCK New Inhibitors for the BPTF Bromodomain Enabled by Structural Biology and Biophysical Assay Development. Org. Biomol. Chem. 2020, 18, 5174–5182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Erijman A; Dantes A; Bernheim R; Shifman JM; Peleg Y Transfer-PCR (TPCR): A Highway for DNA Cloning and Protein Engineering. J. Struct. Biol. 2011, 175, 171–177. [DOI] [PubMed] [Google Scholar]
- (37).Pomerantz WCK; Johnson JA; Ycas PD Applied Biophysics for Bromodomain Drug Discovery. In Topics in Medicinal Chemistry, 2019; Vol. 33, pp 287–337 DOI: 10.1007/7355_2019_79. [DOI] [Google Scholar]
- (38).Mishra NK; Urick AK; Ember SWJ; Scho E; Pomerantz WC Fluorinated Aromatic Amino Acids Are Sensitive 19F NMR Probes for Bromodomain-Ligand Interactions. ACS Chem. Biol. 2014, 9, 2755–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Yu JL; Chen TT; Zhou C; Lian FL; Tang XL; Wen Y; Shen JK; Xu YC; Xiong B; Zhang NX NMR-Based Platform for Fragment-Based Lead Discovery Used in Screening BRD4-Targeted Compounds. Acta Pharmacol. Sin. 2016, 37, 984–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Paul NK; Baksh KA; Arias JF; Zamble DB The Impact of a His-Tag on DNA Binding by RNA Polymerase Alpha-C-Terminal Domain from Helicobacter Pylori. Protein Expression Purif. 2020, 167, No. 105541. [DOI] [PubMed] [Google Scholar]
- (41).Jung M; Philpott M; Müller S; Schulze J; Badock V; Eberspächer U; Moosmayer D; Bader B; Schmees N; Fernández-montalván A; Haendler B Affinity Map of Bromodomain Protein 4 (BRD4) Interactions with the Histone H4 Tail and the Small Molecule Inhibitor JQ1* 2014, 289, 9304–9319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Kalra P; McGraw L; Kimbrough JR; Pandey AK; Solberg J; Cui H; Divakaran A; John K; Hawkinson JE; Pomerantz WCK Quantifying the Selectivity of Protein–protein and Small Molecule Interactions with Fluorinated Tandem Bromodomain Reader Proteins. ACS Chem. Biol. 2020, 15, 3038–3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Chaikuad A; Petros AM; Fedorov O; Xu J; Knapp S Structure-Based Approaches towards Identification of Fragments for the Low-Druggability ATAD2 Bromodomain. Medchemcomm 2014, 5, 1843–1848. [Google Scholar]
- (44).Charier G; Couprie J; Alpha-Bazin B; Meyer V; Quéméneur E; Guérois R; Callebaut I; Gilquin B; Zinn-Justin S The Tudor Tandem of 53BP1: A New Structural Motif Involved in DNA and RG-Rich Peptide Binding. Structure 2004, 12, 1551–1562. [DOI] [PubMed] [Google Scholar]
- (45).Filippakopoulos P; Knapp S The Bromodomain Interaction Module. FEBS Lett. 2012, 586, 2692–2704. [DOI] [PubMed] [Google Scholar]
- (46).Connelly KE; Weaver TM; Alpsoy A; Gu BX; Musselman CA; Dykhuizen EC Engagement of DNA and H3K27me3 by the CBX8 Chromodomain Drives Chromatin Association. Nucleic Acids Res. 2019, 47, 2289–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Katuwawala A; Kurgan L Comparative Assessment of Intrinsic Disorder Predictions with a Focus on Protein and Nucleic Acid-Binding Proteins. Biomolecules 2020, 10, 1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Dyson HJ Roles of Intrinsic Disorder in Protein-Nucleic Acid Interactions. Mol. Biosyst. 2012, 8, 97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Weissman JD; Singh AK; Devaiah BN; Schuck P; LaRue RC; Singer DS The Intrinsic Kinase Activity of BRD4 Spans Its BD2-B-BID Domains. J. Biol. Chem. 2021, 297, No. 101326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Philpott M; Rogers CM; Yapp C; Wells C; Lambert JP; Strain-Damerell C; Burgess-Brown NA; Gingras AC; Knapp S; Müller S Assessing Cellular Efficacy of Bromodomain Inhibitors Using Fluorescence Recovery after Photobleaching. Epigenet. Chromatin 2014, 7, No. 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Runcie AC; Zengerle M; Chan KH; Testa A; Van Beurden L; Baud MGJ; Epemolu O; Ellis LCJ; Read KD; Coulthard V; Brien A; Ciulli A Optimization of a “”Bump-and-Hole” Approach to Allele-Selective BET Bromodomain Inhibition. Chem. Sci. 2018, 9, 2452–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.